

A prospective observational study confirms the value of molecular residual disease (MRD) for patients with stage I–IIIA non–small cell lung cancer after resection, highlighting the negative predictive value of longitudinal undetectable MRD.
Abstract
The efficacy and potential limitations of molecular residual disease (MRD) detection urgently need to be fully elucidated in a larger population of non–small cell lung cancer (NSCLC). We enrolled 261 patients with stages I to III NSCLC who underwent definitive surgery, and 913 peripheral blood samples were successfully detected by MRD assay. Within the population, only six patients (3.2%) with longitudinal undetectable MRD recurred, resulting in a negative predictive value of 96.8%. Longitudinal undetectable MRD may define the patients who were cured. The peak risk of developing detectable MRD was approximately 18 months after landmark detection. Correspondingly, the positive predictive value of longitudinal detectable MRD was 89.1%, with a median lead time of 3.4 months. However, brain-only recurrence was less commonly detected by MRD (n = 1/5, 20%). Further subgroup analyses revealed that patients with undetectable MRD might not benefit from adjuvant therapy. Together, these results expound the value of MRD in NSCLC.
Significance:
This study confirms the prognostic value of MRD detection in patients with NSCLC after definitive surgery, especially in those with longitudinal undetectable MRD, which might represent the potentially cured population regardless of stage and adjuvant therapy. Moreover, the risk of developing detectable MRD decreased stepwise after 18 months since landmark detection.
This article is highlighted in the In This Issue feature, p. 1599
INTRODUCTION
To increase the cure rate of localized non–small cell lung cancer (NSCLC) after radical resection, continuous efforts have been made to improve the adjuvant treatment model in recent decades. Conventional adjuvant chemotherapy and radiotherapy are the main approaches but have limited survival benefits (1–3). Encouragingly, substantial progress has been made since the introduction of tyrosine kinase inhibitors and immune-checkpoint inhibitors (ICI), as reported in the ADAURA and IMpower 010 trials (4, 5). However, a simple one-size-fits-all strategy and long-term treatment period result in unnecessary toxic side effects and a considerable psychological burden to a large proportion of patients. Therefore, accurately predicting disease recurrence risk in patients is a key challenge in this context.
Recently, several techniques have been developed for the detection of molecular residual disease (MRD) in NSCLC, mainly by tracking ultra-low-frequency somatic tumor mutations in cell-free DNA (cfDNA; refs. 6–11). It is now widely recognized that patients with detectable MRD have a worse prognosis than those with undetectable MRD (12–14). However, several important questions remain to be answered. First, much more attention is currently focused on positive MRD signals, which imply cancer persistence or disease progression. However, in theory, negative MRD signals also have important clinical significance. Thus, what are the long-term clinical outcomes of patients with longitudinal undetectable MRD? Second, to what extent can MRD play a predictive role in the adjuvant treatment of NSCLC? Third, to what extent does the presence of nonshedding tumors affect MRD monitoring in NSCLC? Finally, what could be the potential limitations of MRD application in NSCLC, and which hurdles remain to be overcome? Therefore, we conducted this prospective, noninterventional, observational study to elucidate the role of MRD monitoring in patients with stage I to IIIA NSCLC after definite surgical resection.
RESULTS
Patient Characteristics
A total of 261 preoperative blood samples, 256 tumor tissues, and 652 postoperative blood samples were successfully profiled from 261 patients with stage I to III NSCLC (Figs. 1 and 2A). All patients had surgical pathology-proven adenocarcinoma (n = 203, 77.8%), squamous cell carcinoma (n = 33, 12.6%), or others (n = 25, 9.6%), with 104 patients (39.8%) with stage IA, 59 patients (22.6%) with stage IB, 53 patients (20.3%) with stage II, and 45 patients (17.2%) with stage III disease (Fig. 2B). The median age of the patients was 62 (range, 27–84) years, and 37.2% (n = 97) of patients had a history of tobacco use. Of the patients, 66 (25.3%) received perioperative therapy, including chemotherapy in 40.9% (n = 27), targeted therapy in 45.5% (n = 30), and ICIs or ICI-combined chemotherapy in 13.6% (n = 9; Fig. 2B; Supplementary Table S1).
Figure 1.

Flow diagram of patient inclusion in subanalyses with clinical questions answered by each analysis denoted.
Figure 2.

Study schematic and baseline characteristics. A, Study flowchart. Patients with stage I to III NSCLC (tumor diameter ≥2 cm) treated with definitive surgery were enrolled. Peripheral blood samples were collected before surgery and every 3 to 6 months after surgery. B, Heat map plot based on baseline characteristics and preoperative ctDNA tests of each patient. C, Multivariate logistic regression model for preoperative ctDNA detection. Pathologic type (adenocarcinoma) and SUVmax were independently associated with preoperative ctDNA detection. D, Detection rate of preoperative ctDNA in patients with different pathologic types. E, Detection rate of preoperative ctDNA in patients with different groups of SUVmax subgroup. F, Schematic diagram of residual blood sample collection from resected lung lobe. G, Comparison between two peripheral blood samples and residual blood ctDNA analysis of additional 11 patients with stage I NSCLC. AD, adenocarcinoma; Chemo, chemotherapy; SCC, squamous cell carcinoma; TKI, tyrosine kinase inhibitor; TMB, tumor mutation burden.
