

Summary
MASTL is a mitotic accelerator with an emerging role in breast cancer progression. However, the mechanisms behind its oncogenicity remain largely unknown. Here, we identify a previously unknown role and eminent expression of MASTL in stem cells. MASTL staining from a large breast cancer patient cohort indicated a significant association with β3 integrin, an established mediator of breast cancer stemness. MASTL silencing reduced OCT4 levels in human pluripotent stem cells and OCT1 in breast cancer cells. Analysis of the cell-surface proteome indicated a strong link between MASTL and the regulation of TGF-β receptor II (TGFBR2), a key modulator of TGF-β signaling. Overexpression of wild-type and kinase-dead MASTL in normal mammary epithelial cells elevated TGFBR2 levels. Conversely, MASTL depletion in breast cancer cells attenuated TGFBR2 levels and downstream signaling through SMAD3 and AKT pathways. Taken together, these results indicate that MASTL supports stemness regulators in pluripotent and cancerous stem cells.
Subject areas: Cell biology, Stem cells research, Cancer, Proteomics
Graphical abstract
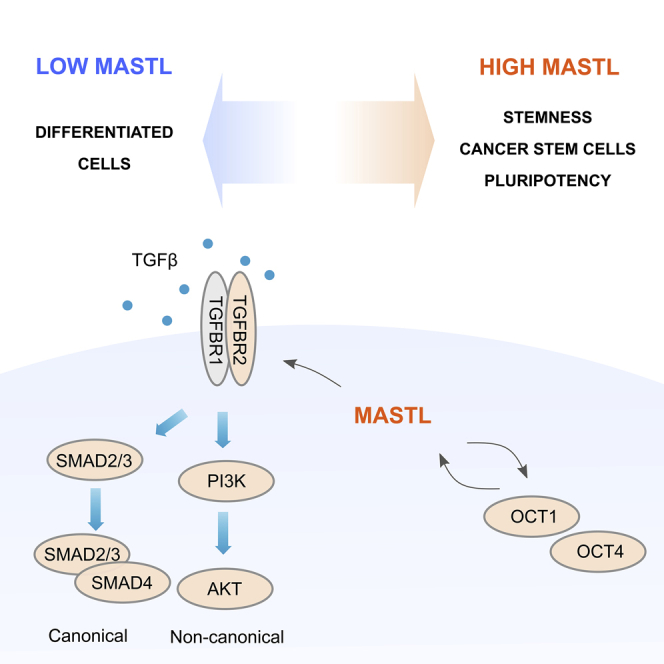
Highlights
-
•
MASTL displays positive correlation with β3 integrin expression in breast tumors
-
•
MASTL is enriched in human pluripotent stem cells and breast cancer stem cells
-
•
Depletion of MASTL leads to decline of stemness indicators OCT1, OCT4, and NANOG
-
•
MASTL supports TGF-β receptor II expression and activation of SMAD3 and AKT
Cell biology; Stem cells research; Cancer; Proteomics
Introduction
Microtubule-associated serine/threonine-protein kinase-like (MASTL; a.k.a. Greatwall (Gwl) kinase) accelerates cell cycle progression through inhibition of the PP2A-B55 phosphatase activity (Vigneron et al., 2016). More recently, MASTL was shown to regulate cell invasion and cell contractility via kinase-independent positive regulation of myocardin-related transcription factor A/serum response factor (MRTF-A/SRF) transcriptional activity (Taskinen et al., 2020). Furthermore, MASTL has emerged as a putative oncogene mediating therapy resistance in several cancer types (Conway et al., 2020; Marzec and Burgess, 2018). Especially in breast cancer, high MASTL expression levels correlate with increased tumor growth and metastasis in vivo (Álvarez-Fernández et al., 2018; Rogers et al., 2018; Vera et al., 2015). However, the mechanisms behind the oncogenicity of MASTL seem to be more complex than mere acceleration of cell growth.
Transforming growth factor (TGF) family proteins can regulate cell proliferation and differentiation, thus playing key roles in development and cancer progression. TGF-β is an evolutionarily conserved secreted protein with widespread expression in both embryonic and adult tissues (Hiepen et al., 2020). The key cell-surface mediator of TGF-β signaling is the type II TGF-β receptor (TGFBR2), which forms a heteromeric receptor with the type I TGF-β receptor (TGFBR1). An activated receptor complex leads to canonical TGF-β signaling via SMAD transcriptional effectors (Massagué, 2012). TGF-β receptor complex can transmit signals also through non-canonical SMAD-independent pathways, such as the mitogen-activated protein kinases (MAPKs), phosphatidylinositide 3-kinase (PI3K), protein kinase B (AKT), TNF receptor-associated factor 4/6 (TRAF4/6), and the Rho family of small GTPases (Zhang, 2009).
As in other cancer types, in mammary carcinogenesis, TGF-β acts initially as a tumor suppressor. However, in later stages of cancer, TGF-β promotes tumor progression, in part by enhancing epithelial to mesenchymal transition (EMT), tumor cell motility, invasiveness, and the capacity to form metastases (Barcellos-Hoff and Akhurst, 2009). TGF-β can enhance tumor progression via its ability to dedifferentiate multiple cell types (Padua and Massagué, 2009). In breast cancer, TGF-β has been reported to enrich the cancer stem cell (CSC) population and promote self-renewal (Asiedu et al., 2011; Bhola et al., 2013; Bruna et al., 2012; Lo et al., 2012; Nie et al., 2016; Scheel et al., 2011) and high expression of cytoplasmic TGF-β in triple-negative breast cancer (TNBC) is associated with high histologic grade, lymph node metastases, and short disease-free survival (Zhang et al., 2017). However, the underlying mechanisms regulating TGF-β signaling and its impact on tumor biology warrants further investigation.
Here, we identify a previously unknown role for MASTL in supporting the levels of major stemness regulators, OCT4 and OCT1, in pluripotent stems cells and breast cancer stem cells, respectively. In addition, we uncover a MASTL-mediated positive regulation of TGFBR2 expression and downstream activation of canonical SMAD2/3 and non-canonical AKT signaling.
Results
MASTL expression correlates with β3-integrin (ITGB3) positive breast cancers
We used immunohistochemistry (IHC) to study MASTL expression in a large patient cohort consisting of 851 patients with breast cancer who took part in the randomized FinHer trial (Joensuu et al., 2009) (Figures 1A–1C and S1). Previous studies have found MASTL protein expression to be associated with basal, Ki-67 positive, high histological grade, and ER-negative (−) breast cancers (Álvarez-Fernández et al., 2018; Rogers et al., 2018), and the IHC analysis results corroborated these findings in the FinHer cohort (Figures 1B and S1A). Furthermore, MASTL expression correlated significantly with steroid hormone receptor negativity and had a significantly stronger association with ER(−), HER2(−) receptor subtype (Figure 1B). Therefore, high MASTL expression is associated with ER(−), HER2(−) breast cancers, a surrogate for triple-negative breast cancers (TNBCs). In addition, a subset of HER2-positive (+) tumors had high MASTL expression (Figure 1B), suggesting that some MASTL-positive patients could benefit from HER2-based treatments.
Figure 1.

MASTL expression correlates with β3-integrin (ITGB3)-positive breast cancers
(A) Immunohistochemical (IHC) staining of MASTL in breast cancer. ROI = region of interest.
(B) Estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2) expression and amplification among MASTL-negative and -positive patients in the FinHer breast cancer trial series. The numbers refer to number of patients and the values in brackets are % of all patients.
(C) Immunohistochemical (IHC) staining of MASTL in histologically normal human breast tissue: ROI = region of interest.
(D) MASTL expression among ITGB3-negative and -positive patients in the FinHer breast cancer series.
(E) Survival of FinHer patients with negative (ITGB3-), intermediate (ITGB3+), or high (ITGB3++) ITGB3 expression. Kaplan-Meier survival-table method and log rank test. See also Figure S1.
In normal breast tissue, we found MASTL to be expressed predominantly in the myoepithelial cells (Figure 1C). Of note, in tissue samples, MASTL was found to have an apparent cytoplasmic expression in addition to the expected nuclear location. In breast cancer, MASTL expression was frequent in the ductal subtype and was associated with poor histological grade of differentiation (Figures 1A and S1A). Interestingly, myoepithelial mammary stem cells isolated from the basal layer of the mammary epithelium have repopulation capacity when transplanted as single cells (Prater et al., 2014). Furthermore, poorly differentiated histological breast cancers are associated with an embryonic stem cell signature, ER negativity, the basal subtype, and poor clinical outcome (Ben-Porath et al., 2008). These results are compatible with a hypothesis that MASTL-positive cells may have stem cell characteristics.
To investigate this possibility, we studied the link between MASTL and β3 integrin (ITGB3), also known as CD61. ITGB3 is a cell-surface adhesion receptor with established roles in the microenvironment, epithelial to mesenchymal transition (EMT), and tumor stemness (reviewed by (Zhu et al., 2019)). There was a clear correlation between ITGB3 and MASTL expression across the whole FinHer cohort (Figure 1D). In addition, similar to MASTL, ITGB3 expression was associated with histological grade 3, the ductal histological type, negative steroid hormone receptor expression, p53 expression, HER2+, high Ki-67 expression (Figure S1B), and poor overall survival (p = 0.004) (Figure 1E).
MASTL expression was also associated with positive p53 staining, which correlates with poor survival in TNBCs (Arjonen et al., 2014; Hashmi et al., 2018; Muller et al., 2011). Despite the strong correlation with hormone receptor negativity and high histological grade, high MASTL expression was associated with the absence of axillary lymph node metastases (Figure S1A) and had no significant association (p = 0.448) with the overall survival among the FinHer patients, who were treated with effective multiagent chemotherapy, endocrine therapy, and trastuzumab (50% of HER2+ patients) in a closely monitored clinical trial (Figure S1C). Importantly, however, high MASTL expression was associated with poor prognosis in other breast cancer patient cohorts (Álvarez-Fernández et al., 2018; Rogers et al., 2018; Wang et al., 2014; Yoon et al., 2018).
