

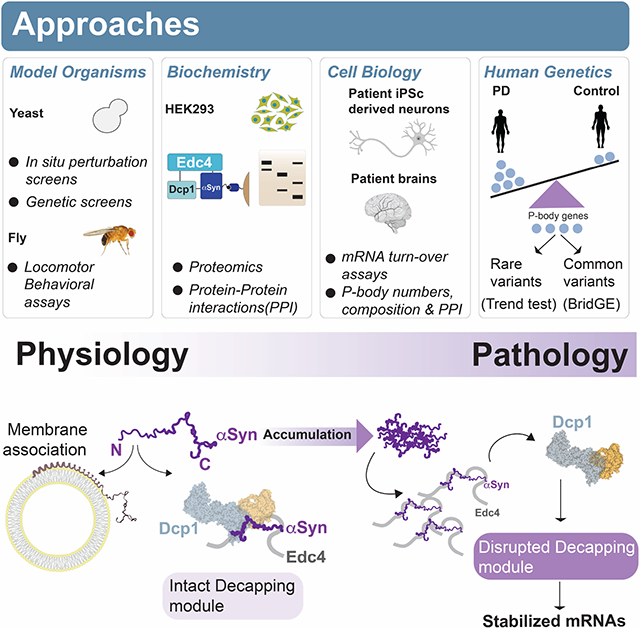
SUMMARY
Alpha-synuclein (αS) is a conformationally plastic protein that reversibly binds to cellular membranes. It aggregates and is genetically linked to Parkinson’s disease (PD). Here, we show that αS directly modulates Processing-bodies (P-bodies), membraneless organelles that function in mRNA turnover and storage. The N-terminus of αS, but not other synucleins, dictates mutually exclusive binding either to cellular membranes or to P-bodies in the cytosol. αS associates with multiple decapping proteins in close proximity on the Edc4 scaffold. As αS pathologically accumulates, aberrant interaction with Edc4 occurs at the expense of physiologic decapping-module interactions. mRNA-decay kinetics within PD-relevant pathways are correspondingly disrupted in PD patient neurons and brain. Genetic modulation of P-body components alters αS toxicity, and human genetic analysis lends support to the disease-relevance of these interactions. Beyond revealing an unexpected aspect of αS function and pathology, our data highlight the versatility of conformationally plastic proteins with high intrinsic disorder.
Graphical Abstract
In Brief
Alpha-synuclein modulates mRNA stability to regulate gene expression with implications for understanding both normal cellular physiology and vulnerability to Parkinson’s disease and related disorders.
INTRODUCTION
Parkinson’s Disease (PD) is the most common neurodegenerative movement disorder (Marras et al., 2018). PD is a multisystem disease associated with a plethora of motor and nonmotor manifestations. Pathologically, these phenotypes are associated with widespread intraneuronal protein aggregates called Lewy bodies (LBs) from the gut to the neocortex. (Goedert et al., 2012; Shulman et al., 2011). The first familial PD cases were attributed to a point mutation (A53>T) at the SNCA locus (Polymeropoulos et al., 1997) encodes a 14kDA protein alpha-synuclein (αS) that was identified as the major component of LBs in PD and related “synucleinopathies” (Spillantini et al., 1997). Notably, human genetic studies also suggested an exquisite sensitivity of neurons to the levels of wild-type αS protein: multiplication of the wild-type SNCA gene locus causes early-onset forms of PD and dementia (Singleton et al., 2003) and common polymorphisms that marginally increase SNCA expression are sufficient to increase risk for late-onset PD(Chang et al., 2017; Soldner et al., 2016).
The function of αS remains elusive. It is vertebrate-specific and highly expressed in neurons, reaching micromolar concentrations in synaptic boutons (Wilhelm et al., 2014). It adopts an amphipathic helical structure when it associates with phospholipid membranes at the cytosolic face of vesicles (Auluck et al., 2010). The most agreed-upon functions of αS include a regulatory role in vesicular trafficking and vesicle fusion (Hunn et al., 2016), chaperoning function for synaptic SNARE complexes(Burré et al., 2018) and in mitochondrial and lysosomal homeostasis(Nguyen et al., 2019). However, αS is found in cell compartments beyond the synapse or membrane, including the nucleus(Kontopoulos et al., 2006; Schaser et al., 2019) and soma (Pei and Maitta, 2019). Diverse biophysical forms are described, ranging from a soluble disordered form to dynamic multimers and self-templating amyloid fibrils (Dettmer et al., 2016; Jarosz and Khurana, 2017). αS knockout mice have relatively modest neurophysiologic deficits and normal lifespan, perhaps reflecting redundancy with two other synuclein family members, beta- and gamma-synuclein, neither of which is definitively associated with PD(Cabin et al., 2002; Greten-Harrison et al., 2010). Moreover, αS is highly expressed in several non-neuronal tissues such as blood, kidney, and adipose tissue(Bozic et al., 2020; Locascio et al., 2015; Uhlen et al., 2015). These findings suggest as-yet-undiscovered functions and interactions for αS.
Previously, we demonstrated considerable overlap between genetic modifiers of αS cytotoxicity and its neighboring proteome in neurons(Chung et al., 2013; Khurana et al., 2017) , thus tying the physiologic function of αS to the toxicity that results from its accumulation (Lam et al., 2020). Beyond vesicle trafficking, the most frequent proteins in these αS “maps” were RNA-binding proteins (RBPs) and ribosomal subunits. While human genetic studies in PD have consistently implicated vesicle-trafficking genes, most of the heritability of PD is as yet unexplained. Larger common and rare-variant analyses(Bandres-Ciga et al., 2020; Chartier-Harlin et al., 2011; Siitonen et al., 2017) increasingly implicate diverse pathways in the disease, including mRNA metabolism(Chang et al., 2017) but the biochemical basis of these genetic risk modifiers and whether these seemingly disparate pathways are inter-connected remains unclear.
Here, we report that αS modulates Processing body (P-body) complexes and mRNA stability in the cytosol. P-bodies are a major hub for RNA metabolism in eukaryotic cells. They comprise RNA and RNA-binding proteins (RBPs) involved in mRNA decapping and degradation, mRNA storage and RNAi silencing (Corbet and Parker, 2020). We find that αS interacts closely with a core set of proteins (Edc4, Dcp1, Dcp2, Xrn1, Edc3) involved in mRNA-decapping and degradation. This interaction is dictated by the N-terminus of αS, the same region involved in membrane anchoring. αS thus dichotomously interacts with either membranes or P-body components. Of these components, αS interacts most strongly with Edc4, the decapping-module scaffold. When αS levels increases to pathological levels, Edc4 increasingly associates with αS, compromising interactions with other decapping-module proteins. This is seen in human neurons harboring familial synucleinopathy mutations, both iPSC-derived and in postmortem brain. Human genetic analyses further tie this pathway to PD risk. In human neurons that express pathologic levels of αS or that are seeded with αS fibrils, mRNAs are globally stabilized. Our data suggest how altered αS membrane interactions and pathology might directly impact other key cytosolic functions, including mRNA metabolism. More broadly, these data shed light on mechanisms through which conformationally plastic proteins like αS can mediate cross-compartment signaling in cells.
RESULTS
P-body genes modify αS toxicity in tractable model organisms
Prior screens in cellular PD models tied αS to multiple RNA-binding protein (RBP) networks (Chung et al., 2017; Khurana et al., 2017). To determine which pathways were most perturbed by αS toxicity and how specific these perturbations were to αS compared to other neurodegeneration (ND) related proteins, we turned to a well-controlled screenable yeast platform called RBP-yTRAP (yeast Transcriptional Reporting of Aggregating Proteins) (Newby et al., 2017). Here, the perturbation on any RBP emerges as a fluorescent output through a synthetic transcription factor. Upon a genetic or chemical challenge, the yTRAP platform measures perturbations of 168 RBPs in intact cells by high-throughput flow cytometry (Fig.1A, Star Methods).
Figure 1. Processing body genes are modifiers of αS mediated toxicity in yeast and Drosophila.

(A) Logic of RBP perturbation screen. The RBP-yTRAP platform (left) is crossed to ND protein expressing yeast (right). Upon induction of ND protein, the mNeonGreen output signal was quantitated by flow cytometry. ZnFs for ZEM module and yTRAP module are different. (AD: Activator domain, ZnF: ZincFinger, RBP: RNA-binding protein, ND: neurodegeneration)
(B) Perturbation screen results. Y axis: Fluorescence linear fold change (induction of ND protein versus no induction), X-axis: RBP sensors (3 biological replicate screens; mean, sd). Four of the most perturbed proteins upon αS induction are marked and the inlet shows the graph for three of these proteins (mean, sd, t-test, ***p<0.001, ****p<0.0001).
(C, D, E) αS toxicity modifier mini-screens guided by Sgn1 yTRAP signal. 39 genes for overexpression/deletion and 24 genes for yTRAP screen. (C, D) Representative plate growth assays of αS toxicity modifiers. (Dotted line indicate a cut within the same plate). (E) yTRAP assay showing αS specific significant perturbation signals (in comparison to Fus) for 11 genes. (n=4 biological replicates, mean, sd, p<0.0005 for all 11 genes, multiple comparison corrected among 24 tested genes). For all the 24 genes tested, see Supp.Fig1H.
(F) Sgn1 guided mini-screens for αS toxicity modifiers converge on complexes within P-body granules. Miniscreens are color-coded.
(G-H) Locomotor based forward genetic screen in flies.(G) RT-qPCR validation of XRN1 knockdown in brains of two RNAi stocks (n=6, mean± sem, two-tailed t-test). (H) Locomotor activity measurements (mean± sd) of flies. Data were analyzed using one-way ANOVA with Tukey post-hoc analysis (n=6).
To detect RBP perturbations in response to ND proteins, we generated yeast strains that express untagged versions of either αS, Tdp-43, Fus and Sod14AV under an estradiol induction system. Fus, Tdp-43 and SOD1 are all implicated in ALS, and Fus and Tdp-43 yeast models have proved informative for understanding aspects of ALS biology (Fushimi et al., 2011; Hayden et al., 2020; Ju et al., 2011). Acute induction of αS, Fus and Tdp-43 each resulted in marked toxicity. Fus-toxicity was matched to that of αS (Supp.Fig.1A) and thus served as a control for ND specifivity. Expression of Sod14AV was non-toxic (Supp.Fig.1A) and was used as a non-toxic control. We crossed the yeast ND models to the yTRAP platform and measured RBP perturbations upon estradiol induction of each ND protein (Fig.1A), performed in biological triplicate after optimization (Supp.Fig.1B,C). Surprisingly, the largest changes in yTRAP sensors were induced by αS expression and not by Fus (Fig.1B). Despite equivalent cytotoxicity of αS and Fus, the respective yTRAP signals were highly distinct (Supp.Fig.1D), implying that the consequences of ND proteotoxicity on RBP biology are highly specific.
Rrt5 and Sgn1 were the most perturbed RBPs upon αS toxicity (Fig.1B, p<0.001). Rrt5 is under-studied with no clear human homolog (Hontz et al., 2009). Sgn1 is a cytoplasmic polyA-binding protein (PABP) family member, which physically and genetically interacts with Pab1/Pabpc1 and translation initiation factors Tif4631/2 (Winstall et al., 2000). Guided by Sgn1’s immediate connection to the polyA tail and translation initiation, we chose 39 genes (TableS1) connected to polyA-tail turnover, translation initiation and mRNA-cap biology and performed targeted-overexpression, deletion and yTRAP screen in the αS toxicity model (Fig.1C-E). In the over-expression screen, eight suppressors of αS toxicity were recovered, including Pab1 and Tif4631/2, known from previous screens (Khurana et al., 2017), but among these suppressors, Pan2/3 polyA ribonucleases were the strongest (Fig.1C, Supp.Fig1E). Notably, these modifiers did not appreciably change αS protein levels (Supp.Fig.1G) and none of them rescued Fus toxicity (Supp.Fig 1F). Dcp2, Not3 and Lsm7 emerged as αS toxicity modifiers in the deletion screen (Fig.1D). The focused yTRAP screen revealed several proteins were specifically perturbed by αS expression (and not by Fus): Edc1 and 2, Not1,3,5,Ccr4 (Ccr-Not complex member), Eif4e (cap-binding protein), Lsm4,6 (Lsm Ring) and the Pan2 nuclease (Fig.1E, Supp.Fig.1H). Taken together, our strongest genetic and yTRAP hits are all involved in regulation of mRNA degradation (summarized in Fig. 1F). In eukaryotes, these machinaries (polyA-tail shortening, decapping, enhancers of decapping and deadenylation) localize to membraneless organelles known as Processing bodies or P-bodies (Decker and Parker, 2012).
To verify this connection in a metazoan model organism with a complex nervous system, we turned to a Drosophila melanogaster synucleinopathy model, in which αS expression in the brain elicits neurodegeneration and loss of locomotor activity(Feany and Bender, 2000; Olsen and Feany, 2019). A recent screen of these phenotypes performed at genome-scale (~5000 genes, Dey and Feany, in preparation) included 34 P-body related genes. Of nine modifier-hits in this pathway, our attention was drawn to a strong hit, the key P-body 5’>3’ ribonuclease PCM/XRN1. We proceeded to secondary validation experiments for this gene (Fig. 1G). Two independent RNAi stocks validated that brain-specific knock down of XRN1 exacerbated the locomotor deficiency of αS expressing flies. The severity of this phenotype correlated with KD efficiency (Fig.1H), and no change in αS protein levels accompanied this modification (Supp. Fig. 1I). Thus, P-body components involved in mRNA degradation are strong and specific modulators of αS-mediated toxicity and this effect is evolutionarily conserved.
αS physically interacts with P-body components with a high affinity towards Edc4
To understand the biochemical basis of these genetic interactions, we turned to HEK293 cells, which endogenously express αS(Lee and Kamitani, 2011). We first epitope-tagged αS with GFP to avoid conformational recognition bias with antibodies (Supp.Fig.2A). We C-terminally tagged αS as the N-terminus is important for its membrane interactions(Fusco et al., 2018). We measured αS-specific protein interactions with quantitative SILAC mass spectrometry (MS) after GFP pulldown in αS-GFP versus control GFP stable lines (Fig.2A,B). Of the top 29 αS interactors (Fig.2C; TableS2), four P-body proteins (Edc3, Edc4, Dcp1, Dcp2) were recovered. These proteins form the ‘decapping module’, which removes the 5’ cap from mRNA (by Dcp2), enabling subsequent 5’ to 3’ mRNA degradation by Xrn1. Other top hits included proteins previously tied to αS and PD biology, such as VapA and VapB (Paillusson et al., 2017).
Figure 2. αS physically interacts with Processing-Body components, specifically the decapping module.

(A) SILAC mass spectrometry (MS) to identify αS-GFP associated proteins.
(B) Expression levels of αS-GFP and GFP-alone control in whole cell lysates (The mid lane is cut).
(C) Decapping module proteins of P-bodies were enriched in αS-GFP pulldowns. Right panel: 29 enriched proteins (αS-GFP log2FC>1 over GFP alone) are re-drawn with adjusted p-values on x-axis (BH correction, p< 0.05).
(D) In-vivo biotin tagging of αS by BirA and isogenic HEK293 cell line generation by FRT integration.
(E) Endogenous P-body components are enriched upon biotinylated αS pulldown. G3BP1 and EIF4E are SG markers. GAPDH and VCP are abundant control proteins.
(F-G) Silver staining of biotinylated αS-Avi pulldowns along with control cell lines showing purity and specificity. The 150 kDA band is Edc4 as revealed by mass-spectrometry. (G) Stable knock-out of Edc4 in stable lines expressing in situ biotinylated αSyn (mock is a non-targeting guide)
(H)Input normalized enrichment of N-terminally nanoluc tagged P-body proteins after biotinylated αSyn pulldown. (n=3, mean,sem). DTL gene is a non P-body control gene.
(I) αSyn and Edc4 interact In vitro in baculovirus mediated Sf9 insect cell expression systems.
(J) Biotinylated αS pulldowns in stable DDX6 knock-out background. Up: IF of P-body markers, Ddx6 and Edc4. Below: αS-Avi pulldown with streptavidin beads (mock;non-targeting gRNA). 2 separate gRNAs were used for stable KO of Ddx6.
The large size of GFP (27 KDa) relative to αS (14 KDa) might alter its interactions. We thus used the small Avi tag (15aa), which can be biotinylated by E.coli BirA ligase (Ioannou et al., 2018). We generated constructs that co-expressed αS-Avi and BirA ligase and stably inserted them into a single FRT site (Fig.2D, HEK293 Flp-In lines). Streptavidin pulldown from extracts yielded highly pure and biotinylation-specific αS enrichments (Supp.Fig.2B, Fig.2F). Probing of endogenous P-body components revealed Edc4, Dcp1, Dcp2, Edc3 co-purifications and slight enrichments for Ddx6 and Lsm14A (Fig.2E). In contrast, the core stress granule components G3bp1 and Eif4e did not co-purify with αS (Fig.2E). Interactions between Edc3 or Edc4 and αS were able to withstand 0.5M salt and RNAse digestions. In contrast, interaction with the RNA helicase Ddx6 was rapidly lost in these conditions (Supp.Fig.2C, D).
In our αS pulldowns, Edc4 showed consistently the highest enrichment among decapping-module proteins and, along with αS, it had the highest peptide count in SILAC MS (Supp.Fig.2E). Silver staining of αS-Avi pulldowns (Fig.2F) revealed a strong 150 KDa band (the expected size of Edc4) which was lost in an EDC4 null background (Fig.2G). Mass-spectrometry analysis confirmed its identity (TableS2). To determine if this affinity was unique to Edc4 among P-body proteins, we tagged ~50 P-body proteins by nanoluciferase and quantified their interactions with αS under over-expression conditions. Multiple interactions were noted (such as Dcp2, Tnrc6a) but Edc4 was the strongest (Fig.2H). We co-expressed αS and Edc4 in Sf9 insect cells with baculovirus-mediated expression. αS could pull down Edc4 in this In vitro system (Fig.2l). αS thus physically associates with decapping-module proteins and, among these, has highest affinity towards Edc4.
Condensed P-body granules are not required for αS interactions
Proteomic analysis of large, condensed P-body granules did not identify αS as a component (Hubstenberger et al., 2017). Furthermore, our immunofluorescence (IF) studies failed to localize αS within microscopically visible “macro” P-bodies (not shown). Interactions between αS and decapping interactions could occur in sub-diffraction limited cytosolic complexes, soluble pools of P-body proteins, or “micro P-bodies.” We fractionated macro and micro P-bodies according to the Hubstenberger protocol (Supp.Fig2F). As expected, decapping proteins were recovered in macro P-bodies. However, αS and the majority of the decapping proteins co-fractioned with micro P-bodies even after solubilization with nucleases (Supp.Fig.2G). When we knocked out Ddx6, essential for P-body condensation(Stefano et al., 2019) (Fig. 2J), condensed macro P-bodies did not form. However, Edc4 and Edc3 protein levels were unchanged (Fig.2J) and αS still interacted with P-body components under these conditions (Fig.2J). Thus, αS associates with the soluble decapping modules.
The αS N-terminus dictates dichotomous interactions with membranes or P-body proteins
We investigated how affinity of αS towards membranes affects its decapping-module interactions. Upon contact with membranes, αS rapidly forms alpha-helices throughout its N-terminal lipid-binding domain (~1-100 aa, NLD) and the acidic C-terminal tail projects from the membrane as a random coil engaged in protein interactions (Snead and Eliezer, 2019) (Fig.3A). Serial truncations of C-terminus of αS did not impair decapping interactions (Supp. Fig3A) so we generated sequential 10 aa deletions of αS spanning the NLD. There was a marked loss in decapping-module binding when the first or second 10 aa segment of αS was deleted (Fig. 3B-C). As the first 20 aa of αS is responsible for its initial anchoring to membranes and propagation of the helical structure along the NLD (Fusco et al., 2014), we hypothesized that the N-terminus of αS dictates a dichotomous binding: αS interacts with either membranes or the decapping module. To investigate this prediction, we performed a thorough mutational analysis of αS.
Figure 3. The N-terminus of αS dichotomously interacts either with decapping-module proteins or with membranes.

All experiments were performed by transient transfection of a single plasmid containing αS-Avi mutant co-expressing BirA ligase in HEK293 cells. αS pulldown was performed by Streptavidin beads and probed for P-body proteins.
(A) A schematic of αS on a bilayer membrane with its acidic C-terminus projecting away from the membrane.
(B) 10 aa deletion-scan of αS-Avi in HEK293 cells in lipid binding domain(1-100 aa). Note the absence of P-body interactions in Δ11-20 aa mutant.
(C) The first 20 aa residues of αS are essential for decapping module interactions (quantified interactions, n=3, mean,sem).
(D) Logic of membrane-averse or membrane-avid stratification of αS surface mutants. Up: αS 11 aa imperfect repeats and the helical view of αS on the right. Below: Familial point mutations in different surfaces are bolded. Combined surface mutants are rationally designed to amplify the effect of the single familial mutation.
(E) Membrane-averse αS G510D and A30P familial mutations (and their extended surface mutants 10 and 11, respectively) do not affect P-body interactions.
(F) Membrane avid αS mutants abrogate P-body interactions (see surface3 and 5).
(G) Increasing membrane avidity of αS by mutating Surface3 threonines to leucine completely diminish decapping module binding.
(H) Non-membrane-bound αS mutant (T44P and A89P) interacts with decapping module interactions through its first 10 aa.
(I) Among synuclein family members, only αS interacts with P-body components.
(J) Replacement of gamma synuclein residues in the first 20aa to alpha synuclein impairs decapping binding.
(K) Proposed model for dichotomous interaction of αS with membranes or P-body components.
Familial PD point mutations in the αS N-terminal sequence alter its membrane-binding propensity, either increasing (E46K) or decreasing (A30P, G51D) affinity (Dettmer, 2018). The structural basis of this membrane avidity is due to the imperfect repeats of 11 aa motifs in the N-terminus(Das and Eliezer, 2019). These repeats topologically align with αS’s 11-3 helicity (11 aa in 3 turns) on the membrane (Fig. 3D, “wheel” representation). Thus, composite point mutations can be rationally designed along αS’s helix surface to exaggerate the biophysical phenotype exerted by the single familial PD point mutations (Dettmer, 2018). We found that the interaction with the decapping module was preserved in the membrane-averse A30P and G51D mutants, as well as their rationally extended surface mutants (Fig. 3E). However, while individual E>K mutants (E35K, E46K, E61K) did not influence P-body binding, the composite highly membrane-avid 3K mutant abrogated it (Fig. 3F, surface3, Lane11).
While no PD mutations have been found in threonine residues deep in the bilayer (Fig.3D, Surface3), substitution of these threonines with hydrophobic (high membrane-affinity) leucines (Perrin et al., 2000) resulted in a complete loss of decapping-module interactions (Fig. 3F; Lane12). A double T>L mutant (T59L, T81L) was sufficient to replicate this loss (Fig.3G) but when exchanged with membrane-repelling hydrophilic residues (T>E and T>K), decapping-module binding was restored to wild-type levels (Supp.Fig.3C). Thus, membrane-bound αS does not appear to interact with decapping module proteins.
It is formally possible that αS requires the N-terminus only to bind to membranes and undergo a conformational change, which in turn allows decapping-module binding in the cytosol. To exclude this possibility, we utilized a mutant of αS that never binds membranes (T44P, A89P) (Burré et al., 2015)and expressed it with or without the first 10 aa (Δ2-10). We observed that this mutant interacted with decapping components but completely lost this interaction when lacking the first 10aa (Fig.3H). Our data suggest that the αS N-terminus directly binds to the decapping-module away from phospholipid membranes.
Among the closely related proteins, alpha-, beta- and gamma-synuclein only αS is definitively linked to PD. Despite the relative conservation at the N-terminus (Supp.Fig3E), only αS binds to decapping proteins (Fig.3I). Substitution of only 5 aa residues that distinguish gamma-synuclein from αS within the first 20 aa resulted in a complete loss of decapping-module binding to αS (Fig.3J). We propose that the very specific sequence of αS N-terminus dictates dichotomous binding either to membranes or to P-body (Fig.3K).
αS interacts with multiple decapping-module proteins on the proximal scaffold of Edc4.
The “proximal” region of the Edc4’s C-terminus (974-1265 aa) orchestrates decapping-module assembly (Fig. 4A) (Chang et al., 2014). Other regions of the protein mediate different nuclear and mitochondrial functions(Gudkova et al., 2012; Hernández et al., 2018). First, we confirmed that there was co-abundance of decapping proteins and αS in cytoplasmic rather than nuclear fractions (Supp.Fig.4D). Next, systematic deletion and truncation analysis of Edc4 indicated that αS does in fact bind to the proximal region of Edc4 (Edc4: 974-1237aa) (Fig.4B,C, Supp.Fig.4A-C). A finer deletion analysis pinpointed a conserved block between aa 1220 and 1240 (Block 4, Fig.4D). Thus, the interactions of αS with Edc4 are likely linked to its canonical P-body function.
Figure 4. αS binds to multiple decapping-module components on the proximal region of the Edc4 scaffold.

(A) Schematic of the question posed.
(B-D) αS-Edc4 interaction mapping. (B) Edc4 and αS interaction occurs on proximal region of Edc4 between amino acids 1166-1265. (C) Edc4 Block4 as necessary for the αS interaction. The blocks were assigned based on Edc4 alignments.
(D) Summary of Edc4 truncations and deletions for αS interaction mapping (Supp.Fig.4 for the corresponding data).
(E) The Edc4-αS binding region modulates recruitment of decapping proteins. HA pulldown of Edc4 variants in HEK293 EDC4-null background in HEK293 cells (See Supp.Fig.4 for western blots).
(F) Edc4, Edc3 and Dcp1 proteins contribute to αS-decapping module interactions. Each bar is the mean of two independent CRISPR line quantification (error= sd). (see Sup.Fig.4J for western blots)
(G) αS is embedded in decapping module at the proximal region of Edc4.
To identify where other decapping-module proteins associate with Edc4 in relationship to the αS interaction site, we pulled down HA-tagged Edc4 (and its deletion mutants) in an EDC4-knockout line. We then probed for other decapping-module proteins. Dcp1-Edc3 binding to Edc4, but not Xrn1, commenced at Edc4 residues 1099-1166 (Fig. 4E). When we extended the Edc4 sequence to include the αS-binding site, Edc3 and Dcp1 bound even more avidly than they did to the full-length Edc4 protein and Xrn1 binding was also detected (Fig.4E). The Edc4 Block4 region was required for the optimal binding of Dcp1 and Edc3 (Fig.4E lower, Supp.Fig.4F). αS therefore interacted with Edc4 exactly where Edc4 binds to other key decapping-module proteins.
To address whether the full integration of αS into the decapping complex involves decapping proteins other than Edc4, we knocked out by CRISPR/Cas9 the three decapping proteins for which we had the best antibodies: Edc4, Dcp1 and Edc3. We then re-evaluated Avi-tagged αS’s interaction with endogenous decapping proteins in these null cell lines (Fig. 4F; Supp.Fig.4G-l). Each decapping-protein knockout reduced interaction of αS with each of the other decapping-module proteins to varying degrees (Fig. 4F). Edc4 was the most critical component (Fig.4F). Thus, the full integration of αS into the decapping module requires other decapping proteins besides Edc4 (Fig.4G).
αS gene dosage modulates P-body biology in human neurons
αS is highly enriched in human neurons, the cell type most consistently implicated in synucleinopathies. Our fly data suggested P-body manipulation in neurons can change a functional motor outcome (Fig.1). We predicted that direct interactions between P-body components and αS occur in human neurons. We turned to the so-called “Iowa kindred” in whom a dominantly inherited triplication of the SNCA locus leads to dementia and parkinsonism associated with LB neuropathology (Hurtig et al., 2000; Singleton et al., 2003). Through somatic-cell reprogramming to iPSCs and differentiation, we and others have identified early neuronal pathologies attributable to αS dosage (Byers et al., 2011; Chung et al., 2013; Mazzulli et al., 2016). We investigated P-body biology in response to altered αS dosage in neurons in pure populations of cortical glutamatergic neurons (“iNs”) induced from iPSCs by forced expression of the Ngn2 transcription factor(Zhang et al., 2013).
