

ABSTRACT
Physiological ageing and tumorigenesis are both associated with epigenomic alterations in human tissue cells, the most extensively investigated of which entails de novo cytosine methylation (i.e., hypermethylation) within the CpG dinucleotides of CpG islands. Genomic regions that become hypermethylated during tumorigenesis are generally believed to overlap regions that acquire methylation in normal tissues as an effect of ageing. To define the extension of this overlap, we analysed the DNA methylomes of 48 large-bowel tissue samples taken from women of different ages during screening colonoscopy: 18 paired samples of normal and lesional tissues from donors harbouring a precancerous lesion and 12 samples of normal mucosa from tumour-free donors. Each sample was subjected to targeted, genome-wide bisulphite sequencing of ~2.5% of the genome, including all CpG islands. In terms of both its magnitude and extension along the chromatin, tumour-associated DNA hypermethylation in these regions was much more conspicuous than that observed in the normal mucosal samples from older (vs. younger) tumour-free donors. 83% of the ageing-associated hypermethylated regions (n = 2501) coincided with hypermethylated regions observed in tumour samples. However, 86% of the regions displaying hypermethylation in precancerous lesions (n = 16,772) showed no methylation changes in the ageing normal mucosa. The tumour-specificity of this latter hypermethylation was validated using published sets of data on DNA methylation in normal and neoplastic colon tissues. This extensive set of genomic regions displaying tumour-specific hypermethylation represents a rich vein of putative biomarkers for the early, non-invasive detection of colorectal tumours in women of all ages.
KEYWORDS: DNA methylation, differentially methylated region, normal colorectal mucosa, sessile serrated lesion, colorectal adenoma, colorectal cancer, ageing
Introduction
Colorectal cancer (CRC) is one of the most frequently diagnosed malignancies throughout the world and it continues to be a major cause of cancer-related mortality (https://gco.iarc.fr/)[1]. And yet, owing to its slow progress, it is also a singularly preventable disease. Although symptoms are generally absent until the disease has reached an advanced stage, the onset of invasive disease is preceded by 5–15 years in which benign CRC precursor lesions can be detected in the colon via colonoscopy and, more recently, via stool or blood assays for biomarkers of colorectal tumours [2–7]. Molecular markers used for this purpose include specific genetic mutations (e.g., those affecting KRAS or BRAF) as well as DNA methylation alterations associated with colorectal neoplasia [8–10]. Like most human cancers, CRCs consistently exhibit diffuse loss of DNA methylation in genomic regions that are methylated in normal colon mucosa. This hypomethylation is accompanied by de novo methylation of genomic regions that are generally unmethylated in normal tissues. Hypermethylation is a more topographically restricted phenomenon that mainly affects CpG islands, including those located in gene promoters. In a few cases, this hypermethylation is associated with loss of expression of the affected gene, an important consideration if that gene has tumour-suppressor properties.
CRC-associated alterations in the DNA methylome can also be detected (albeit in a milder, less extensive form) in the precancerous lesions that give rise to these cancers, i.e., conventional adenomas (cADNs) in most cases but also sessile serrated lesions (SSLs, previously termed SSA/Ps) that develop along the so-called serrated pathway of colorectal tumorigenesis. For example, a subset of sporadic CRCs found in the proximal colon are characterized by the typical gain-of-function BRAF V600E mutation, a conspicuous excess of cytosine methylation involving CpG islands (the so-called ‘CpG island methylator phenotype’ [CIMP] [11–14]), and in many cases hypermethylation-mediated silencing of the DNA mismatch repair gene MLH1 [15,16]. The SSLs that give rise to these cancers already harbour the BRAFV600E mutation as well as an obviously altered DNA methylome [17,18]. The latter, which we refer to as proto-CIMP, is characterized by striking CpG-island hypermethylation that clearly exceeds that observed in cADNs but is less conspicuous (in both the magnitude of the alteration and its extension along the chromatin) than that seen in CIMP(+) CRCs [19]. The transition from SSL to cancer is usually (but not always) caused by the epigenetic silencing of the MLH1 gene. This transition is believed to occur over a relatively short period of time (owing to the rapid accumulation of sources of genetic instability caused by the DNA mismatch repair defect), and it occurs more frequently in women [13,20,21].
Small but significant increases in cytosine methylation within the CpG island encompassing the MLH1 promoter have also been reported by our group in the normal proximal-colon mucosa of elderly women [22]. This finding suggests that, in the cells of an SSL arising in such tissue, the MLH1 promoter might be predisposed to CIMP-dependent silencing, since genomic loci displaying DNA methylation in sporadic cells of healthy ageing tissues tend also to be methylated in tumours arising from these tissues. This phenomenon has been reported also for other genes such as ER, IGF2, N33 and MYOD [23–25].
Normal tissue ageing is accompanied by progressive changes in the CpG-site methylation [26,27]. In several tissues, including the large intestine, differences in this methylome ageing process have been linked to both sex and anatomical site [28–31]. In general, however, loss of methylation is more frequent in lamin-associated domains, which contain repetitive sequences and are gene-poor, whereas hypermethylation mainly affects CpG islands that are typically unmethylated, particularly those located in Polycomb Repressive Complex 2 (PRC2)-targeted genomic regions [26,32–39]. This ‘epigenetic drift’ occurs in human tissues with diverse proliferation profiles (e.g., the small and large intestines, bone marrow, blood, spleen, brain) [27,31,36,40], and it appears to be conserved across species [41–44]. It appears to be unrelated to cellular senescence, proliferation and terminal differentiation, or telomere maintenance and DNA damage signalling. Instead, it has been linked to energy metabolism, the mTOR signalling pathway and hormonal homoeostasis, and it may be subject to modulation by lifestyle interventions [45–51].
Sets of methylation changes (increases as well as decreases) at certain CpG sites have been identified as ‘DNA methylation clocks’ capable of predicting the biological (as opposed to chronological) age of a tissue [28,35,40,52]. Deviations from chronological age include decelerations (associated with a longer, disease-free life expectancy for the tissue) and accelerations (linked to more frequent and/or earlier onset of disease, including cancer) [26,38,45,46,51,53–55]. The lifetime cancer risk for normal tissues correlates positively with their average CpG island methylation values [39]. Consequently, whereas the proliferation rates in the intestinal epithelia of both the small and large bowel is among the highest reported in human tissues, epigenetic ageing rates are higher in the colon [56], where the cancer incidence substantially exceeds that of the small intestine.
Studies conducted in the last two decades to characterize ageing- and/or tumour-associated changes in DNA methylation have generally relied on Illumina arrays that interrogated the methylation status of ~27,000 or ~450,000 of the ~28 million CpG sites of the human genome. While these tools allow reliable determination of the methylation status at specific CpG sites, they provide no details on the extension of such epigenetic change. CpG islands and gene promoters, for example, contain far more CpG sites than those covered by the arrays cited above. Not surprisingly then, few of the CpG sites they captured proved to be hyper- or hypomethylated in ageing normal tissues [27,29,31,36,50,57,58]. And if we consider that methylation changes occur in stretches of neighbouring CpG sites [59,60], it is clear that the array-based findings underestimated the number of CpG sites whose methylation status changes with age.
In this study, we explored human colorectal-cell methylome changes in colonoscopically collected samples of precancerous lesions and normal mucosa from women of different ages. A genome-wide bisulphite sequencing approach was used to assess CpG methylation changes in an area comprising ~2.5% of the genome and characterized by enrichment for CpG islands and shores as well as gene promoters – areas in which hypermethylation is known to occur during tumorigenesis and/or ageing. Our objective was to topographically disentangle ageing- and tumorigenesis-related forms of hypermethylation – more specifically, to identify genomic regions in which aberrant methylation occurs exclusively within the context of colorectal tumorigenesis and not as a result of normal colorectal mucosal ageing. These regions of tumorigenesis-specific hypermethylation should be highly promising candidates for the development of biomarkers for the early detection of colorectal tumours in patients of all ages.
Methods
Fresh colon tissues were prospectively collected during screening colonoscopies performed at Cremona Hospital (Italy) or Zurich Triemli Hospital (Switzerland). To eliminate the possibility of confounding effects related to the sex-related variability in methylome ageing mentioned above [28–31], we restricted our analysis to tissues collected from female screenees. The study protocol was approved by both hospitals’ research ethics committees, and donors provided written consent to tissue collection, testing, and data publication. The samples were numerically coded to protect donors’ rights to confidentiality and privacy.
