

Abstract
Atherosclerosis, the underlying cause of cardiovascular disease (CVD), is a worldwide cause of morbidity and mortality. Reducing ApoB-containing lipoproteins – chiefly, LDL – has been the main strategy for reducing CVD risk. Although supported by large randomized clinical trials, the persistence of residual cardiovascular risk after effective LDL reduction has sparked an intense search for other novel CVD biomarkers and therapeutic targets. Recently, Lox1, an innate immune scavenger receptor, has emerged as a promising target for early diagnosis and CV risk prediction and is also being considered as a treatment target. Lox1 was first described as a 50kDa transmembrane protein in endothelial cells responsible for oxLDL recognition, triggering downstream pathways that intensify atherosclerosis via endothelial dysfunction, oxLDL uptake and apoptosis. Lox1 is also expressed in platelets, where it enhances platelet activation, adhesion to endothelial cells, and ADP-mediated aggregation, thereby favoring thrombus formation. Lox1 was also identified in cardiomyocytes, where it was implicated in the development of cardiac fibrosis and myocyte apoptosis, the main determinants of cardiac recovery following an ischemic insult. Together, these findings have revealed that Lox1 is implicated in all the main steps of atherosclerosis and has encouraged the development of immunoassays for measurement of serum levels of soluble Lox1 (sLox1) to be used as a potential CVD biomarker. Finally, the recent development of synthetic Lox1 inhibitors and neutralizing antibodies with promising results in animal models has made Lox1 a target for drug development. In this review we discuss the main findings regarding the role of Lox1 in the development, diagnosis, and therapeutic strategies for CVD prevention and treatment.
Keywords: Cardiovascular disease, Lox1, atherosclerosis, acute coronary syndrome, myocardial infarction
1. INTRODUCTION
Cardiovascular disease (CVD), chiefly myocardial infarction (MI), is the main cause of mortality worldwide, with an estimated 17 million deaths every year (1, 2). Most often, MI results from occlusion of coronary arteries by thrombi generated over ruptured atherosclerotic plaques (3). Targeting atherosclerosis with lipid-lowering treatments, mainly statins, remains the leading strategy for CVD prevention. Reduction of ApoB-containing lipoproteins results in clinically meaningful reductions in the occurrence of cardiovascular events, as shown in multiple large randomized clinical trials (4). Despite these impressive benefits of lipid lowering therapies, 50–70% of patients who have met their lipid lowering targets remain at high CVD risk. Understanding the drivers for this, so called, “residual CVD risk” is a current focus of intensive research aiming to identify novel CVD biomarkers and molecular targets for intervention (5).
Lectin-type oxidized LDL receptor 1 (Lox1) has recently emerged as a promising biomarker and target for intervention in CVD (6). Lox-1 was first described as a membrane receptor of oxidized LDL (oxLDL) in endothelial cells. Subsequent work showed that Lox1 is also expressed by platelets, cardiomyocytes, macrophages and fibroblasts, with major repercussions for the progression of atherosclerotic disease (7–10) (Figure 1). Growing evidence suggest that Lox1 is involved in all the main steps in the pathogenesis of atherosclerosis. Activation of Lox1 favors oxLDL transcytosis and its retention in subendothelial layer where it may trigger a wide variety of pro-atherogenic events, such as increased foam cell formation, thinning of atherosclerotic plaque fibrous cap, enhanced platelet adhesion/aggregation, thrombus stabilization, endothelial dysfunction and premature senescence. Moreover, following myocardial ischemic insult, expression of Lox1 is markedly amplified, culminating in increased local inflammatory response, cardiomyocyte apoptosis, myocardial fibrosis and decreased myocardial functional recovery (8, 11).
Figure 1.
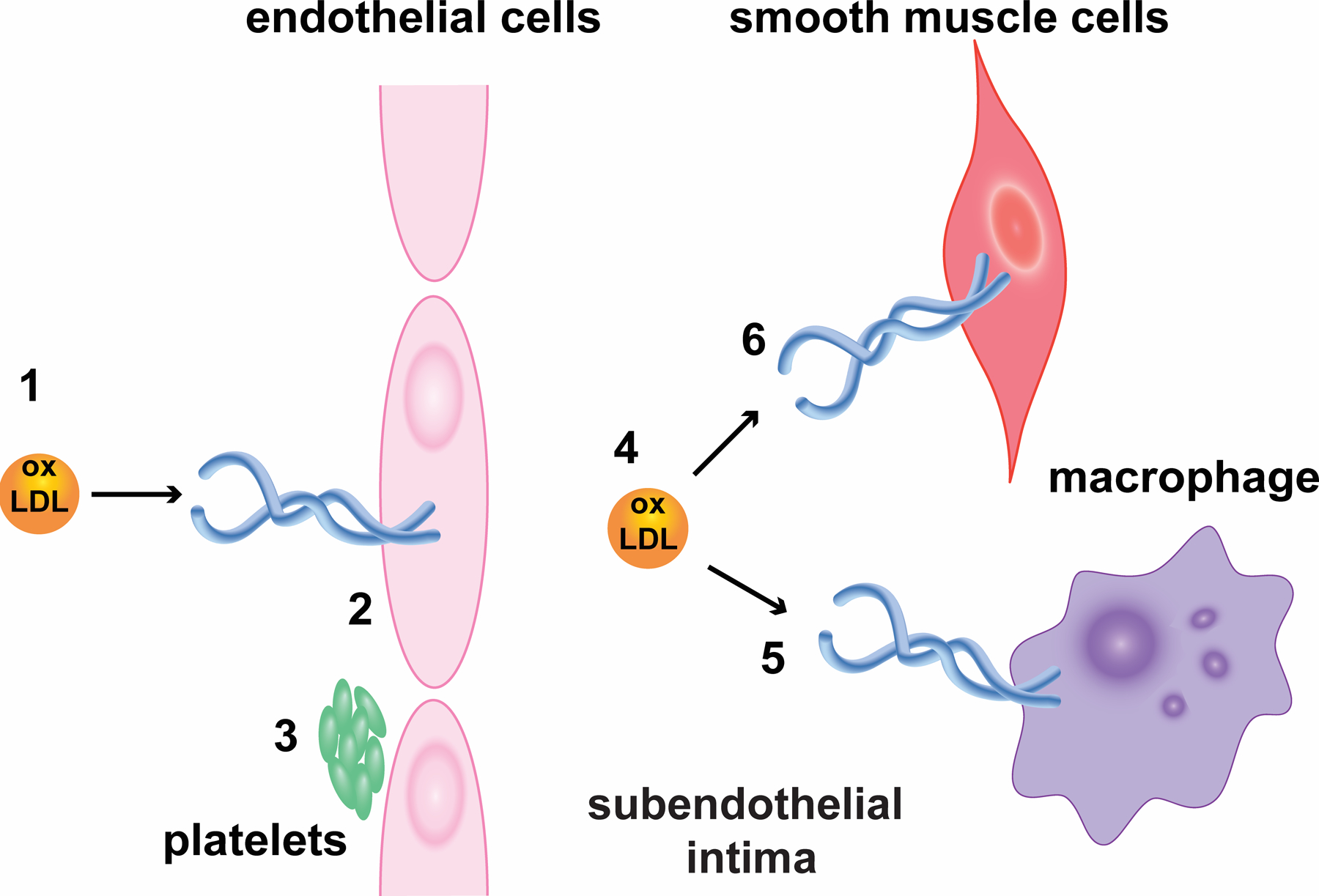
Lox1 sites. The figure shows the expression of Lox1 in different cell types and its activation by oxLDL (1, 4). In endothelial cells (2), activation of Lox1 upregulates the formation of membrane invaginations that promote engulfment of oxLDL and its carriage in sealed vesicles through cytosol for discharge in subendothelial layer. In parallel, oxLDL/Lox1 stimulates the expression of adhesion molecules fueling leukocyte recruitment to atherosclerotic plaque. Furthermore, Lox1 facilitates activation of b-galactosidase and cH2AX, which accelerates the premature senescence of endothelial cells and endothelial dysfunction. Moreover, active Lox1 intensifies platelet (3) adhesion to endothelium, favoring platelet release of growth factors that fuel the progression of atherosclerotic plaque. Besides, Lox1 upregulates platelet activation and ADP-mediated aggregation, both required for occlusive thrombus formation and stability. In macrophages (5), under inflammatory stimuli, Lox1 expression is greatly increased and participates in macrophage lipid engorgement and foam cells formation. Likewise, Lox1 interferes with macrophage calcium metabolisms, supporting its accumulation in atherosclerotic lesions. Finally, Lox1 are expressed in smooth muscle cells (6), where it stimulates metalloproteinases and proapoptotic pathway activation, hence favoring fibrous cap thinning, which turns atherosclerotic plaques vulnerable.
Quantification of serum levels of soluble Lox1 (sLox1), the proteolytic degradation extracellular domain of cell bound Lox1 (Fig 2), has emerged as a promising candidate for earlier diagnosis and risk estimate of CVD. Plasma sLox1 levels are promptly elevated among acute coronary syndrome (ACS) and cerebrovascular disease patients, and higher sLox1 was associated with increased incidence of CVD in recent observational studies (12–15). Furthermore, experimentally treatment with synthetic Lox1 inhibitors improves cardiac recovery and reduces infarct size in animal models, placing Lox1 as a promising therapeutic target for CVD (7). In this review we will first discuss the biochemistry and genetics of Lox-1 and then go on to describe its tissue expression and biological role in different cell types. Finally, we will discuss how Lox-1 relates to the pathogenesis of atherosclerosis and the early studies on its use as a biomarker.
