

Abstract
Phylogenetic analysis of Y-family DNA polymerases suggests that it can be subdivided into several discrete branches consisting of UmuC/DinB/Rev1/Rad30/Rad30A and Rad30B. The most diverse is the DinB family that is found in all three kingdoms of life. Searches of the complete genome of the crenarchaeon Sulfolobus solfataricus P2 reveal that it possesses a DinB homolog that has been termed DNA polymerase IV (Dpo4). We have overproduced and purified native Dpo4 protein and report here its enzymatic characterization. Dpo4 is thermostable, but can also synthesize DNA at 37°C. Under these conditions, the enzyme exhibits misinsertion fidelities in the range of 8 × 10–3 to 3 × 10–4. Dpo4 is distributive but at high enzyme to template ratios can synthesize long stretches of DNA and can substitute for Taq polymerase in PCR. On damaged DNA templates, Dpo4 can facilitate translesion replication of an abasic site, a cis-syn thymine–thymine dimer, as well as acetyl aminofluorene adducted- and cisplatinated-guanine residues. Thus, although phylogenetically related to DinB polymerases, our studies suggest that the archaeal Dpo4 enzyme exhibits lesion-bypass properties that are, in fact, more akin to those of eukaryotic polη.
INTRODUCTION
Our understanding of basic cellular processes like DNA replication and repair have changed dramatically over the past couple of years with the discovery of a plethora of novel DNA polymerases that belong to the UmuC/DinB/Rev1/Rad30 superfamily of proteins (1,2). As this family of proteins are clearly related to each other, yet share little to no similarity to the previously identified polymerase families (3–5), it has recently been suggested that they be called the ‘Y-family’ of DNA polymerases (6).
Phylogenetic analysis suggests that members of the UmuC subfamily are found in bacteria whereas those from the Rad30 branch are found exclusively in eukaryotes (6–8). The DinB subfamily is the most diverse and is found in bacteria, archaea and eukaryotes. Quite remarkably, these DinB orthologs are often well conserved. For example, the DinB-like proteins from humans (DINB1) or mice (Dinb1) share a high degree of similarity to their Escherichia coli counterpart (9,10).
Interestingly, only a couple of archaeal dinB orthologs have been reported in the literature to date. One is the dbh (dinB homolog) gene from Sulfolobus solfataricus P1, which was previously identified using degenerate PCR primers designed to E.coli umuC and dinB sequences (11); the second is a related gene identified as yqjH in Halobacterium species NRC-1 (6,12). In fact, the limited distribution of dinB-like genes in archaea is quite evident as at least 16 archaeal genomes have been completely sequenced and are devoid of any identifiable dinB ortholog.
While searching for aerobic thermoacidophiles at the Pisciarelli hot springs on the outer slope of the Solfatara volcano crater wall near Naples, Italy, Wolfram Zillig and colleagues identified two strains of Sulfolobus that they initially identified as DSM 1616 and DSM 1617 [their designation at the Deutsche Sammlung von Microoganismen (DSM) depository in Germany] (13). The two strains were subsequently called S.solfataricus (13), and designated as subspecies, P1 and P2 (where P stands for Pisciarelli; W.Zillig, personal communication). The two strains are distinct, but are clearly closely related to each other (13). However, much to our surprise, BLAST searches (14,15) of the complete S.solfataricus P2 genome (http://www-archbac.u-psud.fr/Projects/sulfolobus/sulfolobus.html; Genbank accession no. AE006641; 16) using the P1 Dbh protein as a search parameter, revealed a related ortholog that is only 53% identical to the P1 Dbh protein. The P2 DinB-like protein is, therefore, only the third DinB ortholog identified to date in archaea.
The enzymatic properties of DinB-like polymerases have been reported for E.coli (DNA pol IV) (17–19) and from humans [termed pol θ (20) and pol κ (21–25)]. We were therefore interested in comparing the biochemical properties of the archaeal DinB-like enzyme from S.solfataricus P2 to its bacterial and mammalian orthologs. Our expectation was that such characterization would provide insights into the structure–function relationship of the DinB branch of the Y-family of DNA polymerases. Interestingly, the initial characterization of the archaeal polymerase reveals that it is thermostable and while exhibiting some properties similar to DinB enzymes it also unexpectedly shares lesion-bypass properties similar to the distantly related eukaryotic Rad30 (polη) branch of the Y-family of DNA polymerases (6).
MATERIALS AND METHODS
Cloning and overexpression of S.solfataricus P2 DNA polymerase IV
Sulfolobus solfataricus P2 was obtained from the American Type Culture Collection (ATCC, Manasas, VA; catalog number 35092). The P2 dpo4 gene was PCR-amplified by simply resuspending a small aliquot of the lyophilized culture in water and the resulting suspension used directly in PCR reactions. The two primers used for amplification were: ssP2dbhN 5′-GGAATTCATATGATTGTTCTTTTCGTTGATTTG-3′ and ssP2dbhBa 5′-CGCGGATCCTTAAGTATCGAAGAACTTGTCTAATCCTA-3′ that contained NdeI and BamHI restriction enzymes sites, respectively (underlined). A ∼1000 bp PCR fragment was amplified using AmpliTaq polymerase (Roche Molecular, Indianapolis, IN) and the undigested fragment cloned directly into the ‘pGEMT easy’ vector (Promega, Madison, WI). The DNA sequence of the insert was verified and the P2 dpo4 gene was subsequently subcloned into pET22b (Novagen, Madison, WI) as an NdeI–BamHI fragment. Finally, the recombinant plasmid, called p1914, was introduced into RW382 (26), a ΔumuDC595::cat derivative of BL21(λDE3) (27).
Purification of DNA polymerase IV (Dpo4)
An overnight culture of RW382 harboring p1914 was diluted 1:100 in fresh Luria–Bertani media containing 100 µg/ml ampicillin and grown at 37°C. After reaching an OD600 of 0.5, cells were harvested by centrifugation and the cell pellet resuspended in 3 vol of buffer A [75 mM NaCl, 10 mM KHPO4 pH 7, 0.1 mM EDTA, 1 mM dithiothreitol (DTT)]. Cells were lyzed by sonication and the cleared lysate was heat treated at 85°C for 5 min. The heat treatment caused many of the E.coli proteins to denature and these were subsequently removed by centrifugation at 20 000 g for 30 min. The soluble supernatant was applied to a ‘HiQ’ DEAE–Sepharose column (Amersham Pharmacia Biotech, Piscataway, NJ) and bound proteins eluted in a linear 75–1000 mM gradient of NaCl in buffer B (10 mM KHPO4 pH 7, 0.1 mM EDTA, 1 mM DTT). Fractions containing Dpo4 were pooled and concentrated using a Macrosep 30K filter (Pall-Gelman, Ann Arbor, MI) before being applied to a ‘BioGel’ hydroxylapatite column (Bio-Rad, Hercules, CA). Bound proteins were eluted with a 10–1000 mM linear gradient of KHPO4. Pooled fractions containing Dpo4 were subsequently dialyzed against buffer C (Tris–HCl pH 7.5, 20 mM, 50 mM NaCl, 0.1 mM EDTA, 1 mM DTT), and applied to a Superdex 75 gel filtration column (Amersham Pharmacia Biotech). As a final purification step, fractions were concentrated in a microsep 30K filter and applied to a MonoS column (Amersham Pharmacia Biotech). Proteins were eluted using buffer D (20 mM Tris–HCl pH 7.5, 1 M NaCl, 0.1 mM EDTA, 1 mM DTT). Using this protocol, Dpo4 was purified to >95% purity (based upon staining gels with Coomassie Blue R-250) and typically resulted in a yield of ∼700 µg Dpo4 from a 100 ml culture of E.coli cells.
