

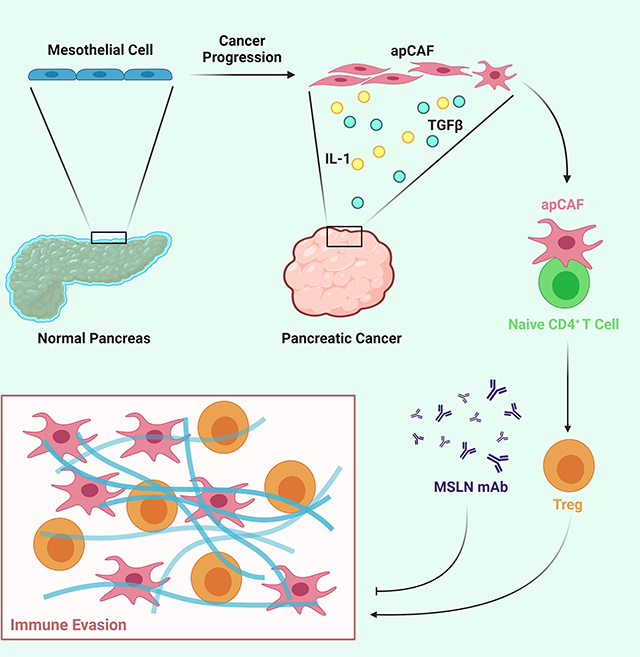
Summary
Recent studies have identified a unique cancer-associated fibroblast (CAF) population termed antigen-presenting CAF (apCAF), characterized by the expression of major histocompatibility complex class II molecules, suggesting a function in regulating tumor immunity. Here by integrating multiple single cell RNA sequencing studies and performing robust lineage tracing assays, we find that apCAFs are derived from mesothelial cells. During pancreatic cancer progression, mesothelial cells form apCAFs by down-regulating mesothelial features and gaining fibroblastic features, a process induced by IL-1 and TGFβ. apCAFs directly ligate and induce naïve CD4+ T cells into regulatory T cells (Tregs) in an antigen-specific manner. Moreover, treatment with an antibody targeting the mesothelial cell marker mesothelin can effectively inhibit mesothelial cell to apCAF transition and Treg formation induced by apCAFs. Taken together, our study elucidates how mesothelial cells may contribute to immune evasion in pancreatic cancer and provides insight on strategies to enhance cancer immune therapy.
Keywords: pancreatic cancer, cancer-associated fibroblast, mesothelial cell, regulatory T cell, mesothelin
Graphical Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDA) is projected to become the second leading cause of cancer-related deaths in the United States by 2030 (Rahib et al., 2014). To date, there is no effective treatment for PDA in part due to the robust desmoplastic reaction that occurs during tumor development (Hosein et al., 2020; Huang and Brekken, 2019). Extensive stroma, which consists of abundant extracellular matrix (ECM) and stromal cells including cancer-associated fibroblasts (CAFs), myeloid cells, lymphocytes and vascular endothelial cells, dominates the PDA microenvironment. CAFs are critical drivers of the desmoplastic reaction, nonetheless, they are also one of the least characterized stromal cells (Chen and Song, 2019; Huang and Brekken, 2020; Kalluri, 2016). Initially, CAFs were considered a group of uniform mesenchymal cells present in the tumor microenvironment (TME) that support tumor progression. It is clear that CAFs can promote PDA progression by producing and remodeling ECM and by secreting chemokines, cytokines, and growth factors (LeBleu and Kalluri, 2018). However, ablation studies demonstrate that CAFs can also limit tumor progression (Ozdemir et al., 2014; Rhim et al., 2014). These conflicting data suggest that CAFs consist of heterogeneous subtypes with different functions. CAF heterogeneity is first characterized in an in vitro PDA organoid co-culture model that identifies two distinct types of CAFs (Ohlund et al., 2017). One has myofibroblastic features (myCAF), while the other has inflammatory features (iCAF). These two CAF populations are also found to be mutually exclusive and coexist in PDA.
To further understand CAF heterogeneity during PDA progression, we have exploited single-cell RNA sequencing (scRNA-seq) to agnostically profile fibroblasts in three stages of PDA (normal pancreas, early and late PDA) derived from KIC (KrasLSL-G12D/+; Ink4afl/fl; Ptf1aCre/+) mice (Hosein et al., 2019). We identified three populations of fibroblasts which we named FB1, FB2 and FB3. All three populations are present in normal pancreas and early-stage PDA, while only FB1 and FB3 are present in late-stage PDA. Among all the fibroblasts populations, FB3 is the most intriguing and unique population as it expresses major histocompatibility complex class II (MHC II) genes, which are known to be specifically expressed by professional antigen presenting cells (APCs). Interestingly, we also found that FB3 is characterized by the expression of mesothelial genes, suggesting that this CAF population may be associated with mesothelial cells. In a later scRNA-seq study in KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Ptf1aCre/+) tumors, a CAF population with a similar gene signature as FB3 was identified and termed antigen-presenting CAFs (apCAFs), due to the expression of MHC II genes (Elyada et al., 2019). Additional scRNA-seq analysis of fibroblasts performed in different stages of KIC tumors (normal pancreas, early and established tumor lesions) compared the gene signature of mesothelial cells identified in normal pancreas with apCAFs and found these two cell populations shared similar gene features (Dominguez et al., 2020), further indicating that apCAFs might be derived from mesothelial cells.
Mesothelial cells are derived from embryonic mesoderm (Koopmans and Rinkevich, 2018). During embryonic development, mesothelial cells form a single layer of epithelial cells called coelomic epithelium, which covers the entire coelomic cavity. Coelomic epithelium has multipotency and contributes to growing organs by differentiating into various cell types including fibroblasts and smooth muscle cells through epithelial-to-mesenchymal transition (EMT). The remaining undifferentiated mesothelial cells mature and form a continuous layer of epithelial cells known as mesothelium in the adult. The mesothelium is traditionally thought to be a passive membrane providing a non-adhesive surface covering body cavities, internal organs and tissues. However, recent studies have found that under certain pathological conditions such as wound healing, adipogenesis, myocardial infarction, peritoneal fibrosis, surgical adhesions and liver injury, mesothelial cells can engage embryonic programs and contribute to the formation of mesenchymal cells (Cao and Poss, 2018; Gupta and Gupta, 2015; Li et al., 2013; Mutsaers et al., 2015; Si et al., 2019; Tsai et al., 2018).
Nonetheless, in cancer, mesothelial cells have not been considered as a functional constituent of the TME. The MHC II-positive CAF populations identified by different scRNA-seq studies underscore the possibility that mesothelial cells may have important functions within the TME and may contribute to the formation of a unique CAF population that can regulate tumor immunity by directly interacting with CD4+ T cells. Here, we investigate the relationship between mesothelial cells and apCAFs, their functions during PDA progression, as well as the signaling mechanisms that drive the formation of apCAFs.
Results
apCAFs are Derived from Mesothelial Cells
We previously profiled multiple genetically engineered mouse models (GEMMs) of PDA (normal pancreas, early KIC, late KIC, late KPC and late KPfC (KrasLSL-G12D/+; Trp53fl/fl; Pdx1Cre/+)) by scRNA-seq and identified three populations of fibroblasts in normal pancreas and early-stage PDA (FB1, FB2 and FB3) and two populations in late-stage PDA (FB1 and FB3) (Hosein et al., 2019). To understand the relationships of fibroblasts among these models, we projected all fibroblasts from normal pancreas, early KIC, late KIC, late KPC and late KPfC onto a single t-distributed stochastic neighbor embedding (tSNE) plot and performed graph-based clustering (Figure 1A). We identified four molecular subtypes of fibroblasts (Figure 1B). Two of the four subtypes represented what we had described as FB1 and FB2 earlier (Hosein et al., 2019), while the previously identified FB3 cluster further divided into two distinct subtypes, which we named FB3A and FB3B here. Interestingly, FB3B expanded specifically in all late-stage GEMMs (Figure 1C). We then investigated the transcriptional profiles of FB3A and FB3B and found that each subtype was characterized by the expression of MHC II pathway (Cd74, H2-Aa, H2-Ab1, H2-Dma, H2-Dmb1, H2-Eb1) and mesothelial cell (Msln, Upk3b, Ezr, Krt19, Nkain4, Lrrn4) genes (Figures 1D and 1E). However, in comparison to FB3A, FB3B showed a lower level of expression of mesothelial marker genes and an increase of inflammatory and myofibroblastic (Il6, Cxcl12, Pdgfra, Acta2, Thy1, Tgfb1) genes (Figures 1D and 1F). These data suggest that FB3A and FB3B are mesothelial cells, with FB3A being normal mesothelial cells and FB3B being mesothelial cells with a fibroblastic phenotype.
Figure 1. Relationship of Normal Mesothelial Cells and apCAFs in Integrated scRNA-seq Analyses.
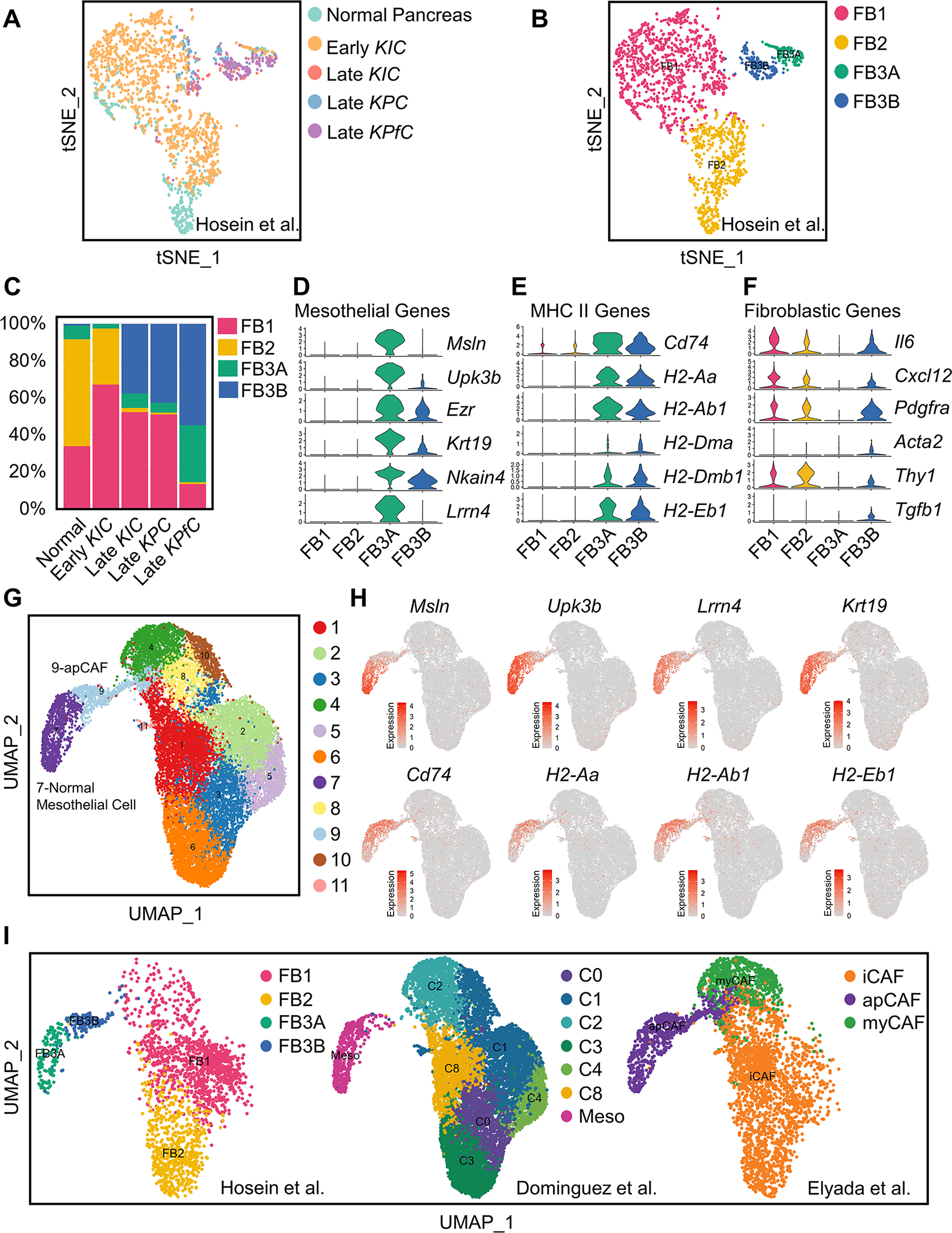
(A-B) All fibroblasts from scRNA-seq of normal pancreas, early KIC, late KIC, late KPC and late KPfC tumor lesions projected onto a tSNE plot with each model (A) or the FB1, FB2, FB3A and FB3B fibroblast populations (B) distinguished by different colors.
(C) Proportions of different fibroblast populations in normal pancreas, early KIC, late KIC, late KPC and late KPfC tumor lesions.
(D-F) Violin plots demonstrating mesothelial (D), MHC II (E) and fibroblastic (F) genes for FB1, FB2, FB3A and FB3B fibroblast populations. The width of the violin plots represents frequency of cells in each region. Values of Y axis indicate log normalized expression of genes.
(G) Graph-based clustering of cells with UMAP was performed with the integrated data and 11 clusters of fibroblasts were identified.
(H) UMAP plots of mesothelial and MHC II genes from the integrated fibroblast data. Color keys indicate log normalized expression.
(I) Distribution of fibroblasts in the integrated data based on the origin of datasets (Dominguez et al., 2020; Elyada et al., 2019; Hosein et al., 2019), including FB1, FB2, FB3A and FB3B from Hosein et al. (Hosein et al., 2019), C0-C8 and normal mesothelial cell from Dominguez et al. (Dominguez et al., 2020) and iCAF, apCAF and myCAF from Elyada et al. (Elyada et al., 2019).
See also Figure S1.
Three scRNA-seq studies including ours have identified mesothelial cell-associated cell populations in PDA: FB3 (Hosein et al., 2019), normal mesothelial cells (Dominguez et al., 2020) and apCAFs (Elyada et al., 2019). To understand the relationship of these mesothelial cell-associated populations (Figures 1A and S1A–S1D), we integrated the three fibroblast datasets and performed graph-based clustering and dimensionality reduction with uniform manifold approximation and projection (UMAP). Fibroblasts from the three datasets fell into 11 distinct clusters (Figures 1G and S1E). To identify the mesothelial clusters, we generated UMAP plots for MHC II pathway (Cd74, H2-Aa, H2-Ab1, H2-Eb1) and mesothelial cell (Msln, Upk3b, Lrrn4, Krt19) genes and found clusters 7 and 9 expressed these signature genes (Figure 1H). We highlighted the distribution of fibroblasts based on the origin of datasets to understand the relationship of FB3A, FB3B, apCAFs and normal mesothelial cells (Figure 1I). We found that cluster 7 in the merged data (Figure 1G) had the same features as FB3A while cluster 9 was the same as FB3B, suggesting that cluster 7 represents normal mesothelial cells and cluster 9 represents the fibroblastic mesothelial cell population. Consistently, the mesothelial cell population identified from normal pancreas by Dominguez et al. (Dominguez et al., 2020) overlapped with cluster 7 (Figure 1I). In addition, we also found that what Elyada et al. (Elyada et al., 2019) termed as apCAFs consisted of normal mesothelial cells and fibroblastic mesothelial cells (Figure 1I). Given that cluster 9 was the population exhibiting CAF features, we defined cluster 9 as apCAFs and cluster 7 as normal mesothelial cells in this study (Figure 1G).