The characteristics of the 48 tissue samples analysed are summarized in Table 1. Normal mucosal biopsies (NM) from the proximal and distal colon were taken from six women with no neoplastic or inflammatory alterations in any area of the gut (three who were <40 years old [younger tumour-free donors or TF-Y] and three other who were >70 [older tumour-free donors or TF-O]). The remaining 18 donors were women whose colonoscopy revealed a precancerous tumour (cADN or SSL) in the proximal colon but no evidence of cancer. In each case, paired samples were taken from the tumour surface (8 cADNs and 10 SSLs) and the normal mucosa located in the same colon segment but >2 cm from the tumour (8 cADN-paired NM and 10 SSL-paired NM). All 18 tumours displayed Paris class Is or IIa morphologic features (i.e., sessile polyps and nonpolypoid lesions that were slightly elevated above the surrounding mucosa) [61], and had maximum diameters of ≥10 mm (to ensure that sufficient tissue was left for the histologic examination). Each tissue sample was promptly placed in a tube filled with AllProtect Tissue Reagent (Qiagen, Hilden, Germany), held at 4°C overnight, and stored at −80°C prior to the DNA extraction step done with Qiagen’s AllPrep Mini Kit.
Table 1.
Characteristics of the 48 tissues analysed in the study
| Donor | Age (y) | Tissue(s) sampled | Colon segment involved | Maximum lesion diameter (mm) | Paris classification # | Pit pattern † | Microscopic appearance | Dysplasia ∆ | BRAF sequence | KRAS sequence | MLH1 staining (IHC) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumour-free | |||||||||||
| Y1 | 33 | NM | CEC & SIG | – | – | – | – | – | – | – | – |
| Y2 | 37 | NM | CEC & SIG | – | – | – | – | – | – | – | – |
| Y3 | 38 | NM | CEC & SIG | – | – | – | – | – | – | – | – |
| O1 | 71 | NM | CEC & SIG | – | – | – | – | – | – | – | – |
| O2 | 78 | NM | CEC & SIG | – | – | – | – | – | – | – | – |
| O3 | 80 | NM | CEC & SIG | – | – | – | – | – | – | – | – |
| Tumour-bearing | |||||||||||
| S1 | 88 | T+ NM | CEC | 15 | 0-IIa | IIIs | SSL | none | c.1799 T > A (V600E) | WT | positive |
| S2 | 68 | T+ NM | ASC | 12 | 0-Is | IIIs, IIIL | SSL | none | c.1799 T > A (V600E) | WT | positive |
| S3 | 61 | T+ NM | ASC | 20 | 0-IIa | II | SSL | dysplasia | c.1799 T > A (V600E) | WT | positive |
| S4 | 71 | T+ NM | ASC | 15 | 0-IIa+IIc | IV, V | SSL | dysplasia | c.1799 T > A (V600E) | WT | positive |
| S6 | 76 | T+ NM | CEC | 25 | 0-IIa | IV | SSL | dysplasia | c.1799 T > A (V600E) | WT | positive |
| S7 | 59 | T+ NM | ASC | 14 | 0-IIa | nr | SSL | dysplasia | c.1799 T > A (V600E) | WT | positive |
| S9 | 69 | T+ NM | ASC | 20 | 0-IIa | II | SSL | none | c.1799 T > A (V600E) | WT | positive |
| S10 | 45 | T+ NM | ASC | 25 | 0-Is | Is | SSL | none | c.1799 T > A (V600E) | WT | positive |
| S15 | 36 | T+ NM | ASC | 15 | 0-IIa | nr | SSL | none | c.1799 T > A (V600E) | WT | positive |
| S17 | 53 | T+ NM | CEC | 15 | 0-IIa | IIIs | SSL | none | c.1799 T > A (V600E) | WT | positive |
| A3 | 78 | T+ NM | ASC | 15 | 0-Is | IV | cADN (TA) | LGD | WT | WT | positive |
| A4 | 64 | T+ NM | ASC | 30 | 0-IIa+Is | IV | cADN (TA) | LGD | WT | c.34 G > T (G12C) | positive |
| A5 | 59 | T+ NM | ASC | 25 | 0-IIa | IIIL | cADN (TA) | LGD | WT | WT | positive |
| A6 | 73 | T+ NM | ASC | 9 | 0-Is | IIIs | cADN (TA) | LGD | WT | WT | positive |
| A7 | 64 | T+ NM | CEC | 20 | 0-Is | IIIL | cADN (TA) | LGD | WT | WT | positive |
| A9 | 73 | T+ NM | ASC | 30 | 0-IIa | IIIL | cADN (TA) | LGD | WT | c.35 G > T (G12V) | positive |
| A11 | 84 | T+ NM | ASC | 40 | 0-IIa | IIIL, IV | cADN (TA) | LGD | WT | c.35 G > T (G12V) | positive |
| A14 | 78 | T+ NM | ASC | 15 | 0-Is | IIIs | cADN (TA) | LGD | WT | WT | positive |
Abbreviations: NM, normal mucosa; T, tumour; CEC, caecum; ASC, ascending; SIG, sigmoid; cADN, conventional adenoma; TA, tubular adenoma; SSL, sessile serrated lesion; LGD, low-grade dysplasia; IHC, immunohistochemistry; nr: not reported.
# Macroscopic appearance of neoplastic lesions was classified according to Paris Endoscopic Classification. The Paris Endoscopic Classification of Superficial Neoplastic Lesions. Gastrointest. Endosc. 2003; 58 (suppl.): S3-S27.
† Morphological analysis of colon crypt patterns according to the Kudo classification. Kudo S et al. Pit pattern in colorectal neoplasia: endoscopic magnifying view. Endoscopy 2001; 33: 367–73.
∆ Low-grade versus high-grade dysplasia as defined by the WHO classification of tumours of the digestive system, editorial and consensus conference in Lyon, France, November 6–9, 1999. IARC.
Genome-wide bisulphite DNA sequencing
DNA extracted from the tissues was analyzed for cytosine methylation using an enrichment protocol that captured CpG-rich genomic loci from all chromosomes, thereby allowing us to analyze an ~80-Mb segment of the genome containing ~2.7 x 106 CpG sites. To this end, we used a pre-designed pool of probes that hybridized to each locus presenting with any patterns of methylation in its CpG sites.
Pre-capture libraries were created according to Kapa Biosystems’ “adapter-ligation-first” protocol, which was provided with the reagents for the SeqCapEpi CpGiant system (Roche, Rotkreutz, Switzerland). Sample DNA (1 µg) and a bisulfite-conversion control consisting of the lambda phage genome were placed in a Covaris microTUBE AFA snap-cap vial (Covaris, Brighton, UK). The DNA sample was sheared for 5 minutes using a Covaris S2 sonicator (duty factor: 10%; peak incident power: 175 watts; cycles per burst: 200) to obtain fragments with an average size of 180 to 220 bp. A 1-µl aliquot of each sample was assessed on an Agilent 2200 TapeStation with D1000 ScreenTape and reagents to verify successful sonication (Agilent, Basel, Switzerland). End repair, A-tailing, ligation of indexed methylated adapters, and dual-Solid Phase Reversible Immobilization size selection were carried out according to the SeqCapEpi CpGiant protocol.
The libraries were bisulfite-converted using the Zymo Research EZ DNA Methylation-Lightning Kit (8 minutes at 98°C, 60 minutes at 54°C). The whole elute from the spin column (22 µl) was then PCR-amplified (2 minutes at 95°C, with 12 cycles of 30 seconds at 98°C, 30 seconds at 60°C, 4 minutes at 72°C and then 10 minutes at 72°C, and held at 4°C) in 30 µl of HiFi HotStart Uracil Ready Mix and Pre-PCR Oligos (Kapa Biosystems). PCR products were cleaned up with AMPure beads and libraries were analyzed on the Agilent 2200 TapeStation (as above).
For each sample, the SeqCapEpi hybridization reaction contained the amplified DNA library, universal and indexed blocking oligos, and the bisulfite capture enhancer. After hybridization (~70 hours), bead-capture, and serial washes, the whole bead-bound DNA libraries were amplified (45 seconds at 98°C, with 16 cycles of 15 seconds at 98°C, 30 seconds at 60°C, 30 seconds at 72°C and then 1 minute at 72°C, and held at 4°C) and sequenced on an Illumina HiSeq2500 instrument (Illumina, San Diego, CA) together with a 10% spike-in of a non-indexed PhiX library (Illumina, San Diego, CA). For each sample, we generated a median of 50 million 125-bp paired-end reads with 30X coverage.
After quality control and coverage filtering, this protocol was used to analyze ~2.4 million CpG sites (2,407,522) across all sequenced samples. Forthy-four percent of these sites were located in CpG islands, 19% were in CpG shores, and 6% were in CpG shelves. The remaining 31% were located outside the above three regions.
Preprocessing of bisulphite sequencing data
Sequencing reads were trimmed with Trim Galore version 0.6.4 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Low-quality reads (default Phred score: 20) were discarded, and five base pairs were removed from both the forward and reverse reads from the 3′ and 5′ orientations. Reads were aligned with the GRCh37/hg19 human reference genome using Bismark, version v0.22.1 [62]. CpG-site methylation calls were generated with the Bismark methylation_extractor to obtain a file containing the total coverage and the number of methylated reads at each CpG site on each strand. The preprocessing steps were implemented as a Snakemake workflow [63], which is available at https://github.com/sorjuela/age_lesions_females. The R package bsseq, version 1.24.1 [64] was used to import the Bismark outputs into R. Sequencing reads from chromosomes X and Y were excluded. CpG sites with fewer than 10 reads in 80% of the samples were removed.