Figure 2:
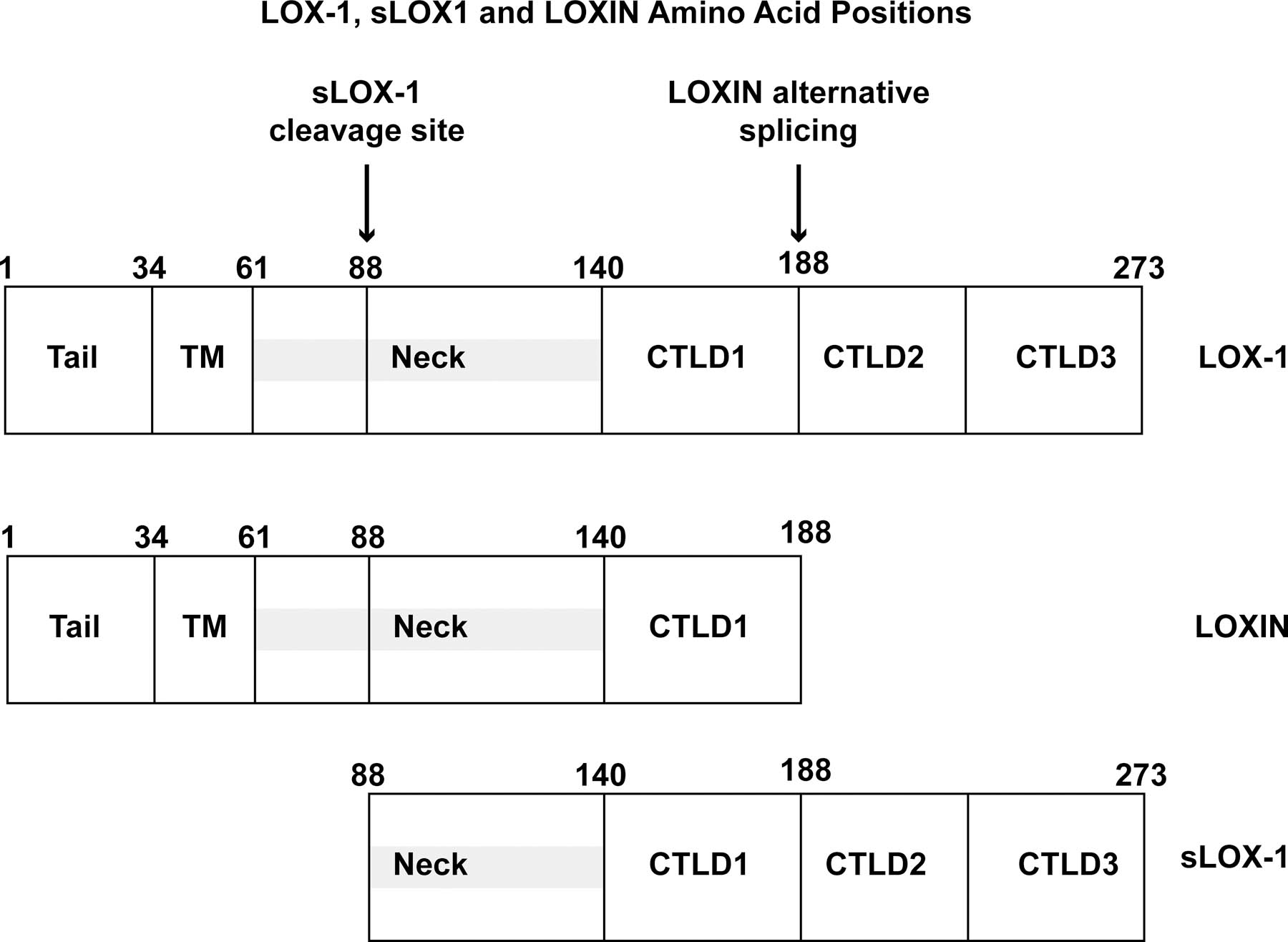
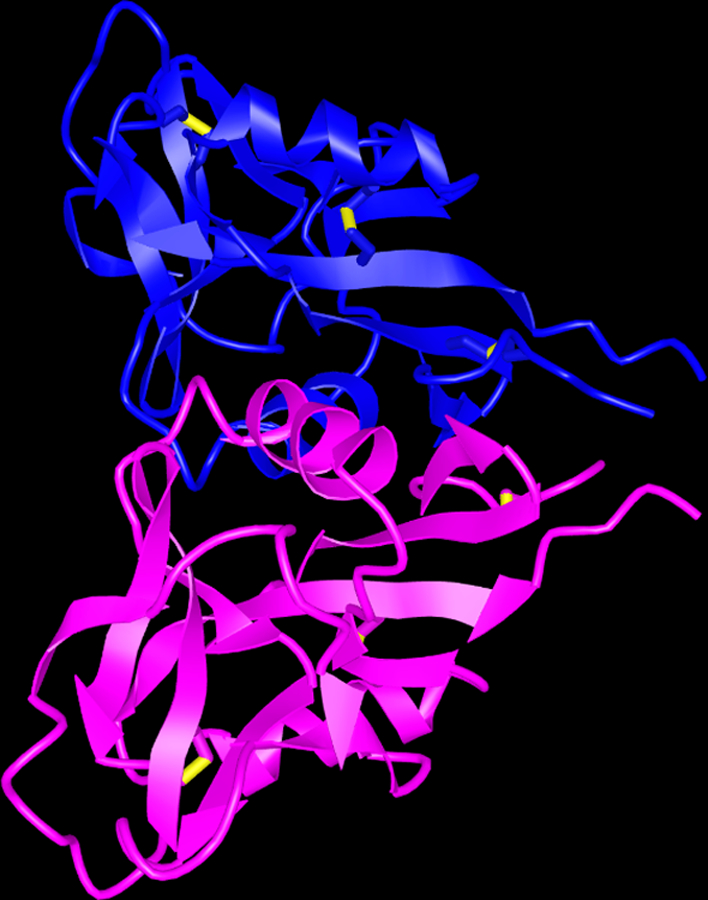
Structure of human Lox1. (A) Diagram of major structural domains of Lox1 and its various cleavage forms. (B). Crystal structure of extracellular domain of Lox1 dimer that forms central hydrophobic tunnel. (22)
1.1. Lox1 overview
Lox1 was first described in 1997 as a 50kDa membrane scavenger receptor involved in internalization of oxLDL by endothelial cells (16). Since then, Lox1 has been localized to endothelial cells, macrophages, platelets, cardiomyocyte and smooth muscle cells (11). Accumulating evidence supports its involvement not only in oxLDL internalization, but also in endothelial dysfunction, atherosclerosis, plaque instability, thrombogenesis and innate immune responses (17). These effects are mediated by the activation of intracellular downstream pathways involving NFkB, MAPK and caspases (17–20). Proteolytic cleavage of its extracellular domain releases sLox-1 in the circulation which can be accurately qualified with recently established and validated assays (21). Because plasma sLox1 levels are thought to reflect cell bound active Lox1, sLox1 is a compelling biomarker for improving cardiovascular risk stratification and timely detection of thromboembolic events.
1.1.1. Lox1 protein structure
Lox1 is a 50kDa Type II transmembrane protein comprised of 273 amino acid chain that structurally belongs to C-type lectin superfamily (16). Lox1 exists as a homodimer that contains a cytoplasmic short N-terminal domain bound to a single hydrophobic transmembrane domain (22, 23). The extracellular region comprises an 80-residue extracellular coiled coil domain (NECK), which is attached to the C-terminal lectin like domain (CTLD) (22). CTLD is a heart-shaped 130-residue coiled structure formed by antiparallel β-sheets flanked by α-helices and stabilized by 3 intrachain disulfide bonds. Importantly, CTLD monomers are linked by 6 highly conserved cysteine residues forming an interchain disulfide bond (23), which promotes the formation of a central hydrophobic tunnel that spans the entire protein (22). The sides of this tunnel are flanked by nonpolar amino acids like tyrosine, thus allowing the tunnel to transfer lipids (22).
The CTLD region of Lox1 is involved in ligand recognition (24). This feature is attributed to a CTLD terminal cluster of positively charged amino acid (termed “basic spine”) involved in binding of negatively charged ligands, such as oxLDL (25). Deletion of CTLD completely abolishes oxLDL binding activity (24). Moreover, deletion or sequential substitution of CTLD basic spine residues by alanine residues abrogates oxLDL binding, supporting that this terminal chain is required for ligand-binding activity (24, 26). Noteworthily, point mutations replacing terminal uncharged by non-charged residues completely abrogates oxLDL binding activity (26). This finding strongly suggests that positively charged residues in CTLD are required for binding of negatively charged oxLDL. Moreover, dynamic structural modeling suggests that replacement of basic spine arginine residues by alanine alters CTLD conformation compromising its binding affinity (27). Overall, these results support that CTLD-mediated ligand-binding involves positively charged C-terminal residues that bind to a variety of negatively charged ligands including oxLDL.
Another functionally important domain of Lox1 is the NECK domain. NECK was first described as an 80-residue α-helical coiled coil structure linked proximately to transmembrane domain and, distally, to CTLD by interchain disulfide bond (28). Importantly, the proximal third of NECK is structurally less stable than the remaining domain and has been identified as the target for proteases responsible for Lox1 juxtamembrane cleavage between Arg88 and Gln89 residues and release of its 34kDa soluble forms (sLox1) into the bloodstream (28, 29). NECK domain cleavage and sLox1 release is mediated, at least in part, by interleukin 18 (IL18) in an ADAM10-dependent fashion (21). Moreover, mutation of the C140 residue of CTDL abrogated C-terminal NECK domain stability, indicating that CTLD integrity is required for the maintenance of coiled-coil NECK structure (28). Of note, NECK is not involved in ligand-binding activity, as deletion of NECK domain had no impact on oxLDL binding affinity (24). Taken together, these results indicate that the NECK domain has a proximal, N-terminal portion, involved in the release of sLox1, and distal portion involved in interactions with the CTLD that increase its stability.
1.1.2. Lox1 Gene
Lox1 is encoded by the single-copy gene ORL1 (OMIM#602601) located in a C-type lectin gene cluster in the p12.3-p13 region of chromosome 12. ORL1 is over 7000bp nucleotides long and contains 6 exons and 5 introns. Each domain is encoded by an exon: Exon 1 encodes 5`-untranslated region (5ÙTR) and cytoplasmic domain, exon 2 encodes cytoplasmic and transmembrane domain, exon 3 encodes the NECK domain, and exons 4–6 encode CTLD and 3ÙTR. Recent findings suggest that ORL1 gene can produce a wide range of protein isoforms dynamically affected by alternative splicing and single nucleotide polymorphisms (SNP) (Figure 2). Expression of these isoforms is regulated by environmental factors, interleukins, transcription factors, and chromatin modulators. Altogether, these factors modulate the expression of specific Lox1 phenotypes with differing effects on atherosclerosis and CVD (30).
