

Abstract
Autophagy represents a fundamental mechanism for maintaining cell survival and tissue homeostasis in response to physiological and pathological stress. Autophagy initiation converges on the FIP200‐ATG13‐ULK1 complex wherein the serine/threonine kinase ULK1 plays a central role. Here, we reveal that the E3 ubiquitin ligase TRIM27 functions as a negative regulatory component of the FIP200‐ATG13‐ULK1 complex. TRIM27 directly polyubiquitinates ULK1 at K568 and K571 sites with K48‐linked ubiquitin chains, with proteasomal turnover maintaining control over basal ULK1 levels. However, during starvation‐induced autophagy, TRIM27 catalyzes non‐degradative K6‐ and K11‐linked ubiquitination of the serine/threonine kinase 38‐like (STK38L) kinase. In turn, STK38L ubiquitination promotes its activation and phosphorylation of ULK1 at Ser495, rendering ULK1 in a permissive state for TRIM27‐mediated hyper‐ubiquitination of ULK1. This cooperative mechanism serves to restrain the amplitude and duration of autophagy. Further evidence from mouse models shows that basal autophagy levels are increased in Trim27 knockout mice and that Trim27 differentially regulates tumorigenesis and metastasis. Our study identifies a key role of STK38L‐TRIM27‐ULK1 signaling axis in negatively controlling autophagy with relevance established in human breast cancer.
Keywords: autophagy, STK38L, TRIM27, tumorigenesis, ULK1
Subject Categories: Cancer, Signal Transduction
E3 ligase TRIM27 and STK38L kinase synergistically control autophagy acting as a rheostat on ULK1.
Introduction
Autophagy is a conserved metabolic recycling pathway whereby proteins, insoluble aggregates, and dysfunctional organelles are engulfed and degraded by lysosomes (Mizushima & Komatsu, 2011). Dysregulated autophagy can lead to a variety of diseases such as cancer and neurodegenerative disorders. Autophagy occurs at low levels under nutrient‐rich conditions where there is constitutive recycling of biomaterials to maintain energy homeostasis (Levine & Kroemer, 2008). Conversely, autophagic flux is increased following stress conditions such as limiting nutrients, hypoxia and growth factor deprivation in order to support cellular activities and maintain cell viability.
Autophagy is a sequential process (Nakatogawa et al, 2009; Yang & Klionsky, 2009) and among autophagy‐related genes, the serine/threonine kinase ULK1 plays a central role in autophagy initiation through a complex formed with ATG13, FIP200, and ATG101 (Ganley et al, 2009; Hosokawa et al, 2009a, 2009b; Mercer et al, 2009; Mizushima, 2010). In yeast, Atg1 complex formation is dynamically regulated by the Tor1 complex, leading to inhibition or initiation of autophagy in a nutrient‐dependent manner. However in mammals, ULK1 constitutively associates with ATG13 and FIP200 irrespective of nutrient conditions and the presence of this complex per se is not sufficient to initiate autophagy (Ganley et al, 2009; Hosokawa et al, 2009a; Wong et al, 2013). This predicates that other factors are involved in initiating autophagy. Indeed, under stress conditions, ULK1 activation is not only induced by AMPK‐mediated phosphorylation but also by mTOR inhibition which leads to the removal of inhibitory mTOR sites on ULK1 (Ganley et al, 2009; Hosokawa et al, 2009a; Kim et al, 2011). Such activation permits ULK1 to phosphorylate downstream targets in the Beclin1 complex to trigger the autophagy cascade (Russell et al, 2013). A number of studies have established that ULK1 is essential for autophagy initiation during starvation, but paradoxically, recent reports have suggested that ULK1 is rapidly degraded upon nutrient‐induced starvation (Allavena et al, 2016; Liu et al, 2016; Nazio et al, 2016). Relatively few studies have disclosed the precise mechanisms involved and how the ULK1 complex is regulated is still largely unknown.
TRIM27 (tripartite motif‐containing 27) belongs to the tripartite motif (TRIM) proteins, a family of more than 70 genes with conserved domain architectures (Hatakeyama, 2017). Most members contain an N‐terminal RING‐finger domain, the signature domain for E3 ubiquitin ligases, along with one or two B‐box zinc finger domains followed by a coiled‐coil domain (Watanabe & Hatakeyama, 2017). TRIM proteins are collectively implicated in a broad range of physiopathological processes, including cancer, immunological diseases, and neurological disorders. Different TRIM family proteins have been variously linked to autophagy where they most often have been shown to influence key upstream pathways that converge on autophagy initiation (Di Rienzo et al, 2020). For example, TRIM37 affects the mTOR pathway to inhibit autophagy (Wang et al, 2018), whereas similarly, the control of intracellular Ca2+ levels by TRIM19 also restrains autophagy through effects on AMPK activity (Missiroli et al, 2016). In some instances, the ubiquitin ligase activity of TRIM proteins directs key targets toward proteasomal degradation as occurs with TRIM28 in the turnover of AMPK (Pineda et al, 2015). Nevertheless, TRIM protein‐mediated ubiquitination does not always result in proteasomal degradation as seen with TRIM50 which activates autophagy through the non‐degradative modification of Beclin‐1 (Fusco et al, 2018). Moreover, not all mechanisms linking TRIM proteins with autophagy have been fully clarified, in particular those directly acting on key autophagy components. Specifically regarding TRIM27, different reports have revealed its functions in cell proliferation, apoptosis, and innate immunity (Zaman et al, 2013; Zheng et al, 2015; Ma et al, 2016); however, its involvement in autophagy has not been reported.
Here, we reveal a regulatory nexus between TRIM27 and the autophagy initiation complex that is critical for controlling autophagy under basal and starvation conditions. We show basal levels of autophagy are maintained through TRIM27‐mediated ubiquitination and proteasomal degradation of ULK1. Following autophagy induction, TRIM27 ubiquitinates and activates the serine/threonine kinase STK38L which then phosphorylates ULK1, delivering ULK1 in a permissive state for hyper‐ubiquitination by TRIM27. We further demonstrate increased basal autophagy in Trim27 knockout mice and establish physiological relevance in the context of breast cancer.
Results
Identification of TRIM27, a novel binding partner of the FIP200‐ATG13‐ULK1 complex
To discover regulators of the FIP200‐ATG13‐ULK1 complex, we used an unbiased mass spectrometry approach to recover endogenous FIP200‐interacting proteins in mouse embryonic fibroblasts (MEF) under control versus starvation conditions. Using FIP200 knockout MEF cells to filter out non‐specific interactors, we identified interactions between FIP200 and previously reported partners including ULK1, ATG13, ATG101 and CCPG1 (Hosokawa et al, 2009b; Wong et al, 2013; Smith et al, 2018) (Fig 1A and Appendix Table S1). An additional 21 novel candidates were selectively retrieved under starvation conditions including two E3 ubiquitin ligases, TRIM27 and NEDD4. Since NEDD4 has previously been implicated in autophagy regulation (Lin et al, 2017; Sun et al, 2017), we focused on verifying the role of TRIM27.
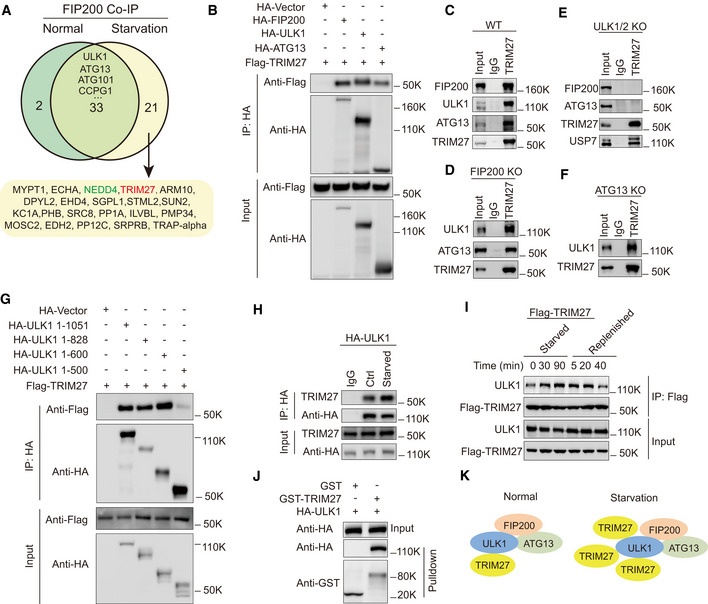
Figure 1. TRIM27 binds directly to ULK1 and this binding is regulated by nutritional starvation.
-
AVenn diagram of FIP200 binding proteins identified in MEF following FIP200 co‐immunoprecipitation analysis under normal versus starvation conditions.
-
BCo‐immunoprecipitation assays in HEK293T cells after co‐transfection with Flag‐TRIM27 and the indicated combinations of empty HA‐vector, HA‐tagged FIP200, ULK1, or ATG13 before immunoprecipitation with anti‐HA antibodies. Immunoprecipitates (top panel) and input samples (bottom panel) were then subjected to Western blotting against anti‐HA or Flag.
-
C–FWT (C), FIP200 knockout (KO) (D), ULK1/2 knockout (E) MEF cells or ATG13 knockout 4T1 cells (F) were subjected to starvation conditions before immunoprecipitation against control Immunoglobulin G (IgG) or anti‐TRIM27 before blotting against the indicated endogenous forms of TRIM27, FIP200, ULK1, ATG13, and USP7.
-
GCo‐immunoprecipitation assays in HEK293T cells after co‐transfection with Flag‐TRIM27 and either full length ULK (HA‐ULK1 1–1,051) or the indicated various truncated forms of ULK1 designated HA‐ULK1 1–828, HA‐ULK1 1–600, and HA‐ULK1 1–500; AA, amino acids.
-
HCo‐immunoprecipitation assays in HeLa cells stably expressing HA‐ULK1 and immunoprecipitation against IgG or HA followed by Western blotting as indicated.
-
IHeLa cells stably expressing Flag‐TRIM27 were starved as indicated or starved for 2h before replenishing with complete medium. Anti‐Flag immunoprecipitates (top panel) and input samples (bottom panel) were then subjected to blotting against Flag and ULK1.
-
JPurified recombinant GST or GST‐TRIM27 was incubated with recombinant HA‐ULK1 and bound ULK1 analyzed by Western blotting.
-
KSchematic diagram of TRIM27 binding to ULK1‐ATG13‐FIP200 complex in normal or starvation conditions.
Data information: For B–J, the experiments were performed at least three times independently with similar results.
Source data are available online for this figure.
To characterize whether TRIM27 bound to FIP200 discretely or as part of the ULK complex, we reconstructed interactions between ectopically expressed Flag‐TRIM27 with individually expressed HA‐tagged forms of FIP200, ULK1 and ATG13. Notably, we found Flag‐TRIM27 was co‐immunoprecipitated with exogenous FIP200, ULK1 and ATG13 (Fig 1B), suggesting their interactions occurred as a complex. These findings were verified in the endogenous context where TRIM27 coprecipitated FIP200, ULK1 and ATG13 in nutrient‐starved MEF (Fig 1C). Repeating the assay in FIP200 knockout cells showed endogenous TRIM27 still bound to ULK1 and ATG13 (Fig 1D and Appendix Fig S1A). Moreover, TRIM27 failed to coprecipitate FIP200 and ATG13 in ULK1/2 double knockout MEF cells, although the previously reported interactor USP7 was recovered (Figs 1E and Appendix Fig S1B). Lastly, TRIM27 was shown to bind to ULK1 in the absence of ATG13 (Figs 1F and Appendix Fig S1C). Together, these data suggested that TRIM27 is associated with the FIP200‐ATG13‐ULK1 complex via interaction with ULK1.
Next, we performed domain mapping to better define the TRIM27‐ULK1 interaction. Interactions were assessed by reciprocal immunoprecipitation between truncated forms of epitope‐tagged ULK1 and TRIM27, respectively. Notably, while the N‐terminal 1–600 amino acids (AA) of ULK1 interacted with TRIM27, 1–500AA did not, thereby localizing the TRIM27‐interaction sequence between 500 and 600AA (Fig 1G). Conversely, the coiled‐coil domain but not the RING‐BOX domain of TRIM27 was required for its interaction with ULK1 (Appendix Fig S1D).
Lastly, given that TRIM27 bound ULK1 in MEF cells under starvation, as independent verification, we then confirmed the association in human HeLa cells, in that HA‐ULK1 bound to more endogenous TRIM27 in response to starvation (Figs 1H and Appendix Fig S1E). Moreover, the interaction between ULK1 and TRIM27 was gradually increased upon starvation but was reduced when the starved cells were replenished with normal growth medium (Fig 1I). Additionally, the interaction between recombinant forms of TRIM27 and ULK1 could be readily observed in glutathione S‐transferase (GST) pulldown assays (Fig 1J). Taken together, our data indicate that TRIM27 binds to components of FIP200‐ATG13‐ULK1 complex via ULK1 and its binding to ULK1 was induced in response to starvation conditions (Fig 1K).
