

Abstract
Plastins/fimbrins are conserved actin-bundling proteins contributing to motility, cytokinesis, and other cellular processes by organizing strikingly different actin assemblies as in aligned bundles and branched networks. We propose that this ability of human plastins stems from an allosteric communication between their actin-binding domains (ABD1/2) engaged in a tight spatial association. Here we show that ABD2 can bind actin three orders of magnitude stronger than ABD1, unless the domains are involved in an equally strong inhibitory engagement. A mutation mimicking physiologically relevant phosphorylation at the ABD1-ABD2 interface greatly weakened their association, dramatically potentiating actin cross-linking. Cryo-EM reconstruction revealed the ABD1-actin interface and enabled modeling of the plastin bridge and domain separation in parallel bundles. We predict that a strong and tunable allosteric inhibition between the domains allows plastins to modulate the cross-linking strength, contributing to remodeling of actin assemblies of different morphologies defining the unique place of plastins in actin organization.
Introduction
Plastins/fimbrins, are conserved actin-bundling proteins involved in cell migration, cytokinesis, and endocytosis1,2. Vertebrates express three tissue-specific plastin isoforms. Plastin 1 (PLS1; a.k.a. I-plastin) is primarily found in brush border microvilli of the intestine and kidneys3,4 and in stereocilia of the inner ear hair cells5,6. Genetic defects in PLS1 gene are linked to hearing loss7,8. Plastin 2 (PLS2; a.k.a. L-plastin, LCP1, LPL) is expressed in hematopoietic cells contributing to immune cell activation, migration, and invasion9–15. Congenital diseases associated with PLS2 mutations are not known, but, intriguingly, PLS2 is ectopically expressed in ~70% of epithelial cancers, where it localizes to the leading edge and invadopodia contributing to metastasis16–20. Plastin 3 (PLS3; a.k.a. T-plastin) is expressed by most solid tissues, where it is enriched at the cell edge21,22, specialized adhesion contacts23, distal parts of filopodia24, sites of endocytosis25, and in the contractile actin cortex formed upon membrane blebbing repair26. Deletions and truncations in PLS3, as well as point mutations perturbing its Ca2+ sensitivity, lead to X-linked congenital osteoporosis with bone fragility22,27. By contributing to endocytosis28 and migration, PLS3 plays neuroprotective roles by lessening the toxicity of pathogenic poly-glutamine-containing proteins29, alleviating symptoms of spinal muscular atrophy30,31, and coordinating neuronal cell migration in embryogenesis32. PLS3 is also an oncogene, whose ectopic expression in hematopoietic cells33,34 and overexpression in cells of solid tissues promote cancer cell proliferation, motility, and drug resistance35,36.
Plastins belong to a superfamily of calponin-homology (CH)-domain-containing actin cytoskeleton organizers, which include filamins, spectrins, α-actinins, IQGAP, and other proteins37. Most members of the superfamily contain a single actin-binding domain (ABD) and must form dimers or higher-order oligomers to tether actin filaments into bundles and meshwork. Plastins are unique in having two ABDs as a monomer (Fig. 1a). Each ABD consists of two CH-domains. All plastins contain an N-terminal regulatory domain (RD) comprised of two EF-hands followed by an EF-hand-binding motif (EBM;38,39) and a disordered linker connecting the regulatory domain to the ABD1-ABD2 core (Fig. 1a). In mammalian plastins, the actin-bundling activity is inhibited upon Ca2+ binding to EF-hands4,30,40, while some fimbrins are insensitive to Ca2+ 41–43.
Figure 1. ABD2 of PLS2 binds to F-actin with nanomolar affinity and nucleates actin.
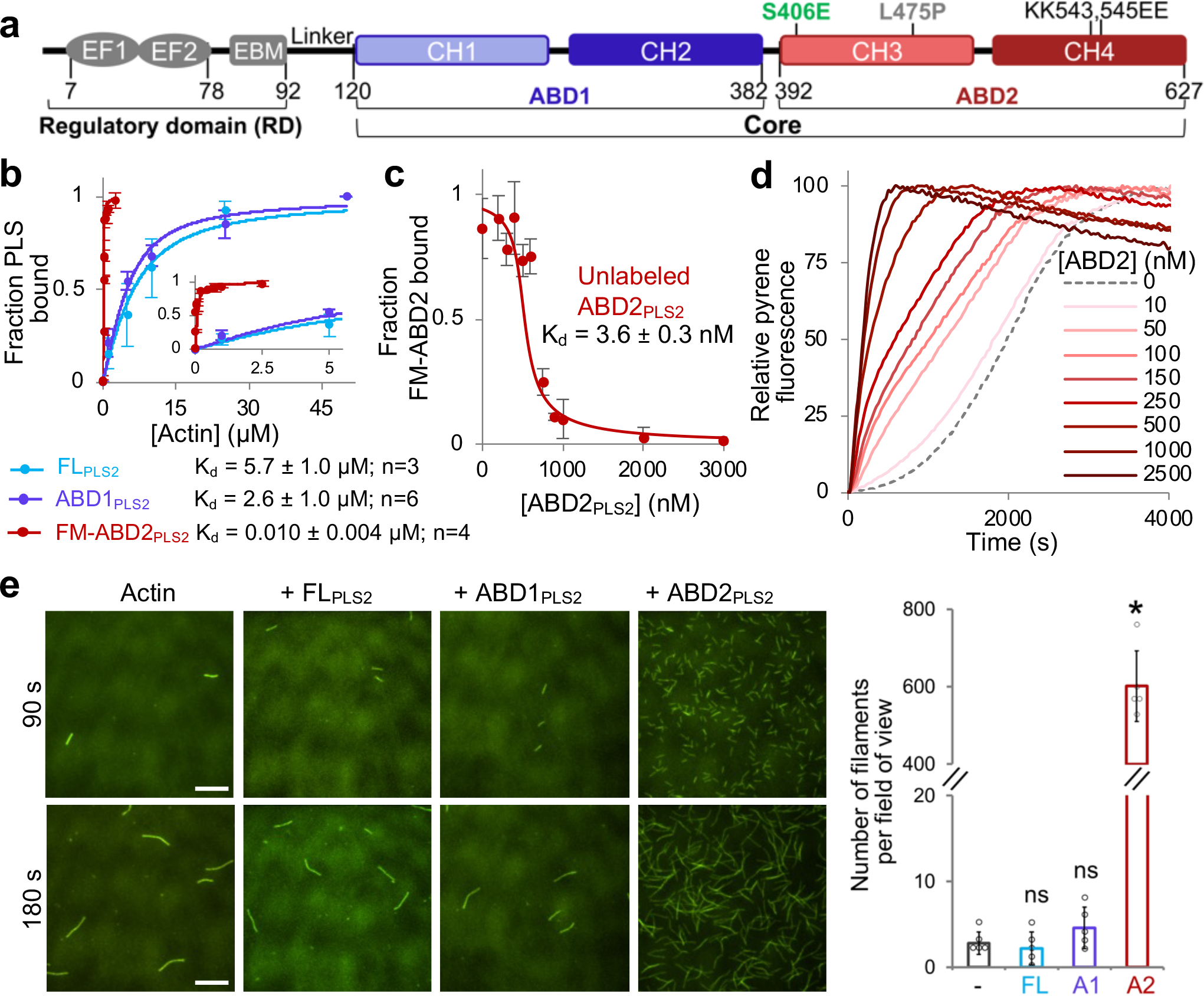
(a) Plastin domain organization. RD, regulatory domain; EF1 and 2, EF-hand motifs; ABD1 and 2, actin-binding domains; CH1–4, calponin-homology motifs. Numbers represent amino acid numbering in the PLS2 sequence.
(b) F-actin binding of full-length PLS2 (FLPLS2) and ABD1PLS2 was assessed by co-sedimentation. Binding of ABD2PLS2 to F-actin was measured by FA. The inset zooms in the 0 – 5 μM range. Error bars are SD of the mean; n - number of repetitions.
(c) F-actin binding of unlabeled ABD2PLS2 was assessed by FA competition. Error bars are SD of the mean; n=3 independent experiments.
(d) Representative normalized fluorescence traces of bulk pyrene-actin polymerization over the range of ABD2PLS2 concentrations.
(e) Representative TIRFM micrographs of actin nucleation by FLPLS2 and individual ABDs (50 nM final concentration) at 90 and 180 s after the initiation of actin polymerization; scale bars are 10 μm. The graph shows the number of actin filaments present in a field of view at 90 s. Error bars are SD of the mean; n=5 individual samples examined over two independent experiments. ANOVA followed by multiple comparison tests (two-tailed Student’s t-tests) with Bonferroni correction was applied: asterisk indicates statistically significant difference (*p<0.0167); individual p-values are 0.58 (FLPLS2 (FL)), 0.18 (ABD1PLS2 (A1)), and *4.6×10−7 (ABD2PLS2 (A2)), each compared to the actin control (−).
Source data are available for graphs in b-e.
The current understanding of the plastin organization is based on crystal structures of the ABD1-ABD2 cores of yeast and plant fimbrins44. For vertebrate plastins, only structures of individual ABD145 and CH4 of ABD2 (PDB:1WJO, 2D85) were resolved by crystallography and NMR, respectively, and the interaction of ABD2 (but not ABD1) with actin was revealed by cryo-electron microscopy (cryo-EM) reconstructions22,46. EM of negatively stained two-dimensional actin arrays assembled on lipid surfaces47 revealed actin filaments assembled in tight parallel bundles with 120-Å spacing between filament centers. Unfortunately, the resolution of the reconstruction (~37 Å) did not allow for revealing the details of ABDs’ interaction with actin. Structures of the EF-hand domain were determined by solution NMR39, but the RD location relative to the ABD1-ABD2 core has not been revealed by high-resolution structural methods. Analysis of PLS3 pathogenic variants linked to hereditary osteoporosis revealed their aberrant sensitivity to Ca2+ and pointed to the localization of RD at the interface between ABD1 and ABD2, in a region enriched by several disordered loops22. Accordingly, it was proposed that the RD in the Ca2+-bound state locks ABD2 in the inhibited conformation without affecting ABD1 binding to actin. In the proposed model, it remained unclear whether the inhibitory effect on ABD2 is imposed strictly by the RD or the inhibition originates from ABD1 and only is regulated by the RD.
Here, we report a structure of ABD1 bound to actin and propose an overarching mechanism of plastin regulation that is mediated by a tunable allosteric inhibition imposed by ABD1 on ABD2. Thus, the inhibitory engagement was strongly weakened by a mutational mimicking of physiologically relevant Ser406 phosphorylation at the ABD1-ABD2 interface enabling a strongly increased bundling potential. We propose that the discovered allosteric inhibition mechanism is a unique feature of plastins that enables regulating the strength of actin cross-linking in a broad range contributing to the assembly and remodeling of various high-order actin structures with different morphological and functional properties.
Results
ABD2 binds to F-actin much stronger than the full-length PLS
ABD1 of human PLS3 (ABD1PLS3) binds F-actin with a Kd in a low-micromolar range, similar to that of full-length (FL) PLS338 (FLPLS3). Similarly, ABD1PLS2 binds to F-actin only slightly better than FLPLS2 (Fig. 1b). Of the three human plastin isoforms, only ABD2PLS2 was soluble, stable, and existed in solution as a monomeric protein (Extended Data Fig. 1a–c). ABD2 from both PLS1 and PLS3 were unstable and could not be purified either alone or as a fusion with a folding-promoting maltose-binding protein48. To our surprise, isolated ABD2PLS2 bound F-actin with a nanomolar Kd, i.e., ~1,500 times stronger than FLPLS2 (Fig. 1b,c), suggesting inhibition of the domain in the context of the full-length protein.
Given that ABD2 binds F-actin at the interface between two long-pitch actin subunits22, we checked whether the binding might stabilize the longitudinal dimer and promote actin nucleation. In pyrenyl-actin polymerization assays, low-nanomolar concentrations of ABD2PLS2 potently increased bulk actin polymerization rate (Fig. 1d). A drastically increased number of filaments in the presence of ABD2PLS2, but neither ABD1PLS2 nor FLPLS2, observed by total internal reflection fluorescence microscopy (TIRFM; Fig. 1e) confirmed that faster polymerization is indeed due to filament nucleation, which was preserved even in the presence of profilin (Extended Data Fig. 1d).
Polymerization of actins with perturbed filament-forming interfaces can be rescued by filament stabilizing factors49,50. Covalent cross-linking of actin monomers by actin cross-linking domain (ACD) toxin yields non-polymerizable oligomers with disturbed longitudinal contacts51. The polymerization of these oligomers can be rescued, however, by enforcing the remaining contacts by phalloidin or reshaping the contacts by cofilin51,52. Similarly to phalloidin, ABD2PLS2 effectively rescued polymerization of ACD-cross-linked actin oligomers (Extended Data Fig. 1e).
ABD1 and ABD2 interact in trans with nanomolar Kd
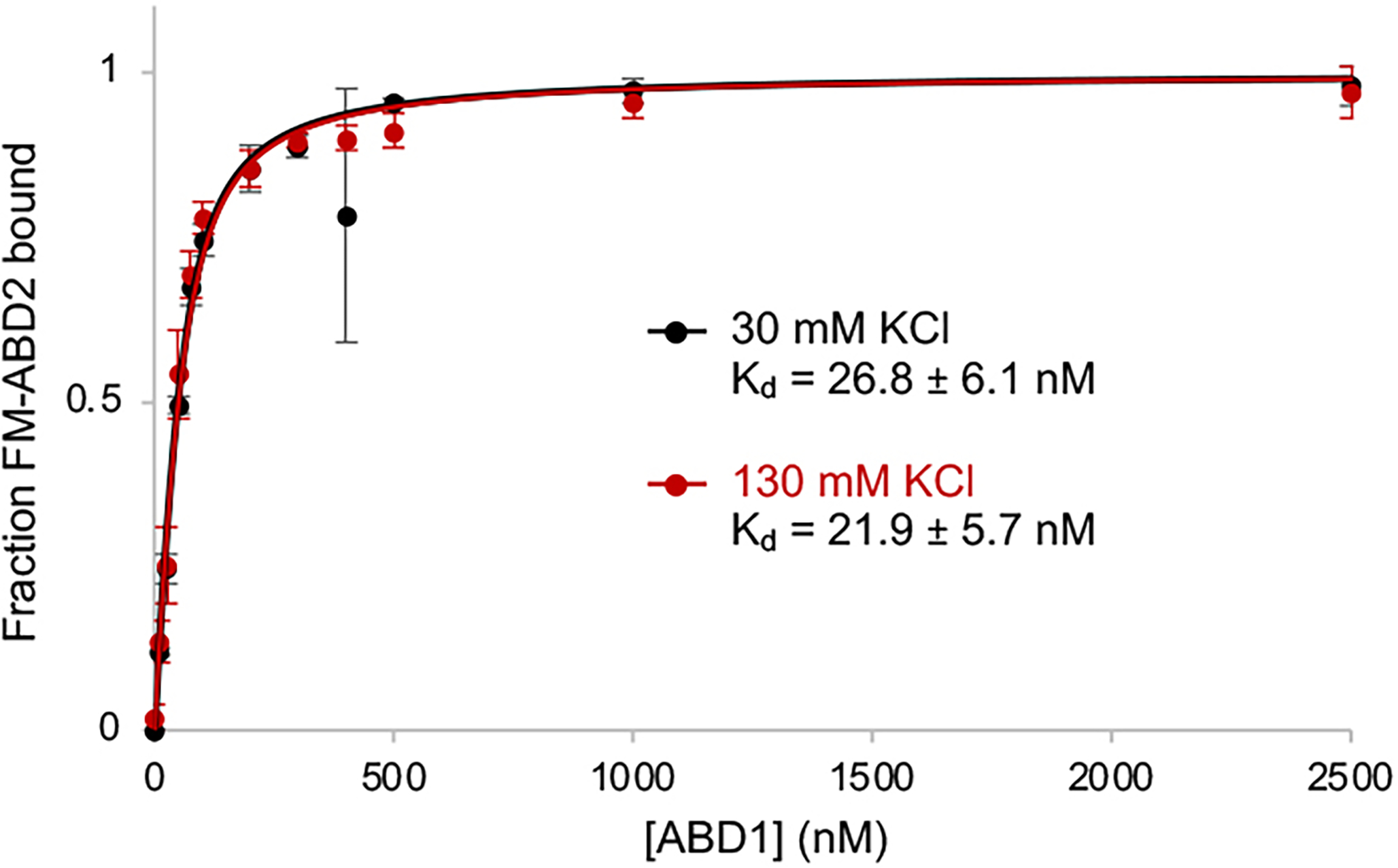
Full-length plastin did not reproduce the potent nucleating ability of ABD2PLS2 (Fig. 1e), suggesting its suppression, potentially by ABD1. Indeed, added in trans, ABD1PLS2 inhibited the nucleating ability of ABD2PLS2 (Fig. 2a). In fluorescent anisotropy (FA) experiments, ABD1PLS2 bound to ABD2PLS2 with a high affinity (nanomolar Kd; Fig. 2b), whereas the ABD1 construct with the regulatory domain (RD-ABD1PLS2) bound ABD2PLS2 even stronger, irrespective of the presence of Ca2+ (Fig. 2c). Therefore, RD contributes to the interaction between the ABDs, in agreement with its mapping to the loop-rich region at the ABD1-ABD2 interface22. The ABD1-ABD2 affinity was nearly identical in 30 and 130 mM KCl (Extended Data Fig. 2), suggesting a dominant role of hydrophobic surfaces in this interaction.
