

Abstract
Sleep disturbances frequently occur in neurodevelopmental disorders such as autism, but the developmental role of sleep is largely unexplored, and a causal relationship between developmental sleep defects and their behavioral consequences in adulthood remains elusive. Here we show that in mice, sleep disruption (SD) in adolescence, but not in adulthood, causes long-lasting impairment in social novelty preference. Furthermore, adolescent SD alters the activation and release patterns of dopaminergic neurons in the ventral tegmental area (VTA) in response to social novelty. This developmental sleep function is mediated by balanced VTA activity during adolescence; chemogenetic excitation mimics whereas silencing rescues the social deficits of adolescent SD. Finally, we show that in Shank3 mutant mice, improving sleep or rectifying VTA activity during adolescence ameliorates adult social deficits. Together, our results identify a critical role of sleep and dopaminergic activity in the development of social interaction behavior.
Keywords: sleep, adolescent development, social novelty preference, VTA, mesocorticolimbic pathway, autism spectrum disorder
Introduction
Virtually all organisms sleep and in humans, sleep takes approximately one third of our lives1,2. The suggested functions of sleep include metabolic restoration, detoxification, and cognitive roles in neural plasticity and memory consolidation, most of which were discovered in adult individuals1–3. On the other hand, sleep architecture demonstrates a clear developmental trajectory. Total sleep time decreases from infancy through adolescence to adulthood4, and compared to adult sleep, electroencephalogram (EEG) of sleep during development contains more components beneficial to memory and cognition, such as slow waves and sleep spindles5–7. In juvenile animals, sleep regulates synaptic plasticity both functionally and structurally3,8–15 and likely plays important roles in developmental wiring of neural circuits. However, whether and how this synaptic function contributes to the developmental shaping of entire circuitry and further elicits long-term behavioral impacts remain elusive. Notably, due to technical limitations, most if not all of the discoveries about sleep functions in development were made in the sensory/motor cortices, whereas evidence has been lacking in the subcortical realm regarding other complex brain functions such as social behavior.
Interestingly, sleep problems including delayed sleep onset, shortened sleep duration and sleep fragmentation are frequently reported (50 – 80%) in autism spectrum disorders (ASDs), schizophrenia (SZ) and other neurodevelopmental disorders, especially in young patients16–21. This implicates a link between sleep disturbances during development and the progression of behavioral abnormalities in these disorders. Specifically, the severity of social communication defects, a core symptom shared by multiple neurodevelopmental disorders17, inversely correlates with sleep duration. However, the underlying causal relationship between social deficits and sleep has not been established and a circuit mechanism not identified. Dopaminergic neurons in the ventral tegmental area (VTADA neurons) together with their projections to the ventral striatum (also known as nucleus accumbens, NAc) and prefrontal cortex (PFC) constitute the mesocorticolimbic pathway, which serves key functions in motivation, emotion, reward-seeking behavior (including that for social reward) and addiction22. Abnormal dopaminergic signaling has been suggested in ASDs based on research in human patients as well as animal models23–26, while previous work from our lab27 and others 28–31 showed that the activity of VTADA neurons is differentially regulated by sleep/wake states. Together these findings place VTA dopaminergic signaling at the nexus of sleep and social behavior, connecting the two essential functions of the brain in the same context.
The VTA-NAc circuit serves a key role in controlling sociability32,33. In contrast, much less is known about the neural underpinnings of another important aspect of social interaction behavior, namely the social novelty preference. In mice, social novelty preference is expressed behaviorally as a shift of interest from the familiar social stimulus to the novel one when both are present. Recent studies indicate that VTA is also involved32, but the circuit mechanism is unknown. More importantly, however, interrogation of either sociability or social novelty preference from a developmental perspective has been lacking. Little is known about how these social circuits are organized through what developmental course and what factors are the key players in the process.
Here we focus on the social novelty preference during social interaction and hypothesize that sleep during development shapes this behavioral preference and its underlying neural circuits. We therefore investigated the impact of developmental sleep disruption (SD) in mice and identified a critical period within adolescence that is most important for same-sex social interactions. We then demonstrate that adolescent SD induces a series of long-lasting functional and structural changes in the VTA dopaminergic circuits important for social interaction behavior, using fiber-photometry, ex vivo electrophysiology and anatomical analyses. Furthermore, using chemogenetic manipulations, we show that a balanced level of VTA activity during adolescence is required for this sleep function and sufficient to shape normal social novelty preference under sleep disruption. Finally, we show that improving NREM sleep or directly reducing VTA activity during adolescent critical period can ameliorate social deficits and restore normal social interaction in the Shank3 InsG3680 autism mouse model.
Results
Adolescent SD impairs social novelty preference in adulthood
We first asked whether social interaction behavior is developmentally shaped, and at which developmental stage. We probed same-sex social interactions in wild-type, C57BL/6J mice at postnatal day 28 (P28), P42, P56 and P84, using a three-chamber social interaction assay with sex- and age-matched stimulus mice from the same genetic background (Extended Data Fig. 1a–c). We found that both sociability (towards a never-before-met stimulus mouse, S1, over a non-social empty mesh cup, E, Trial 1) and social novelty preference (towards a novel stranger mouse, S2, over the familiar S1, Trial 2) developmentally increased through adolescence (P28 – P56) and stabilized or even decreased once the animal entered adulthood (P56 – P84, Extended Data Fig. 1a–c). Thus, we sought to investigate whether sleep disruption (SD) during adolescence impacts social interaction behavior later in adulthood. To disrupt adolescent sleep, we used an automated sleep deprivation system which delivers randomized pushes to the apparatus (Fig. 1a, also see Methods) and prevents mice from sleep. This SD protocol was performed in the early light phase for 4 hours (zeitgeber time, ZT 2 – 6) per day on 5 consecutive days within a mid-adolescent period (P35 – 42, Fig. 1a, see Methods). It effectively abolished both rapid-eye-movement (REM) sleep and non-REM (NREM) sleep (Extended Data Fig. 1d–k, During SD) without inducing permanent changes in sleep architecture (Extended Data Fig. 1d–m, Baseline vs. After SD) or substantial acute stress (Extended Data Fig. 1n). Control littermates (Ctrl) received the same number of pushes at the same age but in early dark phase (ZT 12 – 16) and in a non-randomized manner, which had negligible interference on sleep since they sleep little during the early dark hours. As a result, both Ctrl and SD mice acquired normal sociability when tested at young adult stage (P56, Fig. 1b–i Trial 1, S1 vs. E). However, during Trial 2, while Ctrl animals developed a strong preference towards social novelty, SD mice exhibited an increase in interaction with the familiar mouse S1 and an overall decreased interaction with the novel mouse S2, suggesting loss of social novelty preference (Fig. 1b–i, Trial 2, S2 vs. S1). While the numbers of interaction bouts with each stimulus (Extended Data Fig. 2g) showed consistent changes with total interaction time (Fig. 1g and Extended Data Fig. 2f), no significant difference was found in other parameters including average bout length, entries to each chamber and time in each chamber (Extended Data Fig. 2h, i, j) as well as time spent in each chamber in the habituation phase (Extended Data Fig. 3e) and locomotion during the test trials (Extended Data Fig. 3g,h). This defect in social novelty preference was not due to chronic stress associated with sleep deprivation, as repeated restraint34 in early dark phase during the same adolescent period did not lead to similar defects (Fig. 1j–l). We did not detect a significant Sex effect in any interaction parameters (P > 0.05 for Sex by repeated-measure three-way ANOVA, Extended Data Fig. 2). Thus, we did not treat sex as a biological variable in this type of same-sex social interaction and pooled data from both male-male and female-female interactions together in the rest of the study unless stated otherwise.
Figure 1. Adolescent sleep disruption (SD) induced loss of social novelty preference in adult social interactions.
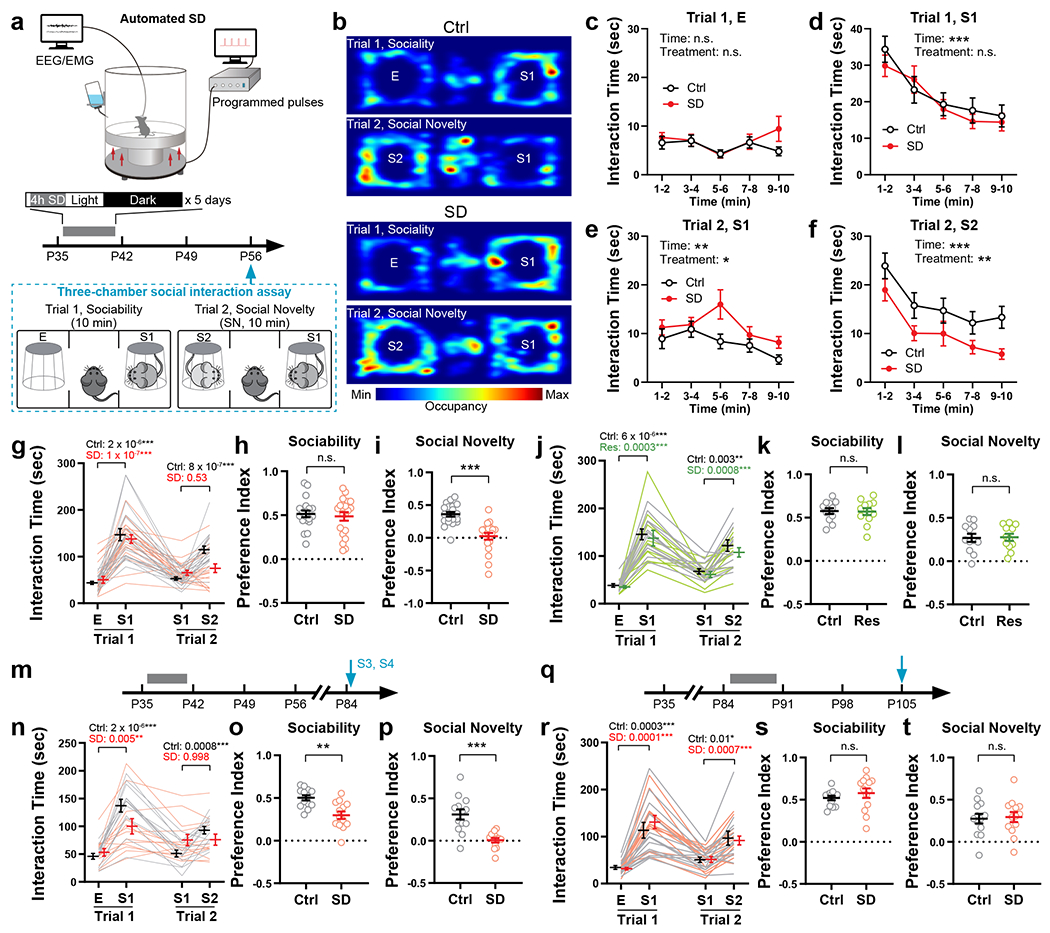
a, Adolescent SD protocol and timeline of experimental procedures. b, Representative occupancy heatmaps of Ctrl and SD mice in the three-chamber apparatus. c-i, Interaction time with each stimulus during the three-chamber test at P56 was quantified. Binned interaction time is shown in c-f, total interaction time is shown in g, and preference indices of sociability and social novelty are shown in h and i, respectively. n = 20 mice (10 males + 10 females). Repeated-measure (RM)-2-way ANOVA, d, Time F(3.66, 139.0) = 16.83, P = 1.2 × 10−10; e, Time F(3.09, 117.3) = 3.98, P = 0.009, Treatment F(1, 38) = 6.27, P = 0.02; f, Time F(3.54, 134.7) = 13.67, P = 1.1 × 10−8, Treatment F(1, 38) = 7.81, P = 0.008; g, Stimulus × Treatment F(3, 114) = 6.23, P = 0.0006, followed by Tukey’s post-tests (P as indicated). Welch’s t-test, h, t(35.99) = 0.46, P = 0.64. Mann-Whitney test, i, P = 4.1 × 10-7. j-l, Mice subjected to adolescent restraint (P35 – 42) were tested using the three-chamber assay at P56. n = 12 mice. j, RM-2-way ANOVA (Stimulus × Treatment F(3, 66) = 0.14, P = 0.94) with Tukey’s post-tests (P as indicated). Welch’s t-tests, k, t(21.47) = 0.13, P = 0.90; l, t(21.44) = 0.11, P = 0.92. m–p, Mice that received adolescent Ctrl or SD (P35 – 42) were tested using the three-chamber assay at P84. n = 13 mice. n, RM-2-way ANOVA (Stimulus × Treatment F(3, 72) = 10.76, P = 6.3 × 10−6) with Tukey’s post-tests (P as indicated). Welch’s t-tests, o, t(21.44)= 3.77, P = 0.001; p, t(16.44) = 4.64, P = 0.0003. q–t, Mice received Ctrl or SD protocol during P84 – 91 and the three-chamber assay was performed at P105. n = 13 mice. r, RM-2-way ANOVA (Stimulus × Treatment F(3, 72) = 0.74, P = 0.53) with Tukey’s post-tests (P as indicated). Welch’s t-tests, s, t(16.97) = 0.92, P = 0.37; t, t(23.93) = 0.22, P = 0.82. Data are shown as mean ± s.e.m. All tests were two-sided. For detailed statistics information, see Supplementary Table 1.
The effect of adolescent SD is long-lasting, as the social novelty preference was still impaired when the mice were re-tested using a new set of stimulus mice (S3, S4) 4 weeks after the initial test (P84, Fig. 1m–p). We note that at this timepoint, the preference index for sociability in SD mice was also lower than the Ctrl group (Fig. 1o), however SD mice still spent almost twice as much time with S1 than E in Trial 1 (Fig. 1n). Thus, we consider SD mice still express sociability. Given that the preference index of sociability at P56 and that at P84 were comparable in Ctrl mice whereas it declined in SD mice (Fig. 1h, o), our interpretation is that adolescent SD may lead to an early decay of sociability in adulthood but does not affect its developmental shaping before P56.
We also performed SD at a later adolescent stage (P42 – 49), which caused similar, but weaker, defect in social novelty preference at P56 (Extended Data Fig. 3a). However, the same SD protocol applied to adult mice (P84 – 91, Fig. 1q), did not cause any defect in either sociability or social novelty preference (Fig. 1r–t), suggesting that social novelty preference requires undisturbed sleep during adolescence, but not during adulthood. Additionally, the impact of adolescent SD was restricted to same-sex social interactions, because SD male mice retained their preference for novel, ovariectomized female conspecifics, indicating SD does not affect sociosexual preference (Extended Data Fig. 3b).
Adolescent SD did not increase restricted and repetitive behavior or anxiety level, as compared to Ctrl mice, the SD group showed similar levels of self-grooming and latency to enter the open arm in elevated plus maze (Extended Data Fig. 3i,j, see Methods). In a novel object recognition assay (Extended Data Fig. 3k, see Methods), SD mice spent less time investigating a non-social novel object compared to the Ctrl group; however, they still spent more time than towards the familiar object (Extended Data Fig. 3m), suggesting attenuated, but still substantial preference for non-social novelty in SD mice (also see Discussion).
To examine whether the social interaction deficit observed in SD mice was due to impaired social recognition or social memory, we performed the two-trial social memory test at P56 – 60. Notably, both groups showed similar decline in investigation time when exposed to the same stimulus mouse after a 30-min interval (Extended Data Fig. 3l, n, 1-stimulus paradigm), suggesting intact recognition and memorization of the stimulus mouse in SD mice. Interestingly, when there was an additional stimulus mouse (S2) in the second trial (Extended Data Fig. 3l,o, 2-stimuli paradigm), Ctrl mice showed further diminished interest in the first stimulus (S1), compared to when there was just S1, despite the 30-min interval. However, in SD mice, this further reduction of S1-interaction was absent regardless of S2 presence (Extended Data Fig. 3p), suggesting an impairment in shifting interest from the familiar stimulus to the novel. Thus, the developmental sleep function we identified here in shaping social interaction is distinct from the adult sleep role in memory consolidation. It likely depends on the circuits underlying preference per se (i.e., the drive to pursue novelty when given a choice) rather than those underlying memory (also see Discussion).
Adolescent SD attenuates VTA activation for social novelty
Which brain structures may underlie the changes in social preference elicited by SD? Given that our behavioral results indicating a defect in the preference for social novelty rather than memory deficits in SD mice (Fig. 1b–i and Extended Data Fig. 3n) and the importance of VTA dopaminergic signaling in regulating both sociability and social novelty preference32,33, we asked whether the activity of VTADA neurons during social interaction was altered by adolescent SD using fiber photometry. DAT-Cre mice subjected to either SD or Ctrl protocol during P35 – 42 received injections of adeno-associated viruses (AAVs) carrying Cre-dependent GCaMP6f and optical fiber implantation to VTA at P42. We recorded population Ca2+ activity of VTADA neurons at P56 – 60 while the animal was engaged in the social interaction test (Fig. 2a, b). Using a Ca2+ transient detection method previously described27, we found no change in the overall activity of VTADA neurons (Fig. 2c and Extended Data Fig. 4b). However, when we aligned the photometric trace to each interaction bout, we found a social novelty-dependent VTA activation pattern in Ctrl mice that was absent in SD mice (Fig. 2d–k). The VTADA neurons in the Ctrl group showed most dramatic activation when the test mouse first encountered the social stimulus (Trial 1 S1, Fig. 2e, l, n), but these S1-induced Ca2+ responses rapidly declined exponentially with more interactions (Fig. 2i). In Trial 2, significant Ca2+ responses recurred and again rapidly declined when novel stranger S2 was encountered whereas no robust response was induced by the familiar S1 (Fig. 2f–l, p). These results are consistent with a previous study showing similar novelty-dependent social responses in VTA under a different experimental design33. However, in SD mice, S1-induced Ca2+ responses were largely attenuated (Fig. 2e, i), but not abolished, and still significantly larger than response to E (Fig. 2o). In contrast, the S2-induced Ca2+ response was completely depleted in SD mice (Fig. 2q). We note that in the SD group, large Ca2+ activities still occurred but did not temporally align with the social interaction events (Extended Data Fig. 4e), suggesting de-coupling of VTA activation and social novelty. As a comparison, the VTA response towards a non-social, but favorable novel stimulus (e.g., a food pellet) did not change in SD mice (Extended Data Fig. 4f–h).
Figure 2. Adolescent SD attenuated the novelty-dependent response pattern of VTADA neurons in social interactions.
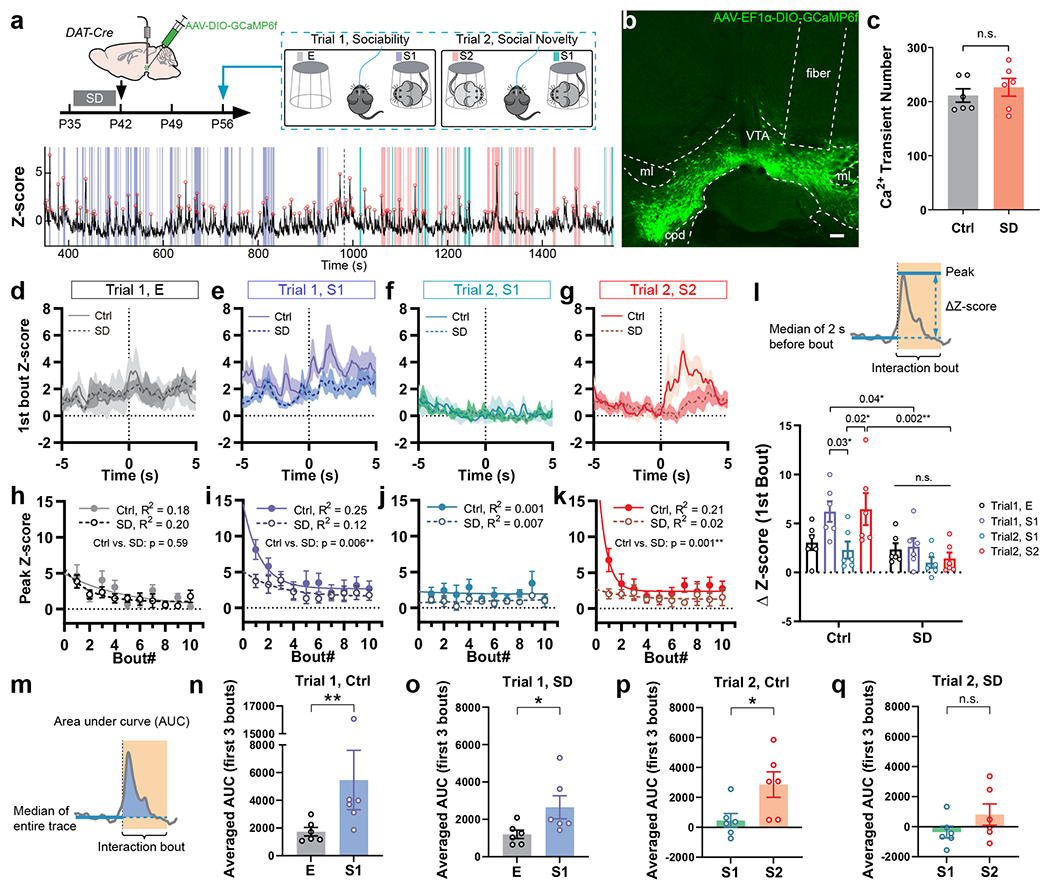
a, Social interaction test with simultaneous fiber photometry was performed on P56 DAT-Cre mice with prior adolescent Ctrl or SD (P35 – 42). Bottom, a representative trace of GCaMP signal recorded. Colored stripes indicate interaction bouts with E (grey), S1 in Trial 1(purple), S1 in Trial 2 (teal) and S2 (pink). Dotted line indicates Trial 2 onset. Red circles indicate transient peaks. b, Representative image showing AAV-delivered GCaMP6f expression and optical fiber placement in the VTA. Scale bar, 100 μm. c, Number of Ca2+ transients detected (Trials 1 + 2). n = 6 mice. Welch’s t-test, t(9.32)= 0.74, P = 0.48. d–g, GCaMP traces during the first interaction bouts with E (d), S1 in Trial 1 (e), S1 in Trial 2 (f) and S2 (g) aligned to the bout onset (time 0 s). n = 6 mice. Shaded area indicates s.e.m. h–k, Peak Z-score of GCaMP signals from first 10 bouts of interactions with E (h), S1 in Trial 1 (i), S1 in Trial 2 (j) and S2 (k) were fitted with one-phase exponential decay model and compared using extra-sum-of-squares F test. n = 6 mice. P is indicated on each graph. In j, P can’t be calculated due to both ambiguous fits. l, ΔZ-score of the first bout of each interaction category. n = 6 mice. RM-2-way ANOVA, Treatment F(1, 10) = 19.27, P = 0.001, followed by Tukey’s post-tests (within each group) and Bonferroni’s post-tests (between groups), post-test P as indicated. m–q Averaged area under curve (AUC) of first 3 interaction bouts of each category. n = 6 mice. Mann-Whitney tests, n, P = 0.009; o, P = 0.04. Welch’s t-tests, p, t(7.77) = 2.47, P = 0.04; q, t(7.82) = 1.43, P = 0.19. Data are shown as mean ± s.e.m. All tests were two-sided. For detailed statistics information, see Supplementary Table 1.
