

Abstract
Topoisomerases play a pivotal role in ensuring DNA metabolisms during replication, transcription and chromosomal segregation. To manage DNA topology, topoisomerases generate break(s) in the DNA backbone by forming transient enzyme-DNA cleavage complexes (TOPcc) with phosphotyrosyl linkages between DNA ends and topoisomerase catalytic tyrosyl residues. Topoisomerases have been identified as the cellular targets of a variety of anti-cancer drugs (e.g. topotecan, irinotecan, etoposide and doxorubicin, and antibiotics (e.g. ciprofloxacin and levofloxacin). These drugs, as well as other exogenous and endogenous agents, convert the transient TOPcc into persistent TOPcc, which we refer to as topoisomerase DNA-protein crosslinks (TOP-DPC) that challenge genome integrity and lead to cell death if left unrepaired. Proteolysis of the bulky protein component of TOP-DPC (debulking) is a poorly understood repair process employed across eukaryotes. TOP-DPC proteolysis can be achieved either by the ubiquitin-proteasome pathway (UPP) or by non-proteasomal proteases, which are typified by the metalloprotease SPRTN/WSS1. Debulking of TOP-DPC exposes the phosphotyrosyl bonds, hence enables tyrosyl-DNA phosphodiesterases (TDP1 and TDP2) to access and cleave the bonds. In this review, we focus on current knowledge of the protease pathways for debulking TOP-DPC and highlighting recent advances in understanding the mechanisms regulating the proteolytic repair pathways. We also discuss the avenues that are being exploited to target the proteolytic repair pathways for improving the clinical outcome of topoisomerase inhibitors.
Keywords: Topoisomerases, DNA-protein crosslinks, Ubiquitin-proteasome pathway, SPRTN, SUMOylation
1. Introduction
DPC and especially those generated by topoisomerases (the TOP-DPC discussed in this review) are a current focus of scientific research due to their prevalence as environmental DNA damage and source of human diseases including cancers and neurodegenerative disorders. They are also a fundamental molecular mechanism for widely used antibacterial and anticancer therapies, which include not only topoisomerase inhibitors (fluoroquinolones, camptothecins, etoposide, doxorubicin to name a few) but also for DNA alkylating agents, platinum derivatives and 5-azacytidine derivatives.
The excision repair of TOP-DPC can be divided in three successive processes (outlined in red in Fig. 1): detection, debulking and excision. The debulking is generally carried out by proteolytic digestion leading to topoisomerase polypeptides remaining covalently linked to the DNA terminus (5’-DpC or 3’-DpC). This is the focus of our review (Sections 4 and 5). An alternative cellular topoisomerase unfolding pathway has recently been discovered and will be discussed in Section 6. The excision pathways for TOP-DpC (DNA-peptide-crosslinks) and non-proteolyzed TOP-DPC by nucleases have been detailed in a complementary review [1] and will only be discussed in Section 9. Both DNA Repair reviews are complementary.
Fig. 1. Pathways for the debulking of TOP-DPC.
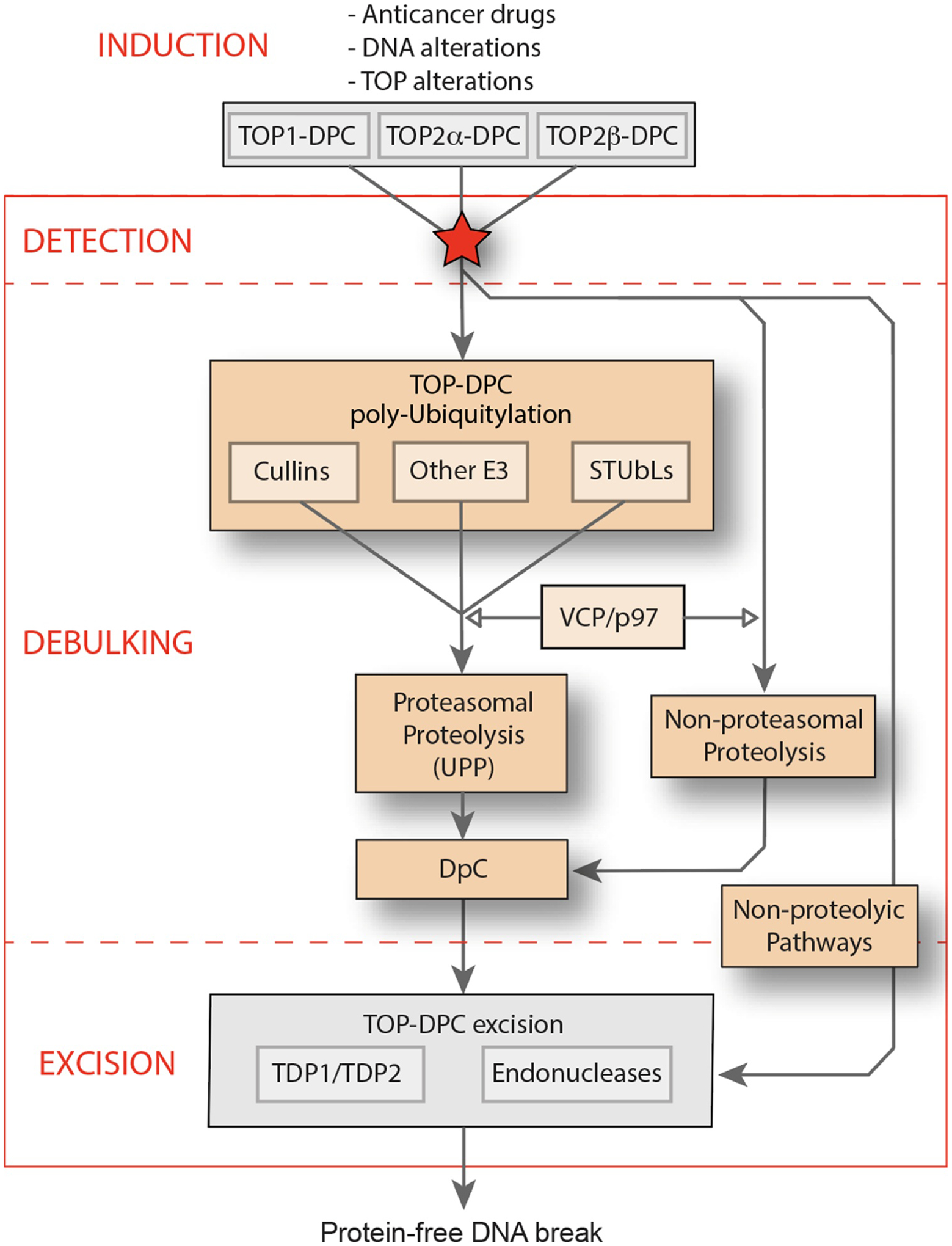
Overall scheme for the induction and excision repair of TOP-DPC. The ubiquitin proteasomal pathway (UPP) is represented in the middle with the different ubiquitin pathways for TOP-DPC (Cullins, SUMO-targeted ubiquitin ligases [STUbLs]). The non-proteasomal pathway is examplifed by Spartan/Wss1. Both the proteasomal and non proteasomal generate DNA peptide crosslinks (DpC). The non-proteolytic pathways shown as the arrow at right includes the endonuclease pathway (examplified by MRE11) and the ZATTZNF451 pathway, which unfolds TOP2-DPC and primes their hydrolytic removal by TDP2 [125]. TOP-DPC excision is detailed in a separate issue of DNA Repair [1].
2. DNA-protein crosslinks (DPC) classification
Covalent attachments of proteins to DNA are among the most frequent and potentially detrimental DNA modifications. DPC represent steric blocks for DNA transactions such as replication and transcription and their translocating helicases and polymerases. In addition, TOP-DPC are always associated with DNA strand breaks, a significant proportion of which are DNA double-strand breaks (DSB) that are also among the most detrimental DNA damaging lesions.
DPC have been classified in two main groups: chemical crosslinks and enzymatic crosslinks [2,3] and in four types based on the mechanisms of their formation and on their chemical structure [4]. Type 1 DPC are not associated with DNA breaks and are both chemical and enzymatic (Fig. 2). They are typically produced by chemical bridges between a base and a nearby chromatin protein as in the case of formaldehyde, acetaldehyde, 1–3 butadiene and heavy metals (chromium and platinum-based chemotherapies including cisplatin, carboplatin or oxaliplatin) [5]. They are also produced enzymatically by DNA incorporation of the anticancer drug 5-azadeoxycytidine, which forms DNA methyltransferase [DNMT] crosslinks. The other DPC are “terminal DPC” in which the DPC is at the end of a DNA break. Type 2 DPC are initiated by a break produced by DNA repair. A typical example is the crosslinking of DNA polymerase β (POLB) at the 5’-end of a break made by AP endonuclease during base excision repair (AP-lyase activity toward 5’-deoxyribose phosphates [5’-dRP]) [6] or the 3’-DPC made by PARP1 [7] (reviewed in [5]). Type 3 DPC are formed by the covalent attachment of type IB topoisomerases (tyrosyl catalytic residues for TOP1 or TOP1MT) or as repair intermediate with the covalent attachment tyrosyl-phosphodiesterase 1 (histidine catalytic residue for TDP1) to the 3’-phosphodiester-end of the broken DNA. Type 4 DPC are formed by covalent attachment of a type IA or IIA topoisomerase (tyrosyl catalytic residues for TOP2α, TOP2β, TOP3α, TOP3β) to the 5’-end of the broken DNA or by the physiological action of the type VI topoisomerase and meiotic recombinase SPO11 as it breaks DNA to form double-strand breaks with its catalytic tyrosine covalently attached to the 5’-end of the DNA by tyrosyl-phosphodiester linkage (Fig. 1d and e).
Fig. 2. Architecture and polarity of TOP-DPC in the context of the enzymatic DPC.
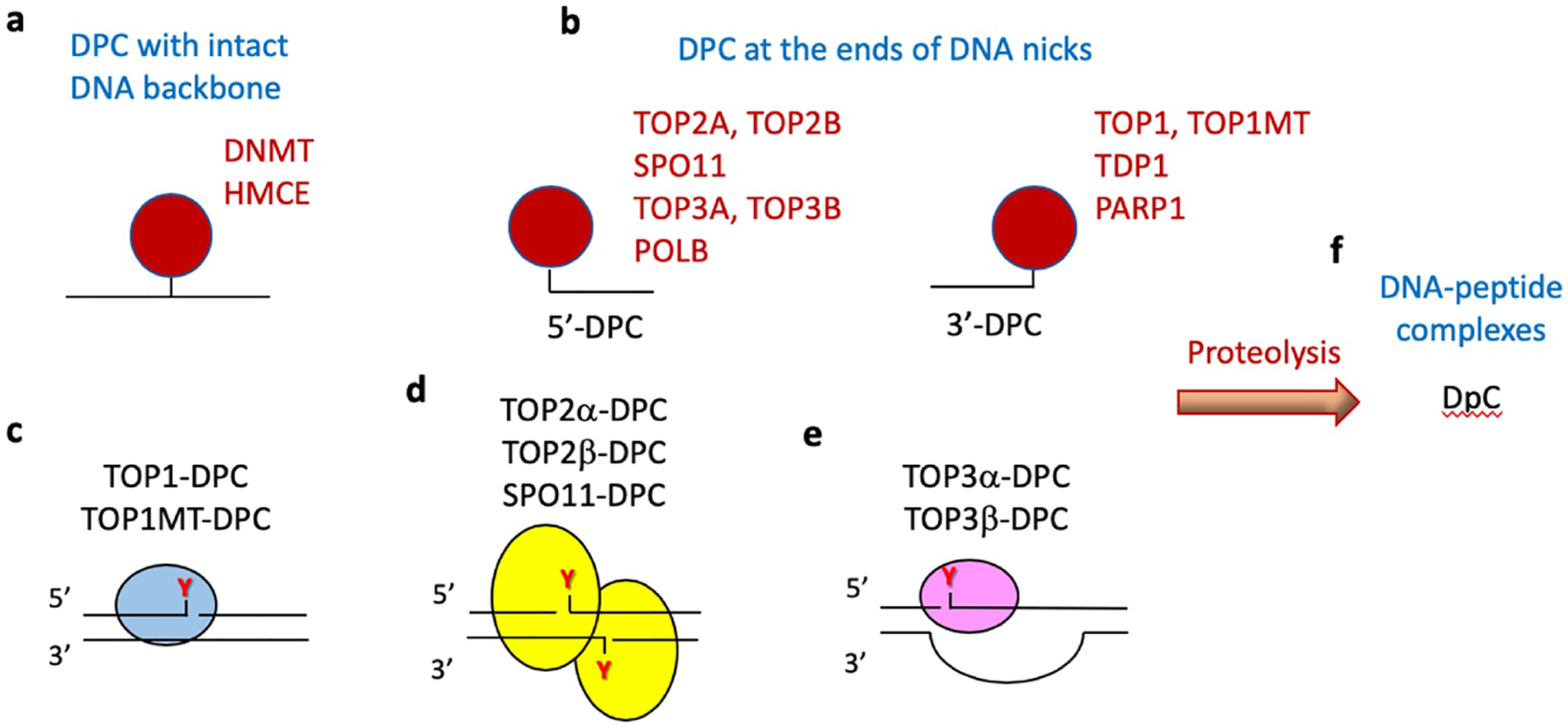
Pathological enzymatic DPC result from abortive enzymatic reactions. Depending on the structure of the DNA at the DPC site, enzymatic DPC can be classified in two groups: (a) DPC with intact DNA backbone as in the case of DNMT (DNA methyltransferase) covalently linked to 5-azacytosine incorporated into the DNA or HMCES (5-hydroxymethylcytosine binding ES specific) at AP sites in single-stranded DNA, and (b) DPC at the end of a nick, or terminal DPC, which can be subclassified as 5’-DPC and 3’-DPC depending whether the DPC is at the 5’- or 3’-terminus of the DNA (see text for details). DNA polymerase β (POLB) forms 5’-DPC whereas tyrosyl-DNA-phosphodiesterase 1 (TDP1) and poly(ADPribose)polymerase 1 (PARP1) form 3’-DPC. TOP-DPC belong to both the 5’- and 3’-DPC subgroups as they generate transient covalent protein-linked DNA breaks (PLDB), termed topoisomerase cleavage complexes (TOPcc) at either the 5’- or 3’-end depending on the topoisomerase. Abortive (trapped) TOPcc form stable TOP-DPC at the end of the broken DNA. c. TOP1 and TOP1MT cleaves one strand of duplex DNA with covalent linkage through their active tyrosine (Y) to the 3’-end. d. TOP2 enzymes (TOP2α [TOP2A] and TOP2β [TOP2B]) and the meiotic recombination topoisomerase-like SPO11 act as homodimers and cleave duplex DNA with covalent linkage through their active tyrosine (Y). e. TOP3 enzymes (TOP3α (TOP3A) and TOP3β [TOP3B]) act as monomers. They reversibly cleave exposed single-stranded DNA regions of the DNA double helix by covalent linkage to the 5’-end using Mg2+. Little is known about the repair of TOP3-DPC (see Fig. 4 and text for details).
Hence, DPC can be classified based on 4 criteria: 1/ whether they are chemical, which can be either direct with a bond between the DNA and the protein as in the case of 5-azadeoxycytine crosslinks to DNMT or indirect through chemical bridges (heavy metal in the case of chromium or cisplatin) [5]; 2/ whether they are enzymatic (Fig. 2) and associated with a break in the DNA backbone (i.e. protein-associated strand breaks [PASB]) as in the case of POLB, topoisomerases and TDP1; 3/ whether they are self-reversible as in the case of POLB, topoisomerases and TDP1 because they represent transient catalytic intermediates; and 4/ whether they are intrinsically irreversible as in the case of the 5-azacytosine-DNMT and SPO11 DPC.
3. Topoisomerase DNA-protein crosslinks (TOP-DPC)
All living cells possess topoisomerases and humans have 6 enzymes encoded by different genes: TOP1, TOP1MT, TOP2A, TOP2B, TOP3A and TOP3B. Topoisomerases have been referred to as the “magicians of the DNA world” [8,9] because they change the topology of the genome without affecting the DNA sequence and without requiring the assistance of protein cofactor [10].
