

Summary
The Ubiquitin-Proteasome System (UPS) is the primary route for selective protein degradation in human cells. The UPS represents an attractive target for novel cancer therapeutics, but the precise UPS genes and substrates important for cancer growth are incompletely understood. Leveraging multi-omics data across more than 9,000 human tumors and 33 cancer types, we found that over 19% of all cancer driver genes impact UPS function. We implicate transcription factors as important substrates, and show that c-Myc stability is modulated by CUL3. Moreover, we developed a deep learning model (deepDegron) to identify mutations that result in degron loss, and experimentally validated predictions that gain-of-function truncating mutations in GATA3 and PPM1D result in increased protein stability. Lastly, we identified UPS driver genes associated with patient prognosis and the tumor microenvironment. This study demonstrates the important role of UPS dysregulation in human cancers and underscores the potential therapeutic utility of targeting the UPS.
eTOC blurb
The mechanisms underlying oncogenic mutations in cancer remain incompletely understood. By leveraging machine learning, Tokheim et al. find ~19% of cancer driver genes impact protein degradation, thus systematically revealing transcription factors as important substrates. They furthermore validate an unconventional role for truncating mutations to increase stability of GATA3 and PPM1D.
Graphical Abstract
Introduction
Cancer is fundamentally a disease of the genome, whereby only certain mutations drive a selective growth advantage for cancer cells, with most mutations being benign passengers that accumulate by chance. From the start of DNA sequencing studies of human tumors (Barbieri et al., 2012; Cancer Genome Atlas Research, 2008; Jones et al., 2008; Wood et al., 2007), it quickly became clear that genes involved in protein degradation were perturbed by mutations in cancer. For example, mutated VHL leads to elevated HIF-1/2a protein abundance, which allows cells to adapt to hypoxic conditions (Iliopoulos et al., 1996; Ivan et al., 2001; Iyer et al., 1998; Jaakkola et al., 2001). The Ubiquitin-Proteasome System (UPS) regulates the degradation of over 80% of proteins in cells (Collins and Goldberg, 2017). UPS dysregulation has been implicated in nearly all of the hallmarks of cancer (Hanahan and Weinberg, 2011), such as USP28 in DNA damage response (Zhang et al., 2006), KEAP1 in oxidative stress (Jaramillo and Zhang, 2013), and FBXW7 in cell proliferation (King et al., 2013; Welcker and Clurman, 2008). Moreover, defects in the UPS have been linked to a variety of other human diseases or disorders (Atkin and Paulson, 2014; Das et al., 2006; Nalepa and Clapp, 2018; Staub et al., 1997); for example, loss-of-function mutations in UBE3A are implicated in Angelman Syndrome, a neurodevelopmental disorder (Buiting et al., 2016). Despite the importance of UPS in human disease and especially cancer, a systems-level understanding of the UPS is still lacking.
The UPS operates through the covalent attachment of ubiquitin (an 8 kDa protein) to lysine residues in substrate proteins, which is achieved through a relay of steps by passing ubiquitin from E1 enzymes to E2 enzymes (Stewart et al., 2016) and, ultimately with the help of E3 ubiquitin ligases, to substrates. While ubiquitination can have many functions, polyubiquitination is often a signal for protein degradation by the 26S proteasome (Collins and Goldberg, 2017). The key step of this process conferring regulatory specificity is performed by the E3 ubiquitin ligases, which are thought to recognize short linear amino acid motifs, known as degrons, on the substrate proteins (Meszaros et al., 2017). With over 600 E3 ubiquitin ligases encoded in the human genome, there are more than 10 million possible E3 ligase-substrate pairs. The transient nature of E3-substrate interactions makes experimental detection of these interactions using co-immunoprecipitation challenging (Ella et al., 2019; Meszaros et al., 2017). In addition, deubiquitinating enzymes (DUBs) act in the opposite direction, preventing degradation by removing ubiquitin from proteins (Reyes-Turcu et al., 2009; Ronau et al., 2016). Although many mechanistic steps of the UPS are well characterized, the regulatory logic of how E3 ubiquitin ligases and DUBs selectively recognize their target protein remains highly incomplete (Deshaies and Joazeiro, 2009). Since it is unclear which genes involved in ubiquitination act in a proteasomal-dependent versus -independent manner, we will include all E1, E2, E3 and DUB enzymes in our subsequent analyses.
A substantial fraction (37–57%) of tumors harbor potentially clinically actionable mutations (Bailey et al., 2018; Zehir et al., 2017). Some of these are in genes that encode the UPS components; for instance, BRCA1 (an E3 ubiquitin ligase) mutant tumors are sensitive to PARP inhibitors through a synthetic-lethal interaction (Robson et al., 2017). Traditionally, clinical actionability has been largely based on classical drug development of small molecules or antibodies that bind to an enzyme or a receptor. Recent developments of protein degrader-based drugs, such as proteolysis targeting chimera (PROTAC) (Sakamoto et al., 2001; Winter et al., 2015), have promised to expand the scope of druggable targets in cancer through a novel mechanism of action. PROTACs that act by co-opting the cell’s normal UPS machinery to degrade specific target proteins are under active drug development, and early PROTAC drugs are undergoing clinical trials in breast and prostate cancers (Scudellari, 2019). However, it is still not well understood how the UPS is usually perturbed in cancers, and how PROTACs or other UPS-targeting drugs could counteract this effect. Thus, a comprehensive characterization of which mutated UPS genes may drive carcinogenesis and their corresponding dysregulated protein substrates is not only important for understanding cancer biology but also of potentially significant therapeutic utility.
Prior studies have either been underpowered to identify significantly mutated genes within the UPS or lacked the capability to identify previously unknown substrates. While Ge and colleagues found 23 mutated genes in the UPS as statistically significant (Ge et al., 2018), their analysis found mostly already known genes and did not consider the affected substrates. Martínez-Jiménez et al. attempted to identify substrates of E3 ligases in cancer (Martínez-Jiménez et al., 2020), but they only analyzed protein expression data for ~200 proteins (Li et al., 2013) and analyzed a handful of E3 ligases with already known degron motifs (Gouw et al., 2018). In contrast, our study considered all the components of the UPS system, including E1 activating enzymes, E2 conjugating enzymes, E3 ligases and deubiquitinating enzymes. In addition, we developed machine learning method to systematically infer degron sequences de novo and identify mutated substrates that escape protein degradation. We aimed to provide the most systematic assessment of the role of protein degradation in human cancers to date, supported by experimental validation of our predictions.
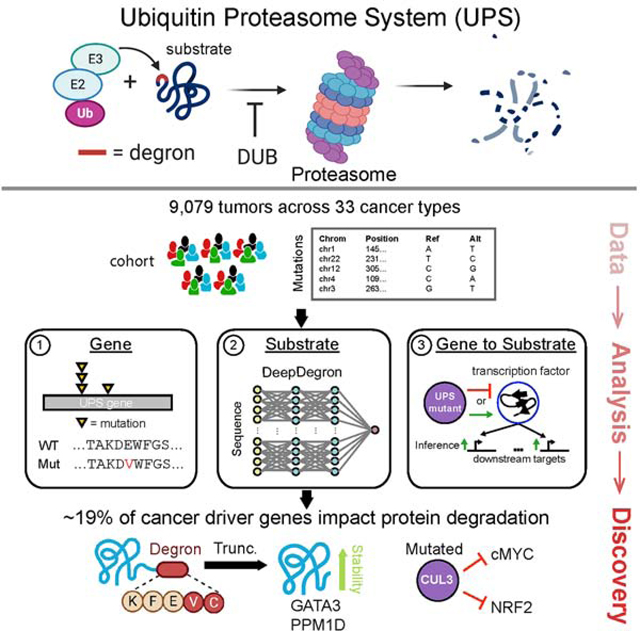
In this study, to dissect the complex regulation of the UPS in cancer, we divided the problem into several steps: identifying mutated UPS genes, identifying mutated substrates, and linking mutated UPS genes to substrates (Figure 1). Towards this aim, we employed integrative computational approaches to identify cancer driver genes in the UPS, associated these with candidate substrates through a multi-omic approach, and leveraged deep learning to model the impact of mutations on degrons. When investigating over 9,000 tumors in 33 cancer types, we found a significantly larger role for UPS dysregulation in carcinogenesis than previously appreciated, comprising approximately 19% of cancer driver genes. Predictions of mutations leading to degron loss in GATA3 and PPM1D were then experimentally validated. Furthermore, UPS alterations are associated with patient prognosis and immune infiltration of the tumor microenvironment (TME). Our results could provide insights to the rational selection of protein degrader drugs to counteract the effects of UPS dysregulation in human cancer.
Figure 1. Study Overview.

Somatic mutations from 33 cancer types in The Cancer Genome Atlas (TCGA) (left) were analyzed to reveal significantly mutated genes in the Ubiquitin-Proteasome System (UPS) and its substrates with a significant enrichment of mutations at known degron-related sites (middle). A machine learning model, deepDegron (bottom right), was then used to find additional degron sites and to implicate the impact of additional mutations. Lastly, leveraging the significantly mutated genes in the UPS pathway, we associated UPS pathway genes with protein abundance or inferred activity of transcription factors to implicate putative substrates (top right).
Results
Expanded landscape of putative cancer driver genes in the Ubiquitin-Proteasome System (UPS)
An understanding of the Ubiquitin-Proteasome System (UPS) requires assessment of both the genes comprising the pathway and the protein substrates that they regulate (Figure 1). To establish a landscape of the former in cancer, we evaluated whether UPS genes (Table S1) were somatically mutated more often than expected by chance across a large cohort of patients’ tumors from The Cancer Genome Atlas (TCGA). The rationale is that driver mutations in a UPS gene would confer a selective growth advantage to a clonal cell population leading to cancer, which leaves a statistically distinguishable signal as compared to mutations that happen by chance. Using the 20/20+ method we previously developed to identify mutated cancer driver genes (Tokheim et al., 2016) (Methods), we found a total of 63 unique UPS genes as putative drivers (q<0.05, Figure 2A, Table S1), covering 28 of 33 cancer types analyzed (Figure 2B). The putative UPS drivers are enriched for curated cancer driver genes in the Cancer Gene Census (p=2e-11, one-tailed Fisher’s exact test) (Sondka et al., 2018), driver genes defined by the TCGA consortium (p=6e-25) (Bailey et al., 2018), and biological processes relevant to cancer (Figure S1D). Moreover, unlike a recent study (Martínez-Jiménez et al., 2020) which only includes E3 ubiquitin ligases, our analysis includes E2 conjugating enzymes, E1 activating enzymes, and deubiquitinases, which led to a greater number of putative UPS driver genes with better agreement with prior literature (Figure S1A-C). Notably, when compared to the results by the TCGA consortium, the UPS putative drivers represented ~16% of all driver genes including 33 genes not previously reported (Figure 2C, Table S1), which suggests a substantial role for the UPS in carcinogenesis. Reflective of occurrence in diverse cancer types, UPS driver genes showed substantial variability in gene dependencies across cell-lineages from CRISPR KO (Figure S1E-G) and contextually co-occurred with other mutations (Figure SH-J). Lastly, we stratified mutated UPS genes by oncogene or tumor suppressor gene scores from 20/20+, and observed the majority to be tumor suppressors (Figure 2A). In some cases, a tumor suppressor gene may also have a high “oncogene” score due to the presence of recurrent hotspot mutations in addition to truncating mutations, suggesting the hotspot mutations have a dominant-negative effect (Davis et al., 2014).
Figure 2. Landscape of cancer driver genes in the Ubiquitin-Proteasome System (UPS).

(A) Driver gene analysis was performed by the 20/20+ method. Scatter plot for each UPS gene (dots) is shown with the maximum oncogene (OG) score (x-axis) and maximum tumor suppressor gene (TSG) score (y-axis) across 33 cancer types and a pan-cancer analysis. Red indicates the gene was found to be statistically significant in at least one analysis.
(B) Fraction of putative cancer driver genes which occur in the UPS pathway (red bar). Dashed line indicates the median across all analyses.
(C) Venn diagram that shows the overlap of putative cancer driver genes in this study (20/20+) with previous studies: TCGA PancanAtlas consortium, ubiquitin pathway analysis by Ge et al., Davoli et al., and of a curated list of cancer driver genes, in general, from the Cancer Gene Census (CGC).
(D) Pie diagram displaying the percentage of UPS driver genes in terms of molecular function.
(E) Lollipop diagram of CUL3 mutations in Head and Neck squamous cell carcinoma in TCGA. Exon-exon junctions are displayed as dashed lines. Color of circles distinguishes the type of mutation, while colored rectangles are uniprot domain annotations of the protein.
(F) Kaplan-meier curves of the relationship between UCHL1 expression and overall patient survival in 4 melanoma datasets.
(G) Lollipop diagram of UCHL1 mutations in TCGA skin cutaneous melanoma cohort. Numbered circles indicate a mutation was found in more than one tumor.
