

Abstract
Coronary artery disease (CAD) is a complex inflammatory disease involving genetic influences across cell types. Genome-wide association studies (GWAS) have identified over 200 loci associated with CAD, where the majority of risk variants reside in noncoding DNA sequences impacting cis-regulatory elements (CREs). Here, we applied single-nucleus ATAC-seq to profile 28,316 nuclei across coronary artery segments from 41 patients with varying stages of CAD, which revealed 14 distinct cellular clusters. We mapped ~320,000 accessible sites across all cells, identified cell type-specific elements, transcription factors, and prioritized functional CAD risk variants. . We identified elements in smooth muscle cell (SMC) transition states (e.g. fibromyocytes) and functional variants predicted to alter SMC and macrophage-specific regulation of MRAS (3q22) and LIPA (10q23), respectively. We further nominated key driver transcription factors such as PRDM16 and TBX2. Together, this single nucleus atlas provides a critical step towards interpreting regulatory mechanisms across the continuum of CAD risk.
Introduction
Coronary artery disease (CAD) is the leading cause of death globally and results from injury to the vessel wall and atherosclerotic plaque buildup. Atherosclerotic coronary arteries are complex due to the propensity of multiple cell types to undergo cell phenotypic switching, including endothelial cells, smooth muscle cells (SMCs), fibroblasts, and various immune cells 1–3. This has hindered efforts to combat the disease process itself, as currently approved therapies only treat the traditional risk factors, such as elevated blood pressure or cholesterol levels. Recent single-cell RNA sequencing (scRNA-seq) analyses have yielded numerous cellular insights into atherosclerosis 4–11. In particular, lineage-traced scRNA-seq approaches have shown that SMCs transdifferentiate to several distinct phenotypes during atherosclerosis: 1) “fibromyocytes” with fibroblast-like signatures7; 2) an intermediate cell state that can become fibrochondrocyte or macrophage-like 9 ; or 3) a transitional state giving rise to multiple plaque cell types10. Together these studies demonstrate that SMC-derived cells can elicit beneficial or detrimental effects depending on the stage of CAD and/or plaque environment. Despite these advances, the underlying cell-specific regulatory mechanisms remain elusive.
As a complex disease, CAD involves an interplay of environmental and genetic factors over the life course. Genome-wide association studies (GWAS) have now identified over 200 independent CAD loci 12–16. Many of these are predicted to function in vessel wall processes such as regulation of vascular remodeling, vasomotor tone, and inflammation 17. The majority of CAD associated single nucleotide polymorphisms (SNPs) reside in non-coding regions and are enriched in cis-regulatory elements (CREs)18, pointing towards regulatory functions19. Since CREs are commonly cell type specific 20,21, understanding CAD regulatory mechanisms at the cellular level is required to fully interpret the functional impact of risk variants. The Assay for Transposase-Accessible Chromatin with sequencing (ATAC-seq) is a widely adopted approach to systematically detect CREs 22 and has been conducted in CAD-relevant cultured human coronary artery SMCs 23–25 and aortic endothelial cells 26. However, cultured cell models often do not fully recapitulate the complex cellular and regulatory landscape in vivo. Thus, single-nucleus ATAC-seq (snATAC-seq) 27,28 of primary human coronary artery samples has the potential to provide a more complete regulatory map to unravel disease mechanisms in vivo.
Single-nucleus epigenomic profiling has recently been applied across various human tissues 29–38, including carotid arteries 8,39 ; however, to date there is still no large reference dataset spanning CAD progression in coronary arteries. In this study, we performed snATAC-based chromatin profiling to uncover ~320,000 candidate CREs in human coronary arteries from 41 patients with varying clinical presentations of CAD. In generating this cell-specific chromatin atlas of the human coronary artery, we identified candidate CREs and transcription factors for the major cell types or transition states in the coronary artery. We then applied these profiles to associate CAD risk variants with specific cell types by linking CREs to target gene promoters. Finally, we employed both allele-specific mapping and sequence-based predictive modeling to resolve genetic regulatory mechanisms that could help inform pre-clinical studies of CAD targets in the vessel wall.
Results
Single-nucleus ATAC-seq profiling in human coronary artery
We performed snATAC-seq on coronary arteries (left anterior descending artery (LAD), left circumflex artery (LCX), or right coronary artery (RCA)) from 41 patients with various presentations of atherosclerosis using a droplet-based protocol 29 (Figure 1a, Supplementary Tables 1–4, Extended Data Figure 1). We isolated nuclei from a total of 44 frozen coronary segments using a protocol optimized for frozen tissues 40. After sequencing, we performed stringent quality control to retain highly informative nuclei (Supplementary Figure 1). The libraries showed the expected insert size distributions and enrichment of reads at transcription start sites (TSS) (Supplementary Figure 1). Aggregating reads from all nuclei approximated bulk coronary ATAC-seq profiles derived from the same patient, further illustrating the quality of the single-nucleus dataset (Supplementary Figure 2).
Figure 1. snATAC-seq profiling of 28,316 nuclei from human coronary arteries reveals cell type chromatin accessibility patterns across 41 individuals.
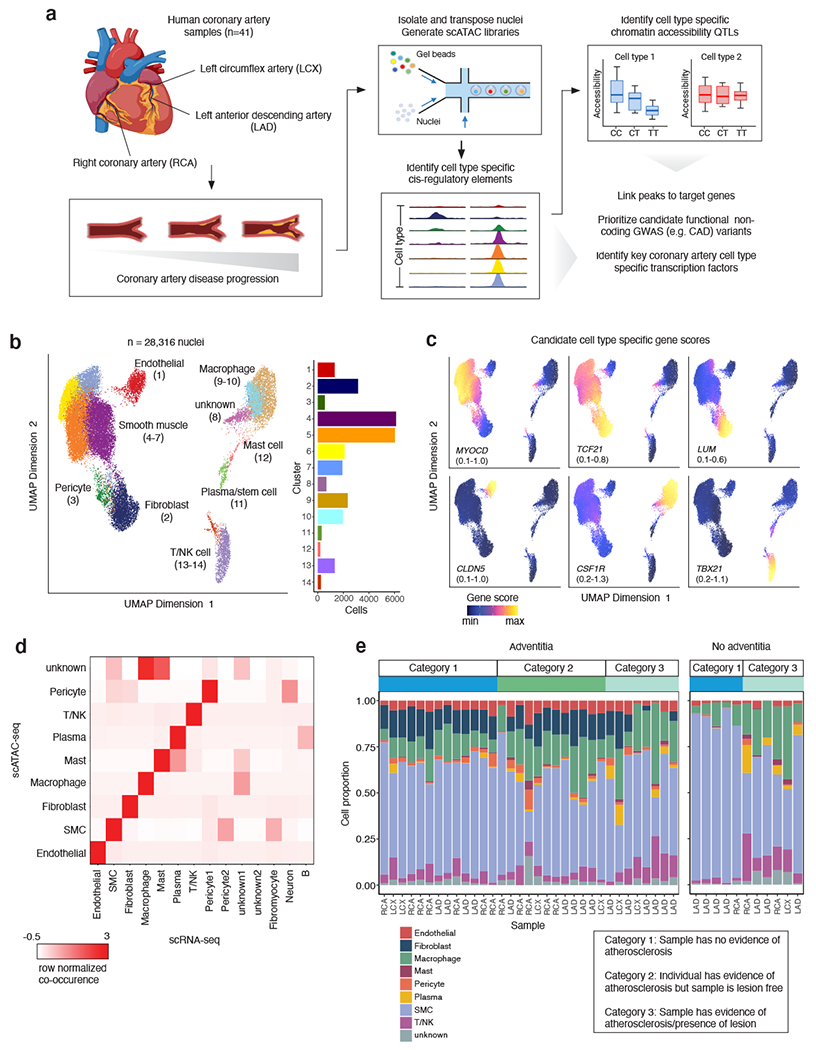
(a) snATAC-seq was performed on nuclei isolated from frozen human coronary artery samples taken from explanted hearts from 41 unique patients. Samples came from segments of either the left anterior descending coronary artery (LAD), left circumflex artery (LCX), or right coronary artery (RCA). After isolation using density gradient centrifugation, nuclei were transposed in bulk and mixed with barcoded gel beads and partitioning oil to generate gel beads in emulsions (GEMs). (b) Uniform manifold approximation and projection (UMAP) and clustering based on single-nucleus chromatin accessibility identifies 14 distinct coronary artery clusters. Each dot represents an individual cell colored by cluster assignment. (c) UMAP plot of Figure 1b colored by gene score for coronary artery cell type marker genes, including myocardin (MYOCD, smooth muscle cells), TCF21 (smooth muscle cells and fibroblasts), LUM (fibroblasts), CLDN5 (endothelial cells), CSF1R (macrophages), and TBX21 (T cells). (d) Heatmap representing the contingency table highlighting correspondence between snATAC-seq and scRNA-seq cell type assignments. (e) Distribution of cell types across all of the snATAC-seq samples, divided by whether or not the corresponding sample had an adventitial layer. Schematic in (a) was created using BioRender.
After filtering, we obtained a total of 28,316 high quality nuclei and identified 14 clusters using iterative Latent Semantic Indexing (LSI) in ArchR41 for dimensionality reduction 29,42 (Figure 1b). Importantly, the identified clusters distinguished biological cell types rather than individual donor or other covariates (e.g., age, sex) (Supplementary Figures 1, 3). We assigned each cluster to a coronary artery cell type using gene activity scores, which infer gene expression based on chromatin accessibility at established marker genes 41. Accessibility at SMC marker genes MYOCD, MYH11, CNN1, TAGLN, and ACTA2 (Figure 1c, Extended Data Figure 2) defined four distinct clusters of SMCs, the most abundant cell type in our dataset (57.8 +/− 17.6% of cells, Figure 1b). We further identified clusters of endothelial cells (CLDN5), fibroblasts (TCF21, LUM), macrophages (CSF1R), and T cells/natural killer cells (TBX21) (Figure 1c). Additional cluster annotations included pericytes, plasma (B) cells, mast cells, and ‘unknown’ immune cells (resembling macrophages/mast cells). Data integration (Methods) showed our gene activity score-based annotations were in high agreement with recently reported scRNA-seq annotations from human coronary artery 7 (Figure 1d, Extended Data Figures 2 and 3). In general, we observed higher immune cell proportions (cells in clusters 8-14) in atheroma and fibrocalcific coronary artery samples (44.1 +/− 18.8%) relative to non-lesion or healthy controls (17.7 +/− 8.3%), which is consistent with the cellular etiology of atherosclerosis progression 43 (Figure 1e, Supplementary Figure 3). In samples devoid of adventitia (n=11), we observed nearly absent fibroblasts as well as depletion of endothelial cells and pericytes (Figure 1e). This is consistent with the expected cell composition of the outer adventitial layer and vasa vasorum 44,45 and supports the specificity of our cell-type annotations.
Characterization of cell type-specific regulatory profiles
We next applied this coronary artery snATAC-seq dataset to characterize cell type-specific cis-regulatory profiles. Using snATAC gene scores we identified 5,121 marker genes across all cell types, which revealed both cell identity and/or disease response genes (Figure 2a, Supplementary Data 1). By aggregating reads from all nuclei we generated a master set of 323,727 peaks (Supplementary Data 2), which mostly map to intronic and intergenic sequences as expected (Extended Data Figure 4 provides peak annotations). Notably, 54% were uniquely accessible in only one or limited cell types (Figure 2b), emphasizing the benefits of single-nucleus profiling to define context-specific regulatory profiles that could be missed in bulk studies.
Figure 2. Human coronary artery cell types display distinct gene regulatory processes.
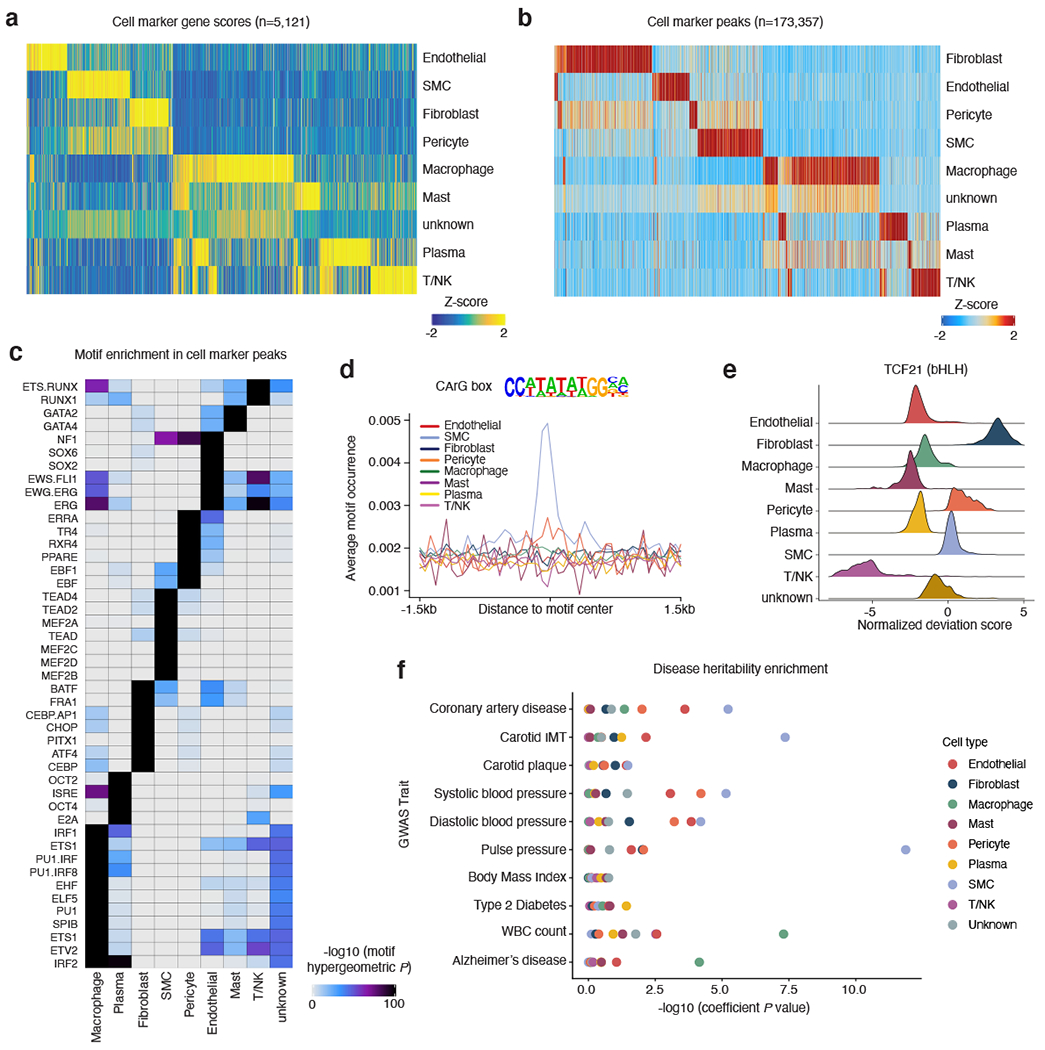
(a) Heatmap of coronary cell type marker genes (n=5,121) across each cell type calculated from snATAC-seq gene scores. Each column represents a unique marker gene. The color represents the normalized gene score of the marker genes in cell types. (b) Heatmap reflecting coronary cell type marker peaks that highlight cis-regulatory elements specific to only one or very limited cell types. Each column represents an individual marker peak. The color represents the normalized marker peak accessibility in cell types. (c) Heatmap of transcription factor motifs enriched in cell type marker peak sequences. The color represents the normalized motif enrichment score calculated in ArchR using HOMER with the hypergeometric test. (d) Representative motif occurrence plot for the CArG box motif. The CArG box motif, which binds myocardin and serum response factor, is highly enriched in smooth muscle cell accessible chromatin. (e) At the individual cell basis, accessible chromatin is highly enriched for the TCF21 motif in fibroblasts, smooth muscle cells, and pericytes. Transcription factor motif deviations (x-axis) were calculated for each cell using chromVAR. The TCF21 deviations for each cell were integrated based on the cell type (y-axis). (f) LD Score Regression (LDSC) reveals differing enrichment of GWAS SNPs for CAD, hypertension, and non-vascular phenotypes within coronary snATAC cell type peaks.
We next compared these marker genes and peaks with cultured human coronary artery SMC super enhancers detected by previous H3K27ac ChIP-seq data 23. SMC ATAC-seq peak clusters (ATAC-seq peaks longer than 10 kb, Methods) showed the highest association with SMC super enhancers (Supplementary Figure 4). These SMC super enhancers showed significantly higher regulatory potential for the identified SMC marker genes compared to the marker genes from all other cell types.
