

Abstract
In this study, we analyzed if Actinomadura sp. RB99 produces siderophores that that could be responsible for the antimicrobial activity observed in co‐cultivation studies. Dereplication of high‐resolution tandem mass spectrometry (HRMS/MS) and global natural product social molecular networking platform (GNPS) analysis of fungus‐bacterium co‐cultures resulted in the identification of five madurastatin derivatives (A1, A2, E1, F, and G1), of which were four new derivatives. Chemical structures were unambiguously confirmed by HR‐ESI‐MS, 1D and 2D NMR experiments, as well as MS/MS data and their absolute structures were elucidated based on Marfey's analysis, DP4+ probability calculation and total synthesis. Structure analysis revealed that madurastatin E1 (2) contained a rare 4‐imidazolidinone cyclic moiety and madurastatin A1 (5) was characterized as a Ga3+‐complex. The function of madurastatins as siderophores was evaluated using the fungal pathogen Cryptococcus neoformans as model organism. Based on homology models, we identified the putative NRPS‐based gene cluster region of the siderophores in Actinomadura sp. RB99.
Keywords: metabolomics, microbial communication, natural products, siderophores
Metabolomic analysis of cocultures of Actinomadura sp. RB99 with fungal symbionts of termites allowed for the MS‐guided isolation of four unreported madurastatin derivatives, including a siderophore‐metal complex thereof.
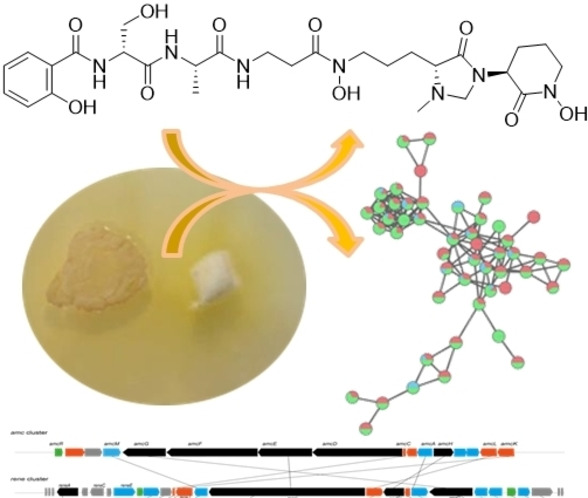
Introduction
Defensive symbiotic microorganisms of fungus farming insects have been shown to produce a diverse set of bioactive natural products to prevent detrimental infestations of the susceptible fungal monoculture.[ 1 , 2 , 3 ] We recently intensified our research efforts on elucidating the defensive functions of Actinobacteria in the fungus‐farming termite Macrotermes natalensis[ 4 , 5 ] and found that most of the actinobacterial isolates produced mixtures of secondary metabolites that are active against the co‐occurring weed fungi. Within these studies, we became particular intrigued by members of the fairly underexplored genus Actinomadura,[ 6 , 7 , 8 ] including Actinomadura sp. RB99 as their culture extracts showed antimicrobial activity against bacterial and fungal tests strains. First metabolic investigations of Actinomadura sp. RB99 resulted in the isolation of a group of cyclic tripeptides, named natalenamides A–C [9] and the polyketide fridamycin. [10] Members of this and phylogenetically related genera have also been recently reported to produce amongst others phenolate‐hydroxamate siderophores to sequester essential metals from the surrounding environment.[ 11 , 12 , 13 ] Intrigued by the structural diversity and important functions of siderophores, we investigated if Actinomadura sp. RB99 has the capabilities to produce siderophores. Here, we report on the successful combination of activity assays, high‐resolution tandem mass spectrometry (HRMS2) and global natural product social molecular networking platform (GNPS) analysis,[ 14 , 15 ] which resulted in the isolation of five madurastatin derivatives (A1, A2, E1, F, and G1) including a siderophore‐metal complex. Furthermore, we were able to deduce their absolute structures from Marfey's analysis, DP4+ probability calculation, and phylogenetic domain analyses of the putative NRPS‐based biosynthetic gene cluster.
Results and Discussion
First, we re‐evaluated the growth modulating activity of Actinomadura sp. RB99 (hence forward named RB99) in co‐cultures with the fungal cultivar Termitomyces sp. 153 or the co‐evolved stowaway fungus Pseudoxylaria sp. X802. Bacterium‐fungus co‐cultures and visible inhibition zones were monitored over time (9, 12 and 14 days). Overall, RB99 showed a strong growth inhibiting activity against Pseudoxylaria sp. X802 while only moderate effects against Termitomyces were observed, which hinted towards a selective inhibition of the weed fungus Pseudoxylaria sp. X802. To evaluate if yet unreported antimicrobial metabolites were present in the exogenous metabolome and whether metal chelating metabolites could be responsible for the observed growth inhibiting effect, we analyzed methanolic extracts obtained from the interaction zone, and the challenged bacterial colony of RB99 (Figure 1A) as well as RB99 cultures grown on different media by ESI‐HRMS2. The acquired MS2‐data was dereplicated using the GNPS platform. [14] The obtained GNPS‐network indicated towards a rich metabolome with several nodes related to yet unreported molecular ions detected in both interaction zone and bacterial mycelium. A small GNPS‐cluster with yet unreported molecular ion peaks (m/z 640.293, 610.283, 608.304, 638.314) sparked our interest. Several nodes contained within the cluster showed m/z differences of Δ m/z=15.994 and 14.015, which indicated structural changes related to one oxygen atom and one methylene moiety. The proposed molecular formula indicated a peptidic backbone with a putative N,O‐ratio characteristic for siderophores (e. g. C27H42N7O10 +, generated for m/z [M+H]+ 624.299).