A multidimensional scaling (MDS) of the distances between the samples was made using the cmdscale function from the stats R package (R Core Team, 2020. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/). The distances were calculated using the arcsine-transformed methylation proportions. The Euclidean distance over the top 10,000 most variable CpG sites was used to summarize the difference between any two of the 48 tissue samples. The rgl R package (Adler D. et al., 2020. rgl: 3D Visualization Using OpenGL. R package version 0.100.54. URL https://CRAN.R-project.org/package=rgl) was used to generate a three-dimensional MDS plot.
Detecting tumorigenesis-, ageing-, and colon segment-associated differential methylation
To identify differentially methylated regions (DMRs) in precancerous colorectal lesions that were tumorigenesis-specific (cADNs, SSLs, or both), we used a two-step approach. First, the R package dmrseq version 1.8.0 [65] was used to analyze the five tissue subset pairs listed in Table 2 (i.e., comparisons i, ii, iii, iv, and v). (The following dmrseq parameters were specified: maxPerms = 20, maxGap = 100, maxGapSmooth = 1000, minNumRegion = 3.) Next, from the DMRs identified as tumorigenesis-associated in the first step (Table 2), we selected those that were tumorigenesis-specific. The initial list consisted on all DMRs identified in comparisons i, ii, and/or iii that displayed no overlap with any of the DMRs identified in comparisons iv and/or v. This list was then filtered further to eliminate those DMRs characterized by a mean methylation level of >0.1 in normal mucosa AND a methylation proportions change (as defined in dmrseq, i.e., the sum of the methylated reads from all samples representing one condition divided by the sum of all reads from all samples representing the same condition) of <0.8 in comparisons i, ii, and iii.
Table 2.
Colorectal tissue-group comparisons used to identify DMRs associated with colorectal tumorigenesis, normal mucosal aging, and anatomic location of normal mucosa
| Comparisons | Tissue samples compared |
Adjustments for: |
DMR associations c |
||
|---|---|---|---|---|---|
| i | SSLs and cADNs (n=18) |
vs. | SSL- and cADN -paired NM (n=18) |
--- | Colorectal tumorigenesis |
| ii | SSLs (n=10) |
vs. | SSL-paired NM (n=10) |
--- | SSL tumorigenesis |
| iii | cADNs (n=8) |
vs. | cADN-paired NM (n=8) |
--- | cADN tumorigenesis |
| iv | TF-O NM samplesa (n=6) |
vs. | TF-Y NM samplesb (n=6) |
Anatomic location of NM | NM aging |
| v | TF NMCEC samples (n=6) |
vs. | TF NMSIG samples (n=6) |
Age group | Anatomic location of NM |
Abbreviations: cADNs, conventional adenomas; DMR, differentially methylated regions; NM, normal mucosa; NMCEC, NM from the cecum; NMSIG, NM from the sigmoid colon; SSLs, sessile serrated lesions; TF, tumor-free; TF-O, older TF donors (>70 y): TF-Y, younger TF donors (<40 y).
aNMCEC and NMSIG samples from the 3 TF-O donors
bNMCEC and NMSIG samples from the 3 TF-Y donors
cTumorigenesis-specific DMRs were those identified in comparisons i, ii, and/or iii but not in comparisons iv or v
Data were explored and analyzed with the R packages GenomicRanges version 1.40.0 [66], SummarizedExperiment version 1.18.2 (Morgan M, Obenchain V, Hester J, Pagès H., 2020; SummarizedExperiment: SummarizedExperiment container, R package version 1.18.2.) and plyranges version 1.8.0 [67]. Plots were generated using the R packages ggplot2 version 3.3.0 [68], GGally version 1.5.0 (Schloerke B, Crowley J, Cook D, Briatte F, Marbach M, Thoen E, et al., 2020. GGally: Extension to ggplot2. https://CRAN.R-project.org/package=GGally) and ComplexHeatmap version 2.4.2 [69]. Heatmaps were drawn by averaging, for each sample, the methylation values of CpG sites overlapping CpG islands or gene promoters (located with the annotation package annotatr version 1.14.0 [70]). UpSet plot DMR counts were calculated using an interval tree algorithm implemented in the ChIPpeakAnno package version 3.22.2 [71].
Preliminary validation of tumorigenesis-specific hypermethylated DMRs
Public datasets generated with Illumina methylation 450k microarrays (GEO:GSE48684 [72], GEO:GSE131013 [73], and COAD-TCGA, which was accessed with the R package curatedTCGAData version 1.12.0 [Ramos M, 2021; curatedTCGAData: Curated Data From The Cancer Genome Atlas (TCGA) as MultiAssayExperiment Objects. R package version 1.12.0]) were used to validate the specificity of the DMRs identified as ageing- and colon-segment independent markers of the DNA hypermethylation that occurs during colorectal tumorigenesis.
Microarray beta values were downloaded and pre-processed using the R packages minfi version 1.34.0 [74] and IlluminaHumanMethylation450kanno.ilmn12.hg19 version 0.6.0 (Hansen KD, 2016; IlluminaHumanMethylation450kanno.ilmn12.hg19: annotation for Illumina’s 450k methylation arrays. R package version 0.6.0). For each sample, we averaged the microarray beta values for all probes that overlapped one of our tumorigenesis-specific DMRs and used this value to create the heatmaps presented in Figure 4. (Only tumorigenesis-specific DMRs containing at least one microarray probe were included in this analysis.) For each sample from the microarray studies, we then calculated the median of all the mean beta values of the probes overlapping with each of our tumorigenesis-specific DMRs. The result was a single beta-value signature for each sample. Using the pROC package version 1.16.2 [75], we tested the ability of this signature to discriminate between tumour samples (cADNs or CRCs – cases) and normal tissue (normal mucosa samples – controls). Receiver operating characteristic (ROC) curves were then plotted and the areas under the curves (AUC) calculated to assess the diagnostic accuracy of the beta value per sample in each microarray dataset. Briefly, we plotted empirical ROC curves with the sensitivity on the y-axis and specificity on the x-axis. These metrics were calculated at each possible threshold, i.e., each possible median beta value. (Note that the x-axis ranges from 100 to 0; therefore, higher specificity values are shown in the left-hand half of the graph.) We also included in the plots the optimal threshold for each dataset, i.e., the point on the ROC curve representing the threshold with the highest possible specificity (first percentage) and sensitivity (second percentage). In addition, we calculated AUC, sensitivity (TPR) and specificity (1-FPR) for each tumorigenesis-specific DMR.
Figure 4.
