

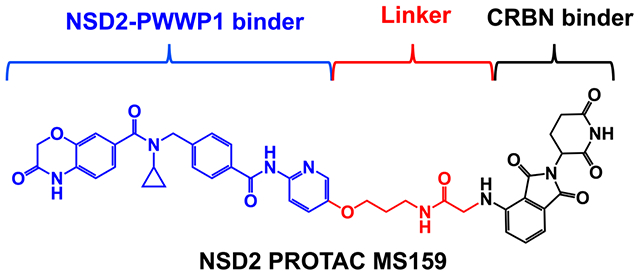
Abstract
Overexpression of nuclear receptor binding SET domain protein 2 (NSD2) is frequent in multiple myeloma (MM). However, existing NSD2 inhibitors are largely ineffective in suppressing MM cell proliferation. Here, we report the discovery of a first-in-class NSD2 proteolysis targeting chimera (PROTAC) degrader, 9 (MS159), and two structurally similar controls, 17 (MS159N1) and 18 (MS159N2), with diminished binding to the cereblon (CRBN) E3 ligase and NSD2, respectively. Compound 9, but not 17 and 18, effectively degraded NSD2 in a concentration-, time-, CRBN-, and proteasome-dependent manner. Compound 9 also effectively degraded CRBN neo-substrates IKZF1 and IKZF3, but not GSPT1. Importantly, compound 9 was much more effective in suppressing the growth in cancer cells than the parent NSD2 binder. Moreover, compound 9 was bioavailable in mice. Altogether, compound 9 and its two controls 17 and 18 are valuable chemical tools for exploring the roles of NSD2 in health and disease.
Graphical Abstract
INTRODUCTION
Nuclear receptor binding SET domain protein 2 (NSD2, also known as WHSC1 and MMSET), which is a protein lysine methyltransferase (PKMT), catalyzes the mono- and dimethylation of histone H3 lysine 36 (H3K36).1,2 It belongs to the NSD subfamily, which also includes NSD1 and NSD3 (also known as WHSC1L1).3,4 NSD2 is associated with diverse human diseases including cancers.5,6 It is implicated in the development of multiple myeloma (MM), predominantly in patients who carry a t(4,14) translocation that leads to aberrant upregulation of this gene.7 A recurrent gain-of-function mutation (E1099K) in the SET domain of NSD2 that increases its catalytic efficiency was reported in pediatric acute lymphoblastic leukemia (ALL) patients.8 Although the NSD subfamily of methyltransferases are attractive therapeutic targets, only limited progress has been made in developing selective inhibitors.9–11
In addition to a conserved catalytic SET domain, NSD proteins also have multiple chromatin-interacting domains such as two PWWP (proline–tryptophan–tryptophan–proline) and five PHD (plant homeodomain) domains as well as a DNA binding HMG-box (high mobility group box).12 It is increasingly evident that these domains play important roles in NSD2 function. However, the roles of these domains, individually or cooperatively, are not yet fully elucidated. Recently, small-molecule antagonists that bind the N-terminal PWWP (PWWP1) domain of NSD313 and NSD214,15 were described. We very recently reported the development of the first-in-class NSD3 small-molecule degrader,16 using the proteolysis targeting chimera (PROTAC) technology.17–19 A PROTAC is a heterobifunctional small molecule, which consists of a ligand for the protein of interest (POI), a ligand for an E3 ligase such as von Hippel–Lindau (VHL) or cereblon (CRBN) to recruit the ubiquitin–proteasome system (UPS) and a linker for connecting the two ligands. PROTACs induce the formation of POI–PROTAC–E3 ligase ternary complexes, leading to the targeted degradation of the POIs by the 26S proteasome.17–19 By employing the PROTAC technology and utilizing the reported ligand of the NSD2 PWWP1 domain as a NSD2 binder, one could develop a NSD2 PROTAC, which would temporally remove not only the chromatin binding function of NSD2 but all of its other functions including the methyltransferase activity. To date, NSD2 PROTAC degraders have not been reported.
Herein, we describe the design, synthesis, and characterization of a first-in-class NSD2 PROTAC degrader. After conducting a brief structure—activity relationship (SAR) study, we identified compound 9 (MS159), which connects the selective NSD2-PWWP1 antagonist UNC693415 to a CRBN E3 ligase ligand, as an effective NSD2 PROTAC degrader. Compound 9 induced NSD2 protein degradation in cells in a concentration-, time-, CRBN-, and proteasome-dependent manner. Importantly, compound 9 inhibited cell growth much more effectively than the NSD2-PWWP1 antagonist UNC6934 in MM cell lines (KMS11 and H929), suggesting that pharmacological degradation of NSD2 could be an effective and superior therapeutic strategy to pharmacological inhibition of the protein–protein interactions (PPIs) between the NSD2-PWWP1 domain and chromatin. Interestingly, compound 9 also effectively degraded CRBN neo-substrates IKZF1 and IKZF3 but not GSPT1. In addition, we developed two close analogues of compound 9, compounds 17 (MS159N1), and 18 (MS159N2) as negative controls. Overall, this study presents a set of novel chemical tools for the research community to further study the roles of NSD2 in pathophysiology.
RESULTS AND DISCUSSION
Design, Synthesis, and SAR Results of NSD2 PROTAC Degraders.
While NSD2 has attracted much attention as a therapeutic target for multiple myeloma and other cancers, unfortunately, efforts targeting its catalytic activity have not yielded potent and selective NSD2 inhibitors to date.9,10 A short while ago, small-molecule antagonists targeting the NSD2-PWWP1 domain with low micromolar affinity were discovered via virtual screening and scaffold hopping approaches.14 More recently, it was reported that structure-based optimization of aforementioned small-molecule PPI inhibitors led to the discovery of UNC6934 (Figure 1A), which selectively binds the NSD2-PWWP1 domain (Kd = 91 ± 8 nM) and disrupts the PPIs between the NSD2-PWWP1 domain and the H3K36 dimethylation (H3K36me2) mark of nucleosome.15 We therefore chose to utilize UNC6934 as a high affinity and selective NSD2 binder to develop a PROTAC degrader of NSD2. The cocrystal structure of the NSD2-PWWP1 domain in complex with UNC6934 (Figure 1B, PDB 6XCG) revealed that this small molecule occupied the methyllysine binding pocket. The cyclopropyl group immersed deep in an aromatic cage of the NSD2-PWWP1 domain and formed a very tight fit with this aromatic cage. While the benzoxazinone group sat in a cavity adjacent to the cyclopropyl binding pocket and made key hydrogen bonding interactions, the pyrimidine ring was solvent-exposed (Figure 1B, marked by the red circle). In addition, a biotin-labeled affinity reagent, which has a comparable binding affinity (Kd = 46 nM) to the NSD2-PWWP1 domain was previously prepared for pull-down studies.15 This affinity reagent contains a phenyl group replacing the pyrimidine ring, and the para position of the phenyl ring was used as the biotin attachment point. On the basis of the insights from the cocrystal structure as well as results of this biotinylated reagent, we designed and synthesized a set of putative NSD2 PROTACs, 1–11, by linking UNC6934 to VHL or CRBN E3 ligase ligands via a diverse set of linkers (Figure 1C). These putative degraders were designed by replacing the pyrimidine group with either a phenyl or pyridine group to release the para position as the tethering site for connecting to a linker and E3 ligase ligand. We introduced a carboxylic acid group at the para position of the phenyl ring (compound 12 in Scheme 1A) or an ether spacer with a terminal amine group at the para position of the pyridine ring (compound 13 in Scheme 1A) and linked these two precursors to VHL or CRBN E3 ligase ligands via a set of carbon or poly(ethylene glycol) (PEG) linkers (summarized in Figure 1C).
Figure 1.
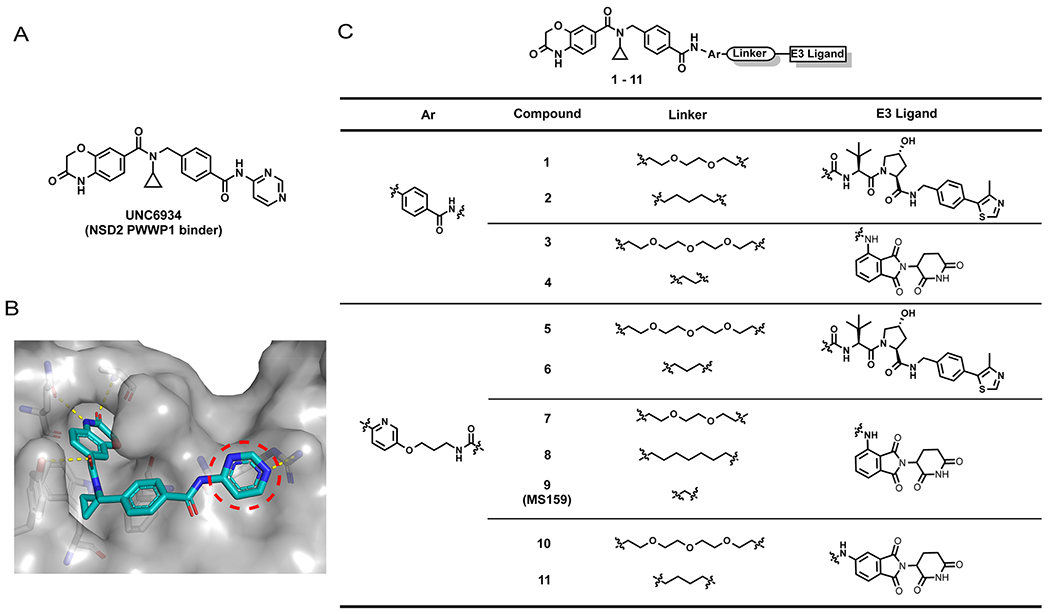
Design of putative NSD2 degraders 1–11. (A) Chemical structure of the NSD2 PWWP1 binder UNC6934. (B) Crystal structure of the NSD PWWP1 domain (gray) in complex with UNC6934 (blue) (PDB 6XCG). The pyrimidine ring of UNC6439 (highlighted by the red dashed circle) is solvent exposed. Key residues of NSD2 are shown in gray. Hydrogen bond interactions between UNC6439 and the key residues of the NSD2 PWWP1 domain are highlighted by yellow dashed lines. (C) Chemical structures of putative NSD2 degraders 1–11.
Scheme 1. Synthesis of compounds 1–11a.
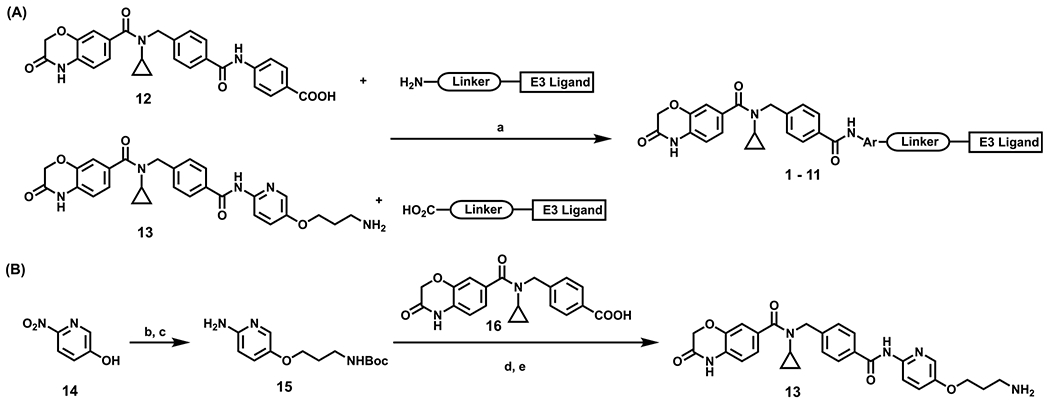
aReaction conditions: (a) HOAT, NMM, EDCI, DMSO, corresponding linker–E3 ligand, rt; (b) Cs2CO3, DMF, tert-butyl (3-bromopropyl)carbamate, 80 °C; (c) Pd/C, H2, MeOH, rt; (d) 16, SOCl2, 40 °C, remove solvent, then 15, pyridine, DMAP, rt; (e) DCM, TFA, rt.
Synthetic routes for the preparation of compounds 1–11 are outlined in Scheme 1. The amide coupling reaction of the known compound 12 with the corresponding terminal amino group-bearing linkers attached to E3 ligase ligands15,20,21 afforded the desired compounds 1–4 (Scheme 1A, top). Similarly, compounds 5–11 were synthesized by following the same amide coupling conditions between compound 13 and the corresponding intermediates featuring a terminal carboxylic acid attached to E3 ligase ligands via linkers (Scheme 1A, bottom). While compound 12 was synthesized following the published procedures,15 synthesis of compound 13 is shown in Scheme 1B. Briefly, a simple nucleophilic substitution reaction between commercially available 6-nitropyridin-3-ol (14) and tert-butyl (3-bromopropyl)carbamate followed by a hydrogenation reaction afforded compound 15. Compound 13 was obtained by a coupling reaction of compound 15 with compound 16, which was synthesized according to published procedures,15 followed by a deprotection reaction (Scheme 1B).
