

Abstract
As levels of acetylcholinesterase (AChE) decrease while levels of butyrylcholinesterase (BChE) increase in later stages of Alzheimer's disease (AD), BChE stands out as a promising target for treatment of AD. Therefore, several benzimidazole-carbamates were designed based on docking studies to inhibit BChE selectively over AChE, while retaining a reasonable solubility. Synthesized molecules exhibit IC50 values from 2.4 μM down to 3.7 nM with an overall highly hBChE-selective profile of the designed compound class. After evaluation of potential neurotoxicity, the most promising compound was further investigated in vivo. Compound 11d attenuates Aβ25–35-induced learning impairments in both spontaneous alternation and passive avoidance responses at a very low dosage of 0.03 mg kg−1, proving selective BChE inhibition to lead to effective neuroprotectivity in AD.
Benzimidazole-based inhibitors of butyrylcholinesterase were designed and tested for their activity and selectivity in vitro, leading to compound (11d) that attenuated Aβ25-35-induced learning impairments in an Alzheimer's disease mouse model.
1. Introduction
Due to an aging population, the number of patients diagnosed with Alzheimer's disease (AD) is expected to grow rapidly. While 5.8 million people in the US are estimated to live with AD in 2020, this number is expected to double by 2050, putting an immense pressure on the health care system.1–3 This is further enhanced by the length of treatment, as people age 65 or older have an average life expectancy of four to eight years after diagnosis, while some survive up to 20 years after diagnosis.4,5 Therefore, total payments for treatment of patients suffering from AD and dementia are estimated at $290 billion, whereby only 67% are paid by Medicare or Medicaid while up to 22% are out of pocket payments, pointing towards an extreme burden for patients and relatives, not only financially but also psychologically.6 As AD patients need long and intensive caregiving, ranging from medication to feeding and dressing etc., AD marks the 6th largest cause of disability adjusted life years (DALYs), a measurement of the burden caused by disease.7
Several drugs have been approved by the U.S. Food and Drug Administration (FDA) to improve the life of AD patients. Unfortunately, none of these drugs can slow down degradation of neurons, leading to mediocre results for long term treatment of patients suffering from AD, since these drugs only enable symptomatic treatment. In disease progression, extracellular deposits of amyloid beta (Aβ) in senile plaques, hyperphosphorylation of tau proteins and oxidative stress have been identified.8–12 But, while the mentioned factors contribute to neurodegeneration, substantial deficits of the neurotransmitter acetylcholine (ACh) due to a loss of cholinergic neurons and cholinergic function, summarized in the “cholinergic hypothesis”, lead to an overall cognitive decline, resulting in memory loss and a significant interference with the ability of the patient to perform everyday activities.9,13,14 Therefore, inhibition of acetylcholinesterase (AChE), responsible for ACh degradation, was focused on for a symptomatic treatment of AD. Unfortunately, AChE inhibitors (donepezil, rivastigmine and galantamine) lead to undesired side effects, ranging from bradycardia to vomiting and nausea.15 Also, with further disease progression, levels of AChE seem to decrease as much as 90%.16,17
In contrast to AChE, levels of butyrylcholinesterase (BChE) increase with disease progression, making it an interesting target for mid- and late-stage medication.22 Several investigations by us could already prove positive effects of BChE inhibition in vivo, since chronic administration leads to pronounced neuroprotectivity with improved parameters in both short- and long-term memory.19,20,23–25
We developed a selective hBChE inhibitor (2), based on a tetracyclic carrier unit combined with a heptyl carbamate, acting as pseudo-irreversible inhibitor of hBChE.26In vivo testing revealed a beneficial, but moderate effect on spontaneous alternation and passive avoidance impairments.23 We also identified BChE/CB2R hybrid compounds based on benzimidazoles acting as reversible inhibitors of BChE (chart 1). Benzimidazole 3, representing the most potent inhibitor of this set, showed an IC50 (hBChE) value of 1.2 μM (chart 1). Additional computational docking and in vivo experiments confirmed the benzimidazole template as suitable lead structure for further development.19,27 Consequently, combination of the benzimidazole scaffold with the pseudoirreversible mode of action of compound 2 was expected to deliver improved results on hBChE inhibition and in vivo behaviour. We therefore designed benzimidazole-carbamates based on our previous SAR studies for reversible inhibitors, while also investigating novel carbamate templates for BChE inhibition.
Chart 1. a) Pseudo-irreversible inhibitors 1–2 of BChE; b) reversible inhibitor 3 of BChE; c) selective CB2R agonist 4.18–21.

2. Results and discussion
2.1. Molecular docking studies
As the orientation of the carrier scaffold is essential to enable the interaction of the nucleophilic Ser198 of hBChE with the carbamate unit, molecular docking studies are necessary to verify the binding mode of prototype compounds.25
As benzimidazole-based inhibitors of BChE were initially designed by Dolles et al. to interact non-covalently with hBChE, we investigated structural modifications on compounds 3 and 4 by a non-covalent docking approach using the docking program GOLD (v2021.1.0).27,28 To confirm the suitability of the 2-benzylbenzo[d]imidazole recognition unit and to analyse the introduction of a novel carbamate structure for hBChE inhibition, we selected compound 11d as an initial example for this series of inhibitors. This structure differs from compound 4 by exchange of the ethoxy group with a small azetidine-carbamate to test suitability of our structural approach with a sterically undemanding motif. The studies were performed with the PDB structure 4BDS (resolution 2.10 Å),29 that corresponds to the hBChE in complex with the non-covalent inhibitor tacrine, which suits this study surpassingly, as we wanted to explore the non-covalent binding mode of this inhibitor series. The γ-oxygen atom of Ser198 was clarified as the center of search space. These docking parameters resulted in highly convergent results and were therefore applied, generating the displayed poses (Chart 2). The carbamate carbonyl of the top-ranked pose of compound 11d is facing the oxyanion hole and well-coordinated by Gly116 (Chart 2C), bringing the carbamate function in the right direction for a nucleophilic attack by the γ-oxygen of Ser198. Compound 11d is oriented towards Asp70 and Tyr332, that form a hydrogen bond and are located right at the opening of the active site notch (Chart 2B). Here, the benzimidazole moiety is placed parallel to the aromatic ring of Tyr332, forming π–π interactions (Chart 2A). The residues Asp70 and Tyr332 are reported to play a key role in the initial recognition of ligands and in substrate trafficking because of their strategically important position at the rim of the pocket.30,31 The benzene ring of compound 11d is well enveloped by a system of aromatic side chains forming a rather lipophilic area consisting of Phe329, Trp82 and His438 (Chart 2A).
Chart 2. Binding position of the best consensus rank of compound 11d. The scaffold of compound 11d is visualized in green, the catalytic serine (S198) is highlighted in pink. Key interaction distances are shown in Å. A) Visualization of interactions of compound 11d with the active site of the hBChE. B) View from the outside of the active site shows the orientation of compound 11d towards the surface and the placement of the carbamate function near the catalytic triade (blue surface). C) Illustration of the carbamate function directing towards the oxyanion hole.