We obtained iPSC reprogrammed from a female Iowan kindred patient (Star Methods) and confirmed the genotype (Supp.Fig.5A). We generated an isogenic allelic series of iPSCs through a CRISPR/Cas9 approach (Fig.5A; Supp.Fig.5B,C). We selected clones in which sequencing across the SNCA locus indicated disruption of 2 alleles or 4 SNCA alleles (Supp.Fig.5D). For scalable, virus-free and consistent generation of neurons, we transposed into the iPSC lines an all-in-one PiggyBac (pB) vector that enabled doxycycline (dox)-inducible expression of Ngn2 (Fig.5A, lower schematic). We made multiple modifications in this vector to ensure stable and high expression-levels of the transgene cargo (Star Methods). A pure population of neurons was generated within two weeks of dox induction (Fig.5C, Supp.Fig.5E). Western blot confirmed in these neurons the presence of pathologic (4-copy parental), wild-type (2-copy knockdown), and null (0-copy knockout) levels of the αS protein (Fig.5B).
Figure 5. αS interacts with Edc4 in human neurons in a dose-dependent manner and modulates P-body number.

(A) Strategy to generate human neurons with differential SNCA copy numbers (see methods for details). Below; all-in-one PiggyBac construct to induce cortical neuron differentiation from iPSCs.
(B) αS protein levels from whole cell extracts of induced neuron(iN) triplication series. Vcp and Gapdh; endogenous markers.
(C) A modified all-in-one PiggyBac to induce cortical neuron differentiation and in situ biotinylation of αS.
(D) IF images of differentiated induced neurons with pan-neuronal markers, Tuj1 and Map2. Edc4 marks the P-body puncta. Below: fivefold magnification of the inlets.
(E) Biotinylated αS pulldown from induced neurons. PiggyBac depicted in (C) was used for neuronal differentiation and αS-Avi biotinylation. Experiment is repeated after 3 and 4 weeks of differentiation.
(F) Endogenous αS interacts with Edc4 in a dose-dependent manner in iPSC-derived neurons. Left: Immuno-Proximity ligation assay (PLA) for αS and Edc4 in 3-week-old neurons with differing SNCA copy numbers. Right: Super plots of PLA experiment quantification; 4 independent replicates across 3 neuronal differentiations (biological replicates) are shown. Imaged fields represent the total number of images taken (each image > 20 neurons). Summary statistics; ratiometric paired t-test (n=4, p=0.0008). Each independent replicate is color-coded.
(G) Edc4 puncta numbers decrease in SNCA triplication compared wild-type (2-copy) levels of SNCA. Left: IF for endogenous Edc4 and Tuj1(pan-neuronal marker) in 3-week-old iNs with differing SNCA copy numbers. Merged channel images are shown. Right: Super plots of IF quantifications; 4 independent replicates across 3 separate neuronal differentiations. Summary statistics; ratiometric paired t-test (n=4, p=0.0196).
(H) IF of Edc4 puncta in neurons derived from an familial SNCA mutation (A53T) line and its isogenically corrected control. Super plots of IF quantifications across two biological replicates. Summary statistics; ratiometric paired t-test (n=2, p=0.0567)
To assess physical interactions between αS and decapping-module proteins, we generated a pB vector that enabled dox-inducible expression of Ngn2 and αS-Avi. BirA ligase was concomitantly expressed under the (postmitotic neuron-specific) synapsin promoter (Fig.5C,E). This pB vector was stably introduced into our SNCA complete knockout iPSC to avoid confounding effects of endogenous αS. Pull-down by streptavidin beads recovered Edc4 and other decapping-module components in the αS-Avi-expressing neurons, but not in the untagged αS condition. Thus, αS also interacts with the decapping module in human neurons (Fig.5E).
To exclude artefacts induced by αS over-expression, we adapted the proximity ligation assay (PLA). PLA utilizes oligonucleotide-hybridized antibodies to detect close protein-protein interactions in situ (Maio et al., 2016). We first confirmed that Edc4 puncta co-localized with Dcp1 (Supp.Fig5F) and that Edc4 generated a true positive PLA signal with Dcp1 (Supp.Fig.6G) in 2copy SNCA iNs. Next, we quantitatively measured PLA signal between Edc4 and αS with automated microscopy. Not only was there a positive PLA signal between Edc4 and αS in iNs, but there was a clear linear relationship between αS levels and the PLA foci counts (Fig.5F, Supp.Fig.5G,H). Thus, αS interacts with Edc4 in situ in human neurons and this interaction is proportionate with αS levels.
While αS interacts with micro rather than macro P-bodies (Fig. 2J), increased αS levels could alter macro P-bodies if an equilibrium exists between micro and macro compartments. Consistent with this possibility, pathologic levels of αS (4-copy) result in decreased P-body numbers compared to wild-type levels of αS (2-copy) (Fig.5G, Supp.Fig.5 I, J). We asked if the same change was induced by a disease-causing point mutation in αS(Soldner et al., 2016) We and others previously reported on cellular pathologies in neurons harboring the A53>T αS mutation(Chung et al., 2013; Khurana et al., 2017; Ryan et al., 2013). We rederived this iPSC line and performed genetic correction with CRISPR/Cas9 (exon-3 targeting guide RNA and repair template; see Star Methods). These lines were also targeted with PiggyBac Ngn2 constructs and transdifferentiated to cortical neurons (Fig. 5A). At 3-4 weeks, exactly as in triplication neurons, P-body numbers decreased in SNCA A53>T mutant neurons compared to their isogenically corrected controls (Fig.5H). Thus, in human neurons, P-body homeostasis is altered by either amplification or disease-associated point mutations of αS.
αS upregulation disrupts Edc4 complexes
αS associates with Edc4 at a key region where multiple decapping-module proteins bind (Fig. 4). Thus, pathologic accumulation of αS could alter mRNA metabolism by directly disrupting P-body composition. Consistent with this idea, in HEK293 cells that expressed αS under a dox-inducible promoter, the endogenous Edc4-Dcp1 interaction (measured by co-IP) decreased as αS levels increased (Fig.6A, Supp.Fig.6A). In an orthogonal assay, we optimized native PAGE to capture high-molecular weight Edc4 and Edc3 protein complexes that are nuclease resistant, closely mimicking the micro P-bodies (Supp.Fig.6B-F and Star Methods). In this assay, the amount of high-molecular weight native Edc4 and Edc3 complexes diminished as αS levels increased (Fig.6B).
Figure 6. Pathologic αS increase disrupts decapping-module composition and mRNA turnover rates in human neurons.

(A) Endogenous Edc4 immunoprecipitation (IP) after dox addition (10 ng/ml) in tetON inducible SNCA and wt HEK293 cells (n=3, mean,sd, two-tailed t-test)
(B) Blue native-PAGE analysis of whole cell extracts from a tetON inducible SNCA HEK293 cells.
(C-D) Superplots of PLA between Dcp1 and Edc4 in SNCA 4-copy and 2-copy neurons. 9 independent replicates across 3 neuronal differentiations, ratiometric paired t-test (n=9, p=0.0004).
(E) Artificial tethering of αS to a nanoluciferase reporter mRNA.
(F) Strategy for ActinomycinD (ActD) pulse experiment to measure decay rate differences in patient derived neurons with different SNCA copy numbers.
(G) Volcano plot for the differential gene expression between SNCA 2 and 4 copy neurons (SNCA ~1.7 fold change in linear scale)
(H) Hypothesis tested in ActD pulse experiment in patient derived triplication neurons. Prediction: Δslope(2copy-4copy) < 0 indicating mRNA stabilization.
(I) Left panel: Slope diagram of transcripts with significant differential expression within the first hour of ActD addition (1024 genes, FDR<0.05, Likelihood ratio test (LRT) of DeSeq2). Right panel: Slopes of transcripts among the right panel that decayed in the 2-copy neurons (n=709, unpaired t-test, p<0.001)
(J-L) Δslope histograms (2copy minus 4copy) for all decaying genes within the first hour of ActD treatment in 2-copy neurons. Above the histogram is the question posed. (n; number of decaying transcripts, p-value: paired one-sided t-test for the hypothesis if Δslope<0, The summary of the test is depicted in the inlet). Histogram binning colors differentiate Δslope=0 boundary.
(M) Model proposed for αS accumulation impact of αS accumulation on decapping module composition and mRNA decay rates.
To connect these findings more closely to human PD pathophysiology, we turned once more to the SNCA triplication series iNs (Fig.5A). We compared the in-situ interactions between Edc4 and Dcp1 (by PLA) in SNCA 4-copy and isogenic SNCA 2-copy neurons. Edc4-Dcp1 PLA signal in SNCA 4-copy neurons was significantly less than the signal in 2-copy neurons (Fig.6C, Supp.Fig.6G). Importantly, Dcp1 and Edc4 protein levels remained unchanged (Fig.6D). Thus, upregulated levels of αS can disrupt decapping-module composition also in a disease-relevant context.
αS upregulation perturbs mRNA decay dynamics
If pathologic levels of αS disrupts endogenous decapping-module integrity, mRNA-decay kinetics could also be disrupted. We tested if αS can modulate mRNA stability by tethering it to an mRNA reporter (λN-boxB system)(Keryer-Bibens et al., 2008). We generated a dual mRNA-tether reporter system that enables a ratiometric luminescence readout of mRNA abundance (Supp.Fig.6H). As expected, tethering of P-body proteins, including Edc4, to λN resulted in decay of the reporter (Fig. 6E; Supp.Fig.6I). Bringing αS proximal to the reporter recapitulated this trend, suggesting αS can mimic bona fide mRNA degradation proteins (Fig.6E).
Turning to the more disease-relevant human neuronal model, we compared genome-wide mRNA decay kinetics between SNCA 4-copy and 2-copy neurons. We confirmed that at baseline (time=0), SNCA was differentially expressed between the lines, consistent with the genotypes (Fig.6G). If the decapping module is compromised, SNCA 4-copy neurons would have compromised mRNA degradation and thus slower mRNA decay rates compared to 2-copy SNCA. We thus measured mRNA levels up to 12 hours from transcriptional inhibition by ActinomycinD (ActD) (Fig. 6F). The decay rate for any mRNA in any time-interval can be represented as the log fold expression change during that interval. In down-sloping (decaying) genes, the difference between 2copy and 4copy log2FC (the “Δslope2c-4c” in Fig. 6H-I) is predicted to be negative.
The most robust alterations in mRNA decay rates between the genotypes occurred in the first hour after ActD addition (Δslopet1-t0; 1204 genes with differential slopes, likelihood ratio test (LRT), FDR<0.05)(Fig.6I, Supp.Fig.6J). Robust enrichments in genes related to protein targeting, mRNA translation initiation and mRNA catabolism was observed when gene-ontology analysis was conducted (Supp. Fig.6K). In the ‘protein targeting” category, aside from ribosomal genes, 9 out 28 genes (AP3D1, CLU, GABARAP, HSPA8, LAMP2, PIKFYVE, RAB8B, SCARB2, VPS13A, VPS13D) have previously been linked to PD (TableS6). More importantly, a preponderance of these transcripts (709/1204) exhibited Δslope2c-4c<0 (Fig.6I, green sector and right panel) in line with our prediction of compromised decapping.
We broadened our analysis to all decaying genes in the SNCA 2-copy neurons (regardless of LRT significance). Slope distributions for these genes were visualized as a histogram where the Δslope2c-4c<0 genes are on the left (red) and the Δslope2c-4c>0, genes are on the right(blue) (Fig.6J). When we compared all the 9561 decaying genes in a pairwise slope analysis, we noted a preponderance of genes exhibiting Δslope2c-4c<0 (Fig.6J, p=5.8 e-09).
We examined mRNA kinetics in the presence or absence of αS pre-formed fibrils (PFFs). PFFs are frequently used to induce αS aggregation pathology (Volpicelli-Daley et al., 2014). Addition of PFFs to the 2-copy neurons phenocopied the same global effect of mRNA stabilization as increasing αS copy-number (Fig.6K, n=10061 genes, p=0.00042). However, addition of PFFs to the 4-copy SNCA neurons had no detectable effect on the kinetics of decaying genes (Fig.6L, n=9647 genes, p=0.99). These global trends were replicated for the second time interval (1-3 hrs) as well, but not for non-decaying (up-regulated) genes(Supp.Fig.6L-N). Thus, pathophysiologic of levels of αS can lead to disruption of the decapping-module and aberrant stabilization of mRNAs (Fig.6M).
αS accumulation in synucleinopathy brain is associated with perturbed mRNA metabolism and decapping-module disruption
iPSC-derived iNs are a disease-relevant model for investigating the early pathologies of synucleinopathy. We extended our investigation of perturbed mRNA metabolism to human postmortem brain tissue. To address whether altered mRNA metabolism occurs as αS accumulates in the brain, we turned to the Religious Orders Study and Memory and Aging Project (Bennett et al., 2018). ROSMAP is a population-based study in which detailed measures of postmortem neuropathology can be directly related to a multitude of clinical and molecular phenotypes. We analyzed mRNA abundance in dorsolateral prefrontal cortex (DLPFC)(Mostafavi et al., 2018). DLPFC is matched to our iPSC cortical neuron model, but also a region with relatively early PD pathology, thus avoiding end-stage neuronal and glial responses.
We assessed statistically significant correlation/anti-correlation with four phenotypes related to PD and synucleinopathy (clinical PD diagnosis, clinical PD severity, presence of LB pathology, degree of Lewy LB pathology; nrange=573-595) (Supp.Fig.7A). GO terms related to RNA metabolism were enriched with respect to each of these four phenotypes (unbiased FDR correction; adjusted p-value<0.05) (Supp.Fig.7A with enrichment shown across different ontologies and specifically for the “degree of LB pathology” phenotype in Fig.7B). These findings suggest that, chronically in the brain, with increasing severity and advancement of αS neuropathology, RNA metabolism is perturbed in addition to well-known PD pathology pathways such as protein folding, mitochondrial-stress and ER protein localization. Interestingly, these pathways were similarly over-represented in our neuronal RNA kinetics experiments (Supp.Fig. 6K).
Figure 7. Postmortem brain and human-genetic analyses further connect P-body perturbations to PD.

(A,B) PLA between Edc4-αSyn in PD patient and control brains and its quantification(D) (*** p<0.001, one way ANOVA with Bonferroni correction, Map2 neuronal marker)
(C,D) PLA between Edc4-Dcp1 in PD patient and control brains and its quantification(E) (*** p<0.001, one way ANOVA with Bonferroni correction)
(E-F) Common variant BridGE analysis in PD and ALS cohorts to infer putative gene-gene interactions among P-body genes and SNCA. 53 P-body genes are grouped according to their functions. At the center, the causal genes are shown, SNCA for PD and TDP-43 for ALS. The map of significant SNP interactions (summarized at the gene level) with P-body genes and TDP-43 (TARDBP) (G) and SNCA (H) are shown. Protective and deleterious interactions are color coded. In (H), the LSM7 SNP GWAS genome-wide hit is indicated. Reduced opacity: The intra P-body interactions, High opacity: SNCA and P-body gene interactions.
(G) Logic of RVTT analysis. Left: Traditional Burden test collapse all mutations irrespective of their frequency. Right: RVTT accounts for trends in qualifying-mutation proportions in case versus controls. Mega-gene represents all genes in the pathway. Orange dots are collapsed qualifying mutations.
(H) Rare Variant Trend test (RVTT) analysis of P-body pathway genes In two different PD cohorts, PPMI and PDBP, RVTT was applied to P-body genes. Two “bags” of P-body genes were tested: CORE genes and DECAPPING genes, which are indicated in the lists. Three separate mutation-types (missense damaging, missense neutral and synonymous) were interrogated in both cohorts.
If the ROSMAP expression profile changes reflect a response to αS accumulation, then disruption of the decapping-module ought to be apparent in human brain, just as we observed it in human synucleinopathy neurons (Fig.6C-D). To match our iPSC analysis, we sourced rare familial synucleinopathy (SNCA duplication and SNCA A53T mutation) brain tissue (TableS7). We performed PLA analysis between αS and Edc4, as well as Edc4 and Dcp1. We selected the frontal cortex to match our iPSC and ROSMAP analyses, and to avoid the confounders noted above. In patient brains, compared to control brains, αS-Edc4 interactions significantly increased when measured by PLA (Fig. 7C,D, Supp.Fig7D). However, as in patient-derived neurons, the Dcp1-Edc4 interaction was concomitantly lost (Fig. 7E-F), without any change in levels of Dcp1 or Edc4 (Supp.Fig.7B, E). Thus, decapping module integrity is also compromised in synucleinopathy patient brains.
Two human genetic analyses identify a disease risk signal related to P-body genes in sporadic PD
To extend our findings beyond familial synucleinopathy to patients with so-called “sporadic” PD, we turned to human genetic analysis. Population-based GWAS studies have been instrumental in identifying causal genetic drivers of disease phenotype, however, most PD heritability remains unexplained(Keller et al., 2012). GWAS have implicated one P-body gene, LSM7, that is in linkage with a genome-wide significant single nucleotide polymorphism (SNP) rs62120679 (Chang et al., 2017). Most previous GWAS analyses have focused on single-variant tests, which identify individual genes contributing to disease risk. While identifying cumulative genetic changes in a pathway is often underpowered in GWAS analysis, directing analysis to a biologically meaningful genetic space, such as a pathway or well-defined network, can increase power (Bandres-Ciga et al., 2020; Fang et al., 2019; Wang et al., 2017). We performed two analyses geared to determine whether cumulative genetic changes in the P-body pathway are associated with PD risk. We considered P-body proteins a good candidate ontological network for such interactions.
To explore the space of common variants in P-body genes and SNCA, we applied a method known as BridGE(Fang et al., 2019; Wang et al., 2017). BridGE can be used to identify enrichment in the statistical signal of association in a predetermined set of biologically interacting genes (e.g., pathways or protein complexes) based on GWAS cohort data. Pathway-level analyses with BridGE can enhance statistical power when the genes included in the pathway have association signals that are undetectable individually.
In seven independent PD case-control cohorts (TableS7; Star Methods), we used BridGE to interrogate SNPs linked to SNCA and a canonical set of 53 P-body genes (Fig.7G,H). We focused on high confidence statistical signals (FDR<0.1) in one cohort and replicated in at least two independent cohorts (sample permutation p-value<0.05) (Star Methods). In total, we identified 76 significant signals for gene pairs among SNCA and P-body genes (TableS7), 23 of which included SNCA (Fig. 7H). These genes comprised multiple pathways, reminiscent of interactions we had identified in our systematic yeast genetic screen (Fig. 1C-F). The three most inter-connected genes (or “hubs”) of the resultant network were SNCA, SMG7 and LSM7. As noted above, LSM7 is in linkage with a SNP previously implicated in PD at genome-wide significance that regulates LSM7 expression (rs62120679)(Chang et al., 2017). Importantly, the result was specific. When we applied the same method to the same set of 53 P-body genes in four ALS cohorts (TableS7), there was only a single significant gene pair that included a P-body gene and TDP-43 and none with FUS, along with reduced intra P-body gene pairs (Fig.7G, TableS7). Thus, biological interactions between SNCA and P-body genes are specifically associated with PD risk.
To assess whether an accumulation of rare variants in key P-body genes might confer risk of PD in humans, we performed a pathway-level rare-variant burden test in two case-control whole-genome sequencing datasets from AMP-PD (Accelerating Medicines Partnership for PD), namely, PDBP and PPMI. After quality filtering, PPMI and PDBP datasets comprised 572 (PD: 391, Control: 181) and 1273 (PD: 823, Control: 450) individuals, respectively (STAR Methods). With these modest sample sizes, traditional rare variant association tests that collapse variants at the gene level lack of statistical power. Moreover, if a standard test (like SKAT-O) is adapted such that genes within a pathway are considered a “mega-gene”, no distinction is made between one variant in that mega-gene or many. To address this issue, we recently developed a pathway-based association test, namely, the rare variant trend test (RVTT), that assesses the relationship of the frequency of qualifying variants in a pathway with disease risk leveraging the Cochran-Armitage test statistic (Bendapudi et al., 2022). If a pathway is implicated in a disease, we are likely to see an accumulation of qualifying rare variants in the pathway in cases compared to controls(Fig.7G). RVTT selects qualifying rare variants using a variable minor allele frequency threshold approach(Price et al., 2010), and measures the strength of association using permutation-based p-values. A strength of the method is the ability to compare variants predicted to be deleterious versus variants predicted to be neutral (e.g., synonymous variants).
We divided genes into CORE and DECAPPING modules (STAR Methods) and used RVTT to test the enrichment of rare qualifying variants in these modules (Fig. 7H). In the PDBP cohort, we found that PD patients were enriched in rare-damaging (perm p-val=0.027) and damaging-missense (perm p-val = 0.013) variants in the core P-body genes compared to controls. In contrast, there was no difference in synonymous (perm p-val=0.557) or neutral (perm p-val=0.489) rare variants. This trend was recapitulated for damaging-missense variants (perm p-val = 0.045) in PPMI, in which there was also no difference in synonymous (perm p-val=0.633) or neutral (perm p-val=0.217) rare variants. Regarding the decapping-module, there was a trend in the larger PDBP dataset (perm p-val= 0.053; synonymous variants perm p-val=0.351) but none noted in PPMI (Fig. 7H; TableS7). These results suggest that a cumulative effect of protein altering variants in key P-body genes potentially contributes to the PD risk even when individual variants and genes do not associate with the disease.
DISCUSSION
Despite decades of intense research, αS has continued to surprise. Its connection to PD and other neurodegenerative synucleinopathies has drawn most attention to its role at the vesicle membrane, particularly at the presynaptic terminal of neurons. Here, we uncover a role for αS in the cytosol as a regulator of P-body homeostasis through binding to the decapping module. Our data provide a way to conceptualize how effects of αS on membrane trafficking can affect P-body function. This is because αS utilizes the same stretch of N-terminal amino acids for membrane anchoring(Fusco et al., 2014) as it does for binding the decapping module (Fig.6). Thus, factors affecting αS residency on the membrane, including membrane composition, fluidity and rigidity, could affect how available αS is for decapping-module interactions. This in turn could lead to alterations of mRNA stability through affecting composition and function of the physiologic decapping module. Interestingly, another small protein, the 7kDa microprotein NBDY, also regulates P-body physiology through an interaction with Edc4 (D’Lima et al., 2016). We posit that αS could act as a ‘cross-compartmental envoy’ and that some of its known genetic interactions with membrane trafficking and mRNA metabolism genes (Khurana et al., 2017; (Dhungel et al., 2015) could be explained through this dual residency. In future studies, it will be worth exploring whether these interactions play a role in synaptic functions, including memory formation, that heavily depend on post-transcriptional gene regulation.
It is worth noting that the classes of mRNAs stabilized with αS accumulation belong to the pathways that we and others have previously tied to αS biology in genetic screens, namely protein trafficking and mRNA metabolism (Supp Fig. 6K), and stabilized genes including protein trafficking genes implicated in PD GWAS (SCARB2 and VPS13) (Fowler et al., 2020; Gonçalves et al., 2016; Khurana et al., 2017; Mitchell et al., 2021; Murphy et al., 2015; Nalls et al., 2019). In postmortem brain, these transcript classes are also altered with αS accumulation (Fig. 7B). Thus, altered P-body homeostasis may in turn feedback to other PD-relevant cellular alterations. It is tempting to speculate that some of these transcriptional changes may even sense and protect against the cytosolic accumulation of αS that occurs in synucleinopathies (Dettmer et al., 2016).
The critical role of the N-terminus of αS in the P-body interaction explains why these interactions might have eluded prior systematic studies of αS interactors: such studies either used only the C-terminal of αS as bait(McFarland et al., 2008) or employed large, potentially disruptive N-terminal tags(Hein et al., 2015). Notably, a recent cancer proteomics study also identified decapping-module interactions with αS, supporting our observations (Kennedy et al., 2020).
The connection between αS and decapping proteins might extend beyond neurons. αS is highly abundant in hematopoietic cells(Barbour et al., 2008) and within CNS glia (Brück et al., 2016; Halliday and Stevens, 2011; Scheiblich et al., 2021) . Therefore, effects of αS on the cellular transcriptome could be highly context dependent. In hematopoietic cells, inflammatory cytokines are heavily regulated at the post-transcriptional level by P-body proteins (Ivanov and Anderson, 2013). The connection of αS to P-bodies may be especially important in the context of viral and inflammatory responses in non-neuronal tissues. P-bodies have been implicated in controlling viral life-cycles from yeast to human cells and they are well-established targets of viral proteins(Beckham and Parker, 2008; Gaete-Argel et al., 2019). Intriguingly, αS has also been directly connected to viral responses in mouse studies (Beatman et al., 2016; Massey and Beckham, 2016). Future studies should investigate whether αS’s immune connections are linked in part to P-body regulation.
The impetus for our investigations arose in genetic and proteome analyses in yeast and flies (Fig. 1). How αS impacts P-body biology in yeast and flies remains speculative for now. In S.cerevisiae, there is no Edc4 homolog, although there is a homolog (Pdc1) in S.pombe. While we cannot rule out that as-yet-unknown bridging factors exist across species between αS and P-bodies, a parsimonious explanation is inherent plasticity in the P-body network. Although decapping-module proteins are conserved from yeast to humans, the module is held together by short linear interaction motifs (SLIMs). Such motifs evolve more rapidly than well-folded domains, enabling a high degree of plasticity(Jonas and Izaurralde, 2013). Indeed, many decapping protein interactions have been reconfigured during evolution(Fromm et al., 2011; She et al., 2008). We hypothesize that the αS-decapping protein interaction surfaces might differ in different species. Our specific genetic approaches, BridGE for common variants and RVTT for rare variants (Fig. 7), are also predicated on this idea. We surmised that, for the P-body pathway, individual genes would be less likely to show signal than a pathway-based approaches. Both methods suggested in different ways that cumulative genetic interactions (BridGE) or rare-variant burden conferred increased risk for PD.
Limitations
Genetic manipulations in this study were performed in model organisms (Fig.1). Direct genetic manipulations of P-body genes in human neurons, albeit nontrivial, will be important to perform in the future. They could establish the dependence of mRNA stability directly on P-body proteins and direct dependence of αS toxicity on P-body genes in more disease-relevant cells. Redundancy and baseline alterations in mRNA stability will be experimental challenges to overcome, as will the development of tractable αS toxicity assays in iPSC neurons. In vitro kinetic competition assays and atomic-resolution structural information will be required to delineate how exactly αS modulates the decapping module enzymatic activities. These studies will likely require reconstitution of decapping-module proteins along with αS in vitro. Importantly, high quality reagents for Xrn1 and Dcp2 (such as nanobodies, knock-ins) are needed to interrogate these important enzymes in synucleinopathies. In future studies, more subtle measurements of mRNA decay kinetics, such as metabolic labeling, could be utilized to avoid secondary effects of ActinomycinD
A thorough evaluation of the effect on P-body physiology and mRNA stability in SNCA knock-out cells is warranted and will strengthen the argument that P-bodies are tied to endogenous αS function. Our investigations should also be extended to different cell types, within and outside the CNS. In this study, our neuronal experiments were performed in tractable iNs and correspondingly postmortem studies forcused on cortex. Although brain cortex is also affected in PD and other synucleinopathies (Foffani and Obeso, 2018), the investigation of P-bodies should be extended to midbrain dopaminergic neurons and other sites of PD pathology.
Our data suggest that αS is more closely associated with soluble micro P-body complexes where active mRNA degradation is thought to take place(Eulalio et al., 2007; Horvathova et al., 2017). How αS is excluded from the condensed P-bodies remains unknown. Furthermore, how common gene ontologies recovered from neuronal decay data and ROSMAP analysis arise and if there are any detectable RNA motifs in the transcriptome affected by αS accumulation should be more thoroughly studied.
Experimentally determined biological interactions, in this case among P-body genes and SNCA (Fig. 1), can increase power in human genetic analyses (Fig. 7). In the BridGE analysis, common SNP variants linked to SNCA and P-body genes, when analyzed as SNP1-SNP2 pairs, reveal association with PD risk. This is missed when SNPs are considered individually. Hubs of the inferred gene-gene interaction network include SNCA and LSM7 (Fig.7E), the latter linked in prior studies to PD through a genome-wide significant SNP (rs6212067) (Chang et al., 2017). Notably, the method assumes gene-gene interactions underlie biological interactions, but it does not definitively establish epistasis, namely that SNP1 modifies the risk induced by SNP2, which would require more mechanistic studies. Single-gene rare-variant studies are even more statistically challenging. While such studies have recently identified suggestive signal in P-body and related genes (Chartier-Harlin et al., 2011; Siitonen et al., 2017), the studies are thoroughly under-powered. RVTT (Fig. 7G-7H) provides a statistically powerful way to quantify association of rare variants in modest datasets (Fig.7H). It has several notable limitations, however. First, it assumes that all rare qualifying variants in the pathway have effects in the same direction. Second, if the pathway under question contains many unimportant genes, RVTT is underpowered to detect a signal. Third, RVTT ignores the edge relationships of the genes within the pathway.