In an attempt to better understand what caused the observed changes in VTA activation pattern, we performed whole-cell patch clamp recordings from VTADA neurons in acute brain slices prepared from young adult DAT-Cre::Ai14 mice with or without prior adolescent SD (Extended Data Fig. 5). We did not find significant differences between groups in spontaneous or evoked firing, hyperpolarization-activated (Ih) currents or cell capacitance of VTADA neurons, nor in the excitatory or inhibitory synaptic transmission onto these neurons (Extended Data Fig. 5). However, we found that VTADA neurons from SD mice had significantly increased membrane resistances compared to the Ctrl group (Extended Data Fig. 5c, i), suggesting plastic adaptations in VTADA neurons that may compensate for changes in circuit dynamics.
Adolescent SD impairs dopamine release in social interaction
The roles of VTADA neurons in processing social reward and regulating social behavior rely on their major projection outputs in the mesocorticolimbic pathway, i.e., the nucleus accumbens (NAc) and prefrontal cortex (PFC). Indeed, NAc neurons, as well as multiple neuronal types in the medial PFC (mPFC), are important players in regulating social interaction33,35–41. To directly measure dopamine release in NAc and examine whether it is targeted by adolescent SD, we utilized the GPCR-activation-based dopamine sensor (GRAB sensor DA2m, Fig. 3a, b)42 and combined it with fiber photometry (Fig. 3a, b). Consistent with the VTA neuronal response pattern (Fig. 2) and the Ca2+ signals recorded from striatal dopaminergic axons33, novelty-dependent dopamine release was detected in the NAc of Ctrl mice during their initial encounters with either S1 or S2 (Fig. 3c–o), followed by a rapid decline in response amplitude as novelty decreased (Fig. 3h, j). Interestingly, dopamine release in SD mice did not decrease during the first a few interaction bouts with S1 in Trial 1, and even slightly increased (Fig. 3d, l–n) showing a significantly slower decline of signal peaks compared to the Ctrl group (Fig. 3h). More importantly, we did not detect dopamine release in response to the novel stimulus S2 (Fig. 3f–l, p). These results align with our behavioral data showing retention of interest in S1 and less interaction with S2 in the SD group for the social novelty trial, suggesting aberrantly elevated dopamine release confined to the first social stimulus and impairment in shifting this rewarding signal to the novel stranger. We also attempted to record dopamine release from mPFC during social interaction; however, these measures were unsuccessful, likely because of sparse dopamine release events in mPFC.
Figure 3. Adolescent SD altered the dopamine release in NAc in response to social stimuli.
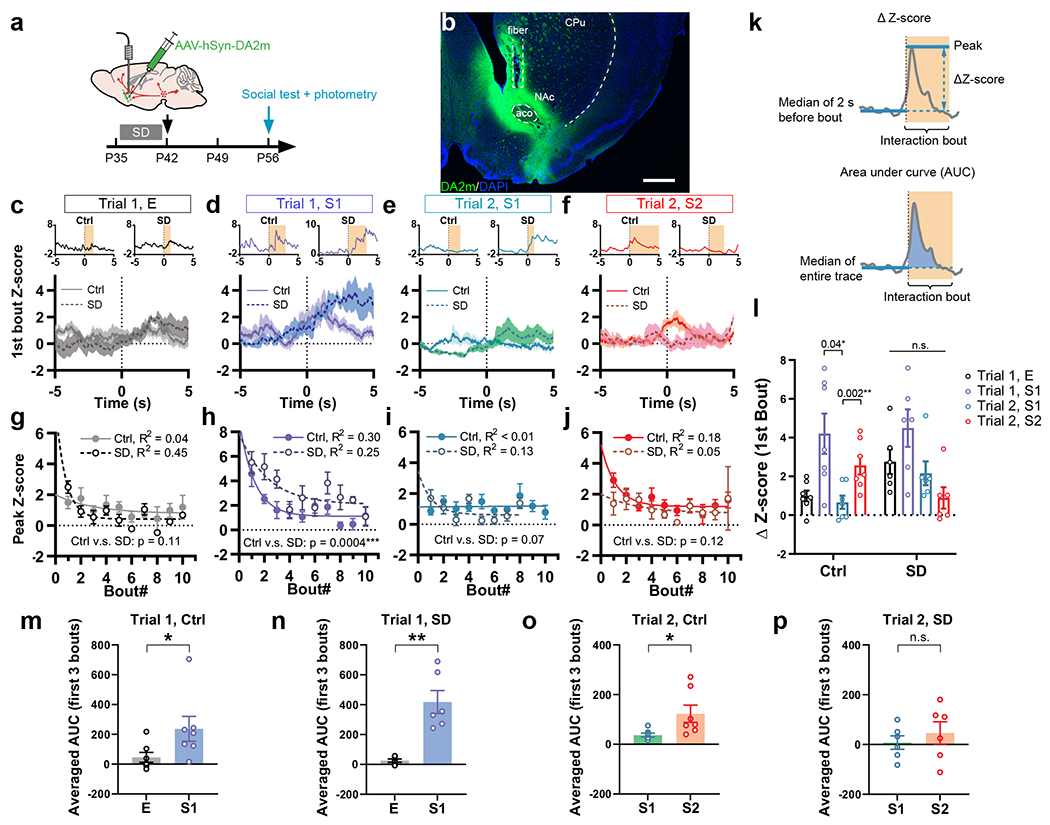
a, Mice with adolescent Ctrl or SD (P35 – 42) received AAVs carrying GRABDA sensor DA2m and optical fiber implantation at P42, and the social interaction test with simultaneous fiber photometry was performed at P56. b, Representative image showing DA2m immunostaining (green) and optical fiber placement to NAc. Scale bar, 500 μm. c–f, DA2m signals during the first interaction bouts with E (c), S1 in Trial 1 (d), S1 in Trial 2 (e) and S2 (f) aligned to the bout onset (time 0 s). n = 7 in Ctrl; 6 in SD. Shaded area indicates s.e.m. Upper insets show representative individual traces. g–j, Peak Z-score of DA2m signals from first 10 bouts of interactions with E (g), S1 in Trial 1 (h), S1 in Trial 2 (i) and S2 (j) were fitted with one-phase exponential decay model and compared using extra-sum-of-squares F test. n = 7 in Ctrl; 6 in SD. P is indicated on each graph. k, Calculation of ΔZ-score and AUC. l, ΔZ-score of the first bout of each interaction category. n = 7 in Ctrl; 6 in SD. RM-2-way ANOVA, Treatment x Stimulus, F(3, 33) = 3.24, P = 0.03, followed by Tukey’s post-tests within each group (P as indicated). m–p, Averaged AUC of first 3 interaction bouts of each category. n = 7 in Ctrl; 6 in SD. Mann-Whitney test, m, P = 0.02. Welch’s t-tests, n, t(16.97) = 0.92, P = 0.004; o, t(6.59) = 2.43, P = 0.048; p, t(8.20) = 0.73, P = 0.48. Data are shown as mean ± s.e.m. All tests were two-sided. For detailed statistics information, see Supplementary Table 1.
Adolescent SD alters VTA projection profile
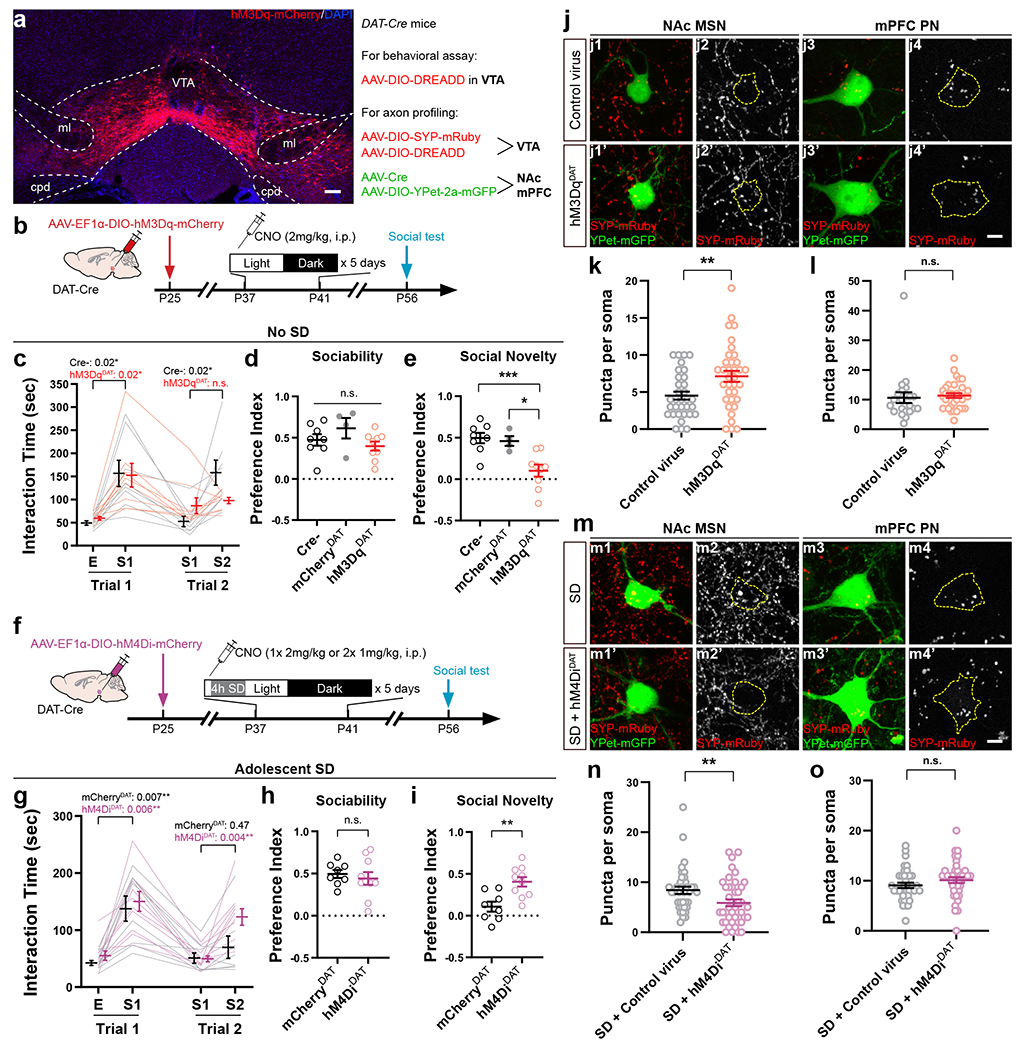
Since adolescent SD leads to long-lasting social defect in adulthood and abnormal dopamine release in NAc, i.e., prolonging during the sociability trial but lost for social novelty, we hypothesized that SD instigates permanent structural changes in the wiring of VTA outputs. Although distinct from conventional synaptic transmission and considered a more diffuse signal, dopamine is released from axonal varicosities with active zones and vesicle docking/release machinery that resemble the presynaptic structure of glutamatergic/GABAergic synapses43. Additionally, each single release event is spatially controlled and only affects the receptors in close vicinity (within a few microns)43. Therefore, we examined the projections from VTADA neurons to NAc and mPFC using a dual-labeling strategy (Fig. 4a). In DAT-Cre mice, AAV-mediated synaptophysin-mRuby (SYP) expression in VTADA neurons allowed visualization of presynaptic boutons of VTA axons while an AAV-hSyn-Cre + AAV-EF1α-DIO-YPet-2a-mGFP mixture injected to NAc/mPFC labeled the structure of target neurons, i.e., medium spiny neurons (MSNs, also known as spiny projection neurons) in NAc as well as pyramidal neurons (PNs) and interneurons (INs) in mPFC (Fig. 4b–d, Extended Data Fig. 6a–c).
Figure 4. Adolescent SD altered the projection profile of VTADA axons in NAc and mPFc.
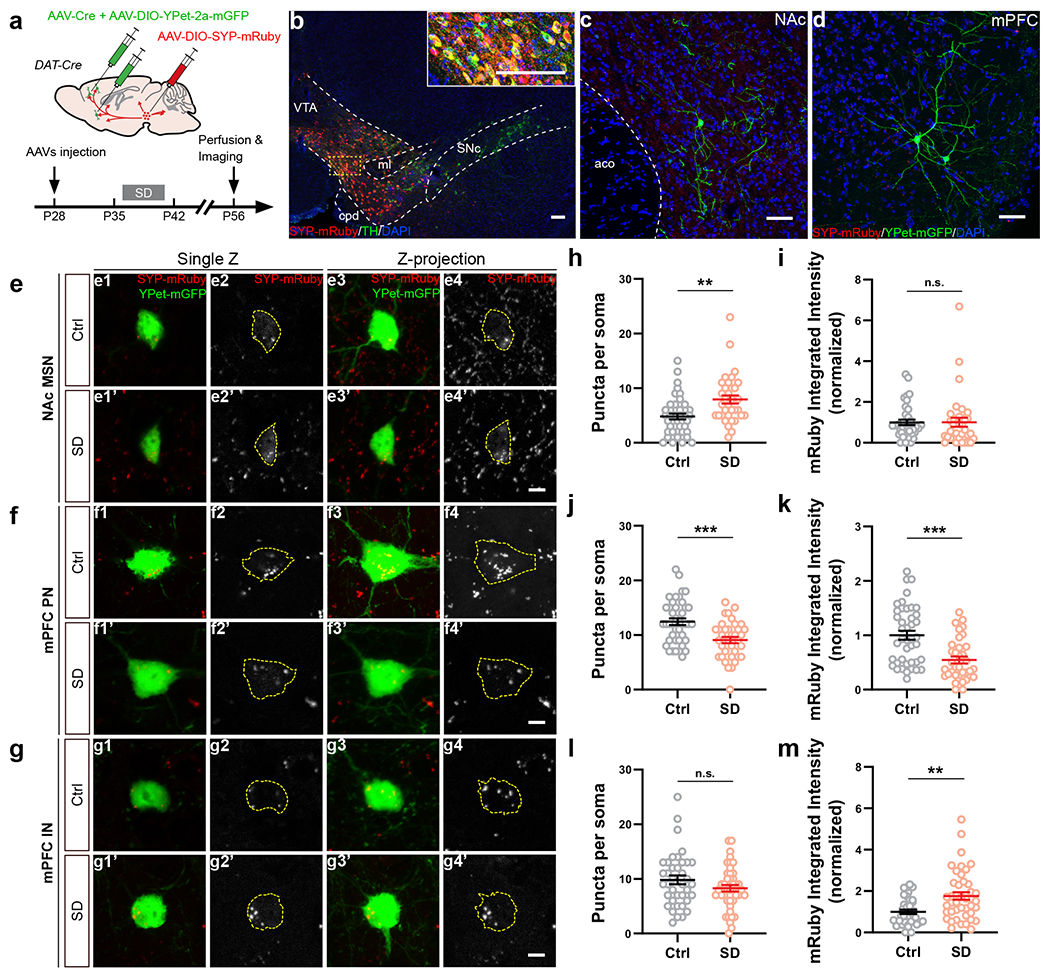
a, Dual-color labeling strategy in DAT-Cre mice. b, Representative images showing the expression of SYP-mRuby (red) in VTA counter-stained with TH (green). Inset, magnified image of the yellow-boxed area. c, d, Representative images showing the labeling of a medium spiny neuron in NAc (c) and a pyramidal neuron in mPFC (d). e–m, Quantification of VTADA axonal termini on the somata of target neurons, i.e., NAc MSNs (e, h, i), mPFC PNs (f, j, k) and INs (g, l, m). Example images (e–g) are shown in single Z plane images (e1–g2’) and maximum intensity projection of Z-stack images (e3–g4’). Dotted contour indicates the soma area. SYP-mRuby puncta within the soma area were counted (h, 43 Ctrl/35 SD neurons from 4 mice each; j, 42 Ctrl/37 SD neurons from 5 mice each; l, 38 Ctrl/44 SD neurons from 5 mice each) and measured for their integrated intensity (i, 39 Ctrl/35 SD neurons from 4 mice each; k, 42 Ctrl/36 SD neurons from 5 mice each; m, 38 Ctrl/43 SD neurons from 5 mice each). Welch’s t-tests, h, t(63.67) = 3.42, P = 0.001; j, t(76.98) = 4.02, P = 0.0001; k, t(73.78) = 4.35, P = 0.00004. Mann-Whitney test, m, P = 0.002. Individual color channels of example images in b–g were adjusted for brightness/contrast before merged. Scale bars, b, 100 μm; c, d, 50 μm. e–m, 5 μm. Data are shown as mean ± s.e.m. All tests were two-sided. For detailed statistics information, see Supplementary Table 1.
We first focused on the SYP-mRuby puncta on the cell body of target neurons. Consistent with our finding that dopamine release in NAc was aberrantly increased (Fig. 3), we found more SYP puncta on the MSN somata in SD mice than in Ctrl mice, suggesting elevated VTA innervation in the NAc of SD mice (Fig. 4e, h, i). On the other hand, mPFC PNs in SD mice showed less peri-soma SYP puncta (Fig. 4f, j) when compared to the Ctrl group, suggesting decreased VTA innervation, whereas no change was found on INs (Fig. 4g, l). Additionally, compared to Ctrl animals, the fluorescence intensity of individual SYP puncta was decreased on PNs (Fig. 3k) but increased on INs in the SD group, suggesting possible alterations in presynaptic vesicle load44. No significant changes were found in VTA innervation that targeted the dendritic portion of MSNs and PNs (Extended Data Fig. 6g–p). Collectively, these morphological characterizations suggest SD-induced bidirectional changes in multiple VTA projection targets essential for social interaction, with VTA innervation increased/strengthened in NAc while decreased/weakened on mPFC PNs (also see Discussion).
Adolescent VTA activity is critical for social novelty preference
VTADA neurons are highly active during wakefulness but mostly silent during sleep at the population level27–31 and hence are overexcited by SD during adolescence (Extended Data Fig. 7). Therefore, we asked whether a balanced level of VTA activity maintained by a coherent sleep/wake cycle during adolescence is critical for shaping social novelty preference. To address this point, we first examined whether directly overexciting VTADA neurons during adolescence is sufficient to cause defects in adult social interaction. We injected AAVs carrying an excitatory Designer Receptor Exclusively Activated by Designer Drugs (DREADD), hM3Dq, into the VTA of DAT-Cre mice (hM3DqDAT) at P25 (Fig. 5a, b). VTA excitation was confirmed by increased locomotor activity 30 min after injection of clozapine-N-oxide (CNO, Extended Data Fig. 8a), suggesting an overall elevated DA tone, and by the preference to drink CNO solution over water in a two-bottle free-choice drinking test (Extended Data Fig. 8b, c). To artificially excite VTADA neurons during adolescence, we delivered daily CNO injections (i.p. 2 mg/kg) 2 hours after light phase onset from P37 to P41 (Fig. 5a). This treatment mildly increased wakefulness and decreased NREM sleep 2 – 3 h after CNO injection, however, nearly 60% of NREM sleep was retained in contrast to the SD protocol (Extended Data Fig. 8k). Control groups (Cre– littermates given hM3Dq viruses and DAT-Cre mice with mCherry control viruses) received the same dose of CNO and developed normal sociability and social novelty preference (Fig. 5c–e and Extended Data Fig. 8d). In comparison, in hM3DqDAT animals, adolescent CNO injections led to loss of social novelty preference without affecting the overall sociability at P56 (Fig. 5c–e). Similar to SD mice, hM3DqDAT animals exhibited no deficit in social memory (Extended Data Fig. 8m).
