

ABSTRACT
Hyperglycemia and type 2 diabetes (T2D) are caused by failure of pancreatic beta cells. The role of the gut microbiota in T2D has been studied, but causal links remain enigmatic. Obese individuals with or without T2D were included from two independent Dutch cohorts. Human data were translated in vitro and in vivo by using pancreatic islets from C57BL6/J mice and by injecting flagellin into obese mice. Flagellin is part of the bacterial locomotor appendage flagellum, present in gut bacteria including Enterobacteriaceae, which we show to be more abundant in the gut of individuals with T2D. Subsequently, flagellin induces a pro-inflammatory response in pancreatic islets mediated by the Toll-like receptor (TLR)-5 expressed on resident islet macrophages. This inflammatory response is associated with beta-cell dysfunction, characterized by reduced insulin gene expression, impaired proinsulin processing and stress-induced insulin hypersecretion in vitro and in vivo in mice. We postulate that increased systemically disseminated flagellin in T2D is a contributing factor to beta-cell failure in time and represents a novel therapeutic target.
KEYWORDS: Gut microbiota, type 2 diabetes, inflammation, beta-cell function, flagellin
Introduction
While obesity is linked to insulin resistance, it is failure of pancreatic beta-cells that drives hyperglycemia and subsequent type 2 diabetes (T2D)1. Although in later stages of T2D insulin secretory rates are lowered, prior to the diagnosis and in earlier phases of the disease, insulin secretion is actually increased.2 Insulin hypersecretion, particularly in the fasted state, is considered harmful as it is associated with impaired proinsulin processing, insulin secretory stress and depletion of intracellular insulin stores,3 further promoting obesity and T2D development.4 Drivers of hyperinsulinemia are still elusive but could relate to islet-exposure to excessive nutrients such as carbohydrates and lipids,5 as well as a chronic low-grade inflammatory response known to be present in beta cells of people with T2D.6 In this regard, an influx of pro-inflammatory macrophages in islets of people with T2D has been noted.7 These macrophages produce pro-inflammatory cytokines such as interleukin (IL)-1β and IL-6, which have been associated with insulin hypersecretion8 and beta-cell failure.7 The triggers that ignite beta-cell inflammation in T2D remain presently unknown.
A known player in the field of glucose metabolism is the intestinal microbiota.9 Several cohort and intervention studies have shown an association between gut microbiota composition and T2D incidence.10 Individuals with obesity and T2D often have lower microbial diversity while showing increased abundance of potentially pathogenic gram-negative bacteria, including Proteobacteria.11 Mechanistic studies have linked metabolites produced by the gut microbiota to impaired glucose metabolism and a pro-inflammatory state.12 In addition to microbial metabolites, structural components of gram-negative bacteria, such as lipopolysaccharide (LPS), a cell-wall component, and flagellin, a structural protein of the bacterial locomotor appendage flagellum, may systemically disseminate in people with T2D.13 These bacterial molecules activate pro-inflammatory pathways by binding to pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs), expressed on epithelial cells and cells of the innate immune system.9
Here, we provide evidence for a novel pathway in which exaggerated systemic dissemination of gut-derived flagellin in T2D induces a pro-inflammatory state in beta cells. This inflammatory response is mediated by flagellin-mediated activation of TLR5 expressed on resident islet macrophages. Functionally, the inflammatory response is associated with impaired insulin gene expression and proinsulin processing while inducing hyperinsulinemia. Collectively, these processes markedly reduce insulin storage, which potentially contribute to beta-cell failure over time.
Results
Fecal Enterobacter cloacae abundance is associated with hyperglycemia in humans
To investigate the link between beta-cell dysfunction and altered gut microbiota, we analyzed fecal samples for microbiota composition using 16S rRNA sequencing in participants enrolled in the Healthy Life in an Urban Setting (HELIUS) study, a prospective cohort in Amsterdam.14 To prevent confounding effects of ethnic differences on gut microbiota composition,15 we analyzed the samples of 803 Dutch origin participants (Table S1). We observed increased abundance of Gram-negative Enterobacteriaceae in people with T2D as compared to normoglycemic controls (Figure 1a, Table S2), confirming a previous report where Enterobacteriaceae were increased in people with T2D.16 Detailed compositional analysis can be found in Balvers et al.17 To reduce the influence of confounding factors, we randomly selected 100 people with T2D and compared them to 50 age-, sex- and BMI-matched normoglycemic controls also recruited within the HELIUS cohort (Table S3). We confirmed an enrichment of Enterobacteriaceae in people with T2D using quantitative polymerase chain reaction (qPCR) (Figure 1b). Furthermore, we observed a positive relation with the long-term glucose marker hemoglobin A1c (HbA1c) and Enterobacteriaceae abundance (Figure 1c).
Figure 1.
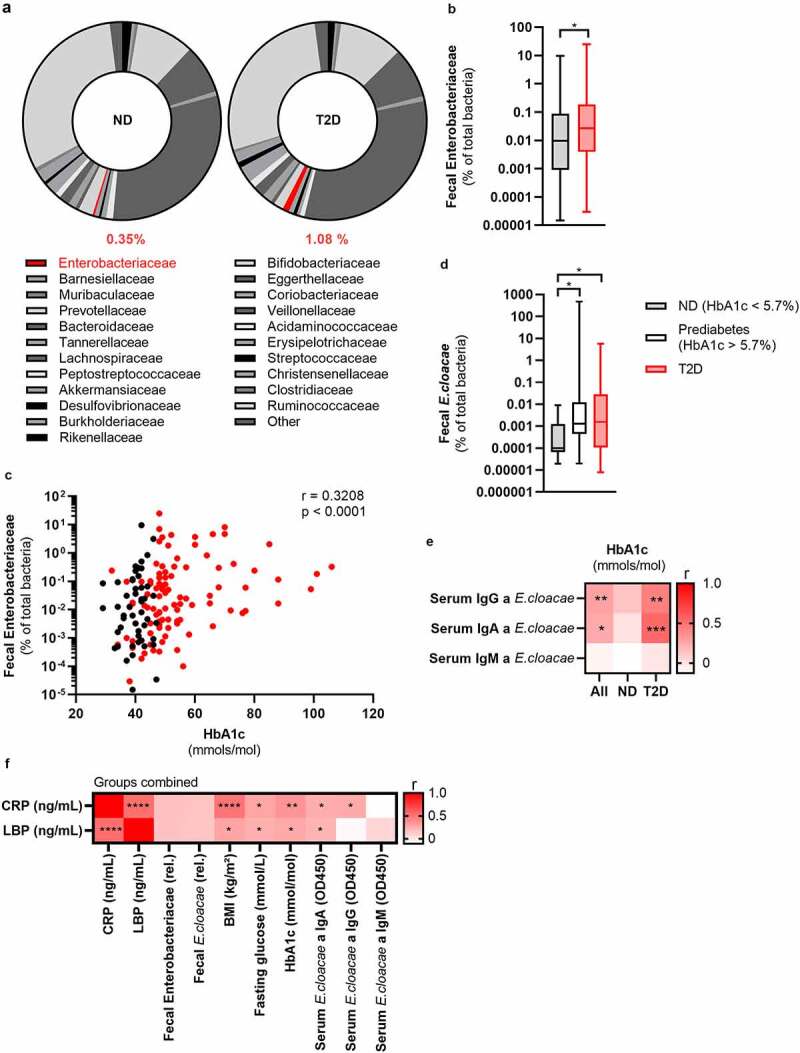
Fecal Enterobacteriaceae correlates with glucose values in the HELIUS cohort.
The fecal 16s rRNA of the gut bacteria from the HELIUS cohort was sequenced and quantified via qPCR. Further, the long-term glucose marker HbA1c and antibodies against Enterobacter cloacae were measured. (a) Fecal microbiota composition of people with or without T2D was measured via 16S rRNA sequencing (all Dutch origin participants, N = 803, % abundance, median). See table S2 for detailed composition. Here, Enterobacteriaceae is highlighted in red. (b) Fecal Enterobacteriaceae (qPCR, normalized to total fecal bacterial DNA) is increased in people with T2D compared to age-BMI-sex matched healthy controls (N = 150, box plot with min and max). (c) Fecal Enterobacteriaceae (qPCR, normalized to total fecal bacteria) positively correlates with the long-term glucose marker hemoglobin A1c (HbA1c) (N = 150). (d) Fecal Enterobacter cloacae (qPCR, normalized to total fecal bacteria) is increased in prediabetes and T2D (N = 150, box plot with min and max). (e) Correlation analysis of serum antibodies against E. cloacae and HbA1c (N = 80). (f) Correlation analysis of inflammatory markers CRP and LBP compared to various characteristics (N = 80). Mann Whitney test (b, d) and Spearman correlation (c, e); *p < .05, **p < .01, ***p < .001. Abbreviations: ND, no diabetes; T2D, type 2 diabetes; HbA1c, glycated hemoglobin; Ig, immunoglobulin; CRP, C-reactive peptide; LBP, Lipopolysaccharide binding protein.