See also Figure S1
The 63 putative UPS driver genes spanned E3 ubiquitin ligases (n=46), E2 conjugating enzymes (n=5), E1 activating enzymes (n=1) and deubiquitinating enzymes (n=11) (Figure 2D). Identified Components of E3 ubiquitin ligases represent not only target recognition subunits but also cullin scaffold proteins (n=5). These included CUL3, which exhibited widely distributed loss-of-function mutations and a recurrent mutation (p.R709) near the activating neddylation site (Figure 2E). While DUBs were fewer in number, expression of driver DUB genes had prognostic value in 16 of 33 cancer types analyzed (Figure S1L, Methods). For example, high expression of UCHL1 was significantly associated with worse overall survival in TCGA metastatic melanoma patients (Figure 2F), consistent with our prediction of UCHL1 being an oncogene in melanoma due to a recurrent H161Y mutation at its active site (Figure 2G). The UCHL1 gene expression association was also replicated in an independent metastatic melanoma cohort (p=0.0002, Cox PH model) (Jayawardana et al., 2015). We reasoned that since UCHL1 expression is associated with poor prognosis in melanoma, it might also be relevant in recent immunotherapy trials in melanoma. Indeed, UCHL1 expression was also associated with worse overall survival in a study of anti-PD-1 treatment (p=0.008) (Hugo et al., 2016) and approached significance in another study with Nivolumab on treatment-naïve patients (p=0.06) (Riaz et al., 2017). This underscores that both E3 ubiquitin ligases and deubiquitinating enzymes might play important roles in cancer progression.
Degron annotations limit the number of significantly mutated UPS substrates
While alterations affecting genes in the UPS pathway would be expected to lead to multiple changes in downstream protein substrates, mutations in the substrates themselves could provide greater specificity for cancer cells by affecting much fewer proteins. We therefore hypothesized that we could identify substrate mutations under positive selection in tumors by finding enriched missense mutations at known degron-related sites (Methods). From the PhosphoSitePlus database (Hornbeck et al., 2015), we found that mutations were enriched in annotated ubiquitination sites in the SF3B1 gene in breast cancer and in the KIT gene in skin cutaneous melanoma (q<0.1, Table S2). Mutations were also enriched at annotated degron sites (Meszaros et al., 2017) located in CTNNB1 (Figure S2A), SPRY1, NFE2L2 (Figure S2B) and EPAS1 (Figure S2C), and phosphodegron sites located in CTNNB1 and CCND1 (q<0.1, Table S2, Figure 3A). An example is CCND1 mutant endometrial tumors (Figure 3B), which as expected showed higher protein expression (Figure 3C, left) and greater cell cycle progression than wildtype tumors (Figure 3C, right). Surprisingly, mutations outside the phosphodegron also displayed a similar trend, largely consisting of truncating mutations that also eliminate the phosphodegron (Figure 3B) while being predicted to escape Nonsense-Mediated Decay (NMD) (Lindeboom et al., 2016). Likewise, CTNNB1 mutant tumors were also associated with a functional effect, including altered transcriptional activity (Figure S2D-E), activation of WNT signaling (Figure S2F) and an altered tumor microenvironment (Figure S2G), consistent with prior reports (Hatzis et al., 2008; Spranger et al., 2015). In total, the significantly mutated genes impacting the UPS, either in the pathway genes directly or on their substrates, to 19% of all cancer driver genes identified by the TCGA PancanAtlas consortium analysis (Bailey et al., 2018). We note, however, that the smaller number of genes with mutations in degrons in cancer is likely due to the considerable sparsity in known degron annotations (Meszaros et al., 2017). Therefore, the true proportion of cancer driver genes impacting UPS is very likely to be higher than 19%.
Figure 3. Somatic mutations are enriched at known degron sites.

(A) Heatmap displaying genes that are enriched for mutations either at literature annotated degron sites (Meszaros et al., 2017), ubiquitination sites (PhosphositePlus), or phosphodegrons (PhosphoSitePlus). Red indicates significant enrichment (q<0.1) for a given gene (y-axis) and cancer type (x-axis) in TCGA.
(B) Lollipop diagram of CCND1 mutations in Uterine Corpus Endometrial Carcinoma (UCEC) in TCGA.
(C) Boxplots showing the association of CCND1 mutations with Cyclin D1 protein abundance (p=4e-8, Wald test) and a marker of cell cycle progression (MKI67, p=0.003) in UCEC. Heatmap shows t-statistics of the association, after adjustment for RNA expression and tumor subtype. Tumor subtypes: CN_LOW=copy number low; MSI=microsatellite instable; POLE=POLE mutated. RPPA=Reverse Phase Protein Arrays.
See also Figure S2
deepDegron infers degron sequences
While a few UPS substrate mutations can be implicated in cancer based on known degrons, systematic investigation requires better degron annotation. To address this challenge, we developed a protein sequence-based model, deepDegron, that leverages data from recently published Global Protein Stability (GPS) analysis of N-terminal and C-terminal sequences from the human proteome (Koren et al., 2018; Timms et al., 2019) to predict degrons (Figure S3). GPS uses fluorescence-activated cell sorting (FACS) to quantify protein stability based on the abundance of a fluorescent reporter protein (GFP, green) fused to a short peptide compared to a control reporter with no fusion partner attached (DsRed, red) (Figure S3A). Because the peptides consisted of known sequences and could contain degrons, we reasoned that deepDegron could learn the sequence rules of degron impact on protein stability. deepDegron is a feed-forward neural network with one input layer, two hidden layers with a ReLU activation function and an output layer (Figure S3B, Methods). Hyperparameters were determined by performance on a leave-out dataset, such as the number of units in each layer, dropout rate, training epochs, and peptide sequence encoding (Figure S3C). On a held-out test set, deepDegron achieved high performance at predicting the results of the GPS assay (Figures 4A and 4B). This was higher than the previously proposed rule-based alternatives (Koren et al., 2018), such as the number of bulky amino acids, the number of acidic residues or top 100 motifs, and better than a combination thereof (logistic regression) (Figures 4A and 4B).
Figure 4. deepDegron accurately predicts the impact of primary sequence on protein stability.

(A) Performance of deepDegron at predicting the stability of C-terminal peptides from the Global Protein Stability (GPS) assay according to the area under the Receiver Operating Characteristic curve (AUC; maximum=1.0, random=0.5) (see “deepDegron data set” in STAR methods).
(B) ROC curve for the N-terminal peptide GPS assay.
(C) Diagram showing that the degron potential score is computed based on the difference between a deepDegron model that uses the position of the amino acids versus one that does not (“Bag of Amino Acids”).
(D) Sequence logo visualizations of select motifs identified by deepDegron (q<0.05, binomial test, Methods).
(E) DeepDegron predicted change in degron potential (delta degron potential) for various mutations of the C-terminal peptide encoded by CHGA.
(F) Correlation between the change in degron potential and the protein stability index according to a saturation mutagenesis study of CHGA.
(G) GPS stability measurements of C-terminal (top) or N-terminal (bottom) peptides derived from the indicated genes, comparing wild-type (gray histograms) and double mutant (red) sequences. X-axis is proportional to GFP / DsRed signal as measured by flow cytometry (see STAR methods); Y-axis is normalized cell count.
Protein stability is likely affected by general biophysical characteristics of the attached peptide in the GPS assay, such as hydrophobicity and intrinsic disorderedness (van der Lee et al., 2014). However, we were most interested in understanding the specific sequence motifs that might mediate degron recognition by specific ubiquitin ligases. Therefore, to infer degrons, we trained two deep learning models: one containing position information from the primary sequence and another without position information (“bag of amino acids” representation, Figure 4C). We hypothesized that the difference between these two models could approximate a degron potential score, where high scores demonstrate position-specific features to be more informative than general degradation properties.
To identify the degron motifs learned by deepDegron, we performed de novo motif enrichment analysis from both the human N- and C-terminome (Table S4, Methods). Our analysis revealed numerous previously known motifs (Figure S4), such as -GG and GA- in C-end and N-end degrons, respectively, but also previously unknown motifs such as C-terminal C[A/G]C[R] and N-terminal [P]LxxR (Figure 4D). While previous models have emphasized the impact of di-amino acid motifs on C- and N-end degrons (Koren et al., 2018; Timms et al., 2019), the discovered motifs suggest that additional complexity might exist with a longer extended degron, albeit with partial degeneracy at these residues as evidenced by the sequence logo plots (Figure 4D). To assess whether deepDegron could accurately predict the impact of mutations on degrons, we evaluated its performance relative to saturation mutagenesis experiments (Koren et al., 2018; Timms et al., 2019). For example, the deepDegron model scored most mutations in the c-end -RG motif as disrupting a degron in the CHGA protein (Figure 4E), as demonstrated by a strong negative change in degron potential when the last two amino acids are mutated. Indeed, when compared to the experimental results, the predicted change in degron potential was, as expected, negatively correlated with protein stability (Figure 4F). Moreover, this negative correlation was observed for all saturation mutagenesis experiments performed on N-terminal and C-terminal peptides (Figures S4E and S4F). Taken together, these results suggest deepDegron is capable of capturing the sequence-level rules of degrons.
To experimentally validate the new degron predictions by the deepDegron model, we used the GPS stability assay. We selected 21 significant degron motifs for testing, comprising 9 predicted N-terminal degrons and 12 predicted C-terminal degrons (STAR methods). GPS was used to examine the stability of the terminal 23-mer peptide derived from each of the 21 proteins, comparing the wild-type sequence to a mutant version containing two point-mutations in the putative degron motif. The precise mutations were chosen such as too maximize the decrease in degron potential as determined by deepDegron (Table S4, STAR methods). Altogether, we found that mutation of 8 out of 12 (67%) C-terminal degrons and 8 out of 9 (89%) N-terminal degrons resulted in protein stabilization (Figure 4G, Figure S4G, Figure S5C, Figure S5N and Table S4G). These results underscored the potential power of deepDegron as a tool for degron discovery.
deepDegron identifies mutations likely disrupting degrons in cancer
Given the strong concordance between deepDegron’s predictions and the available experimental data, we reasoned that we could systematically apply deepDegron to identify mutations that may disrupt degrons in cancer. We thus computed the change in degron potential between the mutated and wildtype sequence in TCGA (delta degron potential), and assessed whether there was enrichment for mutations predicted to disrupt a degron in genes (Methods). Our analysis revealed that mutations in GATA3 and PPM1D had the most significantly disrupted degrons across all cancer types analyzed (q<0.1, Figure 5A, Figure S5A, Table S5). Indeed, for breast cancer in which GATA3 was identified as significant, the change in degron potential (−23) was far more impactful than expected by chance (Figure 5B, Figure S4D).
Figure 5. deepDegron finds C-terminal degrons disrupted by mutations in cancer.

(A) Scatter plot showing the results of the mutational enrichment for C-end degron loss across all analyses (33 cancer types and pan-cancer). P-value resolution is limited to 0.0001.
(B) Example of GATA3 in breast cancer, which shows that the change in degron potential (red) is considerably more negative than the background model (blue).
(C) Lollipop diagram of TCGA mutations in GATA3 for breast cancer. Colored rectangles are Zinc Finger domain 1 (ZnF1) and 2 (ZnF2).
(D) Boxplot showing the association of GATA3 mutations with GATA3 protein abundance in TCGA breast cancer (top left).
(E) Western blot of the protein expression of GATA3 mutants compared to control. F=FLAG tag.
(F) Top, average read coverage profile for peaks. Bottom, overlap of up-regulated ChIP-seq peaks for GATA3 mutants.
(G) Pathway enrichment analysis of up-regulated peaks for GATA3 mutants.
(H) Distribution of expression for genes nearby up-regulated peaks stratified by tumor subtype.
(I) Western blot showing the impact of mutating the GATA3 degron on markers for luminal and basal-like breast cancer.
(J) Western blot analysis of PPM1D (WIP1) mutant versus control. HA=hemagglutinin tag.
See also Figure S5
GATA3 is an essential transcription factor (Figure S5B) that regulates the luminal differentiation of mammary tissue (Kouros-Mehr et al., 2006) and cooperates with ESR1 to mediate estrogen response (Eeckhoute et al., 2007; Theodorou et al., 2013). Heterozygous GATA3 mutations typically occur in the ER+ subtype of breast cancer (Luminal A or Luminal B) and show a clear bias for frameshift and splice site mutations near the 3’ end of the gene (Figure 5C). Notably, the mutations are clustered on the last exon-exon junction such that they are not expected to cause non-sense mediated decay (Lindeboom et al., 2016). According to the deepDegron model, the -AxG sequence (x=any amino acid) at the C-terminus of wild type GATA3 is strongly predictive of its degron potential, and frameshift or splice site mutations would eliminate this motif. Consistent with the predicted loss-of-degron effect for these mutations, we found that GATA3 mutant tumors in TCGA had elevated protein abundance according to Reverse Phase Protein Arrays (RPPA) (p=9e-9, Wald test, Figure 5D). To experimentally confirm the minimal degron region, we generated a double point mutant in the C-terminal -AxG motif of GATA3 and measured protein stability by the GPS assay. Similar to clinical tumor samples, we found the double point mutant of the GATA3 C-terminus had significantly higher protein expression compared to the wildtype sequence (Figure S5C). Moreover, individual substitution of either amino acid led to increased protein expression in the context of the full-length GATA3 protein as assessed by immunoblot, suggesting that both residues are critical for degron recognition (Figure 5E). Given that GATA3 was also substantially upregulated upon treatment with the proteasome inhibitor MG132 (Figure S5D), the identified -AxG motif is likely a degron mediating protein degradation of GATA3 via the UPS. Additionally, RNA expression was not substantially elevated in the mutants (Figure S5E), thus ruling out potential transcriptional effects. These findings were further confirmed in a second cell line (HEK293FT), underscoring the robustness of our finding that mutations lead to degron loss in GATA3 (Figure S5F-H).