Mesothelial Cells Expand and Contribute to Desmoplasia during PDA Progression
To validate the transcriptomic findings, we performed immunohistochemical (IHC) staining with markers that were mesothelial cell specific. However, we noticed that most of the mesothelial markers such as mesothelin were expressed at a low level in apCAFs (Figure S2A). Nonetheless, we identified some markers that were specific for mesothelial cells and fibroblasts and were stable during mesothelial cell to apCAF transition (Figure S2A). One of the markers was podoplanin, which was reported by Elyada et al. as a pan-CAF marker (Elyada et al., 2019). Another marker was cadherin-11. IHC for podoplanin, cadherin-11 and mesothelin in normal mouse pancreatic tissue and early and late KPfC tumors (Figure S2B) revealed that in normal pancreata, the mesothelium was marked by all three proteins. In early tumor lesions, podoplanin and cadherin-11 marked the expanding mesothelial cells that contributed to the desmoplastic reaction in the niche of early tumor lesions (Figure S2B, arrows). In comparison, mesothelin failed to mark the delaminating mesothelial cells in early PDA lesions. Furthermore, we found that in late-stage PDA, the mesothelial layer remained podoplanin and cadherin-11 positive (Figure S2B). To investigate whether mesothelial cells also contributed to stroma formation in other tumor settings, we established an orthotopic model of PDA using a primary mouse PDA cell line derived from a KPfC tumor. Mouse pancreata were harvested one week after implantation, which captured early tumor lesions (Figure S2C). IHC for cadherin-11 revealed that in contrast to the cadherin-11+ mesothelium around the uninvolved normal pancreas (Figure S2C, black arrow), cadherin-11+ mesothelium adjacent to the tumor expanded and contributed to the tumor stroma (Figure S2C, red arrows). Because podoplanin and cadherin-11 also mark other fibroblast populations, we performed multiplex IHC for podoplanin and the MHC II molecule CD74 to mark mesothelial cells in early and late-stage KPfC tumors. We found that podoplanin+ CD74+ mesothelial cells formed a thin membrane lining the edge of pancreas (Figure S2D, left, arrow) and these cells contributed to the TME of early tumor lesions. IHC of late-stage tumors indicated that mesothelial cells develop into a major population of CAFs within the tumor stroma (Figure S2D, right, arrow).
We then harvested three late-stage tumors from KPfC mice (PDA1, PDA2, PDA3), digested the whole tumors into single cell suspension and analyzed the proportion of apCAFs (Figure 2A). We found that all three tumors consisted of a marked population of apCAFs, with a percentage that was comparable to iCAFs and myCAFs. To trace the fate of mesothelial cells during PDA progression, we developed a lineage-tracing assay by exploiting a cell-permeable dye, 5(6)-carboxyfluorescein N-hydroxysuccinimidyl ester (CFSE). Once incorporated within cells, CFSE forms stable covalent bonds with intracellular molecules that will be transferred to daughter cells but not adjacent cells (Parish, 1999). This method has been applied to trace the fate of mesothelial cells in a surgical adhesion mouse model (Tsai et al., 2018). We injected CFSE intraperitoneally into wildtype mice and harvested the pancreata after two days. We found that the mesothelium of normal pancreata was labeled by the cell tracking dye (Figure 2B). In contrast, we performed the same assay in 60-day-old tumor-bearing KIC or KPfC mice and strikingly, we found CFSE-marked cells that infiltrated into the tumor stroma from mesothelial regions (Figure 2B). To ensure the CFSE signal was mesothelium specific, we co-stained the CFSE-labelled tissues with cadherin-11 (Figure 2C). We found that in normal pancreata and PDAs, CFSE+ cells were also cadherin-11+. In addition, we co-stained with the pancreatic cancer cell marker SOX9 and found that CFSE+ cells were distinct from SOX9+ cancer cells (Figure 2C), further supporting that the CFSE signal was specific for mesothelium-derived stromal cells.
Figure 2. Mesothelial Cells Expand and Gain Fibroblastic Feature during PDA Formation.
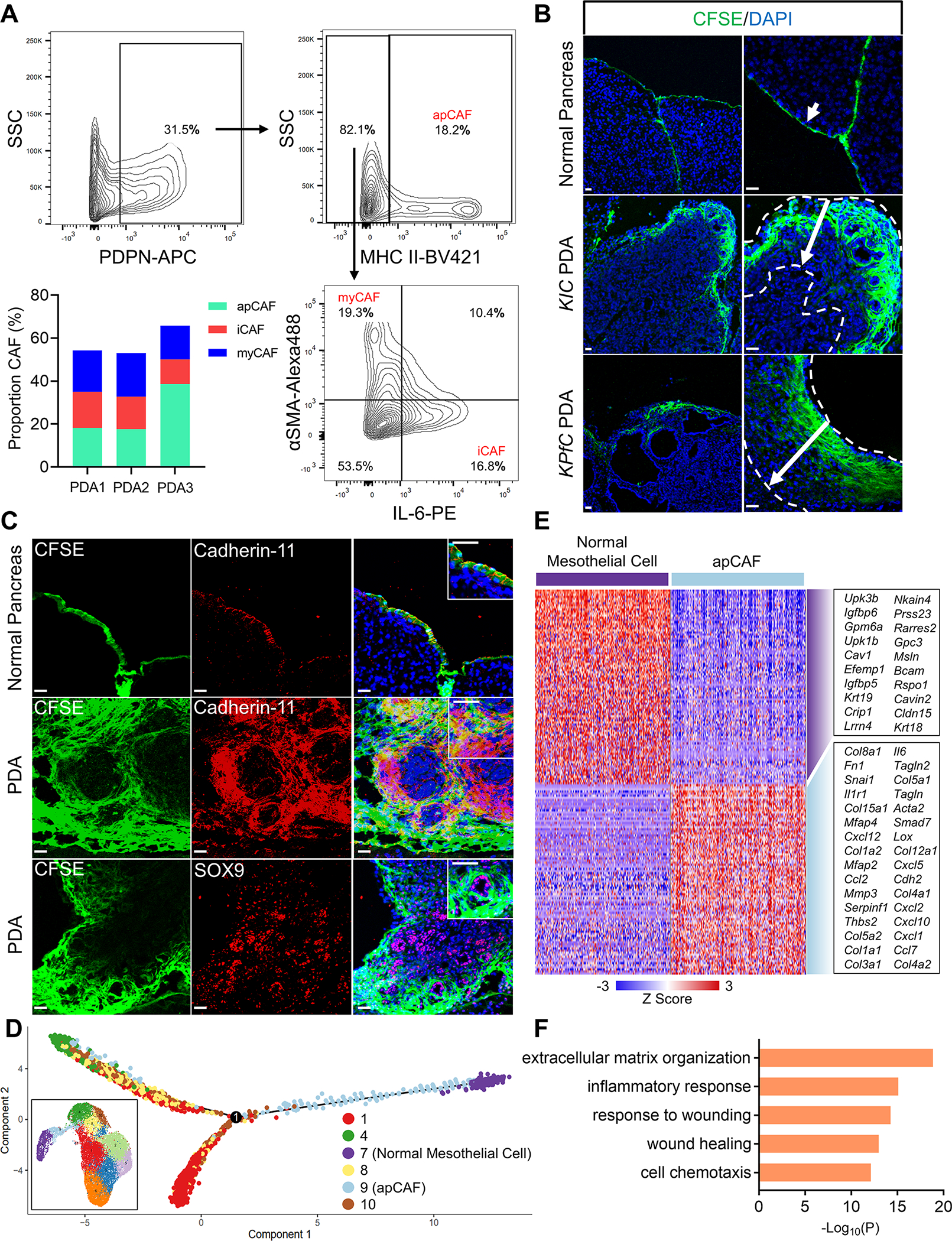
(A) Three 60-day-old KPfC tumors were analyzed for CAFs through flow cytometry (PDA1, PDA2, PDA3). The flow cytometry gating of PDA1 and percentages of CAF subtypes in the three tumors was shown. Podoplanin+ (PDPN+) (total CAFs, 31.5%); PDPN+MHC II+ (apCAFs, 18.2%); PDPN+MHC II−IL-6+ (iCAFs, 16.8%); PDPN+MHC II−αSMA+ (myCAFs, 19.3%).
(B) Mesothelium tracing with CFSE in normal mice, KIC and KPfC mice with PDAs (arrows). (CFSE, green; DAPI, blue). Scale bar 50 μm.
(C) The normal pancreas or KPfC PDA tissues from the CFSE lineage tracing assay (B) were stained for cadherin-11 (red) or SOX9 (red) and overlapped with CFSE (green) and DAPI (blue) staining. Scale bar 50 μm.
(D) Pseudotime analysis with normal mesothelial cells (cluster 7), apCAFs (cluster 9) and other closely associated fibroblast populations (clusters 1, 4, 8, 10) from the integrated data (inset, Figure 1G).
(E) The heatmap displaying the top significant genes (cutoff: P<10−20) for normal mesothelial cells (cluster 7) and apCAFs (cluster 9) from the integrated data (Figure 1G). (F) Top biological processes of Gene Ontology (GO) analysis with the up-regulated gene cluster in apCAFs compared with normal mesothelial cells (E).
Wound-associated Tumor Paracrine Signals Induce the Mesothelial cell-apCAF Transition
In the merged scRNA-seq data of fibroblasts (Figure 1G), the mesothelial cell/apCAF populations had a close relationship with other fibroblast populations (clusters 1, 4, 8, 10). Given that during embryonic development, mesothelial cells can differentiate into fibroblasts and smooth muscle cells (Rinkevich et al., 2012), we performed pseudotime analysis with the normal mesothelial cell (cluster 7), apCAF (cluster 9) and other closely associated fibroblast populations (clusters 1, 4, 8, 10). We found that although normal mesothelial cells gained fibroblastic features and trended towards fibroblasts when they became apCAFs, they had limited contribution to the formation of other CAF populations (Figure 2D), supporting our previous report, which suggested that iCAF and myCAF lineages are derived from resident fibroblasts (Dominguez et al., 2020).
Since apCAFs gained a differential gene signature by down-regulating mesothelial genes and up-regulating fibroblastic genes (Figures 2E and S3A), we performed pathway and functional enrichment analysis with the up-regulated genes in apCAFs, in an effort to identify the biological processes involved in driving such gene signature changes. We found that many of the identified biological processes were related to wounding or inflammatory response (Figure 2F), suggesting that a wound-associated signal from the tumor niche can induce apCAF formation.
To validate the mesothelial cell-apCAF transition and robustly determine the fate of mesothelial cells during PDA progression, we exploited an inducible Cre-loxP system driven by the promoter of the mesothelial cell-specific gene Wt1 (Figure S2A). Similar systems have been used previously to demonstrate mesothelial cells give rise to fibroblasts in other disease settings such as liver and lung fibrosis (Li et al., 2013; Sontake et al., 2018). We established a Wt1CreERT2; R26LSL-tdTomato model (Figure 3A). The Wt1CreERT2/+; R26LSL-tdTomato/+ mice express a Cre-ERT2 fusion protein at the Wt1 gene locus. The CreERT2 excises the LSL sequence from the R26 locus upon tamoxifen (TAM) treatment, which will irreversibly induce tdTomato expression. We injected TAM into the Wt1CreERT2; R26LSL-tdTomato mice and harvested the pancreata 5 days after the last dose of TAM injection (Figure 3B). We found that the mesothelium of normal pancreata expressed tdTomato (Figures 3C–3D, arrows). As a negative control, R26LSL-tdTomato mice without Wt1CreERT2 had no tdTomato expression upon TAM injection (Figure 3E). Next, we traced the fate of mesothelial cells during PDA progression. After labeling mesothelial cells as tdTomato+ in the pancreas of Wt1CreERT2; R26LSL-tdTomato mice, we orthotopically injected a syngeneic PDA cell line derived from KPfC mice. We harvested the mature tumors 3 weeks after the implant (Figure 3B). We found that mesothelial cells substantially expanded in PDA and tdTomato+ cells were found in the periphery and interior of the tumors (Figures 3F–3H). Moreover, we noticed that in normal pancreas, mesothelial cells did not express fibroblastic markers such as αSMA and IL-6 (Figures 3C–3D), while tdTomato+αSMA+ (Figure 3F) or tdTomato+IL-6+ (Figure 3G) cells were seen within the stroma of PDA. This mesothelial lineage tracing experiment provided strong evidence that mesothelial cells gain fibroblastic features and contribute to the stroma formation in PDA.
Figure 3. Lineage Tracing of Mesothelial Cells in PDA with Inducible Transgenic Mouse Model.