Figure 1.

A) Actinomadura sp. RB99 colony (BC) inhibiting growth of garden weed Pseudoxylaria sp. X802 (FC) after 14 d grown on PDA and formation of a zone of inhibition (ZI). B) GNPS based molecular network clusters of target molecular ion features of RB99 co‐cultivated with Pseudoxylaria sp. X802 (14 d, PDA, red nodes represent bacterial colony, blue nodes represent zone of inhibition (ZI). C) GNPS networking features from cultivation on different media (red, ISP2 medium; green, ISP5 sea salt medium; blue, ISP5 medium). D‐E) Extracted ion chromatograms of target molecular ion features when D) RB99 was co‐cultured with Pseudoxylaria sp. X802 and E) RB99 was cultured on different growth media (ISP2, ISP5 sea salt, ISP5), target molecular ion features: m/z 624.2977 (Sum formula C27H42N7O10 +, calcd., 624.2988, all EICs are displayed in 5 ppm range).
Next, we evaluated the abundances of the identified target ions and the relative chrome azurol S (CAS) activity of the chemical extracts obtained from the different cultivation conditions. Overall, we found correlation between the intensity of the detected molecular ion peaks and higher biomass production (Figure 1, for example growth in ISP2 or ISP5‐sea salt medium for several days), while intensities of all chemical features were significantly reduced when RB99 was grown on ISP5 minimal medium (ISP5*). Intriguingly, addition of a complex sea salt mixture significantly restored signal intensities of most molecular ion features (Figure 1E). We then corroborated these findings by testing the metabolite extracts for siderophore activity using the well‐established CAS‐assay (Figures S1 and S2). As the production of the putative siderophores appeared to be mostly dependent on the amount of produced biomass, Actinomadura sp. RB99 was then cultivated in ISP2 medium containing 1 % sea salt for eight days (8 L, 150 rpm, 28 °C). Centrifuged culture supernatant was extracted using the non‐polar copolymer styrene‐divynilbenzene‐based adsorbent resin HP20 and metabolites were eluted using a gradient of MeOH/H2O. The resulting methanolic extract was subjected to solvent partitioning with EtOAc, and high‐resolution tandem mass spectrometry (HR‐MS2) and CAS‐activity guided purification yielded five madurastatins A2 (1), E1 (2), F (3), G1 (4), and A1 (5), including four new derivatives 2–5 (Figures 2 and 3).[ 6 , 10 ]
Figure 2.

Chemical structures of isolated madurastatins A2 (1) and A1 (5), fragmentation patterns and 2D NMR correlations (blue bond: 1H‐1H COSY correlations, pink arrow: HMBC correlations).
Figure 3.

Chemical structures of A) isolated madurastatins E1 (2), F (3), and G1 (4), synthetic madurastatins (4 a and 4 b), and structures of predicted madurastatins H1 (6) and J1 (7) based on HR‐MS2 fragmentation analysis, and B) features of previously reported members of the madurastatin family.
Structural elucidation of madurastatin derivatives
The molecular formula of compound 1 was determined as C27H41N7O10 [m/z 624.3022 [M+H]+, calcd. for C27H42N7O10 +, 624.2993)] and based on ESI (+)‐HRMS‐data dereplication, the isolated compound was tentatively assigned as the siderophore madurastatin A2, which belongs to a compound family named madurastatin[ 16 , 17 , 18 ] and whose core structure has been recently in part revised.[ 19 , 20 , 21 ] The planar structure of 1 was verified by 1D and 2D NMR analysis, a correlating MS/MS pattern and biosynthetic considerations (see below) (Figure 2) and displayed five amino acid spin systems starting from salicylic acid and continued with serine, alanine, β‐alanine, N‐methyl‐N‐hydroxyl‐ornithine and a cyclic N‐hydroxyl‐ornithine moiety (characteristic m/z pattern 131.0815, Figure S12).[ 19 , 20 , 21 ] As the absolute configuration of madurastatin A2 was not yet reported, we performed acid hydrolysis of compound 1 and conversion with Marfey's reagent (1‐fluoro‐2,4‐dinitrophenyl‐5‐L‐alanineamide (L‐FDAA)). For comparison, L/D‐Ser, L/D‐Ala, L/D‐Orn, and N‐methyl‐L‐Orn were also derivatives with L‐FDAA.
LC–MS based comparative analysis of reaction products indicated the absolute configurations as d‐Ser, l‐Ala, l ‐Orn, and N‐methyl‐L‐Orn (Figures S74 and S75) and allowed to propose the absolute configuration of madurastatin A2 for the first time. A detailed analysis of the identified molecular ion signals of the madurastatin GNPS cluster also indicated the presence of a madurastatin A2 homolog containing presumably an oxazoline moiety (apo‐5: m/z 606.2882). However, different purification protocols resulted exclusively in the isolation of the hydrolyzed derivative 1 with matching NMR pattern. A similar phenomenon was observed in a study on gobichelins, [22] in which an intrinsic instability of the oxazoline ring was suggested. To avoid spontaneous hydrolysis of the oxazoline ring, we then conducted the isolation of the corresponding Ga(III)‐complex (Figure 2). Replacement of Fe3+ by titration with Ga(NO3)3 solution resulted in a complex with the molecular formula C27H36N7O9Ga as deduced from HR‐ESI‐MS analysis (m/z 672.1891 [M+H]+, calcd. for C27H37N7O9Ga+, 672.1909) with a Ga isotope ratio (69Ga:71Ga=5 : 3). The residual Fe(III) complex (m/z 659.1984 [M+H]+, calcd. for C27H37N7O9Fe+, 659.1997) with a Fe isotope ratio (54Fe:56Fe=6 : 100) was detected in traces.