To investigate the transcription factors (TFs) potentially driving the regulatory profiles/programs in each coronary artery cell type, we performed HOMER 46 motif enrichment analysis for these marker peaks (Figure 2c, Supplementary Data 3). Top enriched motifs in SMCs (MEF2 family 47, TEAD family 48,49, CArG box binding myocardin/serum response factor (Figure 2d) 50–53) strongly agree with established SMC TFs in the literature. Similarly, we observed enrichment of ETS and SOX family motifs in endothelial cells 54,55, PU.1/SPIB and IRF motifs in macrophages 56, CEBP and AP-1 family members in fibroblasts 57, RUNX family motifs in T cells 58,59, and GATA family motifs in mast cells 60 (Figure 2c). Besides defining established cell type specific TFs, we also discovered a number of lesser known coronary TF motifs (e.g., SIX1/2 in fibroblasts and PRDM1 in immune cells). As a complementary approach, we applied chromVAR 61 on a per-cell level and observed cell-specific motif enrichment (e.g., TCF21 in SMCs and fibroblasts) (Figure 2e). Importantly, TCF21 was also highly enriched in fibromyocytes (Figure 3c), providing epigenomic-based support for this TF previously shown to drive SMC modulation 7,62.
Figure 3. Sub-cluster analysis of smooth muscle cell accessible chromatin identifies fibromyocyte regulatory programs.
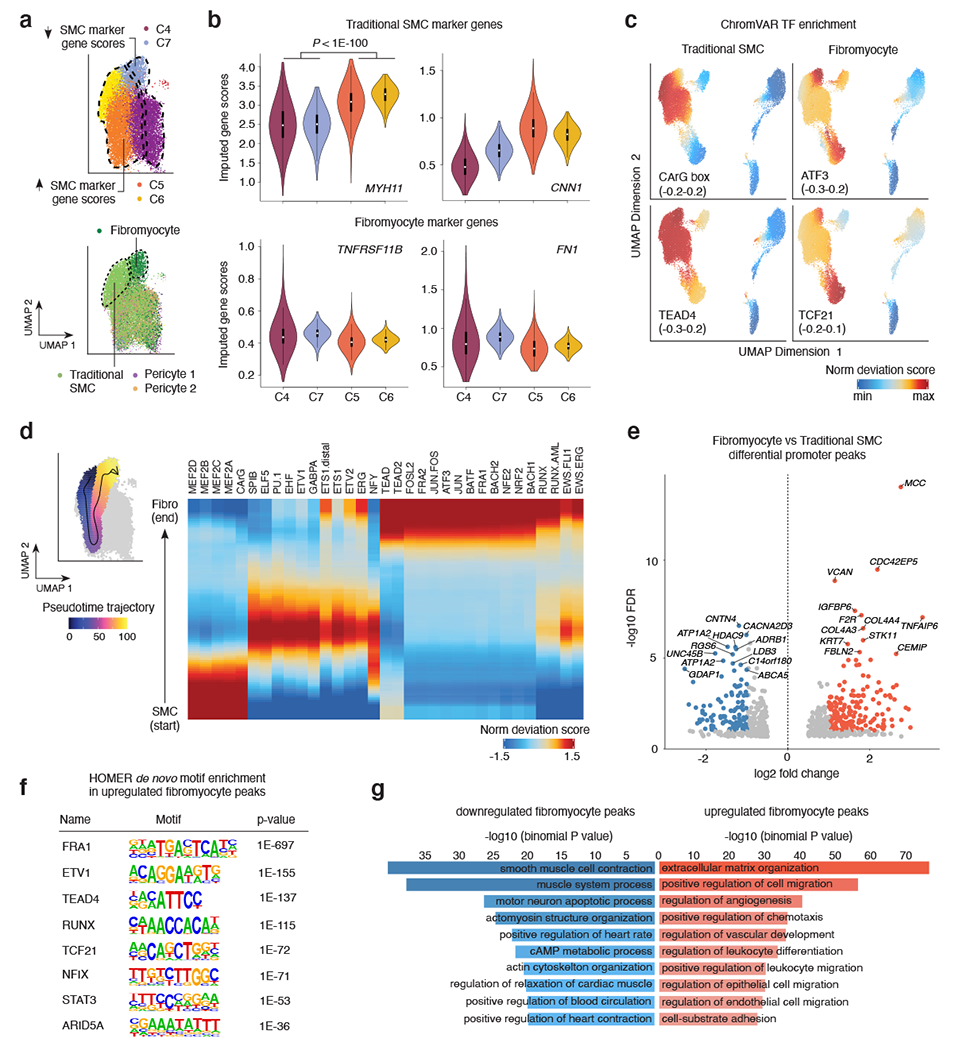
(a) snATAC UMAP for the 4 SMC clusters (C4-C7). The UMAP was colored by snATAC cluster (top) and by cell type labels assigned by scRNA-seq label transfer (bottom). Dashed lines demarcate boundary of cells with increased SMC marker gene scores (clusters C5 and C6), or decreased SMC marker gene scores (C4 and C7). Integrated snATAC/scRNA UMAP highlights both Fibromyocyte SMCs and traditional SMCs (demarcated by dashed lines and colors) within clusters 4-7. “Pericyte 1” and “Pericyte 2” labels from scRNA-seq were also mixed in clusters 4 and 5. (b) Quantification of imputed snATAC gene scores highlights higher chromatin accessibility at differentiated SMC marker genes MYH11 and CNN1 in clusters 5 and 6, and higher accessibility at modulated SMC/Fibromyocyte marker genes TNFRSF11B and FN1 in clusters 4 and 7. P-values were calculated using a one-sided Wilcoxon test. The exact P-values are as follows: MYH11: p=0; CNN1: p=0; TNFRSF11B: p=0; FN1: p=1.7e-260. The N values for nuclei in each cluster are as follows: C4: 6275; C7: 1971; C5: 6134; C6: 1988. (c) ChromVAR transcription factor motif enrichment for differentiated SMC CArG box in traditional SMC and enrichment for ATF3 and TCF21 motifs in modulated SMC/Fibromyocyte and Fibroblast clusters. The TEAD4 motif is enriched in both contractile and modulated SMCs. (d) Left, scatter plot overlay of SMC UMAP depicts the trajectory path from differentiated SMC to modulated SMC/Fibromyocyte sub-clusters (left). Motif enrichment heatmap shows the top enriched motifs across the trajectory pseudo-time (right). Values represent accessibility gene z-scores. (e) Volcano plot of differential peak analysis (subset to promoter peaks) comparing Fibromyocyte and traditional SMCs. Fibromyocyte and SMC annotated cells were defined based on RNA label transferring (Methods) and significant peaks determined by a Wilcoxon-test as implemented in ArchR. Peaks with significant differences at FDR <= 0.05 and Log2 fold change > 1 were colored light red (Fibromyocyte upregulated) and blue (Fibromyocyte downregulated). (f) Top enriched motifs within the total upregulated Fibromyocyte peaks (5,681) detected using HOMER de novo enrichment analysis with the hypergeometric distribution test. P-values shown are unadjusted for multiple comparisons. (g) Functional annotation of Fibromyocyte upregulated (light red) and downregulated (blue) peaks conducted using GREAT with the binomial distribution test. Top enriched biological processes functional terms are listed. P-values shown are unadjusted for multiple comparisons.
In order to determine whether cell type-specific accessible regions were enriched for GWAS variants for CAD and other vessel wall phenotypes, we performed cell type Linkage Disequilibrium (LD) Score Regression (LDSC) 63. CAD and blood pressure GWAS variants were highly enriched in SMC, endothelial cell, and macrophage peaks (Figure 2f), while variants for pulse pressure and intimal-medial thickness (cIMT) were specifically enriched in SMC peaks (Figure 2f). This is in line with the major contribution of SMC in subclinical atherosclerosis compared to more advanced atherosclerosis involving multiple cell types 3,64. In contrast, there was limited enrichment for non-vascular traits (Figure 2f). Overall, this comprehensive set of coronary artery snATAC-seq chromatin profiles provides a rich landscape to unravel cell-specific regulatory mechanisms in healthy conditions and across diverse diseased stages.
Characterization of gene regulatory programs in SMCs
Given the extensive phenotypic plasticity of SMCs, we next investigated differences in the cis-regulatory profiles between contractile and modulated SMCs. While the studies from Alencar et al. 10 and Pan et al. 9 both provide compelling evidence of modulated SMC populations, we expanded upon the Wirka et al. 7 study. This study included human coronary arteries and identified a modulated ‘fibromyocyte population’ (markers TNFRSF11B and FN1). SMCs in our dataset partition into four sub-clusters (Figure 1b), referred to as C4-C7 (Figure 3a). Clusters C5 and C6 have greater accessibility in differentiated SMC genes (MYH11 and CNN1), whereas clusters C4 and C7 have greater accessibility in phenotypically modulated SMC marker genes (TNFRSF11B and FN1) (Figure 3a–b). To address potential noise due to sparsity of snATAC-based gene scores, we also derived integrated RNA scores using mutual nearest neighbor integration and label transfer between identified anchors in the snATAC-seq data and Wirka et al. 7 scRNA-seq data (Methods, Extended Data Figures 2,3). This approach provided higher resolution to clearly delineate the SMC-derived fibromyocyte population based on TNFRSF11B, FN1 and other modulation markers (Figure 3a, Extended Data Figure 2). Consistently, chromVAR based TF enrichment revealed highly enriched motifs for AP-1 family members (e.g. ATF3) and TCF21 in the fibromyocyte cluster, which was depleted of differentiated SMC CArG box motif (Figure 3c, Extended Data Figure 2). Other TFs such as TEAD4 were enriched in all SMC clusters (Figure 3c). Together these results suggest that we can leverage single-nucleus accessibility profiles to understand regulatory drivers of SMC phenotypic modulation.
In a similar approach, we leveraged chromVAR motif deviations to perform trajectory analyses in SMC clusters. By assigning a path of accessibility from differentiated SMCs towards fibromyocytes (Figure 3d), we identified enriched MEF2 and CArG motifs at the start of the trajectory, followed by enrichment of ETS and NFY motifs, then AP-1 and RUNX motifs in fibromyocytes (Figure 3d). Using the scRNA-seq integrated data, we further identified 7,802 differentially accessible peaks (5,681 upregulated and 2,121 downregulated) between cells annotated as fibromyocytes vs. traditional/differentiated SMCs (Supplementary Data 4). In particular, we identified 170 significantly upregulated and 108 downregulated promoter peaks in fibromyocytes (Figure 3e, Supplementary Data 4). Promoters with higher accessibility in fibromyocytes include several extracellular matrix (ECM) genes (e.g., VCAN, COL4A3/4 and TNFAIP6), previously identified using scRNA-seq 7. However, we also revealed a number of candidate fibromyocyte markers such as the Rho GTPase effector gene, CDC42EP5, linked to actin-mediated migration/proliferation 65. Using HOMER de novo motif enrichment, we again observed AP-1 (FRA1), RUNX, TEAD, and TCF21 motifs in upregulated fibromyocyte peaks, but also motifs for inflammatory response factors STAT3 and ARID5A 66 (Figure 3f, Supplementary Data 4). The RUNX motif includes RUNX2, suggesting this fibromyocyte population may include osteochondrogenic cells 67,68. Conversely, MEF2A and CArG box motifs were the top enriched motifs in the downregulated peaks. Genomic region enrichment analysis identified ECM organization and cell migration processes in upregulated fibromyocyte peaks, compared to enrichment for SMC contraction and related processes in downregulated peaks (Figure 3g). Together, these snATAC based results confirm the role of TCF21 and also identify candidate TFs underlying SMC phenotypic modulation during CAD.
Annotation of target cell types and genes at CAD GWAS loci
Noncoding GWAS variants are enriched in CREs and often operate in a cell type-specific manner 18,30,69. We thus prioritized candidate functional CAD GWAS variants 13,15 using a multi-tiered approach. To first identify variants in CREs, we overlapped CAD lead variants (and variants in high linkage disequilibrium (r2 > 0.8; EUR)) from two recent CAD GWAS meta-analyses13,15 with snATAC peaks (Figure 4a). This resolved a subset of variants (+/− 50 bp) overlapping both shared and marker peaks (Figure 4b, Supplementary Data 5), with the majority of CAD SNPs residing within SMC peaks, followed by macrophage, fibroblast, and endothelial cell peaks. Based on these overlaps, we then highlighted target cell types for CAD regulatory variants (Figure 4c). Several top candidate CAD variants map to cell type-specific peaks, including rs3918226 at the NOS3 (endothelial nitric oxide synthase) locus and rs9337951 at the KIAA1462/JCAD (Junctional cadherin 5 associated) locus 70 both within endothelial peaks, and rs7500448 at the CDH13 (T-cadherin) locus within a SMC peak (Figure 4c). Other CAD variants map to peaks shared across SMC, fibroblast and pericyte cell types such as rs1537373 at the 9p21 locus (CDKN2B-AS1/ANRIL), rs2327429 (upstream of TCF21) and rs9515203 at COL4A2 (Figure 4c). The lead variant rs9349379 (PHACTR1/EDN1), disrupting a MEF2 binding site 71, is located within SMC and macrophage peaks (Figure 4c). We identified other CAD variants in macrophage peaks (e.g. rs7296737 at SCARB1 and rs12246441 at TSPAN14) (Extended Data Figure 7). We also prioritized a number of CAD loci within CREs acting through more than one cell type and confirmed SMC-specificity for previously validated loci such as LMOD1 72 (Figure 4d).
Figure 4. Single-nucleus chromatin accessibility further resolves mechanisms for functional CAD GWAS loci.
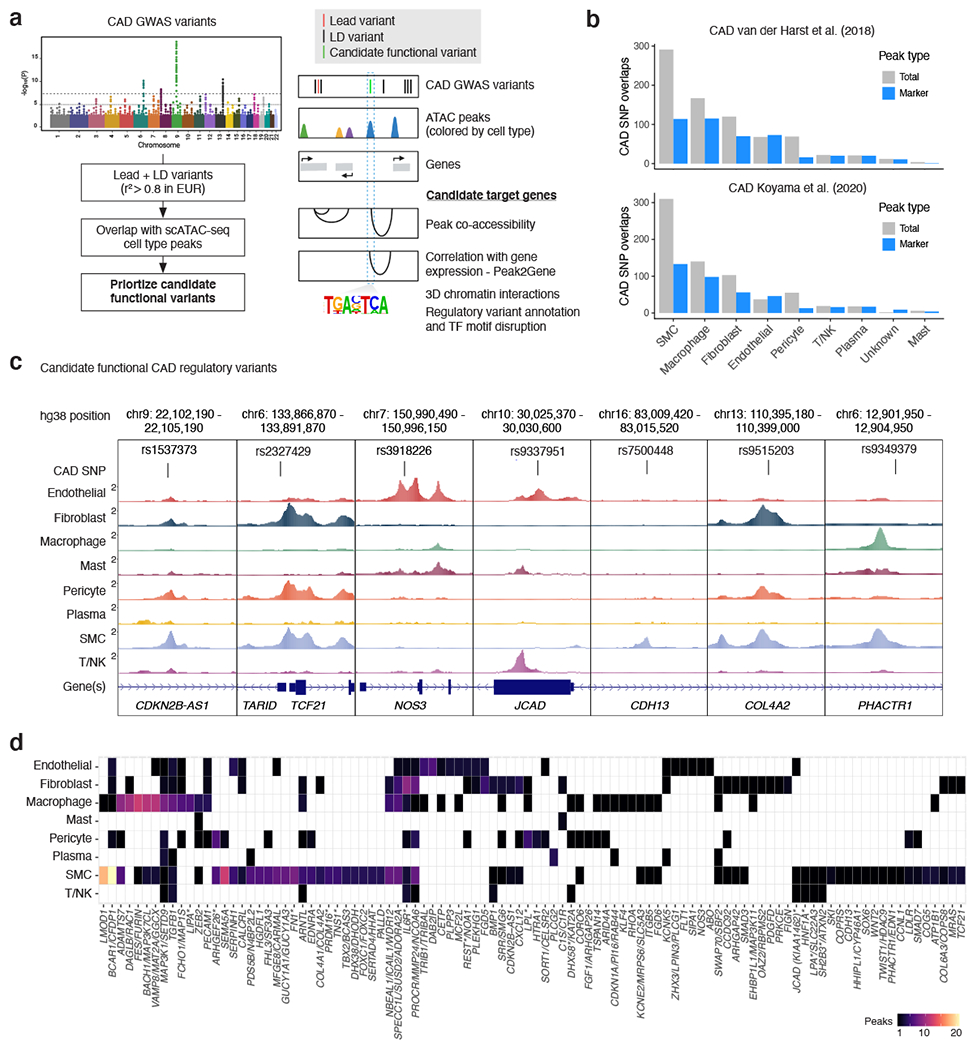
(a) To prioritize candidate CAD-associated GWAS variants we used a multi-tiered strategy, first by taking variants in moderate to high linkage disequilibrium (LD) with the reported lead variants (r2 >= 0.8). We next prioritized variants overlapping snATAC peaks and narrowed down the cell type(s) whereby these variants are potentially functioning. Finally we determined whether candidate variants are within transcription factor motifs and linked to target genes through co-accessibility and links to gene expression through scRNA-seq integration (Peak2Gene). (b) Overlap of LD-expanded (r2 >=0.8; EUR) CAD GWAS variants (+/− 50 bp) with coronary artery cell type peaks (both from the total peak set and marker peaks). LD-expanded SNPs were obtained from two recent CAD GWAS studies (van der Harst et al. 2018 and Koyama et al. 2020) that performed trans-ancestry meta-analysis. (c) Examples of the benefits of snATAC for pinpointing cell types whereby candidate CAD regulatory variants are acting. Highlighted are candidate functional variants at the 9p21 (CDKN2B-AS1/ANRIL), TARID/TCF21, NOS3, KIAA1462/JCAD, CDH13, COL4A2, and PHACTR1 loci. (d) Heatmap showing number of peaks per cell type overlapping CAD GWAS variants for 100 of the CAD loci (van der Harst et al. Circulation Research 2018). Full overlaps of CAD GWAS variants with snATAC peaks are provided in Supplementary Data 5. Schematic in (a) was created using BioRender.