Overall, 2,751 variants were identified in tumor tissues, including 2,150 single-nucleotide variants (SNV), 293 insertions/deletions (indels), 19 fusions, and 289 somatic copy-number variants (Supplementary Fig. S1A). We identified 2,002 driver events (median, 6; range, 0–57) based on previously reported criteria, and most of them (95.6%) were clonal events. A high prevalence of driver events in EGFR, TP53, and LRP1B occurred in patients with adenocarcinoma, whereas more frequent TP53, LRP1B, and CDKN2A mutations were observed in other patients.
Existence of Nonshedding Tumor
Considering the presence of tumors, circulating tumoral DNA (ctDNA) tests before surgery may be used to assess tumor DNA shedding for each individual. Preoperative ctDNA tests were successfully performed for all enrolled patients; however, only 36.4% of them (n = 95) were positive, with an average of four mutations per patient and a median variant allele fraction (VAF) of 0.71%. Analogous to previous studies, the presence of detectable ctDNA before surgery was highly correlated with larger tumor size, tumor stage, and pathologic type. Among patients with PET-CT scan, preoperative ctDNA concentration was highly correlated with the maximum standardized uptake value (SUVmax). Regarding gene mutations, detectable ctDNA was more frequent in patients with TP53, MLL2, MLL3, ATM, FAT1, or SMARCA4 mutations; conversely, an inverse correlation between EGFR mutations and preoperative ctDNA was identified. However, only SUVmax and pathologic type were statistically significant in the multivariate analysis (Fig. 2C–E). Additionally, among the preoperative ctDNA mutations, most of them (76.0%) were matched in the corresponding tumor tissues and were more common as clonal mutations and higher VAF variants defined in tissues (Supplementary Fig. S1B and S1C). The undetectable preoperative ctDNA is associated with a better prognosis for patients [hazard ratio (HR) = 0.21; 95% confidence interval (CI): 0.11–0.38; Supplementary Fig. S1D].
We realized that, when compared with previous data, patients with detectable ctDNA before surgery were underrepresented (36.4%) in our study. Although this might be related to the effect of the above factors on tumor DNA shedding ability, it might also be attributed to the insufficient detection sensitivity of our assay. Hence, to further clarify assay performance and nonshedding of tumors, we compared the ctDNA results with those originating from peripheral and residual blood of another 11 patients with stage I disease (Fig. 2F; Supplementary Fig. S2). Interestingly, both ctDNA tests showed 100% agreement (Fig. 2G). Undetectable ctDNA in residual blood, which confirmed that the tumor was nonshedding, was identified in six patients (54.5%); correspondingly, no positive signals were found in the peripheral blood of these patients. It should be noted that five of them with detectable ctDNA in residual blood also had positive signals in the two peripheral blood samples, and the ctDNA concentration in residual blood was much higher than that in peripheral blood (Fig. 2G). However, three of them were found only once and were positive (P01, P04, and P08), especially those variants with a VAF of less than 0.05%. This suggests that insufficient sensitivity for low-frequency mutations does exist.
Undetectable MRD Defines Potentially Cured Population
As of December 31, 2021, a total of 652 postoperative blood samples were successfully tested for MRD (average 2.5 times per patient, range, 1–6), and the median follow-up period was 19.7 months. A total of 174 ctDNA mutations were found in postoperative blood samples, and 74.6% of them were matched in the corresponding tumor tissues. Sixteen and 25 patients were excluded from the analysis of landmark and longitudinal MRD predictive values, respectively, because of insufficient follow-up (less than half a year; Fig. 1). According to the predefined time point analysis, 245 were classified as landmark time points, 588 were longitudinal time points, and 44 belonged to surveillance time points. The detection of ctDNA-MRD is based on capture-based next-generation sequencing (NGS) approaches. A customized panel was designed to cover frequently mutated genes in lung cancer (Supplementary Table S2). The bioinformatics pipeline integrated tumor genotype–informed and tumor genotype–naïve ctDNA analysis; however, considering the analytic sensitivity and specificity, tumor-informed ctDNA analysis in plasma to track the known genotype identified in tumor tissues was highly preferred (Supplementary Fig. S3).
In the landmark time point analysis, 224 patients (91.4%) had undetectable MRD; among them 65.6% were stage I, 18.8% stage II, and 15.6% stage III, and most of them (n = 194) remained disease-free, with a negative predictive value (NPV) of 86.6%. Conversely, among 21 patients with detectable landmark MRD, 17 had recurrence, and the positive predictive value (PPV) was 81.0%. Moreover, when integrating longitudinal time points, the NPV and PPV were further increased to 96.8% and 89.1%, respectively (Fig. 3A). In other words, 96.8% of patients with longitudinal undetectable MRD were still disease-free at the last follow-up (70.1% of them were stage I, 17.4% stage II, and 12.5% stage III). The NPV of longitudinal MRD detection maintained extremely high levels at different stages: 98.5% in stage I; 88.9% in stage II; and 100% in stage III (Fig. 3B). Consequently, patients with undetectable MRD at landmark or longitudinal time points had better disease-free survival (DFS) than those with detectable MRD [landmark: unreached vs. 12.1 months (4.7–19.5); HR = 0.08; 95% CI, 0.02–0.33; longitudinal: unreached vs. 15.9 months (13.8–18.0); HR = 0.02; 95% CI, 0.01–0.05; Fig. 3C and D]. Moreover, the results remained highly significant when accounting for guarantee-time bias (P < 0.001; HR = 0.03; 95% CI, 0.01–0.06; Supplementary Table S3). Remarkably, compared with landmark single time point detection, the survival curve of patients with longitudinal undetectable MRD was nearly perfect, which ideally corresponded to the cured population; and it is not affected by clinical stages (Fig. 3E). The detection of MRD preceded radiographic progression in 87.2% of patients and by a median of 3.4 months (Fig. 3F).