The multiple topological manipulations carried out by topoisomerases include the relaxation of DNA to avoid supercoiling with the generation of plectonemic and toroidal DNA structures, as well as the removal and introduction of knots and catenanes. The DNA relaxation function is common to all topoisomerases and is critical during DNA replication and transcription to avoid both DNA overwinding (positive supercoils, which antagonizes DNA strand separation) ahead of translocating helicase complexes and DNA underwinding (negative supercoils promoting alternate DNA structures including R-loops, guanosine quartets, cruciforms and z-DNA) in the wake of translocating helicases complexes (TOP1 and TOP2 enzymes). The DNA decatenation function is essential following replication as the daughter DNA molecules are intertwined because of the helical structure of the DNA (TOP2 and TOP3 enzymes). The biological function of knots remains hypothetical [10,11].
Topoisomerases form TOP-DPC to break the DNA backbone and carry out their functions. Hence, TOP-DPC have the following characteristics [10]:
Each DPC is coupled with a DNA break generated by the formation of a topoisomerase tyrosyl-phosphodiester bond at one end of the break (Fig. 2); the polarity of the covalent linkage is a characteristic of the enzyme class: 5’-linkage for the type IA (TOP3α, TOP3β) and type IIA enzymes TOP2α and TOP2 β) and 3’-linkage for the type IB enzymes (TOP1 and TOP1mt). Hence TOP-DPC are protein-linked DNA breaks (PLDB) [also referred to as PASB (protein-associated strand breaks)]. We can also refer to them as terminal breaks, which are 3’-DPC for TOP1 and TOP1MT, and 5’-DPC for TOP2α, TOP3β, TOP3α, TOP3β and SPO11 (Fig. 2c–e)
A significant fraction of the TOP-DPC breaks are double-strand breaks (DSB) (reviewed in [10]) because the topoisomerases form dimeric complexes and cleave the opposite DNA strands in concert in the case of the type IIA enzymes (TOP2α and TOP2β) and SPO11. TOP1 single-strand breaks (SSB) are also converted into single-ended DSB by replication run-off [12] or replication fork breakage after fork reversal [13,14]. In addition, TOP1 SSB can be converted to DSB by transcription-mediated R-loop formation [15] or when they form opposite to a preexisting nick (Table 1) [16–20].
Under normal conditions, with the exception of SPO11, TOP-DPC are intrinsically transient and self-reversible. In other words, once the DNA realigns itself across the break, the tyrosyl covalent bond is eliminated by the nucleophilic attack of the free hydroxyl end of the DNA across the break as it rejoins the DNA backbone and releases the active topoisomerase molecule. These catalytic intermediates are referred to as the topoisomerase cleavage (or cleavable) complexes (TOPcc).
The TOP-DPC (TOP1-DPC, TOP1MT-DPC, TOP2α-DPC, TOP2β-DPC, TOP3α-DPC and TOP3β-DPC) are likely to be the most frequent DPC at any given time in the cell because of the ubiquitous functions of the 6 human topoisomerases as they form constantly and rapidly reversible TOPcc. Circumstantial evidence for the ubiquitous nature of TOP-DPC is the evolution of tyrosyl-DNA phosphodiesterases (TDP1 and TDP2) as specialized enzymes to remove abortive TOP-DPC [21] and the fact that genetic inactivation of TDP1 and TDP2 cause clinical syndromes (SCAN1 and SCAR23 due to the accumulation of TOP-DPC in neurons [22–27]).
TOP-DPC do not form randomly in the genome as they are associated with the diverse functions of the topoisomerases at specific genomic sites during DNA replication, transcription and chromatin remodeling [10]. Moreover, topoisomerases tend to cleave DNA at preferred sequences (albeit with limited sequence selectivity, which allows them to carry out their genome-wide functions [28–32].
TOP-DPC are trapped by widely used anticancer drugs, by DNA structural alterations and by post-translational modifications of the topoisomerase polypeptides. A main mechanism for the trapping of TOP-DPC involves the misalignment of the broken ends within the TOPcc because of the presence of a drug molecule between the broken DNA ends of the TOPcc or because of the misalignment of the ends by DNA alterations (Table 1 and see below).
Table 1.
Inducers of TOP-DPC including anticancer drugs, common DNA alterations and transactions, carcinogenic physical agents and metabolic products and DNA repair defects.
| Inducers | Molecular mechanism | TOP1-DPC | TOP2-DPC | References | |
|---|---|---|---|---|---|
| Anticancer drugs | TOP1 and TOP2 poisons | Interfacial inhibition with a single drug molecule bound at the DNA break-topoisomerase interface |
|
|
[193,194,195,196] |
| Platinum salts | DNA distortion | Cisplatin, carboplatin | Unknown | [197] | |
| Antimetabolites | DNA distortions; ribonucleotide incorporations | Hydroxyurea, Gemcitabine, Cytarabine, Fluorodeoxyruridine | Unknown | [19,20,160,198,199,200,201,202,203] | |
| Others | Distortion of DNA minor groove | Hoechst 33342 Nogalamycin Actinomycin D |
Actinomycin D | [204,205,206,207,208,209] | |
| Carcinogens | Food & environmental products | DNA distortions & topoisomerase alterations | Trapping by carcinogenic base damage, exocyclic adducts, crosslinks, benzo[a] pyrene & crotonaldehyde adducts | Trapping by flavone derivatives, tea and wine products & carcinogenic base adducts | [5,203,210,211,212,213,214,215,216,217,218,219,220,221] |
| UV adducts & IR-induced DNA damage | DNA distortions, base alterations & breaks | Trapping by DNA alterations | Trapping by DNA alterations and enzymatic inhibition | [222,223,224] [16,17,18,218,225] | |
| Endogenous metabolites & DNA alterations | ROS - aldehydes | DNA distortions | DNA alterations | DNA alterations | [215,219,226,227] |
| Abasic sites, mismatches, DNA breaks | DNA distortions | DNA distortions | DNA distortions | [16,17,18,19,20,218,219,225,228,229,230] | |
| Ribonucleotide incorporation in DNA | Chemical reactivity & DNA distortions | Trapping of flanking TOP1cc | Trapping of TOP2cc | [19,20,160,198,199,200] | |
| Transcription | Unknown | Unknown | Enhancers | Promoters | [35,36,37,38,39,40] |
| DNA repair defects | Genetic alterations | Lack of TOP-DPC excision | TDP1, SPRTN, RNase H2 and ATM deficiencies | TDP2 deficiency and MRE11 | [23,25,27,35,129,231,232,233,234] |
4. Induction of trapped (irreversible/abortive) TOP-DPC
Any TOPcc is a potentially irreversible TOP-DPC. Table 1 details the broad range of conditions leading to trapped TOP-DPC, which are also referred to as abortive TOPcc. Matching references are included in the right column of the table. TOP3-DPC have not been included. We have recently reported their existence upon mutation of TOP3B [33].
The most studied pharmacological and therapeutic generators of TOP-DPC are small cytotoxic molecules used as anticancer agents and also used as chemical probes to study the repair of TOP-DPC. These drugs are highly specific for either TOP1 or TOP2 as they form biochemical contacts at the enzyme-DNA interface that prevent DNA rejoining. They are commonly referred to as TOP poisons [34]. The camptothecins selectively poison TOP1cc whereas etoposide and its derivative teniposide only poison both TOP2α and TOP2β but not TOP1 enzymes. Other agents that poison TOPcc have additional DNA effects, which are generally considered as their primary mechanism of action; among them, cisplatin, which distorts the DNA helical structure by forming intra-strand crosslinks or the antimetabolites gemcitabine and cytarabine, which poison TOP1cc by altering the bases incorporated into DNA during replication (Table 1 and references therein).
Chemical and physical carcinogens are a common source of TOP-DPC as they form DNA base adducts (benzo[a]pyrene, aldehydes, nucleophiles), crosslinks (ultraviolet [UV] and ionizing radiations [IR]) and DNA breaks. A recent review gives an detailed overview of the environmental DPC [5].
Endogenous metabolic and DNA repair processes can generate TOP-DPC as in the case of oxygen radicals (reactive oxygen species [ROS]), aldehydes, abasic sites, mismatches, DNA breaks and ribonucleotide incorporation during DNA synthesis (Table 1).
Independent studies have also revealed the presence of TOP-DPC during normal transcription, especially during transcriptional activation by hormones (such as androgens and estrogens) and cellular stress pathways. TOP2βcc have been mapped to promoters and TOP1cc to enhancers. The conceptual question is whether these TOP-DPC are byproducts of the normal TOPcc that adapt the DNA to the transcription processes or whether they are promoted by some unknown factor to anchor the promoters or enhancers to scaffolding nuclear structures [10,35].
Evidence for endogenous TOP-DPC is their accumulation and the development of genomic instability in mice and patients lacking the TOP-DPC repair enzymes, TDP1 and TDP2, which are the tyrosyl-DNA phosphodiesterases that excise TOP1-DPC and Spartan (SPRTN) (SprT-like domain at the N-terminus), a metalloprotease, which proteolyzes DPC during DNA replication [35–40].
5. Debulking the TOP-DPC by the ubiquitin-proteasome pathway (UPP)
5.1. Introduction to the UPP
The 26S proteasome is a large (~2.5–2.6 MDa) and highly sophisticated protease complex that degrades intracellular proteins in both the cytoplasm and nucleus [41–43]. The ATP-driven proteasome-mediated degradation plays a pivotal role in modulating a wide array of cellular processes such as cell cycle progression, transcription, signal transduction, response to cellular stress, immune response as well as DNA damage response and repair [44]. The 26S proteasome comprises two sub-assemblies: the 20S core particle (CP), a hollow cylinder in which proteins are degraded by a chymotrypsin-like protease, a trypsin-like protease and a peptidyl-glutamyl peptide-hydrolyzing (PHGH) protease. At the top and bottom of the core particle are two 19S regulatory particles (RP) that bear multiple ATPase and ubiquitin binding sites that recognize, deubiquitylate, unfold and transfer the substrates to the CP for destruction (Fig. 3) [42].
Fig. 3. The ubiquitin-proteasome system (UPP).
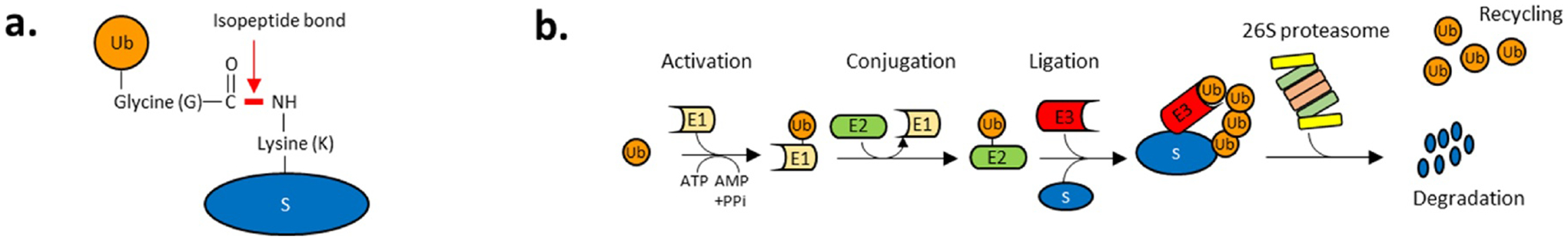
a. Ubiquitin (Ub) is a small protein (~76 amino acid) forming a covalent isopeptide bond between its c-terminal glycine carboxyl group and the NH2 group of a lysine residue on either substrate proteins or ubiquitin itself. b. Substrate ubiquitylation is achieved in three steps: substrate proteins are processed by ATP-activated enzymatic cascades with ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3). Ub is first activated by an E1 in an ATP-dependent manner. The E1 catalyzes the acyl-adenylation of the C-terminus of an ubiquitin molecule using ATP. The activated Ub is then conjugated to E1 through a thioester bond between the c-terminal carboxyl group of ubiquitin and the E1 cysteine sulfhydryl group (activation). E2 enzymes transfers the Ub molecule from E1 to the active site cysteine of an E2 via a trans(thio)esterification reaction (conjugation). E3 ligases allow the attachment of the Ub molecule from E2 to a substrate protein (S) by catalyzing the formation of an isopeptidyl linkage between a lysine residue of the substrate and the c-terminal glycine of the ubiquitin (ligation). Lysine 48-linked polyubiquitylation targets substrates to the 26S proteasome. The 19S regulatory particle of the proteasome docks the ubiquitylated substrate, unfolds its and deubiquitylates the substrate to recycle ubiquitin and transfer it to the 20S core particle where 3 proteolytic subunits degrade the substrate (see Fig. 4).
Proteasomal degradation requires the prior covalent addition of polyubiquitin chains to substrate proteins (ubiquitylation) to direct those ubiquitylated substrates to the proteasome [45]. Ubiquitin (Ub) is a 76 amino acid, 8.5 kDa polypeptide conserved in all eukaryotes. Ubiquitylation is a three-step enzymatic cascade catalyzed sequentially (Fig. 3). First, a ubiquitin activating enzyme (E1) forms a thioester bond between a cysteine of the E1 protein and the C-terminal glycine of ubiquitin in an ATP-dependent reaction. This activation allows one of ~40 ubiquitin-conjugating enzymes (E2s) to catalyze the transfer of Ub from E1 to the active cysteine of E2. In the final step, one of ~600 ubiquitin ligases (E3s) recognizes the target protein and transfers the Ub moiety from the E2 to the substrate by creating an isopeptide bond between a ε-NH2 of a lysine residue of the substrate and the C-terminal glycine of Ub (Fig. 3a). In human, the majority of ubiquitin ligases belongs to the family of RING (Really Interesting New Gene) finger domain proteins, which bind proteins and transfer Ub moieties from the E2 to their targets [46]. RING ligases can act as monomers (TRIM41, c-CBL), homodimers (RNF4), heterodimers (BRCA1-BARD1, RINGA/Bmi1), as multisubunit ligases (APC/C), or in complex with a Cullin family member (Cullin-RBX1/2 ligase complexes also referred to as CRL [Cullin Ring Ligases]) [47].
Ub molecules can form various polymeric chains (polyubiquitylation) with different lysine residues via isopeptidyl linkages in an iterative manner [48]. These chains are linked through one of the seven lysines (K6, K11, K27, K29, K33, K48 and K63) as well as the N-terminal methionine residue (M1) of Ub [49]. Ub chains linked via K48 target proteins for proteasome-mediated degradation [50] (Fig. 3b). K63-linked polyubiquitylation is not associated with proteasomal degradation and has roles in many cellular events including endocytic trafficking, inflammation and DNA repair. The function of other lysine linkages remains largely unknown. Ubiquitylation can be reversed by a class of deubiquitylating enzymes (DUB), which release ubiquitin from substrate proteins by cleaving the isopeptidyl linkage [51]. In addition to the proteasomal DUB subunit PSMD14 (Fig. 3b), which are required to recycle Ub and allow the proteolytic degradation of the polyubiquitylated substrates, there are approximately 79 known DUBs in human cells. They are classified into two main categories: cysteine proteases and zinc metalloproteases [52]. Reversal of ubiquitin modifications by DUB is associated with ubiquitin maturation and editing. Once a polyubiquitin chain is detached from its target protein, it is broken down into single ubiquitin molecules by a DUB to replenish the ubiquitin pool [53].
Ubiquitylation and the UPP perform regulatory roles in the repair of virtually all types of DNA damage [54] including direct or reversal repair by O6-methylguanine DNA methyltransferase (MGMT) [55], mismatch repair (MMR) [56], base-excision repair (BER) [57], nucleotide-excision repair (NER) [58,59], DSB repair and post-replication repair (PRR) by translesion synthesis (TLS) [60], as well as DNA interstrand crosslink (ICL) repair by the Fanconi anemia (FA) pathway [61] and DPC repair [62]. In addition, poly- and mono- ubiquitylation of histones play critical roles in response to DSB by recruiting DNA repair complexes to the damage sites and facilitating cell cycle arrest [63].
5.2. UPP in the repair of TOP1-DPC
The first study suggesting the implication of the UPP for TOP1-DPC showed that CPT induces a time- and concentration-dependent decrease of cellular TOP1 protein [64]. A follow-up study demonstrated that CPT-induced TOP1 downregulation is a result of the 26S proteasome-mediated degradation as a resistance mechanism to CPT [65]. Treatment with CPT reduced cellular TOP1 by 75 % in the mouse mammary carcinoma cell line ts85 (temperature-sensitive for ubiquitin-activating enzyme) within 2–4 hours. TOP1 was ubiquitylated within minutes of CPT treatment at permissive temperature but not at the restrictive temperature for its thermolabile E1 [65]. MG-132 and lactacystin, specific inhibitors of the 26S proteasome, prevented destruction of cellular TOP1, suggesting that the UPP degrades TOP1 in response to TOP1 trapping [65]. A subsequent study [66] demonstrated that cell lines defective in the proteasomal degradation of TOP1 were hypersensitive to CPT, implying that the UPP-mediated degradation of TOP1 is a resistance mechanism to CPT in human cancer cells. Additional studies show that inhibition of ubiquitylation and of the proteasome markedly enhance the levels of drug-induced TOP1-DPC [67,68] and delay the removal of the DPC [68], further demonstrating that the UPP targets TOP1-DPC.