High MASTL levels correlate with OCT1 and breast cancer stemness
Depletion of MASTL in MCF7 and T47D breast cancer cells decreases anchorage-independent cell growth and sensitizes breast cancer stem cells to radiation (Yoon et al., 2018). In addition, MASTL overexpression in non-transformed MCF10A cells promotes anchorage-independent cell growth (Rogers et al., 2018; Vera et al., 2015). ITGB3 is an established marker of breast cancer stem cells (BCSCs) (Lo et al., 2012; Vaillant et al., 2008) and a functional contributor to a breast tumor stemness program (Seguin et al., 2014). Thus, the significant correlation between MASTL and ITGB3 in breast cancer prompted us to hypothesize that MASTL could support breast cancer stemness.
To investigate this, we first studied MASTL expression in breast cancer cell lines using the Cancer Cell Line Encyclopedia (CCLE) database. MASTL had a significantly higher expression in ITGB3-enriched cell lines compared to low (Figure S2A). To study this further, we cultured MDA-MB-231 cells as monolayers or mammospheres, the latter of which can be used to enrich BCSCs (Dontu et al., 2003; Ponti et al., 2005). As expected, mammosphere culture led to upregulation of ITGB3 levels (Figure 2A). MASTL protein levels, as well as the transcriptional stemness regulator OCT1 (Maddox et al., 2012), were also enriched in spheres compared to monolayer cultures, in two different TNBC cell lines (MDA-MB-231 and MDA-MB-436; Figures 2A, 2B, S2B, and S2C). Furthermore, silencing of OCT1 decreased MASTL levels significantly (Figures 2C and 2D), whereas overexpression of OCT1 increased MASTL expression (Figures 2E and 2F). These results imply that OCT1 could regulate MASTL expression. Indeed, analysis of the promoter sequence predicts three possible binding sites for OCT1 in the MASTL promoter, two of them being dual (Figure S2D). Taken together, these results indicate that MASTL is upregulated in TNBC mammospheres, and its expression both correlates with and depends on specific breast CSC markers.
Figure 2.

High MASTL levels correlate with OCT1 and mammosphere formation
(A) Western blotting of β3-integrin (ITGB3), OCT1, MASTL, and GAPDH in MDA-MB-231 cells grown as a monolayer (2D) or as mammospheres.
(B) Relative protein expression of MASTL to GAPDH, experimental setup shown in (A). (n = 3 biologically independent experiments, unpaired t-test, mean ± SD).
(C) Western blotting of OCT1, MASTL, and GAPDH in MDA-MB-231 cells silenced with siControl or siOCT1 for 48 h.
(D) Relative protein expression of MASTL to GAPDH, experimental setup shown in (C), two datapoints collected after 96 h and three after 48 h (n = 5 biologically independent experiments, one sample t-test, mean ± SD).
(E) Western blotting of OCT1, MASTL, and tubulin in MDA-MB-231 cells overexpressing EGFP-control (enhanced green fluorescent protein) or hemagglutinin (HA)-tagged OCT1.
(F) Relative protein expression of MASTL to tubulin, experimental setup shown in (E). (n = 3 biologically independent experiments, one sample t-test, mean ± SD).
(G) Representative flow cytometry histograms of MDA-MB-231 cells treated with DMSO control or DCF-DA (2ʹ,7ʹ-Dichlorofluorescin Diacetate) to measure ROS activity after silencing with siControl (gray), siMASTL#6 (red), or siMASTL#7 (orange) for 48 h.
(H) Geometric Mean of the DCF-DA signal (ROS activity) in the experimental setup described in (G). (n = 3 biologically independent experiments, unpaired t-test, mean ± SD).
(I) Representative images of tetracycline-induced shControl and shMASTL#3 MDA-MB-231 cells grown in mammosphere culture conditions (7 days) and stained with Calcein (Nikon Eclipse Ti-E widefield microscope, Hamamatsu Orca C13440 Flash 4.0 ERG [b/w] sCMOS camera and Plan Apo lambda 20×/0.80, WD 1,000-μm objective).
(J) Average mammosphere size (average ferret diameter of spheres in μm) and spheres/1,000 cells plated, experimental setup shown in (I). (n = 3 biologically independent experiments, eight replicate wells/experiment, unpaired t-test, mean ± SEM).
(K) Western blotting of OCT1, MASTL, and GAPDH in tetracycline-induced (4 days) shControl and shMASTL#3 MDA-MB-231 cells.
(L) Relative protein expression of MASTL to GAPDH, experimental setup shown in (K). (n = 3 biologically independent experiments, one sample t-test, mean ± SD).
(M) Representative flow cytometry histograms of CD44 expression in tetracycline-induced (4 days) shControl and shMASTL#3 MDA-MB-231 cells.
(N) Geometric mean of CD44 expression in the experimental setup described in (M). (n = 5 biologically independent experiments, unpaired t-test, mean ± SD). See also Figure S2.
Low levels of reactive oxygen species (ROS) is another hallmark shared by normal stem cells and CSCs (Diehn et al., 2009). Silencing of MASTL with two independent siRNAs increased ROS activity significantly in MDA-MB-231 cells (Figures 2G and 2H). In addition, MASTL silencing (with inducible shRNA or transient siRNA transfection) significantly decreased the size of the mammospheres formed by MDA-MB-231 and MDA-MB-436 cells (Figures 2I, 2J, S2E–S2H). However, the effect of MASTL silencing on the number of spheres reached significance only in MDA-MB-436 cells, when >60 μm sphere size was used as a cut off. These data are concordant with previous studies performed with MCF7, T47D, and MCF10A breast cell lines (Rogers et al., 2018; Vera et al., 2015; Yoon et al., 2018) demonstrating that MASTL supports anchorage-independent cell growth.
To further study the role of MASTL in supporting stemness, we silenced MASTL with inducible shRNAs for four days and studied the expression of stemness markers OCT1, CD44, and CD24. Silencing of MASTL significantly decreased OCT1 protein expression (western blotting) and CD44 cell-surface levels (flow cytometry) in MDA-MB-231 cells (Figures 2K–2N). CD24 expression was extremely low or could not be detected in MDA-MB-231 cells, in line with previous studies (Feng et al., 2019; Li et al., 2017); Figure S2I). These results imply a functional significance for high MASTL levels in supporting stemness.
MASTL is highly expressed in pluripotent stem cells and supports stemness
To further explore the potential link between MASTL and stemness, we investigated MASTL expression in human pluripotent stem cells (hPSCs). Analysis of gene expression data from pluripotent stem cells indicated significantly higher MASTL expression in pluripotent cells compared to fibroblasts or other somatic controls (Figure 3A). In line with these data, MASTL protein levels were high in hPSCs compared to differentiated fibroblasts (Figure 3B). Furthermore, MASTL expression declined clearly upon cell differentiation to any of the three distinct ecto-, endo-, or mesoderm lineages (Figure 3C). Embryonic body differentiation of hPSCs also correlated with clearly reduced MASTL, NANOG, and SOX2 levels, providing additional support for the notion of high MASTL expression correlating with stemness (Figure 3D).
Figure 3.

MASTL is highly expressed in pluripotent stem cells and supports stemness
(A) Expression of MASTL in the Amazonia database (http://amazonia.transcriptome.eu) (Gene expression Atlas for Embryonic SC, Probe 228468_at, U133P2) (mean + SEM, unpaired t-test).
(B) Western blotting of MASTL, OCT4, vimentin, and GAPDH in hiPSCs and differentiated fibroblasts.
(C) Expression of MASTL in differentiation to embryonic layers day 5 (Gifford et al., 2013), (mean + SEM, unpaired t-test).
(D) Western blotting of MASTL, NANOG, SOX2, and GAPDH in embryonic body (EB) differentiation in addition to phase-contrast images (scale bar 100 μm).
(E–H) Immunostaining of MASTL, NANOG, SSEA-5, and DAPI during reprogramming and quantification of the nuclear MASTL signal intensity in NANOG/SSEA-5 positive and negative cells (n = 3 biologically independent experiments, unpaired t-test, mean + SD). Scale bar, 10 μm.
(I) Western blotting of MASTL, β1-integrin, NANOG, SOX2, and GAPDH in original fibroblasts used for reprogramming and in hiPSCs.
(J) Western blotting of MASTL, OCT4, NANOG, and GAPDH in siControl-, siMASTL#6-, and siMASTL#7-treated hiPSCs for 48 h.
(K and L) Relative protein expression of OCT4 (K) or NANOG (L) to GAPDH, experimental setup shown in (J). (n = 3 biologically independent experiments, unpaired t-test, mean ± SD). See also Figure S3.
In interphase cells, MASTL is known to localize in nuclei prior to the nuclear envelope breakdown (Álvarez-Fernández et al., 2013). As expected, nuclear MASTL was evident in hPSCs compared to fibroblasts based on immunofluorescence (Figure S3A). To further study if MASTL expression is linked to stemness, MASTL levels were studied during reprogramming of fibroblasts to hPSCs with overexpression of KLF4, SOX2, MYC, and OCT4. In reprogramming, nuclear MASTL expression was significantly higher in cells positive for NANOG (pluripotency transcription factor) or SSEA-5 (a cell-surface marker for undifferentiated pluripotent cells) (Figures 3E–3H). Concordantly, MASTL protein levels were higher in reprogrammed cells than in the parental fibroblasts (Figure 3I). Finally, to study if high MASTL levels correlate with stemness marker expression, we investigated the outcome of MASTL depletion in hPSCs. Silencing of the MASTL for 48 h resulted in a significant reduction of OCT4 and NANOG, the key pluripotency driving transcription factors (Figures 3J–3L). Taken together, these data indicate that high MASTL expression correlates with stemness markers in human pluripotent stem cells.