More specifically, alternative splicing of ORL1 yields three LOX-1 mRNA variants: transcript variant 1 (NM_002543), transcript variant 2 (NM001172632) and transcript variant 3 (NM_001172633) (30). Transcript variant 1 contains all 6 exons and leads to the transcription of full-length Lox1 with full oxLDL-binding activity. In contrast, transcript variant 3 lacks exon 5, hence forming an interrupted CTLD-containing protein known as Loxin (30). As discussed above, CTLD integrity is required for ligand-binding (24–27). Henceforth, Loxin is unable to bind oxLDL. Further, it has been reported that Loxin dimerizes with full length Lox1 inhibiting its ligand binding activity (31). It has been sown that this dominant negative action of Loxin suppresses oxLDL/Lox1 signaling, oxLDL-induced positive feedback on Lox1 expression, and downstream proatherogenic processes (30). For example, transfection of human endothelial cells with constructs overexpressing Loxin lead to decreased oxLDL-induced apoptosis (32). Finally, the Loxin allele is relatively abundant in the population (20–30%) and several studies have shown that the presence of this allele is associated with reduced CVD risk (33).
These data suggest that modulation of alternative splicing favoring Loxin over Lox1 expression could be protective. Although this is a promising hypothesis, it lacks confirmation from large, randomized clinical trials, although recent observational studies have reinforced the association of SNPs to ACVD (6, 34–39). Chen et al. found that risk SNPs were independently related to coronary artery disease (CAD), documented as coronary stenosis on angiography in a sub-analysis of the Women’s Ischemia Syndrome Evaluation (WISE) study (6). Furthermore, Guo et al. showed in a case-control study from a Chinese population, that the frequency of risk SNPs is greater among atherosclerotic cerebral infarction subjects, when compared to healthy controls (34). In a similar approach, Tatsuguchi et al. found a 2.8-fold higher chance of presenting risk SNPs among MI individuals in comparison to healthy controls (35). Likewise, Predazzi et al. found that the occurrence of a risk SNP (rs11053646) is related to increased carotid intima media thickness (cIMT) in a large cohort of asymptomatic male individuals, suggesting a role of this polymorphism in the initiating events of ACVD (36). Finally, seminal work by Xu et al. found an association between expression of risk SNPs and progression of left ventricle hypertrophy in hypertensive subjects (37).
1.1.3. Modulation of Lox1 Synthesis
Lox1 synthesis is primed by activation of the ORL1 promoter by transcription factors, chiefly nuclear factor kappa beta (NFkB). Under normal physiological conditions NFkB is present in its inactive form bound to IkB in the cytosol. When oxLDL binds to Lox1, it triggers a sequence of downstream events culminating in release of NFkB from its inhibitor and migration of NFkB to nucleus where it binds to the ORL1 promoter enhancing its transcription. Indeed, exposure of endothelial cells to oxLDL increases the expression of Lox1 in a dose-dependent manner that can be blocked by NFkB inhibitors (40). Moreover, in cultured endothelial cells addition of anti-Lox1 monoclonal antibodies suppresses NFkB activity and expression of pro-inflammatory cytokines and vascular adhesion molecules (10, 19).
Recent evidence also supports that Lox1 synthesis is modulated by statins. Pretreatment of endothelial cells with statins impaired Lox1 expression in a dose- and time-dependent manner (41). Initially it had been assumed that this effect was due to the well-established lipid lowering effect of statins that could impair oxLDL uptake and endothelial dysfunction, abrogating oxLDL amplification of Lox1 expression. However, further experiments demonstrated that statins depletion of cell membrane cholesterol disrupts caveolae and destabilizes Lox-1.(115) In addition, statins bind to Lox1 hydrophobic tunnel impairing Lox1 binding activity and, consequently, the oxLDL/Lox1 axis downstream effects (42, 43). Furthermore, treatment of endothelial cells with statins markedly abrogates oxLDL-induced suppression of eNOS, a key enzyme for endothelial NO production important for endothelial function and atheroprotection (44).
Angiotensin II (AngII) has also been implicated in Lox1 activation. Treatment of endothelial cells with Ang II induces Lox1 expression in a time- and dose-dependent manner (45). Inhibition of NFkB and preincubation with angiotensin receptor blocker Losartan nullifies this effect (45). Similar results were also found when endothelial cells were exposed to shear stress, supporting a role of disturbed blood flow on Lox1 mechanotransduction (46). Although far less explored, a role for other commonly used medications on Lox1 modulation has also been investigated. For example, treatment of endothelial cells with aspirin inhibits oxLDL-induced production of matrix metalloproteinases and suppresses the positive feedback on Lox1 expression (47). Preincubation of endothelial cells with calcium channel blocker nifedipine abrogates oxLDL-induced apoptosis (48). Similarly, treatment of endothelial cells with metformin reduces expression of Lox1, likely due to reduction in ROS-NFkB activation of ORL1 promoter and reduction in advanced glycated end-products (AGE) that are stimuli for Lox1 synthesis (49).
1.2. LOX1 signalling
Ligand binding to Lox1 stimulates a variety of downstream signaling events resulting in alterations of cell behavior in a variety of cell types. (113) (Fig. 3). Binding of oxLDL to Lox1 stimulates RhoA-dependent downregulation of endothelial nitric oxide synthetase (eNos) and Rac-mediated activation of NADPH and production of reactive oxygen species (ROS). Lox1 activation increases phosphorylation of p66shc resulting in increased oxidative stress and ROS production. Superoxide is a potent scavenger of nitric oxide (NO) which is required for endothelium-dependent NO-mediated vasodilation. Further, activated Lox1 stimulates stress activated janus kinase (JNK)-dependent activation of arginase-I which limits L-arginine, the substate for NO production, further reducing NO availability. (114) Lox1 activation also stimulates inflammatory pathways by simulating the activities of NFkB, AP1, and the NLR family, pyrin domain-contain 3 (NLRP3) resulting in increased IL-1β production and increased inflammatory response which, in turn, stimulates Lox1 expression leading to further amplification of Lox1 signaling. Additional signaling pathways induced by Lox1 activation include MAPK, protein kinase C, octamer-binding protein-1, and PI3K/Akt. Below we discuss how these signaling cascades influence the behavior of cells in the cardiovascular system and may contribute to disease.
Figure 3.
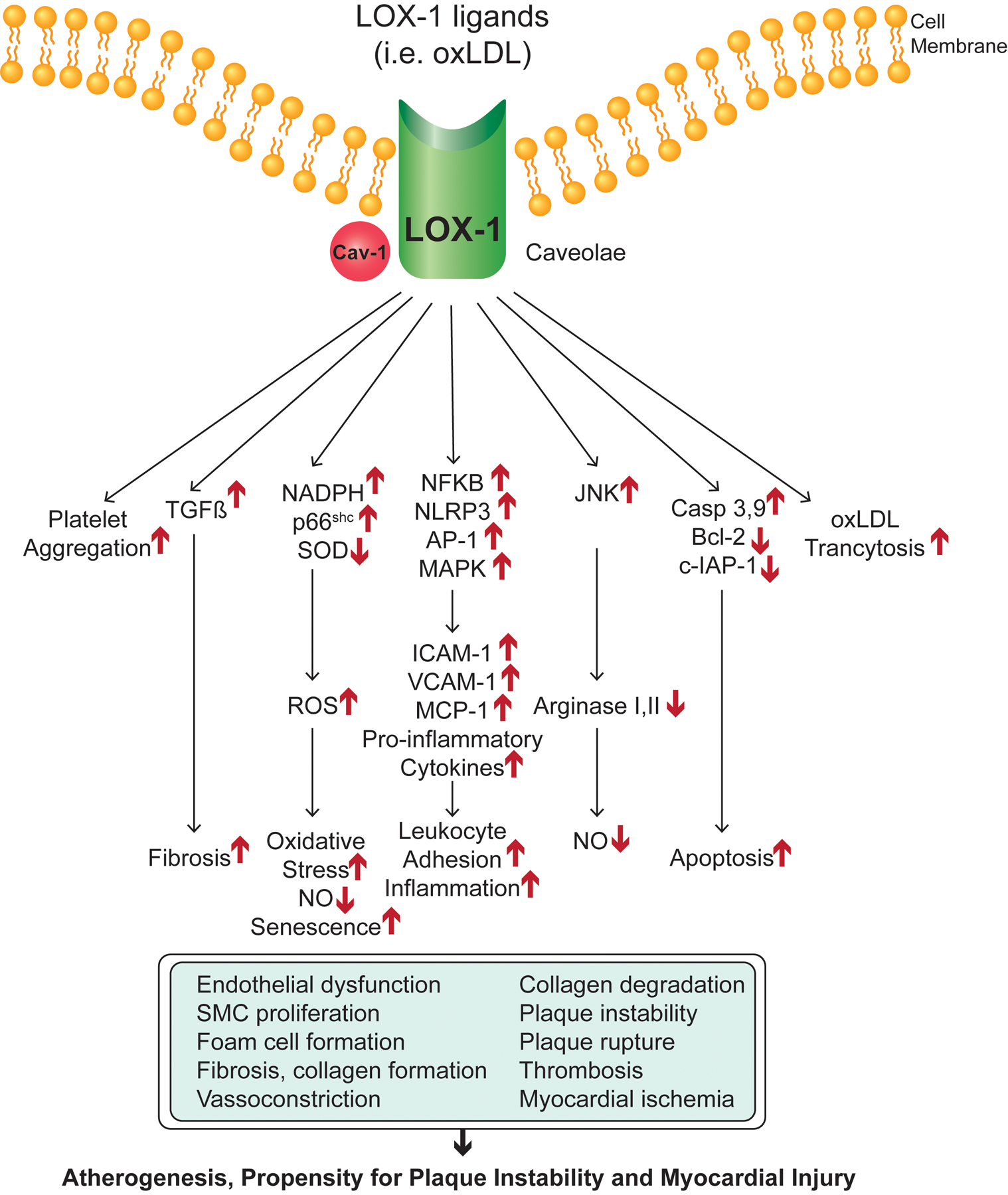
Lox1 signaling pathways. Lox1 representation at the cell membrane, main downstream pathways triggered by activated Lox1 and alterations in biological process and cell behaviors as they relate to CVD.