In sum, we have identified a direct cytosolic role for αS in P-body regulation, mediated by the same N-terminus residues responsible for membrane tethering. This dichotomous function sheds new light on αS biology and the pathophysiology of synucleinopathies, and genetic drivers of disease vulnerability. Our data open up new ways of considering the role of αS role in health and disease, in different cellular compartments and in different cell types. Beyond αS and P-bodies, parallels to the biology of other intrinsically disordered and conformationally plastic proteins will likely be rich and worthy of further exploration.
STAR METHODS
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Vikram Khurana (vkhurana@bwh.harvard.edu)
Material availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
The original mass spectra for the SILAC experiments and the protein sequence database used for searches have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu) and are accessible at ftp://massive.ucsd.edu/MSV000089026/. Bulk RNAseq data is submitted to GEO with the submission number GSE199349 and will be available as of 06.01.2022. BridGE analysis used publicly available GWAS data and obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accessions (phs000196.v3.p1, phs000089.v3.p2, phs000048.v1.p1, phs000126.v2.p1, phs000918.v1.p1, phs000394.v1.p1, phs000127.v2.p1, phs000344.v1.p1, phs000101.v3.p1) and the Accelerating Medicines Partnership Parkinson's disease (AMP PD: https://amp-pd.org/). Rare Variant Trend Test (RVTT) analysis used publicly available data and the data for case-control Parkinson’s disease cohorts were obtained from the AMP-PD Knowledge Platform (https://www.amp-pd.org). AMP PD – a public-private partnership – is managed by the FNIH and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research, the National Institute of Neurological Disorders and Stroke, Pfizer, and Verily. Specifically, we utilized the case-control cohort from the Parkinson’s Disease Biomarker Program (PDBP) consortium which is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. Full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript. Additionally, we used the sporadic PD cases and healthy controls from PPMI sub-cohort of AMP-PD. PPMI (www.ppmi-info.org) is a public-private partnership, that is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, List of full names of all the PPMI funding partners found at www.ppmi-info.org/fundingpartners. The PPMI Investigators have not participated in reviewing the data analysis or content of the manuscript.
The code used for the analysis of the RNAseq data can be found in https://github.com/bwh-bioinformatics-hub/Hallacli2022Cell. (DOI:10.5281/zenodo.6477548). ROSMAP analysis did not create original code but descriptions of clinical and pathological outcomes are available at the Rush Alzheimer’s Disease Centre Research Resource Sharing Hub (https://www.radc.rush.edu). The BridGE software for gene-gene interaction analysis is available at https://github.com/csbio/BridGE/tree/master/BridGE_genes. (DOI: 10.5281/zenodo.6473560).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
Experimental models and subject details
Yeast models
All yeast strains (S. cerevisiae) were on W303 background (W303, Mat a/alpha, can1-100, his3-11,15, leu2-3,112, trp1-1, ura3-1, ade2-1::Ade2). Unless otherwise stated, yeasts were grown at 30°C in standard growth media. Rich media, YPD, was 1% yeast extract, 2% peptone and 2% glucose. Synthetic media was comprised of complete supplement mixture (CSM) media (and its drop out derivatives) together with 6.7g/L yeast nitrogen base and 2% glucose. For solid media, 2% agar was included. RBP yTRAP library was generated previously (Newby et al., 2017) and all yeast strains used and generated in this study are tabulated in Table S1.
Fly models
Drosophila crosses, aging and assays were performed at 25°C. Assays were performed on 10-day-old flies. The human QUAS-wild type α-synuclein line has been described previously (Ordonez et al., 2018). Briefly, human wild-type form of α-synuclein (SNCA) was cloned into the pQUAST vector and flies created by embryo injection (BestGene) to generate the QUAS-α-synuclein WT line. The pan-neuronal nSyb-QF2 and nSyb-GAL4 drivers were used to control expression of transgenic SNCA and transgenic RNAi, respectively. Control flies were of the genotype nSyb-QF2, nSyb-GAL4 /+. Locomotor assays were performed as described in (Olsen and Feany, 2019). XRN1(PCM) RNAi lines are stock1 (HMS01169), stock2 (GLC01410).
Insect cell expression
ExpiSf9™cells were obtained from ThermoFisher as part of the expression system starter kit (A38841). The cells were cultured at 27°C in disposable Erlenmeyer flasks (vented caps) with gentle shaking (95 rpm). The cells were kept at >90% viability measured by cell counting with Trypan blue. Cells were passaged when they reached 10 mil/ml confluency back to 1 mil/ml confluency.
Human cell lines
HEK Flp-In™ T-REx™ 293 cell line (RRID:CVCL_U427, fetal kidney origin, female sex) was purchased from Invitrogen (Catalog No: R78007). The cells were maintained in DMEM (Thermo Fisher: 10566; high glucose, GlutaMAX™ Supplement, minus NaPyruvate), supplemented with heat inactivated 10% Fetal Bovine Serum, Zeocin (100 μg/ml) and Blasticidin (3-5 μg/ml) with 5% CO2 and 37°C. For lentivirus generation, HEK293 FT cells were purchased from Invitrogen (R70007) and maintained in supplemented DMEM media (DMEM, high glucose, GlutaMAX™ Supplement,1X Non-Essential Amino Acids, 1X Sodium Pyruvate, 10% Hi FBS, 0.5 mg/ml Geneticin). Cells were passaged every 4-5 days when they reached to 90% confluency. The cells were checked for mycoplasma contamination every month. Pen/Strep was avoided in DMEM to unmask any bacterial/fungi contamination. Most of the human cell lines were derived from the HEK Flp-In™ T-REx™ 293 (see below).
iPSC
SNCA A53T fibroblasts from a Contursi kindred patient (female gender, age 49) were previously described(Chung et al., 2013). They were collected under BU protocol (#H-27479) and then cultured/reprogrammed/differentiated under MGH protocol (#2009P000775) and MIT COUHES protocol (#0807002834). The line was licensed from Whitehead Institute to Yumanity Therapeutics, and thereafter to BWH. The SNCA triplication iPSC cell line of an ‘Iowa kindred’ patient with early-onset and severe parkinsonism and dementia was obtained from the NINDS Human Cell and Data Repository (Patient code: NDS00201; iPSC line code: ND34391, female gender, age 55). In general, iPS cells were cultured at 37°C with 5% CO2 and checked for mycoplasma every second week. iPS cells were cultured on 6-well plate coated with Matrigel (Corning, Cat. No. 354277) in mTeSR1 medium (StemCell Technologies, Cat. No. 85850) and passaged using Gentle Cell Dissociation Reagent (GCDR, StemCell Technologies, Cat. No. 07174) or PBS-EDTA(1 mM) when they reached a density of 70–90%. For single cell dispersion of iPS cells for neuronal differentiation, Accutase (Gibco, 00-4555-56) was used (see below for neuronal differentiation). For SNCA triplication lines, G-banded karyotyping was performed every 10-12 passages (Cell Line Genetics). TaqMan Copy Number Assay were performed to confirm SNCA triplication in the ND34391 iPSC line. Genomic DNAs were extracted using QIAamp DNA Mini Kit (Qiagen, Cat. No. 51304). Human SNCA copy number primer (Hs02236645_cn), TaqMan Copy Number Reference Assay (Human, RNAse P, ThermoFisher, Cat. No. 4403326) and TaqMan Genotyping Master Mix (ThermoFisher, Cat. No. 4371355) were used in the assay. A control iPSC line (14-20 035iPS courtesy of Matt Huentelman, TGEN), a second SNCA triplication line and a mutant SNCA (A53>T) line were used as controls (The results can be found in Supp.Fig.5A). The piggybac mediated Ngn2 transgenesis, CRISPR mediated isogenic corrections of these lines are described in detail below.
iPSC derived neurons
Patient derived human cortical neurons were derived from a modified Ngn2 transcription factor induction protocol and cultured at 37°C with 5% CO2 and checked for mycoplasma every second week (described below in detail). Neuronal identity was verified by immunofluorescence of Map2 and Tuj1, pan-neuronal markers. Ngn2 induced cortical neurons lacked glial markers (S100B and Vimentin) and a proliferation marker Ki67 (Supp.Fig.5E). Neurons were cultured at 37°C with 5% CO2 and checked for mycoplasma every second week.
ROSMAP Study subjects
Postmortem data analyzed in this study were gathered as part of the Religious Orders Study and Memory and Aging Project (ROS/MAP) (Bennett et al., 2012a, 2012b; Jager et al., 2018), two longitudinal cohort studies of the elderly, one from across the United States and the other from the greater Chicago area. All subjects were recruited free of dementia (mean age at entry=78±8.7 (SD) years) and signed an Anatomical Gift Act allowing for brain autopsy at time of death. Written informed consent was obtained from all ROS/MAP participants and study protocols were approved by the Rush University Institutional Review Board.
Post-mortem brain tissue for PLA analysis
Postmortem brains from familial PD patients (SNCA A53T, SNCA duplication, and control brains) were obtained from Mayo Clinic Florida Brain Bank under IRB 15-009452. The details of these brains are tabulated in TableS7. We thank Dr. Dennis Dickson for generously providing this tissue as part of a collaboration to our colleagues Drs Dennis Selkoe and Tim Bartels, for whom we are grateful for mediation in obtaining these rare reagents.
Method details
Molecular cloning
Gene cloning was done either by traditional cloning (restriction enzyme-ligation), gateway cloning or Gibson cloning method. All geneblocks (double stranded DNA fragments) and primers were purchased from IDT (Integrated DNA Technologies). For traditional cloning ligations, 2X Quick Ligase is used according to manufacturer’s protocol (NEB, M2200). For gateway cloning, LR and BP clonase mix were purchased from Invitrogen and used half the amount of supplier recommended protocol. For Gibson cloning, in-house 1.33X Assembly Master Mix was made as follows: 1.875 mL Assembly Master Mix was generated by mixing 500 μL 5x Gibson Buffer (6 ml of Gibson buffer includes: 3 mL 1M Tris.HCl pH 7.5, 300 μL 1M MgCl2, 60 μL each of 100 mM dNTPs, 300 μL of 1M DTT, 300 μL of 100 mM NAD+, 1.5 g of PEG-8000, added up to 6mL by ddI H2O), 1 μL T5 Exonuclease, 31.2 μL Phusion Polymerase, 250 μL Taq DNA Ligase, 1093 μL ddI H2O. The Assembly Master Mix was aliquoted and stocked at −20°C. Each Gibson reaction was performed mixing 3 μL of the Master mix and 1 μL of backbone/insert constructs and incubated at 50°C for 1 hour. For the propagation of Gateway donor or destination vectors, plasmids were propagated in ccdB resistant strains of E.coli. For the propagation of regular constructs, NEB DH5α strain was used. For highly repetitive DNA containing plasmids, NEB Stable® cells were used. For large (>10 kB) and repetitive DNA containing plasmids- such as PiggyBac constructs- NEB® 10-beta cells were used. All plasmids used and generated in the study are tabulated in Table S2.
Yeast transformation
Overnight yeast cultures were grown to saturation and diluted back to OD600=0.1 in 50 ml rich media for incubation at 30°C until OD600=0.5-0.9. The cells were collected by centrifugation (1,500g, 5 min, RT) and washed with 25 ml dH2O at room temperature (RT). After spin, the pellet was re-suspended in 5 ml SORB buffer (100 mM LiOAc, 10 mM Tris.HCl pH 8.0, 1 mM EDTA, 1M Sorbitol). After another round of spin, the cells were re-suspended in 360 μL SORB buffer + 40 μL salmon sperm DNA (2 mg/ml stock). These competent cells can be kept indefinitely at −80°C. For integrations, 50 μl thawed cells were mixed with 10 μl of linearized (or digested) DNA and six volumes of PEG buffer (100mM LiOAc, 10mM Tris.Cl pH 8.0, 1mM EDTA, 40% PEG 3350) was added on to the cells and briefly vortexed for incubation at RT for 30 minutes. 1/9 volume of DMSO was added and the cells were heat shocked at 42°C for 15 min without shaking. The cells were centrifuged for 2000 rpm for 3 minutes and the supernatant was aspirated. For auxotrophic markers, the cells were plated directly on selective media plates, while for antibiotic markers (Nourseothricin, G418, Hygromycin) cells were recovered in 4ml YPD at 30°C for at least 4 hours.
Generation of Estradiol inducible Neurodegenerative protein expressing (pZ_ND) yeast strains
ZEM module (Zif268 - Estradiol Receptor ligand binding domain - Msn2 activation domain) is constitutively expressed under EIFa promoter. pZ denotes for the recognition element of ZEM module upstream of the ND open reading frame. Coding sequence for ND proteins were taken from human orfeome collection (for SOD(A4V) point mutant was introduced by point mutagenesis kit) and cloned into LEU integration pZ plasmids. For SNCA, two copy was integrated, one in LEU loci and the other in HIS locus. One copy untagged SNCA (wt) was marginally toxic (unpublished observation) whereas GFP tagged one copy SNCA was quite toxic. All toxicity measurements were determined by growth curve analysis at OD600 (explained below).
Design of RNA-binding protein yTRAP sensors
The yTRAP RBP platform is a compendium of 168 well-characterized haploid yeast strains (TableS1). Each strain expresses a specific RBP tagged with a sensor module (yTRAP sensor) and an output module. RBP tagged with yTRAP sensor is expressed on top of the endogenous RBP. yTRAP sensor module is composed of a nuclear localization sequence(NLS), a synthetic Zinc Finger (ZnF) domain and VP64 activation domain(AD). Furthermore, each strain contains the cognate DNA element specific to the synthetic ZnF which drives the expression of bright mNeonGreen reporter. This cognate ZnF promoter along with the mNeonGreen reporter is the ‘output module’. The output module assesses any tagged RBP for its availability to bind the ZnF cognate promoter. This “availability” requires both the tagged protein’s presence in sufficient quantity and its ability to migrate to the nucleus. Since this availability state can be altered through a variety of perturbations (including aggregation, degradation, sequestration, or release from a complex), the yTRAP sensor generates an indiscriminatory integrated perturbation output (Newby et al., 2017).
Generation of RNA-binding protein yTRAP sensors
For each sensor strain, 2 μg of the sensor plasmid (gateway cloned to yTRAP destination vector) was digested with Notl and the digest (without purification) was transformed to W303, Mat a, can1-100, his3-11,15, leu2-3,112, trp1-1, ura3-1, ade2-1::Ade2 (This strain is corrected for Ade2 gene to remove the build-up of red color). Each transformant was re-streaked twice for pure yeast clones.
Growth curve analysis for yeast
Yeast cultures were grown to saturation either overnight or 24 hours in 96 deep well plates. 96 deep well plate selected cultures were grown for two days for saturation in selected media. The cells are diluted 400 times to a rough OD of 0.03. 70 μl of 0.03 OD of cells are transferred to 384 well plates (Nunc-265202) and OD is measured with Biotek Epoch2 microplate reader for at least 45 hours with recordings every 15 minutes at OD 600 nm and 30°C.
Flow cytometry for the yeast
Saturated cultures were diluted hundred-fold to 200 μl fresh synthetic media in a 96 well plate. At the time of measurement, typically 6 hours after the dilution, 10 μg/mL propidium iodide (Sigma Aldrich Cat. No. P4864) was added to media to detect dead cells. All the measurements were acquired by MacsQuant VYB flow cytometer (Miltenyi Biotech) with a 96-well plate platform. A typical run contained 10.000 events with medium flow rate (~700-800 events per second) in the screen mode. The B1 channel (525/50 filter) was used to measure green fluorescence. The Y3 channel (661/20 filter) was used to measure red fluorescence or propidium iodide stain.
Yeast genetics and rescue experiments
Yeast strains for the overexpression experiments were generated by transforming the transgene plasmid (a low-copy, galactose inducible plasmid containing the gene of interest with a URA3 selectable marker). These strains were pinned from solid media into liquid media in 96 well plates and grown to saturation. They were then diluted into media containing raffinose as the carbon source and grown to saturation once again to relieve the repression of the GAL1 promoter. Finally, these cultures were diluted to an OD of 0.2 in fresh media, grown for 6 hours and pinned in serial dilution onto plates containing estradiol and galactose (SG-URA+Estradiol). The plates were incubated for 48 hours and imaged using a scanner. Estradiol concentration was 5 nM for deletion experiment and 3 nM for overexpression experiments. For gene deletions, a KAN cassette was inserted in the open reading frame of the genes and confirmed by PCR.
Yeast image acquisitions
All strains were constructed in the using the W303 genetic background yeast strain. The EIFα-promoter driven ZEM was integrated at the YBR032W locus and pZ-driven SNCA or empty control vector was integrated at the HIS3 locus. The cells bearing an empty vector were differentiated from SNCA-expressing cells with the addition of a constitutively expressed blue fluorescent protein driven by the TDH3 promoter integrated at the LEU2 locus. For the co-culture experiments, cells harboring a single copy of SNCA were mixed at a 2:1 ratio with cells harboring a single copy of the empty plasmid. They were then grown for 6 hours from a starting density of OD600 = 0.08 in synthetically defined complete media containing 1 μM estradiol. All images were acquired with a Nikon Plan Apo 100x oil objective (NA 1.4) using a Nikon Eclipse Ti-E inverted microscope and a CCD camera (Andor technology).
yTRAP perturbation screen details (pZ_ND x RBP-yTRAP)
Neurodegeneration (ND) causative protein expressing yeast strains were mated to yTRAP RBP library in rich media and double selected to include both mating types. The selection was carried out twice and single clones were struck. Before the day of flow cytometry, the cells had been inoculated at least 36 hours prior to reach to saturation. This oversaturation was to normalize variations that might have come from unequal initial dilution for flow cytometry. On the day flow measurements, the cells were diluted 100x fold in synthetic media (SD-CSM) and induced by estradiol (100 nM final concentration). An important detail to consider was to stagger control (uninduced strain) right below induced strain to eliminate time delays during the flow acquisition. After 6 hours of estradiol induction (incubation at 30°C), plates were measured by flow cytometry as detailed above. The measurements were gated against dead cells, large size excluded (FC vs SSC) and median values for GFP signal was chosen as the sample mean indicator. Each screen was carried out three times. Estradiol induced cells were slightly brighter than un-induced cells. To eliminate this intra-plate variation (intra-scaling), 10 darkest sensors were chosen, averaged and the brightness was normalized accordingly. The screen was performed three times. Individual replicate values were calculated from median mNeonGreen signal of 10000 yeast cells measured by flow cytometry.
Quantitative PCR for Drosophila RNAi lines
RNA was isolated from four Drosophila heads using Trizol following previously published protocol (Sarkar et al., 2019). Briefly, 4 drosophila heads were homogenized together in 1 mL of Trizol and incubated for 10 mins. Post incubation 0.2 mL of chloroform was added, incubated for 3 mins and centrifuged at 12,000 g for 15 mins. 0.5 mL of the aqueous layer was transferred to a fresh tube and incubated with 0.7 mL of isopropanol for 10 mins to precipitate the RNA. Post incubation the tubes were centrifuged at 12,000 g for 10 mins, the supernatant was discarded, the pellet was washed two times with 70 % ethanol, and air dried. RNA was dissolved in water and measured using a NanoDrop. Post-RNA isolation, 500 ng of RNA was converted to cDNA using high-capacity cDNA synthesis kit from Applied Biosystems (# 4368814). Quantitative SYBR green PCR assay was performed using SYBR green master mix from Applied Biosystems (#4309155). The following validated primers were synthesized at the Biopolymer Facility at Harvard Medical School; Xrn1_F- GCGAGGAGGTCAAGTTCGG, Xrn1_R- GCTCGTCTGTTCTCAGGGC, Rpl32_F- GACCATCCGCCCAGCATAC, and Rpl32_R- CGGCGACGCACTCTGTT. The fold change in gene expression was determined by the ΔΔCt method, where Ct is the threshold value. Rpl32 was used as a housekeeping gene.
Baculovirus mediated insect cell protein expression
ExpiSF™ expression system (ThermoFisher: A38841) guidelines from the manufacturer’s was followed. In short, a pFASTBacDual expression plasmid was used to express either HA-Edc4 alone(N-terminally tagged) or αS-V5/HA-Edc4. These plasmids were transposed to DH10Bac™ competent cells. Proper integration of pFASTBac plasmids were screened both by blue-white screening and PCR analysis. The bacmid was purified with a modified miniprep protocol, making sure that the large bacmid is not sheared during the isolation. The bacmids were kept at 4°C no more than a week before the infection to avoid degradation. ExpiSf9™cells were cultured at 27°C in disposable Erlenmeyer flasks with vented caps with gentle shaking. The cells were kept at >90% viability measured by cell counting with Trypan blue. The bacmids were transfected to ExpiSf9™cells and the cells were observed for decline of growth and volume expansion. When the cell viability decreased to 60% and the cell diameter enlarged 20% (roughly to 20 um), the media was collected(P0). P0 viruses were directly used for infection (500 ul for 50 ml culture) and expression. After 3 days of infection, the cell pellet was harvested and kept at −80°C. Protein extraction and pulldown were carried exactly as in HEK293 cells (see below) except that cell lysis of Sf9 cells were carried out by repeated freeze-thawing.
Transient transfection of HEK293 derived lines
For the transient transfection of HEK293 cell lines, a total of 5 μg of plasmid(s) (for 10 cm diameter dish) or 2 μg of total plasmids (for 6 well plates) was transfected with Lipofectamine 2000 (Thermo Fisher, Cat. No: 11668) in a 1-3 ratio (3 μL of Lipofectamine2000 per 1 μg of plasmid) according to manufacturer’s protocol. The confluency of the cells ranged between 60-90% on the day of transfection. The post-transfection incubation period with Opti-MEM reduced serum media was 5-6 hours, after which the cell media was changed to regular media. Cell harvesting was carried out 36-48 hours after transfection.
Generation of human HEK293 stable cell lines
For generation isogenic FLP recombinase mediated stable cell lines, 0.3 million HEK Flp-ln™ T-REx™ 293 cells were seeded to each well of 6 well plate two days prior to transfection. On the day of transfection, the cells should be 80-90% confluent. For transfection, 200 ng of FRT containing plasmid (for the negative control, 200 ng plasmid without an FRT site was used) and 2000 ng of pOG44 recombinase plasmid was co-transfected as follows: Plasmids were mixed in OptiMEM media (without selection) to a total volume of 150 μl and mixed well (Part A). 6 μl of Lipofectamine2000 reagent was mixed with 150 μl of OptiMEM, mixed well and incubated at room temperature for 5 minutes (Part B). Part A and B are mixed and incubated for 25 minutes at room temperature. The cells were washed with DPBS and supplemented with 1 ml of OptiMEM (no antibiotics). The PartA-B mix was dropwise applied on to the cells, mixed gently and incubated for 6 hours in the incubator. After 6 hours, the media was changed to DMEM. 24 hours later, the cells were trypsinized and each well was transferred to a 10 cm dish without any selection. Following day, the media was replaced with the selective media (DMEM, high glucose, GlutaMAX™ Supplement,10% HI FBS, Blasticidin (5 μg/ml), Hygromycin (100-200 μg/ml)). We have observed that Hygromycin selection could start in a lower concentration 100 μg/ml and increased over time to 200 μg/ml. The media was replaced every 3-4 days and colonies should be visible within 2-3 weeks. After the control transfection was completely dead and visible colonies were seen in the experimental transfections, the plate was trypsinized and reseeded to a 10 cm dish. When the cells reached confluency, a stock was generated immediately and continued to be passaged with DMEM, Blasticidin (3 μg/ml), Hygromycin (100 μg/ml). The cells were checked for transgenic expression with dox (0.1 μg/ml). Further quality check could be done by Zeocin sensitivity. For the generation of PiggyBac mediated insertions, essentially same protocol was followed except the FLP recombinase was exchanged with PiggyBac Transposase plasmid. All stable cell lines were kept as pooled, no single cell isolation was carried out to avoid cell to cell variability. Stable cell lines generated for this study are tabulated in Table S3.
Stable Pooled CRISPR/Cas9 Knockouts in HEK293 cells
pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene ID: 62988) was a kind gift from Dr. Lei Liu. This plasmid contains Cas9 from S. pyogenes with 2A self-cleaving peptide to Puromycin selection along with U6 promoter sgRNA. In order to track BbsI digestion more clearly, mRuby sequence was inserted in place of mock gRNA sequence. After complete digestion, 698 bp mRuby sequence should be observed for confident assessment of digestion. Guide RNA oligo designs targeted to P-body components were initially chosen from IDT and Synthego algorithms. Our observations after three knock out (KO) experiments (3 guides from IDT, 3 from Synthego) confirmed that Synthego designs yielded better KOs, therefore we chose Synthego designs. Cloning of guide RNAs into the Cas9 vector was followed exactly as the Zhang lab protocols (CRISPR Genome Engineering Toolbox). Briefly, 1 μg backbone was digested using FastDigest BbsI (Fermentas) together with FastAP (Fermentas) and gel purified by Qiagen columns. The oligos (100 μM) were annealed and phosphorylated in a single reaction, using T4 PNK (NEB) in T4 ligation buffer (NEB). The conditions were 37°C 30 min and 95°C ramping down to 25°C with 5°C/min rate. The oligos were diluted 200x fold and ligated to the 50 ng digested backbone with Quick Ligase (NEB). The plasmids were digest and sequence verified. Stable lines were generated as mentioned above with initial 1 μg/ml Puromycin selection and maintaining the cultures in 0.5 μg/ml Puromycin concentration. The cell lines were kept pooled due to biochemical nature of experiments rather than phenotypic assays. Three different guides were used but best two were chosen for the KO experiments for independent verification. The KO was verified by western blotting and immunofluorescence against the targeted protein. For mock knockout control stable lines, original vector was used directly. All guide RNAs used are tabulated in Table S4
Immunofluorescence (IF) in HEK cell lines
Freshly passaged 10000 HEK293 cells were seeded on poly-D-Lysine (Thermo, A3890401) coated 96 well glass bottom microplate (MGB096-1-2-LG-L, 0.17mm, low Glass). Coating was carried out at least three hours and extensively washed with water and 1x PBS before the cells were seeded. After the experiment, cells were fixed with 4% PFA in 1x PBS (16% Paraformaldehyde; EM Grade (EM Sciences; Cat#: 15710; Lot:180910-04) was freshly open and diluted into 10X PBS Buffer; pH7.4 (Invitrogen; Cat#: AM9625; Lot: 00602726) and brought up with ultrapure water (Thermo,10977015) to final 4% PFA and 1x PBS concentration). Fixation was carried out for 15 minutes at room temperature (@RT) after which the wells were carefully washed with 1x PBS three times and final wash was carried out with 1x PBS with 0.02% Sodium Azide mix for storage. The plate was sealed with aluminum tape (Corning® 96-well Microplate Aluminum Sealing Tape, 6570) for storage at 4°C. The cells were permeabilized (PBS 0.5% Triton X-100) 15 minutes and blocked (5% BSA, 0.05% Triton-X100) for 1 hour @RT. Primary antibody incubation (150 μl blocking solution with primary antibody) was carried out in dark 4°C overnight, no shaking. (Please refer to TableS5 for the antibody concentrations). The cells were washed 3 times with 1x PBS for 10 minutes @RT and incubated for 1 hour @RT in dark with secondary Alexa conjugated (488 nm or 594 nm) antibodies (1:1000x diluted in blocking solution). The cells were washed two times with 1x PBS (10 min each) and incubated for 10 minutes with PBS-Hoechst solution (2000x fold dilution to PBS from commercial stock, Hoechst 33342), after which the cells were washed twice with PBS and images were acquired immediately.
Images were acquired with Nikon Eclipse Ti inverted microscope, with 40X Plan Apo dry objective (MRD0045) and Andor Zyla 4.2P sCMOS camera (No binning, 200 MHz readout rate, 12 bit & Gain4 dynamic range). For HEK293 cells, usually 5 Z-stacks were acquired within a 2 μm range. For image processing, FIJI software was used. Separate channel stacks were Z-projected (maximum intensity) and ROIs (Region of interest) were selected from each field manually.