Two processes, transcription and replication trigger the ubiquitylation and proteasomal degradation of TOP1-DPC (Fig. 4a, b). In the first, TOP1-DPC block the translocating RNA polymerase II (Pol II) elongation complex, which elicits a signal for the recruitment of the UPP and the degradation of both the TOP1-DPC and the large subunit of Pol II Rpb1. This transcription-coupled degradation of TOP1-DPC was proposed to expose TOP1-occluded SSB, enabling the recruitment of PARP1 and the repair of the broken DNA [69]. The second cellular process initiating the UPP-mediated degradation of TOP1-DPC is the collision of translocating DNA polymerase complexes (replisomes) with TOP1-DPC (Fig. 4a) [12,14]. CPT-induced cytotoxicity against cancer cells is well established to arise from replication fork collisions [12,14,70–72], and γH2AX, ATM-Ser1981 phosphorylation, Chk2-Ser33/35 phosphorylation, RPA2 phosphorylation, which are markers of DSB are readily induced by CPT in replication-dependent manner [73,74]. Inhibition of the UPP abolishes the induction of all these DSB characteristics, indicating that ubiquitin-mediated proteasomal degradation removes the bulky DPC thereby exposes the otherwise TOP1-concealed DNA breaks to activate DNA damage responses [75]. Implicit from these findings is a model wherein the ongoing replication fork is arrested by TOP1-DPC, which in turn triggers the UPP-mediated repair of TOP1-DPC, leading to the formation of DSB that comprises TOP1-concealed break and the leading strand where replication fork runs off [12].
Fig. 4. Known UPP repair pathways for TOP-DPC.
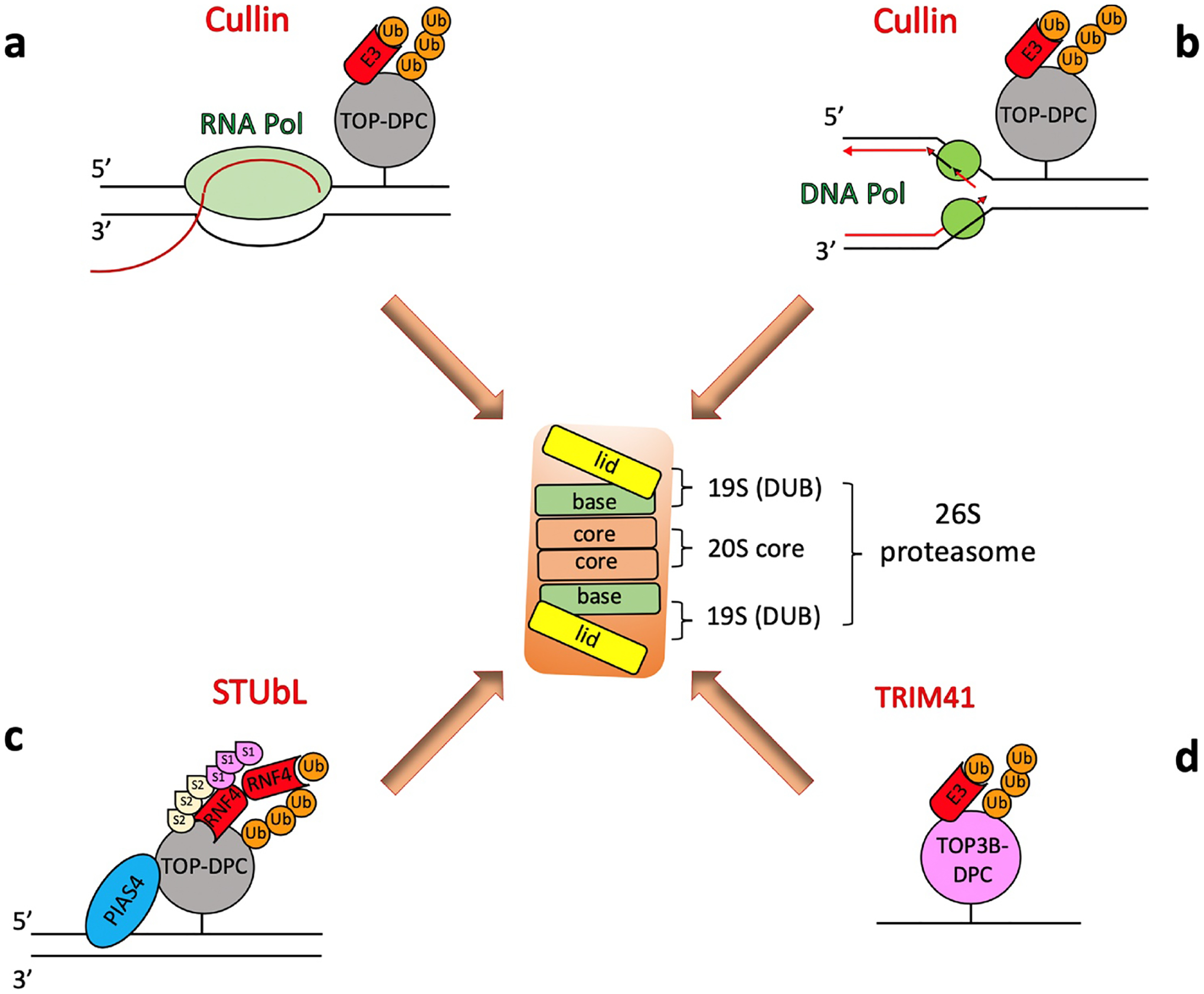
a-b. DNA translocating complexes including transcription (a) and replication (b) trigger the ubiquitylation and subsequent proteasomal degradation of TOP-DPC. Cullin-RING ligases have been implicated as the E3 for both TOP1- and TOP2-DPC. c. An alternative ubiquitylation pathway is by STUbL (SUMO-targeted Ub ligases), which are recruited by SUMOylation of the TOP1- and TOP2-DPC. SUMOylation-dependent ubiquitylation is catalyzed by SUMO ligase PIAS4 and STUbL RNF4 in human cells and Siz1 and Slx5/Slx8 in yeast. d. Recent studies show that TOP3β-DPC are directly ubiquitylated by the E3 ligase TRIM41 (see text for details and reference).
To date, several E3 ligases have been implicated in the UPP for TOP1-DPC (Table 2). The first reported was Cullin3 (CUL3), the scaffolding protein of CRL3 following the discovery that CPT-resistant human cancer cell lines exhibit enhanced TOP1-DPC degradation and high expression of CUL3 [67]. Downregulation of CUL3 not only enhanced TOP1-DPC but also reversed CPT resistance, indicating that CRL3 promotes TOP1-DPC clearance and CPT resistance. A follow-up study confirmed this conclusion by showing that depletion of CUL3 attenuated CPT-induced DSB signals [75]. Apart from CUL3, CUL4B, the scaffolding protein of CRL4 complexes has been linked to TOP1-DPC degradation. Patient-derived lymphoblastoid cell lines deficien for CUL4B were found to display increased sensitivity to CPT and delayed degradation of TOP1 in response to CPT [76]. BRCA1 another ubiquitin ligase has also been reported to ubiquitylate TOP1 in a DNA-PKcs-dependent manner [77,78]. The proposed model is that DNA-PKcs phosphorylates TOP1 at Ser10, which in turn evokes BRCA1 to ubiquitylates TOP1 [78].
Table 2.
Ubiquitin ligases for proteasomal degradation of TOP-DPC.
| Ubiquitin Ligase | Detection/signal | Substrate | TOP-DPC Inducing Agent | References |
|---|---|---|---|---|
| Cullin 3-RING ligase 3 (CRL3) | Unknown | TOP1-DPC | CPT | [67] |
| BRCA1 | Transcription and serine 10 phosphorylation of TOP1 by DNA-PK | TOP1-DPC | CPT | [77,235] |
| Cullin 4B-RING ligase (CRL4B) | Unknown | TOP1-DPC | CPT | [76] |
| Slx5-Slx8 heterodimer | SUMOylation in yeast S. cerevisiae by Siz1 | TOP1- and TOP2-DPC | CPT, etoposide and amsacrine | [68,119,236] |
| RNF4 homodimer (Slx5-Slx8 ortholog) | SUMOylation in human by PIAS4 | TOP1- and TOP2-DPC | CPT and etoposide | [68] |
| SCFβ-TrCP (SKP1-Cullin 1-F box protein (β-TrCP) | Phosphorylation of TOP2β in its degron motif | TOP2β-DPC | Teniposide (VM26) | [123] |
| Bmi1-Ring1A Heterodimer | Unkown | TOP2α-DPC | Teniposide (VM26) | [85] |
Although the “division of labor” between ubiquitin ligases for the repair of TOP1-DPC is presently unclear, it is plausible that different E3 ligases are employed in a context-dependent manner. For example, given the role of CRL4 in replication licensing by degrading the CDT1 and CDKN1A (p21WAF1) in a PCNA-dependent manner [79,80], it is conceivable that CRL4 may be primarily recruited to ubiquitylate TOP1-DPC upon the stalling of replication forks.
5.3. UPP in the repair of TOP2-DPC
Proteasomal degradation of TOP2 was first reported in adenovirus E1A-infected human carcinoma cells [81,82] with the finding that TOP2α was degraded by the proteasome during adenovirus E1A-induced apoptosis. Teniposide was later shown to induce ubiquitin-dependent degradation of TOP2 [83]. The proteasome was found to preferentially degrade TOP2β over TOP2α [84], and inhibition of transcription but not replication prevented teniposide-induced TOP2β degradation, in line with the cardinal role of TOP2β in regulating transcription [10]. A follow-up study in colorectal cancer cell lines found that TOP2α also undergoes proteasome-mediated degradation upon treatment with teniposide, and that TOP2α degradation is blocked by transcription but not replication inhibitors [85]. These findings suggest that interference with transcription triggers the degradation of both TOP2α and TOP2β by the UPP (Fig. 4a; Table 2). TOP2 proteasomal degradation that interfere with replication remained unexplained from this set of results. Also, etoposide-induced TOP2α- and β-DPC were found significanty increased by ubiquitylation and proteasome inhibitors [68,86], and their clearance was blocked UPP inhibition, indicating a direct role of the UPP in the degradation of TOP2-DPC for repair. The TOP2 catalytic inhibitor ICRF-193 (bisdioxopiperazine), which traps TOP2 on DNA in a closed clamp conformation after the breaks are sealed, was also reported to arrest transcription and induce TOP2β degradation [84], supporting the model wherein TOP2 trapping rather than TOP2-linked DSB blocks advancement of RNA Pol II and triggers the UPP. Accordingly, proteasome inhibitors effectively sensitize tumor cells to etoposide and other TOP2 inhibitors [87,88] and improve the efficacy of etoposide in the refractory HT-29 xenograft [87].
Several ubiquitin E3 ligases have been implicated in the ubiquitylation of TOP2α-DPC (Table 2), of which BMI1/RING1A was the first identified for human TOP2α-DPC degradation by ubiquitylation upon exposure to etoposide [85]. APC/C-Cdh1 has also been shown to be required for ubiquitylation and proteasomal degradation of etoposide-induced TOP2α-DPC in human cells [89]. Yet, the mechanism that determine which Ub ligase processes TOP2-DPC remains elusive and warrants further investigations.
5.4. SUMOylation of TOP1-DPC and TOP2-DPC for UPP
Ubiquitin-like proteins (UBLs) act in parallel yet connected pathways with ubiquitin [90]. The Small Ubiquitin-like Modifiers (SUMOs), one of the most studied UBL families, includes four members: SUMO-1, SUMO-2, SUMO-3 and SUMO-4 [91]. Like ubiquitin, SUMO modifications are covalently attached to substrate proteins through an enzymatic cascade catalyzed by E1/E2/E3 enzymes. The two E1 enzymes are SUMO-Activating Enzymes Subunit 1 and 2 (SAE1 and SAE2), an-dUBC9 is the only known SUMO E2 conjugating enzyme in eukaryote. In general, most SUMOylated proteins contain a consensus motif Ψ-K-x-D/E, in which Ψ is a hydrophobic residue, K (lysine), which is used for SUMO conjugation, x indicates any amino acid residue, and D/E either aspartic or glutamic acid, respectively [92]. SUMOylation regulates a wide range of biological processes including subcellular transport, protein-protein interactions, transcriptional regulation, cell cycle progression, apoptosis as well as DNA damage responses and repair (DDR) [91].
SUMOylation is involved in different cellular contexts of DDR [93]. It plays key roles in BER, NER and DSB repair by facilitating the assembly of repair protein complexes at DNA damage foci and by regulating their activities and interactions [94–99]. Recently, SUMO-2/3 modfications have been shown to signal the ACRC/GCNA family that belongs to the family ofSprT proteases (see next section) to target the SUMOylated DPC in response to formaldehyde in C. elegans and human germ cells [100].
Most relevant to our review, SUMOylation is also a signal for the UPP [101]. It recruits the ubiquitin E3 ligase RING-finger protein 4 (RNF4) to SUMOylated proteins, which enables their Ub-mediated proteasomal degradation [102,103]. RNF4 is a SUMO-Targeted Ubiquitin Ligase (STUbL) (Fig. 4c) recruited to SUMOylated substrate proteins by its 3 SIMs (SUMO Interacting Motifs). Its RING-finger domain belongs to the zinc finger (Znf) domain family. It interacts with E2 enzyme(s), Ub and substrates [104]. RNF4 co-localizes with SUMO-1 in PML (ProMyelocytic Leukemia) nuclear bodies [105] and ubiquitylates PML proteins that have been modified by poly-SUMO2/3 chain [102,103,106].
Ubiquitylation by RNF4 is coordinated with the PIAS-type SUMO ligases [96,97,107], which contain a SP-RING domain that structurally resembles the classic RING finger domain of Ub E3 ligases [108]. Unlike RING finger domain proteins that chelate two zinc ions via cystine and histidine residues to interact with the E2 enzymes, the SP-RING domain requires a single zinc ion for the coordination [109]. The SAP (SAF-A/B, Acinus and PIAS) domain of PIAS SUMO E3 enzymes acts as a DNA binding domain. PIAS4- and Ubc9-mediated SUMOylation of DDR proteins such as RPA, 53BP1 and BRCA1 induces their recruitment to DNA damage sites [95,110,111]. Also, PIAS1, 4-mediated SUMOylation at the DSB foci is a prerequisite for RNF4-dependent ubiquitylation of DDR proteins [97], which ensures proteasome-mediated proteolysis of RPA and BRCA1 for DNA repair [97,107]. The disassembly of MDC1 by RNF4 is a prerequisite for the recruitment of downstream DDR factors [112]. Indeed, the removal of RPA is required for the loading of RAD51 and BRCA2 on resected DNA for HR-dependent DSB repair [96].
Human TOP1 can be modified by SUMO-1 both in vitro and in vivo [113–115], and CPT stimulates the SUMO-1 modification of TOP1 [113]. TOP1 SUMO-1 modification also drives the delocalization of TOP1 from nucleoli and nucleoplasm upon exposure to TOP1 inhibitors [116,117], but the consequences of the delocalization remain unknown. In yeast S. pombe, SUMOylation engages the ubiquitylation pathway. The SUMOylation of TOP1 by Pli1 (the ortholog of vertebrate Siz/PIAS) recruits the STUbL Slx8 (the RNF4 ortholog) [118,119].
TOP2 SUMOylation drives TOP2 to centromeres for sister chromatid decatenation in yeast and Xenopus laevis, indicating a role of SUMOylation in regulating TOP2 activity and preserving segregation fidelity under unperturbed condition [120,121]. SUMO-1 modifications of human TOP2 have also been observed in response to teniposide, implying SUMOylation as a response mechanism against TOP2-DPC [122]. The TOP2 catalytic inhibitor ICRF-193 also stimulates SUMO-1 modification of TOP2 [113], suggesting that the trapping of TOP2 (either covalent or non-covalent) on DNA rather than TOP2-mediated DNA lesions induces TOP2-DPC SUMOylation.