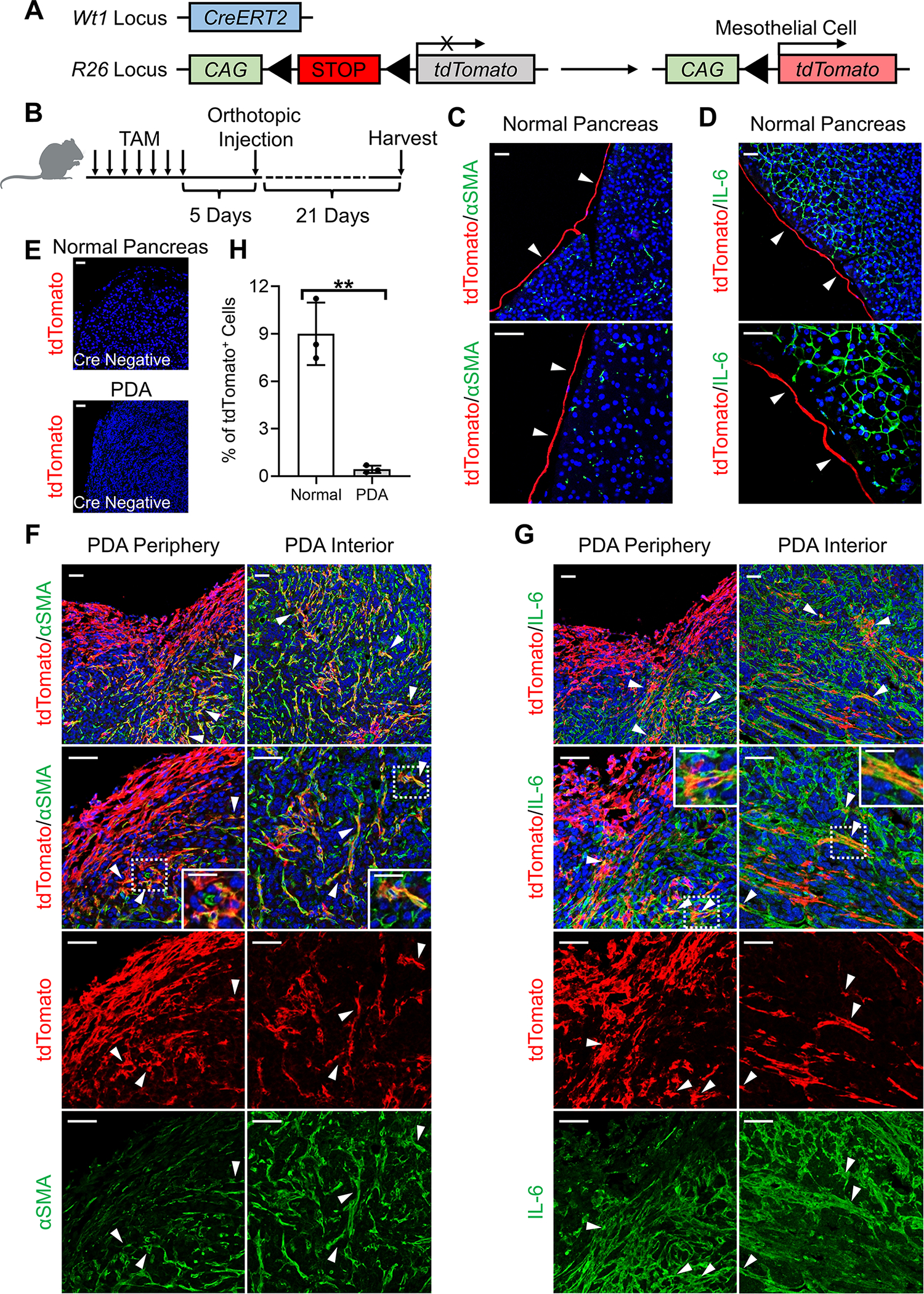
(A-B) Wt1CreERT2; R26LSL-tdTomato model was established to trace the fate of mesothelial cells (A). Seven doses of TAM were injected into the Wt1CreERT2; R26LSL-tdTomato mice to induce the tdTomato expression in mesothelial cells (B). Pancreata were either harvested for analysis or orthotopically injected with the KPfC cell line BMFA3 for tumor formation 5 days after the last dose of TAM injection.
(C-D) Normal pancreata of Wt1CreERT2; R26LSL-tdTomato harvested after TAM injection were stained for tdTomato (red), DAPI (blue), αSMA (green, C) or IL-6 (green, D). Scale bar 50 μm.
(E) R26LSL-tdTomato mice without Wt1CreERT2 were treated with TAM negative control and followed the same scheme as (B). Normal pancreata or PDAs were harvested and stained for tdTomato (red) and DAPI (blue). Scale bar 50 μm.
(F-G) PDAs from Wt1CreERT2; R26LSL-tdTomato mice following (B) were stained for tdTomato (red), DAPI (blue), αSMA (green, F) or IL-6 (green, G). Both periphery and interior of the tumors were shown. Scale bar 50 μm. Scale bars inside the magnification boxes 25 μm. (H) tdTomato+ cells of normal pancreata (C-D) and PDAs (F-G) were quantified, n=3, data shown as mean ± SD, statistical analysis, t-test, **P<0.01.
The Mesothelial Cell-apCAF Transition is Recapitulated by a Mesothelial Cell Line
To examine if a tumor paracrine signal could induce mesothelial cells to differentiate into apCAFs, we established a mouse pancreatic mesothelial cell line. Mesothelium was harvested from the normal pancreata of immortomice (Jat et al., 1991). Mesothelial explants were seeded onto a tissue culture dish and cultured at 33 °C. Five days after seeding, cells started to migrate from the mesothelium (Figure 4A). Once confluent, these cells were subjected to flow cytometry. 95% of the cells were podoplanin+MHC II+ (Figure 4A). They were collected and named pancreatic mesothelial (PanMeso) cells. PanMeso cells carry the thermo-induced large tumor antigen gene. They remain immortalized when they are cultured at 33 °C but undergo senescence within 7 days at 37 °C (Figure S3B). The phenotype of PanMeso cells is stable in vitro. We passaged them to 20 passages and found the expression of podoplanin and MHC II remained stable (Figure S3C). Moreover, the passage 10–20 PanMeso cells (Parental 1–3) were subjected to transcriptomic analysis and found to have a stable mesothelial gene signature with low expression of fibroblastic genes (Figure 4B).
Figure 4. Recapitulating the Mesothelial Cell-apCAF Transition in PanMeso Cells.
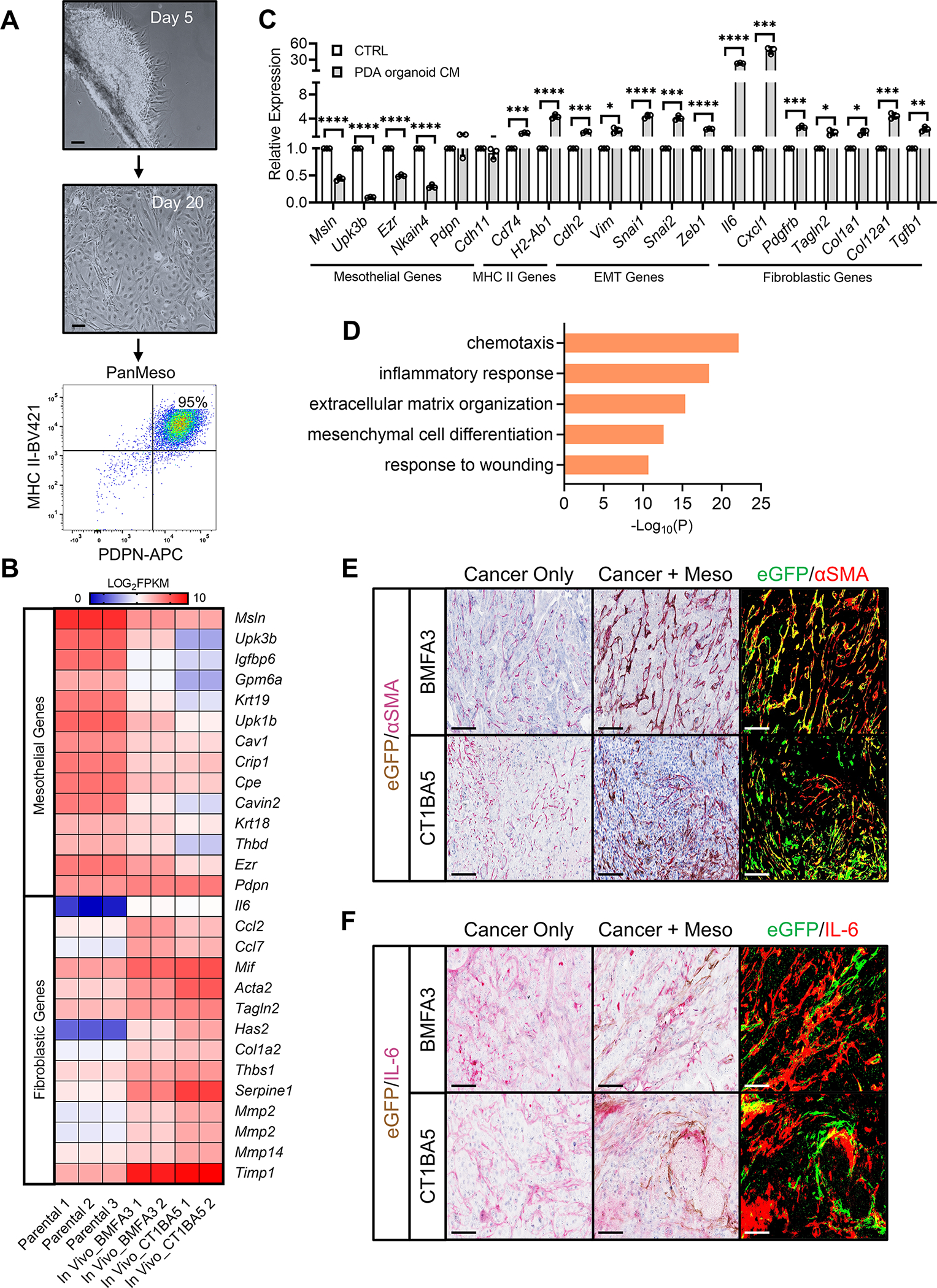
(A) Day 5 and day 20 of pancreatic mesothelium tissues from immortomice were shown. Cells became confluent and were subjected to FACS. Podoplanin+MHC II+ cells were collected (PanMeso cells). Scale bar 50 μm.
(B) The heatmap generated from the RNA-seq data comparing mesothelial genes and fibroblastic genes between parental PanMeso cells and PanMeso cells sorted from BMFA3 or CT1BA5 tumors.
(C) The PanMeso cells were treated with PDA organoid conditioned medium derived from KPfC tumors for 48 hrs. Cells were harvested and subjected to qPCR for mesothelial, MHC II, EMT and fibroblastic genes. n=3, data shown as mean ± SD, statistical analysis, t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
(D) Top biological processes of GO analysis with the up-regulated gene cluster in sorted compared with parental PanMeso cells (B) were shown.
(E-F) The tumors of (B) were fixed and stained for eGFP (brown) and the fibroblastic marker αSMA (red, E) or IL-6 (red, F). eGFP was also highlighted as green and αSMA or IL-6 was highlighted as red by ImageJ (right panel in E and F). Yellow marked the eGFP+ PanMeso cells expressing fibroblastic marker αSMA or IL-6. Scale bar 25 μm.
See also Figure S3.
We treated PanMeso cells with KPfC PDA organoid conditioned medium (CM) and performed qPCR (Figure 4C). Treatment with PDA organoid CM significantly down-regulated mesothelial genes (Msln, Upk3b, Ezr, Nkain4). Consistent with the scRNA-seq data (Figure S2A), the expression of Pdpn and Cdh11 was not decreased by the CM. Moreover, MHC II pathway genes (Cd74, H2-Ab1) were up-regulated by the CM. Interestingly, we found that many EMT-related genes (Cdh2, Vim, Snai1, Snai2, Zeb1) were induced by the CM, suggesting that the embryonic EMT program in mesothelial cells that contributes to the formation of mesenchymal cells in developing organs was reactivated by a tumor organoid-derived paracrine signal (Koopmans and Rinkevich, 2018; Rinkevich et al., 2012). Furthermore, CM induced a fibroblastic transition in the PanMeso cells evidenced by up-regulation of fibroblastic genes including iCAF genes (Il6, Cxcl1, Pdgfrb) and myCAF genes (Tagln2, Col1a1, Col12a1, Tgfb1).
To investigate whether the in vivo TME could induce the apCAF phenotype in PanMeso cells, we co-injected eGFP-PanMeso cells with a primary KPfC cell line (BMFA3 or CT1BA5). As control, we also injected cancer cells alone. Tumors were harvested one month after injection and digested into single cell suspension. GFP+ cells were collected by FACS sorting and subjected to transcriptomic analysis. We found that GFP+ PanMeso cells sorted from the tumors had a down-regulation of mesothelial genes and an up-regulation of fibroblastic genes compared to parental PanMeso cells (Figure 4B). We performed pathway and functional enrichment analysis with the differentially up-regulated genes in the sorted GFP+ cells and found that the biological processes involved were similar to the prior scRNA-seq analysis (Figure 2F), which included wounding and inflammatory response (Figure 4D). We further performed IHC staining with the tumors for eGFP and αSMA or IL-6 (Figures 4E–4F). We found that the TME induced eGFP+ PanMeso cells to express αSMA and IL-6. Taken together, the in vitro and in vivo data using PanMeso cells recapitulated the fibroblastic transition in mesothelial cells and underscored the importance of tumor paracrine signals in promoting this process.
apCAFs Induce Naïve CD4+ T Cells into Regulatory T Cells
Although apCAFs express MHC II molecules and can present antigen to CD4+ T cells, they lack classical co-stimulatory molecules (e.g., CD40, CD80 and CD86) that are necessary to induce full CD4+ T cell activation and clonal expansion following T cell receptor (TCR) ligation (Elyada et al., 2019). Therefore, we sought to understand the function of apCAFs in PDA and determine how the interaction between apCAFs and CD4+ T cells influences T cell phenotype. First, we compared the antigen-presenting capability among APCs, iCAFs, myCAFs, normal mesothelial cells and apCAFs. Normal mesothelial cells from normal pancreata (podoplanin+MHC II+) and APCs (podoplanin−MHC II+, including dendritic cells, macrophages), apCAFs (podoplanin+MHC II+), iCAFs (podoplanin+MHC II−Ly6C+) (Elyada et al., 2019), myCAFs (podoplanin+MHC II−Ly6C−) from KPfC tumors were sorted by FACS (Figures 5A and S4A). We incubated the sorted cells with the ovalbumin (OVA) peptide (OVA 323–339), and then co-cultured them with CD4+ T cells isolated from OT II mice (Barnden et al., 1998). We found that normal mesothelial cells and apCAFs induced T cell expression of early activation markers, CD25 and CD69, in an OVA-specific manner (Figures 5B and 5C), similar to APCs whereas iCAFs and myCAFs could not activate CD4+ T cells. Moreover, this effect was more obvious in apCAFs compared to normal mesothelial cells. To validate this result, we performed a similar assay using PanMeso cells (Figure 5A). PanMeso cells were pre-treated with control medium or PDA organoid CM for 2 days before the incubation with OVA peptide. We found that the formation of apCAFs induced by PDA organoid CM led to an increase of antigen-presenting capability in PanMeso cells (Figures 5B and 5C).
Figure 5. apCAFs Induce Naïve CD4+ T Cells into Tregs.