1H NMR analysis of the complex revealed a similar signal set as observed for compound 1 and the presence of a cyclic oxazoline moiety instead of a d‐serine [HMBC correlation of H2‐9 (δ H 4.69 and 4.43) to C‐7 (δ C 170.46)]. This assumption was supported by comparative MS2‐data analysis that showed the absence of the characteristic fragment ions m/z 121.0283 and 208.0605 and the presence of fragment ion m/z 162.0550 belonging to an oxazoline moiety (Figure 2). Based on the highly similar NMR pattern, biosynthetic considerations (see below) and previous structure elucidation efforts on madurastatins,[ 16 , 17 , 18 , 19 , 20 , 21 ] we assigned compound apo‐5 the name madurastatin A1 and propose a revision of the originally reported structure. [16]
Based on our detailed analysis of the madurastatin‐related GNPS network, three related compounds (2–4) were isolated and structurally identified based on 1D and 2D NMR analysis and HRMS/MS interpretation (Figure 3A). Compound 2 was determined to have the molecular formula of C28H41N7O10 [m/z 636.3017 [M+H]+, Calcd. for C28H42N7O10 +, 636.2993]. Detailed analysis of 1D and 2D NMR spectra revealed almost identical NMR spectra of 1 and 2, except for the presence of an additional methylene [δ H 3.77 (1H, d, J=4.0 Hz, H‐2’a) and 4.34 (1H, d, J=4.0 Hz, H‐2’b); δ C 68.0]. HMBC correlations of H3‐1’/C‐20, H3‐1’/C‐2’, H‐2’/C‐20, H‐2’/C‐21, H‐23/C‐2’, and H‐23/C‐21 suggested the presence of a 4‐imidazolidinone moiety in conjunction with the N‐methyl‐l‐Orn moiety and the compound was named madurastatin E1 (2).
For compound 3, the molecular formula was determined to be C27H38N6O10 [m/z 605.2568 [M−H]−, calcd. for C27H37N6O10, 605.2571]. The 1H and 13C NMR spectra analysis as well as HR‐MS/MS data indicated a similar structure as compound 1 lacking the N‐methyl unit at C‐20 (δ C 36.7) and carrying an additional oxygenated methine at C‐26 [δ H 5.75 (1H, brs); δ C 82.0] with COSY correlations from H‐23 to H‐26 and HMBC correlation from H2‐24 to C‐26)]. To determine the absolute configurations of 2 and 3, a DP4+ probability analysis for the comparison of computationally calculated NMR chemical shifts of 2 a and 2 b with experimental data of 2 suggested that the absolute configuration at C‐20 in 2 was R‐form (Figure S77). Moreover, the calculated 1H and 13C chemical shifts of 3 a and 3 b agreed with experimental values of 3, and thus confirmed the absolute configuration at C‐26 in 3 (Figure S78). In case of compound 4, a molecular formula of C16H21N3O7 [m/z 368.1451 [M+H]+, Calcd. for C16H22N3O7, 368.1458] was proposed, which indicated the presence of salicylate, serine, alanine, and ß‐alanine.
This deduction was verified by 2D NMR analysis (1H‐1H COSY, HSQC, and HMBC correlations of H2‐14/C‐13, H2‐14/C‐16, and H2‐15/C‐16) and 3 J HH vicinal correlations between H2‐14 and H2‐15. To further proof the stereochemistry of compound 4, we also obtained two synthetic isomers of 4 a (d‐serine) and 4 b (l‐serine) and comparative 1D NMR analyses indicated identical chemical shift and J‐coupling pattern between madurastatin G1 (4) and synthetic isomer salicyl‐D‐Ser‐l‐ala‐ß‐Ala‐OH (4 a) (synthesized by Biosyntan GmbH), suggesting again the presence of d‐serine in 4 (Figures S50‐S61). Furthermore, the existence of d‐serine in the structure of 4 was also verified by DP4+ analysis by the comparison of calculated 1H and 13C chemical shifts of 4 a and 4 b with experimental data of 4 (Figure S79). Next, we evaluated the MS2‐fragmentation patterns of other molecular ions found in the madurastatin GNPS cluster and putatively assigned the molecular ion signals as madurastatins H1 (6, m/z 610.2829 [M+H]+, calcd. for C26H40N7O10 + 610.2831) and J1 (7, m/z 640.2947 [M+H]+, calcd. for C27H42N7O11 + 640.2937) based on their individual fragmentation patterns (Figures S17–S22).
To the best of our knowledge about eight other madurastatin derivatives have been reported in the literature and exhibit structural variations such as the oxazoline moiety (C‐7, C‐8) instead of a serine (C‐8) or the presence of a 4‐imidazolidinone ring (C‐20, C‐21) in madurastatins D1 and D2 (Figure 3B).[ 16 , 17 , 18 , 19 , 20 , 21 ] The planar and absolute structures of madurastatins, however, have been topic of constant discussion in recent years and have not been fully resolved yet. While originally proposed to be composed of an aziridine‐containing pentapeptide,[ 16 , 17 , 18 ] a structural revision of the aziridine ring of madurastatin C1 was later suggested by Thorson [19] and then verified by Hall. [20] The absolute stereochemistry of madurastatin C1, however, remains ambiguous as a congener with opposite optical rotation but otherwise identical analytical features were reported. The structures of madurastatins are related to the siderophores gobichelin, [22] and amychelin. [23] While the structure of amychelin was unequivocally determined based on X‐ray and NMR analysis of the metal complex, [23] the structures of gobichelins were proposed based on isotope‐feeding, Marfey's analysis and in silico domain predictions of the biosynthetic pathways.