1.3. LOX1 in Atherosclerosis
Atherosclerosis is an indolent and progressive disease of mid- and large arteries, characterized by the accumulation of fatty material in the vascular intima (50). A prime event in atherosclerosis is subendothelial retention of apoB-containing lipoproteins, chiefly low-density lipoprotein (LDL), a spherical particle formed by ApoB100, phospholipids, free cholesterol and water-insoluble cholesteryl-esters (51). It is now unequivocally demonstrated that elevated plasma LDL is a key risk factor and main driver of atherosclerosis (52). This was first established in Familial Hypercholesterolemia patients with homozygous mutations in the LDL receptor (LDLR), leading to serum levels of LDL as high as 300–600mg/dL and myocardial infarction in early adulthood if not earlier (53). Statins upregulate the expression of hepatic LDLR by blocking endogenous cholesterol synthesis, which accounts for their ability to reduce CV events (54). In fact, for each 1mmol/L (~40mg/dL) reduction in plasma LDL cholesterol (LDL-c), there is an approximate 20% reduction in the incidence of CV events (4, 55, 56). Similarly, PCSK9 inhibitors are believed to reduce CV evens by upregulating LDL receptors and lowering LDL-c (57, 58).
1.3.1. Endothelial cells
Endothelial cells play a primary role in the transport of LDL particles from vascular lumen to the subendothelial intima (59, 60). The precise molecular basis of this event remains under intense debate. Initially, it was suggested that the transport of LDL through endothelial barrier was mediated by LDLR. However, subsequent studies showed significant LDL subendothelial retention even in LDLR−/− models and clinical conditions with suppressed LDLR expression suggesting that receptors other than LDLR are involved in LDL trans-endothelial transport (61, 62). Current knowledge is that LDLR function primarily involves unmodified LDL endocytosis for lysosomal hydrolysis and further provision of cellular cholesterol demand in a tightly regulated manner (63). An alternative hypothesis was that LDL crosses EC barrier through intercellular junctions (paracellular transport). However, this seems unlikely as LDL particle diameters (~20nm) are larger than the usual diameter of intercellular gap junction of 2–3nm (64). Currently, the most widely accepted hypothesis is that LDL is carried through endothelial cells in specialized vesicles and delivered to subendothelial space by exocytosis (65–68). This involves an initial step that is directly influenced by the oxLDL/Lox1 axis: transcytosis.
Transcytosis refers to the internalization of molecules by endothelial cells, their carriage through cytosol in sealed vesicles, and their discharge in the subendothelial space (69). The initial step of transcytosis is the formation of caveolae. Caveolae are 50–100nm membrane invaginations, modelled by its main structural protein, caveolin-1 (70). Besides caveolin-1, caveolae comprises a variety of scavenger receptors, such as CD36 (71), SR-BI (72, 73) and Lox1 (74), that permit endocytosis of macromolecules like insulin (75), albumin (76) and LDL (68). After particle engulfment, the newly formed vesicle is sealed by membrane fission, which is controlled by GTPase protein dynamin (77). The resultant free vesicle traffics through transendothelial channels along microtubules driven by kinesin and dynein ATPase to the basolateral membrane (78–80). Finally, the vesicle coat undergoes fusion with basolateral membrane in a process tightly regulated by soluble NSF attachment protein receptor (SNARE), thus releasing its content to the subendothelial space (81).
Accumulating evidence supports the key role of caveolae formation in atherosclerosis. Overexpression of caveolin1 leads to increased atherosclerotic lesion and leukocyte adhesion molecule expression (65). Similarly, genetic ablation of caveolae increases serum levels of LDL in ApoE−/− mice, indicating impaired lipoprotein uptake from bloodstream (66). While native LDL uptake by caveolae is mediated by SR-BI (72) and CD36 (71), it was only recently shown that oxLDL transcytosis involves binding to Lox1. Kumano et al. for the first time demonstrated that palmitoylation of Lox1 cysteine residues triggers Lox1 recruitment to caveolae where it is colocalized and presents its oxLDL ligand to newly formed vesicles (74). Besides, other experiments demonstrated that oxLDL/Lox1 axis reciprocally potentiates caveolin1 expression, suggesting a positive feedback mechanism that may perpetuate oxLDL transcytosis (68).
Recruitment of monocytes to the endothelial layer through monocyte adhesion molecules is a hallmark of proatherogenic foam cells formation (82). It has been suggested that Lox1 promotes monocyte adhesion to endothelial cells. Li et al. demonstrated that incubation of endothelial cells with oxLDL upregulates the expression of monocyte chemoattractant protein 1 (MCP1) and monocyte adhesion to cell surface while preincubation with antisense Lox1 abolished this effect (19). Similarly, Akhmedov et al. verified that overexpression of LDL in ApoE−/− mice leads to increased expression of VCAM1 and E-selectin, that occurs secondarily to activating phosphorylation of the p65 subunit of NFkB (10). The same study revealed increased synthesis of reactive oxygen species and impaired endothelial function by Lox1 overexpression. Moreover, oxLDL-mediated increase in VCAM1 synthesis was suppressed by inhibition of caveolin1 (68). These results support that oxLDL upregulates the expression of monocyte adhesion molecules by endothelial cells in a caveolae- and Lox1-dependent manner and that Lox1 plays a pivotal role in initiating atherosclerosis as a potent effector of oxLDL transmigration.
A recent novel finding is that Lox1 mediates oxLDL -induced endothelial cell dysfunction and premature senescence, a non-proliferative cell state associated with secretion of a variety of pro-inflammatory factors (i.e. the senescence associated secretory phenotype, SASP) and paracrine senescence propagation to nearby cells (i.e. senescence spreading) (83). Pretreatment of mice with LDL increased senescence-associated nuclear cH2AX deposition and expression of the classic senescence biomarker, b-galactosidase (84). This finding was reproduced in vitro, as exposure of cultured endothelial cells to LDL amplified mitochondrial free-radical release, telomerase activity and was related to endothelial cells dysfunction (84). Abrogation of Lox1 in KO models fully abolished this effect, supporting this additional promising mechanism as a potential therapeutic target in atherosclerosis.
1.3.2. Macrophages
Once retained in subendothelial layer, oxLDL is entrapped in the extracellular matrix and is susceptible to engulfment by macrophages forming lipid-engorged foam cells that prime atherosclerotic plaques (51). Macrophages from vascular intima are converted to cholesterol-rich foam cells by uptake of retained LDL by 3 main mechanisms: LDLR, receptor-independent fluid-phase endocytosis, and scavenger receptors, including Lox1 (82). Although LDLR is ubiquitously expressed in cell surfaces and is responsible for LDL internalization when intracellular cholesterol levels are below physiological demand, foam cells are formed in LDLR−/− models indicating that cholesterol storage in macrophages are more likely driven by LDLR-independent mechanisms (85). Accordingly, Kruth et al. demonstrated that macrophages perform fluid-phase endocytosis by cell membrane fusion with macropinocytotic vacuoles carried with fatty material and LDL particles from vascular intima (86, 87). In contrast to LDLR mechanisms, fluid-phase endocytosis is not limited by intracellular cholesterol levels and uptake of LDL is directly proportional to LDL concentration in the subendothelium.
Normally, the aforementioned mechanisms are responsible for most of the LDL engulfment by macrophages, while Lox1 is responsible for only 5% of oxLDL uptake. In fact, Lox1 is only scarcely expressed in quiescent macrophage but, after exposure to inflammatory stimuli and hyperglycemia, Lox1 expression is dramatically upregulated promoting cellular lipid accumulation and macrophage activation (88). Importantly, Lox1 activation increases NRLP3 inflammasome activation, mtDNA damage and ROS production in active macrophages (89). Likewise, activated Lox1 inhibits calpain1 and increased Ca+2 levels, impairing macrophage migration, contributing to macrophage retention in atherosclerotic lesions (90, 91).
1.3.3. Smooth muscle cells
Smooth muscle cells (SMC) are responsible for deposition of extracellular matrix, mainly formed by collagens, proteoglycans and elastin. These molecules are organized to form a fibrous cap to the lipid core of atherosclerotic lesion. Importantly, extensive lipid cores covered by thin fibrous cap are more prone to rupture (vulnerable plaques), which is the prime event of platelet aggregation and occlusion of coronary artery, the main cause of MI (50).
A growing body of evidence suggest that activation of SMC Lox1 promotes atherosclerotic plaque instability. Exposure of SMCs to oxLDL triggers time- and dose-dependent upregulation of Lox1 expression driven by activation of NFkB and JNK (92). Exposure of cultured SMC to oxLDL, but not native LDL, triggers pro-apoptotic pathways involving Bcl2 and Bax proteins in a dose-dependent manner (93). This effect is neutralized by pretreatment with Lox1 inhibitors, showing that activation of oxLDL/Lox1 axis causes SMC apoptosis (93). Further in vivo experiments with hypercholesterolemic rabbits showed that expression of Lox1 is enhanced in unstable atherosclerotic plaques and colocalizes with metalloproteinase 9 (MMP9) and monocyte chemoattractant protein (MCP1), both of which have been implicated in plaque instability (94). The involvement of Lox1 in plaque destabilization was further reinforced by recent experiments showing that ApoE−/− mice treated with Lox1-siRNA have significantly thicker fibrous caps and reduced macrophage content atherosclerotic plaques compared to control ApoE−/− mice (91). This finding unequivocally demonstrated that activation of Lox1 by oxLDL promotes atherosclerotic plaque instability and plaque rupture.
Beyond its involvement in fibrosis and plaque rupture, recent evidence suggest that Lox1 may also be involved in vascular calcification and arterial stiffness, a common finding in human atherosclerotic lesions suggesting that that osteo and chondrocytic transformation of SMCs and their dedifferentiation from a contractile into a proliferative phenotype are important for the initiation and progression of atherosclerosis (95). Interestingly, it was recently shown that a form of dysfunctional HDL, oxidized HDL (oxHDL), which is recognized and functions as a Lox1 ligand, induces SMC calcification and proliferation (96). These findings raise the possibility that Lox1 blockade may not only suppress atherosclerotic plaque vulnerability, but it may also delay or suppress vascular calcification and improve vascular compliance in disease and aging.
1.3.4. Platelets
Coronary artery occlusion due to thrombus formation over ruptured atherosclerotic plaques remains the main cause ischemia and MI (97). Both the platelet-endothelium adhesion and platelet aggregation that drive occlusive thrombus formation are influenced by Lox1.