We have recently extended these results by showing that SUMOylation engages the ubiquitin-mediated proteasomal degradation of both TOP1- and TOP2-DPC in both S. cerevisiae and human cancer cells [68] (Fig. 4c). TOP1-DPC, TOP2α-DPC and TOP2β-DPC in spite of their structural differences (Fig. 2) are similarly and promptly modified by SUMO-2/3, SUMO-1 and Ub in a sequential order, which leads to their proteasomal degradation. We identified PIAS4 as the SUMO ligase and RNF4 as the STUbL in human cells and Siz1 as the SUMO ligase and Slx5-Slx8 as the STUbL in yeast. The SUMOylation and SUMO-dependent ubiquitylation of TOP-DPC were found replication/transcription-independent [68] (Fig. 4c). One conceivable model for the SUMO-dependent TOP-DPC repair could be that prolonged residence of topoisomerases on chromatin engages the SUMO system for repair. Another explanation is that SUMO may play a role in dissociating non-covalent DNA-bound topoisomerases from chromatin and the failure to release the covalent DNA-bound topoisomerases by SUMO triggers the UPP for destruction.
Given the distinction between TOP1, TOP2α and β in terms of their sizes, catalytic mechanisms and functions, one could imagine that TOP1- and TOP2-DPC are targeted by different ubiquitylation pathways for the repair in different cellular contexts. By contrast to the cullin-RING pathways [67,76,123], the PIAS4-RNF4 axis appears able to engage on both TOP1- and TOP2-DPC independently of DNA transactions and DDR (Fig. 4c). Similarly, we recently found that TOP3β-DPC are directly ubiquitylated by the E3 RING ligase TRIM41 though its recruitment to the TOP3β-DPC [33].
As discussed below (Section 6), in addition to their role in initiating TOP-DPC ubiquitylation, a recent study reported that SUMO-2/3 modifications of TOP2-DPC and binding of ZNF451 (ZATT), a zinc-finger-containing SUMO ligase, changes the conformation of TOP2α-and β-DPC [124,125], which enables TDP2 to gain access to the TOP2-DNA junction through its SUMO-interacting motif (SIM) and to carry out the phosphotyrosyl bond cleavage without requiring prior proteolysis.
6. Debulking of TOP-DPC by non-proteasomal proteases
Mounting evidence show the existence of non-proteasomal pathways for the debulking of TOP-DPC (Table 3; Fig. 1). Unlike the UPP, which digests ubiquitylated TOP-DPC by multiple incisions through its chymotryptic, tryptic and post-acidic subunits inside the proteasome, the non-proteasomal proteases digest the DPC in situ. Also, their catalytic mechanism is different as they are metalloproteases (SPRTN/Wss1), serine (FAM111A) and aspartate (Ddi1) proteases (Table 3).
Table 3.
Non-Proteasomal Proteases for TOP-DPC.
| Proteases | Organisms | TOP1-DPC | TOP2-DPC | Notes | References |
|---|---|---|---|---|---|
| SPRTN | Human Mouse | Yes | Yes | In vitro and in vivo for TOP1-DPC (human, mouse) and TOP2-DPC (human) | [128,129,132] |
| Wss1 | Yeast | Yes | Unknown | In-vitro and in-vivo evidence for TOP1-DPC after CPT treatment, in parallel with TDP1. | [126] |
| GCNA/ACRC | C. elegans, mouse, Drosophila melanogaster, human | Unknown | Yes | Selective for germline and early embryo across species. (C. elegans, Drosophila melanogaster and mouse) | [154,155] |
| Ddi1 | Yeast | Yes | Unknown | In vivo | [146,156] |
| FAM111A | Human | Yes | Unknown | In vivo for TOP1-DPC | [158] |
6.1. Spartan (SPRTN/Wss1)
SPRTN (Wss1 in S. cerevisiae) is the most studied non-proteasomal metalloprotease for TOP-DPC and general DPC. It was initially reported in yeast that the metalloprotease Wss1 (Weak suppressor of smt3) removes TOP1-DPC and formaldehyde (FA)-induced DPC to protect cells against CPT and FA, respectively [126]. Wss1 was shown to cleave TOP1 and other DNA-binding proteins in vitro using its proteolytic activity in a DNA-dependent manner [126]. Genome-wide screen revealed that loss of Wss1 is synthetic lethal with loss of TDP1, and that such synthetic lethality could be reversed by deletion of TOP1 implying the natural occurrence of endogenous TOP1-DPC (see Section 3). Accordingly, yeast cells lacking both Tdp1 and Wss1 accumulate Top1-DPC and exhibit gross genomic rearrangements as well as hypersensitivity to CPT [126]. A concomitant study [127] using a plasmid containing a site-specific DPC (using the DNMT HpaII) identified a replication-coupled protease pathway that repairs the DPC independently of the proteasome in Xenopus egg extract. Subsequently, SPRTN (SprT-like domain at the N-terminus) was identified as the replication-coupled protease for DPC repair in metazoan [128–132].
SPRTN and its yeast functional ortholog Wss1 both contain a N-terminal metalloprotease domain: the SprT domain in SPRTN and the WLM domain in Wss1 [133] (Fig. 5a). However, the SprT domain of SPRTN only shares the active HEXXH motif with the WLM domain of Wss1 (up to 13 % sequence similarity) [134]. SPRTN was initially described as a PCNA interacting protein involved in the regulation of translesion synthesis, preventing UV-induced mutagenesis during replication [135–141]. Later studies demonstrated the importance of the SPRTN protease activity towards TOP-DPC as well as DPC with intact DNA backbone (see Fig. 2a) by showing that the loss of SPRTN in mammalian cells resulted in a failure to repair FA-induced DPC as well as TOP1-DPC and TOP2-DPC [128–132,142]. Biochemical studies confirmed that SPRTN cleaves a range of DPC substrates including TOP1 and TOP2α using its catalytic glutamic acid residue that coordinate a metal Zn2+ cofactor [128,132].
Fig. 5. Repair of TOP-DPC by the non-proteasomal metalloproteases SPRTN/Wss1.
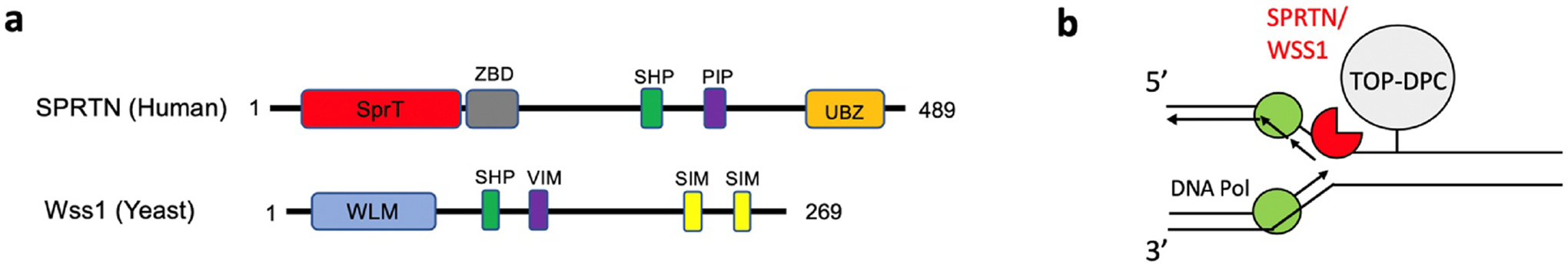
a. Domain/motif architecture of human SPRTN and its yeast ortholog Wss1. ZBD, Zinc binding domain; PIP, PCNA interaction peptide; SHP, p97 or VCP-binding motif; SIM, SUMO interaction motif; SprT, the metalloprotease domain similar to that of the Escherichia coli SprT protein; UBZ, ubiquitin-binding zinc finger; VIM; VCP interaction motif; WLM, Wss1- like metalloprotease domain Schematic of the repair of TOP-DPC by SPRTN/Wss1. b. As a component of the replisome, SPRTN/WSS1 is activated by its binding to ssDNA. It targets TOP-DPC as well as general DPC for proteolysis in association with replication.
SPRTN binds single-stranded DNA (ssDNA), double-stranded DNA (dsDNA) and other DNA structures [62,130–132]. However, SPRTN was found to digest substrates only in the presence of ssDNA but not dsDNA [130,131], implying that a ssDNA segment adjacent to the DPC must be present for SPRTN to efficiently act on the DPC.
SPRTN deficiency in mice results in TOP1-DPC accumulation in the liver at an early age and promotes liver tumorigenesis [129], suggesting a role of SPRTN as tumor suppressor. In human, SPRTN deficiency is the cause of Ruijs-Aalfs syndrome, an autosomal recessive disorder with premature aging, chromosome instability and early onset of hepatocellular carcinoma [143,144].
The mechanisms that control SPRTN protease activity towards DPC remain poorly understood. One reported regulatory mechanism is that dsDNA binding of SPRTN induces its autocleavage thereby preventing undesired cleavage of chromatin-associated proteins under normal conditions [128,131,132,134]. ssDNA binding of SPRTN, on the other hand, primarily stimulates both autocleavage and substrate cleavage [131]. The ssDNA segment adjoining the DPC site was recent found to be generated by accessory DNA helicase RTEL1, which enables the CMG helicase to bypass a DPC on the leading strand template and allows SPRTN to repair the DPC [145]. SPRTN is mono-ubiquitylated under unperturbed condition and de-ubiquitylated upon exposure to DPC-inducing agents. This deubiquitylation appears required for the binding of SPRTN to DNA, implying an ubiquitin switch mechanism controlling the recruitment of SPRTN to chromatin [131]. Future investigations to identify the E3 ligase and particularly the deubiquitylating enzyme for controlling this ubiquitin switch are warranted [146]. Chk1 also appears to phosphorylate SPRTN upon its release from replicative chromatin, which also promotes the recruitment of SPRTN to chromatin for DPC repair and may explain the integration of SPTN activity with the replication stress response [147].
Echoing the finding that SUMOylation primes TOP-DPC for proteasome-dependent degradation by recruiting RNF4 in human cells and Slx5-Slx8 in yeast [1], Wss1 also targets SUMOylated substrates through its c-terminal SUMO-interacting motifs in yeast [148,149] (Fig. 5A). In contrast, in vertebrates, SPRTN does not contain SIMs but instead bears a ubiquitin-binding zinc finger domain (UBZ) [136,137], implying that SPRTN may recognize and target ubiquitylated DPC. The difference between SPRTN and Wss1 is also evidenced by the inability of SPRTN to complement wss1Δ yeast cells [128]. A very recent study, however, shows that SPRTN clears SUMOylated (SUMO-1) DPC during replication in human cells [150]. Due to the lack of SUMO-interacting domain, it remains unclear how SPRTN is recruited by SUMOylation of the DPC.
6.2. ACRC/GCNA
ACRC (acidic repeat-containing protein), also known as GCNA (germ cell nuclear antigen-1), is a metalloprotease that repairs DPC in metazoan in parallel with SPRTN. ACRC is highly expressed during sexual cycles in germ cells while maintaining low levels in somatic and most cancer cells (http://discover.nci.gov/cellminercdb) [151,152]. Phylogenetic analyses showed that ACRC is a SPRTN-related protease carrying the SprT domain [153]. ACRC is recruited to SUMOylated DPC through its SIMs upon FA treatment, [100]. In C. elegans, the ACRC protein ortholog GCNA-1 appears crucial for the cells to withstand DPC-inducing agents [100]. Two recent studies [154,155] provide evidence supporting the role of ACRC/GCNA in limiting DPC accumulation and preventing replication stress in metazoans by showing that ACRC repairs TOP2-DPC in Drosophila while suppressing copy number variation in human germ cell tumors [155]. GCNA colocalizes with TOP2 on condensed chromosomes during mitosis in C. elegans [155] and suppresses Spo11-dependent DNA damage via its SprT protease activity during meiosis in Drosophila [154], implying a role of ACRC/GCNA in repairing both TOP2- and SPO11-DPC (see Fig. 2).
6.3. Ddi1
Ddi1 (DNA-damage inducible 1), a yeast aspartate protease harboring the retroviral protease-like (RVP) domain, acts as an alternative pathway to Wss1-dependent DPC repair in budding yeast [146,156]. Simultaneous inactivation of Ddi1 and Wss1 shows synergistic effect on CPT-induced cytotoxicity [146]. DDi1 is also implicated in the repair of TOP2-DPC based on the finding that the Ddi1Δ cells are hypersensitive to etoposide. Ddi1 appears to function in parallel to Wss1, as the hypersensitivity to etoposide was greatly enhanced in cells lacking both Ddi1 and Wss1 [146]. The recruitment of Ddi1 for DPC repair appears, like Wss1 to dependent on replication. Yet, Ddi1 was recently found to proteolyze polyubiquitylated substrates presumably via its ubiquitin-interacting UBA domain [157], raising the possibility that Ddi1 targets ubiquitylated DPC whereas Wss1 is recruited by DPC SUMOylation in yeast. Further studies are warranted to determine whether DDI1 and DDI2, the human homologs of Ddi1, also act as proteases for both TOP1- and TOP2-DPC.
6.4. FAM111A
The serine protease FAM111A (FAMily with sequence similarity 111 member A) removes TOP1-DPC and PARP1-DNA complexes using its trypsin-like domain to ensure replication fork progression in human cells [158]. FAM111A-deficient cells not only accumulate TOP1-DPC and PARP1-induced DNA damage but are also hypersensitive to CPT and to the PARP inhibitors niraparib and talazoparib [158]. The role of FAM111 in TOP1-DPC repair during replication is highly reminiscent of SPRTN. Moreover, both FAM111A and SPRTN bind ssDNA and undergo autocleavage. One difference between FAM111A and SPRTN is the inability of SPRTN to prevent replication fork stalling from PARP1-DNA complexes, raising the possibility that FAM111A targets chromatin-bound proteins that blocks forks regardless of the chemical properties of the protein-DNA interface whereas SPRTN may only target the covalent DPC during replication.
7. Non-proteolytic debulking of TOP-DPC
Early biochemical studies of the tyrosyl-DNA phosphodiesterases established that neither TDP1 and TDP2 can effectively hydrolyze the topoisomerase tyrosyl phosphodiester covalent bonds when the TOP-DPC are in their native conformation unless the TOP-DPC are first heat-denatured [159,160]. These experiments suggested that the tyrosyl-DNA bonds, which are concealed in the TOP-DPC could be made accessible by unfolding the topoisomerases, and that the proteolytic pathways reviewed above were not the only cellular mechanism for debulking TOP-DPC.
The biological evidence for such a pathway in human cells was provided by a landmark study showing that the binding of ZATTZNF451 (a prototypical member of the SUMO E3/E4 ligase family) to TOP2α and TOP2β and the SUMOylation of TOP2α and TOP2β promotes the binding of TDP2 to the TOP2-DPC and the release of the SUMOylated topoisomerases with formation of clean (protein-free) DSB [125]. This initial discovery and a recent follow-up study show that ZATTZNF451 binding to TOP2α and TOP2β does not require etoposide treatment and that SUMOylated (SUMO2- but not SUMO1-modified) TOP2α and TOP2β are bound to TDP2 and ZATTZNF451 even under baseline conditions (without drug treatment) in HEK293 F cells [125,161]. Notably, based on survival studies and DSB repair (using γH2AX), the ZATTZNF451 pathway appears to overlap only partially with the TDP2 and proteasome pathways, indicating the redundancy of the different TOP2-DPC repair pathways even within a single cell type (human HEK293 F and murine MEF cells) [125,161].
The ZATTZNF451 pathway is not involved in TOP1-DPC [125] in spite of the well-established SUMOylation of the TOP1-DPC [1,68,113]. SUMO-1 modifications that cap the TOP1- and TOP2-DPC SUMO2/3 chains in response to TOP inhibitors [68] may prevent the recruitment of TDP2 to SUMO2 and to the TOP-DPC.
Although this ZATTZNF451 pathway appears well-defined for TOP2α- and TOP2β-DPC, it is likely that it is coordinated with the other debulking pathways, as recently demonstrated by the recruitment of TDP2 to TOP2-DPC both by its “split-SIM” motifs and its N-terminus UBA motif [125,161,162] and the lack of ZATTZNF451 ortholog in C. elegans [125]. It remains unknown whether there exists a ZATTZNF451 functional equivalent for TOP1-DPC repair to obviate the need for a proteolytic debulking step.