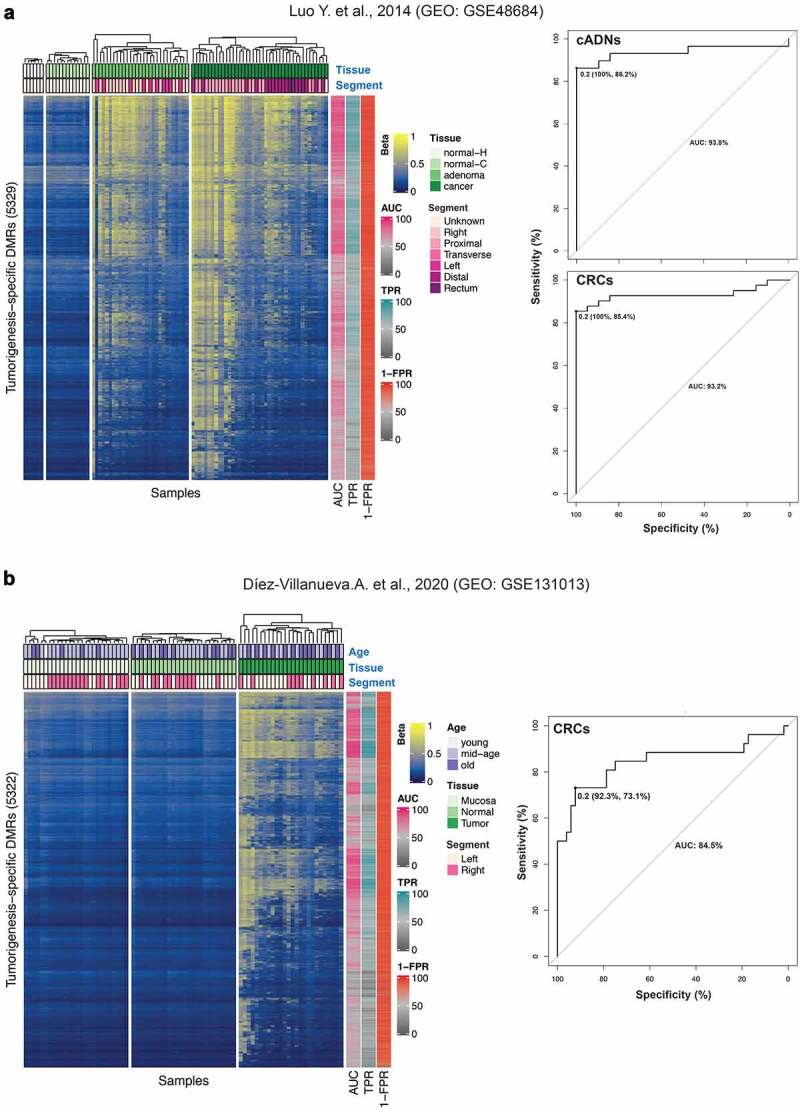
Validation of our tumorigenesis-specific hypermethylated DMRs. Findings were validated against publicly accessible Illumina 450 K microarray datasets. a. The GEO dataset GSE48684 published by Luo et al. [72]. Left: Heatmap of the 89 female colorectal tissue samples included in the dataset. Each row corresponds to one of our tumorigenesis-specific hypermethylated DMRs containing at least 1 of the Illumina microarray probes (total: 5329 of the 5521 hypermethylated DMRs we classified as highly tumorigenesis-specific; see Results). The beta value (from 0, blue, to 1, yellow) reported for each DMR is the average beta across all the microarray probes within our tumorigenesis-specific DMR coordinates (details in the Methods section). Our tumorigenesis-specific DMRs displayed hypermethylation in the tumours (vs. normal mucosa) studied by Luo et al. Metadata available for the GSE48684 dataset includes tissue (normal-H: normal mucosa samples from patients with no history of CRC; normal-C: normal mucosa samples from patients with concurrent CRC; adenoma: cADN; cancer: CRC) and colon segment (Right, Proximal and Transverse: proximal colon; Left and Distal: distal colon). Donor ages were not reported. Right: ROC curves showing the high accuracy of tumour classification as cADN (AUC: 93.8%) or CRC (AUC: 93.2%) based on the median of DMR beta values (optimal cut-off: 0.2; specificity 100%, sensitivity 86.2% for cADNs; specificity 100%, sensitivity 85.4% for CRCs) as described in the Methods section. AUC, sensitivity (TPR) and specificity (1-FPR) for each tumorigenesis-specific DMR are also shown (colour annotation on the right side of the heatmap). b. The GEO GSE131013 dataset published by Díez-Villanueva et al. [73]. Left. Heatmap of the 78 female colorectal tissue samples in this dataset based on the beta values for 5322 of our highly tumorigenesis-specific hypermethylated DMRs (as described for panel A). Our tumorigenesis-specific DMRs displayed hypermethylation in the tumours (vs. normal mucosa) studied by Díez-Villanueva et al. Metadata available for the Díez-Villanueva dataset includes age (young: <40 [1 donor]; middle-age: 40–70, 48 women; old: >70, 29 women), tissue (Mucosa, normal mucosa sample from healthy donors; Normal, normal mucosa sample adjacent to CRC; Tumour, CRC) and colon segment (Left, distal colon; Right, proximal colon). Right: ROC curve analysis of the accuracy of the median of DMR beta values (optimal cut-off 0.2; specificity 92.3%, sensitivity 73.1%) in predicting tissue type (correct classification of CRC – AUC: 84.5%). AUC, sensitivity (TPR) and specificity (1-FPR) for each tumorigenesis-specific DMR are also shown (colour annotation on the right side of the heatmap).
Scripts for all analyses performed in R (version 4.0.0) are publicly accessible at https://github.com/sorjuela/age_lesions_females/tree/master/scripts.
Results
The 48 colorectal tissue samples included in this study are thoroughly described in the Methods section and Tables 1and 2. When all 48 samples were subjected to multidimensional scaling (MDS) based on the 10,000 CpG sites whose methylation status varied most widely in these tissues, the precancerous tumours were neatly segregated from their paired samples of normal mucosa (SSL-paired NM and cADN-paired NM) (dimension 1), and clear separation of SSLs and cADNs was also observed (dimension 2) (Supplementary Figure 1). Unsupervised hierarchical clustering based on the methylation status of the 1000 most variable CpG islands across the 48 colon tissues also showed that the 18 precancerous tumour samples could be clearly distinguished from the 30 NM samples (18 SSL- or cADN-paired NM samples + 12 NM samples from the six tumour-free donors [TF-NM]) (Figure 1a). In both precancerous tumour types, the 1000 CpG islands considered are far more methylated than in any of the four groups of NM samples. Separation of tumours from NM was also evident when we considered the methylation status of the 1000 most variable promoters, although tumorigenesis-associated hypermethylation was encountered less frequently in these genomic elements than in CpG islands (Supplementary Figure2a). In many promoter regions, hypomethylation was also evident in cADNs (compare Figure 1a with Supplementary Figure 2a).
Figure 1.
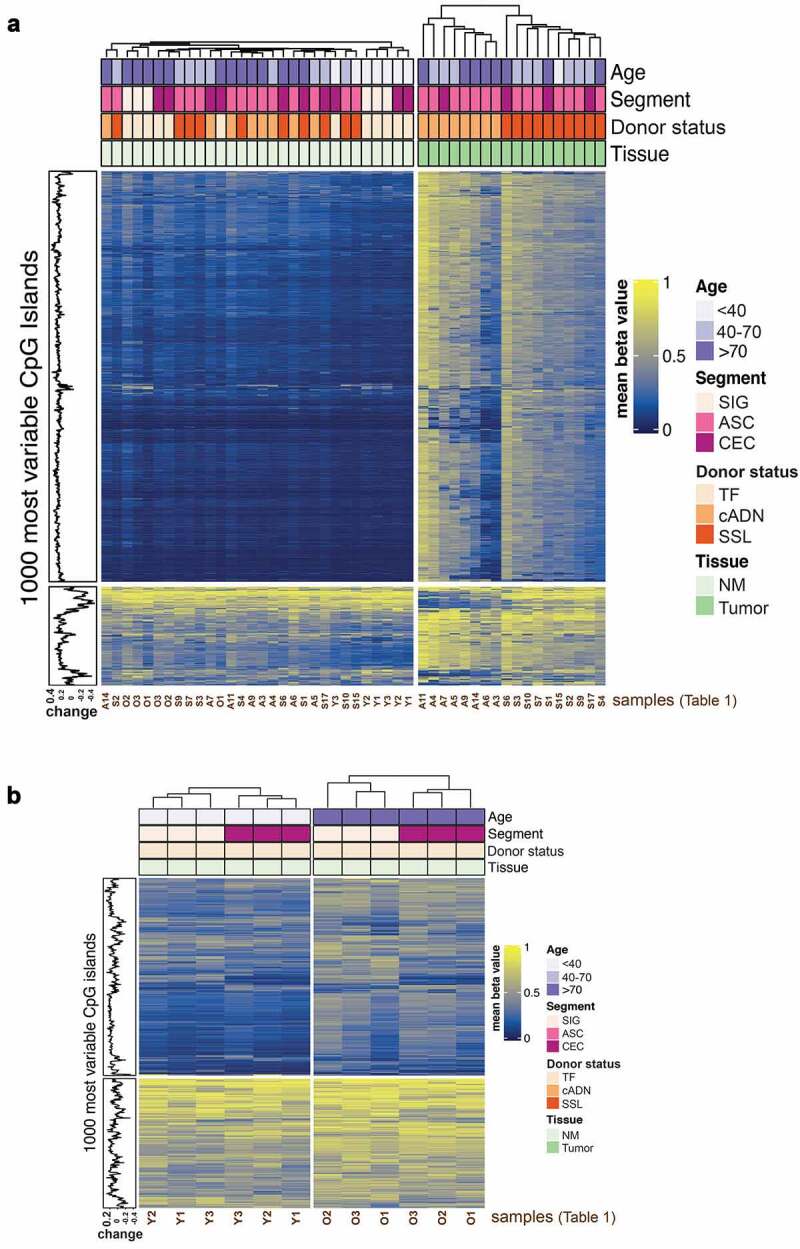
Methylomes of the colorectal tissues analysed. Unsupervised hierarchical clustering was performed based on the methylation status of the 1000 CpG islands displaying the highest variability in the tissues analysed: 18 precancerous tumours from the proximal colon (10 SSLs, 8 cADNs) and 30 NM samples (18 from the SSL or cADN donors, 12 from TF donors). a. Heatmap of all 48 samples. In both precancerous tumour types, the 1000 CpG islands considered are far more methylated than in any of the four groups of NM samples. SSLs and cADNs can also be distinguished from one other. b. Heatmap of the 12 NM samples from TF donors (1 CEC + 1 SIG per donor). Hierarchical clustering was performed within each age group. The CpG islands were more highly methylated in samples from TF donors >70 (TF-O NM) than in those from younger (<40y) TF donors (TF-Y NM). In both age groups, caecal and sigmoid NM samples (TF NMCEC and TF NMSIG) could also be distinguished. Abbreviations: ASC, ascending colon; cADN, conventional adenoma; CEC, caecum; NM, normal mucosa; O, older; SSL, sessile serrated lesion; SIG, sigmoid colon; TF, tumour-free; Y, younger.