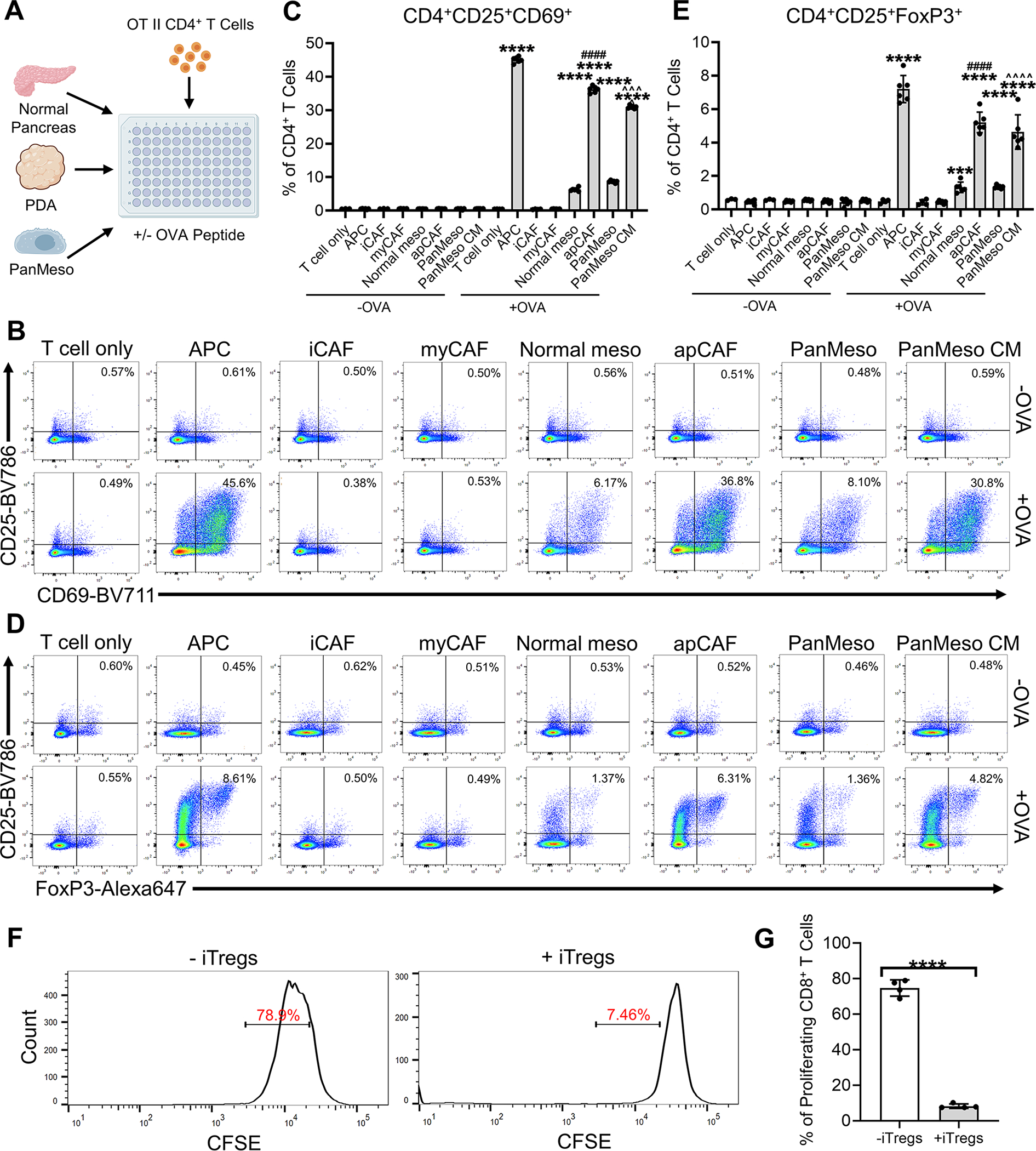
(A) An illustration of the APC/mesothelial cell/CAF-T cell co-culture assay.
(B-C) The CD4+ T cells after being co-cultured (A) were subjected to flow cytometry for the analysis of the early activation markers of TCR ligation CD25 and CD69 and representative plots were shown (B). CD4+CD25+CD69+ T cells of the flow cytometry were quantified (C), n=6, data shown as mean ± SD, statistical analysis, t-test, ****P<0.0001 vs without OVA control. ####P<0.0001 vs normal mesothelial cells. ^^^P<0.001 vs control PanMeso cells.
(D-E) The CD4+ T cells after being co-cultured (A) were subjected to flow cytometry for the analysis of Treg markers CD25 and FoxP3 and representative plots were shown (D). CD4+CD25+FoxP3+ Tregs of the flow cytometry were quantified (E), n=6, data shown as mean ± SD, statistical analysis, t-test, ***P<0.001, ****P<0.0001 vs without OVA control. ####P<0.0001 vs normal mesothelial cells. ^^^^P<0.0001 vs control PanMeso cells.
(F-G) Naïve OT II CD4+ T cells were co-cultured with apCAFs sorted from tumors in the absence or presence of OVA peptide. The uninduced CD4+ T cells (without OVA) and iTregs (with OVA) were then co-cultured with CFSE-labeled CD8+ T cells with subsequent flow cytometry analysis. Representative plots of CFSE-labeled CD8+ T cells were shown (F). Proliferating CD8+ T cells (CFSE low) were quantified (G), n=4, data shown as mean ± SD, statistical analysis, t-test, ****P<0.0001.
See also Figure S4.
The lack of co-stimulatory molecules on professional APCs can lead to T cell anergy or induction of regulatory T cells (Tregs) (Bour-Jordan and Bluestone, 2009; Ferrer et al., 2011; Mikami et al., 2020; Semple et al., 2011). Tregs are a subset of CD4+ T cells that are critical for immune tolerance. They are also a driver of immune evasion in cancer (Wing et al., 2019). Therefore, we tested the hypothesis that apCAFs induce Treg formation. We examined the presence of CD25+FoxP3+ Tregs in the CD4+ T cells co-cultured with OVA-loaded apCAFs. We found that apCAFs induced Treg formation in an antigen-specific manner (Figures 5D and 5E). The Treg-inducing capability of apCAFs was almost as efficient as tumor-associated APCs. In addition, we examined the expression of proliferation marker Ki67+ in Tregs and found that CD25+FoxP3+Ki67+ Tregs were increased by apCAFs (Figures S4B).
To validate the immunosuppressive function of the Tregs induced by apCAFs (iTregs), we performed a Treg suppression assay. First, we co-cultured naïve OT II CD4+ T cells with apCAFs in the absence or presence of OVA peptide. The uninduced CD4+ T cells (without OVA) and iTregs (with OVA) were then co-cultured with CFSE-labeled CD8+ T cells with subsequent flow cytometry analysis. We found that iTregs significantly inhibited the proliferation of CD8+ T cells (Figures 5F–5G), demonstrating CD8+ T cell inhibitory function. Taken together, these data suggest that apCAFs might be a unique immune-regulating CAF population that can induce Treg formation and expansion through antigen-dependent TCR ligation.
apCAFs are Present in Human Tumors and Correlate with Tregs
To demonstrate the human relevance of apCAFs, we collected a cohort of tumor tissues from 33 PDA patients and performed multiplex staining for apCAFs and Tregs. For this experiment, we exploited the marker PDGFRα which is expressed by all CAF populations except normal mesothelial cells (Figure S2A). Therefore, PDGFRα+CD74+ cells specifically highlighted apCAFs (Figure 6A, red arrows) while PDGFRα+CD74− cells highlighted other CAFs. We found that the percentages of apCAFs in human PDA were heterogeneous, ranging from ~0% up to ~35% (Figure 6B). In spite of this heterogeneity, we found apCAFs significantly correlated with Tregs in PDA patients while other CAFs had no correlation (Figures 6C–6D).
Figure 6. apCAFs are Present in Human Tumors and Correlate with Tregs.
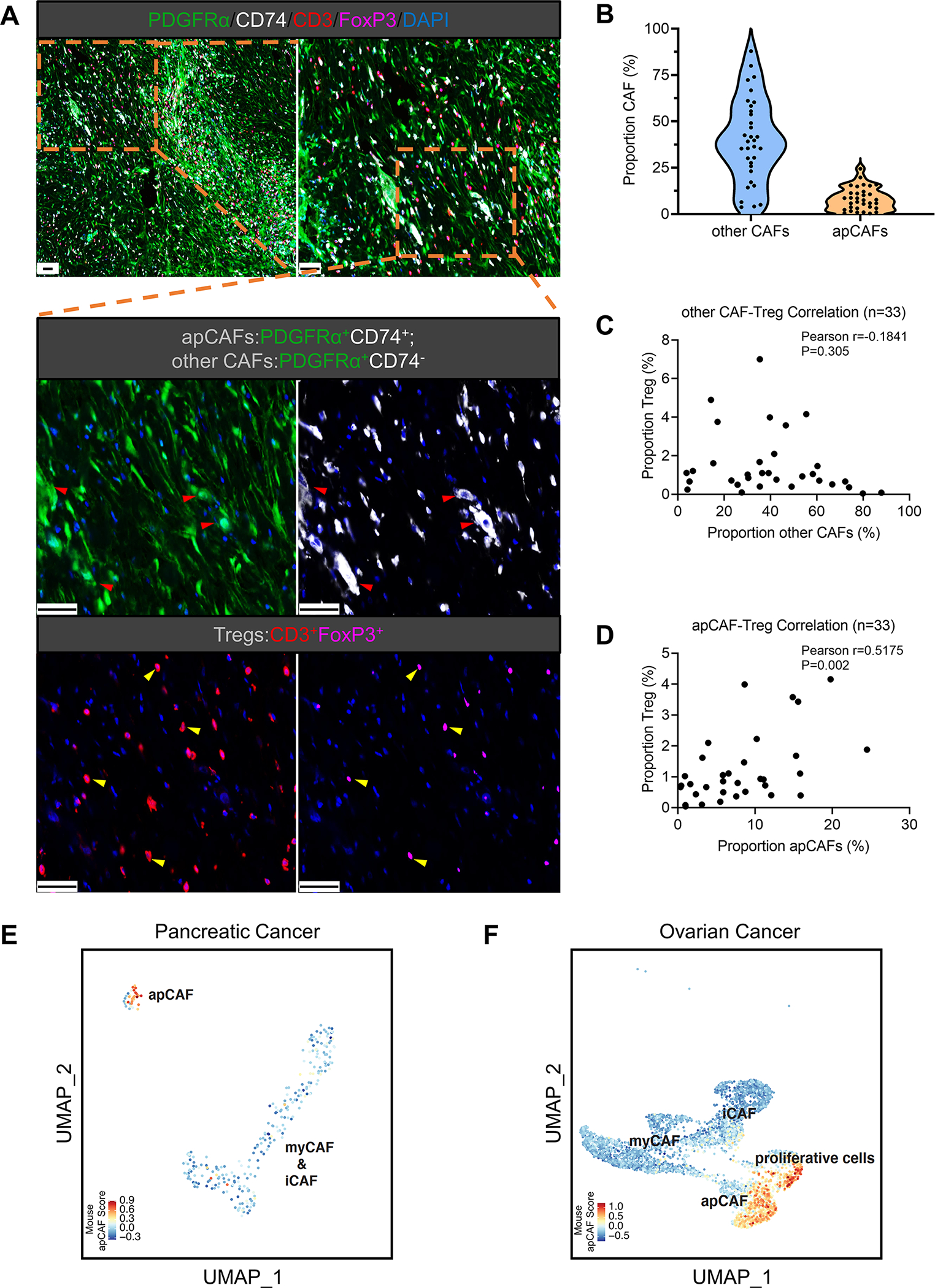
(A-D) A cohort of PDA tissues from 33 patients were subjected to multiplex staining for apCAFs (PDGFRα+CD74+), other CAFs (PDGFRα+CD74−) and Tregs (CD3+FoxP3+). PDGFRα (green), CD74 (white), CD3 (red), FoxP3 (magenta), DAPI (blue). Scale bar 50 μm (A). The staining images were analyzed and the percentages of apCAFs, other CAFs and Tregs were quantified by the HALO image analysis platform. Quantification of other CAFs and apCAFs was shown by the violin plot (B). The width of the violin plot represents frequency of patients (each dot represents one patient) in each region. Pearson correlation analysis was performed between other CAFs & Tregs (C) and apCAFs & Tregs (D).
(E-F) The mouse apCAF gene signature generated from the integrated dataset (Figure 1G) was scored in two human cancer scRNA-seq datasets including PDA (E) and ovarian cancer (F). The fibroblast subpopulations from these two datasets were identified based on the cluster annotations mentioned in the original publications (iCAF, myCAF, apCAF in the PDA dataset; iCAF, myCAF, apCAF and proliferative cells in the ovarian cancer dataset).
See also Figure S5.
Moreover, we evaluated our mouse apCAF gene signature in two human scRNA-seq datasets of PDA and ovarian cancer (Hornburg et al., 2021; Lee et al., 2021). First, we identified the fibroblast subpopulations from these two datasets by using cluster annotations mentioned in the original publications (iCAF, myCAF, apCAF in the PDA dataset; iCAF, myCAF, apCAF and proliferative cells in the ovarian cancer dataset) (Figures 6E–6F). We then utilized the apCAF gene signature derived from our scRNA-seq data in mouse PDA (Figure 1G) and assigned scores for these mouse apCAF signature genes to the fibroblast compartments in the two human scRNA-seq datasets. We found that our mouse apCAF signature overlapped with the apCAF clusters originally defined by Hornburg et al. and Lee et al. (Figures 6E–6F).
In a recent study, MHC II+ CAFs were also found in non-small cell lung cancer (Kerdidani et al., 2022). To examine whether the MHC II+ CAFs in Kerdidani et al., (2022) are similar to pancreatic apCAFs, we scored the signature of the MHC II+ CAFs reported by Kerdidani et al., in the two human scRNA-seq datasets of PDA and ovarian cancer. We found that the signature of lung MHC II+ CAFs from Kerdidani et al., marked apCAFs in PDA but in ovarian cancer it marked iCAFs and only a small portion of apCAFs (Figure S5A–S5B). Additional analysis of the immune compartments indicates that the Kerdidani et al., MHC II+ CAF signature also highlights myeloid cells, which we did not see with our apCAF signature (Figure S5C–S5D). Taken together, these data support the existence of apCAFs in human tumors and also demonstrate that the signature derived from our analysis veritably marks the apCAF population across different species and other tumor types. However, these data also indicate that in some organs, such as lung, apCAFs may not be specific to the mesothelial lineage, and as Kerdidani et al., suggested, alveolar type II cell might be another cell of origin for MHC II+ CAFs in lung.
IL-1 and TGFβ Signaling Pathways are Responsible for the Induction of apCAFs
To identify the signaling pathways that drive the mesothelial cell to apCAF transition, we extracted differential genes that were up-regulated in apCAFs and subjected them to motif enrichment analysis (Figure 7A). The gene profile of apCAFs was driven by NF-κB signaling (Nfkb1, Rela) or TGFβ signaling (Smad1, Smad3, Smad4). These data are highly relevant, given that these two signaling pathways have been demonstrated to be the main drivers of CAF heterogeneity in PDA (Biffi et al., 2019; Dominguez et al., 2020). NF-κB signaling, which can be induced by IL-1, is predicted to be responsible for the formation of the iCAF lineage, while TGFβ is predicted to drive the myCAF lineage during PDA progression. To validate this result and test whether these two signaling pathways were also responsible for driving mesothelial cells to differentiate into apCAFs, we treated PanMeso cells with PDA organoid CM and found that NF-κB signaling and TGFβ signaling were activated (Figure 7B). We also examined if IL-1 and TGFβ drive an apCAF phenotype in mesothelial cells. We treated PanMeso cells with IL-1, TGFβ or a combination of the two factors, and investigated the resulting gene signature by qPCR (Figures 7C and 7D). We found that either IL-1 or TGFβ could significantly down-regulate the expression of mesothelial genes (Msln, Upk3b, Ezr, Nkain4) (Figure 7C). In contrast, Cdh11 and Pdpn were not down-regulated, supporting our previous finding that these two genes are stable markers of mesothelial cell-apCAF and other CAF lineages. In comparison, we found that fibroblastic genes were differentially up-regulated by IL-1 or TGFβ (Figure 7D). For example, some genes were specifically up-regulated by IL-1 (Cxcl1), some were up-regulated by TGFβ (Cxcl12, Col1a1, Col1a2, Col12a1, Tgfb1), some could be up-regulated by either factor (Il6), and some depended on the presence of both factors (Il1a). Taken together, these qPCR data suggest that IL-1 and TGFβ can drive normal mesothelial cells into fibroblasts with an apCAF phenotype.