Next, we sought to evaluate whether GATA3 mutations mediate their effect on breast cancer through elevated protein expression. If so, these mutations should shift a Basal-like breast cancer cell line (MBA-MD-231) towards a gene expression program of estrogen receptor positive (ER+) breast cancers. We therefore compared the genome-wide binding sites of mutated GATA3 to wildtype GATA3 by ChIP-seq (Figures S5I-J, Tables S5B-E). As a control, we created GATA3 constructs that would be stable regardless of point mutation status by adding a FLAG-tag to the C-terminus of GATA3 (Figure 5E). The addition of extra residues blocks the function of the C-end degron because the location at the extreme C-terminus is required (Koren et al., 2018). Notably, GATA3 mutations led to a consistent overall gain in binding compared to wildtype GATA3 (p<1e-16, Fischer’s Exact test), but only without a FLAG-tag control (Figure 5F, Figures S5K-L). Up-regulated binding sites were preferentially near estrogen signaling genes (Figure 5G), but no pathway was enriched in the presence of a FLAG-tag control (FDR<0.1). Moreover, genes closest to up-regulated binding sites displayed substantially higher expression in ER+ compared to Basal-like subtypes of breast cancer (Figure 5H), which was not the case with the FLAG-tag control (Figure S5K-M). Lastly, mutation of the GATA3 degron shifted protein expression biomarkers towards an ER+ state in the basal-like MDA-MB-231 breast cancer cell line (Figure 5I). Taken together, GATA3 mutations in breast cancer, at least in part, mediate their effect by increasing protein stability through elimination of a degron.
Similar to the GATA3 prediction, deepDegron also predicted that truncating mutations in PPM1D will disrupt a C-terminal -VC degron motif (Figure S5N). PPM1D encodes the Ser/Thr phosphatase WIP1 which negatively regulates p53 (Bulavin et al., 2002; Emelyanov and Bulavin, 2015) and was reported to be frequently amplified in breast cancer (Li et al., 2002; Rauta et al., 2006). Consistent with an oncogenic role through negative regulation of TP53, PPM1D is more essential in TP53 wildtype compared to TP53 mutant cell lines from CRISPR screens reported in DepMap (Figure S5O). Furthermore, PPM1D truncating mutations observed in the TCGA were mutually exclusive with TP53 mutations (p=0.04, one-sided Mantel-Haenzel test), suggesting they might redundantly impact the same pathway. Supporting our prediction of a mechanism involving degron loss, a double point mutant of the -VC motif in WIP1 C-terminal peptide displayed elevated protein stability by GPS (Figure S5P). Point mutation of either amino acid residue also led to increased protein expression of full-length WIP1 according to Western blot analysis (Figure 5J), suggesting that both amino acids are critical. Functionally, the higher protein expression of mutant WIP1 resulted in greater dephosphorylation of known downstream targets in the DNA damage response pathway (Figure 5J), including p53 (Lu et al., 2005; Shreeram et al., 2006). Although in vivo evidence of WIP1 protein expression is not available in TCGA, similar truncating mutations have been reported to lead to greater protein stability of WIP1 and to chemotherapy resistance in acute myeloid leukemia (Hsu et al., 2018; Kahn et al., 2018). Our finding of truncating mutations leading to C-end degron loss in WIP1 (PPM1D gene) is consistent with this reported clinical phenomenon.
Integrative analysis of UPS driver genes identifies putative Transcription Factor (TF) substrates
Having analyzed both UPS substrates and UPS driver genes in isolation, we next wanted to explore the pairing of UPS genes with their substrates. One approach is to correlate the presence of putative driver mutations in UPS components with protein abundance measurements of potential substrates from Reverse Phase Protein Arrays (RPPA) (Li et al., 2013), after adjusting for RNA expression and other covariates (Methods). While we could confirm known UPS-substrate relationships, such as the targeting of CCNE1 by FBXW7 (Koepp et al., 2001; Strohmaier et al., 2001), we could only find a small number of associations (Table S6). This is unlikely to be due to incorrect labeling of cancer driver mutations (Methods), as our predictions were significantly correlated with a previous saturation mutagenesis experiment performed on the E3 ubiquitin ligase BRCA1 (Figure S6). Rather, RPPA only contains abundance measurements for a limited number of proteins (n=198).
To expand our analyses further (Figure S7A-B), we reasoned that transcription factors (TF) might be a particularly amenable substrate to analyze, as the RNA expression of a TF’s target genes might serve as a proxy for the TF protein activity (Figure 6A). We generated differential expression profiles comparing tumor samples carrying wildtype vs putative driver mutations in the UPS genes (Methods). RNA expression of the TF was then adjusted for as a covariate, presumably leaving effects of the TF based on the protein-level. TF regulator analysis using RABIT (Jiang et al., 2015) was then performed to infer substrate TFs based on their target genes defined by thousands of uniformly processed TF ChIP-seq profiles from the Cistrome database (Zheng et al., 2019).
Figure 6. UPS-substrate inference finds association with markers of tumor immune microenvironment.

(A) Diagram depicting the strategy for associating UPS genes with putative transcription factor substrates.
(B) Scatterplot showing the significance of each transcription factor (TF) association for a particular UPS genes (x-axis).
(C) Diagram of inferred substrate relationships of KEAP1 and CUL3.
(D) Western blot showing the co-immunoprecipitation of CUL3 with c-Myc.
(E) Western blot showing increased c-Myc protein abundance in CUL3 KO cells.
(F) Quantification of c-Myc protein half-life upon CUL3 KO in Cal27 and Cal33 cells. Cycloheximide (CHX), a protein translation inhibitor, was given at a concentration of 100 μg/ml. Errorbar = +/− 1 SEM.
(G) Enrichment analysis for degron motifs in associated TF’s for 4 E3 ubiquitin ligases that have a previously reported degron motif (Fisher’s exact test).
(H) Heatmap displaying the association (t statistic) of mutations in UPS driver genes with 5 immune-related biomarkers * = FDR<0.1.
(I) Z-score measuring the relative abundance of cancer cells with a gene knockout when they are co-cultured with T cells, where negative values indicate sensitivity to T cell killing.
As a proof-of-principle, we first tested whether a known TF, NFE2L2, could be retrieved by analyzing its own degron mutations. Indeed, NFE2L2 was correctly identified as the top hit (Figure S7A) for explaining the differentially expressed genes in tumors containing NFE2L2 mutations. Applying the method globally to UPS-substrate inference, we found 494 cancer-specific associations (Table S7) as significant at a conserved family wise error rate of 0.05 (Bonferonni method, corresponding to p<7.8e-7). As some could be downstream effects, we decided to focus on the top 100 associations (Figure 6B), where at most 5 associations per UPS gene are shown in Table 1. Importantly, there was no indication of systematic differences in ChIP-seq quality in our significant results (Figure S7C-F, STAR methods), suggesting technical artifacts are likely low.
Table 1.
Mutated UPS driver genes are associated with transcription factor activity.
| UPS gene | Transcription Factor (cancer type) |
|---|---|
| FBXW7 | EP300 (LUSC, CESC); KLF4 (HNSC, LUSC); MYC (READ, BLCA); GRHL2 (HNSC); XBP1 (UCS) |
| KEAP1 | NFE2L2 (PANCAN, LUAD) |
| WWP2 | EP300 (HNSC); KDM4C (HNSC) |
| BAP1 | XBP1 (BRCA, CHOL, MESO); YY1 (LIHC, PANCAN); CDK9 (PANCAN); MITF (UVM); TAF1 (PANCAN) |
| CUL3 | NFE2L2 (PANCAN, LUSC, KIRP); MYC (PANCAN, KIRP, HNSC); BRD4 (KIRP) |
| SPOP | AR (PRAD); EP300 (UCEC); NKX3-1 (PRAD); ARRB1 (PRAD) |
| TBL1XR1 | XBP1 (BRCA); BRD4 (BRCA); MBD2 (BRCA) |
| TRAF3IP2 | MYC (BLCA); EED (BLCA) |
| CYLD | RELA (HNSC); EP300 (HNSC); FOS (HNSC) |
| MYCBP2 | PROX1 (COAD); MAX (COAD); MYC (COAD) |
| ZBTB11 | EED (HNSC) |
| BIRC6 | HNF4A (ESCA); FOS (HNSC); EP300 (HNSC); MAX (ESCA); KDM4C (HNSC) |
| RNF111 | EP300 (HNSC) |
| MAP3K1 | XBP1 (PANCAN); ESR1 (PANCAN); WDR5 (BRCA); EP300 (CESC); GRHL2 (CESC) |
| UBA1 | RUNX1 (LAML) |
| LTN1 | TTF1 (LUAD) |
| FUS | EP300 (BLCA) |
| KMT2B | STAT1 (HNSC); RFX1 (PANCAN); REST (COAD); CDX2 (COAD); HEY1 (PANCAN) |
| BRCA1 | ESR1 (BRCA) |
| EP300 | IRF4 (BRCA); XBP1 (HNSC, CESC); MAX (HNSC); SMARCA4 (PANCAN); KLF5 (CESC) |
| USP9X | XBP1 (PCPG); GRHL2 (HNSC); GTF2B (BRCA); IRF2 (COAD) |
| CUL1 | CDK8 (BLCA) |
| KMT2A | FLI1 (LIHC); NR2F2 (LIHC); FOXO1 (BLCA) |
| SMURF2 | FOXP1 (SKCM) |
| ZBTB7B | MYOD1 (UCS); GABPA (UCS) |
| VHL | STAT1 (KIRC); ARNT (KIRC) |
| CUL2 | SUPT5H (BLCA) |
| CUL7 | CTCF (BRCA) |
| ZBTB3 | MYH11 (LAML) |
| CUL4B | STAT1 (LGG) |
See also Figure S7
Numerous UPS-substrate associations we identified were previously validated in the literature, such as FBXW7 and c-Myc (encoded by the MYC gene) (King et al., 2013), SPOP and AR (An et al., 2014), and BRCA1 and ERα (encoded by the ESR1 gene) (Eakin et al., 2007; Ma et al., 2010). In some cases, while not finding the direct target, our analysis found proteins that were either regulated by the direct target, such as UPS gene CYLD and downstream RELA (Kovalenko et al., 2003), or interaction partners in the same protein complex, such as VHL-ARNT where ARNT forms a dimer with the known VHL target HIF-1α (Tanimoto et al., 2000). For example, as indicated by our results and previous literature (Shibata et al., 2008), KEAP1 directly regulates NRF2 (encoded by the NFE2L2 gene) and is known to form a complex with CUL3, a scaffold protein for many substrate recognition subunits (Figure 6C). As expected, CUL3 mutations also showed modulation of NRF2, but, unlike KEAP1, were associated with MYC or BRD4 in our analysis. This would suggest an effect based on a different substrate recognition subunit. Supportive of this hypothesis, NFE2L2 showed co-essentiality with both KEAP1 (p=2.6e-7, Wald test) and CUL3 (p=0.02, Wald test) in the cancer cell lines from DepMap CRISPR screens, whereas MYC is co-essential with CUL3 (p=0.0004, Wald test) but not with KEAP1. As expected for a direct regulatory relationship, CUL3 and c-Myc co-immunoprecipitated together (Figure 6D), and knockout of CUL3 resulted in elevated protein expression (Figure 6E) and increased protein half-life of c-Myc in CAL27 cells (Figure 6F). The increased c-Myc protein half-life was also reproducible in a second cell line (Figure 6F, right), which suggests the of role of CUL3 in degrading c-Myc is robust. While a direct assessment of the overall sensitivity or specificity of our approach is not possible due to limited known examples, we did find that for the four E3 ubiquitin ligases with reported degron motifs (Meszaros et al., 2017), there was a significant enrichment of degron motifs in our results (p=0.03, Figure 6G). This suggests our analysis overall is enriched for direct targets.
UPS driver genes correlate with altered tumor-immune microenvironment
We noticed that many of the TFs in our analysis are related to interferon response (STAT1, IRF2 and IRF4) or are potentially immunomodulatory (RELA, XBP1 and MYC) (Cubillos-Ruiz et al., 2017; Grivennikov et al., 2010; Kortlever et al., 2017; Wellenstein and de Visser, 2018). We therefore sought to examine whether mutations in our putative UPS driver genes are associated with altered tumor-immune microenvironment. By correlating the tumor mutation status with previous immune-related signatures from the TCGA (Thorsson et al., 2018), we found that 11 UPS genes had a significant correlation (q<0.1, Figure 6G, Table S3). Many of the associations were related to interferon gamma (IFNG) response, so we examined whether they were hits in a previous CRISPR screen of cancer cells co-cultured with T cells (Pan et al., 2018). In this CRISPR screen, knockout of genes in cancer cells that regulate T-cell-mediated killing are expected to impact the fitness of those cancer cells in vitro, leading to altered representation of corresponding guide RNAs (Methods). Indeed, guide RNAs targeting CUL3 (q-value=0.0001) and FBXW7 (q-value=0.002) exhibited significant depletion in the CRISPR screen (Figure 6H), suggesting increased sensitivity to T cell killing. Since CUL3 mutations in human tumors are correlated with weak IFNG response, this would suggest that either CUL3 mutations might only be advantageous for cancer cells in a low IFNG environment or that CUL3 mutations might attenuate cancer cell response to IFNG. In either scenario, we reasoned that an altered cancer cell IFNG response could change the anti-tumor efficacy of cytotoxic T lymphocytes (CTL). Indeed, for head and neck squamous cell carcinoma, we found that a proxy for CUL3-activitiy based on NRF2 (encoded by NFE2L2) protein abundance altered the association between a CTL biomarker and overall patients’ survival (Figure S7H). Future experiments are needed to clarify which CUL3 substrate recognition adaptor protein and its corresponding substrate mediate this effect. In summary, these analyses revealed a potential immunomodulatory role of UPS in IFNG response in cancer.