8. p97/VCP/Cdc48 as a mediator for the debulking of TOP-DPC
p97, also called Cdc48 in yeast or valosin-containing protein (VCP) in higher eukaryotes, is a conserved AAA + ATPase [163–166] known for extracting client proteins from their environment and facilitating their proteasomal degradation by inducing conformational alteration in the proteins through ATP hydrolysis [166,167]. p97 plays a crucial role in disassembly and rearrangement of chromatin-associated protein complexes to ensure DNA replication and DNA repair processes to preserve genome integrity [167]. As an enzymatic complex, p97 cooperates with a variety of protein cofactors that associate with its N-terminal domain or C-terminus via distinct binding motifs/domains to determine substrate specificity and obtain additional enzymatic activities [167].
Cdc48 had been shown to facilitate the activity of Wss1 for the processing SUMOylated substrates in yeast [126], but it remained unknown whether p97/VCP played a direct role in the repair of TOP-DPC until a recent study showed that p97 inhibition leads to an accumulation of TOP1-DPC in human cells [168]. The study identified TEX264, a gyrase inhibitory-like protein, as a p97 cofactor that recruits p97 to SUMO-1 modified TOP1-DPC to facilitate the SPRTN-dependent proteolysis [168]. Given the role of p97 in unfolding their client proteins, it can be speculated that p97 remodels TOP1-DPC on chromatin and exposes their cleavage sites, thereby promoting SPRTN activity towards the DPC (Fig. 1).
p97 is also known to collaborate with the CRL ubiquitin ligases to promote proteasomal degradation of the CRL-ubiquitylated proteins through its cofactors that contain various ubiquitin binding domains [169]. This raises the possibility that p97 may also play a role in facilitating the CRL-dependent degradation of TOP-DPC (Fig. 1). Future studies are warranted to interrogate whether p97 enables TDPs to act on the phosphotyrosyl bonds of TOP-DPC through conformational remodeling as another proteolysis-independent mechanism in addition to ZATTZNF451.
9. Pathway choice for TOP-DPC debulking
To suppress topoisomerase-induced genomic instability, cells have evolved multiple proteolytic mechanisms to efficiently cope with TOP-DPC. Yet, how the cell dictates the pathway choice between proteasome, non-proteasomal proteases and non-proteolytic pathways (Fig. 1) is an important remaining conundrum. A very recent study in Xenopus egg, shows that the proteasome specifically targets polyubiquitylated DNA methyltransferase HpaII-DPC for degradation whereas SPRTN digests the unmodified counterpart during replication [62]. Yet, the replication-associated protease SPRTN is probably not relevant in non-replicative cells as a repair mechanism for TOP-DPC. As mentioned, it has been established that TOP1-DPC as well as TO2β-DPC are subjected to ubiquitylation and targeted by the proteasome upon their detection by RNA polymerase II [69,84], implying the key role of the ubiquitin-proteasome pathway for the repair of TOP-DPC in the context of transcription both in non-replicative and proliferating cells.
Emerging evidence suggests a role of SUMOylation as an early defense mechanism against TOP-DPC and general DPC [68,100]. It is also clear that SUMOylation of topoisomerases is readily detected under physiological conditions [68,125]. Moreover, the SUMOylation of TOP-DPC has recently been demonstrated in yeast as well as human cells to serve as a signal for the ubiquitylation of TOP-DPC [68]. Whether the responsive SUMOylation of TOP-DPC and general DPC plays a role in recruiting SPRTN remains to be clarified. One study in C. elegans and human cells observed that SUMOylation was not required for the enrichment of SPRTN on chromatin for the repair of DPC induced by formaldehyde [100] whereas a more recent study found that SPRTN targets TOP1-DPC for proteolysis upon SUMO-1 modification of the DPC [150], imply a specific role of SUMOylation in regulating the SPRTN-dependent repair of TOP1-DPC.
It is likely that the debulking pathway choice is dependent on cell proliferation and cell cycle molecular component (whether cells are actively replicating such as germ cells or quiescent such as neurons). Cellular differentiation is also critical, and most importantly the extent and levels of TOP-DPC. Indeed, cells may use some pathways for endogenous and low levels of physiological TOP-DPC (associated with transcription, replication or recombination) and additional pathways for acute and high levels TOP-DPC such as those induced by anticancer TOP1 and TOP2 poisons and acute environmental exposure to DNA toxins (see Table 1).
10. Excision of the residual TOP-DpC following proteolysis
The excision step for TOP-DpC (Fig. 1) is reviewed in a complementary publication [1] and will only be summarized and elaborated upon here. Due to the unproteolyzable tyrosyl phosphodiester bond left by the debulking proteases, specialized mechanisms are entailed for the clearance of the remaining DNA-bound topoisomerase peptide (DpC) (Fig. 1). As indicated above, TDP1 and TDP2 serve as precise excision mechanisms to hydrolyze the tyrosine-DNA bond following the debulking step [21]. In addition to the TDP hydrolytic pathway, cells use endonucleases such as MRE11 to remove the TOP-DPC by trimming the DNA strand covalently attached to the TOP-DPC (Fig. 1).
10.1. TDP1
TDP1 belongs to the phospholipase D (PLD) superfamily; it is highly conserved from yeast to humans [170,171] and hydrolyzes the 3’ phosphotyrosyl bond of TOP1-DPC as well as other types of 3’-blocking lesions [21]. TDP1 also exhibits activity against 5’-phosphotyrosyl linkages, albeit much weaker than does TDP2 [160,172], suggesting a role of TDP1 as backup excision mechanism for repairing TOP2-DPC [173]. The resulting “clean” DNA termini with a 3’-phosphate is then further processed, leading to restoration of the DNA by non-homologous end joining (NHEJ) or homologous recombination (HR) [21,174,175]. Given that the phosphotyrosyl bond is completely buried by the bulky DNA-bound topoisomerase, it is conceivable that additional steps are required to unveil the TOP1-occluded bond for access by TDP1. Indeed, in vitro studies have shown that TDP1 act on proteolyzed or denatured TOP1-DPC but not native, full-length TOP1-DPC [176,177], indicating that exposure of the phophotyrosyl bond by proteolysis by the proteasome or other proteases or possibly unfolding of the TOP-DPC are critical for the action of TDP1 [176].
In yeast, Wss1 and Tdp1 compensate each other in the repair of Top1-DPC and only the Δtdp1Δwss1 double-mutant displays elevated Top1-DPC and hypersensitivity to CPT [126]. Although it appears plausible that Wss1 could debulk Top1-DPC to allow Tdp1 to process the remaining Top1-DpC, the redundant roles of Wss1 and Tdp1 in Top1-DPC repair raise the possibility that Tdp1 could be act on denatured full-length TOP1-DPC without the need for proteolysis in yeast or that Tdp1 can be coupled with another protease, such as Ddi1 [146] (see Section 5.2). By contrast, in mammals, a couple of studies [129,132] suggest that SPRTN and TDP1 both work in the same repair pathway of TOP1-DPC as depletion of TDP1 in SPRTN-depleted cells does not cause further hypersensitization and TOP1-DPC accumulation compared to SPRTN knockout alone. The degree of sensitization to CPT in SPRTN depleted mammalian cells is similar to that of TDP1 depleted cells [129,132].
Following the debulking of TOP1-DPC either by the proteasome or SPRTN, the role of NER nucleases ERCC1-XPF has been invoked in the repair of TOP1-DPC in parallel with the TDP1 hydrolysis pathway [178]. Thus, it is conceivable that TDP1 hydrolyzes 3′-phosphotyrosyl bond linked to DNA, leaving a 3′ phosphoryl nick DNA that is eventually repaired by the BER pathway (XRCC1, polynucleotide kinase 3′-phosphatase, POLB and LIGIII). Alternatively, ERCC1-XPF and RPA remove 3′-phosphotyrosyl bound peptides to generate a gap for POLD, FEN1 and LIG1, which are NER factors [179].
Although Tdp1 has been shown to excise Top2-DPC in yeast [180] and DT40 cells [172], and to also excise 5’-DpC in biochemical assays [172,177], studies in human lymphoma cells indicate a lack of involvement in TDP2 knockout TK6 cells [181]. Hence, further studies are warranted to clarify the relevance of TDP1 for TOP2-DPC in different models and circumstances.
10.2. TDP2
Yeast does not express TDP2 and only Tdp1, which appear to perform the function of TDP2 for the Top2-DPC [180]. The polarity of TDP2 is opposite to TDP1; it primarily acts on 5’-phosphotyrosyl linkages for the repair of TOP2-DPC and TOP3-DPC [1,33,182]. In human cells, TDP2 appears to display activity toward the 3’-counterpart, albeit less efficient than TDP1 [172,173]. Yet, genetic studies indicate that TDP2 acts as a backup pathway for TOP1-DPC in the absence of TDP1 [173,181] Akin to TDP1, TDP2 preferentially excises TOP2-DPC fragments after proteolysis or denatured TOP2-DPC in vitro [183], suggesting a role of proteolytic debulking as a precondition for the activity of TDP2, presumably by unveiling the 5’ tyrosine-DNA bond.
As mentioned in Section 6, ZATT (ZNF451) is a SUMO E3 ligase that induces conformational change in TOP2-DPC by either direct binding or SUMO-2/3 modification, enabling TDP2 to gain access to the 5’ tyrosyl bond without the debulking of the TOP2 adduct [125]. Although SPRTN depleted human cells are sensitive to ETP [128,132] to a similar extent to TDP2 depleted cells, the genetic interaction of SPRTN and TDP2 as well as TDP1 still needs to be further investigated. Also, how SPRTN and TDPs (TDP1 and TDP2) are coordinate with each other in counteracting TOP-DPC is still unclear and warrants further investigations.
10.3. MRE11
The Mre11-Rad50-Nbs1 nuclease complex has been demonstrated to play an important role in processing TOP2- as well TOP1-DPC in parallel to TDPs [1,184,185]. It has been proposed that Mre11, in cooperation with its functional partner CtIP, incise the DNA segment adjoining TOP-DPC, imlying that the exposure of the phosphotyrosyl bond is not required for Mre11 to remove TOP-DPC. This model is supported by evidence that Nbs1 promotes Mre11/Rad50 catalyzed endo- and exonucleolytic cleavage of DNA containing 5’-adducts to generate clean double-strand break ends [186] as well as the fact that Mre11 processes the covalent DNA-bound SPO11, a TOP2-like protein that mediates meiotic recombination, without the need for proteases to digest the bulky SPO11 adduct [187,188].
In yeast, the lack of both TDP1 and MRE11 renders cells substantially more sensitive to CPT than either single mutant where the nuclease dead MRE11 shows mild sensitivity and TDP1 KO are insensitive to CPT [189]. In mammalian cells, endogenous TOP2-DPC accumulate in MRE11-deficient cells, but not in TDP2-deficient cells [190]. However, deletion of TDP2 in cells that lack MRE11 markedly increases DNA damage, etoposide sensitivity and TOP2-DPC accumulation [190]. It appears that the clean DSB that are generated by both TDP2- and MRN-dependent repair of TOP2-DPC are ultimately channeled into the NHEJ pathway in vertebrates [190,191]. A very recent study reported that Mre11 is recruited by the ubiquitin E2 enzyme UBC13-mediated K63 polyubiquitylation for the repair of etoposide-induced TOP2-DPC in G1 cells [192]. The substrates of the ubiquitylation were nevertheless not identified in the study. Given that both TOP1- and TOP2- DPC can be modified by ubiquitin through K63 linkage [68], it will be important to determine whether UBC13 specifically targets TOP-DPC for the ubiquitylation to employ the Mre11-dependent repair.
11. Concluding remarks
Many of the pathways described here for the TOP-DPC are common to other DPC, such as the proteasomal and SPRTN pathway. Using topoisomerase inhibitors is a powerful and convenient approach to discover novel pathways for the debulking of DPC prior to their proteolytic processing. While our understanding of the repair mechanisms for TOP-DPC has increased markedly in the last years, many pressing questions remain. In particular, it is undetermined whether the repair machineries are recruited to the sites of TOP-DPC, and/or whether TOP-DPC are relocalized (presumably by chromatin remodeling) to regions (repair factories) with high abundance of the repair proteins together with the proteasome. The mechanisms that determine the division of labor between all the identified protease and debulking pathways also remain largely unknown. The hierarchy, interplay and redundancy of the protease pathways therefore warrant future exploration.
Apart from DPC formed by TOP1 and TOP2, the biology and repair of TOP3-DPC (Induction/detection/debulking and excision) formed by the type IA topoisomerases has only begun to be investigated. While we recently reported a common pathway involving the proteasome and TDP2 and a specific UPP with the E3 ligase TRIM41 for the repair of TOP3β-DPC [33], it is yet unclear whether additional pathways are involved for TOP3β-DPC, and whether TOP3α forms DPC under physiological conditions or in response to exo/endogenous agents and how such TOP3-DPC are processed.
Understanding the debulking pathways in the context of the DDR pathways will help rationalize combination therapies such as combining topoisomerase inhibitors with inhibitors of the proteasome, which are FDA-approved for a small population of cancer patients, or ubiquitin or SUMO or p97/VCP inhibitors, which are clinical development. The question will be to define the synthetic lethality, which is likely due to the redundancy and parallel pathways for the debulking of TOP-DPC.
Acknowledgements
Our studies are supported by the NCI Intramural Program, the Center for Cancer Research at the NIH (Z01-BC-006161 and Z01-BC-006150). We wish to thank all the current and past members of the DTB Laboratory of Molecular Pharmacology for their sustained contributions.
Footnotes
Declaration of Competing Interest
The authors declare that there is no conflict of interest.