DNA templates
Most synthetic oligonucleotides were synthesized by Lofstrand Laboratories (Gaithersburg, MD) using standard techniques and were gel-purified prior to use. The exceptions were the lesion-containing 30mers, which have been described previously (28–30; see below). The undamaged DNA templates used in the replication assays have also been reported previously (31), thereby allowing a direct comparison of replication fidelities to those obtained with different DNA polymerases assayed in earlier studies (31). The primer for the undamaged templates was a 16mer: 5′-CTTGAAAACATAGCGA-3′, that was 5′-labeled with [γ-32P]ATP (5000 Ci/mmol; 1 Ci = 37 GBq) (Amersham Pharmacia Biotech) using T4 polynucleotide kinase (Life Technologies, Gaithersburg, MD). Most reactions utilized a 40mer oligonucleotide template with the sequence: 5′-AGCGTCTTAATCTAAGCTXTCGCTATGTTTTCAAGGATTC-3′, where X was either G, A, T or C. This sequence (X = A) corresponds to nucleotides 4072–4121 of M13mp18. The nucleotides underlined indicate the location of the annealed radiolabeled primer. Where noted, the 40mer oligonucleotide template was replaced with circular single-stranded M13mp18 DNA (New England Biolabs, Beverly, MA).
The oligonucleotide templates containing lesions were synthetic oligonucleotide 30mers (28–30). For the cis-syn and 6-4 dimers the template sequence was 5′-CTCGTCAGCATCTTCATCATACAGTCAGTG-3′, where the two underlined thymines indicate the position of the CPD (cis-syn cyclobutane pyrimidine thymine–thymine dimer) or 6-4PP. The abasic template was 5′-CTCGTCAGCATCTXCATCATACAGTCAGTG-3′, where X represents the location of the synthetic abasic site. The primer for the CPD, 6-4, abasic and undamaged control TT template, was a 16mer with the sequence 5′-CACTGACTGTATGATG-3′. The sequence of the acetyl aminofluorene (AAF) template was 5′-CTCTTCACCTCTAGTCTCCTACACTCAATC-3′ where the underlined guanine indicates the location of the AAF adduct. The 16mer primer for the AAF template was 5′-GATTGAGTGTAGGAGA-3′. The cisplatin template was 5′-CTCGTCACCTCTGGTCTCCTACAGTCAGTG-3′ where the two underlined guanines indicate the location of the 1,2-cisplatinated guanine adduct. The primer for this template was 5′-CACTGACTGTAGGAGA-3′ (30). The undamaged control for the AAF and cisplatin templates was a 30mer, 5′-CTCGTCAGCATCTGCATCATACAGTCAGTG-3′, which was primed using the 16mer CPD/6-4 primer described above.
Replication reactions
Replication reactions were performed essentially as described previously (31,32), except that KCl was omitted from the reaction. Briefly, radiolabeled primer/template (P/T) DNAs were prepared by annealing the respective 5′-32P-labeled 16mer primer to the unlabeled template DNA at a molar ratio of 1:1.5. Standard 10 µl reactions contained 40 mM Tris–HCl pH 8.0, 5 mM MgCl2, 100 µM of each ultrapure dNTP (Amersham Pharmacia Biotech), 10 mM DTT, 250 µg/ml bovine serum albumin (BSA), 2.5% glycerol, 10 nM 5′-[32P]P/T DNA and 10 nM Dpo4. After incubation at 37°C for various times, reactions were terminated by the addition of 10 µl of 95% formamide/10 mM EDTA and the samples heated to 100°C for 5 min. Reaction mixtures (5 µl) were subjected to 20% polyacrylamide/7 M urea gel electrophoresis and replication products visualized by autoradiography or PhosphorImager analysis (Molecular Dynamics, CA).
Kinetic analysis of replication products
Time-course reactions using the standard replication conditions were initially performed so as to ensure that the reaction was in the linear range (usually <20% of primer utilization) (33,34). Subsequent reactions measuring the incorporation of the correct nucleotide were performed for 2 min. The concentration of the correct nucleotide varied from 0.5 to 40 µM and the incorrect nucleotide from 1 to 16 mM. Reactions were initiated by the addition of the appropriate dNTP. Reaction products were separated in a 20% polyacrylamide gel containing 7 M urea and gels dried prior to quantitative PhosphorImager analysis using the ImageQuant software (Amersham Pharmacia Biotech). The apparent Vmax and Km values for each enzyme and nucleotide incorporated were determined from a Hanes–Woolf plot by linear least-squares fit as described previously (34). The catalytic efficiency of nucleotide insertion was calculated as the ratio of Vmax/Km and the frequency of misinsertion were calculated as (Vmax/Km)incorrect/(Vmax/Km)correct, as described previously (34).
Thermostability of Dpo4
The thermostability of the Dpo4 was compared with that of polymerase I (pol I) (New England Biolabs) and Taq polymerase (Roche Molecular) by heating each enzyme to temperatures ranging between 37 and 95°C for 5 min. An aliquot of each enzyme was then used in a standard 5 min replication reaction at 37°C that measured the incorporation of a single nucleotide, dCMP, opposite template G (see replication reactions described above). The amount of enzyme used in the replication reaction was initially determined by making serial dilutions of each enzyme and identifying the minimal amount of enzyme necessary to give 100% extension of the radiolabeled primer at 37°C. For Dpo4, it was 10 nM; for Taq polymerase it was 0.1 U; and for E.coli pol I Klenow fragment it was 0.1 U. The ability of each enzyme to extend the primer was quantitated by ImageQuant software (Molecular Dynamics) and subsequently plotted as a function of the temperature to which the enzyme was initially exposed.
Processivity of Dpo4
To measure the processivity of Dpo4, standard primer extension reactions were performed at 37°C for 5 min in the presence of all four dNTPs each at 100 µM and the template utilized was circular M13mp18. The amount of radiolabeled P/T was kept constant at 10 nM, and the amount of P2 Dpo4 polymerase varied from 200 to 0.5 nM. Reaction products were separated in a 20% polyacrylamide gel containing 7 M urea and gels dried prior to quantitative PhosphorImager analysis.