With these heterobifunctional compounds in hand, we next assessed their effects on reducing the NSD2 protein level in 293FT cells via Western blotting at two concentrations (Figure 2). Among compounds 1–4, compound 1, which is based on the VHL E3 ligase ligand VHL-122,23 with a PEG linker, clearly reduced the NSD2 protein level with a slight hook effect at 10μM (Figures 2 and Supporting Information (SI), S1A), while no obvious NSD2 degradation was observed for compounds 2–4 (Figure 2). We further investigated the effect of compound 1 on NSD2 degradation at lower concentrations and found that compound 1 did not induce NSD2 degradation at 0.1 and 0.5 μM (SI, Figure S1A). We also confirmed the NSD2 degradation effect induced by compound 1 at 2.5 μM and the hook affect at 10 μM (SI, Figure S1A). Among compounds 5–11, compound 9 clearly stood out and effectively induced degradation of NSD2 at both 2.5 and 10 μM without a hook effect. Compound 9 features a (pyridinyl)-oxy-propylamine group attached to the CRBN E3 ligase ligand pomalidomide24 via a very short alkyl linker (Figures 1C and 3A). From this SAR study, we selected compound 9 for advancing to further studies, based on its NSD2 degradation activity without a hook effect.
Figure 2.
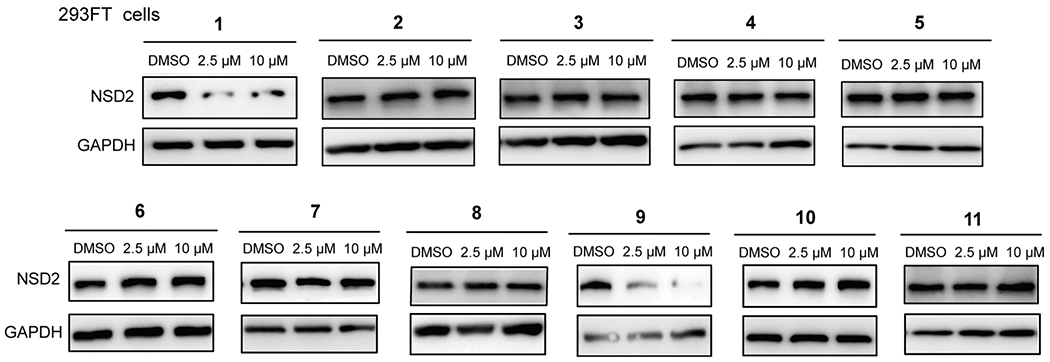
Western blotting analysis of the NSD2 protein level in 293FT cells treated with compounds 1–11. 293FT cells were treated with DMSO or compounds 1–11 at the indicated concentration for 48 h. GAPDH was used as the loading control. WB results are representative of at least two independent experiments.
Figure 3.
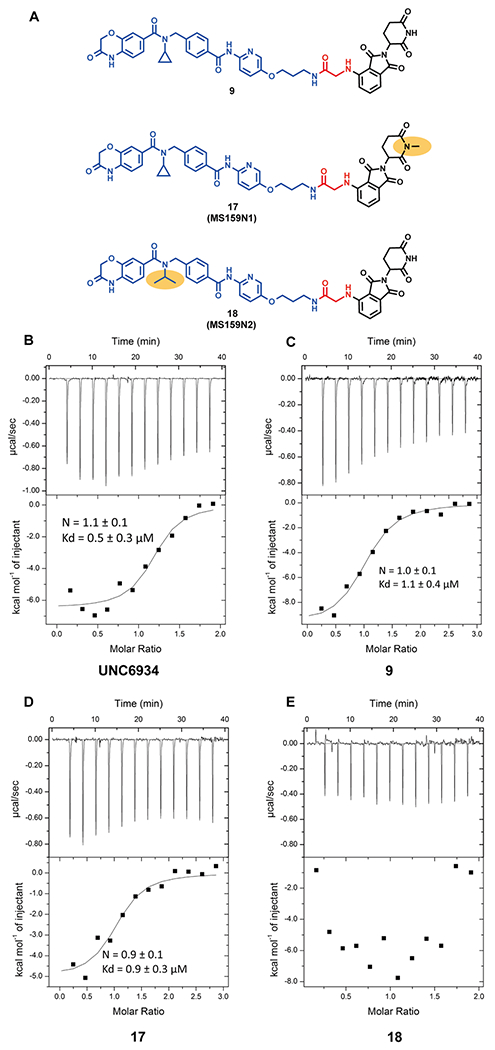
(A) Chemical structures of the NSD2 PROTAC degrader 9 (top), and its negative controls 17 (middle) and 18 (bottom). The yellow ellipse highlights the groups changed from 9. (B-E) Inverse ITC titrations of the NSD2 PWWP1 domain into UNC6934 (B), 9 (C), 17 (D), and 18 (E). The calculated binding affinity values represent the mean ± SD from two independent experiments. The first injection has been removed from the fitting.
To facilitate the assessment and characterization of compound 9, we also designed and synthesized two close analogues of compound 9, compounds 17 and 18, as negative control compounds (Figure 3A). Compound 17 was designed to be devoid of binding to the CRBN E3 ligase while maintaining a similar binding affinity to the NSD2-PWWP1 domain. This was achieved by the addition of a methyl group to the glutarimide moiety of pomalidomide, which is known to abrogate the ability to bind CRBN (Figure 3A, middle).25 On the other hand, compound 18 was designed to reduce its binding to the NSD2-PWWP1 domain by replacing the cyclopropyl group with the isopropyl group (Figure 3A, bottom), while keeping the same linker and CRBN binding moiety. This is based on the previous report that, because the cyclopropyl ring of UNC6934 (Kd = 91 ± 8 nM) fits extremely tightly to the aromatic cage of the binding pocket, swapping it with the sterically bulkier isopropyl group prevents the tight fit, leading to a loss of binding to the NSD2-PWWP1 domain (Kd > 20 μM).15
We next assessed the binding affinities of UNC6934, degrader 9, and the two negative controls 17 and 18 to the NSD2-PWWP1 domain using isothermal titration calorimetry (ITC). Comparing to UNC6934 (Kd = 0.5 ± 0.3 μM, Figure 3B), compound 9 (Kd = 1.1 ± 0.4 μM, Figure 3C), and compound 17 (Kd = 0.9 ± 0.3 μM, Figure 3D) maintained similar binding affinities to the NSD2-PWWP1 domain, validating our design hypothesis. Furthermore, as expected, compound 18, which contains a relatively inactive ligand of the NSD2-PWWP1 domain, did not show any appreciable binding to the NSD2-PWWP1 domain under the experimental conditions tested (Figure 3E).
Synthetic routes for preparation of compounds 17 and 18 are outlined in Scheme 2. Commercially available thalidomide-4-fluoride (19) and its N-methylated derivative 2020 were converted to intermediates 21 and 22, respectively, via a nucleophilic substitution reaction with microwave heating and subsequent removal of the protecting group. An amidecoupling reaction between 13 and 22 afforded 17, while a coupling reaction between intermediates 21 and 24 yielded 18. Intermediate 24 was obtained from the known compound 23, which was prepared according to reported procedures,15 by its conversion to the corresponding acid chloride and then a coupling reaction with 15 (see Scheme 1) followed by tert-butyl carbamate deprotection.
Scheme 2. Synthesis of Negative Controls 17 and 18a.
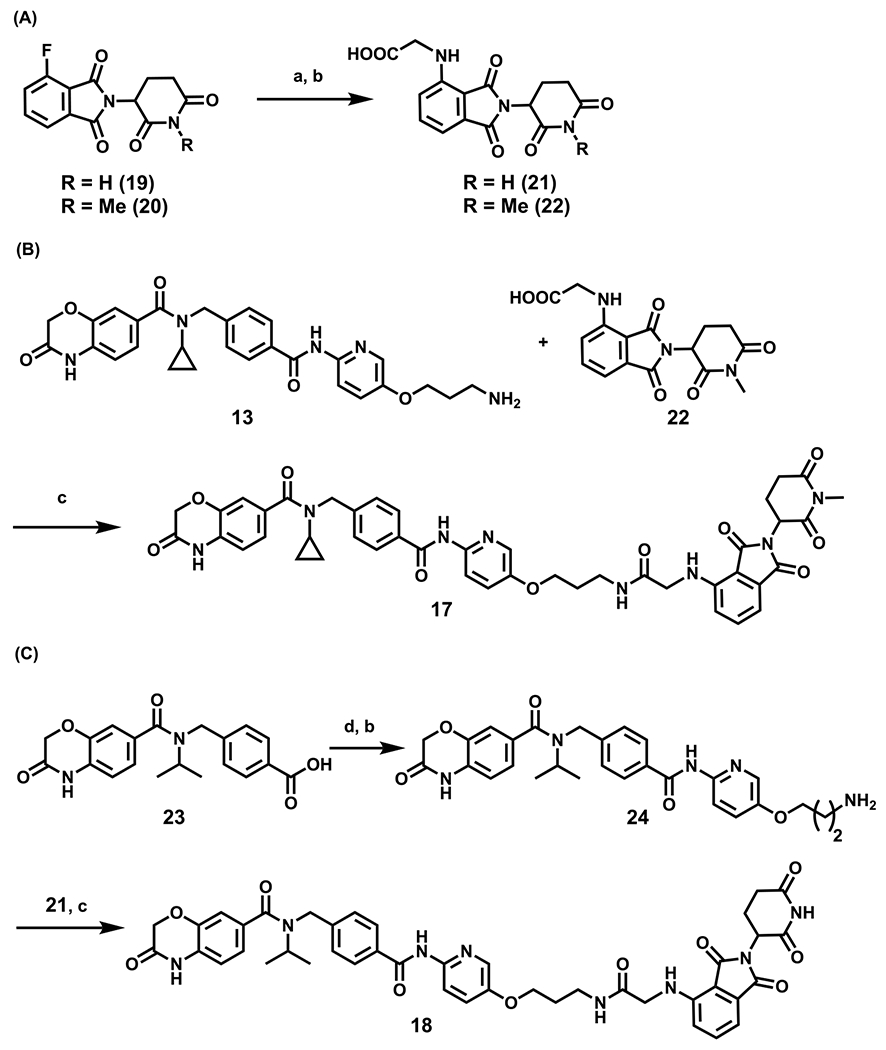
aReaction conditions: (a) NMP, DIPEA, MW, 100 °C, tert-butyl glycinate; (b) DCM, TFA, rt; (c) HOAT, EDCI, NMM, DMSO, rt; (d) SOCl2, 40 °C, then 15, pyridine, DMAP, rt.
Characterization of Compound 9’s NSD2 Degradation Effect in 293FT Cells.
We next evaluated the NSD2 degradation induced by compound 9 in 293FT cells treated with compound 9 in a range of concentrations (0.5–10 μM). As illustrated in Figure 4A,B, compound 9 reduced the NSD2 protein level in a concentration dependent manner, with the DC50 value of 5.2 ± 0.9 μM and Dmax > 82% in 293FT cells after 48 h treatment. A time course study was also performed to determine the kinetics of the NSD2 degradation induced by compound 9. 293FT cells were treated with compound 9 at 5 μM for various duration ranging from 0 to 72 h. We observed some NSD2 degradation as early as 36 h, significant degradation at 48 h, and the maximum degradation effect at 72 h (Figure 4C).
Figure 4.
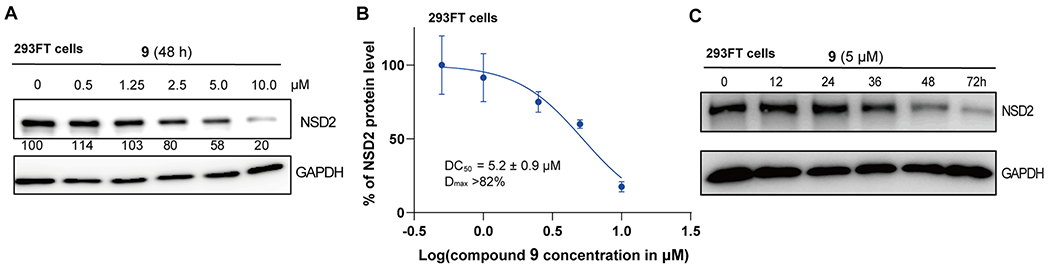
Compound 9 reduces the NSD2 protein level in 293FT cells in a concentration- and time-dependent manner. (A) Compound 9 concentration-dependently induces NSD2 protein degradation. 293FT cells were treated with the indicated concentration of compound 9 for 48 h. Cell lysates were collected, and the protein level of NSD2 was detected by Western blotting with GAPDH as the loading control. The relative strength of the NSD2 signal on the Western blots was measured by densitometry. The WB results are representative of three independent experiments. (B) DC50 and Dmax values of the NSD2 degradation induced by compound 9. Results are presented as the mean ± SD from two independent experiments. (C) Compound 9 reduces the NSD2 protein level in a time-dependent manner. 293FT cells were treated with 5 μM of compound 9 for 0, 12, 24, 36, 48, or 72 h. The NSD2 protein level was detected by Western blotting with GAPDH as the loading control.