Besides their role in thrombus formation, platelets also contribute to progression of atherosclerosis via production of a variety of growth factors that promote atherosclerotic plaque growth (98). Platelets adhere to endothelium, endocytose, and migrate to subendothelial space where they release pro-atherogenic growth factors (99). Very recently, it was demonstrated that expression of Lox1 by endothelial cells is involved in platelet-endothelial adhesion. For example, pretreatment of endothelial cells with Lox1 inhibitors reduces platelet adhesion by half, and significantly abrogates activation of proatherogenic endothelial dysfunction downstream pathways (9). Further, in vivo inhibition of Lox1 diminishes the exposure of tissue factor and plasminogen activator inhibitor type 1 (PAI-1), that primes recruitment of platelets to atherosclerotic plaques favoring thrombus formation (8).
The recent demonstration of Lox1 in the surface of platelets motivated intense search for a potential role of Lox1 in platelet aggregation and thrombogenesis. In vitro models demonstrated that Lox1 is stored in platelet granules and presented to cell surface upon platelet activation by external stimuli, such as oxLDL, shear stress and proinflammatory cytokines (11). Because Lox1 recognizes and binds to surrounding active platelets, it was hypothesized that Lox1 could participate in platelet-platelet interaction, promoting aggregation and formation of stable thrombi. This hypothesis was confirmed by Holy et al. who demonstrated that exposure of cultured platelets to modified LDL induced ADP-mediated platelet aggregation, an effect that was inhibited by pretreatment with Lox1 inhibitors (8). Taken together, these data suggest that while Lox1 from endothelial cell favors platelet adhesion and exposure of tissue factor and PAI-1, presentation of Lox1 by active platelets significantly boosts their ADP-mediated aggregation.
1.4. The effects of LOX1 in myocardial ischemia and fibrosis
Expression of Lox1 has been implicated in the inflammatory response and cardiac remodeling following an ischemic insult. Myocardial ischemia enhances expression of Lox1, which, in turn, promotes cardiomyocyte apoptosis, local inflammation, and fibroblast activation, thereafter, favoring myocardial fibrosis and loss of function (101). These effects are at least partially mediated by the MAPK and TGF-ß1 pathways and may be future targets for MI and HF management (18).
Mice subjected to chronic ischemia induced by left coronary artery (LCA) occlusion had a 2-fold increased expression of Lox1 (7). This effect is abolished in Lox1-KO mice which also showed improved myocardial recovery (i.e. increased ejection fraction and attenuated cavity dilation) and reduced final infarct size in comparison to their wild-type counterparts (7). Further, Lox1 activation induced cardiomyocyte apoptosis, an effect partially mediated by the mitogen-activated protein kinase (MAPK) pathway that was fully reverted by Lox1 inhibition (18). Moreover, in ischemia-reperfusion injury (IRI) experiments, inhibition of Lox1 mitigates the activation of pro-apoptotic caspase 3 and lipid peroxidation products, hence leading to decreased local inflammatory response and final infarct size (100).
The role of Lox1 in myocardial fibrosis has also been largely explored. Recent in vitro studies by Villa et al. demonstrated that oxLDL induces differentiation of cardiac fibroblasts into cardiac myofibroblasts, enhancing collagen type I and fibronectin secretion, boosting myocardial fibrosis. These effects were fully abolished in Lox1-KO mice (101). Similarly, in doxorubicin (Dox)-induced cardiomyopathy models, Lox1-KO mice had decreased synthesis of Lox1, TNFα and interleukin 1 beta (IL-1b) in comparison to wild-type mice. Histologically, this resulted in smaller area of fibrosis and attenuated leukocyte myocardial infiltration in the Lox1-KO mice. Furthermore, echocardiographic studies following Dox injection demonstrated preserved systolic and diastolic function in the Lox1-KO, but not wild-type mice (102). Taken together, these data strongly suggest that Lox1 activation by oxLDL contributes to the progression of fibrosis and cardiac dysfunction following an ischemic insult or induced cardiomyopathy and raise the possibility that drugs that block Lox1 may be effective cardioprotective treatments of these diseases.
1.5. Measurement of serum sLox1 as a biomarker of CVD
As previously stated, a growing body of evidence support that Lox1 contributes to the progression of atherosclerosis and may worsen clinical recovery from ischemic events. Although assays for direct measurement of cellular Lox1 levels are not available, validated assays for sLox1, which is thought to reflect cell bound Lox1, are commercially available and are been used to assess their utility in cardiovascular risk estimation to guide preventive medical strategies in the future (12). In line with this, in a recent prospective cohort with 2437 participants followed for 11 years, those in the highest quartile of sLox1 had a 1.7-fold increased chance of stroke and 2-fold higher risk of coronary heart disease in comparison to the lowest quartile (13). This is consistent with several other studies demonstrating a role for sLox1 in predicting the occurrence and severity of a growing number of atherosclerotic CVD. (107)
As an example, the potential utility of sLox1 plasma levels as a biomarker for early stratification of patients with ACS was recently determined. It is widely accepted that, in ACS patients, timely diagnosis is a prerequisite for opportune reperfusion favoring prognosis and myocardial recovery. However, the available biomarkers of myocardial ischemic injury, mainly CK-MB and troponin T, have their peak values after 2–6h after the index event, what could delay identification of ACS in the early hours. In a recent study, sLox1 measurement was compared to CK-MB and Troponin T in regard to diagnostic accuracy in a small cohort of ACS patients (103). Importantly, using a validated cut-off value of sLox1 (91ng/mL), sLox1 detected STEMI with 89.6% sensitivity and 82.4% specificity, and NSTEMI with 79.5% sensitivity and 82.4% specificity (103). sLox1 sensitivity was greater than its counterparts (93% vs 56% vs 33% for sLox1, troponin T and CK-MB, respectively) at hospital admission, but not at 24h from the index event (89% vs 100% vs 93% for sLox1, troponin T and CK-MB, respectively) (103). These findings were corroborated by further studies demonstrating timely elevation of sLox1 in ACS patients, with sLox1 levels being more prominent among STEMI than NSTEMI individuals and reaching peak levels earlier than troponin (103–105). Moreover, in ACS patients, elevated sLox1, but not troponin T nor C-reactive protein (CPR), were related to a higher frequency of ruptured atherosclerotic plaque documented by optical coherence tomography (OCT) imaging catheters (106). These finding support the contention that measurement of sLox1 may be used to add diagnostic accuracy in early diagnosis and characterization of ACS individuals.
The prognostic value of sLox1 in ACS has also been explored. In a prospective cohort of 948 individuals who underwent primary percutaneous intervention (PCI), high sLox1 was significantly related to the occurrence of major cardiovascular events (MACE) in a 2 years follow-up (107). Further analysis demonstrated that among individuals with stable CAD referred to elective PCI, the risk of periprocedural myocardial infarction was higher among those with elevated baseline sLox1 levels compared to low sLox1 group (108). Similarly, in a cohort study of stable coronary disease, incidence of MACE was 6-fold higher among sLox1 highest tertile in comparison to the lowest tertile (109). This result is in accordance with further findings of 1.96-fold higher frequency of complex lesions among stable CAD patients with elevated baseline sLox1 levels (110).
Similar results have been described for detection of acute stroke. In a recent case-control study, sLox1 was greater among individuals presenting with ischemic or hemorrhagic stroke in the past 3 days, in comparison to healthy controls (15). Furthermore, results from a large cohort including 4703 individuals have found that those in the highest tertile of sLox1 had a 75% higher risk of stroke in comparison to the lowest tertile (111). Moreover, analysis of atherosclerotic plaque from endarterectomized individuals has demonstrated a strong correlation between sLox1 and plaque content of Lox1, oxLDL, metalloproteinases, and proinflammatory cytokines, hence providing a possible explanation for the verified relation between sLox1 and stroke occurrence (111). Likewise, a recent case-control study with individuals referred to carotid endarterectomy has found higher sLox1 among individuals with previous acute ischemic stroke or diagnosed severe carotid atherosclerotic disease in comparison to healthy controls (112). Furthermore, in a prospective cohort of acute ischemic stroke individuals, elevated sLox1 were related to a 2-fold increased risk of poorer long-term functional outcome, and to a higher frequency of moderate stroke (NIHSS>5) and worst degree of intracranial artery stenosis (14). A summary of the main findings from these observational studies using sLox1 as biomarker of CVD is presented in Table 1.
Table 1.