Figure 7. IL-1 and TGFβ are Responsible for Mesothelial Cell-apCAF Transition.
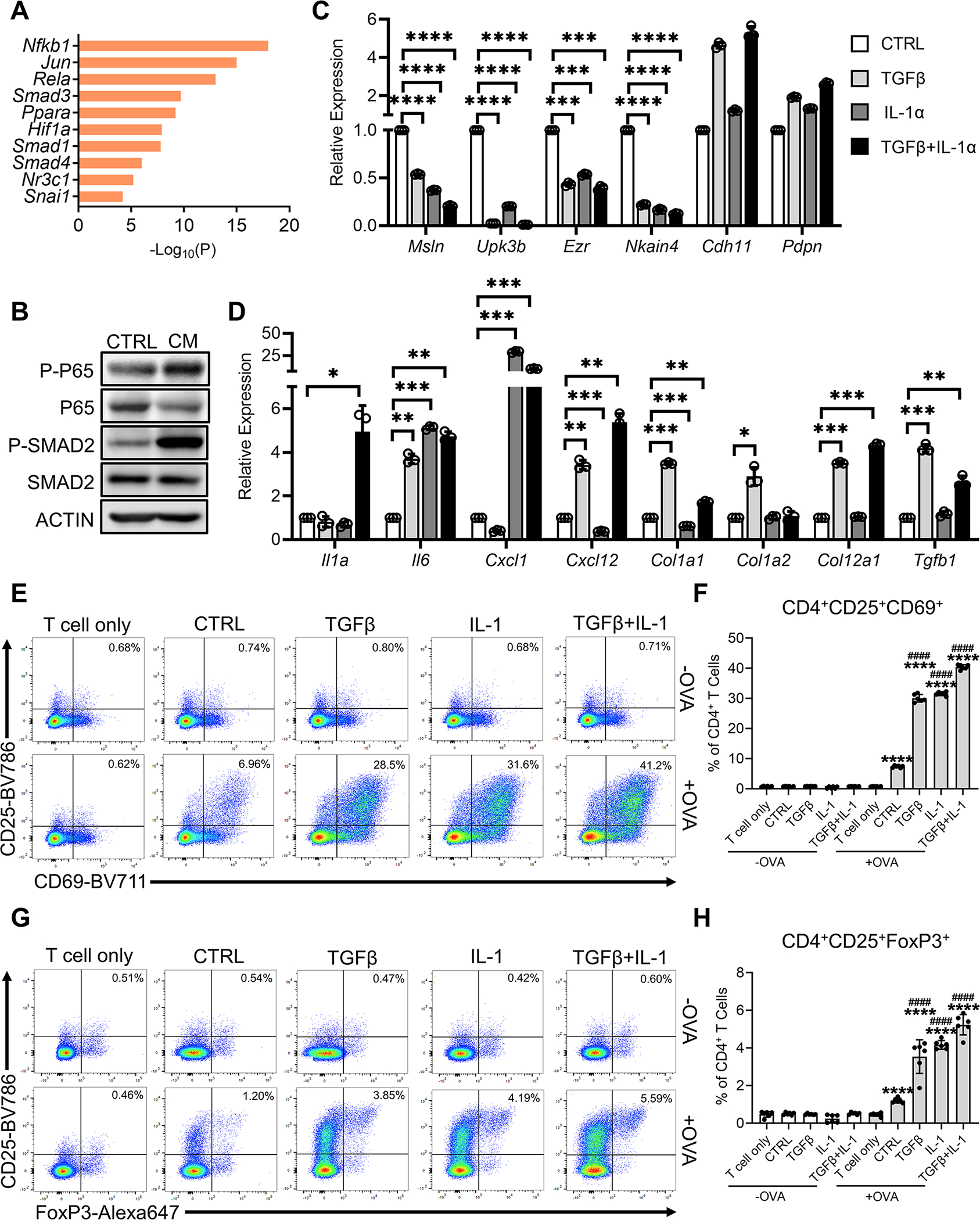
(A) The differential genes that were up-regulated in apCAFs compared to normal mesothelial cells from the integrated data (Figure 1G) were subjected to motif enrichment analysis. Top transcription factors from the analysis are shown.
(B) Western blots with PanMeso cells treated with control medium or PDA organoid conditioned medium (CM) for 48 hrs, probed for the phosphorylation of the NF-κB signaling (P-P65) and TGFβ signaling (P-SMAD2) proteins.
(C-D) The PanMeso cells were treated with IL-1α (5 ng/mL), TGFβ (30 ng/mL) or a combination of IL-1α (5 ng/mL) and TGFβ (30 ng/mL) for 48 hrs and subjected to qPCR for mesothelial (C) and fibroblastic (D) genes. n=3, data shown as mean ± SD, statistical analysis, t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
(E-F) The PanMeso cells were pre-treated with IL-1α (5 ng/mL), TGFβ (30 ng/mL) or a combination of IL-1α (5 ng/mL) and TGFβ (30 ng/mL) for 48 hrs and washed. Cells were then incubated with the OVA peptide (OVA 323–339) for 4 hrs. The PanMeso cells were washed and co-cultured with CD4+ T cells isolated from OT II mice for 18 hrs. The CD4+ T cells after the co-culture were subjected to flow cytometry for the analysis of the early activation markers of TCR ligation CD25 and CD69 and representative plots were shown (E). CD4+CD25+CD69+ T cells of the flow cytometry were quantified (F), n=6, data shown as mean ± SD, statistical analysis, t-test, ****P<0.0001 vs without OVA control. ####P<0.0001 vs control with OVA treatment.
(G-H) The CD4+ T cells after the co-culture were subjected to flow cytometry for the analysis of the Treg markers CD25 and FoxP3 and representative plots were shown (G). CD4+CD25+FoxP3+ Tregs of the flow cytometry were quantified (H), n=6, data shown as mean ± SD, statistical analysis, t-test, ****P<0.0001 vs without OVA control. ####P<0.0001 vs control with OVA treatment.
To examine whether IL-1 and TGFβ affect the function of mesothelial cells, we performed the mesothelial cell-CD4+ T cell co-culture assay using PanMeso cells (Figures 7E–7H). Parental PanMeso cells were pre-treated with IL-1, TGFβ or a combination of the two factors for 2 days and washed before incubation with OVA peptide. CD4+ T cells isolated from OT II mice were then co-cultured with PanMeso cells and subjected to flow cytometry for the analysis of T cell ligation and Treg formation. We found that OVA-specific TCR activation was enhanced by pre-treatment of PanMeso cells with IL-1 or/and TGFβ (Figures 7E and 7F). In addition, IL-1 or/and TGFβ pre-treatment of PanMeso cells significantly enhances Treg-induction (Figures 7G and 7H). Our data suggest that IL-1/NF-κB and TGFβ signaling pathways are responsible for the differentiation of normal mesothelial cells into apCAFs with the capacity to activate CD4+ T cells and promote their differentiation into FoxP3+ Tregs.
Mesothelial Cell to apCAF Transition can be Inhibited by an anti-Mesothelin Antibody
Although IL-1 and TGFβ induce mesothelial cell to apCAF transition, these two signals are important in the formation of iCAF and myCAF populations. They also affect many biological processes in cancer cells and other stromal cells (Derynck et al., 2021; Mantovani et al., 2018). Moreover, TGFβ is also known to be a tumor suppressor (Huang et al., 2019). Therefore, IL-1 and TGFβ may not serve as good targets for specific inhibition of the mesothelial cell to apCAF transition. Recently, two studies reported that mesothelial cells contribute to the fibroblast formation in the mouse models of surgical adhesion and liver fibrosis (Koyama et al., 2017; Tsai et al., 2018). In these two studies, a mouse specific blocking monoclonal antibody (mAb) against the mesothelial cell marker mesothelin (MSLN Ab) was used to inhibit the mesothelial cell to fibroblast transition. In turn, the fibrotic phenotypes were significantly reduced in both disease settings. Therefore, we tested the effect of the same mAb on apCAF formation from mesothelial cells. We treated PanMeso cells with KPfC PDA organoid CM with control Ab or MSLN Ab and then performed qPCR. Treatment with MSLN Ab significantly inhibited the down-regulation of mesothelial genes and the up-regulation of EMT and fibroblastic genes induced by PDA organoid CM (Figure 8A). To validate this effect in vivo, we performed the mesothelial cell lineage tracing study (Figure 3B) with the MSLN Ab treatment. After the TAM induction, Wt1CreERT2; R26LSL-tdTomato mice were treated with control Ab or MSLN Ab, and then implanted with PDA cells orthotopically. Treatment with control Ab or MSLN Ab was maintained during the PDA progression. We found that MSLN Ab treatment significantly reduced the percentage of tdTomato+ cells in TAM-induced Wt1CreERT2; R26LSL-tdTomato mice (Figures 8B and 8C). Moreover, the fibroblastic feature of mesothelial cells marked by αSMA and IL-6 was also significantly decreased by the MSLN Ab (Figures 8B and 8D–8E). These data suggest that the mesothelial cell to apCAF transition can be inhibited by targeting MSLN in vitro and in vivo.
Figure 8. Inhibition of Mesothelial Cell to apCAF Transition by Targeting Mesothelin.
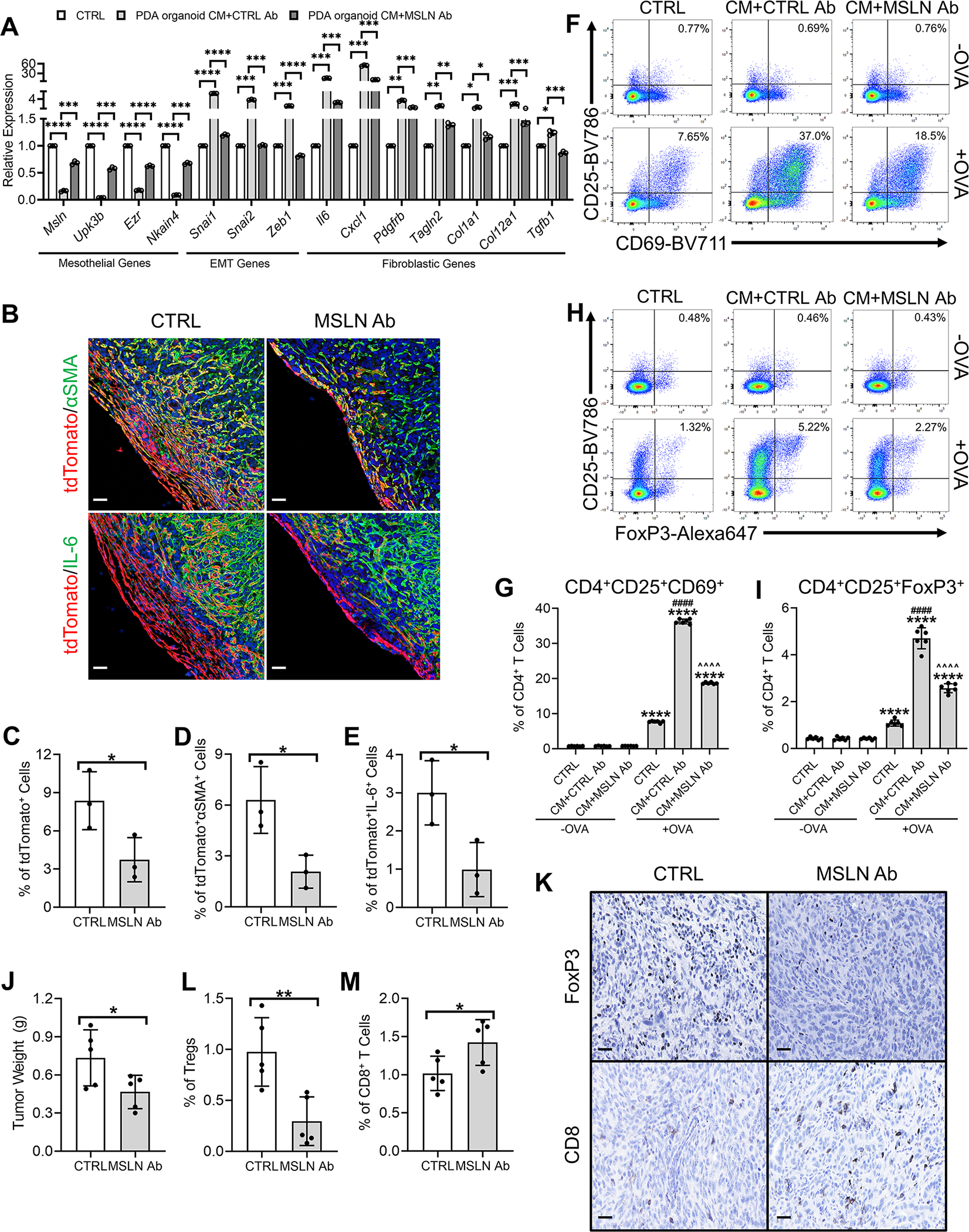
(A) PanMeso cells were treated with KPfC PDA organoid CM with the presence of isotype control Ab (CTRL Ab) or mouse specific mesothelin antibody (MSLN Ab) and subjected to qPCR to examine mesothelial, EMT and fibroblastic genes. n=3, data shown as mean ± SD, statistical analysis, t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
(B-E) Seven doses of TAM were injected into the Wt1CreERT2; R26LSL-tdTomato mice to induce the tdTomato expression in mesothelial cells. 3 days after the last dose of TAM injection, mice were given 1 dose of CTRL Ab or MSLN Ab. The mice were then orthotopically injected with the KPC cell line 6499c4 5 days after the last dose of TAM injection. Control Ab or MSLN Ab treatment were given once per week during the tumor progression. Tumors were harvested 21 days after the orthotopic injection and stained for tdTomato (red), DAPI (blue), αSMA (green) or IL-6 (green) (B). Scale bar 50 μm. tdTomato+ (C), tdTomato+αSMA+ (D) and tdTomato+IL-6+ (E) cells were quantified. n=3, data shown as mean ± SD, statistical analysis, t-test, *P<0.05.
(F-G) PanMeso cells were pre-treated with control medium or PDA organoid CM in the presence of CTRL Ab or MSLN Ab for 48 hrs, washed and incubated with the OVA peptide (OVA 323–339) for 4 hrs. The PanMeso cells were then washed again and co-cultured with CD4+ T cells isolated from OT II mice for 18 hrs. The CD4+ T cells after the co-culture were subjected to flow cytometry for the analysis of the early activation markers of TCR ligation CD25 and CD69 and representative plots were shown (F). CD4+CD25+CD69+ T cells of the flow cytometry were quantified (G), n=6, data shown as mean ± SD, statistical analysis, t-test, ****P<0.0001 vs without OVA control. ####P<0.0001 vs control with OVA treatment. ^^^^P<0.0001 vs PDA organoid CM treatment.