MASTL regulates cell contractility and MRTF-A/SRF signaling in a kinase activity-independent manner (Taskinen et al., 2020). To test the requirement of MASTL kinase activity in pluripotency maintenance, we treated cells with the MASTL kinase inhibitor GKI-1 (Ocasio et al., 2016). Optimization of GKI-1 dosage revealed that hPSCs are very sensitive to the GKI-1 inhibitor compared to differentiated vimentin-positive fibroblast-like cells generated from the original hiPSC line (Figures S3B and S3C). This indicates that higher concentrations of GKI-1 have profound effects on the cell cycle of hPSCs and possibly also their viability. To overcome this and to test if GKI-1 influences pluripotency marker expression, we chose to use the tolerated 3.5 μM dose of GKI-1 as it is close to the demonstrated EC50 of 4.9 μM for human MASTL (Ocasio et al., 2016). 48-h treatment with GKI-1 had no effect on OCT4 or NANOG expression (Figure S3D) suggesting that MASTL-mediated regulation of pluripotency markers might be independent of its kinase activity. However, it is important to acknowledge that the inhibitor concentration used might not be sufficient to suppress MASTL kinase activity completely.
MASTL depletion influences stem-cell-associated surface proteins
Cell-surface proteins are critical for the characterization, isolation, and regulation of stem cells (Kim and Ryu, 2017). We performed stable isotope labeling with amino acids in cell culture (SILAC) surfaceome mass-spectrometry-based proteomics (Diaz-Vera et al., 2017) to determine if MASTL affects stemness by regulating the cell-surface proteome. Heavy and light isotope-labeled endogenous cell-surface proteins of control- or -MASTL-silenced MDA-MB-231 cells were purified by biotin enrichment and subjected to mass spectrometry analysis (Figure 4A). In total, 2,604 proteins were identified. MASTL silencing for 48 h significantly affected the expression of 16 of them (Table S1 and Figure 4B). Functional enrichment analysis with g-Profiler (Raudvere et al., 2019) (https://biit.cs.ut.ee/gprofiler/index.cgi) mapped most of the identified proteins to the cell membrane as expected (Figure 4B). Interestingly, more than half of the differentially expressed proteins were linked to tissue development. Of these, ANO6, FZD6, ICAM1, ATP2A2, SLC20A1, and TGFBR2 were downregulated and INSR, F11R, and EPHA4 were upregulated. Two of the downregulated proteins AN06 and ATP2A2 are linked to calcium signaling (Lytton and MacLennan, 1988; Schroeder et al., 2008), suggesting that this might be an interesting avenue to investigate in the future. Our attention was, however, drawn to TGF-β signaling as it has been linked to stemness (see below). MASTL silencing clearly downregulated TGF-β receptor II (TGFBR2), a central regulator of the TGF-β pathway (Massagué, 2012), and upregulated F11R, which is a junctional protein recently shown to be negatively regulated by TGF-β in breast cancer (Bednarek et al., 2020) (Figure 4B).
Figure 4.

MASTL depletion influences stem-cell-associated surface proteins
(A) Experimental outline for stable isotope labeling with amino acids in cell culture (SILAC)-based proteomics of surface proteins (surfaceome) in heavy and light isotope-labeled siControl- and siMASTL-treated cells.
(B) Significantly altered surface proteins in MASTL-silenced cells (SILAC siMASTL/siControl Ratio (log2) >0.5/<-0.5 and FDR <0.05) and functional enrichment based on g-Profiler (Raudvere et al., 2019). See also Figure S4 and Table S1.
MASTL supports TGFBR2 levels
TGF-β and ITGB3 signaling have been proposed to be essential for BCSCs (Lo et al., 2012). In addition, TGF-β signaling is essential for maintenance of hPSCs (James et al., 2005). The mediator of TGF-β signaling is the heteromeric TGF-β receptor, consisting of TGFBR1, TGFBR2, or TGFBR3 (Massagué, 2012). Of these, only TGFBR2 was significantly downregulated on the cell surface of MASTL-silenced cells. TGFBR2 was significantly decreased also at the mRNA (Illumina transcriptome data) and total protein level (SILAC), further implying a role for MASTL in regulating TGFBR2 (Figure S4A) (Taskinen et al., 2020). We did not detect any differences in TGFBR1 expression (Figure S5A). In addition, no effect was observed on TGFBR3 total protein level (SILAC) (Taskinen et al., 2020), although we did detect a significant decrease at the mRNA level with one of the siRNAs (Figure S5B). The decrease in TGFBR2 following MASTL silencing was validated with qRT-PCR and Western blotting in MDA-MB-231 cells (Figures 5A–5C), where the specificity of the TGFBR2 antibody was verified using two TGFBR2-specific siRNAs (Figure 5D). These data were further supported by the CCLE database where TGFBR2 levels showed significant correlation with MASTL expression (Figure 5E). Conversely, overexpression of MASTL led to a significant increase in TGFBR2 levels in MCF10A cells with low endogenous MASTL expression. Overexpression of the kinase-active (WT) and kinase-dead (G44S) MASTL increased TGFBR2 to the same extent (Figures 5F and 5G) and the MASTL kinase inhibitor (GKI-1; 20 μM) had no influence on TGFBR2 expression (Figures 5H and 5I). These results indicate that MASTL supports TGFBR2 expression in a kinase-independent manner.
Figure 5.

MASTL supports TGFBR2 levels
(A) Relative TGFBR2 mRNA expression in siControl-, siMASTL#6-, and siMASTL#7-treated MDA-MB-231 cells after 48 h of silencing (n = 5 biologically independent experiments, unpaired t-test, mean ± SD).
(B) Western blotting of MASTL, TGFBR2, and GAPDH in siControl-, siMASTL#6-, and siMASTL#7-treated MDA-MB-231 cells after 48 h.
(C) TGFBR2 protein expression relative to GAPDH in siControl-, siMASTL#6-, and siMASTL#7-treated MDA-MB-231 cells after 48 h (n = 3 biologically independent experiments, unpaired t-test, mean ± SD).
(D) Western blotting of MASTL, TGFBR2, and GAPDH in siControl-, siMASTL#6-, siMASTL#7-, siTGFBR2#5-, and siTGFBR2#6-treated MDA-MB-231 cells after 48 h. Cells were stimulated with TGF-β (20 ng/mL) for 45 min immediately before collection.
(E) TGFBR2 expression in breast cancer cell lines with high or low MASTL expression based on CCLE data (unpaired t-test, mean ± SEM).
(F) Western blotting of MASTL, TGFBR2, and GAPDH in EGFP (control), EGFP MASTL wild-type (WT), and EGFP MASTL kinase-dead (G44S) overexpressing MCF10A cells 24 h after transfection.
(G) TGFBR2 protein expression relative to GAPDH in EGFP (control), EGFP MASTL wild-type (WT), and EGFP MASTL kinase-dead (G44S) overexpressing MCF10A cells after 24 h (n = 3 biologically independent experiments, one-sample t-test, mean ± SD).
(H) Western blotting of MASTL, TGFBR2, and GAPDH in DMSO- and GKI-1 (20 μM)-treated MDA-MB-231 cells for 48 h.
(I) TGFBR2 protein expression relative to GAPDH in DMSO- and GKI-1-treated MDA-MB-231 cells after 48 h (n = 3 biologically independent experiments, one-sample t-test, mean ± SD). See also Figure S5.
MASTL controls AKT and SMAD3 activation via TGFBR2
Although the effect of MASTL on TGFBR2 was statistically significant and detected with multiple analysis methods, the difference observed was relatively small. Therefore, we decided to test whether the observed difference would have biological significance. TGF-β is known to activate canonical and non-canonical signaling pathways via TGFBR2 (Zhang, 2009). One of the non-canonical pathways is the PI3K-AKT signaling pathway. As MASTL expression levels have been shown to correlate with AKT activation (Rogers et al., 2018; Vera et al., 2015) and PI3K/AKT signaling has a vital role in pluripotency and CSCs (Yu and Cui, 2016), we decided to investigate the relationship between MASTL, TGF-β, and AKT signaling in detail. MASTL-silenced cells exhibited lower AKT activation in response to TGF-β compared to control cells, in line with decreased TGFBR2 levels following MASTL silencing (Figures 6A and 6B). Importantly, TGF-β treatment of MDA-MB-231 cells led to a significant activation of AKT, which was not influenced by treatment with the MASTL kinase inhibitor (Figures 6C and 6D). These results suggest an important role for MASTL in the TGF-β-mediated activation of the PI3K-AKT signaling pathway, potentially through the regulation of TGFBR2 levels in a kinase-independent fashion.
Figure 6.

MASTL controls AKT and SMAD3 activation via TGFBR2
(A) Western blotting of MASTL, TGFBR2, pAKT (Ser473), AKT, and GAPDH in siControl-, siMASTL#6-, and siMASTL#7-treated MDA-MB-231 cells after 48 h of silencing. Cells were stimulated with TGF-β (20 ng/mL) for 45 min immediately before collection.
(B) pAKT (Ser473) protein levels relative to total AKT, experimental setup shown in A. (n = 3 biologically independent experiments, one sample t-test, mean ± SD).
(C) Western blotting of MASTL, pAKT (Ser473), AKT, and GAPDH in DMSO- and GKI-1 (20μM)-treated (48 h) MDA-MB-231 cells with and without TGF-β stimulation (20 ng/mL, 45min).
(D) pAKT (Ser473) protein levels relative to total AKT, experimental setup shown in (C). (n = 3 biologically independent experiments, t-test with Welch’s correction, mean ± SD).
(E) Western blotting of MASTL, pSMAD3, SMAD3, and GAPDH in siControl- and siMASTL#7-treated MDA-MB-231 cells after 48 h. Cells were TGF-β stimulated 10 ng/mL 24 h before collection.
(F) pSMAD3 protein levels relative to total SMAD3, experimental setup shown in E. (n = 3 biologically independent experiments, unpaired t-test, mean ± SD).
(G) Western blotting of MASTL, NANOG, OCT4, and GAPDH in siControl-, siMASTL#6/7-, and siTGFBR2#5/6-treated hiPSCs after 48 h.
In addition to non-canonical signaling pathways, activated receptor complexes consisting of TGFBR2 and TGFBR1 can activate canonical TGF-β signaling that acts via SMAD transcriptional effectors (Massagué, 2012). Activation of SMAD2/3 is one of the key downstream SMAD proteins driving stemness (Mullen and Wrana, 2017). MASTL silencing significantly reduced TGF-β-induced activation of SMAD3 in MDA-MB-231 cells (Figures 6E and 6F). Furthermore, silencing of TGFBR2 in hiPSCs led to a reduction of pluripotency markers, NANOG and OCT4, which are known to be activated by SMAD2/3 (Mullen et al., 2011; Xu et al., 2008) similarly to MASTL silencing (Figure 6G). These results indicate that MASTL supports TGF-β signaling through upregulation of TGFBR2 and that this signaling axis is important for the maintenance of conventional SMAD and non-conventional PI3K-AKT signaling.