Discussion
While the tumor transcriptome has been extensively studied by RNA-seq, an understanding of how dysregulated pathways lead to altered proteomic states is far less understood. This is in spite of early DNA sequence studies of human cancers implicating driver genes that impact protein degradation through the Ubiquitin-Proteasome System (UPS) (Barbieri et al., 2012). In this study, we addressed this issue by performing a systematic analysis of the UPS and its corresponding substrates in 33 human cancer types. This revealed a much larger role of UPS in cancer than previously appreciated, constituting over 19% of cancer driver genes. Moreover, our study includes the technical innovation of modeling degron loss by deep learning (deepDegron) and associating potential transcription factor substrates of UPS genes by their inferred activity from TF ChIP-seq targets. By considering all components of the whole UPS pathway, de novo degrons from machine learning, and transcription factor substrates, our study increased the significantly mutated UPS genes compared to (Ge et al., 2018) by ~3-fold, and expanded the analysis of UPS substrates by ~4-fold compared to (Martínez-Jiménez et al., 2020). These approaches could also be leveraged by researchers of other diseases to interpret the role of protein degradation.
Our study has several limitations. First, while our analysis of transcription factor substrates of the UPS did identify bona fide direct targets, it was unavoidable to also find transcription factors that are either regulated by or reside in the same protein complex as the actual substrate. Thus, careful considerations should be given to the possibility of related proteins when interpreting results. Second, although our analysis had the power to identify UPS driver genes mutated at low frequencies, for some of these genes there were simply not enough mutations to make confident associations with potential substrates. Larger multi-omic studies or larger tumor profiling cohorts will be better powered to make such connections in the future. Lastly, while our study provides an important advance in trying to systematically understand the UPS in cancer, we are far from a complete landscape. One reason is that due to the lack of systematic protein stability assays, we were not able to infer mutated degrons in the middle of proteins. The other reason is that since mass spectrometry-based proteomics that can assess upwards of 10,000 proteins have only been conducted on limited samples in limited cancer types (Mertins et al., 2016; Zhang et al., 2016), we could only associate potential substrates that are either transcription factors or on the RPPA panel (~200 proteins).
Although truncating mutations are commonly associated with tumor suppressor genes and a loss-of-function effect (Vogelstein et al., 2013), we found that truncating mutations may actually be gain-of-function in oncogenes for more cases than previously appreciated. For example, CCND1, GATA3, and PPM1D have truncating mutations clustered near the 3’end of their respective genes, which are predicted to lead to degron loss and showed evidence for higher protein abundance. This is somewhat surprising as it has been previously suggested that truncated proteins are rapidly degraded by protein quality control mechanisms (Goldberg, 2003). Indeed, clinical databases, such as OncoKB (Chakravarty et al., 2017), have assumed GATA3 truncating mutations as likely loss-of-function, but our evidence would suggest otherwise. Experimental point mutants of the GATA3 degron recapitulated the increased protein abundance seen for truncating mutations. Notably, truncation of the N-terminal part of proteins that lead to degron loss is appreciated for fusion genes, such as TMPRSS2:ETV1 (Vitari et al., 2011) and TMPRSS2:ERG (Gan et al., 2015). It is also possible that truncation of other types of inhibitory sequences could produce similar pro-oncogenic phenotypes, so not all cases of clustered truncating mutations may result in degron loss. Nonetheless, we expect as more degron motifs are discovered, there will be a concordant increase in identifying gain-of-function truncating mutations.
Our finding that most driver genes in the UPS are tumor suppressors suggest that therapeutic targeting of up-regulated substrates may be a more therapeutically efficacious strategy than targeting the UPS driver genes themselves. Indeed, mutations in the E3 ligase SPOP abrogate androgen receptor (AR) protein degradation (An et al., 2014) and targeted therapies against (non-mutated) AR are effective in prostate cancer (Watson et al., 2015). The advent of PROTACs may be a key advance, as unaffected UPS genes could be co-opted to replace the function of mutated tumor suppressor genes. Moreover, this same approach conceptually could be applied to target substrates that have escaped UPS recognition through mutations that result in degron loss. To this end, numerous questions about the UPS remain to be answered, among such: are mutations in UPS driver genes preferentially selected because of their impact on single or multiple substrates? what are all the substrates of each specific UPS driver gene? why are UPS genes a driver in one cancer type but not in another? Future studies with increasing scale of tumor proteome-wide profiles may resolve such questions and capture a comprehensive picture of how the UPS modulates cancer initiation and progression. In the future, improved understanding of the UPS will undoubtedly provide insights guiding the development of novel cancer therapeutics that target protein degradation.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents (including code) should be directed to and will be fulfilled by the Lead Contact, X. Shirley Liu (xsliu@ds.dfci.harvard.edu).
Materials Availability
Materials associated with the paper are available upon request to Lead Contact, X. Shirley Liu (xsliu@ds.dfci.harvard.edu).
Data and Code Availability
Raw gel pictures and data necessary to recreate figures using the Jupyter notebook code (see below) are available on mendeley data: http://dx.doi.org/10.17632/kgfzbpv2w4.1. Raw sequencing data, called peaks and bigwig files for GATA3 ChIP-seq are available GEO (GSE162003). The DeepDegron code is available on github: https://github.com/ctokheim/deepDegron. The code for associating UPS genes with putative transcription factor substrates is also available on github: https://github.com/ctokheim/tf_association. Jupyter notebooks for data analysis are stored on github (https://github.com/ctokheim/Tokheim_Mol_Cell_2020).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Human embryonic kidney 293FT cell line (HEK293FT) was obtained from Thermo Fisher Scientific. HEK293T cells were grown in DMEM supplemented with 10% fetal bovine serum, 2% penicillin/streptomycin, 1% L-glutamine and 100 mM sodium pyruvate according to standard protocol and maintained at 37°C with 5% CO2.
Human breast cancer MDA-MB-231 cell line was obtained from American Type Culture Collection (ATCC). MDA-MB-231 cells were grown in DMEM supplemented with 10% fetal bovine serum, 2% penicillin/streptomycin, 1% L-glutamine and 100 mM sodium pyruvate according to standard protocol and maintained at 37°C with 5% CO2.
Human oral squamous cell carcinoma cell lines CAL27 and CAL33 were kindly provided by Ravi Uppaluri laboratory. Cells were cultured in in DMEM supplemented with 10% fetal bovine serum, 2% penicillin/streptomycin, 1% L-glutamine and 100 mM sodium pyruvate according to standard protocols and maintained at 37°C with 5% CO2. Cell lines were stored in liquid nitrogen at early passages and were cultured within 20 doublings.
METHOD DETAILS
Mutation dataset
We used somatic mutations from 33 cancer types called by the MC3 group in The Cancer Genome Atlas (TCGA) (https://gdc.cancer.gov/about-data/publications/pancanatlas, v0.2.8), which were formed by the consensus of multiple mutation calling algorithms in a unified pipeline (Ellrott et al., 2018). We then filtered the dataset according to quality control metrics for both mutations and tumor samples. Specifically, the following filters were applied: 1) mutations should have passed all QC metrics by the MC3 group (i.e., “PASS” in the “filter” column), except for the allowance of whole genome amplified samples in ovarian cancer and AML where the majority of tumor samples used a whole genome amplification step; 2) tumor samples which failed pathology review were excluded; 3) for statistical power reasons, we excluded hypermutated tumors (Lawrence et al., 2014; Tokheim et al., 2016), defined as having a greater number of mutations than 1.5x the interquartile range above the 3rd quartile (Tukey’s condition) for the respective tumor’s cancer type. Because this procedure also excludes outliers for cancer types with overall low tumor mutation burden, we also required the tumor sample to have greater than 1,000 mutations to be excluded. These filters resulted in 1,457,702 mutations for final analysis.
Gene and Protein Expression Data
Gene expression estimates from RNA-seq were quantified from the RSEM v2 pipeline (Li and Dewey, 2011) of TCGA. The data was downloaded from the Genomic Data Commons website (http://api.gdc.cancer.gov/data/3586c0da-64d0-4b74-a449-5ff4d9136611). RNA expression values were log normalized (i.e. log2(RSEM+1)) and centered with median value of zero per gene. Normalized protein expression from Reverse Phase Protein Arrays (RPPA) was also download from the Genomic Data Commons website (http://api.gdc.cancer.gov/data/fcbb373e28d4-4818-92f3-601ede3da5e1).
Ubiquitin-Proteasome System (UPS) pathway genes
We curated a set of UPS genes from two previous publications (Ge et al., 2018; Meszaros et al., 2017), which included E1 activating enzymes, E2 conjugating enzymes, E3 ubiquitin ligases and deubiquitinating enzymes. We used only those annotated with literature support from Ge et al. and the E3 ubiquitin ligases reported by Meszaros et al. Additionally, we removed a gene, CDH1, that was erroneously labeled as involved with ubiquitination due to conflicting symbols with a known UPS gene (FZR1, known at the protein-level as Cdh1). This resulted in a set of 775 genes for further analysis (Table S1).
Driver gene analysis
To ascertain which genes in the UPS pathway might promote cancer development and progression, we analyzed whether genes in the UPS were significantly mutated in human cancers by the method 20/20+ (Tokheim et al., 2016). 20/20+ was ran using default parameters except for usage of 100,000 simulations, as described previously (https://github.com/KarchinLab/2020plus, v1.2.0) (Bailey et al., 2018), on each of the 33 cancer types individually and all cancer types aggregated together (known as a “pan-cancer” analysis). Briefly, 20/20+ is a random forest method that scores the propensity of a gene to be an oncogene, a tumor suppressor gene or, in general, a cancer driver gene (scores range from 0 to 1). P-values for each score are then computed based on a Monte Carlo simulation procedure that generates a background distribution of mutations accounting for nucleotide sequence context (probabilistic2020 python package, v1.2.0). Here, to increase statistical power to identify lowly mutated driver genes in the ubiquitin pathway, we performed a restricted hypothesis test on only the 775 UPS genes annotated above. Genes were deemed significant at a false discovery rate of 0.05 (Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995)) and those with high effect size (score > 0.5 out of 1.0).
Lollipop diagram visualization
Mutations on protein sequence were visualized using the ProteinPaint tool (https://pecan.stjude.cloud/proteinpaint) (Zhou et al., 2016). Mutations were submitted according to their genomic coordinates and mutations that do not match the default reference transcript used by ProteinPaint are not shown. Height corresponds to the number of mutations while the x-axis represents the codon position along the protein sequence. Protein domains are shown as colored boxes along the protein sequence.
Expression and essentiality analysis of putative driver genes
The putative UPS driver genes were characterized by their tissue expression from GTEx (Consortium et al., 2017) and cancer cell line essentiality in CRISPR screens from DepMap (~500 cell lines) (Meyers et al., 2017). The 63 driver genes were compared to both other UPS genes not found as drivers and all other non-UPS genes using a Mann-Whitney U test. Version 7 of TPM expression values from GTEx were used (https://gtexportal.org/home/). Additionally, CERES scores (Meyers et al., 2017), which quantify how essential a gene is in CRISPR screens, were obtained from the 2019 Q1 data release of DepMap. Negative CERES scores indicate a gene is essential in a particular cancer cell line.
Recent evidence suggests context-specific roles for UPS driver genes (Haigis et al., 2019), such as PARP inhibitors being selectively effective in BRCA1-mutant tumors in traditionally BRCA-associated cancer types (breast, ovary, prostate and pancreas) (Jonsson et al., 2019). We therefore examined the specificity of UPS driver genes in both cell-lineage and genetic mutation contexts. According to the Genotype-Tissue Expression (GTEx) data (Aguet et al., 2017), we noticed that putative UPS driver genes were expressed in most normal tissues, and more broadly expressed than other UPS genes or non-UPS genes (p<0.05, Figure 2H). However, from CRISPR screens across ~500 cancer cell lines from the DepMap (Meyers et al., 2017), UPS driver genes showed significantly higher variability in the gene dependency scores (CERES scores) across cell lines compared to other genes (p<0.05), suggesting substantial cell type-specific essentiality (Figure 2I). One possible explanation for the variable gene essentiality despite widespread expression might, in part, arise from cells uniquely expressing or modifying certain important substrates of the UPS (Figure S1F). This is consistent with previous literature that substrate recognition by E3 ubiquitin-ligases, such as c-CBL and β-TrCP, can depend on signaling pathways which mark degrons by phosphorylation (Zheng and Shabek, 2017).