Figure 2. ABD1 of PLS2 interacts with and suppresses ABD2.
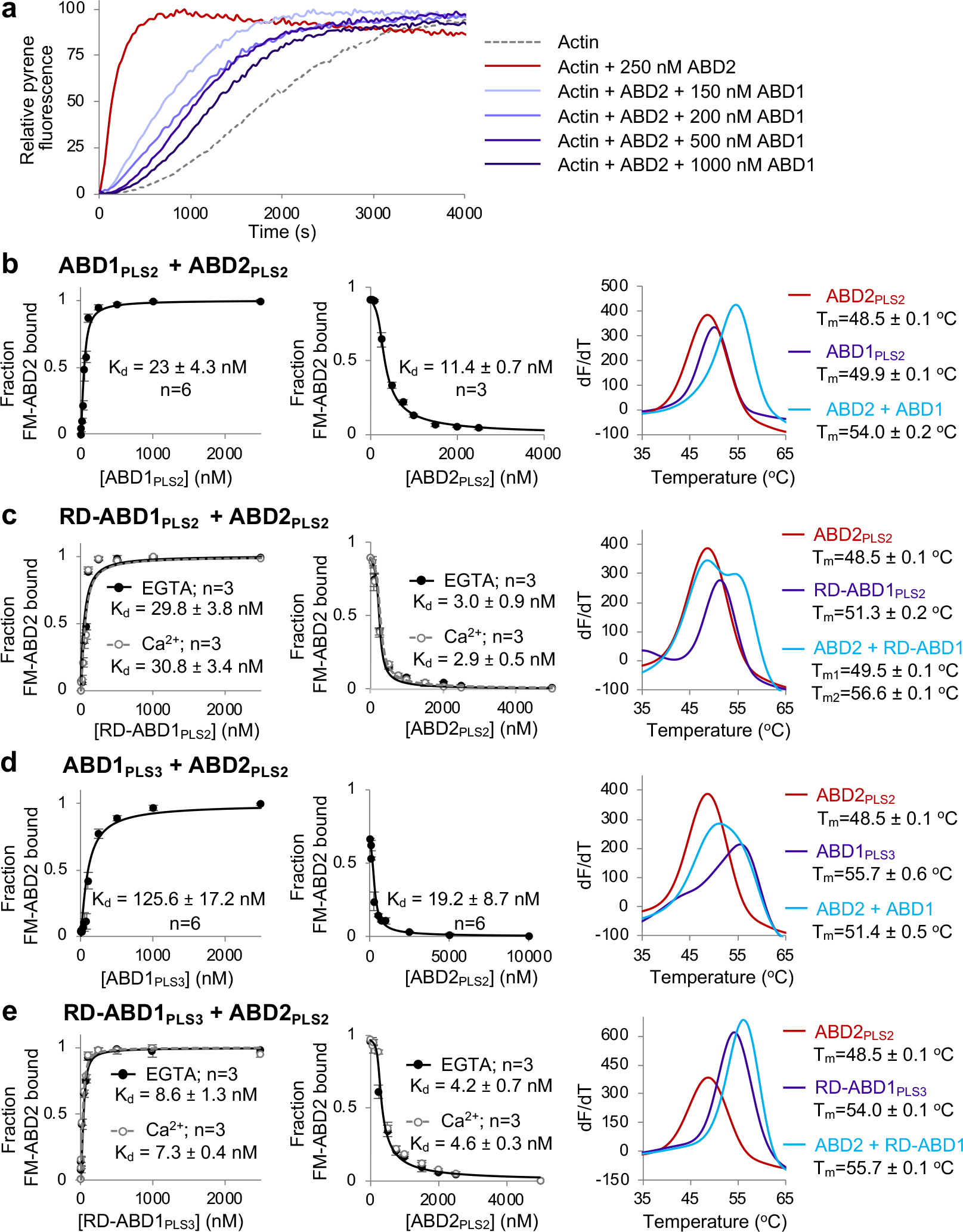
(a) Representative normalized fluorescence traces of ABD2PLS2-accelerated actin polymerization examined over the range of ABD1PLS2 concentrations.
(b-e) Binding of the individual ABDs of PLS2 and PLS3 to each other in trans were measured by fluorescence anisotropy using FM-ABD2PLS2 (left panels) and FA competition assays with unlabeled ABD2PLS2 (middle panels). Error bars are SD of the mean; n - number of repetitions. Melting temperatures (Tms) of ABDs (alone or combined) were determined by DSF (right panels): graphs show first derivatives (−dF/dT) of melting curves (average of n=3 independent experiments).
Source data are available for graphs in a-e.
Formation of protein complexes is often accompanied by an increase in thermostability that can be detected by differential scanning fluorimetry (DSF)53. Accordingly, the in trans ABD1-ABD2 complexes were more stable than individual domains (Fig. 2b,c), not however reaching the stability of FLPLS2 (Figs. 3 and 5b). Due to reduced stability and prompt degradation, neither ABD2PLS1 nor ABD2PLS3 could be characterized. Yet, ABD1PLS3 and RD-ABD1PLS3 bound to ABD2PLS2 with high affinities similar to those of respective PLS2 constructs (Fig. 2d,e), suggesting that the RD-ABD1/ABD2 interface is highly preserved in plastin isoforms.
Figure 3. F-actin binding destabilizes PLS2 due to ABD1-ABD2 disentanglement.
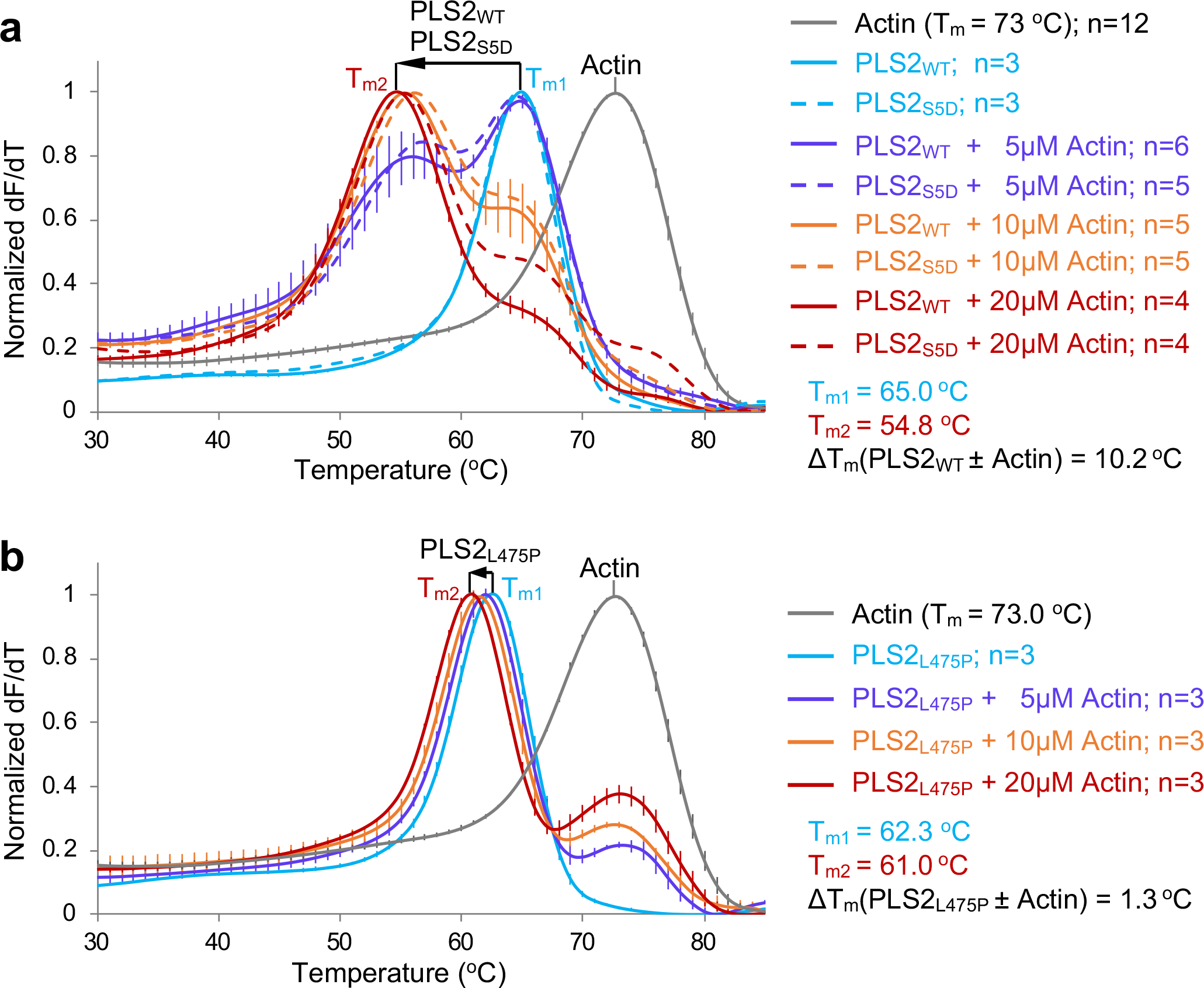
Melting temperatures (Tms) of bundling-competent WT and S5D (a) and bundling-incompetent L475P (b) full-length PLS2 constructs were assessed by DSF in the absence or presence of F-actin. Error bars represent the SE of the mean (n – number of independent experiments). Direction/magnitude of Tm shifts upon binding to actin are shown by arrows. Source data are available for graphs in a,b.
Figure 5. S406E PLS2 mutation mimicking physiologically relevant phosphorylation of S406 releases the inhibition of ABD2 by uncoupling it from ABD1.
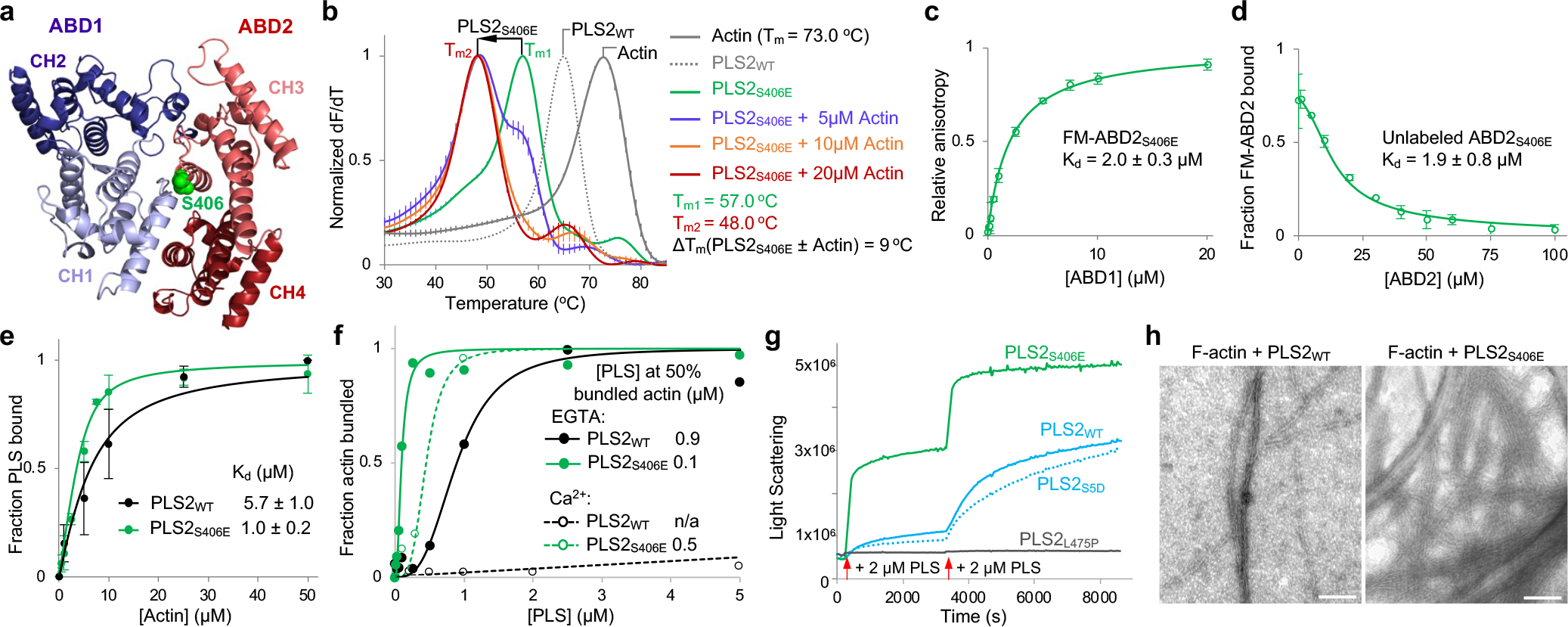
(a) An AlphaFold-model of PLS2 core. S406 is highlighted in green.
(b) The Tms of PLS2S406E in the presence and absence of F-actin were assessed by DSF. Error bars are SE of the mean (n=3 independent experiments).
(c, d) The binding affinity of ABD1 and ABD2S406E to each other were measured by fluorescence anisotropy (c) and confirmed by FA competition with unlabeled ABD2S406E (d). Error bars are SD of the mean; n=3 independent experiments.
(e, f) F-actin binding (e; n=3) and bundling (f; n=2) activities of PLS2WT and PLS2S406E were assessed by high- and low-speed co-sedimentation, respectively. Error bars in e are SD of the mean; n – number of independent experiments.
(g) F-actin bundling by WT, S5D, L475P, and S406E PLS2 was monitored by light scattering. Red arrows indicate additions of the plastin constructs to F-actin.
(h) Representative TEM images of actin bundles formed by PLS2WT and PLS2S406E. Scale bars are 100 nm.
Source data are available for graphs in b-g.
To summarize, ABD1 and ABD2 of plastins interact with each other with high affinity comparable to that of isolated ABD2 to F-actin, suggesting that ABD1/ABD2 interaction suppresses the ABD2 high-affinity binding to F-actin.
F-actin binding weakens ABD1-imposed inhibition of ABD2
Given that in trans the strength of the inhibitory ABD1/ABD2 interaction is nearly matching that of the ABD2/F-actin interaction (Figs. 2b,c and 1b,c), the inhibitory component must be prevailing when ABD1 and ABD2 are covalently linked, reconciling the possibility that the weaker domain ABD1 binds actin first38. Therefore, for bundling to happen, the inhibition must be weakened, e.g., by actin. Since separated ABD1 and ABD2 (Fig. 2b,c) are less stable than the FLPLS2 (Fig. 3a), we should expect destabilization of PLS2 upon F-actin binding due to the anticipated ABD1-ABD2 disentanglement.