(H-I) The CD4+ T cells after the co-culture were subjected to flow cytometry for the analysis of the Treg markers CD25 and FoxP3 and representative plots were shown (H). CD4+CD25+FoxP3+ Tregs of the flow cytometry were quantified (I), n=6, data shown as mean ± SD, statistical analysis, t-test, ****P<0.0001 vs without OVA control. ####P<0.0001 vs control with OVA treatment. ^^^^P<0.0001 vs PDA organoid CM treatment.
(J-M) C57BL/6 mice were treated with one dose of control or MSLN Ab and 6499c4 cells were then injected orthotopically into the mice. Control or MSLN Ab treatment were maintained once per week during the tumor formation. 21 days after orthotopic injection, tumors were harvested, weighed (J) and stained for FoxP3 and CD8 (K). Scale bar 25 μm. Tumor weights (J), FoxP3+ Tregs (L) and CD8+ T cells (M) were quantified. n=5, data shown as mean ± SD, statistical analysis, t-test, *P<0.05, **P<0.01.
See also Figure S6.
We then performed the PanMeso cell-CD4+ T cell co-culture assay (Figure 5A) in the presence of control Ab or MSLN Ab. We found that MSLN Ab significantly inhibited the antigen-presenting and Treg-inducing capabilities of PanMeso cells induced by the PDA organoid CM (Figures 8F–8I). We decided to further investigate the effect of MSLN Ab on Treg formation in a syngeneic mouse model of PDA. To establish such a model with a high baseline number of Tregs, we exploited a library of congenic tumor cell clones derived from KPC mice. Tumors generated from these clones have been characterized thoroughly for the immune landscape in a previous study and recapitulated T cell-inflamed and non-T-cell-inflamed tumor microenvironment (Li et al., 2018). First, we validated three T cell-inflamed (6499c4, 2838c3, 6620c1) and two non-T-cell-inflamed (6419c5, 6694c2) clones by establishing orthotopic tumors with each clone respectively. Tumor tissues were subjected to IHC staining for Tregs and CD8+ T cells (Figure S6). Consistent with the previous report (Li et al., 2018), the three T cell-inflamed clones had substantially higher numbers of Tregs and CD8+ T cells. We also noticed that clone 6499c4 tumors had the highest proportion of Tregs. Therefore, we orthotopically injected 6499c4 tumor cells into C57BL/6 mice and subsequently treated the mice with control Ab or MSLN Ab. Strikingly, we found that treatment with the MSLN Ab significantly decreased tumor weight and reduced Treg abundance while increasing the percentage of CD8+ T cells (Figures 8J–8M). Taken together, these data suggest that targeting MSLN may be a potential strategy to effectively inhibit mesothelial cell to apCAF transition and overcome apCAF-induced immunosuppression.
Discussion
Given that the majority of organs are lined by mesothelium, it is likely that our findings are not limited to PDA. We scored our mouse apCAF signature in a scRNA-seq dataset derived from human ovarian cancer and found it marked two populations of CAFs in this dataset. Two studies in breast cancer reported a CAF-S1 CAF population, which expressed MHC II and was found to be linked to Tregs and immunotherapy resistance (Costa et al., 2018; Kieffer et al., 2020). However, although we have shown apCAFs induce Treg formation in PDA, they may have more complicated immune-regulating functions. For example, MHC II+ CAFs are associated with Tregs and immunotherapy resistance in breast cancer (Costa et al., 2018; Kieffer et al., 2020), but they were also shown to have positive correlation with survival of breast cancer patients (Friedman et al., 2020). In addition, MHC II+ CAFs in lung cancer were shown to be able to enhance cancer immunity through CD4+ T cells (Kerdidani et al., 2022). In a study using mass cytometry to profile CAF lineages in mouse models of PDA, four populations of CD105− CAFs were identified, two populations of which expressed MHC II (Hutton et al., 2021). Interestingly, the four CD105− CAF populations were found to positively correlate with different CD8+ and CD4+ T cell subtypes including Tregs. RNA-seq data of the sorted CD105− CAFs showed a mesothelial gene signature. CD105− cells were then cultured and immortalized by SV40 LT by retroviral vector for subsequent studies. Our data suggest that mesothelial cells/apCAFs are sensitive to senescence in vitro. Therefore, in vitro transduction of non-immortalized CD105− cells may lead to the exclusion of the mesothelial lineage. Although further studies are still needed to determine the identity of the four CD105− populations, these data suggest that apCAFs may be associated with a T cell-inflamed phenotype and likely have important functions in regulating the Treg-CD8+ T cell ratio. Taken together, these studies underscore the importance of apCAFs and highlight that future lineage tracing, ablation and functional studies are needed to determine the contribution of mesothelial cells to CAFs and immune regulation in different cancer types.
Multiple lines of evidence have reported that there may be crosstalk between CAFs and Tregs. For example, the ablation of myCAFs can increase the number of Tregs in PDA (Ozdemir et al., 2014). On the other hand, the depletion of Tregs can lead to a reduction of myCAFs (Zhang et al., 2020). Importantly, in these studies, the mesothelial cell-apCAF lineage was not examined. We predict that the mesothelial-apCAF lineage would be affected by myCAF ablation and thus the results of ablation studies should be re-examined in the context of new information defining CAF subsets more completely. Further studies are needed to elucidate the functional interaction among different CAF populations and the relationship between CAFs and immune cells.
We identified a strategy to inhibit apCAF formation by targeting MSLN. MSLN is a cell-surface glycoprotein expressed by mesothelial cells under physiological conditions. However, MSLN is also expressed by cancer cells in many cancer types, including PDA, ovarian cancer, endometrial cancer, lung cancer, biliary cancer, gastric cancer and breast cancer (Del Bano et al., 2019; Hassan et al., 2016). Due to its high expression in a variety of solid tumors and low expression in other critical tissues MSLN has become an attractive target for cancer therapy. To date, many strategies have been developed to target MSLN+ cancers, including, antibody-drug conjugates (ADCs), chimeric antigen receptor (CAR) T cells and vaccines against MSLN. These strategies are being actively tested preclinically and clinically (Hassan et al., 2016). Here we found that an mAb targeting MSLN effectively inhibited apCAF formation and reduced the Treg/CD8+ T cell ratio. These results suggest that current MSLN targeted therapies may have synergistic effects by targeting MSLN+ cancer cells and apCAFs.
In summary, we have identified mesothelial cells as the origin of apCAFs. Through a fibroblastic transition, mesothelial cells contribute to the formation of a Treg-inducing CAF population, termed apCAFs, which directly contribute to tumor immune regulation. Understanding the process of mesothelial to apCAF transition and the function of apCAFs in additional detail has the potential to reveal therapeutic strategies that could unlock the efficacy of immune therapy for PDA and other cancers that are resistant to immune therapy.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Huocong Huang (huocong.huang@utsouthwestern.edu)
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate new raw scRNA-seq data. The raw scRNA-seq data we used to extend our analyses on fibroblasts and raw RNA-seq data of PanMeso cells have been uploaded to the National Center for Biotechnology Information’s Gene Expression Omnibus database repository (https://www.ncbi.nlm.nih.gov/geo) under accession number GSE125588 and GSE196740. In addition, we consulted other publicly available scRNA-seq datasets, including GSE129455 and GSE156405 from the National Center for Biotechnology Information’s Gene Expression Omnibus database repository, E-MTAB-8483 from ArrayExpress Archive of Functional Genomics Data (https://www.ebi.ac.uk/arrayexpress/) and EGAS00001004935 from EGA European Genome-Phenome Archive (https://ega-archive.org/). Additional information related to the data in this study is available from the lead contact upon request.
Experimental Model and Subject Details
Cell Lines
BMFA3 and CT1BA5 were mouse primary pancreatic cancer cell lines derived from KPfC mice on a C57BL/6 background and isolated as described previously (Huang et al., 2019; Ludwig et al., 2018). KPC cell clones 6419c, 6694c2, 6499c4, 2838c3 and 6620c1 on a C57BL/6 background were obtained from Dr. Ben Stanger (University of Pennsylvania) (Li et al., 2018). PanMeso cells were established as follows. Mesothelium was harvested from normal pancreata of immortomice under the dissecting microscope. The pancreatic mesothelium was seeded onto a tissue culture dish and cultured at 33 °C. Cells were subjected to FACS when they had been cultured for 20 days and became confluent. Podoplanin+MHC II+ cells were collected and named pancreatic mesothelial (PanMeso) cells. All mouse primary pancreatic cancer cell lines were cultured in DMEM (Invitrogen) containing 10% FBS and 1% penicillin/streptomycin (Gibco/Thermo) and maintained at 37°C in an atmosphere of 5% CO2. The PanMeso cells were cultured in the mesothelial cell media (medium 199 (Gibco/Thermo), 10% FBS, 1% penicillin/streptomycin (Gibco/Thermo), 3.3 nM mouse epidermal growth factor (Biolegend), 400 nM hydrocortisone (MilliporeSigma), 870 nM zinc-free bovine insulin (MilliporeSigma), 20 mM HEPES (Thermo Fisher Scientific)), maintained at 33°C, washed with PBS three times and cultured in new mesothelial cell media at 37°C three days before any experiments. To examine cell senescence, β-galactosidase staining kit was used following the kit protocol (Cell Signaling). All cell lines in this study were confirmed to be free of mycoplasma (e-Myco kit, Boca Scientific) before use.
Animals
KIC and KPfC mice were generated as previously described (Hingorani et al., 2003; Hingorani et al., 2005). Mice were sacrificed when they had early-stage tumors (40-day-old) or late-stage (60-day-old) tumors for KIC and KPfC mice. The KIC mice are on a mixed background (C57BL/6 with FVB). The KPfC mice are a pure C57BL/6 genetic background. For staining and lineage tracing assays, 3–6 mice (male and female) were used per group. Normal pancreata were obtained from Cre-negative littermates of the KIC or KPfC mice. 6-week-old mice male and female mice were purchased from The Jackson Laboratory, including C57BL/6J mice (JAX stock #000664), Wt1CreERT2 mice (JAX stock #010912), R26LSL-tdTomato mice (JAX stock #007914), immortomice (JAX stock #032619) and OT II mice (JAX stock #004194). Wt1CreERT2 mice were backcrossed into a C57BL/6 background. All animals were housed in a pathogen-free facility with 24-hr access to food and water. Animal experiments in this study were approved by and performed in accordance with the institutional animal care and use committee at the UTSW Medical Center at Dallas. Mice were euthanized by cervical dislocation under anesthesia.
Patient samples
Unstained paraffin-embedded 5μm tissue sections of 33 PDA patients were obtained with voluntary patient consent from UTSW Tissue Management Shared Resource and MD Anderson Institutional Tissue Bank, which were obtained under a protocol approved by the UTSW or MD Anderson Institutional Review Board. Age and gender of the patients were undisclosed.
Method Details
Plasmids
pLV-eGFP (Addgene Plasmid #36083) was used to expressed eGFP in PanMeso cells.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissues were cut in 5-μm sections. Sections were evaluated by single color or multiplex immunohistochemical analysis following our previously reported protocol (Sorrelle et al., 2019) using antibodies specific for cadherin-11 (LSBio, 1:300), podoplanin (MilliporeSigma, 1:300), mesothelin (LSBio, 1:1000), CD74 (for mouse, Biolegend, 1:300; for human, Cell Signaling, 1:300), SOX9 (MilliporeSigma, 1:500), tdTomato (MyBioSource, 1:1000), αSMA (Biocare Medical, 1:500), IL-6 (LSBio, 1:1000), eGPF (Abcam, 1:200), PDGFRα (Cell Signaling, 1:500), FoxP3 (R&D Systems, 1:500), CD3 (Invitrogen, 1:500), CD8 (Cell Signaling, 1:500). For brightfield images, slides were scanned by NanoZoomer 2.0-HT digital slide scanner (Hamamatsu) and images were obtained with NDP.view2 software (Hamamatsu) and analyzed by ImageJ. For fluorescent images, images were obtained and analyzed with the Zeiss LSM 780 confocal microscope, ZEN software (Zeiss) and ImageJ. For quantification of multiplex staining, slide images were captured at 40X magnification using a Vectra Polaris Slide Scanner (AKOYA Biosciences). Images were deconvoluted and re-stitched using Phenochart and inForm softwares (Akoya Biosciences). Reconstituted images underwent multiplex quantitative analysis using HALO image analysis software (Indica Labs). Density and percentages of signal or cell types were quantified.
RT-PCR
RNA was extracted using RNeasy Mini Kit (Qiagen). cDNAs were synthesized from 1 μg of total RNA using the iScript cDNA synthesis kit (Bio-Rad). The expression of genes was measured by qRT-PCR with SYBR-Green Master Mix (Bio-Rad). For a detailed list of qRT-PCR primer sequences, see Table S1.
Western Blot Analysis
Cells were extracted with radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Nonidet P-40) for SDS-PAGE. Protein concentration was determined using a BCA Protein Assay Kit (Thermo Fisher Scientific). Laemmli sample buffer was then added to the protein lysates and boiled for 10 mins. The proteins were resolved by SDS-PAGE, electrophoretically transferred to nitrocellulose membranes, and blocked in 5% nonfat dry milk or bovine serum albumin. Blocking buffer was then removed, and the membranes were incubated with primary antibodies of SMAD2 (Cell Signaling, 1:1000), Phosph-SMAD2 (Cell Signaling, 1:1000), P65 (Santa Cruz, 1:1000) or Phospho-P65 (Cell Signaling, 1:1000) in TBST (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) for 1 hr, then washed 3 × 10 mins with TBST, and incubated with horseradish peroxidase-conjugated anti-mouse or rabbit secondary antibody (Jackson ImmunoResearch Laboratories) for 1 hr. The secondary antibody was removed by washing 3 × 10 mins with TBST. The membranes were incubated with SuperSignal West Pico substrate (Thermo Fisher Scientific) for the detection of the immunoreactive bands.
Orthotopic Models
BMFA3 or CT1BA5 cells were injected orthotopically (5 × 105 cells) with or without PanMeso cells (5 × 105 cells) in 6-week-old C57BL/6 mice (male and female, n=6/group). To harvest early-stage tumors, mice were sacrificed 1 week after injection. To harvest late-stage tumors, mice were sacrificed 1 month after injection. Tumors were then subjected to digestion, FACS sorting cells for RNA-seq or IHC staining. KPC clones 6419c5, 6694c2, 6499c4, 2838c3 or 6620c1 were injected orthotopically (2 × 105 cells) in 6-week-old C57BL/6 mice (male and female, n=5/group). For therapies, C57BL/6 mice were treated with one dose of control (clone 2A3, BioXCell, 50 μg/dose) or anti-mouse MSLN monoclonal Ab (clone B35, MBL Bio, 50 μg/dose) before tumor implant. Control or MSLN Ab treatment were maintained once dose per week during the tumor formation. 21 days after orthotopic injection, tumors were harvested for staining.