Enterobacter cloacae (E. cloacae), a prominent member of the family of Enterobacteriaceae, was previously shown to be associated with impaired glucose tolerance in humans and mice.18 In line with these studies, in our cohort, levels of E. cloacae directly increased with deterioration of glucose tolerance (Figure 1d). Further, fecal abundance of E. cloacae also positively correlated with HbA1c (Figure S1A). Thus, as a proof-of-concept, we selected E. cloacae for subsequent experiments although we acknowledge that other bacteria of the family Enterobacteriaceae may also associate with glucose (dys)metabolism.
An immune response against Enterobacter cloacae is associated with hyperglycemia in people with type 2 diabetes
An appropriate immune response to opportunistic bacteria is necessary to prevent inflammation. To assess whether there was a systemic immune response to E. cloacae, we measured plasma antibody levels. We observed a numerical increase in IgG titers against E. cloacae in T2D, but otherwise no significant difference between the matched groups with respect to antibodies was noted (Figure S1B). Since obesity per se is a known driver of gut dysbiosis and impaired immune response to antigens, it may explain why we found no differences between the two groups.19,20 However, there was a positive correlation between HbA1c levels and systemic IgG as well as IgA against E. cloacae (Figure 1e), particularly in people with T2D. Further, there was a significant positive correlation between fecal abundance of Enterobacteriaceae and plasma IgG against E. cloacae (Figure S1C). Lastly, C-reactive peptide (CRP) and LPS binding protein (LBP) positively correlated with glucose values, BMI and antibodies against E. cloacae. Further, there was a trend towards a positive correlation between fecal Enterobacteriaceae and E. cloacae with LPB and CRP (p < .18). This is suggestive of an immune response against systemically disseminated bacterial components of E. cloacae.
Enterobacter cloacae induces beta-cell inflammation and dysfunction in vitro
Given the link between beta-cell driven hyperglycemia and fecal presence of E. cloacae as well as systemic antibodies against E. cloacae, we questioned if E. cloacae would be able to alter pancreatic beta-cell function. We isolated pancreatic islets from C57BL6/J mice fed a conventional chow diet. Islets were co-incubated with 106 colony forming units (CFUs) of heat-inactivated E. cloacae or vehicle for 72 hours (Figure 2a-g). We found that beta-cells exposed to heat-inactivated E. cloacae had lower expression of genes involved in insulin production, including the key transcription factors pancreatic duodenal homeobox 1 (Pdx1) and Mafa (Figure 2a). This lowered expression coincided with a higher inflammatory tone (Figure 2 b and c), including upregulation of pro-inflammatory cytokines (Il1b, Il6 and tumor necrosis factor α), the NLRP3 inflammasome, the macrophage marker F4/80, and Tlr2. Interestingly, increased Tlr2 expression was previously reported in pancreatic islets of people with diabetes.21 Heat-inactivated E. cloacae did not affect cell viability since ATP content was not altered (Figure 2d).22 Incubation with heat-inactivated E. cloacae also had functional consequences for beta cells. As such, insulin content was markedly reduced after 72 hours of incubation with E. cloacae (Figure 2e) but was unchanged for pro-insulin content (Figure S2A). In addition, both during low- and high glucose concentrations, beta cells treated with heat-inactivated E. cloacae increased insulin secretion (figure 2f). Lastly, E. cloacae treatment increased fasting proinsulin secretion as well as fasting proinsulin/insulin ratios, indicating disturbed proinsulin processing (Figure 2g; Figure S2B). This highlights that insulin expression and processing is disturbed potentially due to reduced expression of important transcription factors such as Pdx1 and Mafa involved in the insulin production (Figure 2a). We observed similar data in human islets, where heat-inactivated E. cloacae lowered MAFA expression, increased secreted IL-6, and tended to reduce insulin content (Figure S2C-G). Thus, the profile of increased inflammation, reduced insulin gene expression, impaired proinsulin processing and insulin hypersecretion potentially contributes to the detrimental reduction in beta-cell insulin content.
Figure 2.
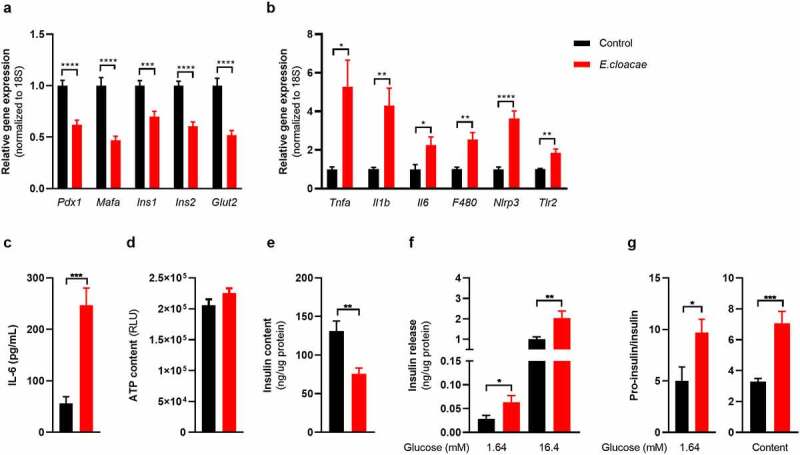
Enterobacter cloacae induces beta-cell inflammation and dysfunction.
Freshly isolated pancreatic islets from healthy C57BL6J mice were treated with heat-inactivated bacteria for 72 h (106 colony forming units/mL). (a) Enterobacter cloacae reduces expression of beta-cell genes, (b) increases expression of inflammatory genes in islets, (c) increases IL-6 secretion by islets, (d) does not change ATP content in islets, (e) reduces insulin content in islets, (f) increases insulin secretion from islets during low- and high glucose conditions, (g) increases the ratio between secreted pro-insulin and insulin, as well as the islet content of pro-insulin relative to insulin. Data shown are mean ± SEM (three representative experiments per panel, n = 9). Unpaired t-test (a, b, c, d, g) and Mann Whitney test (e, f) were used. Significance level: *p < .05, **p < .01, ***p < .001, ****p < .0001. Gene expression was normalized using 18s as a reference gene. Abbreviations: PDX1, pancreatic and duodenal homeobox 1; INS1 and INS2, insulin 1 and 2; NLRP3, NACHT, LRR and PYD domains-containing protein 3; TNF-α, tumor necrosis factor-alpha; IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; TLR2, Toll-like receptor 2; RLU, Relative light unit.
Opportunistic bacteria, but not beneficial bacteria, induce beta-cell inflammation and dysfunction
Next, to address whether the E. cloacae-mediated effects were specific to this bacterial species, we repeated the experiments with Escherichia coli (E. coli), another Gram-negative bacterium from the family Enterobacteriaceae.23 E. coli was also increased in T2D participants of the HELIUS study (Figure S1D). In line with our E. cloacae findings, E. coli reduced expression of insulin genes (Figure S3A) and induced an inflammatory response with increased expression of Il6 (Figure S3B), and IL-6 protein secretion (Figure S3C). E. coli also reduced cellular insulin content (Figure S3D) and increased insulin secretion (Figure S3E-F). In order to rule out an effect of bacterial co-incubation per se, we investigated the effects of two bacteria that have been identified as beneficial for the host. These included the Gram-positive Faecalibacterium prausnitzii16 and Gram-negative Bacteroides ovatus.24 The abundance of F. prausnitzii was decreased in people with T2D (Figure S1F). In contrast to the two opportunistic bacteria, F. prausnitzii and B. ovatus did not affect islet inflammation, insulin content or insulin secretion (Figure S3A-F). These data indicate that only a subset of bacteria induces an inflammatory response and beta-cell dysfunction. Based on previous mouse data linking E. cloacae to impaired glucose tolerance,18 we decided to further scrutinize the effect of this bacterium on beta-cell function as proof-of-concept.
Toll-like receptor-2 and Toll-like receptor-4 deletion do not protect against Enterobacter cloacae-induced beta-cell inflammation and dysfunction
TLR2 and TLR4 are involved in beta-cell replication21 and have been proposed as two key PRRs that mediate the inflammatory response induced by endogenous and exogenous molecules, the latter including bacterial components such as LPS.25 In addition, TLR2 and TLR4 are expressed by pancreatic islet cells.26 Therefore, we isolated islets from TLR2 and TLR4 knockout mice and incubated them with E. cloacae (Figure S4). Despite the absence of TLR2, E. cloacae reduced insulin gene expression (Figure S4A), increased expression of pro-inflammatory cytokines (Figure S4B), increased secreted IL-6 (Figure S4C), and reduced insulin content (Figure S4D). Similarly, TLR4-deficient islet cells were not protected from the effects of E. cloacae, as the expression of insulin genes was still reduced (Figure S4F). Regarding inflammation, E. cloacae incubation did not increase Il6 expression but secretion was still increased despite TLR4 knockout (Figure S4G-H). Insulin content was also reduced by E. cloacae incubation (Figure S4I). Therefore, we concluded that PRRs other than TLR2 and TLR4 likely play roles in E. cloacae-induced beta-cell inflammation and dysfunction.