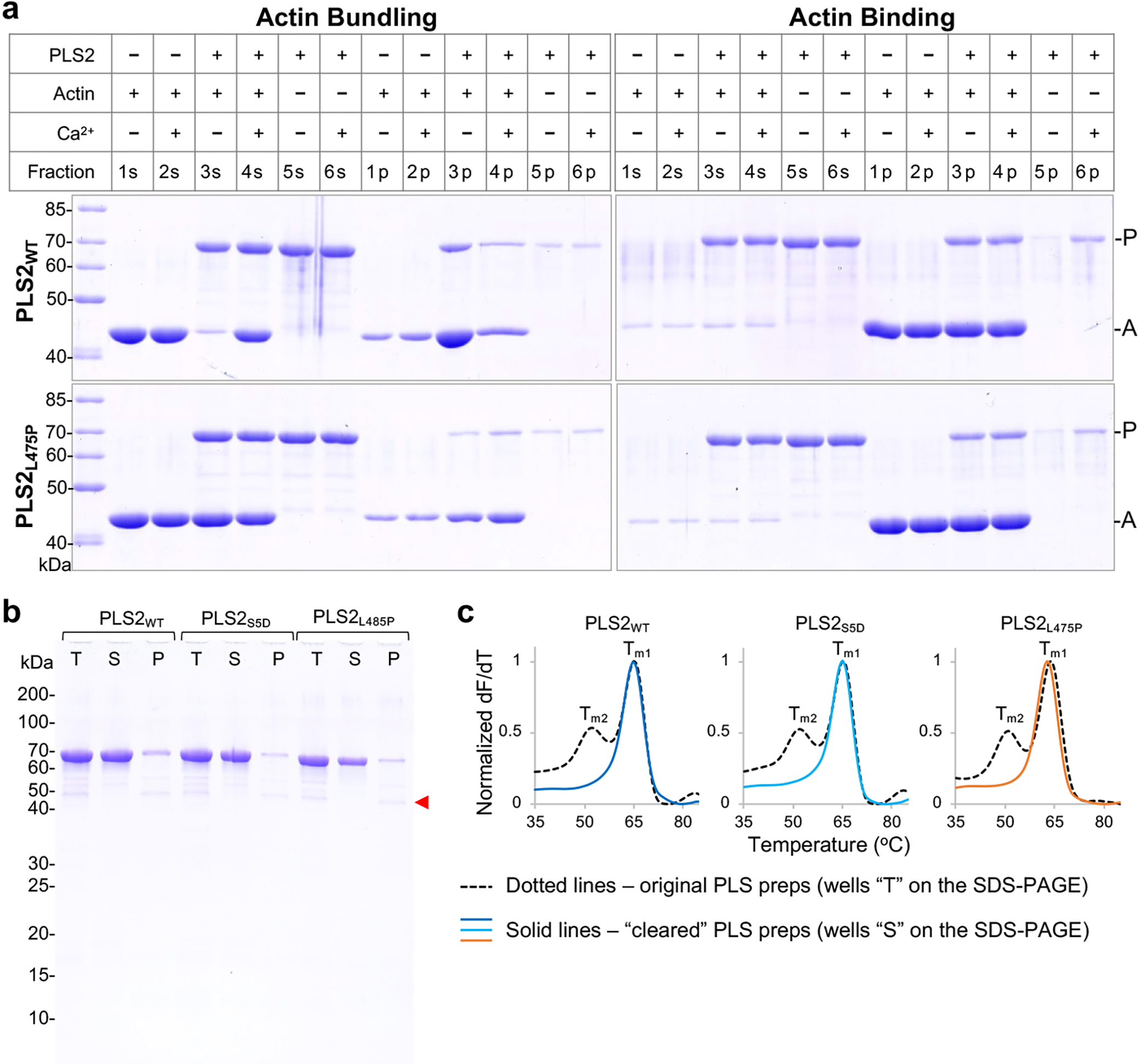
To distinguish the effects of the single-domain binding from those of binding via both domains, we reproduced the osteoporosis PLS3L478P mutation that perturbs the actin-binding interface of ABD222 in PLS2 (PLS2L475P). Similarly to PLS3L478P, PLS2L475P was able to bind (presumably through intact ABD1) but unable to bundle F-actin effectively regardless of the presence of Ca2+ (Extended Data Fig. 3). In agreement with our hypothesis, the Tm of bundling-incompetent PLS2L475P decreased by 1.3 °C upon binding to F-actin (Fig. 3b), suggesting only a minor disengagement of the domains. In contrast, wild-type (WT) PLS2 (PLS2WT) was destabilized by F-actin to a notably greater extent (by ~10 °C; Fig. 3a). Similar destabilization (Fig. 3a) was observed for the S5D PLS2 mutant (PLS2S5D), mimicking an activating Ser5 phosphorylation in the regulatory domain19,20. It is likely that the initial mild disengagement of ABD1-ABD2 upon ABD1 binding to F-actin (as observed in PLS2L475P) is followed by a stronger domain separation in PLS2WT and PLS2S5D due to a competitive binding of ABD2 to F-actin resulting in bundling.
ABD1 binds F-actin similarly to ABD2
While the structure of high-affinity ABD2 bound to F-actin is available22, the attempts to solve a weaker complex of ABD1 with F-actin were unsuccessful46. To address this, we mapped the ABD1/F-actin interface by mutagenesis to identify regions that can be used for cross-linking to F-actin using homobifunctional thiol-reactive reagents as described in the Methods section. Since the Cys-null construct of PLS3 (but not that of PLS2) is fully functional38, the former was used to generate single-Cys mutants of human plastin RD-ABD1. Cross-linking of Q194C RD-ABD1 to K50C on actin using 3,6-dioxaoctane-1,8-diyl bismethanethiosulfonate (MTS-8-MTS) provided the highest yield of the sought product (Extended Data Fig. 4a–d) and was selected for cryo-EM.
Analysis of 155,939 segments of decorated actin filaments yielded a model of ABD1 bound to F-actin at an overall 5.1-Å resolution (Fig. 4a,b). However, the resolution of ABD1 became progressively worse the farther from the actin (Extended Data Fig. 4e). RD was not detected, pointing to its high flexibility. The binding interface of CH1 is oriented towards actin similarly to that of CH3 of ABD2PLS222 and spectrin54 (Fig. 4c). Specifically, the loop 184–189 and the first five residues of the helix 190–206 of CH1 are sufficiently close to make contacts at both sides (subdomains 1 and 3) of the hydrophobic cleft of the “i” actin subunit but also with the 44–48 segment of the D-loop (subdomain 2) of the longitudinally adjacent subunit “i+2”. Also, in subdomain 1 of the “i+2” subunit, the tip of 79–95 helix is in proximity to residues Gln118, Asn213, and His225 of plastin’s CH1. With the exception of Asn213, all these PLS3 residues are different from those determined in a previous model based on a ~30-Å resolution reconstruction of ABD1-decorated actin filaments55.
Figure 4. Cryo-EM reconstruction of ABD1-decorated F-actin.
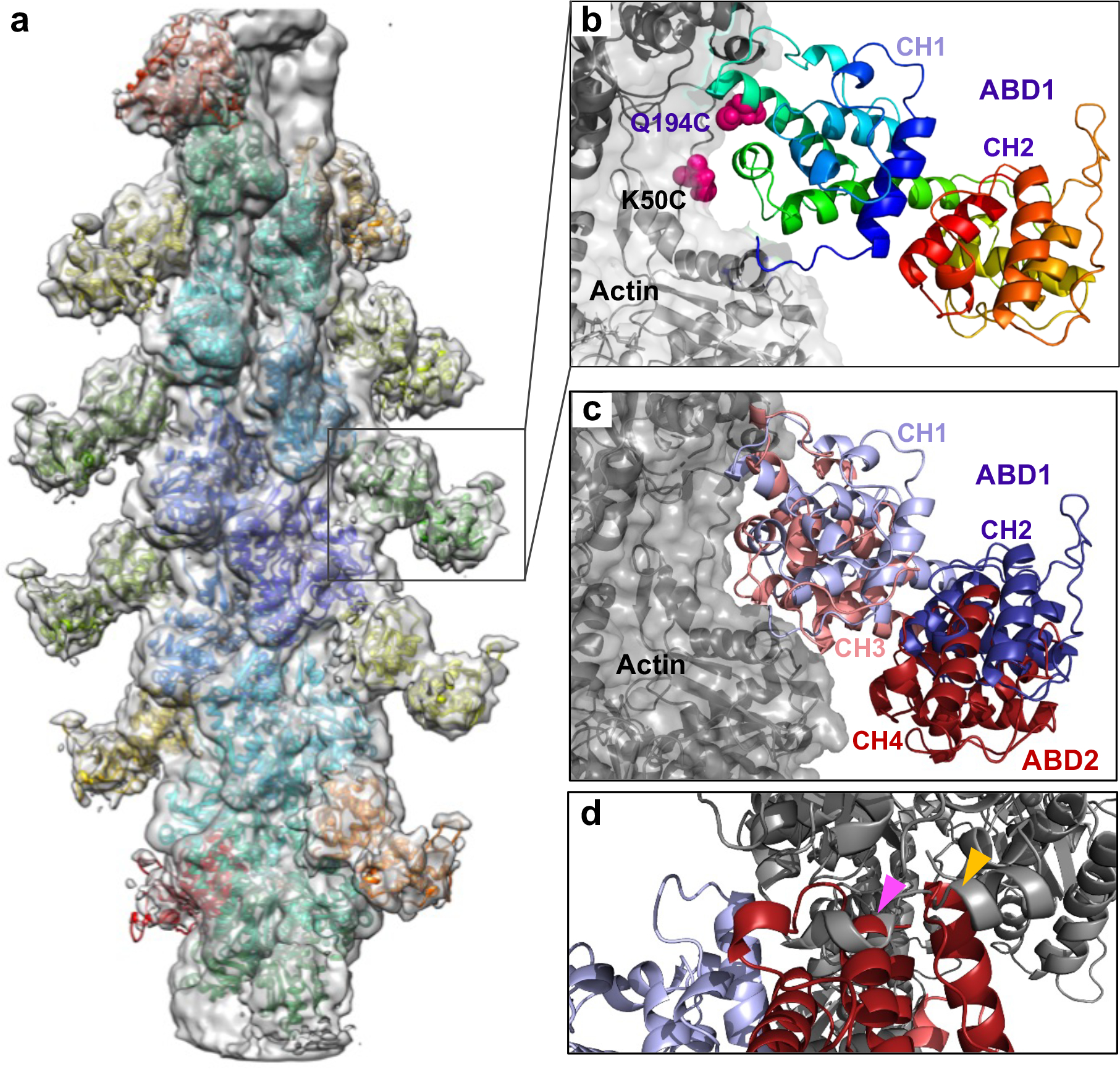
(a) Cryo-EM density map of actin filament decorated by ABD1PLS3 is shown as a grey transparent surface. The model built by fitting a cryo-EM structure of F-actin (PDB: 6ANU) and crystal structure of ABD1 of PLS3 (PDB: 1AOA) is shown as colored ribbons.
(b) Close-up view of the boxed area in a (ABD1 is shown as rainbow-colored ribbon from N-terminus in blue to C-terminus in red; actin is in grey). Chemically cross-linked residues in ABD1 (Q194C) and actin (K50C) are shown as magenta spheres.
(c) Superposition of ABD1 (CH1 in light blue, CH2 in dark blue; present study: EMD-25371) and ABD2 (CH3 in light red, CH4 in dark red; PDB: 6VEC) in the F-actin-bound states.
(d) Alignment of the cryo-EM ABD1/F-actin model with the AlphaFold-generated model of human PLS3 core (https://alphafold.ebi.ac.uk/entry/P13797) revealed clashes between ABD2 and F-actin (indicated by arrowheads): 521–535 ABD2 helix clashes with 359–365 actin helix (magenta arrowhead); 553–562 ABD2 helix clashes with 356–358 actin loop and 359–365 actin helix (yellow arrowhead). Color scheme as in c.
Alignment of the cryo-EM ABD1 structure with an AlphaFold-generated model56 of the PLS3 core (Fig. 4d) and the X-ray structures of yeast and plant fimbrin cores44 revealed clashes between ABD2 and the actin filament. The reorientation of the ABD2 required to eliminate the clash is a likely trigger of the experimentally detected (Fig. 3) disengagement of ABD1 from ABD2, enabling the initial priming of ABD2 for bundling.
A destabilizing mutation at the ABD1-ABD2 interface
Since ABD1-actin affinity is relatively low, only a thermodynamically comparable portion of the potent ABD1-imposed inhibition can be released upon binding to actin, bestowing a weak bundling strength. A more effective uncoupling can result in a more active protein with different properties. To determine whether such modulation can be achieved physiologically, we focused on reported post-translational modifications of PLS2. Ser5, Ser7, and Thr89 phosphorylation has been characterized to various degrees9,13,57,58, while phosphorylation at other sites identified by high-throughput methods (PhosphoSitePlus59) have not been examined. Among these, phosphorylation of Ser40660–64 is of particular interest as it resides at the interface between the ABDs and may influence the ABD1/ABD2 crosstalk (Fig. 5a).
A phospho-mimetic PLS2S406E construct was significantly less thermostable as compared to PLS2WT (Fig. 5b), tentatively reflecting a disengagement of ABD1 from the stabilizing and inhibitory interaction with ABD2. Indeed, the in trans binding of ABD2S406E to ABD1 was more than two orders of magnitude weaker than that of ABD2WT (Kd = 2 μM vs Kd = 11.4 nM; Figs. 5c,d and 2b, respectively). In agreement with the inhibition hypothesis, full-length PLS2S406E showed moderately improved F-actin binding and dramatically reinforced bundling efficiencies (Fig. 5e,f). Although inhibited by Ca2+, bundling of actin by PLS2S406E was still stronger than that by PLS2WT in the absence of Ca2+ (Fig. 5f). The mutation strongly enhanced the rate of bundling and size of bundles (Fig. 5g,h). The release of the inhibition from ABD2 was not, however, complete, as under the conditions when isolated ABD2 was highly potent (i.e., at 50-nM concentration; Fig. 1e), PLS2S406E showed no nucleation (Extended Data Fig. 5a), and nucleated only weakly at high (1-μM) concentration (Extended Data Fig. 5b). Accordingly, binding to F-actin further destabilized PLS2S406E confirming a major role of F-actin in the disengagement of the ABD domains (Fig. 5b). Notably, the transition towards the destabilized PLS2S406E state occurs at lower actin concentrations, compared to PLS2WT/F-actin (Figs. 5b and 3a, respectively), in agreement with the mutant’s higher affinity for F-actin and a much higher bundling ability. Interestingly, phosphorylation analogous to that of PLS2 at S406 was not reported for either PLS1 or PLS3, and the recombinant phospho-mimetic PLS1S407E and PLS3S409E constructs aggregated upon purification. These data strongly support the hypothesis that ABD2 in the normal mode of plastins is potently inhibited by ABD1. This inhibition can be tuned down by uncoupling of the domains upon binding of ABD1 to F-actin and further weakened by physiological modifications (e.g., PLS2 S406 phosphorylation).