Discussion
MASTL is upregulated in various types of cancers and is involved in tumor recurrence (Conway et al., 2020; Marzec and Burgess, 2018). In addition, TGF-β promotes cancer progression and stemness in multiple cancer types (Ikushima and Miyazono, 2010). Here, we show that MASTL supports TGFBR2 expression and affects both canonical SMAD3 and non-canonical PI3K-AKT activation downstream of TGF-β signaling. Furthermore, MASTL influences the stemness regulator OCT1 in cancer cells, and the pluripotency regulator OCT4 in hiPSCs.
The cancer-promoting role of MASTL has been suggested to be linked to AKT signaling, but the exact mechanism has remained unclear. Exogenous overexpression of MASTL triggers AKT activation in an ENSA/PP2A-independent manner (Rogers et al., 2018; Vera et al., 2015). In addition, AKT-mediated proliferation in colorectal cell lines can be attenuated by MASTL silencing (Reshi et al., 2020). Our results show that silencing of MASTL leads to a significant decrease in TGFBR2 levels and TGF-β-induced PI3K-AKT activation. These data provide mechanistic insight into MASTL-dependent AKT activation.
We find that MASTL has a noticeable expression in pluripotent stem cells and is required for the maintenance of pluripotency. The most important signaling pathways maintaining pluripotency are PI3K/AKT and SMAD2/3 signaling (Singh et al., 2012), both of which are positively regulated by TGFBR2. In fact, TGF-β-signaling is one of the essential components necessary to maintain hPSCs (James et al., 2005) as NANOG, which maintains the core pluripotency transcription regulators (OCT4, SOX2, and NANOG) (Xu et al., 2008), is a direct target of TGF-β-mediated SMAD signaling in hPSCs. Therefore, MASTL, through regulation of TGFBR2 levels, can impact stemness via canonical and non-canonical TGF-β signaling. Besides TGF-β, MASTL displays a significant positive correlation with β3 integrin expression in breast tumors. Like TGF-β signaling, β3 integrin is essential for self-renewal of BCSCs and supports breast cancer metastasis (Lo et al., 2012; Parvani et al., 2013).
Our data suggest that MASTL transcription may be regulated by OCT1. Interestingly, OCT1 and OCT4 (products of the POU2F1 and POU5F1 genes) are both members of the POU domain transcription factor family (Vázquez-Arreguín and Tantin, 2016). The classical DNA recognition sequence of these transcription factors is an octamer motif 5′-ATGCAAAT-3′, though several substitutes are tolerated especially in the context of dual binding (Schöler et al., 1989; Vázquez-Arreguín and Tantin, 2016). These elements were found to be present in the MASTL promoter, implying that high MASTL levels in stem cells are likely to be maintained by OCT1 and OCT4, which are both highly expressed in stem cells.
In addition to TGFBR2, Frizzled-6 (FZD6) identified in our cell-surface proteome profiling could contribute to enhanced stemness properties downstream of MASTL. Frizzled-6, a receptor of Wnt ligands, promotes cell motility and metastasis of TNBCs (Corda et al., 2017). Furthermore, Wnt signaling is required for the maintenance of CD44high/CD24low breast stem cell populations (Scheel et al., 2011) and is central for induction and preservation of pluripotency (de Jaime-Soguero et al., 2018). Importantly, MASTL promotes Wnt/β-catenin signaling in colon cancer (Uppada et al., 2018) and TGF-β cooperates with Wnt in breast cancer to induce EMT (Scheel et al., 2011). However, the MASTL-FZD6 link remains to be investigated in the future.
We have recently shown that MASTL regulates actin stress fiber formation and cell contractility in a kinase-independent manner by promoting MRTF-A/SRF-mediated transcription of actomyosin-related genes (Taskinen et al., 2020). Dysregulation of cell contractility and TGF-β signaling is associated with cancer (Melchionna et al., 2021). Interestingly, TGF-β has been shown to trigger nuclear translocation of MRTF-A and -B which coordinately regulate transcription of Slug, a well-known inducer of EMT with SMAD3 (Morita et al., 2007). Furthermore, SRF depletion has been shown to lead to TGFBR2 downregulation (Taylor et al., 2014). Here, we show that MASTL positively regulates TGFBR2 levels in a kinase-independent manner as overexpression of kinase-dead MASTL increases TGFBR2 levels similar to WT, and MASTL kinase inhibition does not influence TGFBR2 levels or AKT activation. This and our previous findings suggest that MASTL supports MRTF-A signaling via TGFBR2 in addition to associating with MRTF-A and retaining it in the nucleus (Taskinen et al., 2020).
In addition, we show that the expression of hPSC pluripotency factors is not sensitive to MASTL kinase inhibition. MASTL inhibitors have been designed for cancer treatment to inhibit cell cycle progression, which is dependent on MASTL kinase activity (Kim et al., 2020; Ocasio et al., 2016). Our data suggest that MASTL may serve another pro-tumorigenic function, by the virtue of promoting TGF-β signaling, that is impervious to its kinase activity-targeting therapeutics. Importantly, several TGF-β inhibitors, with promising results in the treatment of multiple cancers including breast, are under intensive development (Huynh et al., 2019). Therefore, a combination therapy with MASTL and TGF-β inhibitors could prove more effective than either approach alone.
Limitations of the study
Our current results indicate that depletion of MASTL results in downregulation of stemness markers in BCSC and pluripotent stem cells and that increased MASTL levels correlate with more stem-like cell states. MASTL depletion results in downregulation of TGFBR2 and compromises TGF-β signaling, a well-established positive regulator of stemness. This suggests that MASTL supports stemness by facilitating TGFBR2 levels and signaling. However, we have not fully uncovered the mechanism whereby MASTL regulates TGFBR2. In addition, prolonged MASTL depletion inhibits cell cycle progression, limiting our ability to investigate MASTL regulation of stem cell biology beyond three days. Therefore, more follow-on studies are necessary to fully unravel the mechanism of how MASTL regulates TGF-β signaling and stemness in different contexts.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-MASTL (1:1,000 for WB, 1:1,500 for IHC) | Sigma-Aldrich | Cat# HPA027175; RRID: AB_1853591 |
| Rabbit anti-MASTL (1:1,000 for WB) | Abcam | Cat# ab86387; RRID: AB_1925198 |
| Mouse anti-GAPDH (1:20,000 for WB) | HyTest | Cat# 5G4MAB6C5; RRID: AB_1067079 |
| Rabbit anti-Integrin beta 3 (1:200 for WB) | Abcam | Cat# ab179473; RRID: AB_2917988 |
| Rabbit anti-OCT1 (1:1,000 for WB) | Proteintech | Cat# 10387-1-AP; RRID: AB_2167052 |
| Mouse anti-α-tubulin (1:2,000 for WB) | Developmental Studies Hybridoma bank (DSHB) | Cat# 12G10 anti-alpha-tubulin; RRID: AB_1157911 |
| Goat anti-NANOG (1:400 for WB, 1:50 for IF) | R&D Systems | Cat# AF1997; RRID: AB_355097 |
| Mouse anti-SOX2 (1:500 for WB, 1:100 for IF) | R&D Systems | Cat# MAB2018; RRID: AB_358009 |
| Rabbit anti-OCT3/4 (1:1,000 for WB) | Santa Cruz Biotechnology | Cat# sc-9081; RRID: AB_2167703 |
| Mouse anti-Vimentin (1:500 for WB) | Santa Cruz Biotechnology | Cat# sc-6260; RRID: AB_628437 |
| Mouse anti-Integrin β1 (1:1,000 for WB) | BD Biosciences | Cat# 610468; RRID: AB_397840 |
| Mouse anti-SSEA-5 (1:100 for IF) | Millipore | Cat# MABD88; RRID:AB_2917989 |
| Mouse anti-TGFBR2 (1:150 for WB) | Santa Cruz Biotechnology | Cat# sc-17799; RRID: AB_628348 |
| Rabbit anti-pSMAD3 (Ser423/425, 1:500 for WB) | Cell Signaling Technology | Cat# 9520; RRID: AB_2193207 |
| Rabbit anti-SMAD3 (1:500 for WB) | Abcam | Cat# ab28379; RRID: AB_2192903 |
| Rabbit anti-pAKT (Ser473, 1:500 for WB) | Cell Signaling Technology | Cat# 9271; RRID: AB_329825 |
| Rabbit anti-AKT (1:1,000 for WB) | Cell Signaling Technology | Cat# 9272; RRID: AB_329827 |
| Mouse anti-AKT (1:1,000 for WB) | Cell Signaling Technology | Cat# 2920; RRID: AB_1147620 |
| Rabbit anti-Integrin beta 3 (IHC) | Abcam | Cat# ab75872; RRID: AB_2249317 |
| Mouse anti-CD24, Alexa Fluor 647 (1:20 for FACS) | BD Biosciences | Cat# 561644; RRID: AB_10894010 |
| Rat anti-CD44, FITC (1:20 for FACS) | BD Biosciences | Cat# 553133; RRID: AB_2076224 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (1:400 for IF) | Thermo Fisher Scientific | Cat# A10037; RRID: AB_2534013 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (1:400 for IF) | Invitrogen | Cat# A-21202; RRID: AB_141607 |
| IRDye 680RD Donkey anti-Mouse IgG (1:5,000 for WB) | LI-COR Biosciences | Cat# 926-68072; RRID: AB_10953628 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (1:400 for IF) | Invitrogen | Cat# A-21206; RRID: AB_2535792 |
| IRDye 800CW Donkey anti-Rabbit IgG (1:5,000 for WB) | LI-COR Biosciences | Cat# 926-32213; RRID: AB_621848 |
| Chemicals, peptides, and recombinant proteins | ||
| Calcein AM | Invitrogen | C3100MP |
| GKI-1 | MedChemExpress, (Ocasio et al., 2016) | HY-100521 |