These results suggest a pseudoirreversible binding mode of the compounds, as the carbamate moiety is in a suitable position for the transfer of the carbamate function, supporting the design of novel benzimidazole-based carbamates as inhibitors of hBChE.
Due the nature of interaction between the compound 11d and BChE, this finding could provide a molecular picture of enzyme selectivity over AChE. Therefore, we conducted docking studies of 11d at AChE (Fig. S2†). The active sites of hBChE and hAChE do not only differ in the space available around their respective catalytic triades, but also in the amino acids framing the latter, despite the high homology of both active sites.32,33 These differences in the micro-environment of the gorges lead to repulsive effects in the binding of aromatic compounds.34 Bosak et al. have observed poor inhibition rates for an epinephrine-derivative at hAChE in comparison with the hBChE and correlated these findings with a mismatched π–π-interaction of this compound at hAChE.35 For hBChE, we described a π–π-interaction of benzimidazole with Tyr332 and a benzene function well surrounded by aromatic amino acids Phe329 and Trp82 (Chart 2). Substitution of Gln119 in hBChE for Tyr124 in hAChE may lead to the observed repulsive effects at this binding position already described by Saxena et al. (Fig. S2†).33,34 Furthermore, studies with alkaloid scaffolds have shown that π–π-interactions are crucial for the stabilisation of compound:BChE complexes.32 Our data substantiates the findings in this study, leading to the observed selectivity of our inhibitors. The differences in space and orientation of the active sites are more thoroughly discussed in the ESI† (Fig. S2).
2.2. Design and chemistry
With the suitability of a carbamate introduction at the phenolic position hinted by molecular docking studies, two distinct positions of the parent molecule are possible to modify. Dolles et al. previously optimised hBChE inhibition of the 2-benzylbenzo[d]imidazole core structure relying on reversible inhibition, giving a first starting point for structure–activity relationship studies of the new compound class.19 Supporting these studies, an improved hBChE inhibition was achieved modifying the amide moiety at the benzimidazole. While parent molecule 4, carrying a diethylamide, was not able to inhibit the enzyme at all, distinct amide moieties enabled micromolar inhibition of the enzyme. We focused on piperidine, aniline and isopentyl amine moieties to obtain the corresponding amides and subsequently improve hBChE inhibition. Furthermore, introduction of alkylene-linked piperidines led to a comparable improvement.19 As the choice of carbamate is vital for inhibition, when transferring the concept of reversible binding to irreversible binding, several carbamates besides the docked, sterically less demanding azetidine, were introduced, the cyclic pyrrolidine carbamate and two long-chain carbamates (Chart 3).
Chart 3. Design concept based on the 2-benzyl-benzimidazole core.

Modifications of the 2-benzylbenzo[d]imidazole core structure were enabled by two different synthesis routes starting from either 4-fluoro-3-nitrobenzoic acid (Scheme 1) or ethyl 4-fluoro-3-nitrobenzoate (Scheme 2), both commercially available. To introduce a) different carbamates on the phenolic position and b) differently-linked piperidines linked to the benzimidazole core, a seven-step synthesis was utilized to obtain 6 molecules of interest (Scheme 1).
Scheme 1. Synthesis of compounds 11a–f. Reagents and conditions: (i) 1. oxalyl chloride, DMF, DCM, 0 °C to rt, 3 h; 2. diethyl amine, TEA, DCM, 0 °C to rt, 1 h; (ii) primary amine, TEA, EtOH, 55 °C, 3 h or overnight; (iii) H2, Pd/C, EtOH, rt, 3 h; (iv) 2-(4-(benzyloxy)phenyl)acetic acid, HBTU, TEA, DMF, rt, 3 h; (v) acetic acid, 135 °C, 3 h; (vi) H2, Pd/C, MeOH, rt, overnight; (vii) heptyl isocyanate, TEA, DCM, rt, overnight or pyrrolidinecarbonyl chloride, NaH, THF, rt, 12 h or 4-nitrophenyl azetidine-carboxylate, potassium tert-butoxide, 55 °C, 12 h or 1. (3,4-dihydroisoquinolin-2(1H)-yl)hexan-1-amine, 4-nitrophenylchloroformate, TEA, DCM, 4.5 h; 2. 10a, TEA, DCM, rt, overnight.

Scheme 2. Synthesis of compounds 18a–c. Reagents and conditions: (i) isopentyl amine, TEA, EtOH, 55 °C, 3 h; (ii) H2, Pd/C, EtOH, rt, 3 h; (iii) 2-(4-(benzyloxy)phenyl)acetic acid, HBTU, TEA, DMF, rt, 3 h; (iv) 1. acetic acid, 135 °C, 3 h; 2. LiOH, water, THF, reflux, overnight; (v) amine, HBTU, TEA, DMF, rt, overnight; (vi) H2, Pd/C, MeOH, rt, overnight; (vii) heptyl isocyanate, TEA, DCM, rt, overnight.