We next performed a set of mechanism of action (MOA) experiments to establish that the NSD2 degradation induced by compound 9 is indeed dependent on the ubiquitin–proteasome system (UPS) (Figure 5A). We cotreated 293FT cells with 5 μM of compound 9 together with one of the following compounds: either 10 μM of the CRBN E3 ligase ligand pomalidomide, 5 μM of UNC6934, 5 μM of the proteasome inhibitor MG132, or 10 μM of the neddylation inhibitor MLN4924. Compared with the DMSO control, cotreatment with pomalidomide, which competes with compound 9 for binding the CRBN E3 ligase, or cotreatment with the NSD2-PWWP1 binder UNC6934, which competes with compound 9 for binding the NSD2-PWWP1 domain, significantly rescued the compound 9-induced reduction of the NSD2 protein level. Furthermore, cotreatment with MG132, a proteasome inhibitor, or with MLN4924, an inhibitor of NEDD8-activating enzyme inhibitor that blocks cullin neddylation and subsequently inactivates cullin RING E3 ligase complexes, also successfully restored the NSD2 protein level in the compound 9-treated 293FT cells. Collectively, these results indicate that the compound 9-induced degradation of NSD2 is indeed mediated through a NSD2-, CRBN-, and UPS-dependent mechanism.
Figure 5.
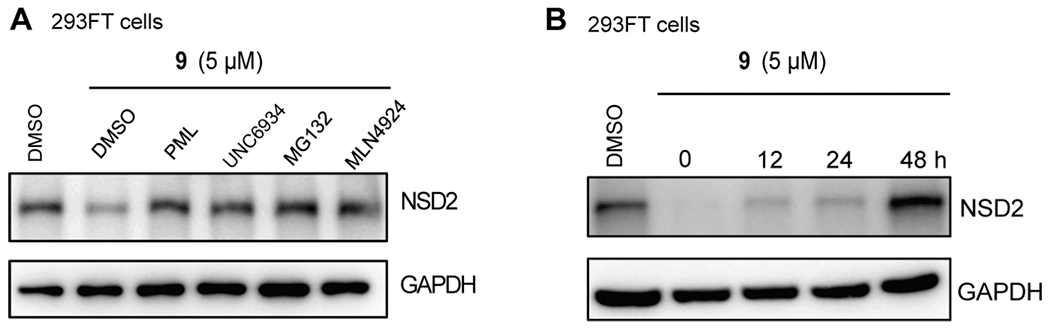
(A) Compound 9 reduces the NSD2 protein level in 293FT cells in a NSD2, CRBN-, and proteasome-dependent manner. 293FT cells were treated with 5 μM of compound 9 for 48 h. During the last 6 h, the cells were cotreated with either 10 μM of pomalidomide (PML), 5 μM of UNC6934, 5 μM of MG132, or 10 μM of MLN4924. The NSD2 protein level was detected by Western blotting with GAPDH as the loading control. (B) NSD2 degradation induced by compound 9 is reversible. 293FT cells were treated with 5 μM of compound 9 for 72 h, and compound 9 was then washed out. The NSD2 protein level was detected by Western blotting at 0, 12, 24, and 48 h post the washout. GAPDH was used as the loading control.
To assess the reversibility of the compound 9-induced NSD2 degradation, we next conducted washout experiments (Figure 5B). In 293FT cells, treatment with compound 9 for 48 h led to a significant reduction of the NSD2 protein level as expected. We then washed out the compound with fresh medium and observed that the NSD2 protein level was slowly and gradually recovered after 12 and 24 h and fully recovered after 48 h, indicating that the NSD2 degradation mediated by compound 9 is reversible (Figure 5B).
Compound 9 Degrades NSD2 and IKZF1/3 in Cancer Cells.
We next evaluated the effect of compound 9 on reducing the NSD2 protein level in two multiple myeloma cell lines, KMS11 and H929. With DMSO as the control, the cancer cells were treated with compound 9, UNC6934, 17, or 18 at 2.5 μM for 72 h. As shown in Figure 6, while UNC6934, compounds 17 and 18 did not induce obvious NSD2 degradation, compound 9 effectively degraded NSD2 in both cell lines. Interestingly, the NSD2 degradation effect mediated by compound 9 was more pronounced in H929 cells compared with KMS11 cells. Since the degradation of CRBN:IMiD neosubstrates, such as GSPT1, IKZF1, and IKZF3, by CRBN-recruiting PROTACs has been previously reported,26,27 we also evaluated compound 9-induced degradation of aforementioned neosubstrates in these two cancer cell lines. It should be noted that, GSPT1, a translation termination factor, is probably the most concerning neo-substrate, due to its critical role in most cells and potential toxicities resulting from its degradation. On the other hand, IKZF1 and IKZF3 are the validated oncotargets of multiple myeloma, and it has been reported that degradation of IKZF1 and IKZF3 along with CDK4 and CDK6 by a CDK4/6 PROTAC displayed notably higher efficacies in killing mantle cell lymphoma cells over CDK4 and CDK6 degradation alone.27 Therefore, it is critical to determine the neo-substrate degradation profile of compound 9. We were pleased to find that compound 9, as well as the control compounds 17 and 18, did not degrade GSPT1 in both KMS11 and H929 cells (Figure 6A,B). On the other hand, compound 9 and its negative control compound 18, which binds CRBN but not the NSD2-PWWP1 domain, clearly reduced the IKZF1 protein level (Figures 6A and 7) as well as the IKZF3 protein level to a lesser extent (Figure 6B) in KMS11 and H929 cells. As expected, UNC6934 and compound 17, the negative control that binds the NSD2-PWWP1 domain but not CRBN, did not display any degradation effect on these CRBN:IMiD neo-substrates. With these results in hand, we also revisited compound 1, a VHL-recruiting NSD2 PROTAC which degraded NSD2 at 2.5 μM. As expected, compound 1 did not degrade GSPT1 or IKZF3 in 293FT cells (SI, Figure S1A). However, this compound was also ineffective in degrading NSD2 in KMS11 and H929 cells (SI, Figure S1B). Taken together, these results demonstrated that compound 9 is an effective degrader of NSD2 as well as IKZF1 and IKZF3 in KMS11 and H929 multiple myeloma cells.
Figure 6.
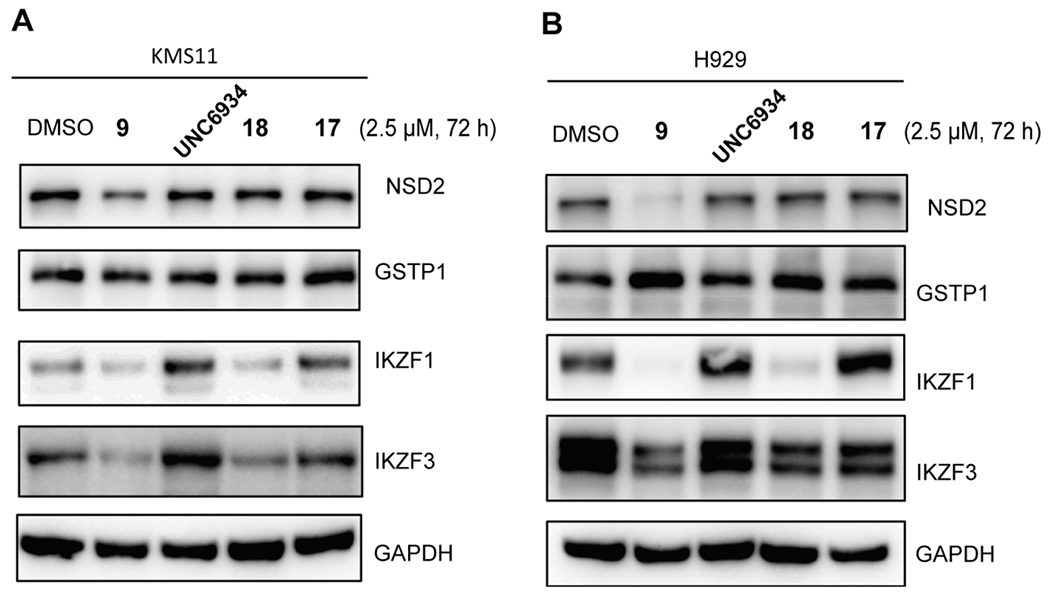
Compound 9 reduces the protein levels of NSD2, IKZF1, and IKZF3, but not GSPT1, while compound 18 degrades IKZF1 and IKZF3 but not NSD2 and GSPT1, and compound 17 and UNC6934 do not degrade any of these proteins, in KMS11 (A) and H929 (B) cells. The cells were treated with DMSO or 2.5 μM of the indicated compound for 72 h. The protein levels were detected by Western blotting with GAPDH as the loading control. The Western blotting results are representative of two independent experiments.
Figure 7.
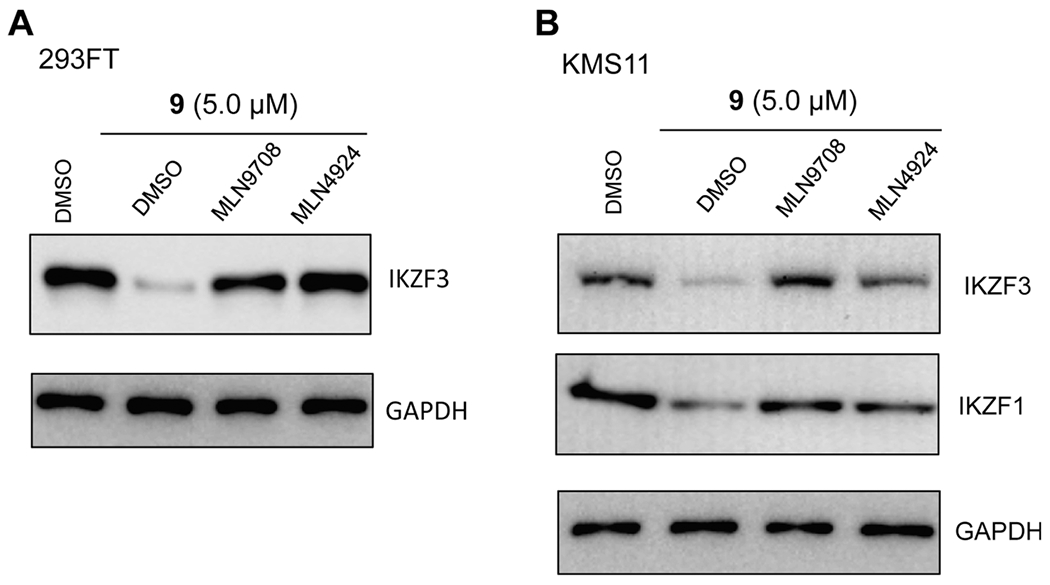
(A) Compound 9 reduces the IKZF3 protein level in 293FT cells in a proteasome-dependent manner. 293FT cells were pretreated with DMSO, 10 nM of MLN9708 or 0.5 μM of MLN4924 for 2 h, followed by a 6 h treatment with 5 μM of compound 9. (B) Compound 9 reduces the IKZF1 and IKZF3 protein levels in KMS11 cells in a proteasome-dependent manner. KMS11 cells were pretreated with DMSO, 10 nM of MLN9708 or 0.5 μM of MLN4924 for 2 h, followed by a 6 h treatment with 5 μM of compound 9. The IKZF1 and IKZF3 protein levels were detected by Western blotting with GAPDH as the loading control. The WB results are representative of at least two independent experiments.
To clarify that the mechanism of IKZF1 and IKZF3 degradation induced by compound 9 is UPS-mediated, we first pretreated 293FT (Figure 7A) and KMS11 (Figure 7B) cells with the proteosome inhibitor MLN9708 (10 nM) or the neddylation inhibitor MLN4924 (0.5 μM), followed by the treatment with 5 μM of compound 9 and then measured the IKZF3 protein level in 293FT cells and the IKZF1 and IKZF3 protein levels in KMS1 cells via Western blotting. In the compound 9-treated 293FT cells, compared with the DMSO control, pretreatment with MLN9708, a proteasome inhibitor, or with MLN4924, a NEDD8-activating enzyme inhibitor that blocks cullin neddylation and subsequently inactivates cullin RING E3 ligase complexes, successfully restored the IKZF3 protein level (IKZF1 expression level was too low in this cell line to obtain clear blots) (Figure 7A). Furthermore, in the compound 9-treated KMS11 cells, compared with the DMSO control, pretreatment with MLN9708 or with MLN4924 also at least partially rescued the IKZF1 and IKZF3 degradation induced by compound 9 (Figure 7B). Collectively, these results indicate that the compound 9-induced degradation of IKZF1/3 is indeed mediated through a UPS-dependent mechanism.
Furthermore, to demonstrate that the IKZF1 and IKZF3 degradation induced by compound 9 is CRBN-dependent, we treated wild-type (WT) 293FT cells (Figure 8, left), as well as 293FT cells with the CRISPR-Cas9-mediated knockout (KO) of CRBN (Figure 8, right), with compound 9 at a range of concentrations and measured the NSD2 and IKZF3 protein levels (the IKZF1 protein level was too low in 293FT cells to obtain clear blots) by Western blotting. While clear degradation of NSD2 and IKZF3 was observed in WT 293FT cells, no NSD2 or IKZF3 degradation was detected in the CRBN KO 293FT cells. These results clearly indicate that the compound 9-induced degradation of IKZF3 as well as NSD2 is indeed dependent on the E3 ligase CRBN.
Figure 8.