Figure 5. Activity level of VTADA neurons during adolescence is critical for social novelty preference.
a, hM3Dq-mCherry expression in VTA. Sale bar, 100 μm. Virus combinations used are listed on the Right. b, Timeline of experimental procedures. CNO was administered at ZT2. c–e, Tree-chamber assay was performed on hM3DqDAT mice and control littermates at P56 – 60 (n = 8 Cre-/4 mCherryDAT/9 hM3DqDAT mice). c, RM-2-way ANOVA (Stimulus × Treatment F(3, 45) = 3.93, P = 0.01) with Tukey’s post-tests (P as indicated). One-way ANOVA, d, F(2, 18) = 1.74, P = 0.20; e, F(2, 18) = 10.58, P = 0.0009, with Sidak’s post-tests, *** P = 0.0009, * P = 0.01. f, Timeline of experimental procedures. CNO was administered prior to SD onset. g–i, Three-chamber assay was performed on hM4DiDAT and mCherryDAT mice at P56 – 60. n = 8 mCherryDAT/10 hM4DiDAT mice. g, RM-2-way ANOVA (Stimulus × Treatment F(3, 48) = 1.95, P = 0.13) with Tukey’s post-tests (P as indicated). Welch’s t-test, h, t(14.60) = 0.61, P = 0.55, i, t(15.51) = 3.59, P = 0.003. j, m, Representative images showing NAc MSNs and mPFC PNs in hM3DqDAT (j) and SD + hM4DiDAT mice (m) and respective controls. Scale bar, 5 μm. Dotted contour indicates the soma area. k, l, n, o, Quantification of SYP-mRuby puncta in hM3DqDAT (k, l) and SD + hM4DiDAT (n, o) mice and control animals. k, n = 33 Control/35 hM3DqDAT MSNs from 4 mice each, Welch’s t-test, t(61.07) = 2.83, P = 0.006. l, n = 22 Control/33 hM3DqDAT PNs from 4 mice each, Mann-Whitney test, P = 0.14. n, n = 38 Control/42 hM4DiDAT MSNs from 4 mice each, Mann-Whitney test, P = 0.008. o, n = 37 Control/39 hM4DiDAT PNs from 4 mice each, Welch’s t-test, t(73.45) = 1.31, P = 0.19. Individual color channels of example images in a, j, and m were adjusted for brightness/contrast before merged. Data are shown as mean ± s.e.m. All tests were two-sided. For detailed statistics information, see Supplementary Table 1.
Since chemogenetic VTA excitation decreased NREM sleep, it is possible that other circuits affected by decreased sleep/increased waking recapitulated the SD behavioral phenotype. To exclude this possibility, we expressed an inhibitory DREADD in VTADA neurons in DAT-Cre mice (hM4DiDAT, Fig. 5f) to specifically offset VTA excitation by adolescent SD. Reduced intake of sucrose following CNO injections validated effective VTA inhibition (Extended Data Fig. 8n). We sleep-deprived both hM4DiDAT mice and mCherryDAT control littermates from P37 to P41 using the same SD protocol described above, and daily CNO injections were given prior to SD onset to suppress VTA activity (Fig. 5f, also see Methods). EEG recording confirmed that even with the CNO injections, hM4DiDAT mice stayed mostly awake during the entire SD session (Extended Data Fig. 8h–j, l). While sleep was still deprived at the behavioral level, inhibition of VTADA neurons during adolescent SD sessions rescued the social novelty preference in hM4DiDAT mice when they reached young adulthood, whereas adolescent SD in mCherryDAT mice again impaired social interaction (Fig. 4g–i). Together, these results demonstrate that adolescent sleep affects VTADA neurons in developmental shaping of social novelty preference, and that a balanced level of VTA activity during the adolescent critical period is necessary and sufficient to mediate this key sleep function. In contrast to the rescue of social novelty preference, sleep-deprived hM4DiDAT mice did not increase their exploration of non-social novel object (Extended Data Fig. 9o), suggesting separate circuits for social and non-social novelty preferences.
In addition to the behavioral results, we also found consistent, bidirectional structural changes of VTA axons at P56 in mice with VTA overexcitation, or inhibition under SD, during prior adolescence. We found increased dopaminergic innervation in the NAc of hM3DqDAT animals compared to control animals only expressing the labeling viruses (Fig. 5a, j, k), while the SD + hM4DiDAT mice showed decreased VTA innervation onto MSNs which would otherwise be elevated by SD (Fig. 5m, n). Interestingly, we did not find significant changes in the VTA axons onto mPFC PNs (Fig. 5l, o), suggesting these projections, much less abundant than those to NAc, may not be causally linked to the role of sleep in shaping social novelty preference.
Adolescent sleep restoration rescues social deficits in InsG3680 mice
Having established that disrupting adolescent sleep persistently impairs social novelty preference, the social novelty response of VTA-NAc circuitry, and VTA projection profile in adulthood, we wondered whether sleep abnormalities spontaneously occur during adolescence in animal models of neurodevelopmental disorders like ASDs and whether restoration of adolescent sleep can rescue the social deficits in these animals. To this end, we utilized a mouse model, the Shank3 InsG3680 knock-in (InsG3680), which carries an ASD-associated guanine insertion at the cDNA position 3680 of Shank345. These homozygous mutant mice exhibit robust defects in social interaction behavior, especially in social novelty preference45 (Extended Data Fig. 9a–c). We first examined the spontaneous sleep architecture over a 24-hour cycle in adolescent InsG3680+/+ mice (P35 – 42) and found more wakefulness and less NREM sleep compared to WT littermates predominantly in the light phase (Extended Data Fig. 9d–h). Power spectrum analyses also revealed increased Alpha power (7 – 12 Hz) during NREM sleep and reduced Theta power (4 – 7 Hz) during REM sleep in homozygous mutants (Extended Data Fig. 9i–l). Additionally, we found increased VTA projections to the NAc of InsG3680+/+ mice compared to WT littermates at P56 by labeling the VTA axons using an AAV-mTH-Cre construct we generated (Extended Data Fig. 9m–t). This VTA axon phenotype resembles that of animals with adolescent SD (Fig. 4h) or aberrant VTA excitation (Fig. 5k).
Next, we sought to correct the adolescent sleep defects in InsG3680 mice. Flupirtine is a selective KCNQ2/3 potassium channel opener and also functions as an NMDA receptor antagonist and GABAA receptor modulator46. It was originally used as a clinical analgesic, but most recently it was found in our lab to significantly increase NREM sleep47. Here we delivered daily Flupirtine injections to adolescent InsG3680+/+ mice (i.p., 30 mg/kg/day × 5 days, P35 – 42, Fig. 5a) in early light phase and found that it significantly increased NREM amount and reduced wakefulness (Fig. 5b, c and Extended Data Fig. 10a, b), leading to a complete compensation of sleep time to a level comparable to that in WT mice (Extended Data Fig. 10c, NREM, Flup, 22481 ± 805.4 s vs. Extended Data Fig. 9g, NREM, WT, 20697 ± 1360 s). More excitingly, in contrast to the homozygous mutants that received vehicle injections (saline/0.3%DMSO), those given Flupirtine during adolescence developed strong preference towards the novel stranger over the familiar stimulus mouse in the social interaction test performed at P56 (Fig. 6d–f), and the preference index for social novelty in the Flupirtine group was similar to WT animals (Fig. 6f, Flup, 0.45 ± 0.08 vs. Extended Data Fig. 9c, Social Novelty WT, 0.43 ± 0.08), suggesting a full rescue of social novelty preference. Alternatively, we used more selective dual orexin receptor 1/2 antagonist (DORA12)48 to block the hypothalamic hypocretin (also known as orexin) signaling which is essential for promoting sleep-to-wake transition and maintaining wakefulness49,50, and achieved similar rescue of adolescent NREM sleep and restoration of adult social novelty preference in InsG3680+/+ mice (Extended Data Fig. 10e–n ).
Figure 6. Adolescent restoration of NREM sleep rescued the social interaction deficit in Shank3 InsG3680+/+ mice.
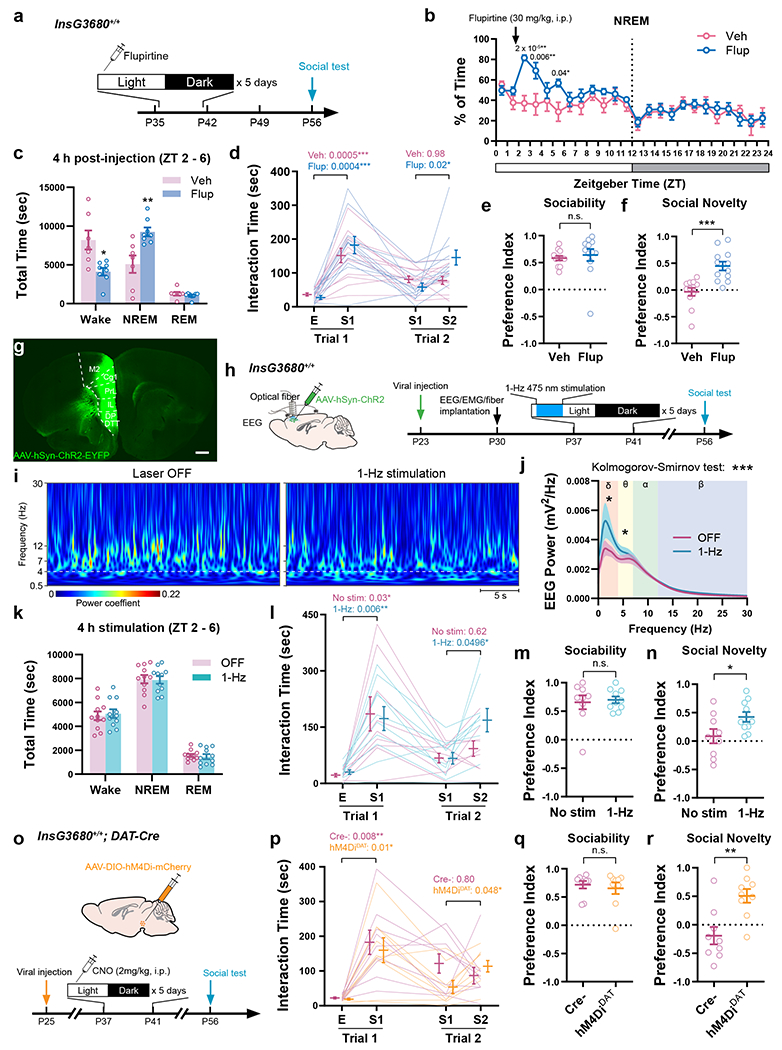
a-c, Adolescent Flupirtine injections in InsG3680+/+ mice. Hourly binned NREM sleep is shown in b, and quantification of time in each state within 4 h following the injection is shown in c. n = 8 in Flup/7 in Veh (0.3% DMSO in saline). b, RM-2-way ANOVA (Time × Treatment F(23, 299) = 2.61, P = 0.0001) with Bonferroni’s post-tests (P as indicated). c, Welch’s t-test, Wake t(7.88) = 3.09, P = 0.02; NREM t(8.85) = 3.33, P = 0.009. d–f, Flup- or Veh-treated InsG3680+/+ mice were tested for social interactions at P56. n = 12 mice. d, RM-2-way ANOVA (Stimulus × Treatment F(3, 66) = 3.95, P = 0.01) with Tukey’s post-tests (P as indicated). Mann-Whitney test, e, P = 0.13, f, P = 0.0001. g, h, AAV-hSyn-ChR2-EYFP was injected to the PFC of InsG3680+/+ mice (g, Scale bar, 500 μm), and 1-Hz light stimulation (ZT 2 – 6) was delivered daily between P37 – 41. i, j, Example heatmap (i) and quantification (j) of EEG power spectrum with or without 1-Hz stimulation. n = 11 mice in j, shaded area indicates s.e.m., Kolmogorov-Smirnov test, P = 0.0001. Paired t-test for frequency band, δ, t(10) = 2.35, P = 0.04; θ, t(10) = 3.03, P = 0.01; α, t(10) = 0.42, P = 0.68; β, t(10) = 1.88, P = 0.09. k, Quantification of time in each state with or without 1-Hz stimulation. n = 11 mice. Paired t-test, Wake, t(10) = 0.45, P = 0.66; NREM, t(10) = 0.19, P = 0.86; REM, t(10) = 0.68, P = 0.51. l–n, InsG3680+/+ mice with or without adolescent 1-Hz stimulation were tested using the three-chamber assay at P56. n = 9 in No stim/10 in 1-Hz. l, RM 2-way ANOVA (Stimulus × Treatment F(3, 51) = 1.72, P = 0.18) with Tukey’s post-tests (P as indicated). Welch’s t-test, m, t(11.51) = 0.31, P = 0.77; n, t(14.52) = 2.21, P = 0.04. o-r, AAV-DIO-hM4Di-mCherry was injected to InsG3680+/+; DAT-Cre+/− mice for adolescent VTA inhibition, and three-chamber assay was performed at P56. n = 9 mice. p, RM-2-way ANOVA (Stimulus × Treatment F(3, 48) = 1.85, P = 0.15) with Tukey’s post-tests (P as indicated). q, Mann-Whitney test, P = 0.86; r, Welch’s t-test, t(15.20) = 3.60, P = 0.003. Data are shown as mean ± s.e.m. All tests were two-sided. For detailed statistics information, see Supplementary Table 1.
Furthermore, we asked whether increasing a particular sleep component without changing the total amount of time in sleep/wake states would improve the social performance of InsG3680+/+ mice in adulthood. To this end, we employed an optogenetic approach to enhance cortical slow wave activity (SWA). SWA propagates throughout the cortex, is expressed as 0.5 – 4 Hz slow oscillations in EEG recording and believed to play important roles in cognition and plasticity7. High-density EEG recording in human subjects revealed the frontal and parietal cortices as the main origins of SWA propagation during adolescence7,51. Thus, to increase SWA in InsG3680+/+ mice, we injected AAV-hSyn-ChR2 into either the frontal (PFC) or parietal cortex (primary somatosensory region, see Methods) at P23, implanted electrodes for EEG/EMG recording and optical fiber for optogenetic stimulation at P30 and applied 1-Hz light stimulation52 using a 473 nm laser during P37 – 41 (ZT 2 – 6, 4 h/day × 5 days, Fig. 6g, h). The 1-Hz stimulation in either cortical location robustly enhanced EEG power predominantly in the Delta range (0.5 – 4 Hz, Fig. 6i, j) without altering the total amount of Wake, NREM or REM states (Fig. 6k). At P56, the mutant mice with adolescent 1-Hz stimulation spent more time interacting with the novel social stimulus (Fig. 6l–n), compared to those that received no stimulation during adolescence, suggesting rescue of social novelty preference.
Since inhibiting VTADA activity concurrently with adolescent SD prevented the defect in social novelty preference (Fig. 5f–i), we wondered if the same adolescent chemogenetic manipulation could also improve the social deficits in InsG3680+/+ mice in adulthood. To this end, we bred the InsG3680+/+; DAT-Cre mice. AAV-DIO-hM4Di-mCherry was injected to the VTA of these mice (InsG3680+/+; hM4DiDAT) as well as InsG3680+/+; Cre– littermates at P25, followed by 5 days of CNO injections in early light phase during P37 – 41 (2 mg/kg/day, i.p., at ZT 2, Fig. 6o). When their social interaction was assayed at P56, the InsG3680+/+; hM4DiDAT animals exhibited normal social novelty preference with significantly increased interaction with S2 and decreased interaction with S1 during Trial 2, as opposed to the lack of such preference in InsG3680+/+; Cre– littermates that received the same CNO treatment (Fig. 6p–r).
Thus, we show that improving NREM sleep (by increasing its total amount or specifically SWA) as well as reducing the VTA activity within the adolescent critical period (P35 – 42) restores social novelty preference in InsG3680+/+ mice.
Discussion
A developmental link between sleep and social interaction
During adolescence neural networks undergo substantial synaptic remodeling and circuit refinement that heavily impacts adult brain functions53–56. Here we show that sleep during a critical period at adolescent stage (P35 – 42, Fig. 1b–i), but not in later adulthood (Fig. 1q–t), shapes social novelty preference, but not sociability, by keeping a balance in dopaminergic activity during adolescence (Fig. 5). However, we did not examine an earlier age window (< P35) since we cannot reliably record EEG in younger pups. Thus, whether sleep at younger age has similar or distinct function remains unclear. The causal link between adolescent sleep and adult social novelty preference is further strengthened by our finding that restoration of only 5 days of NREM sleep, or just the slow wave component, within the adolescent critical period of P35 – 42 is sufficient to fully rescue the social interaction deficits in InsG3680+/+ mice, using pharmacological and optogenetic approaches, respectively (Fig. 6a–n and Extended Data Fig. 10). As the most prominent component and the main feature of NREM sleep, SWA increases from early childhood to adolescence51,57 and then decreases as the brain matures into adulthood5,6, thus is correlated with the development of brain connectivity58. Consistent with this logic, the NREM silencing of VTADA neurons during adolescence was essential for this sleep function, because adolescent inhibition of VTA activity restored the social novelty preference in both SD mice (Fig. 5f–i) and InsG3680+/+ mice (Fig. 6o–r). Together, these pieces of evidence demonstrate a critical role of adolescent NREM sleep in the development of social novelty preference, whereas REM sleep seems dispensable for this sleep function. Our experiments provide further proof-of-principle evidence for potential applications using early interventions targeting NREM sleep and/or NREM-specific circuits as therapeutic means to ameliorate or prevent the progression of social symptoms in neurodevelopmental disorders.
VTA-NAc social circuit as a target of adolescent SD
Many factors may contribute to the behavioral loss of social novelty preference, including lowered general sociability, impaired perception and/or memorization of social stimuli, defects in processing social novelty as a rewarding signal, or a combination of these factors. In our experiments, adolescent SD did not affect the development of sociability (Fig. 1c–i). We also show that SD animals had intact social recognition and social memory at least when facing single social stimulus mouse (Extended Data Fig. 3l,n, 1-stimulus paradigm, also in Fig. 1d, habituation curves to S1 are identical between groups), and the fact that SD mice did investigate the novel stranger when it was initially introduced (Fig. 1f, 1 – 2 min) also suggests intact social perception. It was only when there were two stimulus mice, either in the three-chamber setup (Fig. 1e–g) or in the 2-stimuli social memory paradigm (Extended Data Fig. 3l, o, p), that a shift of interest from the familiar to the novel occurred in Ctrl mice but not in SD mice. Thus, the defect is in the preference per se when making a choice, which suggest the social novelty signal is likely not rewarding in SD animals.
Consistent with this hypothesis, we identified a series of functional and structural alterations in the VTA-NAc circuit, which has a well-established role in reward processing and behavioral reinforcement22. In mice with adolescent SD, the VTA activation patterns were no longer specific to social novelty stimulations (Fig. 2), with many activation events seen outside the social interaction bouts (Extended Data Fig. 4e) while the social novelty-specific Ca2+ responses were largely attenuated (Fig. 2). More direct measurements of dopamine release in the NAc revealed that in contrast to Ctrl animals, the dopamine signal associated with S2 interaction in SD animals was completely abolished while that with S1 interaction was aberrantly strengthened and showed a significantly slower decline in signal peaks as the S1 novelty decreased (Fig. 3). Consistent with this observation, the VTA projections were also elevated in the NAc of SD mice (Fig. 3e, h). Together, these findings suggest a VTA-NAc circuit less tuned to social novelty, more confined to the first social stimulus encountered but incapable of shifting the rewarding signaling to the novel social stimulus.
Additionally, the difference between NAc dopamine signal and VTA Ca2+ activity in SD animals when responding to S1 in Trial 1 (Fig. 2e, i vs. 3d, h) raised the possibility that dopamine release mechanisms from VTA axons might be altered and decoupled from their soma activation in SD animals, which calls for further investigation. One seventh of the dopaminergic neurons also co-release glutamate59, and VTA vGluT2+ neurons have been implicated in sociability defects, although their contribution to social novelty preference was not examined60. In our study, however, sociability is unaffected, and our direct measurement of dopamine release favors that it is indeed dopamine that signals the social novelty and is impaired by adolescent SD, although additional contribution from glutamate cannot be excluded.
It is worth noting that SD revealed a mild reduction in the preference for non-social novelty. This effect is likely mediated through a mechanism independent of VTA circuits and social novelty preference because 1) despite decreased investigation time, SD mice still exhibited a robust preference toward the novel object over the familiar one (Extended Data Fig. 3m), 2) the VTA Ca2+ response and NAc dopamine signal to the non-social stimulus were both similar between Ctrl and SD groups (Fig. 2, 3 and Extended Data Fig. 4f–h), and 3) renormalization of VTA activity in SD mice restored the preference for social novelty (Fig. 5f–i) but not that for non-social novelty (Extended Data Fig.8o). Nonetheless, these results suggest additional developmental functions of sleep in shaping adult behavior that warrant further exploration.
Methods
Animals
All experimental protocols were approved by the Stanford University Animal Care and Use Committee and are in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Shank3 InsG3680 knock-in mice (InsG3680, full name: STOCK Shank3tm3.1Gfng/J; JAX strain 028778; gift of Dr. Guoping Feng, Massachusetts Institute of Technology, U.S.A.)45 were kept on 129S2/SvPasCrl background. DAT-IRES-Cre mice (DAT-Cre, full name: B6.SJL-Slc6a3tm1.1(cre)Bkmn/J; JAX strain 006660, only heterozygotes were used for experiments) were kept on C57BL/6J background. DAT-Cre::Ai14 mice were generated by crossing the DAT-Cre+/+ male with the Ai14+/+ female (full name: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J; JAX strain 007914, gift of Dr. Jun Ding, Stanford University, U.S.A.), which was also kept on C57BL/6J background. To generate the InsG3680+/+; DAT-Cre mice, DAT-Cre+/+ mice (C57BL/6J background) were crossed with InsG3680+/+ mice (129S2/SvPasCrl background), and the InsG3680+/−; DAT-Cre+/− offspring (50% 129S2 / 50% C57 background) were then backcrossed to the InsG3680+/+ parent. Thus, the InsG3680+/+; DAT-Cre+/− offspring (75% 129S2 / 25% C57 background) were used for experiments and the InsG3680+/+; DAT-Cre−/− littermates were used as control. All mice were born and housed at constant temperature (22 ± 1 °C) and humidity (40 – 60%), under a 12/12-hour light–dark cycle (lights-on: 7:00 a.m. – 19:00 p.m., ZT 0 – 12; lights-off: 19:00 p.m. – 7:00 a.m., ZT 12 – 24), with access to food and water ad libitum. The mouse pups were weaned on postnatal day 21 (P21) and subject to experimental procedures after they were P28 or older, except for viral injections which were carried out at P21 – 25 in some experiments (see details below). Mice were group housed in 2 – 5 per cage except for those for EEG/EMG recording, which were housed individually after surgery. Both male and female mice were used in behavioral and ex vivo electrophysiology experiments. However, male samples were more than female samples in experiments with sleep recording, fiber photometry and optogenetic stimulation, because female pups were significantly smaller, and their skulls thinner, than male pups at the time of EEG/fiber implantation (around P30), making them less likely to recover from the surgery. Also, the oestrus cycle tremendously impacts synapse density61, therefore female mice were excluded from our synaptic labeling experiments as well.