| Recombinant human TGF-B1 | R&D systems | 240-B-002 |
| 2′,7′-Dichlorodihydrofluorescein diacetate (DCF-DA) | Sigma-Aldrich | D6883 |
| Critical commercial assays | ||
| Nucleospin RNA kit | Macherey-Nagel | 740955.50 |
| High capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | N/A |
| Histofine® Simple Stain MAX PO (R) kit | Nicherei Biosciences | 414141F |
| Deposited data | ||
| Cancer Cell Line Encyclopedia (CCLE) | https://sites.broadinstitute.org/ccle/ | N/A |
| Surface proteomics data | This paper | PRIDE: PXD019993 |
| Total proteomics data | (Taskinen et al., 2020) | PRIDE: PXD013757 |
| Transcriptome data | (Taskinen et al., 2020) | GEO: GSE131833 |
| Experimental models: Cell lines | ||
| Human: MDA-MB-231 | ATCC | HTB-26; RRID: CVCL_0062 |
| Human: MDA-MB-436 | ATCC | HTB-130; RRID: CVCL_0623 |
| Human: MCF10A | ATCC | CRL-10317; RRID: CVCL_0598 |
| Human: HEL11.4 | Timo Otonkoski (Mikkola et al., 2013) | RRID: CVCL_JE79 |
| Human: HEL62.4 | Timo Otonkoski | N/A |
| Oligonucleotides | ||
| AllStars Neg. Control siRNA | Qiagen | 1027281 |
| Hs_MASTL_6 FlexiTube siRNA (target sequence 5′-ACGCCTTATTCTAGCAAATTA-3′) | Qiagen | SI02653014 |
| Hs_MASTL_7 FlexiTube siRNA (target sequence 5′-CAGGACAAGTGTTATCGCTTA-3′) | Qiagen | SI02653182 |
| Hs_POU2F1_5 Flexitube siRNA (target sequence 5′-GGUCCAACUCGCUGGAACATT-3′) | Qiagen | SI03071908 |
| siTGFBR2#5 (target sequence 5′-AAAGCCTGGTGAGACTTTCTT-3′) | Qiagen | SI00301910 |
| siTGFBR2#6 (target sequence 5′-CTCCAATATCCTCGTGAAGAA-3′) | Qiagen | SI02223179 |
| pLenti-H1-shRNA(Neg control) (RFP-Puro) Sequence: GTCTCCACGCGCAGTACATTT |
AMSBIO | N/A |
| pLenti-H1-shRNA(h MASTL)#3-Rsv (RFP-Puro) Sequence: TCGTTGGGATTTAACACACCA |
AMSBIO | N/A |
| RT-PCR primer for GAPDH Forward: GCCCAATACGACCAAATCC Reverse: AGCCACATCGCTCAGACA Probe: 60 |
Universal Probe Library (Roche) | N/A |
| RT-PCR primer for TGFBR2 | ThermoFisher Scientific | Hs00234253_m1 |
| Recombinant DNA | ||
| pcDNA EGFP-C2 | (Taskinen et al., 2020) | N/A |
| pcDNA EGFP MASTL WT (siRNA resistant) | (Taskinen et al., 2020) | N/A |
| pcDNA EGFP MASTL G44S (siRNA resistant) | (Taskinen et al., 2020) | N/A |
| pCGN-OCT1 | A gift from Winship Herr (Addgene plasmid # 53308; http://n2t.net/addgene:53308) | RRID: Addgene_53308 |
| Software and algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | RRID: SCR_003070 |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | RRID: SCR_002798 |
| Xcalibur | ThermoFisher Scientific | RRID: SCR_014593 |
| Flowing Software | https://bioscience.fi/services/cell-imaging/flowing-software/ | N/A |
| MaxQuant version 1.5.5.1 | (Cox and Mann, 2008) | RRID: SCR_014485 |
| Andromeda search engine | (Cox et al., 2011) | N/A |
| Perseus software version 1.5.5.3 | (Tyanova et al., 2016) | RRID: SCR_015753 |
| Significance B algorithm | (Cox and Mann, 2008) | N/A |
Resource availability
Lead contact
-
•
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Johanna Ivaska (johanna.ivaska@utu.fi).
Materials availability
-
•
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
MDA-MB-231 (female, human triple-negative adenocarcinoma, ATCC) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich) and 1% MEM non-essential amino acid solution (Sigma-Aldrich), in + 37°C, 5% CO2. For passaging, the cells were detached by using trypsin-EDTA solution (T4174, Sigma-Aldrich).
MDA-MB-436 (female, human adenocarcinoma, ATCC) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich), 10 μg/mL insulin (Sigma-Aldrich) and 1% penicillin-streptomycin (Sigma-Aldrich) in + 37°C, 5% CO2. For passaging, the cells were detached by scraping.
MCF10A (female, human epithelial, ATCC) cells were cultured in DMEM/Nutrient Mixture F-12 (Gibco) supplemented with 5% horse serum (Gibco), 100 ng/mL cholera toxin (Sigma), 20 ng/mL human epidermal growth factor (EGF, Sigma-Aldrich), 0.5 μg/mL hydrocortisone (Sigma-Aldrich), 10 μg/mL insulin (Sigma-Aldrich), and 1% penicillin-streptomycin (Sigma-Aldrich) in + 37°C, 5% CO2. For passaging, the cells were detached by using 0.25% trypsin-EDTA in HBSS (Biowest, L0931-100).
Tetracycline-inducible shRNA MDA-MB-231 cells (shControl and shMASTL#3) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich), 1% penicillin-streptomycin (Sigma-Aldrich), 1 μg/mL puromycin (Sigma) and 10 μg/mL blasticidin (Gibco) in + 37°C, 5% CO2. Induction was performed by adding 3 mg/mL tetracycline in the culture medium.
For mammosphere formation, Mammocult Basal media (Stem Cell Technologies, 05620) was used. Briefly, cells were detached with trypsin, separated into single cells with 0.35 μm cell strainer in normal culture media, washed with mammosphere media, and plated on low adhesion plates. For sphere formation assays, 1,000 cells/96-well plate well in 100 ul volume of Mammocult Basal media were plated (8 replicate wells/condition). Fresh media was added on day 2. Tetracycline-induced shRNA silenced MDA-MB-231 cells were imaged at day 7 and siRNA silenced MDA-MB-436 cells at day 5. Before imaging, proliferative spheres were stained with calcein AM (C3100MP) for 1 hour at +37°C. Images were captured with (Nikon Eclipse Ti-E widefield microscope, Hamamatsu Orca C13440 Flash 4.0 ERG [b/w] sCMOS camera and Plan Apo lambda 20×/0.80, WD 1,000-μm objective). Sphere size was analyzed with ImageJ software. Spheres having greater than 60 μm Feret diameter were included in the analysis.
Human induced pluripotent cell (hiPSC) lines HEL11.4 and HEL62.4 were obtained from the University of Helsinki. Cell lines were generated from male (HEL11.4) or female (HEL62.4) fibroblasts using Sendai viruses (Mikkola et al., 2013) and this protocol was also used to study induction of MASTL expression during reprogramming. HiPSCs were grown in feeder-free conditions on Matrigel (MG) (Corning, 354277) in Essential 8 (E8) Basal medium (Life Technologies, A15169-01) supplemented with Essential 8 supplement (Life Technologies, A1517-01) or in mTeSR1 (StemCell Technologies, 85850) at +37°C, 5% CO2 in a humidified incubator. Culture media was changed daily. For passaging, cells were detached using sterile filtered 0.5 mM EDTA (Life Technologies, 15575-038) in PBS for 3 min at room temperature (RT). The parental fibroblast cell line of HEL11.4 were obtained from the University of Helsinki, and were cultured in Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F-12 (DMEM/F-12) (Life Technologies, 10565-018) supplemented with 10% Fetal Bovine Serum (FCS) (Biowest, S1860) at +37°C, 5% CO2 in a humidified incubator. Fibroblasts were passaged using 0.25% trypsin-EDTA in HBSS (Biowest, L0931-100).
For embryonic body (EB) differentiation cells were passaged normally and plated on low adhesion plates. Half of the medium was replaced every second day with full E8 medium. In order to generated differentiated fibroblasts, differentiated EBs were plated on normal culture plates. Attached cells were detached with trypsin and cultured in DMEM (Sigma-Aldrich) supplemented with 10% FBS (Sigma) and 2 mM l-glutamine (Sigma).
Method details
Transfections
Plasmid DNAs were transfected into MCF10A cells by using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s protocol. SiRNAs were transfected into MDA-MB-231 and MDA-MB-436 cells by using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s protocol.
HPSCs were transfected by using Lipofectamine RNAiMAX transfection reagent (Invitrogen) using modified protocol. Briefly, a high concentration (180 nM) of the siRNA was used in the transfection mixture which was added on freshly passaged colonies. Transfection was repeated a day after by adding new transfection mixture with fresh media.
Transductions
Lentiviral transductions of shControl and shMASTL#3 were performed according to the manufacturer’s protocol (AMSBIO).
RT-PCR
Total RNA was extracted using the Nucleospin RNA kit (Macherey-Nagel) and DNase was digested with DNaseI Amplification Grade kit (Invitrogen) according to manufacturers’ instructions. Synthesis of cDNA from RNA was performed using high capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The expression levels of TGFBR2 and GAPDH (endogenous control) were determined by using QuantStudio™ 12K Flex Real-Time PCR System (Thermo Fisher Scientific).
Immunofluorescence & confocal microscopy
Cells were fixed with 4% paraformaldehyde 20 min, washed three times with PBS and permeabilized with 0,5% Triton in PBS at RT for 20 min. Fixed cells were stained with primary antibodies in 30% horse serum in PBS at 4°C overnight, washed with PBS, and incubated with secondary antibodies and 4′6-diamidino-2-phenylindole (DAPI, 1:10,000) in 30% horse serum in PBS at RT for 1h. Cells were washed with PBS and imaged with a 3i CSU-W1 spinning disk confocal microscope, using Slidebook 6 software, Hamamatsu sCMOS Orca Flash4.0×camera and 64×objective. Quantification of MASTL in NANOG/SSEA-5 positive and negative cells was performed using ImageJ (National Institutes of Health).