Polymerase chain reaction with Dpo4
Linear DNA fragments were pre-amplified and diluted aliquots were used as a template in subsequent assays. Reaction volumes (50 µl) contained 30 µM of each primer pair, 40 mM Tris–HCl pH 8.0, 5 mM MgCl2, 10 mM DTT, 60 mM KCl, 2.5% glycerol, 2.5 mM of each ultrapure dNTP and 10 ng of DNA. Reactions employed a manual denaturation at 100°C for 5 min before the addition of Dpo4 DNA polymerase (20–400 ng). One unit of AmpliTaq DNA polymerase (Roche Molecular) was used as a positive control. DNA primers were prepared by Lofstrand Laboratories. The forward and reverse primers were 5′-CGCTGTTGCTCATTTGAGC-3′ and 5′-CGAACCAGGTCAGAGATGGTCAGCC-3′ for the ∼180 bp fragment. The temperature profile used with these primers was 30 cycles of 90°C for 30 s, 55°C for 20 s and 65°C for 60 s. The forward and reverse primers used to amplify the ∼1300 bp fragment were 5′-ATGCCTGTATTTGCCTTGGTGGACTGC-3′ and 5′-CCGGGATCCGCGCTACTTAACTCGCGG-3′. The temperature profile used with these primers was 30 cycles of 90°C for 30 s, 55°C for 20 s and 65°C for 3 min. Products were separated on a 2% agarose gel, stained with ethidium bromide and visualized on a ChemiImager 4000 low light imaging system (Alpha Innotech Corporation, San Leandro, CA).
RESULTS
Identification of the S.solfataricus P2 dpo4 gene
Utilizing the S.solfataricus P1 Dbh protein (GenBank accession no. U52110) as our search query, we performed BLASTP searches (14,15) of archaeal genomes available at the National Center for Biotechnology Information (NCBI) GenBank database, as well as at web sites of various unfinished genome projects. The best match was a protein identified as Sso2448 by the S.solfataricus P2 genome-sequencing project, which is 53% identical and 75% similar to the P1 Dbh protein. After consultation with Drs Yvan Zivanovic and Patrick Forterre at the Université Paris-Sud, France, it has been agreed that the P2 DinB ortholog be termed DNA polymerase IV, or Dpo4 (as S.solfataricus P2 possesses three other B-family polymerases: Dpo1, Dpo2 and Dpo3; Genbank accession no. AE006641; http://www-archbac.u-psud.fr/Projects/sulfolobus/sulfolobus.html; 16). The name Dpo4 is also consistent with E.coli nomenclature where DinB is also known as DNA pol IV (17).
Initially, the fact that the P2 Dpo4 protein is only 53% identical to the P1 Dbh protein was a little disconcerting, as we were expecting a near perfect match. However, the P2 dpo4 gene, like its P1 ortholog (11), appears to be arranged in an operon with a putative ribokinase, and upstream of the ribokinase there is a gene encoding a protein with similarity to E.coli RuvB (compare Genbank accession number U52110 with http://www-archbac.u-psud.fr/Projects/sulfolobus/sulfolobus.html). There is 57% identity between the P1 and P2 ribokinase proteins and 85% identity between the RuvB-like proteins. Whereas the two S.solfataricus strains were isolated in close proximity to each other and were believed to be closely related (13), comparison of the cloned P1 DNA sequences deposited in GenBank and their orthologs in the complete P2 genome reveal that their identity varies between 23 and 100%, with the average identity being ∼71% (F.Boudsocq, unpublished observations). The P1 Dbh and P2 Dpo4 proteins are, therefore, slightly more diverged from each other than most S.solfataricus P1 and P2 proteins. However, like other members of the Y-family of polymerases, the N-terminus of the two proteins is quite well conserved and includes the five motifs identified in other members of the family (7,11,21,29,35,36). The S.solfataricus P2 Dpo4 protein therefore represents only the third archaeal member of the Y-family of polymerases identified to date.
Overproduction and purification of S.solfataricus P2 Dpo4
Many of the recently characterized Y-family DNA polymerases have been purified through the use of recombinant glutathione S-transferase (GST), histidine or maltose-binding protein (MBP) fusions (17,21,23–25,29,31,37–41). The exceptions are E.coli UmuC, which has been purified as a native complex consisting of UmuD′2C (42,43), R46-encoded MucB, which was purified in a denatured form and subsequently renatured (44,45) and E.coli DinB, which was initially purified as histidine and MBP fusions (17,18) but has now been purified in native form (19) (M.F.Goodman, University of Southern California, personal communication). We were therefore interested in attempting to purify native Dpo4 lacking any recombinant tags. This goal has been largely achieved by the fact that the protein is thermostable and can be easily purified from thermolabile contaminants (see below). Indeed, by simply heating the soluble cell extract to 85°C for 5 min caused many of the native E.coli proteins to denature and precipitate. As a result, we obtained a soluble extract that was highly enriched for Dpo4 (Fig. 1, track S). The abundance of Dpo4 in the heat-treated extract allowed us to easily monitor the enzymes purification through the visual inspection of Coomassie Blue stained SDS–PAGE gels. Minor contaminants were subsequently removed by standard chromatographic methods and resulted in preparations of P2 Dpo4 that were >95% pure (Fig. 1). The yield of protein was significant and was in the range of 7–15 mg/l of initial E.coli cell culture. The P2 Dpo4 protein consists of 352 amino acids with a predicted mass of 40 189 Da and an estimated pI of 9.11. However, it should be noted that the protein migrated slightly faster than a marker protein of ∼38 kDa (E.coli RecA protein) on denaturing SDS–polyacrylamide gels (Fig. 1).
Figure 1.

Purification of S.solfataricus Dpo4 protein. Aliquots from various stages of the purification were separated on a 12% polyacrylamide–SDS gel and proteins visualized after staining with Coomassie Brilliant Blue R-250. –, Whole cell extract of RW382; +, whole extract from RW382 harboring p1914 (P2 Dpo4); S, clarified supernatant after heating the soluble cell extract at 85°C for 5 min; Q, pooled HiQ column fractions; H, pooled hydroxylapatite column fractions; M, Dpo4 fractions after Superdex 75 gel-filtration and MonoS columns. The molecular weight of marker proteins (BSA 66 kDa and E.coli RecA 38 kDa) are indicated on the left of the gel.