Developmental sleep disruption
Sleep disruption (SD) was achieved using the automated Sleep Deprivation System (ViewPoint Life Sciences, Inc., Lyon, France) which is composed of a deprivation chamber (PVC cylinder, Height: 46 cm, Width: 30 cm, Weight: 5 Kg) with a shaking platform at the bottom, a controller and a computer with the controlling program installed. Mice were transferred to the deprivation chamber where they had access to food and water ad libitum, and returned to the home-cage daily after SD or Ctrl sessions. Programed electromagnetic pulses were delivered to push the bottom platform of the deprivation chamber to keep the mice awake during the sessions. The parameters of electromagnetic pulses were as follow: SD, a train of randomized 2 – 8 pulses (15-ms duration) delivered at 2 Hz every randomized 0.2 – 0.8 minutes, for 4 hours daily during early light phase (Zeitgeber time, ZT 2 – 6); Ctrl, a train of 5 pulses (15-ms duration) delivered at 2 Hz every 0.5 minutes, for 4 hours daily during early dark phase (ZT 12 – 16). We avoided performing the SD protocol in the very beginning of light phase (ZT 0 – 2) to avoid drift in the animals’ circadian rhythm. The SD or Ctrl protocol was performed for 5 consecutive days between P35 and P42 or between P42 and P49, then the mice were left undisturbed before the behavioral tests were carried out on or after P56. For adult SD, same SD or Ctrl protocol was performed between P84 and P91, and the behavioral tests were done at least 24 h after the last SD session (> P92). For mice with simultaneous EEG/EMG recording, up to 4 mice were sleep-deprived/recorded at the same time, and a divider was placed in the deprivation chamber to separate them. The parameters of SD protocol were deliberately tuned not to induce acute stress, as confirmed by ELISA of plasma corticosterone level after the SD session (Extended Data Fig. 1n). Single housing the animals was required for EEG recording but was shown to potentially decrease later social interaction40,62. We found in our experimental setup, the mice that had implantations and single-housed for EEG recording (including 3 Ctrl and 4 SD mice in Fig. 1c–i) did show overall less interactions with both non-social objects and stimulus mice, but the relative portion of each interaction category (preferences) did not deviate from those group-housed. Thus, we included the mice with adolescent EEG recording in the behavioral analysis and also added additional animals without EEG recording to the data pool, and ratio of EEG/no-EEG animals was counter-balanced between groups. The effects of adolescent SD on social interactions were similar between the automated method and the manual, “gentle-touch” protocol13,14,27(Extended Data Fig. 3f), in which the mice stayed in their home-cages and were monitored by an experimenter (W.-J. B.). If an animal remains motionless for more than 5 seconds, it was gently touched with a soft brush by the experimenter. Novel objects (paper tubes, cotton nestlets, etc) were also added into the home-cage to promote wakefulness but were removed after the SD session. As control, the same numbers of touching and novel objects were given to Ctrl mice during the dark phase. The social preferences in the Ctrl mice (both in the automated method and “gentle-touch” protocol) did not differ from those in mice with normal, undisturbed sleep/wake cycle during the entire adolescence (Extended Data Fig. 3c, d).
Developmental restraint stress
A well-established, noninvasive restraint paradigm was used to generate stress during adolescence34. At the onset of dark phase (ZT12), adolescent mouse pups (WT, C57BL/6J background) were individually placed into a clean 50-ml Falcon conical tube with heads facing the conical tube bottom. Small open holes were pre-made on the tube wall as well as tube bottom for ventilation purposes. Since adolescent mouse pups were smaller in shape compared to adult mice, cotton balls were stuffed into the tube to help restrain the animals without inducing suffocation. After 4 hours of restraint session in early dark phase (ZT12 – 16), mice were immediately released from the restraint tubes and returned to their home-cages. This 4-hour restrain procedure was performed for 5 consecutive days within P35 – 42 age windows. Littermate controls received equivalent amount of handling but no actual restraint in the same age range.
ELISA
For measurement of plasma corticosterone, mice were anesthetized using isoflurane and a small quantity of blood (~ 100 μl) was collected from the retro-orbital sinus on the first and last day of SD immediately after the SD session. Control samples were collected from naïve animals without any manipulation at the same Zeitgeber time. Plasma was separated from the whole blood sample by centrifuge and subjected to ELISA according to manufacturer’s instructions (Enzo, ADI-900-097). Mice used for ELISA were not used in any other experiments, due to potential confounding effects of repeated blood collection.
Surgeries
For all surgeries, the animal received a subcutaneous injection of Buprenorphine SR (1mg/kg) before incision and was anesthetized with a mix of ketamine (100mg/kg) and xylazine (20 mg/kg) injected intraperitoneally (i.p.). The animal was then placed on a stereotaxic rig (David Kopf Instruments, Tujunga, CA) for the following surgical procedures. The animal was given 0.5 ml saline (i.p.) before and after the surgery to prevent dehydration and help recovery. Triple-antibiotic ointments were used post-surgically, and the animal was kept in a cage placed on a heating pad until fully awake.
EEG/EMG electrodes implantation
Cortical EEG and EMG electrodes were implanted at P28 – 30 as described in our previous studies27,48,63. Briefly, stainless steel mini-screws (US Micro Screw) for EEG were implanted to the skull above the frontal (AP +1.5 mm; ML 1 mm) and temporal (AP −2.5 mm; ML 2.5 mm) lobes. Mini-rings made of metal wires (316SS/44T, Medwire) were inserted into neck muscles for EMG recording. The electrodes were previously soldered to a 4-pin connector which was mounted on the skull using Metabond (Parkell) and dental cement.
Viral injection and optical fiber implantation
Adeno-associated viruses (AAV) were purchased from the Stanford Neuroscience Gene Vector and Virus Core (GVVC) unless stated otherwise. Z coordinates were from the skull at bregma unless stated otherwise. For DREEDD virus infusion at P25, 500 nl of AAV-DJ- EF1α-DIO-hM3Dq-mCherry (4.26 × 1012 Vg/ml), AAV-DJ- EF1α-DIO-hM4Di-mCherry (1.45 × 1012 Vg/ml) or AAV-DJ- EF1α-DIO-mCherry (5.00 × 1012 Vg/ml) was unilaterally infused to the VTA (AP −2.9 mm; ML +0.2 mm; DV – 4.3 mm) of DAT-Cre or InsG3680+/+; DAT-Cre mice through a 33-gauge needle (Hamilton 776206) or a glass pipette attached to a microsyringe (Hamilton), which led to bilateral infection of DA neurons in VTA, but not in SNc. For VTA fiber photometry, the needle or glass pipette was advanced in an 8° angle to the VTA (AP −3.1 mm; ML +1.1 mm; DV – 4.25 mm from dura) of DAT-Cre mice at P42, and 500 nl of AAV-DJ-EF1α-DIO-GcaMP6f (1.13 × 1012 Vg/ml) was infused unilaterally. A mono fiber-optic cannula (400 μm in diameter, N.A. = 0.48, 5 mm, Doric Lenses, Inc., Quebec, Canada) was then implanted through the same route of viral injection to above the VTA (8°-angled, AP −3.1 mm; ML +1.1 mm; DV −4.1 mm from dura) and mounted on the skull using Metabond (Parkell) and dental cement. For photometric recording of VTA activity during adolescent sleep and SD, 500 nl of AAV-DJ-EF1α-DIO-GcaMP6f (1.13 × 1012 Vg/ml) was infused (8°-angled, AP −2.9 mm; ML +1.0 mm; DV – 4.1 mm from dura) at P21, and fiber-optic cannula (400 μm in diameter, N.A. = 0.48, 4.5 mm, Doric Lenses, 8°-angled to VTA, AP −2.9 mm; ML +1.0 mm; DV −4.1 mm from dura) along with EEG/EMG electrodes were implanted at P30. For monitoring dopamine release in NAc, 200 nl of AAV9-hSyn-DA2m42 (2.33 × 1013 Vg/ml, Wzbioscience) was infused to NAc (AP +1.2 mm; ML +1.3 mm; DV – 4.5 mm) and fiber-optic cannula (200 μm in diameter, N.A. = 0.39, 4.5 mm, RWD Life Science) were implanted (AP +1.2 mm; ML +1.3 mm; DV – 4.2 mm) at P42. For 1-Hz cortical stimulation, 300 nl of AAV-DJ-hSyn-ChR2 (2.70 × 1012 Vg/ml) was injected to PFC (AP +1.50 mm; ML +0.5 mm; DV – 1.8 mm) or to the primary somatosensory cortex (AP −1.5 mm; ML +1.5 mm; DV – 0.5 ~ 0.6 mm from dura) at P23. Fiber-optic cannula (400 μm in diameter, N.A. = 0.39, 1.5 mm, RWD Life Science) for delivering light stimulation was placed right above the cortical region where the glass pipette entered previously together with the electrodes implanted for EEG/EMG recording at P30.
Viral constructs used for the dual-color labeling of VTA axons are described as follows. pAAV-hSyn-DIO-Synaptophysin-mRuby was made from pAAV hSyn FLEx mGFP-2A-Synaptophysin-mRuby (gift of Dr. Liqun Luo, Addgene #71760)64 by deleting the mGFP-2A. pAAV-EF1α-DIO-Ypet-2a-mGFP65 was a gift from Dr. Xiang Yu (Peking University, China), which combines cytoplasmic Ypet, an improved version of EYFP with largely enhanced brightness66, with membrane bound GFP (mGFP)67, rendering perfect labeling of neuronal morphology in vivo. pAAV-mTH-Cre was generated by replacing the EGFP sequence in pAAV-mTH-GFP (gift of Dr. Viviana Gradinaru, Addgene #99128)68 with the coding sequence of Cre recombinase. For dual-color labeling, AAV-DJ-hSyn-DIO-Synaptophysin-mRuby (700 nl, 6.56 × 1012 Vg/ml) was infused unilaterally to the VTA (AP −3.0 mm; ML +0.2 mm; DV – 4.5 mm) of DAT-Cre mice, and a 1:1 mixture (200 – 300 nl) of AAV-DJ-hSyn-Cre (diluted to ~2 × 109 Vg/ml) and AAV-DJ-EF1α-DIO-Ypet-2a-mGFP (1.77 × 1012 Vg/ml) was infused to ipsilateral NAc (AP +1.2 mm; ML +1.0 mm; DV −4.5mm) and mPFC (AP +1.54 mm; ML +0.3 mm; DV −2.6 mm) of the same animal using glass pipettes at P28 – 30, followed by Ctrl/SD protocol and perfusion at P56 – P60. For labeling in InsG3680+/+ mice (Extended Data Fig. 9m–t), AAV-DJ-hSyn-DIO-Synaptophysin-mRuby (6.56 × 1012 Vg/ml) was mixed with AAV-DJ-mTH-Cre (9.05 × 1012 Vg/ml) at approximately 1:1 titer ratio, and 500 nl of this mixture was injected to unilateral VTA using the same coordinates. For dual labeling in combination with chemogenetic manipulations (Fig. 5), AAV-DJ-hSyn-DIO-Synaptophysin-mRuby (6.56 × 1012 Vg/ml) was mixed with the DREADD virus at approximately 1:1 titer ratio, and 700 nl of this mixture was injected to the VTA of DAT-Cre mice at P25, using the coordinates for younger animals (AP −2.9 mm; ML +0.2 mm; DV – 4.3 mm), to allow proper DREADD expression before CNO treatments started on P37. Control DAT-Cre littermates received only the labeling viruses but not the DREADD virus.
All viral infusion was performed at a rate of 100 nl/min. After each infusion, the needle or glass pipette was kept still for 5 min before slowly withdrawn. Absorbable sutures were used to close the incision if needed.
EEG/EMG recording and photostimulation
After the surgery, the animal was allowed to recover for 1 week and then connected to a flexible recording cable at least 24 hours before the recording began. EEG and EMG recording across a complete 24-hour light-dark cycle was performed on the day before SD started (Baseline), on a single day within the 5-d SD period (During SD), or 24 hours after the last SD day (After SD). For InsG3680 mice, EEG/EMG recordings were carried out between P35 – P42, but mostly on P37 – 38. Flupirtine (Tocris, Cat#. 2867) was prepared at a concentration of 3 mg/ml in saline containing 0.3% DMSO (v/v, vehicle) and administered daily at the dosage of 30 mg/kg (0.1 ml/10g i.p., sonicated prior to use) at 2 h after lights-on (ZT2) for 5 consecutive days between P35 and P42. DORA12 was provided by Merck (One Merck Drive, Whitehouse Station, NJ) through the Merck Investigator Studies Program, dissolved in a mixture of 50% saline and 50% Poly (ethylene glycol) average Mn 400 (PEG400) at a concentration of 2 mg/ml, and administered daily at the dosage of 20 mg/kg (0.1 ml/10g i.p., sonicated prior to use)48 at 2 h after lights-on (ZT2) for 5 consecutive days between P35 and P42. For 1-Hz cortical stimulation, InsG3680+/+ mice with previous viral injection (P23) and fiber-optic/electrode implantation (P30) were connected to the recording cable and the fiber-optic patch cord (400μm Core, 0.39NA, 3m, RWD Life Science) 24 hours before stimulation. The fiber-optic patch cord was connected to a 473 nm blue laser (LaserGlow) beforehand, and the laser power was adjusted such that the intensity of light exiting the patch cord was 8 – 12 mW. For photostimulation, a train of 60 blue light pulses (pulse duration, 15 ms; interval, 1 s) was triggered by a Master-8 pulse generator (A.M.P.I., Israel) and delivered every 2 min for 4 hours in early light phase (ZT 2 – 6) daily from P37 to P41. EEG/EMG signals were amplified through a multi-channel amplifier (Grass Instruments) and collected by VitalRecorder (Kissei Comtec Co.) at sampling rate of 256 Hz filtered between 0 and 120 Hz for offline signal analysis.
Chemogenetic overexcitation and inhibition by CNO injections
Adolescent CNO injections (P37 – 41) were carried out 12 days following the viral injection. For overexcitation of VTADA neurons, DAT-Cre mice previously injected with DREADD or control viruses received an single intraperitoneal injection of CNO (Enzo, BML-NS105-0025) dissolved in saline at the dose of 2 mg/kg body weight at ZT2 (2 h after light phase onset), and the CNO injection was performed daily for 5 consecutive days starting on P37. Although off-target effects are likely negligible at this low dose69, to avoid this issue and to maximize the usage of animals, we injected the same dose of CNO to both Cre– littermates receiving the hM3Dq virus (Cre–) as well as DAT-Cre mice receiving AAV-DIO-mCherry (mCherryDAT) as controls. No difference was found in the social performance between these two types of controls, both showing strong sociality preference and social novelty preference during adult social interactions (Extended Data Fig. 8d). For VTA inhibition, half of the DAT-Cre mice in both hM4DiDAT and mCherryDAT groups received CNO injections at a single dose of 2 mg/kg body weight (i.p.) 30 min before the SD onset (ZT 1.5). The other half of mice received CNO at an initial dose of 1 mg/kg (i.p.) 30 min before the SD onset (ZT 1.5) followed by a second injection of the same dose 2 h later (ZT 3.5), to provide sustained inhibition of VTADA neurons over the whole SD session (ZT 2 – 6). Both injection schemes were performed daily for 5 consecutive days starting on P37 and concurrent with SD, and yielded similar behavioral phenotypes. For adolescent VTA inhibition in InsG3680+/+; DAT-Cre mice, daily injections of CNO (2 mg/kg, i.p.) were applied at ZT2 between P37 – P41 without sleep intervention.
Behavioral assays
All behavioral assays, including fiber photometry during social interaction, were carried out in early dark phase (ZT 12 – 16) in a dark experiment room under dim red illumination to minimize acute disruption to sleep and circadian rhythms. Mice were habituated to the experiment room at least 1 hour before the test started. All behavioral assays except for the drinking test were recorded with a camera mounted on the ceiling of the experiment room.
Three-chamber social interaction
The experimental procedure was as described elsewhere70. In brief, the test was performed in an apparatus of 3 compartment chambers (2 side chambers: 26 cm × 23 cm; middle chamber: 11 cm × 23 cm) with connecting doors. For habituation, the test mouse was placed in the middle chamber and allowed 10 minutes of free exploration of all 3 chambers in the empty apparatus. The mouse was then returned to the middle chamber and doors covered by cardboard. An empty metal mesh cup (10 cm in diameter, Empty, E) was placed randomly in one of the 2 side chambers, serving as the non-social novel object, and another identical mesh cup containing a never-before-met stimulus mouse (Stranger 1, S1) was placed in the opposite chamber. The door covering was then removed, and the test mouse was free to explore and interact with either E or S1 for 10 minutes (Trial 1). After Trial 1 was completed, a second never-before-met mouse (Stranger 2, S2) was placed in the previously empty cup, serving as the social novelty stimulus while the S1 had become familiar. The test mouse was again allowed to explore both stimulus mice for another 10 minutes (Trial 2). The sides where E, S1 or S2 was placed were randomized and counterbalanced across test animals. Stimulus mice were gender-matched and age-matched wild-type mice from the same background with the test mice, except for probing the male-female interactions, where female mice were used and ovariectomized at least one week before the test. The stimulus mice were pre-habituated to the mesh cup so they can sit quietly during the test session. The moving trajectory of test mouse was traced from the video recording, and the total distance traveled during each trial was measured, using a custom Python script.
Two-trial social memory test
The two-trial social memory test was performed at the age of P56 – 60. The experimental procedure was as described elsewhere71 with modifications (Extended Data Fig. 3l). The test mouse was placed in an open arena (black walled, 54 cm × 26 cm) containing an empty mesh cup (for the 1 stimulus paradigm, identical to that used in the three-chamber test) or two empty cups (for the 2 stimuli paradigm) for habituation of 10 minutes. In the 1 stimulus paradigm, a never-before-met stimulus mouse of same sex was then placed into the mesh cup, the test mouse was allowed to explore the stimulus mouse for 5 minutes (Trial 1). The stimulus mouse was then removed, and the test mouse was left alone in the arena for 30 minutes. After this 30-minute interval, the same stimulus mouse was put back in the mesh cup, and the test mouse was allowed another 5 minutes of free interaction (Trial 2). In the 2 stimuli paradigm, the first stimulus mouse (S1) was placed randomly in one of the empty cups for Trial 1, and a second stranger mouse was put in the other empty cup for Trial 2. The behavior of test mice during each trial were video recorded and their interactions with the stimulus mice were later scored.
Novel object recognition (NOR) with reduced memory requirements
NOR assay typically includes a training trial, a delay period of 24 hours for memory consolidation, and a test trial48,72. However, in order to better assay the novelty preference per se, we conducted this assay using a protocol with reduced memory requirements (Extended Data Fig. 3k). After 10 minutes of habituation to the apparatus (black walled open arena, 54 cm × 26 cm), mice were given two identical inanimate objects for exploration for 10 minutes (Training). Each object was placed with equal distance to the walls and corners of the arena with no specific spatial or odor cues. Immediately after this 10-minute training trial, one of the objects was replaced with a novel, inanimate object (Test), and the animal was allowed another 5 minutes of exploration. Animals displaying a dramatic bias for the objects during Training (> 65% investigation with one object) were excluded from the data pool.
Elevated plus maze (EPM)
EPM is a well-established assay measuring anxiety in laboratory rodents73. It was done in an elevated maze with four arms (two open and the other two closed, each arm is 66 cm long and 5 cm wide). Mice were placed at the junction of the open and closed arms, facing the open arm opposite to the experimenter. Mice were allowed to explore the maze for 5 minutes. Due to occurrence of falling from the open arms during our experiments (3 out of 8 in SD group), we quantified the latency to first entry into the open arms instead of total time spent in the open arms.
Open field test
Open field test was performed 30 min after the hM3DqDAT mice and control littermates received the CNO injections (2 mg/kg, i.p.). The test mouse was placed into a standard open field arena (44cm x 44cm total) and allowed to explore unimpeded for 8 min. The trajectory of mouse movement was tracked and total distance traveled was calculated using an open source MATLAB toolbox, Autotyping (https://www.seas.upenn.edu/~molneuro/software.html).
Two-bottle free-choice drinking test
Mice were individually housed at least 4 days before test and were given access to two water bottles in their home-cages on the test day, with one bottle filled with water and the other containing CNO solution (50mg/L, for validating hM3Dq excitation) or Sucrose solution (1% w/v, for validating hM4Di inhibition). The amounts of water and CNO/Sucrose consumed during 24 h (for hM3Dq) or during a 4-h drinking session in early dark phase (for hM4Di) were measured. For sucrose preference, mice were put on water regulation prior to test (2 d of 4-h water access followed by at least 2 d of 2-h water access) to increase the desire for liquid reward. Animal weight was monitored to ensure they did not fall below 80 % of baseline body weight. The drinking test procedure started at least 1 day after other behavioral tests. Failure to show a preference for CNO or a reduced preference for sucrose indicated bad DREADD expression in the VTA, and thus such mice were excluded from the data pool of all experiments.