Lineage-tracing Assay
CellTrace CFSE dye (Thermo Fisher Scientific, 1:1000, 2mL/mouse) was injected intraperitoneally into C57BL/6 mice bearing normal pancreata or late-stage-tumor-bearing KIC or KPfC mice (60-day-old). Normal pancreata or PDAs were harvested 2 days after the injection, snap freezed by liquid nitrogen, and cut into 10-μm frozen sections. Tissues were then fixed by ice-cold 100% methanol for 15 mins at −20°C and rinsed 3 × 5 mins with PBS (1.86 mM NaH2PO4, 8.41 mM Na2HPO4, 175mM NaCl, pH 7.4). For CFSE signal imaging, fixed tissues were directly coverslipped with Prolong Gold Antifade Reagent with DAPI (Cell Signaling). For co-staining, fixed tissues were then blocked with 5% bovine serum albumin in PBST (1.86 mM NaH2PO4, 8.41 mM Na2HPO4, 175mM NaCl, 0.05% Tween 20, pH 7.4) for 1 hr, incubated with diluted primary antibody against cadherin-11 (LSBio, 1:200), SOX9 (MilliporeSigma, 1:200) or α-SMA (Biocare Medical, 1:500) for 1hr, rinsed 3 × 5 mins with PBST, incubated with diluted secondary antibody for 1hr, then rinsed 3 × 5 mins with PBST and coverslipped with Prolong Gold Antifade Reagent with DAPI. For lineage tracing in Wt1CreERT2; R26LSL-tdTomato model, seven doses of TAM were injected into the Wt1CreERT2; R26LSL-tdTomato mice to induce the tdTomato expression in mesothelial cells. TAM (MilliporeSigma, 20 mg/ml) was dissolved in corn oil by shaking overnight at 37° C, and injected intraperitoneally (75 mg/kg body weight). Pancreata were either harvested for analysis or orthotopically injected with BMFA3 cells (5 × 105 cells in PBS) 5 days after the last dose of TAM injection. Tumor tissues were harvested 21 days after implant, stained for tdTomato, αSMA, IL-6 and DAPI following the multiplex staining protocol. Images were obtained and analyzed with the Zeiss LSM 780 confocal microscope, ZEN software (Zeiss) and ImageJ.
Tumor Organoid Conditioned Medium
Mouse PDA organoids were isolated from KPfC mice with late-stage tumors. Tumor tissues were minced and digested at 37°C for 12 hr in a digestion buffer containing 0.012% collagenase XI (MilliporeSigma) and 0.012% dispase (Gibco) in DMEM (Invitrogen) containing 1% FBS. The dissociated organoids were then pelleted, washed with PBS and seeded on petri dish in DMEM (Invitrogen) containing 10% FBS. After 4 days of culture, the medium was filtered through a 0.22 μm ultra-low protein binding filter and collected as conditioned medium.
Sorting of APCs, Mesothelial Cells or CAFs
The digestion buffer was prepared in DMEM with 1% FBS: collagenase type I (45 units/mL, Worthington Biochemical), collagenase type II (15 units/mL, Worthington Biochemical), collagenase type III (45 units/mL, Worthington Biochemical), collagenase type IV (45 units/mL, Gibco, Thermo Fisher Scientific), elastase (0.08 units/mL, Worthington Biochemical), hyaluronidase (30 units/mL, MilliporeSigma), and DNAse type I (25 units/mL, MilliporeSigma). Normal pancreata or tumors were enzymatically digested into a single cell suspension as follow. Freshly dissected tissues were minced in a 10 cm petri dish. Samples were then resuspended in 10mL digestion buffer and incubated on a shaker at 37°C for 60 mins. Then 30 mL of DMEM containing 1% FBS was added, and cells were washed 3 times with PBS before filtering through a 70-μm mesh filter (MilliporeSigma). Single cells were resuspended in PBS with 1% FBS, blocked with anti-mouse CD16/CD32 Fc Block for 15 mins at 4°C (BD Biosciences, clone 2.4G2, 1:50) and then incubated with antibodies against podoplanin (Biolegend, 1:200), I-A/I-E (Biolegend, MHC class II, 1:200) and Ly6C (Biolegend, 1:200) for 30 mins at 4°C. Cells were washed with PBS containing 1% FBS 3 times and subjected to cell sorting on the FACSAria sorter (UTSW flow cytometry core). Sorted cells were collected into ice cold DMEM with 10% FBS.
APC/Mesothelial Cell/CAF-CD4+ T cell Co-culture Assay and Flow Cytometry Analysis
20,000 sorted APCs/iCAFs/myCAFs/normal mesothelial cells/apCAFs or PanMeso cells were seeded in U-bottom 96-well plates and incubated with or without 25 μg/ml OVA peptide (OVA 323–339) (GenScript) in DMEM with 10% FBS for 4 hrs at 37°C. Spleens from 6-to-8-week-old OT II mice were harvested and CD4+ T cells were isolated using a MojoSort mouse CD4+ T cell isolation kit (Biolegend). APCs/mesothelial cells/CAFs were washed 3 times and co-cultured with 50,000 CD4+ T cells in DMEM with 10% FBS for 18 hrs. T cells were then washed twice with PBS and stained with Fixable Viability Stain (BD Biosciences) for 1 hour. The cell suspensions were then washed and stained with antibodies against CD4 (BD Biosciences, 1:100), CD25 (BD Biosciences, 1:100) and CD69 (BD Biosciences, 1:100) for 1 hr at 4°C. Surface-stained cells were fixed, permeabilized (FoxP3 Buffer Set, BD Biosciences), and stained for intracellular markers FoxP3 (BD Biosciences, 1:100) and Ki67 (Biolegend, 1:100). Cells were analyzed using FACS LSRFortessa SORP, and the FlowJo software. For CAF proportion analysis, cell suspension from digested KPfC tumors were stained with antibodies against podoplanin (Biolegend, 1:200), I-A/I-E (Biolegend, MHC class II, 1:200), αSMA (Thermo Fisher Scientific, 1:200) and IL-6 (Biolegend, 1:200).
Treg Suppression Assay
20,000 sorted apCAFs from KPfC tumors were seeded in U-bottom 96-well plates and incubated with or without 25 μg/ml OVA peptide (OVA 323–339) (GenScript) in DMEM with 10% FBS for 4 hrs at 37°C. Spleens from 6-to-8-week-old OT II mice were harvested and CD4+ T cells were isolated using a MojoSort mouse CD4+ T cell isolation kit (Biolegend). apCAFs were washed 3 times and co-cultured with 50,000 CD4+ T cells in DMEM with 10% FBS for 18 hrs. CD4+ T cells were then collected and co-cultured with CD8+ T cells (1:1 ratio) from splenocytes of C57BL/6 mice that were isolated using MojoSort mouse CD8+ T cell isolation kit (Biolegend) and labeled with CellTrace CFSE dye (Thermo Fisher Scientific, 1 μM). After 72 hours, cells were stained with CD8 antibody (BD Biosciences, 1:100) and the percentage of proliferating CD8+ T cells was analyzed by CFSE signal with flow cytometry.
RNA Isolation and Sequencing
RNA of parental and sorted PanMeso cells was extracted using RNeasy Mini Kit (Qiagen). RNA purity and concentration measurement, preparation of RNA library and transcriptome sequencing was conducted by Novogene Co. Genes with adjusted p-value < 0.05 and log2(FoldChange) > 0 were considered as differentially expressed. Heatmap was generated using GraphPad Prism software.
scRNA-seq Data Preprocessing
The scRNA-seq data were analyzed using R package Seurat v3.2.2 (Butler et al., 2018; Stuart et al., 2019). Fibroblasts scRNA-seq data from Hosein et al. were analyzed as previously described (Hosein et al., 2019), and converted to Seurat v3 format using the function UpdateSeuratObject. The scRNA-seq data by Dominguez et al. were obtained from the ArrayExpress database under the accession number E-MTAB-8483, and quality control filtering was performed as described previously (Dominguez et al., 2020). Cell clusters were identified using the first 12 principal components under the clustering resolution of 0.6. Identities of the cell clusters were matched to the previously reported cluster names based on the expression of genes mentioned in Dominguez et al. (Dominguez et al., 2020). The scRNA-seq data by Elyada et al. were obtained from the Gene Expression Omnibus under the accession number GSE129455 (Elyada et al., 2019). Identities of the cell clusters were matched to the previously reported cluster names based on the expression of genes mentioned in Elyada et al. (Elyada et al., 2019).
Integration of Multiple scRNA-seq Datasets
The datasets by Hosein et al., Dominguez et al., and Elyada et al. (Dominguez et al., 2020; Elyada et al., 2019; Hosein et al., 2019) were integrated as described in Seurat v3.2 Vignettes using the Standard Workflow (https://satijalab.org/seurat/v3.2/integration.html). Briefly, top 2,000 variable features were identified for each dataset. Next, integration anchors were identified using the first 30 dimensions, and the datasets were integrated using Seurat function IntegrateData. The integrated dataset was subsequently rescaled, principal component analysis was performed, and dimensional reduction was performed using the first 20 principal components. The cell clusters were identified from the integrated dataset using the first 12 principal components under the clustering resolution of 0.6. Differential expression analysis was performed using the Seurat functions FindMarkers and FindAllMarkers. For log normalized expression of genes in violin and UMAP plots, feature counts for each cell were divided by the total counts of the cell and multiplied by the scaling factor of 100,000. This number was then natural log+1 transformed.
Pseudotime Analysis
R package Monocle2 (v2.16.0) was used to align cells from clusters 1, 4, 7, 8, 9, 10 of the integrated dataset (Qiu et al., 2017). The cells from Elyada et al. (Elyada et al., 2019) were not included in this analysis due to the lack of raw read counts. The top 1,000 differentially expressed genes were used to order cells in the pseudotime trajectories, determined using the Monocle2 function differentialGeneTest. Dimensional reduction was performed by the Monocle2 function reduceDimension using DDRTree method.
Scoring of Mouse apCAF Signature in Human Cancer
We evaluated the presence of apCAFs in two scRNA-seq datasets for human PDA (GSE156405) (Lee et al., 2021) and human ovarian cancer (EGAS00001004935) (Hornburg et al., 2021) generated using 10X Genomics and available as processed CellRanger files. Each of these datasets was stratified into fibroblasts and immune cells and we used cluster annotations and/or markers as mentioned in the publications to identify fibroblast and immune cell subpopulations. Next, we utilized the following apCAF gene expression signatures derived from the analysis of our scRNA-seq data in mouse PDA (1) and another published study in lung (2) (Kerdidani et al., 2022):
GJB3, GJB5, LSR, EZR, KRT19, KRT18, KRT8, SDC4, PTPRF, CD82, NFE2L3, TPI1, ID1, FAM110C
CD74, SLPI, IL6, CFD, C1QA, C1QB, SFTC, CLU, LYZ, HLA-DRA, HLA-DRB1
These two apCAF signatures were scored in the fibroblast and immune cell compartment of the aforementioned human cancer datasets using the AddModuleScore function in Seurat v4.0.4.
Gene Ontology Analysis and Motif Enrichment Analysis
Gene ontology analysis and motif enrichment analysis were performed using Metascape (Zhou et al., 2019). The motif enrichment analysis from Metascape was based on the TRRUST algorithm.