TRIM27 is an E3 ubiquitin ligase targeting ULK1
We next evaluated whether TRIM27 served to ubiquitinate ULK1 using His‐based ubiquitination assays in HEK293T cells. Indeed, co‐expression of TRIM27 selectively promoted the ubiquitination of ULK1, but not FIP200 or ATG13 (Figs 2A and EV1A and B). Depletion of endogenous TRIM27 significantly decreased ULK1 ubiquitination (Fig 2B) and as anticipated, the RING‐finger domain of TRIM27 was required for ULK1 ubiquitination (Fig EV1C). Moreover, GST‐TRIM27 readily ubiquitinated recombinant HA‐ULK1 in vitro (Fig 2C). We also compared the relative ability of TRIM27 to ubiquitinate ULK1 against NEDD4L, another E3 ligase previously shown to target ULK1 (Nazio et al, 2016) and its homologue NEDD4 (Yang & Kumar, 2010). Interestingly, TRIM27 mediated comparatively higher levels of ULK1 ubiquitination than either NEDD4L or NEDD4 (Appendix Fig S2A). Notably, knockdown of TRIM27 did not affect interactions between ULK1 and NEDD4L or NEDD4 (Appendix Fig S2B and C) suggesting TRIM27 does not intrinsically compete with NEDD4L or NEDD4 for ULK1 binding. Thus, together these data establish ULK1 as a direct substrate of TRIM27 E3 ligase.
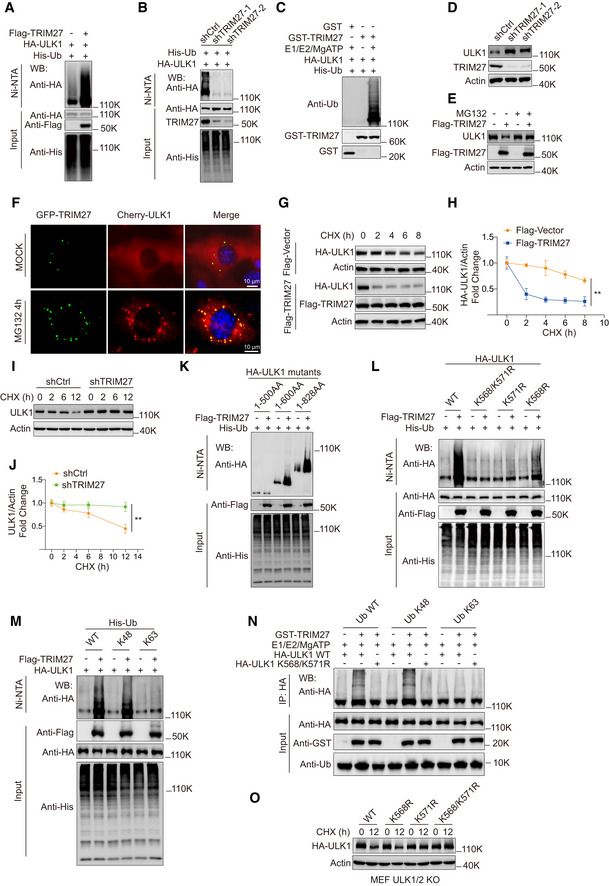
Figure 2. TRIM27 promotes ULK1 degradation via K48‐linked ubiquitination of ULK1 at K568/K571.
-
A, BUbiquitination assays in HEK293T cells after co‐transfection with HA‐ULK1 and His‐Ub along with Flag‐TRIM27 (A) or after knockdown of TRIM27 using independent shRNAs (shTRIM27‐1 and ‐2) (B). Samples recovered with Ni‐NTA (top panel) and input samples (bottom panel) were then subjected to blotting against anti‐HA, Flag or His as indicated.
-
CIn vitro ubiquitination assays performed with the indicated combinations of recombinant GST, GST‐TRIM27, affinity‐purified HA‐ULK1 and His‐Ub in the presence or absence of E1, E2, and ATP. The reactions were analyzed with anti‐Ub, anti‐HA, or anti‐GST.
-
D, EWestern blot analysis of ULK1 levels in HeLa cells stably expressing control or TRIM27 shRNAs (D), or HEK293T cells transfected with empty Flag vector (−) or Flag‐TRIM27 after treatment with or without 10 μM MG132 for 4 h (E).
-
FFluorescence microscopy image of MEFs with stable expression of mCherry‐ULK1 and GFP‐TRIM27 after treating cells with DMSO (MOCK) or 10 μM MG132 for 4 h. Scale bar, 10 μm.
-
G–JHA‐ULK1 levels (G, H), or endogenous ULK1 levels (I, J) in 50 μg/ml cycloheximide (CHX)‐treated HEK293T cells with or without co‐expression of Flag‐TRIM27 (G, H) or TRIM27 RNAi (I, J). Western blots against HA‐ULK1 and actin control (G, I) were subjected to densitometric quantitation (H, J).
-
K, LUbiquitination assays were performed in HEK293T cells as per (A, B) after co‐transfection with full length HA‐ULK1 or the indicated truncated constructs (K) or double (K568/K571R) or single (K568R, K571R) lysine substitution mutants (L) in the presence or absence of Flag‐TRIM27.
-
MUbiquitination of HA‐ULK1 measured as per (A, B) in cells co‐expressing Flag‐TRIM27 with either His‐Ub WT, His‐Ub K48, or His‐Ub K63, respectively.
-
NIn vitro ubiquitination assays performed as per (C) against affinity‐purified HA‐ULK1 WT or the HA‐ULK1 K568/K571R mutant in the presence or absence of GST‐TRIM27.
-
OULK1/2 knockout MEFs stably expressing the indicated HA‐ULK1 constructs were subjected to Western blot analysis against HA or actin before (0 h) and after treatment with CHX for 12 h.
Data information: For (A‐O), the experiments were performed at least three times independently with similar results. All the statistical data are presented as mean ± SD of three independent experiments. Statistical analysis was performed using two‐tailed unpaired Student’s t‐tests (H and J), **P < 0.01.
Source data are available online for this figure.
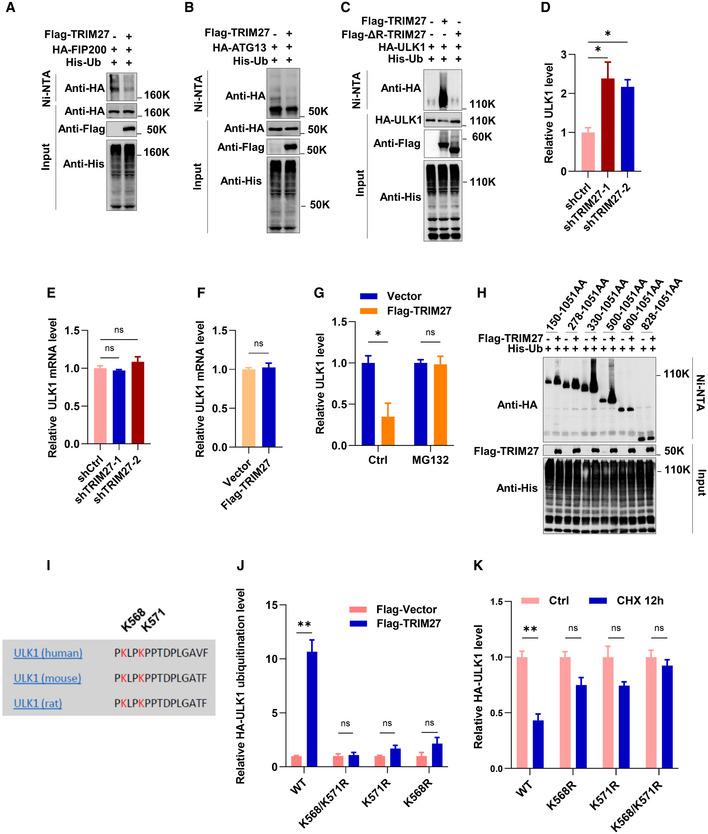
Figure EV1. The ubiquitination of FIP200 and ATG13 is not promoted by TRIM27, and ULK1 mRNA levels are not affected by TRIM27.
-
A, BUbiquitination analysis of HA‐FIP200 (A) or HA‐ATG13 (B) co‐expressed with Flag‐TRIM27 and His‐Ub in HEK293T cells by Western blot.
-
CUbiquitination analysis of HA‐ULK1 co‐expressed with Flag‐TRIM27 or Flag‐ΔR‐TRIM27 and His‐Ub in HEK293T cells.
-
DQuantification of data presented in Fig 2D showing the mean ± SEM of three independent experiments. P‐values were determined by a one‐way ANOVA with Dunnett’s multiple comparisons test, *P < 0.05.
-
ERelative ULK1 mRNA levels in HeLa cells with or without knockdown of TRIM27 using qPCR. Data are mean ± SEM of three independent experiments. P‐values were determined by a one‐way ANOVA with Dunnett’s multiple comparisons test. ns, not significant.
-
FRelative ULK1 mRNA levels in HeLa cells with or without over‐expression of Flag‐TRIM27. Data are mean ± SEM of three independent experiments. P‐values were determined by a two‐tailed unpaired t‐test. ns, not significant.
-
GQuantification of data presented in Fig 2E shows mean ± SEM of three independent experiments. P‐values were determined by an unpaired t‐test with Holm‐Sidak’s method for multiple t‐tests, *P < 0.05. ns, not significant.
-
HUbiquitination analysis of HA‐ULK1 truncation constructs co‐expressed with Flag‐TRIM27 and His‐Ub in HEK293T cells.
-
IAlignment of human and rodent ULK1 aa sequences shows conservation of TRIM27‐mediated ubiquitination sites.
-
JQuantification of data presented in Fig 2L shows mean ± SEM of three independent experiments. P‐values were determined by an unpaired t‐test with Holm‐Sidak’s method for multiple t‐tests, **P < 0.01. ns, not significant.
-
KQuantification of data presented in Fig 2O shows mean ± SEM of three independent experiments. P‐values were determined by an unpaired t‐test with Holm‐Sidak’s method for multiple t‐tests, **P < 0.01. ns, not significant.
Source data are available online for this figure.
We then investigated the consequences of TRIM27‐mediated ULK1 ubiquitination. Knockdown of TRIM27 increased ULK1 protein levels, but did not affect ULK1 mRNA (Figs 2D and EV1D and E). Similarly, TRIM27 overexpression had no effect on ULK1 mRNA expression (Fig EV1F), but notably downregulated ULK1 protein levels, which was blocked by proteasomal inhibition with MG132 (Figs 2E and EV1G), indicating increased proteasomal degradation. In support, MG132 treatment resulted in marked increases in the colocalization of overexpressed mCherry‐ULK1 and exogenous GFP‐TRIM27 in punctate cytoplasmic structures (Fig 2F). Consistently, cycloheximide chase assays revealed that ectopic Flag‐TRIM27 expression destabilized ULK1 protein levels (Fig 2G and H), whereas TRIM27 knockdown increased ULK1 stability (Fig 2I and J). Thus, TRIM27‐mediated ULK1 ubiquitination leads to its proteasomal degradation.
To identify ULK1 ubiquitination sites, we first determined ubiquitination levels in ULK1 C‐terminal and N‐terminal truncation mutants after overexpression of TRIM27 (Figs 2K and EV1H). This analysis suggested that the principal ubiquitination sites exist within 500–600AA of ULK1, a region that contains only two lysine residues (K568 and K571; Fig EV1I). Consequently, single and double substitution mutants were prepared to test whether either or both acted as TRIM27 substrate sites in ubiquitination assays. Notably, the K568R and K571R mutants partially diminished, while the K568/K571R double mutant almost completely abolished ULK1 ubiquitination (Figs 2L and EV1J), indicating that TRIM27 modifies ULK1 at residues K568 and K571.
Polyubiquitylation principally occurs as either K48 or K63‐linked ubiquitin chains (Ohtake et al, 2016). K48‐linked polyubiquitination mostly appears as a universal signal for proteasomal degradation, whereas K63‐linkage is often implicated in protein activity regulation. To characterize TRIM27‐mediated polyubiquitination in ULK1, we repeated the ubiquitination assays using mutant His‐Ub K48 (all lysines mutated to arginine except lysine 48), and His‐Ub K63 (lacking all lysine residues except lysine 63). We found that TRIM27 markedly promoted ULK1 polyubiquitination in the presence of K48, but not K63 His‐Ub in cell‐based (Fig 2M) and in vitro assays (Fig 2N). Furthermore, consistent with TRIM27 acting to decrease ULK1 protein stability, reconstituting ULK1 knockout cells with single or double mutant forms of HA‐ULK1 showed that the K568/K571R ULK1 mutant accumulated in cycloheximide‐treated cells (Figs 2O and EV1K). Collectively, these data show that TRIM27 functions as a negative regulator of ULK1 by promoting K48‐linked polyubiquitination and proteasomal degradation.