Mutational co-occurrence
We analyzed whether non-silent mutations in putative driver genes in the ubiquitin pathway would tend to co-occur in the same tumor samples with mutations in 299 driver genes identified previously by the TCGA (Bailey et al., 2018). We used the Mantel-Haenszel test to identify pairs of genes with an odds ratio significantly different from 1.0 at an FDR threshold of 0.25. To control for the confounding effect of tumor mutation burden, we adjusted for high (>500 mutations; half the hypermutator threshold) and low (<=500 mutations) tumor mutation burden samples in our analysis. In the pan-cancer analysis, we also adjusted for the cancer type of the tumor labeled by TCGA.
Next, we sought to examine whether mutations in UPS driver genes would contextually co-occur or be mutually exclusive with mutations in other driver genes in the same tumor. This revealed 13 of the UPS driver genes with an enriched co-mutational pattern with other driver genes previously identified by TCGA (Bailey et al., 2018) (Figure S1H, Table S1). For example, we found KEAP1-KRAS-STK11 to be co-mutated in lung adenocarcinoma (LUAD) tumors, which have been reported to form a biologically distinct subtype of KRAS mutant LUAD (Skoulidis et al., 2015). Previously, mutations in STK11 have been implicated in a T cell exclusion phenotype for these tumors and ultimately responsible for resistance to immune checkpoint inhibition (Hellmann et al., 2018). Instead, we found that mutation of the E3 ligase KEAP1, regardless of STK11 status, correlates with lower immune infiltration in TCGA (Figure S1I), suggesting that KEAP1 has additional immunomodulatory roles. The interaction with other driver genes might be partially related to UPS driver genes being preferentially situated centrally in a protein-protein interaction network (Figure S1J), a property previously noted for other driver genes (Davoli et al., 2013). In summary, the 63 putative UPS driver genes we identified showed context-specificity with regard to both cell type and genetic mutations.
Global Protein Stability (GPS) Assays
GPS experiments were performed as described in Koren et al., 2018 and Timms et al, 2019. Individual sequences encoding example 23-mer peptides were PCR-amplified from either the N-terminome (Timms et al., 2019) or C-terminome (Koren et al., 2018) oligonucleotide libraries and cloned into lentiviral GPS expression vectors. Lentivirus was packaged through the transfection of HEK-293T cells (ATCC® CRL-3216™) grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (HyClone) and penicillin/streptomycin (Thermo Fisher Scientific). HEK-293T at around 70% confluency were transfected with the GPS vector plus four packaging plasmids (encoding Gag-Pol, Rev, Tat and VSV-G) using PolyJet In Vitro DNA Transfection Reagent (SignaGen Laboratories) as recommended by the manufacturer. The media was changed after 24 hours, and the viral supernatant collected a further 24 hours later. Following centrifugation (800 × g, 5 min) to remove cellular debris, the viral supernatant was applied to target HEK-293T cells. After a further 48 hours, stability measurements were made by flow cytometry using a BD LSRII instrument (Becton Dickinson); at least 10,000 DsRed+ cells were collected in each case. The resulting data were analyzed using FlowJo software.
deepDegron
deepDegron is a feed forward neural network trained on the Global Protein Stability (GPS) assay (Yen et al., 2008), which at proteome-scale measures the conferred stability or instability of peptides when attached to GFP in HEK293T cells. Importantly, the GPS assay also contains an internal control DsRed (located on the same transcript) which does not contain an attached peptide. FACS is then used to sort cells based on the red (DsRed) to green (GFP) ratio into separate bins and subsequently barcodes are sequenced to quantify the representation of peptides in each bin.
Data set.
Data from the GPS assay related to N-terminal (Timms et al., 2019) and C-terminal (Koren et al., 2018) peptides were collected from their respective publications and analyzed separately. In the case of the C-terminal data, we analyzed the full 23-mer peptide screen. While for the N-terminal data, we only analyzed peptides with an initiator methionine (24-mer), but since the methionine was always the same at the first position, we did not include the methionine in our model (23-mer). To establish a classification task for the deepDegron model, we binarized each peptide into two classes based on the mode of the read count distribution across bins in the GPS assay. If a peptide’s modal bin was in the lower half of the red to green ratio it was assigned as instable (class=1) and the remaining were assigned as stable (class=0). If a gene had multiple peptides in the GPS assay, we only used the first occurrence for further analysis.
Neural network.
deepDegron, a two hidden-layer feed forward neural network, was trained using the Keras python package with the tensorflow backend (https://github.com/ctokheim/deepDegron). ReLu activation functions were used for hidden layers and the sigmoid function was used for the final output node, which generally performs well for neural network models (He et al., 2016; Krizhevsky et al., 2012; Simonyan and Zisserman, 2014). Training was performed using the Adam optimizer using the default learning rate, given it has previously been suggested that Adam gives superior results compared to other optimizers (Kingma and Ba, 2014).
Training, validation and test sets.
We randomly separated out 30% of the sequences for purpose of evaluation as a test set. For the remaining 70% of the data, we randomly split again 70% (49% overall) of that data into a training set and 30% (21% overall) as a validation set for hyperparameter selection.
Hyperparameters.
Like most machine learning algorithms, neural networks benefit from fine tuning hyperparameters of the model. Here, we utilized grid search over hyperparameters for both feature engineering and neural network parameters. For feature engineering, we considered position-specific one-hot encoding of various lengths of the peptide from the terminal-ends (l=6, 12, 18 or 23) with the remaining portion of the peptide sequence encoded only in terms of the count of each amino acid type (i.e. position agnostic). This was intended to limit the number of learned parameters of the model, if certain regions of the peptide were more important. Additionally, given previous evidence of the importance of dimer amino acid motifs at the very end of protein sequence (Koren et al., 2018), we also allowed for the one-hot encoding of di-amino acid motifs (di=True or False). For neural network parameters, we considered different number of nodes for each layer (n=8 or 16). Additionally, we considered various levels of dropout regularization (d=0, 0.25 or 0.5) for connections between the input and 1st hidden layer since it contained the greatest number of parameters in the model. Lastly, we also considered the number of epochs used for training (e=20, 40 or 60).
Evaluation.
The optimal hyperparameters were selected according to the highest area under the Receiver Operating Characteristic curve (auROC) on the validation data set. The C-terminal deepDegron hyperparameters that were selected are: n=8, d=0.0, e=20, l=6 and di=True. While the N-terminal deepDegron hyperparameters that were selected are: n=16, d=0.5, e=20, l=6 and di=True.
The deepDegron models were then compared to a Random Forest model (scikit-learn with 1,000 trees as performance usually only increases with this parameter (Oshiro et al., 2012)), which empirically performs well on many machine learning tasks (Caruana and Niculescu-Mizil, 2006), and previously proposed rule-based alternatives (Koren et al., 2018), such as the number of acidic residues (D, E), number of bulky hydrophobic residues (F, W, Y) or the number of top 100 motifs. Evaluations for all models were performed on the held-out test set and compared using the auROC metric.
Degron Potential Calculation
We calculated a degron potential score to correct for protein stability likely reflecting both amino acid order effects (e.g., a degron motif exists) versus general amino acid properties. To do this, in addition to the model outlined in the deepDegron section (Methods), we trained a second model (“bag of amino acids”) containing the same hyperparameters that only has the count of each amino acid in the peptide sequence as features (20 features). We then calculated a degron potential score as the difference in prediction between the position specific model and the “bag of amino acids” model.
Motif Analysis
Motif analysis was conducted by measuring enrichment for sequence motifs among top degron potential scored peptides from deepDegron. First, we ranked all peptide sequences by degron potential score from high to low likelihood of containing a degron. Second, we performed area auROC analyses to calculate at which point the top degron potential sequences would cease to have meaningful enrichment. To determine this cutoff, we computed at various cutoffs a delta auROC score, which we defined as the difference in auROC between the two deepDegron models (position specific versus “bag of amino acid” model) tested on sequences where the top-ranking X and bottom-ranking X sequences were removed. The delta auROC was calculated and plotted over various cutoffs of X ranging from 0 to 8000 with an increment of 20. We then used the elbow-method (Goutte et al., 1999) based on the point of maximal curvature to delineate the transition (X*) from a performance gap existing to nearly equivalent performance. Since curvature is only well defined for continuous functions, we used an algorithmic approximation from the kneed python package with default parameters (v0.4.1, https://github.com/arvkevi/kneed) (Satopaa et al., 2011). Third, we calculated the background probability p that a particular peptide would contain particular motifs of length 2 (with or without gaps) and 3. We only considered motifs within the proximal 6 amino acids to either the N-terminus or the C-terminus, as our performance evaluation above suggested most gains were in this region. Additionally, since the number of possible motifs grows exponentially with motif length, we only considered gapped and position-specific motifs for length 2 motifs. Fourth, using a binomial model with background probability p, we measured whether motifs had significantly more motifs c than expected for the top X* sequences. Fifth, we corrected for multiple hypotheses by the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995) and declared significant motifs at false discovery rate threshold of 0.05. Lastly, to identify potentially extended motifs outside those identified by our analysis, we generated sequence logo visualizations by compiling all the top sequences that contained the motif and inputting these sequences into the WebLogo software (Crooks et al., 2004).
Monte Carlo simulations
To establish a background distribution of mutations, we performed Monte Carlo simulations as described previously (Tokheim et al., 2016). Briefly, for single nucleotide variants, we moved mutations uniformly at random within the same gene but matched the same nucleotide context as the observed mutation (C*pG, CpG*, TpC*, G*pA, A, C, G, T). Indels were moved within the same gene without regard for the flanking sequence, as mutational signatures for indels are less known than for single nucleotide variants (Alexandrov et al., 2013). Based on the simulated mutations, we then recategorized the effect of the variant. For example, a mutation may have originally been a nonsense mutation but when moved to a new position it may be a missense mutation in a known degron site. Test statistics for degron enrichment were then computed and this simulation procedure was repeated 10,000 times. P values were computed based on the resulting empirical distribution, i.e., the fraction of simulations with test statistics that were as or more extreme then the observed value.
Mutation enrichment at known degrons, ubiquitination sites or phosphodegrons
Known degron sites were collected from a recent literature review (Meszaros et al., 2017), while ubiquitination sites and phosphodegrons (phosphorylation sites annotated as involved with “protein degradation”) were obtained from the PhophoSitePlus database (Hornbeck et al., 2015). For each cancer type, we analyzed whether the number of missense mutations found in annotated sites of a gene were higher than expected based on an empirical background distribution established through Monte Carlo simulations (see section above). In the case of the phosphodegron analysis, we also considered the flanking 3 amino acids on either side of the phosphorylation site. Genes were deemed significant at a False Discovery Rate (FDR) of 0.1. Based on manual review of the literature, one significant result (BRAF, ubiquitination site enrichment due to K601E mutations) was excluded from further analysis due to previously literature suggesting a distinct mechanism of action (Yao et al., 2017).
Calculation of degron impact bias
Because known degron sites are limited, we also assessed for genes containing a significant enrichment of mutations predicted to lead to degron loss by deepDegron. First, we computed the change in degron potential (delta degron potential) between the mutated and reference protein sequence for each mutation in the 33 cancer types available from TCGA. Second, we computed a gene-wise test statistic as the sum of delta degron potential for all mutations within a gene. Scores considerably less than zero indicate degron loss. Third, to evaluate the statistical significance, we performed Monte Carlo simulations (described above) to compute a p-value corresponding to seeing a score equal to or lower than the observed value (i.e. degron loss). Like for the known degron case, significant enrichment was defined at an FDR of 0.1 and, additionally, required the delta degron potential to indicate a preferential loss of a degron (delta degron potential below −1).
Selection of deepDegron motifs for experimental validation
To validate deepDegron predictions for degrons, we selected 10 novel motifs for experimental validation. For this we used the GPS assay to compare the protein stability of GFP fused to either the wildtype peptide, or one containing point mutations in the predicted degron motifs. Since some motifs partially overlapped, we prioritized motifs based on statistical significance (q<0.05) and independence from other tested motifs. Motifs were equally divided between predicted C-terminal (-LxRxx, -MxxxV, -CxxR, -VS, and -LxxAx; x=any amino acid) and N-terminal degrons (GxL-, xPL-, RxR-, GxxxA- and RxxP-). To avoid introducing generally stabilizing amino acids that are independent of a degron motif, point mutations were selected based on maximally decreasing the degron potential of the sequence while maintaining the score of the “bag of amino acid” model within a range of 0.1 from the original sequence. The selected double mutants for each motif are listed in Table S4G. The same selection procedure for point mutants was carried out for the degron motifs of GATA3 and PPM1D.
