

Summary
The proper function of the genome relies on spatial organization of DNA, RNA, and proteins, but how transcription contributes to the organization is unclear. Here, we show that Condensates Induced by Transcription Inhibition (CITIs) drastically alter genome spatial organization. CITIs are formed by SFPQ, NONO, FUS, and TAF15 in nucleoli upon inhibition of RNA polymerase II (RNAPII). Mechanistically, RNAPII inhibition perturbs ribosomal RNA (rRNA) processing, releases rRNA-processing factors from nucleoli, and enables SFPQ to bind rRNA. While accumulating in CITIs, SFPQ/TAF15 remain associated with active genes and tether active chromatin to nucleoli. In the presence of DNA doublestrand breaks (DSBs), the altered chromatin compartmentalization induced by RNAPII inhibition increases gene fusions in CITIs and stimulates the formation of fusion oncogenes. Thus, proper RNAPII transcription and rRNA processing prevent the altered compartmentalization of active chromatin in CITIs, suppressing the generation of gene fusions from DSBs.
Graphical Abstract
eTOC Blurb
Yasuhara et al. showed that transcription inhibition and nucleolar stress induce the formation of protein-RNA condensates, which are named CITIs, in nucleoli. CITI formation promotes the localization of active chromatin to nucleoli, increasing gene fusion in the presence of DNA breaks.
Introduction
Spatial organization of chromosomes is critical for the function of the genome (Misteli, 2007). Chromatin forms local loops and longer-distance topologically associating domains (TADs) important for organizing and regulating gene expression within a chromosome (van Steensel and Furlong, 2019). The genome is also regulated by chromosome folding and positioning at higher orders, such as the organization of chromosomal regions in Compartments A and B (Gibcus and Dekker, 2013). The Compartment A is generally associated with open chromatin, high gene expression, early DNA replication, and tends to localize to the nuclear interior. In contrast, the Compartment B is associated with close chromatin, low gene expression, late DNA replication, and is often biased toward the nuclear periphery. Importantly, the large-scale organization of chromosomes in Compartments A and B is often compromised in cancer cells, which may have a tumor-suppressive role (Johnstone et al., 2020), suggesting a link between altered chromatin compartmentalization and tumor evolution.
The spatial organization of chromosomes is regulated not only by DNA, but also by proteins and RNA. Protein compartmentalization driven by multivalent interactions and liquid-liquid phase separation (LLPS) provides a means to regulate protein functions spatially (Banani et al., 2017; Harrison and Shorter, 2017; Shin and Brangwynne, 2017). Membrane-less nuclear organelles including nucleoli, PML bodies, paraspeckles and others are necessary for specific functions of the genome (Banani et al., 2017; Mitrea and Kriwacki, 2016). Notably, compartmentalization of proteins on DNA can regulate the spatial organization of specific genomic loci and regions, conferring distinct functional and regulatory properties (Hnisz et al., 2017). For example, in the context of transcription activation, the phase-separating properties of transcription factors contribute to enhancer-promoter interactions (Sabari et al., 2018). Altered spatial organization of DNA and proteins is associated with changes of transcriptional activity, yet the process of transcription itself is also important for DNA and protein compartmentalization. More than five decades ago, Donna Granick observed that transcription inhibition resulted in a drastic reorganization of nucleoli, giving rise to the structure called “nucleolar necklace” (Granick, 1975a, b; Granick and Granick, 1971). It was thought that transcription inhibition compromises ribosome assembly, leading to aberrant appearance of nucleoli. Interestingly, nucleolar necklaces were observed not only in cells under transcriptional stress, but also in cells under other stresses, suggesting that multiple cellular stresses converge on altered ribosome biogenesis to distort the compartmentalization of nucleoli. Nonetheless, at a molecular level, it remains unclear how transcription inhibition and other cellular stresses affect ribosome biogenesis, how changes of ribosome biogenesis alter the spatial organization of DNA and proteins in nucleoli, and how the changes of nucleoli influence the genome and stress responses.
In addition to nucleolus, paraspeckle is another type of membrane-less organelle containing RNA and RNA-binding proteins (Fox et al., 2018). The long non-coding RNA NEAT1 and two RNA-binding proteins, SFPQ (also known as PSF) and NONO (also known as p54nrb), have important architectural roles in paraspeckles. NEAT1 binds to multiple SFPQ and NONO molecules through its middle domain, allowing SFPQ/NONO to undergo oligomerization (Mao et al., 2011; Yamazaki et al., 2018). The NEAT1-mediated oligomerization of SFPQ/NONO ultimately leads to LLPS, forming paraspeckles in the nucleus. Several other RNA-binding proteins with LLPS properties, such as FUS and TAF15, are also present in paraspeckles (Nakagawa et al., 2018; West et al., 2016). Paraspeckles are shown to regulate gene expression through multiple mechanisms, including nuclear retention of mRNAs, sequestration of RNA-binding proteins, and regulation of microRNA formation (Fox et al., 2018). Outside of paraspeckles, SFPQ and NONO are detected at promoters of genes, where they interact with RNAPII and function as a coactivator or corepressor (Amelio et al., 2007; Emili et al., 2002). While the functions of SFPQ/NONO in paraspeckles and gene expression are well established, it remains unclear whether SFPQ/NONO can undergo LLPS by binding to RNAs other than NEAT1 and influence chromatin organization at higher orders.
In this study, we find that SFPQ and NONO undergo rapid LLPS in nucleoli after RNAPII inhibition, giving rise to Condensates Induced by Transcription Inhibition (CITIs). CITIs contain SFPQ, NONO, FUS, and TAF15, but unlike paraspeckles, they do not contain NEAT1. CITIs are induced not only by RNAPII inhibition, but also by physiological stresses such as UV and cold shock, as well as therapeutic stresses imposed by chemotherapy. Interestingly, the formation of CITIs is associated with the loss of ribosomal RNA (rRNA)-processing factors from nucleoli and a robust binding of SFPQ to rRNA, suggesting that CITIs are rRNA-nucleated condensates. During CITI formation, SFPQ remains associated with active genes and tethers active chromatin to nucleoli, drastically altering the spatial organization of the genome. The localization of active chromatin to CITIs increases the illegitimate fusions of the DNA double-strand breaks (DSBs) in active genes, promoting the formation of fusion oncogenes. These results suggest that proper RNAPII transcription and rRNA processing are important for preventing the LLPS of SFPQ/NONO on rRNA, which could otherwise alter the compartmentalization of active chromatin and increase gene fusions.
Results
Formation of SFPQ/NONO condensates upon RNAPII inhibition
To investigate the effects of transcription inhibition on protein compartmentalization, we first examined the impact of 1 hr treatment with THZ1, an irreversible inhibitor of the CDK7 kinase, which is necessary for RNAPII transcription (Kwiatkowski et al., 2014), on several proteins known to form condensates. Across multiple cell lines (U2OS, HeLa, MRC5, RPE), paraspeckle-associated proteins SFPQ and NONO underwent drastic relocalization upon THZ1 treatment, forming large condensates in the nucleus regardless of the cell cycle stage (Figures 1A and S1A–D). Surprisingly, NEAT1, the key RNA component of paraspeckles, was not detected in the SFPQ condensates after THZ1 treatment (Figure S1E), suggesting that the THZ1-induced SFPQ condensates are distinct from paraspeckles. Furthermore, the SFPQ condensates did not colocalize with SC-35 or PML (Figure S1F), showing that they are also distinct from nuclear speckles and PML bodies.
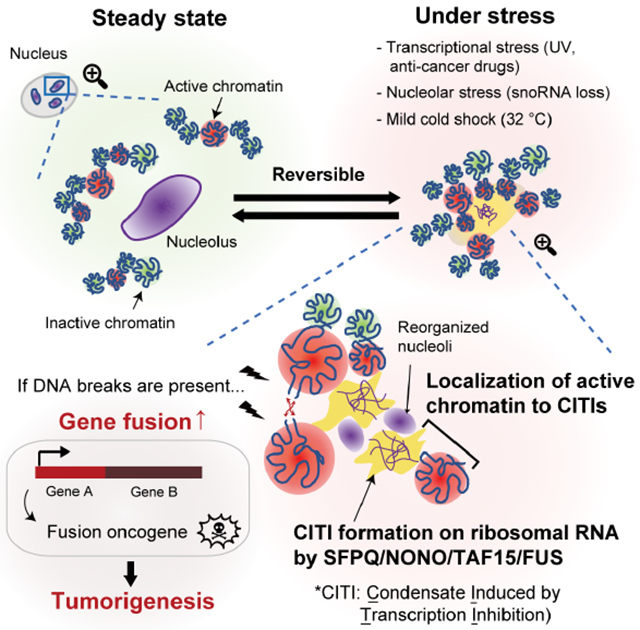
Figure 1. RNAPII Inhibition Induces CITIs.
(A) The effect of THZ1 (1 μM) treatment for 1 hr on the localization of SFPQ and NONO was analyzed by immunofluorescence (IF) in the U2OS cell line. Scale bars, 10 μm. NONO and SFPQ insets (white dashed line) shown at right, with nucleus outlined.
(B, C) The effect of treatment with the indicated drugs (DRB 100 μM for 1 hr, Triptolide 1 μM for 1 hr, Actinomycin D 1 μM for 1 hr, CX5461 10 μM for 3 hr, CPT 1 μM for 1 hr) or UV irradiation (30 J m−3, fixed 1 hr after irradiation) on the localization of SFPQ was analyzed by IF. Scale bars, 10 μm (B). The quantification of the total area of SFPQ condensates per cell upon treatment with the indicated drugs or UV irradiation is shown as mean with 95% confidence interval (CI) (C).
(D, E) The effect of acute degradation of RNAPII or THZ1 treatment on the localization of SFPQ was analyzed by IF after 1 hr auxin induction using the DLD1 mAID-POLR2A-mClover cell line. Scale bars, 10 μm (D). The quantification of the total area of SFPQ condensates per cell is shown as mean with 95% CI (E).
(F, G) The effect of THZ1 treatment on the localization of TAF15 (F) or FUS (G) was analyzed by IF. Scale bars, 10 μm.
(H) Live-cell imaging of GFP-TAF15 or GFP-FUS between 0 to 60 min after THZ1 treatment. See also Video 1 and 2. Scale bar, 10 μm.
Next, we tested whether induction of these SFPQ/NONO condensates was a general feature of transcription inhibition. Similar to THZ1, RNAPII-inhibiting compounds 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) and Triptolide induced SFPQ condensates robustly, but the RNA polymerase I (RNAPI) inhibitor CX5461 (Drygin et al., 2011) did not (Figure 1B and 1C). Actinomycin D, which inhibits all three RNA polymerases (Bensaude, 2011), induced SFPQ condensates that were distinctly smaller and differently distributed, possibly associated with nucleolar caps (Shav-Tal et al., 2005) (Figure 1B and 1C). The topoisomerase I (Top1) inhibitor Camptothecin (CPT), which blocks RNAPII elongation by trapping Top1 on DNA (Baranello et al., 2016; Sordet et al., 2008), and ultraviolet (UV) light, which causes degradation of RNAPII (Lans et al., 2019; Pommier, 2006), also induced SFPQ condensates (Figure 1B and 1C).
To test whether loss of RNAPII protein or its regulators can induce SFPQ condensates independently of the compounds or stresses that inhibit RNAPII, we used a cell line expressing an auxin-induced degron-tagged RNAPII to acutely deplete the polymerase (Nagashima et al., 2019). Acute degradation of RNAPII by 1 hr auxin treatment efficiently induced SFPQ condensates that were comparable in size to those induced by THZ1, recapitulating the effect of THZ1 (Figure 1D and 1E). Furthermore, depletion of CDK7/9, two kinases promoting RNAPII transcription, or PAF1C, a complex promoting RNAPII elongation, also induced SFPQ condensates (Figure S1G). Taken together, these results suggest that loss of RNAPII transcription induces a previously uncharacterized type of SFPQ/NONO condensates in the nucleus, which we have named Condensates Induced by Transcription Inhibition (CITIs).
FUS and TAF15 are components of CITIs
To further understand the compositions of CITIs, we tested other proteins with LLPS properties. Among the three members of the FET (FUS/EWS/TAF15) family of RNA-binding proteins (Harrison and Shorter, 2017; Schwartz et al., 2015), TAF15 and FUS were efficiently localized to CITIs after RNAPII inhibition (Figure 1F, 1G, and S1H). The RNA binding proteins hnRNPA1 and TDP-43 were segregated away from CITIs (Figure S1I and S1J). To analyze the kinetics of CITI formation, we performed live-cell imaging of cells stably expressing GFP-tagged TAF15 and FUS (Video 1, 2, and Figure S1K). CITIs appeared around 20 min after THZ1 treatment and their size rapidly increased until reaching a plateau at 50 min (Figure 1H, Video 1, and 2). These results establish that CITIs are composed of SFPQ/NONO/TAF15/FUS and quickly induced by RNAPII inhibition.
To understand how CITIs are assembled, we tested the functional relationships among CITI components. Knockdown of SFPQ and NONO revealed that they rely on each other to localize to CITIs (Figure S2A and S2B). Although NEAT1 was not detected in CITIs (Figure S1E), SFPQ and NONO did not localize to CITIs efficiently in NEAT1-knockdown cells, suggesting that NEAT1 may prime SFPQ/NONO for CITI formation (Figure S2A–D). NEAT1 knockdown significantly reduced the chromatin binding of SFPQ (Figure S2E–G), raising the possibility that the chromatin-bound fraction of SFPQ contributes to CITI formation. The localization of TAF15 and FUS to CITIs was dependent on SFPQ and NONO (Figure S2H–J), but the localization of SFPQ and NONO to CITIs was independent of TAF15 and FUS (Figure S2K and S2L). These results suggest that SFPQ/NONO are core components of CITIs, whereas TAF15/FUS are likely clients.