Fidelity of S.solfataricus P2 Dpo4
As Dpo4 is phylogenetically related to a growing family of DNA polymerases (6), we expected that it would exhibit intrinsic polymerase activity. Although S.solfataricus species grow optimally at 75–80°C, they are also capable of growing at much lower and higher temperatures (13). Indeed, our preliminary replication assays indicated that the enzyme is active at 37°C. This was particularly useful, as the melting point (Tm) of the primer used in the replication assays is 49°C. Although we could have potentially designed and utilized a P/T with a higher Tm, we preferred to use the P/T with the lower Tm, as it allowed a direct comparison to the distantly related human polι enzyme in the same nucleotide sequence context (31). As a consequence, all replication assays were performed at 37°C unless otherwise stated. Under these conditions, Dpo4 appears able to discriminate between correct and incorrect nucleotides, as in all cases, the most efficient primer extension is observed in the presence of the correct incoming dNTP. However, it should also be noted that misincorporations at each template site are also clearly visible (Fig. 2). In the presence of all four dNTPs, Dpo4 extends primers in a distributive manner to the end of the template. Overall, misincorporation frequencies for Dpo4 are in the range of 8 × 10–3 to 3 × 10–4 (Table 1) and these values are similar to those reported for the related E.coli pol IV and the human polκ (DINB1) enzymes (17,18,20,22,24).
Figure 2.

Ability of S.solfataricus Dpo4 to incorporate nucleotides at various template sites. The extent of (mis)incorporation was measured at each template site in the absence (0) or presence of all four dNTPs (4) or presence of each individual dNTP (100 µM) (G, A, T, C). Reactions were for 5 min at 37°C with 10 nM of Dpo4. The sequence context of each template is given above each panel and the target template nucleotide is indicated in bold.
Table 1. Misinsertion fidelity of P2 Dpo4 on undamaged DNA templates as determined by steady-state kinetic analysis.
| dNTP-template | Vmax/Km | finc |
|---|---|---|
| The data were derived from five different experiments and have standard errors of <20%. The reaction time was 2 min with 5 nM of P2 Dpo4 and replication assays included 60 mM KCl. The nucleotide misincorporation ratio, finc = (Vmax/Km)incorrect/(Vmax/Km)correct. | ||
| dCTP-G | 13.40 | 1 |
| dTTP-G | 0.0057 | 4.24 × 10–4 |
| dATP-G | 0.0064 | 4.76 × 10–4 |
| dGTP-G | 0.0047 | 3.50 × 10–4 |
| dCTP-A | 0.0032 | 5.01 × 10–4 |
| dTTP-A | 6.19 | 1 |
| dATP-A | 0.0131 | 2.12 × 10–3 |
| dGTP-A | 0.0051 | 8.08 × 10–3 |
| dCTP-T | 0.0023 | 3.69 × 10–4 |
| dTTP-T | 0.0035 | 5.62 × 10–4 |
| dATP-T | 6.23 | 1 |
| dGTP-T | 0.0056 | 8.99 × 10–4 |
| dCTP-C | 0.0015 | 4.26 × 10–4 |
| dTTP-C | 0.0015 | 4.26 × 10–4 |
| dATP-C | 0.0083 | 2.36 × 10–3 |
| dGTP-C | 3.52 | 1 |
Processivity of S.solfataricus P2 Dpo4
In the absence of cofactors, the related E.coli pol IV and human polκ enzymes exhibit differences in their intrinsic processivity. Escherichia coli pol IV is strictly distributive and only extends primers by 1 nt (17) whereas human polκ extends primers by ∼25 nt per binding event (22). We were therefore interested in determining the processivity of the archaeal P2 Dpo4 polymerase. To do so, we utilized the ∼7.2 kb circular M13mp18 as a template for replication reactions. In these experiments, the concentration of template was fixed at 10 nM, and the amount of P2 Dpo4 varied over a 400-fold range. At high enzyme to P/T ratios (20-fold excess of enzyme to P/T), Dpo4 gave replication products that were several hundred nucleotides in length (Fig. 3). At equimolar enzyme to P/T ratios, radiolabeled primers were extended in a distributive manner up to ∼50 nt. At much lower enzyme to P/T ratios (20-fold excess of P/T to enzyme), the majority of primers were only extended by 1–2 nt, although a small fraction did appear to be extended by 10–11 bp (Fig. 3). Thus, we conclude that in the absence of additional cofactors, Dpo4 is an essentially distributive enzyme that only extends primers by 1–2 nt per binding event. However, at high enzyme to P/T ratios, dissociation and rebinding of the enzyme to the P/T is robust and can lead to the synthesis of polynucleotide chains of several hundred nucleotides in length. Similar results were obtained in experiments where the radiolabeled P/T and enzyme were kept constant, but an excess of unlabeled P/T was added to the reaction as a ‘trap’ for any Dpo4 molecules that dissociated from the labeled P/T (data not shown).
Figure 3.
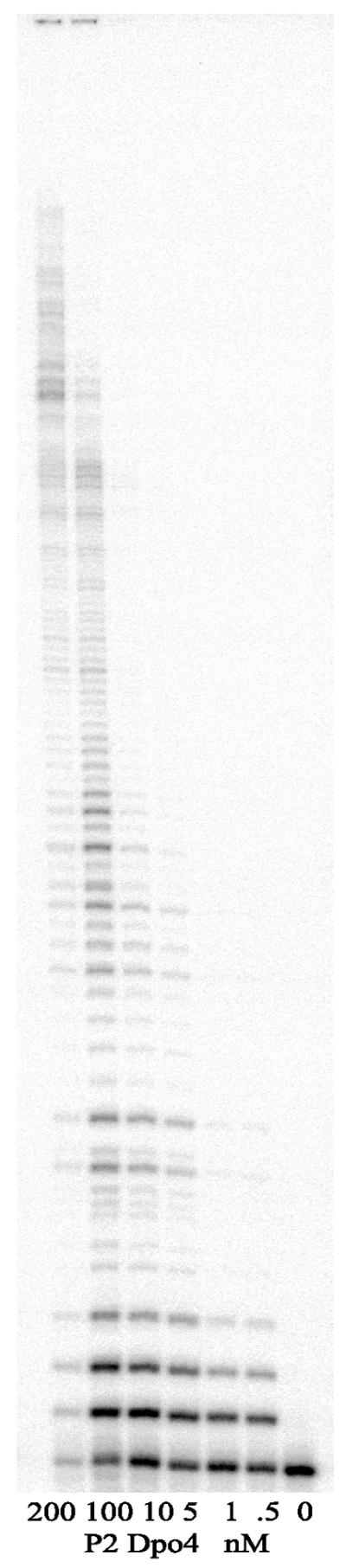
Sulfolobus solfataricus P2 Dpo4 is a distributive polymerase. The ability of Dpo4 to extend a primer annealed to a long (∼7.2 kb) single-stranded DNA template was assayed over a range of enzyme concentrations. The P/T was kept fixed at 10 nM and the enzyme concentration varied from 200 to 0.5 nM as indicated. Reactions contained all four dNTPs (100 µM each) and were performed for 5 min at 37°C. This experiment shows that in the absence of additional cofactors, P2 Dpo4 is a distributive enzyme, primarily synthesizing 1–2 nt per binding event.