Generation of lentiviral expression vectors
Plasmids (hWIP1-FLAG, pHAGE-GATA3) were obtained from Addgene. Overexpression vector pLenti-EF1a-PGK-Puro was kindly provided by Kai Wucherpfennig laboratory. Different forms of wild-type or mutated GATA3/PPM1D sequence were amplified by PCR and subcloned into a pLenti-EF1a-PGK-Puro empty vector via Gibson assembly to generate different overexpression vectors (GATA3 and PPM1D). Next, small amount (1 μl) of the Gibson assembly reactions was transformed into competent cells. Competent cells were incubated on ice for 30 minutes, then subjected to heat shock in a water bath or electroporated by a Gene Pulser Xcell Electroporator (Bio-Rad Laboratories) and returned to ice for 2 minutes. LB media (1 ml) was added to the competent cells and the cells were allowed to recover at 37 °C for 60 minutes on a shaker; subsequently 30 μl of the mixture (LB+ competent cells) was plated on LB-agar plates containing 100 μg/ml ampicillin and incubated at 37 °C overnight (12–16 hours).
Generation of CRISPR/Cas9 Knock-out cells
Construction of lenti-CRISPR/Cas9 vectors targeting AAVS1 (Control) or CUL3 was performed following the protocol associated with the backbone vector lentiCRISPR_V2 (Addgene). The sgRNA sequences used are listed in the Key Resources Table. CAL27 cells were infected with lentivirus expressing sgRNAs targeting AAVS1 or CUL3. After puromycin selection for 3 days, cells were expanded for at least 7 days and collected. CUL3 knockout was verified by western blot analysis.
Viral library production
The pLenti-EF1a-GATA3/pLenti-EF1a-PPM1D expression constructs and the empty pLenti-EF1a-PGK-Puro vector were transfected into the 293FT cell line at 80–90% confluency in 10 cm tissue culture plates. Viral supernatant was collected at 48 and 72 hours post-transfection, filtered via a 0.45 mm filtration unit (Corning). The supernatant was subsequently aliquoted and stored in −80 °C freezer until use.
Viral transduction of cells
Cells were cultured in complete growth medium according to standard protocols. For viral transduction, a total of 3×105 cells were transduced with lentivirus containing gene cDNA construct described above at a high level of multiplicity of infection (MOI) in 10 cm tissue culture plates. After puromycin selection for 3 days, surviving cells were allowed to grow for another 7 days to overexpress specific genes. Immunoblotting and PCR were performed to confirm the expression of specific genes.
Co-immunoprecipitation of CUL3 protein
Human oral squamous cell carcinoma CAL27 cells were lysed in Tris buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 5% glycerol, with protease and phosphatase inhibitors) for 30 min with gentle rocking at 4°C. Cell lysate was spun down by a centrifuge in cold room at 12,000 rpm for 10 minutes and then supernatant was collected and incubated with CUL3 antibody coupled to Protein A/G agarose beads (Pierce Biotechnology) at 4°C overnight (12 hours). Beads were washed extensively in Tris lysis buffer containing 0.5 M NaCl and then eluted in LDS-sample buffer (Invitrogen) containing 1% 2-mercaptoethanol. Cell lysate was supplemented with 4X SDS loading buffer (0.2 M Tris-HCl, 0.4 M DTT, 8.0% SDS, 6 mM Bromophenol blue, 4.3 M Glycerol) and heated at 95 °C for 15 minutes before western blot analysis.
Western Blot of protein expression in human cells
Pellets from 5 × 106 cells were collected and digested by 500 μl RIPA Buffer (Invitrogen). Samples were incubated on ice for at least 15 minutes and centrifuged at 12,000 rpm for 10 minutes at 4°C, then subjected to BCA analysis (Thermo scientific). Approximately 40–60 μg of total protein from each sample was loaded for western blot analysis.
Measurement of protein half-life
Cancer cells (1×106) were seeded onto 100mm petri dishes in complete growth medium according to standard protocols and incubated in a CO2 incubator. After 24 hours incubation, remove the medium and add complete medium with 100μg/ml cycloheximide (CHX; dissolved in DMSO) into each dish. Cells were exposed to cycloheximide for 0, 4, 8 or 12 hours to inhibit the protein synthesis according to the experimental design. Then, cell lysates were collected at different time points and MYC protein levels were examined by western blot using an anti-MYC antibody. Western bands of MYC and β-ACTIN were quantified in triplicates using ImageJ software.
Real-time reverse transcription-PCR
RNA was extracted using RNeasy Plus Mini Kit (Qiagen) from HEK293FT and MDA-MB-231 cells. Then, RNA was reverse transcribed into cDNA using iScriptTM cDNA Synthesis Kit (Bio-Rad Laboratories). Approximately 50 ng cDNA from each sample was mixed with gene-specific primers (Table S5) and SsoAdvancedTM universal SYBR® Green supermix (Bio-Rad Laboratories) following the manufacturer’s protocol. Reactions were performed on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories).
ChIP sequencing of GATA3
MDA-MB-231 cells were plated in 15 cm tissue culture plates and cultured for 3 days. For GATA3 ChIP-sequencing, approximately 1×107 cells per condition were harvested and crosslinked by a two-step fixation, including 2 mM disuccinimidyl glutarate (DSG, Life Technologies) treatment for 45 minutes and followed by 10 minutes fixation using 1% methanol-free formaldehyde at room temperature (Eeckhoute et al., 2007; Singh et al., 2019). Cells were lysed in 1% SDS lysis buffer and sheared to 200–700 bp in size using the Covaris E220 ultrasonicator (PIP 140, DF 5%, CPB 200). Approximately 50 mg of sheared chromatin per condition were diluted and then incubated overnight with 5 ug GATA3 antibody (14074, Cell Signaling). Precipitates were then washed with following buffers: RIPA 0 buffer (0.1% SDS, 10 mM Tris-HCl pH 7.4, 1% Triton-X100, 1 mM EDTA, 0.1% sodium deoxycholate), RIPA 0.3 buffer (0.1% SDS, 1% Triton-X100, 0.1% sodium deoxycholate, 10 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.3 M NaCl) and LiCl buffer (250 mM LiCl, 1 mM EDTA, 5% NP-40, 0.5% sodium deoxycholate, 10 mM Tris-HCl). DNA sequencing libraries were prepared using the Smarter Thruplex DNAseq kit (Takara Bio Inc.) according to the manufacturer’s protocol. Libraries were sequenced on an Illumina HiSeq 2500 with 150 bp paired-end reads.
Data analysis of GATA3 ChIP-seq
Chromatin Immunoprecipitation sequencing (ChIP-seq) of GATA3 was analyzed using the ChiLin pipeline (Qin et al., 2016). Briefly, the Sentieon Bwa-mem aligner was used to map reads to the hg38 reference genome (https://support.sentieon.com/manual/). ChIP-seq peak calling was then performed using MACS2 v2.1.4 (Zhang et al., 2008), with the following parameters: “-SPMR -B -q 0.01 –keep-dup 1”. Mapped reads were then down sampled to 4 million for subsequent quality control analysis. Quality control consisted of five metrics (Table S5): 1) the average read quality according to FastQC (Andrews, 2010); 2) the fraction of uniquely mapped reads; 3) a PCR bottleneck coefficient, which is the fraction of locations with one uniquely mapped read; 4) fraction of reads in peaks according to MACS2 (Zhang et al., 2008) (more, the better); 5) overlap of peaks with DNA hypersensitivity sites. All samples were of adequate quality.
To provide a consistent peak set across multiple samples for downstream analysis, we merged overlapping peaks using bedtools v2.29.2 (Quinlan and Hall, 2010). Differential peak analysis between wildtype GATA3 and mutant GATA3 was then performed using DESeq2 with the default Wald test (Love et al., 2014). Peaks were regarded as significant at Benjamini-Hochberg False Discovery Rate of 0.1 (Table S5). A heatmap visualizing the peaks was then generated using the deeptools package (v3.3.0) (Ramirez et al., 2016). KEGG pathway enrichment of the up-regulated GATA3 peaks was then conducted using Cistrome GO (Li et al., 2019).
Labeling of driver mutations
Even implicated cancer driver genes contain a mixture of driver and passenger mutations when examined across multiple patients’ tumors (Torkamani and Schork, 2008). Therefore, we restricted our subsequent analysis of putative substrates or immune-related biomarkers to likely driver mutations in the implicated set of 63 ubiquitin pathway genes. For tumor suppressor genes, we regarded any loss-of-function mutation (frameshift insertions or deletions, nonsense mutations, splice site mutations, lost start mutations, or lost stop mutations) as likely oncogenic, which is consistent with variant annotation guidelines from curated databases such as OncoKB (Chakravarty et al., 2017). However, the interpretation of missense mutations is often more difficult. We therefore used missense mutations that were previously reported to be drivers by CHASMplus at an FDR of 0.01(Tokheim and Karchin, 2019).
Comparison of CHASMplus to saturation mutagenesis
To understand the accuracy of the driver mutation labeling by CHASMplus, we compared predictions to a recent saturation mutagenesis study (Findlay et al., 2018) of the functional effect of all BRCT and RING domain variants in BRCA1, an E3 ubiquitin ligase. The study used a multiplexed functional assay in a homology-directed repair (HDR) sensitive cell line (HAP1) to measure the impact of BRCA1 mutations. Scores for CHASMplus were obtained from OpenCRAVAT (https://opencravat.org/) (Masica et al., 2017) and then assessed for their spearman correlation with the functional HDR scores. Additionally, CHASMplus scores were assessed for their performance at distinguishing ClinVar labeled pathogenic versus benign variants in BRCA1 based on the area under the Receiver Characteristic Curve. ClinVar labels were obtained from Findlay et al. (n=46).
Quality control of Cistrome ChIP-seq data
First, we examined the overall distribution of 5 quality control (QC) metrics for ChIP-seq from putative substrates identified by Rabit compared to all transcription factors in the Cistrome database. The 5 QC metrics were: 1) the average read quality according to FastQC; 2) the fraction of uniquely mapped reads; 3) a PCR bottleneck coefficient; 4) fraction of reads in peaks according to MACS2 (Zhang et al., 2008) (more, the better); 5) overlap of peaks with DNA hypersensitivity sites. By kernel density estimation, we observed that the putative substrates had a nearly identical distribution of QC scores across all 5 metrics (Figure S7), suggesting that there is no systematic QC problem in our analysis.
Next, we wanted to investigate whether only a few transcription factors might appear as outliers. To do this, we analyzed the number of times a transcription factor appeared in the Rabit result and its corresponding median log-transformed p-value. We reasoned that poor-quality ChIP-seq might consistently, across many analyses, appear as highly significant, possibly due to technical artifacts. Outlier analysis was carried out through robust covariance estimation (scikit learn python package) (Rousseeuw and van Driessen, 1999), assuming a gaussian distribution and a significant contamination rate of 0.05 (Figure S7). After manual examination of the outliers, we identified the genes SCML2 and ZNF274 as having significantly worse ChIP-seq quality than compared to other transcription factors in the Cistrome database (Figure S7). We therefore exclude these two transcription factors from further analysis.
QUANITIFICATION AND STATISTICAL ANALYSIS
Gene ontology enrichment analysis
We performed gene ontology enrichment analysis for putatively identified driver genes using DAVID (Huang da et al., 2009) with the 775 UPS genes as the background. Biological process terms were deemed significant at an FDR of 0.25 (Figure S1).
Overlap with previously implicated driver genes
We compared our putative UPS driver genes to a previous study that found significantly mutated UPS genes (Ge et al., 2018), Davoli et al. (Davoli et al., 2013), the Cancer Gene Census (downloaded January 7, 2017) (Sondka et al., 2018), and the set of driver genes defined by the TCGA PancanAtlas consortium (Bailey et al., 2018). Gene list enrichment was assessed using a one-tailed fisher exact test with a background consisting of all UPS genes.
Boxplots
All boxplots show the distribution quartiles with whiskers representing the quartile +/− 1.5 times the Interquartile Range (IQR),
Protein-protein interaction network and Betweenness Centrality
Protein-protein interaction network data was download from the BioGrid website (v3.5.178) (Oughtred et al., 2019). The betweenness centrality measures how often a node in a network is situated on the shortest path between two other nodes in a network. Nodes with higher betweenness centrality are often hubs within a network. Betweenness centrality was computed for the BioGrid (Oughtred et al., 2019) protein-protein interaction network (downloaded 11/22/2019) using the networkx python package. Formally, for all possible pairs of nodes (s and t) in a network with nodes V, the betweenness centrality of a node (n) is the fraction of shortest paths (σ) that go through that node (Equation 1).
| (equation 1) |
Where σst(n) is the number of shortest paths between node s and t that go through node n and σst is the total number of shortest paths
Association of mutations with protein abundance
Using linear regression, we correlated the mutation status of each of the 63 putative driver genes with protein abundance from Reverse Phase Protein Arrays (RPPA) in TCGA. Only non-silent mutations were considered. A Wald test was performed after adjustment for tumor purity by ABSOLUTE (downloaded from https://gdc.cancer.gov/about-data/publications/pancanatlas) (Carter et al., 2012), tumor subtype (Sanchez-Vega et al., 2018) and RNA expression of the potential substrate (FDR<0.1 and effect size>0.25, Figure S6). The adjustment for RNA expression of potential substrates is important because it helps distinguish between direct UPS effects mediated through the protein-level from upstream effects at the transcriptional level.