Whole cell protein extraction from mammalian cell lines
The cells were harvested by Trypsin-EDTA and washed twice with PBS at room temperature. After transferring to 1.5 ml protein low binding mini tubes (Eppendorf; 22431081), the cell pellets were flash frozen in liquid nitrogen. Removing from cold storage, the cell pellets were immediately topped with RIPA buffer (Thermo Scientific; 89900) supplemented with Complete EDTA-free protease inhibitor cocktail (Sigma-Aldrich; 11873580001) and PhosSTOP phosphatase inhibitor cocktail (Sigma-Aldrich; 4906845001). The cell pellets were kept on ice for 30 minutes and vortexed every 10 minutes to enable complete suspension of pellets to the RIPA buffer. After incubation, the cells were centrifuged at 21000g at tabletop centrifuge for 25 minutes and immediately 10 μL was separated for BCA protein quantification. The rest of the supernatant was mixed with equal volume of 4X NuPAGE™ LDS Sample Buffer (Thermo Fisher; NP0007) supplemented with 40 mM TCEP Bond Breaker (Thermo Fisher; 77720) and boiled for 10 minutes at 65°C. For long term storage, the cell lysates were kept at −80°C. For the quantification of protein concentration, 10 μL of lysate sample (right after extraction) was diluted 4-5 times and measured with Pierce™ BCA Protein Assay Kit according to manufacturer’s guidelines (Thermo Fisher; 23225). For western blotting, 5-20 μg of total protein was loaded on the gel.
Pulldowns in HEK293 cell lines
The cells (iPSC derived neurons or HEK293 cell lines) were harvested by Trypsin-EDTA and washed twice with PBS at room temperature. After transferring to 1.5 ml protein low binding centrifuge tubes, the cell pellets were flash frozen in liquid nitrogen. The pellets were resuspended in 500-1000 μl lysis buffer (50 mM HEPES/KOH pH 7.4, 150 mM KCl, 0.1% Triton X-100, 1 mM EDTA, 10% Glycerol, Complete EDTA-free protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail). The cell pellet was resuspended in lysis buffer and passed through 27-gauge needle 8 times. The lysate was incubated further on ice for 15 minutes and centrifuged at 21000g at a table-top centrifuge for 25 minutes. The supernatant was considered as whole cell lysate. The protein concentration was measured with BCA assay and 50 μg of protein lysate in 50 μL of lysis buffer was mixed with 50 μL 4X NuPAGE™ LDS Sample Buffer supplemented with 40 mM TCEP Bond Breaker to generate 0.5 μg/μl of INPUT sample. For the pull-down of Avi-tagged proteins, MyOneT1 Streptavidin magnetic Dynabeads (Invitrogen; 65601) were used. For every 500 μg of protein extract, 10 μl of MyOneT1 beads was used (This ratio was optimized to minimize background). Before the pulldown, the beads had been equilibrated with once with PBS and twice with lysis buffer. The pulldown was carried out at cold room in a rotator for 30 minutes. The beads were washed three times with wash buffer (50 mM HEPES/KOH pH 7.4, 250 mM KCl, 0.1% Triton X-100, 1 mM EDTA, 10% Glycerol, Complete EDTA-free protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail). After the third wash, the beads were resuspended in 50 μl 2X NuPAGE™ LDS Sample Buffer (supplemented with 20 mM TCEP) by briefly mixing in a thermoshaker. To break the Biotin-Streptavidin interaction, the beads were incubated for 15 minutes at 95°C. The eluate was briefly centrifuged and transferred to a fresh tube. For western blotting, 5-10 μg of INPUT and 20% of final pulldown eluate is loaded on the gel. For silver staining of the gels, SilverQuest™ Silver Staining Kit was used according to the manufacturer’s protocol (Invitrogen; LC6070). For HA pulldowns of Edc4 variants, essentially same protocol was followed except that HA pulldown time was 2 hours with Pierce Anti-HA Magnetic Beads(Life Technologies,88836). For endogenous IP experiments, optimal bead and antibody amounts in the linear range had been determined first. The antibodies were conjugated to magnetic ProtA/G beads(ThermoFisher) and IP was performed for 3 hours as explained above.
For quantitative IP experiments in Fig.2H, HEK293 SNCA-Avi+BirA ligase stable cell line was used. Several P-body genes were gateway cloned (Fig.2H) into a destination vector that generates an N terminally tagged nanoluciferase protein of interest. The cells were transiently transfected in a 24 well format and streptavidin pulldown was performed as described above. 1% of the input was taken and measured with luminescence plate reader and this value was used for normalization of the pulldown value. In the final wash step of pulldown, the beads were transferred to a 96 well (each transfection was divided into 4 well in 96 well) white walled flat glass bottom plate and read for luminescence. The IP measurement was recorded. The pulldown value was normalized to input value.
Western blotting
For SDS-PAGE electrophoresis, NuPAGE™ 4-12% Bis-Tris protein gels were used (Thermo Fisher; NP0321BOX) with MOPS running buffer (Thermo Fisher; NP0001). The gels were transferred to PVDF membranes (Invitrogen; IB24002) with iBlot™ 2 Gel Transfer Device with the preset P0 setting. After the transfer, the membranes were fixed with 0.4% Paraformaldehyde in PBS for 15 minutes at room temperature with slight rocking (This step is especially critical for detection of α-Synuclein protein). The membranes were washed three times with PBS-T (PBS with 0.1% Tween-20) and blocked with blocking buffer (5% BSA in PBS-T) for 30 minutes at room temperature. Primary antibodies are diluted in blocking buffer and incubated overnight at cold room. After three times PBS-T washing, the secondary HRP conjugated antibodies are incubated with modified blocking buffer (1% BSA in PBS-T) for 45 minutes at room temperature with slight rocking. For the detection of Avi-tagged proteins, HRP coupled Streptavidin was used overnight in blocking buffer (1:10000 dilution). SuperSignal™ West Femto Maximum Sensitivity HRP Substrate (Thermo Fisher; 1859022) or Clarity Western ECL Substrate (Biorad; 1705060) was used for signal generation. All the exposures were recorded digitally by iBright™ CL1000 Imaging System. Generally multiple exposures were recorded by signal accumulation mode. All antibodies used are tabulated in TableS5.
Blue Native PAGE analysis and P-body solubilization
Blue Native PAGE analysis was essentially optimized from Invitrogen™ Native PAGE system. NativePAGE™ Sample Prep Kit (BN2008) was purchased and Native cell lysis buffer (1x NativePAGE™ Sample Buffer, 2 mM MgCl2, 1% CHAPS, 1x Complete EDTA-free protease inhibitor cocktail and 1 x PhosSTOP phosphatase inhibitor cocktail) was prepared by using the 4x Sample buffer included in the kit. 1 million cells (HEK293 cells) was resuspended in 70 μl of Native cell lysis buffer and 5U of Benzonase was added to the samples. The cells were incubated for 25 C for 25 minutes in a thermoshaker block with 800 rpm shaking. For the detergent optimization experiments, the Native cell lysis buffer included either 1% DDM, 1% Digitonin, 0.1% Triton X-100 or 0.5 % NP-40 as final concentrations. After nuclease incubation, the lysates were centrifuged at 21130 rcf for 15 minutes at 4C. The supernatants were transferred to a fresh tube and 5 μl is used for BCA protein assay. 20-30 μg of extracts was loaded to NativePAGE™ 3-12% Bis-Tris protein gel along with 20 μl of NativeMark™ Unstained Protein Standard. For the first hour of run (150V, 60 minutes), the inner chamber included Dark Cathode buffer and for the remaining 80 minutes (150V), the inner chamber buffer was exchanged with light cathode buffer. During the whole running, the outer chamber buffer remained the same. The gel was transferred onto Immobilon® 0.2 um PVDF membrane (ISEQ00010) with 1X NuPAGE® Transfer Buffer. The transfer was in cold room, with 400 mA stable current for 120 minutes. After the transfer, the membrane was washed several times with ethanol to remove the coomasie dye until no further washing was possible. The membrane was let dry overnight on bench. The membrane was reactivated with methanol and washed several times with PBS-T (PBS with 0.05% Tween-20) and continued as regular western blot.
Reasoning of Native Page utilization in the context of P-body components:
Our data indicated that the Edc4-αS interaction occurs in soluble sub-complexes, rather than in condensed P-body granules (Fig.2J). We called them micro P-bodies. We reasoned that these sub-complexes could be resolved by native-PAGE electrophoresis when freed from multivalent RNA interactions that might otherwise drive them to P-bodies. Detergent type, nucleases and lysis temperature (Supp.Fig.6B-F) were all important factors for resolving Edc4 complexes and, in particular, we found CHAPS detergent and lysis temperature critical for recovering Edc4 complexes with native-PAGE (Supp.Fig.6B,C). A large proportion of Edc4 was resolved as an approximately 1 megadalton (MDa) size complex with higher bands indicating the presence of larger complexes (Supp.Fig.6D). When we knocked out Edc3 by CRISPR/Cas9, these higher bands diminished (Supp.Fig.6E). In contrast to Edc4, most Edc3 was resolved as half megadalton complexes with some >1MDa complexes observable. These larger complexes were dependent on the presence of Edc4 (Supp.Fig.6F).
RNA tethering assays
RNA tethers are stable RNA hairpin structures exhibiting a high affinity for a cognate protein. We used an RNA tether (5x BoxB elements) with high affinity for LambdaN peptide (λN). Traditionally RNA tethering assays were done with three different plasmids; a reporter with the RNA elements, a second reporter for transfection normalization and the third plasmid containing protein of interest to be tethered. Transient transfection of three different plasmid could be a source of noise, therefore, we sought to reduce the complexity of the system in two ways. 1) We combined reporter and normalizer reporter in the same vector via bi-directional promoter. The design is represented in Supp.Fig.6H. 2) Instead of transient transfection, we made a stable line for the reporter construct. Thus the only variable is the transfected transgene to be tethered. Other modifications included: 1) To both reporters, a degron sequence was included to keep linearity between RNA and protein levels of the reporter. For the normalizer reporter, the firefly luciferase (Luc2), CP degron was used. For the nanoluc reporter, PEST sequence was used. 2) All modules were cloned into a piggyBac (pB) plasmid and the stable cell line was generated with pB transposition with the aid of the pB transposase. Once the cell lines were generated, a gateway destination vector was generated to express C terminally tagged LambdaN tag. To this gateway destination, P-body genes and SNCA gene were LR cloned. The resultant expression vectors were transiently transfected in a 96 well format. Two days before the transfections, 10K reporter cell line was seeded to polyD-lysine coated glass bottom opaque walled sterile plates. The transfections were carried out by Lipofection by Lipo2000 (ThermoFisher) and the measurements were done with NanoGlo Dial Luciferase Reporter Assay System (Promega, N1610). The measurements were recorded in FluoStar Omega (NMG Labtech) reader.
SILAC-MS for αS interactors in HEK293 cells
SILAC (Stable Isotope Labeling) of HEK293 cells:
HEK cells stably expressing matched levels of GFP or αS-GFP were grown in SILAC media containing “light” or “heavy” isotopes for at least 5 cell doubling time to allow complete isotope incorporation into the proteome.
Antibody crosslinking:
~110 μl Biorad AffiPrep Protein A beads were equilibrated into PBST, washed 3 times and resuspended in 500 μl PBST. 55 μg of affinity-purified rabbit anti GFP antibody (generous gift from Dr. Iain Cheeseman, Whitehead Institute) was mixed into the beads PBST slurry and rotated for 1 hour at room temperature. The antibody attached beads were washed 3 times with PBST and 3 times with 0.2 M sodium borate, pH 9. After the final wash, 900 μl of the 0.2M sodium borate, pH 9 was added to bring the final volume to ~ 1ml. Freshly prepared 100 μl of 220 mM dimethylpimelimidate (Sigma, D8388) was added to the tube and rotated gently at room temperature for 30 min. Beads were washed 2 times with 0.2M ethanolamine 0.2M NaCl pH 8.5 and left rotating in the same buffer for 1 hour at room temperature to inactivate the residual crosslinker.
Co-immunoprecipitation:
HEK293 cells expressing either GFP or αS-GFP were lysed with nondenaturing lysis buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 1% Triton X-100 and 2 mM EDTA). The lysate was rotated for 30 min at 4°C and centrifuged at 10,000g for 20 min at 4°C. Supernatant was collected and protein concentrations determined using BCA assay. ~400 mg of lysate was incubated with 50 μl anti-GFP antibody crosslinked beads for 4 hours at 4°C. Beads from GFP or αS-GFP samples were washed once with nondenaturing lysis buffer, mixed together, washed one more time with the same buffer and denatured by boiling for 5 min in 2X LDS buffer.
Mass spectrometry and analysis:
Following elution from anti-GFP antibody beads, proteins were separated on a NuPAGE Novex Bis-Tris 4-12% gel run for 1 hour at 130V. The gel was stained overnight with Coomassie G-250 (Invitrogen) and subsequently destained with water. Lanes for each replicate were manually cut into six gel bands. Processing of gel bands was completed essentially as described in (Hung et al., 2016). Gel bands were destained in 1:1 acetonitrile: 100 mM ammonium bicarbonate pH 8.0 for several hours followed by dehydration with acetonitrile. Dehydrated gel bands were swelled with 100 μL of 10 mM DTT in 100 mM ammonium bicarbonate for one hour while shaking followed by 100 μL of 55 mM iodoacetamide in 100 mM ammonium bicarbonate for 45 minutes in the dark. The iodoacetamide solution was removed and bands were dehydrated with acetonitrile. Approximately 10-50 μL of 20 ng/μL Sequencing Grade Trypsin (Promega, V511X) was added to each sample and digestion was completed overnight with shaking at room temperature. After the overnight digestion, excess trypsin solution was removed from each sample and bands were incubated with 20 μL of extraction solution (60% acetonitrile/0.1% TFA). The extraction solution was collected after 10 minutes, and this process was repeated two additional times. The final peptide extraction was completed by incubating gel bands with 100% acetonitrile for 1-2 minutes. Extracted peptides were dried to completeness in a vacuum concentrator, then reconstituted in 100 μL of 0.1% formic acid and loaded onto C18 StageTips(Empore; 66883-U). The tips were washed twice with 50 μL of 0.1% formic acid, and peptides were eluted with 50% acetonitrile/0.1% formic acid, before drying in a vacuum concentrator.
Dried peptides were reconstituted in 0.1% formic acid/3% acetonitrile and analyzed on a Q Exactive mass spectrometer (Thermo Scientific) coupled online to an Easy-nLC 1000 UPLC-ultra performance liquid chromatography (Proxeon). Online chromatographic separation was completed using a capillary column (360 μm o.d. × 75 μm i.d.) with a 10 μm integrated emitter tip and self-packed with 23 cm of 1.9 μm ReproSil-Pur C18-AQ resin (Dr. Maisch GmbH). The UPLC mobile phase A was 0.1% formic acid/3% acetonitrile and the mobile phase B was 0.1% formic acid/90% acetonitrile. A linear gradient flowing at 200 nL/min and starting at 6% B and increasing to 30% B over 82 minutes was used to separate peptides with a total run time of 120 minutes. The Q Exactive mass spectrometer was operated in the data-dependent mode such that after each MS1 scan, consecutive high energy collision dissociation MS2 scans were recorded on the 12 most abundant precursors. The resolution used to acquire MS1 and MS2 scans was 70,000 and 17,500, respectively. The MS1 advance gain control (AGC) target was set to 3×106 ions and the maximum ion time was set to 10 msec. MS2 scans were recorded with an AGC target of 5×104 ions with a maximum ion time of 120 msec, isolation width of 2.5 m/z, normalized collision energy of 25, dynamic exclusion time of 20 sec, underfill ratio of 5.0%, and peptide match and isotope exclusion enabled. Proteins and peptides were identified and quantified using the MaxQuant software package (version 1.3.0.5) (Cox and Mann, 2008). Data were searched against the UniProt human database. A fixed modification of carbamidomethylation of cysteine and variable modifications of N-terminal protein acetylation, oxidation of methionine. The enzyme specificity was set to trypsin allowing cleavages N-terminal to proline and a maximum of two missed cleavages was utilized for searching. The maximum precursor ion charge state was set to 6. The precursor mass tolerance was set to 20 ppm for the first search. The peptide and protein false discovery rates were set to 0.01 and the minimum peptide length was set to 6. Protein information was obtained from the MaxQuant Protein Groups table. Proteins identified as reverse hits or contaminants were removed from the dataset. The results of the mass spectrometry analysis can be seen in Table S2. For assessment of significance, we z-scored the log ratios for all proteins (TableS2, Column I), followed by a one-sided z-test for proteins with log2 FC > 1. The resulting p-values for these proteins were adjusted for multiple testing using the Benjamini-Hochberg (BH) FDR approach (TableS2, Column K, used for Fig.2C).
SNCA copy-number reduction from triplication SNCA iPSCs
CRISPR design:
Isogenic SNCA knock out series were obtained using the CRISPRs/Cas9 system. Guide RNAs targeting exon 2 (harboring ATG translational start-site of SNCA) of the SNCA gene were designed at http://crispr.mit.edu/ (Supp.Fig.5B). The gRNAs were cloned into PX458 (Addgene, plasmid #62988), a single plasmid containing both sgRNA and the Cas9 (pSpCas9(BB)-2A-GFP, following the protocol (Ran et al., 2013). CRISPRs were then tested in 293T cells and cutting efficiency was determined by Sanger sequencing and TIDE analysis (https://tide.deskgen.com: Supp.Fig.5C). Guide2: tgttttccagtgtggtgtaa, Guide3: gtaaaggaattcattagcca, Guide4: gccatggatgtattcatgaa. Guide RNA 3 was chosen for the final editing of SNCA copy reduction)
Transfection:
iPS cells were cultured to 70% confluency and dissociated into single cells using Accutase (StemCell Technologies, Cat. No, 07920). Cells were washed with DMEM/F12 1:1 medium to remove Accutase. 1.0x106 cells were transfected with 2.5 μg of the CRISPR/Cas9 plasmid PX459 using Lipofectamine 3000 Transfection Reagent (ThermoFisher, Cat. No. L3000015). The DNA-lipid complex was prepared following the Lipofectamine 3000 Reagent Protocol and incubated at room temperature for 15 minutes. The single cells were resuspended to a minimal volume (eg. 1.0x106 cells in about 50 μl medium) and the DNA-lipid complex was added dropwise to the cells. The cells were mixed by gently flicking the tube wall 2-3 times and and then incubated at room temperature for another 10 minutes. 2 ml prewarmed mTeSR1 medium supplemented with 10 μM Rock inhibitors was added. Cells were moved into one well of a Matrigel-coated 6-well plate. 48-hour post transfection, cells were subjected to cell sorting and GFP positive cells were collected and plated at clonal density (5k to 10k cells per 10 cm dish). In about 7-10 days, colonies were picked into a 96-well plate and expanded for genotyping. In total, 60 clones were selected for further analysis.
Genotyping:
DNA for genotyping was extracted using the prepGEM® DNA Extraction Kits (ZyGem, Cat. No. PT10050). PCR genotyping was performed using Phusion Green Hot Start II High-Fidelity DNA Polymerase (ThermoFisher, Cat. No. F537) following the manufacturer's instructions at an annealing temperature of 62 °C. The following screening primers were designed flanking the CRISPR targeted SNCA exon2 site: F 5’TAGCCAAGATGGATGGGAGATG and R 5’CCATCACTCATGAACAAGCACC, which was also used for Sanger sequencing. The indel rate was >80%. 19 clones with indels resulting in significant deletions and/or potential ORF shifts were further investigated via TOPO cloning using TOPO TA Cloning Kit for Subcloning (ThermoFisher, Cat. No. 450641), single clone PCR and Sanger sequencing. iPSC cells after Cas9 editing were G-band karyotyped and checked for BCL2L1 locus copy number, a hotspot on chromosome 20 that confers growth advantage to pluripotent cells.
Evaluation of knock-out level:
Candidate knock out clones were transfected with PiggyBac TRE-NGN2-puromycin, then transdifferentiated to neurons (as described below). SNCA expression level were determined by qRT-PCR using TaqMan primer Hs00240906_m1 and western blotting using a monoclonal Antibody to αS (4B12) (Thermo Fisher, Cat. No. MA1-90346).
SNCA-A53T iPSC reprogramming and genetic correction
The source fibroblasts were from a female Contursi Kindred patient harboring the SNCA A53T mutation female gender, age 49)(Chung et al., 2013). The iPSC were generated from fibroblasts obtained from a patient skin biopsy at Boston University. A53T fibroblasts were collected under BU protocol (#H-27479) and then cultured/reprogrammed/differentiated under MGH protocol (#2009P000775) and MIT COUHES protocol (#0807002834). The line was licensed from Whitehead to Yumanity and thereafter to BWH. The fibroblast to iPSC reprogramming was performed at the Harvard Stem Cell Institute iPS Core following a microRNA-enhanced mRNA reprogramming method by combining Stemgent mRNA Reprogramming kit (00-0071) and Stemgent microRNA Booster Kit (00-0073) based on the manufacturer’s protocol. All the QC was followed according to NYSCF protocols (Pauli et al., 2015). The corrected clone (Corr28) and the original clone was PiggyBac transfected with VK_1018 plasmid all in one piggyBac(see below and TableS3 for the plasmid). Differentiation and assays were performed as described below, similar to SNCA triplication line.
Ngn2 mediated neuronal differentiation of iPSCs
All-in-one PiggyBac plasmids:
The following gene blocks were inserted between the PiggyBac inverted repeats in 5’- >3’ order: 5' PiggyBac ITR, iA4 insulator sequence, bGH poly(A) signal, Blasticidin cassette-2A linker-rtTA4 CDS (2A:self-cleaving peptide), hPGK promotor, UCOE insulator, Tet Response Promotor, Gateway Cloning Cassette, Internal Ribosome Entry Site, NGN2-2A-linker-Puromycin cassette, Woodchuck Proximal Response Element (WPRA), hGH polyA signal, cHS4 insulator, iA2 insulator, 3' PiggyBac ITR. A Thymidine Kinase expression cassette was inserted into the backbone of the plasmid for negative selection of the non-transposition random insertions. Chromatin insulating factors and both positive and negative selection markers improves the functionality of this plasmid. This final plasmid (VK_1018) is used for transgenesis of iPSCs. Please see Fig.5A for the depiction of this construct. A modified version of this construct designed to transfer SNCA-Avi-BirA system was similarly generated. In this latter case, the construct expressed a Tet-On module (4th generation), dox- inducible Ngn2, αS-Avi (or αS only as a negative control) with an internal ribosomal entry site (IRES) element along with a BirA ligase that is expressed in mature neurons under the Synapsin promoter(see Fig.5C for depiction of this construct).
NGN2 transgenesis into iPSCs by PiggyBac vector:
One day before transfection, one confluent well of iPSCs on a 6-well plate was dissociated with Accutase (Invitrogen: E124759) and single-cell dissociated. The iPSCs were transferred to a 15-mL conical tube and centrifuged at 800 rpm for 3 minutes. The iPS cell pellet was re-suspended, counted and seeded at 1.5 million per well in 2 mL/well StemFlex with 10 μM Rock Inhibitor in a new Matrigel-coated 6-well plate. On day of transfection, a transfection mixture was prepared with 2 μg of the PiggyBac construct, 1.5μg transposase plasmid, and 10.5μL TransIT-LT1 Transfection Reagent (Mirus: MIR2304) in 200μL Opti-MEM medium (Gibco: 31985062) per transfection. The transfection mixture was left in the hood for 20 minutes at room temperature. For a well of iPSCs at ~70-80% confluence and in a single layer, old media was replenished with 2mL fresh StemFlex supplemented with 10μM Rock Inhibitor (PEPROTECH: 1293823-50MG). Subsequently, the 200μL transfection mixture was added to the well evenly and dropwise. Transfected cells were incubated at 37°C for 6 hours without disturbance, after which the transfection medium was replaced by 2mL fresh StemFlex supplemented with 10 μM Rock Inhibitor. Media change with 2mL fresh StemFlex supplemented with 10μM Rock Inhibitor was performed daily. Two days post transfection, 5 μg/mL Blasticidin was added to the StemFlex medium to select for cells that successfully incorporated the backbone of the PiggyBac plasmid. The Blasticidin selection was continued for 5 days and media change were performed daily. In cases in which more than 80% cells died within the 5 days, Blasticidin selection was paused until the well become confluent for further expansion. Beyond seven days post transfection, Rock Inhibitor was taken out of the culturing media. Large batches of early-passage Ngn2 iPSC lines were generated and frozen down to minimize genetic drift and instability of PiggyBac-transfected polyclonal lines. From these frozen starter lines, iPSC lines were not passaged more than 3 times before differentiation.
iPSC neuronal differentiation up to Day0:
Our differentiation protocols are described in relation to the day of dox: Day0. 6-well plates were coated with 1mL of 80 μg/mL Matrigel in DMEM/F12 (Gibco: 11330-032) at a minimum of two hours prior to cell seeding. iPSCs previously frozen in 10% DMSO (Santa Cruz: SC-358801) in KnockOut Serum (Gibco: 10828-028) were thawed into 2mL StemFlex (Gibco: A33493-01) with 10 μg/mL Rock Inhibitor (PEPROTECH: 1293823-50MG) per well in the Matrigel-coated 6-well plate. Cells were allowed to recover overnight at 37°C. Complete media change with fresh StemFlex was carried out for iPSCs every day. As iPSCs reached ~70% confluency (usually within one to two days post thawing), Blasticidin (InvivoGen: ANT-BL-1) was added to StemFlex at 5 μg/mL concentration to select for cells with the PiggyBac backbone. To match confluency levels of iPSCs and correct for distinct growth rates across lines, iPSC lines were passaged at different ratios as they reached ~90% to 100% confluency. To passage iPSCs, each well on a 6-well plate was washed with 1mL DPBS-EDTA (1 mM) (Ultrapure 0.5M EDTA, pH8.0 (Invitrogen: 15575-038) DPBS (Gibco: 21-031-CV)). The iPSCs were incubated in another 1mL of 1mM EDTA in DPBS at room temperature for 3 to 4 minutes, depending on the confluency. 1mM EDTA in DPBS was removed and iPSCs were lifted from the wells in 1mL StemFlex and distributed across wells. Immediately before Day 0 of induced cortical neuronal differentiation, control and experimental iPSC lines were kept at similar confluency to control for growth stage and cell cycle.
iPSC neuronal differentiation after Day0:
On Day0 of differentiation, iPSCs were washed with DPBS, then dissociated with Accutase (Invitrogen: E124759) and re-suspended in StemFlex with 10 μg/mL Rock Inhibitor and 0.5 μg/mL dox (Sigma: D9891-5G). The iPSCs were counted and seeded at 10 million per Matrigel-coated 10-cm plate or ~1.25-1.5 million per well of a Matrigel-coated 6-well plate. Starting from Day 1, StemFlex medium was completely removed and replaced with Neurobasal Complete medium with 0.5 μg/mL dox and 5 μg/mL Blasticidin. Neurobasal Complete formulation: 500mL Neurobasal (Gibco: 21103049), 10mL of B27 supplement (Gibco, 17504044), 5mL N2 supplement (Life Technology: 17502-048), 5mL Non-Essential Amino Acids (Gibco: 11140-050), 5mL GlutaMAX (Gibco: 35050-061), and 5mL Penicillin-Streptomycin (Gibco: 15140-122)). From Day 2 to Day 6, complete media change was performed daily with 0.5 μg/mL dox and 5 μg/mL Puromycin (InvivoGen: ANT-PR-1), which selects for cells expressing the Ngn2 construct, in Neurobasal medium. On Day 7, differentiated neurons were lifted with Accutase, counted, and re-plated into final plates. For routine biochemistry, Poly-L-Ornithine/Laminin pre-coated multi-well plates (Corning: 354658 or 354659) were used for differentiation after day 7. For imaging, 96-well glass bottom imaging plates (Brooks: MGB096-1-2-LG-L) were coated with 0.1%(v/v) Poly-ethylenimine in 50 mM Borate buffer (PEI; Fisher: 24765-1, 20X Borate Buffer; Fisher: 28341) overnight at 4°C. Following overnight PEI coating and at least two hours prior to re-plating Day 7 neurons, 96-well plates were washed twice with distilled water and once with 1X DPBS, as PEI is toxic to the cells. Washed plates were further coated with Laminin (Sigma: L2020-1MG) in 1X DPBS with Magnesium and Calcium (Gibco: 14040-133) at 37°C. Final concentration for laminin was at least 5 μg/ml. Neurons were re-plated in the final plates at the same density as the seeding density on Day 0. For 96-well plates, Day 7 neurons were seeded at 30,000-50,000 cells per well (higher densities cause clustering making image analysis difficult).