Since noncoding variants do not always regulate the nearest gene(s), we also linked candidate variants to target promoters through co-accessibility and scRNA-seq integration (Methods, Figure 4a). For instance, the SMC peak-containing variant, rs7500448, shows high co-accessibility with the CDH13 promoter (Extended Data Figure 5), which is also an artery-specific eQTL for CDH13 in GTEx. Another relevant example is rs998584, located within a strong fibroblast peak 3’ of VEGFA, which is highly co-accessible with the VEGFA promoter (Extended Data Figure 5). In an orthogonal approach, we combined co-accessibility and RNA integration to identify peaks where accessibility correlates with target gene expression (referred to as Peak2Gene links). We identified a total of 148,617 Peak2Gene links when aggregating all cell types (Extended Data Figure 5), including for many CAD risk variants (Supplementary Data 5). Together, these single-nucleus chromatin annotations refine candidate regulatory mechanisms at CAD GWAS loci, for future functional validation in the appropriate cell types.
Prioritizing cell type-specific CAD functional variants
Chromatin accessibility quantitative trait locus (caQTL) mapping is a powerful association analysis to resolve candidate GWAS regulatory mechanisms 73–78. We thus calculated caQTLs in our dataset in four major coronary cell types (SMC, macrophages, fibroblasts, and endothelial cells) (Supplementary Data 6). Given our modest sample size (n=41), we used RASQUAL 79 for caQTL mapping to capture both population and allele-specific effects (Methods), as done previously for cultured coronary artery SMCs 80. As expected, the number of QTLs discovered per coronary cell type was proportional to the respective number of annotated nuclei (Figure 5a), with the most belonging to SMCs (1,984 at 5% FDR). 26% of these single cell caQTLs were also observed in coronary artery bulk ATAC-seq libraries (Supplementary Data 7) from the same patients (n=35) with 86% consistent effect size directions (Extended Data Figure 6). To determine whether these caQTLs regulate gene expression, we queried these variants for eQTL signals in GTEx artery tissues (coronary, aorta, and tibial). Out of the 1,984 unique SMC caQTLs (5% FDR), 47% were significant eQTLs (GTEx 5% FDR) in at least one GTEx arterial tissue. Most of the coronary SMC caQTLs that are GTEx eQTLs are shared across all artery types (Figure 5b). We also identified 71% concordant coronary artery caQTL and eQTL effect sizes (Figure 5c), which is consistent with reported findings in human T cells 75.
Figure 5. Identification of genetic variants that regulate chromatin accessibility within coronary artery cell types.
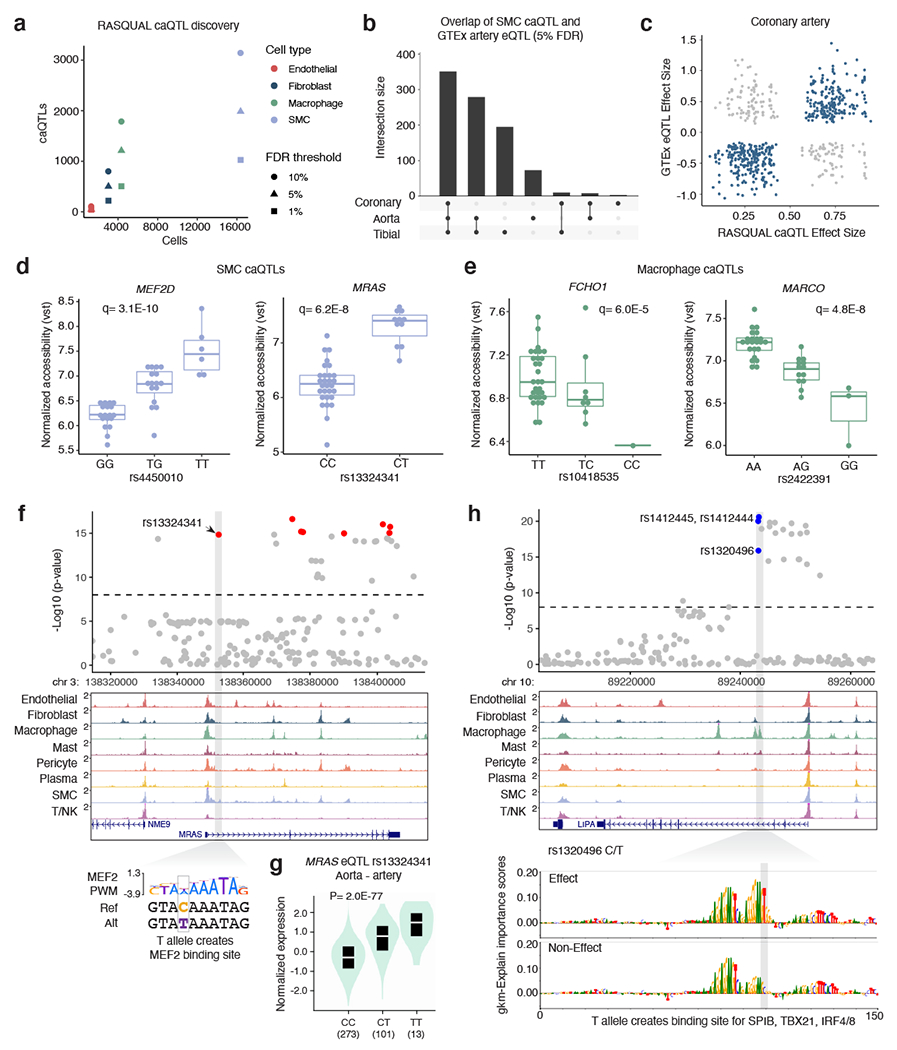
(a) The number of chromatin accessibility quantitative trait loci (caQTLs) identified using RASQUAL (at 10%, 5%, and 1% FDR cutoffs) within a cell type is proportional to the number of annotated cells. The color represents the cell type and shape represents the FDR cutoff. (b) UpSet plot for smooth muscle cells caQTLs that are expression quantitative trait loci in GTEx arterial tissues. Bars represent the intersection size for overlap of eQTLs between coronary artery, aorta, and tibial artery. (c) Comparison of RASQUAL effect sizes with GTEx effect sizes (beta). (d) Boxplots highlighting smooth muscle cell (n=40) normalized accessibility for MEF2D and MRAS caQTL variants and (e) macrophage (n=39) normalized accessibility for FCHO1 and MARCO caQTL variants. Vst: variance stabilizing transformation. The q-values represent the lead caQTL SNP q-value (Benjamini-Hochberg correction) generated from the likelihood-ratio test for the respective peak in RASQUAL. Boxplots (d-e) represent the median and interquartile range (IQR), with upper (75%) and lower (25%) quartiles shown and each dot representing a separate individual. (f) Example genome browser tracks showing CAD-associated caQTL at the MRAS locus in a smooth muscle cell specific peak. The T allele for rs13324341 creates a MEF2 putative binding site. (g) In GTEx artery (aorta shown here, n=387 unique individuals) the T allele for rs13324341 is highly associated with increased MRAS mRNA levels. The cis-eQTL p-value is shown from the GTEx pipeline that performs linear regression between genotype and normalized gene expression levels. Boxplot (black) within the violin plot includes median (white line) and IQR from 25% to 75%. (h) Example of prioritization of functional CAD variants using lsgkm machine learning based prediction. The rs13202496 variant at the LIPA locus (chromosome 10) resides in a strong macrophage peak. The T allele is predicted to create a putative SPIB binding site and increased chromatin accessibility. Feature importance score tracks for effect and non-effect alleles are visualized by gkmExplain (Methods).
We next applied these cell type caQTLs to further dissect CAD related mechanisms in the vessel wall. One example is rs4450010 at the MEF2D migraine/cardiovascular-associated gene within a SMC peak. The rs4450010-T allele creates a TEF1 (TEAD) binding site and correlates with both increased peak accessibility (Figure 5d) and increased MEF2D RNA expression in GTEx arterial tissues (Supplementary Figure 5). Several CAD GWAS variants were significant caQTLs in SMC or macrophages. For example, rs13324341 within intron 1 of MRAS (Muscle RAS oncogene homolog), also in a DNase site 81, is both a SMC caQTL and strong eQTL in GTEx arterial tissues (Supplementary Figure 5). Other top CAD GWAS-overlapping caQTLs include, among others, rs73551705 (BMP1) and rs17293632 (SMAD3) in SMCs and rs72844419 (GGCX) and rs10418535 (FCHO1) in macrophages (Extended Data Figure 6, Figure 5e). At the MRAS locus, the rs13324341 minor allele T (increased CAD risk) creates a MEF2 binding site (Figure 5f) and correlates with both increased accessibility and increased MRAS mRNA levels (Figure 5g). These MEF2D, MRAS, BMP1, SMAD3, and FCHO1 SNPs (or highly linked SNPs, r2 > 0.8) are all significant caQTLs (5% FDR) in bulk coronary artery ATAC-seq data (Supplementary Data 7).
To complement our QTL-based approach, we employed a machine learning-based strategy to assign sequence importance scores to CAD variants (10,117 tested) with effects on chromatin accessibility 82. Across three similar approaches (GkmExplain 83, gkmpredict, deltaSVM 84) we identified 127 high- or moderate-confidence CAD variants with predicted functional effects on chromatin accessibility (Supplementary Data 8). 102 (80%) had functional probability scores > 0.6 in RegulomeDb 2.0 and were annotated by enhancer, promoter, and TF ChIP-seq enrichment as well as motif disruption (Supplementary Data 8). About half of these variants were predicted to be functional in a single cell-type. One representative CAD variant, rs1320496 (LIPA), resides in a strong macrophage-specific peak, with the T allele (increased CAD risk) creating putative binding sites for SPIB, TBX21, and IRF4/8 (Figure 5h). Another intergenic SNP, rs10418535-C/T (between MAP1S and FCHO1), resides in a macrophage-specific peak with Peak2Gene links to FCHO1. The rs10418535-C allele (increased CAD risk) disrupts a PU.1/IRF motif and is predicted to attenuate chromatin accessibility (Extended Data Figure 7). rs10418535 is also a macrophage caQTL with a positive effect for the T allele, consistent with the deltaSVM prediction (Figure 5e, Extended Data Figure 7). Together, we demonstrate cell-type caQTL mapping and machine learning are complementary approaches to pinpoint candidate functional disease risk variants at high resolution.
PRDM16 and TBX2 are top candidate CAD transcription factors
Epigenomic profiles in disease-relevant tissues have been shown to resolve the correct target gene(s) at GWAS risk loci, which are often incorrectly annotated to the nearest gene 85. Importantly, our data nominates many targets of CAD risk variants in the vessel wall, including two previously unannotated TFs as candidate causal genes at their respective loci (Supplementary Data 5). The first locus on chr17 harbors dozens of tightly linked CAD variants within peaks in the BCAS3 gene (Extended Data Figure 8). However, Peak2Gene analysis demonstrates stronger links between these peaks and TBX2 expression in coronary arteries. At the second locus on chr1, several linked CAD variants are located within SMC peaks 5’ of the ACTRT2 gene and are highly correlated with PRDM16 and LINC00982 (PRDM16 divergent transcript) expression, but not other genes at the locus (Figure 6a). This locus also harbors an independent missense CAD-associated SNP (rs2493292; p.Pro634Leu) in exon 9 of PRDM16, suggesting both noncoding and coding effects on PRDM16 expression (Figure 6a). These results are consistent with activity-by-contact enhancer-gene mapping of CAD SNPs in coronary artery in ENCODE (Supplementary Table 6), differential expression (Supplementary Table 7) and cis-eQTLs (TBX2) in artery tissues (Supplementary Data 10), supporting PRDM16 and TBX2 as target CAD genes.
Figure 6. PRDM16 is a CAD-associated key driver transcriptional regulator in SMCs.
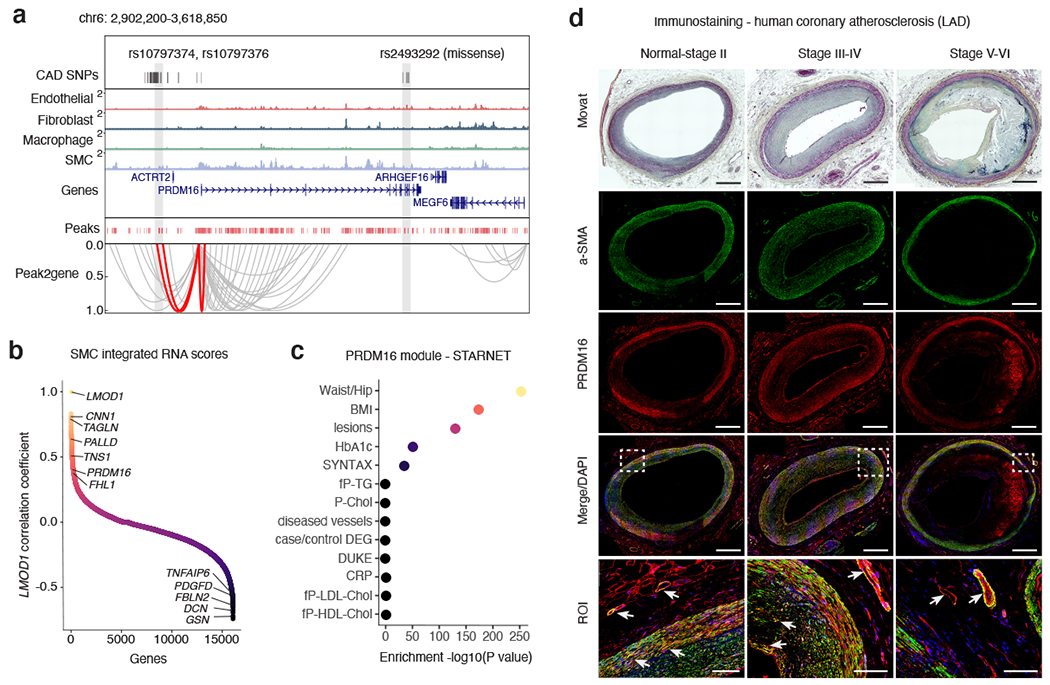
(a) Genome browser track highlighting the association between CAD associated SNPs and SMC marker genes through co-accessibility (peak2gene) detected by snATAC-seq data (Methods). The red loops represent the association between PRDM16 promoter and CAD associated SNPs. (b) Correlation coefficients of snATAC/scRNA integration scores gene expression levels between LMOD1 and genome-wide coding genes in SMCs. Genes were ranked by Pearson’s correlation coefficient with LMOD1. Representative positive and negative correlated SMC gene names are labeled. (c) Clinical trait enrichment for PRDM16 containing module in subclinical mammary artery in STARNET gene regulatory network datasets. Pearson’s correlation p-values (gene-level) were aggregated for each co-expression module using a two-sided Fisher’s exact test. Case/control differential gene expression (DEG) enrichment was estimated by a hypergeometric test. (d) Movat pentachrome staining and PRDM16 (red) and alpha-smooth muscle actin (a-SMA) (green) immunofluorescence staining of atherosclerotic human coronary artery segments - left anterior descending (LAD) from normal-Stage I, Stage III-IV, and Stage V-VI lesions based on Stary classification stages. Whole slide images captured from 20x confocal microscopy stitched tiles. PRDM16/a-SMA co-staining (see arrows) depicted in yellow from merged images. DAPI (blue) marks nuclei. n = 4 per group. Scale bar = 1 mm, except for region of interest (ROI): scale bar = 100 μm.