Siderophore properties
The opportunistic‐pathogenic fungus Cryptococcus neoformans is known to infect immunocompromised individuals thereby causing pulmonary cryptococcosis and cryptococcal meningitis. [24] C. neoformans itself is unable to synthesize siderophores by its own, and thus scavenges siderophores produced by co‐occurring microorganisms.[ 25 , 26 ] Previous studies also showed that two deletion strains, one lacking a gene encoding a ferroxidase CFO1 (cfo1Δ) and one lacking a gene coding for a iron permease CFT1 (cft1Δ),[ 27 , 28 ] were only able to grow in the presence of a chelating siderophore (e. g. FeIII‐loaded siderophore ferroxamine (Figure 4A)).
Figure 4.
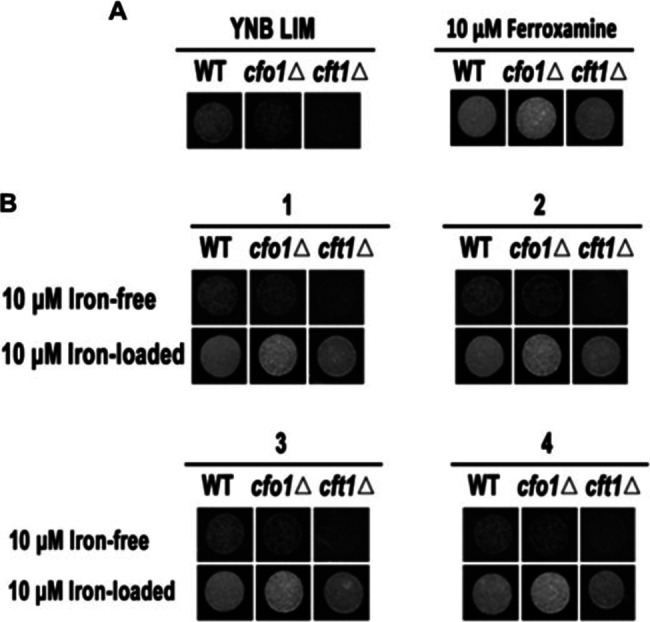
Siderophore activities of the compounds 1–4 using C. neoformans as model organism. A) Wild‐type C. neoformans and mutant strains cfo1Δ and cft1Δ were grown on iron‐depleted medium with or without siderophore (ferroxamine) and growth was monitored two days after incubating at 30 °C. B) Mutant strains were grown in iron‐depleted medium containing 10 μM of compounds 1–4 (in the absence (Iron‐free) or presence of Fe(III) (Iron‐loaded). Growth was monitored two days after incubating at 30 °C.
These physiological characteristics were exploited to test the siderophore activity of madurastatins. As depicted in Figure 4, only the wild‐type strain barely grew in the presence of iron‐free madurastatins 1–4, while all three strains (wild‐type, cfo1Δ, and cft1Δ) fully recovered growth in the presence of FeIII‐chelating madurastatins 1–4 supporting their role as siderophores (Figure 4B).
Biosynthetic gene cluster and pathway analysis
To identify the biosynthetic gene cluster region encoding for madurastatins and to support the deduced stereochemical assignments, we then performed an antiSMASH (version 5.0.0) [29] analysis of the RB99 genome followed by a MultiGeneBLAST study.[ 4 , 30 ] Only one likely gene cluster region was identified (rene), which showed overall similarities to the amychelin gene cluster amc (37 %), the gobichelin cluster (33 %), and the recently identified mad clusters related to madurastatin C1 and D (31 and 41 %) (Figure 5). [21]
Figure 5.

A) Comparison of biosynthetic gene clusters based on amino acid identity (amc (amychelin), rene (madurastatins) and mad (madurastatins)). B) Proposed biosynthetic pathway for the formation of madurastatins (blue: proposed structures obtained via a functional Cyc1 domain; abbreviations: T, thiolation domain; Cyc, Heterocyclization domain; A, adenylation domain; LCL Condensation domain; DCL epimerization and condensation domain).
The cluster region encoded for 45 genes of which 17 genes (reneA‐reneU) were predicted to encode for enzymes belonging to the core biosynthesis (approx. length 27.3 kbp).[ 31 , 32 ] Next to regulation‐related gene sequences (reneF, reneT), the cluster encoded for a NRPS (reneA, reneL reneN, reneQ), a putative L‐ornithine N5 monooxygenase (reneJ), a putative aspartate 1‐decarboxylase responsible for β‐amino acid biosynthesis (reneO), [33] a salicylate synthase (reneM), a putative S‐adenosyl‐methionine (SAM)‐dependent methyltransferase (RB99_01616), tailoring enzymes (reneI, reneJ, reneM), and exporter proteins (reneE, reneG, reneK, reneP, reneR, reneS, reneU). We then mined the antiSMASH database and performed a detailed phylogenetic analysis of the encoded and available adenylation (A)‐domains (total of 1125A‐domains) and 310 condensation (C)‐domains (Figures S75–S76, Table S11–S12) using the web‐based bioinformatics platform Galaxy, [34] which overall supported the observed amino acid sequence of madurastatins. As depicted in Figure 5, the biosynthesis of madurastatins presumably commences with loading of the salicylate moiety to the thiolation domain of the putative phenyloxazoline synthase ReneQ (83 % identity to Mad63), which carries a heterocyclization domain catalyzing the condensation of salicylate and L‐ or D‐serine to a phenyloxazoline ring. As we were not able to detect any dedicated epimerase‐related sequence within the NRPS, it remains speculative at this point whether or not ReneQ also catalyzes the conversion of L‐serine to the detected D‐serine or if the C‐domain is selective for D‐Ser due to yet unknown reasons. Future in vitro experiments will be necessary to distinguish between these options. In a next step, ReneL (78 % identity to Mad31), which contains four sets of C, A, T domains and one N‐methylation (nMT) domain, presumably extends the growing chain by incorporating L‐alanine (glycine or serine) (C2‐LCL), β‐alanine (C3‐LCL) and N‐hydroxy L‐ornithine (C4‐DCL) based on predictions of domains of known NRPS systems (Table S8–S9).