First, 4-fluoro-3-nitrobenzoic acid was converted to the corresponding acid chloride and reacted with dimethylamine, forming N,N-diethyl-4-fluoro-3-nitrobenzamide 5. Subsequent nucleophile aromatic substitution with isopentyl amine or the respective aminoalkylpiperidines yielded N,N-diethyl-4-(isoalkylamino)-3-nitrobenzamides 6a–c in high yields. Reduction of the nitro group was carried out using hydrogen and palladium on carbon as catalyst, making no further purification besides simple filtration necessary. The corresponding anilines 7a–c were then coupled with benzyl protected 4-hydroxyphenylacetic acid. In this reaction step 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and triethylamine (TEA) were used to form the active ester that was reacted with anilines 7a–c to obtain diamides 8a–c. Cyclisation to the benzimidazoles 9a–c were carried out by refluxing in acetic acid. Then, deprotection of the benzyl ether using hydrogen and palladium on carbon yielded the key intermediates 10a–c with a free phenol for subsequent functionalisation. To obtain the desired carbamates 11a–f, different procedures were applied. For aryl–OCO–NH heptyl carbamates 11a, 11e and 11f the phenols 10a–c were treated with TEA and reacted with heptyl isocyanate in yields from 70 to 86%, applying mild conditions, which are sufficient due to high reactivity of the isocyanates. Due to a lack of commercially available precursors for direct conversion, azetidine was first treated with 4-nitrophenyl chloroformate to yield the corresponding 4-nitrophenyl dialkylcarbamate, which could be isolated. Subsequent treatment with phenol 10a, using potassium tert-butoxide, yielded 11d in a yield of 76%.
A similar procedure was carried out for the synthesis of compound 11b, but both steps were carried out by a two-step, one-pot procedure due to the higher reactivity of the monoalkyl-active ester, yielding only 9% of product using TEA as base. This clearly points out the advantage of using the isolated active ester, as the in situ conversion often leads to undesired side products, that can be avoided applying a two-step procedure. Unfortunately, especially 4-nitrophenyl carbamates from primary amines are sensitive to several purification procedures, e.g. extraction and column chromatography. Less activated carbamates are therefore expected to enable higher yields. To obtain pyrrolidine derivative 11c, pyrrolidine carbonyl chloride and sodium hydride were used, yielding 77% of the desired product.
Modifications at the amide require another synthetic strategy, starting from ethyl 4-fluoro-3-nitrobenzoate, which was treated with isopentyl amine to yield aniline 12 (Scheme 2). Subsequent reduction of the nitro group using palladium on carbon gave dianiline 13 in high yields, followed by coupling of the primary amine with 2-(4-(benzyloxy)phenyl)acetic acid to obtain the corresponding amide 14. Cyclisation to yield the benzimidazole bicyclus was carried out by refluxing in acetic acid. Without further purification, the mixture was basified by LiOHaq and refluxed to cleave the ester group. The obtained carboxylic acid 15 was then coupled with either aniline, piperidine or isopentyl amine using HBTU as coupling agent, yielding the corresponding amides 16a–c. Deprotection of the phenol using palladium on carbon yielded the precursors 17a–c for subsequent carbamylation by heptyl isocyanate to obtain compounds 18a–c (Scheme 2).
2.3. Pharmacological profile
Inhibition of cholinesterases
All target compounds were tested for their ability to inhibit hBChE, as well as hAChE, to evaluate the influence of different carbamate units introduced at the phenol moiety, as well as changes at the benzimidazole core. The use of human enzyme for both cholinesterases is vital due to a different activity profile when compared to commonly applied eqBChE or eeAChE, as reported by Dolles et al.19,27
Introduction of a heptyl carbamate (11a) led to a similar inhibition of hBChE, when compared to parent molecule 2, but a loss in selectivity over hAChE. The tetrahydroisoquinoline residue of compound 11b, which was used to investigate the pseudo-irreversible kinetics of such compounds by Hoffmann et al., led to a lower inhibition, but a gain in selectivity over hAChE, when compared to its unfuctionalized heptyl derivative 11a.23 The introduction of small, cyclic carbamates based on azetidine and pyrrolidine showed a superior and selective hBChE inhibition of the sterically less demanding azetidine carbamate 11d, exhibiting an one-digit nanomolar IC50, while compound 11c inhibits hBChE by a factor of 20 lower. It should be mentioned that selectivity was regained by introduction of small, cyclic carbamate units, when compared to inhibitor 11a (Table 1).
In vitro assessment of benzimidazole-carbamates.

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | IC50 (hBChE) [nM] (pIC50 ± SEM) or inhibition @ 10 μM [%] | IC50 (hAChE) [nM] (pIC50 ± SEM) or inhibition @ 10 μM [%] | SI (IC50 (hAChE)/IC50 (hBChE)) | Solubility @ rt [μg mL−1] |
| 2 | 47.6 (7.3 ± 0.04) | 39% | >100 | <1.0 | |||
| Tacrine | 46.8 (7.3 ± 0.02) | 188 (6.7 ± 0.06) | 4.0 | n.d. | |||
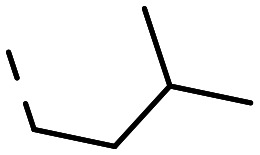
| 11a |

|
|

|
13.2 (7.9 ± 0.02) | 175 (6.8 ± 0.03) | 13 | <1.0 |
| 11b |

|

|

|
76.5 (7.1 ± 0.03) | 9522 (5.0 ± 0.06) | 105 | n.d.a |
| 11c |

|

|

|
139 (6.9 ± 0.02) | 16% | >10 | 3.9 |
| 11d |

|

|

|
6.9 (8.2 ± 0.02) | 711 (6.2 ± 0.05) | 103 | 7.2 |
| 11e |

|

|

|
91.4 (7.0 ± 0.03) | 10 120 (5.0 ± 0.05) | 111 | <1.0 |
| 11f |

|

|

|
2405 (5.6 ± 0.1) | 2148 (5.7 ± 0.03) | 0.9 | 67 |
| 18a |

|

|

|
30.6 (7.5 ± 0.03) | 58.0 (7.2 ± 0.04) | 1.9 | <1.0 |
| 18b |

|

|

|
3.7 (8.4 ± 0.04) | 55% | >500 | <1.0 |
| 18c |

|

|

|
15.5 (7.8 ± 0.02) | 49% | >500 | n.d.b |
Partial decomposition during saturation experiment was observed.
Compound was insoluble in methanol; n.d. not determined.
As the introduction of piperidine moieties for reversible, benzimidazole-based inhibitors led to improved binding to BChE, small alkyl chains connecting piperidine to the benzimidazole core were also investigated.19 Unfortunately, both compounds, 11e (ethylene) and 11f (butylene) showed decreased hBChE inhibition with compound 11f also losing selectivity over hAChE (Table 1).
Subsequently, the amide was investigated, as this position is sensitive to modifications regarding reversible inhibitors. All three amides, derived from piperidine (18a), isopentyl amine (18b) and aniline (18c) showed excellent one- to two-digit nanomolar hBChE inhibition, with 18b exhibiting an IC50 value of 3.7 nM. Selectivity was hereby only lost for piperidine derivative 18a (Table 1).
Solubility
Solubility increases with reduction of the aliphatic part of the carbamate. While heptyl carbamates 2 and 11a are soluble to an amount of less than 1 μg mL−1 in phosphate-buffered saline (PBS), pyrrolidine-carbamate 11c is soluble at 3.9 μg mL−1, which was determined by UV quantification comparing the saturated solution with a calibration curve, giving a calibration equation for each compound. This is then further improved by azetidine-carbamate 11d with 7.2 μg mL−1. For piperidine derivatives, the butylene linked compound 11f possesses the highest solubility of all tested compounds, while the shorter ethylene linker of compound 11e shows lower solubility. Unfortunately, all introduced amides (18a–c) could not improve solubility when compared to heptyl-carbamate 11a.
Kinetic investigations into binding mode of compound 11d
As compound 11d was promising for further investigations due to its selectivity and one-digit nanomolar inhibition of hBChE, as well as its good solubility, we investigated its binding mode for hBChE in more detail.
Predominantly, carbamate-based inhibitors of BChE act in a pseudoirreversible manner, and the inhibition process is divided into three steps: reversible formation of an enzyme inhibitor complex (EC-X) described by the equilibrium constant Kc, carbamate transfer onto the enzyme forming EC described by rate constant k3 and decarbamylation of the enzyme described by rate constant k4 (Chart 4).
Chart 4. Kinetic characterisation of compound 11d, acting as a pseudoirreversible inhibitor of hBChE.