Day7 seeding medium composition was Neurobasal complete medium supplemented with 0.5 ug/ml dox and 10 uM Rock inhibitor. On Day 8, an equal volume of Neurobasal complete medium supplemented with BDNF(Peprotech: 450-02), GDNF(Peprotech: 450-10), cAMP(Sigma: D0260-100MG), Laminin, and AraC was added (final concentrations were: 2x Laminin; 2 ug/ml, 2x cAMP; 1mM, 2x BDNF; 20 ng/ml, 2x GDNF; 20 ng/ml, 2x AraC; 1uM). From Day8, media change was performed every three days. On Day 11, a final complete media change was performed with 1:1 Neurobasal/Neurobasal Plus medium supplemented with BDNF, GDNF, cAMP and Laminin. From Day 14, half media changes with Neurobasal Plus medium supplemented with BDNF, GDNF, cAMP, and Laminin were carried out until Day 56 (Week 8) at the latest. Note that when Neurobasal plus medium was used, B27 Plus supplement (Gibco, A3582801) was used to make the complete media. Neurons in 6-well or 24-well plates were washed and harvested with 1X DPBS. Those neurons were pelleted at 400g, flash frozen in liquid nitrogen, and stored at −80°C for subsequent assays.
Immunofluorescence (IF) in iPSC-derived human neurons
Day 21 (three-week-old) neurons seeded on PEI and Laminin coated 96-well glass bottom microplates were fixed in 4% PFA (16% Paraformaldehyde; EM Grade (EM Sciences; Cat#: 15710; Lot:180910-04) in 1X DPBS with 20% Sucrose (10X DPBS, Invitrogen: AM9625) for 15 minutes at room temperature, washed twice with 1X DPBS, and stored in 0.02% NaAzide in 1X DPBS and sealed with aluminum tape (Corning® 96-well Microplate Aluminum Sealing Tape, 6570) at 4°C if not immediately proceeded to subsequent IF and imaging. To start the IF process, the fixed neurons were further washed three times with 1X DPBS for 5 minutes each. The neurons were then permeabilized (PBS 0.5% Triton X-100) for 15 minutes and blocked (5% BSA, 0.05% Triton-X100 (Sigma: T8787)) for 1 hour @RT. Primary antibody incubation (200 μl blocking solution with primary antibody) was carried out in dark 4°C overnight, no shaking. The neurons were washed 3 times with 1X DPBS for 5 minutes each @RT and incubated for 1 hour @RT in dark with secondary Alexa conjugated (488 nm or 594 nm) antibodies (1:1000x diluted in blocking solution). The neurons were washed once with 1X DPBS and incubated for 10 minutes with PBS-Hoechst solution (2000x fold dilution to PBS from commercial stock, Hoechst 33342, Invitrogen H3570) at @RT, and then further washed twice for 5 minutes each. The plate was sealed with aluminum tape and imaged immediately. The images were acquired by Nikon Eclipse Ti inverted microscope, with 40X Plan Apo dry objective (MRD0045) and Andor Zyla 4.2P sCMOS camera (No binning, 200 MHz readout rate, 12 bit & Gain4 dynamic range). For cultured cortical neurons, 7 Z-stacks were acquired within a 2 μm range. For image processing, FIJI software was used. Separate channel stacks were Z-projected (maximum intensity) and ROIs (Region of interest) were selected from each field manually unless otherwise stated.
Proximity Ligation Assay (PLA) in iPSC derived human neurons
Duolink™ In Situ Orange Starter Kit Mouse/Rabbit (Sigma: DUO92102) was used for this assay. Same 96-well plate was used and identical IF procedures were performed until the permeabilization step, after which the permeabilized neurons were blocked in Duolink™ Blocking Solution (80 μL/well; vortex well before use) for one hour at 37°C (in a Tissue Culture incubator). Primary antibodies were diluted in the Duolink™ Antibody Diluent (vortex well before use) to normal IF concentrations (refer to antibody Table S5 for details) and 80 μL of corresponding diluted primary antibody solution was added to each well. For PLA negative controls only one of the primary antibodies was processed. The plate was sealed tight with aluminum tape to prevent dehydration and incubate at 4°C overnight. 1X Wash Buffers A and B were made according to Duolink™ manufacturer protocol. In short, each wash buffer pouch was dissolved in 1L autoclaved MiliQ water. The wash buffers were stored in dark at 4°C and were brought to RT before use. Following overnight primary antibody incubation, the neurons were washed twice with 1X Wash Buffer A (200 μL/well) @RT for 5 minutes each. PLUS and MINUS PLA Probes were diluted 1:5 in pre-vortexed Duolink™ Antibody Diluent accordingly. The neurons were incubated in the diluted PLA probe solution for one hour at 37°C and subsequently washed twice with 1X Wash Buffer A @RT. 5X Ligation Buffer was thawed and diluted to 1X with Ultrapure distilled water (Invitrogen: 10977-015) immediately before use and Ligase was added to the freshly made 1X Ligation Buffer at 1:40 dilution. The neurons were incubated in the ligase solution for 30 minutes at 37°C. Following the ligation step, the neurons were washed twice with 1X Wash Buffer A @RT. 5X Amplification Buffer was thawed and diluted to 1X with UltraPure distilled water immediately before use and Polymerase was added to the freshly made 1X Ligation Buffer at 1:80 dilution. The neurons were incubated in the ligase solution for 100 minutes at 37°C. After amplification, the neurons were washed twice with 1X Wash Buffer B (200 μL/well) @RT for 10 minutes each and further washed once with 0.01X Wash Buffer B (diluted in UltraPure distilled water) @RT for 2 minutes. The neurons were then incubated with PBS-Hoechst solution (2000x fold dilution to PBS from commercial stock, Hoechst 33342, Invitrogen) at @RT for 10 minutes, and further washed with 1X DPBS twice for 5 minutes each. The plate was sealed with aluminum tape and imaged immediately.
High content image acquisition for iPSC derived neurons
High content image acquisition (for IF and PLA) was performed on the aluminum tape-sealed, glass-bottom 96-welll plates with the High Content (HC) Module of Nikon Eclipse Ti inverted microscope, with 40X Plan Apo dry objective (MRD0045) and Andor Zyla 4.2P sCMOS camera (No binning, 200 MHz readout rate, 12 bit & Gain4 dynamic range), which matches manual acquisition settings using the same Nikon microscope. Z-stack settings were also kept the same as used in manual acquisitions for cortical neurons and HEK293 cells, respectively. A new project was set up for each well so that there was no plate movement across wells. For consistency, same exposure time and Zyla settings was kept identical for all acquired channels across each experimental group. ~40-70 images (positions or field of image) were acquired for each well in a regular pattern and in a meandering direction within a centrally restricted area of the well to avoid edge effect. X/Y distance between positions was wide enough with no overlap to ensure that each position was well isolated and statistically independent for subsequent analysis. The Nikon Perfect Focus System (PFS) was used for all HC acquisitions to ensure every image taken was well in focus, while autofocus was kept off to avoid potential confusions and variabilities. Z-position and PFS offset were set up for each well manually. PFS was set to automatically turn on as the plate moving across positions and to stay off during acquisition. FIJI macros (custom written) were used to analyze HC images. file and to perform intensity projection (Extended Depth of Focus, i.e., EDF) for individual channels. For either PLA puncta counting or P-body puncta counting in neurons, each imaging field went through these processes;
High content data was split into individual imaging fields
z-Stacks were projected with EDF (Extended Depth of Focus).
A cell mask is created based on Hoechst staining and 5um perimeter of the nucleus. For neurons, we could not find a reliable neurite mask. The high density of the neurons, their clustering in the later days of culturing made individual neurite masking impossible. The most reliable counts of P-bodies or their PLA signal, in our estimation, were the ones around the nucleus. Furthermore, the neuritic debris could also auto fluoresce, making it harder to differentiate signals from false positives.
Very bright nucleus signals were omitted as they represent dead cells
Puncta ROIs were counted within the cell mask
The data per each image is always represented as total puncta counts / Cell ROI counts. Therefore, the data is represented as the average density of puncta per image.
Generation of αS PFFs (Preformed fibrils)
For PFF generation, Michael J.Fox Foundation guidelines were followed. Purified and lyophilized αS monomers were a kind gift from Tim Bartels. The lyophilized monomers (1 mg) were resuspended in filtered 150 ul PBS and centrifuged for 10 minutes at full speed to get rid of the residual large aggregates. The supernatant was transferred to a fresh protein-low binding tube. The protein concentration was measured both by BCA assay and nanodrop (parameters; E/1000= 5960.00 and MW = 14.50) with several technical repeats and geometric mean of two assays were accepted as the protein concentration (3.518 mg/ml). The resuspended protein was put in a thermo-shaker at 37°C with heated lid option to avoid condensation and shook for 1 week at constant speed of 1000 rpm. The aggregation was visually checked after the third day each day for a typical cloud formation indicating protein condensation. After a week, the fibrils were aliquoted in small volumes to protein low binding tubes, dried on dry ice and kept at −80°C until further use.
For the fibril application on the cells, the PFFs were first sonicated to generate toxic protofibrils with the following settings: (Bioruptor, 4°C, High Setting, 10 cycle, 30 seconds ON, 30 seconds OFF). To check the status of sonicated and unsonicated PFFs, TEM images were taken. For TEM imaging, 4μl of the sample was adsorbed for 1 minute to a carbon coated grid (EMS www.emsdiasum.com order# CF400-CU) that had been made hydrophilic by a 20 or 30 second exposure to a glow discharge (25mA). Excess liquid was wicked on a filter paper (Whatman #1). The grid was floated briefly on a drop of water (to wash away phosphate or salt), wicked on a Whatman filter paper and then stained with 0.75% uranyl formate (EMS catalog # 22451) or 1% Uranyl Acetate (EMS catalog # 22400) for 20-30 seconds. After removing the excess stain with a filter paper, the grids were examined in a JEOL 1200EX Transmission electron microscope or a TecnaiG2 Spirit BioTWIN and images were recorded with an AMT 2k CCD camera.
ActinomycinD Pulse time course in iPSC induced neurons
The experiment was performed on SNCA 4 copy and SNCA 2-copy (CRISPR corrected isogenic line, as described in detail above). The genotypes of the cell lines were ensured by western blot. ActD (final concentration 10 μg/ml) was applied on day21 neurons. Time points at 0 hour (no addition of ActD), 1,3,6 and 12 hours were taken for RNA isolation. In total 60 samples (2 genotypes X 5 time points X 2 treatment (PFF) X 3 replicates) were processed for RNA isolation with Trizol method. Each sample comes from a well of 24 well plate with ~50K neurons. For the PFF treatment, Day14 neurons were treated with 5 ug/ml sonicated PFF. Each well was incubated with 1 ml of Trizol and rotated on an orbital shaker for 10 minutes. The Trizol was kept at −80°C until processing. The RNA from Trizol mix was recovered by PureLink RNA Mini kit (Invitrogen, cat#: 12183020) with on column DNAse digestion. Additionally, initial viscosity was removed by Qiashredder. The purity, concentration and the integrity of the RNA samples were confirmed with NanoDrop and TapeStation. 1 μl of total RNA was diluted 20-fold and measured with Agilent TapeStation 2200 according to manufacturer’s protocol using regular RNA Screen Tapes (Agilent: 5067-5576) or measured with nanodrop. 12 samples were taken to estimate the RIN value of the general quality of the samples (all RIN values were above 9.5). For library preparation, 300 ng of the total RNA was used from each sample. Sample libraries were prepared for RNA-Seq using KAPA Biosystems KAPA mRNA Hyperprep Kit, according to manufacturer’s directions. Briefly, total RNA (300 ng per sample) was enriched for polyadenylated sequences using oligo-dT magnetic bead capture. The enriched mRNA fraction was then fragmented, and first-strand cDNA was generated using random primers. Strand specificity was achieved during second-strand cDNA synthesis by replacing dTTP with dUTP, which quenches the second strand during amplification. The resulting cDNA was A-Tailed and ligated with indexed adapters. Finally, the library was amplified using a DNA Polymerase which cannot incorporate past dUTPs, effectively quenching the second strand during PCR. Indexing was achieved using the KAPA Dual Indexing plate.
RNA sequencing data analysis pipeline
RNA-seq raw files in FASTQ format were processed in a customized pipeline. For each sample, we first filtered out reads that failed vendor check or are too short (<15nt) after removing the low-quality ends or possible adaptor contamination by using fastq-mcf with options of “-t 0 -x 10 -l 15 -w 4 -q 10 -u”. We then checked the quality using FastQC for the remaining reads. Reads were then mapped to the human genome (GRCh38/hg38) using STAR (Dobin and Gingeras, 2015) (v 2.7.3) by allowing up to 10% mismatches per pair and 100 multiple hits. Gene expression levels were quantified in both FPKM (Fragments Per Kilobase of transcript per Million mapped reads) and in raw reads count. FPKM were estimated using cufflinks (Trapnell et al., 2012) (v2.2.1) by enabling multi-mapped read correction and masking out the reads mapped to the mitochondrial genome, rRNAs, and tRNAs. Raw reads counts were estimated by htseq-count (Anders et al., 2015)with the parameters of “-m intersection-strict -t exon -i gene_id -s no”. We used GENCODE (v37) as gene annotation and GRCh38.primary_assembly.genome.fa downloaded from EBI FTP as reference genome.
Sample QC based on RNA-seq data
We made RLE plot to detect sample outliers. In brief, for each gene and each sample, ratios are calculated between the expression of a gene and the median expression of this gene across all samples of the experiment. For each sample, these relative expression values are displayed as a box plot. Since it is assumed that in most experiments only relatively few genes are differentially expressed, the boxes should be similar in range and be centered close to 0. We also calculated pair-wise Spearman correlations of gene expression quantification across samples and measured the median correlation (D-statistics) for each sample. Samples with D-statistics markedly different from the rest of samples were deemed outliers. Moreover, we investigated the variance among the samples by running PCA based on the top 500 most variable genes.
Condition-specific time-series RNA-Seq analysis
To examine genes that show condition-specific expression pattern along the time points, we performed LRT (likelihood ratio test) by removing the condition:timepoint interaction term in the reduced model (e.g. full model as ~ condition + timepoint + condition:timepoint and reduced model as ~ condition + timepoint). The design above controls for differences between condition at time=0, and then tests to see if there are any differences at one or more time points after time 0. We tested this globally by including all time points and locally by only including two consecutive timepoints. Condition-specific time-series LRT tests were conducted for both genotypes (e.g., SNCA 2 copies vs. 4 copies) and PFF treatment (e.g., PFF+ vs. PFF). To test if any globe difference in slope between conditions (e.g., PFF+ vs. PFF−), we performed paired one-sided t-test of the slope between two consecutive time points for all genes. The statistical tests were conducted using DESeq2 (Love et al., 2014) (v1.30.1) in R (4.0.3).
Immunofluorescence assays in Post-mortem Brains
Acquisition of brain samples :
Post-mortem brain samples were acquired from Mayo Clinic. Two patients with PD diagnosis (male, 63, for SNCA gene duplication; male,57, for SNCA A53T) and two healthy control cases free from PD diagnosis (male,81 and male,78) were investigated. The details of the cases could be found in TableS7. Brain tissue was fixed in formalin and processed for paraffin embedding. Sections were cut at 6 micron and stored at room temperature for microscopy analysis.
Antibodies:
Anti-MAP2 antibody (Millipore, AB5543) used at 1:1000 concentration, anti-Edc4 (Abcam, ab72408) used at 1:200 concentration, anti-Dcp1a (Abnova, H00055802-M06), used at 1:200 concentration, anti-SNCA (BD Transduction, 610787), used at 1:500 concentration.
Immunofluorescence assay:
Immunofluorescence staining of the proteins of interest were performed in human brain tissue sections as previously described by Keeney et al.(Keeney et al., 2021). Briefly, human brain tissue sections were deparaffinized and, to quench autofluorescence, the sections were incubated in sudan black for 5 min at room temperature. The sections were then permeabilized and incubated with specific primary antibodies (see above for antibody dilutions) to the proteins to be detected. Fluorescent secondary antibodies were added to the reaction and incubated. Confocal microcopy for the detection of the proteins of interest was performed in MAP2-positive cells
Proximity Ligation Assay(PLA):
PLA (DUO92004-100RXN; DUO92002-100RXN; DUO92007-100RXN, Sigma-Aldrich) was performed in paraffin-embedded fixed tissue prepared for immunofluorescence, as described above. To detect neurons in brain, an immunofluorescence staining for MAP2 was performed before PLA, then samples were incubated with specific primary antibodies to the proteins of interest for PLA. Secondary antibodies conjugated with oligonucleotides were added to the reaction and incubated. Ligation solution, consisting of two oligonucleotides and ligase, was added. In this assay, the oligonucleotides hybridize to the two proximity ligation probes and join to a closed loop if they are in close proximity. Amplification solution, consisting of nucleotides and fluorescently labeled oligonucleotides, were added together with polymerase. The oligonucleotide arm of one of the proximity ligation probes acts as a primer for “rolling-circle amplification” using the ligated circle as a template, and this generates a concatemeric product. Fluorescently labeled oligonucleotides hybridize to the rolling circle amplification product. The proximity ligation signal was visible as a distinct fluorescent spot and was analyzed by confocal microscopy. Control experiments were run in parrallel by primary antibodies deletion. For more details and step-by-step protocol see(Keeney et al., 2021).
Fluorescence measurements:
Quantitative fluorescence measurements were made with an Olympus upright 3-laser scanning confocal microscope, taking care to ensure that images contained no saturated pixels. For quantitative comparisons, all imaging parameters (e.g., laser power, exposure, pinhole) were held constant across specimens. Olympus Fluoview 1000 Software was used for the analysis.
Statistical analyses:
Each result presented here was derived from 4 cohorts of human cortex specimen (2 controls, SNCA A53T mutation, SNCA duplication mutation). 4 – 5 sections for each cohort were analyzed. Each number is representative of the fluorescence intensity detected in ROIs plotted on single neurons (MAP 2 positive). For comparisons of multiple experimental conditions, 1-way ANOVA was used, and if significant overall, post hoc corrections (Bonferroni or Sidak) for multiple pairwise comparisons were made (GraphPad PRISM). P-values less than 0.05 were considered significant. All graphs show the statistical distribution of each point.
Religious Orders Study and Memory and Aging Project (ROS/MAP) analysis
Study subjects:
Postmortem data analyzed in this study were gathered as part of the Religious Orders Study and Memory and Aging Project (ROS/MAP) (Bennett et al., 2012a, 2012b; Jager et al., 2018), two longitudinal cohort studies of the elderly, one from across the United States and the other from the greater Chicago area. All subjects were recruited free of dementia (mean age at entry=78±8.7 (SD) years) and signed an Anatomical Gift Act allowing for brain autopsy at time of death. Written informed consent was obtained from all ROS/MAP participants and study protocols were approved by the Rush University Institutional Review Board.
Clinical evaluation:
A clinical diagnosis of Parkinson’s disease was determined at time of death by a neurologist with expertise in dementia, blinded to all postmortem data. To reach consensus on particular subjects, case conferences were held including one or more neurologists and a neuropsychologist. In addition, a global parkinsonian summary score was derived as the average of four separate domains calculated from the 26-item modified version of the motor section of the United Parkinson’s Disease Rating Scale (including bradykinesia, gait, rigidity, and tremor scores).
Neuropathological evaluation:
All subjects’ brains were examined postmortem by a board-certified neuropathologist blinded to clinical data. Brains were removed in a standard fashion as previously described (Bennett et al., 2006). Each brain was cut into 1cm coronal slabs. Slabs from one hemisphere, and slabs from the other hemisphere not designated for rapid freezing, were fixed for at least three days in 4% paraformaldehyde. Tissue blocks from eight brain regions were processed, embedded in paraffin, cut into either 6 micron or 20μm sections, and mounted on glass slides. Lewy body pathology was measured by immunostaining, as described(Schneider et al., 2012). Briefly, Lewy bodies in substantia nigra and cortex were separately identified, with only intracytoplasmic Lewy bodies indicating positive staining. To simplify the staging of pathology, the McKeith criteria (McKeith, 2006) were modified such that nigral predominant Lewy body pathology included cases with Lewy bodies in the substantia nigra without evidence of Lewy bodies in the limbic or neocortical regions. Limbic-type Lewy body disease included cases with either anterior cingulate or entorhinal positivity without neocortical Lewy body pathology. Neocortical-type Lewy body pathology required Lewy bodies in either midfrontal, temporal, or inferior parietal cortex with either nigral or limbic positivity, but often with both. Based on this, each subject was assigned to one of four mutually exclusive categories: 0 = no, 1 = nigral-predominant, 2 = limbic-type or 3 = neocortical-type Lewy body pathology. An additional binary indicator of Lewy bodies, present vs. absent overall, was also derived. Finally, the degree of neuronal loss in the substantia nigra was quantified in the mid to rostral midbrain near or at the exit of the 3rd nerve using H&E stain using a four-staged scale (0=none/rare/scattered, 1=mild, 2=moderate, 3=severe), as described(Buchman et al., 2012). Further descriptions of clinical and pathological outcomes are available at the Rush Alzheimer’s Disease Centre Research Resource Sharing Hub (https://www.radc.rush.edu).
Sample preparation and RNA sequencing:
Tissue for RNA sequencing (RNAseq) was dissected from dorsolateral prefrontal cortex (DLPFC) using a strand-specific dUTP method (Levin et al., 2010) with poly-A selection (Adiconis et al., 2013). Five micrograms of total RNA as measured by RiboGreen at a concentration of 50 nanogram/microliter with RNA Integrity Number (RIN) score of 5 or better were submitted for cDNA library construction. Sequencing was carried out using the Illumina HiSeq2000 with 101 bp paired end reads for a targeted coverage of 50M paired reads.
RNA sequencing data processing and quality control:
Ten batches of paired-end (101bp reads) RNAseq data were processed separately using the same pipeline, as previously published(Felsky et al., 2020). Briefly, 1) fastq file quality control was performed using FastQC v0.11.5 (default parameters), 2) STAR v2.5.3a (Dobin et al., 2013) was used to align reads (GRCh38.91 reference), 3) RSEM v1.2.31(Li and Dewey, 2011) was used to quantify expression from aligned BAM files, and 4) multiqc v1.5 (Ewels et al., 2016) was used to aggregate quality metrics from fastqc and Picard tools V2.17.4 (http://broadinstitute.github.io/picard/), 5) quality reports were examined for each batch and exclusion of samples was initially carried out according to manual identification of outlying samples primarily considering low numbers of aligned reads, excess GC coverage bias, high percentage of read duplicates, and abnormal distribution of read assignments across genomic annotations. For Post-quantification QC, expected counts were aggregated across all batches and used as input to limma (v3.38.3)(Ritchie et al., 2015) voom in R (v3.5.2) (Law et al., 2014). First, any genes with a median expected count lower or equal to 15 were excluded. Second, principal components analyses were performed on each batch separately, and subjects flagged for pruning if deviating from the sample median of any of the first 5 principal components by more than 3 interquartile ranges. Third, gene expected counts were log2 transformed with an offset of 0.5 and each gene evaluated for extreme outlying observations separately. For each gene, any log2(expected count) value deviating from the sample median by 3 interquartile ranges was coerced to its nearest most extreme value within that range. This resulted in a final set of 17 675 genes and 596 subjects for differential expression analyses.
Differential expression analysis:
For each PD-related outcome, the R (v3.6.3) limma package ‘lmFit’ function was used to model log2(expected counts) as a linear function including sequencing batch, age at death, sex, RIN, postmortem interval, percent duplicated reads, and median 3’ bias as covariates. Robust linear modeling was used (using Huber M estimators), allowing for a high number (10,000) of iterations to reach convergence. Significance for effects of pathological variables on gene expression was determined using empirical Bayes moderation (the ‘eBayes’ function).
Rank-Based Gene Ontology (GO) enrichment of differential expression results:
For each set of differential expression summary statistics, genes were ranked based on their moderated t-statistics. AUC-based enrichment was then performed using the R tmod package (version 0.46.2) (3rd and Domaszewska, 2016)to test for GO term enrichment including categories of biological processes, cellular components, and molecular functions(Ashburner et al., 2000). Thus, an AUC greater than 0.5 indicates a GO group enriched for overexpression, and an AUC less than 0.5 indicates a group enriched for underexpression in relation to each outcome measure. Annotations were limited to those including between 10-200 tested genes. GO annotations were extracted from the org.Hs.eg.db and GO.db (version 3.10.0) R packages, using the full background of genes passing QC for differential expression. unbiased FDR correction was used (q<0.05) to determine significant enrichment. To minimize redundancy between these broad categories, we limited our final aggregation of GO terms to biological processes only and condensed semantically-overlapping groups using the REVIGO(Supek et al., 2011) tool (dispensability <0.1) to enhance interpretability.
Human Genetics; BridGE analysis in PD and ALS cohorts
Validating P-body biological interactions in human genome-wide association studies:
To evaluate biological interactions that are associated with disease risk, we adapted the recently published method, BridGE (Fang et al., 2019). BridGE enables identification of statistical associations with disease risk through collectively analyzing SNPs linked to gene pairs of interest (in this case taken from a set including P-body genes and SNCA). The rationale is that association can be more readily detected by aggregating multiple individual SNP-SNP interactions mapping to the corresponding two genes than by considering SNPs within the genes taken individually. The software for this analysis is available at https://github.com/csbio/BridGE/tree/master/BridGE_genes.
GWAS data:
We obtained seven GWAS cohorts for PD including a total of 11,830 PD cases and 11,030 PD controls, and four GWAS cohorts for ALS disease including a total of 3,275 ALS cases and 11,556 ALS controls (TableS7). Six of the PD cohorts are downloaded from the dbGaP website, under phs000196.v3.p1 (PD-NGRC) (Hamza et al., 2010), phs000089.v3.p2 (PD-NIA)(Fung et al., 2006; Simon-Sanchez et al., 2007), phs000048.v1.p1 (PD-LEAPS) (Maraganore et al., 2005), phs000918.v1.p1 (PD-IPDGC) (Auer et al., 2012; (IPDGC) and (WTCCC2), 2011; Nalls et al., 2014, 2015), phs000126.v2.p1 (PD-CIDR) (Nichols et al., 2005), phs000394 (PD-APDGC)(Beecham et al., 2015). The last PD cohort is from The Accelerating Medicines Partnership program for Parkinson's disease (PD-AMP). The ALS cohorts are also downloaded from the dbGaP web site, under phs000127.v2.p1 (ALS-Irish)(Cronin et al., 2008), phs000344.v1.p1 (ALS-Finland)(Laaksovirta et al., 2010), and phs000101.v3.p1(Chiò et al., 2009). For the phs000101.v3.p1 dataset, we merged data genotyped with the Illumina HumanHap550 and the Human610 platforms and analyzed it as a single cohort (ALS-NIH-HumanHap). The last ALS cohort was formed from those samples from phs000101.v3.p1 that were genotyped with Illumina OmniExpress (ALS-NIH-OmniExpress).
P-body gene set:
We included the following 53 P-body-related genes in our analysis: PNRC2, DCP2, EDC3, EDC4, DCP1B, DCP1A, NBDY, 4E-T, DDX6, EIF4ENIF1, LSM14A, LSM14B, PATL1, IGF2BP2, PAN2, PAN3, CNOT1, CNOT10, CNOT11, CNOT2, CNOT3, CNOT4, CNOT6, CNOT6L, CNOT7, CNOT8, CNOT9, XRN1, LSM1, LSM2, LSM3, LSM4, LSM5, LSM6, LSM7, AGO2, TNRC6A, TNRC6B, TNRC6C, MOV10, FMR1, AGO1, ZCCHC3, SMG7, UPF1, UPF2, APOBEC3G, APOBEC3F, ZCCHC11, SYNCRIP, HNRNPU, IGF2BP1, DHX9
GWAS data processing:
We followed a standard GWAS data processing procedure. First, we removed the SNPs with a minor allele frequency less than 0.05, missing genotype rate greater than 0.02, or those for which a Hardy-Weinberg equilibrium test resulted in p < 10−6. We also removed samples that had a missing genotype rate greater than 0.02, or with genetic relatedness to other samples (proportion IBD score greater than 0.2). We also removed outlier samples based on multi-dimensional scaling (MDS) analysis using the European ancestry from 1000 genome project as reference (Fang et al., 2019). Then, we matched each case with a control to control confounding due to population structure as previously described in (Fang et al., 2019). The number of cases and controls after data processing for each individual cohort can be found in Table S7. We binarized the data based on two genetic models, recessive and dominant. We did not consider an additive genetic model since BridGE previously discovered very few significant pathway-level interactions when using an additive model (Fang et al., 2019).
SNP-to-gene mapping:
A SNP is assigned to a gene based on its physical location in the genome. We assigned each SNP to any upstream and downstream gene within 50kb.