Analysis of the 30 NM samples revealed only small differences related to donor age (<40; 40–70; >70 years) or anatomical location (caecum vs. sigmoid) (Supplementary Figure 3a and Supplementary Figure 3b), confirming that the methylation changes associated with tumorigenesis are more frequent and marked than those related to NM ageing in different colonic segments. However, the methylation changes associated with donor age and anatomical location were more evident when we limited our comparison to the TF-O NM (NM of tumour-free older women) and TF-Y NM (NM of tumour-free younger women) samples (Figure 1b and Supplementary Figure 2b).
Unsupervised analysis was also performed to better explore methylation values and changes in CpG islands that emerged from each tissue group comparison. The methylation values for CpG sites within CpG islands were considerably higher in cADNs and SSLs than in donor-matched samples of NM (Figure 2a). In the group of normal mucosal samples from the tumour-free donors, those from women >70 displayed slightly higher methylation levels than the samples from women <40. As for the NM samples from the tumour-bearing donors, methylation values were higher in those from cADN donors than in SSLs donors, which is consistent with the average ages of these two subgroups (71.6 vs 62.6 years, respectively; Table 1). In Figure 2b, the methylation changes occurring at each CpG site within CpG islands during tumorigenesis (in SSLs or cADNs) are compared with changes associated with ageing (NM from TF donors >70 vs. <40) and with the methylation differences between sigmoid and caecal NM samples from the TF women. The plots in Figure 2b show that in both precancerous lesions, but especially SSLs, the predominant alteration involves hypermethylation, and most of these changes occur in both tumour types (positive trend in plot i). Hypermethylation is also detectable in the NM samples from the older TF donors (y axes in plots ii), but the level is much lower than that seen in the tumours (x axes in plots ii). Most of this ageing-associated hypermethylation is also present in tumours (positive trend in plots ii). Finally, the methylation differences observed between sigmoid and caecal NM were very small. In this analysis as well, the hypermethylation changes occurring in CpG islands were more prominent than those seen in gene promoters (Supplementary Figure 4).
Figure 2.
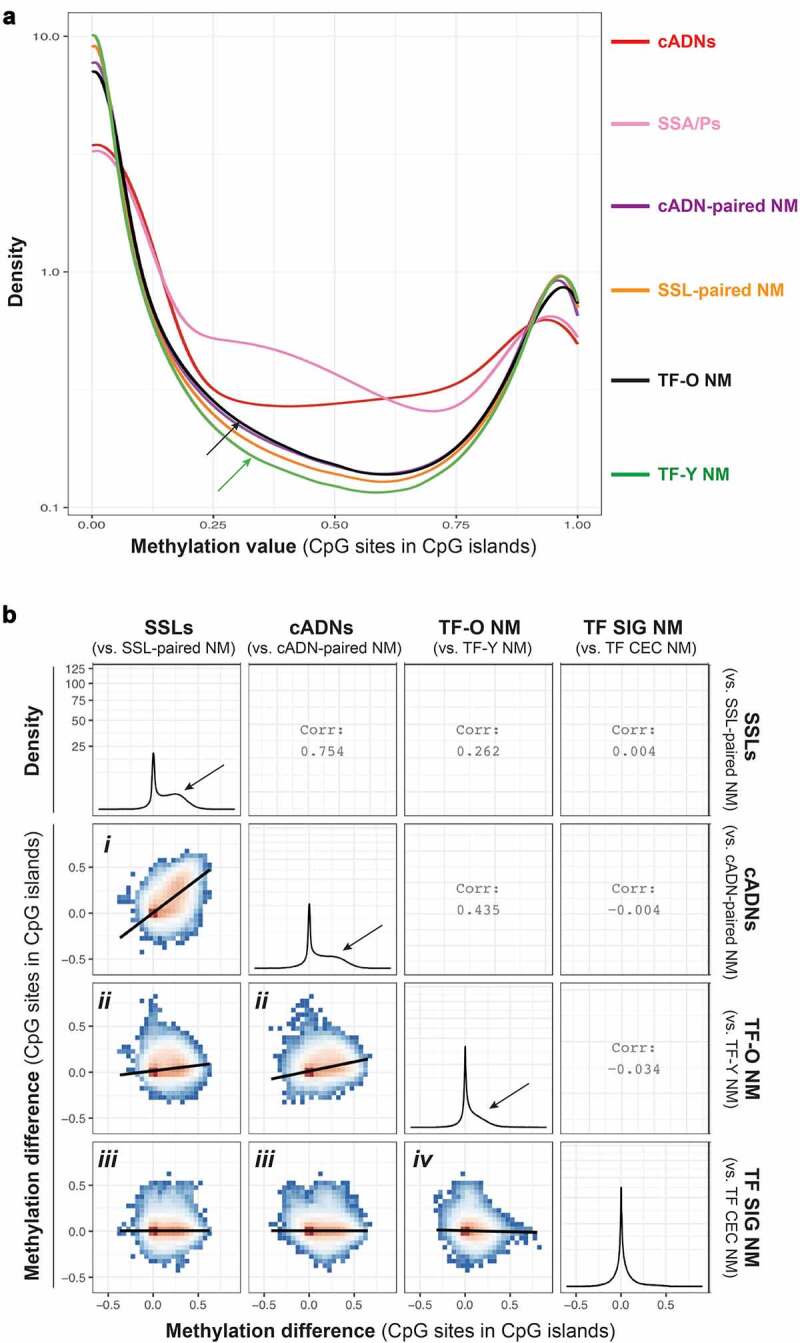
The CpG island hypermethylation phenotype in tumours and ageing NM. a. Density plot showing the methylation value at CpG sites within CpG islands in tumours (cADNs and SSLs) and in NM samples from cADN donors, SSL donors, older (>70) TF donors (TF-O), and younger (<40) TF donors (TF-Y). Methylation levels in each tumour type clearly exceed levels in any of the four classes of NM. Levels in TF-O NM (black arrow) were only slightly higher than those in TF-Y NM (green arrow). Methylation values were calculated for the 1,060,602 CpG sites remaining after the exclusion of sites displaying 0 methylation in all tissue samples. The y-axis was log10-transformed. b. Six ggpairs plots comparing the methylation changes identified in tumours (i.e., methylation difference between SSLs, or cADNs, and matched samples of NM) with those associated with NM ageing (NM samples from TF donors >70 vs. TF donors <40) and with changes associated with the anatomical location of the NM sample of TF donors (SIG vs. CEC). Methylation difference on axes refers to the methylation proportions change, as defined by dmrseq (see Methods). Methylation differences were calculated for 653,484 CpG sites in CpG islands. Colours in ggpairs plots represent the density of points per square, e.g., red bins contain the highest density of points. Regression lines are drawn over each ggpairs plot. Numbers in empty quadrants correspond to the Pearson correlations (Corr) of each ggpairs plot. As for the four density plots, the y-axis was square root-transformed. Black arrows in these plots indicate the magnitude of hypermethylation in tumours and in the TF-O NM samples. (Abbreviations as defined in Figure 1.).
As for hypermethylated DMRs (i.e., genomic regions with several, concomitantly-hypermethylated CpG sites), those identified in tumours were far more numerous and displayed higher levels of hypermethylation than those associated with NM ageing or anatomical location (Figure 3). We identified 16,581 DMRs in SSLs and 10,795 in cADNs that were hypermethylated relative to donor-matched samples of normal mucosa. The total number of CpG sites in these DMRs were 348,829 in SSLs and 237,160 in cADNs. Far fewer hypermethylated DMRs were associated with donor age (3002 regions; 72,316 CpG sites) or colon segment (1162 regions; 13,625 CpG sites) (Figure 3b). The vast majority (95%) of the ageing-associated DMRs were hypermethylated in older compared with younger TF women, while the rest showed the opposite direction (i.e., more methylated in younger). As for the colonic segments, the majority (71%) were hypermethylated in sigmoid versus caecum, while the rest were more methylated in caecum than sigmoid. Supplementary Figure 5 shows the genomic regions that contain the hypermethylated DMRs in precancerous lesions (versus paired NM) and in TF-O NM (versus TF-Y NM).
Figure 3.