CITIs are formed around the FC and DFC of nucleoli
In the time-lapse analysis of TAF15 and FUS (Figure 1H, Video 1, and 2), we noticed that TAF15/FUS condensates first appeared in the nuclear regions where TAF15/FUS levels were low before RNAPII inhibition, which raised a question about the nature of the regions in which CITIs are formed. To understand whether CITIs are formed in specific compartments of the nucleus, we used laser scanning confocal microscopy, including a spinning disk confocal microscope with optical pixel reassignment (“SoRa”) providing a lateral resolution of ~120 nm, to analyze SFPQ/TAF15 and several nucleolar markers. Morphologically, nucleolus consists of three compartments: fibrillar center (FC), dense fibrillar component (DFC), and granular component (GC). Our confocal imaging data show that the nuclear regions where TAF15/FUS levels were low before RNAPII inhibition were occupied by NPM1 (a GC marker) (Figure 2A). After THZ1 treatment, TAF15 formed shell-like structures ~1 μm across that perfectly colocalize with SFPQ. Furthermore, TAF15 condensates surrounded but did not colocalize with scattered foci of RPA194 (a FC marker) and DKC1 (a DFC marker) (Figure 2A). NPM1 did not substantially overlap with TAF15, but the TAF15 structures were formed at the peripheries of NPM1-rich regions that were 1-3 μm in diameter (Figure 2A). Three-dimensional visualization of TAF15, DKC1, and NPM1 and quantification of their spatial relationships clearly demonstrate that CITIs occupy the space between the reorganized FC/DFC and GC of nucleoli (Figure 2B–D, S2M, and Video 3).
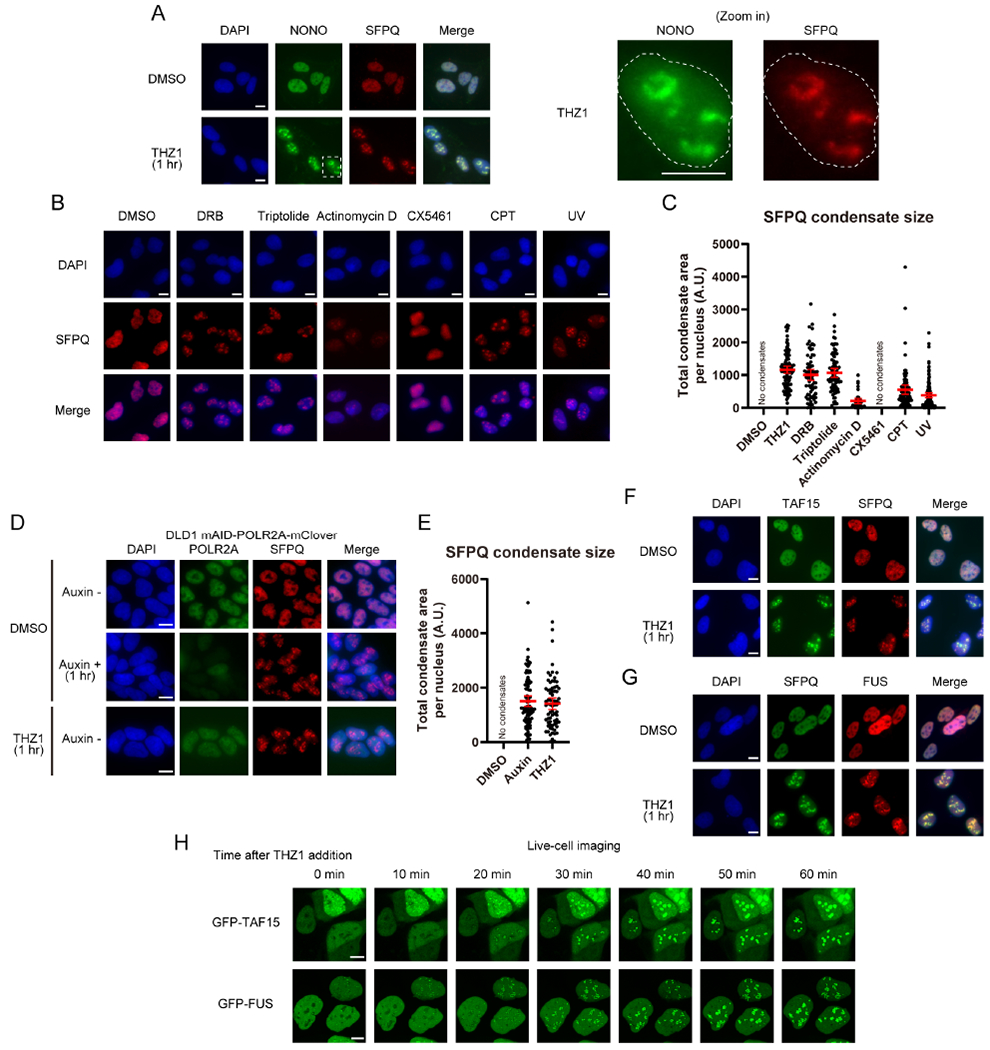
Figure 2. CITIs are Formed around FC and DFC in Nucleoli.
U2OS cells expressing GFP-TAF15 were untreated or treated for 1 hr with THZ1 and stained for GFP-TAF15 (green) and two other proteins associated with different nucleolar compartments or SFPQ.
(A) Three-color SoRa imaging of representative cells. The color of each protein is shown on the top. Scale bar, 5 μm.
(B) SoRa volumetric renderings of z-stacks of a single representative nucleus. Green, TAF15; magenta, DKC1; blue, NPM1. See also Video 3 and Figure S2M.
(C, D) The DKC1 or NPM1 fraction of volume inside CITIs (C) and fraction of NPM1 surface area adjacent to TAF15 or DKC1 (D) were quantified from the SoRa volumetric renderings.
(E) Two-color STORM imaging of representative CITIs. Top row shows the individual localizations for each channel as points. Bottom row shows the reconstructed image. Scale bar, 100 nm.
(F) Map of localizations of each channel for a representative region of interest (ROI) for Clus-DoC analysis (top row). Map of degree of GFP-TAF15 localizations in the ROI colored by DoC score (middle row). Distribution of DoC scores for GFP-TAF15 to the other channel across 4-6 nucleolar regions per sample (bottom row, mean with SD, n=4-6). Scale bar, 5 μm.
Using a stochastic optical reconstruction microscope (STORM) providing a lateral resolution of ~20 nm, we confirmed that TAF15 colocalizes with SFPQ extensively (Figure 2E). Notably, the clusters of TAF15 signals are intermingled with clusters of RPA194 (FC) and DKC1 (DFC), confirming that CITIs are formed around FC and DFC. Indeed, colocalization analysis by the Clus-DoC algorithm (Pageon et al., 2016) revealed that the DoC scores for TAF15-SFPQ are mostly positive and close to 1 (a high degree of colocalization), and those for TAF15-DKC1 and TAF15-RPA194 are largely negative (proximal but separate), whereas those for TAF15-NPM1 are mostly 0 (more than 500 nm apart or no correlation) or negative (proximal but separate) (Figure 2F). Together, these results suggest that CITIs are primarily formed around the FC and DFC compartments, and they are adjacent to but do not substantially overlap with the GC compartment.
CITIs display features of LLPS
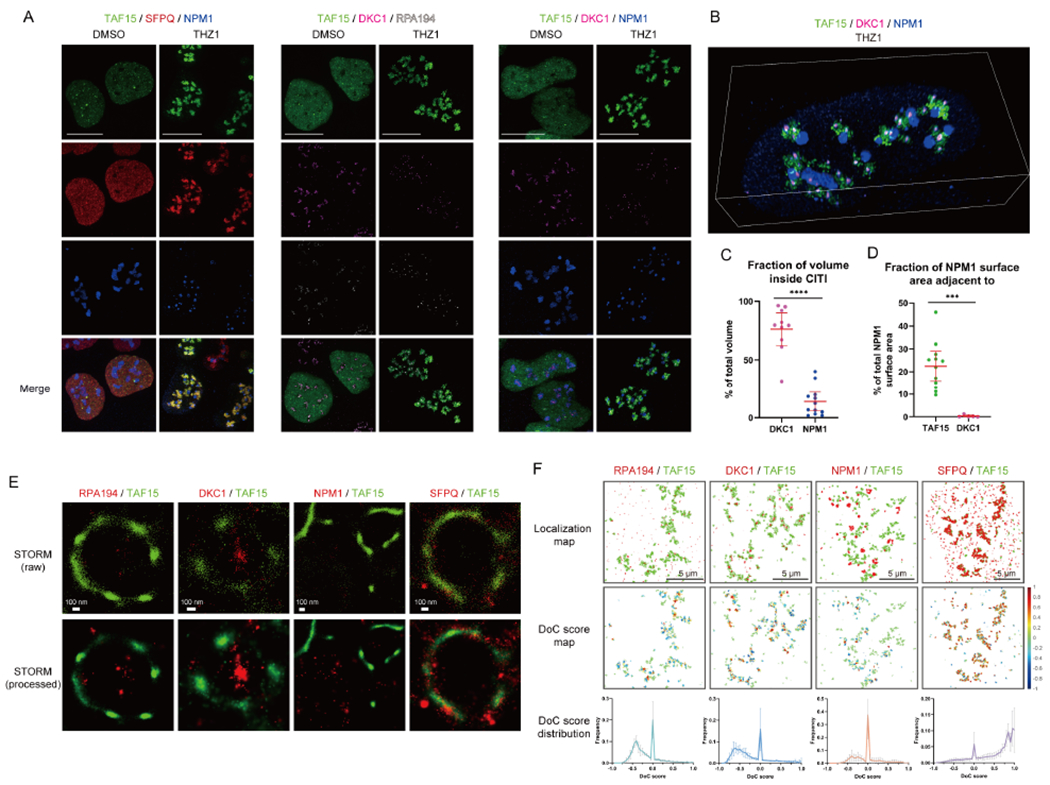
To understand the molecular dynamics in CITIs, we performed fluorescence recovery after photobleaching (FRAP) analysis on exogenously expressed GFP-TAF15 and endogenously tagged GFP-SFPQ (Figure 3A). The fluorescence of GFP-TAF15 and GFP-SFPQ recovered efficiently in both CITIs and nucleoplasm after photobleaching (Figure 3B). The diffusion coefficient of GFP-TAF15 in CITIs was 0.234 ± 0.028 μm2 s−1, and that of GFP-SFPQ was 0.483 ± 0.036 μm2 s−1 (Figure 3C), which are comparable to those of BRD4 and MED1 in LLPS condensates (Sabari et al., 2018). Since BRD4 and MED1 condensates are susceptible to 1,6-hexanediol, which disrupts hydrophobic protein-protein and protein-RNA interactions (Kroschwald et al., 2017; Sabari et al., 2018), we tested the effect of this compound on TAF15 localization. The THZ1-induced TAF15 condensates were significantly decreased by 1,6-hexanediol (Figure 3D and 3E). Thus, TAF15 localizes to CITIs through hydrophobic interactions and displays features associated with LLPS. Notably, CITIs were rapidly disassembled after DRB washout (Figure 3F), showing that CITIs are reversible.
Figure 3. CITI is a Dynamic Structure.
(A) Representative images from the FRAP analysis of THZ1-treated GFP-TAF15 expressing cells. The bleached area is indicated with a yellow square. Scale bar, 1 μm.
(B) The relative fluorescent recovery of GFP-TAF15 or GFP-SFPQ after bleaching of condensates (CITIs) or diffused areas (nucleoplasm) was analyzed (mean with SD, n=10 or 11 for data points of the cells from each category, representative results from three biological replicates). The fitted curves for each category are shown as a dotted line.
(C) The diffusion coefficient was calculated from the FRAP analysis (mean with SEM, n=3 for data points of three biological replicates).
(D, E) The effect of 1,6-hexanediol treatment (1.5%, 15 min) on TAF15 in CITIs was analyzed by IF (D). Scale bars, 10 μm. Since the nuclear size was shrunk by 1,6-hexanediol treatment, the size of CITIs was normalized to the nucleus size (E). The results are shown as mean with 95% CI.
(F) The effect of DRB wash out on CITIs was analyzed by IF. Scale bars, 10 μm. The quantification of CITI positive cells at each time point is shown.
SFPQ binds to rRNA to form CITIs
Given that SFPQ is an RNA-binding protein, we asked whether SFPQ accumulates on RNA transcripts in nucleoli after RNAPII inhibition. To visualize the overall changes of transcription in cells undergoing CITI formation, we treated cells with 5-Ethynyl Uridine (EU), which is incorporated into elongating transcripts. In the absence of THZ1, EU was efficiently incorporated in the nucleus and formed foci in regions with high transcription activity (Figure S3A). After THZ1 treatment, the overall EU incorporation was drastically reduced, but residual EU incorporation was detected and colocalized with CITIs (Figure S3A). Depletion of UBF, a key factor for rRNA transcription, significantly reduced the size of CITIs (Figure S3B). Furthermore, the RNAPI inhibitor CX5461 also impaired CITI formation (Figure S3C). Even when cells were treated with CX5461 after CITI formation, the size of CITIs was still reduced (Figure S3D). Thus, nucleoli become the main sites of remaining transcription activity after RNAPII inhibition, and RNAPI activity is required for both the formation and maintenance of CITIs.