Western blot
Cells were first washed with ice-cold PBS on ice and lysed with TXLB (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton-X, 0.5% glycerol, 1% SDS) supplemented with complete protease and phosphatase inhibitor tablets (Sigma-Aldrich). Cell lysates were boiled for 5 min and sonicated prior to measurement of protein concentrations with DC Protein assay (Bio-Rad). Protein concentrations were balanced by adding the required amount of TXLB. SDS sample buffer was added on samples, and the samples were boiled for 5 min prior to loading on precast Tris-Glycine-eXtendet SDS-PAGE gels with a 4–20% gradient (Bio-Rad). After separation, the proteins were transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% milk in TBST (Tris-buffered saline with 0,1% Tween 20) for 1h at RT. Primary antibodies were diluted in StartingBlock (PBS) Blocking Buffer (37538, Thermo Scientific), and membranes were incubated in the antibody dilutions at +4°C overnight. Membranes were washed three times with TBST prior to incubating in fluorophore-conjugated Odyssey secondary antibodies (LI-COR Biosciences) for 1 h at RT. Membranes were washed three times with TBST, and scanned with Odyssey infrared system (LI-COR Biosciences). Band intensities were quantified using ImageJ.
GKI-1 sensitivity
Cells were plated the day before the exposure to the GKI-1 inhibitor (MedChemExpress, HY-100521). After 24 hours of incubation, sample wells were fixed with ice cold methanol for 20 min, washed with water, stained with 0.2% Crystal violet in 10% ethanol for 5 min and washed carefully with water. Inverted plates were dried overnight and the wells were scanned. Signal intensity was measured with ImageJ.
The FinHer trial, immunohistochemistry
Breast cancer tissue samples were collected from women who had undergone breast surgery and who agreed to participate in the FinHer trial (Joensuu et al., 2006, 2009). In FinHer, 1,010 women with axillary node-positive cancer or high-risk node-negative cancer were randomly assigned to receive three cycles of docetaxel or vinorelbine, followed by (in both groups) three cycles of fluorouracil, epirubicin, and cyclophosphamide (FEC). The women with HER2 (ERBB2)-positive cancer were further assigned to receive or not to receive nine weekly trastuzumab infusions concomitantly with chemotherapy. Presence or absence of amplified ERBB2 was verified using chromogenic in situ hybridization (CISH). An ethics committee at the Helsinki University Central Hospital approved the use of archival formalin-fixed paraffin-embedded tumour tissue for the purposes of the present study (permission HUS 106/13/03/02/2015). The FinHer trial participants provided informed consent for the FinHer trial participation and another consent for the use of breast cancer tissue for research related to the trial prior to initiation of the trial treatments.
Breast tumour tissue was available from 851 (84%) out of the 1,010 patients for immunohistochemistry. Tissue microarrays were constructed from representative tumour tissue regions, and 5 μm sections were cut on SuperFrost+ slides (Menzel-Gläser). The sections were deparaffinized and endogenous peroxidase activity was blocked with 1% hydrogen peroxide. Antigen retrieval was carried out in sodium citrate (10 mmol/L, pH 6.0) in Antigen Retriever 2100 (Aptum Biologics Ltd., Southampton, UK) for MASTL, and in Tris-EDTA buffer (10 mmol/L, pH 9.0) in water bath (+98°C, 30 minutes) for ITGB3 immunostaining. Antibodies were diluted in PowerVision Universal IHC blocking diluent (Leica Biosystems, UK; dilutions: MASTL (HPA027175), 1:1,500; ITGB3, 1:250) and incubated on slides either at +4°C overnight (MASTL) or 1 hour at room temperature (ITGB3). Binding of the primary antibodies were detected by using Histofine® Simple Stain MAX PO kit for rabbit antibodies (Nichirei Biosciences Inc., Japan) and visualized by using 3,3′-diaminobenzidine (ImmPACT™ DAB, Vector Laboratories, Burlingame, CA, USA) following the manufacturers’ recommendations. The slides were counterstained with Mayer’s haematoxylin. Intensities of cytoplasmic MASTL, and ITGB3 expression in tumour cells were scored as negative, weak or high immunostaining.
Frequency tables were analyzed using the chi square test. Overall survival was calculated from the date of randomization to the date of death. Survival between groups was compared using Kaplan–Meier survival-table method and log-rank test. All p values are two-sided.
Surfaceome proteomics
For quantitative MS analysis MDA-MB-231 cells were labelled by culturing them in DMEM without Arg/Lys (A14431-01), 200 μM L-glutamine, 100 U/mL penicillin streptomycin, (110 mg/L - 1 mM) sodium pyruvate and 10% 10 kDa dialyzed serum (F0392, Sigma). The media for Light labelling was supplemented with Arg 0 (84 mg/L, A6969, Sigma) and Lys 0 (146 mg/L, L8662, Sigma) and for Heavy labelling with Arg10 (CNLM-539-H, Cambridge Isotope Laboratories) and Lys8 (CNLM-291-H, Cambridge Isotope Laboratories). Incorporation of the isotopes was confirmed by MS analysis after six passages. SiControl and siMASTL#6 samples silenced for 48 h were prepared two times with light and heavy labelled cells obtaining four experimental replicates. For each independent experiment, siControl and siMASTL silenced samples were mixed using a label-swap replication strategy.
To label cell surface, culture plates were washed carefully with cold PBS on ice, after 0.13 mg/mL of Sulfo-NHS-SS-Biotin (21331, Thermo Scientific) in PBS was added and incubated 60 min at +4°C in slow rocker. Excess of biotin was carefully washed with cold PBS for three times after cells were lysed into NDLB buffer (200 mM NaCl, 75 mM Tris, 15 mM NaF, 1.5 mM Na3VO4, 7.5 mM EDTA and 7.5mM EGTA, 1.5% Triton X-100, 0.75% NP-40 Igepal CA-630, protease inhibitors (1183617001 Sigma). Samples were syringed with needle and cleared by centrifugation (13,000 rpm, 10 min, +4°C). Equal protein amounts of heavy and light lysates were combined, and biotinylated proteins were enriched with streptavidin agarose beads (16-126 Millipore) 60 min, +4°C, rotation. After multiple washes with cold NDLB-buffer and PBS, proteins were eluted into DTT-buffer (0.1 M DTT, 0.1 M Tris pH 7.5) following incubation of 20 min, 1,000 rpm mixer, RT. Supernatants were collected by centrifugation of 60 s, 13,000 rpm, boiled with loading buffer and eluted proteins were then separated by SDS-PAGE, stained with Coomassie blue.
All gel lanes were subdivided into 5 slices, each washed twice with 50 mM ammonium bicarbonate, and 50 mM ammonium bicarbonate with 50% acetonitrile. Proteins in the gel bands were then reduced using dithiothreitol (10 mM at 54°C for 30 minutes) and subsequently alkylated with iodoacetamide (55 mM at room temperature for 45 minutes). Gel pieces were washed again with 50 mM ammonium bicarbonate, with 50 mM ammonium bicarbonate with 50% acetonitrile; and finally dehydrated using acetonitrile before drying in a SpeedVac. Trypsin (5 μg/mL trypsin gold; Promega in 25 mM ammonium bicarbonate) was added and incubated overnight at +35°C. Tryptic peptides were extracted from gel pieces with two 50% (v/v) acetonitrile/water washes and subsequently dried in a SpeedVac.
Mass spectrometry analysis
Digested peptides were desalted using StageTip (Rappsilber et al., 2007) and separated by nanoscale C18 reverse-phase liquid chromatography using an EASY-nLC II 1200 (Thermo Fisher Scientific) coupled to a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific). Elution was carried out using a binary gradient with buffer A (2% acetonitrile) and B (80% acetonitrile), both containing 0.1% formic acid. Samples were loaded into a 50 cm fused silica emitter (New Objective) packed in-house with ReproSil-Pur C18-AQ, 1.9 μm resin (Dr Maisch GmbH). Packed emitter was kept at 50°C by means of a column oven (Sonation) integrated into the nanoelectrospray ion source (Thermo Fisher Scientific).
The gradient used started at 2% of buffer B, was maintained constant for 3 minutes, and increased to 20% over 70 minutes and then to 41% over 20 minutes. Finally, a column wash was performed at 90% of B for 12 minutes for a total gradient duration of 105 minutes. The eluting peptide solutions were automatically (online) electrosprayed into the mass spectrometer via a nanoelectrospray ion source (Sonation). An Active Background Ion Reduction Device (ABIRD) was used to decrease ambient contaminant signal level.
Data was acquired using Xcalibur software (Thermo Fisher Scientific) and acquisition was carried out in positive ion mode using data dependent acquisition. A full scan (FT-MS) over mass range of 375–1,500 m/z was acquired at 60,000 resolution at 200 m/z, with a target value of 3,000,000 ions for a maximum injection time of 20 ms. Higher energy collisional dissociation fragmentation was performed on the 15 most intense ions, for a maximum injection time of 50 ms, or a target value of 50,000 ions. Multiply charged ions having intensity greater than 50,000 counts were selected through a 1.4 m/z window and fragmented using normalised collision energy of 27. Former target ions selected for MS/MS were dynamically excluded for 13 s.
ROS activity
Cells were detached with trypsin and suspended into full culture media. 10 μM DCF-DA (2′,7′-Dichorofluorecindiacetate [D6883, Sigma]) was added following incubation of 30 min at +37°C. Formation of fluorescence was measured using LSR Fortessa (BD) and analyzed using Flowing Software (GeoMean fluorescence intensity).
Flow cytometry
Tetracycline-induced shRNA silencing in MDA-MB-231 cells was performed by adding 3 mg/mL tetracycline in the culture medium (96 h). Cells were detached using non-enzymatic cell dissociation solution (C5914, Sigma), and 200,000 cells were re-suspended in 100 μL of ice-cold 4% FBS in PBS. Cells were incubated with Alexa Fluor 647 conjugated CD24 and FITC conjugated CD44 antibodies on ice for 20 min, washed once and re-suspended in 300 μL 4% FBS in PBS. Cells were filtered and analysed using BD LSRFortessa cell analyser (BD Biosciences).