Release of ABD2 inhibition alters cellular PLS distribution
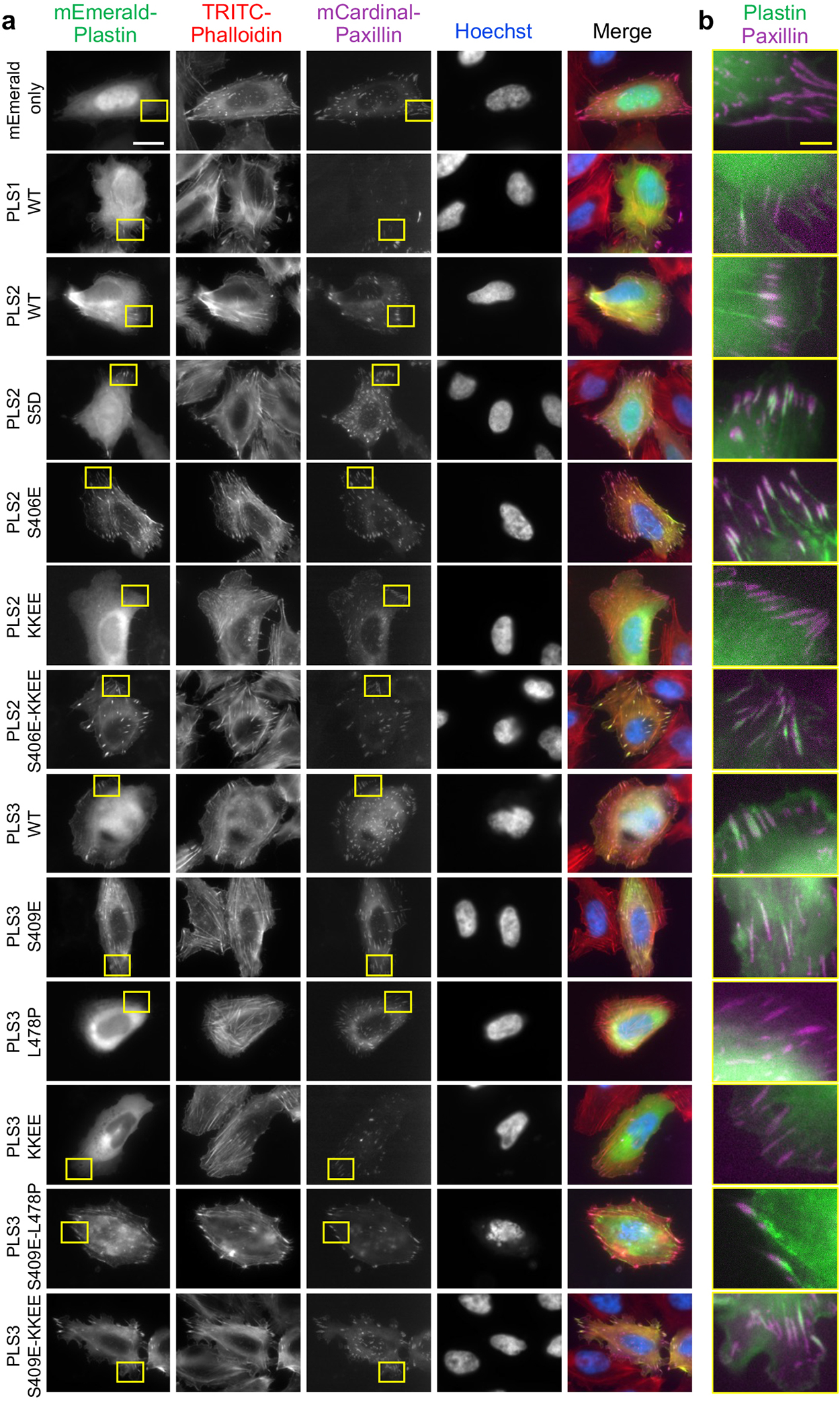
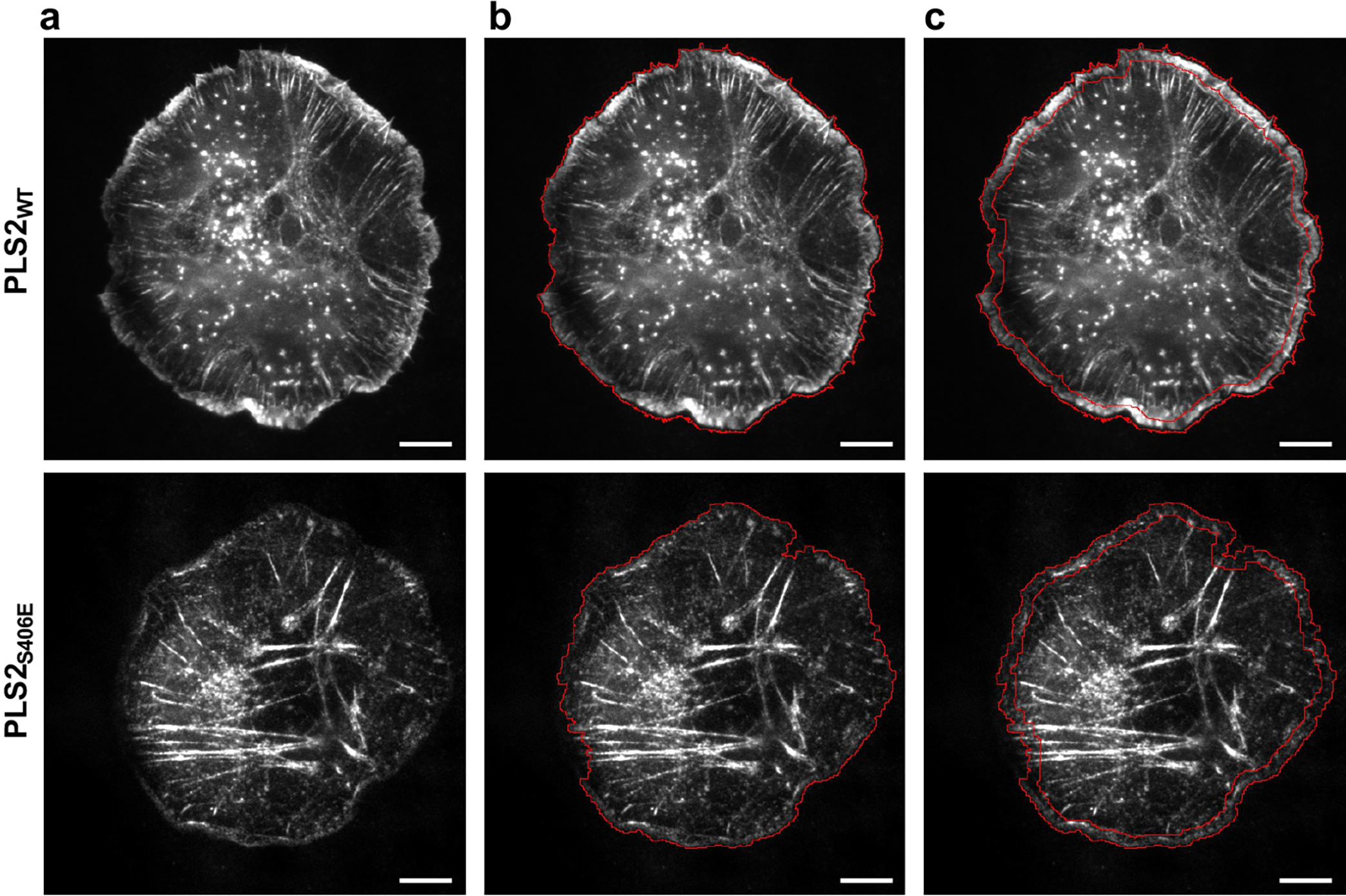
In a cellular context, recombinant WT PLS1, PLS2 and PLS3 were enriched at the cell leading edge and localized to actin-rich structures in the lamellipodia, filopodia, focal adhesions, and, to a lesser degree, stress fibers in Xenopus laevis XTC fibroblasts (Fig. 6a,b) and human U2OS osteosarcoma cells (Extended Data Fig. 6). When tracked by single-molecule speckle (SiMS) TIRFM65,66, all plastin isoforms undergo retrograde flow in the lamellipodia in a similar to actin mode (rates ~0.025 nm/s; Fig. 6e and Supplementary Video 1). In contrast, PLS2S406E was depleted from the lamellipodia but highly enriched at focal adhesions and stress fibers, whose morphology was enhanced and perturbed (Fig. 6a–d and Extended Data Fig. 6 and 7). Recycling of plastin from focal adhesions to the lamellipodial branched actin network requires unperturbed inherent Ca2+ sensitivity22. The localization of PLS2S406E at the focal adhesions, therefore, is logically explained by its enhanced bundling capacities even in the presence of Ca2+ notably exceeding those of PLS2WT under no Ca2+ conditions (Fig. 5f). Since PLS2S406E cannot be adequately inhibited by Ca2+, it likely gets depleted from the lamellipodial actin networks via the retrograde flow leading to its concomitant accumulation at the focal adhesions and stress fibers in the lamella.
Figure 6. Intracellular localization of human plastins.
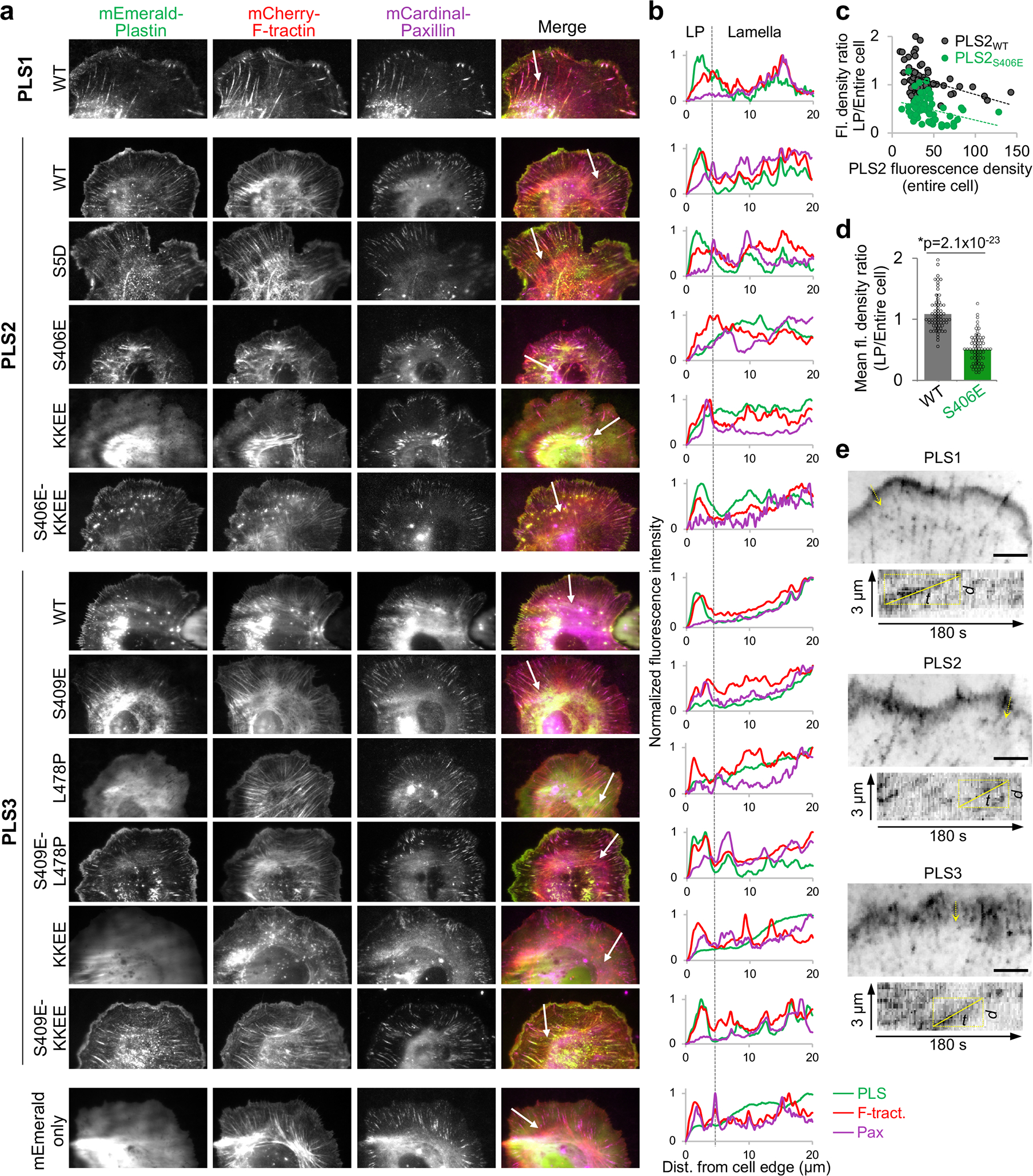
(a, b) XTC cells co-transfected with mEmerald-tagged plastin constructs, mCherry-F-tractin and mCardinal-paxillin (actin and focal adhesion markers, respectively) were imaged by TIRFM (a). Fluorescence intensity profiles (b) for all three channels (green - PLS, red - F-tractin, magenta - paxillin (Pax)) were generated along the white arrows shown in a, which also serve as scale bars (20 μm). LP is lamellipodia.
(c) Fluorescence densities of mEmerald-PLS2WT and mEmerald-PLS2S406E in lamellipodia (LP; 0–2μm from the cell edge) were normalized to those in the entire cell and plotted against the total cell fluorescence density for each cell. Number of cells quantified over two independent transfections are n=64 (PLS2WT) and n=67 (PLS2S406E). (d) Ratios of fluorescence densities (mean ± SD) for the same set of cells as in (c). Two-tailed Student’s t-test was applied: asterisk indicates statistically significant difference (*p<0.05).
(e) Average intensity projections and kymographs of SiMS TIRFM images of XTC cells transfected with mEmerald-tagged WT plastin isoforms show retrograde flow (see also Supplementary Video 1). Scale bars are 5 μm.
Source data are available for graphs in c,d.
Weakening the actin-binding surface of ABD2PLS2 by charge-inversing KK542,545EE mutations (KKEE), leading to actin-bundling deficiency in vitro38, resulted in a diffuse localization of the mutated PLS2 and PLS3 proteins (Fig. 6a,b). Such localization is similar and consistent with that of a PLS3 congenital osteoporosis variant with the actin-binding interface of ABD2 impaired by the L478P mutation22, corroborating that binding via both domains (i.e., the bundling ability) is essential for plastin localization to actin-rich structures in the cell. Introduction of the ABD1-ABD2 disengaging mutation S406E on the KK542,545EE background (S406E-KKEE) restored normal localization of PLS2 to F-actin-rich structures (Fig. 6a,b), suggesting that a weakened ABD2 interface can be compensated by attenuating the inhibition from ABD1. Curiously, while being unstable upon expression in E. coli, PLS3S409E localized to stress fibers in eukaryotic cells similarly to PLS2S406E (Fig. 6 and Extended Data Fig. 6), suggesting that the mutational destabilization of ABD2PLS3 is compensated in the cell either by the presence of actin or other cellular factors.
Discussion
While the ability of plastins to organize aligned bundles (e.g., in microvilli, cochlear stereocilia, and filopodia) is understandable and shared with other compact actin bundlers (e.g., fascin), their affinity towards complex networks of poorly aligned actin filaments (e.g., at the leading edge, in podosomes, immune synapses, and cuticular plates of hair cells) is puzzling. Indeed, filaments in such meshes are typically short, while the concentration of overlapping and properly oriented filaments suitable for cross-linking is low. In a large family of CH-domain actin organizers, plastins are unique in containing two ABDs non-identical in their properties38,46,47,67 in intimate association with each other44. In the present study, we discovered an allosteric inhibition between the ABDs and mechanisms of its physiological attenuation resulting in dramatic changes in properties of plastins demonstrated at the molecular and cellular levels. We propose that the allosteric autoinhibition is the key feature of plastins/fimbrins underlying their versatile involvement in various cellular processes and association with both well and poorly aligned filament bundles (Fig. 7a,b).
Figure 7. Hypothetical experimental model of actin bundling by plastins.
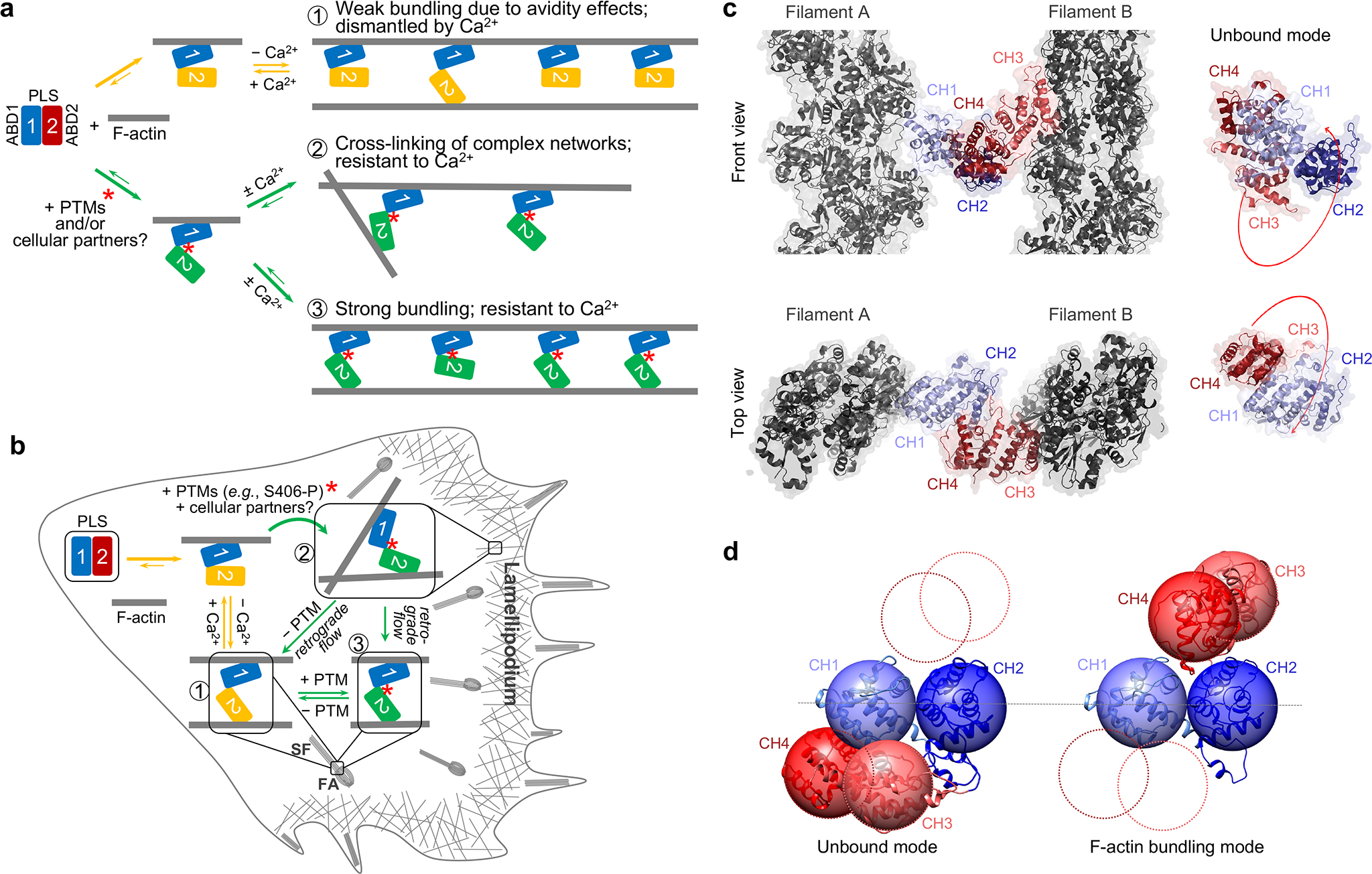
(a, b) In a regular operation mode (yellow arrows), ABD2 is strongly inhibited by ABD1. Binding of ABD1 to F-actin favors slight release of the inhibition resulting in a weak bundling mode optimal for organized bundles, where many plastin molecules are engaged in cumulative weak interactions in the F-actin bundle (1). Binding of Ca2+ to RD keeps ABD2 in the inhibited state. PTMs (e.g., phosphorylation of S406) and/or partners further weaken the ABD1-ABD2 contacts, allowing strong bundling (green arrows). This bundling mode may enable cross-linking in sparse actin meshes (2) and Ca2+-resistant organized bundles (3). Under physiological conditions, recycling of plastins from focal adhesions (FA) and stress fibers (SF) to lamellipodial branched networks is governed by spatio-temporally controlled PTMs and Ca2+. Constitutively strong bundlers PLS2S406E and PLS3S409E cannot be recycled leading to their accumulation at FA and SF with concomitant depletion from the lamellipodial networks (b).