The required N‐hydroxy‐L‐ornithine is likely oxidized by ReneJ prior to the incorporation as reported for coelichelin [35] and madurastatins. [21] The encoded methyltransferase (nMT) is assumed to catalyze the mono and di‐N‐methylation of L‐ornithine; however the timing of the methylation remains unknown. The last module of ReneL is composed of the C4‐LCL and a T‐domain, but lacks the necessary A‐ and thioesterase domain that could catalyze the release of the siderophore from the NRPS. Thus, ReneL likely requires the support by a trans acting A‐domain (ReneA) that incorporates ornithine and/or N‐hydroxy‐L‐ornithine moiety into the growing peptide chain. [36] Similar to the proposed amychelin biosynthesis, a designated hydrolase (homolog to AmcB) could catalyze the cyclization and chain release; [23] however, a spontaneous non‐enzymatic amidation yielding the madurastatin core structure cannot be excluded at this stage. Finally, additional oxidation steps of the N‐methyl group might cause the formation of the imidazolidinone moiety (e. g. subsequent substitution reaction) or N‐hydroxylation steps are likely responsible for the formation of derivatives 2 and 3.
Conclusion
In summary, we analyzed co‐cultures and growth condition of Actinomadura sp. RB99 and identified five madurastatins A2 (1), E1 (2), F (3), G1 (4), and A1 (5) including a metal complex thereof. While production levels of madurastatins were only partially dependent on culture conditions, their active secretion into co‐culture interaction zones and their siderophore activities suggest their importance for survival of the producing organisms. The abundance of homologous gene cluster sequences within members of the genus Actinomadura and potentially beyond further supports the importance of madurastatins as actinobacterial siderophore class. Further studies related to their intrinsic metal chelating properties in combination with the analysis of their evolutionary pathways will uncover their success story as actinobacterial siderophore class.
Experimental Section
The datasets supporting this article have been uploaded as part of the ESI and contains details for chemical procedures, 1D and 2D NMR of described compounds, as well as HRMS data, cultivation procedures and bioassay data. This material is available free of charge.
Co‐cultivation studies: A circular colony of Actinomadura sp. RB99 was streaked out in PDA media and grown for 7 days at room temperature. Cocultures with Pseudoxylaria sp. X802 or sp. X187 were inoculated with agar plugs (∼0.3 cm×0.3 cm) in approximately 2–3 cm distance to the bacterial colony. In case of Termitomyces, a mycelial suspension was used to inoculate agar plates. Fungal growth was observed for 14 days in triplicates. Fungal and bacterial monocultures acted as controls. Distance of fungal mycelium growth was measured after 9, 12, 14 days and the rate of inhibition was calculated based on the ratio of growth (R=A/T) facing away (A) and towards (T) the bacterial pellet in the direct connecting axis.
CAS activity assay: Using a glass beaker, 6 mL of 10 mM hexadecyltrimethylammonium bromide (HDTMA/CTAB) were diluted with H2O up to a volume of 5 mL. While stirring, a mixture consisting of 150 μL FeCl3 (1 mM) previously dissolved in 10 mM HCl and 750 μL aqueous chrome azurol S (2 mM, CAS) was slowly added. In another beaker, 0.43 g of anhydrous piperazine was dissolved in 1 mL H2O. 625 μL HCl (10 N) was very slowly added to the piperazine solution and the resulting pH was checked to be approximately 5.6. The buffered piperazine solution was carefully added while stirring to the Fe‐CAS‐HDTMA solution. And the resulting CAS reagent was stored in the dark at 4 °C. The CAS solution was tested using 10 mM EDTA, observing a color change from blue‐green to orange.
Screening of siderophore activity: For siderophore activity tests, 200 μL of CAS solution was added to 10 μL clear sample extract (1 mg/mL, MEOH) using 1 mM EDTA as positive and pure MeOH as negative controls. Mixtures were incubated in the dark and absorbance at λ Abs=630 nm was monitored for up to one hour using a UV‐VIS spectrometer.
MS2 and GNPS‐based discovery of madurastatin congeners: A molecular network was created using the online workflow (https://ccms‐ucsd.github.io/GNPSDocumentation) on the GNPS website (http://gnps.ucsd.edu). The data was filtered by removing all MS/MS fragment ions within +/−17 Da of the precursor m/z. MS/MS spectra were window filtered by choosing only the top 6 fragment ions in the +/−50 Da window throughout the spectrum. The precursor ion mass tolerance was set to 0.02 Da and a MS/MS fragment ion tolerance of 0.02 Da. A network was then created where edges were filtered to have a cosine score above 0.7 and more than 6 matched peaks. Further, edges between two nodes were kept in the network if and only if each of the nodes appeared in each other's respective top 10 most similar nodes. Finally, the maximum size of a molecular family was set to 100, and the lowest scoring edges were removed from molecular families until the molecular family size was below this threshold. The spectra in the network were then searched against GNPS’ spectral libraries. The library spectra were filtered in the same manner as the input data. All matches kept between network spectra and library spectra were required to have a score above 0.7 and at least 6 matched peaks.