Both PRDM16 and TBX2 are snATAC SMC marker genes and remarkably PRDM16 is one of the top SMC marker genes along with known SMC gene LMOD1 72 (Supplementary Data 1). Given the similar gene score enrichment of PRDM16 and LMOD1 in SMC, we ranked PRDM16 by correlating all SMC gene scores and integrated RNA scores with LMOD1 (Figure 6b). Interestingly, PRDM16 was modestly correlated with traditional SMC markers, and negatively correlated with fibromyocyte marker genes. This may implicate PRDM16 as a SMC injury-response gene as opposed to a SMC identity marker gene. In additional scRNA-seq datasets, PRDM16 and TBX2 are enriched in mural cells (SMCs and pericytes) in both human coronary artery 7 and mouse aorta 9 (Supplementary Figure 6). To gain further insight into these two TF genes, we queried the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) gene regulatory networks across 7 cardiometabolic tissues (n=600), which revealed both PRDM16 and TBX2 as significant key driver genes in artery tissues (Supplementary Table 8, Extended Data Figure 8). In subclinical artery, the PRDM16-regulated module was highly enriched for the presence of atherosclerotic lesions and CAD severity, as well as metabolic clinical traits (Figure 6c, Extended Data Figure 8). Finally, we confirmed PRDM16 protein expression via immunofluorescence of normal, subclinical and advanced atherosclerotic coronary artery segments, with alpha-smooth muscle actin (a-SMA) and LMOD1 as positive controls (Figure 6d, Extended Data Figure 9). Similar to a-SMA, PRDM16 localized to the SMCs in the medial layer and small vessels in the vasa vasorum in healthy arteries, however expression was more restricted to the vasa vasorum and endothelium in diseased arteries (Figure 6d, Extended Data Figure 9). While we highlight these two examples, particularly PRDM16, this coronary dataset can be similarly utilized to prioritize mechanisms at many other CAD loci.
Discussion
In this study we have generated a single-nucleus atlas of human coronary artery chromatin accessibility for over 40 patients encompassing healthy and atherosclerotic samples, which captures gene regulation in vivo. Over half of the 323,767 identified cis-regulatory elements (54%) are unique to a specific cell type or limited number of cell types, underscoring the power of single-nucleus epigenomics for resolving unique cell type regulatory processes. Our snATAC results also provide direct insights into SMC phenotypic modulation. More specifically, we discovered accessible regions, genes and putative TF motifs that may drive the transition of native SMCs towards modulated SMCs (e.g. fibromyocytes). Finally, using an integrative statistical genetics and machine learning approach we prioritized cell-specific candidate regulatory variants and mechanisms underlying CAD loci.
There are now over 200 genetic loci associated with CAD risk, primarily located within noncoding genomic regions 13,15. This single-nucleus coronary artery epigenomic landscape provides a valuable resource to disentangle the target cell type(s), candidate causal genes and variants at the expanding number of CAD risk loci in diverse populations. For example we highlight a top caQTL rs13324341 at the MRAS locus which alters a MEF2 binding site in SMCs. Given the role of MRAS in Noonan syndrome-associated cardiomyocyte hypertrophy 86, these results may provide clues into SMC growth responses during CAD. We also highlight top predicted CAD regulatory variants acting in one or more cell types (e.g. rs1320496 at LIPA in macrophages). This dataset can also be leveraged to interrogate GWAS loci for related common vascular diseases (e.g., hypertension or coronary artery calcification).
By taking into account co-accessibility and scRNA-seq integration, we systematically link CAD risk variants to target gene promoters. This is critical given that GWAS variants are estimated to only target the nearest gene ~50% of the time. For example, using this approach we nominate two TF genes, PRDM16 and TBX2, as top candidate genes at their respective loci. PRDM16 is a top SMC marker gene from snATAC gene scores, however it may not be limited to marking SMC identity. PRDM16 (MEL1) is a TF known for roles in metabolism and controlling brown fat-to-skeletal muscle switches 87–89. However, PRDM16 is enriched in GTEx arterial tissues and was identified as a key driver gene in STARNET artery tissue, consistent with our snATAC data. PRDM16 regulates TGFβ signaling 90 through direct interactions with Smad91 and SKI92 proteins, both of which are associated with CAD 13. PRDM16 may play key roles in endothelial cells in arterial flow recovery 93. Similarly, TBX2 is enriched in GTEx arterial tissues and SMC clusters in our dataset, consistent with prior studies showing that Tbx2 activates SRF 94. TBX2 also links to relevant CAD pathways such as BMP, TGFβ, and FGF signaling 95. Functional follow-up studies to investigate target binding sites and affected SMC processes for these TFs may reveal additional mechanisms of disease risk.
While this study provides high-resolution insights into coronary artery gene regulatory signatures using primary human tissue samples, there are some known limitations. Given the lack of available lineage-tracing snATAC datasets, we cannot fully annotate intermediate cell types or precisely resolve their origins and fates during atherosclerosis 9,10. For example, some SMC-derived cells may be incorrectly annotated in T cell clusters, consistent with Alencar et al. 10 and Hansson et al. 96. Also, it is worth noting that we captured more nuclei from subclinical lesions compared to advanced atherosclerotic lesions, which potentially reflects higher difficulty in nuclei extraction for diseased samples. Finally, given our modest sample size for QTL based studies, we were underpowered to discover a large number of caQTLs for less abundant cell types (e.g. endothelial, T cells) or less frequent transition states. Future studies that can capture more nuclei per individual, especially in diseased coronary samples, will facilitate identification of additional context-specific regulatory mechanisms. Decreasing costs and adoption of single-nucleus and spatial sequencing technologies may further improve discovery of regulatory variants and mechanisms through multi-modal and integrative approaches 97,98.
In summary, we provide an atlas of chromatin accessibility in both healthy and atherosclerotic human coronary arteries. These cell type-specific epigenomic profiles characterise cis-regulatory programs at base-pair resolution to further our understanding of cell plasticity and heritable disease risk in the coronary vessel wall. We anticipate this will provide a valuable resource for the field, and act as a key next step toward functionally interrogating causal disease processes and informing pre-clinical studies to treat atherosclerosis.
Methods
Ethics Statement
All research described herein complies with ethical guidelines for human subjects research under approved Institutional Review Board (IRB) protocols at Stanford University (#4237 and #11925) and the University of Virginia (#20008), for the procurement and use of human tissues and information, respectively.
Coronary artery tissues and human subjects
Freshly explanted hearts from orthotopic heart transplantation recipients were procured at Stanford University under approved IRB protocols and written informed consent. Participants were not compensated for this study. Hearts were arrested in cardioplegic solution and rapidly transported from the operating room to the adjacent lab on ice. The proximal 5–6 cm of three major coronary vessels (left anterior descending (LAD), left circumflex (LCX), and right coronary artery (RCA)) were dissected from the epicardium on ice, trimmed of surrounding adipose (and in some samples the adventitia), rinsed in cold phosphate buffered saline, and rapidly snap frozen in liquid nitrogen. Coronary artery samples were also obtained at Stanford University (from Donor Network West and California Transplant Donor Network) from non-diseased donor hearts rejected by surgeons for heart transplantation and procured for research studies. All hearts were procured after written informed consent from legal next-of-kin or authorized parties for the donors. Reasons for rejected hearts include size incompatibility, comorbidities or risks for cardiotoxicity. Hearts were arrested in cardioplegic solution and transported on ice following the same protocol as hearts used for transplant. Explanted hearts were generally classified as ischemic or non-ischemic cardiomyopathy and prior ischemic events and evidence of atherosclerosis was obtained through retrospective review of electronic health records at Stanford Hospital and Clinics. The disease status of coronary segments from donor and explanted hearts was also evaluated by gross inspection at the time of harvest (for presence of lesions), as well as histological analysis of adjacent frozen tissues embedded in OCT blocks. Frozen tissues were transferred to the University of Virginia through a material transfer agreement and Institutional Review Board approved protocols. All samples were then stored at −80°C until day-of-processing.
Coronary artery sample processing and nuclei isolation
We performed single-nucleus ATAC on four coronary artery samples per day. For nuclear isolation we used a similar protocol to Omni-ATAC 40 that was optimized for frozen tissues and reported lower mitochondrial reads. We used approximately 50 mg of tissue per sample and the full nuclear isolation protocol is provided in Supplemental Methods. After the iodixanol gradient step we then carefully took the band containing the nuclei (setting the pipette volume to 100 μl) and added the nuclei to 1.3 mL of cold Nuclei Wash Buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.1% Tween-20) in a 1.5 mL Lo-Bind microcentrifuge tube. The microcentrifuge tube was inverted gently 5 times, nuclei gently mixed by pipetting (setting the pipette volume to 1 mL), and contents passed through a 40 μm Falcon cell strainer (Corning) into a new 1.5 mL Lo-Bind microcentrifuge tube (Eppendorf). Nuclei were pelleted by centrifugation for 5 minutes at 500 g at 4°C and supernatant carefully removed. Finally, this nuclei pellet was gently resuspended in 100 μl of the Nuclei Buffer provided with the kit (diluted from 20X Stock to 1X working concentration with nuclease-free water) by gently pipetting up and down. Samples and nuclei were kept on ice for all steps of the nuclear isolation. For each sample we measured the nuclei concentration by taking the mean of two separate counts using Trypan blue (Thermo Fisher) and the Countess II instrument (Thermo Fisher). Post cell lysis we generally observed less than 5% Live cells when visualizing with the Countess, consistent with proper lysis.
Single-nucleus ATAC library preparation
We used the 10x Genomics Chromium Single Cell ATAC Kit for all snATAC-seq experiments (additional details provided in Supplemental Methods). The full protocols for the single nucleus ATAC-seq data generation are available at the following link: https://support.10xgenomics.com/single-cell-atac.
Single-nucleus ATAC library sequencing
Single-nucleus ATAC libraries were shipped on dry ice to the Genome Core Facility at the Icahn School of Medicine at Mt. Sinai (New York, New York) for sequencing on an Illumina NovaSeq 6000. 40 libraries were sequenced using a NovaSeq S1 flow cell (100 cycles, 2 x 50 bp) and 4 libraries were sequenced using a NovaSeq S Prime (SP) flow cell (100 cycles, 2 x 50 bp).
Raw snATAC data processing and quality control
In-house single-nucleus ATAC-seq (snATAC) data was pre-processed using the 10x Genomics pipeline (Cell Ranger ATAC version 1.2.0 41) using the hg38 genome and default parameters. Samples from different patients were pre-processed separately. Individual cells with high quality were kept for downstream analysis (TSS enrichment >=7, unique barcode number >= 10,000 and doublet ratio < 1.5). QC measurements and filtering were conducted using ArchR (v1.0.1) 41.
Clustering of coronary artery snATAC data
snATAC reads from different individuals were combined at the single nucleus level and then mapped to each 500 bp bin across the hg38 reference genome for dimensionality reduction and clustering. The dimensionality reduction was conducted using a latent semantic indexing (LSI) algorithm and the 25,000 bins with the highest signal variance across individual cells were selected as input. The top 30 dimensions were selected for cell clustering. Cell clusters were identified by a shared nearest neighbor (SNN) modularity optimization-based clustering algorithm from the Seurat (v4.0.0) 99 package). Batch effect removal was conducted using the Harmony (v1.0) 100 package. We did not observe any improvement after batch correction using Harmony 100.
Single-nucleus ATAC gene scores and cell-type specific genes
The chromatin accessibility within a gene body as well as proximally and distally from the TSS was used to infer gene expression via computation of a “Gene Score” using the default method in ArchR (v1.0.1). The gene score profiles for all cells were subsequently used to generate a gene score matrix. The gene score matrix was also integrated with single-cell RNA-seq (scRNA) expression data (described below). Finally, a cell type annotation for each cluster was assigned using gene scores for cell type marker genes and later validated or further refined through scRNA-seq label transfer (Figure 1b–d).
Cell-type specific marker genes in our snATAC data (genes with significantly higher chromatin accessibility in a cluster than in other clusters) were identified using Wilcoxon rank-sum test and the genes with (Benjamini-Hochberg) adjusted p-value <= 0.01 and fold change >=2 were selected. The z-normalized gene scores for the cell-type specific genes were plotted as heatmap (Figure 2a).
scRNA-seq processing and snATAC integrative analysis
We integrated the coronary snATAC-seq dataset (28,316 nuclei) with previously published human coronary artery single-cell RNA-seq (scRNA-seq) from Wirka et al. 7. The preprocessed scRNA-seq data was downloaded from Gene Expression Omnibus (GEO) (accession GSE131780) and processed using Seurat 99 as described in the study. Genes expressed in less than 5 cells were filtered out. Cells with <= 500 or >= 3500 genes were also trimmed from the dataset as they likely represent defective cells or doublet/multiplet events. Moreover, cells containing >= 7.5% of reads mapping to the mitochondrial genome were discarded as low quality/dying cells often exhibit high levels of mitochondrial contamination. Upon discarding poor quality cells, 11,756 high quality cells and 19,965 genes remained for further analysis. Read counts were normalized using Seurat’s global-scaling method that normalizes gene expression measurements for each cell by the total expression, multiplies them by a 10000-scaling factor and log-transforms them. Upon finding the 2000 most variable genes in the data, dimensionality reduction was performed using PCA. The top 10 PCs were further used for UMAP visualization and cell clustering (using a shared nearest neighbor (SNN) modularity optimization-based algorithm in the Seurat package). The cluster specific genes (marker genes) for each cluster were identified with Seurat default method. The cell types of clusters were assigned according to the comparison between the cluster specific genes and the cell-type specific gene lists provided in the previous study (Supplementary Table 6) 7.
The cell type annotated scRNA expression matrix was then integrated with the snATAC gene score matrix (described in the above section) using the “addGeneIntegrationMatrix” function from ArchR, which identifies corresponding cells across datasets or “anchors” using Seurat’s mutual nearest neighbors (MNN) algorithm. To scale this step across thousands of cells, the total number of cells was divided into smaller groups and alignments were performed in parallel. Cell type labels within the Seurat scRNA-seq object metadata were transferred to the corresponding mutual nearest neighbors in the snATAC-seq data along with their gene expression signature. The output of the integration step resulted in snATAC-seq cells having both a chromatin accessibility and gene expression profile. After integration, snATAC cells were re-annotated in UMAP space using the scRNA transferred labels and these defined groups were used for downstream analyses as an alternative annotation in addition to the marker gene-based annotation. The scRNA transferred labels were also used in the Fibromyocyte vs. SMC differential analysis (Figure 3h–j).
Cell-type specific peak and TF motif enrichment
Genome-wide chromatin accessible regions for each “pseudo bulk” sample (reads from the same cluster were combined as a new sample) were detected using the “addReproduciblePeakSet” function in ArchR (with parameters extsize=100, cutOff=0.01, extendSummits=200). 323,767 chromatin accessible regions (peaks) were detected thereafter. The cell-type specific peaks (marker peaks) for each cluster/cell-type were identified using a similar strategy as identification of cell-type specific genes (with parameters FDR <= 0.01 & Log2FC >= 1). This resulted in a total of 173,357 cell-type specific peaks for different cell types. The enriched motifs for each cell type were predicted using the “addMotifAnnotations” function in the ArchR package based on the HOMER 46 motif database (v4.11) . The chromatin accessibility variability and deviation of transcription factors was estimated by chromVAR (1.12.0) 61 with genome-wide motif sites provided as potential binding sites.
Peak pathway annotation
To perform functional annotation of cell-type marker peaks (Extended Data Figure 4), we used GREAT 101 (v4.0.4) with default parameters. The top 5 functional annotation terms (from the Gene Ontology (GO) Biological Processes database) for each cell type were displayed as a dot plot. The colors and sizes of the dots represent -log10(FDR) (from the hypergeometric gene-based test) and the percentage of associated genes, respectively.