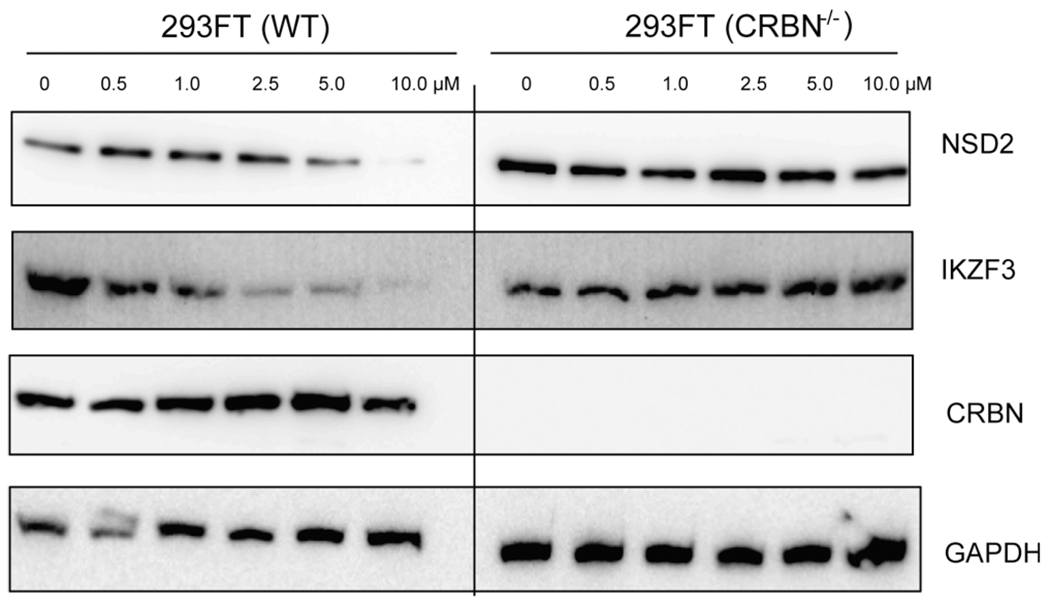
Compound 9 reduces the NSD2 and IKZF3 protein levels in 293FT cells in a CRBN-dependent manner. The NSD2 and IKZF3 protein levels were reduced in WT 293FT cells treated with compound 9 for 48 h in a concentration-dependent manner (left), while the NSD2 and IKZF3 protein levels were not affected by the treatment of compound 9 for 48 h in 293FT cells with the CRISPR-Cas9-mediated KO of CRBN (right). The NSD2, IKZF3, and CRBN protein levels were detected by Western blotting with GAPDH as the loading control. The WB results are representative of at least two independent experiments.
Taken together, these results demonstrated that compound 9 effectively degraded NSD2 as well as IKZF1 and IKZF3 in a UPS- and CRBN-dependent manner.
Next, we assessed selectivity of compound 9 against a panel of 20 protein lysine and arginine methyltransferases. As illustrated in Figure 9, compound 9 did not inhibit any of the 20 methyltransferases including NSD1/2/3 at 10 μM. This selectivity result is consistent with that of the parent NSD2-PWWP1 antagonist UNC6934, which also did not inhibit any of these methyltransferases.15
Figure 9.
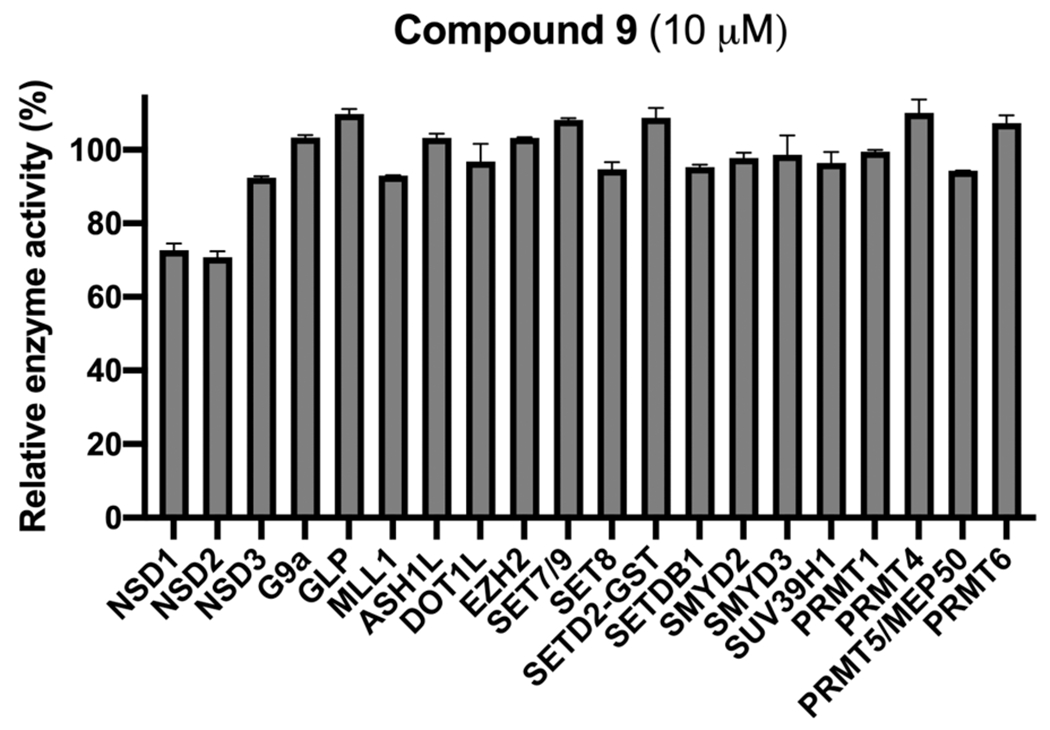
Selectivity of compound 9 (at 10 μM) against 20 protein lysine and arginine methyltransferases.
Compound 9 Suppresses the Growth of Multiple Myeloma Cells.
We next assessed antiproliferative effects of compound 9 in multiple myeloma cells. We treated KMS11 and H929 cells with compound 9 for 8 days and used compounds 17 and 18 as well as UNC6934 as controls. As illustrated in Figure 10A,B, we found that compound 9 effectively inhibited the growth in both KMS11 and H929 multiple myeloma cells. On the other hand, the parent NSD2-PWWP1 antagonist UNC6934 and the negative control 17, which binds the NSD2-PWWP1 domain but does not degrade NSD2 and IKZF1/3, showed little effect on cell growth inhibition in both cell lines. These results clearly indicate that the cell growth inhibition effect of compound 9 is due to its degradation effect, not its effect on blocking PPIs between the NSD2-PWWP1 domain and chromatin. Interestingly, compound 18, which degrades IKZF1/3 but does not bind the NSD2-PWWP1 domain and does not degrade NSD2 (Figures 3E and 6), displayed some growth inhibition in both cell lines, especially in KMS11 cells, suggesting that degradation of IKZF1/3, the oncoproteins of multiple myeloma, contributes to compound 9’s cell growth inhibition effect.
Figure 10.
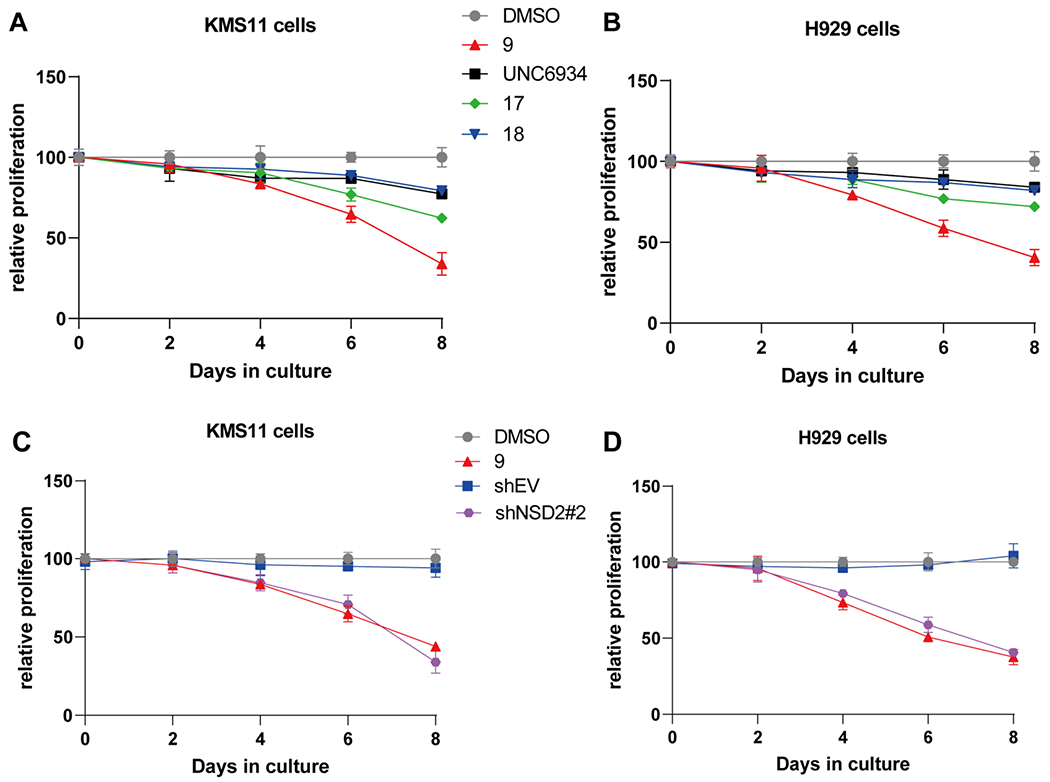
Compound 9 inhibits the proliferation in multiple myeloma cells. (A,B) The effect of compounds 9, 17, and 18 as well as UNC6934 on inhibiting the proliferation of KMS11 (A) and H929 (B) cells. The cells were treated with DMSO, 9, 17, 18, or UNC6934 at 2.5 μM for 8 days. (C,D) The effect of compound 9 (2.5 μM), relative to DMSO, and shRNA-mediated KD of NSD2 (sh#2), relative to transduction of empty vector (shEV), on inhibiting the proliferation in KMS11(C) and H929 (D) cells. The relative cell viabilities to the corresponding control group are shown as the mean ± SD (n = 3).
Furthermore, we investigated the effect of NSD2 KD on the growth of KMS11 and H929 multiple myeloma cells. We first assessed cell proliferation after shRNA-mediated knockdown (KD) of NSD2, relative to transduction of empty vector (shEV), in KMS11 and H929 cells (Figure 10C,D and SI, Figure S2). We also compared the effect of NSD2 KD with that of compound 9 treatment (2.5 μM, 8 days) (Figure 10C,D). These experiments clearly show that (1) the growth of KMS11 and H929 cells is dependent on NSD2, and (2) the effect of compound 9 on cell growth inhibition largely phenocopies that of NSD2 KD in these two cell lines.
Taken together, these results show that our NSD2 and IKZF1/3 PROTAC degrader 9 is much more effective in suppressing the growth of multiple myeloma cells than the NSD2-PWWP1 antagonist UNC6934. These results also suggest that (1) the NSD2 degradation induced by compound 9 is the major contributor to its cell growth inhibition effect in these multiple myeloma cell lines, and (2) the degradation of IKZF1/3 also contributes to compound 9’s cell growth inhibition effect, but in a lesser degree.
Compound 9 Is Bioavailable in Mice.
Lastly, we evaluated in vivo mouse PK properties of compound 9. Following a single intraperitoneal (IP) administration of compound 9 at 50 mg/kg, concentrations in plasma from male Swiss albino mice over a period of 8 h were determined. We found that a single IP injection resulted in the maximum plasma concentration of 1.35 ± 0.08 μM attained at 0.5 h post dosing (Figure 11). At 8 h post dosing, the concentration of compound 9 was still around 0.98 ± 0.04 μM. Importantly, the compound was well tolerated by the treated mice, and no clinical signs were observed in the PK study. Overall, these results suggest that compound 9 could be a valuable chemical tool for investigating the effects of dual NSD2 and IKZF1/3 degradation in vivo.
Figure 11.
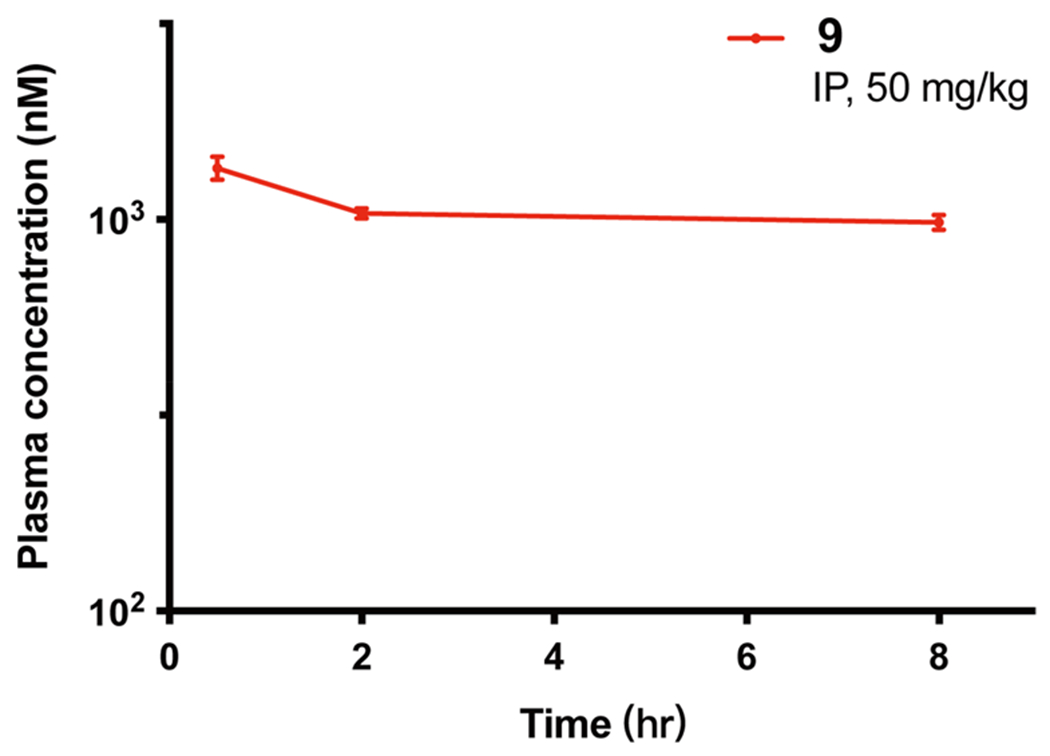
Plasma concentrations of compound 9 over 8 h, following a single 50 mg/kg IP injection in male Swiss albino mice. The compound concentration shown at each time point is the mean ± SD from three test mice.