Time‐resolved analysis of siderophore production in different media: To analyze the influence of media composition and cultivation‐time on the production of putative siderophore Actinomadura sp. RB99 was grown under three different media conditions in duplicates (full medium ISP2, minimal medium ISP5 and altered minimal media ISP5 with sea salt). Cultures were extracted after two, four and eight days of growth. Samples were analyzed at a concentration of 75 μg/mL and peak integration (XIC, 5 ppm) was used for quantification.
Isolation of siderophores: Actinomadura sp. RB99 was grown in 50 mL ISP‐2 broth for seven days at 30 °C (pre‐culture) and used to inoculate 100 ISP‐2 agar plates. Plates were incubated for 10 days at 30 °C, cut into small pieces, consolidated, and immersed overnight in MeOH. The MeOH phase was filtered and evaporated in vacuo. The resultant MeOH extract (11 g) was dissolved in distilled water (700 mL) and then solvent‐partitioned with EtOAc (700 mL) three times, providing 4.5 g of residue. The EtOAc‐soluble fraction (4.5 g) was loaded onto a silica gel column for open column chromatography and fractionated with a gradient solvent system of CH2Cl2‐MeOH (50 : 1 to 0 : 1, v/v) to afford seven fractions (A–G). Fraction E (110 mg) was subjected to preparative reversed‐phase HPLC (Phenomenex Luna C18, 250×21.2 mm i.d., 5 μm) using MeOH‐H2O (1 : 9–1 : 0, v/v, gradient system, flow rate: 5 mL/min) to give four subfractions (E1‐E4). Compound 4 (2.0 mg, t R=29.0 min) was purified from subfraction E3 (20 mg) by semi‐preparative reversed‐phase HPLC eluting 28 % MeOH/H2O (isocratic system, flow rate: 2 mL/min). Fraction F (490 mg) was separated by using preparative reversed‐phase HPLC (Phenomenex Luna C18, 250×21.2 mm i.d., 5 μm) using CH3CN‐H2O (0.5 : 9.5–1 : 0, v/v, gradient system, flow rate: 5 mL/min) to give four subfractions (F1–F4). Subfraction F3 (50 mg) was isolated by semi‐preparative reversed‐phase HPLC eluting 30 % MeOH/H2O (isocratic system, flow rate: 2 mL/min), affording compounds 2 (2.3 mg, t R=50.0 min) and 3 (1.4 mg, t R=55.0 min). Five subfractions (G1–G5) were acquired from fraction G (300 mg) using preparative reversed‐phase HPLC (Phenomenex Luna C18, 250×21.2 mm i.d., 5 μm) using CH3CN‐H2O (1 : 9‐1 : 0, v/v, gradient system, flow rate: 5 mL/min). Compound 1 (4.3 mg, t R=15.0 min) was isolated from subfraction G2 (45 mg) by semi‐preparative reversed‐phase HPLC eluting 19 % MeOH/H2O (isocratic system, flow rate: 2 mL/min). Next, the resultant MeOH extract was dissolved in 10 % MeOH/90 % H2O and loaded on a conditioned SPE−C18 cartridge (5 g/45 mL), and then fractioned by step‐gradient of MeOH and H2O mixture (20 mL/fraction). The resultant fractions were concentrated under reduced pressure and submitted to LCMS analysis. The fraction eluted at 40 % MeOH contained the apo‐siderophore (1) with the m/z at 624.2977 [M+H]+ as well as a Fe‐siderophore complex with an m/z of 659.1983 [M+H]+. Then, the 40 % MeOH fraction was subjected to Sephadex LH20 purification and metabolites eluted using 50 % MeOH. MS‐guided analysis indicated that the Fe‐siderophore complex was enriched in Fr. 12. This fraction was titrated with a 100 mM Ga(NO3)3 aq. solution to exchange Fe3+ with Ga3+ yielding a siderophore complex with an m/z of 672.1894 [M+H]+. The complex was separated by semipreparative HPLC (Phenomenex Synergi‐HydroRP, 250×10 mm i.d., 5 μm) using CH3CN/0.1 % FA gradient (0–5 min, 10 % CH3CN/90 % H2O (0.1 % FA); 5–40 min, 10 % CH3CN/90 % H2O (0.1 % FA)‐28 % CH3CN/72 % H2O (0.1 % FA), flow rate at 2 mL/min), affording compound 5 (1.0 mg; t R=20.0 min).
Madurastatin A2 (1): Amorphous powder; [α] −12.1 (c 0.05, MeOH); UV (MeOH) λmax (log ϵ) 202 (4.23), 238 (1.31), 299 (0.76) nm; 1H (800 MHz) and 13C NMR (200 MHz), see Table S4 and Table S5, respectively; positive‐mode HR‐ESI‐MS m/z 624.3022 [M+H]+ (Calcd. for C27H42N7O10 +, 624.2993).
Madurastatin E1 (2): Amorphous powder; [α] −8.6 (c 0.04, MeOH); UV (MeOH) λmax (log ϵ) 202 (4.20), 238 (1.25), 299 (0.73) nm; 1H (800 MHz) and 13C NMR (200 MHz), see Table S4 and Table S5, respectively; positive‐mode HR‐ESI‐MS m/z 636.3017 [M+H]+ (Calcd. for C28H42N7O10 +, 636.2993).