Fiber photometry
Mice were subjected to the social interaction test with simultaneous fiber photometry recording at least 2 weeks after virus injection and optical fiber implantation. Fiber photometry was performed as previously described27,63,74. In brief, blue light from a 470-nm excitation LED (M470F3, Thorlabs, NJ, USA) controlled by a custom MATLAB program (MathWorks, Natick, MA, USA) and a multifunction data acquisition device (NI USB-6259, National Instruments, Austin, TX, USA) reflected off a dichroic mirror (MD498, Thorlabs) to excite GCaMP6f signal through a fiber-optic patch cord (MFP_400/460/1100, N.A. = 0.48, length = 3 m, Doric Lenses) attached to the cannula implant on the mouse skull. GCaMP6f emission fluorescence passed through the same patch cord, a GFP emission filter (MF525-39, Thorlabs) and a focusing lens (LA1540-A, Thorlabs), and was detected by a photodetector (Model 2151, Newport, Irvine, CA, USA) and amplified by a lock-in amplifier (Model SR830, Stanford Research Systems, Sunnyvale, CA, USA). Amplified signals were sampled at 500 Hz using a custom MATLAB code and the data acquisition device (NI USB-6259, National Instruments). Alternatively, photometry was done in a Neurophotometrics FP3002 system (Neurophotometrics LLC, San Diego, CA, USA) using a sampling frequency of 50 Hz (For dopamine sensor experiments and VTA photometry with food stimulation). Prior to testing, mice were habituated to the patch cord for 30 min daily for 3 days. To allow full mobility of the fiber patch cord in freely moving animals, social interaction testing was performed in a rectangular open arena (54 cm × 26 cm) which was similar in size with the three-chamber apparatus but without compartment walls, with the mesh cups containing the stimulus objects or mice placed randomly on opposite sides of the short axis (Fig. 2a). This alternative testing apparatus did not change the behavioral pattern of Ctrl mice, nor the effect of SD (Extended Data Fig. 4a). After being connected to the patch cord, the animal was allowed 5 min of habituation to the arena. The photometry recording then captured 5 min of baseline activity, 10 min of Trial 1 (E vs. S1), and 10 min of Trial 2 (S1 vs. S2). Upon the start of recording, the photometer sent a signal to light up a red LED for 500 ms, which was captured by the camera and later used to synchronize the video recording of animal behavior with the photometry trace. For food stimulation test, animals on food restriction 24 hour prior to the test were given a favorable food pellet (hazelnut) in their home cages while the VTA GCaMP6f signal was being recorded. Two to three trials were recorded for each animal, and a non-appetitive, neutral object (e.g., a Q-tip) was used as control.
For photometric recording of VTA activity during adolescent sleep and SD, fiber photometry was performed as described above simultaneously with EEG/EMG recording. For each animal, a 1-h session in home-cage before or after SD was recorded to sample enough spontaneous Wake, NREM and REM bouts, and 3 sessions in the sleep-deprivation chamber (10 – 20 min each) were also recorded.
VTA slice preparation and patch clamp recordings
DAT-Cre::Ai14 mice were deeply anesthetized with a 7.5/0.05 mg (37.5/0.25 mg/ml) ketamine/dexdomitor mixture and euthanized via transcardial perfusion with ice-cold choline dissection solution (in mM: 99 choline Cl, 25 NaHCO3, 1.0 NaH2PO4, 2.5 KCl, 25 D-glucose, 10 Na L-ascorbate, 3 Na pyruvate, 10 MgCl2, 0.5 CaCl2) continuously bubbled with a 95% oxygen/5% carbon dioxide gas (carbogen). 220–250 μm midbrain slices were cut in the horizontal plane in choline dissection solution on a vibrating microtome (Leica VT1200 S). Slices were also manually cut down the midline, resulting in four separate usable slices containing the VTA. Slices incubated in artificial cerebrospinal fluid (aCSF, composition in mM: 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.0 NaH2PO4, 1.0 MgCl2, 2.0 CaCl2, and 25 glucose; 305-315 mOsm) for 10 min at 37 °C, then kept in oxygenated aCSF at room temperature until used for recordings.
Slices were transferred to a recording chamber and continuously perfused with carbogenated aCSF heated to 29 – 31°C. Patch pipettes (1.5 – 7 MΩ) were used to record from fluorescently labeled medial and lateral VTA neurons. Ninety seconds elapsed between break in and the start of recordings. For intrinsic excitability and passive membrane property recordings, an internal solution containing (in mM) 120 K gluconate, 20 KCl, 2 MgCl2, 2 Na2ATP, 0.5 NaGTP, 20 HEPES, 0.5 EGTA (pH 7.2–7.3, 295–305 mOsm) and neurobiotin (2%, Vector Laboratories). All intrinsic excitability recordings were done in current clamp, except for the Ih current test, which used steps from −40 mV to −120 mV in voltage clamp. Ih currents are a hallmark of many ventral tegmental dopaminergic neurons75. Spontaneous activity was measured without current injection, while rheobase was measured with a ramp of current (+100 pA over 5 s) from approximately −70 mV; evoked firing was measured during incremental current steps at 0.1 Hz (−20 to 170 pA, 800 ms duration, 10 pA steps). For synaptic transmission studies, an internal solution containing (in mM): 130 CsCl, 2 MgCl2, 10 HEPES, 2 Na2ATP, 0.4 Na3GTP, and 0.6 EGTA (pH 7.2–7.3, 295–305 mOsm) was used and neurons were voltage-clamped at −70 mV. To measure evoked input, a bipolar stimulating electrode was placed rostral to the VTA. Excitatory inputs were pharmacologically isolated using picrotoxin (100 μM), and inhibitory inputs were isolated by including NBQX (10 μM) and APV (20 μM) in the bath. The coefficient of variation (CV) was calculated as the standard deviation/mean amplitude of 30 evoked postsynaptic currents (PSCs), and the paired pulse ratio (PPR) was calculated using two PSCs evoked 50 ms apart, with the equation PSC2/PSC1. Excitatory and inhibitory miniature postsynaptic currents (mEPSCs and mIPSCs) were pharmacologically isolated as described above with the addition of TTX (500 nM) to block action potential firing. All recordings were obtained with a 4 kHz bessel filter and digitized at 10 kHz. Liquid junction potentials were calculated using JPCalc software (P. Barry, University of New South Wales, Sydney, Australia; modified for Molecular Devices) and we report corrected junction potentials. Measures were taken from recordings using Clampfit. MiniAnalysis was used to analyze mPSC recordings.
Immunohistochemistry and image acquisition.
Mice were deeply anaesthetized with ketamine/xylazine mixture and perfused with phosphate buffered saline (PBS), followed by cold 4% paraformaldehyde in PBS. The brains were then post-fixed in 4% PFA for 4 h, followed by dehydration in 30% sucrose for at least 48 h. Coronal sections were made at 30 μm for VTA immunohistochemistry and at 50 μm for imaging the VTA projections in NAc and mPFC, using a cryostat (Leica). The following reagents were used for immunostaining: anti-TH antibody (Aves Labs, #TYH, chicken, 1:1000), anti-GFP antibody (for DA2m immunostaining, Abcam, # ab13970, chicken, 1:1000), Streptavidin Alexa Fluor™ 488-conjugate (Invitrogen, #S11223, 1:1000), goat anti-chicken secondary antibody Alexa Fluor 647 (Invitrogen, #A-21449, 1:1000), goat anti-chicken secondary antibody Alexa Fluor 488 (Invitrogen, #A-11039, 1:1000). All brain sections and stained acute slices were imaged on a Zeiss LSM710 confocal microscope (Carl Zeiss, Jena, Germany) using ZEN Black (Zeiss). For images of VTA, tiled scan was performed using a 10× Plan-Apochromat objective (N.A. = 0.45) at 1× optical zoom and 4 μm Z interval, or using a 20× Plan-Apochromat objective (N.A. = 0.8) at 1× optical zoom and 1 μm Z interval. For images of dual-color labeling, the cell bodies of NAc medium spiny neurons, mPFC pyramidal neurons and interneurons as well as their dendritic segments located 40 – 120 μm from the cell body were imaged using a 63× oil immersion Plan-Apochromat objective (N.A. = 1.4) at 2× optical zoom and 0.5 μm Z interval. Over 90% of the neurons in NAc were medium spiny neurons. The pyramidal neurons in mPFC can be identified morphologically by its triangular soma and distinct apical dendrite extending towards cortical pia, while the interneurons often show non-pyramidal, round/oval somata and much simpler dendritic arbor. All images used for quantitative analysis (Fig. 4e–g, 5j and 5m) were acquired using the same microscope setting.
Quantification and Statistical Analysis
Behavioral scoring
All behavioral scoring was performed blinded to the experimental conditions, which was coded by computer-generated random numbers (https://www.random.org/) at the time of testing. For social interaction, the time spent in direct interactions between the test mouse with either E, S1, and S2 (e.g., sniffing, touching of nose, head or forelimbs with an orientation towards the wire cup) in each trial, as well as number of interaction bouts, number of entries to each chamber and time spent in each chamber during each trial and the habituation phase were measured manually either using a stopwatch or in BORIS (University of Torino, Italy)76. Average length of interaction bout was calculated by dividing the total interaction time with number of interaction bouts. For InsG3680 mice, due to the large within-group variation, the interaction time with E, S1, or S2 was normalized to total interaction time (E + S1 + S2) during the entire test session (20 min) for each animal. Preference indices for sociality or social novelty (SN) were calculated as follows, PISociality = (tS1 – tE)/(tS1 + tE); PISN = (tS2 – tS1)/(tS2 + tS1), where tE, tS1 and tS2 are the time of interaction with E, S1 and S2, respectively, within the given trial. The time spent in self-grooming by the test mouse was also quantified for the entire test session from the video recording (Trial 1 + 2, 20 min in total). For the social memory test, same criteria for social interaction were used.
Fiber photometry data analysis
Analysis was performed in MATLAB R2017b (MathWorks) using custom scripts. Photometry datasets were first corrected for baseline decay. Z-score was then calculated from the detrended signal F as Z-score = (F – mean(Fbaseline)) / s.d.(Fbaseline), where Fbaseline is from the 5-min baseline recording immediately before Trial 1. The converted Z-score dataset was then synchronized to the video recording (25 fps) of the test by the last frame in which the red LED was on. The interaction bouts with social or non-social objects were scored from the recorded video blinded to the experimental conditions using BORIS76 or a custom MATLAB code, both generated timestamps of the start and end of each interaction bout. The bout onset was defined as the initiation of actual physical contact (e.g., nose, head or forelimbs touching the wire cup). Interaction behaviors less than 0.2 s apart from each other were considered as the same interaction bout, and interaction bouts that last less than 1 s were excluded from analysis. The timestamps were further used to capture the Z-score data of each interaction bout, and the peak Z-score within bout as well as median of Z-score during the 2 s immediately before bout onset were measured. ΔZ-score was calculated as peak Z-score subtracted by median of Z-score 2 s pre-onset. Z-score Area Under Curve (AUC) was calculated within the interaction bout using the trapz function in MATLAB and relative to the median of entire Z-score dataset. The peak Z-scores from the first 10 bouts of each interaction category were fitted to one-phase exponential decay curve in GraphPad Prism 9 (GraphPad Software). Detection of total transients in GCaMP signals was performed using the Z-score dataset, by a method reported in our previous study 27. In brief, we applied two low-pass filters (0 – 4 Hz; 0 – 40 Hz) to the dataset, respectively, and calculated the derivative trace of the squared difference of these two filtered datasets. Transients were identified by thresholding both this derivative trace and the original Z-score trace at mean + 1 s.d. For analyzing the VTA activity during adolescent sleep and sleep deprivation, detrended photometry data (in Z-score) were split into Wake, NREM, REM and SD states according to the EEG/EMG data, and the frequency as well as the mean amplitude of Ca2+ transients were calculated for each state.
EEG/EMG analysis
Raw EEG/EMG data were converted in SleepSign (Kissei Comtec Co.), exported to MATLAB (MathWorks) and manually scored using custom scripts77. Wake, NREM, and REM episodes longer than 5 s were quantified based on previously described criteria49. In brief, wakefulness is characterized by desynchronized small-amplitude EEG and extensive EMG activity. NREM is characterized by synchronized, large-amplitude, delta (0.25 – 4 Hz)-dominant EEG and reduced EMG activity compared to wakefulness. REM is featured by an EEG of high frequency (4 – 9 Hz-dominant) and relatively uniform amplitude smaller than that in NREM and often associated with a flat EMG (muscle atonia). Power-frequency distributions between 0.5 – 30 Hz frequency range of NREM and REM episodes were analyzed using the power spectral density (PSD) function in the Matlab scripts, and the relative power of Delta (0.5 – 4 Hz), Theta (4 – 7 Hz), Alpha (7 – 12 Hz) and Beta (12 – 30 Hz) bands was calculated as fraction of total power (0.5 – 30 Hz) for each individual animal.
Image analysis
All morphological analyses were performed in Fiji (NIH, Bethesda, MD, USA) and blinded to the conditions with images coded with random numbers at the time of data acquisition. For quantifying the soma SYP-mRuby signal, green channel (YPet-mGFP) images were used to determine the soma area, and the SYP-mRuby puncta colocalizing with the green fluorescence were manually counted in the Z-stack images using the Cell Counter plugin in Fiji. The contact by a SYP-mRuby punctum forms a hollow spot in the YPet-mGFP fluorescence of the postsynaptic cell (a “pit” on postsynaptic membrane, Extended Data Fig. 6d–f) in most cases (> 90 %), suggesting these are synapse-like contacts made by varicosities of VTA axons, and hence potential dopamine release sites, rather than axons passing by or random overlap of fluorescence signals43. For measuring SYP-mRuby intensity, the Z-stack image was projected by maximum intensity and a region of interest (ROI) was determined by soma fluorescence of YPet-mGFP. The integrated intensity of SYP-mRuby puncta within this soma ROI was then thresholded and measured in Fiji. Quantifications of spine density and % spines in contact with SYP-mRuby signal was as previously reported54. Briefly, dendritic segments (typically 40 – 70 μm in length) were traced using the Simple Neurite Tracer plugin, and the total spine number on the segment, as well as the number of those overlapping with SYP-mRuby puncta (mRuby+), were counted using the Cell Counter plugin in Fiji. Spine density was calculated as total spine number / segment length, and mRuby+ % = mRuby+ number / total spine number. Protrusions longer than 5 μm were considered as filopodia and excluded from the analysis. SYP-mRuby intensity on spines was measured the same way as soma SYP-mRuby intensity, except that ROIs were drawn by hand for spine-contacting SYP-mRuby puncta. The integrated intensity of SYP-mRuby puncta was normalized to the Ctrl group for each cohort of animals. Because so few of SYP-mRuby puncta were found in contact with spines of cortical neurons, their intensity was not measured.
All images were analyzed as acquired with no post-acquisition modifications. For example images, brightness/contrast adjustment within linear ranges were made for individual channels in Fiji when necessary. For SYP-mRuby example images in Fig. 4e–g, 5j, and 5m, background was subtracted for demonstration purposes before brightness/contrast adjustments within linear ranges in Fiji. Control and experiment conditions were adjusted with the same parameters.
Statistics
Datasets were checked for normality and statistical tests were carried out using GraphPad Prism 9 (GraphPad software). All tests used were two-tailed. Welch’s t-tests were used for comparison between 2 conditions, except for paired comparisons of data obtained from the same animal, where paired t-tests were used. Alternatively, for datasets that did not follow normal distribution, Mann-Whitney U-tests (for unpaired comparisons) and Wilcoxon matched-pairs signed rank test (for paired comparisons) were used. One-way ANOVA followed by Sidak’s, Dunnett’s, or Tukey’s multiple comparison tests were used for comparison of 3 or more conditions, depending on what conditions to be compared post-hoc. For experiments with two independent variables, two-way ANOVA followed by Bonferroni’s or Tukey’s post-tests were used, depending on which factor to be tested. Three-way ANOVA was used to compare gender effect together with that of treatment and stimulus (or time). Repeated measures were incorporated when appropriate (RM-ANOVA). The non-linear regression and simple linear regression functions in GraphPad Prism 9 (GraphPad software) were used for fitting the photometry data. We note that GraphPad Prism 9 failed to compare the fitted curves in Fig. 2j due to ambiguous fitting. Preference indices, sleep states or EEG wavebands are considered categories but not independent variables, therefore t-tests, Mann-Whitney U-tests or one-way ANOVA were used for comparisons within each category. Nested t-tests or two-way RM-ANOVA were used for slice electrophysiology data. A detailed list of statistical tests used for each figure panel, including numbers of male and female samples as well as the exact n, t, F and P values, is summarized in Supplementary Table 1. All data are presented as mean ± s.e.m. unless otherwise indicated in the figure legends. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications in the field 27,37,48,74,78,79. * P < 0.05; ** P < 0.01; *** P < 0.001; n.s., not significant, P > 0.05.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The data that support the findings of this study are available within this paper and its Supplementary Information. Source data are provided with this paper.
Code availability
All custom code used in this study is available from the corresponding authors upon reasonable request.
Extended Data
Extended Data Fig. 1. Developmental shaping of social preferences and adolescent sleep disruption (SD).
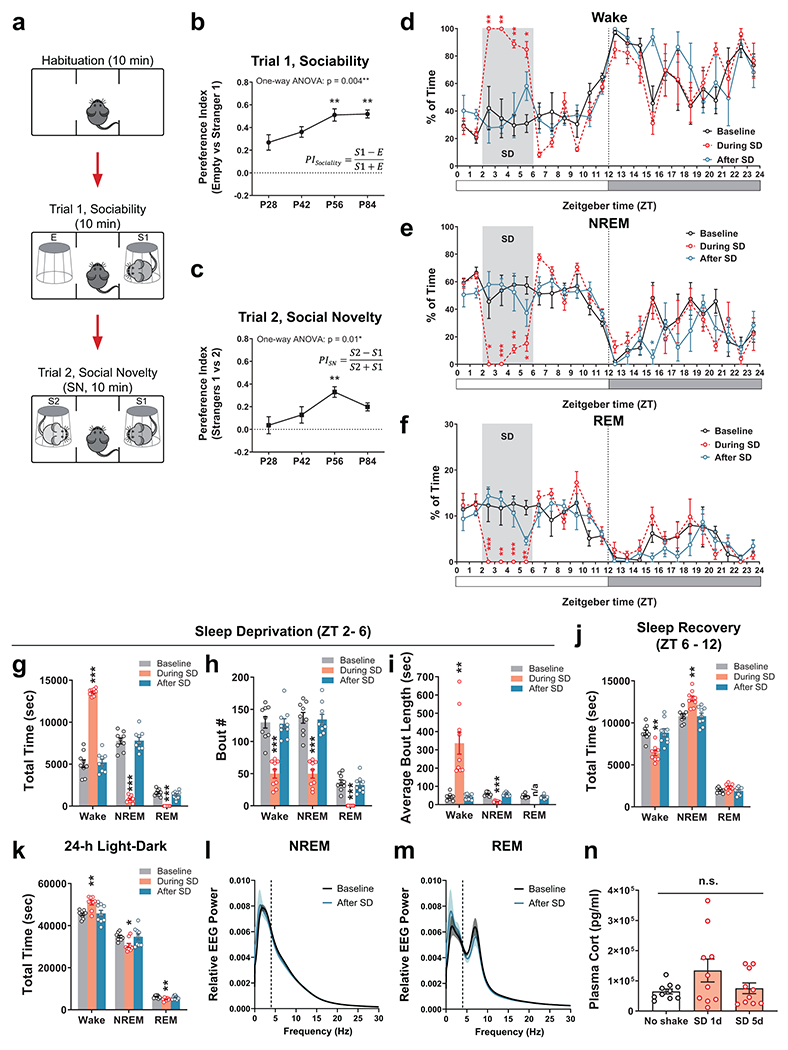
a, Schematics of the three-chamber social interaction assay. b, c, Naïve WT C57BL/6J mice showed age-dependent increases of sociability (b) and social novelty preference (c) in three-chamber social interactions over the developmental course (n = 10 each). Preference indices were calculated as indicated in each graph. One-way ANOVA, b, F (3, 36) = 5.33, P = 0.004; c, F (3, 36) = 4.23, P = 0.01, Dunnett’s post-hoc comparisons to P28, b, P56, ** P = 0.007, P84, ** P = 0.005; c, P56, ** P = 0.004. d – f, Hourly percentage of Wake (d), NREM (e) and REM (f) sleep in adolescent mice (P35 – 42) over a 24-hr light-dark cycle before (Baseline, black), during (red), and after (blue) the 5 days with daily SD sessions (ZT 2 – 6, indicated by the grey stripe). The 3 groups of data were from the same mice (n = 4). RM 2-way ANOVA followed by Bonferroni’s post-tests, Wake, Time × Treatment F (46, 207) = 3.18, P = 9.5 × 10−9, post-test During SD vs. Baseline, ZT2-3 ** P = 0.008, ZT3-4 ** P = 0.001, ZT4-5 ** P = 0.005, ZT5-6 * P = 0.02; NREM, Time × Treatment F (46, 207) = 2.90, P = 1.4 × 10−7, post-test During SD vs. Baseline, ZT2-3 * P = 0.01, ZT3-4 *** P = 0.0007, ZT4-5 ** P = 0.007, ZT5-6 * P = 0.03; After SD vs. Baseline, ZT15-16 * P = 0.02; REM, Time × Treatment F (46, 207) = 3.23, P = 5.9 × 10−9, post-test During SD vs. Baseline, ZT2-3 *** P = 0.0008, ZT3-4 ** P = 0.002, ZT4-5 *** P = 0.0004, ZT5-6 ** P = 0.02. g – k, Total amount of time (g), bout number (h) and average bout length (i) of Wake, NREM and REM states between ZT 2 – 6, total amount of 3 states during the following 6 hrs (ZT 6 – 12, j) and over the 24-hour light-dark cycle (k) (n = 9). One-way ANOVA within each state, g, Wake, F (1.48, 11.83) = 230.2, P = 0.000000001; NREM, F (1.28, 10.24) = 167.7, P = 0.00000005; REM, F (1.78, 14.21) = 79.99, P = 0.00000003; h, Wake, F (1.72, 13.75) = 54.78, P = 0.0000005; NREM, F (1.87, 14.94) = 70.18, P = P = 0.00000003; REM, F (1.42, 11.38) = 58.42, P = 0.000003; i, Wake, F (1.01, 8.07) = 24.47, P = 0.001; NREM, F (1.40, 11.18) = 67.54, P = P=0.000002; j, Wake, F (1.60, 12.81) = 11.30, P = 0.002; NREM, F (1.49, 11.94) = 11.41, P = 0.003; REM, F (1.48, 11.87) = 5.01, P = 0.03; k, Wake, F (1.93, 15.40) = 10.27, P = 0.002; NREM, F (1.80, 14.40) = 7.72, P = 0.006; REM, F (1.93, 15.43) = 7.59, P = 0.005. Dunnett’s multiple comparisons to Baseline, * P < 0.05; ** P < 0.01; *** P < 0.001. Unpaired t-test for REM in i, P = 0.31. l, m, EEG power spectrum of NREM (l) and REM (m) sleep was not altered after 5 days of SD compared to Baseline (n = 9). Both P > 0.05 by Kolmogorov-Smirnov test. n, Plasma corticosterone level of adolescent mice (P35 – 42) immediately after the SD session on 1st day and 5th day of SD compared to naïve mice of same age receiving no shake (n = 10 each). One-way ANOVA, F (2, 27) = 2.30, P = 0.12, followed by Tukey’s multiple comparisons test, all n.s. Data are shown as mean ± s.e.m. All tests were two-sided. n.s., not significant.