Toll-like receptor-5 deletion protects against Enterobacter cloacae-induced beta-cell dysfunction
Several members of Enterobacteriaceae, including E. cloacae, express flagellins as both virulence and motility factors.27 Bacterial flagellin is recognized by TLR5,28 which is expressed by various cell types including epithelial cells and monocytes. We measured the effects of E. cloacae in islets from TLR5 knockout mice. TLR5 deletion partially protected islets from beta-cell dysfunction with preserved expression of insulin genes (Figure 3a). TLR5 deficiency did not reduce the effects of E. cloacae on the expression of Il6 (Figure 3b), although it did reduce the secretion of IL-6 as compared to WT islets (Figure 3c). In addition, in TLR5 knockout islets, E. cloacae did not reduce insulin content (Figure 3d), while similar insulin secretion rates were observed as from WT islets (Figure 3e).
Figure 3.
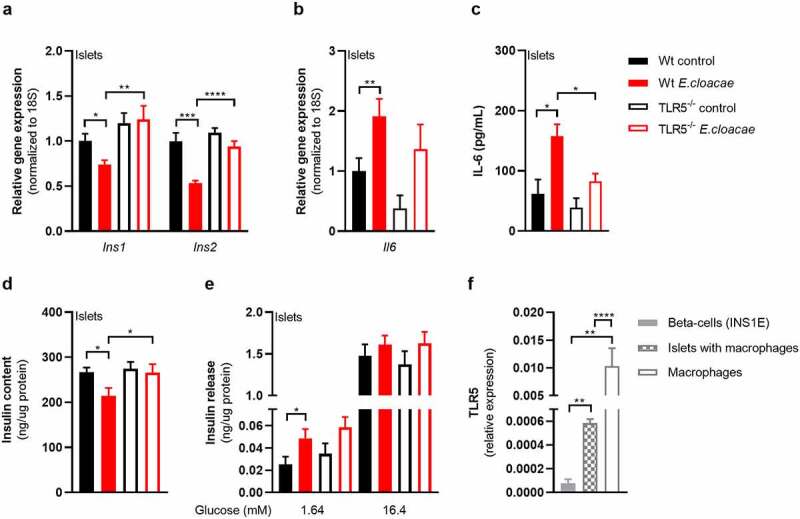
TLR5 mediates beta-cell dysfunction in pancreatic islets.
Freshly isolated pancreatic islets from C57BL6J TLR5−/− mice (10 weeks old) were treated with heat-inactivatedEnterobacter cloacae (106 CFUs/mL) for 72 h. (a) TLR5 knockout protects from E. cloacae-induced insulin expression loss, (b) prevents the effect of E. cloacae on pancreatic islet inflammation, (c) partially protects from E. cloacae induced IL-6 secretion, (d) protects from E. cloacae-induced insulin content loss. (e) E. cloacae induces insulin hypersecretion at low glucose condition in wild-type islets. (f) Clonal beta cells express low levels of TLR5 compared to pancreatic islets and macrophages. Data shown are mean ± SEM (three representative experiments per panel, n = 9). Unpaired t-test (a-c, f) and Mann Whitney test (d, e) were used for statistical analysis. Significance level: *p < .05, **p < .01, ***p < .001, ****p < .0001. Abbreviations: INS1 and INS2, insulin 1 and 2; IL-6, Interleukin 6; TLR5, Toll-like receptor 5.
To further assess the role of TLR5 in mediating the effects of E. cloacae, we co-incubated WT primary mouse islets with E. cloacae with or without the TLR5 inhibitor TH1020. As TH1020 proved toxic to cells after prolonged incubation, we studied the islets after 6h of treatment. In line with TLR5 knockout islets, TH1020 reduced the effects of E. cloacae on insulin gene expression and partially reduced the E. cloacae-induced expression of Il6 (Figure S5A-B). Due to the toxic effects of TH1020, particularly on Glut2 expression, we did not measure glucose-stimulated insulin secretion.
Macrophages mediate Enterobacter cloacae-induced beta-cell inflammation and dysfunction via TLR5 activation
As TLR5 is barely expressed in beta cells (figure 3f), we concluded that other islet-associated cells in the pancreas, particularly resident islet macrophages, could mediate the observed effects of TLR5 activation. Indeed, macrophages as well as pancreatic islets containing macrophages had higher TLR5 expression than pure beta cells (figure 3f). Macrophages are important for pancreatic islet physiology,29 but can induce beta-cell dysfunction when activated towards a pro-inflammatory phenotype.30 We thus depleted macrophages from murine pancreatic islets using clodronate-liposomes,31 followed by incubation with E. cloacae (Figure 4). Macrophage markers were significantly reduced in pancreatic islets after treatment (Figure S6A). Reduction of islet macrophages resulted in the maintenance of insulin gene transcription following E. cloacae treatment (Figure 4a). Further, expression of IL-6 was slightly reduced (Figure 4b) and its secretion was almost abolished (Figure 4c). Insulin content (Figure 4d) was not affected by E. cloacae in islets lacking macrophages. Insulin secretion was slightly increased (Figure 4e). In line with a role for islet resident macrophages, we found that pure beta cells (INS1E clonal cell line) did not show inflammation or beta-cell dysfunction following E. cloacae treatment (Figure S6B-E). Further, human islet organoids that consist of pure human endocrine cells did not show signs of beta-cell dysfunction upon E. cloacae treatment (Figure S6F-G). These results collectively indicate that islet-resident macrophages mediate E. cloacae-induced islet-cell inflammation and dysfunction.
Figure 4.
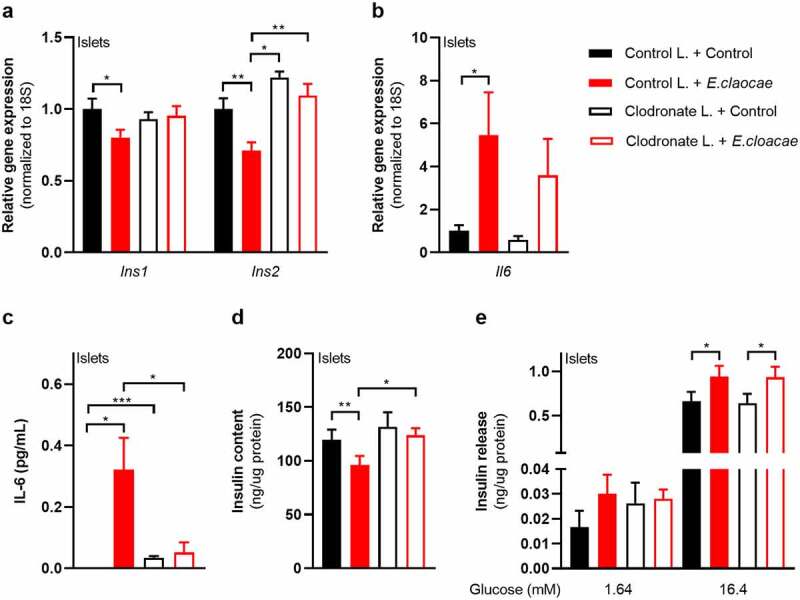
Macrophages mediate beta-cell dysfunction in pancreatic islets.
Macrophage-depleted islets from wild-type C57BL6J mice were treated with heat-inactivated Enterobacter cloacae (106 CFUs/mL) for 72h. (a) Macrophage depletion in pancreatic islets protects from E. Cloacae- induced reduction in insulin gene expression. (b) E. cloacae increases beta-cell inflammation. (c) E. cloacae increases beta-cell IL-6 secretion, which is prevented by macrophage depletion in pancreatic islets. (d) E. cloacae reduces insulin content, which is prevented by macrophage depletion. (e) E. cloacae induces insulin hypersecretion both in WT and in macrophage depleted islets. Data shown are mean ± SEM (three representative experiments per panel, n = 9). Unpaired t-test was used for statistical analysis (a-e). Significance level: *p < .05, **p < .01, ***p < .001, ***p < .001, ****p < .0001. Abbreviations: INS1 and INS2, insulin 1 and 2; IL-6, Interleukin 6.
Bacterial flagellin induces beta-cell inflammation and dysfunction
Flagellin is the ligand for TLR5,32 and therefore we hypothesized that this bacterial component could be the driving force behind the E. cloacae-induced phenotype. We isolated flagellin from E. cloacae and incubated pancreatic islets with it (Figure S5C-G). It induced inflammation and pancreatic beta-cell dysfunction. However, the isolated flagellin showed several impurities. Therefore, we continued with a pure commercial flagellin, which was derived from a very similar bacteria, namely Salmonella typhimurium (both from the family Enterobacteriaceae).