Madurastatin F (3): Amorphous powder; [α] −6.4 (c 0.03, MeOH); UV (MeOH) λmax (log ϵ) 202 (4.25), 239 (1.24), 298 (0.76) nm; 1H (800 MHz) and 13C NMR (200 MHz), see Table S4 and Table S5, respectively; negative‐mode HR‐ESI‐MS m/z 605.2568 [M−H]− (Calcd. for C27H37N6O10 −, 605.2571).
Madurastatin G1 (4): Amorphous powder; [α] −7.7 (c 0.03, MeOH); UV (MeOH) λmax (log ϵ) 202 (4.09), 239 (1.15), 298 (0.74) nm; 1H (800 MHz) and 13C NMR (200 MHz), see Table S4 and Table S5, respectively; positive‐mode HR‐ESI‐MS m/z 368.1451 [M+H]+ (Calcd. for C16H22N3O7 +, 368.1458).
Madurastatin A1 (5, Ga3+‐complex): Orange powder; [α] +269.2 (c 0.01, 50 % MeOH); UV (MeOH) λmax (log ϵ) 202, 239, 298 nm; 1H (600 MHz) and 13C NMR (150 MHz), see Table S6; positive‐mode HR‐ESI‐MS m/z 672.1891 [M+H]+ (Calcd. for C27H37N7O9Ga+, 672.1909).
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2019R1A5A2027340 and 2021R1A2C2007937). This study was also supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT and Future Planning 2019R1A4A1024764. We acknowledge support by the Postdoctoral Research Program of Sungkyunkwan University (2021) and under the framework of international cooperation program managed by the National Research Foundation of Korea (2020K2A9A2A06037042). We are deeply thankful for funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Project‐ID 239748522 – CRC 1127 (project A6). We thank Mrs. Heike Heinecke (Hans Knöll Institute) for recording NMR spectra. Open Access funding enabled and organized by Projekt DEAL.
S. R. Lee, F. Schalk, J. W. Schwitalla, H. Guo, J. S. Yu, M. Song, W. H. Jung, Z. W. de Beer, C. Beemelmanns, K. H. Kim, Chem. Eur. J. 2022, 28, e202200612.
GNPS=global natural product social molecular networking platform.
Contributor Information
Prof. Christine Beemelmanns, Email: Christine.Beemelmanns@leibniz-hki.de.
Prof. Ki Hyun Kim, Email: khkim83@skku.edu.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Ramadhar T. R., Beemelmanns C., Currie C. R., Clardy J., J. Antibiot. 2014, 67, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beemelmanns C., Guo H., Rischer M., Poulsen M., Beilstein J. Org. Chem. 2016, 12, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmidt S., Kildgaard S., Guo H., Beemelmanns C., Poulsen M., Nat. Prod. Rep. 2022, 39, 231–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benndorf R., Guo H., Sommerwerk E., Weigel C., Garcia-Altares M., Martin K., Hu H., Kuefner M., de Beer Z. W., Poulsen M., Beemelmanns C., Antibiotics 2018, 7, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murphy R., Benndorf R., De Beer Z. W., Vollmers J., Kaster A. K., Beemelmanns C., Poulsen M., mSphere 2021, 6, e01233–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo H., Benndorf R., Leichnitz D., Klassen J. L., Vollmers J., Görls H., Steinacker M., Weigel C., Dahse H. M., Kaster A. K., de Beer Z. W., Poulsen M., Beemelmanns C., Chem. Eur. J. 2017, 23, 9338–9345. [DOI] [PubMed] [Google Scholar]
- 7. Benndorf R., Martin K., Küfner M., de Beer Z. W., Vollmers J., Kaster A. K., Beemelmanns C., Int. J. Syst. Evol. Microbiol. 2020, 70, 5255–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo H., Schwitalla J. W., Benndorf R., Baunach M., Steinbeck C., Görls H., de Beer Z. W., Regestein L., Beemelmanns C., Org. Lett. 2020, 22, 2634–2638. [DOI] [PubMed] [Google Scholar]
- 9. Lee S. R., Lee D., Yu J. S., Benndorf R., Lee S., Lee S. D., Huh J., de Beer Z. W., Kim Y. H., Beemelmanns C., Kang K. S., Kim K. H., Molecules 2018, 23, 3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoon S.-Y., Lee S. R., Hwang J. Y., Benndorf R., Beemelmanns C., Chung S. J., Kim K.-H., Nutrients 2019, 11, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kramer J., Özkaya Ö., Kümmerli R., Nat. Rev. Microbiol. 2020, 18, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miethke M., Marahiel M. A., Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukuda T. T. H., Helfrich E. J. N., Mevers E., Melo W. G. P., Van Arnam E. B., Andes D. R., Currie C. R., Pupo M. T., Clardy J., ACS Cent. Sci. 2021, 7, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang M., Carver J. J., Phelan V. V., Sanchez L. M., Garg N., Peng Y., Nguyen D. D., Watrous J., Kapono C. A., Luzzatto-Knaan T., Porto C., Bouslimani A., Melnik A. V., Meehan M. J., Liu W. T., Crüsemann M., Boudreau P. D., Esquenazi E., Sandoval-Calderón M., Kersten R. D., Pace L. A., Quinn R. A., Duncan K. R., Hsu C. C., Floros D. J., Gavilan R. G., Kleigrewe K., Northen T., Dutton R. J., Parrot D., Carlson E. E., Aigle B., Michelsen C. F., Jelsbak L., Sohlenkamp C., Pevzner P., Edlund A., McLean J., Piel J., Murphy B. T., Gerwick