A recent study showed that the intergenic spacer regions of rDNA repeats are aberrantly transcribed by RNAPI upon RNAPII inhibition, giving rise to sense intergenic non-coding RNAs (sincRNAs), which disrupt nucleolar organization (Abraham et al., 2020). However, knockdown of sincRNAs did not affect CITI formation (Figure S3E), suggesting that CITI formation is independent of sincRNAs. To test whether SFPQ binds to rRNA in nucleoli during CITI formation, we performed RNA immunoprecipitation (RIP) of SFPQ. After 1 hr of THZ1 treatment, the binding of SFPQ to RNAPII transcripts (NEAT1, GAPDH) was largely unchanged (Figure 4A). However, using primers that specifically detect the premature rRNA (pre-rRNA), we found that the binding of SFPQ to pre-rRNA was drastically increased (Figure 4A). Furthermore, an SFPQ mutant that lacks RNA-binding domains failed to localize to the CITIs formed by endogenous SFPQ/TAF15 (Figure 4B). Thus, the binding of SFPQ to rRNA, which localizes SFPQ to nucleoli/CITIs, is drastically increased by RNAPII inhibition, providing an explanation for the accumulation of SFPQ around the FC and DFC of nucleoli.
Figure 4. Perturbation of rRNA Processing Induces CITIs.
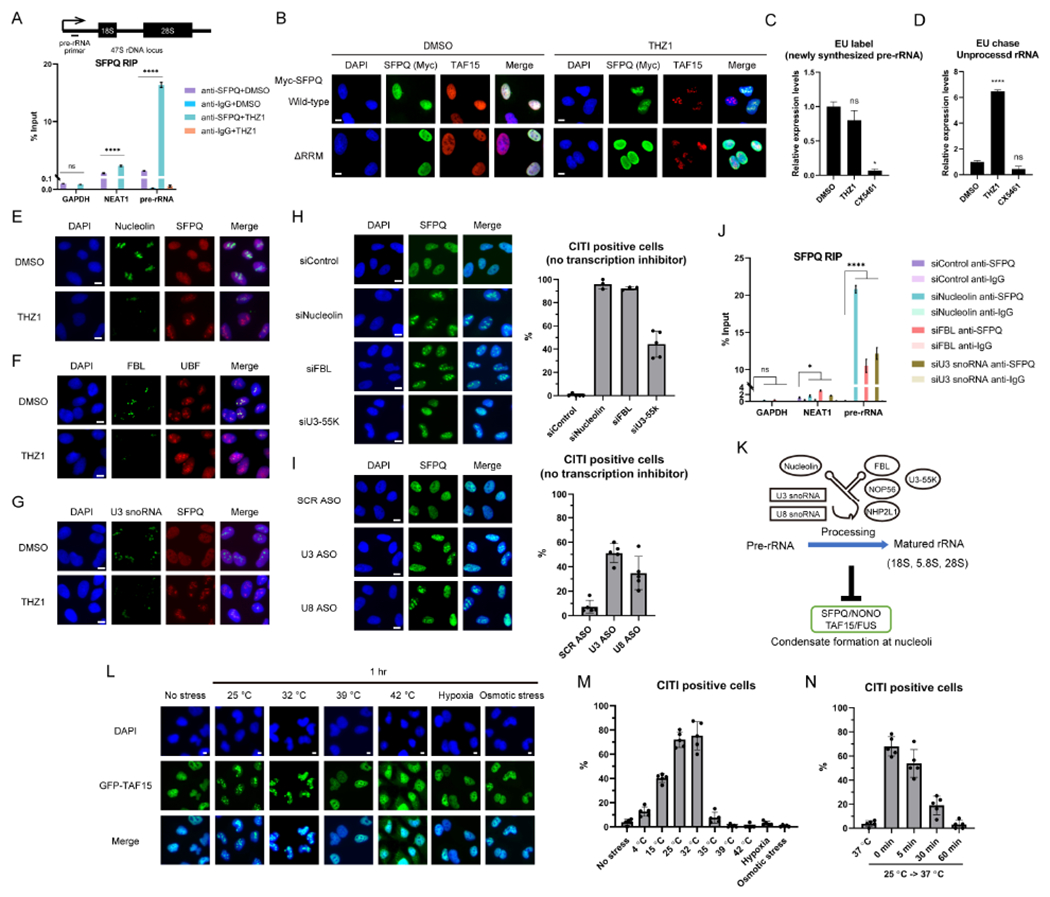
(A) The effect of THZ1 on the rRNA binding of SFPQ was analyzed by RIP (mean with SD, n=3 for measurements from one representative experiment of two biological replicates). The position of the primers for detection of pre-rRNA is shown above.
(B) A SFPQ mutant that lacks RNA-binding domains (ΔRRM) was exogenously expressed and its localization was analyzed by IF. It is of note that the ΔRRM mutant did not localize to CITIs formed by endogenous SFPQ/TAF15. Scale bars, 10 μm.
(C) The newly-synthesized RNA was labeled by EU in the presence of THZ1 or CX5461 for 1 hr. After biotin labeling and pulldown by streptavidin beads, the pre-rRNA levels in EU-labeled RNA were quantified by qPCR (mean with SD, n=3 for measurements from one representative experiment of three biological replicates).
(D) The EU-labeled (1 hr) newly-synthesized RNA was chased for another 1 hr in the presence of THZ1 or CX5461 and analyzed as in (C).
(E-G) The effects of THZ1 treatment on the localization of Nucleolin (E), FBL (F), or U3 snoRNA (G) and CITIs were analyzed by IF. Scale bars, 10 μm.
(H, I) The localization of SFPQ after knockdown (KD) of Nucleolin, FBL, U3-55K (H) or U3, U8 snoRNAs (I) was analyzed by IF. Scale bars, 10 μm. The quantification of CITI positive cells is shown.
(J) The effects of Nucleolin, FBL, or U3 snoRNA KD on the rRNA binding of SFPQ was analyzed as in (A).
(K) A model for the relationship between rRNA processing and condensate formation at nucleoli.
(L, M) The effect of cold/heat shock, hypoxia, or osmotic stress on TAF15 localization was analyzed by IF (L). Scale bars, 10 μm. The quantification of CITI positive cells is shown (M).
(N) The reversibility of CITIs induced by mild cold shock (25 °C) was tested as in Figure 3F.
Perturbance of rRNA processing induces CITIs
We next tested whether rRNA transcription and/or processing are affected upon RNAPII inhibition. We used qPCR to quantify the pre-rRNA labeled with EU and found that only CX5461, but not THZ1, reduced pre-RNA synthesis (Figure 4C). When pre-rRNA was first labeled with EU and then exposed to THZ1 or CX5461, unprocessed pre-rRNA remained at a much higher level in THZ1-treated cells than in untreated or CX5461-treated cells (Figure 4D). These results suggest that THZ1 does not affect pre-rRNA synthesis but blocks its processing, which is consistent with previous studies (Abraham et al., 2020; Bensaude, 2011). To investigate whether rRNA processing is compromised in CITIs, we analyzed several rRNA-processing factors in nucleoli. Pre-rRNA is modified and processed into matured rRNAs by Nucleolin, an rRNA-binding protein, together with box C/D and H/ACA snoRNAs and their associating factors, such as Fibrillarin (FBL) and DKC1 (Boisvert et al., 2007). One hour of THZ1 treatment did not reduce the protein levels of Nucleolin, components of box C/D snoRNPs (FBL, NOP56, NHP2L1), and the box H/ACA snoRNPs component DKC1 (Figure S3F). However, Nucleolin and box C/D snoRNP components, but not DKC1, were released from nucleoli after THZ1 treatment (Figure 4E, 4F, S3G, and S3H). Among the snoRNAs involved in rRNA processing, the U3/U8 box C/D snoRNPs are critical in the early steps of rRNA processing and transcribed by RNAPII (Langhendries et al., 2016). U3 snoRNA was depleted from the nucleoli after THZ1 treatment (Figure 4G). The levels of U3/U8 and other box C/D snoRNAs transcribed by RNAPII were generally reduced by THZ1 (Figure S3I). Notably, the THZ1-induced release of Nucleolin from nucleoli occurred even in SFPQ knockdown cells (Figure S3J), showing that the loss of rRNA-processing factors is not a consequence of CITI formation.
We next asked whether a loss of rRNA-processing factors is sufficient to induce CITIs. Knockdown of Nucleolin, FBL, and U3-55K (U3 snoRNA-specific binding protein) induced CITIs without RNAPII inhibition (Figure 4H). Of note, NEAT1- and SFPQ-positive paraspeckles and NEAT1-negative CITIs coexisted in FBL-knockdown cells (Figure S3K), showing that the disassembly of paraspeckles is not required for CITI formation. Depletion of U3 or U8 snoRNA with anti-sense oligos (ASOs) also induced CITIs (Figure 4I). Similar to the effects of RNAPII inhibition (Figure 4A), depletion of Nucleolin, FBL, or U3 snoRNA significantly increased SFPQ binding to rRNA (Figure 4J). These results suggest that rRNA-processing factors such as Nucleolin and box C/D snoRNPs are critical to prevent SFPQ from binding rRNA and forming condensates in nucleoli (Figure 4K).
To understand whether the CITI formation induced by defective rRNA processing is a physiologically relevant event, we asked if any cellular stress altering rRNA processing can induce CITIs. Mild cold shock reduces rRNA processing by affecting the RNase function in nucleoli (Knight et al., 2016). Exposing cells to mild cold shock (25 °C and 32 °C) for 1 hr, but not heat shock (39 °C or 42 °C), hypoxia, and osmotic stress, induced CITIs (Figure 4L and 4M). The CITIs induced at 25 °C completely disappeared after cells were returned to 37 °C for 1 hr (Figure 4N), consistent with the reversibility of CITI observed after transient RNAPII inhibition (Figure 3F). Together, these results suggest that the perturbation of rRNA processing by RNAPII inhibition or physiological stresses induces CITIs.
SFPQ and TAF15 remain associated with active genes while forming CITIs
A number of RNA-binding proteins involved in transcription regulation associate with chromatin (Van Nostrand et al., 2020; West et al., 2014), prompting us to perform chromatin immunoprecipitation sequencing (ChIP-seq) analysis on SFPQ and TAF15. SFPQ and TAF15 signals are enriched at numerous loci in the genome, whereas no enrichment was detected at these sites in IgG controls and input DNA (Figure S4A). Importantly, concomitant with the accumulation of SFPQ in CITIs after THZ1 treatment, SFPQ ChIP-seq signals were significantly increased at the rRNA coding region of rDNA repeats (Figure S4B). Consistent with the roles of SFPQ and TAF15 in transcription regulation (Hosokawa et al., 2019; Kwon et al., 2013; Ma et al., 2016), specific SFPQ and TAF15 signals are generally higher near transcription start sites (TSSs) compared to other genomic regions (Figure S4C and S4D). The levels of SFPQ and TAF15 at TSSs positively correlated with the transcriptional levels of the genes (Figure S4E). Peak analysis confirmed that SFPQ peaks are enriched at TSSs ± 1Kb of expressed genes (Figure S4F). Using Drosophila chromatin as a spike-in control for quantifying ChIP-seq signals, we found that the SFPQ signals at TSSs were largely retained after THZ1 treatment, whereas those in other regions were slightly reduced (Figure S4G and S4H). Thus, during CITI formation, SFPQ and TAF15 remain associated with TSSs of active genes, raising a question as to whether these gene loci are tethered to CITIs by SFPQ and TAF15.
Mislocalization of Compartment A to nucleoli upon RNAPII inhibition
To test whether specific chromosomal regions are tethered to CITIs after RNAPII inhibition, we performed Tyramide Signal Amplification sequencing (TSA-seq) (Chen et al., 2018; Zhang et al., 2020) using antibodies against SFPQ (a CITI marker after THZ1 treatment) or UBF (a constitutive nucleolar marker) (Figure 5A, S5A, and S5B). In this assay, the chromosomal regions proximal to SFPQ/UBF are preferentially modified by peroxidase-mediated deposition of Biotin-Tyramide, allowing us to identify the genomic regions proximal to CITIs/nucleoli. We first tested whether the THZ1-induced SFPQ enrichment in nucleoli can be detected by TSA-seq. The SFPQ TSA-seq signals were significantly increased in the rRNA coding region of rDNA repeats after THZ1 treatment (Figure 5B and 5C). Using biotinylated Drosophila DNA as a spike-in control, we confirmed that SFPQ TSA-seq signals are increased in the rRNA coding region after THZ1 treatment (Figure S5C and S5D). Together, these data suggest that the proximity of SFPQ to the rDNA repeats is increased after THZ1 treatment, corroborating with the increase of SFPQ staining in nucleoli after THZ1 treatment and the increased binding of SFPQ to rRNA.
Figure 5. CITIs Localize Active Chromatin to Nucleoli upon RNAPII Inhibition.
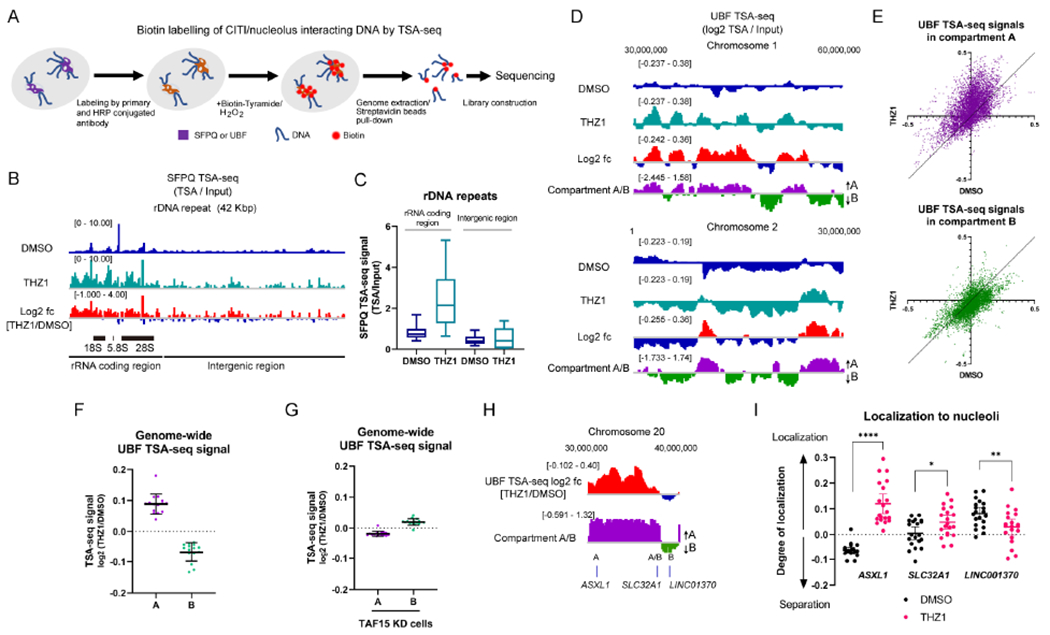
(A) The experimental scheme of the SFPQ/UBF TSA-seq.