Both flagellin and flagellin-bearing E. cloacae activated TLR5 in a human embryonic kidney (HEK) reporter cell line (Figure 5a), which was dose-dependently inhibited by the TLR5 inhibitor TH1020. Furthermore, flagellin induced a pro-inflammatory response in human macrophages, which was reduced when co-incubated with TH1020 (Figure 5b). Residual inflammation might be mediated by NLRC4, which is the second receptor for flagellin.33 Additionally, flagellin impaired insulin gene expression (Figure 5c), induced beta-cell inflammation (Figure 5d-e) and reduced insulin content (figure 5f) while increasing insulin release from pancreatic islets (Figure 5g), thus resembling the phenotype induced by E. cloacae.
Figure 5.
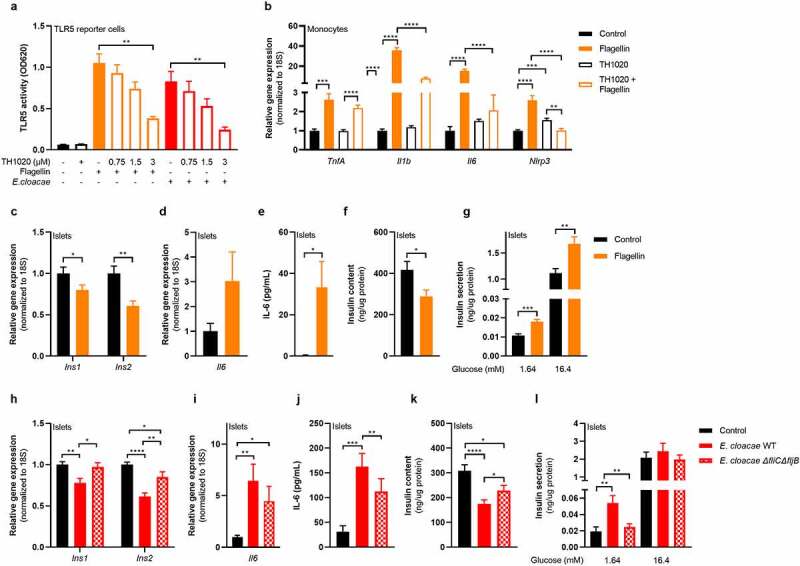
Flagellin induces beta-cell dysfunction in pancreatic islets.
HEK TLR5 reporter cell line (a), human peripheral monocytes (b) and pancreatic islets (c-l) from C57BL6J mice were incubated with flagellin (100 ng/mL) or E. cloacae (106 CFUs/mL) for 72h. E. cloacae wild-type (a, h-l) or flagellin knockout E. cloacae (h-l) was used. (a) Flagellin and E. cloacae activate TLR5 in HEK reporter cell line. TLR5 inhibitor TH1020 inhibits receptor activity dose-dependently. (b) Flagellin increases expression of inflammatory genes in monocytes, which is reduced by TLR5 inhibitor TH1020. (c) Flagellin reduces insulin gene expression, (d) increases expression of inflammatory IL-6, (e) increases secreted IL-6, (f) reduces insulin content, (g) induces insulin hypersecretion in islets. (h) Knockout of flagellin in E. cloacae partially protects against loss of insulin gene expression,(i) partially protects against inflammatory response, (j) partially protects against enhanced IL-6 release, (k) protects against loss of insulin stores, (l) protects against impaired secretory insulin response. Data shown are mean ± SEM (3 independent experiments, n = 9). Unpaired t-test was used for statistical analysis: *p < .05, **p < .01, ***p < .001, ****p < .0001. Abbreviations: INS1 and INS2, insulin 1 and 2; IL-6, Interleukin 6; FliBC−/−, flagellin gene B and C knockout.
To further dissect the role of flagellin in the beta-cell deteriorating effects mediated by E. cloacae, we generated an E. cloacae-flagellin strain (ΔfliCΔfljB) lacking both flagellin genes fliC and fliB.34 E. cloacae ΔfliCΔfljB reduced insulin expression less than the wild-type (Figure 5h). In addition, expression of inflammatory cytokines and secreted IL-6 were lower in islets exposed to the ΔfliCΔfljB strain as compared to islets exposed to wild-type E. cloacae (Figure 5i-j). Residual inflammation might come from other bacterial molecules such as LPS. Islets had higher insulin content after incubation with the ΔfliCΔfljB strain as compared to wild-type E. cloacae (Figure 5k). Finally, the ΔfliCΔfljB strain did not induce fasting insulin hypersecretion (Figure 5l). Collectively, these results suggest that flagellin, as part of the flagellum carried by bacteria belonging to Enterobacteriaceae, plays a pivotal role in beta-cell inflammation and beta-cell dysfunction via TLR5 activation on resident islet macrophages.
Flagellin treatment augments insulin secretion in mice
To translate beta-cell dysfunction inducing effects of flagellin to an in vivo situation, we injected flagellin intraperitoneally into diet-induced obese (DIO) C57BL6J mice twice weekly for four weeks (Figure 6a). Flagellin injection did not alter body weight or fasting glucose (Figure 6b-c). However, flagellin-treated mice had lower glucose levels during an intraperitoneal glucose tolerance test compared to the placebo group (Figure 6d-e), which was driven by increased insulin secretion (figure 6f) since insulin sensitivity did not differ between groups (Figure 6g-h, Figure S7). Similar to the in vitro experiments, there was a higher inflammatory tone in the pancreas of flagellin-treated mice as shown by higher Il1b expression (Figure 6i) and a trend towards more inflammation in pancreatic islets isolated from flagellin-treated mice (Figure 6k). Although insulin content did not differ between groups, insulin release tended to increase during glucose-stimulated insulin secretion ex vivo in islets isolated from the flagellin group (Figure 6j, m). While glucose lowering by increased insulin secretion could be perceived as beneficial, it is important to realize that insulin hypersecretion is related to T2D development over time. As this concerns a process of long duration, prolonged treatment with flagellin is needed to obtain a significant hyperglycemic phenotype. However, our results suggest that flagellin-induced transcriptional and functional alterations in beta-cell function, as observed in vitro, can be conceptually replicated in vivo in a mouse model.
Figure 6.
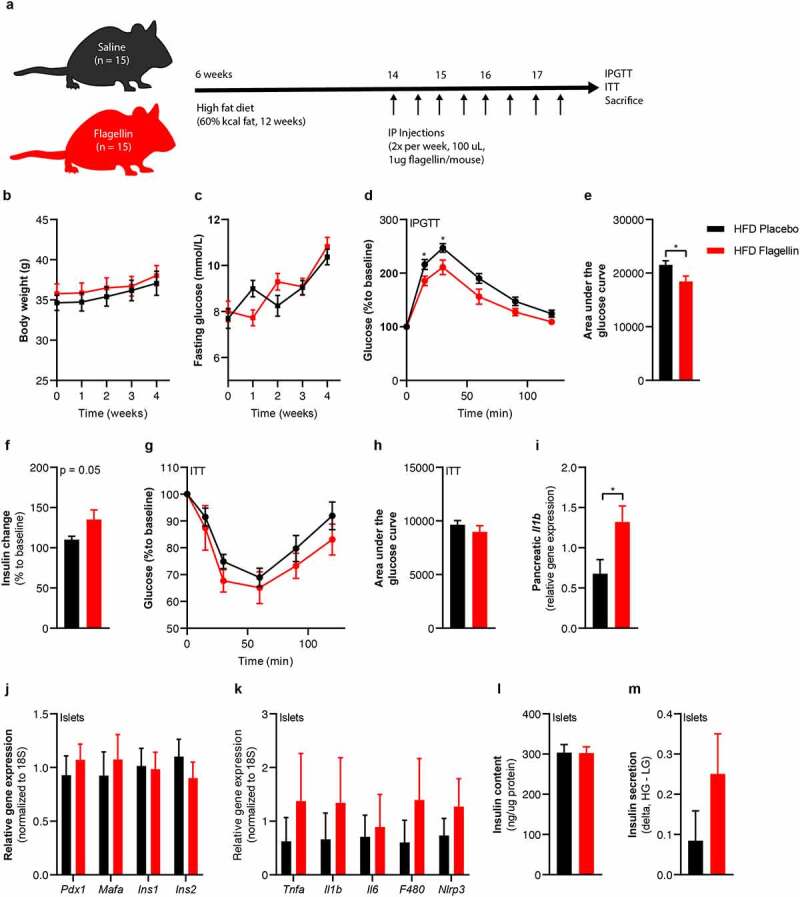
Flagellin injection in mice changes glucose tolerance.