Transcription factor substrate analysis
Conceptually, alterations in UPS genes should be able to explain the downstream target gene expression of a transcription factor by modulation through protein abundance or activity (Figure S7). To analyze this, first, we computed the differential expression between tumor samples containing putative driver mutations in a gene of interest versus those that did not (t test), while adjusting for tumor purity by ABSOLUTE (downloaded from https://gdc.cancer.gov/about-data/publications/pancanatlas) (Carter et al., 2012) and tumor subtypes (Sanchez-Vega et al., 2018). The generated differential expression profile was then analyzed by Rabit (Jiang et al., 2015) to associate top transcription factor (TF) regulators. Rabit infers transcriptional regulators based on TF binding sites using thousands of ChIP-seq profiles from the Cistrome database (Zheng et al., 2019) while adjusting for background covariates such as CpG density. For computational tractability reasons, we then corrected for transcription factor RNA expression only for the top 10 hits according to p-value, by repeating the above analysis but with the TF RNA expression included as a covariate. A second round of Rabit analysis was then conducted using the TF adjusted differential expression profiles. While results were only carried out for the top 10 hits in each analysis, multiple testing correction (Bonferroni method) was carried out with consideration of all TFs as possible (family wise error rate < 0.05). Note, analysis was only performed for the cancer types implicated by driver analysis for the specific UPS gene. Code used for this analysis is available on GitHub (https://github.com/ctokheim/tf_association).
Gene co-essentiality analysis from DepMap
The correlation between two gene’s dependency scores (CERES score) from CRISPR screens in DepMap was analyzed through a linear regression model. The cell culture type (adherent, suspension, etc.) and a CRISPR quality control metric (SSMD of control genes) was added as covariates. The statistical significance of the correlation was assessed by a Wald test.
Correlation with immune-related gene expression signatures
Using a linear regression model, we correlated the mutation status (see section: labeling of driver mutations) of each identified UPS driver gene or significantly mutated substrate with at least 5 putative driver mutations to several immune-related gene expression biomarkers from Thorson et al (signatures: leukocyte fraction, IFNG response, TGFB response, macrophage regulation and wound healing) (Thorsson et al., 2018). A t test was used to assess significance after adjusting for tumor subtypes and the non-silent mutation rate of a tumor. Associations were deemed significant at an FDR threshold of 0.1.
Correlation with T cell co-culture CRISPR screen
Data from a previous T cell co-culture CRISPR screen (Pan et al., 2018) across two conditions were used to assess whether UPS genes correlated with immune-related gene expression signatures might affect T cell mediated killing of cancer cells. The two conditions used in the screen were: 1) Pmel T cells which recognize endogenously expressed gp100 antigen on a B16 melanoma cell line while in the presence of IFNG compared to a non-antigen-specific T cell; 2) OT1 T cells that recognize B16 cells with media supplemented with or without the ovalbumin antigen. The log fold change of the single guide RNA (sgRNA) and the estimate of significance (z-score) were obtained through the TIDE website (http://tide.dfci.harvard.edu/) (Jiang et al., 2018). The z-scores from the two conditions (Pmel and OT1) were combined using Stouffer’s method to generate a meta-analysis z-score and corresponding p-value.
Association with overall patient survival
Curated overall survival information for TCGA was obtained from the genomic data commons (https://gdc.cancer.gov/about-data/publications/pancanatlas) (Liu et al., 2018). Using a Cox proportional-hazard model, a Wald test was used to assess the statistical significance of any association with survival. Tumor purity and subtype were included as covariates. Kaplan-Meier curves were generated using the TIDE website (http://tide.dfci.harvard.edu/) (Jiang et al., 2018).
Supplementary Material
Table S1: Mutated UPS genes identified as significant putative driver genes by 20/20+. Related to Figure 2.
Table S2: Enrichment analysis of mutations in degron-related sites. Related to Figure 3.
Table S3: Correlation of degron mutations and mutated UPS genes with immune-related biomarkers. Related to Figure 3 and Figure 6.
Table S4: deepDegron predictions and motif analysis for GPS screen. Related to Figure 4.
Table S5: Results of genes with significant enrichment of degron disrupting mutations by deepDegron. Related to Figure 5.
Table S6: Correlation of mutations in UPS driver genes with protein abundance (RPPA). Related to Figure 6.
Table S7: Mutated UPS genes associated with altered activity of transcription factors. Related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-mouse/human GATA3 | Cell Signaling Technology | Cat#5852S |
| Rabbit monoclonal anti-human PPM1D/WIP1 | Abcam | Cat#ab31270 |
| Rabbit monoclonal anti-human Phospho-p53 (Ser15) | Cell Signaling Technology | Cat#9284S |
| Rabbit monoclonal anti-human p53 | Cell Signaling Technology | Cat#9282S |
| Mouse monoclonal anti-human Phospho-ATM (Ser1981) | Cell Signaling Technology | Cat#4526S |
| Rabbit monoclonal anti-human/mouse ATM | Cell Signaling Technology | Cat#2873S |
| Mouse monoclonal anti-human c-Myc | Santa Cruz Biotechnology | Cat#SC-40 |
| Rabbit monoclonal anti-human/mouse CUL3 | Cell Signaling Technology | Cat#2759S |
| Rabbit monoclonal anti-human/mouse β-Actin | Cell Signaling Technology | Cat#4970S |
| Rabbit monoclonal anti-human IgG XP Isotype Control | Cell Signaling Technology | Cat#3900S |
| Mouse monoclonal anti DYKDDDDK Tag | Cell Signaling Technology | Cat#8146S |
| Rabbit monoclonal anti-HA-tag | Cell Signaling Technology | Cat#3724S |
| Goat anti-Mouse IgG Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#31430 |
| Donkey anti-Rabbit IgG Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#31458 |
| Rabbit monoclonal anti-human/mouse KIT | Cell Signaling Technology | Cat#3074 |
| Rabbit monoclonal anti-human/mouse CDH1 | Cell Signaling Technology | Cat#13116 |
| Rabbit monoclonal anti-human FOXA1 | Cell Signaling Technology | Cat#53528 |
| Mouse monoclonal anti-human KRT18 | Sigma Aldrich | Cat#WH0003875M1 |
| Rabbit monoclonal anti-human/mouse TP63 | Abcam | Cat#ab124762 |
| Rabbit monoclonal anti-human/mouse JAG1 | Cell Signaling Technology | Cat#70109 |
| Rabbit monoclonal anti-human FOXA1 | Cell Signaling Technology | Cat#53528 |
| Mouse monoclonal anti-human KRT14 | Santa Cruz | Cat#sc-53253 |
| Bacterial and Virus Strains | ||
| XL10-Gold Ultracompetent Cells | Agilent | Cat#200314 |
| Endura ElectroCompetent Cells | Lucigen | Cat#60242-2 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PBS | GIBCO | Cat#14190250 |
| DMEM, high glucose, pyruvate | GIBCO | Cat#11995065 |
| Lonza BioWhittaker L-Glutamine (200mM) | Lonza | Cat#BW17605E |
| Fetal bovine serum | VWR | Cat#9706 |
| Penicillin-Streptomycin | GIBCO | Cat#15140122 |
| PolyJet In Vitro DNA Transfection Reagent | SignaGen Laboratories | Cat#SL100688 |
| E-Gel Low Range Quantitative DNA Ladder | Invitrogen | Cat#NP0008 |
| E-Gel EX Agarose Gels, 2% | Life Technologies | Cat#G402002 |
| NuPAGE 3–8% Tris-Acetate Protein Gels, 1.5 mm, 10-well | Life Technologies | Cat#EA0378BOX |
| NuPAGE™ LDS Sample Buffer | Life Technologies | Cat#NP0008 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat#32106 |
| Precision Plus Protein™ Dual Color Standards | Bio-Rad Laboratories | Cat#161-0394 |
| X-tremeGENE™ HP DNA Transfection Reagent | Sigma-Aldrich | Cat#6366236001 |
| Polybrene | Sigma-Aldrich | Cat#107689-10G |
| Puromycin dihydrochloride | Thermo Fisher Scientific | Cat#A1113803 |
| BamHI-HF | New England Biolabs | Cat#R3136S |
| EcoRI-HF | New England Biolabs | Cat#R3101S |
| FastDigest Esp3I | Thermo Fisher Scientific | Cat#FD0454 |
| Q5 DNA Polymerase | New England Biolabs | Cat#M0491L |
| Nuclease-Free Water | Ambion | Cat#AM9938 |
| Pierce™ Homobifunctional Cross Linkers | Life Technologies | Cat#20593 |
| 2-Mercaptoethanol | Sigma Aldrich | Cat#M6250-10ML |
| Dynabeads™ Protein A | Thermo Fisher Scientific | Cat#10004D |
| Dynabeads™ Protein G | Thermo Fisher Scientific | Cat#10002D |
| EDTA | Sigma Aldrich | Cat#E8008-100ML |
| Protease/Phosphatase Inhibitor Cocktail (100X) | Cell Signaling Technology | Cat#5872S |
| Quick-Load 1 kb Plus DNA Ladder | New England Biolabs | Cat#N0469S |
| LB Broth | Mp Biomedicals | Cat#244610 |
| L-Broth Agar Large Capsules | Mp Biomedicals | Cat#MP 113001236 |
| RIPA buffer | Invitrogen | Cat#R0278 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Life Technologies | Cat#28906 |
| Opti-MEM I Reduced Serum Medium, no phenol red | Thermo Fisher Scientific | Cat#11058021 |
| Cycloheximide powder | Cell Signaling Technology | Cat#2112 |
| Critical Commercial Assays | ||
| QIAprep Spin Miniprep Kit | QIAGEN | Cat#27106 |
| RNeasy Plus Mini Kit | QIAGEN | Cat#74134 |
| QIAquick PCR Purification Kit | QIAGEN | Cat#28104 |
| QIAquick gel extraction kit | QIAGEN | Cat#28704 |
| Gibson Assembly Master Mix | New England Biolabs | Cat#E2611L |
| iScript cDNA Synthesis Kit | Bio-Rad Laboratories | Cat#1708891 |
| SsoAdvanced Univ SYBR Grn Suprmx | Bio-Rad Laboratories | Cat#1725272 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32854 |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32855 |
| GenElute™ HP Plasmid Maxiprep Kit | Sigma-Aldrich | Cat#NA0410-1KT |
| Ampure xp | Beckman Coulter | Cat#A63881 |
| BCA Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| SMARTer® ThruPLEX® DNA-Seq Kit | Takara Bio | Cat#R400675 |
| Experimental Models: Cell Lines | ||
| Human: HEK293FT | Thermo Fisher Scientific | Cat#R70007 |
| Human: MDA-MB-231 | American Type Culture Collection | Cat#ATCC-HTB-26 |
| Human: CAL27 | Ravi Uppaluri lab | N/A |
| Human: CAL33 | Ravi Uppaluri lab | N/A |
| Deposited Data | ||
| GATA3 ChIP-Seq | This paper | GSE162003 |
| Original gel images | This paper | doi:10.17632/kgfzbpv2w4.1 |
| Oligonucleotides | ||
| Primers for PCR, see Table S5 | Invitrogen | N/A |
| Recombinant DNA | ||
| hWIP1-FLAG | Addgene | Addgene Plasmid #28105 |
| pHAGE-GATA3 | Addgene | Addgene Plasmid #116747 |
| lentiCRISPR v2 puro | Addgene | Addgene Plasmid #98290 |
| pMD2.G | Addgene | Addgene Plasmid #12259 |
| psPAX2 | Addgene | Addgene Plasmid #12260 |
| pHAGE-CMV-DsRed-IRES-GFP | Koren et al., 2018 | N/A |
| pHAGE-SFFV-GFP-IRES-DsRed | Timms et al., 2019 | N/A |
| pLenti-EF1a-PGK-Puro | Kai Wucherpfennig lab | N/A |
| pLenti-EF1a-GATA3-WT | This paper | N/A |
| pLenti-EF1a-GATA3-A442M | This paper | N/A |
| pLenti-EF1a-GATA3-G444E | This paper | N/A |
| pLenti-EF1a-GATA3-H400 | This paper | N/A |
| pLenti-EF1a-GATA3-WT-Fg | This paper | N/A |
| pLenti-EF1a-GATA3-A442M-Fg | This paper | N/A |
| pLenti-EF1a-GATA3-G444E-Fg | This paper | N/A |
| pLenti-EF1a-GATA3-H400-Fg | This paper | N/A |
| pLenti-EF1a-PPM1D-WT | This paper | N/A |
| pLenti-EF1a-PPM1D-V604Q | This paper | N/A |
| pLenti-EF1a-PPM1D-C605W | This paper | N/A |
| pLenti-EF1a-PPM1D-L450 | This paper | N/A |
| pLenti-EF1a-PPM1D-WT-HA | This paper | N/A |
| pLenti-EF1a-PPM1D-V604Q-HA | This paper | N/A |
| pLenti-EF1a-PPM1D-C605W-HA | This paper | N/A |
| pLenti-EF1a-PPM1D-L450-HA | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com |
| DeepDegron | This Paper | https://github.com/ctokheim/deepDegron |
| Transcription factor inference | This Paper | https://github.com/ctokheim/tf_association |
| Other | ||
| Corning Filter System (0.45um) | Corning Life Sciences | Cat#431096 |
| milliTUBE 1 ml AFA Fiber | Covaris Inc. | Cat#520130 |
| NITROCEL MEMB 0.45um | Bio-Rad Laboratories | Cat#1620115 |
| Multiplate™ 96-Well PCR Plates | Bio-Rad Laboratories | Cat#MLL9601 |
| QUBIT ASSAY TUBES SET | Life Technologies | Cat#Q32856 |
| Microseal ‘B’ Adhesive Seals | Bio-Rad Laboratories | Cat#MSB-1001 |
Highlights.