Quantification and Statistical Analysis
All purchased mice in this study had similar age. Male and female mice were included in similar numbers for each animal experiment. All data are reported as mean ± SD. Statistical analysis was performed with a 2-tailed t-test using GraphPad Prism software and R language (https://www.R-project.org/). P < 0.05 was considered statistically significant. All in vitro experiments were performed with at least three biological replicates. Illustrations were created with BioRender.com.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken Anti-GFP | Abcam | Cat# ab13970 |
| BV421 Rat Anti-CD4 (GK1.5) | BD Biosciences | Cat# 562891 |
| BV786 Rat Anti-CD8 (53-6.7) | BD Biosciences | Cat# 563332 |
| BV786 Rat Anti-CD25 (PC61) | BD Biosciences | Cat# 564023 |
| BV711 Hamster Anti-CD69 (H1.2F3) | BD Biosciences | Cat# 740664 |
| Rat Anti-Mouse CD16/CD32 Fc Block (2.4G2) | BD Biosciences | Cat# 553141 |
| Alexa Fluor 647 Rat anti-FoxP3 (MF23) | BD Biosciences | Cat# 560401 |
| Mouse Anti-Smooth Muscle Actin (1A4) | Biocare Medical | Cat# 001 |
| Rat IgG2a Isotype Control (2A3) | BioXCell | Cat# BE0089 |
| PE Anti-Ly6C (HK1.4) | Biolegend | Cat# 128008 |
| Rat Anti-CD74 (CLIP) | Biolegend | Cat# 151002 |
| Brilliant Violet 421 Rat Anti-I-A/I-E (M5/114.15.2) | Biolegend | Cat# 107632 |
| PE Anti-IL-6 (MP5-20F3) | Biolegend | Cat# 504504 |
| PE Anti-Ki-67 (16A8) | Biolegend | Cat# 652404 |
| APC Anti-Podoplanin (8.1.1) | Biolegend | Cat# 127410 |
| Rabbit Phospho-NF-κB p65 (Ser536) (93H1) | Cell Signaling | Cat# 3033S |
| Rabbit Anti-CD74 (D5N3I) | Cell Signaling | Cat# 77274S |
| Rabbit Anti-CD8 (D4W2Z) | Cell Signaling | Cat# 98941S |
| Rabbit Anti-PDGFRα (D13C6) | Cell Signaling | Cat# 5241S |
| Mouse Anti-Smad2 (L16D3) | Cell Signaling | Cat# 3103S |
| Rabbit Anti-Phospho-Smad2 (Ser465/467) (138D4) | Cell Signaling | Cat# 3108S |
| Mouse Anti-CD3 | Invitrogen | Cat# PA1-29547 |
| Peroxidase AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L) | Jackson ImmunoResearch | Cat# 703-035-155 |
| Peroxidase AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 715-035-150 |
| Peroxidase AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 711-035-152 |
| Peroxidase AffiniPure Goat Anti-Syrian Hamster IgG (H+L) | Jackson ImmunoResearch | Cat# 107-035-142 |
| Rabbit Anti-Cadherin-11 | LSBio | Cat# LS-B2308 |
| Rabbit Anti-IL-6 | LSBio | Cat# LS-C809503 |
| Rabbit Anti-Mesothelin | LSBio | Cat# LS-C407883 |
| Rat Anti-Mesothelin (B35) | MBL Bio | Cat# D233-3 |
| Rabbit Anti-Actin | MilliporeSigma | Cat# A2066 |
| Syrian Hamster Anti-Podoplanin (8.1.1) | MilliporeSigma | Cat# MABT1512 |
| Rabbit Anti-Sox9 | MilliporeSigma | Cat# AB5535 |
| Goat Anti-tdTomato | MyBioSource | Cat# MBS448092 |
| Rabbit Anti-FoxP3 | R&D Systems | Cat# MAB8214 |
| Rabbit Anti-NFκB p65 | Santa Cruz | Cat# sc-109 |
| Alexa 488 Anti-Smooth Muscle Actin (1A4) | Thermo Fisher Scientific | Cat# 53-9760-82 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A-11030 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 546 | Thermo Fisher Scientific | Cat# A-11035 |
| Bacterial and Virus Strains | ||
| pLV-eGFP | Addgene | Cat# 36083 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse EGF | Biolegend | Cat# 585608 |
| OVA Peptide (323-339) | GenScript | Cat# RP10610 |
| Collagenase XI | MilliporeSigma | Cat# C7657 |
| DNase I | MilliporeSigma | Cat# D4527 |
| Hyaluronidase | MilliporeSigma | Cat# H3506 |
| Hydrocortisone | MilliporeSigma | Cat# H0888 |
| Tamoxifen | MilliporeSigma | Cat# T5648 |
| Recombinant Human TGFβ1 (CHO derived) | PeproTech | Cat# 100-21C |
| Recombinant Mouse IL-1 alpha/IL-1F1 Protein | R&D Systems | Cat# 400-ML-005/CF |
| Gibco Dispase | Thermo Fisher Scientific | Cat# 17-105-041 |
| Collagenase IV | Thermo Fisher Scientific | Cat# 17104019 |
| Collagenase I | Worthington Biochemical | Cat# LS004196 |
| Collagenase II | Worthington Biochemical | Cat# LS004176 |
| Collagenase III | Worthington Biochemical | Cat# LS004182 |
| Elastase | Worthington Biochemical | Cat# LS002279 |
| Critical Commercial Assays | ||
| Fixable Viability Stain | BD Biosciences | Cat# 564406 |
| Mouse FoxP3 Buffer Set | BD Biosciences | Cat# 560409 |
| MojoSort mouse CD4 T cell isolation kit | Biolegend | Cat# 480033 |
| MojoSort mouse CD8 T cell isolation kit | Biolegend | Cat# 480035 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| iTaq Universal SYBR® Green Supermix | Bio-Rad | Cat# 1725121 |
| ProLong Gold Antifade Reagent with DAPI | Cell Signaling | Cat# 8961S |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | Cat# 9860S |
| Opal 520 Reagent Pack | PerkinElmer | Cat# FP1487001KT |
| Opal 570 Reagent Pack | PerkinElmer | Cat# FP1488001KT |
| Opal 620 Reagent Pack | PerkinElmer | Cat# FP1495001 KT |
| Opal 690 Reagent Pack | PerkinElmer | Cat# FP1497001 KT |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| CellTrace CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34554 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34577 |
| ImmPRESS AP Horse Anti-Rabbit IgG Polymer Detection Kit | Vector Laboratories | Cat# MP-5401 |
| ImmPRESS AP Horse Anti-Mouse IgG Polymer Detection Kit | Vector Laboratories | Cat# MP-5402 |
| ImmPRESS HRP Horse Anti-Goat IgG Polymer Detection Kit | Vector Laboratories | Cat# MP-7405 |
| ImmPRESS HRP Goat Anti-Mouse IgG Polymer Detection Kit | Vector Laboratories | Cat# MP-7452 |
| ImmPRESS HRP Goat Anti-Rabbit IgG Polymer Detection Kit | Vector Laboratories | Cat# MP-7451 |
| Biological Samples | ||
| Unstained paraffin-embedded tissue sections of PDA | UTSW Tissue Management Shared Resource | https://www.utsouthwestern.edu/departments/simmons/research/shared-resources/tissue-management.html |
| Unstained paraffin-embedded tissue sections of PDA | MD Anderson Institutional Tissue Bank | https://www.mdanderson.org/research/research-resources/core-facilities/institutional-tissue-bank.html |
| Experimental Models: Cell Lines | ||
| Mouse Cell Line: BMFA3 | Huang et al., 2019 | N/A |
| Mouse Cell Line: CT1BA5 | Huang et al., 2019 | N/A |
| Mouse Cell Line: PanMeso | This Paper | N/A |
| Mouse Cell Line: 6419c5 | Li et al., 2018 | N/A |
| Mouse Cell Line: 6694c2 | Li et al., 2018 | N/A |
| Mouse Cell Line: 6499c4 | Li et al., 2018 | N/A |
| Mouse Cell Line: 2838c3 | Li et al., 2018 | N/A |
| Mouse Cell Line: 6620c1 | Li et al., 2018 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: KIC (KrasLSL-G12D/+;Ink4afl/fl;Ptf1aCre/+) | Hingorani et al., 2005 | N/A |
| Mouse: KPfC (KrasLSL-G12D/+;Trp53fl/fl;Pdx1Cre/+) | Hingorani et al., 2003 | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Mouse: Immortomouse (Tg(H2-K1-tsA58)6Kio/LicrmJ | The Jackson Laboratory | Cat# 032619 |
| Mouse: OT II (B6.Cg-Tg(TcraTcrb)425Cbn/J) | The Jackson Laboratory | Cat# 004194 |
| Mouse: Wt1tm2(cre/ERT2)Wtp/J | The Jackson Laboratory | Cat# 010912 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | Cat# 007914 |
| Deposited Data | ||
| Single-cell RNA sequencing | Hosein et al., 2019 | GEO: GSE125588 |
| Single-cell RNA sequencing | Dominguez et al., 2020 | ArrayExpress: E-MTAB-8483 |
| Single-cell RNA sequencing | Elyada et al., 2019 | GEO: GSE129455 |
| Single-cell RNA sequencing | Lee et al., 2021 | GEO: GSE156405 |
| Single-cell RNA sequencing | Hornburg et al., 2021 | EGA European Genome-Phenome Archive: EGAS00001004935 |
| Bulk RNA sequencing | This Paper | GEO: GSE196740 |
| Software and Algorithms | ||
| R package Seurat (v3.2.2) | Butler et al., 2018 Stuart et al., 2019 | N/A |
| R package Seurat (v4.0.4) | Hao et al., 2021 | N/A |
| R package Monocle2 (v2.16.0) | Qiu et al., 2017 | N/A |
| R language | R-project | https://www.R-project.org/ |
| FlowJo software (v10.7) | BD Biosciences | https://www.flowjo.com/ |
| Graphpad Prism 8.0 software | GraphPad Software, Inc. | http://www.graphpad.com/scientific-software/prism/ |
| ZEN Imaging Software | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Vectra Polaris Slide Scanner | Akoya Biosciences | https://resources.perkinelmer.com/lab-solutions/resources/docs/APP_Vectra-Polaris_013270_01.pdf |
| Phenochart | Akoya Biosciences | https://www.akoyabio.com/support/software/phenochart-whole-slide-viewer/ |
| inForm | Akoya Biosciences | https://www.akoyabio.com/phenoimager/software/inform-tissue-finder/ |
| HALO image analysis software | Indica Labs | https://indicalab.com/news/indica-labs-release-halo-the-next-generation-image-analysis-platform/ |
| NDP.view2 software | Hamamatsu Photonics | https://www.hamamatsu.com/us/en/product/type/U12388-01/index.html |
| Metascape | Zhou et al., 2019 | https://metascape.org/ |
Acknowledgements
This work was supported by NIH K99 (CA252009) to HH; NIH R01 (CA243577) and U54 (CA210181 Project 2), the Effie Marie Cain Fellowship, and the Jean Shelby Fund for Cancer Research at Communities Foundation of Texas to RAB. RC and GM were supported by the UT Southwestern TARDIS Physician-Scientist Fellowship funded by the Burroughs Wellcome Fund. We acknowledge helpful input from other members of the Brekken laboratory and Drs. Michael Dellinger, Eric N. Olson and Rana Gupta. We thank Dr. Ben Stanger for provision of PDA cell lines. We also thank UT Southwestern core facilities supported in part by the Cancer Center Support Grant (P30 CA142543), core facilities used included the McDermott Center Next-Generation Sequencing Core, the Live Cell Imaging Core and the Flow Cytometry Core.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnden MJ, Allison J, Heath WR, and Carbone FR (1998). Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 76, 34–40. [DOI] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson DA (2019). IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 9, 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour-Jordan H, and Bluestone JA (2009). Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev 229, 41–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, and Poss KD (2018). The epicardium as a hub for heart regeneration. Nat Rev Cardiol 15, 631–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Song E (2019). Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov 18, 99–115. [DOI] [PubMed] [Google Scholar]
- Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, et al. (2018). Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 33, 463–479 e410. [DOI] [PubMed] [Google Scholar]
- Del Bano J, Flores-Flores R, Josselin E, Goubard A, Ganier L, Castellano R, Chames P, Baty D, and Kerfelec B (2019). A Bispecific Antibody-Based Approach for Targeting Mesothelin in Triple Negative Breast Cancer. Front Immunol 10, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Turley SJ, and Akhurst RJ (2021). TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol 18, 9–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 10, 232–253. [DOI] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer IR, Wagener ME, Song M, Kirk AD, Larsen CP, and Ford ML (2011). Antigen-specific induced Foxp3+ regulatory T cells are generated following CD40/CD154 blockade. Proc Natl Acad Sci U S A 108, 20701–20706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman G, Levi-Galibov O, David E, Bornstein C, Giladi A, Dadiani M, Mayo A, Halperin C, Pevsner-Fischer M, Lavon H, et al. (2020). Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat Cancer 1, 692–708. [DOI] [PubMed] [Google Scholar]
- Gupta OT, and Gupta RK (2015). Visceral Adipose Tissue Mesothelial Cells: Living on the Edge or Just Taking Up Space? Trends Endocrinol Metab 26, 515–523. [DOI] [PubMed] [Google Scholar]
- Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, and Pastan I (2016). Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J Clin Oncol 34, 4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hornburg M, Desbois M, Lu S, Guan Y, Lo AA, Kaufman S, Elrod A, Lotstein A, DesRochers TM, Munoz-Rodriguez JL, et al. (2021). Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 39, 928–944 e926. [DOI] [PubMed] [Google Scholar]
- Hosein AN, Brekken RA, and Maitra A (2020). Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol 17, 487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, Maitra A, Olson E, Verma U, and Brekken RA (2019). Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, and Brekken RA (2019). The Next Wave of Stroma-Targeting Therapy in Pancreatic Cancer. Cancer Res 79, 328–330. [DOI] [PubMed] [Google Scholar]
- Huang H, and Brekken RA (2020). Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am J Physiol Cell Physiol 319, C233–C243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhang Y, Gallegos V, Sorrelle N, Zaid MM, Toombs J, Du W, Wright S, Hagopian M, Wang Z, et al. (2019). Targeting TGFbetaR2-mutant tumors exposes vulnerabilities to stromal TGFbeta blockade in pancreatic cancer. EMBO Mol Med 11, e10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, Karim S, Paulus-Hock V, Watt D, Steele N, et al. (2021). Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 39, 1227–1244 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, and Kioussis D (1991). Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A 88, 5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kerdidani D, Aerakis E, Verrou KM, Angelidis I, Douka K, Maniou MA, Stamoulis P, Goudevenou K, Prados A, Tzaferis C, et al. (2022). Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J Exp Med 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, Lameiras S, Albergante L, Bonneau C, Guyard A, et al. (2020). Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov 10, 1330–1351. [DOI] [PubMed] [Google Scholar]
- Koopmans T, and Rinkevich Y (2018). Mesothelial to mesenchyme transition as a major developmental and pathological player in trunk organs and their cavities. Commun Biol 1, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, Wang P, Liang S, Iwaisako K, Liu X, Xu J, Zhang M, Sun M, Cong M, Karin D, et al. (2017). Mesothelin/mucin 16 signaling in activated portal fibroblasts regulates cholestatic liver fibrosis. J Clin Invest 127, 1254–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, and Kalluri R (2018). A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Bernard V, Semaan A, Monberg ME, Huang J, Stephens BM, Lin D, Rajapakshe KI, Weston BR, Bhutani MS, et al. (2021). Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-world Pancreatic Cancer Biopsies. Clin Cancer Res 27, 5912–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, 178–193 e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang J, and Asahina K (2013). Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc Natl Acad Sci U S A 110, 2324–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KF, Du W, Sorrelle NB, Wnuk-Lipinska K, Topalovski M, Toombs JE, Cruz VH, Yabuuchi S, Rajeshkumar NV, Maitra A, et al. (2018). Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res 78, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Barajon I, and Garlanda C (2018). IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev 281, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikami N, Kawakami R, Chen KY, Sugimoto A, Ohkura N, and Sakaguchi S (2020). Epigenetic conversion of conventional T cells into regulatory T cells by CD28 signal deprivation. Proc Natl Acad Sci U S A 117, 12258–12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers SE, Birnie K, Lansley S, Herrick SE, Lim CB, and Prele CM (2015). Mesothelial cells in tissue repair and fibrosis. Front Pharmacol 6, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214, 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish CR (1999). Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol Cell Biol 77, 499–508. [DOI] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM (2014). Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich Y, Mori T, Sahoo D, Xu PX, Bermingham JR Jr., and Weissman IL (2012). Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat Cell Biol 14, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple K, Nguyen A, Yu Y, Wang H, Anasetti C, and Yu XZ (2011). Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 117, 3096–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si M, Wang Q, Li Y, Lin H, Luo D, Zhao W, Dou X, Liu J, Zhang H, Huang Y, et al. (2019). Inhibition of hyperglycolysis in mesothelial cells prevents peritoneal fibrosis. Sci Transl Med 11. [DOI] [PubMed] [Google Scholar]
- Sontake V, Kasam RK, Sinner D, Korfhagen TR, Reddy GB, White ES, Jegga AG, and Madala SK (2018). Wilms’ tumor 1 drives fibroproliferation and myofibroblast transformation in severe fibrotic lung disease. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrelle N, Ganguly D, Dominguez ATA, Zhang Y, Huang H, Dahal LN, Burton N, Ziemys A, and Brekken RA (2019). Improved Multiplex Immunohistochemistry for Immune Microenvironment Evaluation of Mouse Formalin-Fixed, Paraffin-Embedded Tissues. J Immunol 202, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JM, Sinha R, Seita J, Fernhoff N, Christ S, Koopmans T, Krampitz GW, McKenna KM, Xing L, Sandholzer M, et al. (2018). Surgical adhesions in mice are derived from mesothelial cells and can be targeted by antibodies against mesothelial markers. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- Wing JB, Tanaka A, and Sakaguchi S (2019). Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 50, 302–316. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, Halbrook CJ, Menjivar RE, Kemp SB, Sirihorachai VR, et al. (2020). Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov 10, 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new raw scRNA-seq data. The raw scRNA-seq data we used to extend our analyses on fibroblasts and raw RNA-seq data of PanMeso cells have been uploaded to the National Center for Biotechnology Information’s Gene Expression Omnibus database repository (https://www.ncbi.nlm.nih.gov/geo) under accession number GSE125588 and GSE196740. In addition, we consulted other publicly available scRNA-seq datasets, including GSE129455 and GSE156405 from the National Center for Biotechnology Information’s Gene Expression Omnibus database repository, E-MTAB-8483 from ArrayExpress Archive of Functional Genomics Data (https://www.ebi.ac.uk/arrayexpress/) and EGAS00001004935 from EGA European Genome-Phenome Archive (https://ega-archive.org/). Additional information related to the data in this study is available from the lead contact upon request.