Figure 3.

MRD monitoring after surgery. A, The NPV and PPV of undetectable and detectable MRD at landmark and longitudinal time points, respectively. B, NPV and PPV of undetectable and detectable MRD at longitudinal time points across different stages. C, Kaplan–Meier analysis of DFS stratified by landmark MRD status: detectable (n = 21) versus undetectable (n = 224). D, Kaplan–Meier analysis of DFS stratified by longitudinal MRD status: detectable (n = 46) versus undetectable (n = 190). E, Kaplan–Meier analysis of DFS stratified by longitudinal MRD status across different stages. F, Kaplan–Meier analysis of time to MRD detection and time to imaging recurrence from the end of treatment for all patients. G, Flowchart of occurrence time of MRD or recurrent events in stage II/III patients; the peak timeframe of detectable MRD occurrence was 12 to 18 months after surgery. H, Estimated hazard rate curves for event occurrences in stage II/III patients with landmark-undetectable MRD (n = 85, events were defined as detectable MRD or disease recurrence).
Temporal MRD Detection Pattern
To further elucidate the temporal dynamic patterns during the MRD monitoring process, we analyzed the occurrence time of MRD or recurrent events in stage II/III patients, who had a higher risk of disease recurrence (Fig. 3G). As can be seen, a total of 85 patients had landmark undetectable MRD to start with. In the first six months, three cases developed recurrence without detectable MRD, and other patients had undetectable MRD. A total of 18 patients subsequently developed detectable MRD. The peak timeframe during which MRD was detected was 12 to 18 months after surgery (n = 8/18, 44.4%; Fig. 3G). And the hazard rate curve estimated for occurrences of detectable MRD or disease recurrences in these 85 patients showed a peak at 18 months. Hence, when patients maintained undetectable MRD for more than 18 months, the risk of developing detectable MRD or recurrence decreased stepwise (Fig. 3H). This information may be useful in representing the potentially cured population, regardless of stage.
Sensitivity
As of December 31, 2021, 47 patients (18.0%) had confirmed recurrence (Fig. 1). From the landmark time point, the sensitivity was 36.2% (95% CI, 21.9%–50.4%); integrating longitudinal time points increased the sensitivity to 87.2% (95% CI, 77.3%–97.1%). The sensitivity of the surveillance time points was 86.4% (95% CI, 75.8%–96.9%; Fig. 4A). The timing and receipt of perioperative therapy, surgery, and serial blood collection for each patient with recurrence are shown in Fig. 4B. Of note, six of them (12.8%) had undetectable MRD during the entire follow-up period. Undetectable MRD was significantly associated with the site of recurrence, because four of them had only metastatic brain diseases. The overall sensitivity of patients with brain-only metastasis was 20% (1 in 5).
Figure 4.

Sensitivity analysis and treatment history of 47 patients with recurrence. A, Sensitivity analysis of MRD detection at landmark, longitudinal, and surveillance time points. B, Treatment and MRD test history of all 47 patients with recurrence. Chemo, chemotherapy; TKI, tyrosine kinase inhibitor.
Predictive Value for Adjuvant Therapy
Because MRD status reflected the tumor load, we next explored the MRD predictive value for adjuvant therapy (Fig. 1). Fifty-five patients received adjuvant therapy in this cohort, including chemotherapy (n = 23), targeted therapy (n = 26), and ICIs with or without chemotherapy (n = 6). All these patients had additional MRD time points (preadjuvant time points) after surgery and before adjuvant therapy, and 10 of them had detectable MRD (18.2%). We further compared these 10 patients with other patients with landmark-detectable MRD. Adjuvant therapy was found to confer a survival benefit for patients with detectable MRD (P = 0.022; HR = 0.34; 95% CI, 0.12-0.88; Fig. 5A and B). However, this observation was preliminary due to the limited sample size. Additionally, it is worth noting that patients without adjuvant therapy tended to be older and disease was at an earlier stage (Supplementary Table S4). Conversely, 45 patients had undetectable MRD at the preadjuvant time point. We also compared these patients with other landmark-undetectable MRD patients and found that adjuvant therapy could not improve DFS (Fig. 5C). Similar results were further confirmed between the two groups after propensity score matching to balance baseline variables (Fig. 5D and E; Supplementary Table S5). This implies an extremely low tumor load for patients with undetectable MRD, and adjuvant therapy may be unnecessary for these patients.
Figure 5.

MRD predictive value on adjuvant therapy. A, Heat map plot based on baseline characteristics of patients with detectable MRD at preadjuvant and landmark time points (n = 23). B, Kaplan–Meier analysis of DFS stratified by adjuvant therapy for patients with detectable MRD at preadjuvant and landmark time points: with adjuvant therapy (n = 10) versus without (n = 13). C, Kaplan–Meier analysis of DFS stratified by adjuvant therapy for patients with undetectable MRD at preadjuvant and landmark time points: with adjuvant therapy (n = 45) versus without (n = 182). D, Heat map plot based on baseline characteristics of patients with undetectable MRD at preadjuvant and landmark time points after propensity score matching (PSM). E, After PSM, Kaplan–Meier analysis of DFS stratified by adjuvant therapy for patients with undetectable MRD at preadjuvant and landmark time points: with adjuvant therapy (n = 22) versus without (n = 22). F, Dynamic changes of ctDNA frequency before and after adjuvant therapy for patients with detectable MRD (n = 10), and the DFS of these patients. The arrow represents five of them who still maintained disease-free status. AD, adenocarcinoma; Chemo, chemotherapy; SCC, squamous cell carcinoma; TKI, tyrosine kinase inhibitor.