(B, C) The genome view of SFPQ TSA-seq at the rDNA repeat unit aligned with the log 2 ratio of THZ1 and DMSO (log2 fc) (B). The quantification of signals at the rRNA coding region and intergenic region within the repeat unit is shown in (C). Representative results from two biological replicates are shown.
(D) Two representative genome views of UBF TSA-seq aligned with Compartment A/B prediction scores. The log 2 ratio of THZ1 and DMSO is also aligned. The TSA-seq signals were smoothed in a 1-Mb window so that the scale of changes fitted with that of the Compartment A/B profile. Representative results from two biological replicates are shown.
(E, F) The effect of THZ1 treatment on the genome-wide UBF TSA-seq signals were analyzed with the scatter plots (DMSO vs THZ1) (E) or the dot plots of the log2 ratio of THZ1 and DMSO (mean with SD, n=15 for data points from individual chromosomes) (F) in either Compartment A or B.
(G) The genome-wide UBF TSA-seq signals in TAF15 KD cells at either Compartment A or B were quantified as in (F).
(H, I) The log2 ratio of UBF TSA-seq signals (THZ1/DMSO) and compartment A/B prediction scores at ASXL1, SLC32A1, and LINC01370 (H). Colocalization between these gene loci and the DFC region of nucleoli identified by DKC1 were analyzed by FISH in untreated and THZ1-treated cells (I). The degree of localization was calculated by the divergence from background frequency and compared (mean with 95% CI, n=20 for data points from two biological replicates).
Next, we analyzed SFPQ TSA-seq signals outside of the rDNA repeats. Prior to THZ1 treatment, SFPQ displayed a profile correlated with Compartment A (Figure S5E and S5F), which is consistent with the binding of SFPQ to active genes (Figure S4B–H). After THZ1 treatment, we observed a general reduction of SFPQ TSA-seq signals with the spike-in control (Figure S5F and S5G), which is consistent with the reduced SFPQ staining outside of nucleoli (Figure S5A). However, when analyzed by depth normalization, the distribution of SFPQ TSA-seq signals remained largely unchanged after THZ1 treatment, and SFPQ remained proximal to Compartment A (Figure S5H–J). Thus, although the overall proximity of SFPQ to active chromatin is reduced by RNAPII inhibition, it stays bound to active genes as shown in ChIP-seq and remains proximal to Compartment A as shown in TSA-seq.
If Compartment A is tethered to nucleoli after RNAPII inhibition, the genomic regions associated with SFPQ should be increasingly proximal to the nucleolar marker UBF. Indeed, the UBF TSA-seq signals were significantly increased by THZ1 in Compartment A, but not in Compartment B (Figure 5D–F). Notably, recent Hi-C studies showed that Compartments A and B are not grossly altered by RNAPII inhibition (Jiang et al., 2020; Kloetgen et al., 2020), excluding the possibility that the changes observed in TSA-seq are due to the alterations of Compartments A and B. Furthermore, the increase of UBF signals in Compartment A was abolished by TAF15 knockdown (Figure 5G). Because TAF15 is recruited to CITIs by SFPQ/NONO (Figure S2K and S2L), this result suggests that TAF15 tethers Compartment A to CITIs.
To visualize the localization of Compartment A to CITIs, we used fluorescent in situ hybridization (FISH) to analyze several loci on chromosome 20 (Chr 20), including the ASXL1 gene in Compartment A, the SLC32A1 gene at a Compartment A/B boundary, and the LINC01370 gene in Compartment B (Figure 5H and S6A). The colocalization of ASXL1 with the nucleolar marker DKC1 was significantly increased by THZ1 treatment, whereas the colocalization of SLC32A1 and LINC001370 with DKC1 was only modestly increased or decreased (Figure 5I). The colocalization of ASXL1 with CITIs was observed when CITIs were induced by knockdown of FBL or U3 snoRNA (Figure S6B). Additional loci in Compartment A on chromosomes 2 and 7 colocalized with CITIs at higher frequencies than the loci in Compartment B on the same chromosomes (Figure S6C and S6D). Knockdown of TAF15 or FUS reduced the ASXL1-CITI colocalization to the level of SLC32A1-CITI colocalization, but it did not further decrease the SLC32A1-CITI colocalization (Figure S6E). Together, these results further support the notion that RNAPII inhibition preferentially localizes Compartment A to nucleoli.
Defective RNAPII transcription associates with gene fusions in tumors
Induced chromosomal proximity is an important mechanism driving gene fusions (Mani et al., 2009; Mitelman et al., 2007). The tethering of Compartment A to CITIs raises the possibility that compromised RNAPII transcription increases gene fusions at active genes. To test this possibility, we analyzed the gene fusions and RNA-seq data of all breast cancer, sarcoma, and lung cancer samples in The Cancer Genome Atlas (TCGA) database. The median expression level of each gene was calculated from all tumor samples of each cancer type, and all genes or the subset of genes implicated in fusion events (Yoshihara et al., 2015) were plotted according to their expression levels (Figure S7A). The distribution of the genes implicated in fusions was strikingly biased toward high expression (Figure S7A), suggesting that gene fusions are more likely to occur in highly expressed genes.
Knowing that gene fusions preferentially occur in active genes, we then asked whether the transcription factors regulating RNAPII are positively or negatively associated with gene fusions. For each transcription factor of interest, tumor samples were divided into high- and low-expression groups, and the numbers of gene fusions in the two groups were compared. High expression of the factors promoting the initiation or elongation of RNAPII transcription was associated with low frequency of gene fusion, whereas high expression of the factors inhibiting RNAPII elongation was associated with high frequency of gene fusion (Figure 6A). Thus, at highly expressed genes, compromised RNAPII transcription is associated with increased gene fusions in tumors.
Figure 6. Defective RNAPII Transcription Promotes Gene Fusions.
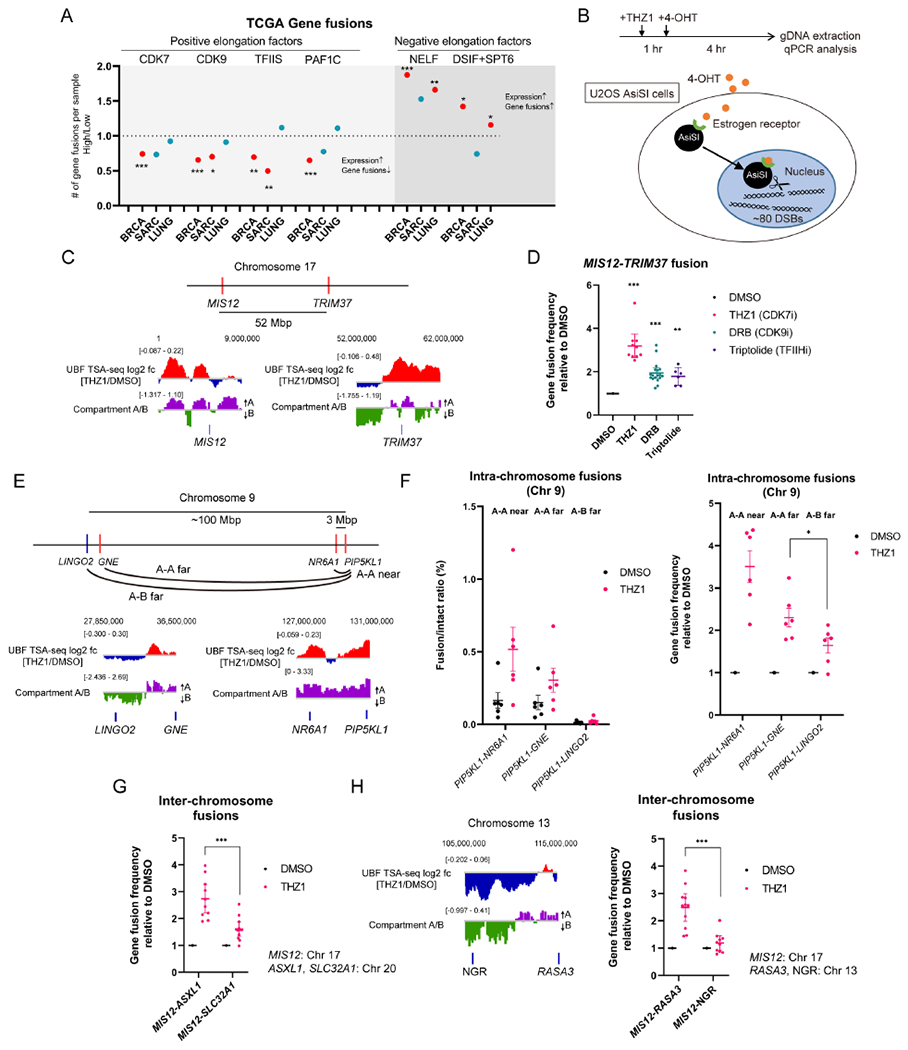
(A) The correlation between the indicated gene or complex expression levels and the number of gene fusions per sample was analyzed in the indicated TCGA datasets. The results were shown as the ratio of the average of gene fusions in high to low groups. A significant ratio over 1 indicates that the high expression of the gene of interest is associated with a higher number of gene fusions, while a ratio below 1 indicates that the low expression of the gene of interest is associated with the higher number of gene fusions. The significant correlation is shown in red. See also Table S2.
(B) A schematic representation of the system for chromosomal translocation detection using the U2OS AsiSI cell line. The timeline of experiments is shown.
(C) The approximate position of MIS12 and TRIM37 on Chr 17 and the log2 ratio of UBF TSA-seq signals (THZ1/DMSO) and Compartment A/B scores at these loci are shown.
(D) The effect of THZ1, DRB, or Triptolide treatment on the frequency of MIS12-TRIM37 fusion was analyzed (mean with 95% CI, n=10, 18, 6, left to right, for data points from more than four biological replicates).
(E) The approximate position of PIP5KL1, NR6A1, GNE, and LINGO2 on Chr 9 and the log2 ratio of UBF TSA-seq signals (THZ1/DMSO) and Compartment A/B scores at these loci are shown.
(F) The effect of THZ1 treatment on the frequency of intra-chromosome gene fusion between Compartment A-A (PIP5KL1-NR6A1, PIP5KL1-GNE) and Compartment A-B (PIP5KL1-LINGO2) was analyzed (mean with SEM, n=6 for data points of independent experiments).
(G) The effect of THZ1 treatment on the frequency of inter-chromosome gene fusion was compared between MIS12-ASXL1 and MIS12-SLC32A1 (mean with SEM, n=10, 12, left to right, for data points of independent experiments).
(H) The effect of THZ1 treatment on MIS12-RASA3 fusion and MIS12-NGR fusion was analyzed as in (G). (mean with SEM, n=11, 10, left to right, for data points of independent experiments). The approximate position of RASA3 and NGR on Chr 13 and the log2 ratio of UBF TSA-seq signals (THZ1/DMSO) and Compartment A/B scores at these loci are shown left.
RNAPII inhibition promotes gene fusion in Compartment A
To directly assess the effects of RNAPII inhibition on gene fusion, we used 4-OHT to induce nuclear import of the estrogen receptor (ER)-tagged AsiSI nuclease, which generates ~80 site-specific DNA double-strand breaks (DSBs) across the genome (Figure 6B) (Clouaire et al., 2018; Cohen et al., 2018; Iacovoni et al., 2010). The fusion between two AsiSI-cleaved genes on Chr 17, MIS12 (near a Compartment A/B boundary) and TRIM37 (in Compartment A), was measured by qPCR (Figure 6C and 6D). The MIS12-TRIM37 fusion was significantly increased by THZ1, DRB, and Triptolide (Figure 6D). Notably, the numbers of γH2AX foci (a marker of DSBs) and the proportion of cells with significant levels of RAD51 foci (a marker for homologous recombination) were unchanged by THZ1 or SFPQ knockdown up to 4 hr (the timepoint for detecting gene fusions) (Figure S7B and S7C), suggesting that DSB formation and repair are not significantly affected by RNAPII inhibition and the presence or absence of CITIs. In contrast to RNAPII inhibitors, neither Actinomycin D nor puromycin increased the MIS12-TRIM37 fusion (Figures S7D and S7E). These results suggest that selective inhibition of RNAPII, but not general inhibition of RNA polymerases or protein translation, increases gene fusion.
To test whether fusions of the genes in Compartment A are preferentially increased by RNAPII inhibition, we compared the frequency of three distinct intra-chromosome gene fusions between AsiSI-cleaved genes on Chr 9: PIP5KL1-NR6A1 (~3 Mb apart, Compartment A-A near), PIP5KL1-GNE (~100 Mb apart, Compartment A-A far), and PIP5KL1-LINGO2 (~100 Mb apart, Compartment A-B far) (Figure 6E). The frequencies of the Compartment A-A fusions were higher than that of the Compartment A-B fusion (Figure 6F, left). Moreover, the Compartment A-A fusions were more significantly stimulated by THZ1 treatment than the Compartment A-B fusion (Figure 6F, right). Given that the distances from PIP5KL1 to GNE and LINGO2 are similar, these data suggest that the difference in fusion frequency stems from the distinct compartmentalization of GNE and LINGO2.