TRIM27 negatively regulates autophagy
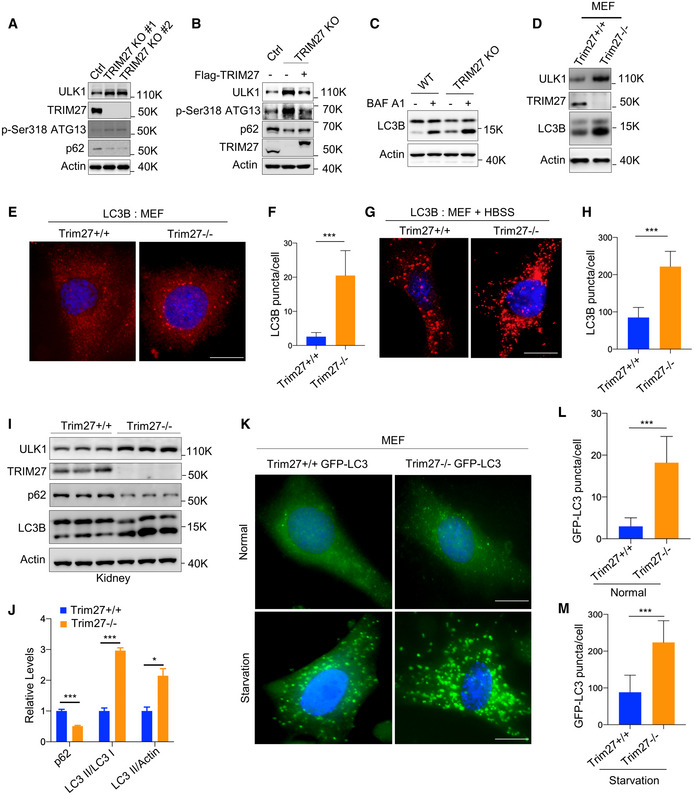
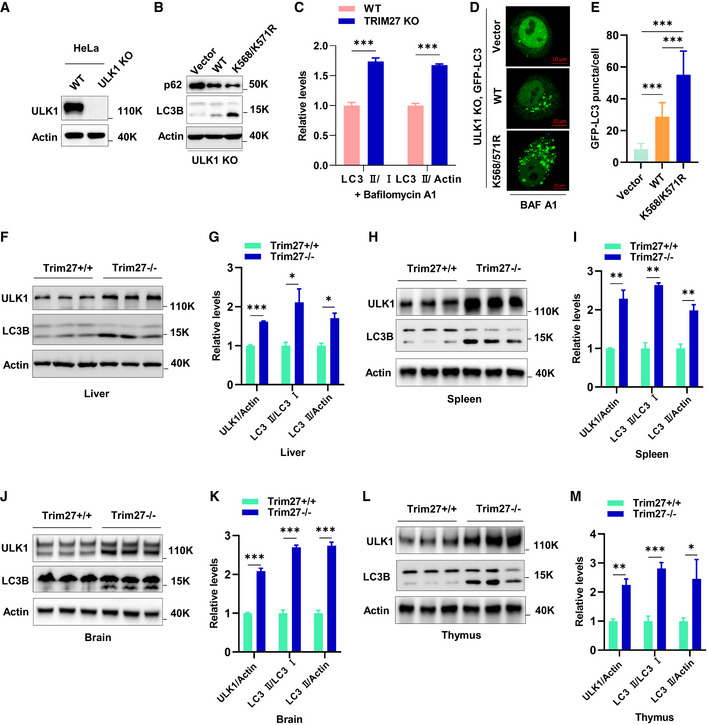
Given the essential role of ULK1 in autophagy initiation, we sought to determine how TRIM27 affects autophagy. As anticipated from the preceding experiments, deletion of TRIM27 in HeLa cells using CRISPR‐Cas9 increased ULK1 levels with accompanying rises in Ser318‐phosphorylated ATG13 levels which is a known ULK1 substrate (Fig 3A). Moreover, autophagy flux was increased in TRIM27‐depleted cells as indicated by reduced p62 levels and these effects were reversed when the knockout cells were reconstituted with exogenous TRIM27 (Fig 3B). Consistently, the re‐introduction of ULK1 in ULK1‐KO HeLa cells showed that p62 degradation was enhanced by the K568/K571R mutant, suggesting heightened autophagy flux (Fig EV2A and B). Furthermore, TRIM27 depletion resulted in significant increases in the LC3‐II/LC3‐I ratio (a readout of autophagy) in the presence and absence of Bafilomycin A, an inhibitor that blocks autophagic flux by inhibiting autophagosome‐lysosome fusion (Figs 3C and EV2C). Lastly, imaging of the GFP‐LC3 autophagy reporter in ULK1‐KO HeLa cells in the presence of Bafilomycin A showed that ectopic expression of the K568/K571R ULK1 mutant significantly increased the numbers of GFP‐LC3 puncta (autophagosomes) compared to wildtype ULK1 (Fig EV2D and E). Therefore, TRIM27‐mediated regulation of ULK1 serves to inhibit autophagy.
Figure 3. TRIM27 is a negative regulator of autophagy.
-
AWestern blot analysis of autophagy marker proteins (p62 and Ser318 phosphorylated ATG13) in WT HeLa cells and two independent TRIM27 KO clones. ULK1 and TRIM27 levels were measured, along with actin as a loading control.
-
BTRIM27 KO HeLa cells were reconstituted with Flag‐TRIM27 and Western blotting was performed as per (A).
-
CLC3 conversion was measured by Western blot in WT or TRIM27 KO HeLa cells with or without pretreatment with 200 nM bafilomycin A1 (BAF A1).
-
DWestern blot analysis of indicated proteins in MEFs generated from Trim27+/+ or Trim27−/− mice.
-
E–HImmunofluorescence analysis of endogenous LC3 in MEFs generated from Trim27+/+ or Trim27−/− mice cultured in fed (E) or starved conditions (G). Quantification of LC3B puncta Fed conditions (F), starved conditions (H) (n = 50 cells per condition). Data are mean ± SD in F and H. Scale bar, 10 μm.
-
I, JWestern blot analysis of autophagy markers in the kidneys of 3‐month‐old Trim27+/+ and Trim27−/− mice. Each lane represents a different mouse (I). Quantification of p62, LC3‐II/LC3‐I ratio, and LC3‐II/actin ratio in I (J). Data are mean ± SEM for three mice per genotype.
-
KMicroscopy analysis of GFP‐LC3 puncta in MEFs generated from indicated genotype mice in normal or starvation conditions. Scale bar, 10 μm.
-
L, MQuantitation of data presented in (K) shows mean number of GFP‐LC3 puncta per cell ± SD (n = 50 cells per condition).
Data information: (F, H, J, L and M) Statistical analysis was performed using two‐tailed unpaired Student’s t‐tests, *P < 0.05, ***P < 0.001.
Source data are available online for this figure.
Figure EV2. Knockout of TRIM27 leads to an increase of autophagy in multiple tissues.
-
AVerification of ULK1 knockout in HeLa cells by Western blot.
-
Bp62 or LC3 levels in HeLa ULK1 KO cells co‐expressing vector control, HA‐ULK1 WT, or HA‐ULK1 K568/K571R.
-
CQuantification of LC3‐II/LC3‐I ratio, and LC3‐II/actin ratio with Bafilomycin A1 treatment related to Fig 3C. Data are mean ± SEM for three independent experiments. P‐values were determined by an unpaired t‐test with Holm‐Sidak’s method for multiple t‐tests, ***P < 0.001.
-
DRepresentative images of GFP fluorescence localization in HeLa ULK1 KO cells co‐expressing GFP‐LC3 together with vector control, HA‐ULK1 wildtype, or HA‐ULK1 K568/K571R, respectively.
-
EQuantitation of data presented in (D) shows mean number of GFP‐LC3 puncta per cell ± SD (n = 30 cells per condition). P‐values were determined by a one‐way ANOVA with Tukey’s multiple comparisons test, ***P < 0.001.
-
F, GWestern blot analysis of ULK1 accumulation and LC3 conversion in the liver of 3‐month‐old Trim27+/+ and Trim27−/− mice. Each lane represents a different mouse (F). Quantification of ULK1, LC3‐II/LC3‐I, and LC3‐II/actin ratios in F (G).
-
H, IWestern blot analysis of ULK1 accumulation and LC3 conversion in the spleen of 3‐month‐old Trim27+/+ and Trim27−/− mice. Each lane represents a different mouse (H). Quantification of ULK1, LC3‐II/LC3‐I, and LC3‐II/actin ratios in H (I).
-
J, KWestern blot analysis of ULK1 accumulation and LC3 conversion in the brain of 3‐month‐old Trim27+/+ and Trim27−/− mice. Each lane represents a different mouse (J). Quantification of ULK1, LC3‐II/LC3‐I, and LC3‐II/actin ratios in J (K).
-
L, MWestern blot analysis of ULK1 accumulation and LC3 conversion in the thymus of 3‐month‐old Trim27+/+ and Trim27−/− mice. Each lane represents a different mouse (L). Quantification of ULK1, LC3‐II/LC3‐I, and LC3‐II/actin ratios in L (M).
Data information: (G, I, K and M) Data are mean ± SEM for three mice per genotype. P‐values were determined by an unpaired t‐test with Holm‐Sidak’s method for multiple t‐tests, *P < 0.05, **P < 0.01; ***P < 0.001.
Source data are available online for this figure.
We next generated Trim27 knockout (Trim27−/−) mice to further explore the role of TRIM27 in autophagy (Appendix Fig S3A–C). Consistently we found that basal autophagy levels were increased in Trim27−/− MEF compared to wild‐type (WT) MEF controls as reflected by increases in LC3‐II protein abundance (Fig 3D) along with increased autophagosome numbers (Fig 3E and F). As expected, starvation increased autophagosomes, but their numbers were greatly accentuated in Trim27−/− MEF (Fig 3G and H). Strikingly, comparisons of kidney tissues from mice against those from littermate controls demonstrated significant increases in LC3 conversion ratios (LC3‐II/LC3‐I) and downregulation of p62 (Figs 3I and J). Moreover, further analysis of liver, spleen, brain, and thymus lysates also showed increased accumulation of ULK1 protein levels in major organs of Trim27 knockout mice (Fig EV2F–M), suggesting high basal levels of autophagy in the absence of TRIM27. Lastly, to provide an independent demonstration of these findings, we derived MEF cells from Trim27−/− mice crossed with GFP‐LC3 autophagy reporter mice. Analysis of these cells confirmed that TRIM27 knockout increased both basal and starvation‐induced autophagy (Fig 3K–M). Together, our data indicate a crucial role of TRIM27 as a negative regulator of autophagy.
STK38L negatively regulates starvation‐induced autophagy via promoting binding between TRIM27 and ULK1
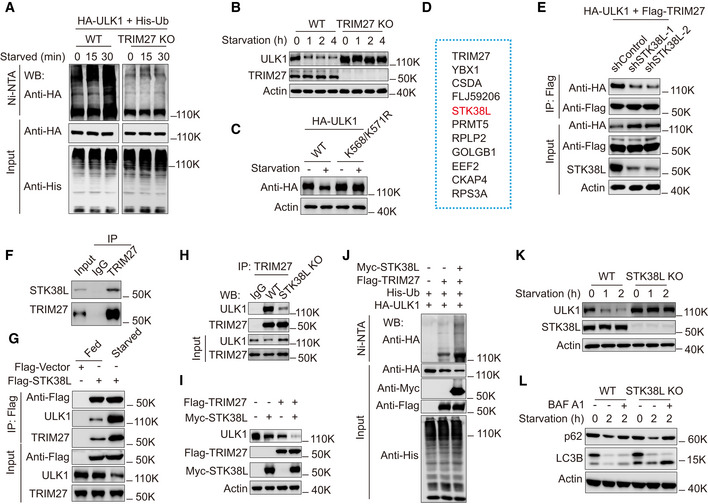
Previous reports have shown that ULK1 undergoes rapid degradation during nutrient starvation (Liu et al, 2016; Nazio et al, 2016) and consistently we found that starvation downregulated ULK1 in HeLa cells (Appendix Fig S4A). Our prior experiments established increased binding of TRIM27 with ULK1 under starvation conditions (Fig 1I), but it remained to be determined to what extent TRIM27 was involved in the starvation‐induced downregulation of ULK1. Toward this, we found that ULK1 ubiquitination was greatly enhanced during starvation and these increases were blocked by TRIM27 depletion (Fig 4A). Consistently, TRIM27 depletion markedly delayed ULK1 degradation (Fig 4B) and moreover, the K568/K571R HA‐ULK1 mutant was resistant to cell starvation‐induced degradation when used to reconstitute ULK1 KO cells (Fig 4C). Thus, the enhanced ubiquitination of ULK1 by TRIM27 is triggered by nutrient starvation and is responsible for the stimulus‐specific degradation of ULK1.