Measuring association through SNP-SNP pairwise analysis:
Genetic associations are identified by considering the joint presence of SNPs in genes, taken pairwise, among which biological interactions are known to occur. These associations are not identifiable by considering individual SNP contributions to phenotype. We applied a modified version of the hygeSSI interaction score originally proposed in BridGE (Fang et al., 2019). Given a binary-coded SNP pair (Sx, Sy) and a binary class label C, the modified-hygeSSI measures the genetic interaction between two SNPs Sx and Sy as follows:
Where P11, P10, P01, and P00 are hypergeometric test p-values that represent the strength of the genotype-phenotype association for corresponding binarized genotype combinations 11, 10, 01, and 00. α is a nominal p-value threshold (α = 0.05).
Construction of SNP-SNP interaction networks:
For each SNP pair, we assume that biological interactions will be reflected through different types of gene-gene interactions including recessive-recessive (RR), recessive-dominant (RD), dominant-dominant (DD), as well as a combined interaction score that is the maximum of the three. We then built a SNP-SNP interaction network for each of the RR, RD, DD, and combined interactions. More details on network construction are described in (Fang et al., 2019).
Measuring gene-gene interactions and evaluating significance:
For each pair of genes evaluated, we measured putative gene-gene interactions as follows. First, a gene-gene interaction density score was summarized by averaging all SNP-SNP pairwise interaction scores associated with the two genes, excluding any SNPs that are linked to both genes. We also removed interactions for gene pairs that show evidence of Linkage Disequilibrium (LD). Specifically, if two SNPs X and Y are in LD (r2 > 0.2), and they show evidence of shared interactions (genetic interaction profile cosine similarity > 0.5), we assigned a genetic interaction score of 0 to all X or Y-associated SNP pairs. Second, we computed a p-value associated with the density of SNP-SNP interactions observed between each pair of genes by comparing the observed distribution of SNP-SNP scores with a background distribution consisting of all SNP-SNP interactions including either of the genes a using Wilcoxon rank-sum Test. Third, we permuted the case-control labels 1000 times to generate a null distribution of the two scores for each gene-gene interaction (the density and the rank-sum p-value) to derive a single, empirical p-value per gene pair. Finally, we apply the Benjamini–Hochberg method on these empirical p-values to control FDR, accounting for all gene pairs tested. The resulting FDR per gene pair is the focus of our results.
Replicating putative gene-gene interactions:
For each disease cohort, we discovered gene-gene interactions from each of the SNP-SNP interaction networks constructed based on different genetic models (RR, RD, DD, combined). Replicated gene-gene interactions were defined as interactions that satisfy FDR <= 0.1 in at least one of the SNP-SNP interaction networks in one cohort and have a permutation p-value <= 0.05 in at least two other cohorts. We report such interactions for each disease.
Discovery of pathway-level associations among SNCA and P-body genes in PD:
At a cutoff of FDR < 0.1, under our model assumptions, we found interactions among SNCA and a total of 28 P-body genes, taken pairwise, across the seven analyzed PD cohorts (Table S7). The number of discoveries from each individual cohort can be found in Table S7. Among these discovered interactions, 23 of them could be replicated in at least two independent cohorts, and 20 of them were associated with increased risk for PD. This degree of connectivity was statistically significant when we compared it with the number of replicated interactions derived from 1000 sample permutations in which we repeated the entire discovery process with the same gene-pairs (permutation p-value=0.001).
Analogous analysis between FUS and P-body genes in ALS:
At a cutoff of FDR < 0.1, we found analogous interactions between FUS and a total of 14 P-body genes across the four analyzed ALS cohorts (Table S7). The number of discoveries from each individual cohort can be found in Table S7. None of these 14 interactions could be replicated in two or more additional cohorts. Three of these 14 increased-risk interactions could be replicated in one independent cohort.
Analogous analysis between TARDBP and P-body genes in ALS:
At a cutoff of FDR < 0.1, we found analogous interactions between TARDBP and a total 18 P-body genes across the four analyzed ALS cohorts (Table S7). The number of discoveries from each individual cohort can be found in Table S7. Among these discovered interactions, only 1 decreased-risk interaction meets our replication criteria in at least two independent datasets (permutation p-value=0.09). Four of these 18 interactions could be replicated in one independent cohort.
BridGE acknowledgements:
The authors thank the NIH-designated data repository (dbGaP: The database of Genotypes and Phenotypes), and the submitting investigators who submitted data from the original studies to the dbGap repository: phs000196.v3.p1 (Dr. Haydeh Payami), phs000089.v3.p2 (Dr. Robert Brown, Dr. David Simon, Dr. Dennis Dickson, Dr. Matthew Farrer, Dr. Emily Gorbold, Dr. Ira Shoulson, Dr. John A. Hardy, Dr. Laura Marsh, Dr. Michael Okun, Dr. Ted Roghstein, Dr. Zbigniew Wszolek), phs000048.vl.pl (Dr. Demetrius M. Maraganore), phs000126.v2.p1(Dr. Tatiana Foroud, Dr. Richard H. Myers), phs000918.v1.p1 (Dr. Andrew B. Singleton, Dr. Mike A. Nalls), phs000394.v1.p1 (Dr. Jeffery Vance), phs000127.v2.p1 (Dr. Orla Hardiman, Dr. Brendan Loftus, Dr. Daniel G. Bradley, Dr. Alex Evans), phs000344.v1.p1 (Dr. Bryan J. Traynor, Dr. Pentti J. Tienari), phs000101.v3.p1 (Dr. Bryan J. Traynor), and their primary funding organizations including National Institutes of Health, National Institute of Neurological Disorders and Stroke, National Institutes of Health, National Institute on Aging, Michael J. Fox Foundation, International Parkinson Disease Genomics Consortium, USAMRAA, Muscular Dystrophy Association, Science Foundation Ireland, Microsoft Research, the ALS Association, The Helsinki University Central Hospital, the Finnish Academy, the Finnish Medical Society Duodecim, Kuopio University. The authors also thank the AMP PD, a public-private partnership, which is managed by the FNIH and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research, the National Institute of Neurological Disorders and Stroke, Pfizer, and Verily.
Rare variant trend test (RVTT)
To answer whether an enrichment of rare variants in key P-body genes might confer risk of PD in human population, we applied a pathway-based association test, namely, the rare variant trend test (RVTT)(Bendapudi et al., 2022) to case-control PD datasets from AMP-PD. Traditionally, rare variant association tests collapse variants at the gene level by presence or absence to identify risk genes in a case-control cohort. However, due to relatively small sample sizes of PD cohorts and the burden of multiple hypotheses correction, the statistical power of such tests remains low. One promising way to increase the power of these tests is to assess association at the pathway level combining variants in multiple genes. But gene-level collapsing tests are not readily applicable to pathways. They look at the presence or absence of a variant, whereas most individuals, irrespective of their disease status, are likely to carry at least one variant in a large pathway.
The advantage of RVTT over traditional burden tests is that instead of considering only the presence or absence of variants in a pathway, it looks at the frequency of qualifying variants in the pathway. Under the null hypothesis, the test assumes that there is no linear trend in the binomial proportions of cases and controls across increasing number of rare variants in the pathway of interest. The alternative hypothesis indicates the presence of a linear trend. RVTT leverages Cochran-Armitage statistic (Cochran WG, 1954) to quantify this trend. Qualifying rare variants are selected using a variable threshold approach (Price et al., 2010). And the strength of significance is indicated by permutation-based p-values.
Analysis of rare variant burden of P-body genes in PD with RVTT
We analyzed the whole exome sequencing data from AMP-PD (2020 release) repository maintained by the NIH to assess the rare variant burden of P-body genes in PD. The two largest independent cohorts within AMP-PD are PDBP (Parkinson’s Disease Biomarker Program) and PPMI (Parkinson’s Progressive Marker Initiative), offering an opportunity for cross-validation within the same dataset. We excluded any individual belonging to the genetic registry, genetic cohort, SWEDD, and prodromal categories. We included only individuals of European ancestry in both datasets to remove any potential confounding effect of population structure. For each of the cohorts, our analysis started with a joint-called variant call format (VCF) file (assembly version: hg38). We kept only the variants that were bi-allelic, had a missing genotype rate of <= 10%, had a minimum read depth of >= 10x, and were from the “PASS” category (i.e., variants that passed standard quality filters defined by the GATK pipeline). After variant and sample level quality control, PPMI and PDBP datasets comprised 572 (PD: 391, Control: 181) and 1273 (PD: 823, Control: 450) individuals, respectively.
We annotated the functional consequences of the variants using SnpEff (v 4.3t) software. We excluded all non-coding, intronic, and intergenic variants, except the splice region ones from further analysis. We classified the remaining variants as synonymous, missense, splice donor, splice acceptor, splice region, stop-gained, stop-lost, start-lost, frameshift, in-frame insertion, and in-frame deletion variants. We termed the set of stop-gained, stop-lost, start-lost, frameshift, in-frame insertion, in-frame deletion, and splice region variants as loss-of-function (LoF) variants. Next, we annotated the vcf file with the gnomAD minor allele frequencies of variants and the in-silico predictions of deleteriouness of the missense variants by PolyPhen2 and SIFT from the dbNSFP (v4.0a) database using the SnpSIFT (v 4.3t) software. All missense variants that were predicted to be either “P” or “D” by PolyPhen2 or “deleterious” by SIFT were considered as “damaging missense” variants for our analysis. All “damaging missense” variants and “LoF” variants were considered together as “damaging” variants.
Since RVTT is a pathway-based test of association, we divided the P-body genes into two modules(i) CORE module, and (ii) DECAPPING module. The CORE module contains DDX6, EIF4ENIF1, LSM14A, LSM14B, PATL1, LSM1, LSM2, LSM3, LSM4, LSM5, LSM6, and LSM7 genes. The DECAPPING module contains PNRC2, DCP2, EDC3, EDC4, DCP1B, DCP1A, and NBDY genes.
RVTT was applied to test the rare variant burden of these P-body gene modules in both the PDBP and PPMI datasets. The test selected qualifying rare variants by varying the MAF cutoff up to 0.05 (Variable-Threshold approach: Price et al., 2010). We quantified the strength of association using permutation-based p-values (10,000 permutations). Four variant categories (damaging, damaging missense, neutral missense, and synonymous) were interrogated in both cohorts. The validity of significant results was confirmed by demonstrating that there is an accumulation of damaging and damaging missense variants in patients versus controls, but no statistically significant difference in proportions of synonymous and neutral missense variants within the same pathways in cases vs. controls.
Quantification and statistical analysis
Unless otherwise stated, plots were generated by GraphPad Prism. R packages used in ROSMAP analysis or BriDGE analysis are indicated in the respective methods section along with the statistical tests. All Python and related libraries were acquired through Anaconda distribution. Graphs in Supp.Fig.1H and Supp.Fig.5G were generated in the Jupyter Notebook by using Matplotlib, Seaborn, Pandas libraries. Unless otherwise noted, statistical tests were done in GraphPad Prism. Unpaired t-test was used to compare two groups. For multiple t-test comparisons, multiple hypothesis corrections were performed as indicated in the respective figure legends. For image analysis, FIJI distribution of ImageJ was used. For counting Edc4 or PLA puncta custom FIJI macros were written and these macros are available upon request.
Supplementary Material
Figure S1 (Related to Figure1). Details of perturbation screen (yTRAP) methodology and yeast genetics.
(A) Logistic growth curves of liquid cultures (measured by optical density at 600 nm absorbance) of yeast ND strains upon estradiol induction for 40 hrs. Measurements were taken every 15 minutes in four independent cultures. Dots represent mean (+/− SD, errors bars were usually invisible due to high reproducibility of OD measurements). Gray curves indicate no estradiol addition, orange curves indicate estradiol addition and thus ND protein expression.
(B) Optimization of estradiol induction (Neurodegenerative gene expression) interval: Diploids of αS x yTRAP strains (16 yTRAP sensors) were measured by flow cytometry for indicated time points. They were normalized to a ‘dark’ sensor which doesn’t give fluorescent output. Note that after 6 hours, the signals revert because the cells approach the end of log phase. We chose 6 hours as the maximum allowed time for toxicity.
(C) The SOD1(4AV) mutant was selected as the negative control for the screen. Selected sensors were crossed to a non-ND expressing strain (ZEM only) and SOD1(4AV) mutant. After 6 hours of induction no significant effect was measured between the sensors. Thus, instead of ZEM only strain, we chose to express a non-toxic SOD1(4AV) gene as our negative control.
(D) αS expression induced more perturbation in the RBP proteome than FUS protein. Correlation plots, along with linear regression line, of screen signals were shown for FUS versus SOD1 (Pearson correlation score is shown in the label) and FUS versus SNCA.
(E) Rescue of αS mediated toxicity with Sgn1 related genes. 39 proteins (TableS1) were overexpressed in αS expressing background. 10 rescue events were shown with duplicates for αS. SG: Stress granule related genes
(F) Rescue of αS mediated toxicity with the proteins in (E) were not recapitulated for FUS mediated toxicity.
(G) αS protein expression is not changed upon modifier expression in the mini overexpression screen. Western blots (2 biological replicates) were quantified from whole cell lysates from.
(H) Heatmap of yTRAP perturbation scores upon αS expression belonging to 24 genes were plotted (averaged log2 fold changes were normalized to Fus expression). Each row is an individual RBP sensor. Significant discoveries are indicated with asterics (p<0.005, two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 1%). These 11 discoveries were re-plotted in Fig.1E.
(I) Western blot of whole cell extraction from fly brains for αS levels. These fly lines were used for the locomotor activity assay shown in Fig.1H
Figure S2 (Related to Figure2). αS purification optimization in human cells
(A) IF signals with various commercial αS antibodies in HEK293 cell lines. Different αS antibodies are indicated above in wild-type HEK293 cells. Merge picture indicates 488 nm αS (green) channel and DAPI (magenta) stain. Scale bar = 10 μm
(B) Bead optimization for streptavidin pulldown. 500 μg of HEK293 Flp-In αS-Avi (+/− BirA ligase) was incubated with 10, 20 or 50 μl MyOneT1 magnetic streptavidin beads. Filled triangles indicate endogenous biotinylated proteins. 10 μl beads/500 μg of extract was considered optimal and used for subsequent analyses.
(C) αS and decapping module interactions can withstand high ionic strength. Increasing KCl concentrations were used in washing step of streptavidin pulldown. Edc4 and Edc3 can withstand up to 0.5M KCl, whereas the Ddx6 interaction is lost rapidly.
(D) αS and decapping module interactions are RNA-independent. During the wash step, the strep beads were incubated with Rnase cocktail for 15 minutes at room temperature. 3 independent pulldowns were blotted with the indicated antibodies.
(E) Mass spectrometry counts belonging to Fig.2A-C. Decapping modules are significantly enriched in αS-GFP pulldowns.
(F) Schematic of protocol for macro P-body pelleting according to Hubstenberger,2017 protocol. Macro P-bodies represent large RNP complexes that can be pelleted at 10000g, whereas the supernatant contains smaller complexes. Those smaller complexes can be further solubilized with nuclease digestions to enrich protein-held-together complexes.
(G) Many components of P-bodies can be found largely in non-pelleted smaller complexes. After gentle cell lysis and removing cell nucleus at 800g, macro-P-bodies (‘condensed granules) were pelleted at 10000g. The un-pelleted lighter P-body components were further solubilized by various nucleases (Benzonase, RQ1 Dnase, a cold optimal cryonase; non-specific nuclease). The nuclease digestion after 10000g pelleting was repeated both at 4°C and 25°C for the nucleases to function optimally.
(H) Ddx6 CRISPR KO western blot from whole cell lysates of HEK293 cells with three different guides.
(I) IF of Ddx6 CRISPR KO lines and dissolution of P-bodies. The first two panels (Ddx6 and Edc4) were also highlighted in Fig.2J
Figure S3 (Related to Figure3). αS mutational analysis indicate negative correlation with decapping module interactions and membrane binding
(A) Stepwise truncations of αS-Avi up to amino acid 100 was transfected into HEK293 cells and pulled down with streptavidin beads. Two technical replicates per each pulldown are shown.
(B) Representative western blotting of 10aa deletion mutants used in quantification of main Fig.3C.
(C) Repulsion of αS away from the membrane restores P-body binding. T59 and T81 in αS were exchanged for hydrophilic residues (E or K) and P-body binding (normalized to wild-type αS) was measured in 3 independent pulldowns.
(D) Representative western blotting of various αS mutants used in quantification of main Fig.3H-J
(E) Protein alignment of synuclein protein family. Imperfect repeats are demarcated.
Figure S4 (Related to Figure4). Decapping module proteins along with αS come in close proximity at the proximal region of Edc4.
(A) αS binds to the C-terminus of Edc4 after the low complexity linker (aa 535-974) region. The indicated large truncations of HA-tagged Edc4 were individually transiently transfected into αS-Avi(+BirA ligase) cell line and pulled down with streptavidin beads. Two independent pulldowns are shown side by side. Note that αS can pulldown endogenous Edc4.
(B) Serial deletions at the C terminus of Edc4 show that αS can only bind to Edc4 further down the aa1166.
(C) Proximal region deletion (Δ974-1265) on Edc4 abrogates αS-Edc4 interactions.
(D) Cellular fractionation experiments in HEK293 cells indicate a large portion of αS in cytosolic pools. Laminin is a nucleocytoplasmic marker, histone H3 is a nuclear marker. GAPDH is cytoplasmic.
(E) Western blots for Fig.4E. N terminally HA tagged Edc4 variants were transiently transfected in Edc4 CRISPR KO background and HA pulldown was performed to check decapping module interactions.
(F) Same as in (E). HA-Edc4(ΔBlock4) was investigated for decapping module interactions. The quantification of three different biological replicates were shown in Fig.4E
(G) Edc4, Edc3 and Dcp1a CRISPR knockout (KO) cell lines in wild-type HEK293 background. Whole cell extract western blots for two independent cell lines per KO are shown. Mock refers to a non-targeting guide RNA. These guides were chosen for KO in αS-Avi+BirA ligase background.
(H) Establishment of an anti-p70 (mouse) antibody as a surrogate Edc4 antibody in HEK293 cells. Mock and Edc4 KO cell lines were probed with anti-Edc4 and anti-p70 for immunofluorescence. Anti-p70 marks a nuclear protein but also strongly cross-reacts with Edc4-positive P-bodies in cytoplasm as has previously been shown in other cells(Kedersha and Anderson, 2007). The p70 cytoplasmic signal is not present with Edc4 KO. This antibody was used in combination with other anti-decapping protein antibodies to overcome species-limitation of antibodies.
(I) IF of P-body components Edc4, Dcp1, Edc3 and Xrn1, their knockdown confirmation and their P-body localization. Mouse host antibodies (α-Dcp1, α-p70 for Edc4) are stained together with rabbit host antibodies (α-Edc3, α-Xrn1, α-Edc4) where possible. Single channels are gray-scaled and channel color is represented in the antibody name. The “merge” channel is the red/green channel combined along with DAPI stain pseudo-colored as magenta). The DAPI channel is not individually shown. The occasional structures with fibrillar appearance shown with the Edc3 are possibly antibody artifacts because GFP-labeled Edc3 did not show any fibrillar pattern in any genetic background (data not shown).
(J) Representative western blot for main Fig.4F. Decapping module-αS interactions were investigated in Edc4, Edc3 and Dcp1 KO HEK293 backgrounds.
Figure S5 (Related to Figure5). Quality controls of iPSC editing, PLA in neurons and IF quantifications
(A) TaqMan Copy Number Assay was performed to confirm triplication at the SNCA locus (4-copy total) in in the female NINDS ND34391 iPSC line. Copy number is compared to a control iPSC line (TGEN 14-20 035iPS), and previously published iPSC, a male triplication line (WIBR-SNCA-Tripl) and a female A53T line(Chung et al., 2013). WIBR-SNCA-Tripl(1) was reprogrammed with floxable lentiviral Yamanaka factors (as described in Chung et al. 2013). WIBR-SNCA-Tripl(2) was reprogrammed with a nonintegrating method (mRNA).
(B-C) gRNA design and selection of Guide 3 on the basis of TIDE analysis. (C) Details of TIDE analysis performed in HEK293 cells
(D) G-band karyotype of original ND34391 iPSC and the 2-copy derivative edited clone.
(E) Ngn2 induced cortical neurons show no glial or proliferation markers. 3-week-old wild-type SNCA level neurons (2-copy) were stained with various neuronal and glial markers. Vimentin and s100b are glial markers and they show no observable signal. Ki67 is a proliferation marker, which is absent in post-mitotic neurons.
(F) Edc4 and Dcp1 mark P-bodies in induced neurons. Edc4 and p-70 antibody cross-reacts in neurons. The known cross-reaction of p-70 antibody with Edc4 was confirmed in patient derived iNs, validating the Edc4 antibody. In iPSC derived neurons (SNCA wild type levels from the triplication series), Edc4 co-localizes with Dcp1 in punctate structures, prominently in cytoplasm.
(G) Quality control of PLA assay in induced neurons. PLA with only secondary probes was performed in week3 of iPSC derived neurons from wild-type levels of SNCA (2-copy) from the triplication series. Scale bar is 10 μm. No signal is detected, which indicates marginal false positive rates for the PLA.
(H) Edc4-αS PLA in SNCA triplication iN series. Each dot is the average PLA signal per neuron. Number of images taken are shown in the bars. Each individual point is an image field (number of fields in parentheses). Error bars show SEM. Two-tailed unpaired t-test results are shown.
(I) Decrease in P-body numbers in SNCA triplication lines is robust across days in culture and neuronal density. Edc4 puncta (representing P-bodies) counts for week 3 and 4 in an independent differentiation of the triplication iPSC series with different seeding densities of neurons. IF experiment was performed as in Fig.5H. Each dot is the average Edc4 puncta per cell in a field of observation. Several fields were measured with a Nikon microscope (high-content analysis package) and puncta were called and quantitated with custom FIJI macros. Dead cells were not counted. Both 100K and 50K/ well seeding are shown. Edc4 puncta numbers deviate from wild type neurons in SNCA triplication and SNCA KO (one-way ANOVA results are shown for each group).
(J) Outlier elimination principle from Edc4 puncta count analysis. Due to clustering of neurons, certain image fields yield aberrant IF patterns. These are readily revealed by plotting Edc4 total puncta and averaged Edc4 puncta per neuron against cell numbers (DAPI counts). Outliers, denoted by boxes, were removed from the analysis.
Figure S6 (Related to Figure6). Blue Native-PAGE optimization for P-body components and mRNA decay assay details
(A) Left: Dox induction curve in HEK293 Tet::SNCA line and normalized quantification of SNCA levels. 10 ng/ml of dox is deemed optimal for induction. Right: Edc4 immunoprecipitation (IP) optimization for Fig.6A. Bead and antibody concentrations were optimized to capture the mid-linear range of Dcp1 pulldown. Lane6 conditions were deemed optimal for immunoprecipitation.
(B) Lysis temperature is critical for resolving soluble Edc4 complexes. Three contrasting conditions were tested (4°C versus 25°C; 5 units versus 10 units of benzonase; low versus high RNAse concentration. The major determinant that emerged was temperature. 25°C room temperature improved resolution of Edc4 complexes, possibly due to enhanced kinetics of nucleases. 5 units of benzonase at 25°C was chosen for all the following experiments.
(C) CHAPS is the choice of detergent for cell lysis for Blue Native-PAGE in HEK293 cells. 5 different detergents were used with indicated concentrations and blue native-PAGE was performed followed by Edc4 blot. Note DDM disrupts Edc4 to 0.5 MDa band whereas 1% CHAPS reveal higher order Edc4 complexes more clearly than other detergents.
(D) Blue Native-PAGE of P-body components with 1% CHAPS and 5 Units of Benzonase (25 minutes at 25°C lysis). Note >1 MDa large Edc4 complex(es) along with smaller Edc3, Dcp1a complexes. 60 kDa αS is the unstructured monomer and higher order structures are either various membrane bound conformations or higher-order oligomers of αS.
(E) In blue native-PAGE gels, soluble Edc4 complexes are resolved as two high molecular weight (HMW) species that are dependent and independent of Edc3. Blue native-PAGE gels from whole cell extracts were run for HEK293 wildtype, Edc4 null and Edc3 null background with optimized parameters. Edc4 was probed and two exposures are shown.
(F) Soluble Edc3 mostly forms 0.5 MDa complexes along with low-abundance Edc4-dependent ~1 MDa complexes. Experiments were performed as in (E) and probed for Edc3. Note that high exposure reveals Edc4-dependent Edc3 complexes.
(G) Quality control for PLA assay in differentiated neurons (SNCA 2-copy) to measure in situ Dcp1 and Edc4 interactions. No signal is detected unless both primary antibodies were used.
(H) Schematic depiction of a piggybac vector constructed for generating a stable HEK-293 cell line expressing the tethering reporter (used for Fig.6E). A normalizer module with puromycin selection (separated by 2A self-cleaving peptide) is transcribed bidirectionally with the reporter module. Reporter module is a ultrabright nanoluciferase (nanoluc) with 4x Lambda boxes in 3’UTR which is recognized by λN peptide conjugated protein of interest (POI). The normalizer module is an orthogonal firefly luciferase (Luc2). Both luciferases contain strong degrons (CP and PEST) to keep linearity between RNA and protein levels. Tether output is calculated as the ratio between nanoluc and firefly luciferase luminescence.
(I) Different P-body components are tethered to the reporter to check the validity of the assay. 4 independent experiments are shown (centrality: mean, spread: SEM).
(J) Histograms of log fold changes of whole transcriptome during the 12-hour ActinomycinD (ActD) pulse between SNCA 2-copy and SNCA 4-copy differentiated neurons. The major differences between the two genotypes are obvious in the first 3 hour but subsides in later time points.
(K) Go enrichment analysis for the 1204 genes that were LRT significant in ActD time course analysis described in Fig.6I. The ‘nuclear transcribed mRNA catabolic process, NMD’ category is highlighted as the same GO category was recovered in the ROSMAP data (Supp.Fig.7B). ‘Protein targeting’ is highlighted due to their relevance to PD pathology.
(L-N) Above: Delta slope histograms for decaying genes within the first and third hour of ActD treatment for differentiated neurons. The number of genes and p value for a paired t-test (looking at Δ slope<0 condition) is shown in each histogram. This figure is the extension of main Fig.6J-L. The conditions plotted are; SNCA 2copy versus 4 copy for(L), SNCA 2copy versus SNCA 2-copy +PFF for (M) and SNCA 4-copy versus SNCA 4-copy +PFF for (N). Below: Same analysis but for up-slope genes (i.e., up-regulated genes) for two-time intervals. The differential slope affect was not observed for up-regulated genes except for SNCA 4-copy versus SNCA 4-copy +PFF condition. Significant p-values are bolded.
Figure S7 (Related to Figure7). P-body biology in human disease
(A) Differential Gene Expression (DGE) analysis from ROS/MAP data. Four clinical and neuropathological phenotypes related to PD and synucleinopathy are shown (Column:Pathology). mRNA expression data from dorsolateral prefrontal cortex (DLPFC) were analyzed for significant correlation or anti-correlation in relation to these phenotypes (adjusted p-value <0.05). mRNA metabolism related pathways (GO ID and Description columns) are shown for each of these PD phenotypes, along with the adjusted p values. Direction indicates correlation or anti-correlation.
(B) GO enrichment analysis of a specific PD phenotype (Degree of LB pathology) from ROSMAP data. Bar graph of enrichment scores(−log10p) are shown for genome-wide significant GO categories.
(C) IF signals of Edc4 and Dcp1 from post-mortem brain images that were used for measuring PLA signals of Edc4-Dcp1(Fig7A-D).
(D) Quantification of signals in (C), see STAR methods for details.
(E) PLA controls in post-mortem brains. For Edc4-Dcp1 PLA control, either of the first antibody is omitted.
(F) PLA control in post-mortem brains. Quantification of PLA between αS and Edc4 and when either of the antibody is omitted. Quantification as in Fig.7D and E.
(G) PLA quantifications of Dcp1-Edc4 and with the omission of Edc4 antibody for the control brains.