References
- [1].Sun Y, Saha S, Wang W, Saha LK, Huang SN, Pommier Y, Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC), DNA Repair (Amst) 89 (2020) 102837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhang H, Xiong Y, Chen J, DNA-protein cross-link repair: what do we know now? Cell Biosci. 10 (2020) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stingele J, Bellelli R, Boulton SJ, Mechanisms of DNA-protein crosslink repair, Nat. Rev. Mol. Cell Biol 18 (2017) 563–573. [DOI] [PubMed] [Google Scholar]
- [4].Ide H, Nakano T, Salem AMH, Shoulkamy MI, DNA-protein cross-links: formidable challenges to maintaining genome integrity, DNA Repair (Amst) 71 (2018) 190–197. [DOI] [PubMed] [Google Scholar]
- [5].Kojima Y, Machida YJ, DNA-protein crosslinks from environmental exposure: mechanisms of formation and repair, Environ. Mol. Mutagen (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Daskalova SM, Bai X, Hecht SM, Study of the lyase activity of human DNA polymerase beta using analogues of the intermediate schiff base complex, Biochemistry 57 (2018) 2711–2722. [DOI] [PubMed] [Google Scholar]
- [7].Prasad R, Horton JK, Dai DP, Wilson SH, Repair pathway for PARP-1 DNA-protein crosslinks, DNA Repair (Amst) 73 (2019) 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang JC, Cellular roles of DNA topoisomerases: a molecular perspective, Nat. Rev. Mol. Cell Biol 3 (2002) 430–440. [DOI] [PubMed] [Google Scholar]
- [9].Bizard AH, Hickson ID, The many lives of type IA topoisomerases, J. Biol. Chem (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pommier Y, Sun Y, Huang SN, Nitiss JL, Roles of eukaryotic topoisomerases in transcription, replication and genomic stability, Nat. Rev. Mol. Cell Biol 17 (2016) 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Valdes A, Segura J, Dyson S, Martinez-Garcia B, Roca J, DNA knots occur in intracellular chromatin, Nucleic Acids Res 46 (2018) 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y, Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5’-phosphorylated DNA double-strand breaks by replication runoff, Mol. Cell. Biol 20 (2000) 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Regairaz M, Zhang Y-W, Fu H, Agama KK, Tata N, Agrawal S, Aladjem MI, Pommier Y, Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes, J. Cell Biol 195 (2011) 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M, Topoisomerase I poisoning results in PARP-mediated replication fork reversal, Nat. Struct. Mol. Biol 19 (2012) 417–423. [DOI] [PubMed] [Google Scholar]
- [15].Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, Kohn KW, Bonner WM, Pommier Y, Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks, EMBO Rep 10 (2009) 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pommier Y, Jenkins J, Kohlhagen G, Leteurtre F, DNA recombinase activity of eukaryotic DNA topoisomerase I; effects of camptothecin and other inhibitors, Mutat. Res 337 (1995) 135–145. [DOI] [PubMed] [Google Scholar]
- [17].Pourquier P, Pilon A, Kohlhagen G, Mazumder A, Sharma A, Pommier Y, Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps: importance of DNA end phosphorylation and camptothecin effects, J. Biol. Chem 272 (1997) 26441–26447. [DOI] [PubMed] [Google Scholar]
- [18].Wang X, Henningfeld KA, Hecht SM, DNA topoisomerase I-mediated formation of structurally modified DNA duplexes. Effects of metal ions and topoisomerase I inhibitors, Biochemistry 37 (1998) 2691–2700. [DOI] [PubMed] [Google Scholar]
- [19].Kim N, Huang SY, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks-Robertson S, Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I, Science 332 (2011) 1561–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang SY, Ghosh S, Pommier Y, Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites, J. Biol. Chem 290 (2015) 14068–14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pommier Y, Huang SY, Gao R, Das BB, Murai J, Marchand C, Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2), DNA Repair (Amst) 19 (2014) 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW, Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1, Nature 434 (2005) 108–113. [DOI] [PubMed] [Google Scholar]
- [23].Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, Northrop JL, Salih MA, Inoue K, Armstrong DL, Champoux JJ, Takashima H, Boerkoel CF, Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? EMBO J 26 (2007) 4732–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ, SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity, EMBO J 24 (2005) 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR, Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy, Nat. Genet 32 (2002) 267–272. [DOI] [PubMed] [Google Scholar]
- [26].Walton C, Interthal H, Hirano R, Salih MAM, Takashima H, Boerkoel CF, Spinocerebellar ataxia with axonal neuropathy, Diseases of DNA Repair, Springer-Verlag, Berlin, Berlin, 2010, pp. 75–83. [DOI] [PubMed] [Google Scholar]
- [27].Gomez-Herreros F, Schuurs-Hoeijmakers JH, McCormack M, Greally MT, Rulten S, Romero-Granados R, Counihan TJ, Chaila E, Conroy J, Ennis S, Delanty N, Cortes-Ledesma F, de Brouwer AP, Cavalleri GL, El-Khamisy SF, de Vries BB, Caldecott KW, TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function, Nat. Genet 46 (2014) 516–521. [DOI] [PubMed] [Google Scholar]
- [28].Capranico G, Binaschi M, DNA sequence selectivity of topoisomerases and topoisomerase poisons, Biochim. Biophys. Acta 1400 (1998) 185–194. [DOI] [PubMed] [Google Scholar]
- [29].Porter SE, Champoux JJ, Mapping in vivo topoisomerase I sites on simian virus 40 DNA: asymmetric distribution of sites on replicating molecules, Mol. Cell. Biol 9 (1989) 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jaxel C, Capranico G, Wassermann K, Kerrigan D, Kohn KW, Pommier Y, DNA sequence at sites of topoisomerase I cleavage induced by camptothecin in SV40 DNA, in: Potmesil M, Kohn KW (Eds.), DNA Topoisomerases in Cancer, Oxford University Press, New York, Oxford, 1991, pp. 182–195. [Google Scholar]
- [31].Pommier Y, Capranico G, Orr A, Kohn KW, Local base sequence preferences for DNA cleavage by mammalian topoisomerase II in the presence of amsacrine or teniposide, Nucleic Acids Res 19 (1991) 5973–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Capranico G, Kohn KW, Pommier Y, Local sequence requirements for DNA cleavage by mammalian topoisomerase II in the presence of doxorubicin, Nucleic Acids Res 18 (1990) 6611–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Saha S, Huang S-Y, Jo U, Zhang H, Tse-Dinh Y-C, Pommier Y, Topoisomerase 3B (TOP3B) DNA and RNA cleavage complexes and pathway to repair TOP3B-linked RNA and DNA breaks, bioRxiv (2020) 2020.2003.2022.002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pommier Y, Marchand C, Interfacial inhibitors: targeting macromolecular complexes, Nat. Rev. Drug Discov 11 (2011) 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morimoto S, Tsuda M, Bunch H, Sasanuma H, Austin C, Takeda S, Type II DNA topoisomerases cause spontaneous double-strand breaks in genomic DNA, Genes (Basel) 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Puc J, Kozbial P, Li W, Tan Y, Liu Z, Suter T, Ohgi KA, Zhang J, Aggarwal AK, Rosenfeld MG, Ligand-dependent enhancer activation regulated by Topoisomerase-I activity, Cell 160 (2015) 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription, Science 312 (2006) 1798–1802. [DOI] [PubMed] [Google Scholar]
- [38].Ju BG, Rosenfeld MG, A breaking strategy for topoisomerase IIbeta/PARP-1-dependent regulated transcription, Cell Cycle 5 (2006) 2557–2560. [DOI] [PubMed] [Google Scholar]
- [39].Lis JT, Kraus WL, Promoter cleavage: a topoIIbeta and PARP-1 collaboration, Cell 125 (2006) 1225–1227. [DOI] [PubMed] [Google Scholar]
- [40].Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, Nelson WG, Yegnasubramanian S, Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements, Nat. Genet 42 (2010) 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dahlmann B, Kopp F, Kuehn L, Niedel B, Pfeifer G, Hegerl R, Baumeister W, The multicatalytic proteinase (prosome) is ubiquitous from eukaryotes to archaebacteria, FEBS Lett 251 (1989) 125–131. [DOI] [PubMed] [Google Scholar]
- [42].Tomko RJ Jr., M. Hochstrasser, Molecular architecture and assembly of the eukaryotic proteasome, Annu. Rev. Biochem 82 (2013) 415–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Collins GA, Goldberg AL, The logic of the 26S proteasome, Cell 169 (2017) 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Adams J, The proteasome: structure, function, and role in the cell, Cancer Treat. Rev 29 (Suppl 1) (2003) 3–9. [DOI] [PubMed] [Google Scholar]
- [45].Varshavsky A, The ubiquitin system, an immense realm, Annu. Rev. Biochem 81 (2012) 167–176. [DOI] [PubMed] [Google Scholar]
- [46].Chasapis CT, Spyroulias GA, RING finger E(3) ubiquitin ligases: structure and drug discovery, Curr. Pharm. Des 15 (2009) 3716–3731. [DOI] [PubMed] [Google Scholar]
- [47].Morreale MK, Balon R, Beresin EV, Coverdale JH, Brenner A, Guerrero A, Louie AK, Roberts LW, Providing psychiatric care for an expanding population of Cancer survivors: imperatives for psychiatric education and leadership, Acad. Psychiatry 41 (2017) 1–3. [DOI] [PubMed] [Google Scholar]
- [48].Kravtsova-Ivantsiv Y, Ciechanover A, Non-canonical ubiquitin-based signals for proteasomal degradation, J. Cell. Sci 125 (2012) 539–548. [DOI] [PubMed] [Google Scholar]
- [49].Swatek KN, Komander D, Ubiquitin modifications, Cell Res 26 (2016) 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yau R, Rape M, The increasing complexity of the ubiquitin code, Nat. Cell Biol 18 (2016) 579–586. [DOI] [PubMed] [Google Scholar]
- [51].Reyes-Turcu FE, Ventii KH, Wilkinson KD, Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes, Annu. Rev. Biochem 78 (2009) 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mevissen TET, Komander D, Mechanisms of deubiquitinase specificity and regulation, Annu. Rev. Biochem 86 (2017) 159–192. [DOI] [PubMed] [Google Scholar]
- [53].Clague MJ, Urbe S, Komander D, Breaking the chains: deubiquitylating enzyme specificity begets function, Nat. Rev. Mol. Cell Biol 20 (2019) 338–352. [DOI] [PubMed] [Google Scholar]
- [54].Vlachostergios PJ, Patrikidou A, Daliani DD, Papandreou CN, The ubiquitin-proteasome system in cancer, a major player in DNA repair. Part 1: post-translational regulation, J. Cell. Mol. Med 13 (2009) 3006–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Srivenugopal KS, Yuan XH, Friedman HS, Ali-Osman F, Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea, Biochemistry 35 (1996) 1328–1334. [DOI] [PubMed] [Google Scholar]
- [56].Hernandez-Pigeon H, Quillet-Mary A, Louat T, Schambourg A, Humbert O, Selves J, Salles B, Laurent G, Lautier D, hMutS alpha is protected from ubiquitin-proteasome-dependent degradation by atypical protein kinase C zeta phosphorylation, J. Mol. Biol 348 (2005) 63–74. [DOI] [PubMed] [Google Scholar]
- [57].Edmonds MJ, Parsons JL, Regulation of base excision repair proteins by ubiquitylation, Exp. Cell Res 329 (2014) 132–138. [DOI] [PubMed] [Google Scholar]
- [58].Reed SH, Gillette TG, Nucleotide excision repair and the ubiquitin proteasome pathway–do all roads lead to Rome? DNA Repair (Amst) 6 (2007) 149–156. [DOI] [PubMed] [Google Scholar]
- [59].Lommel L, Ortolan T, Chen L, Madura K, Sweder KS, Proteolysis of a nucleotide excision repair protein by the 26 S proteasome, Curr. Genet 42 (2002) 9–20. [DOI] [PubMed] [Google Scholar]
- [60].Chen J, Bozza W, Zhuang Z, Ubiquitination of PCNA and its essential role in eukaryotic translesion synthesis, Cell Biochem. Biophys 60 (2011) 47–60. [DOI] [PubMed] [Google Scholar]
- [61].Garner E, Smogorzewska A, Ubiquitylation and the Fanconi anemia pathway, FEBS Lett 585 (2011) 2853–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Raschle M, Walter JC, Duxin JP, Replication-coupled DNA-Protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts, Mol. Cell 73 (2019) 574–588 e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cao J, Yan Q, Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer, Front. Oncol 2 (2012) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Beidler DR, Cheng YC, Camptothecin induction of a time- and concentration-dependent decrease of topoisomerase I and its implication in camptothecin activity, Mol. Pharmacol 47 (1995) 907–914. [PubMed] [Google Scholar]
- [65].Desai SD, Liu LF, Vazquez-Abad D, D’Arpa P, Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin, J. Biol. Chem 272 (1997) 24159–24164. [DOI] [PubMed] [Google Scholar]
- [66].Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF, Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells, Cancer Res 61 (2001) 5926–5932. [PubMed] [Google Scholar]
- [67].Zhang HF, Tomida A, Koshimizu R, Ogiso Y, Lei S, Tsuruo T, Cullin 3 promotes proteasomal degradation of the topoisomerase I-DNA covalent complex, Cancer Res 64 (2004) 1114–1121. [DOI] [PubMed] [Google Scholar]
- [68].Sun Y, Jenkins LMM, Su YP, Nitiss KC, Nitiss JL, Pommier Y, A conserved SUMO-Ubiquitin pathway directed by RNF4/SLX5-SLX8 and PIAS4/SIZ1 drives proteasomal degradation of topoisomerase DNA-protein crosslinks, bioRxiv (2019) 707661. [Google Scholar]
- [69].Desai SD, Zhang H, Rodriguez-Bauman A, Yang JM, Wu X, Gounder MK, Rubin EH, Liu LF, Transcription-dependent degradation of topoisomerase I-DNA covalent complexes, Mol. Cell. Biol 23 (2003) 2341–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hsiang Y-H, Lihou MG, Liu LF, Arrest of DNA replication by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin, Cancer Res 49 (1989) 5077–5082. [PubMed] [Google Scholar]
- [71].Holm C, Covey JM, Kerrigan D, Pommier Y, Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells, Cancer Res 49 (1989) 6365–6368. [PubMed] [Google Scholar]
- [72].Cliby WA, Lewis KA, Lilly KK, Kaufmann SH, S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function, J. Biol. Chem 277 (2002) 1599–1606. [DOI] [PubMed] [Google Scholar]
- [73].Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y, Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA-double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes, J. Biol. Chem 278 (2003) 20303–20312. [DOI] [PubMed] [Google Scholar]
- [74].Shao R-G, Cao C-X, Zhang H, Kohn KW, Wold MS, Pommier Y, Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes, EMBO J 18 (1999) 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lin CP, Ban Y, Lyu YL, Liu LF, Proteasome-dependent processing of topoisomerase I-DNA adducts into DNA double strand breaks at arrested replication forks, J. Biol. Chem 284 (2009) 28084–28092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kerzendorfer C, Whibley A, Carpenter G, Outwin E, Chiang SC, Turner G, Schwartz C, El-Khamisy S, Raymond FL, O’Driscoll M, Mutations in Cullin 4B result in a human syndrome associated with increased camptothecin-induced topoisomerase I-dependent DNA breaks, Hum. Mol. Genet 19 (2010) 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sordet O, Larochelle S, Nicolas E, Stevens EV, Zhang C, Shokat KM, Fisher RP, Pommier Y, Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription- and BRCA1-dependent degradation of topoisomerase I, J. Mol. Biol 381 (2008) 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ando K, Shah AK, Sachdev V, Kleinstiver BP, Taylor-Parker J, Welch MM, Hu Y, Salgia R, White FM, Parvin JD, Ozonoff A, Rameh LE, Joung JK, Bharti AK, Camptothecin resistance is determined by the regulation of topoisomerase I degradation mediated by ubiquitin proteasome pathway, Oncotarget 8 (2017) 43733–43751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Havens CG, Walter JC, Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase, Genes Dev 25 (2011) 1568–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li JM, Jin J, CRL ubiquitin ligases and DNA damage response, Front. Oncol 2 (2012) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nakajima T, Ohi N, Arai T, Nozaki N, Kikuchi A, Oda K, Adenovirus E1A-induced apoptosis elicits a steep decrease in the topoisomerase II alpha level during the latent phase, Oncogene 10 (1995) 651–662. [PubMed] [Google Scholar]
- [82].Nakajima T, Morita K, Ohi N, Arai T, Nozaki N, Kikuchi A, Osaka F, Yamao F, Oda K, Degradation of topoisomerase IIalpha during adenovirus E1A-induced apoptosis is mediated by the activation of the ubiquitin proteolysis system, J. Biol. Chem 271 (1996) 24842–24849. [DOI] [PubMed] [Google Scholar]
- [83].Mao Y, Desai SD, Ting CY, Hwang J, Liu LF, 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes, J. Biol. Chem 276 (2001) 40652–40658. [DOI] [PubMed] [Google Scholar]