Trajectory analysis
The trajectory analysis was performed using the “addTrajectory” function in ArchR and specifying the cluster order (cluster 6 – cluster 5 – cluster 7, Figure 3f). We further visualized trajectory-dependent changes of (summarized) ATAC-seq motif signals using the “plotTrajectoryHeatmap” function in ArchR (Figure 3g).
Peak to gene linkage and co-accessibility analysis
We leveraged the integrated scRNA and snATAC data in order to explore correlations between co-accessible regions and gene expression. These candidate gene regulatory interactions were predicted using the “getPeak2GeneLinks” function with default parameters in ArchR. The peak2gene loops were collected and plotted using the Sushi package 102 with red color highlighting SNP-associated loops and grey color for other loops (Figure 6a–b). For the loops around VEGFA and CDH13 promoter (Extended Data Figure 5), the loops were predicted using the ArchR “addCoAccessibility” function with the additional parameter “maxDist=1e6”.
Differential analysis between SMCs and fibromyocytes
Details for differential analyses between SMCs and fibromyocytes are provided in Supplementary Methods.
LD score regression
We used the LDSC package (https://github.com/bulik/ldsc) to perform LD Score Regression using our single-nucleus ATAC peaks 63. We first downloaded GWAS summary statistics for: CAD 13; carotid intima-media thickness (cIMT) 103; carotid artery plaque 103; diastolic blood pressure (DBP), systolic blood pressure (SBP), and pulse pressure (PP) from the Million Veterans Program (MVP) 104; Alzheimer’s Disease 105; type 2 diabetes (UK Biobank) 106; body mass index (BMI) (UK Biobank) 106; and White Blood Cell (WBC) count (UK Biobank) 106. The UK Biobank summary statistics were downloaded from https://alkesgroup.broadinstitute.org/UKBB/. We used the provided munge_sumstats.py script to convert these GWAS summary statistics to a format compatible with ldsc. For each coronary artery cell type we lifted over bed file peak coordinates from hg38 to hg19. We then used these hg19 bed files to make annotation files for each cell type. We performed LD Score Regression according to the cell type-specific analysis tutorial (https://github.com/bulik/ldsc/wiki/Cell-type-specific-analyses).
CAD GWAS datasets
For comparison with CAD GWAS data we primarily used summary statistics from van der Harst et al. 13 that performed GWAS in UK Biobank subjects followed by replication in CARDIoGRAMplusC4D. For overlap of cell type peaks we also used SNPs from this GWAS and a recent CAD GWAS (Koyama et al. Nature Genetics 2021) that performed trans-ancestry meta-analysis 15. We obtained the list of lead SNPs (p < 5 x 10−8) from GWAS catalog (https://www.ebi.ac.uk/gwas) for the van der Harst study (accessions: GCST005194-GCST005196) and Supplementary Table 8 of Koyama et al. We used these lead variants as input to LDlinkR (https://ldlink.nci.nih.gov, https://github.com/CBIIT/LDlinkR) and subsequently kept variants with r2 >= 0.8 using EUR population.
Overlap of CAD GWAS SNPs with cell type peaks
We first used the UCSC liftover tool to convert the LDlinkR output of LD-expanded CAD SNPs from hg19 to hg38 coordinates. To account for CAD variants directly adjacent to a peak we considered a 100 bp window with the SNP lying within the center. We used BEDOPS 107 (v2.4.37) to extend the SNP position 50 bp in each direction. Next we used bedtools (v2.26.0) 108 to intersect these SNP coordinates with peak bed files from each cell type. We considered 1) peaks from the total peak set annotated to that cell type and 2) cell type marker peaks.
Genomic DNA sequencing
We isolated genomic DNA for each patient using the Qiagen DNeasy Blood and Tissue Kit. Approximately 20-25 mg of frozen left ventricle or coronary artery was placed in a 1.5 mL microcentrifuge tube and tissue lysed using lysis buffer, proteinase K, heating at 56°C for 1-3 hours or overnight, and intermittent vortexing. We then followed the kit instructions and genomic DNA eluted using 100 μl of Buffer AE (TE buffer). Genomic DNA samples were diluted to concentrations of between 5 ng/μl and 15 ng/μl in skirted 96 well PCR plates using TE buffer. Plates were sealed and shipped to Gencove (New York, USA) for 0.4X low-pass genomic DNA sequencing.
Genotype phasing, imputation, and genomic liftover
We obtained and downloaded the low-pass 0.4x whole-genome sequencing files (unphased) from the Gencove website. These were all provided in human genome build b37. We phased and imputed to the 1000 Genomes reference panel using Beagle (v5.1) 109,110. We then used Picard to liftover the phased autosomal VCFs from b37 to hg19, then hg19 to hg38 (“Picard Toolkit.” 2019. Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/; Broad Institute). Approximately 43,000 variants could not be mapped after liftover and were subsequently discarded. This left approximately 38 million total variants (10.1 million variants with minor allele frequency >1%).
Chromatin accessibility QTL preprocessing
To identify cell type-specific chromatin accessibility QTLs (caQTLs) we focused on four coronary cell types: smooth muscle cells, endothelial cells, fibroblasts, and macrophages. We first extracted cell-type assigned reads from our ArchR analysis in bam format for each snATAC library. For each individual cell type we excluded individuals with less than 20 cells from caQTL analysis. We ended up with SMC bam files for 40 patients, endothelial cell bam files for 37 patients, fibroblast bam files for 26 patients (due to some samples lacking adventitia), and macrophage bam files for 39 patients. To obtain region sets we took the peak set across all cell types and converted these peaks from bed to saf format. We used these peak coordinates in saf format and cell type bam files as input for featureCounts111 (v1.6.4) with the -p flag for paired-end mode. This subsequently generated raw count matrices for SMC, endothelial, fibroblast, and macrophage cells. For each cell type we only retained peaks with an average of 5 read counts across individuals.
We used RASQUAL79 (v1.0) to calculate caQTLs, which leverages differences in read counts between individuals as well as allele-differences within an individual at heterozygous sites 79. To simplify preparation of RASQUAL input files we used rasqualTools (https://github.com/kauralasoo/rasqual/tree/master/rasqualTools) to prepare compatible snATAC read count, metadata, and sample specific offset files. To calculate sample offsets we adjusted for library size as well the GC content of each peak.
For each individual we obtained genotypes from the low-pass whole genome sequencing that was performed by Gencove. We used VCFtools 112 to filter for variants with at least 5% minor allele frequency and select patients with corresponding snATAC-seq libraries. We used RASQUAL to create allele-specific vcf files (createASVCF.sh) for each cell type, which contains genotype information plus counts for reference and alternative alleles. We bypassed the qcFilterBam part of the createASVCF.sh script due to incompatibility and memory issues with our snATAC bam files. However, our bam files extracted from each cell type were previously filtered using ArchR and contain high quality cells and reads.
Calculation of chromatin accessibility QTLs
For each snATAC peak we tested association for all variants within a +/− 10 kb window. We ran RASQUAL using the -t flag to output only the top associated SNP for each peak. For all RASQUAL runs we adjusted for age, sex, and the first three principal components of ancestry in the covariate file (-x flag). To obtain a null distribution of q values we performed 5 separate permutation runs for each cell type using the --random-permutation flag to break the relationship between genotype and peak accessibility.
To adjust for multiple testing, we performed two FDR (false discovery rate) corrections. First, for each peak we obtained a q value corresponding to the SNP level FDR (Benjamini-Hochberg method) for that peak.. Next, the permutation test in RASQUAL adjusts for genome-wide multiple testing. For each peak we averaged the q values across the 5 RASQUAL permutation runs. This produced two vectors: one with real RASQUAL q values and one from the permuted q values for each peak. By comparing the real and permuted vectors of q values we were then able to calculate the q value corresponding to either 10% FDR, 5%, or 1% FDR. For plotting RASQUAL caQTL results as boxplots, we took raw count files for each cell type, adjusted for library size and performed variance stabilizing transformation in DESeq2 113 (v1.26.0).
Overlap of caQTLs with GTEx eQTLs
We used the QTlizer R package 81 to query the significant SMC caQTL rsIDs for eQTL signals in GTEx v8. We only retained GTEx eQTL signals at 5% FDR and subsequently filtered for relevant arterial tissues (coronary artery, aorta, tibial artery).
Publicly available gene expression data
Gene expression levels, expression quantitative loci (eQTL) data, and eQTL boxplots were obtained from the Genotype-Tissue Expression (GTEx) v8 portal website (https://www.gtexportal.org/home/). Differential gene expression data from publicly available Gene Expression Omnibus (GEO) in cardiovascular relevant systems was obtained via the HeartBioPortal (https://heartbioportal.com/).
Functional variant sequence-based predictive modeling
We first downloaded CAD GWAS summary statistics from van der Harst et al.13 and retained variants passing the genome-wide threshold (p < 5 x 10−8). This resulted in 10,117 variants that were tested. The variant scoring analysis (Figure 5h) was conducted using the lsgkm package (https://github.com/kundajelab/lsgkm) 82,114 and the GkmExplain package (https://github.com/kundajelab/gkmexplain) 83. For a given cell type (e.g. SMC), the reads from all the individual cells assigned to the cell type were first collected as a pseudo bulk sample. The pseudo bulk ATAC-seq peaks were detected with MACS2 115 (paired end mode, with additional parameter -q 0.01). In the model building step, peaks were split 10-fold for cross-validation. For each fold, the top 60000 peaks with highest −log10(q-value) were selected as the training set. The +/− 500 bp sequence from the peak summits were used as a positive set, while sequences from a 1000 bp region outside of peaks with matching GC-content were used as a negative set. The importance score of all the positions around the target SNP (up to +/− 100 bp) were plotted as sequence logo (Figure 5h).
STARNET gene regulatory network analysis
Based on STARNET multi-tissue gene expression data (bulk RNA-seq), tissue-specific and cross-tissue co-expression modules were inferred using WGCNA 116 as previously described 117. Enrichment for clinical trait associations was computed by aggregating Pearson’s correlation p values by co-expression module using Fisher’s method. Enrichment for differentially expressed genes was calculated using the hypergeometric test, with differentially expressed genes called by DESeq2 (+/− 30% change, FDR < 0.01) with adjustment for age and gender. The gene regulatory network was inferred among PRDM16 and TBX2 co-expressed genes using GENIE3 118 with potential regulators restricted to eQTL genes or known transcription factors. To identify hub genes in the network, weighted key driver analysis (wKDA) was carried out using the Mergeomics R package 119.
Immunofluorescence of human coronary artery tissues
Human coronary artery tissues were obtained as described above. Briefly, coronary artery segments were isolated from healthy and subclinical atherosclerotic left main and right coronary artery branches. Tissues were embedded in OCT blocks, snap-frozen in liquid nitrogen and stored at −80°C. Tissue blocks were cryosectioned at −20°C and 6 μm thickness and processed for immunostaining. Sections were rehydrated in PBS at room temperature (RT) and fixed in 10% neutral buffered formaldehyde for 10 min at RT, followed by PBS washes, protein blocking in casein buffer for 1 hour at RT, and incubated overnight at 4°C with anti-LMOD1 rabbit polyclonal antibody (Proteintech, 15117-1-AP; 1:100), alpha-smooth muscle actin (a-SMA) mouse monoclonal antibody (clone 1A2; Agilent/Dako, M0851; 1:100) or anti-PRDM16 rabbit polyclonal antibody (Abcam, ab106410; 1:2000) or no antibody negative control (PBS), with optimal dilutions determined by titrations with control tissues. Sections were washed in PBS and incubated with donkey anti-rabbit Alexa Fluor 555 conjugated secondary antibody (Thermo Fisher, A31572; 1:150) or donkey anti-mouse Alexa 488 conjugated secondary antibody (Thermo Fisher, A21202; 1:150) for 30 min at RT, washed in PBS, and stained with DAPI (1:500) and coverslipped using aqueous mounting media.
Whole slide images were captured at 25X magnification using a Zeiss LSM 880 Indimo, AxioExaminer confocal microscope with a Plan-Apochromat 25×/0.8 M27 objective in Line Sequential unidirectional mode. Signal corresponding to the DAPI (channel 1) and the protein of interest (channel 2) were obtained using lasers (respective wavelength of excitation of 405 and 561nm); a PMT and filters were used to collect the fluorescence emitted respectively at 410-480nm and 561-597nm. Images in both channels were merged with the Zeiss ZEN 3.3 Lite software (version 3.3.89). Brightness, gamma, and contrast were uniformly adjusted. Corresponding regions of interest of the sections immunostained with both antibodies were numerically magnified. Whole slide images were reconstructed from tiles acquired in brightfield using a high resolution HV-F203SCL Hitachi camera mounted on a Axio Scan microscope using a Plan-Apochromat 10X/0.3 objective.
For histology analysis, adjacent sections were stained with hematoxylin and eosin and Movat pentachrome as previously described 120. Images were captured using a Zeiss 183 Axio Scan Z1 at 20X magnification. The resulting czi files were visualized for staining using Zeiss ZEN 3.3 Lite software (version 3.3.89).
Histological analysis and quantitation of atherosclerosis
Please refer to Supplemental Methods
Sample size
No sample size calculations were performed a priori. Sample size (n=41) was determined based on the availability of tissue materials. However we also confirmed with a power analysis calculator that this sample size has 95% power to detect low frequency cell types (5-10%) based on the average number of cells captured per sample. Additional descriptions of post-hoc power calculations are provided in Supplementary Figure 7 and Supplementary Methods.
Genome annotations and browser tracks
All sequence alignments and annotations are with respect to the hg38 human reference genome. For each cell type we created bigWig files from aggregated cells and created custom tracks that were uploaded to the UCSC Genome Browser and viewed using hg38.
Statistical analyses
The statistical tests performed are listed in the respective figure legends or sections of the Methods. Data collection and analyses were performed with authors blind to the precise disease stages of the samples.
Extended Data
Extended Data Fig. 1.
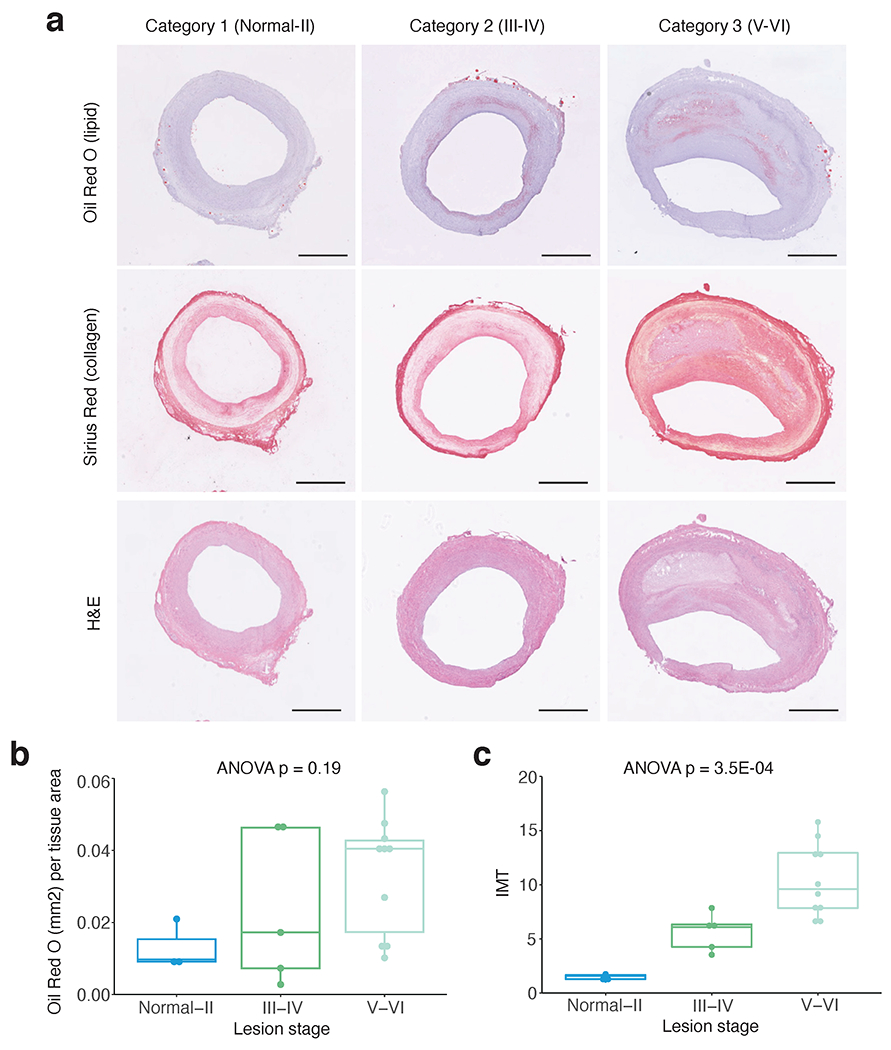
Histological characterization of human coronary artery sections. (a) Representative histology staining of adjacent frozen human coronary artery sections at different disease categories used for snATAC profiling. Category 1 reflects normal to Stary atherosclerosis stage I/II lesions with adaptive intimal thickening and early lipid (Oil Red O (ORO)) and collagen (Sirius Red) accumulation in the subintimal layer. Category 2 reflects Stary stage III/IV early/intermediate atheroma lesions with increased lipid and collagen accumulation and proliferation (Hematoxylin & Eosin (H&E)). Category 3 reflects Stary stage V/VI advanced fibroatheroma or complex lesions with more severe lipid and collagen deposition as well as lipid core and thin media layer. (b) Whole slide quantitative results of ORO area (mm2) normalized to overall tissue area and (c) Sirius Red based quantitation of intima-media thickness (IMT) with maximum intima and average media width captured from >6 automatically defined measurements (Methods). (a-c) Similar results were observed from n=3, n=5, and n=10 independent donor samples per lesion stage, respectively. One-way ANOVA p-values after Tukey post-hoc test are shown for comparisons across lesion stages. Boxplots (b-c) represent the median and interquartile range (IQR) with upper (75%) and lower (25%) quartiles shown. Scale bars = 1 mm.