Figure 4. STK38L negatively regulates starvation‐induced autophagy via promoting binding between TRIM27 and ULK1.
-
AUbiquitination analysis of HA‐ULK1 in HeLa derivatives generated in Fig 1(H) co‐expressing His‐Ub with or without TRIM27 knockout, the cells were treated with starvation for the indicated time.
-
BWestern blot analysis of ULK1 levels in HeLa cells with or without TRIM27 knockout. The cells were treated with starvation for the indicated time. The lysates were analyzed using anti‐ULK1, anti‐TRIM27, or anti‐Actin.
-
CHA‐ULK1, HA‐ULK1 K568/K571R were stably expressed in ULK1 knockout HeLa cells, the cells were treated with starvation, the lysates were analyzed by Western blot using anti‐HA, or anti‐Actin.
-
DList of potential Top 10 proteins that bind to TRIM27 under starvation conditions identified by mass spectrometry.
-
ECo‐immunoprecipitation of Flag‐TRIM27 with HA‐ULK1 in HEK293T cells with or without STK38L knockdown, the cells were treated with starvation. Flag‐TRIM27 was immunoprecipitated using anti‐Flag, the bound HA‐ULK1 was detected using anti‐HA.
-
FCo‐immunoprecipitation of endogenous TRIM27 with endogenous STK38L in HeLa cells.
-
GCo‐immunoprecipitation of Flag‐STK38L with ULK1, or TRIM27 in HEK293T cells with or without starvation treatment. Flag‐STK38L was immunoprecipitated using anti‐Flag, the immunoprecipitates and lysates were analyzed with anti‐ULK1, anti‐TRIM27.
-
HCo‐immunoprecipitation of endogenous TRIM27 with endogenous ULK1 in HeLa cells with or without STK38L knockout. The cells were treated with starvation before lysis.
-
IWestern blot analysis of ULK1 levels in starved HeLa cells expressing either Myc‐STK38L, or Flag‐TRIM27, or both. The lysates were analyzed using anti‐ULK1, anti‐Flag, anti‐Myc, or anti‐actin.
-
JUbiquitination analysis of HA‐ULK1 in HEK293T cells co‐expressing Flag‐TRIM27 and His‐Ub, or together with Myc‐STK38L.
-
KWestern blot analysis of ULK1 levels in HeLa cells with or without STK38L knockout, the cells were treated with starvation for the indicated time.
-
LWestern blot analysis of p62 or LC3 levels in HeLa cells with or without STK38L knockout, the cells were treated with starvation for the indicated time. Bafilomycin A1 (BAF A1) was used to inhibit the fusion between autophagosomes and lysosomes.
Source data are available online for this figure.
We further considered the mechanisms responsible for driving interactions between TRIM27 and ULK1 in response to starvation. Mindful of the important role of regulation by posttranslational modifications, mass spectrographic analysis of TRIM27‐interacting proteins identified the protein kinase STK38L among the top ten interacting proteins (Fig 4D and Appendix Table S5). Within the top 50 candidates, we also detected JAK1 kinase, previously shown to bind to TRIM27 (Zhang et al, 2018). Notably, knockdown of STK38L, but not JAK1 significantly inhibited the interaction between TRIM27 and ULK1 under starvation conditions (Figs 4E and Appendix Fig S4B–E). We therefore focused our investigations on understanding how the STK38L‐TRIM27 interaction might impinge on the regulation of ULK1.
We confirmed co‐immunoprecipitation between endogenous TRIM27 and STK38L (Fig 4F) and moreover, demonstrated that ectopic Flag‐STK38L co‐precipitated both TRIM27 and ULK1 with their recovery levels increasing after starvation (Fig 4G). Additionally, STK38L depletion markedly diminished the levels of interaction between TRIM27 and ULK1 (Figs 4H and Appendix Fig S4F and G). Intriguingly, STK38L overexpression decreases endogenous ULK1 levels to some extent, recapitulating the effects of TRIM27 overexpression whereas co‐expression of STK38L and TRIM27 markedly downregulated ULK1 levels (Fig 4I). These observations were consistent with STK38L promoting ULK1 ubiquitination, and indeed, the addition of STK38L increased the levels of TRIM27‐mediated ULK1 ubiquitination (Fig 4J). Conversely, depletion of STK38L greatly delayed the starvation‐induced degradation of ULK1 (Fig 4K). Together these data support the notion that STK38L mediates interactions between TRIM27 and ULK1, leading to the ubiquitination and degradation of ULK1 under starvation conditions.
Lastly, examination of autophagy in control and STK38L KO cells showed that the absence of STK38L promoted increases in LC3 conversion in Bafilomycin A1‐treated cells under starvation (Fig 4L), indicating that STK38L exerts an inhibitory effect on autophagy flux. Taken together, these data propose that STK38L functions as a mediator of the TRIM27‐ULK1 interaction, acting to constrain autophagic flux by controlling the levels of ULK1. Nevertheless, the precise mechanisms involved required further investigation.
STK38L mediates phosphorylation of ULK1 at Serine 495
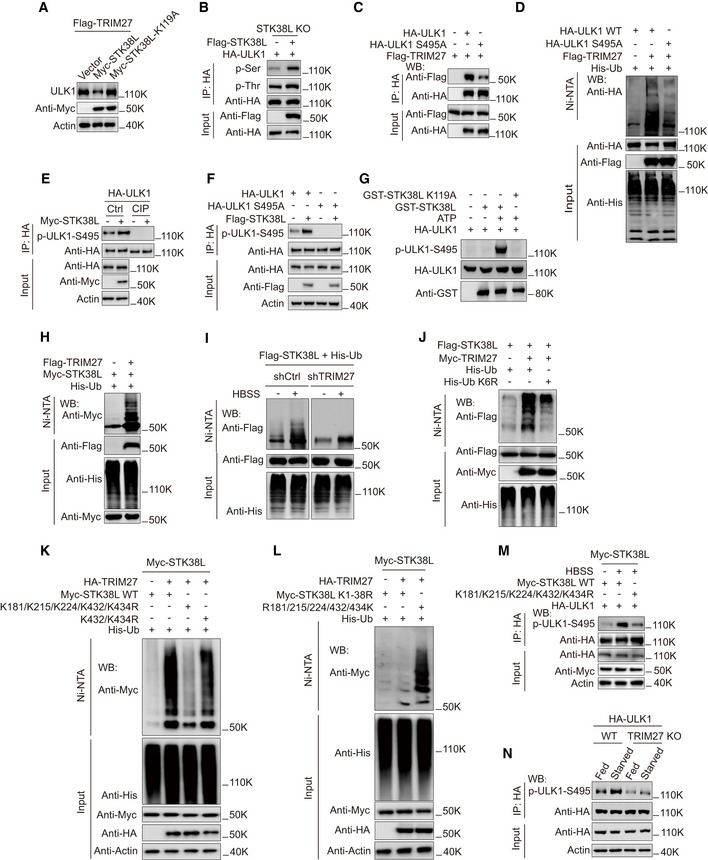
Since STK38L is fundamentally a serine/threonine kinase, we tested whether its kinase activity was required to regulate ULK1 stability. Notably, use of the kinase‐dead K119A STK38L mutant (Stegert et al, 2005) failed to promote ULK1 degradation during starvation (Figs 5A and EV3A), indicating that STK38L‐mediated ULK1 degradation depends on kinase activity. Nevertheless, whether STK38L directly phosphorylated ULK1 or alternatively regulated TRIM27 by phosphorylation needed to be clarified. This point was resolved by measuring total serine/threonine phosphorylation levels where the ectopic expression of STK38L promoted phosphorylation increases in ULK1 (Fig 5B) but not TRIM27 (Fig EV3B). Moreover, the phosphorylation of ULK1 by STK38L could be readily reproduced using in vitro kinase assays (Fig EV3C). Thus, ULK1 is directly phosphorylated by STK38L and we next sought to identify the phosphorylation site(s) involved.
Figure 5. TRIM27‐mediated ubiquitination activates STK38L to phosphorylate ULK1.
-
AWestern blot analysis of ULK1 levels in HeLa cells stably expressing Flag‐TRIM27, the cells were transfected with either Myc‐vector, Myc‐STK38L, or Myc‐STK38L‐K119A. Then the cells were treated with starvation before lysis.
-
BPhosphorylation of HA‐ULK1 in HeLa STK38L knockout cells with or without expression of Flag‐STK38L. HA‐ULK1 was immunoprecipitated and then analyzed by Western blot using anti‐phosphoserine or anti‐phosphothreonine, and the cell lysates were analyzed using anti‐HA and anti‐Myc.
-
CCo‐immunoprecipitation of Flag‐TRIM27 with HA‐ULK1 or HA‐ULK1‐S495A in HEK293T cells. HA‐ULK1 or HA‐ULK1 S495A was immunoprecipitated using anti‐HA, the immunoprecipitates and lysates were analyzed with anti‐HA or anti‐Flag.
-
DUbiquitination analysis of HA‐ULK1 WT or HA‐ULK1 S495A in HEK293T cells co‐expressing Flag‐TRIM27 and His‐Ub.
-
EPhosphorylation of HA‐ULK1 in HEK293T cells co‐expressing Myc‐STK38L with or without CIP treatment. HA‐ULK1 was immunoprecipitated with anti‐HA and analyzed by Western blot using a specific antibody against p‐ULK1‐S495 (Ser495).
-
FPhosphorylation of HA‐ULK1 or HA‐ULK1 S495A in HEK293T cells co‐expressing Myc‐STK38L. HA‐ULK1 or HA‐ULK1 S495A was immunoprecipitated with anti‐HA and analyzed by Western blot using a specific antibody against p‐ULK1‐S495 (Ser495).
-
GIn vitro kinase assay using GST‐STK38L, or GST‐STK38L K119A (a kinase‐inactive mutant) and HA‐ULK1 as substrate. GST‐STK38L or GST‐STK38L K119A was pulled down using glutathione Sepharose beads. HA‐ULK1 was immunoprecipitated from HEK293T using anti‐HA and treated with lambda phosphatase, the reaction product was analyzed by Western blot using p‐ULK1‐S495 (Ser495).
-
HUbiquitination analysis of Myc‐STK38L in HEK293T cells co‐expressing Flag‐TRIM27 and His‐Ub.
-
IUbiquitination analysis of Flag‐STK38L in HEK293T cells co‐expressing His‐Ub with or without TRIM27 knockdown, cells were cultured in fed or starved conditions as indicated.
-
JUbiquitination analysis of Flag‐STK38L in HEK293T cells co‐expressing Myc‐TRIM27 with either His‐Ub (wild‐type) or His‐Ub K6R.
-
KUbiquitination analysis of Myc‐STK38L WT, Myc‐STK38L‐K432R+K434R or Myc‐STK38L‐K181R+K215R+K224R+K432R+K434R (Myc‐STK38L‐5KR) mutant in HEK293T cells co‐expressing HA‐TRIM27 and His‐Ub.
-
LUbiquitination analysis of Myc‐STK38L K1‐38R mutant (All lysines were mutated to arginines), or Myc‐STK38L‐R181/215/224/432/434K (All lysines were mutated to arginines except the indicated five lysine residues) mutant in HEK293T cells co‐expressing HA‐TRIM27 and His‐Ub.
-
MPhosphorylation of HA‐ULK1 S495(Ser495) in HEK293T cells co‐expressing Myc‐STK38L WT, or Myc‐STK38L‐5KR mutant in K. The cells were cultured in fed or starved conditions as indicated and HA‐ULK1 was immunoprecipitated using anti‐HA, the immunoprecipitates and lysates were analyzed with a specific antibody against p‐ULK1‐S495 (Ser495), anti‐Myc, anti‐HA, or anti‐actin.
-
NPhosphorylation of HA‐ULK1 in HeLa cells with or without TRIM27‐knockout, cells were cultured in fed or starved conditions as indicated, HA‐ULK1 was immunoprecipitated using anti‐HA, the immunoprecipitates and lysates were analyzed with a specific antibody against p‐ULK1‐S495 (Ser495), anti‐HA, or anti‐actin.
Source data are available online for this figure.