- [84].Xiao H, Mao Y, Desai SD, Zhou N, Ting CY, Hwang J, Liu LF, The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Alchanati I, Teicher C, Cohen G, Shemesh V, Barr HM, Nakache P, Ben-Avraham D, Idelevich A, Angel I, Livnah N, Tuvia S, Reiss Y, Taglicht D, Erez O, The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of Top2alpha cleavage complex - a potentially new drug target, PLoS One 4 (2009) e8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Sciascia N, Wu W, Zong D, Sun Y, Wong N, John S, Wangsa D, Ried T, Bunting SF, Pommier Y, Nussenzweig A, Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity, Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ogiso Y, Tomida A, Lei S, Omura S, Tsuruo T, Proteasome inhibition circumvents solid tumor resistance to topoisomerase II-directed drugs, Cancer Res 60 (2000) 2429–2434. [PubMed] [Google Scholar]
- [88].Lee KC, Bramley RL, Cowell IG, Jackson GH, Austin CA, Proteasomal inhibition potentiates drugs targeting DNA topoisomerase II, Biochem. Pharmacol 103 (2016) 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Eguren M, Alvarez-Fernandez M, Garcia F, Lopez-Contreras AJ, Fujimitsu K, Yaguchi H, Luque-Garcia JL, Fernandez-Capetillo O, Munoz J, Yamano H, Malumbres M, A synthetic lethal interaction between APC/C and topoisomerase poisons uncovered by proteomic screens, Cell Rep 6 (2014) 670–683. [DOI] [PubMed] [Google Scholar]
- [90].Gareau JR, Lima CD, The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition, Nat. Rev. Mol. Cell Biol 11 (2010) 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Flotho A, Melchior F, Sumoylation: a regulatory protein modification in health and disease, Annu. Rev. Biochem 82 (2013) 357–385. [DOI] [PubMed] [Google Scholar]
- [92].Yeh ET, SUMOylation and De-SUMOylation: wrestling with life’s processes, J. Biol. Chem 284 (2009) 8223–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jackson SP, Durocher D, Regulation of DNA damage responses by ubiquitin and SUMO, Mol. Cell 49 (2013) 795–807. [DOI] [PubMed] [Google Scholar]
- [94].Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M, Crystal structure of thymine DNA glycosylase conjugated to SUMO-1, Nature 435 (2005) 979–982. [DOI] [PubMed] [Google Scholar]
- [95].Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP, Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks, Nature 462 (2009) 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yin Y, Seifert A, Chua JS, Maure JF, Golebiowski F, Hay RT, SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage, Genes Dev 26 (2012) 1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Galanty Y, Belotserkovskaya R, Coates J, Jackson SP, RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair, Genes Dev 26 (2012) 1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sarangi P, Zhao X, SUMO-mediated regulation of DNA damage repair and responses, Trends Biochem. Sci 40 (2015) 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Akita M, Tak YS, Shimura T, Matsumoto S, Okuda-Shimizu Y, Shimizu Y, Nishi R, Saitoh H, Iwai S, Mori T, Ikura T, Sakai W, Hanaoka F, Sugasawa K, SUMOylation of xeroderma pigmentosum group C protein regulates DNA damage recognition during nucleotide excision repair, Sci. Rep 5 (2015) 10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Borgermann N, Ackermann L, Schwertman P, Hendriks IA, Thijssen K, Liu JC, Lans H, Nielsen ML, Mailand N, SUMOylation promotes protective responses to DNA-protein crosslinks, EMBO J 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Miteva M, Keusekotten K, Hofmann K, Praefcke GJ, Dohmen RJ, Sumoylation as a signal for polyubiquitylation and proteasomal degradation, Subcell. Biochem 54 (2010) 195–214. [DOI] [PubMed] [Google Scholar]
- [102].Tatham MH, Geoffroy MC, Shen L, Plechanovova A, Hattersley N, Jaffray EG, Palvimo JJ, Hay RT, RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation, Nat. Cell Biol 10 (2008) 538–546. [DOI] [PubMed] [Google Scholar]
- [103].Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, Zhou J, Zhu J, Raught B, de The H, Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway, Nat. Cell Biol 10 (2008) 547–555. [DOI] [PubMed] [Google Scholar]
- [104].Plechanovova A, Jaffray EG, McMahon SA, Johnson KA, Navratilova I, Naismith JH, Hay RT, Mechanism of ubiquitylation by dimeric RING ligase RNF4, Nat. Struct. Mol. Biol 18 (2011) 1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hakli M, Karvonen U, Janne OA, Palvimo JJ, SUMO-1 promotes association of SNURF (RNF4) with PML nuclear bodies, Exp. Cell Res 304 (2005) 224–233. [DOI] [PubMed] [Google Scholar]
- [106].Percherancier Y, Germain-Desprez D, Galisson F, Mascle XH, Dianoux L, Estephan P, Chelbi-Alix MK, Aubry M, Role of SUMO in RNF4-mediated promyelocytic leukemia protein (PML) degradation: sumoylation of PML and phospho-switch control of its SUMO binding domain dissected in living cells, J. Biol. Chem 284 (2009) 16595–16608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Vyas R, Kumar R, Clermont F, Helfricht A, Kalev P, Sotiropoulou P, Hendriks IA, Radaelli E, Hochepied T, Blanpain C, Sablina A, van Attikum H, Olsen JV, Jochemsen AG, Vertegaal AC, Marine JC, RNF4 is required for DNA double-strand break repair in vivo, Cell Death Differ 20 (2013) 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rabellino A, Andreani C, Scaglioni PP, The role of PIAS SUMO E3-Ligases in Cancer, Cancer Res 77 (2017) 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Hochstrasser M, SP-RING for SUMO: new functions bloom for a ubiquitin-like protein, Cell 107 (2001) 5–8. [DOI] [PubMed] [Google Scholar]
- [110].Bologna S, Altmannova V, Valtorta E, Koenig C, Liberali P, Gentili C, Anrather D, Ammerer G, Pelkmans L, Krejci L, Ferrari S, Sumoylation regulates EXO1 stability and processing of DNA damage, Cell Cycle 14 (2015) 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Shima H, Suzuki H, Sun J, Kono K, Shi L, Kinomura A, Horikoshi Y, Ikura T, Ikura M, Kanaar R, Igarashi K, Saitoh H, Kurumizaka H, Tashiro S, Activation of the SUMO modification system is required for the accumulation of RAD51 at sites of DNA damage, J. Cell. Sci 126 (2013) 5284–5292. [DOI] [PubMed] [Google Scholar]
- [112].Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, Powell S, Zhang J, Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation, J. Biol. Chem 283 (2008) 31608–31616. [DOI] [PubMed] [Google Scholar]
- [113].Mao Y, Sun M, Desai SD, Liu LF, SUMO-1 conjugation to topoisomerase I: a possible repair response to topoisomerase-mediated DNA damage, Proc. Natl. Acad. Sci. U. S. A 97 (2000) 4046–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Horie K, Tomida A, Sugimoto Y, Yasugi T, Yoshikawa H, Taketani Y, Tsuruo T, SUMO-1 conjugation to intact DNA topoisomerase I amplifies cleavable complex formation induced by camptothecin, Oncogene 21 (2002) 7913–7922. [DOI] [PubMed] [Google Scholar]
- [115].Yang M, Hsu CT, Ting CY, Liu LF, Hwang J, Assembly of a polymeric chain of SUMO1 on human topoisomerase I in vitro, J. Biol. Chem 281 (2006) 8264–8274. [DOI] [PubMed] [Google Scholar]
- [116].Rallabhandi P, Hashimoto K, Mo YY, Beck WT, Moitra PK, D’Arpa P, Sumoylation of topoisomerase I is involved in its partitioning between nucleoli and nucleoplasm and its clearing from nucleoli in response to camptothecin, J. Biol. Chem 277 (2002) 40020–40026. [DOI] [PubMed] [Google Scholar]
- [117].Mo YY, Yu Y, Shen Z, Beck WT, Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein, J. Biol. Chem 277 (2002) 2958–2964. [DOI] [PubMed] [Google Scholar]
- [118].Steinacher R, Osman F, Lorenz A, Bryer C, Whitby MC, Slx8 removes Pli1-dependent protein-SUMO conjugates including SUMOylated topoisomerase I to promote genome stability, PLoS One 8 (2013) e71960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Heideker J, Prudden J, Perry JJ, Tainer JA, Boddy MN, SUMO-targeted ubiquitin ligase, Rad60, and Nse2 SUMO ligase suppress spontaneous Top1-mediated DNA damage and genome instability, PLoS Genet 7 (2011) e1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Takahashi Y, Yong-Gonzalez V, Kikuchi Y, Strunnikov A, SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II, Genetics 172 (2006) 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Azuma Y, Arnaoutov A, Dasso M, SUMO-2/3 regulates topoisomerase II in mitosis, J. Cell Biol 163 (2003) 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mao Y, Desai SD, Liu LF, SUMO-1 conjugation to human DNA topoisomerase II isozymes, J. Biol. Chem 275 (2000) 26066–26073. [DOI] [PubMed] [Google Scholar]
- [123].Shu J, Cui D, Ma Y, Xiong X, Sun Y, Zhao Y, SCF(beta-TrCP)-mediated degradation of TOP2beta promotes cancer cell survival in response to chemotherapeutic drugs targeting topoisomerase II, Oncogenesis 9 (2020) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zagnoli-Vieira G, Caldecott KW, TDP2, TOP2, and SUMO: what is ZATT about? Cell Res 27 (2017) 1405–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Schellenberg MJ, Lieberman JA, Herrero-Ruiz A, Butler LR, Williams JG, Munoz-Cabello AM, Mueller GA, London RE, Cortes-Ledesma F, Williams RS, ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links, Science 357 (2017) 1412–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S, A DNA-dependent protease involved in DNA-protein crosslink repair, Cell 158 (2014) 327–338. [DOI] [PubMed] [Google Scholar]
- [127].Duxin JP, Dewar JM, Yardimci H, Walter JC, Repair of a DNA-protein crosslink by replication-coupled proteolysis, Cell 159 (2014) 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lopez-Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic-Terzic I, Terzic J, Dikic I, SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Maskey RS, Flatten KS, Sieben CJ, Peterson KL, Baker DJ, Nam HJ, Kim MS, Smyrk TC, Kojima Y, Machida Y, Santiago A, van Deursen JM, Kaufmann SH, Machida YJ, Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis, Nucleic Acids Res 45 (2017) 4564–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Morocz M, Zsigmond E, Toth R, Enyedi MZ, Pinter L, Haracska L, DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA, Nucleic Acids Res 45 (2017) 3172–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjaer S, Tainer JA, Skehel JM, Groll M, Boulton SJ, Mechanism and regulation of DNA-Protein crosslink repair by the DNA-Dependent metalloprotease SPRTN, Mol. Cell 64 (2016) 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, Drobnitzky N, Freire R, Amor DJ, Lockhart PJ, Kessler BM, McKenna GW, Gileadi O, Ramadan K, Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-Protein crosslink repair, Mol. Cell 64 (2016) 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Yang X, Li Y, Gao Z, Li Z, Xu J, Wang W, Dong Y, Structural analysis of Wss1 protein from saccharomyces cerevisiae, Sci. Rep 7 (2017) 8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Li F, Raczynska JE, Chen Z, Yu H, Structural insight into DNA-Dependent activation of human metalloprotease spartan, Cell Rep 26 (2019) 3336–3346 e3334. [DOI] [PubMed] [Google Scholar]
- [135].Ghosal G, Leung JW, Nair BC, Fong KW, Chen J, Proliferating cell nuclear antigen (PCNA)-binding protein C1orf124 is a regulator of translesion synthesis, J. Biol. Chem 287 (2012) 34225–34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Davis EJ, Lachaud C, Appleton P, Macartney TJ, Nathke I, Rouse J, DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage, Nat. Struct. Mol. Biol 19 (2012) 1093–1100. [DOI] [PubMed] [Google Scholar]
- [137].Mosbech A, Gibbs-Seymour I, Kagias K, Thorslund T, Beli P, Povlsen L, Nielsen SV, Smedegaard S, Sedgwick G, Lukas C, Hartmann-Petersen R, Lukas J, Choudhary C, Pocock R, Bekker-Jensen S, Mailand N, DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks, Nat. Struct. Mol. Biol 19 (2012) 1084–1092. [DOI] [PubMed] [Google Scholar]
- [138].Centore RC, Yazinski SA, Tse A, Zou L, Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response, Mol. Cell 46 (2012) 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Juhasz S, Balogh D, Hajdu I, Burkovics P, Villamil MA, Zhuang Z, Haracska L, Characterization of human Spartan/C1orf124, an ubiquitin-PCNA interacting regulator of DNA damage tolerance, Nucleic Acids Res 40 (2012) 10795–10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kim MS, Machida Y, Vashisht AA, Wohlschlegel JA, Pang YP, Machida YJ, Regulation of error-prone translesion synthesis by Spartan/C1orf124, Nucleic Acids Res 41 (2013) 1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Machida Y, Kim MS, Machida YJ, Spartan/C1orf124 is important to prevent UV-induced mutagenesis, Cell Cycle 11 (2012) 3395–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ, Spartan deficiency causes genomic instability and progeroid phenotypes, Nat. Commun 5 (2014) 5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Abugable AA, Morris JLM, Palminha NM, Zaksauskaite R, Ray S, El-Khamisy SF, DNA repair and neurological disease: from molecular understanding to the development of diagnostics and model organisms, DNA Repair (Amst) 81 (2019) 102669. [DOI] [PubMed] [Google Scholar]
- [144].Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J, Cabrera E, Freire R, Pope K, Nahid A, Norris F, Leventer RJ, Delatycki MB, Barbi G, von Ameln S, Hogel J, Degoricija M, Fertig R, Burkhalter MD, Hofmann K, Thiele H, Altmuller J, Nurnberg G, Nurnberg P, Bahlo M, Martin GM, Aalfs CM, Oshima J, Terzic J, Amor DJ, Dikic I, Ramadan K, Kubisch C, Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features, Nat. Genet 46 (2014) 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Sparks JL, Chistol G, Gao AO, Raschle M, Larsen NB, Mann M, Duxin JP, Walter JC, The CMG helicase bypasses DNA-Protein cross-links to facilitate their repair, Cell 176 (2019) 167–181 e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Serbyn N, Noireterre A, Bagdiul I, Plank M, Michel AH, Loewith R, Kornmann B, Stutz F, The aspartic protease Ddi1 contributes to DNA-Protein crosslink repair in yeast, Mol. Cell (2019). [DOI] [PubMed] [Google Scholar]
- [147].Halder S, Torrecilla I, Burkhalter MD, Popovic M, Fielden J, Vaz B, Oehler J, Pilger D, Lessel D, Wiseman K, Singh AN, Vendrell I, Fischer R, Philipp M, Ramadan K, SPRTN protease and checkpoint kinase 1 cross-activation loop safeguards DNA replication, Nat. Commun 10 (2019) 3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Balakirev MY, Mullally JE, Favier A, Assard N, Sulpice E, Lindsey DF, Rulina AV, Gidrol X, Wilkinson KD, Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Mullen JR, Chen CF, Brill SJ, Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase, Mol. Cell. Biol 30 (2010) 3737–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Vaz B, Ruggiano A, Popovic M, Rodriguez-Berriguete G, Kilgas S, Singh AN, Higgins GS, Kiltie AE, Ramadan K, SPRTN protease and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability, bioRxiv (2020) 2020.2002.2014.949289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, Iorio F, Sousa FG, Elloumi F, Aladjem MI, Thomas A, Sander C, Kohn KW, Benes CH, Garnett M, Reinhold WC, Pommier Y, CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of Cancer cell lines, iScience 10 (2018) 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Carmell MA, Dokshin GA, Skaletsky H, Hu YC, van Wolfswinkel JC, Igarashi KJ, Bellott DW, Nefedov M, Reddien PW, Enders GC, Uversky VN, Mello CC, Page DC, A widely employed germ cell marker is an ancient disordered protein with reproductive functions in diverse eukaryotes, Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Fielden J, Ruggiano A, Popovic M, Ramadan K, DNA protein crosslink proteolysis repair: from yeast to premature ageing and cancer in humans, DNA Repair (Amst) 71 (2018) 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Bhargava V, Goldstein CD, Russell L, Xu L, Ahmed M, Li W, Casey A, Servage K, Kollipara R, Picciarelli Z, Kittler R, Yatsenko A, Carmell M, Orth K, Amatruda JF, Yanowitz JL, Buszczak M, GCNA preserves genome integrity and fertility across species, Dev. Cell 52 (2020) 38–52 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Dokshin GA, Davis GM, Sawle AD, Eldridge MD, Nicholls PK, Gourley TE, Romer KA, Molesworth LW, Tatnell HR, Ozturk AR, de Rooij DG, Hannon GJ, Page DC, Mello CC, Carmell MA, GCNA interacts with spartan and topoisomerase II to regulate genome stability, Dev. Cell 52 (2020) 53–68 e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Svoboda M, Konvalinka J, Trempe JF, Grantz Saskova K, The yeast proteases Ddi1 and Wss1 are both involved in the DNA replication stress response, DNA Repair (Amst) 80 (2019) 45–51. [DOI] [PubMed] [Google Scholar]