To prove pseudoirreversible mode of inhibition and characterise compound 11d for further investigations kinetically, we determined Kc, k3 and k4. To investigate carbamylation, experiments similar to the IC50 determination were carried out at different time points to calculate Kc and k3 assuming first order kinetics.20,23,36,37 Decarbamylation was quantified by dilution experiments, as described before.20,23,25,37,38 hBChE was incubated with an high amount of inhibitor to assure complete inhibition (<10% enzyme activity). After dilution of the enzyme solution, reversible inhibitors show immediate restoration of the enzyme activity, while pseudoirreversible inhibitors show slow recovery of enzyme activity over time.20
We calculated a Kc value of 3.0 μM and a k3 value of 8.9 min−1, expressing a comparatively fast carbamylation.23,37 Pseudoirreversible mode of action was shown and quantified by dilution experiments, showing a half-life of 56.3 min of the carbamylated enzyme expressed by a k4 value of 0.74 h−1.
Off-target affinity on cannabinoid receptors
Due to a high structural resemblance of investigated ligands to selective hCB2R agonist 4, radioligand binding studies were done for compounds 11a, 11c and 11d, as these compounds were only modified at the ethoxy group, when compared to compound 4. Both 11a and 11c showed negligible binding to hCB2R (resp.; 33% and <10% at 10 μM). Compound 11d exhibits an pIC50 (hCB2R) of 5.3 (Fig. S1†) in a CB2R competition assay against [3H]CP55940, probably due to the sterically less demanding azetidine. Taking the nanomolar binding to hBChE into account, the ligand can be described as selective over both hAChE and hCB2R.
2.4. Toxicity and in vivo evaluation
Due to its one-digit nanomolar inhibition of hBChE, its selectivity over hAChE and CB2R, as well as its improved solubility, when compared to the heptyl carbamates, inhibitor 11d was chosen for further investigation.
To examine potential neurotoxic potential of inhibitor 11d prior to the in vivo application, murine hippocampal neuronal (HT22) cells were treated with carbamate 11d and its expected cleavage product 10a in a MTT assay (Chart 5).
Chart 5. Neurotoxicity studies on neuronal HT-22 cells for compounds 11d and 10a. Levels of significance: *p < 0.05; **p < 0.01; ***p < 0.001.

Both compounds show a neglectable decrease in cell viability at higher concentrations, but no neurotoxicity at tested doses.
Consequently, in vivo testing of compound 11d for potential pro-cognitive effects was carried out using an established AD mice model.19,23,24,39,40 BChE knockout experiments had already confirmed excellent suitability of the used experimental setup to validate the effect of BChEIs on Aβ25–35 induced learning performances in mice.38,41 Cognitive dysfunction was hereby induced by singular intracerebroventricular injection of neurotoxic Aβ25–35 prior to treatment with compound 11d for seven days. The compound was solubilised in saline using 18% of DMSO, utilising the better solubility of inhibitor 11d when compared to parent compound 2, which had to be solubilised in 60% of DMSO in saline, and injected once daily intraperitoneally.23
Weight loss after ICV injection was below 2 mg, while mice recovered fast afterwards, leading to an overall weight gain of 1–3 g from day 2 to day 10. Memory of the treated mice was then examined using two complementary tests, spontaneous alternation and passive avoidance. Spontaneous alternation, an index of spatial working memory, was measured in a Y-maze on day 8. Long-term memory was assessed using a step-through type passive avoidance procedure, wherein training was carried out on day 9 and retention on day 10. Results of both experiments confirmed Aβ25–35-induced learning impairments for both short- and long-term memory response. Regarding spontaneous alternation, inhibitor 11d attenuated learning impairments significantly at a dose as low as 0.03 mg kg−1 (Chart 6A). In passive avoidance, compound 11d also attenuated learning impairments at 0.03 mg kg−1 (Chart 6B), showing excellent correlation between both experiments.
Chart 6. Effect of inhibitor 11d on Aβ25–35 induced learning impairments in mice: spontaneous alternation performance (A) and retention latency in the passive avoidance test (B). Mice were treated with Aβ25–35 (9 nmol, intracerebroventricularly (ICV)) or vehicle (3 μL of ddH2O, V1) on day 1, then received either vehicle (DMSO 18% in saline, V2) or compound 11d (0.01–0.3 mg kg−1) intraperitoneally (IP) between day 1 to 7. Mice were then tested for spontaneous alternation on day 8 and passive avoidance on day 9 (training) and day 10 (test). Group size is shown beneath each bar graph. Data show ± SEM (A) or median and interquartile range (B). ANOVA: F(5,84) = 6.583; p < 0.0001 in (A); Kruskal–Wallis ANOVA: H = 12.41; p = 0.0295 in (B). ***p < 0.001 vs. vehicle/vehicle-treated group; ***p < 0.001 vs. V1/V2-treated group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs. Aβ25–35/V2 treated group; Dunnett's test in (A); Dunn's test in (B).