(a) Six-week-old mice (n = 15) were fed a high fat diet (60% kcal fat) for 12 weeks. In the last 4 weeks of the diet, the mice were injected with either 1 µg flagellin in 100 µL saline or saline alone twice weekly. (b) Flagellin injections do not change the body weight, (c) do not change fasting plasma glucose concentrations. (d, e) Flagellin-injected mice have improved glucose tolerance compared to placebo-treated mice (lower area under the glucose curve). (f) Fold change (15 min to baseline) of plasma insulin is greater in flagellin-treated mice compared to placebo (n = 10). (g, h) Insulin sensitivity is not affected by flagellin injection (0.75 IU/kg). The relative change in glucose to baseline is shown. (i) Flagellin increases pancreatic Il1b expression. (j) Flagellin injections do not affect beta-cell gene expression (n = 5), (k) numerically increase markers of beta cell inflammation (n = 5), (l). do not affect insulin content (n = 5) (m), numerically increase insulin release from beta cells (n = 5). Data shown are mean ± SEM. Unpaired t-test (b-m) was used *p < .05, **p < .01, ***p < .001, ****p < .0001. Abbreviations: INS1 and INS2, insulin 1 and 2; NLRP3, NACHT, LRR and PYD domains-containing protein 3; IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; TNF-α, tumor necrosis factor-alpha; ITT, insulin tolerance test.
Systemic flagellin dissemination relates to beta-cell dysfunction in humans
Fecal flagellin has been reported to be increased in obese people compared to lean controls.20 Similarly, we observed that obese mice had a higher flagellin load compared to lean mice (Figure S8). We predicted fecal flagellin gene abundance in the 150 HELIUS participants by inference from 16S rRNA profiles using PICRUSt (Table S3) (Figure 7a). This prediction suggested an increased fecal flagellin abundance in T2D (Figure 7b). Next, we measured bacterial flagellin in the human blood circulation of the HELIUS cohort. While there was no difference between the matched obese groups, we did observe a positive correlation between serum flagellin load and HbA1c in T2D (Figure 7c). Further, the inflammatory marker CRP correlated with fecal flagellin and serum flagellin (Figure 7d). We hypothesized that increased flagellin reaches the circulation following a meal, which was previously shown to drive translocation of endotoxins.35 We therefore measured postprandial plasma flagellin, C-peptide and plasma glucose in 80 matched participants of our bariatric surgery cohort36 comprising obese normoglycemic and obese T2D people during a mixed-meal test (MMT) (Figure 7a, Table S4). C-peptide was chosen as it reflects insulin secretion rates and is not affected by potential differences in clearance by the liver, as is the case for plasma insulin concentrations. The MMT additionally allowed us to study the relationship between meal-induced flagellin and beta-cell response to an MMT. Participants with T2D had hyperglycemia following the MMT (Figure 7e-f) and lower C-peptide concentrations (Figure 7g-h) compared to normoglycemic humans with obesity. In both groups, flagellin increased during the MMT (Figure 7i). Postprandial area under the curve (AUC) for flagellin correlated with AUC C-peptide (Figure 7j), highlighting the link between bacterial flagellin and beta-cell insulin secretion in human beta-cell physiology. Interestingly, this correlation is mostly driven by individuals without diabetes (ND r= 0.6, p= .001; T2D r= 0.069, p= .736), potentially due to the presence of beta-cell failure in T2D.
Figure 7.
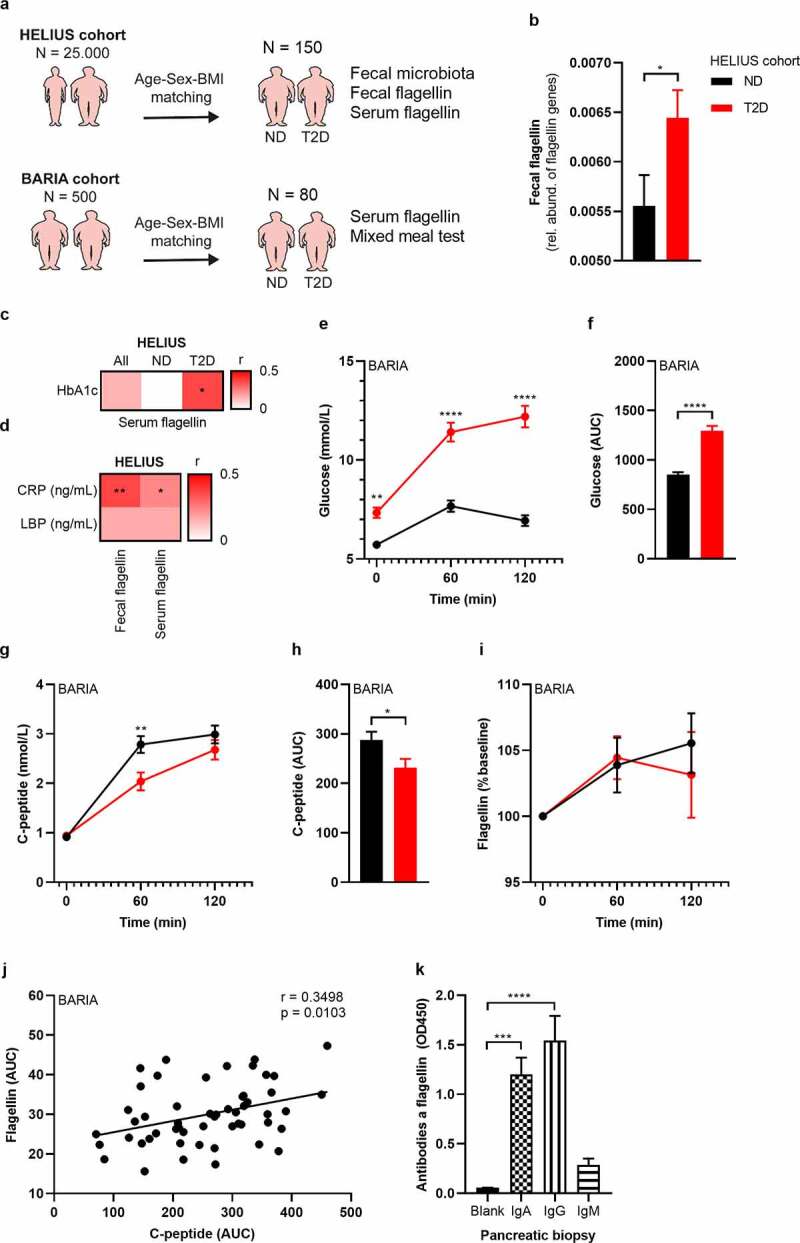
Fecal and serum flagellin is associated with glucose intolerance in humans.
(a) 150 people were randomly selected from the HELIUS cohort.14 Participants with T2D were matched with normoglycemic controls according to age, sex and BMI. In addition, 80 participants were selected from the bariatric surgery cohort (BARIA).36 (b) Fecal flagellin genes are increased in T2D, as inferred from 16S rRNA gene profiles (HELIUS). (c) Correlation matrix between serum flagellin and glucose marker HbA1c (HELIUS). (d) Correlation matrix of CRP and LBP with fecal flagellin and serum flagellin (HELIUS). (e-f) Plasma glucose concentrations during a mixed meal test (MMT, BARIA). (g, h) Plasma C-peptide concentrations during the MMT (BARIA). (i) Serum flagellin increased during the MMT (BARIA). (j) A positive correlation between serum flagellin AUC and plasma C-peptide AUC during the MMT (BARIA) exists. (k) Antibodies against bacterial flagellin were measured in homogenized human pancreatic biopsies via ELISA. Data shown are mean ± SEM. Unpaired t-test (b-h) and Spearman correlation (i) were used: *p < .05, **p < .01, ***p < .001, ****p < .0001. Abbreviations: HELIUS, Healthy Life in an Urban Setting; AUC, Area under the curve; MMT, mixed meal test; Ig, immunoglobulin; T2D, type 2 diabetes; BMI, body mass index; ND, no diabetes; CRP, c-reactive peptide; LBP, LPS binding protein.
Finally, to support the hypothesis that systemically disseminated flagellin causes the observed beta-cell phenotype, we collected pancreatic biopsies from people with T2D undergoing pancreatic surgery for benign lesion. We collected five biopsies of the head of the pancreas. Recent antibiotics use (<3 months) was an exclusion criterion (Table S5). While it was technically not possible to measure flagellin in these biopsies due to interference of the pancreatic enzymes with the flagellin assay, we observe the presence of antibodies against flagellin, supporting the notion that the well-vascularized pancreatic islets are exposed to flagellin (Figure 7k). A previous study indicated that a functional immune response is essential to control flagellin expressing bacteria,37 which seems to be reduced in obese humans.20
Discussion
In this study, we reveal a novel pathway by which flagellin, a structural component of bacteria residing in the gut, systemically disseminates following food ingestion. In pancreatic islets that are abundantly vascularized,38 we propose that flagellin activates the innate immune system and induces an inflammatory response following binding to TLR5 receptors expressed by resident islet macrophages. This leads to beta-cell dysfunction, characterized by impaired insulin gene expression, impaired insulin processing, insulin hypersecretion and reduced insulin content. This study provides a new insight into the link between gut microbiota composition and T2D.