We also compared two sets of inter-chromosomal gene fusions between MIS12 and AsiSI-cleaved genes in different compartments: ASXL1 (in Compartment A on Chr 20) and SLC32A1 (at a Compartment A/B boundary on Chr 20) (Figures 5H and 6G), RASA3 (in Compartment A on Chr 13) and NGR (in Compartment B on Chr 13) (Figure 6H). The gene fusions between MIS12-ASXL1 and MIS12-RASA3 were more significantly stimulated by THZ1 compared to MIS12-SLC32A1 and MIS12-NGR (Figures 6G and 6H), suggesting that genes in Compartment A are more prone to gene fusion. Notably, induction of DSBs at ASXL1 and SLC32A1 did not alter their localization to CITIs, suggesting that the tethering of active chromatin to CITIs is not driven by DSBs (Figure S7F). Thus, the localization of Compartment A to nucleoli upon RNAPII inhibition is associated with increased frequencies of intra-and inter-chromosomal gene fusions in this compartment.
Gene fusions occur in CITIs
To test whether CITIs contribute to the induction of gene fusions by RNAPII inhibition, we analyzed the effects of 1,6-hexanediol and CX5461, both of which inhibit CITI formation (Figures 3D, 3E, S3C, and S3D). Both 1,6-hexanediol and CX5461 reduced the THZ1-induced increase of the MIS12-TRIM37 fusion (Figures 7A and 7B). Furthermore, knockdown of SFPQ and NONO reduced the MIS12-TRIM37 fusion (Figures 7C and 7D), suggesting that the key components of CITIs play a critical role in the induction of gene fusions by THZ1. Consistently, the levels of SFPQ or NONO expression positively correlated with gene fusions in about half of the cancer types analyzed (Figure S7G). In contrast to SFPQ/NONO, knockdown of BRD4, a factor involved in enhancer-promoter interactions, or SRSF2, a nuclear speckle factor, did not affect the MIS12-TRIM37 fusion (Figures S7H and S7I). The MIS12-TRIM37 fusion was also reduced by knockdown of TAF15 and FUS (Figure 7E). These results are consistent with the roles of TAF15/FUS in tethering Compartment A to CITIs upon RNAPII inhibition (Figures 5G and S6E), which is associated with increased gene fusion.
Figure 7. CITIs Mediate Oncogenic Gene Fusions.
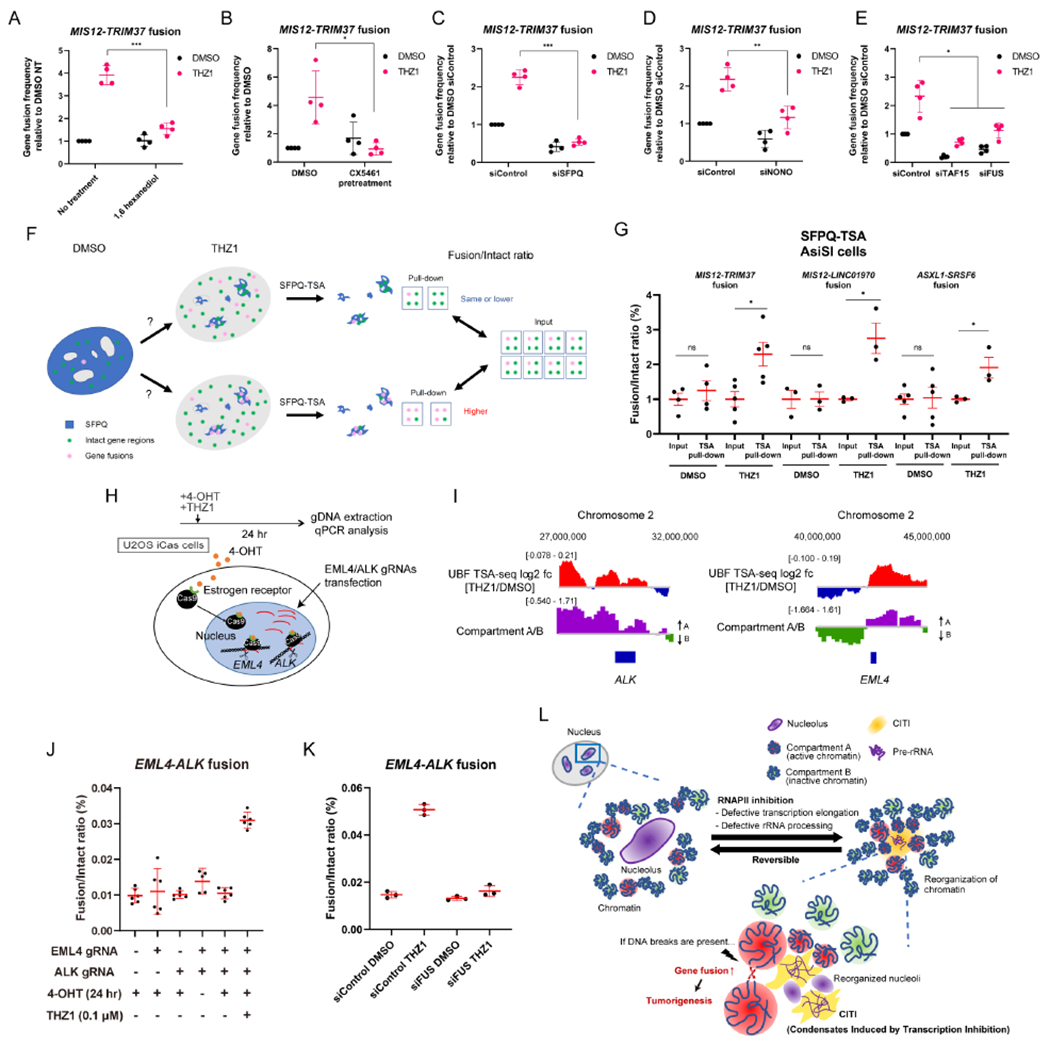
(A-B) The effect of pretreatment with 1,6 hexanediol (A) or CX5461 (pretreatment at 1 μM for 24 hr) (B) on the frequency of MIS12-TRIM37 fusion was analyzed (mean with SD, n=4 for data points from one representative experiment of three biological replicates).
(C-E) The effect of SFPQ KD (C), NONO KD (D), or TAF15/FUS KD (E) on the frequency of MIS12-TRIM37 fusion was analyzed (mean with SD, n=4 for data points from one representative experiment of three biological replicates).
(F) Two hypothetical outcomes of gene fusion detection by SFPQ TSA labelling. The total fusion events within the whole nucleus are detected in the input sample, while the fusion events at CITIs are detected in the pull-down sample.
(G) Gene fusions occurring in CITIs were detected by SFPQ TSA labelling. The intact/fusion ratio was compared between the input and pull-down samples in untreated and THZ1-treated cells. Three different gene fusions (MIS12-TRIM37, MIS12-LINC01970, ASXL1-SRSF6) were tested (mean with SEM, n=3-4 for data points of biological replicates).
(H) A schematic representation of the system for chromosomal translocation detection using the U2OS iCas cell line. The timeline of experiments is shown.
(I) The approximate position of ALK and EML4 on Chr 2 and the log2 ratio of UBF TSA-seq signals (THZ1/DMSO) and Compartment A/B scores at these loci are shown.
(J) The frequency of EML4-ALK fusion were analyzed by qPCR in the cells transfected with the indicated gRNAs and treated as indicated (mean with 95% CI, n=5-6 for data points from two biological replicates).
(K) The effect of FUS KD on the frequency of EML4-ALK fusion was analyzed (mean with SD, n=3 from one representative experiment of three biological replicates).
(L) A model for how defective RNAPII transcription leads to localization of Compartment A, but not B, to CITIs and promotes oncogenic gene fusions. See the text for details.
To directly test whether the increase of gene fusion upon RNAPII inhibition occurs in CITIs, we used SFPQ-TSA to capture the DNA proximal to SFPQ and qPCR to quantify gene fusions in the SFPQ-proximal DNA. If gene fusions occur randomly in the nucleus, the fusion/intact ratio should be similar in input DNA and the SFPQ-proximal DNA (Figure 7F, upper scenario). In contrast, if gene fusions preferentially occur in CITIs, the fusion/intact ratio should be higher in the SFPQ-proximal DNA (Figure 7F, lower scenario). For all the gene fusions between DSBs in Compartment A that we tested (MIS12-TRM37, MIS12-LINC01970, and ASXL1-SRSF6), the fusion/intact ratios were similar between input and SFPQ-proximal DNA in the absence of THZ1 (Figures 7G and S7J). However, in the presence of THZ1, the ratios were higher in the SFPQ-proximal DNA than in input DNA (Figure 7G), strongly suggesting that these fusions preferentially occur in CITIs. Thus, we propose that CITIs are an environment favoring gene fusion.
RNAPII inhibition increases oncogenic gene fusion
Finally, we asked whether the effects of RNAPII inhibition on gene fusion are relevant to the formation of fusion oncogenes. The EML4-ALK fusion is one of the most prevalent oncogenes in cancer (Bayliss et al., 2016; Lin et al., 2009). To conditionally generate DSBs in EML4 and ALK genes, we created a cell line (U2OS iCas) in which the ER-tagged Cas9 (ER-Cas9) is imported into the nucleus upon 4-OHT treatment (Figures 7H and S7K) (Liu et al., 2016). Both EML4 and ALK genes are in Compartment A, and both loci increasingly interacted with nucleoli upon THZ1 treatment in UBF TSA-seq (Figure 7I). When DSBs were induced at EML4 and ALK, only background levels of the EML4-ALK fusion were observed (Figures 7J, lanes 4 and 5, and S7L). However, when the cells carrying DSBs at EML4 and ALK were exposed to THZ1, a striking induction of EML4-ALK fusion was detected (Figures 7J, lane 6, and S7L). This induction of EML4-ALK fusion by THZ1 was abolished by knockdown of FUS (Figure 7K), which tethers Compartment A to CITIs. Together, these results demonstrate that RNAPII inhibition potentiates the formation of a fusion oncogene, suggesting that compromised RNAPII transcription and/or rRNA processing in cancer cells may contribute to the generation of fusion oncogenes.
Discussion
This study uncovers an unexpected role of RNAPII-mediated transcription in maintaining the proper compartmentalization of DNA and proteins in the nucleus (Figure 7L). SFPQ, NONO, TAF15, and FUS are the major components in both paraspeckles and CITIs (Naganuma et al., 2012; Sasaki et al., 2009; West et al., 2016; Yamazaki et al., 2018), suggesting that paraspeckles and CITIs are condensates formed by a similar group of LLPS proteins. Notably, paraspeckles and CITIs are different in their RNA scaffolds; while paraspeckles are assembled on NEAT1, CITIs are assembled on rRNA upon dissociation of rRNA processing factors. Given that depletion of rRNA processing factors is sufficient to induce CITIs without affecting RNAPII transcription, we speculate that RNAPII inhibition induces CITIs by perturbing rRNA processing. RNAPII inhibition may affect rRNA processing in several ways. First, RNAPII transcribes the U3/U8 box C/D snoRNAs, which are critical for rRNA processing. Second, ongoing RNAPII transcription is important for the transcription-coupled processing of box C/D snoRNAs and the assembly of snoRNPs (Richard and Kiss, 2006). Finally, the reduction in box C/D snoRNPs at nucleoli indirectly affects the binding of other factors to pre-rRNA. It is possible that the efficient binding of rRNA processing factors to pre-rRNA in nucleoli is mediated by LLPS (Yao et al., 2019). RNAPII inhibition may disrupt the existing mechanism of LLPS in nucleoli, thereby allowing CITIs to form on rRNA through an alternative LLPS mechanism.
While our results clearly demonstrate the formation of CITIs, the physiological functions of CITIs remains unknown. We show that CITIs are induced by physiologically relevant stresses such as mild cold shock and UV. CITIs are also induced by reduction in factors promoting RNAPII transcription, which occurs in cancer cells. Finally, CITIs are induced by a number of cancer therapeutics that inhibit RNAPII transcription. All these results suggest that the formation of CITIs is a general response to transcriptional and nucleolar stresses, including those generated in physiological, oncogenic, and therapeutic conditions. It is important to note that CITIs are dynamic and readily reversible. The formation of CITIs may provide a transient and reversible mechanism to cope with cellular stresses and promote efficient recovery. The exact functions of CITIs in the stress responses still need to be investigated in future studies.
It is interesting to note that nucleolar stress is commonly observed in cancer cells due to the fast cell cycling and proliferation (Pelletier et al., 2018; Sharifi and Bierhoff, 2018). The oncogenic stress induced by oncogene overexpression can also be a source of nucleolar stress (Nishimura et al., 2015). Furthermore, RNAPII transcription is often compromised in cancer cells due to alterations in the transcriptional complex, chromatin structure, and RNA processing (Modur et al., 2018). It is conceivable that the CITI-mediated response may help cancer cells cope with nucleolar and transcription problems, and disruption of the CITI-mediated response may sensitize cancer cells to these stresses. In addition, transcription elongation has recently emerged as an attractive target for cancer therapy (Liang et al., 2018). Inhibitors of several kinases involved in transcription elongation, such as CDK7, CDK9, and CDK12/13, are being tested in clinical trials (Chou et al., 2020). The cancer cells defective in the CITI-mediated response may be particularly vulnerable to problems in RNAPII transcription and therefore sensitive to transcription inhibitors.