Extended Data Fig. 2. Comparisons of same-sex social interaction behavior between sexes.
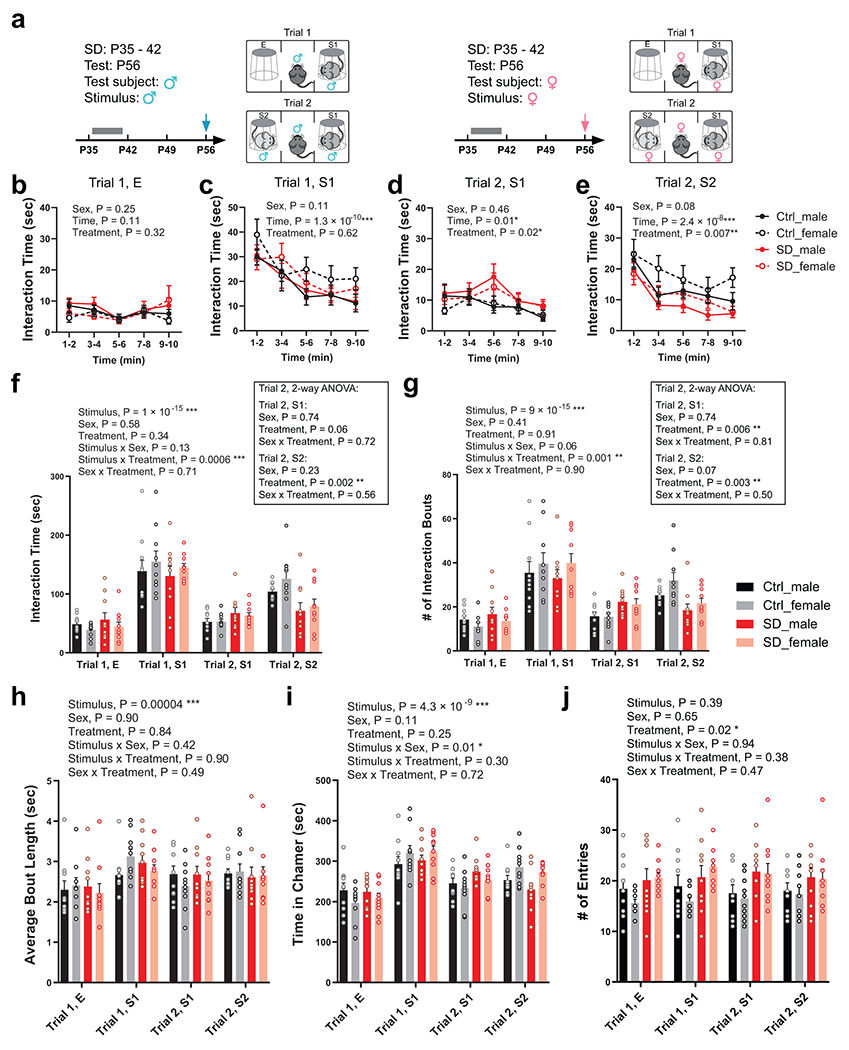
a, Male (left) and female (right) test mice received Ctrl or SD between P35 – 42 and the three-chamber assay were performed using gender-matched stimulus mice, respectively, at P56. b – e, Interaction time with the empty cup (E, b), stranger 1 (S1) in Trial 1(c), S1 in Trial 2 (d), and stranger 2 (S2) in Trial 2 (e) during the three-chamber test was binned every 2 min and presented for male and female test mice separately. n = 10 mice each. RM 3-way ANOVA. P value of each variable (Sex, Time and Treatment) was indicated in the graph. f – j, Total interaction time (f), number of interaction bouts (g), average length of interaction bouts (h), total time spent in each chamber (i), and number of entries of each chamber (j) were presented separately for male-male interactions and female-female interactions and compared directly considering Sex, Stimulus and Treatment as 3 independent variables using RM 3-way ANOVA. n = 10 mice each. ANOVA P values are indicated in the graphs. In all parameters, Sex does not show a significant contribution to total variation (P > 0.05). Additional 2-way ANOVA was performed for total interaction time (f) and number of interaction bouts (g) in Trial 2, S1 and S2 categories (box insets) followed by Bonferroni’s post-tests comparing sexes within treatment. ANOVA P values were indicated in the box insets. All post-tests, male vs. female, P > 0.05. Data are shown as mean ± s.e.m. All tests used were two-tailed. Data of male and female subjects in this Extended Data Figure were combined and presented in Fig. 1c–i. Data are shown as mean ± s.e.m. All tests were two-sided.
Extended Data Fig. 3. Behavioral probing of adolescent SD mice.
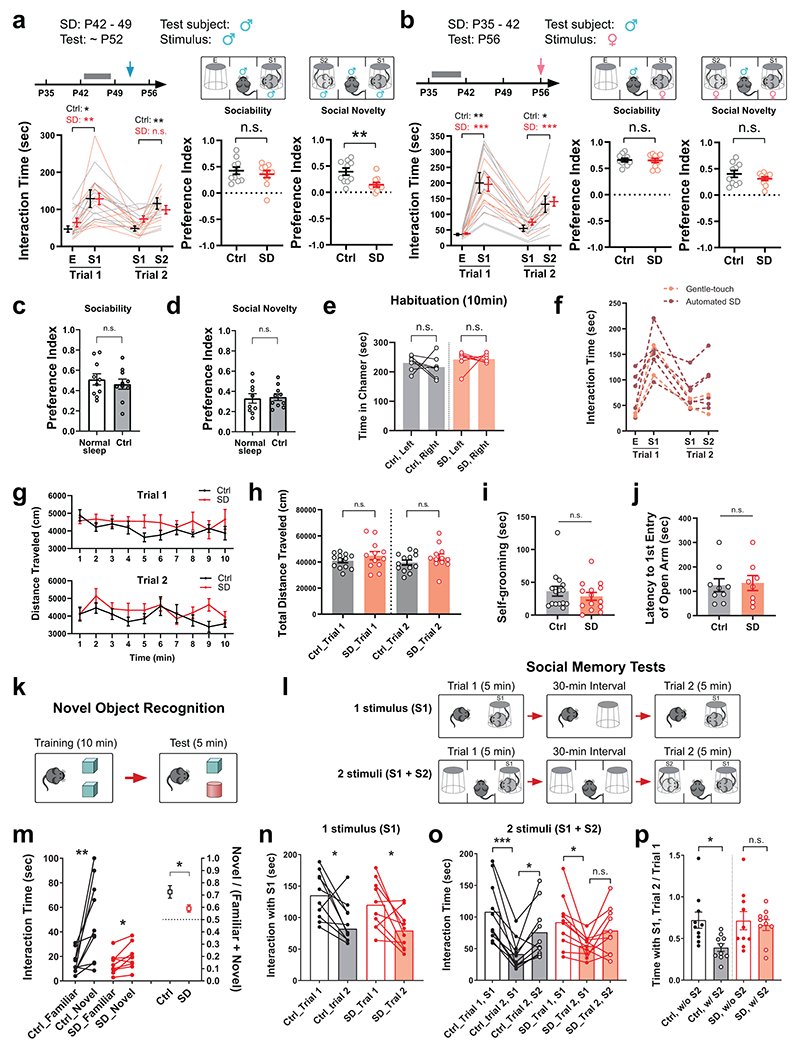
a, Male mice received Ctrl or SD between P42 – 49 and were tested at P52 – 56 using the three-chamber assay with male stimulus mice (n = 10 each). Interaction time, RM 2-way ANOVA, Stimulus × Treatment F (3, 54) = 1.20, P = 0.32, Tukey’s post-test, Trial 1 Ctrl * P = 0.03, SD ** P = 0.004, Trial 2 Ctrl ** P = 0.009, SD P = 0.11. Preference indices, Welch’s t-test, ** P = 0.007. b, Male test mice received Ctrl or SD between P35 – 42 and the three-chamber assay using female stimulus mice at P56 (n = 10 each). Interaction time, RM 2-way ANOVA, Stimulus × Treatment F (3, 54) = 0.28, P = 0.84, Tukey’s post-test, Trial 1 Ctrl ** P = 0.002, SD *** P = 0.0003, Trial 2 Ctrl * P = 0.046, SD *** P = 0.0002; Preference indices, Welch’s t-test, both P > 0.05. c, d, Sociability preference (c) and social novelty preference (d) were not significantly different between mice receiving Ctrl protocol during adolescence and naïve mice with normal, undisturbed sleep. Only male mice were included in comparisons. Ctrl data were the same as that in Extended Data Fig. 2 (n = 10 each). Welch’s t-test. e, Ctrl and SD mice spent equal time in the two side chambers during the habituation phase of three-chamber assay (n = 7 each). Paired t-test. f, Comparison of social interaction performance between male mice sleep-deprived using the gentle-touch method with those deprived by automated SD in 3 cohorts that were wean at approximately same time. Samples were combined and included in the SD group in Fig. 1g. g, h, Mice receiving previous Ctrl or SD (n = 13 in Ctrl; 12 in SD) at P35 – 42 showed similar locomotion dynamics in the three-chamber apparatus (g) and comparable total distance traveled during the whole 20-min session of three-chamber assay (h). g, RM 2-way ANOVA followed by Bonferroni’s post-test, all n.s. h, Welch’s t-test. i, Time spent in self-grooming during the three-chamber assay was similar between groups (n = 15 in Ctrl; 14 in SD). Welch’s t-test P = 0.42. j, Mice receiving SD at P35 – 42 showed similar latency to their first entry to the open arm when placed in an elevated plus maze at P70 – 84 (n = 9 in Ctrl; 8 in SD). Welch’s t-test P = 0.83. The two mice showing latency of 300 sec did not enter the open arm at all during the whole 5-min session. k, The novel object recognition test with reduced memory requirement. The Test trial was performed immediately after 10 min of Training session. m, Interaction time with each object during the Test trial was plotted to the left Y-axis, and the ratio of time spent with the novel object over total interaction time was plotted to the right Y-axis. n = 10 in Ctrl; 9 in SD. Left, paired t-test, Ctrl, ** P = 0.008, SD, * P = 0.03. Right, Welch’s t-test, * P = 0.04. l, The social memory tests. Upper, the 1 stimulus paradigm, where the same stimulus mouse S1 was re-presented to the test mouse in Trial 2 after a 30-min interval; Lower, the 2 stimuli paradigm in which Trial 2 contains a novel stimulus mouse S2 in addition to S1. n, Interaction time with the single stimulus mouse in each trial of the 1 stimulus test. n = 10 each. Paired t-test, both * P = 0.02. o, Interaction time with the 2 stimulus mice during the 2 stimulus test. n = 10 each. RM one-way ANOVA, Ctrl, F (1.96, 17.66) = 18.94, P < 0.0001; SD, F (1.68, 15.16) = 4.44, P = 0.04, Tukey’s post-test, Ctrl, S1, Trial 1 vs trial 2, *** P = 0.0007; Trial 2, S1 vs. S2, * P = 0.03, SD, S1, Trial 1 vs. Trial 2, * P = 0.03. p, Comparisons of ratio of S1 interaction (Trial 2/Trial 1) between paradigms. n = 10 each. Welch’s t-test, Ctrl, ** P = 0.009; SD, P = 0.68. Data are shown as mean ± s.e.m. All tests were two-sided.
Extended Data Fig. 4. Fiber photometry recording of VTADA activity during social interactions.
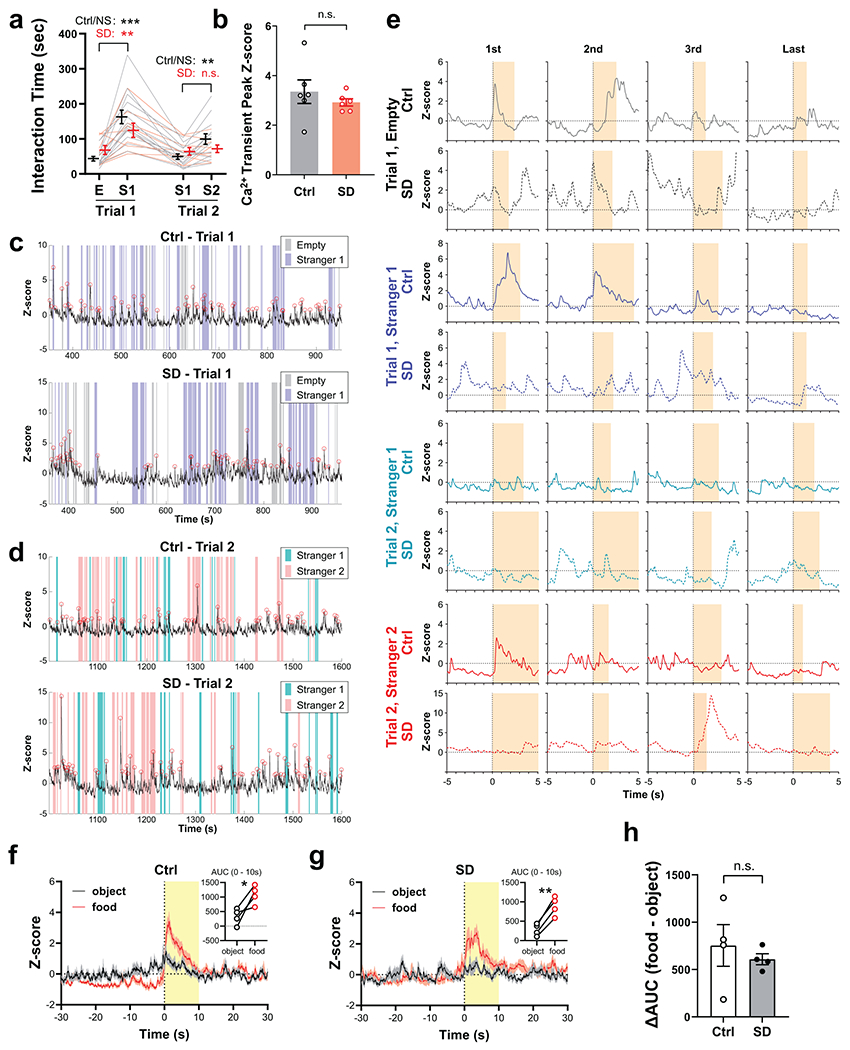
a, Social performance of DAT-Cre mice in the fiber photometry setup (NS, normal sleep. n = 14 in Ctrl/NS; 9 in SD, including mice with poor GCaMP signals). RM 2-way ANOVA, Stimulus × Treatment F (3, 63) = 4.49, P = 0.006, followed by Tukey’s post-test, Trial 1, *** P < 0.0001, ** P = 0.004; Trial 2, ** P = 0.002. b, The mean amplitude of Ca2+ transients detected during the 20 min session (Trials 1 + 2) were similar between Ctrl and SD groups (n = 6 mice, transient peak Z-score were first averaged within each animal). Welch’s t-test, P = 0.41. c, d, Representative traces of GCaMP signals in Ctrl and SD mice during Trial 1 (c) and Trial 2 (d) of the social interaction assay. Colored stripes indicate interaction bouts. Red circles on trace indicate Ca2+ transients detected. e, Example GCaMP traces of the first, second, third and last interaction bout of each category. Time 0 s indicates bout onset, and yellow stripes indicate bout duration. f, g, Ctrl and SD mice in their home-cages were given a food pellet, and the VTA GCaMP6f signals were recorded and aligned to the time point when they first contacted the food pellets. GCaMP6f traces of 10 – 12 trials from n = 4 mice (2 – 3 trials each animal). across all trials were averaged and shown for Ctrl (f) and SD (g) animals. Shaded area indicates s.e.m. Insets show area under curve (AUC) of GCaMP6f signals within 0 – 10 s upon contact with the food pellet or a neutral object of similar size (e.g., a Q-tip head), n = 4 mice, paired t-test, Ctrl, * P = 0.04; SD, ** P = 0.002. h, Relative changes in AUC, n = 4 mice, Welch’s t-test, P = 0.56. Data are shown as mean ± s.e.m. All tests were two-sided.
Extended Data Fig. 5. Adolescent SD increased the membrane resistance but did not affect the intrinsic excitability of or synaptic inputs to VTADA neurons.
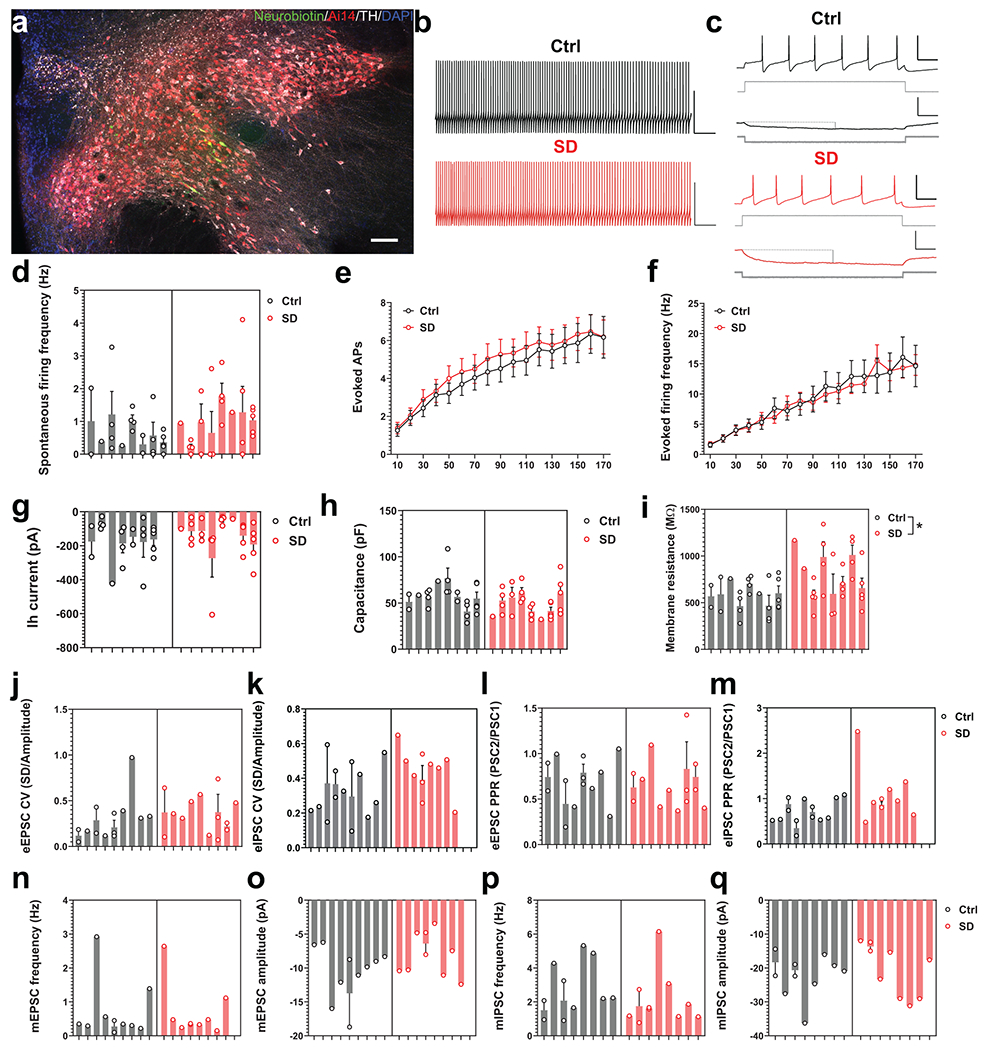
a, Representative image of a horizontal acute brain slice from a DAT-Cre::Ai14 mouse. VTADA neurons were filled with neurobiotin during whole-cell recording. The slices were subsequently fixed in 4% PFA and stained with Streptavidin (Alexa FluorTM 488 conjugate, Invitrogen) and antibodies against TH. Scale bar, 100 μm. b, Representative traces of spontaneous firing measured in a cell from a Ctrl (top, black) and SD animal (bottom, red; scale 50 mV and 5 s). c, Evoked firing in response to depolarizing current (90 pA, upper square pulses in gray) in a Ctrl (top black trace) and SD animal (red top trace; scale bars 50 mV and 0.1 s). The bottom two traces and current steps in Ctrl and SD conditions illustrate the hyperpolarizing response to a −20 pA step used for measurements of membrane resistance (indicated by the connected gray dashed [baseline] and vertical solid lines [deflection amplitude]). d, There were no observed differences in spontaneous firing frequency (n = 23 Ctrl and 27 SD neurons from 8 mice in each group, t(14) = 0.6394, P = 0.5329; data transformed with a square root, nested t-test) between the control (gray) and sleep-deprived animals (red). e, f, There were no effects of sleep deprivation (red) on the number of action potentials (APs) fired (n = 23 Ctrl and 26 SD neurons, F(16, 752) = 0.1899, P = 0.9998; two-way repeated measure ANOVA) or the firing frequency (n = 23 Ctrl and 26 SD neurons, F(16, 752) = 0.4933, P = 0.9511; two-way repeated measure ANOVA) in response to current pulse injections after adolescent sleep deprivation. g, h, There were no observed differences in hyperpolarization-activated (Ih) currents (n = 22 neurons from 7 Ctrl mice and 27 neurons from 8 SD mice, t(13) = 0.6605, P = 0.5204; nested t-test) or capacitance (n = 23 Ctrl and 27 SD neurons from 8 mice in each group, t(14) = 1.364, P = 0.1941; nested t-test) between Ctrl (left, gray) and SD animals (right, red). i, Membrane resistance was significantly increased in SD animals (M = 769.7 ± 54.32 MΩ; red) relative to Ctrl (M = 574.8 ± 36.01 MΩ; gray; n = 23 Ctrl and 27 SD neurons from 8 mice in each group, t(14) = 2.396, *P = 0.0311; nested t-test). j, k, SD did not have a significant effect on the coefficient of variation (CV) of rostrally evoked excitatory (n = 13 neurons from 9 mice in each group, t(24) = 0.9020, P = 0.3760; data transformed with square root, nested t-test) or inhibitory synaptic inputs (n = 10 neurons from 8 SD mice and 13 neurons from 10 Ctrl mice, t(21) = 1.795, P = 0.0870; nested t-test) to VTADA neurons. l, m, No observed differences between Ctrl (gray) and SD animals (red) in the paired pulse ratio (PPR) of rostrally evoked excitatory (n = 13 neurons from 9 mice in each group, t(24) = 0.0684, P = 0.9460; nested t-test) or inhibitory inputs (n = 10 neurons from 8 SD mice and 13 neurons from 10 Ctrl mice, t(16) = 1.927, P = 0.0720; data transformed with square root, nested t-test) to VTADA neurons. n, o, SD had no effect on the frequency (n = 10 neurons from 9 Ctrl mice and 9 neurons from 8 SD mice, t(15) = 0.0400, P = 0.9686; nested t-test) or amplitude (n = 10 neurons from 9 Ctrl mice and 9 neurons from 8 SD mice, t(17) = 1.544, P = 0.1410; nested t-test) of miniature excitatory postsynaptic currents (mEPSCs) in VTADA neurons. p, q, No significant differences in the amplitude (n = 10 neurons from 8 mice in each group, t(14) = 0.4431, P = 0.6645; nested t-test) or frequency (n = 10 neurons from 8 mice in each group, t(14) = 0.9301, P = 0.3681; nested t-test) of miniature inhibitory postsynaptic currents (mIPSCs) recorded in VTADA neurons from Ctrl (gray) or SD (red) animals. For all bar graphs, each bar represents 1 animal while the individual data points are from neurons recorded from that animal. Data are shown as mean ± s.e.m. All tests were two-sided.