Beta-cell dysfunction is the key abnormality that leads to the development of hyperglycemia and T2D. Beta-cell dysfunction is characterized by inappropriate insulin secretion, which can either be excessive or insufficient, upon exposure to glucose or other nutrients.39 The role of hyperinsulinemia in T2D development has received ample attention. While initially reported to be a response against obesity-related insulin resistance, research has indicated that increased insulin secretion can develop in the absence of insulin resistance implying primary beta-cell pathology.40
Induction of hyperinsulinemia has been shown to promote obesity,4 while prevention of hyperinsulinemia by pancreas-specific genetic knockout of insulin expression prevented obesity, improved insulin sensitivity and did not result in overt hyperglycemia.4 With respect to the pancreatic islets, a chronic demand on beta cells to produce insulin is detrimental. As such, a prolonged increase in insulin secretory rates have been related to endoplasmic reticulum (ER) stress, depletion of intracellular insulin stores and beta-cell apoptosis.41 Pancreatic islets from individuals with T2D have lower insulin content compared to healthy controls.42 In mice, hyperglycemia leads to insulin content loss.43 Reversibly, strategies that induce beta-cell rest are linked to improved beta-cell function over time.44
Current evidence relates overnutrition of carbohydrates and non-esterified fatty acids to beta-cell dysfunction.45 Another factor concerns a low-grade inflammatory response46 and ER stress.47 Beta-cell inflammation is a known hallmark of islets in people with T2D,48 and a central role in this regard has been proposed for islet macrophages.6 Macrophages are essential for normal beta-cell function and physiology,29 however, macrophages with a pro-inflammatory phenotype have been linked to beta-cell dysfunction.31 Triggers for activation of pro-inflammatory macrophages are uncertain but may involve hyperglycemia,49 dyslipidemia50 and human islet amyloid polypeptide (hIAPP).51 Here, we show that E. cloacae and flagellin induce inflammation in macrophages. Depleting pancreatic islets from macrophages reduced the deleterious effect on beta cells; however, we need to acknowledge that we only tested the compounds on monocyte-derived macrophages and not isolated islet resident macrophages.
In the present study we show an infectious stimulus triggering inflammation and beta-cell dysfunction: flagellin derived from intestinal microbiota. While Gram-negative bacteria are known to produce canonical flagellin, which is studied here, several other intestinal Firmicutes species are motile and have been described to contain flagella, including several Roseburia, Clostridium, and Lactobacillus spp.52 However, the flagellins of these latter, Gram-positive bacteria have not been well characterized and some are glycosylated resulting in attenuated TLR5 signaling efficiency.53
A first link towards flagellin came from the observation that the fecal abundance of the family of Enterobacteriaceae, specifically E. cloacae, was increased in people with T2D and that the fecal abundance of Enterobacteriaceae and E. cloacae correlated with glucose intolerance in humans. Administration of E. cloacae by oral gavage to mice fed a high-fat diet has previously been shown to induce glucose intolerance.18 We used one strain of E. cloacae that affected glucose tolerance in mice and induced beta-cell dysfunction. We speculate that the whole E. cloacae complex and several other pro-inflammatory bacteria belonging to the family of Enterobacteriaceae have similar effects, however that still needs to be tested. A proposed mechanism by which gut microbiota may influence host metabolism is by escaping immune control and translocating to extra-intestinal tissues.23 While this has been shown for adipose tissue,54 translocation of intestinal bacteria into the pancreas was also suggested to trigger the influx of immune cells and islet inflammation.55 Here, we used E. cloacae as a proof-of-concept bacterium. Several other bacteria belonging to the group of Enterobacteriaceae, such as E. coli, have similar effects on the human metabolism. Further, we used heat-inactivated bacteria that might induce a different immune response than viable bacteria
In patients undergoing pancreatoduodenectomy, pancreatic fluid contained bacterial DNA, with a similar composition, density and diversity as bile and jejunal fluid,56 suggesting direct translocation from the small intestine into pancreatic juice. Others also suggest a bacteriome57 and mycobiome58 in pancreatic tissue of cancer patients. In line with these data, we observe a correlation between systemic antibodies against E. cloacae and hyperglycemia, which may suggest translocation of at least parts of this bacteria to extraintestinal sites.59 Nevertheless, translocation of whole bacteria remains rather controversial9 since there are major challenges related to the sequencing of small amounts of bacterial DNA in extraintestinal tissues.60
Heat-inactivated E. cloacae induced a detrimental beta-cell phenotype with insulin hypersecretion, induction of ER stress markers (elevated PI/I ratio), inflammation and reduced beta-cell insulin content. TLR4 can detect bacterial LPS and is involved in beta-cell dysfunction.21 Surprisingly, TLR4 knockout did not protect from bacteria-induced insulin expression loss, but did reduce the expression and secretion of IL-6. IL-6 is involved in glucose tolerance and beta-cell dysfunction, but its function is far from understood.61 On the other hand, deletion of TLR5, of which flagellin is the dominant ligand, protected against the insulin expression loss after incubation with flagellum-bearing E. cloacae. We speculate that an imbalance of TLR5 signaling drives metabolic diseases.
Flagellum is a virulence factor that enables bacteria to move within the intestine and even adhere to the intestinal wall, a process called encroachment.20 Flagellin is expressed by a variety of bacteria. Here, we used a commercially available pure flagellin from Salmonella typhimurium to avoid impurities from other bacterial components such as LPS. Both bacteria, S. typhimurium and E. cloacae, belong to the family Enterobacteriaceae. We see similar effects for both flagellin types. Flagellin fully reproduced the beta-cell phenotype of E. cloacae. A causal role for flagellin was observed in E. cloacae with flagellin knockout, where the effects on inflammation, hypersecretion and reduced insulin content were strongly diminished. Strengthening the role of flagellin, mice that were injected with flagellin exhibited a similar beta-cell phenotype. However, we cannot deduce whether this effect was directly on beta cells or a secondary response on other cells.
Flagellin, like the widely studied LPS, may translocate after food ingestion. Importantly, flagellin is able to pass the epithelial barrier62 and is a potent stimulus of the mucosal immune response.37 Plasma flagellin levels also positively correlated with HbA1c in our cohort, while the meal-related flagellin increment was associated with higher C-peptide release in obese humans with or without diabetes. Further, we found positive correlations between systemic inflammatory markers CRP and LPB with BMI, glucose values and antibodies against E. cloacae, suggesting that this bacteria induces inflammation in the host.
We acknowledge a number of limitations of this study. First, although the concept of translocation of flagellin to the systemic circulation seems to be plausible to explain beta-cell inflammation,55 we can only speculate if bacterial flagellin is transported via the blood circulation toward the pancreas. More sensitive methods are necessary to quantify small amounts of bacterial components such as flagellin in extra-intestinal tissues.20 Administration of single bacteria, for example flagellin bearing E. cloacae as well as our knockout strain, in mice would help to answer whether flagellin induces glucose intolerance. However, single strains do not engraft in the SPF murine microbiome and likely do not show an effect.18
Second, we provide evidence that people with T2Ds have a higher fecal and circulating flagellin load compared to normoglycemic individuals, with flagellin loads correlating with hyperglycemia. However, such a correlation does not show causation. With our data, we cannot decipher the role of flagellin in prediabetes and overt T2D. We can propose that flagellin induces insulin hypersecretion in early stages of the disease and longitudinal studies need to be done to show that this results in hypoinsulinemia and hyperglycemia over time. Insulin hypersecretion remains a controversial topic. Here we show that flagellin injection increases insulin secretion, beta-cell inflammation, but also reduces glucose levels. Therefore, resembling pre-diabetic states. However, achieving a full hyperglycemia in mice is only possible with genetic modulation and chemical treatment. Following these mice for a longer period would potentially show a more severe phenotype; however, not tested here. Together, we present a novel pathway linking bacterial flagellin from the Gram-negative E. cloacae in a TLR5-macrophage-dependent manner to beta-cell inflammation and beta-cell dysfunction, suggesting a new mechanism linking gut microbiota and T2D prevalence and opening up potential avenues for novel therapies.