Our results reveal that the effects of transcription on gene fusion are complex. On the one hand, high transcriptional activity has a negative impact on genome stability because highly expressed genes are prone to form gene fusions. Highly expressed genes may have a higher chance of DSB formation owing to transcription-replication conflicts and topoisomerase-generated DSBs (Chiarle et al., 2011; Dellino et al., 2019; Gomez-Gonzalez and Aguilera, 2019; Klein et al., 2011), which potentially increases the chance of gene fusions. On the other hand, transcription and transcribed RNAs have positive roles in promoting accurate DNA repair. Others and we have shown that transcription promotes homologous recombination in actively transcribed regions, which ensures the accurate repair of DSBs (Aymard et al., 2014; Keskin et al., 2014; Ohle et al., 2016; Ouyang et al., 2021; Yasuhara et al., 2018). Our results in this study also suggest that efficient transcription prevents gene fusions from DSBs, which occurs through inaccurate DSB repair. Thus, transcription is a double-edged sword that affects genomic integrity both positively and negatively. In future studies, it is important to consider the opposing effects of transcription on DNA repair in various physiological, pathological and therapeutic contexts.
Limitation of the Study
Our results still do not fully explain how CITIs are formed after RNAPII inhibition. How rRNA processing factors are rapidly lost from nucleoli after RNAPII inhibition still needs to be further explained. Moreover, we still don’t understand how the SFPQ/TAF15 associated with active chromatin are tethered to CITIs or interact with CITIs. Although our TSA-qPCR experiments show that the several gene fusions between DSBs in Compartment A are increased in CITIs, a comprehensive view of the interactions among DSBs in CITIs is still lacking. Finally, while the formation of CITIs is clearly associated with an increase of gene fusions, the causality between these molecular events still awaits further investigations.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lee Zou (lee.zou@mgh.harvard.edu).
Materials Availability
Materials generated in this study are available upon reasonable request.
Data and Code Availability
The sequencing datasets generated during this study are publicly available at GEO (GSE166623) as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Cell Lines
In this study, the U2OS cell line was used unless otherwise stated. The U2OS, HeLa, and MRC5 cell lines were purchased from ATCC. The RPE cell line was from Clontech. The U2OS AsiSI cell line was a generous gift from Dr. Legube. The DLD1 mAID-POLR2A-mClover cell line was a generous gift from Dr. Maeshima. U2OS and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 4 mM L-glutamine, and 1% penicillin/streptomycin. MRC5 and RPE cell lines were cultured in DMEM/F-12 medium supplemented with 10% FBS, 0.25% sodium bicarbonate, and 1% penicillin/streptomycin. All cell lines were cultured in a 37 °C incubator at 5% CO2.
METHOD DETAILS
Generation of Cell Lines
For the U2OS cell lines stably expressing GFP-TAF15 or GFP-FUS, U2OS cells were transfected with the pEGFP-C1 plasmid containing cDNA of TAF15 or FUS and selected with 500 μg mL−1 G418. The GFP-SFPQ cell line was developed using pX330-GFP-SFPQ and pUC57-GFP-SFPQ-RT plasmids (generated by Dr. Archa Fox, Addgene #97084, #97090). The U2OS iCas cell line was created by transfecting the iCas plasmid (gifted from Dr. Meng How Tan, Addgene #84232) to U2OS cells and selecting with orange fluorescent protein expression.
RNA Interference
Cells were transfected with the indicated siRNAs using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions and analyzed 48 or 72 hr after transfection. For NEAT1 knockdown, an equal amount mixture of two siRNA oligos was used. Oligonucleotides used in this study are listed in Table S3.
Immunofluorescence
The cells were pre-extracted with the CSK buffer (10 mM PIPES, pH 7.0, 100 mM NaCl, 300 mM Sucrose, and 3 mM MgCl2 containing 0.5% Triton X-100) for 5 min, and fixed with 3% paraformaldehyde (PFA) for 10 min. The cells were incubated with primary antibodies for 1 hr at 37 °C, followed by 1 hr incubation with secondary antibodies. The coverslips were mounted with VECTASHIELD (Vector Laboratories, H-1000) after counterstaining with 4’,6-diamidino-2-phenylindole (DAPI). For quantification, the CITI area in each nucleus was defined and measured using Image J. For SoRa and STORM, cells were grown to between 70–90% confluence on chambered coverslips and fixed as described above. Cells were blocked with 5% normal donkey serum (EMD Millipore), 0.02% (v/v) Triton X-100 in PBS for 4 hr at room temperature or overnight at 4 °C. Primary antibodies were applied at 1:500 dilutions in blocking buffer and incubated overnight at 4 °C. Secondary antibodies (donkey anti-rabbit IgG or anti-mouse IgG (Jackson Immuno Research)) were labeled with Alexa Fluor 647 (Thermo Fisher Scientific) for a dye ratio of ~1:1 as previously described (Schmider et al., 2019). An anti-GFP nanobody (NanoTag N0301-At488-L, Lot 03191102) stoichiometrically conjugated 1:1 with ATTO 488 (Thermo Fisher Scientific) was also used. Secondary antibodies were added at 3 μg mL−1 each in blocking buffer and incubated for 2 hr at room temperature in the dark. All subsequent steps were performed in the dark. Cells were washed with PBS 6 times for 5 min each. Antibody stacks were crosslinked by 4% PFA in PBS for 15 min. Remaining fixative was quenched and washed with 50 mM glycine in PBS twice for 5 min each, followed by PBS twice for 5 min each. Stained cells were stored at 4 °C for up to 2 weeks before imaging.
Optical Pixel Reassignment Spinning Disk Confocal Microscopy
The slide was mounted on the stage with type F immersion oil (refractive index = 1.515) on a Nikon Ti2 Eclipse inverted microscope. The microscope was equipped with a 60× 1.40 NA APO objective on a Nikon CSU-W1 SoRa system including 405 nm, 488 nm, 561 nm, and 647 nm (all 100 mW) lasers, a quadband excitation-emission filter, and a Photometrics Prime BSI camera. Nikon Elements 5.21.03 was used for image acquisition and processing. Z-stacks of images were obtained with a 100 nm step covering the entire cell volume. Each image was obtained with 200 ms exposure using FITC or Cy5 emission filters. Each channel was collected sequentially from longest wavelength to shortest. The images were denoised with Nikon Denoise.ai and deconvolved with Landweber algorithm. The processed image was converted to a volumetric rendering and maximum intensity projection.
Direct Stochastic Optical Reconstruction Microscopy
STORM imaging was essentially performed as described before (Bauer et al., 2021). Imaging buffer containing 10 mM cysteamine (2-mercaptoethylamine, Sigma-Aldrich), 3 U mL−1 pyranose oxidase from Coriolus sp. (Sigma-Aldrich P4234), and 90 U ml−1 catalase was freshly prepared in STORM buffer (10% (w/v) glucose, 10 mM sodium chloride, and 50 mM Tris-HCl pH 8). Cysteamine stock solution was previously titrated to pH 8 and aliquots frozen. Precipitate in pyranose oxidase/catalase 100x enzyme stock solution was cleared by centrifugation at over 14,000×g prior to use. PBS was replaced with the imaging buffer and the slide was mounted on the stage with type F immersion oil (refractive index = 1.515) on a Nikon Ti2 Eclipse inverted microscope. The microscope was equipped with a 100× 1.49 NA APO-TIRF objective with automatic correction collar and a Nikon NSTORM system including 405 nm (20 mW), 488 nm (70 mW), 561 nm (70 mW), and 647 nm (125 mW) lasers, a quadband excitation-emission filter, and a Hamamatsu ORCA Flash4.0 V2 S-CMOS camera. Nikon Elements 5.02 was used for image acquisition. A 10×10 tiled (with 15% overlap) widefield fluorescence image (~790×790 μm2) was obtained with 1 s exposure using GFPHQ, TexasRedHYQ, or Cy5HYQ filter cubes, from which random individual cells were selected for STORM imaging. At least 11000, 256x256 pixel (160 nm pixel−1) frames were collected with 10 ms exposure time at 100% laser power with lasers in highly inclined and laminated optical sheet (HILO) configuration (Tokunaga et al., 2008). Each channel was collected sequentially from longest wavelength to shortest. Localizations were identified with the Nikon NSTORM algorithm for downstream analysis, and with the overlapping points option for image reconstruction.
Immunofluorescence-Fluorescence in situ Hybridization
The THZ1-treated cells stained as described above were post-fixed with 1% PFA for 10 min and incubated at room temperature with 0.1 N HCl for 10 min. After 3 times wash, the cells were treated with RNase A (100 μg mL−1) in 2 × SSC for 1 hr. After equilibration with 50% formamide in 2 × SSC for 2 hr, the coverslips were incubated at 85 °C for 3 min with hybridization mix consisting of 2 μL of FISH probe and 8 μL of hybridization buffer (Empire Genomics), and incubated overnight at 37 °C. On the following day, cells were washed 3 times with 2 × SSC at 37 °C and 3 times with 0.1 × SSC at 60 °C before counterstained with DAPI. Ten images containing ~10 cells (~30-50 FISH foci in total) were randomly taken using a Nikon Eclipse 90i microscope (Nikon). For quantification, after defining the CITI/nucleolar area and the location of FISH foci within each image, the distance between each focus with any CITIs/nucleoli in the same cell was automatically calculated using a distance map function of Image J. The distance equal to 0 was considered as colocalization. The proportion of FISH foci that colocalize with CITIs or nucleoli in each image was normalized to the background frequency of colocalization calculated from the equation: (CITI or nucleolar area) / (total nuclear area). The normalized localization frequency was defined as ‘degree of localization’ in which positive values indicate localization to CITI/nucleoli whereas negative values indicate separation from CITI/nucleoli. The ‘degree of localization’ from one image was shown as one dot in the plots. In various experiments, twenty to thirty images from two to three biological replicates were analyzed per condition. Collectively, ~200-300 cells and ~600-1500 FISH foci are analyzed, and ~20-30 data points are generated per condition.
RNA in situ Hybridization
The cells were fixed with 4% PFA for 10 min and permeabilized with 70% ethanol for 1 hr at 4 °C. After washed with the wash buffer (2 × SSC, 10% formamide) for 5 min, cells were incubated at 37 °C overnight in the hybridization solution (2 × SSC, 100 mg mL−1 dextran sulfate, 10% formamide), containing 125 nM of the Stellaris NEAT1 RNA FISH probes (LGC Biosearch Technologies). The coverslips were washed with the wash buffer at 37 °C for 30 min and with 2 × SSC at room temperature for 5 min before counterstained with DAPI.
Live-Cell Imaging and FRAP Analysis
For live-cell imaging, the cells were plated on 12-well glass-bottom plates (MatTek). Immediately after the addition of the medium containing THZ1 (1 μM), cells were imaged every 2 min using an LSM 710 confocal microscope equipped with a ×63 oil objective and an incubation chamber supplemented with humidity and 5% CO2 (Zeiss). For FRAP analysis, areas of approximately 1.5 μm × 1.5 μm in size in CITIs or the nucleoplasm were bleached with a 488 nm laser at maximum power. The recovery of fluorescence was recorded every 2 sec after bleaching. The fluorescent recovery from the level of the first image right after bleaching was measured. The curves obtained from more than ten cells per experiment were fitted with a one-exponential non-linear regression model using a GraphPad Prism 8 software to calculate the diffusion coefficient.
Formaldehyde Crosslinking RNA Immunoprecipitation
U2OS cells (10 million) with DMSO or THZ1 treatment were harvested and suspended in 10 mL 1% formaldehyde in PBS for 10 min at 37 °C. Crosslinking was stopped by the addition of glycine to a final concentration of 0.25 M followed by incubation at room temperature for 5 min. After pelleting cells at 500 × g for 5 min, the cell pellet was resuspended in 1 mL RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% Sodium Deoxycholate, 1 mM PMSF, 2 mM RVC, Protease and Phosphatase Inhibitor Cocktail followed by sonication (15 min, 30 sec on and 30 sec off) at 4 °C. After centrifuging at 13,000 rpm for 10 min at 4 °C, the supernatant was pre-cleared with 15 μL Protein G Dynabeads and 20 μg mL−1 yeast tRNA at 4 °C for 30 min. The pre-cleared lysate was incubated with 20 μL beads that were pre-coated with 2 μg anti-SFPQ antibody or anti-mouse IgG for 3 hr at 4 °C. The beads were washed three times with washing buffer I (50 mM Tris pH 7.4, 1 M NaCl, 1% NP-40, 1% Sodium Deoxycholate), and three times with washing buffer II (50 mM Tris pH 7.4, 1 M NaCl, 1% NP-40, 1% Sodium Deoxycholate, 1 M Urea). The complex was eluted from beads by adding 150 μL elution buffer (100 mM Tris pH 8.0, 10 mM EDTA, 1% SDS). To reverse crosslinking, 6 μL of 5 M NaCl and 2 μL of 10 mg mL−1 proteinase K were added into the RNA samples and incubated at 42 °C for 1 hr followed by another hour incubation at 65 °C. RNA was extracted by Trizol and then reverse transcription was performed with High-Capacity RNA-to-cDNA™ Kit, followed by RT-qPCR analysis.