Then, we further analyzed the dynamic changes of ctDNA frequency for the 10 patients with detectable MRD who received adjuvant therapy. Five of them did not achieve ctDNA clearance (not-cleared group), two of them experienced temporary ctDNA clearance but subsequently tested ctDNA-positive (cleared then raised group), and three of them maintained ctDNA clearance (cleared group). Patients from the cleared group achieved the longest DFS, and those from the other two groups did not appear to differ from each other in DFS (Fig. 5F). Hence, it may be suggested that ctDNA cleared at a single time point cannot entirely reflect the efficacy of adjuvant therapy.
DISCUSSION
To our knowledge, this prospective study represents the largest sample size for ctDNA-MRD detection in NSCLC, which demonstrated an excellent correlation between MRD and clinical outcome. In particular, those with longitudinal undetectable MRD may represent a potentially cured population. Second, we specifically described the nonshedding vulnerability of ctDNA in NSCLC. Third, we preliminarily explored the predictive value of MRD for adjuvant therapy, which indicated the nonessentiality of adjuvant therapy for patients with undetectable MRD.
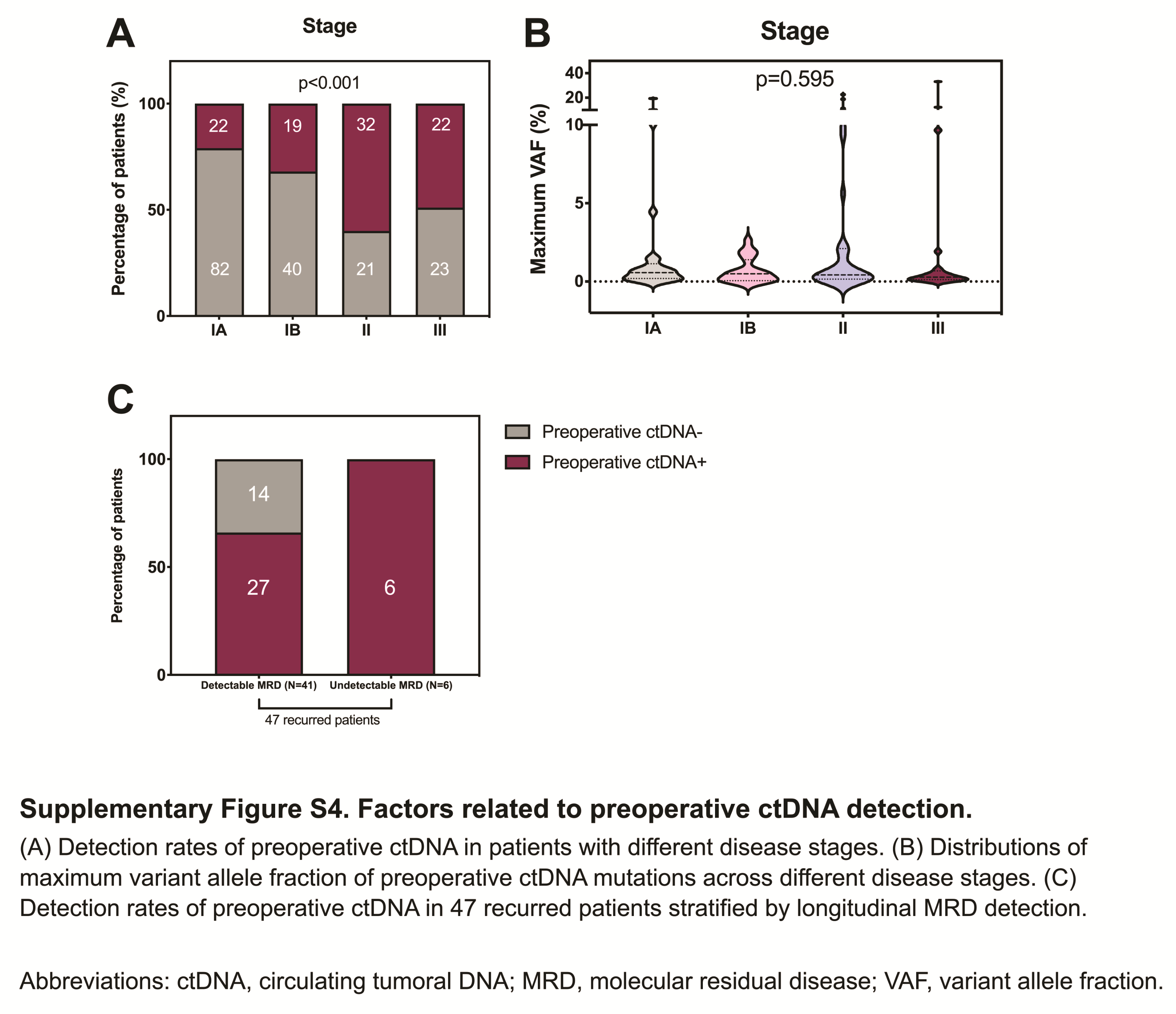
Overall, the detection rate of preoperative ctDNA was 36.4% in the overall population, which is relatively lower than that reported in published studies (15). Several aspects must be considered. First, 62.5% of the enrolled patients had stage I disease in this study. The positive rates of preoperative ctDNA were 21.2% in stage IA, 32.2% in stage IB, 60.4% in stage II, and 48.9% in stage III cases (Supplementary Fig. S4A). However, the maximum VAF in these cases was unrelated to the tumor–node–metastasis stage (Supplementary Fig. S4B). Second, in the independent cohort of patients with stage I NSCLC, six of them (54.4%) were found with no positive ctDNA signal from the residual blood of the resected lung lobe. This implies a certain degree of nonshedding of the tumor. More importantly, undetectable ctDNA in preoperative blood does not seem to affect its usefulness for monitoring disease recurrence. Among a total of 41 recurrent patients with longitudinal detectable MRD, 14 (34.1%) had a negative preoperative ctDNA test. In contrast, all recurred patients with undetectable MRD showed positive ctDNA tests before surgery (6/6, 100%; Supplementary Fig. S4C).