3. Conclusion
In summary, a novel carbamate-based class of hBChE inhibitors could be synthesized in a seven-step procedure. Starting from 2-benzylbenzo[d]imidazoles with micromolar BChE inhibition, docking results led to the development of several nanomolar hBChE inhibitors.19 Introduction of an azetidine-carbamate leads to compounds that possess a superior solubility and improved inhibition of hBChE. The non-neurotoxic compound 11d could further underline its potential therapeutic potential in vivo, as complete recovery of recognition after Aβ25–35 induced learning impairments was observed at low doses, starting at 0.03 mg kg−1. Further in vivo investigation in a more complex environment, e.g. the Hamlet test could give a better insight into the different pro-cognitive properties of this novel compound class.42
4. Experimental section
All reagents were purchased from SigmaAldrich, ABCR and Fluorochem and used without further purification. THF was dried by refluxing over sodium under an argon atmosphere and DCM was dried over magnesium sulfate. Thin-layer chromatography was performed on silica gel 60 (alumina foils with fluorescent indicator 254 nm). For detection, staining by potassium permanganate or Ninhydrin and UV light (254 and 366 nm) were used. For column chromatography, silica gel 60 (particle size 0.040–0.063 mm) was used. Nuclear magnetic resonance spectra were recorded with a Bruker AV-400 NMR instrument (Bruker, Karlsruhe, Germany) in deuterated solvents, and chemical shifts are expressed in ppm relative to the solvent residues used for NMR. Purity was determined by HPLC (Shimadzu Products), containing a DGU-20A3R degassing unit, a LC20AB liquid chromatograph, and a SPD-20AUV/vis detector. UV detection was measured at 254 nm. Mass spectra were obtained by a LCMS 2020 (Shimadzu Products). As a stationary phase, a Synergi 4U fusion-RP (150 mm × 4.6 mm) column was used, and as a mobile phase, a gradient of MeOH/water with 0.1% formic acid was used. Parameters: A = water, B = MeOH, V(B)/(V(A) + V(B)) = from 5% to 90% over 10 min, V(B)/(V(A) + V(B)) = 90% for 5 min, V(B)/(V(A) + V(B)) = from 90% to 5% over 3 min. The method was performed with a flow rate of 1.0 mL min−1. Compounds were only used for biological evaluation if the purity was ≥95% and were dried under high vacuum (<0.1 mbar) beforehand.
All synthesised compounds were confirmed by NMR (1H and 13C) and LCMS analysis, and their spectra, as well as their synthetic procedures according to Schemes 1 and 2, are provided in ESI.†
Cholinesterase inhibition
hBChE (E.C. 3.1.1.8, from humans) was kindly provided by Oksana Lockridge from the University of Nebraska Medical Center. hAChE, ATC and BTC iodines, as well as DTNB, were obtained from Sigma-Aldrich (Steinheim, Germany) and Bio-Techne (Minneapolis, USA). A phosphate solution was prepared by dissolving NaH2PO4 in water (55 mM) and adjusting a pH 8.0 by adding 0.1 M NaOH (buffer A). A 10 mM DNTB stock solution was diluted with buffer to 0.3 mM (buffer B). All experiments were conducted at 25 °C.
IC50 determination
The stock solutions of the test compounds were prepared in DMSO (4 mM) and diluted with buffer A to a starting concentration of 10 or 100 μM, depending on the activity of the compound. Further desired dilutions were prepared on 96-well plate (>1% DMSO) using buffer B. Then 130 μL of twelve different concentrations of compound dilution and 10 μL of the respective enzyme (≈2.5 units per mL) were incubated for 20 min. Afterward, 3 μL of either ATC or BTC solution (45 mM in water) were rapidly added in each well using a multidispenser and enzyme activity was immediately observed via UV (λ = 412 nm) for 3 min at 7 points with an interval of 30 seconds. The enzyme activity of each concentration was calculated using the slope. IC50 values were calculated using GraphPad Prism 5 software by plotting enzyme activity against logarithmic inhibitor concentration. Values are presented as means ± SEM of at least three independent determinations. 100% enzyme activity was calculated on every 96-well plate using buffer B.
Determination of Kc and k3
To determine Kc and k3, a similar setup, as for the calculation of IC50 values, was used. At least five different concentrations were measured at six different time points. The obtained enzyme activities (in percent) were plotted against time and fitted to eqn (I) to determine rate constant kobs using GraphPad Prism 5.
| A = A0·e−kobs·t + A∞ | I |
A is the enzyme activity at time t, A0 is the enzyme activity at time t = 0, and A∞ is the enzyme activity at infinite time. The obtained kobs values were plotted against the concentration using GraphPad Prism 5 to obtain Kc and k3.
Decarbamoylation
For measurement of decarbamoylation kinetics, a high amount of enzyme was incubated with a suitable amount of inhibitor for 1 h. The concentration of inhibitor was chosen so that the enzyme was inhibited to >90%. After incubation, the solution was diluted 1000 fold so that no enzyme was carbamylated anymore, and enzyme activity was measured at several (at least six) time points as described above. To determine full enzyme activity, a batch of the enzyme was treated with buffer instead of inhibitor solution. The enzyme activity in percent was plotted against the time after dilution to give the first-order rate constant k4 using the software GraphPad Prism 5.
All procedures used to determine the binding properties of the inhibitors on ChEs have been established before.19,20,24,26,27,37,43–48
Solubility experiments
For the assessment of the solubility, the continuous shake flask protocol was applied.49 Therefore, 1–2 mg of the analysed compound was dissolved in MeOH at 10 mg mL−1. 8 μL of this solution was diluted in 792 μL of Dulbecco's phosphate buffered saline and shaken overnight. After centrifugation (13.400 rpm, 2 min) and filtration, a sample was analysed using the HPLC method mentioned underneath.