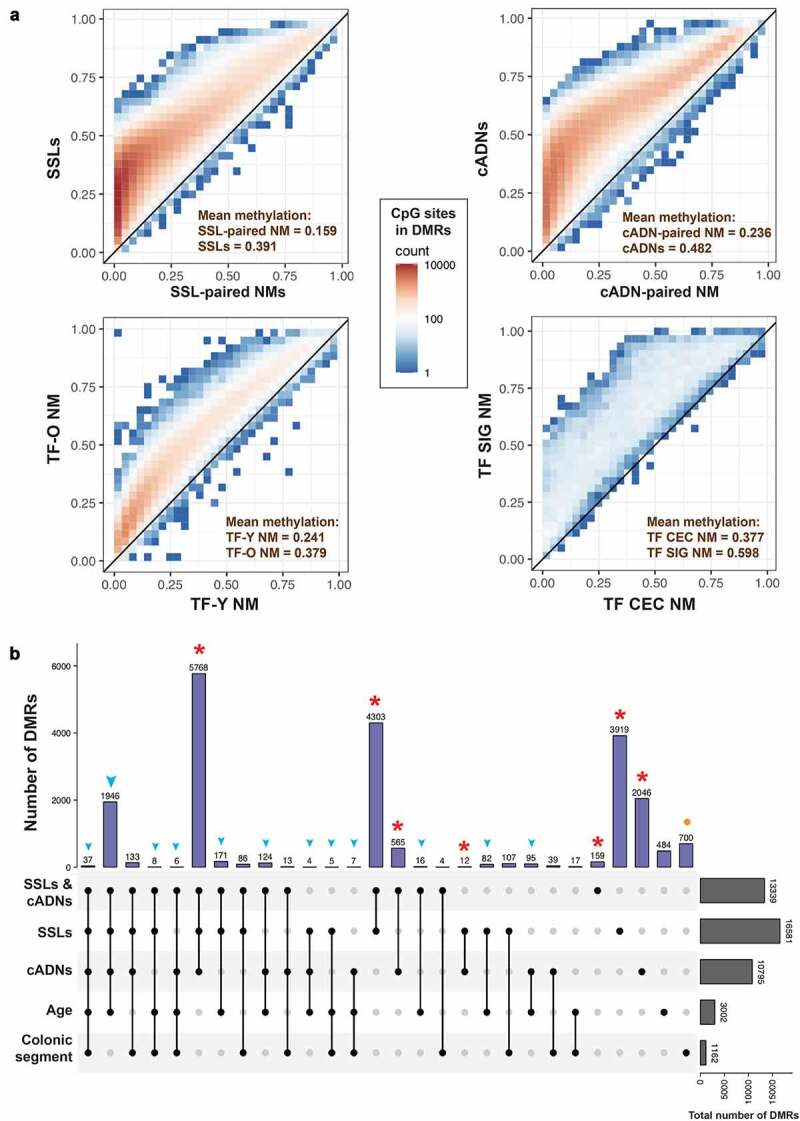
Hypermethylated differentially methylated regions (DMRs) related to early colorectal tumorigenesis, normal mucosa ageing, and/or anatomical location of normal mucosa. a. Scatter plots showing the number of methylated CpG sites within the identified hypermethylated DMRs (colour gradient) and mean methylation levels at these sites in precancerous tumours (SSLs or cADNs) versus SSL-paired NM and cADN-paired NM, respectively; in TF-O NM samples vs. TF-Y NM samples; and in TF SIG NM vs. TF CEC NM. b. UpSet plot showing the tumorigenesis-, ageing-, and colon segment-specificities of the hypermethylated DMRs. The vast majority of the hypermethylated DMRs in tumours (in SSLs, cADNs, or both, red asterisks) were tumorigenesis-specific, i.e., regions that displayed no ageing-related or colon segment-related changes in the NM. In contrast, over 80% of the ageing-associated DMRs overlapped tumorigenesis-associated DMRs (blue arrowheads). Most regions that were differentially methylated in SIG vs. CEC NM samples displayed no methylation alterations associated with ageing or tumorigenesis (orange dots). (Abbreviations as defined in Figure 1.).
A total of 16,772 hypermethylated DMRs were tumorigenesis-specific, i.e., they were present in one (3919 regions in SSLs; 2046 regions in cADNs) or both types (10,807 regions) of precancerous lesions vs. donor-paired normal mucosa but displayed no methylation changes in the normal mucosa that could be attributed to ageing or colon segment (columns with red asterisks in Figure 3b). These DMRs were stringently filtered (q value <0.05; baseline methylation in the normal mucosa of TF women <0.1; methylation proportions change in tumours >0.8, see Methods for detailed description) to identify the 5521 DMRs most promising clinical biomarkers of adenomatous and/or serrated colorectal tumorigenesis in patients of all ages (Supplementary Table 1).
Only 484 of the ageing-associated DMRs did not show any methylation change in tumours or specific colonic segments, while more than 80% of them were hypermethylated also in tumours (blue arrowhead in Figure 3b). As for the segment-associated DMRs, most of them (60%; n = 700 in Figure 3b) appear not to undergo methylation changes during ageing or tumorigenesis (orange dot in Figure 3b). Supplementary Figure 6 shows examples of hypermethylated DMRs found to be tumorigenesis-specific, ageing-specific, or associated with both ageing and tumorigenesis.
We then attempted to validate our tumorigenesis-specific hypermethylated DMRs against published DNA methylation datasets that included a sufficiently high number of colorectal tissue samples of normal mucosa and tumours from women. Only two available datasets met the latter requirements: both were generated with Illumina methylation 450 K microarray technology (rather than the more powerful bisulphite sequencing approach we used), and neither included any SSLs. The first set (published by Luo et al., GEO:GSE48684) [72] comprised data on 89 colorectal tissue samples from women, including 19 of normal mucosa (6 from patients with no history of CRC, 13 from donors with concurrent CRC), 29 cADNs, and 41 CRCs. Donor age was not provided for any of the tissue samples. The second set (GEO:GSE131013, published by Díez-Villanueva et al.) [73] included data on 78 samples, all from women (26 normal mucosa samples from CRC-free colons, 26 normal mucosa samples adjacent to CRCs, and 26 CRCs). Most donors were 40–70 years of age, and only one normal mucosa sample was from a woman under 40. The beta methylation values of array probes that overlapped our tumorigenesis-specific hypermethylated DMRs were higher in tumours than in normal mucosa samples of both of these two datasets (Figure 4ab), whereas normal tissue samples from healthy colons or those collected near a CRC showed very similar methylation patterns. Importantly, the cADNs and CRCs of the two datasets tended to be correctly classified, i.e., clearly distinguished from normal mucosa samples (ROC curves in Figure 4a and b) based on the median of all the DMR’s beta values as a single signature per sample (details in Methods section). These results indicate that the hypermethylated DMRs we identified as tumorigenesis-specific should retain that specificity in other datasets including middle-aged and older women. Similar results were obtained using an additional external dataset (from the TCGA project) that includes CRC and NM samples from both women and men (Supplementary Figure 7).
Discussion
In this study, we identified thousands of genomic regions that undergo hypermethylation early in the course of colorectal tumorigenesis, even though their methylation status in the normal colon mucosa do not appear to change with normal ageing. The tumorigenesis-specificity of most of these DMRs was confirmed when we compared our findings with data from two previously published studies [72,73]. These results support our belief that these hypermethylated DMRs are promising candidates for development as biomarkers for the early, non-invasive detection of colorectal tumours. However, the datasets we used for this preliminary validation included a limited number of normal mucosal samples from younger women. Moreover, they were generated by microarray analysis, which is a less powerful method than bisulphite sequencing for exploring the DNA methylome. A formal bisulphite sequencing-based validation study – similar to our own but comprising a larger number of normal and neoplastic colorectal tissue samples from both women and men – is needed to pinpoint the tumour-specific DMRs with the highest potential for development as blood- or stool-based biomarkers for diagnosing colorectal tumours.
Our present findings are restricted to precancerous rather than invasive colorectal tumours. We have previously shown that SSL- and/or cADNs-associated DMRs are almost invariably present in CIMP(+) and/or CIMP(-) CRCs as well [19] . In some cases, the advanced stages of tumorigenesis are accompanied by spread of the hypermethylation to involve more extensive tracts of the chromatin than those affected in precancerous lesions, and this might result in some degree of overlap with one or more ageing-associated DMRs. However, the hypermethylated DNA tracts we report here as ‘tumorigenesis-specific’ prior to transformation retain this specificity after the lesion has progressed to the more advanced stages of tumorigenesis.
In 1999, Toyota and Issa [24] proposed that colorectal tumorigenesis was associated with two forms of CpG-island hypermethylation: one, which they referred to as type A, took place in normal colorectal epithelial cells as a part of the physiological ageing process and was thought to be a possible facilitator of colorectal tumorigenesis in general; the second (type C) occurred exclusively in CIMP(+) CRCs. This model finds support in our recent identification (based on microarray-based methylation analysis) of two large sets of hypermethylated CpG sites [49]. The first comprised sites aberrantly methylated only in CIMP(+) CRCs and their serrated-pathway precursors. Sites in the second set displayed hypermethylation in both CIMP(-) and CIMP(+) CRCs and also in their non-malignant precursors (adenomas and serrated tumours, respectively), and this set was enriched for CpG sites previously shown to undergo ageing- and lifestyle-dependent hypermethylation in normal colorectal epithelial cells [50]. These findings do not exclude the possibility that certain ageing-associated changes might sometimes be exploited for the purposes of tumorigenesis, particularly during the development of CIMP(+) tumours. Indeed, an event of this type probably occurs in the MLH1 promoter in some later-stage SSLs, as discussed on the Introduction. As tumorigenesis advances, the proto-CIMP(+) typical of SSLs in the proximal colon [19] appears to favour increasingly extensive methylation of the MLH1 promoter, a region that may already display milder, age-related methylation in a small number of normal epithelial cells from the proximal colons of ageing women [22,50]. This epigenetic shift is not, however, an invariable occurrence during serrated-pathway tumorigenesis: in approximately 40% of CIMP(+) CRCs, MLH1 expression is preserved [76].