Thermostability of the Dpo4
As S.solfataricus naturally grows at moderately high temperatures (13), we assumed that the enzyme would be thermostable. Indeed, our purification protocol relies on the fact that the enzyme is not denatured after heating crude cell lysates at 85°C for 5 min (Fig. 1). The thermostability of the highly purified Dpo4 enzyme was more accurately determined by heating aliquots at a variety of temperatures for 5 min. After this time, we assayed the ability of the enzyme to incorporate a single dCMP opposite template G, which is the most catalytically favorable incorporation for the enzyme (Table 1). The thermostability of each enzyme was compared with two well-characterized enzymes, Taq polymerase (Roche Molecular) and E.coli pol I Klenow fragment (New England Biolabs), under the same assay conditions. As seen in Figure 4, Dpo4 remains active over a wide range of temperatures and under these assay conditions was indistinguishable from the thermostable Taq polymerase. In contrast, the activity of pol I Klenow fragment diminished rapidly after heating to temperatures of >65°C and was abolished above 75°C (Fig. 5). The S.solfataricus P2 Dpo4 enzyme is, therefore, the first characterized thermostable member of the Y-family of DNA polymerases.
Figure 4.

Thermostability of S.solfataricus P2 Dpo4. Aliquots of P2 Dpo4 (triangle), Taq polymerase (circle) and E.coli pol I Klenow fragment (diamond) were heated at the temperature indicated for 5 min. The effect of such treatment on polymerase activity was subsequently determined by assaying the respective enzyme’s ability to extend a radiolabeled primer via the correct incorporation of dCMP opposite template G. Replication assays were performed at 37°C and 100% incorporation occurred when all of the primer was extended by 1 nt.
Figure 5.
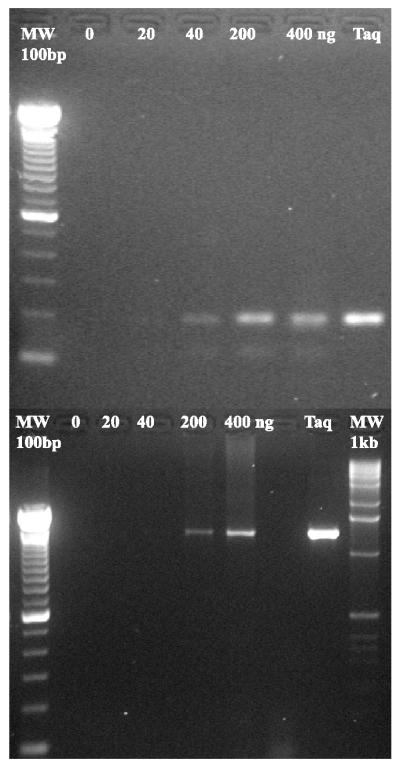
PCR amplification DNA fragments by S. solfataricus P2 Dpo4. (Upper) Amplification of an ∼180 bp fragment by P2 Dpo4 or Taq polymerase. The exact amplification conditions are given in the Materials and Methods. The P2 enzyme concentration was varied from 20 to 400 ng. Track MW represents 100 bp molecular weight markers (Life Technologies). (Lower) Amplification of a ∼1300 bp DNA fragment. Enzyme concentrations are identical to those indicated above and amplification conditions are also given in the Materials and Methods. Track MW represents 1 kb molecular weight markers (Life Technologies).
PCR reactions with S.solfataricus P2 Dpo4
Given that the Dpo4 is thermostable and at high enzyme to P/T ratios can extend primers by several hundred nucleotides, we were interested in determining if the enzyme might be able to substitute for Taq polymerase in polymerase chain reactions (PCR). Indeed, a short linear fragment of ∼200 bp was amplified with ∼70% yield of that obtained with Taq polymerase (Fig. 5, upper panel). We have also been able to amplify a fragment of ∼1300 bp, but as expected the efficiency of the reaction was less efficient than that observed for Taq polymerase under the same conditions (Fig. 5, lower panel). Thus, we conclude that under certain conditions, S.solfataricus P2 Dpo4 can substitute for Taq polymerase in PCR. In general, DNA polymerases used in PCR are derived from either family-A or family-B type polymerases, and we believe that this is the first report of such an activity for a member of the Y-family of polymerases. We have not measured the fidelity of the PCR reaction directly, but if we assume that it is similar to the fidelity of the primer extension reactions performed at 37°C (Table 1), misincorporation frequencies would be in the range of 10–3 to 10–4 making it slightly more error-prone than Taq polymerase.
Ability of P2 Dpo4 to bypass DNA lesions
The Y-family polymerases are best characterized by their ability to facilitate translesion replication of a variety of DNA lesions (reviewed in 2,8,46). Interestingly, the DinB branch of the family appears unable to replicate through UV-induced lesions such as a cis-syn thymine–thymine dimer, or a 6-4 pyrimidine–pyrimidone lesion (18,20,21,23). They are, however, able to replicate through synthetic abasic sites and certain bulky adducts like acetyl aminofluorene adducted guanine residues (AAF-G) (18,21,23,25,47). In these instances, bypass appears to occur via a P/T slippage mechanism, rather than via direct translesion replication, as in vitro replication products are often 1–2 bp shorter than expected. We were therefore interested in determining the ability of the Sulfolobus DinB-like polymerases to bypass a cis-syn thymine–thymine dimer, a 6-4 photoproduct, an abasic site, a 1–2 cisplatinated guanine and an AAF-G lesion (30). Surprisingly, Dpo4 was able to bypass all lesions tested (Fig. 6). In time-course experiments, the lesion most easily bypassed was the abasic site, with close to 100% primer extension by 30 min (Fig. 7).
Figure 6.

Time-course experiments showing the ability of S.solfataricus P2 Dpo4 to bypass various DNA lesions. CPD, cis-syn cyclobutane pyrimidine thymine–thymine dimer: 6-4, 6-4 pyrimidine–pyrimidone thymine–thymine dimer; Abasic, synthetic abasic site; Cis-Pt, 1.2-cisplatinated guanine; AAF, acetyl aminofluorene guanine. The T-template is an undamaged control for the CPD, 6-4 and Abasic lesions. The G-template is the undamaged control for the Cis-Pt and AAF templates. Replication reactions were performed in the presence of 10 nM Dpo4 and 100 µM dNTPs for the times noted.
Figure 7.

Quantitation of primer elongation shown in Figure 6. Filled circle, undamaged T; filled diamond, undamaged G: open triangle, abasic site; filled square, cisplatin-G; open circle, cis-syn TT; filled triangle, 6-4 TT; open square, AAF-G.
Perhaps the most interesting observation is that unlike other DinB-like polymerases, Dpo4 is able to insert bases opposite the CPD and 6-4 lesions. Especially in the case of the CPD, these can be further elongated to generate full-length replication products (Fig. 6). Faint bypass products were also observed with the 6-4 lesion, indicating that it is not an absolute block to Dpo4-dependent replication. Dpo4 was also able to extend primers annealed to the cisplatinated and AAF-G templates, usually by the incorporation of a single nucleotide opposite the first adducted G, although it is also evident that in both cases, a small amount of full-length product can be seen at longer time points, indicating that complete lesion bypass has occurred (Fig. 6).