(c, d) Model of actin bundle cross-linked by plastin based on cryo-EM reconstructions of ABD1 (EMD-25371) and ABD2 (PDB: 6VEC). CH1 is light blue, CH2 is dark blue, CH3 is light red, CH4 is dark red, actin is grey. The domain reorientation required for parallel, in-register actin bundle is illustrated in d (see also Supplementary Video 2): the AlphaFold model of plastin core is on the left, the model for a crossbridge in actin bundle is on the right.
All human plastins bind actin with similar affinities in a micromolar range38. Despite the low affinity of full-length plastins to actin, we found that ABD2 of PLS2 alone binds actin tightly with a low-nanomolar Kd (Fig. 1b,c). This high affinity, however, is balanced by an equally strong inhibitory interaction with ABD1 (Fig. 2). Our data support a sequential binding of the domains to actin38 and favors the idea that the engagement is primed by ABD1. Here we demonstrate that the initial binding attenuates, albeit slightly, the allosteric inhibition (Fig. 3), allowing weak binding of ABD2 to an adjacent filament providing such is available in proximity. Under these conditions, ABD2 remains largely inhibited by a still very strong interaction with ABD1. We propose that this weak bundling mode is optimal for bundling of highly organized parallel actin arrays (e.g., in microvilli and filopodia) where the adequate bundling strength is achieved through avidity (Fig. 7a,b). In this mode, plastins are highly sensitive to the inhibition and dissociation by Ca2+, enabling effective control of their localization22 via respective signaling.
Such weak binding is not effective, however, for the organization of less ordered bundles such as those found in the branched networks associated with the lamellipodia or endocytic machinery. Additional attenuation of the allosteric auto-inhibition required in this case can be bestowed by a post-translational modification (PTM), e.g., PLS2 S406 phosphorylation. Since PLS2S406E is a notably better bundler (Fig. 5f–h), it can cross-link poorly aligned filaments in single nodes essential for organization of meshed networks. Alternatively, attenuation can be achieved via interaction with proteins or other molecules that bind at the ABD1-ABD2 interface, i.e., at a cryptic site obscured by this strong interaction. As the plastin bridges progress with retrograde actin flow towards lamella (Supplementary Video 1), their recycling to the lamellipodial leading edge is orchestrated by Ca2+ 22, which reaches the highest concentration at the interface between lamella and lamellipodium68. Ca2+, however, is unable to recycle phosphorylation-mimicking PLS2S406E and PLS3S409E with the attenuated ABD1-ABD2 inhibition, leading to their depletion from the lamellipodia and accumulation in aligned bundles of focal adhesions and stress fibers (Fig. 6 and Extended Data Fig. 6) in agreement with a low Ca2+-sensitivity of PLS2S406E in vitro (Fig. 5f). Therefore, our model suggests that the attenuation must be released by either removal of the PTM or dissociation from a hypothetical lamellipodial partner (Fig. 7a,b). In differentiated cells (e.g., enterocytes and inner ear hair cells) similar mechanisms can be employed to produce stable actin bundles of microvilli and stereocilia resistant to the presence of Ca2+.
Is the extent of domain separation in plastins proposed here supported by literature data? In physiological bundles, where the role of plastin/fimbrin is unambiguously established (e.g., intestinal microvilli), actin filaments are found in the parallel alignment. Accordingly, 2D-actin arrays assembled in the presence of plastins are also parallel47. By constraining the experimentally determined distance between centers of actin filaments at 12 nm47,69 and using our cryo-EM reconstructions of actin decorated by ABD1 (Fig. 4) and ABD222, we modeled how ABDs of plastin are expected to separate in a parallel, in-register actin bundle (Fig. 7c,d). As illustrated in Supplementary Video 2, the domain reorientation required for such assembly is rather dramatic in agreement with a substantial loss of thermal stability by plastins in the bundling mode (Fig. 3a and 5b). Such strong separation of the domains supports a possibility for plastins to be engaged in the organization of various types of actin assemblies, including antiparallel bundles (as in the contractile ring) and actin networks with angled filament orientations (as in lamellipodia and endocytic patches). Experimentally, plastins/fimbrins have been found associated with both types of structures22,41,67,70. Furthermore, in vitro, at least Saccharomyces pombe yeast fimbrin can form actin bundles in both parallel and antiparallel orientations with a similar likelihood67.
The postulated domain separation is consistent with recognized mechanisms of mechanosensing that operate via i) dynamic “catch” and “slip” bonds71 and ii) exposure of cryptic sites in cytoskeletal proteins72,73. Indeed, indirect evidence support plastin’s role in mechanotransduction. Thus, lamellipodia, endocytic patches, focal adhesions, and the contractile ring (i.e., structures associated with plastins/fimbrins) are the elements whose functionality depends on mechanical forces. PLS3 in mature osteocytes is highly enriched in the bifurcation sites in the osteocyte processes74, i.e., in regions critical for the shear-stress mediated bone remodeling. Similarly, the presence of PLS3 in microvilli and inner ear stereocilia correlates with their well-recognized mechanotransduction properties75. Plastins have been implied in inactivation of integrins9,76,77, key mediators of mechanotransduction from the matrix to the cytoskeleton.
When the current manuscript was in revision, a preprint was published that independently confirmed our biochemical data on the domain separation upon binding of plastin to actin by a machine learning-assisted cryo-EM reconstruction78. While the two studies are complementary and supportive on important aspects of plastin’s physiology, there is a disagreement on which of the domains binds to actin first, which should be clarified in subsequent studies.
Since ABD2 nucleates new actin filaments (Fig. 1d,e and 67), the allosteric inhibition of ABD2 may also serve to prevent undesirable ABD2-induced nucleation of actin filaments. Yet, ABD2-imposed nucleation on the side of existing filaments could strengthen the meshwork of branched filaments and reinforce membrane protrusions necessary to cover matrix gaps21 and, therefore, remains an intriguing possibility that deserves a separate investigation.
The unique properties bestowed by the allosterically controlled protein design are likely to be shared by all plastins/fimbrins. If so, their ABDs should interact with each other with a strength comparable to that of the stronger binding domain with actin, whereas the identity of the stronger domain and the sequence in which the domains interact with actin may vary. The only other plastin/fimbrin characterized to a comparable to PLS2 level is S. pombe Fim167. While Fim1 is a stronger bundler, none of its domains has an affinity in the low-nanomolar range. Isolated ABD2Fim1 binds actin weaker (Kd ~ 650 nM) than ABD2PLS2 (Kd ~ 4 nM; Fig. 1c), while RD-ABD1Fim1 binds much tighter (Kd ~ 220 nM67) than RD-ABD1PLS3 (Kd ~ 4–5 μM38). Yet, considering that ~ 200-nM Kd is near the detection limit for co-sedimentation experiments, its reassessing by more sensitive approaches would be desirable.
In summary, we have characterized a novel mode of plastin regulation wherein ABD1 is always available to bind F-actin, while binding of ABD2 is secondary and a subject of regulation by the inhibitory influence of ABD1, PTMs, and Ca2+. We found that this regulation can be tuned by phospho-mimicking PLS2S406E, where S406 is a physiological PLS2 phosphorylation site. This result is of particular interest as the phosphorylation of S406 was detected in cancers61–64. In the context of the recognized link of PLS2 to cancer metastasis, enforced actin bundling upon S406 phosphorylation may prove to contribute to the malignant phenotype of cancer cells and thus deserves careful investigation. Future studies should clarify whether all members of the plastin family are similarly controlled by the allosteric inhibition, investigate which cellular plastin partners can regulate plastin function and localization by tuning the engagement between ABD1 and ABD2, and test the hypothesis that exposure of cryptic sites upon bundling/domain separation is responsible for the implied mechanosensing abilities of plastins/fimbrins.
Methods:
Protein expression and purification
Actin was purified from skeletal muscle acetone powder from rabbit (Pel-Freez Biologicals, Rogers, AR) or chicken (prepared in-house from flash-frozen chicken breasts (Trader Joe’s, USA)) as previously described79 and stored on ice in G-buffer: 5 mM tris(hydroxymethyl)aminomethane hydrochloride (TRIS-HCl), pH 8.0, 0.2 mM CaCl2, 0.2 mM ATP, 5 mM β-mercaptoethanol (β-ME). Actin was used within one month with dialysis against fresh G-buffer after two weeks of storage.
All plastin constructs used in this study are described in Supplementary Table 1. To produce recombinant plastin constructs, they were cloned into pColdI vector (Takara Bio USA, Mountain View, CA) modified to include a tobacco etch virus (TEV) protease recognition site following the N-terminal 6xHis-tag38. Multi-site-directed mutagenesis was carried out using the QuikChange Lightning Multi-Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Proteins were expressed in and purified from BL21-CodonPlus(DE3)pLysS Escherichia coli (Agilent Technologies, Santa Clara, CA) as described38 and dialyzed against PLS buffer: 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), pH 7.0, 30 mM KCl, 2 mM MgCl2, 0.5 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 mM dithiothreitol (DTT), and 0.1 mM phenylmethylsulfonyl fluoride (PMSF). Purified proteins were flash-frozen in liquid nitrogen and stored at −80 °C.
Human profilin-1 (PFN1) was expressed in BL21-CodonPlus(DE3)-RP E. coli (Agilent Technologies, Santa Clara, CA) and purified as previously described80,81.
ACD from Aeromonas hydrophila (ACDAh) were purified as previously published82. Expression in BL21(DE3)pLysS was induced by IPTG (overnight at 15 °C) and ACD constructs were purified using TALON metal affinity resin (Takara Bio USA, Mountain View, CA) by standard procedure.
Labeling proteins with fluorescent probes
Alexa-488-actin was prepared by labeling 2 mg/ml G-actin in G-buffer devoid of reducing agents with 1.2-molar excess of Alexa Fluor 488-maleimide (Thermo Fisher Scientific, Waltham, MA) for 4 h at 4 °C, followed by dilution to 1 mg/ml and polymerization with 2 mM MgCl2 and 100 mM KCl overnight at 4 °C. To prepare pyrene-actin, 2 mg/ml G-actin was first polymerized in G-buffer devoid of reducing agents supplemented with 2 mM MgCl2 and 100 mM KCl at 25 °C for 30 min and then diluted to 1 mg/ml in F-buffer: 5 mM Tris-HCl, pH 8.0, 0.2 mM ATP, 0.2 mM CaCl2, 1 mM MgCl2, 100 mM KCl. N-(1-pyrene)iodoacetamide (Thermo Fisher Scientific, Waltham, MA) was added to a final concentration of 40 μM, and the labeling was carried out overnight with mixing at 4 °C. Both labeling reactions were quenched with 10 mM β-ME, and labeled F-actins were pelleted at 100,000 × g in a Ti-70 rotor in an Optima XPN 90 ultracentrifuge (Beckman Coulter, Brea, CA) for 90 min followed by three rounds of dialysis of the resulted pellets against G-buffer. All labeled and unlabeled G-actins were further purified by size-exclusion chromatography on Sephacryl S200-HR (GE Healthcare, Chicago, IL), stored on ice in G-buffer, and used within 4 weeks with dialysis to G-buffer after two weeks of storage.
ABD2PLS2 was labeled with fluorescein-5-maleimide (FM; Thermo Fisher Scientific, Waltham, MA). Before labeling, the protein was incubated for 1 h on ice in the presence of 10 mM DTT. The reducing agent was removed by passing twice through a NAP-5 desalting column (GE Healthcare, Chicago, IL) equilibrated with G-buffer lacking β-ME. FM was added at a 1.5-molar excess to protein and incubated overnight on ice. FM-ABD2 was passed through a NAP-5 column equilibrated with G-buffer to remove excess dye.
Fluorescent anisotropy (FA) binding assays
G-actin was switched from Ca2+- to Mg2+-bound state by the addition of 0.1 mM MgCl2, and 0.5 mM EGTA and 10 min incubation on ice. Polymerization was induced by adding 2 mM MgCl2, 30 mM KCl, and 10 mM HEPES, pH 7.0 and incubating for 30 min at room temperature. F-actin was stabilized by a 1.2-molar excess of phalloidin (Sigma-Aldrich, St. Louis, MO). All reactions were carried out in PLS buffer supplemented with 0.2 mM ATP. For actin binding experiments, FM-ABD2 was used at 100 nM, and F-actin was added at concentrations from 0 to 2500 nM. Reactions were incubated for 30 min at 25 °C before measurement. Parallel (||) and perpendicular (|) intensities were measured on an Infinite M1000 Pro plate reader (Tecan US Inc, Morrisville, NC) with λex = 470 nm and λem = 519 nm using i-Control v.1.11 software. Anisotropy was calculated as:
| (1) |
Binding affinities were determined by fitting the data to the binding isotherm equation:
| (2) |
where Kd is the dissociation constant, P is the concentration of plastin, and A is the concentration of F-actin.
Competition anisotropy experiments were carried out with 100 nM FM-ABD2 and 500 nM F-actin. Unlabeled ABD2 was added from 0 to 3000 nM. The dissociation constant of the unlabeled protein was determined by fitting the data to the following equation83:
| (3) |
where A is the concentration of F-actin, P is the concentration of ABD2, Kd1 is the dissociation constant for FM-ABD2 binding to actin, and Kd2 is the dissociation constant for unlabeled ABD2 binding to actin. Data fitting analysis was done using Origin v.9 software (OriginLab Corporation, Northampton, MA).
Anisotropy experiments for ABD binding in trans were carried out similarly. The concentration of FM-ABD2 was 50 nM, and ABD1 constructs were added at concentrations from 0 to 2500 nM. In the trans-ABD competition assays, FM-ABD2 was at 50 nM, and ABD1 was kept at 250 nM. The concentration of unlabeled ABD2 varied from 0 to 5000 nM.
F-actin binding and bundling co-sedimentation assays
In F-actin binding experiments carried out in PLS buffer supplemented with 0.2 mM ATP, plastin constructs were used at a final concentration of 5 μM and F-actin (polymerized as above without the addition of phalloidin) was added from 0 to 50 μM. Actin-bundling experiments contained 2 μM F-actin and plastin concentrations from 0 to 1 μM. Reactions were incubated overnight at 4 °C followed by 1 h at room temperature. Binding reactions were spun at 300,000 × g for 30 min at 25 °C using Optima MAX-TL ultracentrifuge (Beckman Coulter, Brea, CA). Bundling reactions were spun at 17,000 × g for 15 min at 25 °C. Supernatants and pellets were separated and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Gels were stained with Coomassie Brilliant Blue, and band intensities were quantified by densitometry using ImageJ v.2.3 software84,85. Dissociation constants were determined by fitting to the binding isotherm equation (2).
Bundling efficiency was quantified by fitting the data to the Hill equation:
| (4) |
where n is the Hill coefficient, and KAn is the concentration of plastin at 50% actin bundled.