For the calibration equation, 1–2 mg of the analysed compound was dissolved in MeOH and diluted to concentrations of at least five concentrations between 1 μg mL−1 to 100 μg mL−1. The peak areas and the concentrations were plotted against each other to give the calibration curve.
Data are representative of three independent experiments.
HPLC method: a Synergi 4U fusion-RP (150 mm × 4.6 mm) column was used, and as a mobile phase, a gradient of MeOH/water with 0.1% formic acid was used. Parameters: A = water, B = MeOH, V(B)/(V(A) + V(B)) = from 5% to 90% over 10 min, V(B)/(V(A) + V(B)) = 90% for 5 min, V(B)/(V(A) + V(B)) = from 90% to 5% over 3 min. The method was performed with a flow rate of 1.0 mL min−1.
Neurotoxicity
HT22 cells were grown in Dulbecco's modified Eagle medium (DMEM, Sigma Aldrich, Munich Germany) supplemented with 10% (v/v) fetal calf serum (FCS) and 1% (v/v) penicillin–streptomycin. BV-2 cells were grown in low glucose DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FCS and 1% (v/v) penicillin–streptomycin. Cells were subcultured every two days and incubated at 37 °C with 5% CO2 in a humidified incubator.
Compounds were dissolved in dimethyl sulfoxide (DMSO, Sigma Aldrich, Munich, Germany) as stock solutions and diluted further into culture medium.
For determination of cell viability, a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma Aldrich, Munich, Germany) assay was used. MTT solution (4 mg mL−1 in PBS) was diluted 1 : 10 with medium and added to the wells after removal of the old medium. Cells were incubated for 3 hours and then lysis buffer (10% SDS) was applied. The next day, absorbance at 560 nm was determined with a multiwell plate photometer (Tecan, SpectraMax 250).
In vivo studies
Protocols for behavioural experiments were established before.19,24,39–41,50,51 In this work, compound 11d was tested for its neuroprotective properties in the in vivo pharmacological model of Alzheimer's disease induced by intracerebroventricular (ICV) injection of the oligomerized Aβ25–35 peptide in the mouse. Parent compounds 2 and 4 were already tested under the same conditions.19,23 Compounds were injected intraperitoneally (IP) between days 1 and 7. The oligomerized Aβ25–35 peptide was injected ICV on the first day of the study, and behavioural evaluation was performed between days 8 and 10.
Animals
Male swiss mice, 6 weeks old, weighing 29–38 g, were purchased from JANVIER (Saint Berthevin, France) and kept in for housing and experiments in the animal facility of the University of Montpellier (CECEMA, Office of Veterinary Services agreement # B-34-172-23). Upon arrival, animals were divided into groups and housed with access to food and water ad libitum, except during behavioural experiments. Mice were kept in a temperature- and humidity-controlled animal facility on a 12 h/12 h light/dark cycle (lights off at 07:00 PM). All animal procedures were conducted in strict adherence to the European Union directive of September 22, 2010 (2010/63/UE) and the ARRIVE guidelines. The project was authorized by the French National Ethic Committee (APAFIS #148515034).
Preparation of injection solutions
Compound 11d was weighed, dissolved in pure DMSO at a concentration of 3.33 mg mL−1, and diluted into final test concentrations with saline. The maximal percentage of DMSO in saline was 18%. Vehicle solution used for control groups was 18% DMSO in saline. General behaviour of the mice in the home cage was checked visually after injections. In particular, weight gain was checked every day. ICV injections affected animals on day 1, but then weight gain was regular and not significantly different from control. All treatments showed good compound and vehicle solution tolerability. Moreover, animals were tested in behavioural tests 24 h after the last compound/vehicle administration.
Amyloid peptide preparation and intracerebroventricular (ICV) injection
Mice were anesthetized with isoflurane (2.5%) and ICV injected with the Aβ25–35 peptide (9 nmol per mouse), according to the previously described method.41,42,51
Molecular docking studies
Ligand structures were built using MOE (Molecular Operating Environment, version 2021.11, Chemical Computing Group, Canada) and energy-minimized with the forcefield MMFF94x (root-mean-square (rms) gradient = 0.001 kcal mol−1 Å−1). Using the crystal structure of human butyrylcholinesterase (hBchE) in complex with tacrine (resolution 2.10 Å, PDB: 4BDS),29 the enzyme was prepared in MOE by removing all non-protein molecules and protonating the structure at pH = 7, corresponding to the pH of the biological assay.
Docking studies were carried out with GOLD (Genetic Optimisation for Ligand Docking, CCDC Software, England, version v2021.1.0). The docking protocol was based on our previous studies of hBChE inhibitors,23 with the exception that we used of a non-covalent approach here. Fifty independent docking runs were carried out using the knowledge-based scoring function ASP (Astex Statistical Potential).52 The maximum and minimum number of operations were set to 106 and 105 with an autoscale factor of 5. As the demethylated carbamate function is supposed to be transferred to the catalytic Ser198 as seen and suggested by several studies,23,46,53 a constraint was set for the distance between the carbonyl-carbon of the carbamate function and the sidechain oxygen of Ser198 of 2.5–4.0 Å (spring constant = 5.0). The binding site was defined by a radius of 20 Å around the γ-oxygen of Ser198. The results were structurally clustered using an RMS deviation of 2.0 Å as cutoff. The docking result showed high convergence with 33 out of 50 docking poses assigned to one cluster. The pose with the highest score by ASP and member the largest cluster was therefore displayed (Chart 2) in PyMOL (version 2.4.1).54