Extended Data Fig. 6. Axonal projections of VTADA neurons in NAc and mPFC.
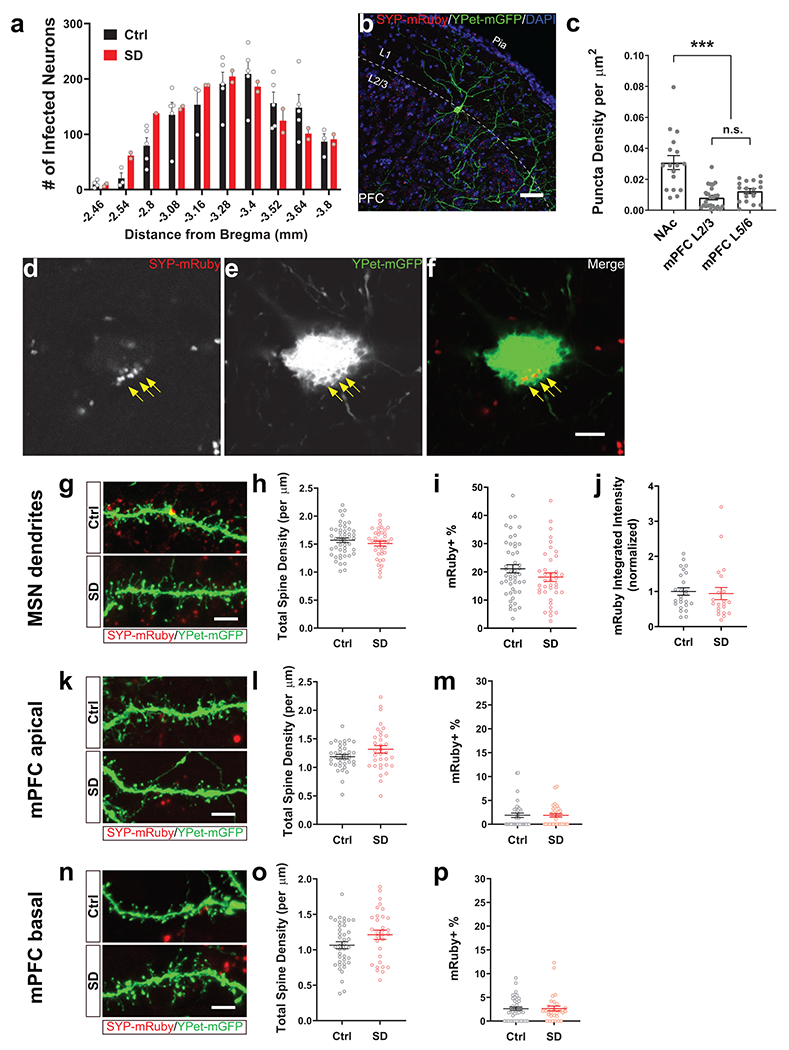
a, Numbers of infected VTADA neurons in DAT-Cre mice (n = 5 in Ctrl; 2 in SD). b, Representative image showing layer 1 – 3 of mPFC (anterior cingulate area). Only very few SYP-mRuby puncta were present in layer 1 compared to deeper layers. Scale bar, 50 μm. c, Quantification of areal density of SYP-mRuby puncta in NAc as well as layer 2/3 and layer 5/6 of mPFC. 17 – 24 fields were imaged in each region from 3 animals, and SYP-mRuby puncta were counted. F (2, 55) = 19.79, P < 0.0001 by one-way ANOVA, followed by Tukey’s post-test, *** P < 0.0001. d-f, A single plane of Z-stack confocal image in separate and merged channel views showing the soma of a mPFC pyramidal neuron (same neuron in Fig. 4f, Ctrl). Arrows indicate SYP-mRuby puncta and corresponding “pits” on the cytoplasmic membrane of the target neuron. Scale bar, 5 μm. g, k, n, Representative examples of MSN dendrites in NAc (g) and apical oblique dendrites (k) or basal dendrites (n) from mPFC pyramidal neurons in Ctrl and SD animals. Scale bar, 5 μm. h-j, Total spine density of NAc MSNs (h, n = 48 neurons from 5 mice in Ctrl; 39 neurons from 4 mice in SD), percentage of dendritic spines in contact with SYP-mRuby puncta (i, n = 48 neurons from 5 mice in Ctrl; 39 neurons from 4 mice in SD) and normalized integrated intensity of SYP-mRuby puncta that colocalized with labeled spines (j, n = 25 neurons from 4 mice in Ctrl; 21 neurons from 3 mice in SD) were not changed by adolescent SD. l, m, Total spine density (l) and percentage of SYP-mRuby-contacting spines (m) on apical oblique dendrites of mPFC pyramidal neurons (n = 37 neurons from 5 mice in Ctrl; 33 neurons from 4 mice in SD). o, p, Total spine density (o) and percentage of SYP-mRuby-contacting spines (p) on basal dendrites of mPFC pyramidal neurons (n = 40 neurons from 6 mice in Ctrl; 31 neurons from 4 mice in SD). h – p, all P > 0.05 by Welch’s t-test. Data are shown as mean ± s.e.m. All tests were two-sided.
Extended Data Fig. 7. VTADA neurons were overexcited by SD during adolescence.
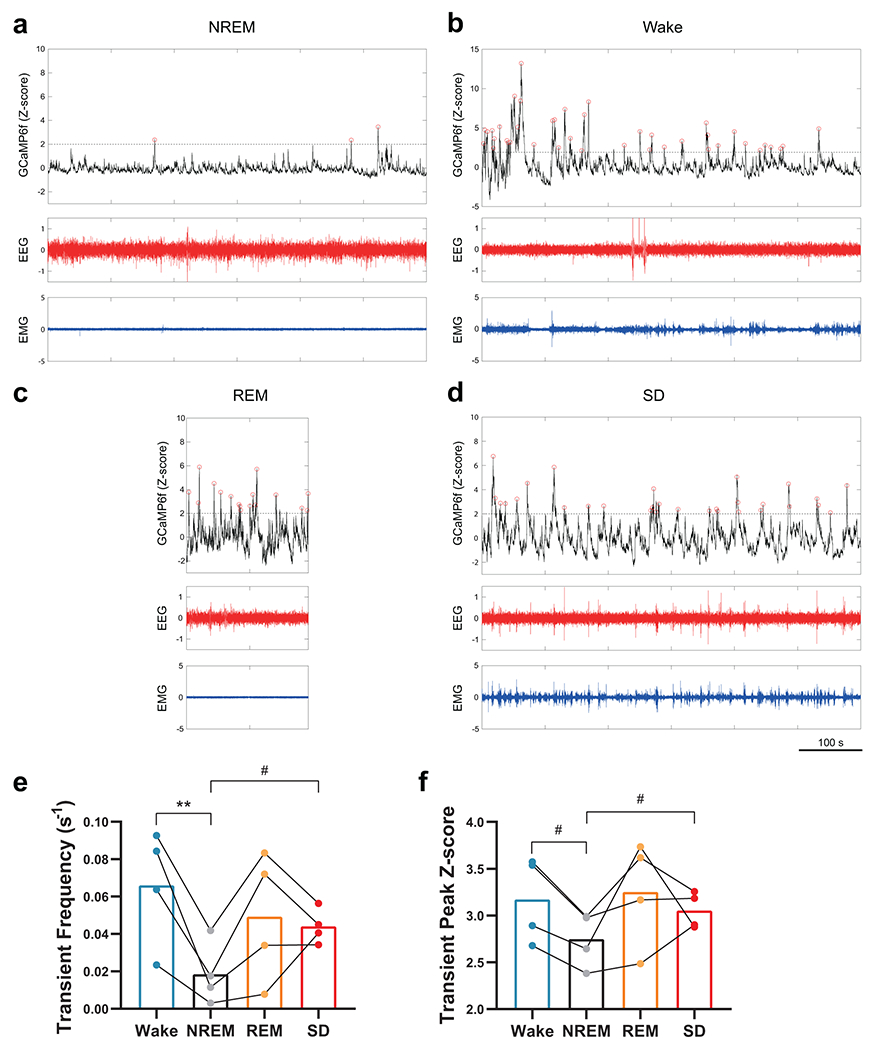
DAT-Cre mice received AAV-DJ-EF1α-DIO-GCaMP6f injections in VTA at P21 and implantations for fiber photometry and EEG/EMG recording at P30. Simultaneous fiber photometry and EEG/EMG recordings were performed at P37 – 38. a – d, Representative GCaMP (top, black), EEG (middle, red) and EMG (bottom, blue) signals during spontaneous NREM sleep (a), wake (b), REM sleep (c) and SD sessions (d). Red circles on GCaMP traces indicate Ca2+ transients detected. e, f, Quantification of frequency (e) and amplitude (peak Z-score, f) of Ca2+ transients detected in each state. n = 4 mice. e, RM one-way ANOVA, F (3, 9) = 5.68, P = 0.02, followed by Dunnett’s multiple comparisons to NREM, ** P = 0.007; NREM vs. SD, # P = 0.01 by paired t-test. f, RM one-way ANOVA, F (3, 9) = 2.57, P = 0.12; NREM vs. Wake, # P = 0.02, NREM vs SD, # P = 0.02, by paired t-test. Data are shown as mean ± s.e.m. All tests were two-sided.
Extended Data Fig. 8. CNO administration in adolescent hM3DqDAT and hM4DiDAT mice.
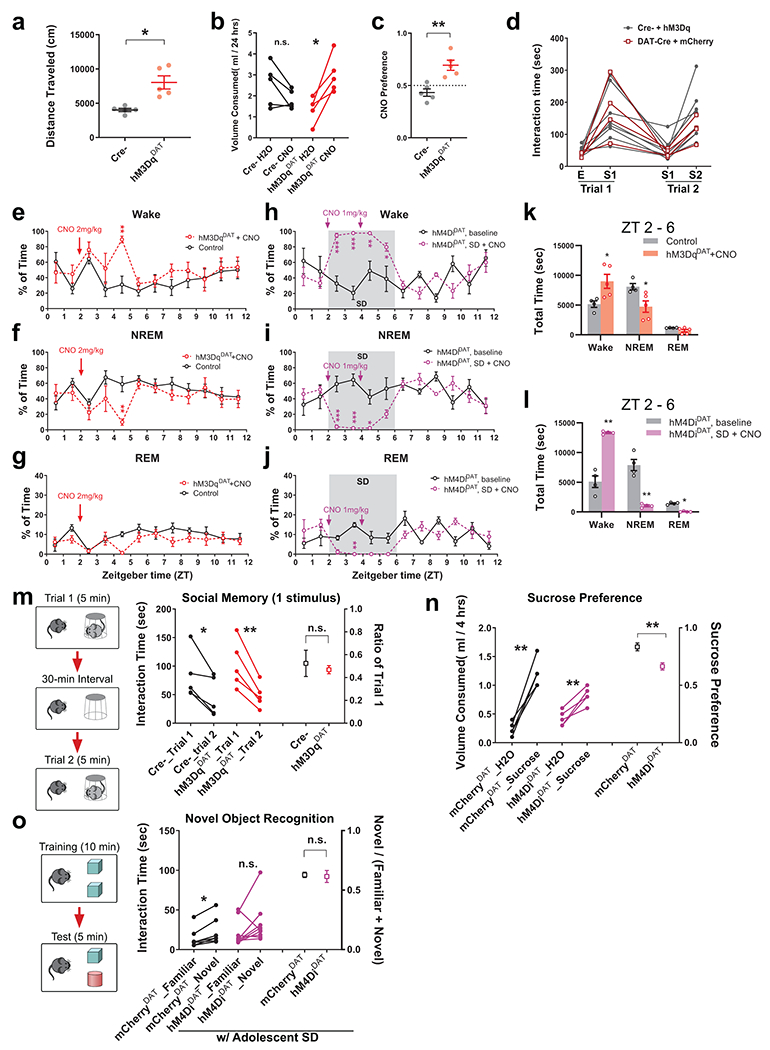
a, Cre- and hM3DqDAT mice were placed in an open-field arena 30 min following the i.p. injection of CNO (2 mg/kg). Total distance traveled in the arena during an 8-min session was measured (n = 5). * P = 0.01 by Welch’s t-test. b, c, The 2-bottle free-choice drinking test. Cre- and hM3DqDAT mice were kept in a cage where they have free access to the bottles containing H2O and CNO solution (50 mg/L), respectively, on opposite sides of the cage. The volume of H2O and CNO solution consumed over 24 hours were recorded. Compared to Ctrl mice, SD mice drank more CNO solution than H2O (b), and the preference for CNO (c), measured by the portion of CNO volume consumed over the total liquid consumed (H2O + CNO), was increased in the SD group (n = 5). b, * P = 0.01 by paired t-test; c, ** P = 0.003 by Welch’s t-test. d, Social performance of Cre- and mCherryDAT animals in the three-chamber assay at P56. e – g, DAT-Cre mice were injected with AAV-DJ-EF1α--DIO-hM3Dq in VTA at P21 and EEG recording was performed at P35 – 42. CNO (2mg/kg, i.p.) or saline was administered to the animal at ZT2. Hourly percentage of Wake (e), NREM (f) and REM (g) states during the 12-hr light phase are shown. Control group includes one mCherryDAT mouse with CNO injection and three hM3DqDAT mice with saline injections (total n = 4); hM3DqDAT + CNO, n = 5. RM 2-way ANOVA followed by Bonferroni’s post-test, e, f, both ** P = 0.006. h – j, DAT-Cre mice were injected with AAV-DIO-hM4Di in VTA at P21 and EEG recording was performed at P35 – 42. Mice were subjected to SD protocol during ZT 2 – 6, and CNO (1mg/kg, i.p.) was administered to the animal at ZT2 and ZT4. Compared to the baseline recording (1 day before SD + CNO treatment) where the animals did not receive SD or CNO, the SD protocol can still effectively deprive both NREM (i) and REM (j) sleep even with the presence of CNO (n = 4). RM 2-way ANOVA followed by Bonferroni’s post-test, h, ZT 2 – 3 *** P = 0.0005, ZT 3 – 4 *** P < 0.0001, ZT 4 – 5 ** P = 0.009, ZT 5-6 * P =0.04; i, ZT 2 – 3 *** P = 0.0002, ZT 3 – 4 *** P < 0.0001, ZT 4 – 5 * P = 0.01; j, ZT 3 – 4 ** P = 0.001. k, l, Quantification of each state during the 4 hours after the CNO injection in hM3DqDAT mice (k) and in hM4DiDAT mice with concurrent SD (l) as well as respective controls. k, n = 4 in control; 5 in hM3DqDAT + CNO, Wake, * P = 0.03; NREM, * P = 0.02 by Welch’s t-test. l, n = 4 mice each, Wake, ** P = 0.003; NREM, ** P = 0.005 by Welch’s t-test. REM, * P = 0.03 by Mann-Whitney test. m, No significant difference was found in the social memory test between Cre– and hM3DqDAT mice after adolescent CNO injections (n = 5). Left, * P = 0.02, ** P = 0.005 by paired t-test. Right, P = 0.67 by Welch’s t-test. n, The DAT-Cre mice that received AAV-DJ-EF1α-DIO-hM4Di-mCherry (hM4DiDAT) or mCherry control virus (mCherryDAT) on water regulation were given a single injection of CNO (2 mg/kg) and subsequently allowed access to both H2O and sucrose solution (1%, w/v) for 4 hours. The mCherryDAT mice showed strong preference to the sucrose, whereas the hM4DiDAT mice, although still consuming more sucrose solution than water, showed a significantly lowered sucrose preference, which was calculated as the volume of sucrose solution consumed divided by the total liquid volume consumed (H2O + sucrose, n = 5). Left, mCherryDAT, ** P = 0.001, hM4DiDAT, ** P = 0.005 by paired t-test. Right, ** P = 0.006 by Welch’s t-test. o, Novel object recognition test with reduced memory requirement was performed on adult mCherryDAT mice and hM4DiDAT mice (P56 – 60) with prior adolescent SD and CNO injections (P37 – 41). n = 7 in mCherryDAT; 9 in hM4DiDAT. Left, Wilcoxon matched-pairs signed rank test, mCherryDAT, * P = 0.02; hM4DiDAT, P = 0.10. Right, Welch’s t-test, P = 0.81. Data are shown as mean ± s.e.m. All tests were two-sided. n.s., not significant.
Extended Data Fig. 9. Adolescent sleep defects and adult in Shank3 InsG3680 mice.
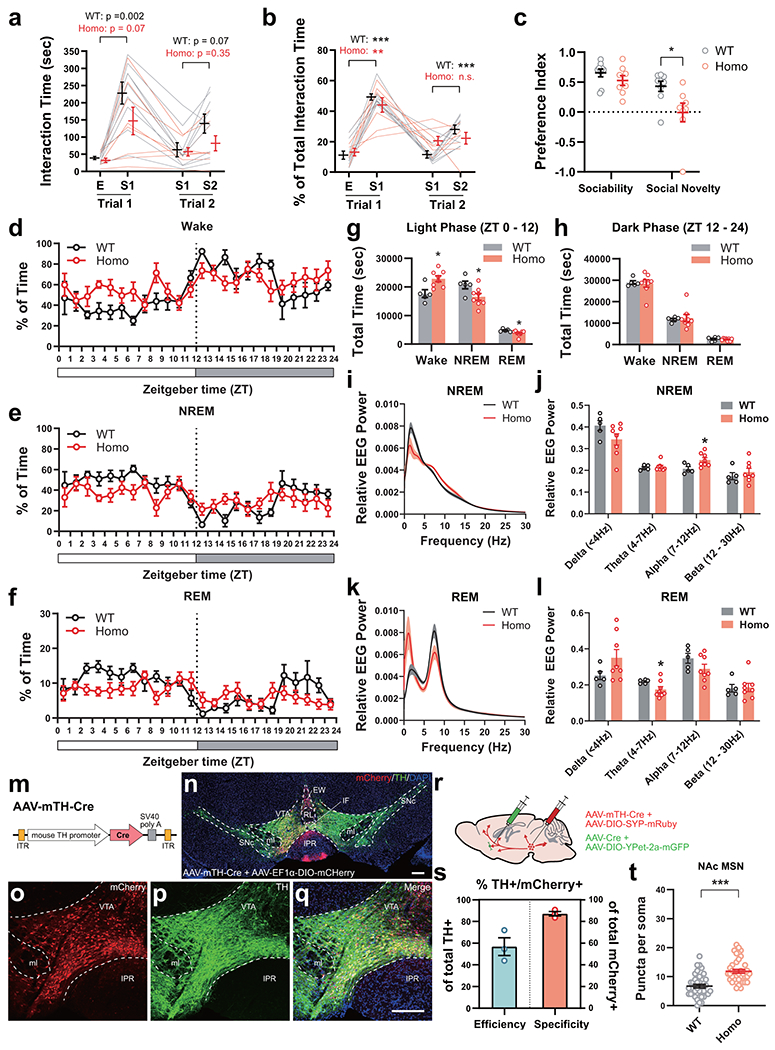
a – c, Three-chamber social test was performed in InsG3680+/+ mutants (Homo, n = 8) and WT littermates (n = 9) at P56. Absolute interaction time and percentage of each interaction category are shown in a and b, respectively. Preference indices are shown in c. a, RM 2-way ANOVA, Stimulus × Genotype F (3, 45) = 2.18, P = 0.10 followed by Tukey’s post-test, P values from the post-test are indicated on the graph; b, RM 2-way ANOVA, Stimulus × Genotype F (3, 45) = 1.97, P = 0.13, Tukey’s post-test, *** P < 0.0001, ** P = 0.004, * P = 0.01; c, Sociability, Mann-Whitney test, P = 0.13, Social Novelty, Welch’s t-test, * P = 0.04. d – f, Hourly percentage of Wake (d), NREM (e) and REM (f) states over the 24-hr light-dark cycle at P35 – 42 in adolescent InsG3680 mice (n = 5 in WT; 8 in Homo). g, h, Amount of Wake, NREM and REM states in InsG3680 mice at P35 – 42 (n = 5 in WT; 8 in Homo) in the light phase (g) and the dark phase (h). Welch’s t-test, g, Wake, * P = 0.02 (t = 3.06, df = 8.24), NREM, * P = 0.04 (t = 2.32, df = 9.26), REM, * P = 0.03 (t = 2.46, df = 10.86). h, all P > 0.05. i – l, EEG Power spectrum of NREM (i) and REM(k) sleep at P35 – 42 (n = 5 in WT; 8 in Homo). Kolmogorov-Smirnov test, i, P = 0.12; k, P = 0.39. Relative EEG powers of each frequency band are shown in j for NREM and l for REM. Welch’s t-test, j, Delta, P = 0.10, Theta, P = 0.63, Alpha, * P = 0.02, Beta, P = 0.43; l, Delta, P = 0.08, Theta, * P = 0.03, Alpha, P = 0.15, Beta, P = 0.90. m, The pAAV-mTH-Cre construct. Cre expression is under control of mouse tyrosine hydroxylase (TH) promoter. n – q, Representative images of a InsG3680 mouse which received injection of AAV-DJ-mTH-Cre + AAV-DJ-EF1α-DIO-mCherry in VTA and after two weeks of viral expression. Red, mCherry fluorescence; Green, TH immunostaining; Blue, DAPI. Scale bars, 200 μm. r, Dual labeling strategy in InsG3680 mice. s, Quantification showing the efficiency (% of TH/mCherry double positive neurons in total TH+ neurons) and specificity (% of double positive neurons in total mCherry+ neurons) of dopaminergic neuron labeling within the VTA area using co-injection of AAV-DJ-mTH-Cre and AAV-DJ-EF1α-DIO-mCherry (or -hM4Di-mCherry). n = 3 animals. t, The VTA axonal terminals in NAc were examined in WT (n = 40 neurons from 4 animals) and Homo (n = 38 neurons from 4 animals) InsG3680 mice at P70, using the dual labeling strategy with AAV-mTH-Cre. Welch’s t-test, P = 0.0000003. Data are shown as mean ± s.e.m. All tests were two-sided. n.s., not significant.