MRD has excellent prognostic value in lung cancer, although previous studies included insufficient sample sizes (9, 12–14). It is worth noting that 75% to 90% of patients with longitudinal undetectable MRD remained disease-free in these studies, indicating that they may have been cured. Identifying and avoiding overtreatment of this potentially cured population is a significant clinical concern. Therefore, we highlighted the NPV of MRD detection in this study. Patients with longitudinal undetectable MRD could maintain an extremely high disease-free rate (96.8%), which ideally defines the potentially cured patient population. More importantly, the dynamic changes of MRD monitoring showed that the peak timeframe of detectable MRD occurrence was during 12 to 18 months after surgery. Hence, patients who had longitudinal undetectable MRD over 18 months may represent the cured population. The question that follows is this: What is the efficacy of adjuvant therapy for patients with detectable and undetectable MRD? In other words, can patients with undetectable MRD be guided to avoid adjuvant therapy? In a phase III randomized trial (IMvigor010) by Powles and colleagues (16), adjuvant atezolizumab was compared with observation after surgical resection for urothelial cancer, and only patients with detectable MRD benefited from adjuvant atezolizumab (DFS, HR = 0.58; 95% CI, 0.43–0.79). In our study, the proportion of patients with stage IB to III NSCLC who received perioperative therapy was 42.0% (13.6% in stage IB, 49.1% in stage II, and 71.1% in stage III), which was similar to previous studies (17–19). And the exploratory analysis appears to reflect the potential predictive value of MRD in adjuvant therapy. However, because of the limited sample size and slightly unbalanced baseline features, it is difficult to draw a reliable conclusion based on the subgroup of detectable MRD. Nevertheless, we did observe the nonessentiality of adjuvant therapy for patients with undetectable MRD after propensity score matching. Further prospective clinical trials are necessary to confirm the predictive value of MRD.
As our data showed, the sensitivity of MRD monitoring is limited in patients with brain-only recurrence. Only one patient had detectable MRD among a total of five patients with brain-only metastases (n = 1/5, 20%). Similar results were also observed in the study by Garcia-Murillas and colleagues on breast cancer; brain-only metastasis was less commonly detected by ctDNA (n = 1/6, 17%; ref. 20). Thus, overcoming the blood–brain barrier for MRD monitoring merits further investigation. Because cerebrospinal fluid (CSF) has been proven to be a comparable or superior detection medium to peripheral blood for central nervous system primary or metastatic tumors (21–23), high-depth ctDNA testing of CSF may be worthwhile, as is the case for the CAPP-seq technique on urine (uCAPP-seq; ref. 24), which could accurately monitor recurrent disease in patients with bladder cancer after radical treatment.
Notably, 24% of the preoperative cfDNA mutations and 25.4% of postoperative cfDNA mutations were not found in the matched tumor tissues. It is challenging to exclude the false positives from these unique cfDNA variants. We believe that these mutations are mainly derived from tumors. In our bioinformatics filter pipeline, matched genomic DNA from the white blood cells and the frequency of the variants in public or internal databases are two important control references. And most of the mutations are reported as driver genes or cancer-related genes in the databases of the Catalogue of Somatic Mutations in Cancer (COSMIC) and The Cancer Genome Atlas (TCGA; Supplementary Fig. S5). None of them are associated with the clonal hematopoiesis–related genes (i.e., DNMT3A, TET2, and PPM1D), as reported in the previous studies (25, 26). On the other hand, ctDNA is thought to reflect the systemic mutation profile. The fact that more unique mutations were detected from ctDNA than tumor tissue was also reported in other studies (27–29). Moreover, the tissue local sampling itself cannot represent the whole picture of the tumor, as intratumoral heterogeneity may also contribute to the inconsistency (30).
The present study had certain limitations. First, the follow-up duration was relatively short (median follow-up duration, 19.7 months). This led to only 47 cases of recurrence in our study. Second, MRD follow-up was not strictly conducted every 3 to 6 months after landmark detection. Nevertheless, the prognostic value of MRD was confirmed with sufficient statistical power in this study.
In conclusion, in this prospective study, we confirmed the prognostic value of ctDNA-based MRD detection in patients with NSCLC after radical resection. We also highlighted the value of undetectable MRD, which could be used to define the potentially cured population in localized NSCLC. Moreover, subgroup analysis suggested that adjuvant therapy may be unnecessary for patients with undetectable MRD; we found that cases with brain-only recurrence were a major unmet challenge for MRD monitoring in NSCLC.