Clinical studies using Lox1 as a predictor of CVD
| Groups | ||||||||
|---|---|---|---|---|---|---|---|---|
| Study | Year | Design | Population | n | Experimental | Control | Endpoint | Outcomes |
| Zhao et al (107) | 2019 | Prospective cohort | CAD individuals undergoing PCI | 984 | MACE | No event | sLox1 | sLox1 and occurrence of MACE in 2 years following PCI are positively related [HR of 1.278 (1.019–1.604), p=0.034] |
| Balin et al (108) | 2012 | Case-control | Elective single vessel PCI individuals. | 214 | PCI-RPMI (+) | PCI-RPMI (−) | sLox1 | Circulating levels of sLOX1 were higher among those with PCI-RPMI [(167 ± 89 vs. 99 ± 68 pg/mL; p < 0 0.001)]. |
| Zhao et al (109) | 2019 | Prospective cohort | Stable CAD patients | 833 | sLox1 > 0.91ng/ml | sLox1 < 0.48ng/mL | MACE in 2 years | Highest tertile of sLox1 were related to a 2-fold increased risk of MACE in 2 years (OR 2.07, 95%CI 1.52 – 2.82; P < 0.001) |
| Zhao et al (110) | 2011 | Cross-sectional | CAD individuals undergoing PCI | 180 | Complex lesions | Simple lesions | sLox1 | sLox1 were significantly higher among individuals with complex coronary lesions [0.914 vs 0.426 ng/mL] |
| Markstad (111) | 2019 | Case-control | Participants of the MDC study | 4703 | Lowest sLox1 tertile | Highest tertile sLox1 | Stroke | Highest tertile of sLox1 was related to a 1.75-fold increased risk of stroke compared to lowest tertile |
| Li et al. (14) | 2018 | Prospective cohort | Acute ischemic stroke individuals | 272 | Poor functional recovery | Good functional recovery | sLox1 | sLOX1 level was significantly higher among individuals with poor functional recovery in 1 year following stroke |
| Inoue et al. (13) | 2010 | Case-control | Japan residents with no prior history of stroke nor CHD | 2295 | Top quartile sLOX1 | Lowest sLox1 quartile | Stroke and CHD | Top sLox1 levels quartile had a 1.74-fold and a 2.09-fold increased incidence of stroke and CHD, respectively. |
| Yokota et al. (15) | 2016 | Case-control | Suita study cohort individuals | 377 | Stroke individuals | Age- and sex-matched healthy controls | sLox1 levels | sLox1 levels were significantly higher in the stroke individuals compared to healthy controls |
| Kobayashi (106) | 2013 | Case-control | ACS individuals subjected to OCT to evaluate culprit plaque morphology | 148 | Ruptured plaque (R-ACS) | Stable angina pectoris | sLox1 levels | sLOX1 levels were significantly higher among R-ACS patients in comparison to stable angina pectoris individuals. |
| NCT03654313 | 2019 | Phase 1 Randomized, Blinded, Placebo-controlled Study | Type 2 diabetes individuals, aged 18 to 65 years old | 88 | MEDI6570 | Placebo | Number of subjects with adverse events and serious adverse events | To evaluate the safety, tolerability, PK and immunogenicity of single and multiple ascending doses of MEDI6570 in subjects with Type 2 Diabetes Mellitus |
| EudraCT: 2020-000840-75 | 2019 | Phase IIB, Randomized, Double blinded, Placebo controlled | Individuals with prior MI, persistent inflammation or elevated BNP | MEDI6570 | Placebo | Non-calcified plaque volume change | To evaluate the effect of MEDI6570 on plaque volume change and markers of heart failure in comparison to placebo | |
CAD, coronary artery disease; PCI, percutaneous coronary intervention; MACE, major acute cardiovascular events; sLox1, serum Lox1; PCI-RPMI, percutaneous coronary intervention-related periprocedural myocardial infarction; CHD, coronary heart disease; ACS, acute coronary syndrome; OCT, optimal coherence tomography
1.6. LOX-1 as a potential target for clinical trials
The promising results from preclinical studies with Lox1 inhibitors sparked a great interest in translation of these findings to clinical scenario. Two ongoing clinical trials are testing an anti-Lox1 neutralizing (MEDI6570®, AstraZeneca, Cambridge, UK) in the clinic. A phase I clinical trial (Clinicaltrials.gov number: NCT03654313) is assessing the safety of increasing doses of MEDI6570® in subjects with type 2 diabetes mellitus while in a phase IIB clinical trial (EudraCT Number: 2020–000840-75), is testing whether MEDI6570® reduces non-calcified atherosclerotic plaque volume or improves biomarkers of heart failure in patients with prior MI. The results from these and future clinical trials may help to pave the ground for LOX-1 inhibitors as novel therapeutic targets for CVD.
2. Conclusions
Over the past years, in the face of an evolving body of evidence, Lox1 has emerged as a compelling target for prediction, diagnosis, and treatment of CVD. Initially described in endothelial cells as an oxLDL receptor with deleterious downstream effects on endothelial function and LDL transcytosis. Current knowledge suggests that Lox1 participates in all main steps of atherosclerosis, favoring the progression of atherosclerotic CVD. Identification of a measurable soluble form of Lox1 (sLox1) derived from the cleavage of its NECK domain, has sparked a whole new horizon of opportunities regarding CV risk prediction and early diagnosis of CVD. In parallel, mounting evidence from animal models supports that Lox1 inhibition improves myocardial recovery after an ischemic insult. In the future, extrapolation of these results to clinical trials could offer a new therapeutic target for the treatment of CVD.
7. Highlights.
Lox1 is a transmembrane protein expressed in endothelial cells, platelets, macrophages, smooth muscle cells and cardiomyocytes. Activation of Lox1 by oxidized low density lipoprotein is implicated in the progression of atherosclerosis, myocardial fibrosis and endothelial dysfunction.
Lox1 serum levels are positively related to the incidence of acute coronary syndrome, stroke and atherosclerotic plaque instability. Moreover, following a myocardial infarction Lox1 peaks more rapidly than CK-MB and troponin, hence displaying a potential role in early detection of myocardial necrosis.
Animal experiments using synthetic Lox1 inhibitors found reduction of final infarct size, better myocardial recovery and plaque regression. Translation of these findings to clinical trials places Lox1 as a potential therapeutical target.
List of Abbreviations
| Abbreviation | Meaning |
|---|---|
| Lox 1 | Lectin-like oxLDL receptor 1 |
| CVD | Cardiovascular disease |
| MI | Myocardial infarction |
| CTLD | C-terminal lectin like domain |
| IL18 | Interleukin 18 |
| 5`UTR | 5`-untranslated region |
| SNP | Single nucleotide polymorphisms |
| ACVD | Atherosclerotic cardiovascular disease |
| cIMT | Carotid intima media thickness |
| NFkB | Nuclear factor kappa B |
| AngII | Angiotensin II |
| LDLR | Low density lipoprotein receptor |
| SR-BI | Scavenger receptor B type I |
| SNARE | Soluble NSF attachment protein receptor |
| mtDNA | Mitochondrial DNA |
| SMC | Smooth muscle cells |
| EC | Endothelial cells |
| MMP | Matrix Metalloproteinase |
| MCP | Monocyte chemoattractant protein |
| siRNA | Silencing RNA |
| PAI-1 | Plasminogen activator inhibitor type 1 |
| KO | Knock-out |
| TGF-ß1 | Transforming growth factor beta 1 |
| MAPK | mitogen-activated protein kinase |
| IRI | Ischemia-reperfusion injury |
| CK-MB | Creatine Kinase MB |
| sLOX1 | Serum Lox1 |
| ACS | Acute coronary syndrome |
| STEMI | ST-elevation myocardial infarction |
| NSTEMI | Non- ST-elevation myocardial infarction |
| CRP | C-reactive protein |
| OCT | Optical coherence tomography |
| PCI | Primary percutaneous intervention |
| MACE | Major acute cardiovascular events |
| NIHSS | NIH stroke scale |
Footnotes
Disclosure:
Authors declare no conflict of interest.
6. References
- 1.Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ Res 2016;118:535–46. [DOI] [PubMed] [Google Scholar]
- 3.Reed GW, Rossi JE, Cannon CP. Acute myocardial infarction. Lancet 2017;389:197–210. [DOI] [PubMed] [Google Scholar]
- 4.Fulcher J, O’Connell R, Voysey M, Emberson J, Blackwell L, Mihaylova B, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015;385:1397–405. [DOI] [PubMed] [Google Scholar]
- 5.Dhindsa DS, Sandesara PB, Shapiro MD, Wong ND. The Evolving Understanding and Approach to Residual Cardiovascular Risk Management. Front Cardiovasc Med 2020;7:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q, Reis SE, Kammerer C, Craig WY, LaPierre SE, Zimmer EL, et al. Genetic variation in lectin-like oxidized low-density lipoprotein receptor 1 (LOX1) gene and the risk of coronary artery disease. Circulation 2003;107:3146–51. [DOI] [PubMed] [Google Scholar]
- 7.Lu J, Wang X, Wang W, Muniyappa H, Hu C, Mitra S, et al. LOX-1 abrogation reduces cardiac hypertrophy and collagen accumulation following chronic ischemia in the mouse. Gene Ther 2012;19:522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holy EW, Akhmedov A, Speer T, Camici GG, Zewinger S, Bonetti N, et al. Carbamylated Low-Density Lipoproteins Induce a Prothrombotic State Via LOX-1: Impact on Arterial Thrombus Formation In Vivo. J Am Coll Cardiol 2016;68:1664–76. [DOI] [PubMed] [Google Scholar]
- 9.Kakutani M, Masaki T, Sawamura T. A platelet-endothelium interaction mediated by lectin-like oxidized low-density lipoprotein receptor-1. Proc Natl Acad Sci U S A 2000;97:360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akhmedov A, Rozenberg I, Paneni F, Camici GG, Shi Y, Doerries C, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J 2014;35:2839–48. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Kakutani M, Naruko T, Ueda M, Narumiya S, Masaki T, et al. Activation-dependent surface expression of LOX-1 in human platelets. Biochem Biophys Res Commun 2001;282:153–8. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann A, Brunssen C, Wolk S, Reeps C, Morawietz H. Soluble LOX-1: A Novel Biomarker in Patients With Coronary Artery Disease, Stroke, and Acute Aortic Dissection? J Am Heart Assoc 2020;9:e013803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue N, Okamura T, Kokubo Y, Fujita Y, Sato Y, Nakanishi M, et al. LOX index, a novel predictive biochemical marker for coronary heart disease and stroke. Clin Chem 2010;56:550–8. [DOI] [PubMed] [Google Scholar]