CONCLUSION
Although the PROTAC technology has been successfully applied to the generation of numerous small-molecule degraders, very few small-molecule degraders of protein methyltransferases (also known as histone methyltransferases) have been reported.19 Recently, UNC6934, a small molecule that selectively binds to NSD2-PWWP1 domain and antagonizes its interaction with H3K36me2, was reported.15 However, this NSD2-PWWP1 domain antagonist is incapable of inhibiting the catalytic activity of NSD2 and ineffective in suppressing the growth of multiple myeloma cells. Here, through a brief SAR study, we discovered a first-in-class NSD2 PROTAC degrader, compound 9, which is an E3 ligase CRBN-recruiting heterobifunctional small molecule that effectively reduced the NSD2 protein level in cells in a concentration-, time-, NSD2-, CRBN-, and UPS-dependent manner. In addition, compound 9 was able to degrade CRBN:IMiD neo-substrates IKZF1 and IKZF3 in addition to NSD2, but not GSPT1, in two multiple myeloma cell lines. Importantly, compound 9 was much more effective than the parent NSD2-PWWP1 antagonist, UNC6934, in inhibiting the proliferation in KMS11 and H929 multiple myeloma cells, suggesting that pharmacological degradation of NSD2 and IKZF1/3 is a superior therapeutic strategy to pharmacological antagonism of the NSD2-PWWP1 and chromatin PPIs. Furthermore, compound 9 was bioavailable in a mouse PK study. Moreover, we developed two structurally similar control compounds, 17, which binds the NSD2-PWWP1 domain but does not degrade NSD2 or IKZF1/3, and 18, which binds CRBN and degrades IKZF1/3 but does not bind the NSD2-PWWP1 domain and does not degrade NSD2. It should be noted that these two control compounds are structurally, minimally altered compared to compound 9. Therefore, they have very similar physicochemical properties and should maintain similar properties such as cell permeability to that of compound 9. Compound 17 acts similarly as UNC6934. It binds NSD2, but cannot degrade NSD2 and IKZF1/3. On the other hand, compound 18 acts similarly as pomalidomide. It degrades the CRBN:IMiD neosubstrates IKZF1 and IKZF3 but does not bind and degrade NSD2. Collectively, compound 9 and its controls 17 and 18 represent a set of valuable chemical tools for the scientific community to further investigate NSD2 functions in health and disease.
EXPERIMENTAL SECTION
Chemistry General Procedures.
All chemical reagents were purchased from commercial vendors and used without further purification. The flash column chromatography was conducted using a Teledyne ISCO CombiFlash Rf+ instrument. This instrument was also equipped with a variable-wavelength UV detector and a fraction collector. RediSep Rf Gold C18 columns were used for purification. High-performance liquid chromatography (HPLC) spectra for compounds were acquired using an Agilent 1200 series system with a DAD detector. Chromatography was performed on a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). Ultra performance liquid chromatography (UPLC) spectra for compounds were acquired using a Waters Acquity I-Class UPLC system with a PDA detector. Chromatography was performed on a 2.1 mm μ 30 mm Acquity UPLC BEH C18 1.7 μm column with water containing 3% acetonitrile, 0.1% formic acid as solvent A, and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.8 mL/min. The gradient program was as follows: 1–99% B (1–1.5 min), and 99–1% B (1.5–2.5 min). High-resolution mass spectra (HRMS) data were acquired in the positive ion mode using with Agilent G1969A API-TOF or with a Waters Acuity Premiere XE TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker DRX-500 spectrometer with 500 MHz for proton (1H NMR) or a Bruker DRX-600 spectrometer with 600 MHz for proton (1H NMR) or 151 MHz for carbon (13C NMR). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed using an Agilent Prep 1200 series with UV detector set to 220 nm. Samples were injected into a Phenomenex Luna 75 mm × 30 mm, 5 μm, C18 column at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% of acetonitrile (A) in H2O (with 0.1% TFA) (B) to 100% of acetonitrile (A). All final compounds had >95% purity using the UPLC and HPLC methods described above. Compounds 12,15 16,15 20,20 21,21 and 2315 are previously reported compounds and were prepared according to published procedures.
N-Cyclopropyl-N-(4-((4-((2-(2-(3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropoxy)ethoxy)ethyl)-carbamoyl)phenyl)carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (1).
To a solution of compound 12 (24 mg, 0.05 mmol), (2S,4R)-1-((S)-2-(3-(2-(2-aminoethoxy)ethoxy)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide28 (29 mg, 0.05 mmol), HOAT (14 mg, 0.1 mmol), and EDCI (20 mg, 0.1 mmol) in DMSO (3 mL) was added NMM (25 mg, 0.25 mmol). The resulting mixture was stirred at room temperature overnight and then purified by preparative HPLC (10–100% MeCN/0.1% TFA in H2O) to afford compound 1 (32 mg, 61% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.89 (s, 1H), 10.43 (s, 1H), 8.98 (s, 1H), 8.57 (t, J = 6.1 Hz, 1H), 8.43 (t, J = 5.6 Hz, 1H), 7.98–7.95 (m, 2H), 7.93 (d, J = 9.4 Hz, 1H), 7.90–7.83 (m, 4H), 7.47 (d, J = 7.9 Hz, 2H), 7.42 (d, J = 8.3 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.22–7.17 (m, 1H), 7.16 (s, 1H), 6.94 (d, J = 8.1 Hz, 1H), 5.12 (s, 1H), 4.73 (s, 2H), 4.63 (s, 2H), 4.56 (d, J = 9.4 Hz, 1H), 4.47–4.40 (m, 2H), 4.38–4.33 (m, 1H), 4.23 (dd, J = 15.8, 5.5 Hz, 1H), 3.71–3.56 (m, 4H), 3.56–3.51 (m, 4H), 3.53–3.47 (m, 2H), 3.47–3.39 (m, 2H), 2.81 (s, 1H), 2.58–2.50 (m, 1H), 2.45 (s, 3H), 2.37 (dt, J = 14.6, 6.2 Hz, 1H), 2.08–2.01 (m, 1H), 1.91 (ddd, J = 12.9, 8.6, 4.6 Hz, 1H), 0.94 (s, 9H), 0.56 (d, J = 6.5 Hz, 2H), 0.49 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C56H65N8O11S+, 1057.4488; found, 1057.4503.
N-Cyclopropyl-N-(4-((4-((6-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)carbamoyl)phenyl)-carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (2).
Following the procedures for synthesis of 1, compound 2 was synthesized with (2S,4R)-1-((S)-2-(6-amino-hexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide28 (27 mg, 0.05 mmol) as the substrate (28 mg, 55% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.89 (s, 1H), 10.43 (s, 1H), 8.99 (s, 1H), 8.57 (t, J = 6.1 Hz, 1H), 8.35 (t, J = 5.6 Hz, 1H), 7.96 (d, J = 8.0 Hz, 2H), 7.89–7.82 (m, 5H), 7.47 (d, J = 7.9 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.22–7.18 (m, 1H), 7.16 (d, J = 1.8 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 5.13 (s, 1H), 4.73 (s, 2H), 4.63 (s, 2H), 4.56 (d, J = 9.4 Hz, 1H), 4.47–4.41 (m, 2H), 4.36 (tt, J = 4.3, 2.6 Hz, 1H), 4.22 (dd, J = 15.8, 5.4 Hz, 1H), 3.71–3.63 (m, 2H), 3.28–3.21 (m, 2H), 2.81 (s, 1H), 2.45 (s, 3H), 2.32–2.23 (m, 1H), 2.19–2.11 (m, 1H), 2.08–2.01 (m, 1H), 1.95–1.88 (m, 1H), 1.61–1.47 (m, 4H), 1.35–1.26 (m, 2H), 0.94 (s, 9H), 0.56 (d, J = 6.7 Hz, 2H), 0.49 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C55H63N8O9S+, 1011.4433; found, 1011.4475.
N-Cyclopropyl-N-(4-((4-((2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)-carbamoyl)phenyl)carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (3).
Following the procedures for synthesis of 1, compound 3 was synthesized with 4-((2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione28 (23 mg, 0.05 mmol) as the substrate (20 mg, 44% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.10 (s, 1H), 10.89 (s, 1H), 10.43 (s, 1H), 8.41 (t, J = 5.6 Hz, 1H), 7.99–7.94 (m, 2H), 7.90–7.83 (m, 4H), 7.58 (dd, J = 8.6, 7.1 Hz, 1H), 7.47 (d, J = 7.6 Hz, 2H), 7.22–7.17 (m, 1H, 7.17–7.11 (m, 2H), 7.04 (d, J = 7.0 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.60 (d, J = 6.3 Hz, 1H), 5.06 (dd, J = 12.8, 5.4 Hz, 1H), 4.73 (s, 2H), 4.63 (s, 2H), 3.61 (t, J = 5.5 Hz, 2H), 3.58–3.51 (m, 10H), 3.49–3.44 (m, 2H), 3.46–3.39 (m, 2H), 2.89 (ddd, J = 17.0, 13.9, 5.4 Hz, 1H), 2.81 (s, 1H), 2.63–2.51 (m, 2H), 2.07–1.99 (m, 1H), 0.56 (d, J = 6.8 Hz, 2H), 0.49 (s, 2H). UPLC > 95%, tR = 1.22 min. MS (ESI) m/z: [M + H]+ calcd for C48H50N7O12+, 916.3512; found, 916.3537.
N-Cyclopropyl-N-(4-((4-((2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)carbamoyl)phenyl)carbamoyl)-benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (4).
Following the procedures for synthesis of 1, compound 4 was synthesized with 4-((2-aminoethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione29 (16 mg, 0.05 mmol) as the substrate (25 mg, 63% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.89 (s, 1H), 10.44 (s, 1H), 8.63 (t, J = 5.5 Hz, 1H), 7.99–7.94 (m, 2H), 7.87 (q, J = 8.9 Hz, 4H), 7.60 (dd, J = 8.6, 7.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.6 Hz, 1H), 7.22–7.18 (m, 1H), 7.16 (d, J = 1.8 Hz, 1H), 7.04 (d, J = 7.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.86 (t, J = 6.1 Hz, 1H), 5.07 (dd, J = 12.9, 5.5 Hz, 1H), 4.73 (s, 2H), 4.63 (s, 2H), 3.50 (dq, J = 34.8, 7.7, 6.4 Hz, 4H), 2.89 (ddd, J = 17.1, 13.9, 5.4 Hz, 1H), 2.82 (s, 1H), 2.63–2.51 (m, 2H), 2.03 (dtd, J = 12.9, 5.3, 2.4 Hz, 1H), 0.58–0.54 (m, 2H), 0.49 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C42H38N7O9+, 784.2726; found, 784.2715.
N-Cyclopropyl-N-(4-((5-(((S)-19-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-5,17-dioxo-8,11,14-trioxa-4,18-diazahenicosyl)-oxy)pyridin-2-yl)carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo-[b][1,4]oxazine-7-carboxamide (5).
Following the procedures for synthesis of 1, compound 5 was synthesized with (S)-15-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-16,16-dimethyl-13-oxo-4,7,10-trioxa-14-azaheptadecanoic acid30 (33 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (34.7 mg, 60% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.88 (s, 1H), 10.64 (s, 1H), 8.99 (s, 1H), 8.57 (t, J = 6.1 Hz, 1H), 8.12–8.08 (m, 2H), 8.02 (d, J = 8.0 Hz, 2H), 7.97–7.90 (m, 2H), 7.48 (dd, J = 9.1, 3.0 Hz, 1H), 7.46–7.39 (m, 4H), 7.39 (d, J = 8.2 Hz, 2H), 7.22–7.18 (m, 1H), 7.16 (s, 1H), 6.94 (d, J = 8.1 Hz, 1H), 4.71 (s, 2H), 4.62 (s, 2H), 4.56 (d, J = 9.4 Hz, 1H), 4.47–4.39 (m, 2H), 4.38–4.33 (m, 1H), 4.23 (dd, J = 16.0, 5.8 Hz, 1H), 4.06 (t, J = 6.3 Hz, 2H), 3.68 (dd, J = 10.6, 4.1 Hz, 1H), 3.65–3.59 (m, 2H), 3.62–3.54 (m, 4H), 3.53–3.43 (m, 8H), 3.23 (q, J = 6.5 Hz, 2H), 2.81 (s, 1H), 2.58–2.52 (m, 1H), 2.45 (s, 3H), 2.40–2.32 (m, 1H), 2.32 (t, J = 6.4 Hz, 2H), 2.07–2.01 (m, 1H), 1.94–1.83 (m, 3H), 0.94 (s, 9H), 0.55 (d, J = 6.8 Hz, 2H), 0.48 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C60H74N9O13S+, 1160.5121; found, 1160.5165.