We have also determined the Dpo4-dependent nucleotide (mis)incorporation pattern at each of the lesions (Fig. 8). If one assumes that in this qualitative assay, the extent of primer elongation in the presence of a single dNTP is a good measure of the enzyme’s fidelity, one observes that Dpo4 generally inserts the correct nucleotide at each respective DNA lesion. Thus, the CPD is bypassed by the incorporation of two A residues, and the AAF-G and cisplatinated G by the incorporation of one or two cytosines, respectively. Although a complete bypass of the 6-4 lesion was limited, most efficient extension of the radiolabeled primer also occurred in the presence of dATP. Like many polymerases, Dpo4 appears to follow the ‘A-rule’ (48) when encountering an abasic site. We have confirmed that the qualitative assay shown in Figure 8 is a good measure of the enzyme’s fidelity by quantitative steady-state kinetic analyses of nucleotide incorporation at the 3′-T of the CPD, the 3′-T of the 6-4 lesion, and the abasic site (Table 2).
Figure 8.

Ability of S.solfataricus P2 Dpo4 to incorporate nucleotides at various damaged template sites. The extent of (mis)incorporation was measured at each template site in the absence (0) or presence of all four dNTPs (4) or presence of each individual dNTP (100 µM) (G, A, T, C). Reactions were for 5 min at 37°C with 10 nM of Dpo4. The sequence context of each template is given above each panel. The extent of primer utilization (expressed as a percentage of the 0 dNTP control) is given below each track. From this qualitative assay, one observes that Dpo4 prefers to incorporate the correct nucleotide at each lesion. These observations have been confirmed by steady-state kinetic analysis (Table 2).
Table 2. Misinsertion fidelity of P2 Dpo4 on damaged DNA templates as determined by steady-state kinetic analysis.
| DNA lesion | dNTP | Vmax / Km (µM–1 min–1) | Relfinc |
|---|---|---|---|
| The data were derived from four different experiments and have standard errors of <20%. The reaction time 5 min with 10 nM of P2 Dpo4. The relative frequency of misincoporation, Relfinc = (Vmax/Km)incorrect/(Vmax/Km)correct at each template site. For the CPD, the 6-4 and abasic site the correct base was assumed to be A. Incorporation at AAF-G and cisplatin-G was also determined, but only the correct incorporation of C opposite each lesion was measurable, and as a consequence no relative frequency of incoporation was determined. | |||
| CPD-TT | dGTP | 0.00042 | 1.2 × 10–2 |
| dATP | 0.03431 | 1 | |
| dTTP | 0.00030 | 8.7 × 10–3 | |
| dCTP | 0.00023 | 6.7 × 10–3 | |
| 6-4-TT | dGTP | 0.00007 | 2.4 × 10–1 |
| dATP | 0.00029 | 1 | |
| dTTP | 0.00007 | 2.4 × 10–1 | |
| dCTP | 0.00010 | 3.4 × 10–1 | |
| Abasic site | dGTP | 0.00411 | 3.6 × 10–2 |
| dATP | 0.11294 | 1 | |
| dTTP | 0.00120 | 1.1 × 10–2 | |
| dCTP | 0.00391 | 3.0 × 10–2 |
DISCUSSION
The Y-family of DNA polymerases are found in all three kingdoms of life. A recent search revealed at least 52 orthologs (6). However, more members of the family are likely to be identified as the list of completely sequenced genomes increases. Phylogenetic analysis of the archaeal Y-family polymerases places them in the broad DinB branch of an unrooted tree (6). To date, only three archaeal dinB-like genes have been reported in the literature. These include a homolog in S.solfataricus P1 (11), S.solfataricus P2 (16) and Halobacterium NRC-1 (12). Characterization of the S.solfataricus P2 DinB-like polymerase reported here reveals properties consistent with its phylogenetic analysis, but also provides insights into several unique features of the enzyme.
Unlike other DinB-like polymerases, Dpo4 is active over a wide range of temperatures (Fig. 4). When we initiated the biochemical studies on Dpo4, we were unsure of which temperature we should employ for the replication reactions. On one hand, S.solfataricus P2 grows optimally between 70 and 80°C, so replication reactions at such temperatures might be considered more physiological. However, previous studies on the fidelity and lesion-bypass properties of various Y-family polymerases have all utilized templates designed to work optimally at 37°C. As Dpo4 appears to retain full activity at the lower temperature, we chose the latter temperature for our initial characterization of the enzyme, as it allows a direct comparison between various Y-family enzymes within the exact same DNA sequence context (for example compare refs 21,28–32). Experiments are currently in progress with modified DNA templates that permit replication and characterization of Dpo4 at much higher temperatures.
In general, P2 Dpo4 exhibited misincorporation frequencies in the range of 10–3 to 10–4, which is similar to that reported previously for E.coli pol IV and human polκ (18,20,22,24). The most efficient catalytic reaction performed by the enzyme is the correct incorporation of dCMP opposite template G, which is 2–4-fold more efficient than the incorporation of any other correct nucleotide. In comparison, E.coli pol IV also shows a slight preference for incorporating C opposite G (18), as does human polκ (24). However, it should also be noted that in another sequence context, incorporation of A opposite T was slightly favored by human polκ (20). In the exact same sequence context used here, the distantly related human polι inserts T opposite A by a factor of 25–100-fold better than any other correct nucleotide (31). Thus, the catalytically efficient insertion of C opposite G may be a feature that is common to DinB-like polymerases.
The ability of Dpo4 to bypass DNA lesions is unique compared with other members of the DinB polymerase family. In fact, its ability to bypass lesions by generally inserting the correct complementary nucleotide opposite a variety of damaged bases is much more akin to eukaryotic polη (28–30,37,49,50) than to DinB-like polymerases. Although found in archaea, Y-family DNA polymerases clearly have a limited distribution and to date, have only been identified in three strains of archaea (11,12,16), two of which (S.solfataricus P1 and P2) are closely related. Presumably, such enzymes provide a selective advantage to S.solfataricus and Halobacterium species that is not necessary in other archaea. Sulfolobus solfataricus grows optimally at close to 80°C and as a consequence, its genome is much more likely to undergo depurination/depyrimidination than organisms which grow at lower temperatures. It may be of no passing coincidence therefore that the lesion most readily bypassed by Dpo4 is an abasic site (Figs 6 and 7).