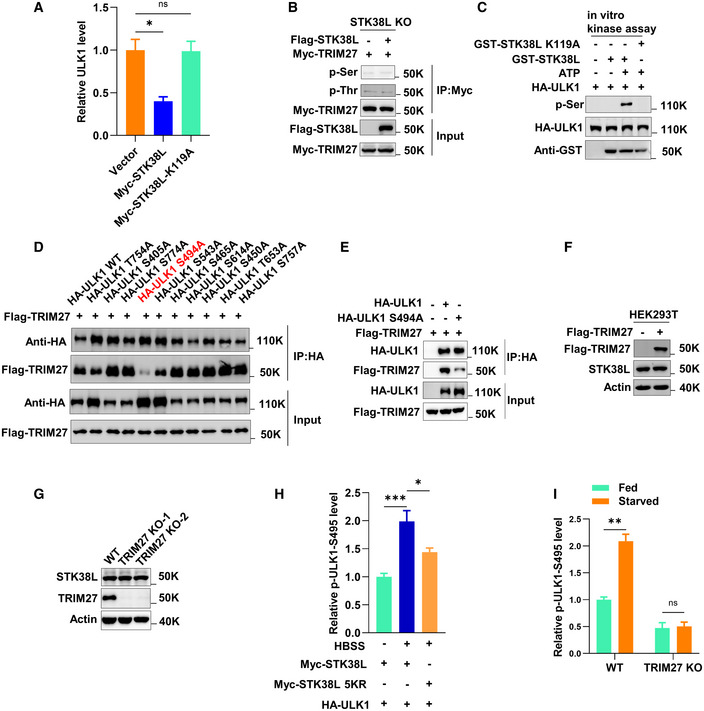
Figure EV3. The phosphorylation modification of ULK1 S494 (mouse) mediated by STK38L promotes the binding of TRIM27 to ULK1.
-
AQuantification of data presented in Fig 5A shows mean ± SEM of three independent experiments. P‐values were determined by a one‐way ANOVA with Dunnett’s multiple comparisons test, *P < 0.05, ns, not significant.
-
BPhosphorylation of Myc‐TRIM27 in HeLa STK38L knockout cells with or without expression of Flag‐STK38L. Myc‐TRIM27 was immunoprecipitated and then analyzed by Western blot using anti‐phospho‐serine or –threonine antibodies. The presence of Myc‐TRIM27 and Flag‐STK38L were revealed using anti‐Myc and anti‐Flag antibodies as indicated.
-
CIn vitro kinase assay using GST‐STK38L, or GST‐STK38L‐K119A (a kinase‐inactive mutant) and HA‐ULK1 as substrate. GST‐STK38L or GST‐STK38L‐K119A was pulled down using glutathione Sepharose beads. Recombinant HA‐ULK1 was immunopurified from HEK293T using anti‐HA and treated with lambda phosphatase. Reaction products were analyzed by Western blot using anti‐phospho‐serine along with anti‐HA and anti‐GST to confirm inputs.
-
DCo‐immunoprecipitation between Flag‐TRIM27 and the indicated HA‐ULK1 mutants from co‐transfected HEK293T cells.
-
ECo‐immunoprecipitation between Flag‐TRIM27 and HA‐ULK1 or HA‐ULK1 S494A in HEK293T cells.
-
FWestern blot measuring STK38L levels in HEK293T with or without over‐expression of Flag‐TRIM27.
-
GWestern blot measuring STK38L levels in HeLa with or without knockout of TRIM27.
-
HQuantification of data presented in Fig 5M shows mean ± SEM of three independent experiments. P‐values were determined by a one‐way ANOVA with Dunnett’s multiple comparisons test, *P < 0.05, ***P < 0.001.
-
IQuantification of data presented in Fig 5N shows mean ± SEM of three independent experiments. P‐values were determined by an unpaired t‐test with Holm‐Sidak’s multiple comparisons test, **P < 0.01, ns, not significant.
Source data are available online for this figure.
As a primary screening strategy, we hypothesized that ablating ULK1 phosphorylation would prevent or reduce the association between ULK1 and TRIM27. Phosphoproteomics analysis identified ten phosphorylated serine and threonine residues in tryptic peptides derived from mouse ULK1 (Appendix Fig S5A and B). Based on this result we prepared individual serine/threonine mutant HA‐ULK1 constructs and conducted co‐immunoprecipitation experiments in HEK293T cells transfected with Flag‐TRIM27. Most serine/threonine ULK1 mutants recovered similar amounts of Flag‐TRIM27 with the exception of the S494A mutant (Fig EV3D and E). Notably, the same residue is conserved in mouse/human ULK1 and similarly, mutation of the equivalent Ser495 residue reduced the association between human ULK1 and TRIM27 (Fig 5C). Highlighting the importance of Ser495, the S495A ULK1 mutant was considerably less ubiquitinated than WT ULK1 when HEK293T cells were co‐transfected with Flag‐TRIM27 (Fig 5D).
To enable further investigation, we developed affinity‐purified antibodies against phosphorylated Ser495 (p‐ULK1‐S495). Notably, p‐ULK1‐S495 reactivity signals showed robust increases when co‐expressed with WT STK38L but all signals were abolished upon phosphatase treatment (Fig 5E). Moreover, no p‐ULK1‐S495 reactivity was seen against the S495A HA‐ULK1 mutant (Fig 5F). Lastly, to verify that STK38L was directly responsible for ULK1 Ser495 phosphorylation, we performed in vitro kinase assays using recombinant forms of GST‐STK38L and HA‐ULK1. As expected, p‐ULK‐S495 antibodies detected strong phosphorylation signals in ULK1 when co‐incubated with GST‐STK38L but not in the presence of the K119A mutant GST‐STK38L (Fig 5G). Together these data indicated that Ser495 is a bona fide phosphorylation site in ULK1 catalyzed by STK38L.
Cooperative regulation between TRIM27 and STK38L is required to mediate ULK1 degradation in response to starvation
The preceding experiments established that ULK1 but not TRIM27 was a substrate of STK38L kinase activity but nevertheless, their physical interaction raised the alternate possibility that STK38L was a substrate of TRIM27. Indeed, we observed that cell starvation conditions induced TRIM27‐dependent ubiquitination of STK38L (Fig 5H and 5I). Notably, STK38L protein levels remained stable during starvation and after TRIM27 ectopic expression or knockout (Figs 4K and EV3F and G), suggesting that STK38L was modified by a non‐degradative type of ubiquitination. Resolving this point was significant given that ubiquitination can regulate kinase activity, for example as has been demonstrated for Akt (Hunter, 2007; Yang et al, 2010).
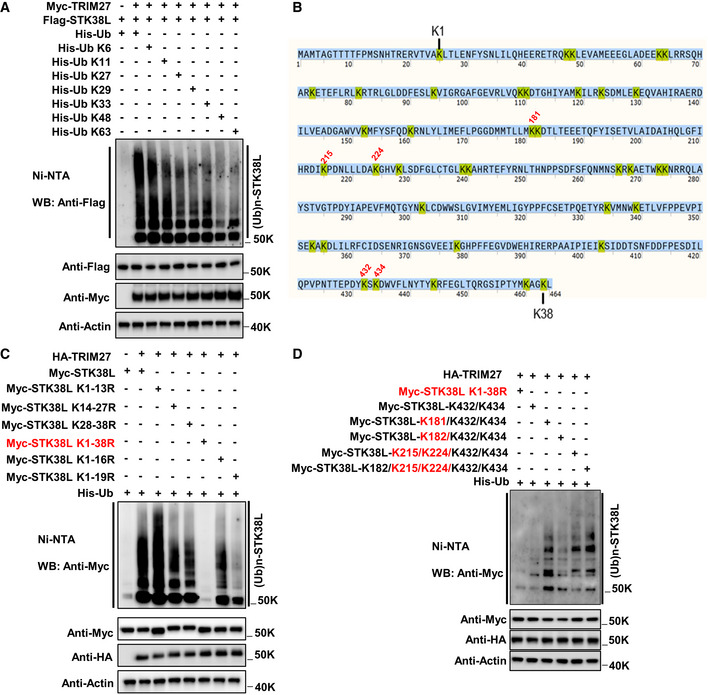
To test this notion, ubiquitination assays were performed in HEK293T cells co‐expressing Myc‐TRIM27 and Flag‐STK38L. Assays of cells transfected with His‐tagged ubiquitin mutants (K6, K11, K27, K29, K33, K48 and K63) where all but one lysine had not been mutated showed that TRIM27‐mediated ubiquitination of STK38L was not K48‐or K63‐linked but rather predominately involved K6‐linkage and to a lesser extent, K11 (Fig EV4A). Follow‐up experiments confirmed that substituting WT His‐ubiquitin for the K6R mutant greatly diminished the total polyubiquitination signals in STK38L (Fig 5J).
Figure EV4. Identification of TRIM27‐mediated ubiquitination sites in STK38L.
-
AUbiquitination analysis of Flag‐STK38L in HEK293T cells co‐expressing Myc‐TRIM27 together with either His‐Ub WT, or the His‐Ub mutants as indicated, respectively. Total cell lysates were subject to ubiquitination analysis involving recovery of His‐labelled proteins by Ni‐NTA followed by Western blot against Flag (top panel). Transfection and loading efficiency were determined by Western blot of input samples (bottom panels).
-
BThe amino acid sequence of STK38L contains a total of 38 lysine residues (yellow) ordered from the N‐ to C‐terminus. Five numbered lysine residues in red (K181, K215, K224, K432 and K434) relate to subsequent findings in panels (C) and (D).
-
CIdentification of STK38L ubiquitination sites. Ubiquitination analysis of Myc‐STK38L or the indicated K to R substitution mutants in HEK293T cells co‐expressing HA‐TRIM27 and His‐Ub before analysis of STK38L ubiquitination. Myc‐STK38L K1‐38R represents the Myc‐STK38L mutant with all lysine (K) residues mutated to arginine (R). K1‐K13R represents the mutant that the 1st to 13th lysine residues of STK38L all mutated to arginine. The other mutant designations were similarly named. As anticipated, mutation of all lysines (K1‐38R) ablated the total ubiquitination signals in the presence of TRIM27 while mutation of lysines 1–19 also showed a large decrease in STK38L ubiquitination. Lesser effects observed with other mutant constructs indicated the ubiquitination sites were primarily located between lysine residues 16–19 and in the C‐terminal region. Some ubiquitination signals for K1‐19R indicate other potential sites exist.
-
DRefinement of the principal C‐terminal ubiquitination sites in STK38L. Myc‐STK38L mutants co‐expressing HA‐TRIM27 and His‐Ub were co‐transfected with individual Myc‐STK38L mutants into HEK293T cells before analysis of STK38L ubiquitination. Myc‐STK38L‐K432/K434 represents the Myc‐STK38L mutant only containing K432 and K434 lysine residues, while all other lysine (K) residues mutated to arginine (R). The other mutants were similarly named. Interpretation of the data indicates the major ubiquitination sites as K181, K215, K224, K432 and K434.
Source data are available online for this figure.
In combination with these findings, we also sought to identify the ubiquitin‐modified sites within STK38L using an empirical screening approach. As anticipated, mutating all 38 lysine residues in STK38L abolished ubiquitination while other mutant constructs exhibited partial loss of ubiquitination, localizing the substrate sites to lysines 16–19 (K181, K182, K215 and K224, respectively) and within lysines 28–38 (Fig EV4B and C). Further iterative interrogation arrived at five residues (K181, K215, K224, K432 and K434) which contributed to varying extents to the TRIM27‐mediated ubiquitination of STK38L (Fig EV4D). In support, the levels of STK38L ubiquitination stimulated by starvation conditions were greatly when all five of the aforementioned lysine residues were substituted for arginine (5KR mutant; Fig 5K), but signals were significantly enhanced in a mutant construct where all but these five lysine residues were mutated to arginine (Fig 5L).
Lastly, we sought to synthesize the functional relationships between TRIM27, STK38L and ULK1. First, we observed that the starvation‐induced increases in ULK1 Ser495 phosphorylation were not observed with the 5KR mutant (Figs 5M and EV3H), indicating that STK38L ubiquitination serves to activate its kinase activity. Consistent with this notion, ULK1 Ser495 phosphorylation changes under starvation conditions required the presence of TRIM27 (Figs 5N and EV3I), highlighting the importance of TRIM27‐mediated activation of STK38L in the phosphorylation of ULK1.
TRIM27 promotes mammary tumorigenesis
The preceding sections established the conserved role of TRIM27 as a negative regulator of autophagy. Nevertheless, it remained important to ascertain the significance of the TRIM27‐ULK1 axis in either physiological or pathological contexts. For this, we noted that TRIM27 was reported to be upregulated in several human cancer types including breast cancer.