Bulk pyrenyl-actin polymerization assays
Ca2+-ATP G-actin was added to a black nonbinding 384-well plate (Corning Inc, Corning, NY) to a final concentration of 2.5 μM (5% pyrenyl-labeled). Next, F-actin was switched from Ca2+-bound to Mg2+-bound state by adding EGTA and MgCl2 to final concentrations of 0.5 and 1 mM, respectively. Following incubation for 1 min at room temperature, polymerization was initiated by adding 0.33 volumes of 3x initiation buffer: 30 mM MOPS, pH 7.0, 0.6 mM ATP, 1.5 mM DTT, 3 mM MgCl2, and 90 mM KCl. At the same time, PLS constructs were added to the desired final concentrations indicated in the figures. In inhibition experiments, ABD2 was maintained at 250 nM, and increasing concentrations of ABD1 were added. Pyrene fluorescence was monitored on an Infinite M1000 Pro plate reader (Tecan US Inc, Morrisville, NC) with λex = 365 nm and λem = 407 nm using i-Control v.1.11 software.
Reconstituted actin polymerization by TIRFM
For in vitro reconstituted TIRFM experiments, Ca2+-ATP G-actin was switched to Mg2+-ATP G-actin by 2 min incubation in 0.05 mM MgCl2 and 0.2 mM EGTA. Mg2+-ATP G-actin (final concentration 0.9 μM, 33% Alexa-488-labeled) in the absence or presence of 1.5 μM profilin (PFN1) was mixed with 50 nM or 1 μM of PLS constructs in the final reaction buffer (pH 7.0): 10 mM imidazole, 0.2 mM EGTA, 1 mM MgCl2, 50 mM KCl, 0.25 mM ATP, 10 mM ascorbic acid, 2.5 mM 3,4-protocatechuic acid (PCA), 0.1% bovine serum albumin, 0.6% methylcellulose-400cP, and 0.1 μM protocatechuate-3,4-dioxygenase (PCD)86. Immediately upon mixing, reactions were transferred to a flow chamber and imaged using Nikon Eclipse Ti-E microscope equipped with a TIRF illumination module and a perfect focus system (Nikon Inc, Melville, NY) using NIS Elements-AR v.4.3 software.
Differential scanning fluorimetry (DSF)
Heat-induced protein denaturation curves were obtained using a CFX Connect Real-Time PCR system (Bio-Rad Laboratories, Hercules, CA) using Bio-Rad CFX Manager v.3.1 software as described previously82,87. Reactions were carried out in PLS buffer supplemented with 0.2 mM ATP (when actin was present) and SYPRO Orange dye (1x final concentration; Invitrogen, Carlsbad, CA). Plastin constructs were added to a final concentration of 5 μM, while F-actin (stabilized by phalloidin at equimolar concentration) was added at 5 μM, 10 μM, and 20 μM. Reactions containing F-actin were incubated overnight at 4 °C prior to measurement. Reactions containing ABD mixtures were incubated for 30 min at 4 °C before measurement. Melting temperatures (Tms) were determined by calculating the max of the first derivative (−dF/dT) of the fluorescence traces (fluorescence vs temperature).
Light scattering assays
Light scattering assays were conducted as described previously38 using PTI QM-400 (Horiba Scientific, Canada) with excitation and emission wavelengths set to 330 nm. The light scattering signals were recorded using FelixGX v.4.9 Horiba PTI software.
Transmission electron microscopy
TEM was performed as described in 52. Samples of F-actin bundled by plastins were applied to carbon-coated grids (Ted Pella, Redding, CA) for 60 s and negatively stained with 1% (w/v) uranyl acetate. The grids were examined using a Tecnai-12 electron microscope (Philips, Netherlands) at an accelerating voltage of 80 keV.
Sedimentation velocity analytical ultracentrifugation
SV-AUC was conducted as described previously88 using a ProteomeLab XL-I analytical ultracentrifugation system (Beckman Coulter, Brea, CA). Absorption at 280 nm data were collected and analyzed using SEDFIT v.16–1c software using a continuous sedimentation coefficient distribution model c(S).
In vitro actin cross-linking by ACD toxin
ACD-catalyzed actin cross-linking was performed as described in 52 using 5 μM G-actin and 10 nM ACDAh toxin. The reactions were initiated by the addition of 2 mM MgCl2. Actin oligomers were polymerized in the presence of ABD2 at 1:1 molar ratio to actin and pelleted by ultracentrifugation at 300,000 × g for 30 min at 4 °C in a TLA100 rotor (Beckman Coulter, Brea, CA). Supernatant and pellet fractions were resolved on 12% SDS-polyacrylamide gels.
Cell culture, transfections, and microscopy
Xenopus laevis XTC cells were cultured in 70% Leibovitz’s L-15 medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS), L-glutamine, and penicillin-streptomycin at 23 °C and ambient CO2. Human U2OS cells were cultured at 37°C with 5% CO2 in a humidified incubator and were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, L-glutamine, and penicillin-streptomycin. XTC cells were obtained from Dr. Watanabe (Kyoto University Graduate School of Medicine, Kyoto, Japan) and were not further authenticated. The source of U2OS cells is unknown but their identity was verified by STR profiling using microsatellite genotyping (Genomics Shared Resource, OSU Comprehensive Cancer Center) with 100% match compared to The Cellosaurus cell line database89. All cells were mycoplasma-negative as determined by PCR per published protocol90.
Plastin constructs (Supplementary Table 1) were cloned using NEBuilder (New England Biolabs, Ipswich, MA) into pcDNA3.1 with mEmerald fused at the C-termini. The plasmids pmCherry-β-actin (Addgene #54967, RRID:Addgene_54967) and pmCardinal-paxillin (Addgene #56171, RRID:Addgene_56171) were gifts from Michael Davidson91,92. Transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA). Transfected XTC cells were plated on polylysine-coated coverslips (Neuvitro Corporation, Vancouver, WA) in Attofluor chambers (Thermo Fisher Scientific, Waltham, MA) in serum-free L-15 medium and imaged 30 min post-plating. TIRFM images were obtained using a Nikon Eclipse Ti-E inverted microscope (Nikon Instruments Inc., Melville, NY) equipped with a TIRF module, a perfect focus system, Nikon CFI Plan Apochromat λ 100x oil objective (NA 1.45), and an iXon Ultra 897 EMCCD camera (Andor Technology, Belfast, UK) using NIS Elements-AR v.4.3 software.
To quantify the distribution of plastins between the lamellipodia and that in the entire (Fig. 6c,d), XTC cells expressing either mEmerald-PLS2-WT or mEmerald-PLS2-S406E were imaged as described above. Cells were “fragmented” in ImageJ v.2.3 (threshold and mask tools), and the obtained masks were “eroded” by 12 px = 2 μm (using Process/Binary/Options/Erode ImageJ tool); the resulting masks were used to generate selections for measuring i) the total cell fluorescence and ii) lamellipodial fluorescence, i.e., fluorescence in the 2-μm-wide band at the cell edge (Extended Data Fig. 7).
For SiMS TIRFM, cells expressing low levels of plastins were selected, and time-lapse imaging (with 2-s intervals) was performed on small cell areas using a field diaphragm to avoid photodamage of the nucleus66,80,93. Average intensity projections and kymographs were generated using ImageJ v.2.3 software. Retrograde flow of PLS constructs was estimated by kymograph analysis in ImageJ using Measure tool (bounding rectangle option): velocities were calculated by dividing the traveled distance (d) by the time (t) using the bounding rectangle for the line drawn to trace the tracks of interest on the kymographs (Fig. 6e).
U2OS cells were transfected as described above, fixed/permeabilized for 15 min in phosphate-buffered saline containing 4% formaldehyde and 0.1% Triton X-100, counter-stained with TRITC–phalloidin and Hoechst dye (Thermo Fisher Scientific, Waltham, MA), and imaged using wide-field epifluorescence, Nikon CFI Plan Apochromat λ 60x oil objective (NA 1.40), and DS-QiMc camera on Nikon Eclipse Ti-E microscope using NIS Elements-AR v.4.3 software.
Immunoblotting
MTS-8-MTS-cross-linked RD-ABD1/F-actin samples were resolved on 9% SDS-PAGE and transferred to nitrocellulose. Western blotting was performed using anti-actin (1:2000; #MA5–11869, ThermoFisher Scientific, Waltham, MA) and anti-PLS3 (1:1000; #SAB2700266, Millipore-Sigma, Burlington, MA) primary antibodies followed by anti-mouse (#A4416) and anti-rabbit (#A0545) secondary antibodies conjugated to horseradish peroxidase (both are from Millipore-Sigma and used at 1:10,000). WesternBright Sirius chemiluminescent horseradish peroxidase substrate (Advansta, San Jose, CA) was used for signal detection in Omega Lum G imaging system (Aplegen, Pleasanton, CA) using OmegaLum G Image Capture software v.2.1.
Cryo-EM reconstruction
Cys-null PLS3 RD-ABD1 (Supplementary Table 1) was mutated to introduce a single Cys residue at positions 194 (Q194C), 216 (A216C), 226 (A226C), or 229 (A229C) using QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). The mutated constructs were purified as described above. Pichia pastoris expression vector pPICZc carrying human β-actin fused with thymosin b4 and 6xHis-tag was a gift from Mohan Balasubramanian (Addgene #111146, RRID:Addgene_111146). β-Actin cDNA was mutated to introduce K50C and C374A for labeling with a cross-linking reagent. The Cys-mutant actin was purified as previously described94. In order to cross-link PLS3 RD-ABD1 to actin, each protein was reduced in the presence of 10 mM DTT for 1 h on ice before being passed twice through a NAP-5 column equilibrated with G-buffer lacking β-ME. Actin was polymerized at 3 μM as described above in the absence of DTT for 1 h at room temperature. Polymerized actin was treated with a 1.2-fold excess of a cross-linking reagent, either N,N′-1,2-phenylenedimaleimide (oPDM), N,N′-1,4-phenylenedimaleimide (pPDM), or 3,6-dioxaoctane-1,8-diyl bismethanethiosulfonate (MTS-8-MTS) (Toronto Research Chemicals, North York, ON, Canada) for 15 min before addition of 25 μM PLS3 RD-ABD1 and incubated overnight on ice. The cross-linking efficiency was verified by SDS-PAGE and Western blotting using anti-actin and anti-PLS3 antibody (see above).
A 1.5-μL sample of cross-linked RD-ABD1/F-actin was applied to discharged lacey carbon grids and frozen with a Leica EM GP plunge freezer. Movies were collected in a Titan Krios at 300 keV equipped with a Falcon III direct electron detector, sampling at 1.4 Å/pixel using EPU v.2 Titan Krios software (FEI). The defocus range was set to 1.5–2.5 μm, with a total dose of ~55 electrons/Å2. MotionCor v.2 was used to motion-correct and dose-weight all the movies, followed by constant transfer function (CTF) estimation of the aligned images using the CTFFIND v.3 program95. Images with sparse CTF estimation were eliminated. The e2helixboxer program in the EMAN v.2.39 software package96 was used for boxing 256-px-long filaments (total 155,939 boxed segments). RELION v.3.0 was used for the following helical reconstruction procedure. The overall resolution of the final reconstruction was determined by the Fourier shell correlation (FSC) between two independent half maps, which was 5.1 Å at FSC = 0.143. The model of F-actin (PDB: 6ANU) and ABD1 of PLS3 (PDB: 1AOA) were fit into the cryo-EM map, followed by examination and adjustment in COOT v.0.8.9.297. The manually curated model was then real-space refined in PHENIX v.1.1998. The cryo-EM map was deposited with accession code EMD-25371 in the Electron Microscopy Data Bank (EMDB).
Statistics and Reproducibility
Graphical data was plotted and analyzed using Origin v.9 (OriginLab Corporation, Northampton, MA), Microsoft Excel for Mac v.16.59 (Microsoft Corporation, Redmond, WA), and JMP Pro v.15.2 (JMP, Cary, NC). All graph data is presented as mean ± standard deviation (SD) or standard error (SE) as indicated in the figure legends. Number of repetitions (n) is indicated in the figures/figure legends. One-way analysis of variance (ANOVA) followed by multiple comparison tests (Student’s t-tests with a two-tailed distribution) with Bonferroni correction was applied to determine statistically significant differences; individual p-values are indicated in the figure legends, where appropriate. Representative microscopy images and representative fluorescence/light scattering traces were obtained from at least two independent experiments. Cryo-EM map was generated by averaging together 155,939 256-px-long filament segments.
Data Availability:
The cryo-EM map of ABD1PLS3/F-actin complex was deposited with accession code EMD-25371 in the EMDB. Figures using protein structures were generated using PDB accession numbers: 6ANU, 1AOA, and 6VEC, and AlphaFold entry: https://alphafold.ebi.ac.uk/entry/P13797. Source data are provided with this paper.
Extended Data
Extended Data Figure 1. ABD2PLS2 exists in solution as a monomer and rescues polymerization of ACD-crosslinked actin oligomers.
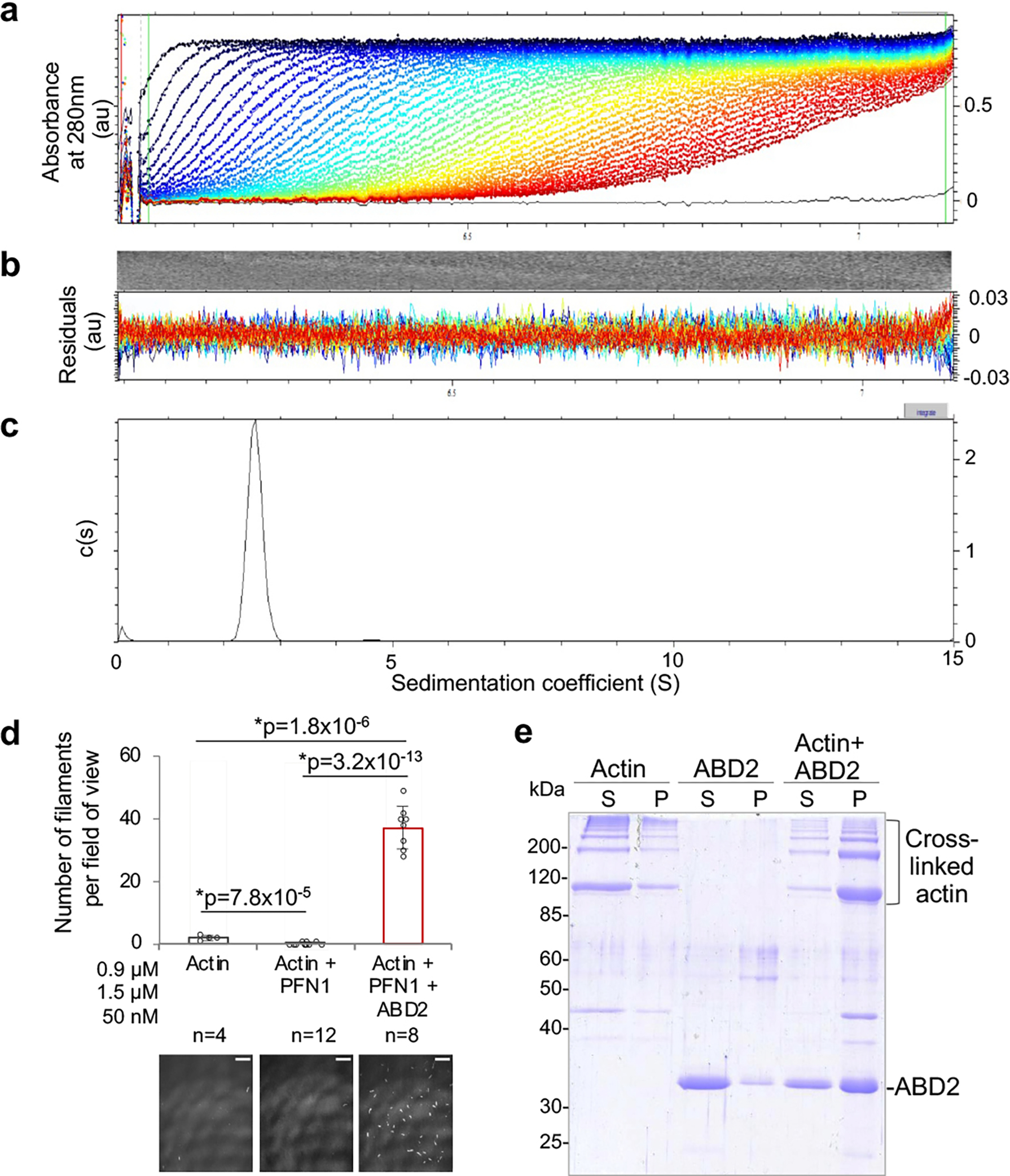
(related to Figure 1). (a-c) Sedimentation velocity analytical ultracentrifugation (SV-AUC) analysis of ABD2PLS2. Raw sedimentation profiles of absorbance at 280 nm versus radius (a) and residual plots (b) are shown. The distribution of sedimentation coefficients (c) indicates the presence of only monomeric species of ABD2PLS2 protein.