METHODS
Study Design and Patients
This prospective observational study recruited patients with stage I to III NSCLC (tumor diameter ≥2 cm) treated with definitive surgery from March 2019 to January 2021 at the Guangdong Provincial People's Hospital and Guangdong Lung Cancer Institute, with or without neoadjuvant or adjuvant therapy (supplementary protocol). This study was approved by the Institutional Review Board of the Guangdong Provincial People's Hospital [approval no. 2018319H (R1)]. Written informed consent was obtained from all patients.
cfDNA Panel Design
A 338-gene panel (Supplementary Table S2) covering a 550-Kbp genome was constructed specifically for lung cancer, colorectal cancer, and hepatocellular carcinoma, using a panel design concept similar to that of CAPP-seq (6). The most common driver mutations across all three cancer types were considered first. Then, actionable sensitive and resistant mutations or gene alterations related to the effects of immunotherapy were added. Finally, frequently mutated regions were selected based on cancer data sets from 48,353 Geneplus-sequenced patients with cancer and other open-access databases, such as COSMIC (http://cancer.sanger.ac.uk/cosmic) and TCGA (https://cancergenome.nih.gov/). The final panel with minimal size maximized the coverage of mutations in the intentional cancer population, contributing to economic ultra-deep sequencing.
Sample Collection
We collected 20 mL of peripheral blood in two 10-mL Streck tubes before and after surgery, at preset time points. Preoperative blood samples were collected up to 3 days before surgery. The landmark time point was defined as 1 month (±7 days) after surgery for patients who did not receive adjuvant therapy. For patients who received adjuvant chemotherapy, the preadjuvant time point was defined as at least 1 week after surgery and before adjuvant therapy, and the landmark time point was defined as 1 month (±7 days) after the last cycle of chemotherapy. For patients who received adjuvant long-term EGFR inhibitors or ICIs, the landmark time point was defined as 1 month (±14 days) after surgery and before the adjuvant therapy. Longitudinal time points were defined as every 3 to 6 months since the landmark detection. The surveillance time point was defined as within 6 months of clinical recurrence. Because imaging follow-up and blood sample collection were prospectively performed during the same period, the surveillance time point could be the same day as clinical recurrence.
To further confirm the nonshedding of lung tumors before surgery, another 11 patients with stage I NSCLC were enrolled from July 2021 to August 2021. During surgery, 5 to 10 mL of blood was immediately collected in a Streck tube through the pulmonary vein stump of the resected lung lobe (Fig. 2F).
Sample Processing and DNA Extraction
Peripheral blood samples were collected in Streck tubes. Within 3 days, the sample was separated by centrifugation at 1,600 × g for 10 minutes, and the supernatant was transferred to microcentrifuge tubes, centrifuged again at 16,000 × g for 10 minutes to remove cell debris, and then frozen at −80°C. Circulating cfDNA was extracted from 2 to 10 mL (median, 7 mL) of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen).
Germline genomic DNA was isolated from peripheral blood lymphocytes (PBL) using the QIAamp DNA Blood Mini Kit (Qiagen). Matched tumor DNA was extracted from fresh frozen tissues or formalin-fixed, paraffin-embedded (FFPE) tumor tissue specimens using the QIAamp DNA Mini Kit (Qiagen) and ReliaPrep FFPE gDNA Miniprep System (Promega), respectively. The concentration and fragment length of cfDNA were determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.).
Library Construction
For germline genomic DNA and tumor DNA, 400 to 800 ng DNA was sheared into fragments at a 200 to 250 bp peak with a Covaris S2 ultrasonicator (Covaris, Inc), and indexed NGS libraries were prepared using NEBNext Ultra DNA Library Prep Kit for Illumina (NEB). cfDNA (median, 52 ng) was used for library construction, and unique identifiers (UID) were tagged on each double-stranded DNA to distinguish authentic somatic mutations from artifacts, improving the ability to precisely track individual plasma molecules.
Target Region Capture and NGS
For tumor genomic and matched germline DNA libraries, a previously reported custom-designed panel (Integrated DNA Technologies, Inc.) covering ∼1.5 Mbp of the genome and targeting 1,021 cancer-related genes was used for hybridization enrichment (31). Plasma and its paired genomic germline DNA libraries were hybridized to another custom-designed biotinylated oligonucleotide probe (Integrated DNA Technologies) covering 550 kb of human sequences. The indexed libraries were sequenced using a 100-bp paired-end configuration on a DNBSEQ-T7RS sequencer (MGI Tech) or Gene+Seq-2000 sequencing system (GenePlus-Suzhou), producing 5, 40, and 5 Gb data for PBLs, plasma, and fresh specimens/FFPE, respectively.
Raw Data Processing
The sequenced reads were mapped to the reference human genome (GRCh37) using the default parameters in BWA version 0.6.2 after removing adaptor and low-quality reads. Duplicated reads were marked and removed using MarkDuplicates tool in Picard (version 4.0.4.0; Broad Institute) for tumor and germline genomic DNA. For cfDNA, duplicated reads were identified by UID and the position of template fragments to eliminate errors introduced by PCR or sequencing using realSeq (v3.1.0 Geneplus-Beijing, inhouse). Local realignment around SNVs and indels, as well as quality control assessment, were performed using GATK (version 3.4.46; Broad Institute).
Tumor Somatic Variant Calling
Tumor somatic SNVs and small indels were profiled using realDcaller (v1.8.1 Geneplus-Beijing, in-house) and TNscope (v3.8.0 Sentieon Inc.). CNVKit was used to detect copy-number alterations. The structural variations were analyzed using the self-developed algorithm NCsv (version 0.2.3 Geneplus-Beijing, inhouse). Detailed variant calling and filter strategies were reported previously (32). Tumor tissues were available for 260 patients and sequenced to a median depth of 1,214 ×, with a median of six mutations (range, 1–93) detected.
ctDNA-MRD Detection
After sequencing errors were polished by UID, SNV calling was performed using a custom bioinformatics pipeline optimized for ultra-low-frequency mutation calling. SNV and indel calling was carried out mainly by realDcaller, while TNscope (Sentieon Inc.) was used as auxiliary software to improve the detection of long indels. Upon annotation completion, variants were filtered according to the following criteria: (i) the variants present in matched genomic DNA were removed; (ii) the single-nucleotide polymorphisms at >1% population allele frequency in ExAc or 1000 Genomes Project were filtered; (iii) the variant positional depth was at least >300 ×, and (iv) for background error removal, a set of ∼500 healthy individual plasma samples were sequenced to construct a background estimate VAF distribution model for each target SNV.