Methods
Participants
For the current study, we included all 803 people of Dutch descent with available data on the gut microbiome from the HELIUS cohort in Amsterdam, the Netherlands.14 For details, regarding the HELIUS study (recruitment, data collection) in general, and regarding this selection in particular, see Deschasaux et al.15 For serum analysis on LPS and flagellin, 150 participants were randomly selected and diabetic participants (n = 100) were age-BMI-sex matched to healthy non-diabetic controls (n = 50). Diabetic participants were selected according to one of the following criteria (at least one): self-reported diagnosis of T2D, use of antidiabetic medication, fasting blood glucose >7.0 mmol/L and HbA1c >48 mmol/mol. All participants did not use antibiotics for the last 3 months.
The HELIUS data are owned by the Amsterdam UMC, location AMC in Amsterdam, The Netherlands. Any researcher can request the data by submitting a proposal to the HELIUS Executive Board. The HELIUS Executive Board will check proposals for compatibility with the general objectives, ethical approvals and informed consent forms of the HELIUS study.
To validate our results in the HELIUS cohort, we included 40 T2D participants and 40 non-diabetic, age, sex and BMI matched controls of the BARIA cohort. For details, see Van Olden et al.36 The BARIA study aims to assess how microbiota and their metabolites affect transcription in key tissues and clinical outcome in obese subjects and how baseline anthropometric and metabolic characteristics determine weight loss and glucose homeostasis after bariatric surgery.
Human pancreatic islets were obtained from Prodo Labs (US). Donor characteristics can be found in Table S7.
Bacteria
Enterobacter cloacae NCDC 279–56 and Escherichia coli K12 were purchased from German Collection of Microorganisms and Cell Cultures (DSMZ, Germany). They were cultured in Luria broth base (Invitrogen, US) and on LB Agar (Invitrogen, US) at 37°C, overnight, before being used for experiments. Faecalibacterium prausnitzii A2-165 was cultured in YCFA media anaerobically and Bacteroides ovatus 3_8_47FAA in YZFAA media anaerobically. Both were a generous gift from Willem de Vos (Wageningen).
Animals
Ten-week-old male C57BL/6 J mice were purchased from Charles River (France) and maintained under specific pathogen-free conditions in the S-building of the Amsterdam UMC, location AMC. TLR2 KO, TLR4 KO, TLR5 KO and C57BL/6 J DIO mice were purchased from Jackson Laboratory (JAX); control animals on C57BL/6 J background were used from JAX facilities instead of Charles River. All animals were socially housed, under a 12 h light/dark cycle until 12–14 weeks and sacrificed for pancreatic islet isolation. Only male mice were included in this study.
For flagellin injection, male obese C57BL6/J mice were ordered from JAX at 13 weeks (n = 15) and acclimatized for 1 week. Power was calculated according to insulin secretion after LPS injection63 with nQuery software (version 8.5.1). Mice were maintained on a high fat diet (60% kcal fat, ResearchDiets). Three to four mice were housed per cage. At week 14, mice were intraperitoneal injected twice weekly with 1 µg flagellin (Invivogen) in 100 uL saline for 4 weeks. Cages were randomly assigned to the treatments. Control mice were injected with100 µL saline. Body weight was measured before and during injections. Animals were fasted for 4h to measure fasting glucose. A intraperitoneal glucose tolerance (IPGTT) test was performed after the last injection (1.5 g/kg glucose in saline). Insulin was measured from the tail vein at baseline and 15 minutes in a subset of animals (n = 10) due to practical limitations. Insulin tolerance test was performed with 0.75 IU/kg (NovoRapid insulin). Glucose was measured in the tail vein blood every 15–30 minutes during 2 hours. Mice were killed with the aid of isoflurane and cardiac puncture. Organs were snap frozen. From a subset of mice (n = 5) due to practical limitations, pancreatic islets were isolated as described above, rested for 3 h in full media and RNA was harvested as well as GSIS was performed. Animal work was performed in accordance with the Central Commission for Animal Experiments (CCD, The Netherlands).
Heat-inactivation of bacteria
The optical density of the bacterial culture was measured at 600 nm (OD600) and diluted to 1E9 colony forming units (CFUs) per mL. Bacteria were centrifuged at 8000 xg for 5 minutes and resuspended in 1 mL sterile phosphate buffered saline (PBS). All bacteria were heat-inactivated at 70°C for 30 min and stored at −80°C in small aliquots for further use.
Pancreatic surgery
Individuals who are scheduled for pancreatic surgery (e.g., pylorus-preserving pancreatoduodenectomy or Whipple’s procedure), because of pancreatic carcinoma, were asked to donate healthy tissue surrounding the tumor. Tissue was harvested under surgical conditions, snap frozen in liquid nitrogen and stored at −80°C until further analysis.
Pancreatic islet isolation
Mice were anaesthetized with 2.5 mg pentobarbital (diluted in sterile saline) per mouse and sacrificed via cervical dislocation. After clamping the Ampulla of Vatar, the pancreas was injected intraductally with approximately 3 mL of collagenase XI (1000 U/ml) in HBSS (without calcium chloride) and placed in 50 mL tubes with an additional 2 mL of collagenase solution. The pancreas was incubated at 37°C for 13 minutes followed by gentle shaking to obtain a homogeneously dispersed pancreas. Digestion was stopped with cold HBSS supplemented with 1 mM CaCl2. Islets were washed two times in cold HBSS with CaCl2 by centrifuging 185 xg for 30 seconds. Next, islets were filtered through a 70 μM prewetted cell strainer. After flushing two times with 10 mL of HBSS with CaCl2, the strainer was turned upside-down over a Petri dish and rinsed with 16 mL of islet media (RPMI 1640 with GlutaMAXTM 1x, 10% FBS and P/S 1x) to collect the islets into the dish. Islets were handpicked under the Nikon SMZ800 microscope into a fresh Petri dish with islet media. Islets were rested overnight to recover from isolation procedure. Fresh pancreatic islets were incubated with 106 CFUs of heat-inactivated bacteria for 72 h at 37°C in full RPMI (1x Glutamax, 10% FBS, Pen/Strep). Islets were picked out of the media and used for GSIS (10 islets per replicate, in triplicates) or RNA isolation (50 islets per replicate, in triplicate). Media was saved at −80°C.
Plasmid construction
Overlap extension PCR64 was used to generate pRE118-pheS-ΔfliC and pRE118-pheS-ΔfljB constructs (pRE118-pheS was a gift from Christopher Hayes of UC Santa Barbara). For pRE118-pheS-ΔfliC construct, two PCR fragments were amplified using E. cloacae genomic DNA as the template. Primer pairs used to amplify the PCR fragments are ecFliC-P1 (5’-GATGATGGTGATGGTACGCGTGGTACCGGTAGTCGCT-3’) plus ecFliC-P2 (5’-GGTTTCTAGGGTCGGTGCCTTAACACTCA-3’), and ecFliC-P3 (5’- CACCGACCCTAGAAACCCTGTCTCTGCTGCGTTAA-3’) plus ecFliC-P4 (5’-GACAGTGAGCTCGCATCGTTAACGCGTCTTCACCAA-3’), respectively. This results in a 789-bp fragment containing the upstream of fliC and a 750-bp fragment containing the downstream of the fliC, respectively. These two PCR fragments were then mixed and used as the template for a secondary PCR (with primer pairs ecFliC-P1 containing a KpnI restriction enzyme site and ecFliC-P4 containing a SacI restriction enzyme site). The 16-bp overlapping sequence in primers ecFliC-P2 and ecFliC-P3 allows the amplification of a 1,539-bp PCR product. This PCR product was digested with KpnI and SacI, and directly cloned into the E. cloacae suicide vector pRE118-pheS (Kanr).
The pRE118-pheS-ΔfliC construct was generated the same as above. Primer pairs used to amplify the PCR fragments are FljB-P1 (5’-GCACGTCTAGAGTGACCTTTATCGTCATCTCACCGT-3’) plus FljB-P2 (5’-GTACCCAGCTGAGTCTGGGATTTGTTCAGGTTGTT-3’), and FljB-P3 (5’- AGACTCAGCTGGGTACTGCTGCGTTAATCTGCGTTA-3’) plus FljB-P4 (5’-GACAGTGAGCTCGTACAGCTATTCGCTGCATAACGA-3’), respectively. This results in a 955-bp fragment containing the upstream of fljB and a 950-bp fragment containing the downstream of the fljB, respectively. These two PCR fragments were then mixed and used as the template for a secondary PCR (with primer pairs FljB-P1 containing a XbaI restriction enzyme site and FljB-P4 containing a SacI restriction enzyme site). The 16-bp overlapping sequence (underlined) in primers FljB-P2 and FljB-P3 allows the amplification of a 1,905-bp PCR product. This PCR product was digested with XbaI and SacI, and directly cloned into the E. cloacae suicide vector pRE118-pheS.