Table S1. Yeast strains used in this study, specifically new yTRAP strains, Related to Fig.1. (Tab1)
Yeast genetic mini-screens screens, Related to Fig.1 (Tab2)
Table S2. SILAC MS results of aSyn-GFP purification, Related to Fig.2 (Tab1)
Mass Spectrometry analysis of alpha-Synuclein-Avi purifications in triplicates, Related to Fig.2 (Tab2)
Table S3. Plasmids used in this study, Related to Fig.3 and other figures (Tab1)
HEK293 stable cell lines generated used in this study, Related to Fig.3 and other figures (Tab2)
Table S4. Guide RNA oligos for CRISPR/Cas9 KO, Related to Fig.4 and other figures
Table S5. Antibodies used in this study and their and application details, Related to Fig.5 and other figures
Table S6. GO enrichment analysis of differentially decayed genes in PD patient neurons, Related to Fig.6 and 7(Tab1)
High enrichment of SNCA and PD relevant genes in Protein Targeting category for differentially decayed genes, Related to Fig.6 and 7 (Tab2)
Table S7. Differential Gene expression analysis of ROSMAP data, Related to Fig.7 (Tab1)
GWAS cohorts used in BridGE analysis, Related to Fig.7 (Tab2)
BRIDGE analysis of SNCA and P-BODY interactions in PD COHORTS, Related to Fig.7 (Tab3)
BridGE analysis of TARDBP(TDP-43) (or FUS) and P-BODY interactions in ALS cohorts, Related to Fig.7 (Tab4)
Bag of genes used in RVTT and p-values, Related to Fig.7 (Tab5)
Post-mortem brains from Mayo Clinic, that are used in PLA assay, Related Fig.7 to(Tab6)
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-EDC4 Antibody, rabbit polyclonal | Abcam | Cat# ab72408, RRID:AB_1268717 |
| Anti-DCP1A mouse monoclonal antibody (M06), clone 3G4 | Abnova | Cat# H00055802-M06, RRID:AB_530021 |
| Anti-EDC3 antibody, rabbit polyclonal | Abcam | Cat# ab168851, RRID:AB_2715533 |
| Anti-Human Dcp2 rabbit Polyclonal Antibody | Abcam | Cat# ab28658, RRID:AB_731858 |
| Anti-XRN1 Antibody, rabbit polyclonal | Bethyl | Cat# A300-443A, RRID:AB_2219047 |
| Anti-G3BP antibody, monoclonal antibody | Abcam | Cat# ab56574, RRID:AB_941699 |
| Anti-p70 S6 kinase a (H-9), mouse monoclonal antibody (cross-reacts with Edc4) | Santa Cruz Biotechnology | Cat# sc-8418, RRID:AB_628094 |
| Anti-VCP antibody, mouse monoclonal | Abcam | Cat# ab11433, RRID:AB_298039 |
| Anti-Human DDX6 Polyclonal, rabbit polyclonal | Novus | Cat# NB 200-191, RRID:AB_523228 |
| Anti-HA Epitope Tag Antibody, rabbit polyclonal | Novus | Cat# NB600-363, RRID:AB_10001504 |
| Anti-Lamin B1 Antibody, rabbit polyclonal | Abcam | Cat# ab16048, RRID:AB_10107828 |
| Anti-Histone H3 Antibody, rabbit polyclonal | Cell Signaling Technology | Cat# 9715, RRID:AB_331563 |
| Anti-LSM14A/RAP55 Antibody, rabbit polyclonal | Thermo Fisher Scientific | Cat# A305-102A, RRID:AB_2631497 |
| Anit-eIF4E (P-2) antibody, mouse monoclonal | Santa Cruz Biotechnology | Cat# sc-9976, RRID:AB_627502 |
| Anti-Tubulin beta 3 (TUBB3) antibody, TUJ1 clone, mouse monoclonal | BioLegend | Cat# 801201, RRID:AB_2313773 |
| Anti-GAPDH (GA1R) antibody, mouse monoclonal | Thermo Fisher Scientific | Cat# MA5-15738, RRID:AB_10977387 |
| Anti-alpha Synuclein Monoclonal Antibody (4B12), used for western blots | Thermo Fisher Scientific | Cat# MA1-90346, RRID:AB_1954821 |
| Anti-Human Alpha -synuclein (211) Monoclonal, used for PLA assays in induced neurons | Santa Cruz Biotechnology | Cat# sc-12767, RRID:AB_628318 |
| Anti-SNCA antibody (Clone 42/α-Synuclein (RUO), aka SYN1), used in brain PLA experiments | BD Biosciences | Cat# 610787, RRID:AB_398108 |
| Anti-MAP2 antibody, rabbit polyclonal | Millipore | Cat# AB5543, RRID:AB_571049 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Post-mortem brain samples for PLA analysis are detailed in TableS7 | Mayo Clinic Florida Brain Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Human recombinant brain-derived neurotrophic factor (BDNF) | Peprotech | 450-02 |
| Human recombinant glia-derived neurotrophic factor (BDNF) | Peprotech | 450-10 |
| Pierce High Sensitivity Streptavidin-HRP | Life Technologies | 21130 |
| Poly-D-Lysine | Thermo Fisher | A3890401 |
| Complete EDTA-free protease inhibitor cocktail | Sigma Aldrich | 11873580001 |
| PhosSTOP phosphatase inhibitor cocktail | Sigma Aldrich | 4906845001 |
| RIPA Lysis Buffer | Thermo Fisher | 89900 |
| TCEP Bond Breaker | Thermo Fisher | 77720 |
| MyOneT1 Streptavidin magnetic Dynabeads | Invitrogen | 65601 |
| cAMP | Sigma | D0260 |
| Poly-Ethylenimine (PEI) | Thermo Fisher | 24765-1 |
| Pierce Anti-HA Magnetic Beads | Life Technologies | 88836 |
| Lipofectamine 2000 | Thermo Fisher | 11668 |
| B27 PLUS supplement | Life Technologies | A35828-01 |
| Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | 354230 |
| Neurobasal Media | Gibco | 21103049 |
| N2 supplement | Life Technology | 17502-048 |
| Rock Inhibitor (Y-27632 DiHydrochloride) | Peprotech | 1293823 |
| TransIT-LT1 Transfection Reagent | Mirus | MIR2304 |
| B27 supplement | Gibco | 17504044 |
| Laminin | Sigma | L2020 |
| Cytosine B-D-Arabinofuranoside Hydrochloride (Arac) | Sigma-Aldrich | C6645 |
| Critical commercial assays | ||
| ExpiSf™ Expression System Starter Kit | Thermo Fisher | A38841 |
| QIAamp DNA Mini Kit | Qiagen | 51304 |
| TaqMan SNCA copy number assay (Hs02236645_cn) | Thermo Fisher | 4400291 |
| BCA Protein Assay Kit | Pierce | 23225 |
| SilverQuest™ Silver Staining Kit | Invitrogen | LC6070 |
| Duolink™ In Situ Orange Starter Kit Mouse/Rabbit | Sigma Aldrich | DUO92102 |
| PureLink RNA Mini Kit | Invitrogen | 12183020 |
| KAPA mRNA HyperPrep Kit | KAPA Biosystems | KR1352 – v4.17 |
| NativePAGE™ Sample Prep Kit | Invitrogen | BN2008 |
| NanoGlo Dual Luciferase Reporter Assay System | Promega | N1610 |
| Superscript™ IV VILO™ (SSIV VILO) Master Mix | Life Technologies | 11756050 |
| Deposited data | ||
| SILAC MS data for alpha-Synuclein interaction | This study | ftp://massive.ucsd.edu/MSV000089026/. |
| Bulk RNAseq from Ngn2 induced neurons | This study | Geo; GSE199349 |
| BridGE PD Cohort1; PD-NGRC | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000196.v3.p1 |
| BridGE PD Cohort2; PD-NIA | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000089.v3.p2 |
| BridGE PD Cohort3; PD-LEAPS | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000048.v1.p1 |
| BridGE PD Cohort4; PD-CIDR | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000126.v2.p1 |
| BridGE PD Cohort5; PD-IPDGC | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000918.v1.p1 |
| BridGE PD Cohort6; PD-APDGC | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000394.v1.p1 |
| BridGE PD Cohort7; PD-AMP | the Accelerating Medicines Partnership | https://amp-pd.org/ |
| BridGE ALS Cohort1; ALS-Irish | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000127.v2.p1 |
| BridGE ALS Cohort2; ALS-Finland | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000344.v1.p1 |
| BridGE ALS Cohort3; ALS-NIH-HumanHap | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000101.v3.p1 |
| BridGE ALS Cohort4; ALS-NIH-OmniExpress | http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap | phs000101.v3.p1 |
| PDBP (Parkinson’s Disease Biomarker Program) used for RVTT analysis | https://amp-pd.org/ | AMP-PD 2020 Release |
| PPMI (Parkinson’s Progressive Marker Initiative) used for RVTT analysis | https://amp-pd.org/ | AMP-PD 2020 Release |
| Experimental models: Cell lines | ||
| HEK Flp-In™ T-REx™ 293 Cell Line | Thermo Fisher | R78007, RRID:CVCL_U427 |
| SNCA triplication iPSC, Iowa Kindred | NINDS Human Cell and Data Repository | Patient code: NDS00201; iPSC line code: ND34391 |
| All cell lines generated/used in TableS3 | This study | N/A |
| Experimental models: Organisms/strains | ||
| D. melanogaster: RNAi for XRN1: XRN1HMS01169 (STOCK1) | Bloomington Drosophila Stock Center | BDSC:34690 FBst0034690 RRID: BDSC_34690 |
| D. melanogaster: RNAi for XRN1: XRN1GLC01410 (STOCK2) | Bloomington Drosophila Stock Center | BDSC:44442 FBst0044442 RRID: BDSC_44442 |
| D. melanogaster: QUAS-a-synuclein(WT) | Ordonez et al, 2018, Sarkar et al, 2021 | N/A |
| Newly generated S.cerevisiae strains are found in Table S1 | This study | N/A |
| Oligonucleotides | ||
| Fly qPCR probes; XRN1 Forward: GCGAGGAGGTCAAGTTCGG | This study | N/A |
| Fly qPCR probes; XRN1 Reverse: GCTCGTCTGTTCTCAGGGC | This study | N/A |
| Fly qPCR probes; Rpl32 Forward: GACCATCCGCCCAGCATAC | This study | N/A |
| Fly qPCR probes; Rpl32 Reverse: CGGCGACGCACTCTGTT | This study | N/A |
| Guide RNA oligos used in this study: TableS4 | This study | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | (Ran et al., 2013) | Addgene:62988 |
| Plasmids used in this study: TableS3 | This study | N/A |
| Software and algorithms | ||
| MaxQuant (version 1.3.0.5) | Cox and Mann, 2008 | N/A |
| FastQC (version 0.11.5) | Andrews, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR (version 2.5.3a) for ROSMAP STAR (version 2.7.3) for RNA decay analysis | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| RSEM (version 1.2.31) | Li and Dewey, 2011 | https://deweylab.github.io/RSEM/ |
| Multiqc (version 1.5) | Ewels et al., 2016 | https://multiqc.info/ |
| Picard Toolkit (version 2.17.4) | Broad Institute | http://broadinstitute.github.io/picard/ |
| R statistical software (version 3.6.3) | R Core Team, 2021 | https://www.r-project.org/ |
| “limma” (R package) (version 3.38.3) | Ritchie et al., 2015 | https://pubmed.ncbi.nlm.nih.gov/25605792/ |
| “voom” (R package) (version 3.5.2) | Law et al., 2014 | https://pubmed.ncbi.nlm.nih.gov/2448524/ |
| “tmod” (R package) (version 0.46.2) | Weiner, 2020 | https://cran.r-project.org/web/packages/tmod/index.html |
| GO.db (R package) (version 3.10.0) | Carlson, 2019 | https://www.bioconductor.org/packages/release/data/annotation/html/GO.db.html |
| REVIGO | Supek et al., 2011 | |
| RNA-seq analysis source code | This paper | DOI:10.5281/zenodo.6477548 |
| The BridGE software analysis | Fang et al., 2019; Wang et al., 2017 | DOI:10.5281/zenodo.6473560 |
| Custom FIJI macros | This study | N/A |
| Other | ||
| Q Exactive mass spectrometer | Thermo Scientific | N/A |
| Easy-nLC 1000 UPLC | Proxeon | N/A |
| 96-well glass bottom microplate for imaging | Brooks Life Science Systems | MGB096-1-2-LG-L |
| Poly-L-Ornithine/Laminin 6-Well Clear Flat Bottom Tc-Treated Plate | Corning | 354658 |
Highlights.
αSynuclein (αS) toxicity linked to P-bodies through yeast, fly and human genetics
Physiologic binding of αS N-terminus to either membranes or P-body decapping module
Pathologic αSyn accumulation disrupts the decapping module in PD neurons and brain
Pathologic αSyn disrupts mRNA stability in PD iPSC neurons
Acknowledgments
We thank all the members of ARCND community for their critical input on the paper, especially Drs Dennis Selkoe, Greg Petsko, Ulf Dettmer, Charles Jennings, Vijay Kuchroo and Tracy Young-Pearse. We thank Ulf Dettmer for the SNCA G51D construct, Lei Liu for PX459 plasmid, Lai Ding at BWH NeuroTechnology Studio for FIJI macros, Luigi Warren (Cellular Reprogramming) for reprogramming of iPSC. NGS was performed at the Whitehead Genome Technology core (overseen by Summet Gupta). We thank Dr. Alexander Drew for helping with quality control of the PPMI and PDBP sub-cohorts of AMP-PD. EH was supported by the HSFP (LT000717/2015-L), NIH (R21NS112858) and Aligning Science Across Parkinson’s Disease(ASAP-000472). DF is supported by the Koerner New Scientist Program from the Koerner Family Foundation. We thank Dr. Leon French for assistance with computational code for enrichment term consolidation for the ROSMAP analysis. WW and CLM are supported by the Biomarkers Across Neurodegenerative Diseases Grant from the Alzheimer’s Association, Alzheimer’s Research UK (ARUK), The Michael J. Fox Foundation for Parkinson’s Research (MJFF), and the Weston Brain Institute (Weston) (BAND-19-615151). VK is a NYSCF Stem Cell Robertson Investigator (NYSCF-R-149), a George C. Cotzias Fellow of the American Parkinson’s Disease Association and both awards supported this work. Additional grants to VK that supported this work include those from Aligning Science Across Parkinson’s Inititiative (ASAP-000472), Michael J. Fox Foundation, NIH (R21NS112858 and R01NS109209) and DOD (W81XWH-19-1-0695). The authors also thank the Accelerating Medicines Partnership Parkinson's disease (AMP PD), a public-private partnership, which is managed by the Foundation for the National Institutes of Health (FNIH) and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research, the National Institute of Neurological Disorders and Stroke, Pfizer, and Verily.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
VK is a co-founder of and senior advisor to Dacapo Brainscience and Yumanity Therapeutics, companies focused on CNS diseases. CYC and XJ contributed to this work as employees of Yumanity Therapeutics.
REFERENCES
- Andrews S (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data; Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Carlson M (2019). GO.db: A set of annotation maps describing the entire Gene Ontology. R package version 3.8.2. [Google Scholar]
- J.W., and Domaszewska T (2016). tmod: an R package for general and multivariate enrichment analysis. Peerj Prepr 4, e2420v1. 10.7287/peerj.preprints.2420v1. [DOI] [Google Scholar]
- Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. (2013). Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nature Methods 10, 623–629. 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Publishing Group; 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer PL, Johnsen JM, Johnson AD, Logsdon BA, Lange LA, Nalls MA, Zhang G, Franceschini N, Fox K, Lange EM, et al. (2012). Imputation of Exome Sequence Variants into Population- Based Samples and Blood-Cell-Trait-Associated Loci in African Americans: NHLBI GO Exome Sequencing Project. Am J Hum Genetics 91, 794–808. 10.1016/j.ajhg.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Caraveo G, and Lindquist S (2010). α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Bi 26, 211–233. 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Bandres-Ciga S, Saez-Atienzar S, Kim JJ, Makarious MB, Faghri F, Diez-Fairen M, Iwaki H, Leonard H, Botia J, Ryten M, et al. (2020). Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol 140, 341–358. 10.1007/s00401-020-02181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, Fox M, Goldstein JM, Soriano F, Seubert P, et al. (2008). Red Blood Cells Are the Major Source of Alpha-Synuclein in Blood. Neurodegener Dis 5, 55–59. 10.1159/000112832. [DOI] [PubMed] [Google Scholar]
- Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, and Beckham JD (2016). Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J Virol 90, 2767–2782. 10.1128/jvi.02949-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham CJ, and Parker R (2008). P Bodies, Stress Granules, and Viral Life Cycles. Cell Host Microbe 3, 206–212. 10.1016/j.chom.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beecham GW, Dickson DW, Scott WK, Martin ER, Schellenberg G, Nuytemans K, Larson EB, Buxbaum JD, Trojanowski JQ, Deerlin VMV, et al. (2015). PARK10 is a major locus for sporadic neuropathologically confirmed Parkinson disease. Neurology 84, 972–980. 10.1212/wnl.0000000000001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendapudi PK, Nazeen S, Ryu J, Söylemez O, Rouaisnel B, Colling M, Pasko B, Robbins A, Bouzinier M, Tomczak L, et al. (2022). Pathway-based Rare Variant Burden Analysis Identifies a Role for the Complement System in an Extreme Phenotype of Sepsis with Coagulopathy. Medrxiv 2022.02.24.22271459. 10.1101/2022.02.24.22271459. [DOI] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, and Wilson RS (2006). Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, and Wilson RS (2012a). Overview and findings from the religious orders study. Current Alzheimer Research 9, 628–645. 10.2174/156720512801322573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, and Wilson RS (2012b). Overview and findings from the rush Memory and Aging Project. Current Alzheimer Research 9, 646–663. 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, and Schneider JA (2018). Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease 64, S161–S189. 10.3233/jad-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozic M, Caus M, Rodrigues-Diez RR, Pedraza N, Ruiz-Ortega M, Garí E, Gallel P, Panadés MJ, Martinez A, Fernández E, et al. (2020). Protective role of renal proximal tubular alpha- synuclein in the pathogenesis of kidney fibrosis. Nature Communications 11, 1–16. 10.1038/s41467-020-15732-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brück D, Wenning GK, Stefanova N, and Fellner L (2016). Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol Dis 85, 262–274. 10.1016/j.nbd.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman AS, Shulman JM, Nag S, Leurgans SE, Arnold SE, Morris MC, Schneider JA, and Bennett DA (2012). Nigral pathology and parkinsonian signs in elders without Parkinson disease. Annals of Neurology 71, 258–266. 10.1002/ana.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, and Südhof TC (2015). Definition of a molecular pathway mediating α-synuclein neurotoxicity. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 35, 5221–5232. 10.1523/jneurosci.4650-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, and Südhof TC (2018). Cell Biology and Pathophysiology of α-Synuclein. Csh Perspect Med 8, a024091–29. 10.1101/cshperspect.a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B, Cord B, Nguyen HN, Schüle B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, and Palmer TD (2011). SNCA Triplication Parkinson’s Patient’s iPSC-derived DA Neurons Accumulate α-Synuclein and Are Susceptible to Oxidative Stress. PLoS ONE 6, e26159–13. 10.1371/journal.pone.0026159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, et al. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. 22, 8797–8807. 10.1523/jneurosci.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-T, Bercovich N, Loh B, Jonas S, and Izaurralde E (2014). The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res 42, 5217–5233. 10.1093/nar/gku129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, Brug M. van der, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 7, 1–10. 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin M-C, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, Kachergus J, Milnerwood AJ, Tapia L, Song M-S, et al. (2011). Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genetics 89, 398–406. 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A, Schymick JC, Restagno G, Scholz SW, Lombardo F, Lai S-L, Mora G, Fung H-C, Britton A, Arepalli S, et al. (2009). A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Human Molecular Genetics 18, 1524–1532. 10.1093/hmg/ddp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, et al. (2013). Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Yi S, Sahni N, Loh KH, Auluck PK, Baru V, Udeshi ND, Freyzon Y, Carr SA, et al. (2017). In Situ Peroxidase Labeling and Mass-Spectrometry Connects Alpha-Synuclein Directly to Endocytic Trafficking and mRNA Metabolism in Neurons. Cell Syst 4, 242–250.e4. 10.1016/j.cels.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet GA, and Parker R (2020). RNP Granule Formation: Lessons from P-Bodies and Stress Granules. Cold Spring Harb Sym 84, 040329. 10.1101/sqb.2019.84.040329. [DOI] [PubMed] [Google Scholar]
- Cronin S, Berger S, Ding J, Schymick JC, Washecka N, Hernandez DG, Greenway MJ, Bradley DG, Traynor BJ, and Hardiman O (2008). A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 17, 768–774. 10.1093/hmg/ddm361. [DOI] [PubMed] [Google Scholar]
- Das T, and Eliezer D (2019). Membrane interactions of intrinsically disordered proteins_ The example of alpha-synuclein. BBA - Proteins and Proteomics 1867, 879–889. 10.1016/j.bbapap.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, and Parker R (2012). P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Csh Perspect Biol 4, a012286–a012286. 10.1101/cshperspect.a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U (2018). Rationally Designed Variants of α-Synuclein Illuminate Its in vivo Structural Properties in Health and Disease. Frontiers in Neuroscience 12, 239–14. 10.3389/fnins.2018.00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Selkoe D, and Bartels T (2016). New insights into cellular α-synuclein homeostasis in health and disease. Curr Opin Neurobiol 36, 15–22. 10.1016/j.conb.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Dhungel N, Eleuteri S, Li L, Kramer NJ, Chartron JW, Spencer B, Kosberg K, Fields JA, Stafa K, Adame A, et al. (2015). Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron 85, 76–87. 10.1016/j.neuron.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, and Gingeras TR (2015). Mapping RNA-seq Reads with STAR. Curr Protoc Bioinform 51, 11.14.1–11.14.19. 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Schweizer D, and Izaurralde E (2007). P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol 27, 3970–3981. 10.1128/mcb.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Wang W, Paunic V, Heydari H, Costanzo M, Liu X, Liu X, VanderSluis B, Oately B, Steinbach M, et al. (2019). Discovering genetic interactions bridging pathways in genome-wide association studies. Nature Communications 10, 1–18. 10.1038/s41467-019-12131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, and Bender WW (2000). A Drosophila model of Parkinson’s disease. Nature 404, 394–398. 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Felsky D, Sariya S, Santa-Maria I, French L, Schneider JA, Bennett DA, Mayeux R, Jager PLD, and Tosto G (2020). The Caribbean-Hispanic Alzheimer’s Brain Transcriptome Reveals Ancestry-Specific Disease Mechanisms. Biorxiv 2020.05.28.122234. 10.1101/2020.05.28.122234. [DOI] [Google Scholar]
- Foffani G, and Obeso JA (2018). A Cortical Pathogenic Theory of Parkinson’s Disease. Neuron 99, 1116–1128. 10.1016/j.neuron.2018.07.028. [DOI] [PubMed] [Google Scholar]
- Fromm SA, Truffault V, Kamenz J, Braun JE, Hoffmann NA, Izaurralde E, and Sprangers R (2011). The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. The EMBO Journal 31, 279–290. 10.1038/emboj.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung H-C, Scholz S, Matarin M, Simón-Sánchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, et al. (2006). Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. The Lancet Neurology 5, 911–916. 10.1016/s1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- Fusco G, Gopinath T, Vostrikov V, Vendruscolo M, Simone AD, Dobson CM, and Veglia G (2014). Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nature Communications 5, 1–8. 10.1038/ncomms4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G, Sanz-Hernandez M, and Simone AD (2018). Order and disorder in the physiological membrane binding of α-synuclein. Curr Opin Struc Biol 48, 49–57. 10.1016/j.sbi.2017.09.004. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Long C, Jayaram N, Chen X, Li L, and Wu JY (2011). Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2, 141–149. 10.1007/s13238-011-1014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaete-Argel A, Márquez CL, Barriga GP, Soto-Rifo R, and Valiente-Echeverría F (2019). Strategies for Success. Viral Infections and Membraneless Organelles. Frontiers in Cellular and Infection Microbiology 9, 369–28. 10.3389/fcimb.2019.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini M-G, Tredici KD, and Braak H (2012). 100 years of Lewy pathology. Nat Rev Neurol 9, 1–12. 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, et al. (2010). αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc National Acad Sci 107, 19573–19578. 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkova D, Panasyuk G, Nemazanyy I, Zhyvoloup A, Monteil P, Filonenko V, and Gout I (2012). EDC4 interacts with and regulates the dephospho-CoA kinase activity of CoA synthase. FEBS Letters 586, 3590–3595. 10.1016/j.febslet.2012.08.033. [DOI] [PubMed] [Google Scholar]
- Halliday GM, and Stevens CH (2011). Glia: Initiators and progressors of pathology in Parkinson’s disease. Movement Disord 26, 6–17. 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, et al. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nature Genetics 42, 781–785. 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden E, Chen S, Chumley A, Xia C, Zhong Q, and Ju S (2020). A Genetic Screen for Human Genes Suppressing FUS Induced Toxicity in Yeast. G3 Genes Genomes Genetics 10, g3.401164.2020. 10.1534/g3.120.401164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015). A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 163, 712–723. 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Hernández G, Ramírez MJ, Minguillón J, Quiles P, Garibay G.R. de, Aza-Carmona M, Bogliolo M, Pujol R, Prados-Carvajal R, Fernández J, et al. (2018). Decapping protein EDC4 regulates DNA repair and phenocopies BRCA1. Nature Communications 9, 967–11. 10.1038/s41467-018-03433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hontz RD, Niederer RO, Johnson JM, and Smith JS (2009). Genetic identification of factors that modulate ribosomal DNA transcription in Saccharomyces cerevisiae. Genetics 182, 105–119. 10.1534/genetics.108.100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvathova I, Voigt F, Kotrys AV, Zhan Y, Artus-Revel CG, Eglinger J, Stadler MB, Giorgetti L, and Chao JA (2017). The Dynamics of mRNA Turnover Revealed by Single-Molecule Imaging in Single Cells. Mol Cell 68, 615–624.e10. 10.1016/j.molcel.2017.09.030. [DOI] [PubMed] [Google Scholar]
- Hubstenberger A, Courel M, Bénard M, Souquere S, Ernoult-Lange M, Chouaib R, Yi Z, Morlot J-B, Munier A, Fradet M, et al. (2017). P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol Cell 68, 1–30. 10.1016/j.molcel.2017.09.003. [DOI] [PubMed] [Google Scholar]
- Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, and Ting AY (2016). Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nature Protocols 11, 456–475. 10.1038/nprot.2016.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunn BHM, Cragg SJ, Bolam JP, Spillantini M-G, and Wade-Martins R (2016). Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci 38, 1–11. 10.1016/j.tins.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM-Y, Clark CM, Glosser G, Stern MB, Gollomp SM, et al. (2000). Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54, 1916–1921. 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- (IPDGC), I.P.D.G.C., and (WTCCC2), W.T.C.C.C. 2 (2011). A Two-Stage Meta-Analysis Identifies Several New Loci for Parkinson’s Disease. Plos Genet 7, e1002142. 10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, and Anderson P (2013). Post-transcriptional regulatory networks in immunity. Immunological Reviews 253, 253–272. 10.1111/imr.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager PLD, Yang H-S, and Bennett DA (2018). Deconstructing and targeting the genomic architecture of human neurodegeneration. Nat Neurosci 21, 1–8. 10.1038/s41593-018-0240-z. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, and Khurana V (2017). Specification of Physiologic and Disease States by Distinct Proteins and Protein Conformations. Cell 171, 1001–1014. 10.1016/j.cell.2017.10.047. [DOI] [PubMed] [Google Scholar]
- Jonas S, and Izaurralde E (2013). The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Gene Dev 27, 2628–2641. 10.1101/gad.227843.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, Brown RH, Lindquist S, et al. (2011). A Yeast Model of FUS/TLS-Dependent Cytotoxicity. Plos Biol 9, e1001052. 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, and Anderson P (2007). Mammalian Stress Granules and Processing Bodies. Methods Enzymol 431, 61–81. 10.1016/s0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Keeney M, Hoffman E, Greenamyre J, and Maio RD (2021). Measurement of LRRK2 Kinase Activity by Proximity Ligation Assay. Bio-Protocol 11, e4140. 10.21769/bioprotoc.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MF, Saad M, Brás J, Bettella F, Nicolaou N, Simón-Sánchez J, Mittag F, Büchel F, Sharma M, Gibbs JR, et al. (2012). Using genome-wide complex trait analysis to quantify “missing heritability” in Parkinson’s disease. Hum Mol Genet 21, 4996–5009. 10.1093/hmg/dds335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SA, Jarboui M-A, Srihari S, Raso C, Bryan K, Dernayka L, Charitou T, Bernal-Llinares M, Herrera-Montavez C, Krstic A, et al. (2020). Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRASG13D. Nat Commun 11, 499. 10.1038/s41467-019-14224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keryer-Bibens C, Barreau C, and Osborne HB (2008). Tethering of proteins to RNAs by bacteriophage proteins. Biol Cell 100, 125–138. 10.1042/bc20070067. [DOI] [PubMed] [Google Scholar]
- Khurana V, Peng J, Chung CY, Auluck PK, Fanning S, Tardiff DF, Bartels T, Koeva M, Eichhorn SW, Benyamini H, et al. (2017). Genome-Scale Networks Link Neurodegenerative Disease Genes to α-Synuclein through Specific Molecular Pathways. Cell Syst 4, 157–170.e14. 10.1016/j.cels.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, and Feany MB (2006). α-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Human Molecular Genetics 15, 3012–3023. 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai S-L, Myllykangas L, Sulkava R, Jansson L, Hernandez DG, Gibbs JR, et al. (2010). Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurology 9, 978–985. 10.1016/s1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam I, Hallacli E, and Khurana V (2020). Proteome-Scale Mapping of Perturbed Proteostasis in Living Cells. Cold Spring Harbor Perspectives in Biology 12, a034124–26. 10.1101/cshperspect.a034124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology 15, R29–17. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, and Kamitani T (2011). Improved Immunodetection of Endogenous α-Synuclein. Plos One 6, e23939–8. 10.1371/journal.pone.0023939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, and Regev A (2010). Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods 7, 709–715. 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323–16. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locascio JJ, Eberly S, Liao Z, Liu G, Hoesing AN, Duong K, Trisini-Lipsanopoulos A, Dhima K, Hung AY, Flaherty AW, et al. (2015). Association between α-synuclein blood transcripts and early, neuroimaging-supported Parkinson’s disease. Brain 138, 2659–2671. 10.1093/brain/awv202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maio RD, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, et al. (2016). α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Science Translational Medicine 8, 342ra78–342ra78. 10.1126/scitranslmed.aaf3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, Andrade M. de, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PVK, Frazer KA, Cox DR, and Ballinger DG (2005). High-Resolution Whole-Genome Association Study of Parkinson Disease. Am J Hum Genetics 77, 685–693. 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras C, Beck JC, Bower JH, Roberts E, Ritz B, Ross GW, Abbott RD, Savica R, Eeden SKVD, Willis AW, et al. (2018). Prevalence of Parkinson’s disease across North America. Npj Parkinson’s Disease 4, 1–7. 10.1038/s41531-018-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AR, and Beckham JD (2016). Alpha-Synuclein, a Novel Viral Restriction Factor Hiding in Plain Sight. DNA and Cell Biology 35, 643–645. 10.1089/dna.2016.3488. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L, and Krainc D (2016). α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proceedings of the National Academy of Sciences of the United States of America 113, 1931–1936. 10.1073/pnas.1520335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland MA, Ellis CE, Markey SP, and Nussbaum RL (2008). Proteomics Analysis Identifies Phosphorylation-dependent α-Synuclein Protein Interactions. Mol Cell Proteomics 7, 2123–2137. 10.1074/mcp.m800116-mcp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG (2006). Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop, pp. 417–423. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein H-U, Patrick E, Komashko V, et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21, 811–819. 10.1038/s41593-018-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Brás J, Sharma M, et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46, 989–993. 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, Saad M, Jansen I, Guerreiro R, Lubbe S, et al. (2015). NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 36, 1605.e7–1605.e12. 10.1016/j.neurobiolaging.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby GA, Kiriakov S, Hallacli E, Kayatekin C, Tsvetkov P, Mancuso CP, Bonner JM, Hesse WR, Chakrabortee S, Manogaran AL, et al. (2017). A Genetic Tool to Track Protein Aggregates and Control Prion Inheritance. Cell 171, 966–968.e18. 10.1016/j.cell.2017.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Wong YC, Ysselstein D, Severino A, and Krainc D (2019). Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends in Neurosciences 42, 140–149. 10.1016/j.tins.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Pankratz N, Hernandez D, Paisán-Ruíz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, et al. (2005). Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 365, 410–412. 10.1016/s0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- Olsen AL, and Feany MB (2019). Glial α-synuclein promotes neurodegeneration characterized by a distinct transcriptional program in vivo. Glia 67, 1933–1957. 10.1002/glia.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ, Noble W, Hanger DP, and Miller CCJ (2017). α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol 134, 129–149. 10.1007/s00401-017-1704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, Tsankov A, Shang L, Krumholz K, Jagadeesan P, et al. (2015). Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods 12, 885–892. 10.1038/nmeth.3507. [DOI] [PubMed] [Google Scholar]
- Pei Y, and Maitta RW (2019). Alpha synuclein in hematopoiesis and immunity. Heliyon 5, e02590. 10.1016/j.heliyon.2019.e02590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, and George JM (2000). Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. The Journal of Biological Chemistry 275, 34393–34398. 10.1074/jbc.m004851200. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Price AL, Kryukov GV, Bakker P.I.W. de, Purcell SM, Staples J, Wei L-J, and Sunyaev SR (2010). Pooled Association Tests for Rare Variants in Exon-Resequencing Studies. Am J Hum Genetics 86, 832–838. 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 43, e47–e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, et al. (2013). Isogenic Human iPSC Parkinson’s Model Shows Nitrosative Stress-Induced Dysfunction in MEF2-PGC1α Transcription. Cell 155, 1351–1364. 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Rokad D, Malovic E, Luo J, Harischandra DS, Jin H, Anantharam V, Huang X, Lewis M, Kanthasamy A, et al. (2019). Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Science Signaling 12, eaat9900. 10.1126/scisignal.aat9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman TA, Luna E, et al. (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Scientific Reports 9, 1–19. 10.1038/s41598-019-47227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L, Eikens F, Odainic A, Spitzer J, Griep A, et al. (2021). Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 10.1016/j.cell.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, and Bennett DA (2012). Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135, 3005–3014. 10.1093/brain/aws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She M, Decker CJ, Svergun DI, Round A, Chen N, Muhlrad D, Parker R, and Song H (2008). Structural Basis of Dcp2 Recognition and Activation by Dcp1. Molecular Cell 29, 337–349. 10.1016/j.molcel.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, Jager PLD, and Feany MB (2011). Parkinson’s disease: genetics and pathogenesis. Pathology Mech Dis 6, 193–222. 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- Siitonen A, Nalls MA, Hernández D, Gibbs JR, Ding J, Ylikotila P, Edsall C, Singleton A, and Majamaa K (2017). Genetics of early-onset Parkinson’s disease in Finland: exome sequencing and genome-wide association study. Neurobiol Aging 53, 195.e7–195.e10. 10.1016/j.neurobiolaging.2017.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Scholz S, Fung H-C, Matarin M, Hernandez D, Gibbs JR, Britton A, Vrieze F.W. de, Peckham E, Gwinn-Hardy K, et al. (2007). Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet 16, 1–14. 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Snead D, and Eliezer D (2019). Intrinsically disordered proteins in synaptic vesicle trafficking and release. The Journal of Biological Chemistry 294, 3325–3342. 10.1074/jbc.rev118.006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, and Jaenisch R (2016). Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99. 10.1038/nature17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, and Goedert M (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stefano BD, Luo E-C, Haggerty C, Aigner S, Charlton J, Brumbaugh J, Ji F, Jiménez IR, Clowers KJ, Huebner AJ, et al. (2019). The RNA Helicase DDX6 Controls Cellular Plasticity by Modulating P-Body Homeostasis. 1–31. 10.1016/j.stem.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, and Šmuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800. 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Tissue-based map of the human proteome. Science (New York, NY) 347, 1260419–1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, and Lee VM-Y (2014). Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite–like aggregates. Nat Protoc 9, 2135–2146. 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu ZZ, Costanzo M, Boone C, Lange CA, and Myers CL (2017). Pathway-based discovery of genetic interactions in breast cancer. PLoS Genetics 13, e1006973–29. 10.1371/journal.pgen.1006973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claßen GA, Krauss M, Haucke V, et al. (2014). Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–1028. 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- Winstall E, Sadowski M, Kühn U, Wahle E, and Sachs AB (2000). The Saccharomyces cerevisiaeRNA-binding Protein Rbp29 Functions in Cytoplasmic mRNA Metabolism. The Journal of Biological Chemistry 275, 21817–21826. 10.1074/jbc.m002412200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to Figure1). Details of perturbation screen (yTRAP) methodology and yeast genetics.