Extended Data Fig. 2.
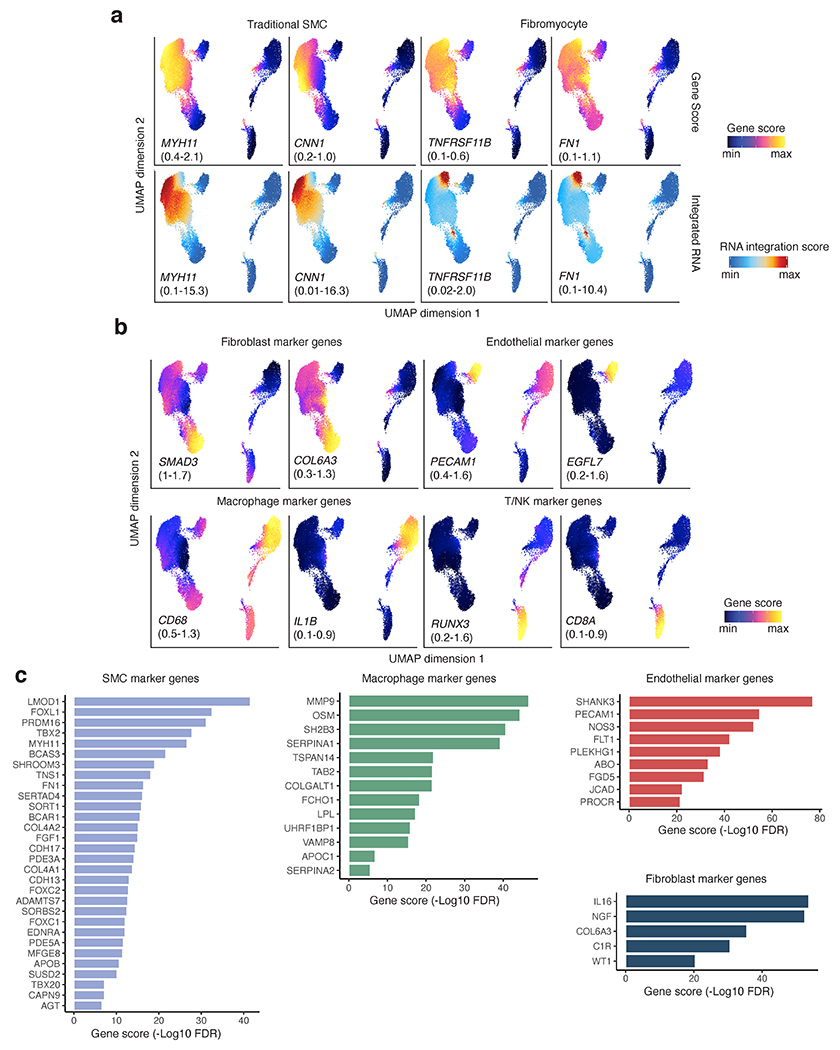
Coronary artery cell type marker genes from snATAC gene scores. (a) Representative UMAP plots of snATAC imputed gene activity scores and integrated RNA scores for SMC and fibromyocyte marker genes. (b) UMAP plots of imputed gene scores for additional cell type marker genes and CAD GWAS genes. (c) Top candidate genes at CAD GWAS loci with cell type enriched chromatin accessibility. Negative Log10 FDR enrichment values shown for CAD GWAS marker genes.
Extended Data Fig. 3.
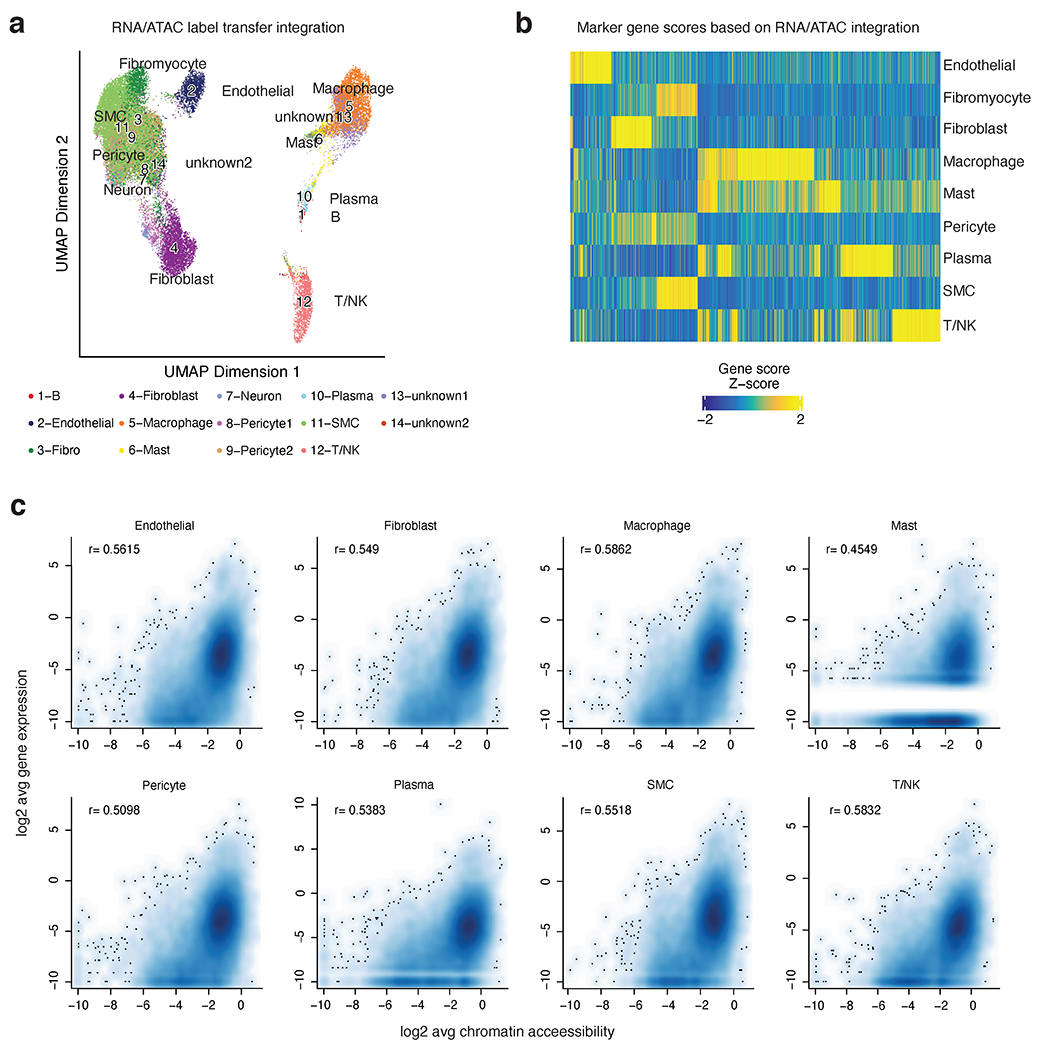
Integration of human coronary artery snATAC-seq data with human coronary artery scRNA-seq (from Wirka et al. Nat Med 2019). (a) UMAP showing projection of scRNA-seq cluster labels onto cells in the snATAC-seq dataset. Colors represent the assigned cellular identities from scRNA-seq label transfer. Detailed parameters of the snATAC-seq/scRNA-seq integration are provided in the Methods section. (b) Heatmap of marker gene scores after ArchR RNA/ATAC integration highlights 4,649 marker features. (c) Correlation of cell type specific scRNA and snATAC promoter accessibility (pseudo bulk reads from ATAC signal centered on TSS (+/− 3kb) for each gene). Log2 transformed data is represented as scatter plots and Pearson correlation coefficients are shown for each cell type. White lines represent missing gene counts from scRNA-seq dataset, which is most apparent in the low abundant Mast cells.
Extended Data Fig. 4.
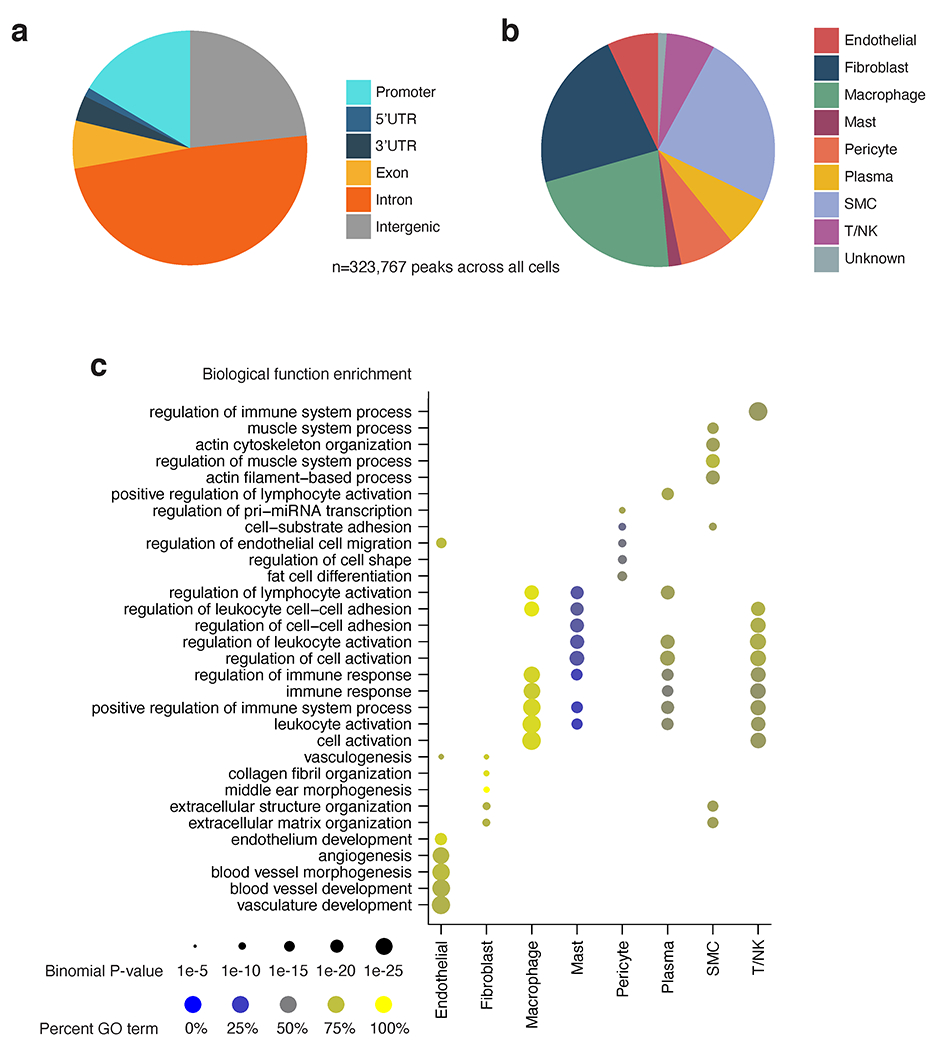
Coronary artery snATAC peak cell type and functional annotation. (a) Pie chart showing genomic annotations of the consensus set of coronary peaks across all cell types (n=323,767). Peaks were annotated using the ChIPseeker R/Bioconductor package (Yu et al. Bioinformatics 2015). (b) Pie chart of cell type annotation for peaks in the consensus peak set (n=323,767) according to ArchR (Granja et al. Nature Genetics 2021). Peaks were annotated with a cell type according to the group from which each peak originated according to ArchR’s iterative overlap procedure. (c) Functional enrichment analysis of cell type marker peaks using GREAT.
Extended Data Fig. 5.
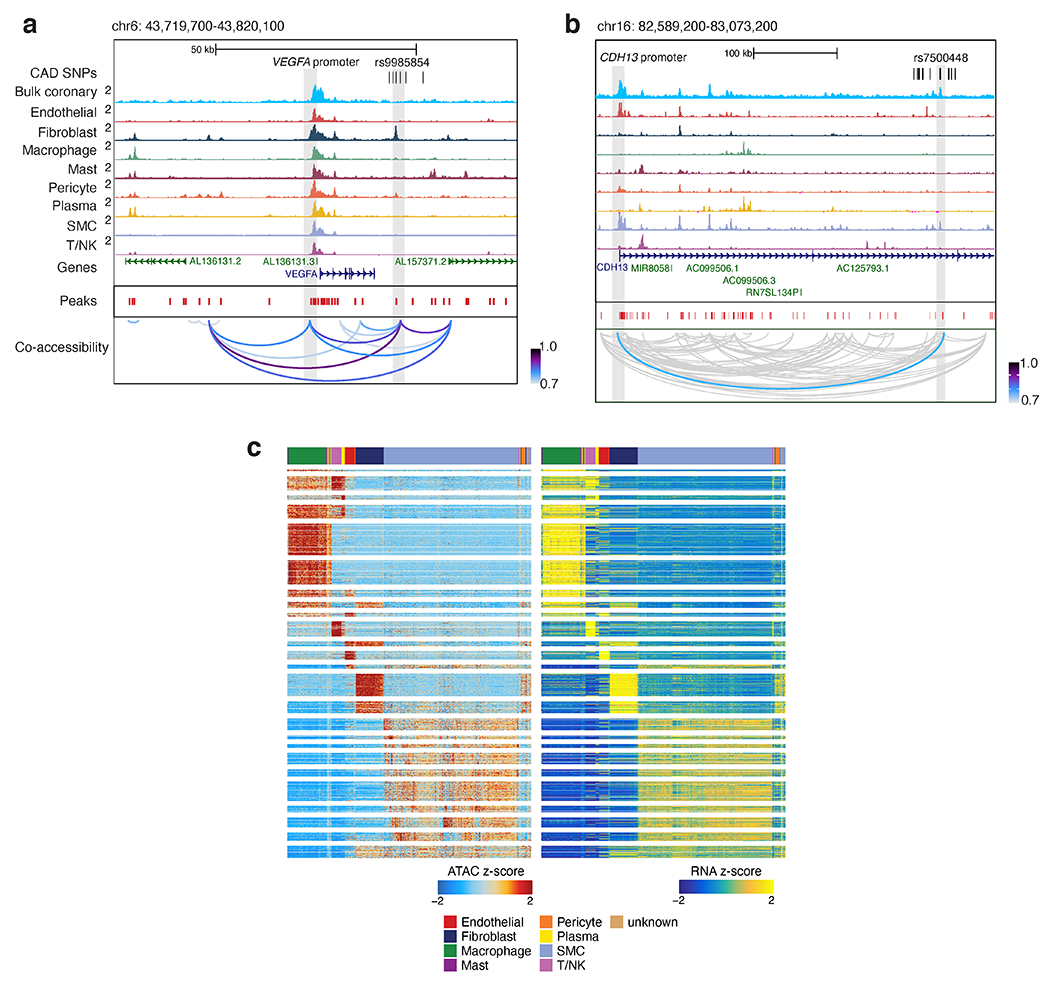
snATAC-seq co-accessibility and integration with scRNA-seq link putative regulatory elements to target promoters. (a) Genome browser tracks highlighting CAD-associated SNPs located within peaks linked to the VEGFA promoter peak through co-accessibility. Chromosome coordinates are hg38 genome build. (b) Genome browser tracks highlighting the intronic CAD SNP rs7500448 located in a smooth muscle cell peak in the CDH13 gene linked to the CDH13 promoter peak through co-accessibility. (c) Heatmap summary of ArchR Peak2Gene links (n=148,617) at 10 kb resolution where chromatin accessibility is highly correlated with target gene expression. Shown on the left are Z-scores for ATAC peak accessibility and on the right are Z-scores for RNA expression.