Like E.coli pol IV, S.solfataricus Dpo4 appears to be distributive in the absence of stimulatory cofactors. However, the processivity of E.coli pol IV is greatly enhanced in the presence of the β-clamp and γ-clamp loading complex (18,19). In contrast, human polκ appears to be much more involved in processing (22), and is not stimulated by human PCNA (25). Sulfolobus solfataricus P2 possesses multiple PCNA orthologs (16,51), and it will be of interest to determine whether their inclusion in Dpo4-dependent replication assays will affect the ability of the enzyme to synthesize DNA processively. Despite the intrinsic distributive nature of the enzyme, the activity of Dpo4 is sufficiently robust that long stretches of DNA can be synthesized at high enzyme to template ratios. This fact, combined with the thermostability of Dpo4, allowed us to use the enzymes as a substitute for Taq polymerase in PCR reactions. The low-fidelity and distributive nature of Dpo4 suggests that it is unlikely to pose as a significant alternative to Taq polymerase in many general PCR reactions, but one might envisage that such an activity might be useful on damaged DNA templates that cannot be amplified by thermostable family-A or family-B polymerases.
Finally, the availability of reasonably large quantities (∼7 mg of purified protein per liter of E.coli culture) of a thermostable member of the Y-family of DNA polymerases, provides an excellent opportunity for crystallographic structure–function studies. Indeed, the structure of Dpo4 in a ternary complex with template DNA and incoming nucleotide has recently been solved at a resolution of 1.7 Å (52). Based upon the biochemical properties of the Dpo4 enzyme reported here, structure–function studies on the polymerase should not only provide insights into bacterial and archaeal enzymes, but eukaryotic DNA polymerases such as human pols η, ι and κ.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Wolfram Zillig, Stephen Bell and members of the Section on DNA Replication, Repair and Mutagenesis for their helpful comments and suggestions during the course of this work. Special thanks go to John McDonald, Ekaterina Frank and Agnès Tissier for invaluable assistance during the initial stages of this project. This work was supported by the NIH Intramural Research Program.
REFERENCES
- 1.Hubscher U., Nasheuer,H.-P. and Syvaoja,J.E. (2000) Eukaryotic DNA polymerases, a growing family. Trends Biochem. Sci., 25, 143–147. [DOI] [PubMed] [Google Scholar]
- 2.Goodman M.F. and Tippen,B. (2000) The expanding polymerase universe. Nature Rev. Mol. Cell. Biol., 1, 101–109. [DOI] [PubMed] [Google Scholar]
- 3.Ito J. and Braithwaite,D.K. (1991) Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res., 19, 4045–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braithwaite D.K. and Ito,J. (1993) Compilation, alignment and phylogenetic relationships of DNA polymerases. Nucleic Acids Res., 21, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cann I.K. and Ishino,Y. (1999) Archaeal DNA replication: identifying the pieces to solve a puzzle. Genetics, 152, 1249–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohmori H., Friedberg,E.C., Fuchs,R.P.P., Goodman,M.F., Hanaoka,F., Hinkle,D., Kunkel,T.A., Lawrence,C.W., Livneh,Z., Nohmi,T., Prakash,L., Prakash,S., Todo,T., Walker,G.C., Wang,Z. and Woodgate,R. (2001) The Y-family of DNA polymerases. Mol. Cell.., 8, 7–8. [DOI] [PubMed] [Google Scholar]
- 7.McDonald J.P., Rapic-Otrin,V., Epstein,J.A., Broughton,B.C., Wang,X., Lehmann,A.R., Wolgemuth,D.J. and Woodgate,R. (1999) Novel human and mouse homologs of Saccharomyces cerevisiae DNA polymerase η. Genomics, 60, 20–30. [DOI] [PubMed] [Google Scholar]
- 8.Friedberg E.C., Feaver,W.J. and Gerlach,V.L. (2000) The many faces of DNA polymerases: strategies for mutagenesis and for mutational avoidance. Proc. Natl Acad. Sci. USA, 97, 5681–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogi T., Kato,T.,Jr, Kato,T. and Ohmori,H. (1999) Mutation enhancement by DINB1, a mammalian homologue of the Escherichia coli mutagenesis protein DinB. Genes Cells, 4, 607–618. [DOI] [PubMed] [Google Scholar]
- 10.Gerlach V.L., Aravind,L., Gotway,G., Schultz,R.A., Koonin,E.V. and Friedberg,E.C. (1999) Human and mouse homologs of Escherichia coli DinB (DNA polymerase IV), members of the UmuC/DinB superfamily. Proc. Natl Acad. Sci. USA, 96, 11922–11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kulaeva O.I., Koonin,E.V., McDonald,J.P., Randall,S.K., Rabinovich,N., Connaughton,J.F., Levine,A.S. and Woodgate,R. (1996) Identification of a DinB/UmuC homolog in the archeon Sulfolobus solfataricus. Mutat. Res., 357, 245–253. [DOI] [PubMed] [Google Scholar]
- 12.Ng W.V., Kennedy,S.P., Mahairas,G.G., Berquist,B., Pan,M., Shukla,H.D., Lasky,S.R., Baliga,N.S., Thorsson,V., Sbrogna,J. et al. (2001) Genome sequence of Halobacterium species NRC-1. Proc. Natl Acad. Sci. USA, 97, 12176–12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zillig W., Stetter,K.O., Wunderl,S., Schulz,W., Priess,H. and Scholz,I. (1980) The Sulfolobus-‘caldariella’ group: taxonomy on the basis of the structure of DNA-dependent RNA polymerases. Arch. Microbiol., 125, 259–269. [Google Scholar]
- 14.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 15.Tatusov R.L., Altschul,S.F. and Koonin,E.V. (1994) Detection of conserved segments in proteins: iterative scanning of sequence databases with alignment blocks. Proc. Natl Acad. Sci. USA, 91, 12091–12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.She Q., Singh,R.K., Confalonieri,F., Zivanovic,Y., Allard,G., Awayez,M.J., Chan-Weiher,C.C., Clausen,I.G., Curtis,B.A., De Moors,A. et al. (2001) The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc. Natl Acad. Sci. USA, 98, 7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner J., Gruz,P., Kim,S.R., Yamada,M., Matsui,K., Fuchs,R.P.P. and Nohmi,T. (1999) The dinB gene encodes an novel Escherichia coli DNA polymerase (DNA pol IV) involved in mutagenesis. Mol. Cell., 4, 281–286. [DOI] [PubMed] [Google Scholar]
- 18.Tang M., Pham,P., Shen,X., Taylor,J.-S., O’Donnell,M., Woodgate,R. and Goodman,M. (2000) Roles of E.coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature, 404, 1014–1018. [DOI] [PubMed] [Google Scholar]