Quantification and statistical analysis
Mass spectrometry data analysis
The MS Raw files were processed with MaxQuant software (Cox and Mann, 2008) version 1.5.5.1 and searched with Andromeda search engine (Cox et al., 2011), querying UniProt (UniProt Consortium, 2010) Homo sapiens (09/07/2016; 92,939 entries). The database was searched requiring specificity for trypsin cleavage and allowing maximum two missed cleavages. Methionine oxidation and N-terminal acetylation were specified as variable modifications, and Cysteine carbamidomethylation as fixed modification. The peptide, protein and site false discovery rate (FDR) was set to 1%.
For quantitation in MaxQuant, multiplicity was set to 2 and Arg0/Arg10, Lys0/Lys8 were used for ratio measurement of SILAC labelled peptides. Only unique peptides were used for protein group quantification. MaxQuant output was further processed and analysed using Perseus software version 1.5.5.3 (Tyanova et al., 2016). The common reverse and contaminant hits (as defined in MaxQuant output) were removed. Only protein groups identified with at least one uniquely assigned peptide were used for the analysis.
To identify regulated proteins, the two-sided Significance B algorithm (Cox and Mann, 2008) with a threshold value of 0.05 (Benjamini–Hochberg FDR used for truncation) was applied to each replicate sample. Protein abundance changes were considered significant if they passed the Significance B test in at least three replicates out of the four analysed. Identified proteins were further sorted for proteins having > 0.5 median siMASTL/siControl log2 fold change, consistent direction of change and significance B < 0.05 in three out of four replicate values.
Statistical analysis
All of the quantifications were presented as mean values (± SD or ±SEM) from 3-5 more biologically independent experiments, unless stated otherwise. Statistical analysis was performed using unpaired t-test, one-sample t-test, chi square test or log-rank test, acquired by GraphPad Prism software. p-values < 0.05 were considered significant in all experiments. All of the statistical details in each experiment can be found from the main text, figure legends or method details.
Acknowledgments
We thank Prof. Timo Otonkoski and the Biomedicum Stem Cell Center, University of Helsinki, for providing hiPSC lines HEL11.4 and HEL62.4. We thank Jenni Siivonen and Petra Laasola for technical assistance. The Ivaska laboratory is acknowledged for lively discussions and critical feedback on the project. Hellyeh Hamidi for editing the manuscript. The Cell Imaging and Cytometry core facility (Turku Bioscience Center, University of Turku and Åbo Akademi University and Biocenter Finland), EuroBioimaging node in Turku, and the Mass spectrometry unit of the Beatson Institute for Cancer Research are acknowledged for services, instrumentation, and expertise. This study has been supported by the Academy of Finland (E.N. 297079 and J.I. 312517), Academy of Finland CoE for Translational Cancer Biology (J.I.), an ERC CoG grant 615258 (J.I.), InFLAMES Flagship Programme of the Academy of Finland (decision number: 337530), the Sigrid Jusélius Foundation (J.I.), the Finnish Cancer Organization (J.I. and E.N.), Finnish Cultural Foundation (E.N., M.E.T., and A.I.) and Cancer Research UK, A17196 (S.L.) and Stand Up to Cancer campaign for Cancer Research UK A29800 (S.Z.). We also acknowledge EMBO for a short-time fellowship (E.N.).
Author contributions
Conceptualization, E.N. and J.I.; Investigation, E.N., M.E.T., S.L., A.I., M.P., J.W., and H.J.; Formal Analysis, E.N., M.E.T., S.L., A.I., H.S., and H.J.; Writing – Original Draft, E.N. and J.I. Writing-editing; E.N., S.L., M.E.T., J.I., A.I., and H.J.; Visualization, E.N. and M.E.T.; Supervision, J.I., J.C.N., S.Z., and J.Is.; Funding Acquisition, J.I.
Declaration of interests
The authors declare no competing financial interests.
Published: June 17, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104459.
Contributor Information
Elisa Närvä, Email: elisa.narva@utu.fi.
Johanna Ivaska, Email: johanna.ivaska@utu.fi.
Supplemental information
Detailed list of proteins detected with Stable Isotope Labeling with Amino acids in Cell culture (SILAC)-based proteomics of surface proteins (surfaceome) in heavy and light isotope labeled siControl- and siMASTL-treated cells. Protein IDs, gene names, unique peptides, sequence coverage, intensity, H/L ratio, and Significance B of all identified proteins (sheet 1) and significant hits (sheet 2).
Data and code availability
-
•
Surface proteomics data have been deposited at the Proteomics Identifications database (PRIDE) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Álvarez-Fernández M., Sánchez-Martínez R., Sanz-Castillo B., Gan P.P., Sanz-Flores M., Trakala M., Ruiz-Torres M., Lorca T., Castro A., Malumbres M. Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc. Natl. Acad. Sci. U S A. 2013;110:17374–17379. doi: 10.1073/pnas.1310745110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Fernández M., Sanz-Flores M., Sanz-Castillo B., Salazar-Roa M., Partida D., Zapatero-Solana E., Ali H.R., Manchado E., Lowe S., VanArsdale T., et al. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2018;25:828–840. doi: 10.1038/s41418-017-0024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjonen A., Kaukonen R., Mattila E., Rouhi P., Högnäs G., Sihto H., Miller B.W., Morton J.P., Bucher E., Taimen P., et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J. Clin. Invest. 2014;124:1069–1082. doi: 10.1172/JCI67280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiedu M.K., Ingle J.N., Behrens M.D., Radisky D.C., Knutson K.L. Tgfβ/tnfα-mediated epithelial–mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71:4707–4719. doi: 10.1158/0008-5472.CAN-10-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff M.H., Akhurst R.J. Transforming growth factor-beta in breast cancer: too much, too late. Breast Cancer Res. 2009;11:202. doi: 10.1186/bcr2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek R., Selmi A., Wojkowska D., Karolczak K., Popielarski M., Stasiak M., Salifu M.O., Babinska A., Swiatkowska M. Functional inhibition of F11 receptor (F11R/junctional adhesion molecule-A/JAM-A) activity by a F11R-derived peptide in breast cancer and its microenvironment. Breast Cancer Res. Treat. 2020;179:325–335. doi: 10.1007/s10549-019-05471-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I., Thomson M.W., Carey V.J., Ge R., Bell G.W., Regev A., Weinberg R.A. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhola N.E., Balko J.M., Dugger T.C., Kuba M.G., Sánchez V., Sanders M., Stanford J., Cook R.S., Arteaga C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A., Greenwood W., Le Quesne J., Teschendorff A., Miranda-Saavedra D., Rueda O.M., Sandoval J.L., Vidakovic A.T., Saadi A., Pharoah P., et al. TGFβ induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat. Commun. 2012;3:1055. doi: 10.1038/ncomms2039. [DOI] [PubMed] [Google Scholar]
- Conway J.R.W., Närvä E., Taskinen M.E., Ivaska J. Kinase-independent functions of MASTL in cancer: a new perspective on MASTL targeting. Cells. 2020;9:E1624. doi: 10.3390/cells9071624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corda G., Sala G., Lattanzio R., Iezzi M., Sallese M., Fragassi G., Lamolinara A., Mirza H., Barcaroli D., Ermler S., et al. Functional and prognostic significance of the genomic amplification of frizzled 6 (FZD6) in breast cancer: FZD6 is frequently amplified in breast cancer. J. Pathol. 2017;241:350–361. doi: 10.1002/path.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- de Jaime-Soguero A., Abreu de Oliveira W., Lluis F. The pleiotropic effects of the canonical Wnt pathway in early development and pluripotency. Genes. 2018;9:93. doi: 10.3390/genes9020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Vera J., Palmer S., Hernandez-Fernaud J.R., Dornier E., Mitchell L.E., Macpherson I., Edwards J., Zanivan S., Norman J.C. A proteomic approach to identify endosomal cargoes controlling cancer invasiveness. J. Cell Sci. 2017;130:697–711. doi: 10.1242/jcs.190835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N., Qian D., Lam J.S., Ailles L.E., Wong M., et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G., Abdallah W.M., Foley J.M., Jackson K.W., Clarke M.F., Kawamura M.J., Wicha M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Huang S., An G., Wang G., Gu S., Zhao X. Identification of new cancer stem cell markers and signaling pathways in HER-2-positive breast cancer by transcriptome sequencing. Int. J. Oncol. 2019;55:1003–1018. doi: 10.3892/ijo.2019.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford C.A., Ziller M.J., Gu H., Trapnell C., Donaghey J., Tsankov A., Shalek A.K., Kelley D.R., Shishkin A.A., Issner R., et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashmi A.A., Naz S., Hashmi S.K., Hussain Z.F., Irfan M., Khan E.Y., Faridi N., Khan A., Edhi M.M. Prognostic significance of p16 & p53 immunohistochemical expression in triple negative breast cancer. BMC Clin. Pathol. 2018;18:9. doi: 10.1186/s12907-018-0077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiepen C., Mendez P.-L., Knaus P. It takes two to tango: endothelial TGFβ/BMP signaling crosstalk with mechanobiology. Cells. 2020;9:1965. doi: 10.3390/cells9091965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh L.K., Hipolito C.J., Ten Dijke P. A perspective on the development of TGF-β inhibitors for cancer treatment. Biomolecules. 2019;9:E743. doi: 10.3390/biom9110743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H., Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat. Rev. Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- James D., Levine A.J., Besser D., Hemmati-Brivanlou A. TGFβ/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- Joensuu H., Kellokumpu-Lehtinen P.-L., Bono P., Alanko T., Kataja V., Asola R., Utriainen T., Kokko R., Hemminki A., Tarkkanen M., et al. Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N. Engl. J. Med. 2006;354:809–820. doi: 10.1056/NEJMoa053028. [DOI] [PubMed] [Google Scholar]