The ubiquitin-proteasome system (UPS) represents ~19% of mutated cancer driver genes
A machine learning approach, deepDegron, reveals de novo degron motifs
Truncating mutations in GATA3 and PPM1D increase protein expression via degron loss
ChIP-seq data can help infer transcription factor substrates of mutated UPS in cancer
Acknowledgements
This study is partially supported by the Breast Cancer Research Foundation (BCRF-20-100) to X.S.L. The study was also supported by the NIH grant (AG11085) to S.J.E. C.T. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRQ-04-20). R.T.T. is supported by a Pemberton-Trinity Fellowship and a Sir Henry Wellcome Postdoctoral Fellowship (201387/Z/16/Z). S.J.E. is an Investigator with the Howard Hughes Medical Institute.
Declaration of Interests
S.J.E is a member of the Molecular Cell advisory board. X.S.L. is a cofounder, board member, and consultant of GV20 Oncotherapy and its subsidiaries, SAB of 3DMedCare, consultant for Genentech, and stockholder of BMY, TMO, WBA, ABT, ABBV, and JNJ, and receives research funding from Takeda and Sanofi. Other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit Y, and Geman D (1997). Shape Quantization and Recognition with Randomized Trees. Neural Computation 9, 1545–1588. [Google Scholar]
- An J, Wang C, Deng Y, Yu L, and Huang H (2014). Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 6, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data (Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom: ). [Google Scholar]
- Atkin G, and Paulson H (2014). Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. (2012). Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57, 289–300. [Google Scholar]
- Breiman L (2001). Random Forests. Mach Learn 45, 5–32. [Google Scholar]
- Buiting K, Williams C, and Horsthemke B (2016). Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol 12, 584–593. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, et al. (2002). Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 31, 210–215. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana R, and Niculescu-Mizil A (2006). An empirical comparison of supervised learning algorithms. Paper presented at: Proceedings of the 23rd international conference on Machine learning (ACM; ). [Google Scholar]
- Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. (2017). OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, and Goldberg AL (2017). The Logic of the 26S Proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT, Laboratory DA, Coordinating Center -Analysis Working, G., Statistical Methods groups-Analysis Working, G., Enhancing, G.g., Fund, N.I.H.C., Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz JR, Bettigole SE, and Glimcher LH (2017). Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 168, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D, and Petsko GA (2006). Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc Natl Acad Sci U S A 103, 4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, and Clurman BE (2014). Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, and Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, and Joazeiro CA (2009). RING domain E3 ubiquitin ligases. Annu Rev Biochem 78, 399–434. [DOI] [PubMed] [Google Scholar]
- Eakin CM, Maccoss MJ, Finney GL, and Klevit RE (2007). Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci U S A 104, 5794–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, and Brown M (2007). Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res 67, 6477–6483. [DOI] [PubMed] [Google Scholar]
- Ella H, Reiss Y, and Ravid T (2019). The Hunt for Degrons of the 26S Proteasome. Biomolecules 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6, 271–281e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelyanov A, and Bulavin DV (2015). Wip1 phosphatase in breast cancer. Oncogene 34, 4429–4438. [DOI] [PubMed] [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, et al. (2015). SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell 59, 917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Leighton JS, Wang Y, Peng X, Chen Z, Chen H, Sun Y, Yao F, Li J, Zhang H, et al. (2018). Integrated Genomic Analysis of the Ubiquitin Pathway across Cancer Types. Cell Rep 23, 213–226e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. [DOI] [PubMed] [Google Scholar]
- Goutte C, Toft P, Rostrup E, Nielsen F, and Hansen LK (1999). On clustering fMRI time series. Neuroimage 9, 298–310. [DOI] [PubMed] [Google Scholar]
- Gouw M, Michael S, Sámano-Sánchez H, Kumar M, Zeke A, Lang B, Bely B, Chemes LB, Davey NE, and Deng Z (2018). The eukaryotic linear motif resource–2018 update. Nucleic acids research 46, D428–D434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, and Karin M (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hatzis P, van der Flier LG, van Driel MA, Guryev V, Nielsen F, Denissov S, Nijman IJ, Koster J, Santo EE, Welboren W, et al. (2008). Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol Cell Biol 28, 2732–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Zhang X, Ren S, and Sun J (2016). Deep residual learning for image recognition. Paper presented at: Proceedings of the IEEE conference on computer vision and pattern recognition. [Google Scholar]
- Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, Ni A, Novik JB, Mangarin LMB, Abu-Akeel M, et al. (2018). Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 33, 843–852e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, Zhang J, Heffernan TP, Gera S, Kovacs JJ, et al. (2018). PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 23, 700–713e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr., and Goldberg MA (1996). Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A 93, 10595–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, and Kaelin WG Jr. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. [DOI] [PubMed] [Google Scholar]
- Jaramillo MC, and Zhang DD (2013). The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardana K, Schramm SJ, Haydu L, Thompson JF, Scolyer RA, Mann GJ, Muller S, and Yang JY (2015). Determination of prognosis in metastatic melanoma through integration of clinico-pathologic, mutation, mRNA, microRNA, and protein information. Int J Cancer 136, 863–874. [DOI] [PubMed] [Google Scholar]
- Jiang P, Freedman ML, Liu JS, and Liu XS (2015). Inference of transcriptional regulation in cancers. Proc Natl Acad Sci U S A 112, 7731–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn JD, Miller PG, Silver AJ, Sellar RS, Bhatt S, Gibson C, McConkey M, Adams D, Mar B, Mertins P, et al. (2018). PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132, 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A, Shi J, Vakoc C, et al. (2013). The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 153, 1552–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma DP, and Ba J (2014). Adam: A method for stochastic optimization. arXiv preprint arXiv:14126980. [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, and Elledge SJ (2001). Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294, 173–177. [DOI] [PubMed] [Google Scholar]
- Koren I, Timms RT, Kula T, Xu Q, Li MZ, and Elledge SJ (2018). The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons. Cell 173, 1622–1635 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros-Mehr H, Slorach EM, Sternlicht MD, and Werb Z (2006). GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127, 1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, and Courtois G (2003). The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424, 801–805. [DOI] [PubMed] [Google Scholar]
- Krizhevsky A, Sutskever I, and Hinton GE (2012). Imagenet classification with deep convolutional neural networks. Paper presented at: Advances in neural information processing systems. [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, Yang JY, Broom BM, Verhaak RG, Kane DW, et al. (2013). TCPA: a resource for cancer functional proteomics data. Nat Methods 10, 1046–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang Y, Peng Y, Austin RJ, van Eyndhoven WG, Nguyen KC, Gabriele T, McCurrach ME, Marks JR, Hoey T, et al. (2002). Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet 31, 133–134. [DOI] [PubMed] [Google Scholar]
- Li S, Wan C, Zheng R, Fan J, Dong X, Meyer CA, and Liu XS (2019). Cistrome-GO: a web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res 47, W206–W211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom RG, Supek F, and Lehner B (2016). The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 48, 1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Nannenga B, and Donehower LA (2005). PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 19, 1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, Tomita YA, and Rosen EM (2010). BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol Endocrinol 24, 76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Jiménez F, Muiños F, López-Arribillaga E, Lopez-Bigas N, and Gonzalez-Perez A (2020). Systematic analysis of alterations in the ubiquitin proteolysis system reveals its contribution to driver mutations in cancer. Nature Cancer 1, 122–135. [DOI] [PubMed] [Google Scholar]
- Masica DL, Douville C, Tokheim C, Bhattacharya R, Kim R, Moad K, Ryan MC, and Karchin R (2017). CRAVAT 4: Cancer-Related Analysis of Variants Toolkit. Cancer Res 77, e35–e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B, Kumar M, Gibson TJ, Uyar B, and Dosztanyi Z (2017). Degrons in cancer. Sci Signal 10. [DOI] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalepa G, and Clapp DW (2018). Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer 18, 168–185. [DOI] [PubMed] [Google Scholar]
- Oshiro TM, Perez PS, and Baranauskas JA (2012). How many trees in a random forest? Paper presented at: International workshop on machine learning and data mining in pattern recognition (Springer; ). [Google Scholar]
- Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R, et al. (2019). The BioGRID interaction database: 2019 update. Nucleic Acids Res 47, D529–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, and Kallioniemi A (2006). The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat 95, 257–263. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, and Wilkinson KD (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al. (2017). Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 377, 523–533. [DOI] [PubMed] [Google Scholar]
- Ronau JA, Beckmann JF, and Hochstrasser M (2016). Substrate specificity of the ubiquitin and Ubl proteases. Cell research 26, 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw PJ, and van Driessen K (1999). A Fast Algorithm for the Minimum Covariance Determinant Estimator. Technometrics 41, 212–223. [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satopaa V, Albrecht J, Irwin D, and Raghavan B (2011). Finding a” kneedle” in a haystack: Detecting knee points in system behavior. Paper presented at: 2011 31st international conference on distributed computing systems workshops (IEEE). [Google Scholar]
- Scudellari M (2019). Protein-slaying drugs could be the next blockbuster therapies. Nature 567, 298–300. [DOI] [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, and Hirohashi S (2008). Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A 105, 13568–13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, et al. (2006). Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell 23, 757–764. [DOI] [PubMed] [Google Scholar]
- Simonyan K, and Zisserman A (2014). Very deep convolutional networks for large-scale image recognition. arXiv preprint arXiv:14091556. [Google Scholar]
- Singh AA, Schuurman K, Nevedomskaya E, Stelloo S, Linder S, Droog M, Kim Y, Sanders J, van der Poel H, Bergman AM, et al. (2019). Optimized ChIP-seq method facilitates transcription factor profiling in human tumors. Life Sci Alliance 2, e201800115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, and Rotin D (1997). Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 16, 6325–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MD, Ritterhoff T, Klevit RE, and Brzovic PS (2016). E2 enzymes: more than just middle men. Cell research 26, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, and Reed SI (2001). Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413, 316–322. [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, Pereira T, and Poellinger L (2000). Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J 19, 4298–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorou V, Stark R, Menon S, and Carroll JS (2013). GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res 23, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms RT, Zhang Z, Rhee DY, Harper JW, Koren I, and Elledge SJ (2019). A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokheim C, and Karchin R (2019). CHASMplus Reveals the Scope of Somatic Missense Mutations Driving Human Cancers. Cell Syst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, and Karchin R (2016). Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci U S A 113, 14330–14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkamani A, and Schork NJ (2008). Prediction of cancer driver mutations in protein kinases. Cancer Res 68, 1675–1682. [DOI] [PubMed] [Google Scholar]
- van der Lee R, Lang B, Kruse K, Gsponer J, Sanchez de Groot N, Huynen MA, Matouschek A, Fuxreiter M, and Babu MM (2014). Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep 8, 1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Leong KG, Newton K, Yee C, O’Rourke K, Liu J, Phu L, Vij R, Ferrando R, Couto SS, et al. (2011). COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature 474, 403–406. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, and Clurman BE (2008). FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8, 83–93. [DOI] [PubMed] [Google Scholar]
- Wellenstein MD, and de Visser KE (2018). Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48, 399–416. [DOI] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113. [DOI] [PubMed] [Google Scholar]
- Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, Drosten M, Zhao H, Cecchi F, Hembrough T, et al. (2017). Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, and Elledge SJ (2008). Global protein stability profiling in mammalian cells. Science 322, 918–923. [DOI] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Zaugg K, Mak TW, and Elledge SJ (2006). A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 126, 529–542. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen CH, Brown M, Zhang X, Meyer CA, et al. (2019). Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 47, D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, Li Y, Zhang Z, Rusch MC, Parker M, et al. (2016). Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 48, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mutated UPS genes identified as significant putative driver genes by 20/20+. Related to Figure 2.
Table S2: Enrichment analysis of mutations in degron-related sites. Related to Figure 3.
Table S3: Correlation of degron mutations and mutated UPS genes with immune-related biomarkers. Related to Figure 3 and Figure 6.
Table S4: deepDegron predictions and motif analysis for GPS screen. Related to Figure 4.
Table S5: Results of genes with significant enrichment of degron disrupting mutations by deepDegron. Related to Figure 5.
Table S6: Correlation of mutations in UPS driver genes with protein abundance (RPPA). Related to Figure 6.
Table S7: Mutated UPS genes associated with altered activity of transcription factors. Related to Figure 6.
Data Availability Statement
Raw gel pictures and data necessary to recreate figures using the Jupyter notebook code (see below) are available on mendeley data: http://dx.doi.org/10.17632/kgfzbpv2w4.1. Raw sequencing data, called peaks and bigwig files for GATA3 ChIP-seq are available GEO (GSE162003). The DeepDegron code is available on github: https://github.com/ctokheim/deepDegron. The code for associating UPS genes with putative transcription factor substrates is also available on github: https://github.com/ctokheim/tf_association. Jupyter notebooks for data analysis are stored on github (https://github.com/ctokheim/Tokheim_Mol_Cell_2020).