Patients with available tumor tissues were sequenced to identify their specific tumor variants. Based on whether the allele was identified in matched tumor tissue, two different methods were used to call plasma cfDNA variants. For tissue-derived variants in plasma, the variants showed a statistically significant difference in background errors, which was considered reliable. Meanwhile, tumor-specific driver mutations required at least two good support reads, and for other nonrecurrent variants, a minimum supporting read of four. For cfDNA variants not occurring in matched tumor tissue, if the following stringent conditions were met, they were considered to be true somatic mutations: (i) for hotspot mutations, ≥4 high-quality support reads, or for non-hotspots, at least ≥8 support reads; and (ii) clonal hematopoiesis were filtered through deep sequencing of paired white blood. These test performance validation experiments have been conducted on standards and validated at 30 ng DNA input amount; the limit of detection achieves 0.1% with analytic sensitivity at 97.9% for SNV or small indel detection. The approach also values these non–tumor-derived mutations due to tumor heterogeneity, tumor evolution, and resistance mutations. A plasma sample with at least one variant detected was defined as ctDNA-positive (Supplementary Fig. S3).
Statistical Analysis
The primary outcomes of the study were the detection of MRD and DFS, as assessed by standard radiographic imaging. DFS was measured from the day of definitive surgery to the first radiographic recurrence or death. Analysis of the PPV and NPV was completed for patients with at least half a year of follow-up since the first detectable or undetectable MRD. The Kaplan–Meier method was used to describe the survival outcomes. A log-rank test was used for hazard ratios, and all P values were based on two-sided testing with statistically significant differences at P ≤ 0.05. One-to-one propensity score matching was applied to reduce selection bias in patients with undetectable MRD who received adjuvant therapy. Dynamic hazard rates of detectable MRD or disease recurrences were assessed to provide a temporal pattern of MRD monitoring; methods have been described in detail elsewhere (33). The overall analysis process is shown in Fig. 1. Statistical analysis was performed using SPSS 26.0 (IBM Corp.) and GraphPad PRISM 8.0. (GraphPad Software).
Data Availability
Sequencing data required to reproduce these findings have been deposited in the EVA database (accession number: PRJEB52694).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We are grateful to the patients and families involved in this study. This work was supported by the Key Lab System Project of Guangdong Science and Technology Department, Guangdong Provincial Key Lab of Translational Medicine in Lung Cancer (Grant No. 2017B030314120 to Y.-L. Wu), Guangdong Provincial People's Hospital Scientific Research Funds for Leading Medical Talents in Guangdong Province (Grant No. KJ012019426 to Y.-L. Wu), Guangdong Provincial People's Hospital Young Talent Project (Grant No. GDPPHYTP201902 to W.-Z. Zhong, Grant No. KY012021189 to S.-Y. Liu) and China Postdoctoral Science Foundation (Grant No. 2021M701422 to S.-Y.M. Liu). The funding sources had no role in the preparation of this manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
X.-C. Zhang reports personal fees from Burning Rock, Inc., CSTONE Pharmaceuticals, Inc., Novartis, AstraZeneca, and Illumina outside the submitted work. Q. Zhou reports other support from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, MSD, Pfizer, Roche, and Sanofi outside the submitted work. W.-Z. Zhong reports other support from AstraZeneca, Bristol Myers Squibb, MSD, Roche, and Innovent outside the submitted work. Y.-L. Wu reports grants and personal fees from AstraZeneca and Boehringer Ingelheim, grants from Bristol Myers Squibb, and personal fees from Beigen, Eli Lilly, MSD, Hengrui, Pfizer, Roche, and Sanofi outside the submitted work. No disclosures were reported by the other authors.
Authors’ Contributions
J.-T. Zhang: Formal analysis, writing–original draft. S.-Y. Liu: Conceptualization. W. Gao: Data curation, software, writing–original draft. S.-Y.M. Liu: Writing–review and editing. H.-H. Yan: Data curation, formal analysis, supervision. L. Ji: Formal analysis. Y. Chen: Methodology. Y. Gong: Resources, data curation. H.-L. Lu: Data curation. J.-T. Lin: Data curation. K. Yin: Data curation. B.Y. Jiang: Data curation. Q. Nie: Data curation. R.-Q. Liao: Data curation. S. Dong: Investigation. Y. Guan: Resources. P. Dai: Resources. X.-C. Zhang: Methodology. J. Yang: Investigation. H.-Y. Tu: Investigation. X. Xia: Resources, methodology. X. Yi: Resources, supervision. Q. Zhou: Supervision. W.-Z. Zhong: Supervision, funding acquisition. X.-N. Yang: Supervision. Y.-L. Wu: Supervision, funding acquisition, writing–review and editing.