(d) Actin nucleation activity of ABD2PLS2 was tested by TIRFM in the presence of profilin (PFN1). Error bars represent the SD of the mean calculated from two independent experiments each containing four replicates. ANOVA followed by multiple comparison tests with Bonferroni correction was applied: asterisks indicate statistically significant difference (*p<0.017). Scale bars are 10 μm.
(e) ABD2PLS2 rescues polymerization of ACD-cross-linked actin oligomers. G-actin was covalently cross-linked by addition of ACD toxin in the absence (Actin) or presence (Actin +ABD2) of ABD2. Following ACD treatment, cross-linked actin was allowed to polymerize by addition of Mg2+ and KCl and subjected to ultracentrifugation to separate non-polymerized soluble (S) and polymerized pellet (P) fractions on SDS-PAGE. ABD2 alone sample (ABD2) treated identically served as a negative control.
Extended Data Figure 2. ABD1/ABD2 interaction is not affected by salt.
(related to Figure 2). The ABD1-ABD2 affinity in the presence of 30 and 130 mM KCl was determined by fluorescence anisotropy assays. Error bars represent the SD of the mean; n=3.
Extended Data Figure 3. L475P mutation diminishes F-actin bundling ability of PLS2.
(related to Figure 3). (a) F-actin bundling and binding by PLS2WT and PLS2L475P was assessed by low- (bundling) and high- (binding) speed sedimentation. Low-speed co-sedimentation (panel Actin Bundling) detected F-actin bundles formed only in the presence of PLS2WT in the absence of Ca2+ (PLS2WT Actin Bundling 3s/3p vs 4s/4p), while PLS2L475P was unable to bundle F-actin regardless of Ca2+ [F-actin remained mainly in the supernatant (PLS2L475P Actin Bundling 3s/3p and 4s/4p)]. In high-speed co-sedimentation assays (panel Actin Binding), both constructs were co-pelleted with F-actin in the absence (Actin Binding 3s/3p) and presence of Ca2+ (Actin Binding 4s/4p), implying that L475P mutation does not affect F-actin binding mediated through ABD1.
(b,c) In DSF experiments, PLS constructs (lines “T” on SDS-PAGE (b)) produced two melting peaks (Tm1 and Tm2, dotted lines on DSF graphs (c)) due to contamination with lower molecular weight fragments (red arrow on SDS-PAGE (b)). The contaminating fragments were neither removable by ion-exchange chromatography (due to similar pIs) nor by gel filtration (due to insufficiently different sizes). Pre-heating of the original proteins to 55 °C followed by ultracentrifugation at 300,000g for 30 min at 4 °C removed lower molecular weight contaminants and a minor fraction of the full-length protein as a precipitate (lines “P” on SDS-PAGE (b)). The supernatants (lines “S” on SDS-PAGE (b)) containing cleared FL PLS constructs displayed single DSF peaks (Tm1) but were otherwise functionally indistinguishable from the originally prepped proteins (c). The cleared samples (lines “S” on SDS-PAGE (b)) were used in DSF experiments (Figure 3).
Extended Data Figure 4. Cryo-EM reconstruction of ABD1/F-actin.
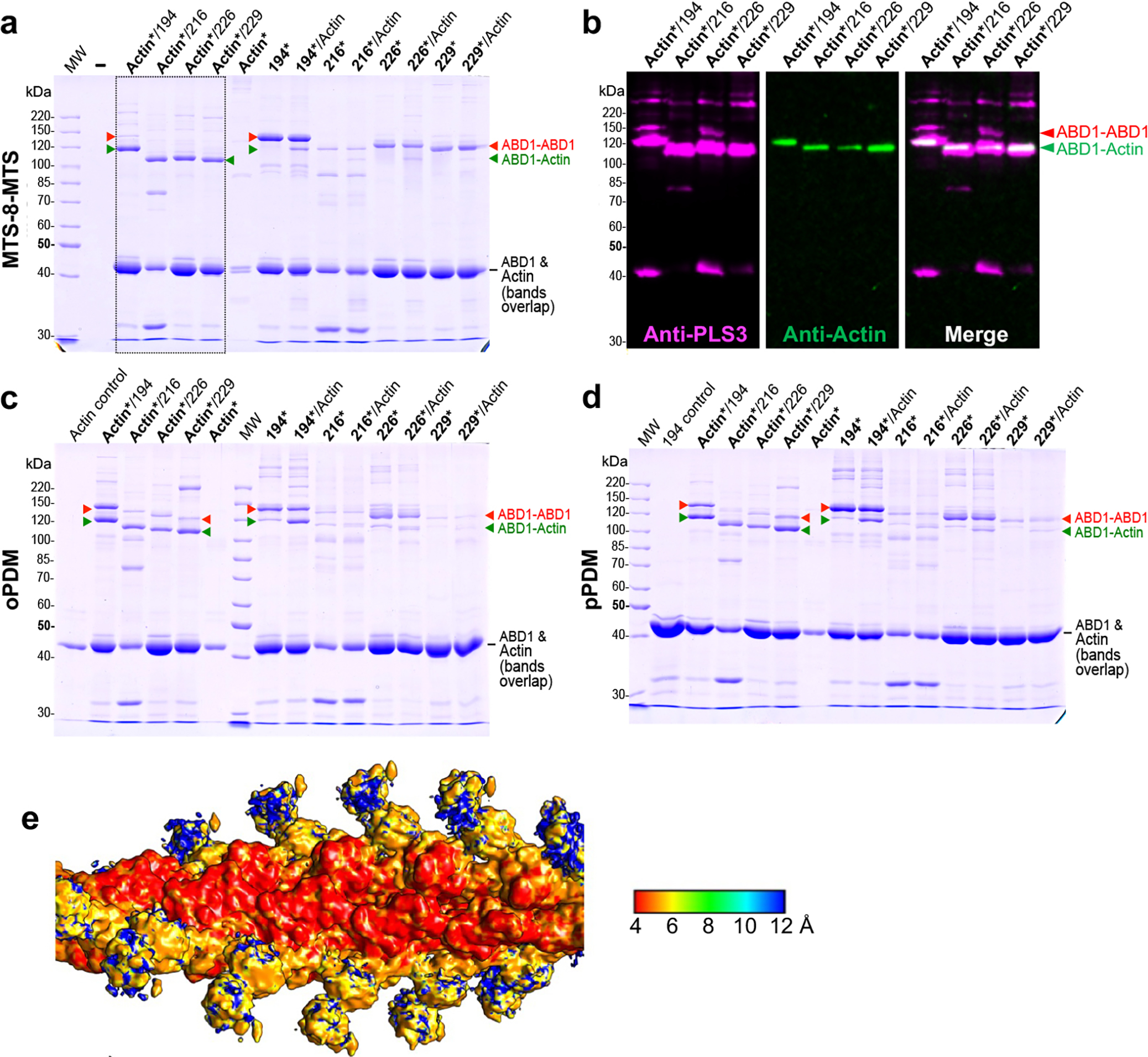
(related to Figure 4). (a-d) Preparation of ABD1/F-actin sample for cryo-EM. To produce F-actin decorated with ABD1, recombinant human β-Actin carrying K50C and C374A mutations was purified from Pichia pastoris and polymerized as described in online Methods. Individual Cys residues were introduced on the Cys-null RD-ABD1PLS3 background (Supplementary Table 1) at the indicated positions resulting in constructs containing single cysteines (Q194C, A216C, L226C, or G229C). Asterisks indicate activation of either actin (Actin*) or RD-ABD1 constructs (194*, 216*, 226*, or 229*) using a corresponding cross-linking reagent [MTS-8-MTS (a), oPDM (c), or pPDM (d)]. Following the activation, 2.5 μM actin was mixed with 10-molar excess of an RD-ABD1 construct. The resulting cross-linked samples were resolved on 9% SDS-PAGE (a, c, d). Note that non-crosslinked actin (42 kDa) and RD-ABD1 constructs (43 kDa) have similar molecular weights resulting in similar mobility on SDS-PAGE. Anti-actin and anti-PLS3 western blotting was performed to confirm identity of the resulting bands; only the immunoblots for the samples boxed in a are shown in b. Red arrowheads indicate RD-ABD1/RD-ABD1 cross-links. Green arrowheads indicate successful formation of RD-ABD1/F-actin cross-links. The cross-linking of MTS-8-MTS-activated actin with the RD-ABD1 (boxed in a) was the most efficient as compared to other tested reagents/combinations, and the sample of MTS-8-MTS-activated actin cross-linked with RD-ABD1Q194C was subjected to cryo-EM.
(e) ABD1/F-actin density map is colored according to the resolution in angstroms.
Extended Data Figure 5. S406E phospho-mimetic mutation moderately increases nucleation activity of PLS2.
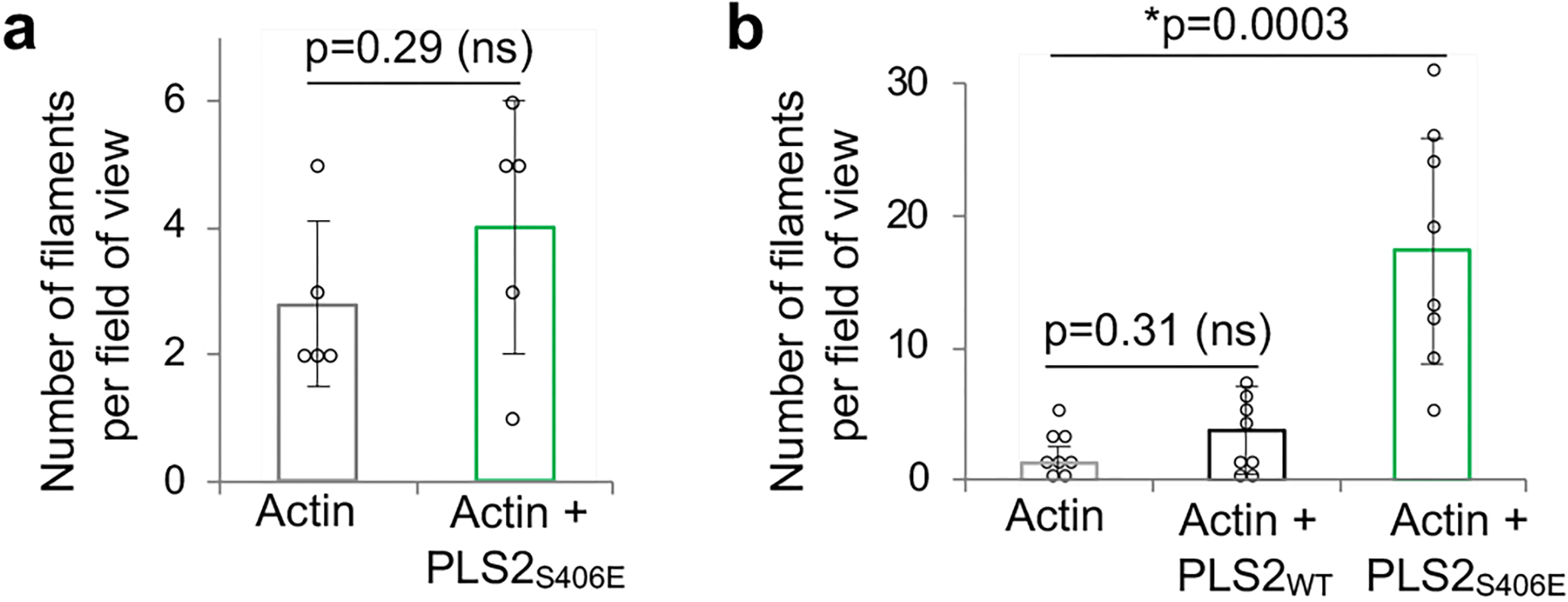
(related to Figure 5). Actin nucleation activity of PLS2S406E at 50-nM (a) and 1-μM (b) was tested by TIRFM. Error bars represent the SD of the mean calculated from two independent experiments each containing 4 replicates. ANOVA followed by multiple comparison tests with Bonferroni correction was applied: asterisk indicates statistically significant difference (*p<0.025) compared to the actin control.
Extended Data Figure 6. Intracellular localization of human plastins.
(related to Figure 6). U2OS cells transiently co-transfected with the indicated mEmerald-tagged plastin constructs and a focal adhesion marker mCardinal-paxillin were fixed and counter-stained with TRITC-phalloidin and Hoechst. Boxed areas in (a) are enlarged in (b). Scale bars are 20 μm in (a) and 5 μm in (b).
Extended Data Figure 7. Cell fragmenting for evaluating the subcellular distribution of plastins.
(related to Figure 6). (a) Transiently transfected XTC cells with mEmerald-tagged PLS2-WT and PLS2-S406E. The entire area of the cells (b) and lamellipodia (c) were outlined using masks as described in Methods. Scale bars are 10 μm.
Supplementary Material
Acknowledgements:
We thank Dr. Fenbin (Jerry) Wang (University of Virginia) for the assistance in modeling parallel actin filaments linked by a plastin bridge and Songyu Dong (OSU) and Lucas Runyan (OSU) for the assistance in protein purification. This work was supported by the National Institute of General Medical Sciences of the NIH under award numbers R01GM114666 (to DSK), R35GM122510 (to EHE), and a 2018 Pelotonia Graduate Fellowship Award at The Ohio State University Comprehensive Cancer Center (to CLS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing Interests Statement:
The authors declare that they have no conflict of interest.