(A) Logistic growth curves of liquid cultures (measured by optical density at 600 nm absorbance) of yeast ND strains upon estradiol induction for 40 hrs. Measurements were taken every 15 minutes in four independent cultures. Dots represent mean (+/− SD, errors bars were usually invisible due to high reproducibility of OD measurements). Gray curves indicate no estradiol addition, orange curves indicate estradiol addition and thus ND protein expression.
(B) Optimization of estradiol induction (Neurodegenerative gene expression) interval: Diploids of αS x yTRAP strains (16 yTRAP sensors) were measured by flow cytometry for indicated time points. They were normalized to a ‘dark’ sensor which doesn’t give fluorescent output. Note that after 6 hours, the signals revert because the cells approach the end of log phase. We chose 6 hours as the maximum allowed time for toxicity.
(C) The SOD1(4AV) mutant was selected as the negative control for the screen. Selected sensors were crossed to a non-ND expressing strain (ZEM only) and SOD1(4AV) mutant. After 6 hours of induction no significant effect was measured between the sensors. Thus, instead of ZEM only strain, we chose to express a non-toxic SOD1(4AV) gene as our negative control.
(D) αS expression induced more perturbation in the RBP proteome than FUS protein. Correlation plots, along with linear regression line, of screen signals were shown for FUS versus SOD1 (Pearson correlation score is shown in the label) and FUS versus SNCA.
(E) Rescue of αS mediated toxicity with Sgn1 related genes. 39 proteins (TableS1) were overexpressed in αS expressing background. 10 rescue events were shown with duplicates for αS. SG: Stress granule related genes
(F) Rescue of αS mediated toxicity with the proteins in (E) were not recapitulated for FUS mediated toxicity.
(G) αS protein expression is not changed upon modifier expression in the mini overexpression screen. Western blots (2 biological replicates) were quantified from whole cell lysates from.
(H) Heatmap of yTRAP perturbation scores upon αS expression belonging to 24 genes were plotted (averaged log2 fold changes were normalized to Fus expression). Each row is an individual RBP sensor. Significant discoveries are indicated with asterics (p<0.005, two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 1%). These 11 discoveries were re-plotted in Fig.1E.
(I) Western blot of whole cell extraction from fly brains for αS levels. These fly lines were used for the locomotor activity assay shown in Fig.1H
Figure S2 (Related to Figure2). αS purification optimization in human cells
(A) IF signals with various commercial αS antibodies in HEK293 cell lines. Different αS antibodies are indicated above in wild-type HEK293 cells. Merge picture indicates 488 nm αS (green) channel and DAPI (magenta) stain. Scale bar = 10 μm
(B) Bead optimization for streptavidin pulldown. 500 μg of HEK293 Flp-In αS-Avi (+/− BirA ligase) was incubated with 10, 20 or 50 μl MyOneT1 magnetic streptavidin beads. Filled triangles indicate endogenous biotinylated proteins. 10 μl beads/500 μg of extract was considered optimal and used for subsequent analyses.
(C) αS and decapping module interactions can withstand high ionic strength. Increasing KCl concentrations were used in washing step of streptavidin pulldown. Edc4 and Edc3 can withstand up to 0.5M KCl, whereas the Ddx6 interaction is lost rapidly.
(D) αS and decapping module interactions are RNA-independent. During the wash step, the strep beads were incubated with Rnase cocktail for 15 minutes at room temperature. 3 independent pulldowns were blotted with the indicated antibodies.
(E) Mass spectrometry counts belonging to Fig.2A-C. Decapping modules are significantly enriched in αS-GFP pulldowns.
(F) Schematic of protocol for macro P-body pelleting according to Hubstenberger,2017 protocol. Macro P-bodies represent large RNP complexes that can be pelleted at 10000g, whereas the supernatant contains smaller complexes. Those smaller complexes can be further solubilized with nuclease digestions to enrich protein-held-together complexes.
(G) Many components of P-bodies can be found largely in non-pelleted smaller complexes. After gentle cell lysis and removing cell nucleus at 800g, macro-P-bodies (‘condensed granules) were pelleted at 10000g. The un-pelleted lighter P-body components were further solubilized by various nucleases (Benzonase, RQ1 Dnase, a cold optimal cryonase; non-specific nuclease). The nuclease digestion after 10000g pelleting was repeated both at 4°C and 25°C for the nucleases to function optimally.
(H) Ddx6 CRISPR KO western blot from whole cell lysates of HEK293 cells with three different guides.
(I) IF of Ddx6 CRISPR KO lines and dissolution of P-bodies. The first two panels (Ddx6 and Edc4) were also highlighted in Fig.2J
Figure S3 (Related to Figure3). αS mutational analysis indicate negative correlation with decapping module interactions and membrane binding
(A) Stepwise truncations of αS-Avi up to amino acid 100 was transfected into HEK293 cells and pulled down with streptavidin beads. Two technical replicates per each pulldown are shown.
(B) Representative western blotting of 10aa deletion mutants used in quantification of main Fig.3C.
(C) Repulsion of αS away from the membrane restores P-body binding. T59 and T81 in αS were exchanged for hydrophilic residues (E or K) and P-body binding (normalized to wild-type αS) was measured in 3 independent pulldowns.
(D) Representative western blotting of various αS mutants used in quantification of main Fig.3H-J
(E) Protein alignment of synuclein protein family. Imperfect repeats are demarcated.
Figure S4 (Related to Figure4). Decapping module proteins along with αS come in close proximity at the proximal region of Edc4.
(A) αS binds to the C-terminus of Edc4 after the low complexity linker (aa 535-974) region. The indicated large truncations of HA-tagged Edc4 were individually transiently transfected into αS-Avi(+BirA ligase) cell line and pulled down with streptavidin beads. Two independent pulldowns are shown side by side. Note that αS can pulldown endogenous Edc4.
(B) Serial deletions at the C terminus of Edc4 show that αS can only bind to Edc4 further down the aa1166.
(C) Proximal region deletion (Δ974-1265) on Edc4 abrogates αS-Edc4 interactions.
(D) Cellular fractionation experiments in HEK293 cells indicate a large portion of αS in cytosolic pools. Laminin is a nucleocytoplasmic marker, histone H3 is a nuclear marker. GAPDH is cytoplasmic.
(E) Western blots for Fig.4E. N terminally HA tagged Edc4 variants were transiently transfected in Edc4 CRISPR KO background and HA pulldown was performed to check decapping module interactions.
(F) Same as in (E). HA-Edc4(ΔBlock4) was investigated for decapping module interactions. The quantification of three different biological replicates were shown in Fig.4E
(G) Edc4, Edc3 and Dcp1a CRISPR knockout (KO) cell lines in wild-type HEK293 background. Whole cell extract western blots for two independent cell lines per KO are shown. Mock refers to a non-targeting guide RNA. These guides were chosen for KO in αS-Avi+BirA ligase background.
(H) Establishment of an anti-p70 (mouse) antibody as a surrogate Edc4 antibody in HEK293 cells. Mock and Edc4 KO cell lines were probed with anti-Edc4 and anti-p70 for immunofluorescence. Anti-p70 marks a nuclear protein but also strongly cross-reacts with Edc4-positive P-bodies in cytoplasm as has previously been shown in other cells(Kedersha and Anderson, 2007). The p70 cytoplasmic signal is not present with Edc4 KO. This antibody was used in combination with other anti-decapping protein antibodies to overcome species-limitation of antibodies.
(I) IF of P-body components Edc4, Dcp1, Edc3 and Xrn1, their knockdown confirmation and their P-body localization. Mouse host antibodies (α-Dcp1, α-p70 for Edc4) are stained together with rabbit host antibodies (α-Edc3, α-Xrn1, α-Edc4) where possible. Single channels are gray-scaled and channel color is represented in the antibody name. The “merge” channel is the red/green channel combined along with DAPI stain pseudo-colored as magenta). The DAPI channel is not individually shown. The occasional structures with fibrillar appearance shown with the Edc3 are possibly antibody artifacts because GFP-labeled Edc3 did not show any fibrillar pattern in any genetic background (data not shown).
(J) Representative western blot for main Fig.4F. Decapping module-αS interactions were investigated in Edc4, Edc3 and Dcp1 KO HEK293 backgrounds.
Figure S5 (Related to Figure5). Quality controls of iPSC editing, PLA in neurons and IF quantifications
(A) TaqMan Copy Number Assay was performed to confirm triplication at the SNCA locus (4-copy total) in in the female NINDS ND34391 iPSC line. Copy number is compared to a control iPSC line (TGEN 14-20 035iPS), and previously published iPSC, a male triplication line (WIBR-SNCA-Tripl) and a female A53T line(Chung et al., 2013). WIBR-SNCA-Tripl(1) was reprogrammed with floxable lentiviral Yamanaka factors (as described in Chung et al. 2013). WIBR-SNCA-Tripl(2) was reprogrammed with a nonintegrating method (mRNA).
(B-C) gRNA design and selection of Guide 3 on the basis of TIDE analysis. (C) Details of TIDE analysis performed in HEK293 cells
(D) G-band karyotype of original ND34391 iPSC and the 2-copy derivative edited clone.
(E) Ngn2 induced cortical neurons show no glial or proliferation markers. 3-week-old wild-type SNCA level neurons (2-copy) were stained with various neuronal and glial markers. Vimentin and s100b are glial markers and they show no observable signal. Ki67 is a proliferation marker, which is absent in post-mitotic neurons.
(F) Edc4 and Dcp1 mark P-bodies in induced neurons. Edc4 and p-70 antibody cross-reacts in neurons. The known cross-reaction of p-70 antibody with Edc4 was confirmed in patient derived iNs, validating the Edc4 antibody. In iPSC derived neurons (SNCA wild type levels from the triplication series), Edc4 co-localizes with Dcp1 in punctate structures, prominently in cytoplasm.
(G) Quality control of PLA assay in induced neurons. PLA with only secondary probes was performed in week3 of iPSC derived neurons from wild-type levels of SNCA (2-copy) from the triplication series. Scale bar is 10 μm. No signal is detected, which indicates marginal false positive rates for the PLA.
(H) Edc4-αS PLA in SNCA triplication iN series. Each dot is the average PLA signal per neuron. Number of images taken are shown in the bars. Each individual point is an image field (number of fields in parentheses). Error bars show SEM. Two-tailed unpaired t-test results are shown.
(I) Decrease in P-body numbers in SNCA triplication lines is robust across days in culture and neuronal density. Edc4 puncta (representing P-bodies) counts for week 3 and 4 in an independent differentiation of the triplication iPSC series with different seeding densities of neurons. IF experiment was performed as in Fig.5H. Each dot is the average Edc4 puncta per cell in a field of observation. Several fields were measured with a Nikon microscope (high-content analysis package) and puncta were called and quantitated with custom FIJI macros. Dead cells were not counted. Both 100K and 50K/ well seeding are shown. Edc4 puncta numbers deviate from wild type neurons in SNCA triplication and SNCA KO (one-way ANOVA results are shown for each group).
(J) Outlier elimination principle from Edc4 puncta count analysis. Due to clustering of neurons, certain image fields yield aberrant IF patterns. These are readily revealed by plotting Edc4 total puncta and averaged Edc4 puncta per neuron against cell numbers (DAPI counts). Outliers, denoted by boxes, were removed from the analysis.
Figure S6 (Related to Figure6). Blue Native-PAGE optimization for P-body components and mRNA decay assay details
(A) Left: Dox induction curve in HEK293 Tet::SNCA line and normalized quantification of SNCA levels. 10 ng/ml of dox is deemed optimal for induction. Right: Edc4 immunoprecipitation (IP) optimization for Fig.6A. Bead and antibody concentrations were optimized to capture the mid-linear range of Dcp1 pulldown. Lane6 conditions were deemed optimal for immunoprecipitation.
(B) Lysis temperature is critical for resolving soluble Edc4 complexes. Three contrasting conditions were tested (4°C versus 25°C; 5 units versus 10 units of benzonase; low versus high RNAse concentration. The major determinant that emerged was temperature. 25°C room temperature improved resolution of Edc4 complexes, possibly due to enhanced kinetics of nucleases. 5 units of benzonase at 25°C was chosen for all the following experiments.
(C) CHAPS is the choice of detergent for cell lysis for Blue Native-PAGE in HEK293 cells. 5 different detergents were used with indicated concentrations and blue native-PAGE was performed followed by Edc4 blot. Note DDM disrupts Edc4 to 0.5 MDa band whereas 1% CHAPS reveal higher order Edc4 complexes more clearly than other detergents.
(D) Blue Native-PAGE of P-body components with 1% CHAPS and 5 Units of Benzonase (25 minutes at 25°C lysis). Note >1 MDa large Edc4 complex(es) along with smaller Edc3, Dcp1a complexes. 60 kDa αS is the unstructured monomer and higher order structures are either various membrane bound conformations or higher-order oligomers of αS.
(E) In blue native-PAGE gels, soluble Edc4 complexes are resolved as two high molecular weight (HMW) species that are dependent and independent of Edc3. Blue native-PAGE gels from whole cell extracts were run for HEK293 wildtype, Edc4 null and Edc3 null background with optimized parameters. Edc4 was probed and two exposures are shown.
(F) Soluble Edc3 mostly forms 0.5 MDa complexes along with low-abundance Edc4-dependent ~1 MDa complexes. Experiments were performed as in (E) and probed for Edc3. Note that high exposure reveals Edc4-dependent Edc3 complexes.
(G) Quality control for PLA assay in differentiated neurons (SNCA 2-copy) to measure in situ Dcp1 and Edc4 interactions. No signal is detected unless both primary antibodies were used.
(H) Schematic depiction of a piggybac vector constructed for generating a stable HEK-293 cell line expressing the tethering reporter (used for Fig.6E). A normalizer module with puromycin selection (separated by 2A self-cleaving peptide) is transcribed bidirectionally with the reporter module. Reporter module is a ultrabright nanoluciferase (nanoluc) with 4x Lambda boxes in 3’UTR which is recognized by λN peptide conjugated protein of interest (POI). The normalizer module is an orthogonal firefly luciferase (Luc2). Both luciferases contain strong degrons (CP and PEST) to keep linearity between RNA and protein levels. Tether output is calculated as the ratio between nanoluc and firefly luciferase luminescence.
(I) Different P-body components are tethered to the reporter to check the validity of the assay. 4 independent experiments are shown (centrality: mean, spread: SEM).
(J) Histograms of log fold changes of whole transcriptome during the 12-hour ActinomycinD (ActD) pulse between SNCA 2-copy and SNCA 4-copy differentiated neurons. The major differences between the two genotypes are obvious in the first 3 hour but subsides in later time points.
(K) Go enrichment analysis for the 1204 genes that were LRT significant in ActD time course analysis described in Fig.6I. The ‘nuclear transcribed mRNA catabolic process, NMD’ category is highlighted as the same GO category was recovered in the ROSMAP data (Supp.Fig.7B). ‘Protein targeting’ is highlighted due to their relevance to PD pathology.
(L-N) Above: Delta slope histograms for decaying genes within the first and third hour of ActD treatment for differentiated neurons. The number of genes and p value for a paired t-test (looking at Δ slope<0 condition) is shown in each histogram. This figure is the extension of main Fig.6J-L. The conditions plotted are; SNCA 2copy versus 4 copy for(L), SNCA 2copy versus SNCA 2-copy +PFF for (M) and SNCA 4-copy versus SNCA 4-copy +PFF for (N). Below: Same analysis but for up-slope genes (i.e., up-regulated genes) for two-time intervals. The differential slope affect was not observed for up-regulated genes except for SNCA 4-copy versus SNCA 4-copy +PFF condition. Significant p-values are bolded.
Figure S7 (Related to Figure7). P-body biology in human disease
(A) Differential Gene Expression (DGE) analysis from ROS/MAP data. Four clinical and neuropathological phenotypes related to PD and synucleinopathy are shown (Column:Pathology). mRNA expression data from dorsolateral prefrontal cortex (DLPFC) were analyzed for significant correlation or anti-correlation in relation to these phenotypes (adjusted p-value <0.05). mRNA metabolism related pathways (GO ID and Description columns) are shown for each of these PD phenotypes, along with the adjusted p values. Direction indicates correlation or anti-correlation.
(B) GO enrichment analysis of a specific PD phenotype (Degree of LB pathology) from ROSMAP data. Bar graph of enrichment scores(−log10p) are shown for genome-wide significant GO categories.
(C) IF signals of Edc4 and Dcp1 from post-mortem brain images that were used for measuring PLA signals of Edc4-Dcp1(Fig7A-D).
(D) Quantification of signals in (C), see STAR methods for details.
(E) PLA controls in post-mortem brains. For Edc4-Dcp1 PLA control, either of the first antibody is omitted.
(F) PLA control in post-mortem brains. Quantification of PLA between αS and Edc4 and when either of the antibody is omitted. Quantification as in Fig.7D and E.
(G) PLA quantifications of Dcp1-Edc4 and with the omission of Edc4 antibody for the control brains.
Table S1. Yeast strains used in this study, specifically new yTRAP strains, Related to Fig.1. (Tab1)
Yeast genetic mini-screens screens, Related to Fig.1 (Tab2)
Table S2. SILAC MS results of aSyn-GFP purification, Related to Fig.2 (Tab1)
Mass Spectrometry analysis of alpha-Synuclein-Avi purifications in triplicates, Related to Fig.2 (Tab2)
Table S3. Plasmids used in this study, Related to Fig.3 and other figures (Tab1)
HEK293 stable cell lines generated used in this study, Related to Fig.3 and other figures (Tab2)
Table S4. Guide RNA oligos for CRISPR/Cas9 KO, Related to Fig.4 and other figures
Table S5. Antibodies used in this study and their and application details, Related to Fig.5 and other figures
Table S6. GO enrichment analysis of differentially decayed genes in PD patient neurons, Related to Fig.6 and 7(Tab1)
High enrichment of SNCA and PD relevant genes in Protein Targeting category for differentially decayed genes, Related to Fig.6 and 7 (Tab2)
Table S7. Differential Gene expression analysis of ROSMAP data, Related to Fig.7 (Tab1)
GWAS cohorts used in BridGE analysis, Related to Fig.7 (Tab2)
BRIDGE analysis of SNCA and P-BODY interactions in PD COHORTS, Related to Fig.7 (Tab3)
BridGE analysis of TARDBP(TDP-43) (or FUS) and P-BODY interactions in ALS cohorts, Related to Fig.7 (Tab4)
Bag of genes used in RVTT and p-values, Related to Fig.7 (Tab5)
Post-mortem brains from Mayo Clinic, that are used in PLA assay, Related Fig.7 to(Tab6)
Data Availability Statement
The original mass spectra for the SILAC experiments and the protein sequence database used for searches have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu) and are accessible at ftp://massive.ucsd.edu/MSV000089026/. Bulk RNAseq data is submitted to GEO with the submission number GSE199349 and will be available as of 06.01.2022. BridGE analysis used publicly available GWAS data and obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accessions (phs000196.v3.p1, phs000089.v3.p2, phs000048.v1.p1, phs000126.v2.p1, phs000918.v1.p1, phs000394.v1.p1, phs000127.v2.p1, phs000344.v1.p1, phs000101.v3.p1) and the Accelerating Medicines Partnership Parkinson's disease (AMP PD: https://amp-pd.org/). Rare Variant Trend Test (RVTT) analysis used publicly available data and the data for case-control Parkinson’s disease cohorts were obtained from the AMP-PD Knowledge Platform (https://www.amp-pd.org). AMP PD – a public-private partnership – is managed by the FNIH and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research, the National Institute of Neurological Disorders and Stroke, Pfizer, and Verily. Specifically, we utilized the case-control cohort from the Parkinson’s Disease Biomarker Program (PDBP) consortium which is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. Full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript. Additionally, we used the sporadic PD cases and healthy controls from PPMI sub-cohort of AMP-PD. PPMI (www.ppmi-info.org) is a public-private partnership, that is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, List of full names of all the PPMI funding partners found at www.ppmi-info.org/fundingpartners. The PPMI Investigators have not participated in reviewing the data analysis or content of the manuscript.
The code used for the analysis of the RNAseq data can be found in https://github.com/bwh-bioinformatics-hub/Hallacli2022Cell. (DOI:10.5281/zenodo.6477548). ROSMAP analysis did not create original code but descriptions of clinical and pathological outcomes are available at the Rush Alzheimer’s Disease Centre Research Resource Sharing Hub (https://www.radc.rush.edu). The BridGE software for gene-gene interaction analysis is available at https://github.com/csbio/BridGE/tree/master/BridGE_genes. (DOI: 10.5281/zenodo.6473560).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request