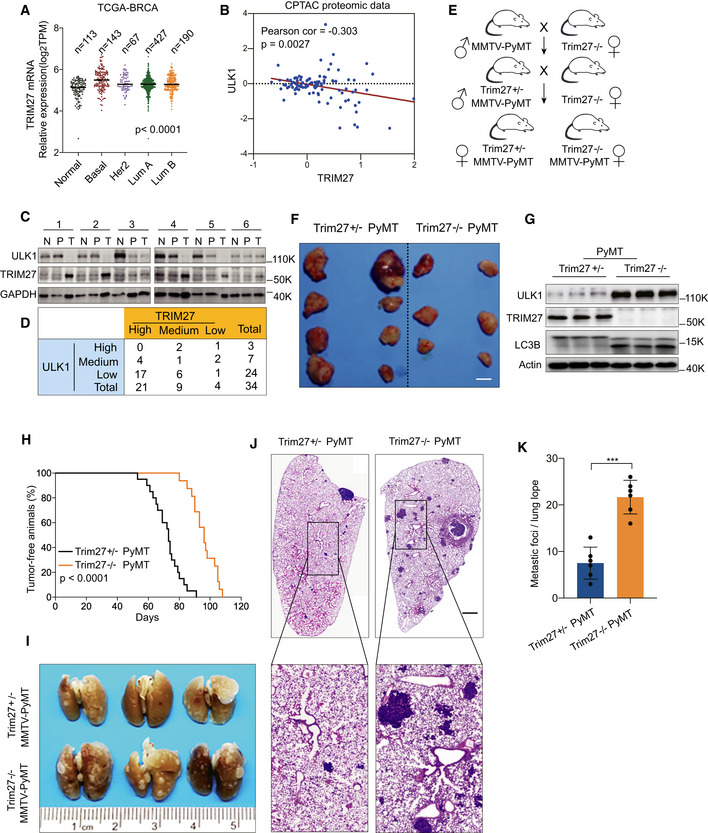
Bioinformatics interrogation of TCGA breast cancer data corroborated previous findings that TRIM27 mRNA was significantly upregulated in breast cancer subtypes compared to normal mammary tissues (Fig 6A). In contrast, ULK1 mRNA levels were not significantly changed in breast tumors compared to normal tissues (Appendix Fig S6A). Previous reports had indicated that ULK1 protein levels were downregulated in breast cancer (Tang et al, 2012), and protein level assessment using CPTAC proteomics data (https://proteomic.datacommons.cancer.gov/pdc/) indicated a negative correlation between TRIM27 and ULK1 protein levels, but not in mRNA levels in breast cancer tissues (Figs 6B and Appendix Fig S6B). We further supplemented these data with analysis of 34 paired human breast cancer and normal adjacent tissues. Strikingly, 21 tumor samples overexpressed TRIM27 when compared to non‐malignant tissues and the majority of these cases also showed low ULK1 levels (17/24 tissues, Fig 6C and D). This inverse relationship is consistent with the mechanism disclosed by our biochemical experiments and suggests that the TRIM27‐ULK1 axis contributes to tumorigenesis in breast cancer.
Figure 6. Knockout of Trim27 inhibits breast tumorigenesis but promotes lung metastasis in PyMT mice.
-
AComparison of the relative mRNA levels of TRIM27 in normal breast tissues (Normal, n = 113) and indicated breast tumors (Basal, n = 143; Her2, n = 67; LumA, n = 427; LumB, n = 190) in the TCGA database. The number of biological replicates are shown as indicated. P < 0.0001 (one‐way ANOVA).
-
BAnalysis of the correlation between ULK1 and TRIM27 expression in the CPTAC proteomics database. P = 0.0027 (Pearson’s correlation method).
-
C, DWestern blot analysis of ULK1 and TRIM27 levels from tumor tissues (T), Pericarcinomatous tissues (P), and their matched surrounding normal tissues (N). GAPDH was used as a loading control (C). A total of 34 samples of human breast cancer were classified into nine groups based on the expression of ULK1 and TRIM27 in tumor tissues compared with surrounding normal tissues (D).
-
ESchematic diagram of Trim27−/− PyMT and Trim27+/− PyMT mice breeding, Trim27+/− PyMT mice as a littermate control.
-
FRepresentative tumors in MMTV‐PyMT mice of the indicated Trim27 genotypes. Scale bar, 5 mm.
-
GWestern blot analysis of indicated proteins in Trim27−/− PyMT or Trim27+/− PyMT mice in primary breast tumors. Actin was used as a loading control.
-
HKaplan–Meier tumor‐free survival curves of Trim27+/− PyMT (median = 75 days, number = 20), or Trim27−/− PyMT mice (median = 95 days, number = 16). P < 0.0001 (Mantel–Cox test).
-
I–KPulmonary surface nodules and hematoxylin and eosin (H&E) staining in Trim27+/− PyMT mice and Trim27−/− PyMT mice (I and J). Scale bar, 1 mm (K). Bar chart showing quantification for the number of metastatic nodules per lung lope (Trim27+/−, n = 6 mice; Trim27−/− n = 6 mice), when compared at the same final timepoint. Each symbol represents a mouse; Data are presented as mean ± SD from three independent experiments; ***P < 0.001 (unpaired student's t‐test).
Source data are available online for this figure.
To test this hypothesis, we employed the transgenic PyMT mouse model that develops spontaneous mammary tumors to test the effects of TRIM27 on tumor initiation and metastasis. We generated MMTV‐PyMT, Trim27−/− mice by crossing Trim27−/− mice with FVB background Trim27+/−, MMTV‐PyMT mice (Fig 6E and Appendix Fig S6C). This experimental design allowed us to compare Trim27−/− and Trim27+/− littermates. Trim27+/− PyMT female mice developed palpable mammary tumors after 8 weeks that were multifocal and involved the entire mammary fat pad. PyMT‐Trim27−/− mice had a significantly decreased tumor burden (Fig 6F) compared to PyMT‐Trim27+/− mice. Indicative of increased basal autophagy, the Trim27−/− tumors displayed dramatically increased ULK1 and LC3‐II levels compared to the Trim27+/− tumors (Fig 6G). Moreover, mammary tumor latency was increased in Trim27−/− PyMT littermates (Fig 6H). In conjunction with these experiments, we also examined pulmonary metastasis in the PyMT mice when mice were analyzed 3 weeks after tumor appearance. In contrast, we observed that the number of metastatic foci and overall lung tumor burden in the Trim27−/− PyMT mice were dramatically increased (Fig 6I–K).
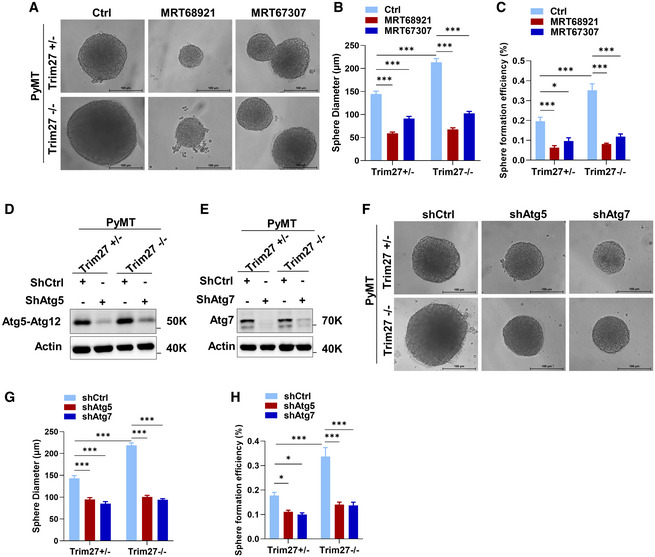
To further investigate whether these phenotypes were manifested through Trim27‐mediated effects on autophagy or through other processes, we examined the effects of TRIM27 knockout and autophagy inhibition on the growth of PyMT‐derived tumor cells in 3D organoid culture. We found that organoid growth, as judged by organoid size and clonogenicity, was relatively increased in Trim27−/− mammary tumor cells compared to Trim27+/− cells. Notably, this growth advantage was nullified under small molecule inhibition of Ulk1 with MRT68921 or MRT67307 (Figs EV5A–C and Appendix Fig S7). Similarly, this growth advantage was reversed by knockdown of the autophagy components Atg5 and Atg7, rendering the growth and clonogenicity of both Trim27+/− and Trim27−/− cells to the same basal growth state (Fig EV5D–H). Together this provides strong evidence that growth differences between Trim27−/− and Trim27+/− cells result from inherently different rates of autophagy flux. Further in vivo assessment of experimental lung metastasis using 4T1 cells showed that inhibition of Ulk1 with MRT68921 significantly inhibited lung colonization (Appendix Fig S8A–D), supporting the notion that increases in autophagy through TRIM27 knockout could contribute to increased metastasis. Together this proposes that TRIM27 promotes mammary luminal tumorigenesis but inhibits systemic metastasis at least partially through autophagy.
Figure EV5. Organoid growth promotion in TRIM27 knockout tumors and metastasis in vivo is dependent on autophagy.
-
ARepresentative morphology of Trim27+/−, Trim27−/− PyMT organoids in 3D sphere formation assays treated with vehicle (DMSO), 100 nM MRT68921 or MRT67307. Scale bar, 100 μm.
-
B, CQuantification of sphere diameter (B) (n = 24) and sphere formation efficiency (diameter ≥ 50 μm) (C) in the indicated groups from three independent pairs of tumors. Data are mean ± SEM; P‐values were determined by a two‐way ANOVA with Tukey’s multiple comparisons test, *P < 0.05, ***P < 0.001.
-
D, EVerification of Atg5 (D) and Atg7 (E) knockdown in PyMT Trim27+/− and Trim27−/− tumor cells by Western blot.
-
FRepresentative morphology of Trim27+/−, Trim27−/− PyMT organoids in 3D sphere formation assays transfected with control or Atg5 or Atg7 knockdown vectors. Scale bar, 100 μm.
-
G, HQuantification of sphere diameter (G) (n = 24) and sphere formation efficiency (diameter ≥ 50 μm) (H) in the indicated groups from three independent pairs of tumors. Data are mean ± SEM; P‐values were determined by a two‐way ANOVA with Tukey’s multiple comparisons test, *P < 0.05, ***P < 0.001.
Source data are available online for this figure.
Discussion
Autophagy must be tightly regulated since imbalances in autophagic flux can lead to cytotoxicity, not to mention the development of various pathologies. Here we reveal the contribution of the E3 ligase TRIM27 in autophagy regulation, both to maintain basal autophagy as well as restrain excessive autophagy under harsh starvation conditions. The latter requires cooperation between TRIM27 and the serine/threonine kinase STK38L whereby TRIM27 ubiquitinates and activates STK38L, allowing STK38L to phosphorylate Ser495 in ULK1, delivering ULK1 in a permissive state for hyper‐ubiquitination by TRIM27. Thus, our study establishes TRIM27 as a novel negative regulator of autophagy, joining KLHL20 and Nedd4L as an elite group of E3 ubiquitin ligases that regulate ULK1 (Liu et al, 2016; Nazio et al, 2016). However, unlike KLHL20 and NEDD4L, depletion of which exhibits neonatal (Woik et al, 2014) or perinatal lethality in mice (Boase et al, 2011), we found that TRIM27 knockout mice are developmentally normal, with no obvious phenotypes after 18 months. As a member of a large gene family with several closely related genes (Hatakeyama, 2017) this may reflect some redundancy among the Trim gene family.
The essential partner of TRIM27 in this mechanism is STK38L, also known as NDR2. STK38L, together with its homolog STK38 (NDR1) and the LATS1/2 kinases, belongs to a small gene family generally considered to participate in cell growth and division (Hergovich, 2016). Mouse knockout models have suggested that NDR1 and NDR2 have redundant functions during embryogenesis (Schmitz‐Rohmer et al, 2015). However, while STK38 was reported to positively regulate autophagy by binding to Beclin1 (Joffre et al, 2015), our study proposes that STK38L acts to terminate autophagy. The distinct subcellular localization of STK38 in the nucleus versus the cytoplasmic location of STK38L was reported to account for their different functions (Abe et al, 2017) but conversely human STK38 has been shown to be primarily cytoplasmic (Hergovich et al, 2005), highlighting the need to consider cellular context. Whether STK38 and STK38L act exclusively as activator and inhibitor of autophagy, respectively, also needs further analysis.
Dysfunctional autophagy has been closely associated with the development of many human diseases including cancers (Mizushima & Levine, 2020). Under normal physiology, autophagy exerts tumor suppressor functions (White, 2015; Singh et al, 2018), whereas in the context of malignancy, autophagy has been highlighted as a therapeutic target (Amaravadi et al, 2016, 2019; Levy et al, 2017). While no relationship was observed between TRIM27 and ULK1 mRNA levels in human breast cancers, high TRIM27 protein levels were clearly associated with decreased ULK1 expression. These observations generally suggest that the post‐translational regulation of ULK1 by TRIM27 is active in many breast tumors and we noted a general trend that high TRIM27‐expressing tumors had accelerated disease progression. Notably, the involvement of TRIM‐family proteins in cancer has long been scrutinized (Hatakeyama, 2011) with TRIM27 being no exception (Zoumpoulidou et al, 2012). Along with breast cancer, TRIM27 overexpression has been reported in numerous carcinoma types (Zoumpoulidou et al, 2012) and pro‐tumorigenic roles have been elucidated in Trim27‐deficient mouse models including chemically‐induced skin cancer (Zoumpoulidou et al, 2012) and colitis (inflammation‐associated) intestinal cancer (Zhang et al, 2018). Building on the consensus of these investigations, we found that TRIM27 promotes murine mammary tumorigenesis. Nevertheless, these studies raise an important point to consider, specifically what mechanism(s) can explain the contribution of TRIM27 to tumorigenesis.