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. P. S. performed synthesis, in vitro testing on ChEs and animal experiments under supervision of T. M. T. Z. performed docking studies on AChE and BChE. J. H. performed neurotoxicity experiments. T. M. was responsible for supervision of animal experiments, provided support on their execution and conducted some additional animal experiments. M. D. was accountable for the oversight and development of the whole project. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
P. Spatz and M. Decker gratefully acknowledge the International Doctoral Program “Receptor Dynamics” by the Elite Network of Bavaria (Elitenetzwerk Bayern) for awarding a Ph.D. position to P. Spatz, financial support and funding of travel costs. The BayFrance (Franco-Bavarian University cooperation, FK03-2020) is gratefully acknowledged for funding of travel costs. M. Decker acknowledges funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG DE1546/10-1). T. Zimmermann acknowledges funding by GRK 2243. We thank Christoph Sotriffer for providing a working space for molecular docking studies and his helpful suggestions on the manuscript. hBChE was kindly donated by Oksana Lockridge. We thank the CECEMA-UM animal facility for animal housing and experiments.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00087c
References
- Citron M. Nat. Neurosci. 2002;5:1055–1057. doi: 10.1038/nn940. [DOI] [PubMed] [Google Scholar]
- Hebert L. E. Beckett L. A. Scherr P. A. Evans D. E. Alzheimer Dis. Assoc. Disord. 2001;15:169–173. doi: 10.1097/00002093-200110000-00002. [DOI] [PubMed] [Google Scholar]
- Matthews K. A. Xu W. Gaglioti A. H. Holt J. B. Croft J. B. Mack D. McGuire L. C. Alzheimer's Dementia. 2019;15:17–24. doi: 10.1016/j.jalz.2018.06.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom S. E. Hubbard R. A. Crane P. K. Haneuse S. J. Bowen J. McCormick W. C. McCurry S. Larson E. B. Am. J. Public Health. 2015;105:408–415. doi: 10.2105/AJPH.2014.301935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd S. Barr S. Roberts M. Passmore A. P. Int. J. Geriatr. Psychiatry. 2013;28:1109–1124. doi: 10.1002/gps.3946. [DOI] [PubMed] [Google Scholar]
- Hurd M. D. Martorell P. Delavande A. Mullen K. J. Langa K. M. N. Engl. J. Med. 2013;368:1326–1334. doi: 10.1056/NEJMsa1204629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad A. H. Ballestros K. Echko M. Glenn S. Olsen H. E. Mullany E. Lee A. Khan A. R. Ahmadi A. Ferrari A. J. Kasaeian A. Werdecker A. Carter A. Zipkin B. Sartorius B. Serdar B. Sykes B. L. Troeger C. Fitzmaurice C. Rehm C. D. Santomauro D. Kim D. Colombara D. Schwebel D. C. Tsoi D. Kolte D. Nsoesie E. Nichols E. Oren E. Charlson F. J. Patton G. C. Roth G. A. Hosgood H. D. Whiteford H. A. Kyu H. Erskine H. E. Huang H. Martopullo I. Singh J. A. Nachega J. B. Sanabria J. R. Abbas K. Ong K. Tabb K. Krohn K. J. Cornaby L. Degenhardt L. Moses M. Farvid M. Griswold M. Criqui M. Bell M. Nguyen M. Wallin M. Mirarefin M. Qorbani M. Younis M. Fullman N. Liu P. Briant P. Gona P. Havmoller R. Leung R. Kimokoti R. Bazargan-Hejazi S. Hay S. I. Yadgir S. Biryukov S. Vollset S. E. Alam T. Frank T. Farid T. Miller T. Vos T. Barnighausen T. Gebrehiwot T. T. Yano Y. Al-Aly Z. Mehari A. Handal A. Kandel A. Anderson B. Biroscak B. Mozaffarian D. Dorsey E. R. Ding E. L. Park E. K. Wagner G. Hu G. Chen H. Sunshine J. E. Khubchandani J. Leasher J. Leung J. Salomon J. Unutzer J. Cahill L. Cooper L. Horino M. Brauer M. Breitborde N. Hotez P. Topor-Madry R. Soneji S. Stranges S. James S. Amrock S. Jayaraman S. Patel T. Akinyemiju T. Skirbekk V. Kinfu Y. Bhutta Z. Jonas J. B. Murray C. J. L. J. Am. Med. Assoc. 2018;319:1444–1472. doi: 10.1001/jama.2018.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen Y. Am. J. Clin. Nutr. 2000;71:621–629. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- Francis P. T. Palmer A. M. Snape M. Wilcock G. C. J. Neurol., Neurosurg. Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K. A. Alafuzoff I. Attems J. Beach T. G. Cairns N. J. Crary J. F. Dickson D. W. Hof P. R. Hyman B. T. Jack, Jr. C. R. Jicha G. A. Knopman D. S. Kovacs G. G. Mackenzie I. R. Masliah E. Montine T. J. Nelson P. T. Schmitt F. Schneider J. A. Serrano-Pozo A. Thal D. R. Toledo J. B. Trojanowski J. Q. Troncoso J. C. Vonsattel J. P. Wisniewski T. Acta Neuropathol. 2015;129:757–762. doi: 10.1007/s00401-015-1407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery W. R. Free Radic. Biol. Med. 1997;23:134–147. doi: 10.1016/S0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Pohanka M. Curr. Med. Chem. 2014;21:356–364. doi: 10.2174/09298673113206660258. [DOI] [PubMed] [Google Scholar]
- Ferreira-Vieira T. H. Guimaraes I. M. Silva F. R. Ribeiro F. M. Curr. Neuropharmacol. 2016;14:101–115. doi: 10.2174/1570159X13666150716165726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry, Jr. A. V. Buccafusco J. J. J. Pharmacol. Exp. Ther. 2003;306:821–827. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- Ali T. B. Schleret T. R. Reilly B. M. Chen W. Y. Abagyan R. PLoS One. 2015;10:1–10. doi: 10.1371/journal.pone.0144337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T. Brockner M. K. Lange M. Bigl V. Neurochem. Int. 1992;21:381–396. doi: 10.1016/0197-0186(92)90189-X. [DOI] [PubMed] [Google Scholar]
- Giacobini E. Int. J. Geriatr. Psychiatry. 2003;18:1–5. doi: 10.1002/gps.935. [DOI] [PubMed] [Google Scholar]
- Bolognesi M. L. Bartolini M. Cavalli A. Andrisano V. Rosini M. Minarini A. Melchiorre C. J. Med. Chem. 2004;47:5945–5951. doi: 10.1021/jm049782n. [DOI] [PubMed] [Google Scholar]
- Dolles D. Hoffmann M. Gunesch S. Marinelli O. Moller J. Santoni G. Chatonnet A. Lohse M. J. Wittmann H. J. Strasser A. Nabissi M. Maurice T. Decker M. J. Med. Chem. 2018;61:1646–1663. doi: 10.1021/acs.jmedchem.7b01760. [DOI] [PubMed] [Google Scholar]
- Sawatzky E. Al-Momani E. Kobayashi R. Higuchi T. Samnick S. Decker M. ChemMedChem. 2016;11:1540–1550. doi: 10.1002/cmdc.201600223. [DOI] [PubMed] [Google Scholar]
- Pagé D. Balaux E. Boisvert L. Liu Z. Milburn C. Tremblay M. Wei Z. Woo S. Luo X. Cheng Y. X. Yang H. Srivastava S. Zhou F. Brown W. Tomaszewski M. Walpole C. Hodzic L. St-Onge S. Godbout C. Salois D. Payza K. Bioorg. Med. Chem. Lett. 2008;18:3695–3700. doi: 10.1016/j.bmcl.2008.05.073. [DOI] [PubMed] [Google Scholar]