- 14.Li XM, Jin PP, Xue J, Chen J, Chen QF, Luan XQ, et al. Role of sLOX-1 in intracranial artery stenosis and in predicting long-term prognosis of acute ischemic stroke. Brain Behav 2018;8:e00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokota C, Sawamura T, Watanabe M, Kokubo Y, Fujita Y, Kakino A, et al. High Levels of Soluble Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 in Acute Stroke: An Age- and Sex-Matched Cross-Sectional Study. J Atheroscler Thromb 2016;23:1222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997;386:73–7. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Mehta JL, Haider N, Zhang X, Narula J, Li D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res 2004;94:370–6. [DOI] [PubMed] [Google Scholar]
- 18.Hu C, Dandapat A, Sun L, Khan JA, Liu Y, Hermonat PL, et al. Regulation of TGFbeta1-mediated collagen formation by LOX-1: studies based on forced overexpression of TGFbeta1 in wild-type and lox-1 knock-out mouse cardiac fibroblasts. J Biol Chem 2008;283:10226–31. [DOI] [PubMed] [Google Scholar]
- 19.Li D, Mehta JL. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 2000;101:2889–95. [DOI] [PubMed] [Google Scholar]
- 20.Li D, Williams V, Liu L, Chen H, Sawamura T, Antakli T, et al. LOX-1 inhibition in myocardial ischemia-reperfusion injury: modulation of MMP-1 and inflammation. Am J Physiol Heart Circ Physiol 2002;283:H1795–801. [DOI] [PubMed] [Google Scholar]
- 21.Mitsuoka H, Kume N, Hayashida K, Inui-Hayashiada A, Aramaki Y, Toyohara M, et al. Interleukin 18 stimulates release of soluble lectin-like oxidized LDL receptor-1 (sLOX-1). Atherosclerosis 2009;202:176–82. [DOI] [PubMed] [Google Scholar]
- 22.Park H, Adsit FG, Boyington JC. The 1.4 angstrom crystal structure of the human oxidized low density lipoprotein receptor lox-1. J Biol Chem 2005;280:13593–9. [DOI] [PubMed] [Google Scholar]
- 23.Xie Q, Matsunaga S, Niimi S, Ogawa S, Tokuyasu K, Sakakibara Y, et al. Human lectin-like oxidized low-density lipoprotein receptor-1 functions as a dimer in living cells. DNA Cell Biol 2004;23:111–7. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Narumiya S, Masaki T, Sawamura T. Conserved C-terminal residues within the lectin-like domain of LOX-1 are essential for oxidized low-density-lipoprotein binding. Biochem J 2001;355:289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohki I, Ishigaki T, Oyama T, Matsunaga S, Xie Q, Ohnishi-Kameyama M, et al. Crystal structure of human lectin-like, oxidized low-density lipoprotein receptor 1 ligand binding domain and its ligand recognition mode to OxLDL. Structure 2005;13:905–17. [DOI] [PubMed] [Google Scholar]
- 26.Shi X, Niimi S, Ohtani T, Machida S. Characterization of residues and sequences of the carbohydrate recognition domain required for cell surface localization and ligand binding of human lectin-like oxidized LDL receptor. J Cell Sci 2001;114:1273–82. [DOI] [PubMed] [Google Scholar]
- 27.Falconi M, Biocca S, Novelli G, Desideri A. Molecular dynamics simulation of human LOX-1 provides an explanation for the lack of OxLDL binding to the Trp150Ala mutant. BMC Struct Biol 2007;7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishigaki T, Ohki I, Utsunomiya-Tate N, Tate SI. Chimeric structural stabilities in the coiled-coil structure of the NECK domain in human lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1). J Biochem 2007;141:855–66. [DOI] [PubMed] [Google Scholar]
- 29.Biocca S, Arcangeli T, Tagliaferri E, Testa B, Vindigni G, Oteri F, et al. Simulative and experimental investigation on the cleavage site that generates the soluble human LOX-1. Arch Biochem Biophys 2013;540:9–18. [DOI] [PubMed] [Google Scholar]
- 30.Rizzacasa B, Morini E, Pucci S, Murdocca M, Novelli G, Amati F. LOX-1 and Its Splice Variants: A New Challenge for Atherosclerosis and Cancer-Targeted Therapies. Int J Mol Sci 2017;18:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biocca S, Filesi I, Mango R, Maggiore L, Baldini F, Vecchione L, et al. The splice variant LOXIN inhibits LOX-1 receptor function through hetero-oligomerization. J Mol Cell Cardiol 2008;44:561–70. [DOI] [PubMed] [Google Scholar]
- 32.Veas C, Jara C, Willis ND, Pérez-Contreras K, Gutierrez N, Toledo J, et al. Overexpression of LOXIN Protects Endothelial Progenitor Cells From Apoptosis Induced by Oxidized Low Density Lipoprotein. J Cardiovasc Pharmacol 2016;67:326–35. [DOI] [PubMed] [Google Scholar]
- 33.Vecchione L, Gargiul E, Borgiani P, Predazzi I, Mango R, Romeo F, et al. Genotyping OLR1 gene: a genomic biomarker for cardiovascular diseases. Recent Pat Cardiovasc Drug Discov 2007;2:147–51. [DOI] [PubMed] [Google Scholar]
- 34.Guo X, Xiang Y, Yang H, Yu L, Peng X, Guo R. Association of the LOX-1 rs1050283 Polymorphism with Risk for Atherosclerotic Cerebral Infarction and its Effect on sLOX-1 and LOX-1 Expression in a Chinese Population. J Atheroscler Thromb 2017;24:572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatsuguchi M, Furutani M, Hinagata J, Tanaka T, Furutani Y, Imamura S, et al. Oxidized LDL receptor gene (OLR1) is associated with the risk of myocardial infarction. Biochem Biophys Res Commun 2003;303:247–50. [DOI] [PubMed] [Google Scholar]
- 36.Predazzi IM, Norata GD, Vecchione L, Garlaschelli K, Amati F, Grigore L, et al. Association between OLR1 K167N SNP and intima media thickness of the common carotid artery in the general population. PLoS One 2012;7:e31086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X, Hou X, Liang Y, Li F, Pang L, Huang G, et al. The gene polymorphism of LOX1 predicts the incidence of LVH in patients with essential hypertension. Cell Physiol Biochem 2014;33:88–96. [DOI] [PubMed] [Google Scholar]
- 38.Mango R, Clementi F, Borgiani P, Forleo GB, Federici M, Contino G, et al. Association of single nucleotide polymorphisms in the oxidised LDL receptor 1 (OLR1) gene in patients with acute myocardial infarction. J Med Genet 2003;40:933–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trabetti E, Biscuola M, Cavallari U, Malerba G, Girelli D, Olivieri O, et al. On the association of the oxidised LDL receptor 1 (OLR1) gene in patients with acute myocardial infarction or coronary artery disease. Eur J Hum Genet 2006;14:127–30. [DOI] [PubMed] [Google Scholar]
- 40.Feng Y, Cai ZR, Tang Y, Hu G, Lu J, He D, et al. TLR4/NF-κB signaling pathway-mediated and oxLDL-induced up-regulation of LOX-1, MCP-1, and VCAM-1 expressions in human umbilical vein endothelial cells. Genet Mol Res 2014;13:680–95. [DOI] [PubMed] [Google Scholar]
- 41.Li D, Chen H, Romeo F, Sawamura T, Saldeen T, Mehta JL. Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther 2002;302:601–5. [DOI] [PubMed] [Google Scholar]
- 42.Biocca S, Iacovelli F, Matarazzo S, Vindigni G, Oteri F, Desideri A, et al. Molecular mechanism of statin-mediated LOX-1 inhibition. Cell Cycle 2015;14:1583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matarazzo S, Quitadamo MC, Mango R, Ciccone S, Novelli G, Biocca S. Cholesterol-lowering drugs inhibit lectin-like oxidized low-density lipoprotein-1 receptor function by membrane raft disruption. Mol Pharmacol 2012;82:246–54. [DOI] [PubMed] [Google Scholar]
- 44.Mehta JL, Li DY, Chen HJ, Joseph J, Romeo F. Inhibition of LOX-1 by statins may relate to upregulation of eNOS. Biochem Biophys Res Commun 2001;289:857–61. [DOI] [PubMed] [Google Scholar]
- 45.Li DY, Zhang YC, Philips MI, Sawamura T, Mehta JL. Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ Res 1999;84:1043–9. [DOI] [PubMed] [Google Scholar]
- 46.Lee JY, Chung J, Kim KH, An SH, Kim M, Park J, et al. Fluid shear stress regulates the expression of Lectin-like oxidized low density lipoprotein receptor-1 via KLF2-AP-1 pathway depending on its intensity and pattern in endothelial cells. Atherosclerosis 2018;270:76–88. [DOI] [PubMed] [Google Scholar]
- 47.Mehta JL, Chen J, Yu F, Li DY. Aspirin inhibits ox-LDL-mediated LOX-1 expression and metalloproteinase-1 in human coronary endothelial cells. Cardiovasc Res 2004;64:243–9. [DOI] [PubMed] [Google Scholar]
- 48.Sugano M, Tsuchida K, Makino N. Nifedipine prevents apoptosis of endothelial cells induced by oxidized low-density lipoproteins. J Cardiovasc Pharmacol 2002;40:146–52. [DOI] [PubMed] [Google Scholar]
- 49.Ouslimani N, Mahrouf M, Peynet J, Bonnefont-Rousselot D, Cosson C, Legrand A, et al. Metformin reduces endothelial cell expression of both the receptor for advanced glycation end products and lectin-like oxidized receptor 1. Metabolism 2007;56:308–13. [DOI] [PubMed] [Google Scholar]
- 50.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers 2019;5:56. [DOI] [PubMed] [Google Scholar]
- 51.Miller YI, Choi SH, Fang L, Tsimikas S. Lipoprotein modification and macrophage uptake: role of pathologic cholesterol transport in atherogenesis. Subcell Biochem 2010;51:229–51. [DOI] [PubMed] [Google Scholar]
- 52.Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouhairie VE, Goldberg AC. Familial hypercholesterolemia. Cardiol Clin 2015;33:169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016;388:2532–61. [DOI] [PubMed] [Google Scholar]
- 56.Josan K, Majumdar SR, McAlister FA. The efficacy and safety of intensive statin therapy: a meta-analysis of randomized trials. CMAJ 2008;178:576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med 2017;376:1713–22. [DOI] [PubMed] [Google Scholar]
- 58.Melendez QM, Krishnaji ST, Wooten CJ, Lopez D. Hypercholesterolemia: The role of PCSK9. Arch Biochem Biophys 2017;625–626:39–53. [DOI] [PubMed] [Google Scholar]
- 59.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 2007;116:1832–44. [DOI] [PubMed] [Google Scholar]
- 60.Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002;417:750–4. [DOI] [PubMed] [Google Scholar]
- 61.Zhao Y, Yang Y, Xing R, Cui X, Xiao Y, Xie L, et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis 2018;271:26–35. [DOI] [PubMed] [Google Scholar]
- 62.Lu R, Yuan T, Wang Y, Zhang T, Yuan Y, Wu D, et al. Spontaneous severe hypercholesterolemia and atherosclerosis lesions in rabbits with deficiency of low-density lipoprotein receptor (LDLR) on exon 7. EBioMedicine 2018;36:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sposito AC, Zimetti F, Barreto J, Zanotti I. Lipid trafficking in cardiovascular disease. Adv Clin Chem 2019;92:105–40. [DOI] [PubMed] [Google Scholar]
- 64.Bossé Y, Feitosa MF, Després JP, Lamarche B, Rice T, Rao DC, et al. Detection of a major gene effect for LDL peak particle diameter and association with apolipoprotein H gene haplotype. Atherosclerosis 2005;182:231–9. [DOI] [PubMed] [Google Scholar]
- 65.Fernández-Hernando C, Yu J, Dávalos A, Prendergast J, Sessa WC. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol 2010;177:998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frank PG, Lee H, Park DS, Tandon NN, Scherer PE, Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol 2004;24:98–105. [DOI] [PubMed] [Google Scholar]