Most of the evidence linking TRIM27 to cancer is associative and few studies have revealed in‐depth mechanisms. Nevertheless, the aforementioned colitis study showing that TRIM27 binds JAK1 to promote JAK1–STAT3 complex formation and activation (Zhang et al, 2018) provides some intriguing parallels with our work. While TRIM27 has been reported to function within the cell nucleus (Kato et al, 2014), the TRIM27‐JAK1–STAT3 association occurred within retromer‐containing endosomes which are essential cargo transporters involved in vesicular trafficking (Zhang et al, 2018). Notably, the retromer has been connected with early endocytosis and autophagy (Cui et al, 2019), although whether the JAK1–STAT3 retromer‐containing endosomes were related to autophagy was not defined. We also recovered JAK1 in our proteomics screen along with STK38L, although JAK1 was not required to support the TRIM27‐ULK1 interaction. This proposes that TRIM27 exerts functional control over different kinases, both positively and negatively, in the context of endosomal compartments. Conceivably, specificity may be achieved through the proteins existing in different cellular compartments but additionally, heterodimerization among TRIM family members has also been proposed to contribute to their specificity (Watanabe & Hatakeyama, 2017). It seems likely that both mechanisms are important.
We have previously shown that autophagy blockade inhibited mouse mammary tumorigenesis in MMTV‐PyMT and MMTV‐Neu mouse models using conditional deletion of FIP200 or conditional knock‐in of an autophagy‐deficient FIP200 mutant (Wei et al, 2011; Chen et al, 2016; Hao et al, 2021). Nevertheless, TRIM27 knockout accompanied by elevated autophagy was associated with increased tumor latency and decreased primary lesion mass. At the same time, we made the intriguing observation that the rates of spontaneous lung metastasis were increased in Trim27−/− mice. Given the multiple targets of TRIM27, it is possible that the decreased mammary tumorigenesis could be caused by the reduced signaling of other TRIM27 targets as observed previously (Zhang et al, 2018). Certainly, 3D experiments undertaken with PyMT cells showed that the in vivo latency of tumors did not inherently result from growth deficiency in the Trim27−/− cells compared to their Trim27+/− counterparts. Rather, this supports the notion that Trim27 acts to provide a permissive state for the transformation event, although further studies will be necessary to investigate this intriguing hypothesis. Nonetheless, our in vivo lung metastasis model showing that Ulk1 inhibition decreased metastasis provides good evidence linking the role of autophagy and the effects of Trim27 in inhibiting metastasis. Indeed, the notion that increased autophagy may facilitate cancer metastasis is well established in different model systems (Mowers et al, 2017). It is also important to note that TRIM27 was deleted in all body tissues and it is therefore possible that changes in autophagy in non‐cancer cells may influence the model findings. Regardless, similar with other studies, our findings suggest that ULK1 protein levels are decreased in breast cancer (Tang et al, 2012), providing general support that autophagy could be downregulated in bulk tumor populations.
Lastly, implications outside of cancer must be considered. Constitutively increased basal autophagy contributes to a longer life span in a beclin1 mutant knock‐in mice (Fernandez et al, 2018) whereas low levels of basal autophagy can promote neurodegenerative diseases, proposing autophagy modulation as a therapeutic strategy in a range of neurodegenerative conditions. A previous study showed that TRIM27 levels were significantly upregulated in brain tissues of Parkinson's disease patients. Notably, depletion of TRIM27 significantly alleviated the severity of Parkinson's symptoms in a mouse model by inhibiting NF‐κB signaling (Liu et al, 2014). Whether TRIM27 is involved in Parkinson's disease pathogenesis via effects on autophagy remains to be determined but it nonetheless represents a promising therapeutic target for controlling neurodegeneration.
Materials and Methods
Antibodies
All antibodies used in this study are listed in Appendix Table S2.
Animal experiments
Trim27−/− mice on the C57BL/6 background were generated by Cyagen Bioscience Incorporation using the CRISPR/Cas9 method with genotypes routinely confirmed by PCR. Trim27−/− mice were crossed with GFP‐LC3–transgenic mice to generate GFP‐LC3, Trim27−/− mice. For the mouse mammary tumor model, Trim27−/− mice were crossed with MMTV‐PyMT mice as previously described (Okamoto et al, 2020). The resulting mice were used for the experiments after backcrossing from the C57BL/6 to the FVB background for more than six generations. All animal experiments were carried out in accordance with the protocols of IACUC of the University of Science and Technology of China (PXHG‐WM2018112917).
Tissue samples
Thirty‐four pairs of breast cancer and adjacent normal tissues were collected from the patients attending the Fourth Affiliated Hospital of China Medical University, China. None of the patients received prior chemotherapy or radiotherapy. Samples were immediately stored in RNAlater until subsequent analysis with all tissues verified by histopathological confirmation in the pathology department. Sample collection was approved by the Research Ethics Committee of Fourth Affiliated Hospital of China Medical University with written informed consent obtained for all patients (Approval EC‐2021‐KS‐041). The experiments involving human participants conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Cell culture and treatments
Wild‐type mouse embryonic fibroblasts (MEFs), FIP200 knockout MEFs (FIP200−/−), ULK1/2 knockout MEFs, HEK293T, and HeLa cells were cultured in DMEM supplemented with 10% FBS, l‐Glutamine (2 mM), penicillin (10 Units/ml) and streptomycin (0.1 mg/ml). Amino acid starvation was conducted by incubating the cells with Hank’s Balanced salt solution containing calcium and magnesium (Thermo Fisher Scientific). Primary tumor cells were obtained 3 weeks after the appearance of palpable lesions in the Trim27+/− and Trim27−/−, MMTV‐PyMT mice. Isolated lesions were mechanically dissociated into small pieces followed by incubation with 1 mg/ml collagenase (Servicebio), DNase (10 mg/ml) and gentamycin (20 ng/ml) at 37°C for 1 h. Thereafter, cell suspensions were cultured in DMEM/F12 (Gibco) supplemented with 10% FBS, 20 ng/ml EGF, 10 µg/ml insulin, and 1% penicillin/streptomycin.
Organotypic spheroid cultures
Organotypic tumor spheroids were established from monolayer cultures of PyMT‐derived tumor lesions. After trypsin dissociation, 3,000 cells/well were seeded into 96‐well Ultra‐Low Attachment Plates (Corning) and cultured in sphere media (DMEM/F12 with B27 (1×, Gibco), 20 ng/ml EGF, 10 ng/ml bFGF, 5 µg/ml insulin, 0.4% BSA and 1% penicillin/streptomycin). The indicated treatments were initiated at the time of plating. Tumor spheroids of diameter > 50 µm were counted after 2–3 weeks of culture.
Cell transfection and gene knockout
Stable expression of full‐length or mutant forms of ULK1, STK38L, and TRIM27 was achieved using lentiviral mediated transduction. The indicated epitope‐tagged constructs were cloned into the Lenti‐EFS‐Flag‐puro vector and transfected together with psPAX2 and pmd2.g into HEK293T to derive lentiviral particles as previously described (Zhang et al, 2021). Filtered (0.45 mm) lentiviral supernatants were added to target cells and puromycin‐resistant populations were used for the experiments. For the generation of ULK1, TRIM27, and STK38L knockout cells, gRNAs were designed using the online CRISPR design tool (http://crispr.mit.edu) and individual sgRNA sequences (Appendix Table S4) were cloned into LentiCRISPR v2 vector (Addgene Plasmid 52961). Transfections were performed using Lipofectamine 2000 (Thermo Fisher) and puromycin‐selected cells were sorted into 96‐well plates for further selection. Gene overexpression and knockout were validated by Western blot. All vectors used in this study are listed in Appendix Table S3.
Real‐Time quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted using TRIzol according to the manufacturer’s instructions before cDNA synthesis using the PrimeScriptTM RT reagent kit (TaKaRa) containing gDNA eraser (DNase). The qPCR reactions were performed using the specified primers (Appendix Table S4) with the One‐Step PrimeScript RT‐PCR kit (TaKaRa). The relative expression was determined using the ΔΔCt method with normalization against β‐actin.
Western blotting
Whole cell extracts were prepared in lysis buffer containing 2% SDS, 62.5 mM Tris pH 6.8, 10% glycerol, and 5% β‐mercaptoethanol. Protein samples were heated at 95°C for 10 min and analyzed by SDS‐PAGE on 8% or 12% gels (45 min, 200 V) (BioRad Laboratories), followed by immunoblotting on nitrocellulose membranes using a wet blotting system (90 min, 250 mA) (BioRad Laboratories). Membranes were incubated with primary and secondary antibodies diluted in 5% milk/TBST solutions, and immunocomplexes were developed using SuperSignal™ West Pico PLUS (Thermo fisher) and visualized using an Azure c600 Western blot imager (Azure Biosystems).
Protein identification using mass spectrometry
The indicated cells were lysed with IP buffer containing 20 mM Tris‐HCl pH 7.4, 137 mM NaCl, 10% (v/v) glycerol, 1% (v/v) NP‐40, supplemented with a protease and phosphatase inhibitor cocktail (Thermofisher). After clarification by centrifugation at 13,000 rpm for 15 min at 4°C, the supernatants were either incubated with control immunoglobulin G (IgG) or primary antibodies pre‐conjugated to protein G Dynabead (10004D, Thermo Fisher). After mixing end‐over‐end for 4 h at 4°C, the beads were collected using a DynaMag magnet and washed five times with IP buffer. The samples were eluted by heating in LDS sample buffer and subjected to SDS–PAGE gel followed by SYPRO Ruby Gel Stain (S‐12000, Thermofisher). Thereafter, gel bands were excised, destained, trypsinized, and subjected to MS analysis. Peptides were ionized with the Nano spray Flex Ion Source (Thermo) and introduced into a Q Exactive mass spectrometer (Thermo). Abundant species were fragmented in high‐energy collision‐induced dissociation (HCD) mode. Data analysis was performed using Proteome Discoverer 1.3 (Thermo) which incorporated the Mascot (Matrix Science) algorithm. The Swissprot protein database was used against mouse sequences. The MS experiment and data processing were performed at the Wayne State University proteomic core facilities.
Ubiquitination assays
HEK293T cells were cotransfected with His‐ubiquitin, Flag‐TRIM27, and substrate constructs before harvesting whole cell lysates after 48 h using 8 M urea lysis buffer at room temperature. The samples were then sonicated before performing pull‐down assays using Pierce Ni‐NTA magnetic agarose beads (Thermo Fisher) according to the manufacturers’ manual. Eluted proteins were separated by SDS–PAGE and further analyzed by Western blotting. Alternatively, for in vitro ubiquitination assays, 100 ng of recombinant GST or GST‐TRIM27 protein was mixed with 500 ng purified HA‐ULK1, 150 nM UBE1 (E1 enzyme), 500 nM UBE3D3 (E2 enzyme), 5 mM ATP, and 150 μΜ ubiquitin. The reactions were incubated at 37°C for 2 h in reaction buffer (Tris‐HCl pH 7.5, 5 mM MgCl2, 50 μM ZnCl2, and 1 mM DTT). The reactions were terminated by boiling in SDS sample loading buffer and analyzed by Western blotting.
In vitro kinase assays
HA‐ULK1 proteins were immunoprecipitated using anti‐HA antibodies coupled to magnetic beads (Thermo Fisher, 88836) from the indicated cells using mild lysis buffer (10 mM Tris‐HCl at pH 7.5, 2 mM EDTA, 100 mM NaCl, 1% NP‐40, 50 mM NaF, supplemented with protease inhibitor cocktail). The bead‐bound proteins were treated with lambda phosphatase, washed extensively in RIPA buffer followed by mild lysis buffer and then equilibrated in kinase buffer (20 mM HEPES at pH 7.4, 1 mM EGTA, 0.4 mM EDTA and 5 mM MgCl2, supplemented with 0.05 mM dithiothreitol). The kinase reactions (30 μl) were initiated by the addition of 10 μM ATP and incubated at 37°C for 1 h. Thereafter, reactions were quenched by the addition of 5×SDS sample buffer followed by heating at 95°C for 5 min before analysis by Western blotting.
Epifluorescence and confocal microscopy
Cells were seeded on glass coverslips and allowed to attach before conducting the indicated treatments. For immunofluorescence staining, the cells were fixed with cold methanol for 10 min and blocked with 1% BSA for 1 h at room temperature. Thereafter, cells were incubated with primary antibodies overnight at 4°C, followed by incubation with the appropriate Alexa fluor‐labeled secondary antibodies (Life Technologies) for 1 h at room temperature. Otherwise, the cells were fixed in 4% formaldehyde solution for imaging fluorescent reporter proteins. All samples were mounted using ProLong Diamond Antifade mountant with DAPI (Cat#P36971, Life Technologies) and visualized using epifluorescence or confocal microscopy (Nikon) as indicated.
GST protein expression and purification
GST‐tagged TRIM27, STK38L, and STK38L‐K119A mutants were expressed in Escherichia coli BL21 by induction with 100 mM Isopropyl β‐D‐1‐thiogalactopyranoside for 12 h at 25°C and affinity purified using glutathione Sepharose beads (Beyotime). After elution using glutathione, the purified GST proteins were dialyzed against 20 mM Tris–HCl, pH 8.0 and 10% glycerol at 4°C overnight.