N1-(3-((6-(4-((N-Cyclopropyl-3-oxo-3,4-dihydro-2H-benzo[b]-[1,4]oxazine-7-carboxamido)methyl)benzamido)pyridin-3-yl)oxy)-propyl)-N5-((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-glutaramide (6).
Following the procedures for synthesis of 1, compound 6 was synthesized with 5-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentanoic acid31 (26 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (25.5 mg, 49% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.89 (s, 1H), 10.64 (s, 1H), 8.99 (s, 1H), 8.57 (t, J = 6.1 Hz, 1H), 8.12–8.06 (m, 2H), 8.02 (d, J = 8.2 Hz, 2H), 7.91–7.86 (m, 2H), 7.48 (dd, J = 9.2, 3.0 Hz, 1H), 7.48–7.41 (m, 4H), 7.39 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.2 Hz, 1H), 7.17–7.14 (m, 1H), 6.94 (d, J = 8.1 Hz, 1H), 4.71 (s, 2H), 4.62 (s, 2H), 4.54 (d, J = 9.4 Hz, 1H), 4.48–4.40 (m, 2H), 4.39–4.33 (m, 1H), 4.22 (dd, J = 15.8, 5.5 Hz, 1H), 4.07 (t, J = 6.2 Hz, 2H), 3.71–3.63 (m, 2H), 3.21 (dq, J = 8.9, 6.6 Hz, 2H), 2.82 (s, 1H), 2.56 (t, J = 5.4 Hz, 1H), 2.45 (s, 3H), 2.28–2.20 (m, 1H), 2.20–2.12 (m, 1H), 2.12–2.01 (m, 3H), 1.95–1.82 (m, 3H), 1.77–1.67 (m, 2H), 0.94 (s, 9H), 0.56 (d, J = 6.8 Hz, 2H), 0.48 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C55H64N9O10S+, 1042.4491; found, 1042.4513.
N-Cyclopropyl-N-(4-((5-(3-(3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)propanamido)-propoxy)pyridin-2-yl)carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (7).
Following the procedures for synthesis of 1, compound 7 was synthesized with 3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)-propanoic acid21 (22 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (26.5 mg, 57% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.88 (s, 1H), 10.63 (s, 1H), 8.09 (d, J = 8.7 Hz, 2H), 8.03–7.99 (m, 2H), 7.91 (t, J = 5.6 Hz, 1H), 7.57 (dd, J = 8.6, 7.0 Hz, 1H), 7.47 (dd, J = 9.1, 3.0 Hz, 1H), 7.42 (d, J = 7.8 Hz, 2H), 7.22–7.17 (m, 1H), 7.17–7.11 (m, 2H), 7.03 (d, J = 7.0 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 6.59 (s, 1H), 5.06 (dd, J = 12.8, 5.4 Hz, 1H), 4.71 (s, 2H), 4.62 (s, 2H), 4.05 (t, J = 6.3 Hz, 2H), 3.60 (q, J = 5.9 5.3 Hz, 4H), 3.56–3.52 (m, 2H), 3.52–3.42 (m, 4H), 3.22 (q, J = 6.5 Hz, 2H), 2.88 (ddd, J = 17.0, 13.9, 5.4 Hz, 1H), 2.83–2.80 (m, 1H), 2.62–2.52 (m, 2H), 2.31 (t, J = 6.4 Hz, 2H), 2.06–1.99 (m, 1H), 1.85 (p, J = 6.6 Hz, 2H), 0.55 (d, J = 6.8 Hz, 2H), 0.48 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C48H51N8O12+, 931.3621; found, 931.3616.
N-Cyclopropyl-N-(4-((5-(3-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octanamido)propoxy)pyridin-2-yl)-carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (8).
Following the procedures for synthesis of 1, compound 8 was synthesized with 8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octanoic acid32 (20 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (22 mg, 50% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.88 (s, 1H), 10.63 (s, 1H), 8.10 (d, J = 9.2 Hz, 2H), 8.03–7.99 (m, 2H), 7.86 (t, J = 5.7 Hz, 1H), 7.57 (dd, J = 8.6, 7.1 Hz, 1H), 7.47 (dd, J = 9.0, 3.1 Hz, 1H), 7.42 (dt, J = 8.3, 3.5 Hz, 2H), 7.20 (d, J = 8.1 Hz, 1H), 7.16 (d, J = 1.8 Hz, 1H), 7.08 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 7.0 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.52 (s, 1H), 5.05 (dd, J = 12.8, 5.4 Hz, 1H), 4.71 (s, 2H), 4.62 (s, 2H), 4.06 (t, J = 6.2 Hz, 2H), 3.28 (q, J = 6.5 Hz, 2H), 3.21 (q, J = 6.5 Hz, 2H), 2.88 (ddd, J = 17.0, 13.9, 5.4 Hz, 1H), 2.84–2.77 (m, 1H), 2.63–2.52 (m, 2H), 2.11–2.04 (m, 2H), 2.06–1.99 (m, 1H), 1.86 (p, J = 6.5 Hz, 2H), 1.56 (q, J = 7.1 Hz, 2H), 1.50 (hept, J = 7.7, 6.9 Hz, 2H), 1.36–1.20 (m, 6H), 0.56 (d, J = 6.8 Hz, 2H), 0.48 (s, 2H). UPLC > 95%, tR = 1.41 min. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C49H53N8O10+, 913.3879; found, 913.3918.
N-Cyclopropyl-N-(4-((5-(3-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetamido)propoxy)pyridin-2-yl)-carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (9).
Following the procedures for synthesis of 1, compound 9 was synthesized with (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine (21)21 (17 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (21.8 mg, 53% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.11 (s, 1H), 10.88 (s, 1H), 10.69 (s, 1H), 8.21 (t, J = 5.7 Hz, 1H), 8.08 (d, J = 8.3 Hz, 2H), 8.2 (d, J = 7.9 Hz, 2H), 7.58 (t, J = 7.8 Hz, 1H), 7.49 (dd, J = 9.1, 3.0 Hz, 1H), 7.43 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 8.1 Hz, 1H), 7.16 (s, 1H), 7.07 (d, J = 7.1 Hz, 1H), 6.99–6.96 (m, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 5.08 (dd, J = 12.8, 5.4 Hz, 1H), 4.72 (s, 2H), 4.62 (s, 2H), 4.06 (t, J = 6.2 Hz, 2H), 3.96 (s, 2H), 3.29 (q, J = 6.4 Hz, 2H), 2.90 (ddd, J = 16.7, 13.7, 5.4 Hz, 1H), 2.82 (s, 1H), 2.63–2.52 (m, 2H), 2.07–2.00 (m, 1H), 1.90 (p, J = 6.6 Hz, 2H), 0.56 (d, J = 7.0 Hz, 2H), 0.48 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 173.3, 170.5, 169.2, 169.0, 167.8, 165.7, 165.3, 152.2, 146.3, 145.9, 143.0, 142.9, 136.7, 134.7, 133.3, 132.5, 132.2, 128.9, 128.7, 127.6, 124.8, 122.4, 117.9, 116.3, 115.9, 115.6, 111.4, 110.4, 67.2, 66.5, 50.7, 49.0, 45.7, 36.0, 31.5, 31.2, 29.2, 22.6, 10.0. HPLC > 95%, tR = 5.21 min. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C43H41N8O10+, 829.2940; found, 829.2920.
N-Cyclopropyl-N-(4-((5-((1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)-12-oxo-3,6,9-trioxa-13-azahexadecan-16-yl)oxy)pyridin-2-yl)carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (10).
Following the procedures for synthesis of 1, compound 10 was synthesized with 3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)-ethoxy)ethoxy)ethoxy)propanoic acid33 (24 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (29.2 mg, 62% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.06 (s, 1H), 10.88 (s, 1H), 10.64 (d, J = 4.0 Hz, 1H), 8.12–8.08 (m, 2H), 8.04–8.00 (m, 2H), 7.93 (t, J = 5.7 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.51–7.45 (m, 1H), 7.42 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 8.3 Hz, 1H), 7.18–7.16 (m, 1H), 7.16 (s, 1H), 7.01 (d, J = 2.2 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.89 (dd, J = 8.4, 2.2 Hz, 1H), 5.03 (dd, J = 12.8, 5.5 Hz, 1H), 4.71 (s, 2H), 4.62 (s, 2H), 4.06 (t, J = 6.2 Hz, 2H), 3.63–3.57 (m, 4H), 3.56–3.49 (m, 4H), 3.52–3.44 (m, 4H), 3.35 (t, J = 5.5 Hz, 2H), 3.22 (q, J = 6.6 Hz, 2H), 2.88 (ddd, J = 16.8, 13.8, 5.4 Hz, 1H), 2.81 (s, 1H), 2.61–2.51 (m, 2H), 2.35–2.28 (m, 2H), 2.05–1.96 (m, 1H), 1.86 (p, J = 6.5 Hz, 2H), 0.56 (d, J = 6.9 Hz, 2H), 0.49–0.46 (m, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C50H55N8O13+, 975.3883; found, 975.3887.
N-Cyclopropyl-N-(4-((5-(3-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)pentanamido)propoxy)pyridin-2-yl)-carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (11).
Following the procedures for synthesis of 1, compound 11 was synthesized with 5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)pentanoic acid20 (18 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (30.1 mg, 70% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.06 (s, 1H), 10.88 (s, 1H), 10.64 (s, 1H), 8.13–8.07 (m, 2H), 8.04–7.98 (m, 2H), 7.92 (t, J = 5.7 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1h), 7.48 (dd, J = 9.1, 3.1 Hz, 1H), 7.42 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 8.0 Hz, 1H), 7.18–7.15 (m, 1H), 7.17–7.14 (m, 1H), 6.97–6.91 (m, 2H), 6.84 (ddd, J = 7.8, 5.6, 2.1 Hz, 1H), 5.03 (dd, J = 12.8, 5.4 Hz, 1h), 4.71 (s, 2H), 4.62 (s, 2H), 4.06 (t, J = 6.3 Hz, 2H), 3.22 (q, J = 6.5 Hz, 2h), 3.16 (t, J = 6.8 Hz, 2H), 2.87 (ddd, J = 16.8, 13.9, 5.4 Hz, 1H), 2.81 (s, 1H), 2.63–2.53 (m, 2H), 2.13 (t, J = 7.2 Hz, 2H), 2.03–1.95 (m, 1h), 1. 90–1.80 (m, 2h), 1.65–1.51 (m, 4H), 0.55 (d, J = 6.8 Hz, 2h), 0.48 (s, 2H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C46H47N8O10+, 871.3410; found, 871.3439.
N-(4-((5-(3-Aminopropoxy)pyridin-2-yl)carbamoyl)benzyl)-N-cyclopropyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxa-mide (13).
A mixture of 4-((N-cyclopropyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamido)methyl)benzoic acid (16)15 (366 mg, 1 mmol, 1 equiv) in SOCl2 (5 mL) was stirred at 40 °C for 1 h. Upon completion, SOCl2 was removed under reduced pressure to give the intermediate, which was used without further purification.
A mixture of the above intermediate, DMAP (12.2 mg, 0.1 mmol, 0.1 equiv), and compound 15 (267 mg, 1 mmol, 1 equiv) in pyridine (10 mL) was stirred at room temperature overnight. Upon completion, the solvent was removed under reduced pressure. The mixture was purified by reverse phase chromatography (10–100% MeCN/0.1% TFA in H2O) to give the desired intermediate (449 mg, 73% yield). 1H NMR (600 MHz, methanol-d4) δ 8.07 (s, 1H), 8.04 (d, J = 7.9 Hz, 2H), 7.93 (d, J = 9.2 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.57–7.53 (m, 2H), 7.25–7.18 (m, 2H), 6.98 (d, J = 8.1 Hz, 1H), 4.86 (s, 2H), 4.64 (s, 2H), 4.16 (t, J = 6.1 Hz, 2H), 3.27 (t, J = 6.7 Hz, 2H), 2.90–2.85 (m, 1H), 2.07–1.97 (m, 2H), 1.45 (s, 9H), 0.68–0.65 (m, 2H), 0.59–0.56 (m, 2H). UPLC > 95%, tR = 1.70 min. MS (ESI) m/z: [M + H]+ calcd for C33H38N5O7+, 616.3; found, 616.4.