Extended Data Fig. 6.
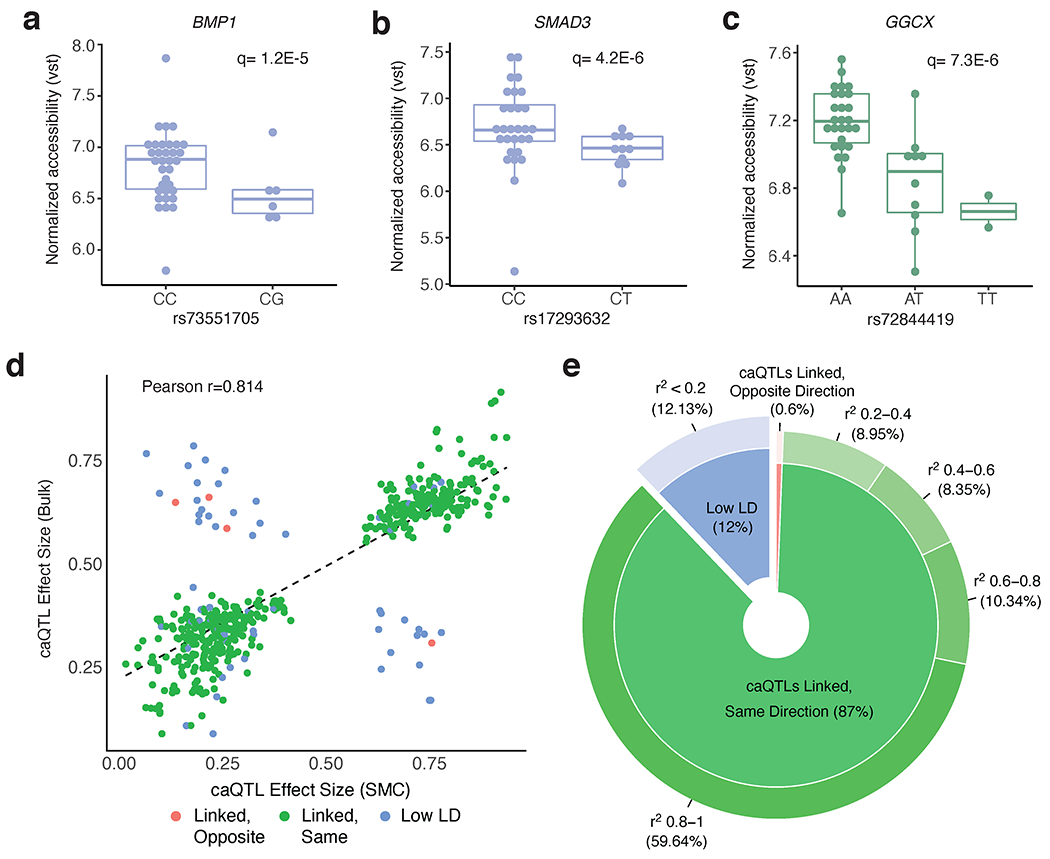
Additional CAD-associated variants that are coronary artery chromatin. accessibility QTLs (caQTLs). (a-b) Smooth muscle cell caQTL boxplots for variants at the BMP1 (rs73551705) and SMAD3 (rs17293632) CAD loci (n=40 unique individuals). (c) Macrophage caQTL boxplot for the rs72844419 variant at the GGCXCAD locus (n=39). Chromatin accessibility reads were normalized using variance stabilizing transformation (vst) in DESeq2. Boxplots represent the median and interquartile range (IQR), while the whisker represent up to 1.5 X IQR. (d-e) Comparison of effect size directions between smooth muscle cell caQTLs (5% FDR) and bulk coronary artery caQTLs (5% FDR), as visualized in scatter plot (d) and donut plot (e). For this analysis, 503 caQTL peaks are shared between both datasets (peaks with a corresponding significant caQTL variant). The rsID reported in the SMC caQTL results (n=40 individuals) was compared with the rsID reported in the bulk caQTL results (n=35 individuals). Two variants were considered to be in linkage disequilibrium (LD) if the r2 value between them was between 0.2 and 1 (in EUR population). If variants had an r2 value < 0.2 (in EUR population), the variants were considered to be in low LD (blue). For the caQTL effect size direction, we considered the RASQUAL Pi statistic. The RASQUAL Pi statistic can range from 0-1, where Pi < 0.5 reflects lower peak accessibility for the alternative allele and Pi > 0.5 reflects higher accessibility for the alternative allele. The effect sizes for linked variants go in the same direction (green) if the Pi values in SMCs and bulk coronary artery are both < 0.5 or both > 0.5. Linear regression line and Pearson correlation coefficient shown in (d).
Extended Data Fig. 7.
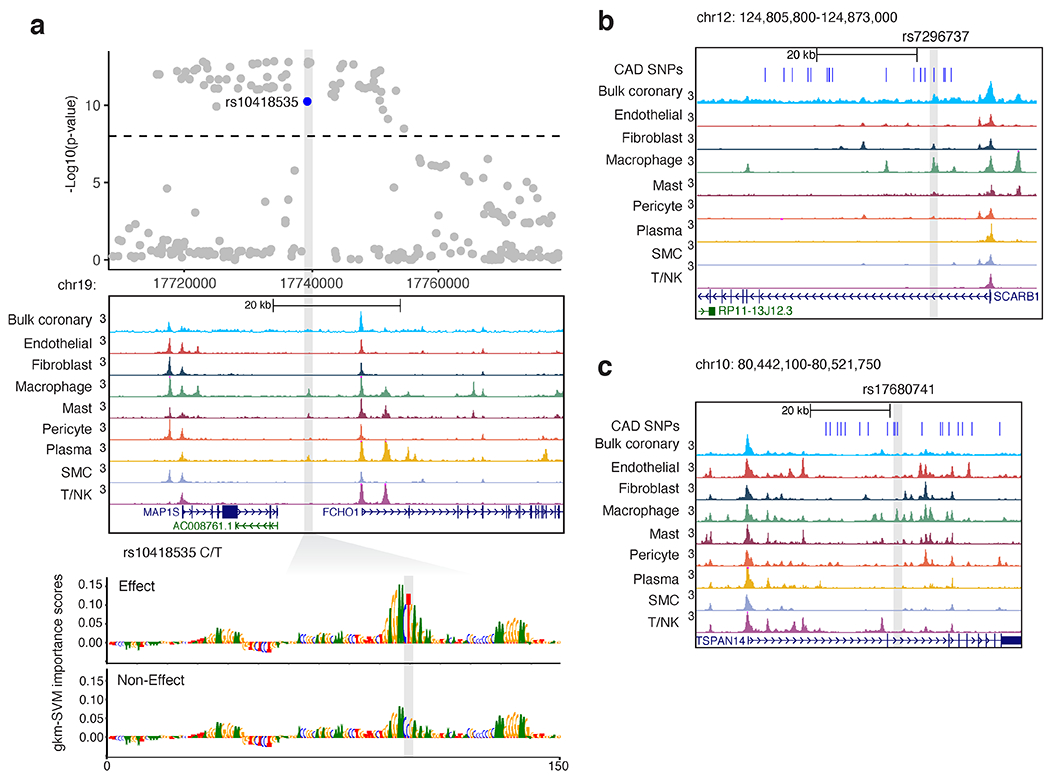
Examples of candidate CAD functional variants within macrophage accessible chromatin. (a) CAD GWAS locus MAP1S/FCH01 on chromosome 19 depicting multiple genome-wide significant variants (above dashed line). Log normalized P-values determined by linear mixed models and adjusted for genome-wide multiple testing as described by van der Harst et al, Circulation 2018. Highlighted variant rs10418535 is located within a macrophage/immune cell ATAC peak as shown in the genome browser tracks. gkm-SVM importance scores show the predicted effects of the T allele to form a functional binding site, while the C allele (non-effect) is predicted to disrupt TF binding. (b) Genome browser view showing 95% credible CAD SNPs (blue), highlighting rs7296737 located within a strong macrophage marker peak in the first intron of SCARB1 on chr12. (c) Genome browser view highlighting top credible CAD SNP rs17680741 residing in macrophage marker peak in the second intron of TSPAN14 on chr10.
Extended Data Fig. 8.
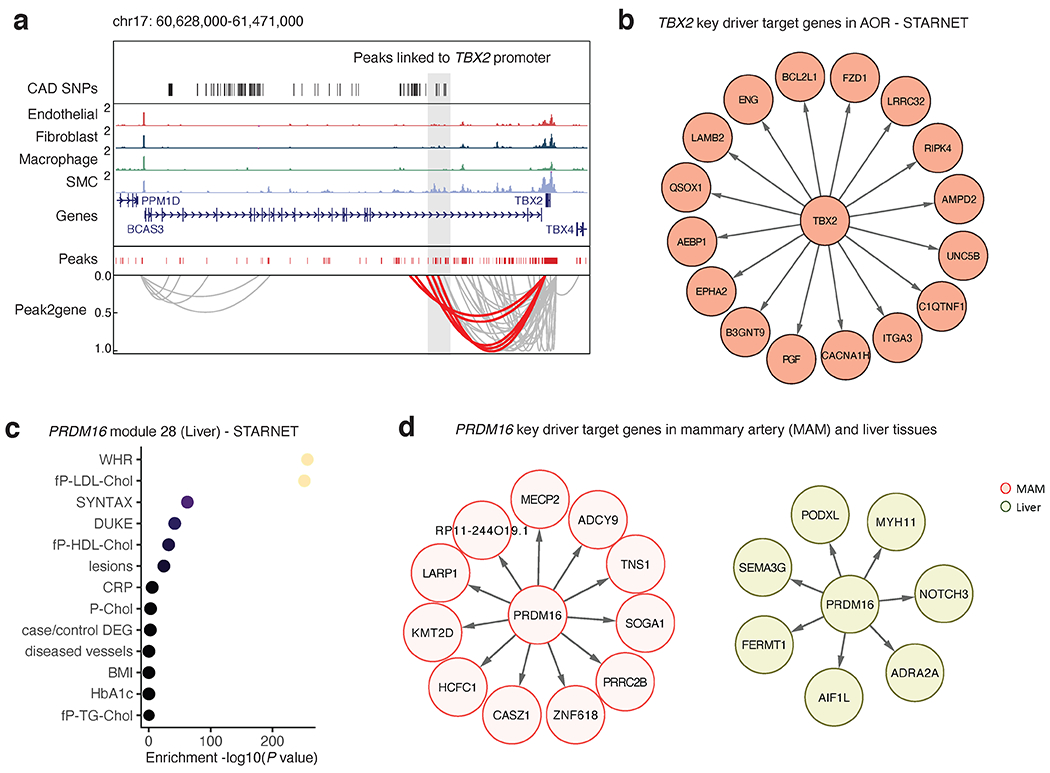
Co-accessibility and gene regulatory analyses prioritize transcriptional regulators TBX2 and PRDM16. (a) Genome browser track highlighting the association between CAD associated SNPs and SMC marker genes through co-accessibility (peak2gene) detected by snATAC-seq data (Methods). The red loops represent the association between TBX2 promoter and CAD associated SNPs. (b) Network visualization of TBX2 key driver target genes in STARNET atherosclerotic aortic root (AOR) tissue. (c) Clinical trait enrichment for PRDM16 module 28 in STARNET liver tissue. Pearson’s correlation p-values (gene-level) were aggregated for each co-expression module using a two-sided Fisher’s exact test. Case/control differential gene expression (DEG) enrichment was estimated by a hypergeometric test. (d) Network visualization of PRDM16 key driver target genes in STARNET mammary artery (MAM) and liver tissues.
Extended Data Fig. 9.
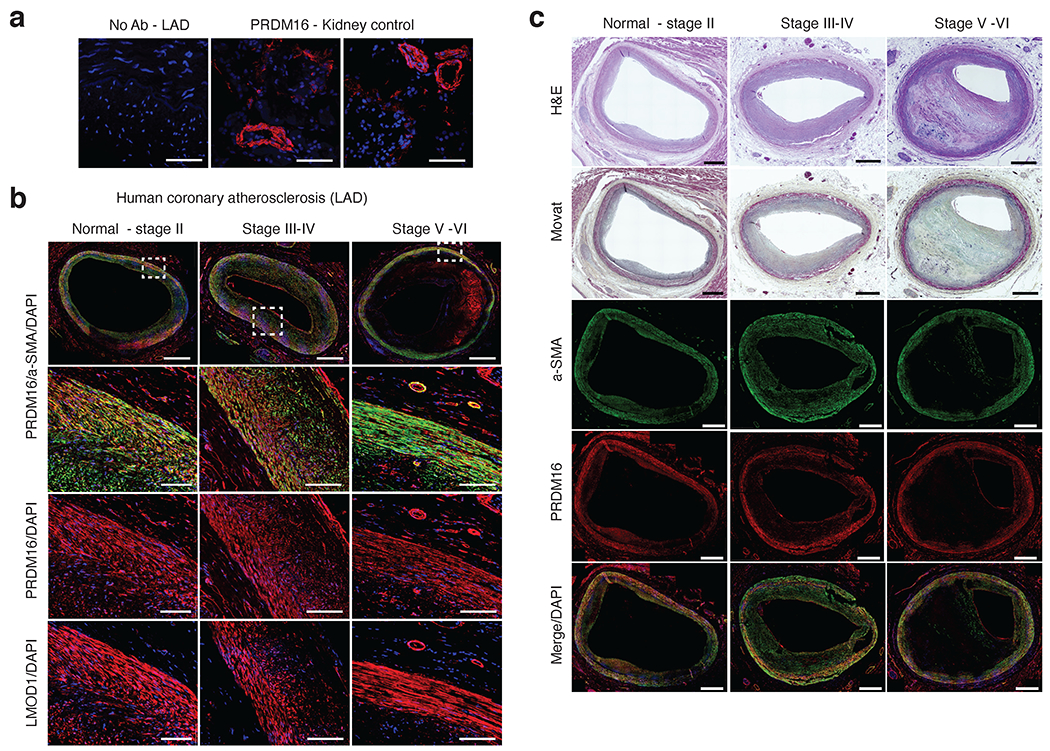
Immunostaining of PRDM16 protein in coronary atherosclerosis sections. (a) Representative negative control (no primary antibody) immunofluoresence (IF) staining in human coronary artery - left anterior descending (LAD). Positive staining of rabbit anti-PRDM16 in vessels in control kidney tissues. Similar results were observed from n = 4 independent donor samples per tissue. Scale bar = 100 um. (b) Representative IF staining of PRDM16 and LMOD1 in atherosclerotic human coronary artery (LAD) segments from normal-Stage II, Stage III-IV, and Stage V-VI lesions based on Stary classification stages. Red = PRDM16 or LMOD1, Green = alpha smooth muscle actin (a-SMA) and blue = DAPI (nuclei). Scale bar = 1mm (whole slide) or 100 um (highlighted regions of interest). (c) Representative hematoxylin & eosin (H&E) and MOVAT histology staining of distinct human coronary artery segments with similar lesion stages as (b). Scale bar = 1mm. (b-c) Similar results were observed from n = 4 (Normal-stage II), n=6 (Stage III-IV), and n=6 (Stage V-VI) independent donor samples per group.