Extended Data Fig. 10. Effects of Flupirtine and DORA12 treatments on adolescent sleep and adult social interactions in Shank3 InsG3680 mice.
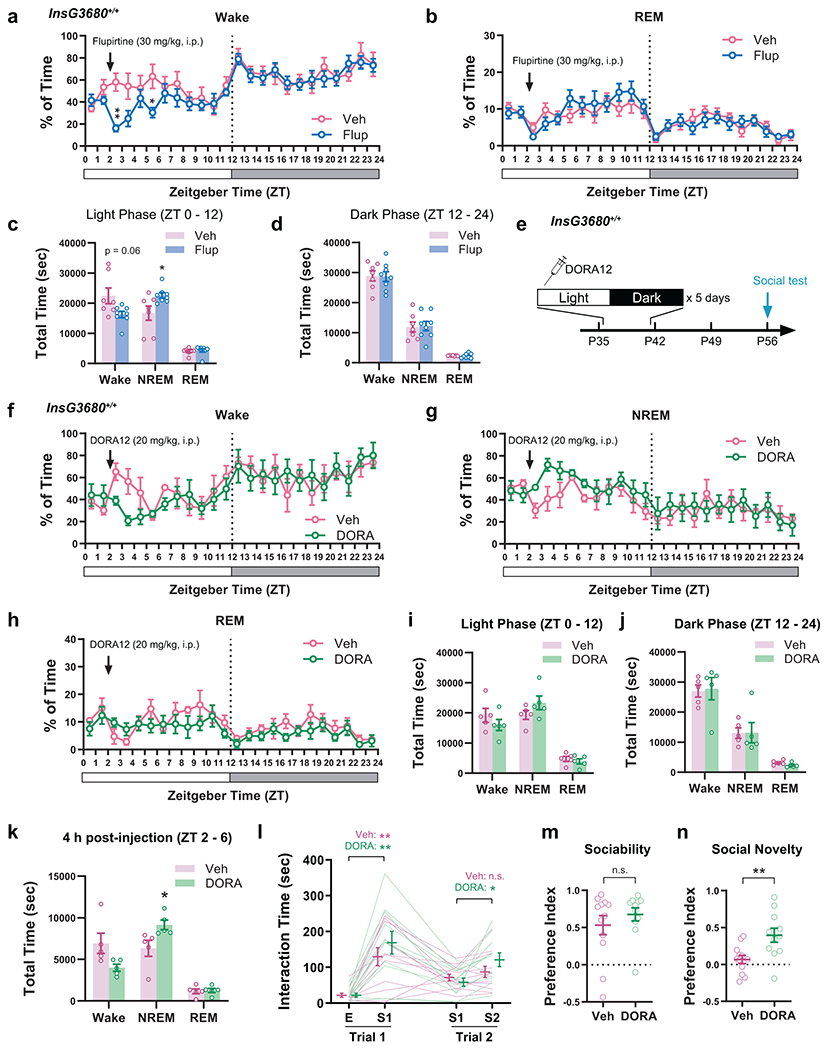
a – d, Hourly percentage of Wake (a) and REM (b) state over a 24-hr light-dark cycle and the quantification of each state during the light (c) or dark (d) phase in adolescent homozygous InsG3680 mice with Flupirtine treatment (n = 7 in Veh; 8 in Flup). RM 2-way ANOVA, a, Wake, Time x Treatment, F (23, 299) = 1.74, P = 0.02; b, REM, Time x Treatment, F (23, 299) = 0.90, P = 0.61, Bonferroni’s post-test, Wake ** P = 0.002, * P = 0.04. c, d, Welch’s t-test, NREM in Light Phase, * P = 0.048. e – h, Hourly percentage of Wake (f), NREM (g) and REM (h) state over a 24-hr light-dark cycle in adolescent homozygous InsG3680 mice with DORA treatment. Arrows indicate injections of DORA12 or vehicle (50% PEG400 in saline) at ZT2. n = 5 each. RM 2-way ANOVA with Bonferroni’s post-test. i – k, Quantification of each state during the light (i) or dark (j) phase and 4 hours following the injection of DORA12 or vehicle (k). n = 5 each. Welch’s t-test, k, NREM, * P = 0.04. l – n, Homozygous InsG3680 mice received daily DORA12 (20 mg/kg, i.p.) or vehicle (50% PEG400 in saline) for 5 consecutive days during P37 – 41, and three-chamber social interaction assay was performed at P56. n = 12 in Veh; 11 in DORA. Interaction time (l), RM 2-way ANOVA, Stimulus × Treatment F (3, 63) = 1.43, P = 0.24, Tukey’s post-test, Trial 1, Veh, ** P = 0.004, DORA, ** P = 0.003; Trial 2, Veh, P = 0.45, DORA, * P = 0.03. Preference indices of sociability (m) and social novelty (n), Welch’s t-test, m, t = 0.93, df = 19.22, P = 0.36, n, t = 3.01, df = 16.29, ** P = 0.008. Data are shown as mean ± s.e.m. All tests were two-sided. n.s., not significant.
Supplementary Material
Acknowledgements
We thank Drs. Guoping Feng and Jun Ding for transgenic mice and Dr. Xiang Yu for constructs. We thank Ayesha Khan, Drs. Kimberly J. Jennings, Keith R. Murphy and Yue Sun for assistance. We thank Drs. Andrew Olson and Gordon Wang, and the Stanford Wu Tsai Neuroscience Microscopy Service (NIH NS069375) for technical support. We thank Merck for providing the DORA12 compound. We acknowledge all de Lecea lab members for critical feedback. This work was supported by Human Frontier Science Program fellowship LT000338/2017-L (W.-J.B.) and National Institutes of Health grants R01 MH102638 (L.d.L.), R01 MH087592 (L.d.L.), R01 MH116470 (L.d.L.), R01 DA011289 (J.A.K.), and F32 NS123008 (C.L.B).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Frank MG & Heller HC The Function(s) of Sleep. Handb Exp Pharmacol 253, 3–34, doi: 10.1007/164_2018_140 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Anafi RC, Kayser MS & Raizen DM Exploring phylogeny to find the function of sleep. Nature reviews. Neuroscience 20, 109–116, doi: 10.1038/s41583-018-0098-9 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Tononi G & Cirelli C Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34, doi: 10.1016/j.neuron.2013.12.025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huber R & Born J Sleep, synaptic connectivity, and hippocampal memory during early development. Trends Cogn Sci 18, 141–152, doi: 10.1016/j.tics.2013.12.005 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Campbell IG & Feinberg I Longitudinal trajectories of non-rapid eye movement delta and theta EEG as indicators of adolescent brain maturation. Proc Natl Acad Sci U S A 106, 5177–5180, doi: 10.1073/pnas.0812947106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vivo L et al. Developmental patterns of sleep slow wave activity and synaptic density in adolescent mice. Sleep 37, 689–700, 700A-700B, doi: 10.5665/sleep.3570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timofeev I et al. Spatio-temporal properties of sleep slow waves and implications for development. Curr Opin Physiol 15, 172–182, doi: 10.1016/j.cophys.2020.01.007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank MG, Issa NP & Stryker MP Sleep enhances plasticity in the developing visual cortex. Neuron 30, 275–287 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Dumoulin Bridi MC et al. Rapid eye movement sleep promotes cortical plasticity in the developing brain. Sci Adv 1, e1500105, doi: 10.1126/sciadv.1500105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaffery JP, Lopez J, Bissette G & Roffwarg HP Rapid eye movement sleep deprivation revives a form of developmentally regulated synaptic plasticity in the visual cortex of post-critical period rats. Neurosci Lett 391, 96–101, doi: 10.1016/j.neulet.2005.08.044 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Shaffery JP, Sinton CM, Bissette G, Roffwarg HP & Marks GA Rapid eye movement sleep deprivation modifies expression of long-term potentiation in visual cortex of immature rats. Neuroscience 110, 431–443 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Maret S, Faraguna U, Nelson AB, Cirelli C & Tononi G Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nature neuroscience 14, 1418–1420, doi: 10.1038/nn.2934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang G et al. Sleep promotes branch-specific formation of dendritic spines after learning. Science 344, 1173–1178, doi: 10.1126/science.1249098 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Ma L, Yang G & Gan WB REM sleep selectively prunes and maintains new synapses in development and learning. Nature neuroscience 20, 427–437, doi: 10.1038/nn.4479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Vivo L et al. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355, 507–510, doi: 10.1126/science.aah5982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaskie RE, Graziano B & Ferrarelli F Schizophrenia and sleep disorders: links, risks, and management challenges. Nat Sci Sleep 9, 227–239, doi: 10.2147/NSS.S121076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veatch OJ et al. Shorter sleep duration is associated with social impairment and comorbidities in ASD. Autism Res 10, 1221–1238, doi: 10.1002/aur.1765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattai AA et al. Sleep disturbances in childhood-onset schizophrenia. Schizophr Res 86, 123–129, doi: 10.1016/j.schres.2006.04.020 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Robinson-Shelton A & Malow BA Sleep Disturbances in Neurodevelopmental Disorders. Current psychiatry reports 18, 6, doi: 10.1007/s11920-015-0638-1 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Carmassi C et al. Systematic Review of Sleep Disturbances and Circadian Sleep Desynchronization in Autism Spectrum Disorder: Toward an Integrative Model of a Self-Reinforcing Loop. Front Psychiatry 10, 366, doi: 10.3389/fpsyt.2019.00366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esbensen AJ & Schwichtenberg AJ Sleep in Neurodevelopmental Disorders. Int Rev Res Dev Disabil 51, 153–191, doi: 10.1016/bs.irrdd.2016.07.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bissonette GB & Roesch MR Development and function of the midbrain dopamine system: what we know and what we need to. Genes Brain Behav 15, 62–73, doi: 10.1111/gbb.12257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marotta R et al. The Neurochemistry of Autism. Brain Sci 10, doi: 10.3390/brainsci10030163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chevallier C, Kohls G, Troiani V, Brodkin ES & Schultz RT The social motivation theory of autism. Trends Cogn Sci 16, 231–239, doi: 10.1016/j.tics.2012.02.007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paval D A Dopamine Hypothesis of Autism Spectrum Disorder. Dev Neurosci 39, 355–360, doi: 10.1159/000478725 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D & Cohen RM Low medial prefrontal dopaminergic activity in autistic children. Lancet 350, 638, doi: 10.1016/s0140-6736(05)63326-0 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR & de Lecea L VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nature neuroscience 19, 1356–1366, doi: 10.1038/nn.4377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lena I et al. Variations in extracellular levels of dopamine, noradrenaline, glutamate, and aspartate across the sleep--wake cycle in the medial prefrontal cortex and nucleus accumbens of freely moving rats. J Neurosci Res 81, 891–899, doi: 10.1002/jnr.20602 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Dahan L et al. Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacology 32, 1232–1241, doi: 10.1038/sj.npp.1301251 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Yu X et al. GABA and glutamate neurons in the VTA regulate sleep and wakefulness. Nature neuroscience 22, 106–119, doi: 10.1038/s41593-018-0288-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oishi Y et al. Activation of ventral tegmental area dopamine neurons produces wakefulness through dopamine D2-like receptors in mice. Brain Struct Funct 222, 2907–2915, doi: 10.1007/s00429-017-1365-7 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Bariselli S et al. Role of VTA dopamine neurons and neuroligin 3 in sociability traits related to nonfamiliar conspecific interaction. Nature communications 9, 3173, doi: 10.1038/s41467-018-05382-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunaydin LA et al. Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551, doi: 10.1016/j.cell.2014.05.017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimprich A et al. A robust and reliable non-invasive test for stress responsivity in mice. Frontiers in behavioral neuroscience 8, 125, doi: 10.3389/fnbeh.2014.00125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakajima M, Gorlich A & Heintz N Oxytocin modulates female sociosexual behavior through a specific class of prefrontal cortical interneurons. Cell 159, 295–305, doi: 10.1016/j.cell.2014.09.020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li K, Nakajima M, Ibanez-Tallon I & Heintz N A Cortical Circuit for Sexually Dimorphic Oxytocin-Dependent Anxiety Behaviors. Cell 167, 60–72 e11, doi: 10.1016/j.cell.2016.08.067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao W et al. Gamma Oscillation Dysfunction in mPFC Leads to Social Deficits in Neuroligin 3 R451C Knockin Mice. Neuron 97, 1253–1260 e1257, doi: 10.1016/j.neuron.2018.02.001 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Liu L et al. Cell type-differential modulation of prefrontal cortical GABAergic interneurons on low gamma rhythm and social interaction. Sci Adv 6, eaay4073, doi: 10.1126/sciadv.aay4073 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang WC, Zucca A, Levy J & Page DT Social Behavior Is Modulated by Valence-Encoding mPFC-Amygdala Sub-circuitry. Cell Rep 32, 107899, doi: 10.1016/j.celrep.2020.107899 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamuro K et al. A prefrontal-paraventricular thalamus circuit requires juvenile social experience to regulate adult sociability in mice. Nature neuroscience 23, 1240–1252, doi: 10.1038/s41593-020-0695-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolen G, Darvishzadeh A, Huang KW & Malenka RC Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501, 179–184, doi: 10.1038/nature12518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun F et al. Next-generation GRAB sensors for monitoring dopaminergic activity in vivo. Nat Methods 17, 1156–1166, doi: 10.1038/s41592-020-00981-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C, Goel P & Kaeser PS Spatial and temporal scales of dopamine transmission. Nature reviews. Neuroscience 22, 345–358, doi: 10.1038/s41583-021-00455-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiedenmann B & Franke WW Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 41, 1017–1028, doi: 10.1016/s0092-8674(85)80082-9 (1985). [DOI] [PubMed] [Google Scholar]
- 45.Zhou Y et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 89, 147–162, doi: 10.1016/j.neuron.2015.11.023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szelenyi I Flupirtine, a re-discovered drug, revisited. Inflamm Res 62, 251–258, doi: 10.1007/s00011-013-0592-5 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Li SB et al. Hyperexcitable arousal circuits drive sleep instability during aging. Science 375, eabh3021, doi: 10.1126/science.abh3021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li SB, Nevarez N, Giardino WJ & de Lecea L Optical probing of orexin/hypocretin receptor antagonists. Sleep 41, doi: 10.1093/sleep/zsy141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K & de Lecea L Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 450, 420–424, doi: 10.1038/nature06310 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li SB, Giardino WJ & de Lecea L Hypocretins and Arousal. Current topics in behavioral neurosciences, doi: 10.1007/7854_2016_58 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Schoch SF et al. Across-night dynamics in traveling sleep slow waves throughout childhood. Sleep 41, doi: 10.1093/sleep/zsy165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wisor JP, Rempe MJ, Schmidt MA, Moore ME & Clegern WC Sleep slow-wave activity regulates cerebral glycolytic metabolism. Cereb Cortex 23, 1978–1987, doi: 10.1093/cercor/bhs189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhatt DH, Zhang S & Gan WB Dendritic spine dynamics. Annual review of physiology 71, 261–282, doi: 10.1146/annurev.physiol.010908.163140 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Bian WJ, Miao WY, He SJ, Qiu Z & Yu X Coordinated Spine Pruning and Maturation Mediated by Inter-Spine Competition for Cadherin/Catenin Complexes. Cell 162, 808–822, doi: 10.1016/j.cell.2015.07.018 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Moyer CE & Zuo Y Cortical dendritic spine development and plasticity: insights from in vivo imaging. Curr Opin Neurobiol 53, 76–82, doi: 10.1016/j.conb.2018.06.002 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Laviola G, Macri S, Morley-Fletcher S & Adriani W Risk-taking behavior in adolescent mice: psychobiological determinants and early epigenetic influence. Neurosci Biobehav Rev 27, 19–31, doi: 10.1016/s0149-7634(03)00006-x (2003). [DOI] [PubMed] [Google Scholar]
- 57.Kurth S et al. Traveling Slow Oscillations During Sleep: A Marker of Brain Connectivity in Childhood. Sleep 40, doi: 10.1093/sleep/zsx121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buchmann A et al. EEG sleep slow-wave activity as a mirror of cortical maturation. Cereb Cortex 21, 607–615, doi: 10.1093/cercor/bhq129 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Kim HJ et al. Systematic analysis of expression signatures of neuronal subpopulations in the VTA. Mol Brain 12, 110, doi: 10.1186/s13041-019-0530-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krishnan V et al. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature 543, 507–512, doi: 10.1038/nature21678 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 61.Woolley CS & McEwen BS Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol 336, 293–306, doi: 10.1002/cne.903360210 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Makinodan M, Rosen KM, Ito S & Corfas G A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360, doi: 10.1126/science.1220845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li SB et al. Hypothalamic circuitry underlying stress-induced insomnia and peripheral immunosuppression. Sci Adv 6, doi: 10.1126/sciadv.abc2590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beier KT et al. Circuit Architecture of VTA Dopamine Neurons Revealed by Systematic Input-Output Mapping. Cell 162, 622–634, doi: 10.1016/j.cell.2015.07.015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu H et al. Social touch-like tactile stimulation activates a tachykinin 1-oxytocin pathway to promote social interactions. Neuron, doi: 10.1016/j.neuron.2021.12.022 (2022). [DOI] [PubMed] [Google Scholar]
- 66.Nguyen AW & Daugherty PS Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol 23, 355–360, doi: 10.1038/nbt1066 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Zacharias DA, Violin JD, Newton AC & Tsien RY Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916, doi: 10.1126/science.1068539 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Chan KY et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nature neuroscience 20, 1172–1179, doi: 10.1038/nn.4593 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gomez JL et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507, doi: 10.1126/science.aan2475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J & Woodgett JR Assessment of social interaction behaviors. J Vis Exp, doi: 10.3791/2473 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winslow JT Mouse social recognition and preference. Curr Protoc Neurosci Chapter 8, Unit 8 16, doi: 10.1002/0471142301.ns0816s22 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Rolls A et al. Optogenetic disruption of sleep continuity impairs memory consolidation. Proc Natl Acad Sci U S A 108, 13305–13310, doi: 10.1073/pnas.1015633108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walf AA & Frye CA The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2, 322–328, doi: 10.1038/nprot.2007.44 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giardino WJ et al. Parallel circuits from the bed nuclei of stria terminalis to the lateral hypothalamus drive opposing emotional states. Nature neuroscience 21, 1084–1095, doi: 10.1038/s41593-018-0198-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ungless MA & Grace AA Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci 35, 422–430, doi: 10.1016/j.tins.2012.02.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olivier Friard MG BORIS: a free, versatile open-source event-logging software for video/audio coding and live observations. Methods in Ecology and Evolution 7, 1325–1330, doi: 10.1111/2041-210X.12584 (2016). [DOI] [Google Scholar]
- 77.Scheffzuk C et al. Selective coupling between theta phase and neocortical fast gamma oscillations during REM-sleep in mice. PLoS One 6, e28489, doi: 10.1371/journal.pone.0028489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hung LW et al. Gating of social reward by oxytocin in the ventral tegmental area. Science 357, 1406–1411, doi: 10.1126/science.aan4994 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sgritta M et al. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 101, 246–259 e246, doi: 10.1016/j.neuron.2018.11.018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within this paper and its Supplementary Information. Source data are provided with this paper.