The above intermediate (400 mg, 0.65 mmol) was dissolved in DCM (6 mL) and TFA (3 mL). The resulting mixture was stirred at room temperature for 2 h and purified by reverse phase chromatography (10–100% MeCN/0.1% TFA in H2O) to give compound 13 (330 mg, 98% yield). 1H NMR (600 MHz, methanol-d4) δ 8.11 (s, 1H), 8.05–7.99 (m, 3H), 7.78 (d, J = 9.1 Hz, 1H), 7.55–7.52 (m, 2H), 7.22 (d, J = 8.1 Hz, 1H), 7.19 (s, 1H), 6.98 (d, J = 8.1 Hz, 1H), 4.85 (s, 2H), 4.63 (s, 2H), 4.25 (t, J = 5.8 Hz, 2H), 3.20 (t, J = 7.2 Hz, 2H), 2.89–2.84 (m, 1H), 2.25–2.18 (m, 2H), 0.72–0.63 (m, 2H), 0.61–0.51 (m, 2H). UPLC > 95%, tR = 1.17 min. MS (ESI) [M + H]+ calcd for C28H30N5O5+, 516.2; found, 516.3.
tert-Butyl (3-((6-Aminopyridin-3-yl)oxy)propyl)carbamate (15).
A mixture of 6-nitropyridin-3-ol (280 mg, 2.0 mmol, 1 equiv), Cs2CO3 (980 mg, 3.0 mmol, 1.5 equiv), and tert-butyl (3-bromoethyl)carbamate (570 mg, 2.4 mmol, 1.2 equiv) in DMF (10 mL) was stirred at 80 °C overnight. The reaction was monitored by UPLC. Upon completion, the mixture was diluted with H2O and extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give the desired intermediate, which was used without further purification. To a solution of above intermediate in MeOH (20 mL) was added Pd/C (100 mg). The resulting mixture was stirred under H2 atmosphere at room temperature for 3 h. The mixture was filtered to remove the Pd/C and purified by reverse phase chromatography (10–100% MeCN/0.1% TFA in H2O) to give compound 15 (380 mg, 71% yield). 1H NMR (600 MHz, DMSO-d6) δ 7.64–7.60 (m, 1H), 7.20–7.15 (m, 1H), 6.87 (s, 1H), 6.50–6.45 (m, 1H), 5.86 (s, 2H), 3.90–3.84 (m, 2H), 3.09–3.02 (m, 2H), 1.80–1.73 (m, 2H), 1.37 (s, 9H). UPLC > 95%, tR = 0.94 min. MS (ESI) [M + H]+ calcd for C13H22N3O3+, 268.2; found, 268.2.
N-Cydopropyl-N-(4-((5-(3-(2-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetamido)propoxy)pyridin-2-yl)carbamoyl)benzyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (17).
Following the procedures for synthesis of 1, compound 17 was synthesized with compound 22 (17 mg, 0.05 mmol) and compound 13 (26 mg, 0.05 mmol) as the substrates (17.3 mg, 41% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.88 (s, 1H), 10.65 (s, 1H), 8.22 (t, J = 5.7 Hz, 1H), 8.09 (d, J = 8.8 Hz, 2H), 8.02 (d, J = 8.0 Hz, 2H), 7.59 (dd, J = 8.5, 7.1 Hz, 1H), 7.47 (dd, J = 9.1, 3.0 Hz, 1H), 7.43 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 8.1 Hz, 1H), 7.16 (s, 1H), 7.13–7.05 (m, 1H), 6.97 (s, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 5.15 (dd, J = 13.0, 5.4 Hz, 1H), 4.71 (s, 2H), 4.62 (s, 2H), 4.06 (t, J = 6.3 Hz, 2H), 3.95 (s, 2H), 3.29 (q, J = 6.5 Hz, 2H), 3.03 (s, 3H), 2.96 (ddd, J = 17.2, 13.9, 5.4 Hz, 1H), 2.81 (s, 1H), 2.76 (ddd, J = 17.2, 4.5, 2.7 Hz, 1H), 2.60–2.51 (m, 1H), 2.08–2.1 (m, 1H), 1.90 (p, J = 6.6 Hz, 2H), 0.56 (d, J = 6.8 Hz, 2H), 0.48 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 172.3, 170.3, 169.2, 169.2 167.8, 165.7, 165.3, 152.2, 146.3, 145.9, 143.0, 142.8, 136.7, 134.9, 133.3, 132.5, 132.3, 128.9, 128.7, 127.6, 124.6, 122.4, 117.9, 116.2 115.9, 115.6, 111.5, 110.3, 67.2, 66.5, 49.6, 45.7, 36.0, 31.6, 29.2 27.1, 21.9, 10.0. HPLC > 95%, tR = 5.35 min. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C44H43N8O10+, 843.3097; found, 843.3122.
N-(4-((5-(3-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetamido)propoxy)pyridin-2-yl)carbamoyl)benzyl)-N-isopropyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (18).
Following the procedures for synthesis of 1, compound 18 was synthesized with (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindo-lin-4-yl)glycine (21)21 (17 mg, 0.05 mmol) and compound 24 (26 mg, 0.05 mmol) as the substrates (19.2 mg, 46% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.10 (s, 1H), 10.86 (s, 1H), 10.61 (s, 1H), 8.21 (t, J = 5.7 Hz, 1H), 8.11–8.06 (m, 2H), 7.98 (d, J = 8.0 Hz, 2H), 7.61–7.56 (m, 1H), 7.46 (dd, J = 9.1, 3.1 Hz, 1H), 7.43 (s, 2H), 7.07 (d, J = 7.0 Hz, 1H), 7.07–7.02 (m, 2H), 6.97 (s, 2H), 6.88 (d, J = 8.5 Hz, 1H), 5.08 (dd, J = 12.8, 5.4 Hz, 1H), 4.64–4.61 (m, 4H), 4.06 (t, J = 6.3 Hz, 2H), 3.95 (s, 2H), 3.29 (q, J = 6.4 Hz, 2H), 2.90 (ddd, J = 16.7, 13.7, 5.4 Hz, 1H), 2.60–2.57 (m, 1H), 2.63–2.52 (m, 2H), 2.07–2.00 (m, 1H), 1.89 (p, J = 6.4 Hz, 2H), 1.16–1.08 (m, 6H). 13C NMR (151 MHz, DMSO-d6) δ 173.3, 170.7, 170.5, 169.2, 169.0, 167.8, 165.7, 165.2, 152.2, 146.3, 146.0, 136.7, 135.1, 132.5, 128.4, 127.0, 124.3, 121.0, 117.9, 116.2, 114.7, 111.4, 110.4, 67.2, 66.5, 49.0, 45.7, 36.0, 31.5, 31.2, 29.2, 22.6, 21.2. HPLC > 95%, tR = 4.13 min. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C43H43N8O10+, 831.3097; found, 831.3115.
(2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-glycine (22).
A mixture of 4-fluoro-2-(1-methyl-2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (20)20 (58 mg, 0.2 mmol), DIPEA (50 mg, 0.4 mmol), and tert-butyl glycinate (32 mg, 0.24 mmol) in NMP (1.5 mL) was stirred in a microwave instrument at 100 °C. Upon completion, the mixture was purified by reverse phase chromatography (10–100% MeCN/0.1% TFA in H2O) to give the corresponding intermediate. The intermediate was dissolved in DCM/TFA (2 mL/1 mL). The resulting mixture was stirred at room temperature for 2 h and then purified by reverse phase chromatography (10–100% MeCN/0.1% TFA in H2O) to afford compound 22 (27.9 mg, 41% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.95–12.91 (m, 1H), 7.64–7.57 (m, 1H), 7.10 (d, J = 13.9 Hz, 1H), 7.04–6.98 (m, 1H), 6.89–6.86 (m, 1H), 5.14 (dd, J = 13.5, 5.8 Hz, 1H), 4.11 (d, J = 5.7 Hz, 2H), 3.03 (s, 3H), 3.00–2.88 (m, 1H), 2.79 (s, 1H), 2.09–2.06 (m, 2H). UPLC > 95%, tR = 1.03 min. MS (ESI) [M + H]+ calcd for C16H16N3O6+, 346.1; found, 346.1.
N-(4-((5-(3-Aminopropoxy)pyridin-2-yl)carbamoyl)benzyl)-N-isopropyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazine-7-carboxamide (24).
Compound 24 was synthesized according to the procedures for preparing compound 13 with compound 2315 as the starting material. 1H NMR (600 MHz, methanol-d4) δ 8.11 (s, 1H), 8.05–7.97 (m, 3H), 7.74 (d, J = 9.2 Hz, 1H), 7.55 (s, 2H), 7.11 (s, 2H), 7.03 (s, 1H), 4.77 (s, 2H), 4.66 (s, 2H), 4.25 (t, J = 5.7 Hz, 2H), 3.20 (t, J = 7.3 Hz, 2H), 2.25–2.18 (m, 2H), 1.21 (s, 6H). UPLC > 95%, tR = 1.35 min. MS (ESI) [M + H]+ calcd for C28H32N5O5+, 518.2; found, 518.6.
Cell Culture.
Human MM lines used in the study are NCI-H929 (American Tissue Culture Collection [ATCC], CRL-9068) and KMS11 (a gift of K. C. Anderson). These lines were cultured in the RPMI 1640 base medium supplemented with 10% of FBS and 1% of penicillin plus streptomycin. 293FT cells (Thermo Fisher Scientific, R70007) were cultured in DMEM base medium supplemented with 10% of FBS and 1% of penicillin plus streptomycin. A 293FT-derivative line with CRBN KO (CRBN−/−) was provided by J. Brander and W. Kaelin (Dana Farber Cancer Institute). Authentication of cell line identities was ensured by the Tissue Culture Facility (TCF) of UNC Lineberger Comprehensive Cancer Center, with the genetic signature profiling and fingerprinting analysis. Every month, a routine examination for potential mycoplasma contamination was carried out by using the commercially available detection kits from Lonza.
Chemicals and Antibodies.
Pomalidomide (catalogue no. 36471) was purchased from AstaTech. MLN9708 (catalogue no. HY-10452) was purchased from MedChemExpress. MG132 (catalogue no. S2619) and MLN4924 (catalogue no. S7109) were purchased from Selleck Chemical. Antibodies used for Western blot included mouse anti-NSD2 (Abcam, ab75359), rabbit anti-GAPDH (Cell Signaling Technology, no. 5174), rabbit anti-IKZF1 (Cell Signaling Technology, no. 14859), rabbit anti-IKZF3 (Cell Signaling Technology, no. 15103), rabbit anti-GSTP1 (Thermo Fisher Scientific, no. PA5-29558), and rabbit anti-CRBN (Cell Signaling Technology, no. 71810). The HRP-linked secondary antimouse IgG antibody (no. 7076) and antirabbit IgG antibody (no. 7074) were purchased from Cell Signaling Technology.
Western Blots.
The cultured cells were collected, rinsed in cold phosphate-buffered saline (PBS), and suspended in buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1% NP-40. After brief sonication and centrifugation, the soluble fractions of cell lysates were mixed with 2× SDS-PAGE loading buffer and boiled for 5 min, followed by loading onto a SDS-PAGE gel for immunoblotting analysis.
RNA Interference-Mediated Gene Knockdown (KD).
Lentiviral pLKO.1-based shRNA vectors for KD of human NSD2 (TRCN0000274182 [sh#1] and TRCN0000019818 [sh#2]) were purchased from Sigma. Viral particles were produced with pLKO-puro-shNSD2 and the packaging plasmids, psPAX2 (Addgene no. 12260) and pMD2.G (Addgene no. 12259) in 293FT cells. After the virus infection was performed in target cells, the infected cells were selected with 1 μ/mL of puromycin.
Cell Growth Inhibition Assay.
Cell growth inhibition assay was performed as described previously.34 In brief, 0.5 million cells per well were seeded in triplicate into 24-well plates, subjected to treatment with various final concentrations of compound. Fresh medium containing compound was changed every 2 days. All flow-growing cells were periodically diluted to keep the cell density less than 1 × 106/mL. Cell numbers were counted by an automated TC10 cell counter (BioRad) every 2 days.
Protein Expression and Purification.
Construct of the NSD2-PWWP1 domain (residues 208–369) was cloned in pDEST15 vector from GenScript. The plasmid containing the NSD2-PWWP1 domain with N-terminal GST tag and a TEV protease cleavage site was transformed and expressed in Escherichia coli BL21 (DE3) cells. The transformed cells were grown in TB medium at 37 °C until OD600 reached 3.0. After cooling down to 15 °C, 0.4 mM of IPTG was added and incubated for overnight. The harvested cells were resuspended in lysis buffer (50 mM phosphate buffer pH 7.5, 1 M NaCl, 5% glycerol) with 0.2 mM ABSEF and 1 mM TCEP and sonicated to lyse cells. The lysate was clarified by centrifugation and loaded onto a 5 mL GST HiTrap (GE healthcare) column using a buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM TCEP). The bound GST-tagged NSD2-PWWP1 protein was eluted with a buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM TCEP, 10 mM GSH), and then the eluent was dialyzed in a TEV protease reaction buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5% glycerol, 1 mM TCEP) to cleave the GST tag. After TEV cleavage in room temperature for an hour, the protein was further purified by size-exclusion HiLoad 26/600 Superdex column (GE healthcare) with a buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 1 mM TCEP, 5% glycerol) and all of fractions were monitored by SDS-PAGE. Then, pure fractions were collected and concentrated to 10 mg/mL for further use.
Isothermal Titration Calorimetry (ITC).