- [157].Yip MCJ, Bodnar NO, Rapoport TA, Ddi1 is a ubiquitin-dependent protease, Proc. Natl. Acad. Sci. U. S. A 117 (2020) 7776–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kojima Y, Machida Y, Palani S, Caulfield TR, Radisky ES, Kaufmann SH, Machida YJ, FAM111A protects replication forks from protein obstacles via its trypsin-like domain, Nat. Commun 11 (2020) 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Yang S-W, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA, A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases, Proc. Natl. Acad. Sci. U. S. A 93 (1996) 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Gao R, Schellenberg MJ, Huang SY, Abdelmalak M, Marchand C, Nitiss KC, Nitiss JL, Williams RS, Pommier Y, Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2-DNA and -RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2), J. Biol. Chem 289 (2014) 17960–17969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Schellenberg MJ, Appel CD, Riccio AA, Butler LR, Krahn JM, Liebermann JA, Cortes-Ledesma F, Williams RS, Ubiquitin stimulated reversal of topoisomerase 2 DNA-protein crosslinks by TDP2, Nucleic Acids Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Rao T, Gao R, Takada S, Al Abo M, Chen X, Walters KJ, Pommier Y, Aihara H, Novel TDP2-ubiquitin interactions and their importance for the repair of topoisomerase II-mediated DNA damage, Nucleic Acids Res 44 (2016) 10201–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Xia D, Tang WK, Ye Y, Structure and function of the AAA+ ATPase p97/Cdc48p, Gene 583 (2016) 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].van den Boom J, Meyer H, VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling, Mol. Cell 69 (2018) 182–194. [DOI] [PubMed] [Google Scholar]
- [165].Meyer H, Bug M, Bremer S, Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system, Nat. Cell Biol 14 (2012) 117–123. [DOI] [PubMed] [Google Scholar]
- [166].Franz A, Ackermann L, Hoppe T, Ring of change: CDC48/p97 drives protein dynamics at Chromatin, Front. Genet 7 (2016) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Hanzelmann P, Schindelin H, The interplay of cofactor interactions and post-translational modifications in the regulation of the AAA+ ATPase p97, Front. Mol. Biosci 4 (2017) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Fielden J, Wiseman K, Torrecilla I, Li S, Hume S, Chiang SC, Ruggiano A, Narayan Singh A, Freire R, Hassanieh S, Domingo E, Vendrell I, Fischer R, Kessler BM, Maughan TS, El-Khamisy SF, Ramadan K, TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts, Nat. Commun 11 (2020) 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Shi W, Ding R, Zhou PP, Fang Y, Wan R, Chen Y, Jin J, Coordinated actions between p97 and Cullin-RING ubiquitin ligases for protein degradation, Adv. Exp. Med. Biol 1217 (2020) 61–78. [DOI] [PubMed] [Google Scholar]
- [170].Interthal H, Pouliot JJ, Champoux JJ, The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Pouliot JJ, Yao KC, Robertson CA, Nash HA, Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes, Science 286 (1999) 552–555. [DOI] [PubMed] [Google Scholar]
- [172].Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y, Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells, J. Biol. Chem 287 (2012) 12848–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Zeng Z, Sharma A, Ju L, Murai J, Umans L, Vermeire L, Pommier Y, Takeda S, Huylebroeck D, Caldecott KW, El-Khamisy SF, TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1, Nucleic Acids Res 40 (2012) 8371–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Kawale AS, Povirk LF, Tyrosyl-DNA phosphodiesterases: rescuing the genome from the risks of relaxation, Nucleic Acids Res 46 (2018) 520–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y, Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions, DNA Repair (Amst) 2 (2003) 1087–1100. [DOI] [PubMed] [Google Scholar]
- [176].Debethune L, Kohlhagen G, Grandas A, Pommier Y, Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase, Nucleic Acids Res 30 (2002) 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Interthal H, Chen HJ, Champoux JJ, Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages, J. Biol. Chem 280 (2005) 36518–36528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Takahata C, Masuda Y, Takedachi A, Tanaka K, Iwai S, Kuraoka I, Repair synthesis step involving ERCC1-XPF participates in DNA repair of the Top1-DNA damage complex, Carcinogenesis 36 (2015) 841–851. [DOI] [PubMed] [Google Scholar]
- [179].Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y, Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells, Nucleic Acids Res 39 (2011) 3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Nitiss KC, Malik M, He X, White SW, Nitiss JL, Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 8953–8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Tsuda M, Kitamasu K, Hosokawa S, Nakano T, Ide H, Repair of trapped topoisomerase II covalent cleavage complexes: novel proteasome-independent mechanisms, Nucleosides Nucleotides Nucleic Acids 39 (2020) 170–184. [DOI] [PubMed] [Google Scholar]
- [182].Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW, A human 5’-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage, Nature 461 (2009) 674–678. [DOI] [PubMed] [Google Scholar]
- [183].Gao R, Schellenberg MJ, Huang SY, Abdelmalak M, Marchand C, Nitiss KC, Nitiss JL, Williams RS, Pommier Y, Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2.DNA and Top2.RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2), J. Biol. Chem 289 (2014) 17960–17969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsutsui K, Takeda S, Sasanuma H, Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes, Mol. Cell 64 (2016) 1010. [DOI] [PubMed] [Google Scholar]
- [185].Hartsuiker E, Neale MJ, Carr AM, Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA, Mol. Cell 33 (2009) 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Deshpande RA, Lee JH, Arora S, Paull TT, Nbs1 converts the human Mre11/Rad50 nuclease complex into an Endo/Exonuclease machine specific for Protein-DNA adducts, Mol. Cell 64 (2016) 593–606. [DOI] [PubMed] [Google Scholar]
- [187].Keeney S, Spo11 and the formation of DNA double-strand breaks in meiosis, Genome Dyn. Stab 2 (2008) 81–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Paiano J, Wu W, Yamada S, Sciascia N, Callen E, Paola Cotrim A, Deshpande RA, Maman Y, Day A, Paull TT, Nussenzweig A, ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis, Nat. Commun 11 (2020) 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Deng C, Brown JA, You D, Brown JM, Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae, Genetics 170 (2005) 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsutsui K, Takeda S, Sasanuma H, Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes, Mol. Cell 64 (2016) 580–592. [DOI] [PubMed] [Google Scholar]
- [191].Gomez-Herreros F, Romero-Granados R, Zeng Z, Alvarez-Quilon A, Quintero C, Ju L, Umans L, Vermeire L, Huylebroeck D, Caldecott KW, Cortes-Ledesma F, TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo, PLoS Genet 9 (2013) e1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Akagawa R, Trinh HT, Saha LK, Tsuda M, Hirota K, Yamada S, Shibata A, Kanemaki MT, Nakada S, Takeda S, Sasanuma H, UBC13-mediated ubiquitin signaling promotes removal of blocking adducts from DNA double-strand breaks, iScience 23 (2020) 101027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Pommier Y, Leo E, Zhang H, Marchand C, DNA topoisomerases and their poisoning by anticancer and antibacterial drugs, Chem. Biol 17 (2010) 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Pommier Y, Marchand C, Interfacial inhibitors: targeting macromolecular complexes, Nat. Rev. Drug Discov 11 (2012) 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Pommier Y, Drugging topoisomerases: lessons and challenges, ACS Chem. Biol 8 (2013) 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Nitiss JL, Targeting DNA topoisomerase II in cancer chemotherapy, Nat. Rev. Cancer 9 (2009) 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].van Waardenburg RC, de Jong LA, van Eijndhoven MA, Verseyden C, Pluim D, Jansen LE, Bjornsti MA, Schellens JH, Platinated DNA adducts enhance poisoning of DNA topoisomerase I by camptothecin, J. Biol. Chem 279 (2004) 54502–54509. [DOI] [PubMed] [Google Scholar]
- [198].Sekiguchi J, Shuman S, Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I, Mol. Cell 1 (1997) 89–97. [DOI] [PubMed] [Google Scholar]
- [199].Wang Y, Knudsen BR, Bjergbaek L, Westergaard O, Andersen AH, Stimulated activity of human topoisomerases IIalpha and IIbeta on RNA-containing substrates, J. Biol. Chem 274 (1999) 22839–22846. [DOI] [PubMed] [Google Scholar]
- [200].Wang Y, Thyssen A, Westergaard O, Andersen AH, Position-specific effect of ribonucleotides on the cleavage activity of human topoisomerase II, Nucleic Acids Res 28 (2000) 4815–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Liao ZY, Sordet O, Zhang HL, Kohlhagen G, Antony S, Gmeiner WH, Pommier Y, A novel polypyrimidine antitumor agent FdUMP[10] induces thymineless death with topoisomerase I-DNA complexes, Cancer Res 65 (2005) 4844–4851. [DOI] [PubMed] [Google Scholar]
- [202].Pourquier P, Gioffre C, Kohlhagen G, Urasaki Y, Goldwasser F, Hertel LW, Yu S, Pon RT, Gmeiner WH, Pommier Y, Gemcitabine (2’,2’-difluoro-2’-deoxycytidine), an antimetabolite that poisons topoisomerase I, Clin. Cancer Res 8 (2002) 2499–2504. [PubMed] [Google Scholar]
- [203].Pourquier P, Takebayashi Y, Urasaki Y, Gioffre C, Kohlhagen G, Pommier Y, Induction of topoisomerase I cleavage complexes by 1-b-D-arabinofur-anosylcytosine (Ara-C) in vitro and in ara-C-treated cells, Proc. Natl. Acad. Sci. U. S. A 97 (2000) 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Jin S, Kim JS, Sim SP, Liu A, Pilch DS, Liu LF, LaVoie EJ, Heterocyclic bibenzimidazole derivatives as topoisomerase I inhibitors, Bioorg. Med. Chem. Lett 10 (2000) 719–723. [DOI] [PubMed] [Google Scholar]
- [205].Sim SP, Pilch DS, Liu LF, Site-specific topoisomerase I-mediated DNA cleavage induced by nogalamycin: a potential role of ligand-induced DNA bending at a distal site, Biochemistry 39 (2000) 9928–9934. [DOI] [PubMed] [Google Scholar]
- [206].Chen AY, Yu C, Gatto B, Liu LF, DNA minor groove-binding ligands: a different class of mammalian topoisomerase I inhibitors, Proc. Natl. Acad. Sci. U. S. A 90 (1993) 8131–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Chen AY, Yu C, Bodley A, Peng LF, Liu LF, A new mammalian DNA topoisomerase I poison Hoechst 33342: cytotoxicity and drug resistance in human cell cultures, Cancer Res 53 (1993) 1332–1337. [PubMed] [Google Scholar]
- [208].Trask DK, Muller MT, Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D, Proc. Natl. Acad. Sci. U. S. A 85 (1988) 1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Wassermann K, Markovits J, Jaxel C, Capranico G, Kohn KW, Pommier Y, Effects of morpholinyl doxorubicins, doxorubicin, and actinomycin D on mammalian DNA topoisomerases I and II, Mol. Pharmacol 38 (1990) 38–45. [PubMed] [Google Scholar]
- [210].Pourquier P, Bjornsti MA, Pommier Y, Induction of topoisomerase I cleavage complexes by the vinyl chloride adduct 1,N6-ethenoadenine, J. Biol. Chem 273 (1998) 27245–27249. [DOI] [PubMed] [Google Scholar]
- [211].Pommier Y, Laco GS, Kohlhagen G, Sayer JM, Kroth H, Jerina DM, Position-specific trapping of topoisomerase I-DNA cleavage complexes by intercalated benzo[a]- pyrene diol epoxide adducts at the 6-amino group of adenine, Proc. Natl. Acad. Sci. U. S. A 97 (2000) 10739–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Pommier Y, Kohlhagen G, Laco GS, Kroth H, Sayer JM, Jerina DM, Different effects on human topoisomerase I by minor groove and intercalated deoxyguanosine adducts derived from two polycyclic aromatic hydrocarbon diol epoxides at or near a normal cleavage site, J. Biol. Chem 277 (2002) 13666–13672. [DOI] [PubMed] [Google Scholar]
- [213].Pourquier P, Waltman JL, Urasaki Y, Loktionova NA, Pegg AE, Nitiss JL, Pommier Y, Topoisomerase I-mediated cytotoxicity of N-methyl-N’-nitro-N- nitrosoguanidine: trapping of topoisomerase I by the O6-methylguanine, Cancer Res 61 (2001) 53–58. [PubMed] [Google Scholar]
- [214].Antony S, Theruvathu JA, Brooks PJ, Lesher DT, Redinbo M, Pommier Y, Enhancement of camptothecin-induced topoisomerase I cleavage complexes by the acetaldehyde adduct N2-ethyl-2’-deoxyguanosine, Nucleic Acids Res 32 (2004) 5685–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Dexheimer TS, Kozekova A, Rizzo CJ, Stone MP, Pommier Y, The modulation of topoisomerase I-mediated DNA cleavage and the induction of DNA-topoisomerase I crosslinks by crotonaldehyde-derived DNA adducts, Nucleic Acids Res 36 (2008) 4128–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Khan QA, Kohlhagen G, Marshall R, Austin CA, Kalena GP, Kroth H, Sayer JM, Jerina DM, Pommier Y, Position-specific trapping of topoisomerase II by benzo[a]pyrene diol epoxide adducts: implications for interactions with intercalating anticancer agents, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 12498–12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Velez-Cruz R, Riggins JN, Daniels JS, Cai H, Guengerich FP, Marnett LJ, Osheroff N, Exocyclic DNA lesions stimulate DNA cleavage mediated by human topoisomerase IIalpha in vitro and in cultured cells, Biochemistry 44 (2005) 3972–3981. [DOI] [PubMed] [Google Scholar]
- [218].Deweese JE, Osheroff N, The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing, Nucleic Acids Res 37 (2009) 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Sabourin M, Osheroff N, Sensitivity of human type II topoisomerases to DNA damage: stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions, Nucleic Acids Res 28 (2000) 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Trabectedin: ET 743, ecteinascidin 743., Yondelis, Drugs R D 4 (2003) 75–81. [DOI] [PubMed] [Google Scholar]
- [221].Austin CA, Patel S, Ono K, Nakane H, Fisher LM, Site-specific DNA cleavage by mammalian DNA topoisomerase II induced by novel flavone and catechin derivatives, Biochem. J 282 (1992) 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Lanza A, Tornatelli S, Rodolfo C, Scanavini MC, Pedrini AM, Human DNA topoisomerase I-mediated cleavages stimulated by ultraviolet light-induced DNA damage, J. Biol. Chem 271 (1996) 6978–6986. [DOI] [PubMed] [Google Scholar]
- [223].Subramanian D, Rosenstein BS, Muller MT, Ultraviolet-induced DNA damage stimulates topoisomerase I-DNA complex formation in vivo: possible relationship with DNA repair, Cancer Res 58 (1998) 976–984. [PubMed] [Google Scholar]
- [224].Corbett AH, Zechiedrich EL, Lloyd RS, Osheroff N, Inhibition of eukaryotic topoisomerase II by ultraviolet-induced cyclobutane pyrimidine dimers, J. Biol. Chem 266 (1991) 19666–19671. [PubMed] [Google Scholar]
- [225].Pourquier P, Ueng LM, Kohlhagen G, Mazumder A, Gupta M, Kohn KW, Pommier Y, Effects of uracil incorporation, DNA mismatches, and abasic sites on cleavage and religation activities of mammalian topoisomerase I, J. Biol. Chem 272 (1997) 7792–7796. [DOI] [PubMed] [Google Scholar]
- [226].Pourquier P, Ueng L-M, Fertala J, Wang D, Park H-J, Essigman JM, Bjornsti M-A, Pommier Y, Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages, J. Biol. Chem 274 (1999) 8516–8523. [DOI] [PubMed] [Google Scholar]
- [227].Lesher DT, Pommier Y, Stewart L, Redinbo MR, 8-Oxoguanine rearranges the active site of human topoisomerase I, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 12102–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Kingma PS, Corbett AH, Burcham PC, Marnett LJ, Osheroff N, Abasic sites stimulate double-stranded DNA cleavage mediated by topoisomerase II, J. Biol. Chem 270 (1995) 21441–21444. [DOI] [PubMed] [Google Scholar]
- [229].Kingma PS, Osheroff N, Topoisomerase II-mediated DNA cleavage and religation in the absence of base pairing. Abasic lesions as a tool to dissect enzyme mechanism, J. Biol. Chem 273 (1998) 17999–18002. [DOI] [PubMed] [Google Scholar]
- [230].Kingma PS, Osheroff N, Apurinic sites are position-specific topoisomerase II poisons, J. Biol. Chem 272 (1997) 1148–1155. [DOI] [PubMed] [Google Scholar]
- [231].Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ, TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo, EMBO J 26 (2007) 4720–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Gomez-Herreros F, Zagnoli-Vieira G, Ntai I, Martinez-Macias MI, Anderson RM, Herrero-Ruiz A, Caldecott KW, TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription, Nat. Commun 8 (2017) 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Zagnoli-Vieira G, Bruni F, Thompson K, He L, Walker S, de Brouwer APM, Taylor R, Niyazov D, Caldecott KW, Confirming TDP2 mutation in spinocerebellar ataxia autosomal recessive 23 (SCAR23), Neurol. Genet 4 (2018) e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsusui K, Takeda S, Sasanuma H, Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes, Mol. Cell 64 (2016) 580–592. [DOI] [PubMed] [Google Scholar]
- [235].Sordet O, Larochelle S, Nicolas E, Stevens EV, Zhang C, Shokat KM, Fisher RP, Pommier Y, Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription- and BRCA1-dependent degradation of topoisomerase I, J. Mol. Biol 381 (2008) 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Wei Y, Diao LX, Lu S, Wang HT, Suo F, Dong MQ, Du LL, SUMO-targeted DNA translocase Rrp2 protects the genome from Top2-Induced DNA damage, Mol. Cell 66 (2017) 581–596 e586. [DOI] [PubMed] [Google Scholar]