- 19.Wagner J., Fujii,S., Gruz,P., Nohmi,T. and Fuchs,R.P. (2001) The β clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep., 1, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson R.E., Prakash,S. and Prakash,L. (2000) The human DINB1 gene encodes the DNA polymerase Polθ. Proc. Natl Acad. Sci. USA, 97, 3838–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohashi E., Ogi,T., Kusumoto,R., Iwai,S., Masutani,C., Hanaoka,F. and Ohmori,H. (2000) Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev., 14, 1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 22.Ohashi E., Bebenek,K., Matsuda,T., Feaver,W.J., Gerlach,V.L., Friedberg,E.C., Ohmori,H. and Kunkel,T.A. (2000) Fidelity and processivity of DNA synthesis by DNA polymerase κ, the product of the human DINB1 gene. J. Biol. Chem., 275, 39678–39684. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y., Yuan,F., Wu,X., Wang,M., Rechkoblit,O., Taylor,J.S., Geacintov,N.E. and Wang,Z. (2000) Error-free and error-prone lesion bypass by human DNA polymerase κin vitro. Nucleic Acids Res., 28, 4138–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Yuan,F., Xin,H., Wu,X., Rajpal,D.K., Yang,D. and Wang,Z. (2000) Human DNA polymerase κ synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res., 28, 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerlach V.L., Feaver,W.J., Fischhaber,P.L. and Friedberg,E.C. (2001) Purification and characterization of pol κ, a DNA polymerase encoded by the human DINB1 gene. J. Biol. Chem., 276, 92–98. [DOI] [PubMed] [Google Scholar]
- 26.McDonald J.P., Frank,E.G., Levine,A.S. and Woodgate,R. (1998) Intermolecular cleavage of the UmuD-like mutagenesis proteins. Proc. Natl Acad. Sci. USA, 95, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorf,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 28.Masutani C., Araki,M., Yamada,A., Kusumoto,R., Nogimori,T., Maekawa,T., Iwai,S. and Hanaoka,F. (1999) Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J., 18, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masutani C., Kusumoto,R., Yamada,A., Dohmae,N., Yokoi,M., Yuasa,M., Araki,M., Iwai,S., Takio,K. and Hanaoka,F. (1999) The XPV (Xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature, 399, 700–704. [DOI] [PubMed] [Google Scholar]
- 30.Masutani C., Kusumoto,R., Iwai,S. and Hanaoka,F. (2000) Mechanisms of accurate translesion synthesis by human DNA polymerase η. EMBO J., 19, 3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tissier A., McDonald,J.P., Frank,E.G. and Woodgate,R. (2000) polι, a remarkably error-prone human DNA polymerase. Genes Dev., 14, 1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 32.Tissier A., Frank,E.G., McDonald,J.P., Iwai,S., Hanaoka,F. and Woodgate,R. (2000) Misinsertion and bypass of thymine–thymine dimers by human DNA polymerase ι. EMBO J., 19, 5259–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boosalis M.S., Petruska,J. and Goodman,M.F. (1987) DNA polymerase insertion fidelity. Gel assay for site-specific kinetics. J. Biol. Chem., 262, 14689–14696. [PubMed] [Google Scholar]
- 34.Creighton S., Bloom,L.B. and Goodman,M.F. (1995) Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading and lesion bypass efficiencies. Methods Enzymol., 262, 232–256. [DOI] [PubMed] [Google Scholar]
- 35.Johnson R.E., Prakash,S. and Prakash,L. (1999) Requirement of DNA polymerase activity of yeast Rad30 protein for its biological function. J. Biol. Chem., 274, 15975–15977. [DOI] [PubMed] [Google Scholar]
- 36.Johnson R.E., Washington,M.T., Prakash,S. and Prakash,L. (1999) Bridging the gap: a family of novel DNA polymerases that replicate faulty DNA. Proc. Natl Acad. Sci. USA, 96, 12224–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson R.E., Prakash,S. and Prakash,L. (1999) Efficient bypass of a thymine–thymine dimer by yeast DNA polymerase, polη. Science, 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- 38.Reuven N.B., Arad,G., Maor-Shoshani,A. and Livneh,Z. (1999) The mutagenesis protein UmuC is a DNA polymerase activated by UmuD’, RecA and SSB and Is specialized for translesion replication. J. Biol. Chem., 274, 31763–31766. [DOI] [PubMed] [Google Scholar]
- 39.Maor-Shoshani A., Reuven,N.B., Tomer,G. and Livneh,Z. (2000) Highly mutagenic replication by DNA polymerase V (UmuC) provides a mechanistic basis for SOS untargeted mutagenesis. Proc. Natl Acad. Sci. USA, 97, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson R.E., Washington,M.T., Haracska,L., Prakash,S. and Prakash,L. (2000) Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature, 406, 1015–1019. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y., Yuan,F., Wu,X. and Wang,Z. (2000) Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase ι. Mol. Cell. Biol., 20, 7099–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruck I., Woodgate,R., McEntee,K. and Goodman,M.F. (1996) Purification of a soluble UmuD’C complex from Escherichia coli: cooperative binding of UmuD’C to single-stranded DNA. J. Biol. Chem., 271, 10767–10774. [DOI] [PubMed] [Google Scholar]
- 43.Tang M., Bruck,I., Eritja,R., Turner,J., Frank,E.G., Woodgate,R., O’Donnell,M. and Goodman,M.F. (1998) Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD’2C mutagenic complex and RecA. Proc. Natl Acad. Sci. USA, 95, 9755–9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarov-Blat L. and Livneh,Z. (1998) The mutagenesis protein MucB interacts with single strand DNA binding protein and induces a major conformational change in its complex with single-stranded DNA. J. Biol. Chem., 273, 5520–5527. [DOI] [PubMed] [Google Scholar]
- 45.Goldsmith M., Sarov-Blat,L. and Livneh,Z. (2000) Plasmid-encoded MucB protein is a DNA polymerase (pol RI) specialized for lesion bypass in the presence of MucA’, RecA and SSB. Proc. Natl Acad. Sci. USA, 97, 11227–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodgate R. (1999) A plethora of lesion-replicating DNA polymerases. Genes Dev., 13, 2191–2195. [DOI] [PubMed] [Google Scholar]
- 47.Napolitano R., Janel-Bintz,R., Wagner,J. and Fuchs,R.P. (2000) All three SOS-inducible DNA polymerases (Pol II, pol IV and pol V) are involved in induced mutagenesis. EMBO J., 19, 6259–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strauss B.S. (1991) The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays, 13, 79–84. [DOI] [PubMed] [Google Scholar]
- 49.Johnson R.E., Kondratick,C.M., Prakash,S. and Prakash,L. (1999) hRAD30 mutations in the variant form of Xeroderma pigmentosum. Science, 285, 263–265. [DOI] [PubMed] [Google Scholar]
- 50.Johnson R.E., Washington,M.T., Prakash,S. and Prakash,L. (2000) Fidelity of Human DNA polymerase η. J. Biol. Chem., 275, 7447–7450. [DOI] [PubMed] [Google Scholar]
- 51.Iwai T., Kurosawa,N., Itoh,Y.H. and Horiuchi,T. (2000) Phylogenetic analysis of archaeal PCNA homologues. Extremophiles, 4, 357–364. [DOI] [PubMed] [Google Scholar]
- 52.Ling H., Boudsocq,F., Woodgate,R. and Yang,W. (2001) Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell, 107, 91–102. [DOI] [PubMed] [Google Scholar]