ChIP-Seq and Data Processing
ChIP assays were carried out on 5 million U2OS cells treated with DMSO or THZ1, following the procedures described previously (Mikkelsen et al., 2007). In brief, chromatin from formaldehyde-fixed cells was fragmented to a size range of 200–700 base pair (bp) with a Branson 250 sonicator. Then 30 ng Drosophila spike-in chromatin (Active Motif, 53083) was added to solubilized chromatin and chromatin complex was immunoprecipitated with 2 μg Spike-in antibody (Active Motif, 61686) and antibodies against SFPQ, TAF15, or normal mouse/rabbit IgG at 4 °C overnight. Antibody–chromatin complexes were pulled down with protein G Dynabeads, washed, and then eluted. After cross-link reversal and RNase A and proteinase K treatment, immunoprecipitated DNA was extracted with AMP Pure beads. ChIP DNA was quantified with Qubit. ChIP DNA samples and input samples (2 ng) were used to prepare sequencing libraries with Ultralow V2 DNA-Seq Library Preparation Kit, and DNA samples were sequenced with the Nextseq 500 Illumina genome analyzer. Sequences were separately aligned to either the human genome (hg19) or the D. melanogaster (dm6) using bwa (Li and Durbin, 2009). Sequences were also aligned to the human rDNA complete repeating unit (GenBank: U13369.1) (Zentner et al., 2011). Aligned reads were then filtered to exclude PCR duplicates and extended to 200 bp to approximate fragment sizes. The total number of sequences which uniquely mapping to the D. melanogaster genome is counted for each paired sample and used to calculate correction factors. Correction factors were then used for the scaling of human sequences (Egan et al., 2016). For the analysis of SFPQ or TAF15 association with genes, the gene list was downloaded from the UCSC table browser and formatted in a bed file so that each gene appears once in the table. The genes whose expression data were missing in the U2OS RNA-seq data were excluded. In total 19,710 genes (including both expressed (>0) and non-expressed (=0) genes) were sorted according to the RNA-seq values in the bed file. For peak calling, the MACS2 algorithm was used with the broadpeak option (Zhang et al., 2008). To create the plots and heatmaps, the computeMatrix and plotHeatmap functions of deepTools were used (Ramirez et al., 2016). The metrics for each experiment are shown in Table S1.
TSA-Mediated Labelling and Sequencing
For UBF labelling, U2OS cells (10-20 million) were fixed with 1.5% paraformaldehyde in PBS for 10 min and permeabilized with 0.5% Triton-X in PBS for 5 min at room temperature. For SFPQ labelling, cells were pre-extracted with CSK buffer for 5 min and fixed with 1.5% paraformaldehyde in PBS for 10 min. Cells were incubated with UBF antibody (1:100) for 2 hr at 37 °C or SFPQ antibody (1:1,000) overnight at 4 °C, followed by incubation with mouse/rabbit-HRP secondary antibody for 1 hr at 37 °C. After washing cells with 0.5% Tween-20 in PBS several times, cells were incubated with 25 μM Tyramide-Biotin, 0.0015% H2O2, 50% Sucrose in PBS for 30 min at room temperature. For imaging analysis, the target proteins were labelled with 1 μM Tyramide-Biotin (Zhang et al., 2020), followed by visualization using fluorescent labelled Streptavidin. After sequential wash with 0.5% Tween-20 in PBS, PBS, and 10mM Tris-HCl 10 mM EDTA, cells were scraped in lysis buffer (10mM Tris-HCl, 10 mM EDTA, 1% SDS, 100 mM NaCl) and incubated at 65 °C overnight to reverse the crosslink. After genomic DNA (gDNA) extraction and sonication, 20-30 μg of gDNA per condition was subjected to pull-down by Dynabeads M-280 Streptavidin for 2 hr at room temperature in rotation. For the spike-in control, Drosophila gDNA (Zyagen, GD-290) was biotinylated using PHOTOPROBE Biotin for Nucleic Acid Labeling kit (Vector Laboratory, SP-1000) according to the manufacturer’s instructions. The biotinylated Drosophila gDNA and extracted gDNA after TSA labeling were mixed at a ratio of 1:1,000 and subjected to pulldown. The pull-down DNA was eluted from beads by incubating with 95% formamide, 1 mM EDTA for 3 min at 95 °C and purified using QIAquick PCR Purification Kit, followed by library construction or qPCR. The library construction, sequencing, mapping of reads, and scaling by spike-in control were performed for both pull-down DNA and Input samples in parallel as described in the ChiP-seq section. Aligned reads were normalized by the corresponding Input sample and averaged in a 1-Mb window for each 100-bp bin. The Compartment A/B was predicted by calculating eigenvectors according to the publicly available Hi-C dataset in U2OS cell line (Kang et al., 2020). Chr 6, 9, 13, 15, 18, 19, 21, X, and Y were excluded from the genome-wide analysis because the prediction of Compartment A/B was not efficient in these chromosomes. To normalize the variability among chromosomes, the TSA signals were normalized by the median of the signals per chromosome. The metrics for each experiment are shown in Table S1.
Database Analysis
The RNA-sequence data for each sample provided by TCGA project were obtained from the Genomic Data Commons Data Portal. The gene fusion data for each sample were obtained from TCGA Fusion Gene Data Portal. For the analysis of the distribution of the expression levels, the median of expression levels for all the genes annotated by ENSG ID (N=60483) was calculated from all the samples in the dataset (BRCA N=1109, LUNG N=1086, SARC N=498) based on RNA-seq data. The genes whose median expression was 0 were excluded from the analysis (the number of genes included in the analysis: BRCA N=31724, LUNG N=32192, SARC N=28720). The genes that underwent gene fusions in any samples whose median expression was not 0 (BRCA N=4624, LUNG N=3653, SARC N=2178) were listed, and the distribution of their median expression together with that of all genes was plotted. The proportion of fusion genes in each expression bin was also calculated. For the analysis of the correlation between gene expression and gene fusion, the expression low and high groups were divided so that the ratio (high/low) of median expression of the gene was the lowest value or not less than 1.5. The mean of gene fusions per sample in the high group was normalized by that of the low group in the figures. The original data before normalization are shown in Table S2.
Gene Fusion Assay
The U2OS AsiSI cell line was treated with 4-hydroxytamoxifen (4-OHT, 300 nM) for 4 hr before gDNA extraction. The inhibitors were added 1 hr before 4-OHT treatment unless otherwise stated. For EML4-ALK fusion assay, the DNA fragment containing the gRNA sequence targeting EML4 or ALK locus was inserted into pLH-sgRNA1 vector (gifted from Dr. Thoru Pederson, Addgene #75388). One day after transfection with gRNAs, U2OS iCas cells were treated with 4-OHT (1 μM) and THZ1 (0.1 μM) and incubated for 24 hr before genomic DNA extraction. The gDNA was extracted using PureLink Genomic DNA Mini Kit and about 500 ng of gDNA was used as a template for the qPCR reactions, which typically produced Ct values around 27-30 for a fusion product. In most cases, two flanking primers per DNA break site were designed. The fusion products were quantified by calculating the fusion/intact ratio: the signals from primer sets across two DNA break sites (fusion) were normalized to the signals from the primer sets flanking the DNA break site (intact).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise stated, the results were tested by two-tailed Welch’s t-test. The results of the test were shown in the graph as ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****p < 0.0001. In the box plots, the median is shown in the center line, the 75/25 percentiles at the box limits and the 90/10 percentile at the whiskers as determined by GraphPad Prism 8 software. Three-dimensional analysis of SoRa images was done using the General Analysis 3 module of Nikon Elements 5.21.03 as follows: 1) objects identified per cell using manually selected thresholds (settings: 1x smoothing, 2x cleaning, fill holes); 2) objects circularly dilated with radii 0.05 μm, 0.1 μm, and 0.2 μm for DKC1, TAF15, and NPM1, respectively; 3) volumetric intersection between objects found; 4) get surface areas and volumes; 5) sum the surface areas and average the volumes; 6) divide the intersection surface area by 2 to approximate the external surface area.
Supplementary Material
Video 1. Formation of TAF15 Condensates upon RNAPII Inhibition, related to Figure 1
The cells expressing GFP-TAF15 were imaged every 2 min from 0 min (immediately after THZ1 addition to the medium) to 60 min. The movie is rendered at 7 frames per second. Scale bar, 10 μm.
Video 2. Formation of FUS Condensates upon RNAPII Inhibition, related to Figure 1
The cells expressing GFP-FUS were imaged every 2 min from 0 min (immediately after THZ1 addition to the medium) to 60 min. The movie is rendered at 7 frames per second. Scale bar, 10 μm.
Video 3. 3-D Visualization of TAF15, DKC1, and NPM1 Localization, related to Figure 2
The 3-D rendering video of a 3-color IF image taken by SoRa. TAF15, green; DKC1, purple; NPM1, blue.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-TAF15 | abcam | Cat# ab134916; RRID:AB_2614922 |
| Rabbit Anti-SFPQ | abcam | Cat# ab177149; RRID:N/A |
| Rabbit Anti-Nucleolin | abcam | Cat# ab22758; RRID:AB_776878 |
| Mouse Anti-SFPQ | abcam | Cat# ab11825; RRID:AB_298607 |
| Mouse Anti-PML | abcam | Cat# ab96051; RRID:AB_10679887 |
| Rabbit Anti-FBL | Cell Signaling | Cat# 2639; RRID:AB_2278087 |
| Rabbit Anti-DKC1 | Gene Tex | Cat# GTX109000; RRID:AB_11165396 |
| Rabbit Anti-TDP-43/TARDBP | Novus Biologicals | Cat# NB110-55376SS; RRID:AB_838957 |
| Rabbit Anti-Phospho-RNA polymerase II CTD Ser2 | Novus Biologicals | Cat# NB100-1805; RRID:AB_10001499 |
| Mouse Anti-SC-35 | Novus Biologicals | Cat# NB100-1774; RRID:AB_10128431 |
| Mouse Anti-NOP56 | Novus Biologicals | Cat# NBP2-36778; RRID:N/A |
| Mouse Anti-hnRNPA1 | Novus Biologicals | Cat# NB100-672SS; RRID:AB_10003087 |
| Mouse Anti-EWSR1 | Novus Biologicals | Cat# NBP1-92686SS; RRID:AB_11008755 |
| Mouse Anti-Nucleolin | Novus Biologicals | Cat# NBP2-44612; RRID:N/A |
| Rabbit Anti-NONO | Proteintech | Cat# 11058-1-AP; RRID:AB_2152167 |
| Rabbit Anti-NHP2L1 | Proteintech | Cat# 15802-1-AP; RRID:AB_2251452 |
| Mouse Anti-FUS/TLS | Santa Cruz | Cat# sc-47711; RRID:AB_2105208 |
| Mouse Anti-UBF | Santa Cruz | Cat# sc-13125; RRID:AB_671403 |
| Mouse Anti-RPA194 | Santa Cruz | Cat# sc-48385; RRID:AB_675814 |
| Mouse Anti-U3-55k | Santa Cruz | Cat# sc-515661; RRID:N/A |
| Mouse Anti-NPM1 | Sigma-Aldrich | Cat# B0556; RRID:AB_2154872 |
| Mouse Anti-MYC | MBL | Cat# M192-3; RRID:N/A |
| Anti-GFP FluoTag-Q ATTO488 | NanoTag | N0301-At488-L; RRID:N/A |
| Anti-mouse IgG | Santa Cruz | Cat# sc-2025; RRID:AB_737182 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# SML1666 |
| THZ1 | Sigma-Aldrich | Cat# 532372 |
| DRB | Sigma-Aldrich | Cat# D1916 |
| Triptolide | Sigma-Aldrich | Cat# T3652 |
| Actinomycin D | Sigma-Aldrich | Cat# A1410 |
| Formaldehyde | Sigma-Aldrich | Cat# F8775 |
| Biotinyl tyramide | Sigma-Aldrich | Cat# SML2135 |
| Hydrogen peroxide solution | Sigma-Aldrich | Cat# H1009 |
| RVC | New England Biolabs | Cat# S1402S |
| CX5461 | Selleck Chemicals | Cat# S2684 |
| Trizol | Thermo Fisher Scientific | Cat# 15596026 |
| Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78445 |
| yeast tRNA | Thermo Fisher Scientific | Cat# AM7119 |
| Protein G Dynabeads | Thermo Fisher Scientific | Cat# 10004D |
| RNase A | TaKaRa | Cat# 740505 |
| Proteinase K | Thermo Fisher Scientific | Cat# 25530049 |
| AMP Pure beads | Beckman Coulter | Cat# A63881 |
| SPRISelect beads | Beckman Coulter | Cat# B23317 |
| RNAiMAX | Thermo Fisher Scientific | Cat# 13778150 |
| Critical Commercial Assays | ||
| PureLink™ Genomic DNA Mini Kit | Thermo Fisher Scientific | Cat# K182002 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28106 |
| Ultralow V2 DNA-Seq Library Preparation Kit | NuGEN | Cat# 0344NB-A01 |
| High-Capacity RNA-to-cDNA™ Kit | Applied Biosystems | Cat# 4387406 |
| Deposited Data | ||
| SFPQ/TAF15 ChIP-seq | This paper | GEO: GSE166623, Table S1 |
| UBF/SFPQ TSA-seq | This paper | GEO: GSE166623, Table S1 |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | HTB-96 |
| RPE | Clontech | N/A |
| HeLa | ATCC | CCL-2 |
| MRC5 | ATCC | CCL-171 |
| U2OS AsiSI | Gifted from Dr. Legube | N/A |
| U2OS GFP-TAF15 | This study | N/A |
| U2OS GFP-FUS | This study | N/A |
| U2OS GFP-SFPQ | This study | N/A |
| U2OS iCas | This study | N/A |
| DLD1 mAID-POLR2A-mClover | Gifted from Dr. Maeshima | N/A |
| Oligonucleotides | ||
| See Table S3 | This paper | N/A |
| Plasmids | ||
| pX330-GFP-SFPQ | Gifted from Dr. Archa Fox | Addgene #97084 |
| pUC57-GFP-SFPQ-RT | Gifted from Dr. Archa Fox | Addgene #97090 |
| iCas | Gifted from Dr. Meng How Tan | Addgene #84232 |
| pLH-sgRNA1 | Gifted from Dr. Thoru Pederson | Addgene #75388 |
| pEGFP-C1-TAF15 | This study | N/A |
| pEGFP-C1-FUS | This study | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| FIJI | Schindelin et al., 2012 | https://fiji.sc/ |
| Galaxy | Afgan et al., 2018 | https://usegalaxy.org/ |
| Integrative Genomics Viewer | Robinson et al. 2011 | http://software.broadinstitute.org/software/igv/ |
| GraphPad Prism 8 | GraphPad Software, Inc | https://www.graphpad.com/scientificsoftware/prism/ |
| NIS elements viewer | Nikon | https://www.microscope.healthcare.nikon.com/ |
Highlights.