- Joensuu H., Bono P., Kataja V., Alanko T., Kokko R., Asola R., Utriainen T., Turpeenniemi-Hujanen T., Jyrkkiö S., Möykkynen K., et al. Fluorouracil, epirubicin, and cyclophosphamide with either docetaxel or vinorelbine, with or without trastuzumab, as adjuvant treatments of breast cancer: final results of the FinHer trial. J. Clin. Orthod. 2009;27:5685–5692. doi: 10.1200/JCO.2008.21.4577. [DOI] [PubMed] [Google Scholar]
- Kim W.-T., Ryu C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017;50:285–298. doi: 10.5483/BMBRep.2017.50.6.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A.-Y., Yoon Y.N., Leem J., Lee J.-Y., Jung K.-Y., Kang M., Ahn J., Hwang S.-G., Oh J.S., Kim J.-S. MKI-1, a novel small-molecule inhibitor of MASTL, exerts antitumor and radiosensitizer activities through PP2A activation in breast cancer. Front. Oncol. 2020;10:571601. doi: 10.3389/fonc.2020.571601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Ma H., Zhang J., Zhu L., Wang C., Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017;7:13856. doi: 10.1038/s41598-017-14364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo P.-K., Kanojia D., Liu X., Singh U.P., Berger F.G., Wang Q., Chen H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin–TGFβ signaling. Oncogene. 2012;31:2614–2626. doi: 10.1038/onc.2011.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J., MacLennan D.H. Molecular cloning of cDNAs from human kidney coding for two alternatively spliced products of the cardiac Ca2+-ATPase gene. J. Biol. Chem. 1988;263:15024–15031. doi: 10.1016/s0021-9258(18)68141-4. [DOI] [PubMed] [Google Scholar]
- Maddox J., Shakya A., South S., Shelton D., Andersen J.N., Chidester S., Kang J., Gligorich K.M., Jones D.A., Spangrude G.J., et al. Transcription factor Oct1 is a somatic and cancer stem cell determinant. PLoS Genet. 2012;8:e1003048. doi: 10.1371/journal.pgen.1003048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec K., Burgess A. The oncogenic functions of MASTL kinase. Front. Cell Dev. Biol. 2018;6:162. doi: 10.3389/fcell.2018.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionna R., Trono P., Tocci A., Nisticò P. Actin cytoskeleton and regulation of TGFβ signaling: exploring their links. Biomolecules. 2021;11:336. doi: 10.3390/biom11020336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola M., Toivonen S., Tamminen K., Alfthan K., Tuuri T., Satomaa T., Natunen J., Saarinen J., Tiittanen M., Lampinen M., et al. Lectin from Erythrina cristagalli supports undifferentiated growth and differentiation of human pluripotent stem cells. Stem Cells Dev. 2013;22:707–716. doi: 10.1089/scd.2012.0365. [DOI] [PubMed] [Google Scholar]
- Morita T., Mayanagi T., Sobue K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007;179:1027–1042. doi: 10.1083/jcb.200708174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen A.C., Orlando D.A., Newman J.J., Lovén J., Kumar R.M., Bilodeau S., Reddy J., Guenther M.G., DeKoter R.P., Young R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147:565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen A.C., Wrana J.L. TGF-β family signaling in embryonic and somatic stem-cell renewal and differentiation. Cold Spring Harb. Perspect. Biol. 2017;9:a022186. doi: 10.1101/cshperspect.a022186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P.A.J., Vousden K.H., Norman J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011;192:209–218. doi: 10.1083/jcb.201009059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z., Wang C., Zhou Z., Chen C., Liu R., Wang D. Transforming growth factor-beta increases breast cancer stem cell population partially through upregulating PMEPA1 expression. Acta Biochim. Biophys. Sin. 2016;48:194–201. doi: 10.1093/abbs/gmv130. [DOI] [PubMed] [Google Scholar]
- Ocasio C.A., Rajasekaran M.B., Walker S., Le Grand D., Spencer J., Pearl F.M.G., Ward S.E., Savic V., Pearl L.H., Hochegger H., Oliver A.W. A first generation inhibitor of human Greatwall kinase, enabled by structural and functional characterisation of a minimal kinase domain construct. Oncotarget. 2016;7:71182–71197. doi: 10.18632/oncotarget.11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padua D., Massagué J. Roles of TGFβ in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- Parvani J.G., Galliher-Beckley A.J., Schiemann B.J., Schiemann W.P. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Mol. Biol. Cell. 2013;24:3449–3459. doi: 10.1091/mbc.E12-10-0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti D., Costa A., Zaffaroni N., Pratesi G., Petrangolini G., Coradini D., Pilotti S., Pierotti M.A., Daidone M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- Prater M.D., Petit V., Alasdair Russell I., Giraddi R.R., Shehata M., Menon S., Schulte R., Kalajzic I., Rath N., Olson M.F., et al. Mammary stem cells have myoepithelial cell properties. Nat. Cell Biol. 2014;16:942–950. doi: 10.1038/ncb3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshi I., Nisa M.U., Farooq U., Gillani S.Q., Bhat S.A., Sarwar Z., Nabi N., Fazili K.M., Andrabi S. AKT regulates mitotic progression of mammalian cells by phosphorylating MASTL, leading to protein phosphatase 2A inactivation. Mol. Cell Biol. 2020;40:e00366-18. doi: 10.1128/MCB.00366-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S., McCloy R.A., Parker B.L., Gallego-Ortega D., Law A.M.K., Chin V.T., Conway J.R.W., Fey D., Millar E.K.A., O’Toole S., et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene. 2018;37:4518–4533. doi: 10.1038/s41388-018-0295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C., Eaton E.N., Li S.J., Chaffer C.L., Reinhardt F., Kah K.-J., Bell G., Guo W., Rubin J., Richardson A.L., Weinberg R.A. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöler H.R., Balling R., Hatzopoulos A.K., Suzuki N., Gruss P. Octamer binding proteins confer transcriptional activity in early mouse embryogenesis. EMBO J. 1989;8:2551–2557. doi: 10.1002/j.1460-2075.1989.tb08393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin L., Kato S., Franovic A., Camargo M.F., Lesperance J., Elliott K.C., Yebra M., Mielgo A., Lowy A.M., Husain H., et al. An integrin β3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014;16:457–468. doi: 10.1038/ncb2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.M., Reynolds D., Cliff T., Ohtsuka S., Mattheyses A.L., Sun Y., Menendez L., Kulik M., Dalton S. Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell. 2012;10:312–326. doi: 10.1016/j.stem.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskinen M.E., Närvä E., Conway J.R.W., Hinojosa L.S., Lilla S., Mai A., De Franceschi N., Elo L.L., Grosse R., Zanivan S., et al. MASTL promotes cell contractility and motility through kinase-independent signaling. J. Cell Biol. 2020;219:e201906204. doi: 10.1083/jcb.201906204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A., Tang W., Bruscia E.M., Zhang P.-X., Lin A., Gaines P., Wu D., Halene S. SRF is required for neutrophil migration in response to inflammation. Blood. 2014;123:3027–3036. doi: 10.1182/blood-2013-06-507582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium The universal protein resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppada S.B., Gowrikumar S., Ahmad R., Kumar B., Szeglin B., Chen X., Smith J.J., Batra S.K., Singh A.B., Dhawan P. MASTL induces Colon Cancer progression and Chemoresistance by promoting Wnt/β-catenin signaling. Mol. Cancer. 2018;17:111. doi: 10.1186/s12943-018-0848-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant F., Asselin-Labat M.-L., Shackleton M., Forrest N.C., Lindeman G.J., Visvader J.E. The mammary progenitor marker CD61/β3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008;68:7711–7717. doi: 10.1158/0008-5472.CAN-08-1949. [DOI] [PubMed] [Google Scholar]
- Vázquez-Arreguín K., Tantin D. The Oct1 transcription factor and epithelial malignancies: old protein learns new tricks. Biochim. Biophys. Acta. 2016;1859:792–804. doi: 10.1016/j.bbagrm.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera J., Lartigue L., Vigneron S., Gadea G., Gire V., Del Rio M., Soubeyran I., Chibon F., Lorca T., Castro A. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. Elife. 2015;4:e10115. doi: 10.7554/eLife.10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S., Robert P., Hached K., Sundermann L., Charrasse S., Labbé J.-C., Castro A., Lorca T. The master Greatwall kinase, a critical regulator of mitosis and meiosis. Int. J. Dev. Biol. 2016;60:245–254. doi: 10.1387/ijdb.160155tl. [DOI] [PubMed] [Google Scholar]
- Wang L., Luong V.Q., Giannini P.J., Peng A. Mastl kinase, a promising therapeutic target, promotes cancer recurrence. Oncotarget. 2014;5:11479–11489. doi: 10.18632/oncotarget.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R.-H., Sampsell-Barron T.L., Gu F., Root S., Peck R.M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R., Thomson J.A. NANOG is a direct target of TGFβ/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008;3:196–206. doi: 10.1016/j.stem.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y.N., Choe M.H., Jung K.-Y., Hwang S.-G., Oh J.S., Kim J.-S. MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer. 2018;18:716. doi: 10.1186/s12885-018-4600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.S.L., Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–3060. doi: 10.1242/dev.137075. [DOI] [PubMed] [Google Scholar]
- Zhang M., Wu J., Mao K., Deng H., Yang Y., Zhou E., Liu J. Role of transforming growth factor-β1 in triple negative breast cancer patients. Int. J. Surg. 2017;45:72–76. doi: 10.1016/j.ijsu.2017.07.080. [DOI] [PubMed] [Google Scholar]
- Zhang Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C., Kong Z., Wang B., Cheng W., Wu A., Meng X. ITGB3/CD61: a hub modulator and target in the tumor microenvironment. Am. J. Transl. Res. 2019;11:7195–7208. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed list of proteins detected with Stable Isotope Labeling with Amino acids in Cell culture (SILAC)-based proteomics of surface proteins (surfaceome) in heavy and light isotope labeled siControl- and siMASTL-treated cells. Protein IDs, gene names, unique peptides, sequence coverage, intensity, H/L ratio, and Significance B of all identified proteins (sheet 1) and significant hits (sheet 2).
Data Availability Statement
-
•
Surface proteomics data have been deposited at the Proteomics Identifications database (PRIDE) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.