Previous research has shown that hypermethylation at certain sites does occur exclusively within the context of normal ageing [26,28,49]. Our data indicate that such ageing-specific hypermethylation is relatively uncommon: as shown in Figure 3b, it accounted for only 15% of the ageing-associated hypermethylated DMRs present in the female colon tissues we studied. Over 80% of the DMRs exhibiting ageing-associated hypermethylation overlapped DMRs associated with tumorigenesis – that is, they acquired additional methylation in precancerous tumour cells, which probably involves increases in the number of methylated CpG sites per DMR and in the number of cells with methylated alleles. This finding suggests an abrupt acceleration of the methylome-ageing clock. This clock reportedly ticks much more slowly after puberty, undergoing a ~ 24-fold decrease relative to the rapid ageing typical of the early postnatal years. Consequently, the methylation levels in a given tissue are normally quite similar in young adults (30–40 years old) and the elderly (aged 70–80), with a difference of less that 0.1 units on a methylation scale of 0 to 1 [45,50]. But in tumours arising in such tissues, methylome ageing is strikingly accelerated (on average, by ~30 years). In the colon, the most dramatic acceleration is seen in CIMP(+) CRCs with the oncogenic BRAFV600E mutation and a hypermethylated MLH1 promoter [35]. However, the upregulation was already quite obvious in the precancerous lesions we studied, where the mean increase in methylation levels over those found in matched samples of normal mucosa exceeded ~0.2 on the 0–1 scale cited above (Figure 3a).
About 30% to ~80% of the hypermethylated CpG sites previously identified as ageing-related are reportedly located in genomic regions bound by the PRC2 complex in embryonic stem cells, regions that often contain the promoters of genes with known roles in embryonic cell differentiation [50,57,77,78]. PRC2 catalyzes the trimethylation of lysine 27 of histone H3 in nucleosomes located at ~2000 gene promoters in embryonic stem cells [79,80]. This epigenetic mark represses the gene’s expression but keeps it ‘poised’ for transcriptional re-activation, i.e., a state of equilibrium between self-renewal and lineage commitment. Normal ageing and tumorigenesis both tend to be associated with methylation of the DNA in these nucleosomes [32–34], which reduces the region’s affinity for PRC2 binding [51,81]. Some investigators suspect that this epigenetic switch locks certain developmentally relevant genes into a more strongly repressed state, which promotes tumorigenesis by inhibiting normal tissue differentiation [38,45,51,82], but thus far, definitive proof for this hypothesis is lacking.
In contrast to ageing-associated DMRs, 16,772 of the tumour-associated DMRs we identified were hypermethylated exclusively in tumours. Only 2883 occurred in regions that also underwent methylation as a function of ageing and/or the anatomical location of the tissue (Figure 3b). This finding argues against the widely held view that the hypermethylome in tumour cells is merely a more advanced version of the ageing-related hypermethylation that occurs in the normal mucosa. The fact that PRC2-targeted regions are reportedly over-represented among both ageing- and tumorigenesis-associated hypermethylated DMRs does not imply that these two DMR subsets are predominantly overlapping. The burst of de novo CpG-island methylation that takes place in early-stage colorectal tumorigenesis does indeed affect some DNA tracts that undergo the slowly progressing methylation seen with normal ageing, but the tumorigenesis-associated process of DNA methylation is far more extensive and ultimately involves a much larger portion of gene-regulatory genomic regions. In all probability, this second wave of hypermethylation is fuelled by molecular mechanisms distinct from those believed to promote ageing-associated methylation (see Introduction section), that is, genetic and epigenetic alterations arising in early phases of colorectal tumorigenesis, such as oncogenic mutation of the KRAS protein or the BRAFV600E mutation. These two mutations are mutually exclusive features of lesions representing incipient colorectal tumorigenesis and suspected to be the triggers of the DNA hypermethylation waves that characterize the CIMP(-) and CIMP(+) tumorigenic processes, respectively [83,84]. Downregulation of the TET1 and TET2 DNA demethylases has also been proposed as a mechanism underlying hypermethylation in the CIMP(+), BRAF600E-driven, serrated pathway of colorectal tumorigenesis [49]. Interestingly, CIMP positivity in a subset of glioblastomas has been causally linked to mutations in the IDH1 or IDH2 genes, which encode an enzyme needed for the production of alpha-ketoglutarate, an essential cofactor of TET DNA demethylases (and of some histone lysine demethylases, as well) [85]. Debate continues over the temporal sequence of the genetic and epigenetic changes that take place during the initial phase of colorectal tumorigenesis (e.g., BRAFV600F and proto-CIMP in serrated colorectal lesions) and their reciprocal effects on one another [84,86,87]. Answers to these questions will ultimately require genetic and epigenetic analyses of minute, very early-stage neoplastic lesions, which are currently feasible only with single-cell omics technologies.
In conclusion, the de novo methylation of promoter CpG islands associated with early-stage colorectal tumorigenesis exceeds that associated with normal ageing in both magnitude and extension. The hypermethylation ‘burst’ that occurs during neoplasia is more pervasive in SSLs, but it also takes place in cADNs, the more common CRC precursors. In both types of precancerous lesion, it is generally associated with distinct oncogenic activation events that are not observed during ageing of the normal colorectal mucosa. The thousands of tumorigenesis-specific hypermethylated regions documented in this report can be exploited to develop biomarkers for the early diagnosis of colorectal tumours.
Supplementary Material
Acknowledgments
We thank Marian Everett Kent for editing the manuscript.
Funding Statement
Swiss National Science Foundation grant no. 310030_179477/1 to S.O., S.S. and G.M.; and Novartis Foundation for medical-biological research, grant no. 17B086 to H.R.P.
Authors’ contributions
G.M., S.O. and H.R.P. designed research; F.C., M.S. and F.B. performed tissue sample and clinical data collection; H.R.P. processed tissue samples for DNA methylation analysis; S.O. and M.D.R. analyzed data; S.O., S.S. and G.M. wrote the paper.
Disclosure statement
The authors declare that they have no competing interests.
Availability of data and material
The raw data generated and analyzed during the current study are available in the ArrayExpress repository (accession numbers: E-MTAB-6951 and E-MTAB-6952 for the tissue samples of women with precancerous lesions, and E-MTAB-9665 for the tissue samples of tumor-free women).