Statistical analysis
Statistical significance was determined using GraphPad Prism 6, using unpaired Student's two‐tailed t‐test, one‐way ANOVA or two‐way ANOVA according to test requirements. Statistical analyses between 2 groups were pre‐assessed for normal distribution using Shapiro‐Wilk test and the variance similarity between the 2 groups were statistically compared using F‐test. No statistical methods were used to determine the sample sizes. Unless expressly noted, experiments were not randomized, and the investigators were not blinded to group allocations and the assessment of outcomes. Data are presented as the mean ± SEM or as mean ± SD (error bars). *P < 0.05; **P < 0.01; and ***P < 0.001 denote statistically significant changes.
Author contributions
Mian Wu: Conceptualization; Resources; Formal analysis; Supervision; Funding acquisition; Visualization; Project administration. Yi Yang: Data curation; Formal analysis; Investigation; Methodology; Writing—original draft. Yifu Zhu: Data curation; Formal analysis; Investigation; Methodology. Shuai Zhou: Data curation; Investigation; Methodology. Peipei Tang: Data curation; Investigation; Methodology. Ran Xu: Investigation; Methodology. Yuwei Zhang: Resources; Formal analysis; Visualization. Dongping Wei: Resources; Validation; Methodology. Jian Wen: Resources; Formal analysis. Rick F Thorne: Formal analysis; Validation; Visualization; Methodology; Writing—review & editing. Xu Dong Zhang: Resources; Formal analysis; Writing—review & editing. Jun‐Lin Guan: Conceptualization; Formal analysis; Supervision; Writing—review & editing. Lianxin Liu: Resources; Formal analysis; Supervision; Funding acquisition; Visualization; Project administration. Song Chen: Conceptualization; Resources; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Project administration; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
SC, JLG, LXL and MW designed the research; YY and YFZ performed most of the experiments; SZ, PPT and RX assisted with most of the cloning and mouse experiments; LXL, XDZ, DPW and JW provide reagents and resources; YY, YWZ, JLG, RFT, XDZ, SC and MW carried out data analysis. YY, RFT and SC wrote the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dr. Sharon Tooze for the general gift of ULK1/2 knockout MEF cells. We also thank Dr. Paul Stemmer in the Proteomics Core Facility of the Institute of Environmental Health Sciences at Wayne State University for the mass spectrometry analysis. We are thankful to Dr. Longping Wen at the University of Science and Technology of China for providing the GFP‐LC3 reporter transgenic mice. This work was supported by the National Natural Science Foundation of China (81820108021, U2004138, 81773132, 31871437, 81970153, 81872393, 81702616), Excellent Youth Foundation of Henan Scientific Committee (21230040016) and National Key R&D Program of China (2019YFA0709300).
The EMBO Journal (2022) 41: e109777.
Contributor Information
Lianxin Liu, Email: liulx@ustc.edu.cn.
Mian Wu, Email: wumian@ustc.edu.cn.
Song Chen, Email: schen@zzu.edu.cn.
Data availability
This study includes no data deposited in external repositories.
References
- Abe S, Nagai T, Masukawa M, Okumoto K, Homma Y, Fujiki Y, Mizuno K (2017) Localization of protein kinase NDR2 to peroxisomes and its role in ciliogenesis. J Biol Chem 292: 4089–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena G, Boyd C, Oo KS, Maellaro E, Zhivotovsky B, Kaminskyy VO (2016) Suppressed translation and ULK1 degradation as potential mechanisms of autophagy limitation under prolonged starvation. Autophagy 12: 2085–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30: 1913–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Kimmelman AC, Debnath J (2019) Targeting autophagy in cancer: recent advances and future directions. Cancer Discov 9: 1167–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boase NA, Rychkov GY, Townley SL, Dinudom A, Candi E, Voss AK, Tsoutsman T, Semsarian C, Melino G, Koentgen F et al (2011) Respiratory distress and perinatal lethality in Nedd4‐2‐deficient mice. Nat Commun 2: 287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang C, Yeo S, Liang CC, Okamoto T, Sun S, Wen J, Guan JL (2016) Distinct roles of autophagy‐dependent and ‐independent functions of FIP200 revealed by generation and analysis of a mutant knock‐in mouse model. Genes Dev 30: 856–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Carosi JM, Yang Z, Ariotti N, Kerr MC, Parton RG, Sargeant TJ, Teasdale RD (2019) Retromer has a selective function in cargo sorting via endosome transport carriers. J Cell Biol 218: 615–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo M, Romagnoli A, Antonioli M, Piacentini M, Fimia GM (2020) TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ 27: 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang WC et al (2018) Disruption of the beclin 1‐BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558: 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco C, Mandriani B, Di Rienzo M, Micale L, Malerba N, Cocciadiferro D, Sjottem E, Augello B, Squeo GM, Pellico MT et al (2018) TRIM50 regulates Beclin 1 proautophagic activity. Biochim Biophys Acta Mol Cell Res 1865: 908–919 [DOI] [PubMed] [Google Scholar]
- Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284: 12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M, Yeo SK, Turner K, Harold A, Yang Y, Zhang X, Guan JL (2021) Autophagy blockade limits HER2+ Breast cancer tumorigenesis by perturbing HER2 trafficking and promoting release Via small extracellular vesicles. Dev Cell 56: 341–355.e5 [DOI] [PubMed] [Google Scholar]
- Hatakeyama S (2011) TRIM proteins and cancer. Nat Rev Cancer 11: 792–804 [DOI] [PubMed] [Google Scholar]
- Hatakeyama S (2017) TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci 42: 297–311 [DOI] [PubMed] [Google Scholar]
- Hergovich A (2016) The roles of NDR protein kinases in hippo signalling. Genes 7: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A, Bichsel SJ, Hemmings BA (2005) Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol Cell Biol 25: 8259–8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N et al (2009a) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N (2009b) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5: 973–979 [DOI] [PubMed] [Google Scholar]
- Hunter T (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell 28: 730–738 [DOI] [PubMed] [Google Scholar]
- Joffre C, Dupont N, Hoa L, Gomez V, Pardo R, Goncalves‐Pimentel C, Achard P, Bettoun A, Meunier B, Bauvy C et al (2015) The pro‐apoptotic STK38 kinase is a new beclin1 partner positively regulating autophagy. Curr Biol 25: 2479–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Enomoto A, Watanabe T, Haga H, Ishida S, Kondo Y, Furukawa K, Urano T, Mii S, Weng L et al (2014) TRIM27/MRTF‐B‐dependent integrin beta1 expression defines leading cells in cancer cell collectives. Cell Rep 7: 1156–1167 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132: 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, Thorburn A (2017) Targeting autophagy in cancer. Nat Rev Cancer 17: 528–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Dai Q, Meng H, Sun A, Wei J, Peng K, Childress C, Chen M, Shao G, Yang W (2017) The HECT E3 ubiquitin ligase NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy. J Cell Sci 130: 3839–3850 [DOI] [PubMed] [Google Scholar]
- Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF et al (2016) Cul3‐KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell 61: 84–97 [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhu M, Lin L, Fan X, Piao Z, Jiang X (2014) Deficiency of Trim27 protects dopaminergic neurons from apoptosis in the neurotoxin model of Parkinson's disease. Brain Res 1588: 17–24 [DOI] [PubMed] [Google Scholar]
- Ma Y, Wei Z, Bast RC Jr, Wang Z, Li Y, Gao M, Liu Y, Wang X, Guo C, Zhang L et al (2016) Downregulation of TRIM27 expression inhibits the proliferation of ovarian cancer cells in vitro and in vivo. Lab Invest 96: 37–48 [DOI] [PubMed] [Google Scholar]
- Mercer CA, Kaliappan A, Dennis PB (2009) A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5: 649–662 [DOI] [PubMed] [Google Scholar]
- Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafa R, Magri E, Raimondi A, Lanza G, Tacchetti C et al (2016) PML at mitochondria‐associated membranes is critical for the repression of autophagy and cancer development. Cell Rep 16: 2415–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22: 132–139 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B (2020) Autophagy in human diseases. N Engl J Med 383: 1564–1576 [DOI] [PubMed] [Google Scholar]
- Mowers EE, Sharifi MN, Macleod KF (2017) Autophagy in cancer metastasis. Oncogene 36: 1619–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10: 458–467 [DOI] [PubMed] [Google Scholar]
- Nazio F, Carinci M, Valacca C, Bielli P, Strappazzon F, Antonioli M, Ciccosanti F, Rodolfo C, Campello S, Fimia GM et al (2016) Fine‐tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol 215: 841–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K (2016) The K48–K63 branched ubiquitin chain regulates NF‐kappaB signaling. Mol Cell 64: 251–266 [DOI] [PubMed] [Google Scholar]
- Okamoto T, Yeo SK, Hao M, Copley MR, Haas MA, Chen S, Guan JL (2020) FIP200 suppresses immune checkpoint therapy responses in breast cancers by limiting AZI2/TBK1/IRF signaling independent of its canonical autophagy function. Cancer Res 80: 3580–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, Potts PR (2015) Degradation of AMPK by a cancer‐specific ubiquitin ligase. Cell 160: 715–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz‐Rohmer D, Probst S, Yang ZZ, Laurent F, Stadler MB, Zuniga A, Zeller R, Hynx D, Hemmings BA, Hergovich A (2015) NDR kinases are essential for somitogenesis and cardiac looping during mouse embryonic development. PLoS One 10: e0136566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS, Arfuso F, Yap CT, Goh BC, Sethi G et al (2018) Dual role of autophagy in hallmarks of cancer. Oncogene 37: 1142–1158 [DOI] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S (2018) CCPG1 Is a non‐canonical autophagy cargo receptor essential for ER‐phagy and pancreatic ER proteostasis. Dev Cell 44: 217–232.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegert MR, Hergovich A, Tamaskovic R, Bichsel SJ, Hemmings BA (2005) Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20‐like kinase MST3. Mol Cell Biol 25: 11019–11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A, Wei J, Childress C, Shaw JH, Peng KE, Shao G, Yang W, Lin Q (2017) The E3 ubiquitin ligase NEDD4 is an LC3‐interactive protein and regulates autophagy. Autophagy 13: 522–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Deng R, Luo RZ, Shen GP, Cai MY, Du ZM, Jiang S, Yang MT, Fu JH, Zhu XF (2012) Low expression of ULK1 is associated with operable breast cancer progression and is an adverse prognostic marker of survival for patients. Breast Cancer Res Treat 134: 549–560 [DOI] [PubMed] [Google Scholar]
- Wang W, Xia Z, Farre JC, Subramani S (2018) TRIM37 deficiency induces autophagy through deregulating the MTORC1‐TFEB axis. Autophagy 14: 1574–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Hatakeyama S (2017) TRIM proteins and diseases. J Biochem 161: 135–144 [DOI] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL (2011) Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 25: 1510–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E (2015) The role for autophagy in cancer. J Clin Invest 125: 42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woik N, Dietz CT, Schaker K, Kroll J (2014) Kelch‐like ECT2‐interacting protein KLEIP regulates late‐stage pulmonary maturation via Hif‐2alpha in mice. Dis Model Mech 7: 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PM, Puente C, Ganley IG, Jiang X (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9: 124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Kumar S (2010) Nedd4 and Nedd4‐2: closely related ubiquitin‐protein ligases with distinct physiological functions. Cell Death Differ 17: 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Wu CY, Wu J, Lin HK (2010) Regulation of Akt signaling activation by ubiquitination. Cell Cycle 9: 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ (2009) An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 335: 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman MM, Nomura T, Takagi T, Okamura T, Jin W, Shinagawa T, Tanaka Y, Ishii S (2013) Ubiquitination‐deubiquitination by the TRIM27‐USP7 complex regulates tumor necrosis factor alpha‐induced apoptosis. Mol Cell Biol 33: 4971–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Shen L, Zhu Y, Xu R, Deng Z, Liu X, Ding Y, Wang C, Shi Y, Bei L et al (2021) KDM6A promotes imatinib resistance through YY1‐mediated transcriptional upregulation of TRKA independently of its demethylase activity in chronic myelogenous leukemia. Theranostics 11: 2691–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HX, Xu ZS, Lin H, Li M, Xia T, Cui K, Wang SY, Li Y, Shu HB, Wang YY (2018) TRIM27 mediates STAT3 activation at retromer‐positive structures to promote colitis and colitis‐associated carcinogenesis. Nat Commun 9: 3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Hou J, Zhou Y, Yang Y, Xie B, Cao X (2015) Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res 25: 1121–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoumpoulidou G, Broceno C, Li H, Bird D, Thomas G, Mittnacht S (2012) Role of the tripartite motif protein 27 in cancer development. J Natl Cancer Inst 104: 941–952 [DOI] [PubMed] [Google Scholar]