- 67.Pavlides S, Gutierrez-Pajares JL, Iturrieta J, Lisanti MP, Frank PG. Endothelial caveolin-1 plays a major role in the development of atherosclerosis. Cell Tissue Res 2014;356:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun SW, Zu XY, Tuo QH, Chen LX, Lei XY, Li K, et al. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol Sin 2010;31:1336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tuma P, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev 2003;83:871–932. [DOI] [PubMed] [Google Scholar]
- 70.Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol 2012;2:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang J, Chu W, Crandall I. Lipoprotein binding preference of CD36 is altered by filipin treatment. Lipids Health Dis 2008;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Armstrong SM, Sugiyama MG, Fung KY, Gao Y, Wang C, Levy AS, et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res 2015;108:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019;569:565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumano-Kuramochi M, Xie Q, Kajiwara S, Komba S, Minowa T, Machida S. Lectin-like oxidized LDL receptor-1 is palmitoylated and internalizes ligands via caveolae/raft-dependent endocytosis. Biochem Biophys Res Commun 2013;434:594–9. [DOI] [PubMed] [Google Scholar]
- 75.Wang H, Liu Z, Li G, Barrett EJ. The vascular endothelial cell mediates insulin transport into skeletal muscle. Am J Physiol Endocrinol Metab 2006;291:E323–32. [DOI] [PubMed] [Google Scholar]
- 76.Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem 2001;276:48619–22. [DOI] [PubMed] [Google Scholar]
- 77.Oh P, McIntosh DP, Schnitzer JE. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol 1998;141:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pelkmans L, Püntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002;296:535–9. [DOI] [PubMed] [Google Scholar]
- 79.Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell 2005;16:3919–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martenson CH, Odom A, Sheetz MP, Graham DG. The effect of acrylamide and other sulfhydryl alkylators on the ability of dynein and kinesin to translocate microtubules in vitro. Toxicol Appl Pharmacol. 1995;133:73–81. [DOI] [PubMed] [Google Scholar]
- 81.Predescu SA, Predescu DN, Shimizu K, Klein IK, Malik AB. Cholesterol-dependent syntaxin-4 and SNAP-23 clustering regulates caveolar fusion with the endothelial plasma membrane. J Biol Chem 2005;280:37130–8. [DOI] [PubMed] [Google Scholar]
- 82.Orekhov AN. LDL and foam cell formation as the basis of atherogenesis. Curr Opin Lipidol 2018;29:279–84. [DOI] [PubMed] [Google Scholar]
- 83.Childs BG, Li H, van Deursen JM. Senescent cells: a therapeutic target for cardiovascular disease. J Clin Invest 2018;128:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang YC, Lee AS, Lu LS, Ke LY, Chen WY, Dong JW, et al. Human electronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018;17:e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 2013;13:709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, et al. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem 2005;280:2352–60. [DOI] [PubMed] [Google Scholar]
- 87.Kruth HS, Huang W, Ishii I, Zhang WY. Macrophage foam cell formation with native low density lipoprotein. J Biol Chem 2002;277:34573–80. [DOI] [PubMed] [Google Scholar]
- 88.Schaeffer DF, Riazy M, Parhar KS, Chen JH, Duronio V, Sawamura T, et al. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res 2009;50:1676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ding Z, Liu S, Wang X, Dai Y, Khaidakov M, Deng X, et al. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: implications in atherogenesis. Cardiovasc Res 2014;103:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Ding Z, Lin J, Guo Z, Mehta JL. LOX-1 in macrophage migration in response to ox-LDL and the involvement of calpains. Biochem Biophys Res Commun 2015;467:135–9. [DOI] [PubMed] [Google Scholar]
- 91.Yang HY, Bian YF, Zhang HP, Gao F, Xiao CS, Liang B, et al. LOX‑1 is implicated in oxidized low‑density lipoprotein‑induced oxidative stress of macrophages in atherosclerosis. Mol Med Rep 2015;12:5335–41. [DOI] [PubMed] [Google Scholar]
- 92.Sun Y, Chen X. Ox-LDL-induced LOX-1 expression in vascular smooth muscle cells: role of reactive oxygen species. Fundam Clin Pharmacol 2011;25:572–9. [DOI] [PubMed] [Google Scholar]
- 93.Kataoka H, Kume N, Miyamoto S, Minami M, Morimoto M, Hayashida K, et al. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2001;21:955–60. [DOI] [PubMed] [Google Scholar]
- 94.Ishino S, Mukai T, Kume N, Asano D, Ogawa M, Kuge Y, et al. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability--analysis in hypercholesterolemic rabbits. Atherosclerosis 2007;195:48–56. [DOI] [PubMed] [Google Scholar]
- 95.Brinkley TE, Nicklas BJ, Kanaya AM, Satterfield S, Lakatta EG, Simonsick EM, et al. Plasma oxidized low-density lipoprotein levels and arterial stiffness in older adults: the health, aging, and body composition study. Hypertension 2009;53:846–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang W, Zhu T, Wu W, Ge X, Xiong X, Zhang Z, et al. LOX-1 mediated phenotypic switching of pulmonary arterial smooth muscle cells contributes to hypoxic pulmonary hypertension. Eur J Pharmacol 2018;818:84–95. [DOI] [PubMed] [Google Scholar]
- 97.Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018;138:e618–e51. [DOI] [PubMed] [Google Scholar]
- 98.von Hundelshausen P, Schmitt MM. Platelets and their chemokines in atherosclerosis-clinical applications. Front Physiol 2014;5:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Badimon L, Padró T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur Heart J Acute Cardiovasc Care 2012;1:60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li D, Williams V, Liu L, Chen H, Sawamura T, Romeo F, et al. Expression of lectin-like oxidized low-density lipoprotein receptors during ischemia-reperfusion and its role in determination of apoptosis and left ventricular dysfunction. J Am Coll Cardiol 2003;41:1048–55. [DOI] [PubMed] [Google Scholar]
- 101.Villa M, Cerda-Opazo P, Jimenez-Gallegos D, Garrido-Moreno V, Chiong M, Quest AF, et al. Pro-fibrotic effect of oxidized LDL in cardiac myofibroblasts. Biochem Biophys Res Commun 2020;524:696–701. [DOI] [PubMed] [Google Scholar]
- 102.Yokoyama C, Aoyama T, Ido T, Kakino A, Shiraki T, Tanaka T, et al. Deletion of LOX-1 Protects against Heart Failure Induced by Doxorubicin. PLoS One 2016;11:e0154994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kobayashi N, Hata N, Kume N, Seino Y, Inami T, Yokoyama S, et al. Soluble lectin-like oxidized low-density lipoprotein receptor-1 as an early biomarker for ST elevation myocardial infarction: time-dependent comparison with other biomarkers: time-dependent comparison with other biomarkers. Circ J 2011;75:1433–9. [DOI] [PubMed] [Google Scholar]
- 104.Lee AS, Wang YC, Chang SS, Lo PH, Chang CM, Lu J, et al. Detection of a High Ratio of Soluble to Membrane-Bound LOX-1 in Aspirated Coronary Thrombi From Patients With ST-Segment-Elevation Myocardial Infarction. J Am Heart Assoc 2020;9:e014008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hayashida K, Kume N, Murase T, Minami M, Nakagawa D, Inada T, et al. Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: a novel marker for early diagnosis. Circulation 2005;112:812–8. [DOI] [PubMed] [Google Scholar]
- 106.Kobayashi N, Takano M, Hata N, Kume N, Yamamoto M, Yokoyama S, et al. Soluble lectin-like oxidized LDL receptor-1 (sLOX-1) as a valuable diagnostic marker for rupture of thin-cap fibroatheroma: verification by optical coherence tomography. Int J Cardiol 2013;168:3217–23. [DOI] [PubMed] [Google Scholar]
- 107.Zhao ZW, Xu YW, Li SM, Guo JJ, Sun JM, Hong JC, et al. Baseline Serum sLOX-1 Concentrations Are Associated with 2-Year Major Adverse Cardiovascular and Cerebrovascular Events in Patients after Percutaneous Coronary Intervention. Dis Markers 2019;2019:4925767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balin M, Celik A, Kobat MA, Baydas A. Circulating soluble lectin-like oxidized low-density lipoprotein receptor-1 levels predict percutaneous coronary intervention-related periprocedural myocardial infarction in stable patients undergoing elective native single-vessel PCI. J Thromb Thrombolysis 2012;34:483–90. [DOI] [PubMed] [Google Scholar]
- 109.Zhao ZW, Xu YW, Li SM, Guo JJ, Yi T, Chen LL. Higher serum lectin-like oxidized low-density lipoprotein receptor-1 in patients with stable coronary artery disease is associated with major adverse cardiovascular events: A multicentre pilot study. Biochem Med (Zagreb) 2019;29:010705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao ZW, Zhu XL, Luo YK, Lin CG, Chen LL. Circulating soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are associated with angiographic coronary lesion complexity in patients with coronary artery disease. Clin Cardiol 2011;34:172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Markstad H, Edsfeldt A, Yao Mattison I, Bengtsson E, Singh P, Cavalera M, et al. High Levels of Soluble Lectinlike Oxidized Low-Density Lipoprotein Receptor-1 Are Associated With Carotid Plaque Inflammation and Increased Risk of Ischemic Stroke. J Am Heart Assoc 2019;8:e009874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Skarpengland T, Skjelland M, Kong XY, Skagen K, Holm S, Otterdal K, et al. Increased Levels of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 in Ischemic Stroke and Transient Ischemic Attack. J Am Heart Assoc 2018;7: e006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tian K, Ogura S, Little PJ, Xu SW, Sawamura T. Targeting LOX-1 in atherosclerosis and vasculopathy: current knowledge and future perspectives. Ann N Y Acad Sci 2019;1443:34–53. [DOI] [PubMed] [Google Scholar]
- 114.Hein TW, Xu X, Ren Y, Xu W, Tsai SH, Thengchaisri N, et al. Requisite roles of LOX-1, JNK, and arginase in diabetes-induced endothelial vasodilator dysfunction of porcine coronary arterioles. J Mol Cell Cardiol 2019;131:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gioia M, Vindigni G, Testa B, Raniolo S, Fasciglione GF, Coletta M, et al. Membrane Cholesterol Modulates LOX-1 Shedding in Endothelial Cells. PLoS One 2015;10:e0141270. [DOI] [PMC free article] [PubMed] [Google Scholar]