References (main):
- 1.Shinomiya H Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. International journal of cell biology 2012, 1–8, doi: 10.1155/2012/213492 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delanote V, Vandekerckhove J & Gettemans J Plastins: versatile modulators of actin organization in (patho)physiological cellular processes. Acta pharmacologica Sinica 26, 769–779, doi: 10.1111/j.1745-7254.2005.00145.x (2005). [DOI] [PubMed] [Google Scholar]
- 3.Grimm-Günter EM et al. Plastin 1 binds to keratin and is required for terminal web assembly in the intestinal epithelium. Molecular Biology of the Cell 20, 2549–2562, doi: 10.1091/mbc.E08-10-1030 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin CS, Shen W, Chen ZP, Tu YH & Matsudaira P Identification of I-plastin, a human fimbrin isoform expressed in intestine and kidney. Molecular and Cellular Biology 14, 2457–2467, doi: 10.1128/mcb.14.4.2457 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drenckhahan D et al. Three different actin filament assemblies occur in every hair cell: each contains a specific actin crosslinking protein. Journal of Cell Biology 112, 641–651 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sobin A & Flock A Immunohistochemical identification and localization of actin and fimbrin in vestibular hair cells in the normal guinea pig and in a strain of the waltzing guinea pig. Acta Oto-Laryngologica 96, 407–412 (1983). [DOI] [PubMed] [Google Scholar]
- 7.Diaz-Horta O et al. Novel variant p.E269K confirms causative role of PLS1 mutations in autosomal dominant hearing loss. Clin Genet 96, 575–578, doi: 10.1111/cge.13626 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Morgan A et al. Mutations in PLS1, encoding fimbrin, cause autosomal dominant nonsyndromic hearing loss. Hum Mutat 40, 2286–2295, doi: 10.1002/humu.23891 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Jones SL, Wang J, Turck CW & Brown EJ A role for the actin-bundling protein L-plastin in the regulation of leukocyte integrin function. Proceedings of the National Academy of Sciences of the USA 95, 9331–9336 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma T, Sadashivaiah K, Madayiputhiya N & Chellaiah MA Regulation of sealing ring formation by L-plastin and cortactin in osteoclasts. Journal of Biological Chemistry 285, 29911–29924, doi: 10.1074/jbc.M109.099697 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morley SC The actin-bundling protein L-plastin: a critical regulator of immune cell function. International journal of cell biology 2012, 1–10, doi: 10.1155/2012/935173 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morley SC et al. The actin-bundling protein L-plastin dissociates CCR7 proximal signaling from CCR7-induced motility. Journal of Immunology 184, 3628–3638, doi: 10.4049/jimmunol.0903851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todd EM, Deady LE & Morley SC The actin-bundling protein L-plastin is essential for marginal zone B cell development. Journal of Immunology 187, 3015–3025, doi: 10.4049/jimmunol.1101033 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wabnitz GH et al. Costimulation induced phosphorylation of L-plastin facilitates surface transport of the T cell activation molecules CD69 and CD25. European journal of immunology 37, 649–662, doi: 10.1002/eji.200636320 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Wabnitz GH et al. Sustained LFA-1 cluster formation in the immune synapse requires the combined activities of L-plastin and calmodulin. European journal of immunology 40, 2437–2449, doi: 10.1002/eji.201040345 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Lin CS, Park T, Chen ZP & Leavitt J Human Plastin Genes: comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. Journal of Biological Chemistry 268, 2781–2792 (1993). [PubMed] [Google Scholar]
- 17.Chaijan S, Roytrakul S, Mutirangura A & Leelawat K Matrigel induces L-plastin expression and promotes L-plastin-dependent invasion in human cholangiocarcinoma cells. Oncology letters 8, 993–1000, doi: 10.3892/ol.2014.2239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foran E, McWilliam P, Kelleher D, Croke DT & Long A The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. International Journal of Cancer 118, 2098–2104, doi: 10.1002/ijc.21593 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Klemke M et al. Phosphorylation of ectopically expressed L-plastin enhances invasiveness of human melanoma cells. International Journal of Cancer 120, 2590–2599, doi: 10.1002/ijc.22589 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Riplinger SM et al. Metastasis of prostate cancer and melanoma cells in a preclinical in vivo mouse model is enhanced by L-plastin expression and phosphorylation. Molecular Cancer 13, 1–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garbett D et al. T-Plastin reinforces membrane protrusions to bridge matrix gaps during cell migration. Nat Commun 11, 4818, doi: 10.1038/s41467-020-18586-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwebach CL et al. Osteogenesis imperfecta mutations in plastin 3 lead to impaired calcium regulation of actin bundling. Bone research 8, 21, doi: 10.1038/s41413-020-0095-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N et al. Actin-bundling protein plastin 3 is a regulator of ectoplasmic specialization dynamics during spermatogenesis in the rat testis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29, 3788–3805, doi: 10.1096/fj.14-267997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue F, Janzen DM & Knecht DA Contribution of Filopodia to Cell Migration: A Mechanical Link between Protrusion and Contraction. Int J Cell Biol 2010, 507821, doi: 10.1155/2010/507821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagiwara M et al. Interaction of activated Rab5 with actin-bundling proteins, L- and T-plastin and its relevance to endocytic functions in mammalian cells. Biochemical and biophysical research communications 407, 615–619, doi: 10.1016/j.bbrc.2011.03.082 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Charras GT, Hu CK, Coughlin M & Mitchison TJ Reassembly of contractile actin cortex in cell blebs. The Journal of cell biology 175, 477–490, doi: 10.1083/jcb.200602085 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwebach CL, Kudryashova E & Kudryashov DS Plastin 3 in X-Linked Osteoporosis: Imbalance of Ca(2+)-Dependent Regulation Is Equivalent to Protein Loss. Front Cell Dev Biol 8, 635783, doi: 10.3389/fcell.2020.635783 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hosseinibarkooie S et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am J Hum Genet 99, 647–665, doi: 10.1016/j.ajhg.2016.07.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ralser M et al. Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Human molecular genetics 14, 2893–2909, doi: 10.1093/hmg/ddi321 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Lyon AN et al. Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Human molecular genetics 23, 1990–2004, doi: 10.1093/hmg/ddt595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oprea GE et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524–527, doi: 10.1126/science.1155085 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morandell J et al. Cul3 regulates cytoskeleton protein homeostasis and cell migration during a critical window of brain development. Nat Commun 12, 3058, doi: 10.1038/s41467-021-23123-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Begue E et al. Inducible expression and pathophysiologic functions of T-plastin in cutaneous T-cell lymphoma. Blood 120, 143–154, doi: 10.1182/blood-2011-09-379156 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Velthaus A et al. The Actin Binding Protein Plastin-3 Is Involved in the Pathogenesis of Acute Myeloid Leukemia. Cancers (Basel) 11, 1663, doi: 10.3390/cancers11111663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xin Z et al. PLS3 predicts poor prognosis in pancreatic cancer and promotes cancer cell proliferation via PI3K/AKT signaling. J Cell Physiol 235, 8416–8423, doi: 10.1002/jcp.29685 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Ma Y et al. Plastin 3 down-regulation augments the sensitivity of MDA-MB-231 cells to paclitaxel via the p38 MAPK signalling pathway. Artif Cells Nanomed Biotechnol 47, 685–695, doi: 10.1080/21691401.2019.1576707 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Korenbaum E & Rivero F Calponin homology domains at a glance. J Cell Sci 115, 3543–3545, doi: 10.1242/jcs.00003 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Schwebach CL, Agrawal R, Lindert S, Kudryashova E & Kudryashov DS The Roles of Actin-Binding Domains 1 and 2 in the Calcium-Dependent Regulation of Actin Filament Bundling by Human Plastins. Journal of Molecular Biology 429, 2490–2508, doi: 10.1016/j.jmb.2017.06.021 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Ishida H, Jensen KV, Woodman AG, Hyndman ME & Vogel HJ The Calcium-Dependent Switch Helix of L-Plastin Regulates Actin Bundling. Sci Rep 7, 40662, doi: 10.1038/srep40662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Namba Y, Ito M, Zu Y, Shigesada K & Maruyama K Human T Cell L-Plastin Bundles Actin Filaments in a Calcium Dependent Manner. Journal of Biochemistry 112, 503–507 (1992). [DOI] [PubMed] [Google Scholar]
- 41.Shirayama S & Numata O Tetrahymena fimbrin localized in the division furrow bundles actin filaments in a calcium-independent manner. J Biochem 134, 591–598, doi: 10.1093/jb/mvg183 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Kovar DR, Staiger CJ, Weaver EA & McCurdy DW AtFim1 is an actin filament crosslinking protein from Arabidopsis thaliana. Plant J 24, 625–636, doi: 10.1046/j.1365-313x.2000.00907.x (2000). [DOI] [PubMed] [Google Scholar]
- 43.Nakano K, Satoh K, Morimatsu A, Ohnuma M & Mabuchi I Interactions among a fimbrin, a capping protein, and an actin-depolymerizing factor in organization of the fission yeast actin cytoskeleton. Mol Biol Cell 12, 3515–3526, doi: 10.1091/mbc.12.11.3515 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klein MG et al. Structure of the actin crosslinking core of fimbrin. Structure 12, 999–1013, doi: 10.1016/j.str.2004.04.010 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Goldsmith SC et al. The structure of an actin-crosslinking domain from human fimbrin. Nat Struct Biol 4, 708–712, doi: 10.1038/nsb0997-708 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Galkin VE, Orlova A, Cherepanova O, Lebart MC & Egelman EH High-resolution cryo-EM structure of the F-actin-fimbrin/plastin ABD2 complex. Proceedings of the National Academy of Sciences of the USA 105, 1494–1498, doi: 10.1073/pnas.0708667105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volkmann N, DeRosier D, Matsudaira P & Hanein D An Atomic Model of Actin Filaments Cross-linked by Fimbrin and Its Implications for Bundle Assembly and Function. Journal of Cell Biology 153, 947–956 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dyson MR, Shadbolt SP, Vincent KJ, Perera RL & McCafferty J Production of soluble mammalian proteins in Escherichia coli: identification of protein features that correlate with successful expression. BMC Biotechnol 4, 32, doi: 10.1186/1472-6750-4-32 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kudryashov DS et al. Cofilin cross-bridges adjacent actin protomers and replaces part of the longitudinal F-actin interface. J Mol Biol 358, 785–797, doi: 10.1016/j.jmb.2006.02.029 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Kudryashov DS, Phillips M & Reisler E Formation and destabilization of actin filaments with tetramethylrhodamine-modified actin. Biophys J 87, 1136–1145, doi: 10.1529/biophysj.104.042242 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kudryashov DS et al. Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc Natl Acad Sci U S A 105, 18537–18542, doi: 10.1073/pnas.0808082105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith H et al. Rounding Out the Understanding of ACD Toxicity with the Discovery of Cyclic Forms of Actin Oligomers. Int J Mol Sci 22, 718, doi: 10.3390/ijms22020718 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niesen FH, Berglund H & Vedadi M The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols 2, 2212–2221, doi: 10.1038/nprot.2007.321 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Avery AW et al. Structural basis for high-affinity actin binding revealed by a beta-III-spectrin SCA5 missense mutation. Nat Commun 8, 1350, doi: 10.1038/s41467-017-01367-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanein D et al. An atomic model of fimbrin binding to F-actin and its implications for filament crosslinking and regulation. Nat Struct Biol 5, 787–792, doi: 10.1038/1828 (1998). [DOI] [PubMed] [Google Scholar]
- 56.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589, doi: 10.1038/s41586-021-03819-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu X et al. Mst1 Kinase Regulates the Actin-Bundling Protein L-Plastin To Promote T Cell Migration. J Immunol 197, 1683–1691, doi: 10.4049/jimmunol.1600874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin CS, Lau A & Lue TF Analysis and mapping of plastin phosphorylation. DNA Cell Biol 17, 1041–1046, doi: 10.1089/dna.1998.17.1041 (1998). [DOI] [PubMed] [Google Scholar]
- 59.Hornbeck PV et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Research 40, D261–270, doi: 10.1093/nar/gkr1122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huttlin EL et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189, doi: 10.1016/j.cell.2010.12.001 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klammer M et al. Phosphosignature Predicts Dasatinib Response in Non-small Cell Lung Cancer. Molecular and Cellular Proteomics 11, 651–668, doi:10.1074/ (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mertins P et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62, doi: 10.1038/nature18003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mertins P et al. Ischemia in Tumors Induces Early and Sustained Phosphorylation Changes in Stress Kinase Pathways but Does not Affect Global Protein Levels. Molecular and Cellular Proteomics 13, 1690–1704, doi:10.1074/ (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou H et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res 12, 260–271, doi: 10.1021/pr300630k (2013). [DOI] [PubMed] [Google Scholar]
- 65.Watanabe N & Mitchison TJ Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 295, 1083–1086, doi: 10.1126/science.1067470 (2002). [DOI] [PubMed] [Google Scholar]
- 66.Kudryashova E et al. Actin Cross-Linking Toxin Is a Universal Inhibitor of Tandem-Organized and Oligomeric G-Actin Binding Proteins. Curr Biol 28, 1536–1547 e1539, doi: 10.1016/j.cub.2018.03.065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skau CT et al. Actin filament bundling by fimbrin is important for endocytosis, cytokinesis, and polarization in fission yeast. Journal of Biological Chemistry 286, 26964–26977, doi: 10.1074/jbc.M111.239004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsai FC & Meyer T Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr Biol 22, 837–842, doi: 10.1016/j.cub.2012.03.037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song J et al. A cryo-tomography-based volumetric model of the actin core of mouse vestibular hair cell stereocilia lacking plastin 1. J Struct Biol 210, 107461, doi: 10.1016/j.jsb.2020.107461 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leite J et al. Equatorial Non-muscle Myosin II and Plastin Cooperate to Align and Compact F-actin Bundles in the Cytokinetic Ring. Front Cell Dev Biol 8, 573393, doi: 10.3389/fcell.2020.573393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu C, Chen Y & Ju LA Dynamic bonds and their roles in mechanosensing. Curr Opin Chem Biol 53, 88–97, doi: 10.1016/j.cbpa.2019.08.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martino F, Perestrelo AR, Vinarsky V, Pagliari S & Forte G Cellular Mechanotransduction: From Tension to Function. Front Physiol 9, 824, doi: 10.3389/fphys.2018.00824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anderson CA, Kovar DR, Gardel ML & Winkelman JD LIM domain proteins in cell mechanobiology. Cytoskeleton (Hoboken) 78, 303–311, doi: 10.1002/cm.21677 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kamioka H, Sugawara Y, Honjo T, Yamashiro T & Takano-Yamamoto T Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 19, 471–478, doi: 10.1359/JBMR.040128 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Weinbaum S, Duan Y, Satlin LM, Wang T & Weinstein AM Mechanotransduction in the renal tubule. Am J Physiol Renal Physiol 299, F1220–1236, doi: 10.1152/ajprenal.00453.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tseng HY et al. LCP1 preferentially binds clasped alphaMbeta2 integrin and attenuates leukocyte adhesion under flow. J Cell Sci 131, doi: 10.1242/jcs.218214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wabnitz GH et al. LFA-1 cluster formation in T-cells depends on L-plastin phosphorylation regulated by P90(RSK) and PP2A. Cell Mol Life Sci 78, 3543–3564, doi: 10.1007/s00018-020-03744-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mei L et al. Structural mechanism for bi-directional actin crosslinking by T-plastin. bioRxiv, 2021.2012.2007.471696, doi: 10.1101/2021.12.07.471696 (2021). [DOI] [Google Scholar]
References (Methods section only):
- 79.Spudich JA & Watt S The Regulation of Rabbit Skeletal Muscle Contraction. Journal of Biological Chemistry 246, 4866–4871 (1971). [PubMed] [Google Scholar]
- 80.Heisler DB et al. ACTIN-DIRECTED TOXIN. ACD toxin-produced actin oligomers poison formin-controlled actin polymerization. Science 349, 535–539, doi: 10.1126/science.aab4090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu J & Pollard TD Profilin binding to poly-L-proline and actin monomers along with ability to catalyze actin nucleotide exchange is required for viability of fission yeast. Mol Biol Cell 12, 1161–1175, doi: 10.1091/mbc.12.4.1161 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kudryashova E, Heisler D, Zywiec A & Kudryashov DS Thermodynamic properties of the effector domains of MARTX toxins suggest their unfolding for translocation across the host membrane. Mol Microbiol 92, 1056–1071, doi: 10.1111/mmi.12615 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Nolen BJ & Pollard TD Structure and biochemical properties of fission yeast Arp2/3 complex lacking the Arp2 subunit. Journal of Biological Chemistry 283, 26490–26498, doi: 10.1074/jbc.M802607200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682, doi: 10.1038/nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schindelin J, Rueden CT, Hiner MC & Eliceiri KW The ImageJ ecosystem: An open platform for biomedical image analysis. Molecular Reproduction and Development 82, 518–529, doi: 10.1002/mrd.22489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aitken CE, Marshall RA & Puglisi JD An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J 94, 1826–1835, doi: 10.1529/biophysj.107.117689 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kudryashova E et al. Human defensins facilitate local unfolding of thermodynamically unstable regions of bacterial protein toxins. Immunity 41, 709–721, doi: 10.1016/j.immuni.2014.10.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Purde V, Busch F, Kudryashova E, Wysocki VH & Kudryashov DS Oligomerization Affects the Ability of Human Cyclase-Associated Proteins 1 and 2 to Promote Actin Severing by Cofilins. Int J Mol Sci 20, 5647, doi: 10.3390/ijms20225647 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bairoch A The Cellosaurus, a Cell-Line Knowledge Resource. J Biomol Tech 29, 25–38, doi: 10.7171/jbt.18-2902-002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Uphoff CC & Drexler HG Detection of Mycoplasma contamination in cell cultures. Curr Protoc Mol Biol 106, 28 24 21–14, doi: 10.1002/0471142727.mb2804s106 (2014). [DOI] [PubMed] [Google Scholar]
- 91.Chu J et al. Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat Methods 11, 572–578, doi: 10.1038/nmeth.2888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rizzo MA, Davidson MW & Piston DW Fluorescent protein tracking and detection: fluorescent protein structure and color variants. Cold Spring Harb Protoc 2009, pdb top63, doi: 10.1101/pdb.top63 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Watanabe N Fluorescence single-molecule imaging of actin turnover and regulatory mechanisms. Methods Enzymol 505, 219–232, doi: 10.1016/B978-0-12-388448-0.00020-6 (2012). [DOI] [PubMed] [Google Scholar]
- 94.Hatano T et al. Rapid production of pure recombinant actin isoforms in Pichia pastoris. Journal of Cell Science 131, jcs213827, doi: 10.1242/jcs.213827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mindell JA & Grigorieff N Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology 142, 334–347, doi: 10.1016/s1047-8477(03)00069-8 (2003). [DOI] [PubMed] [Google Scholar]
- 96.Tang G et al. EMAN2: an extensible image processing suite for electron microscopy. Journal of Structural Biology 157, 38–46, doi: 10.1016/j.jsb.2006.05.009 (2007). [DOI] [PubMed] [Google Scholar]
- 97.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- 98.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM map of ABD1PLS3/F-actin complex was deposited with accession code EMD-25371 in the EMDB. Figures using protein structures were generated using PDB accession numbers: 6ANU, 1AOA, and 6VEC, and AlphaFold entry: https://alphafold.ebi.ac.uk/entry/P13797. Source data are provided with this paper.