ITC experiments were performed with compounds (UNC6934, 9, 17, and 18) and purified NSD2-PWWP1 using a MicroCal iTC200 (Malvern) in 50 mM Tris-HCl pH 6.8, 150 mM NaCl, and 1% DMSO at 25 °C. After an initial 0.4 μL injection, 12 injections from the syringe solution (250 μM of NSD2-PWWP1) were titrated into 300 μL of the cell solution (25 μM of compounds) with stirring at 750 rpm. The data were fitted by single binding site model using Microcal Origin 7.0 (Malvern). The reported values represent the mean ± SD from two independent measurements.
Selectivity Assay.
Selectivity assays against 20 human protein methyltransferases were performed by Reaction Biology Corp using miniaturized radioisotope-based filter binding assay (HotSpot). This biochemical assay monitors the transfer of a 3H-labeled methyl group from the cofactor SAM to the substrate. Twenty methyltransferases (NSD1, NSD2(full length), NSD3, G9a, GLP, MLL1 complex, ASH1L, DOT1L, EZH2 complex, SETD7/9, SETD8, SETD2-GST, SETDB1, SMYD2, SMYD3, SUV39H1, PRMT1, PRMT4, PRMT5/MEP50 complex, and PRMT6) were used as the enzymes. The concentration of the substrate core histone proteins was 5 μM, or 0.05 mg/mL of nucleosomes from HeLa/chicken or core histones from chicken. The concentration of the cofactor SAM was 1 μM. Compound 9 was tested at 10 μM in duplicate.
Mouse PK Study.
A standard in vivo PK study was conducted for compound 9 using three male Swiss Albino mice. The mice were administered intraperitoneally with solution formulation of compound 9 at a 50 mg/kg dose. The formulation vehicle was 5% v/v NMP, 45% v/v propylene glycol, and 50% v/v PEG-400. Then 60 μL of blood samples were collected from each mouse at 0.5, 2, and 8 h. Plasma was harvested by centrifugation of blood and stored at −70 ± 10 °C until analysis. Plasma samples were quantified by fit-for-purpose LC–MS/MS method (LLOQ 5.14 ng/mL for plasma).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the U.S. National Institutes of Health grants R01GM122749 (to J.J.), P30CA196521 (to J.J.), R01CA211336 (to G.G.W.), R01CA215284 (to G.G.W), an endowed professorship from the Icahn School of Medicine at Mount Sinai (to J.J.), and grants/awards from Gabrielle’s Angel Foundation for Cancer Research (to G.G.W.), When Everyone Survives (WES) Leukemia Research Foundation (to G.G.W.), and UNC Lineberger Cancer Center UCRF Stimulus Initiative Grants (to G.G.W. and L.C.). G.G.W. is an American Cancer Society (ACS) Research Scholar, a Leukemia and Lymphoma Society (LLS) Scholar, and an American Society of Hematology (ASH) Scholar in Basic Science. We thank Drs. Minkui Luo, Deyao Li, and Ke Wang for their generous help with obtaining HRMS results at the MSKCC Analytical Core Facility. This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504.
ABBREVIATIONS USED
- ALL
acute lymphoblastic leukemia
- CRBN
cereblon
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMSO
dimethyl sulfoxide
- EDCI
N-(3-(dimethylamino)propyl)-N’-ethylcarbodiimide hydrochloride
- H3K36
histone H3 lysine 36
- HMG-box
high mobility group box
- HOAT
1-hydroxy-7-azabenzotriazole
- MeOH
methanol
- MM
multiple myeloma
- NMM
N-methylmorpholine
- NMP
N-methyl pyrrolidone
- NSD2
nuclear receptor binding SET domain protein 2
- PEG
poly(ethylene glycol)
- PHD
plant homeodomain domain
- PK
pharmacokinetic
- PKMT
protein lysine methyltransferase
- POI
protein of interest
- PPI
protein–protein interaction
- PROTAC
proteolysis targeting chimera
- PWWP
proline–tryptophan--tryptophan–proline
- rt
room temperature
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
- TPD
targeted protein degradation
- VHL
von Hippel–Lindau
- UPS
ubiquitin–proteasome system
- WHSC1
Wolf–Hirschhorn syndrome candidate 1
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00807.
Western blotting analysis of compound 1 in 293FT, KMS11, and H929 cells, immunoblots for NSD2 and tubulin after shRNA-mediated KD of NSD2, and 1H NMR, 13C NMR, and HPLC spectra of compounds 9, 17, and 18 (PDF)
Molecular formula strings and biological data (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c00807
The authors declare the following competing financial interest(s): The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc. J.J. is a cofounder and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc.
Contributor Information
Fanye Meng, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Chenxi Xu, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Kwang-Su Park, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
H. Ümit Kaniskan, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Gang Greg Wang, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Department of Biochemistry and Biophysics and Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Kuo AJ; Cheung P; Chen K; Zee BM; Kioi M; Lauring J; Xi Y; Park BH; Shi X; Garcia BA; Li W; Gozani O NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44 (4), 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Li J; Ahn JH; Wang GG Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell. Mol. Life Sci 2019, 76 (15), 2899–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bhat KP; Kaniskan HÜ; Jin J; Gozani O Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discovery 2021, 20, 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kaniskan HÜ; Martini ML; Jin J Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev 2018, 118 (3), 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vougiouklakis T; Hamamoto R; Nakamura Y; Saloura V The NSD family of protein methyltransferases in human cancer. Epigenomics 2015, 7 (5), 863–74. [DOI] [PubMed] [Google Scholar]
- (6).Husmann D; Gozani O Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol 2019, 26 (10), 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Keats JJ; Reiman T; Maxwell CA; Taylor BJ; Larratt LM; Mant MJ; Belch AR; Pilarski LM In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101 (4), 1520–1529. [DOI] [PubMed] [Google Scholar]
- (8).Jaffe JD; Wang Y; Chan HM; Zhang J; Huether R; Kryukov GV; Bhang HE; Taylor JE; Hu M; Englund NP; Yan F; Wang Z; Robert McDonald E 3rd; Wei L; Ma J; Easton J; Yu Z; deBeaumount R; Gibaja V; Venkatesan K; Schlegel R; Sellers WR; Keen N; Liu J; Caponigro G; Barretina J; Cooke VG; Mullighan C; Carr SA; Downing JR; Garraway LA; Stegmeier F Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet 2013, 45 (11), 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Morishita M; Mevius DEHF; Shen Y; Zhao S; di Luccio E BIX-01294 inhibits oncoproteins NSD1, NSD2 and NSD3. Med. Chem. Res 2017, 26 (9), 2038–2047. [Google Scholar]
- (10).Coussens NP; Kales SC; Henderson MJ; Lee OW; Horiuchi KY; Wang Y; Chen Q; Kuznetsova E; Wu J; Chakka S; Cheff DM; Cheng KC; Shinn P; Brimacombe KR; Shen M; Simeonov A; Lal-Nag M; Ma H; Jadhav A; Hall MD High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J. Biol. Chem 2018, 293 (35), 13750–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Huang H; Howard CA; Zari S; Cho HJ; Shukla S; Li H; Ndoj J; Gonzalez-Alonso P; Nikolaidis C; Abbott J; Rogawski DS; Potopnyk MA; Kempinska K; Miao H; Purohit T; Henderson A; Mapp A; Sulis ML; Ferrando A; Grembecka J; Cierpicki T Covalent inhibition of NSD1 histone methyltransferase. Nat. Chem. Biol 2020, 16 (12), 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bennett RL; Swaroop A; Troche C; Licht JD The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harbor Perspect. Med 2017, 7 (6), a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bottcher J; Dilworth D; Reiser U; Neumuller RA; Schleicher M; Petronczki M; Zeeb M; Mischerikow N; Allali-Hassani A; Szewczyk MM; Li F; Kennedy S; Vedadi M; Barsyte-Lovejoy D; Brown PJ; Huber KVM; Rogers CM; Wells CI; Fedorov O; Rumpel K; Zoephel A; Mayer M; Wunberg T; Bose D; Zahn S; Arnhof H; Berger H; Reiser C; Hormann A; Krammer T; Corcokovic M; Sharps B; Winkler S; Haring D; Cockcroft XL; Fuchs JE; Mullauer B; Weiss-Puxbaum A; Gerstberger T; Boehmelt G; Vakoc CR; Arrowsmith CH; Pearson M; McConnell DB Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol 2019, 15 (8), 822–829. [DOI] [PubMed] [Google Scholar]
- (14).Ferreira de Freitas R; Liu Y; Szewczyk MM; Mehta N; Li F; McLeod D; Zepeda-Velazquez C; Dilworth D; Hanley RP; Gibson E; Brown PJ; Al-Awar R; James LI; Arrowsmith CH; Barsyte-Lovejoy D; Min J; Vedadi M; Schapira M; Allali-Hassani A Discovery of small-molecule antagonists of the PWWP domain of NSD2. J. Med. Chem 2021, 64 (3), 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dilworth D; Hanley RP; Ferreira de Freitas R; Allali-Hassani A; Zhou M; Mehta N; Marunde MR; Ackloo S; Carvalho Machado RA; Khalili Yazdi A; Owens DDG; Vu V; Nie DY; Alqazzaz M; Marcon E; Li F; Chau I; Bolotokova A; Qin S; Lei M; Liu Y; Szewczyk MM; Dong A; Kazemzadeh S; Abramyan T; Popova IK; Hall NW; Meiners MJ; Cheek MA; Gibson E; Kireev D; Greenblatt JF; Keogh M-C; Min J; Brown PJ; Vedadi M; Arrowsmith CH; Barsyte-Lovejoy D; James LI; Schapira M Pharmacological targeting of a PWWP domain demonstrates cooperative control of NSD2 localization. Nat. Chem. Biol 2022, 18 (1), 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xu C; Meng F; Park KS; Storey AJ; Gong W; Tsai YH; Gibson E; Byrum SD; Li D; Edmondson RD; Mackintosh SG; Vedadi M; Cai L; Tackett AJ; Kaniskan HÜ; Jin J; Wang GG A NSD3-targeted PROTAC suppresses NSD3 and cMyc oncogenic nodes in cancer cells. Cell Chem. Biol 2022, 29, 386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nalawansha DA; Crews CM PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol 2020, 27 (8), 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dale B; Cheng M; Park KS; Kaniskan HÜ; Xiong Y; Jin J Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21 , 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Han XR; Chen L; Wei Y; Yu W; Chen Y; Zhang C; Jiao B; Shi T; Sun L; Zhang C; Xu Y; Lee MR; Luo Y; Plewe MB; Wang J Discovery of selective small molecule degraders of BRAF-V600E. J. Med. Chem 2020, 63 (8), 4069–4080. [DOI] [PubMed] [Google Scholar]
- (21).Qiu X; Sun N; Kong Y; Li Y; Yang X; Jiang B Chemoselective synthesis of lenalidomide-based PROTAC library using alkylation reaction. Org. Lett 2019, 21 (10), 3838–3841. [DOI] [PubMed] [Google Scholar]
- (22).Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 2015, 11 (8), 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Galdeano C; Gadd MS; Soares P; Scaffidi S; Van Molle I; Birced I; Hewitt S; Dias DM; Ciulli A Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fischer ES; Bohm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith RE; Harper JW; Jenkins JL; Thoma NH Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512 (7512), 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ishoey M; Chorn S; Singh N; Jaeger MG; Brand M; Paulk J; Bauer S; Erb MA; Parapatics K; Muller AC; Bennett KL; Ecker GF; Bradner JE; Winter GE Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol 2018, 13 (3), 553–560. [DOI] [PubMed] [Google Scholar]
- (27).Jiang B; Wang ES; Donovan KA; Liang Y; Fischer ES; Zhang T; Gray NS Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem., Int. Ed. Engl 2019, 58 (19), 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hu J; Wei J; Yim H; Wang L; Xie L; Jin MS; Kabir M; Qin L; Chen X; Liu J; Jin J Potent and selective mitogen-activated protein kinase kinase 1/2 (MEK1/2) heterobifunctional small-molecule degraders. J. Med. Chem 2020, 63 (24), 15883–15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wei J; Hu J; Wang L; Xie L; Jin MS; Chen X; Liu J; Jin J Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J. Med. Chem 2019, 62 (23), 10897–10911. [DOI] [PubMed] [Google Scholar]
- (30).Cheng M; Yu X; Lu K; Xie L; Wang L; Meng F; Han X; Chen X; Liu J; Xiong Y; Jin J Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem 2020, 63 (3), 1216–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sun N; Ren C; Kong Y; Zhong H; Chen J; Li Y; Zhang J; Zhou Y; Qiu X; Lin H; Song X; Yang X; Jiang B Development of a Brigatinib degrader (SIAIS117) as a potential treatment for ALK positive cancer resistance. Eur. J. Med. Chem 2020, 193, 112190. [DOI] [PubMed] [Google Scholar]
- (32).Zhang X; Thummuri D; Liu X; Hu W; Zhang P; Khan S; Yuan Y; Zhou D; Zheng G Discovery of PROTAC BCL-XL degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem 2020, 192, 112186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Qi J; Armstrong S; Wu L Compounds, compositions, and methods for protein degradation. World WO2020264172, 2020. [Google Scholar]
- (34).Xu BW; On DM; Ma AQ; Parton T; Konze KD; Pattenden SG; Allison DF; Cai L; Rockowitz S; Liu SC; Liu Y; Li FL; Vedadi M; Frye SV; Garcia BA; Zheng DY; Jin J; Wang GG Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015, 125 (2), 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.