SFPQ, NONO, TAF15, and FUS form condensates in nucleoli upon RNAPII inhibition
RNAPII inhibition compromises rRNA processing and enables SFPQ to bind rRNA
SFPQ associates with active genes and localizes active chromatin to nucleoli
Oncogenic gene fusions increase in condensates induced by transcription inhibition
Acknowledgments
We thank Dr. N. Dyson and members of the Zou and Dyson labs for discussions. We thank Dr. Maeshima for the DLD1 mAID-POLR2A-mClover cell line. T.Y. is a JSPS Scientist for Joint International Research (JP20KK0339) and supported by a JSPS overseas fellowship (201860233). M.N.R. is supported by the Thomas F. and Diana L. Ryan MGH Research Scholar Award. L.Z. is the James & Patricia Poitras Endowed Chair in Cancer Research. This work is supported by grants from the NIH (CA263934 and CA218856 to L.Z.; AR065538 to R.J.S. and CA193520 to R.J.S. and N.C.B.; DK007540 to R.J.S. and N.C.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
All authors declare no competing interests. L.Z. is a member of the advisory board of Molecular Cell.
References
- Abraham KJ, Khosraviani N, Chan JNY, Gorthi A, Samman A, Zhao DY, Wang M, Bokros M, Vidya E, Ostrowski LA, et al. (2020). Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 585, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Gruning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, Orth AP, Busby J, Hogenesch JB, and Conkright MD (2007). A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl. Acad. Sci. USA 104, 20314–20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. (2014). Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol 21, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Wojtowicz D, Cui K, Devaiah BN, Chung HJ, Chan-Salis KY, Guha R, Wilson K, Zhang X, Zhang H, et al. (2016). RNA Polymerase II Regulates Topoisomerase 1 Activity to Favor Efficient Transcription. Cell 165, 357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer NC, Yang A, Wang X, Zhou Y, Klibanski A, and Soberman RJ (2021). A cross-nearest neighbor/Monte Carlo algorithm for single-molecule localization microscopy defines interactions between p53, Mdm2, and MEG3. J. Biol. Chem 296, 100540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss R, Choi J, Fennell DA, Fry AM, and Richards MW (2016). Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell. Mol. Life Sci 73, 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaude O (2011). Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert FM, van Koningsbruggen S, Navascues J, and Lamond AI (2007). The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol 8, 574–585. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, and Belmont AS (2018). Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J. Cell Biol 217, 4025–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Quigley DA, Robinson TM, Feng FY, and Ashworth A (2020). Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 10, 351–370. [DOI] [PubMed] [Google Scholar]
- Clouaire T, Rocher V, Lashgari A, Arnould C, Aguirrebengoa M, Biernacka A, Skrzypczak M, Aymard F, Fongang B, Dojer N, et al. (2018). Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 72, 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Puget N, Lin YL, Clouaire T, Aguirrebengoa M, Rocher V, Pasero P, Canitrot Y, and Legube G (2018). Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun 9, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellino GI, Palluzzi F, Chiariello AM, Piccioni R, Bianco S, Furia L, De Conti G, Bouwman BAM, Melloni G, Guido D, et al. (2019). Release of paused RNA polymerase II at specific loci favors DNA double-strand-break formation and promotes cancer translocations. Nat. Genet 51, 1011–1023. [DOI] [PubMed] [Google Scholar]
- Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. (2011). Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, Maile TM, Busby J, Henry C, Kelly TK, et al. (2016). An Alternative Approach to ChIP-Seq Normalization Enables Detection of Genome-Wide Changes in Histone H3 Lysine 27 Trimethylation upon EZH2 Inhibition. PLoS One 11, e0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, and Ingles CJ (2002). Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA 8, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Nakagawa S, Hirose T, and Bond CS (2018). Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci 43, 124–135. [DOI] [PubMed] [Google Scholar]
- Gibcus JH, and Dekker J (2013). The hierarchy of the 3D genome. Mol. Cell 49, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalez B, and Aguilera A (2019). Transcription-mediated replication hindrance: a major driver of genome instability. Genes Dev. 33, 1008–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granick D (1975a). Nucleolar necklaces in chick embryo fibroblast cells. I. Formation of necklaces by dichlororibobenzimidazole and other adenosine analogues that decrease RNA synthesis and degrade preribosomes. J. Cell Biol 65, 398–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granick D (1975b). Nucleolar necklaces in chick embryo fibroblast cells. II. Microscope observations of the effect of adenosine analogues on nucleolar necklace formation. J. Cell Biol 65, 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granick S, and Granick D (1971). Nucleolar necklaces in chick embryo myoblasts formed by lack of arginine. J. Cell Biol 51, 636–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison AF, and Shorter J (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem. J 474, 1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa M, Takeuchi A, Tanihata J, lida K, Takeda S, and Hagiwara M (2019). Loss of RNA-Binding Protein Sfpq Causes Long-Gene Transcriptopathy in Skeletal Muscle and Severe Muscle Mass Reduction with Metabolic Myopathy. iScience 13, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, and Legube G (2010). High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 29, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Huang J, Lun K, Li B, Zheng H, Li Y, Zhou R, Duan W, Wang C, Feng Y, et al. (2020). Genome-wide analyses of chromatin interactions after the loss of Pol I, Pol II, and Pol III. Genome Biol. 21, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone SE, Reyes A, Qi Y, Adriaens C, Hegazi E, Pelka K, Chen JH, Zou LS, Drier Y, Hecht V, et al. (2020). Large-Scale Topological Changes Restrain Malignant Progression in Colorectal Cancer. Cell 182, 1474–1489 e1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Shokhirev MN, Xu Z, Chandran S, Dixon JR, and Hetzer MW (2020). Dynamic regulation of histone modifications and long-range chromosomal interactions during postmitotic transcriptional reactivation. Genes Dev. 34, 913–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, and Storici F (2014). Transcript-RNA-templated DNA recombination and repair. Nature 515, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. (2011). Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, Chen X, Hu H, Bakogianni S, Wang J, et al. (2020). Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Gene 52, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JR, Bastide A, Peretti D, Roobol A, Roobol J, Mallucci GR, Smales CM, and Willis AE (2016). Cooling-induced SUMOylation of EXOSC10 down-regulates ribosome biogenesis. RNA 22, 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, and Simon A (2017). Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters, 10.19185/matters.201702000010. [DOI] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, and McKnight SL (2013). Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhendries JL, Nicolas E, Doumont G, Goldman S, and Lafontaine DL (2016). The human box C/D snoRNAs U3 and U8 are required for pre-rRNA processing and tumorigenesis. Oncotarget 7, 59519–59534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lans H, Hoeijmakers JHJ, Vermeulen W, and Marteijn JA (2019). The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol 20, 766–784. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Smith ER, Aoi Y, Stoltz KL, Katagi H, Woodfin AR, Rendleman EJ, Marshall SA, Murray DC, Wang L, et al. (2018). Targeting Processive Transcription Elongation via SEC Disruption for MYC-Induced Cancer Therapy. Cell 175, 766–779 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, Pandita A, Tang J, and Modrusan Z (2009). Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol. Cancer Res 7, 1466–1476. [DOI] [PubMed] [Google Scholar]
- Liu KI, Ramli MN, Woo CW, Wang Y, Zhao T, Zhang X, Yim GR, Chong BY, Gowher A, Chua MZ, et al. (2016). A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing. Nat. Chem. Biol 12, 980–987. [DOI] [PubMed] [Google Scholar]
- Ma C, Karwacki-Neisius V, Tang H, Li W, Shi Z, Hu H, Xu W, Wang Z, Kong L, Lv R, et al. (2016). Nono, a Bivalent Domain Factor, Regulates Erk Signaling and Mouse Embryonic Stem Cell Pluripotency. Cell Rep. 17, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, Palanisamy N, and Chinnaiyan AM (2009). Induced chromosomal proximity and gene fusions in prostate cancer. Science 326, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YS, Sunwoo H, Zhang B, and Spector DL (2011). Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol 13, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2007). Beyond the sequence: cellular organization of genome function. Cell 128, 787–800. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, and Mertens F (2007). The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–245. [DOI] [PubMed] [Google Scholar]
- Mitrea DM, and Kriwacki RW (2016). Phase separation in biology; functional organization of a higher order. Cell Commun. Signal 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modur V, Singh N, Mohanty V, Chung E, Muhammad B, Choi K, Chen X, Chetal K, Ratner N, Salomonis N, et al. (2018). Defective transcription elongation in a subset of cancers confers immunotherapy resistance. Nat. Commun 9, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma T, Nakagawa S, Tanigawa A, Sasaki YF, Goshima N, and Hirose T (2012). Alternative 3’-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 31, 4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima R, Hibino K, Ashwin SS, Babokhov M, Fujishiro S, Imai R, Nozaki T, Tamura S, Tani T, Kimura H, et al. (2019). Single nucleosome imaging reveals loose genome chromatin networks via active RNA polymerase II. J. Cell Biol 218, 1511–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Yamazaki T, and Hirose T (2018). Molecular dissection of nuclear paraspeckles: towards understanding the emerging world of the RNP milieu. Open Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Kumazawa T, Kuroda T, Katagiri N, Tsuchiya M, Goto N, Furumai R, Murayama A, Yanagisawa J, and Kimura K (2015). Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. 10, 1310–1323. [DOI] [PubMed] [Google Scholar]
- Ohle C, Tesorero R, Schermann G, Dobrev N, Sinning I, and Fischer T (2016). Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 167, 1001–1013 e1007. [DOI] [PubMed] [Google Scholar]
- Ouyang J, Yadav T, Zhang JM, Yang H, Rheinbay E, Guo H, Haber DA, Lan L, and Zou L (2021). RNA transcripts stimulate homologous recombination by forming DR-loops. Nature 594, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pageon SV, Nicovich PR, Mollazade M, Tabarin T, and Gaus K (2016). Clus-DoC: a combined cluster detection and colocalization analysis for single-molecule localization microscopy data. Mol. Biol. Cell 27, 3627–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Thomas G, and Volarevic S (2018). Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat. Rev. Cancer 18, 51–63. [DOI] [PubMed] [Google Scholar]
- Pommier Y (2006). Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, and Kiss T (2006). Integrating snoRNP assembly with mRNA biogenesis. EMBO Rep. 7, 590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YT, Ideue T, Sano M, Mituyama T, and Hirose T (2009). MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 106, 2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmider AB, Vaught M, Bauer NC, Elliott HL, Godin MD, Ellis GE, Nigrovic PA, and Soberman RJ (2019). The organization of leukotriene biosynthesis on the nuclear envelope revealed by single molecule localization microscopy and computational analyses. PLoS One 14, e0211943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Cech TR, and Parker RR (2015). Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem 84, 355–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi S, and Bierhoff H (2018). Regulation of RNA Polymerase I Transcription in Development, Disease, and Aging. Annu. Rev. Biochem 87, 51–73. [DOI] [PubMed] [Google Scholar]
- Shav-Tal Y, Blechman J, Darzacq X, Montagna C, Dye BT, Patton JG, Singer RH, and Zipori D (2005). Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell 16, 2395–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357. [DOI] [PubMed] [Google Scholar]
- Sordet O, Larochelle S, Nicolas E, Stevens EV, Zhang C, Shokat KM, Fisher RP, and Pommier Y (2008). Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription- and BRCA1-dependent degradation of topoisomerase I. J. Mol. Biol 381, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, and Sakata-Sogawa K (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen JY, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Furlong EEM (2019). The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol 20, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, and Kingston RE (2014). The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 55, 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Mito M, Kurosaka S, Takumi T, Tanegashima C, Chujo T, Yanaka K, Kingston RE, Hirose T, Bond C, et al. (2016). Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization. J. Cell Biol 214, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S , Pierron G, and Hirose T (2018). Functional Domains of NEAT1 Architectural IncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 70, 1038–1053 e1037. [DOI] [PubMed] [Google Scholar]
- Yao RW, Xu G, Wang Y, Shan L, Luan PF, Wang Y, Wu M, Yang LZ, Xing YH, Yang L, et al. (2019). Nascent Pre-rRNA Sorting via Phase Separation Drives the Assembly of Dense Fibrillar Components in the Human Nucleolus. Mol. Cell 76, 767–783 e711. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Kato R, Hagiwara Y, Shiotani B, Yamauchi M, Nakada S, Shibata A, and Miyagawa K (2018). Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 175, 558–570. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, and Verhaak RG (2015). The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 34, 4845–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Saiakhova A, Manaenkov P, Adams MD, and Scacheri PC. (2011). Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 39, 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang Y, Chen Y, Gholamalamdari O, Wang Y, Ma J, and Belmont AS (2020). TSA-seq reveals a largely conserved genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. Genome Res. 31, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Formation of TAF15 Condensates upon RNAPII Inhibition, related to Figure 1
The cells expressing GFP-TAF15 were imaged every 2 min from 0 min (immediately after THZ1 addition to the medium) to 60 min. The movie is rendered at 7 frames per second. Scale bar, 10 μm.
Video 2. Formation of FUS Condensates upon RNAPII Inhibition, related to Figure 1
The cells expressing GFP-FUS were imaged every 2 min from 0 min (immediately after THZ1 addition to the medium) to 60 min. The movie is rendered at 7 frames per second. Scale bar, 10 μm.
Video 3. 3-D Visualization of TAF15, DKC1, and NPM1 Localization, related to Figure 2
The 3-D rendering video of a 3-color IF image taken by SoRa. TAF15, green; DKC1, purple; NPM1, blue.
Data Availability Statement
The sequencing datasets generated during this study are publicly available at GEO (GSE166623) as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.