L., Liaw C. C., Yang Y. L., Humpf H. U., Maansson M., Keyzers R. A., Sims A. C., Johnson A. R., Sidebottom A. M., Sedio B. E., Klitgaard A., Larson C. B., Boya C. A. P., Torres-Mendoza D., Gonzalez D. J., Silva D. B., Marques L. M., Demarque D. P., Pociute E., O'Neill E. C., Briand E., Helfrich E. J. N., Granatosky E. A., Glukhov E., Ryffel F., Houson H., Mohimani H., Kharbush J. J., Zeng Y., Vorholt J. A., Kurita K. L., Charusanti P., McPhail K. L., Nielsen K. F., Vuong L., Elfeki M., Traxler M. F., Engene N., Koyama N., Vining O. B., Baric R., Silva R. R., Mascuch S. J., Tomasi S., Jenkins S., Macherla V., Hoffman T., Agarwal V., Williams P. G., Dai J., Neupane R., Gurr J., Rodríguez A. M. C., Lamsa A., Zhang C., Dorrestein K., Duggan B. M., Almaliti J., Allard P. M., Phapale P., Nothias L. F., Alexandrov T., Litaudon M., Wolfender J. L., Kyle J. E., Metz T. O., Peryea T., Nguyen D. T., VanLeer D., Shinn P., Jadhav A., Müller R., Waters K. M., Shi W., Liu X., Zhang L., Knight R., Jensen P. R., Palsson B., Pogliano K., Linington R. G., Gutiérrez M., Lopes N. P., Gerwick W. H., Moore B. S., Dorrestein P. C., Bandeira N., Nat. Biotechnol. 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andreu V. P., Augustijn H. E., vanden Berg K., van der Hooft J. J. J., Fischbach M. A., Medema M. H., mSystems 2021, 6, e00937–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harada K., Tomita K., Fujii K., Masuda K., Mikami Y., Yazawa K., Komaki H., J. Antibiot. 2004, 57,125–35. [DOI] [PubMed] [Google Scholar]
- 17. Mazzei E., Iorio M., Maffioli S. I., Sosio M., Donadio S., J. Antibiot. 2012, 65, 267–269. [DOI] [PubMed] [Google Scholar]
- 18. Kawahara T., Itoh M., Izumikawa M., Sakata N., Tsuchida T., Shin-ya K., J. Antibiot. 2014, 67, 577–580. [DOI] [PubMed] [Google Scholar]
- 19. Shaaban K. A., Saunders M. A., Zhang Y., Tran T., Elshahawi S. I., Ponomareva L. V., Wang X., Zhang J., Copley G. C., Sunkara M., Kharel M. K., Morris A. J., Hower J. C., Tremblay M. S., Prendergast M. A., Thorson J. S., J. Nat. Prod. 2017, 80, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tyler A. R., Mosaei H., Morton S., Waddell P. G., Wills C., McFarlane W., Gray J., Goodfellow M., Errington J., Allenby N., Zenkin N., Hall M. J., J. Nat. Prod. 2017, 80, 1558–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan J.-X., Chevrette M. G., Braun D. R., Harper M. K., Currie C. R., Bugni T. S., Org. Lett. 2019, 21, 6275–6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Y., Unger M., Ntai I., McClure R. A., Albright J. C., Thomson R. J., Kelleher N. L. J. M., MedChemComm 2013, 4, 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seyedsayamdost M. R., Traxler M. F., Zheng S.-L., Kolter R., Clardy J., J. Am. Chem. Soc. 2011, 133, 11434–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao Y., Lin J., Fan Y., Lin X., Annu. Rev. Microbiol. 2019, 73, 17–42. [DOI] [PubMed] [Google Scholar]
- 25. Jung W. H., Sham A., Lian T., Singh A., Kosman D. J., Kronstad J. W., PLoS Pathog. 2008, 4, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bairwa G., Hee Jung W., Kronstad J. W., Metallomics 2017, 9, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jung W. H., Hu G., Kuo W., Kronstad J. W., Eukaryotic Cell 2009, 8, 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raymond-Bouchard I., Carroll C. S., Nesbitt J. R., Henry K. A., Pinto L. J., Moinzadeh M., Scott J., Moore M. M., Eukaryotic Cell 2012, 11, 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blin K., Shaw S., Kautsar S. A., Medema M. H., Weber T., Nucleic Acids Res. 2020, 49, D639-D643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Medema M. H., Takano E., Breitling R., Mol. Biol. Evol. 2013, 30, 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barry S. M., Challis G. L., Curr. Opin. Chem. Biol. 2009, 13, 205–215. [DOI] [PubMed] [Google Scholar]
- 32. Khan A., Singh P., Srivastava A., Microbiol. Res. 2018, 212–213, 103–111. [DOI] [PubMed] [Google Scholar]
- 33. Williamson J. M., Brown G. M., J. Biol. Chem. 1979, 254, 8074–8082. [PubMed] [Google Scholar]
- 34. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Grüning B. A., Guerler A., Hillman-Jackson J., Hiltemann S., Jalili V., Rasche H., Soranzo N., Goecks J., Taylor J., Nekrutenko A., Blankenberg D., Nucleic Acids Res. 2018, 46, W537-W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pohlmann V., Marahiel M., Org. Biomol. Chem. 2008, 6, 1843–1848. [DOI] [PubMed] [Google Scholar]
- 36. Maruyama C., Toyoda J., Kato Y., Izumikawa M., Takagi M., Shin-ya K., Katano H., Utagawa T., Hamano Y., Nat. Chem. Biol. 2012, 8, 791–797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.