- Greig N. H. Utsuki T. Yu Q. Zhu X. Holloway H. W. Perry T. Lee B. Ingram D. K. Lahiri D. K. Curr. Med. Res. Opin. 2001;17:159–165. doi: 10.1185/03007990152673800. [DOI] [PubMed] [Google Scholar]
- Hoffmann M. Stiller C. Endres E. Scheiner M. Gunesch S. Sotriffer C. Maurice T. Decker M. J. Med. Chem. 2019;62:9116–9140. doi: 10.1021/acs.jmedchem.9b01012. [DOI] [PubMed] [Google Scholar]
- Scheiner M. Dolles D. Gunesch S. Hoffmann M. Nabissi M. Marinelli O. Naldi M. Bartolini M. Petralla S. Poeta E. Monti B. Falkeis C. Vieth M. Hubner H. Gmeiner P. Maitra R. Maurice T. Decker M. J. Med. Chem. 2019;62:9078–9102. doi: 10.1021/acs.jmedchem.9b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner M. Sink A. Hoffmann M. Vrigneau C. Endres E. Carles A. Sotriffer C. Maurice T. Decker M. J. Am. Chem. Soc. 2022;144:3279–3284. doi: 10.1021/jacs.1c13492. [DOI] [PubMed] [Google Scholar]
- Darras F. H. Kling B. Heilmann J. Decker M. ACS Med. Chem. Lett. 2012;3:914–919. doi: 10.1021/ml3001825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolles D. Nimczick M. Scheiner M. Ramler J. Stadtmüller P. Sawatzky E. Drakopoulos A. Sotriffer C. Wittmann H.-J. Strasser A. Decker M. ChemMedChem. 2016;11:1270–1283. doi: 10.1002/cmdc.201500418. [DOI] [PubMed] [Google Scholar]
- Jones G. Peter Willett P. Glen R. C. Leach A. R. Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Nachon F. Carletti E. Ronco C. Trovaslet M. Nicolet Y. Jean L. Renard P. Y. Biochem. J. 2013;453:393–399. doi: 10.1042/BJ20130013. [DOI] [PubMed] [Google Scholar]
- Masson P. Froment M.-T. Bartels C. F. Lockridge O. Eur. J. Biochem. 1996;235:36–48. doi: 10.1111/j.1432-1033.1996.00036.x. [DOI] [PubMed] [Google Scholar]
- Rosenberry T. L. Brazzolotto X. Macdonald I. R. Wandhammer M. Trovaslet-Leroy M. Darvesh S. Nachon F. Molecules. 2017;22:1–21. doi: 10.3390/molecules22122098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosak A. Ramic A. Smidlehner T. Hrenar T. Primozic I. Kovarik Z. PLoS One. 2018;13:1–18. doi: 10.1371/journal.pone.0205193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald I. R. Martin E. Rosenberry T. L. Darvesh S. Biochemistry. 2012;51:7046–7053. doi: 10.1021/bi300955k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A. Redman A. M. G. Jiang X. Lockridge O. Doctor B. P. Chem.-Biol. Interact. 1999;119–120:61–69. doi: 10.1016/S0009-2797(99)00014-9. [DOI] [PubMed] [Google Scholar]
- Bosak A. Gazic Smilovic I. Sinko G. Vinkovic V. Kovarik Z. J. Med. Chem. 2012;55:6716–6723. doi: 10.1021/jm300289k. [DOI] [PubMed] [Google Scholar]
- Feaster S. R. Lee K. Baker N. Hui D. Y. Quinn D. M. Biochemistry. 1996;35:16723–16734. doi: 10.1021/bi961677v. [DOI] [PubMed] [Google Scholar]
- Sawatzky E. Wehle S. Kling B. Wendrich J. Bringmann G. Sotriffer C. A. Heilmann J. Decker M. J. Med. Chem. 2016;59:2067–2082. doi: 10.1021/acs.jmedchem.5b01674. [DOI] [PubMed] [Google Scholar]
- Scheiner M. Hoffmann M. He F. Poeta E. Chatonnet A. Monti B. Maurice T. Decker M. J. Med. Chem. 2021;64:9302–9320. doi: 10.1021/acs.jmedchem.1c00534. [DOI] [PubMed] [Google Scholar]
- Gunesch S. Hoffmann M. Kiermeier C. Fischer W. Pinto A. F. M. Maurice T. Maher P. Decker M. Redox Biol. 2020;29:101378. doi: 10.1016/j.redox.2019.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann J. Fayez S. Scheiner M. Hoffmann M. Oerter S. Appelt-Menzel A. Maher P. Maurice T. Bringmann G. Decker M. Chem. – Eur. J. 2020;26:7299–7308. doi: 10.1002/chem.202001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T. Strehaiano M. Simeon N. Bertrand C. Chatonnet A. Behav. Brain Res. 2016;296:351–360. doi: 10.1016/j.bbr.2015.08.026. [DOI] [PubMed] [Google Scholar]
- Crouzier L. Gilabert D. Rossel M. Trousse F. Maurice T. Neurobiol. Learn. Mem. 2018;149:118–134. doi: 10.1016/j.nlm.2018.02.014. [DOI] [PubMed] [Google Scholar]
- Chen X. Tikhonova I. G. Decker M. Bioorg. Med. Chem. 2011;19:1222–1235. doi: 10.1016/j.bmc.2010.12.034. [DOI] [PubMed] [Google Scholar]
- Chen X. Wehle S. Kuzmanovic N. Merget B. Holzgrabe U. Konig B. Sotriffer C. A. Decker M. ACS Chem. Neurosci. 2014;5:377–389. doi: 10.1021/cn500016p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras F. H. Kling B. Sawatzky E. Heilmann J. Decker M. Bioorg. Med. Chem. 2014;22:5020–5034. doi: 10.1016/j.bmc.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Gentzsch C. Chen X. Spatz P. Kosak U. Knez D. Nose N. Gobec S. Higuchi T. Decker M. Mol. Imaging Biol. 2021;23:505–515. doi: 10.1007/s11307-021-01584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch C. Hoffmann M. Ohshima Y. Nose N. Chen X. Higuchi T. Decker M. ChemMedChem. 2021;16:1427–1437. doi: 10.1002/cmdc.202000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner M. Sink A. Spatz P. Endres E. Decker M. ChemPhotoChem. 2020;5:149–159. doi: 10.1002/cptc.202000119. [DOI] [Google Scholar]
- Austin R. P. Bennion C. Bonnert R. V. Cheema L. Cook A. R. Cox R. J. Ebden M. R. Gaw A. Grime K. Meghani P. Nicholls D. Phillips C. Smith N. Steele J. Stonehouse J. P. Bioorg. Med. Chem. Lett. 2015;25:1616–1620. doi: 10.1016/j.bmcl.2015.01.067. [DOI] [PubMed] [Google Scholar]
- Lahmy V. Meunier J. Malmstrom S. Naert G. Givalois L. Kim S. H. Villard V. Vamvakides A. Maurice T. Neuropsychopharmacology. 2013;38:1706–1723. doi: 10.1038/npp.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T. Lockhart B. P. Privat A. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- Mooij W. T. Verdonk M. L. Proteins. 2005;61:272–287. doi: 10.1002/prot.20588. [DOI] [PubMed] [Google Scholar]
- Pejchal V. Stepankova S. Pejchalova M. Kralovec K. Havelek R. Ruzickova Z. Ajani H. Lo R. Lepsik M. Bioorg. Med. Chem. 2016;24:1560–1572. doi: 10.1016/j.bmc.2016.02.033. [DOI] [PubMed] [Google Scholar]
- The Pymol Molecular Graphics System, Version 2.4.1, Schrödinger, LLC [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.