Supplementary Material
Supplementary Table 1 Supplementary Tables 1-9
Supplementary Data 1. Top coronary artery cell type snATAC marker peaks and genes
Supplementary Data 2. Consensus set of human coronary artery snATAC-seq peaks across all cell types
Supplementary Data 3. Transcription factor motif enrichment within cell type peaks
Supplementary Data 4. Differentially accessible regulatory elements and functional annotation between traditional smooth muscle cells and fibromyocytes
Supplementary Data 5. CAD GWAS variants overlapping coronary artery snATAC-seq accessible chromatin sites
Supplementary Data 6. Chromatin accessibility QTLs within individual coronary artery cell types, calculated using RASQUAL
Supplementary Data 7. Chromatin accessibility QTLs from bulk coronary artery ATAC-seq data, calculated using RASQUAL
Supplementary Data 8. Machine learning prediction and annotation results of functional CAD variants for individual coronary artery cell types
Supplementary Data 9. Sample size estimations for top fibromyocyte genes comparing traditional smooth muscle cells and fibromyocytes
Supplementary Data 10. List of PRDM16 and TBX2 eQTLs in atherosclerosis-relevant human gene expression datasets
Acknowledgements
This work was supported by grants from: the National Institutes of Health (R01HL148239 and R00HL125912 to C.L.M.; R35GM133712 to C.Z.; R01HL141425 to A.V.F; R01HL125863 to J.LM.B.; R01HL130423, R01HL135093, and R01HL148167-01A1 to J.C.K.; R35HL144475, R01 HL125224, R01HL134817 and R01HL139478 to T.Q. and R01HL123370 to N.J.L.), the American Heart Association (20POST35120545 to A.W.T; A14SFRN20840000 to J.LM.B; 19EIA34770065 to N.J.L.), the Swedish Research Council and Heart Lung Foundation (2018-02529 and 20170265 to J.LM.B.), and the Fondation Leducq (‘PlaqOmics’ 18CVD02 to N.J.L., J.LM.B., A.V.F. and C.L.M.). We thank Dr. Peter Chiu, Paul Chang and Michael Wong at Stanford University for surgical assistance and research donor heart procurement. We thank Tiffany Koyano at Stanford University for assistance in extracting clinical information. We thank all of the transplant recipients and heart donors, family members, study coordinators and transplant tissue procurement team at Stanford. We thank Dr. Boxiang Liu and Dr. Natsuhiko Kumasaka for helpful discussions on QTL scripts. Finally, we thank Dr. Pat Pramoonjago and Sheri VanHoose at University of Virginia for histological support and all staff of the core facilities for tissue processing, library construction and sequencing assistance.
Competing Interests Statement
Dr. Björkegren is a shareholder in Clinical Gene Network AB who have an invested interest in STARNET. Dr. Finn at CVPath also acknowledges receiving financial support from the following entities: 4C Medical, 4Tech, Abbott Vascular, Ablative Solutions, Absorption Systems, Advanced NanoTherapies, Aerwave Medical, Alivas, Amgen, Asahi Medical, Aurios Medical, Avantec Vascular, BD, Biosensors, Biotronik, Biotyx Medical, Bolt Medical, Boston Scientific, Canon, Cardiac Implants, Cardiawave, CardioMech, Cardionomic, Celonova, Cerus, EndoVascular, Chansu Vascular Technologies, Childrens National, Concept Medical, Cook Medical, Cooper Health, Cormaze, CRL, Croivalve, CSI, Dexcom, Edwards Lifesciences, Elucid Bioimaging, eLum Technologies, Emboline, Endotronix, Envision, Filterlex, Imperative Care, Innovalve, Innovative, Cardiovascular Solutions, Intact Vascular, Interface Biolgics, Intershunt Technologies, Invatin, Lahav, Limflow, L&J Bio, Lutonix, Lyra Therapeutics, Mayo Clinic, Maywell, MDS, MedAlliance, Medanex, Medtronic, Mercator, Microport, Microvention, Neovasc, Nephronyx, Nova Vascular, Nyra Medical, Occultech, Olympus, Ohio Health, OrbusNeich, Ossio, Phenox, Pi-Cardia, Polares Medical, Polyvascular, Profusa, ProKidney, LLC, Protembis, Pulse Biosciences, Qool Therapeutics, Recombinetics, Recor Medical, Regencor, Renata Medical, Restore Medical, Ripple Therapeutics, Rush University, Sanofi, Shockwave, SMT, SoundPipe, Spartan Micro, Spectrawave, Surmodics, Terumo Corporation, The Jacobs Institute, Transmural Systems, Transverse Medical, TruLeaf, UCSF, UPMC, Vascudyne, Vesper, Vetex Medical, Whiteswell, WL Gore, Xeltis. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. All other authors declare that they have no competing interests relevant to the contents of this paper to disclose.
Footnotes
Code availability
Our results make use of published software tools with detailed parameters included in the Methods. All custom scripts used to generate these results are available on GitHub (https://github.com/MillerLab-CPHG/Coronary_snATAC and https://github.com/MillerLab-CPHG/coronary_histology).
Data availability
All raw and processed single-nucleus chromatin accessibility sequencing datasets are made available on the Gene Expression Omnibus (GEO) database (accessions: GSE175621 and GSE188422). The processed and analyzed snATAC data will also be made available on the PlaqView single-cell data portal (https://www.plaqview.com). All caQTL data are available in the Supplementary Data. Low-pass whole genome sequencing based genotyping data are available on dbGaP (accession: phs002855.v1.p1). The human coronary artery scRNA-seq dataset we used in this study from Wirka et al. (Nature Medicine 2019) is available through GEO (accession: GSE131778). The mouse atherosclerosis scRNA-seq dataset from Pan et al. (Circulation 2020) is available through GEO (accession: GSE155513). The reprocessed and analyzed human and mouse datasets are also available on PlaqView. Gene expression levels, expression quantitative loci (eQTL) data, and eQTL boxplots were obtained from the Genotype-Tissue Expression (GTEx) v8 portal website (https://www.gtexportal.org), GEO and HeartBioPortal (www.heartbioportal.com). Gene regulatory network analysis data from the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) are available at http://starnet.mssm.edu.
References
- 1.Libby P Inflammation in atherosclerosis. Nature 420, 868–874 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Souilhol C, Harmsen MC, Evans PC & Krenning G Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res 114, 565–577 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Stary HC et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 92, 1355–1374 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Winkels H et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res 122, 1675–1688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cochain C et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res 122, 1661–1674 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Fernandez DM et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med 25, 1576–1588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wirka RC et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med 25, 1280–1289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Depuydt MAC et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res 127, 1437–1455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan H. et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 142, 2060–2075 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alencar GF et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 142, 2045–2059 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci USA 117, 15818–15826 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikpay M et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet 47, 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Harst P & Verweij N Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res 122, 433–443 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson CP et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet 49, 1385–1391 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Koyama S et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet 52, 1169–1177 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Aragam KG et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. medRxiv (2021) doi: 10.1101/2021.05.24.21257377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erdmann J, Kessler T, Munoz Venegas L & Schunkert H A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc. Res 114, 1241–1257 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards SL, Beesley J, French JD & Dunning AM Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet 93, 779–797 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinz S, Romanoski CE, Benner C & Glass CK The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol 16, 144–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller CL et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun 7, 12092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu B, Gloudemans MJ, Rao AS, Ingelsson E & Montgomery SB Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet 51, 768–769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Q et al. Molecular mechanisms of coronary disease revealed using quantitative trait loci for TCF21 binding, chromatin accessibility, and chromosomal looping. Genome Biol 21, 135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stolze LK et al. Systems Genetics in Human Endothelial Cells Identifies Non-coding Variants Modifying Enhancers, Expression, and Complex Disease Traits. Am. J. Hum. Genet 106, 748–763 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corces MR et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiou J et al. Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk. Nat. Genet 53, 455–466 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiou J et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hocker JD et al. Cardiac cell type-specific gene regulatory programs and disease risk association. Sci. Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muto Y et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun 12, 2190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domcke S et al. A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rai V et al. Single-cell ATAC-Seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol. Metab 32, 109–121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziffra RS et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morabito S et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet 53, 1143–1155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Örd T et al. Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ. Res 129, 240–258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Granja JM et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet 53, 403–411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Granja JM et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 37, 1458–1465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Virmani R, Burke AP, Farb A & Kolodgie FD Pathology of the vulnerable plaque. J. Am. Coll. Cardiol 47, C13–8 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Mulligan-Kehoe MJ & Simons M Vasa vasorum in normal and diseased arteries. Circulation 129, 2557–2566 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Virmani R et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol 25, 2054–2061 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Creemers EE, Sutherland LB, Oh J, Barbosa AC & Olson EN Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol. Cell 23, 83–96 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Maeda T, Gupta MP & Stewart AFR TEF-1 and MEF2 transcription factors interact to regulate muscle-specific promoters. Biochem. Biophys. Res. Commun 294, 791–797 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Almontashiri NAM et al. 9p21.3 Coronary Artery Disease Risk Variants Disrupt TEAD Transcription Factor-Dependent Transforming Growth Factor β Regulation of p16 Expression in Human Aortic Smooth Muscle Cells. Circulation 132, 1969–1978 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Yoshida T et al. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res 92, 856–864 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Du KL et al. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol. Cell. Biol 23, 2425–2437 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J, Kitchen CM, Streb JW & Miano JM Myocardin: a component of a molecular switch for smooth muscle differentiation. J. Mol. Cell. Cardiol 34, 1345–1356 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Wang D-Z et al. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci USA 99, 14855–14860 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meadows SM, Myers CT & Krieg PA Regulation of endothelial cell development by ETS transcription factors. Semin. Cell Dev. Biol 22, 976–984 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stamatovic SM, Keep RF, Mostarica-Stojkovic M & Andjelkovic AV CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J. Immunol 177, 2651–2661 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Zhang DE, Hetherington CJ, Chen HM & Tenen DG The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol. Cell. Biol 14, 373–381 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cui L et al. Activation of JUN in fibroblasts promotes pro-fibrotic programme and modulates protective immunity. Nat. Commun 11, 2795 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kitoh A et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity 31, 609–620 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Ono M et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446, 685–689 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Masuda A et al. Essential role of GATA transcriptional factors in the activation of mast cells. J. Immunol 178, 360–368 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagao M et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ. Res 126, 517–529 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulik-Sullivan B et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tabas I & Lichtman AH Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 47, 621–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farrugia AJ et al. CDC42EP5/BORG3 modulates SEPT9 to promote actomyosin function, migration, and invasion. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nyati KK, Agarwal RG, Sharma P & Kishimoto T Arid5a regulation and the roles of arid5a in the inflammatory response and disease. Front. Immunol 10, 2790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin M-E, Chen T, Leaf EM, Speer MY & Giachelli CM Runx2 expression in smooth muscle cells is required for arterial medial calcification in mice. Am. J. Pathol 185, 1958–1969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin M-E et al. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc. Res 112, 606–616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nott A et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu S et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur. Heart J 40, 2398–2408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beaudoin M et al. Myocardial Infarction-Associated SNP at 6p24 Interferes With MEF2 Binding and Associates With PHACTR1 Expression Levels in Human Coronary Arteries. Arterioscler. Thromb. Vasc. Biol 35, 1472–1479 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nanda V et al. Functional regulatory mechanism of smooth muscle cell-restricted LMOD1 coronary artery disease locus. PLoS Genet 14, e1007755 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benaglio P et al. Mapping genetic effects on cell type-specific chromatin accessibility and annotating complex trait variants using single nucleus ATAC-seq. BioRxiv (2020) doi: 10.1101/2020.12.03.387894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calderon D et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet 51, 1494–1505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gate RE et al. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat. Genet 50, 1140–1150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bryois J et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun 9, 3121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khetan S et al. Type 2 Diabetes-Associated Genetic Variants Regulate Chromatin Accessibility in Human Islets. Diabetes 67, 2466–2477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Currin KW et al. Genetic effects on liver chromatin accessibility identify disease regulatory variants. Am. J. Hum. Genet 108, 1169–1189 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumasaka N, Knights AJ & Gaffney DJ Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet 48, 206–213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu B et al. Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am. J. Hum. Genet 103, 377–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Munz M et al. Qtlizer: comprehensive QTL annotation of GWAS results. Sci. Rep 10, 20417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghandi M, Lee D, Mohammad-Noori M & Beer MA Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol 10, e1003711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shrikumar A, Prakash E & Kundaje A GkmExplain: fast and accurate interpretation of nonlinear gapped k-mer SVMs. Bioinformatics 35, i173–i182 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee D et al. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet 47, 955–961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nasser J et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Higgins EM et al. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2, e91225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seale P et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Seale P et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab 6, 38–54 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kajimura S et al. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 460, 1154–1158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu D et al. PRDM16 Upregulation Induced by MicroRNA-448 Inhibition Alleviates Atherosclerosis via the TGF-β Signaling Pathway Inactivation. Front. Physiol 11, 846 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 91.Warner DR et al. PRDM16/MEL1: a novel Smad binding protein expressed in murine embryonic orofacial tissue. Biochim. Biophys. Acta 1773, 814–820 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Takahata M et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J. Biol. Chem 284, 3334–3344 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Craps S et al. Prdm16 supports arterial flow recovery by maintaining endothelial function. Circ. Res (2021) doi: 10.1161/CIRCRESAHA.120.318501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barron MR et al. Serum response factor, an enriched cardiac mesoderm obligatory factor, is a downstream gene target for Tbx genes. J. Biol. Chem 280, 11816–11828 (2005). [DOI] [PubMed] [Google Scholar]
- 95.Shirai M, Imanaka-Yoshida K, Schneider MD, Schwartz RJ & Morisaki T T-box 2, a mediator of Bmp-Smad signaling, induced hyaluronan synthase 2 and Tgfbeta2 expression and endocardial cushion formation. Proc Natl Acad Sci USA 106, 18604–18609 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hansson GK, Jonasson L, Holm J & Claesson-Welsh L Class II MHC antigen expression in the atherosclerotic plaque: smooth muscle cells express HLA-DR, HLA-DQ and the invariant gamma chain. Clin. Exp. Immunol 64, 261–268 (1986). [PMC free article] [PubMed] [Google Scholar]
- 97.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma S et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 99.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Phanstiel DH, Boyle AP, Araya CL & Snyder MP Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics 30, 2808–2810 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Franceschini N et al. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat. Commun 9, 5141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Giri A et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet 51, 51–62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sudlow C et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neph S et al. BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Browning SR & Browning BL Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet 81, 1084–1097 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Browning BL, Zhou Y & Browning SR A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet 103, 338–348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Danecek P et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee D LS-GKM: a new gkm-SVM for large-scale datasets. Bioinformatics 32, 2196–2198 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Talukdar HA et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst 2, 196–208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huynh-Thu VA, Irrthum A, Wehenkel L & Geurts P Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shu L et al. Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics 17, 874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Otsuka F et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis 241, 772–782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Supplementary Tables 1-9
Supplementary Data 1. Top coronary artery cell type snATAC marker peaks and genes
Supplementary Data 2. Consensus set of human coronary artery snATAC-seq peaks across all cell types
Supplementary Data 3. Transcription factor motif enrichment within cell type peaks
Supplementary Data 4. Differentially accessible regulatory elements and functional annotation between traditional smooth muscle cells and fibromyocytes
Supplementary Data 5. CAD GWAS variants overlapping coronary artery snATAC-seq accessible chromatin sites
Supplementary Data 6. Chromatin accessibility QTLs within individual coronary artery cell types, calculated using RASQUAL
Supplementary Data 7. Chromatin accessibility QTLs from bulk coronary artery ATAC-seq data, calculated using RASQUAL
Supplementary Data 8. Machine learning prediction and annotation results of functional CAD variants for individual coronary artery cell types
Supplementary Data 9. Sample size estimations for top fibromyocyte genes comparing traditional smooth muscle cells and fibromyocytes
Supplementary Data 10. List of PRDM16 and TBX2 eQTLs in atherosclerosis-relevant human gene expression datasets
Data Availability Statement
All raw and processed single-nucleus chromatin accessibility sequencing datasets are made available on the Gene Expression Omnibus (GEO) database (accessions: GSE175621 and GSE188422). The processed and analyzed snATAC data will also be made available on the PlaqView single-cell data portal (https://www.plaqview.com). All caQTL data are available in the Supplementary Data. Low-pass whole genome sequencing based genotyping data are available on dbGaP (accession: phs002855.v1.p1). The human coronary artery scRNA-seq dataset we used in this study from Wirka et al. (Nature Medicine 2019) is available through GEO (accession: GSE131778). The mouse atherosclerosis scRNA-seq dataset from Pan et al. (Circulation 2020) is available through GEO (accession: GSE155513). The reprocessed and analyzed human and mouse datasets are also available on PlaqView. Gene expression levels, expression quantitative loci (eQTL) data, and eQTL boxplots were obtained from the Genotype-Tissue Expression (GTEx) v8 portal website (https://www.gtexportal.org), GEO and HeartBioPortal (www.heartbioportal.com). Gene regulatory network analysis data from the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) are available at http://starnet.mssm.edu.