References
- 1. Pignon JP, Tribodet H, Scagliotti GV, Douillard JY, Shepherd FA, Stephens RJ, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol 2008;26:3552–9. [DOI] [PubMed] [Google Scholar]
- 2. Burdett S, Rydzewska L, Tierney JF, Fisher DJ, PORT Meta-analysis Trialist Group. A closer look at the effects of postoperative radiotherapy by stage and nodal status: updated results of an individual participant data meta-analysis in non–small-cell lung cancer. Lung Cancer 2013;80:350–2. [DOI] [PubMed] [Google Scholar]
- 3. Le Pechoux C, Pourel N, Barlesi F, Faivre-Finn C, Lerouge D, Zalcman G, et al. LBA3_PR An international randomized trial, comparing postoperative conformal radiotherapy (PORT) to no PORT, in patients with completely resected non–small cell lung cancer (NSCLC) and mediastinal N2 involvement: primary end-point analysis of LungART (IFCT-0503, UK NCRI, SAKK) NCT00410683. Ann Oncol 2020;31:S1178. [Google Scholar]
- 4. Wu YL, Tsuboi M, He J, John T, Grohe C, Majem M, et al. Osimertinib in resected EGFR-mutated non–small-cell lung cancer. N Engl J Med 2020;383:1711–23. [DOI] [PubMed] [Google Scholar]
- 5. Felip E, Altorki N, Zhou C, Csőszi T, Vynnychenko I, Goloborodko O, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet 2021;398:1344–57. [DOI] [PubMed] [Google Scholar]
- 6. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zviran A, Schulman RC, Shah M, Hill STK, Deochand S, Khamnei CC, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat Med 2020;26:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chae YK, Oh MS. Detection of minimal residual disease using ctDNA in lung cancer: current evidence and future directions. J Thorac Oncol 2019;14:16–24. [DOI] [PubMed] [Google Scholar]
- 11. Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol 2019;16:409–24. [DOI] [PubMed] [Google Scholar]
- 12. Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non–small-cell lung cancer. Nat Cancer 2020;1:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu B, Guo W, Zhang F, Lv F, Ji Y, Peng Y, et al. Dynamic recurrence risk and adjuvant chemotherapy benefit prediction by ctDNA in resected NSCLC. Nat Commun 2021;12:6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol 2018;15:577–86. [DOI] [PubMed] [Google Scholar]
- 16. Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 2021;595:432–7. [DOI] [PubMed] [Google Scholar]
- 17. Chouaid C, Danson S, Andreas S, Siakpere O, Benjamin L, Ehness R, et al. Adjuvant treatment patterns and outcomes in patients with stage IB-IIIA non-small cell lung cancer in France, Germany, and the United Kingdom based on the LuCaBIS burden of illness study. Lung Cancer 2018;124:310–6. [DOI] [PubMed] [Google Scholar]
- 18. Buck PO, Saverno KR, Miller PJ, Arondekar B, Walker MS. Treatment patterns and health resource utilization among patients diagnosed with early stage resected non-small cell lung cancer at US community oncology practices. Clin Lung Cancer 2015;16:486–95. [DOI] [PubMed] [Google Scholar]
- 19. Wu YL, John T, Grohe C, Majem M, Goldman JW, Kim SW, et al. Postoperative chemotherapy and outcomes from ADAURA: osimertinib as adjuvant therapy for resected EGFR-Mutated NSCLC. J Thorac Oncol 2021;17:423–33. [DOI] [PubMed] [Google Scholar]
- 20. Garcia-Murillas I, Chopra N, Comino-Méndez I, Beaney M, Tovey H, Cutts RJ, et al. Assessment of molecular relapse detection in early-stage breast cancer. JAMA Oncol 2019;5:1473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li YS, Jiang BY, Yang JJ, Zhang XC, Zhang Z, Ye JY, et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann Oncol 2018;29:945–52. [DOI] [PubMed] [Google Scholar]
- 22. De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun 2015;6:8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seoane J, De Mattos-Arruda L, Le Rhun E, Bardelli A, Weller M. Cerebrospinal fluid cell-free tumour DNA as a liquid biopsy for primary brain tumours and central nervous system metastases. Ann Oncol 2019;30:211–8. [DOI] [PubMed] [Google Scholar]
- 24. Dudley JC, Schroers-Martin J, Lazzareschi DV, Shi WY, Chen SB, Esfahani MS, et al. Detection and surveillance of bladder cancer using urine tumor DNA. Cancer Discov 2019;9:500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 2019;25:1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thompson JC, Yee SS, Troxel AB, Savitch SL, Fan R, Balli D, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res 2016;22:5772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang J, Adams HP, Yao L, Yaung S, Lal P, Balasubramanyam A, et al. Concordance of genomic alterations by next-generation sequencing in tumor tissue versus cell-free DNA in stage I–IV non–small cell lung cancer. J Mol Diagn 2020;22:228–35. [DOI] [PubMed] [Google Scholar]
- 29. Guo Q, Wang J, Xiao J, Wang L, Hu X, Yu W, et al. Heterogeneous mutation pattern in tumor tissue and circulating tumor DNA warrants parallel NGS panel testing. Mol Cancer 2018;17:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jamal-Hanjani M, Wsilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non–small-cell lung cancer. N Engl J Med 2017;376:2109–21. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Yao Y, Xu Y, Li L, Gong Y, Zhang K, et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat Commun 2021;12:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin G, Li C, Li PS, Fang WZ, Xu HP, Gong YH, et al. Genomic origin and EGFR-TKI treatments of pulmonary adenosquamous carcinoma. Ann Oncol 2020;31:517–24. [DOI] [PubMed] [Google Scholar]
- 33. Xu ST, Xi JJ, Zhong WZ, Mao WM, Wu L, Shen Y, et al. The unique spatial-temporal treatment failure patterns of adjuvant gefitinib therapy: a post hoc analysis of the ADJUVANT trial (CTONG 1104). J Thorac Oncol 2019;14:503–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data required to reproduce these findings have been deposited in the EVA database (accession number: PRJEB52694).