Consent for publication
Not applicable
Ethics approval and consent to participate
Ethics approval was received from the ethics committees at both Cremona Hospital, Italy (Comitato Etico d’Area Cremona Mantova Lodi, protocollo 13,728/16) and Triemli Hospital, Zurich, Switzerland (KEK-ZH-Nr: 2015-0068 and BASEC-Nr.: 2015-00185)
List of abbreviations
- AUC:
area under the curve;
- cADN:
conventional adenoma;
- CIMP:
CpG island methylator phenotype;
- DMR:
differentially methylated region;
- MDS:
multidimensional scaling;
- NM:
normal mucosa;
- PRC2:
polycomb repressive complex 2;
- ROC:
receiver operating characteristic;
- SSL:
sessile serrated lesion;
- TF-O:
older tumor-free donors;
- TF-Y:
younger tumor-free donors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Arndt V, Feller A, Hauri D, et al. Swiss cancer report 2015. Neuchatel, Switzerland: Federal Statistical Office; 2016; [Google Scholar]
- [2].Winawer SJ. Natural history of colorectal cancer. Am J Medicine. 1999;106:3–6. [DOI] [PubMed] [Google Scholar]
- [3].Lash RH, Genta RM, Schuler CM.. Sessile serrated adenomas: prevalence of dysplasia and carcinoma in 2139 patients. J Clin Pathol. 2010;63:681. [DOI] [PubMed] [Google Scholar]
- [4].Hassan C, Quintero E, Dumonceau J-M, et al. Post-polypectomy colonoscopy surveillance: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2013;45:842–864. [DOI] [PubMed] [Google Scholar]
- [5].Carr PR, Weigl K, Edelmann D, et al. Estimation of absolute risk of colorectal cancer based on healthy lifestyle, genetic risk, and colonoscopy status in a population-based study. Gastroenterology. 2020;159:129–138.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mendelsohn RB, Winawer SJ, Ahnen DJ. Incidence of colorectal cancer matters. Gastroenterology. 2019;158:1191–1195. [DOI] [PubMed] [Google Scholar]
- [7].Kaltenbach T, Anderson JC, Burke CA, et al. Endoscopic removal of colorectal lesions—recommendations by the US multi-society task force on colorectal cancer. Gastroenterology. 2020;158:1095–1129. [DOI] [PubMed] [Google Scholar]
- [8].Potter NT, Hurban P, White MN, et al. Validation of a real-time PCR–based qualitative assay for the detection of methylated SEPT9 DNA in human plasma. Clin Chem. 2014;60:1183–1191. [DOI] [PubMed] [Google Scholar]
- [9].Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal-cancer screening. New Engl J Medicine. 2014;370:1287–1297. [DOI] [PubMed] [Google Scholar]
- [10].Wolf AMD, Fontham ETH, Church TR, et al. Colorectal cancer screening for average‐risk adults: 2018 guideline update from the American cancer society. Ca Cancer J Clin. 2018;68:250–281. [DOI] [PubMed] [Google Scholar]
- [11].Toyota M, Ho C, Ahuja N, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999;59:2307–2312. [PubMed] [Google Scholar]
- [12].Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci. 1998;95:6870–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Truninger K, Menigatti M, Luz J, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology. 2005;128:1160–1171. [DOI] [PubMed] [Google Scholar]
- [14].Hinoue T, Weisenberger DJ, Lange CPE, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marra G, Schär P. Recognition of DNA alterations by the mismatch repair system. Biochem J. 1999;338:1–13. [PMC free article] [PubMed] [Google Scholar]
- [16].Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Bio. 2006;7:335–346. [DOI] [PubMed] [Google Scholar]
- [17].Rex DK, Ahnen DJ, Baron JA, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012;107:1315–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bettington M, Walker N, Clouston A, et al. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–386. [DOI] [PubMed] [Google Scholar]
- [19].Parker HR, Orjuela S, Oliveira AM, et al. The proto CpG island methylator phenotype of sessile serrated adenomas/polyps. Epigenetics. 2018;13:1088–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Snover DC. Diagnostic and reporting issues of preneoplastic polyps of the large intestine with early carcinoma. Ann Diagn Pathol. 2018;39:1–14. [DOI] [PubMed] [Google Scholar]
- [21].Pai RK, Bettington M, Srivastava A, et al. An update on the morphology and molecular pathology of serrated colorectal polyps and associated carcinomas. Modern Pathol. 2019;32:1390–1415. [DOI] [PubMed] [Google Scholar]
- [22].Menigatti M, Truninger K, Gebbers J-O, et al. Normal colorectal mucosa exhibits sex- and segment-specific susceptibility to DNA methylation at the hMLH1 and MGMT promoters. Oncogene. 2009;28:899–909. [DOI] [PubMed] [Google Scholar]
- [23].Ahuja N, Li Q, Mohan AL, et al. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–5494. [PubMed] [Google Scholar]
- [24].Toyota M, Issa J-PJ. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol. 1999;9:349–357. [DOI] [PubMed] [Google Scholar]
- [25].Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. [DOI] [PubMed] [Google Scholar]
- [26].Issa J-P. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014;124:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Maegawa S, Gough SM, Watanabe-Okochi N, et al. Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res. 2014;24:580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kaz AM, Wong C-J, Dzieciatkowski S, et al. Patterns of DNA methylation in the normal colon vary by anatomical location, gender, and age. Epigenetics. 2014;9:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Singmann P, Shem-Tov D, Wahl S, et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenet Chromatin. 2015;8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luebeck GE, Hazelton WD, Curtius K, et al. Implications of epigenetic drift in colorectal neoplasia. Cancer Res. 2019;79:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ohm JE, McGarvey KM, Yu X, et al. A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. [DOI] [PubMed] [Google Scholar]
- [34].Widschwendter M, Fiegl H, Egle D, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. [DOI] [PubMed] [Google Scholar]
- [35].Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. Plos Genet. 2009;5:e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Michalak EM, Burr ML, Bannister AJ, et al. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Bio. 2019;20:573–589. [DOI] [PubMed] [Google Scholar]
- [38].Klutstein M, Nejman D, Greenfield R, et al. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–3450. [DOI] [PubMed] [Google Scholar]
- [39].Klutstein M, Moss J, Kaplan T, et al. Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc Natl Acad Sci. 2017;114:2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Horvath S, Zhang Y, Langfelder P, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maegawa S, Hinkal G, Kim HS, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Beerman I, Bock C, Garrison BS, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12:413–425. [DOI] [PubMed] [Google Scholar]
- [43].Petkovich DA, Podolskiy DI, Lobanov AV, et al. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 2017;25(954–960.e6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tm S, MJ B, A-k S, et al. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017;18:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Raj K, Horvath S. Current perspectives on the cellular and molecular features of epigenetic ageing. Exp Biol Med. 2020;245:1532–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Niehrs C, Calkhoven CF. Emerging role of C/EBPβ and epigenetic DNA methylation in ageing. Trends Genet. 2020;36:71–80. [DOI] [PubMed] [Google Scholar]
- [47].Zhang W, Qu J, Liu G-H, et al. The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Bio. 2020;21:137–150. [DOI] [PubMed] [Google Scholar]
- [48].Xie W, Kagiampakis I, Pan L, et al. DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell. 2018;33(309–321.e5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Noreen F, Küng T, Tornillo L, et al. DNA methylation instability by BRAF-mediated TET silencing and lifestyle-exposure divides colon cancer pathways. Clin Epigenetics. 2019;11:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Noreen F, Röösli M, Gaj P, et al. Modulation of age- and cancer-associated DNA methylation change in the healthy colon by aspirin and lifestyle. J Natl Cancer Inst. 2014;106(7):dju161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jung M, Pfeifer GP. Aging and DNA methylation. Bmc Biol. 2015;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang Z, Wong A, Kuh D, et al. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016;17:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen BH, Marioni RE, Colicino E, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY). 2016;8:1844–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA Methylation . Clocks in aging: categories, causes, and consequences. Mol Cell. 2018;71:882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yu M, Hazelton WD, Luebeck GE, et al. Epigenetic aging: more than just a clock when it comes to cancer. Cancer Res. 2020;80:367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sk L, Nachun D, Mg M, et al. DNA methylation analysis validates organoids as a viable model for studying human intestinal aging. Cell Mol Gastroenterol Hepatol. 2020;9:527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Teschendorff AE, Menon U, Gentry-Maharaj A, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Steegenga WT, Boekschoten MV, Lute C, et al. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Omaha). 2014;36:9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Affinito O, Palumbo D, Fierro A, et al. Nucleotide distance influences co-methylation between nearby CpG sites. Genomics. 2020;112:144–150. [DOI] [PubMed] [Google Scholar]
- [60].Zhang W, Spector TD, Deloukas P, et al. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 2015;16:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Update on the Paris classification of superficial neoplastic lesions in the digestive tract. Endoscopy. 2005;37:570–578. [DOI] [PubMed] [Google Scholar]
- [62].Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Köster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2522. [DOI] [PubMed] [Google Scholar]
- [64].Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Korthauer K, Chakraborty S, Benjamini Y, et al. Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics. 2018;20:367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lawrence M, Huber W, Pagès H, et al. Software for computing and annotating genomic ranges. Plos Comput Biol. 2013;9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lee S, Cook D, Lawrence M. plyranges: a grammar of genomic data transformation. Genome Biol. 2019;20:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wickham H. ggplot2, elegant graphics for data analysis. Switzerland: Springer-Verlag; 2016. [Google Scholar]
- [69].Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. [DOI] [PubMed] [Google Scholar]
- [70].Cavalcante RG, Sartor MA. annotatr: genomic regions in context. Bioinformatics. 2017;33:2381–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhu LJ, Gazin C, Lawson ND, et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. Bmc Bioinformatics. 2010;11:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Luo Y, Wong C-J, Kaz AM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014;147(418–429.e8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Díez-Villanueva A, Sanz-Pamplona R, Carreras-Torres R, et al. DNA methylation events in transcription factors and gene expression changes in colon cancer. Epigenomics. 2020;12:1593–1610. [DOI] [PubMed] [Google Scholar]
- [74].Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Phipps AI, Limburg PJ, Baron JA, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148(77–87.e2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Rakyan VK, Down TA, Maslau S, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Easwaran H, Johnstone SE, Neste LV, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012;22:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bracken AP, Dietrich N, Pasini D, et al. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Gene Dev. 2006;20:1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mohn F, Weber M, Rebhan M, et al. Targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30:755–766. [DOI] [PubMed] [Google Scholar]
- [81].Reddington JP, Sproul D, Meehan RR. DNA methylation reprogramming in cancer: does it act by re‐configuring the binding landscape of Polycomb repressive complexes? Bioessays. 2014;36:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Easwaran H, Tsai H-C, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Serra RW, Fang M, Park SM, et al. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife. 2014;3:e02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fang M, Ou J, Hutchinson L, et al. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG island methylator phenotype. Mol Cell. 2014;55:904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Malta TM, de Souza CF, Sabedot TS, et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 2017;20:608–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Liu Y, Sethi NS, Hinoue T, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33(721–735.e8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tao Y, Kang B, Petkovich DA, et al. Aging-like spontaneous epigenetic silencing facilitates wnt activation, stemness, and braf V600E-induced tumorigenesis. Cancer Cell. 2019;35(315–328.e6). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data generated and analyzed during the current study are available in the ArrayExpress repository (accession numbers: E-MTAB-6951 and E-MTAB-6952 for the tissue samples of women with precancerous lesions, and E-MTAB-9665 for the tissue samples of tumor-free women).