Generation of E. cloacae mutant strains
pRE118-pheS-ΔfliC and pRE118-pheS-ΔfljB constructs were transformed into E. coli MFD(λ pir). E. coli MFD(λ pir) carrying these constructs and WT E. cloacae were grown overnight in LB, and then mixed at a ratio of 4:1 (donor vs recipient strains). To make fljB fliC, E. coli MFD(λ pir) carrying pRE118-pheS-ΔfljB and ΔfliC were grown overnight in LB, and then mixed at a ratio of 4:1. Fifty microliters of the mixture was spotted onto LB agar plate containing diaminopimelic acid (DAP, 0.3 mM) and incubated at 37°C overnight. This was followed by scraping the cell mixtures in PBS and plating onto LB agar containing streptomycin (100 μg/ml) and kanamycin (50 μg/ml). The resulting single-crossover mutants were grown statically in LB at 37°C overnight, and further counter selected on M9 minimal medium agar plates containing 0.4% (w/v) glucose and 0.1% (w/v) p-chlor-ophenylalanine.65 Kanamycin sensitive colonies were screened by colony PCR. The ΔfliC deletion mutant was confirmed by PCR with primers ecFliC-check-F (5’-GCGTTTCTGATGGCGTTCTGAA-3’) and ecFliC-check-R (5’-GCTCGAACTTGTTCATCCCGATT-3’). The predicted size of WT and mutant bands is 1201-bp and 362-bp, respectively. The ΔfljB deletion mutant was confirmed by PCR with primers FljB-check-F (5’-GCAGAACAACCTGAACAAATCCCA-3’) and FljB-check-R (5’-GACACGTTTACGCCGGTTCACTAT-3’). The predicted size of WT and mutant bands is 1811-bp and 387-bp, respectively.
Confirming mutants with a swimming motility assay
WT and mutant E. cloacae strains (ΔfljB, ΔfliC, ΔfljBΔfliC) were grown statically in 2 μl of LB at 30°C for 18 h. Two microliters of these cultures were spotted onto semi-solid nutrient broth (BD) agar plates containing 0.3% agar. After incubating the plates at 37°C for 4 h, pictures showing the swimming motility were taken (Figure S9).
Glucose stimulated insulin secretion
Pancreatic islets or β-cell lines were washed in a 12 well plate 2x with 500 uL low glucose Krebs-Ringer buffer (KRB; 132 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 1 mM MgSO4, 2.5 mM CaCl2, 5 mM NaHCO3, 10 mM HEPES, 0.25% BSA, 1.64 mM glucose) and starved in 500 uL low glucose KRB for 1 h. Islets were split into 10 islets per well in a 12 well plate (triplicates) and incubated for 1 hours in 500 uL low glucose KRB. The same islets were transferred into 500 uL high glucose KRB (16.4 mM) for 1 h. Finally, islets were washed 2x with 1 mL PBS and lysed with 150 uL RIPA buffer. Islet lysate was spun at 14.000 xg for 10 min at 4°C, and the supernatant was stored at −20°C until further use.
DNA isolation
Genomic DNA was isolated from overnight bacteria cultures with QIAamp® DNA Blood Mini Kit (Qiagen, Germany) according to manufacturer's instructions.
Fecal DNA was extracted from 150 mg fecal material and the sorted fractions using a repeated bead beating protocol (method 5).66 DNA was purified using Maxwell RSC Whole Blood DNA Kit. 16S rRNA gene amplicons were generated as described below.
RNA isolation and cDNA synthesis
RNA was isolated with TriPureTM isolation reagent (Roche). Cells were separated from the culture media, and 300 uL TriPureTM was added. After lysis, 60 uL chloroform was added, the mixture was vigorously shaken for 15 seconds and incubated for 3 minutes at room temperature. Next, samples were spun for 15 minutes at 12.000 xg (4°C) and the aqueous phase was mixed with 190 uL isopropanol with 0.44 uL GlycoBlueTM. After an overnight incubation at −20°C, samples were spun at 12.000 xg for 10 minutes (4°C) and the pellet was 2x washed with 1 mL of 75% ethanol (7.500 xg, 5 minutes, 4°C). Next, the pellet was dried at room temperature for 10 minutes, 18 uL RNase free H2O was added and incubated at 56°C for 10 minutes. RNA concentration was measured with Nanodrop. cDNA synthesis was performed with SensiFASTTM cDNA Synthesis Kit according to manufacturer's instructions.
PCRs
Gene expression was measured via real-time quantitative PCR (RT-qPCR) with the aid of PCR machine (BioRad, US). SensiFASTTM SYBR® No-ROX Kit was used according to manufacturer's instructions. For each well, 7.5 ng cDNA and 1 μM primer mix were used in a 10 uL PCR mix. For primers, see Table S6. Temperatures are used as following, if not stated differently: 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds and 60°C for 30 seconds with a plate reading, followed by a melt curve with increment of 0.5°C every 5 seconds starting from 65°C to 95°C. TLR5 expression was measured in cDNA from pure beta cells (rat INS1E cells), murine pancreatic islets and human monocytes. Three different sets of primers were used for each species and normalized to 18S reference gene (see primer list, Table S6).
Fecal bacteria were measured via quantitative PCR with the aid of PCR machine (BioRad, US). SensiFASTTM SYBR® No-ROX Kit was used according to manufacturer's instructions. For each well, 10 ng genomic DNA and 300 nM primer mix were used in a 10 uL PCR mix. For primers, see Table S6. For the total bacteria in feces, EUBAC primers and temperature settings were used as stated.67 For Enterobacteriaceae detection, En-lsu3 was used as described.68 Primers for Enterobacter cloacae was designed for the V3V4 regions. Temperatures as described above were used. Standard amplicons were made with genomic DNA from E. coli or E. cloacae and Taq DNA Polymerase (Qiagen, Germany) according to manufacturer’s instructions. Amplicons were cleaned with QIAquick PCR purification Kit (Qiagen, Germany). Copy numbers were calculated according to the standard curve.
Cell lines
HEK-BlueTM hTLR5 cells were used according to manufacturer's instructions. For LPS and flagellin detection in the blood circulation, 20 uL serum was used per well (96 well plate) and mixed with 180 uL of 1.4x1E5 cells/mL in detection media. Cells were incubated for 16 hours and the supernatant was read at OD655. INS-1E cells were cultured in RPMI 1640 media (5% FBS, 1x P/S, 1x HEPES, 50 μM β-mercaptoethenol, 1x sodium pyruvate) and passaged with 0.25% Trypsin-EDTA. Cells were seeded in a 12 well plate at 75.000 cells/mL, rested overnight and incubated with heat-inactivated bacteria for 72 hours.
Antibody analysis
Bacteria were grown overnight, and the optical density was measured at OD600. Bacteria were diluted to have 1E9 CFUs/mL and washed with 1 mL sterile PBS (8000 xg, 5 minutes, 4°C). Bacteria were sonicated on ice at 30% amplitude for 20 × 30 seconds cycles with 60 seconds intervals. NuncTM MicroWellTM 96-well microtiter plates (ThermoScientificTM, US) were coated with 200.000 sonicated bacteria per well (100 uL) overnight at 4°C. Plates were washed 3x with 300 uL per well of PBS and blocked with 150 uL PBS with 1% BSA for 2 hours at room temperature. Plates were washed again with PBS, and 100 uL of 250x diluted serum samples (PBS/BSA) was added and incubated for 4 hours at room temperature. Plates were washed 3x with PBS with 0.05% Tween 20 and 100 uL of secondary antibody (2000x diluted HRP anti-human IgG; 50.000x diluted HRP anti-human IgM; 20.000x diluted HRP anti-human IgA; in PBS/Tween 20) was added for 2 hours at room temperature. Plates were washed with PBS again, and 100 uL of TMB was added for 15 minutes. The reaction was stopped with 50 uL of 0.5 M HCl and read at OD450.
Pancreatic biopsies were 10x diluted according to tissue weight and homogenized in ultrapure water (Invitrogen, US) with the aid of a sterile metal bead. The homogenate was spun NuncTM MicroWellTM 96-well microtiter plates (ThermoScientificTM, US) were coated overnight (4°C) with 100 ng per well of flagellin from Salmonella typhimurium (Invivogen, US). The plates was washed 3x with 300 uL PBS/Tween 20. Afterward, 100 uL of homogenized pancreas was added and incubated for 1 h at 37°C. The plates was washed 3x with 300 uL PBS/Tween 20. Secondary antibodies and TMB were added as described above.
ELISA
Insulin was measured in low glucose KRB, high glucose KRB and cell lysate from GSIS experiments with ALPCO mouse ultrasensitive insulin ELISA according to manufacturer's instructions. Concentrations were normalized to total protein content measured via QuantiProTM BCA Assay Kit. IL-6 concentrations were measured in cell supernatants via ELISA MAXTM Deluxe Set Mouse IL-6 and IL-6 human uncoated ELISA kit according to manufacturer's instructions. Proinsulin was measured with Mercodia Rat/Mouse Proinsulin ELISA (Schweden). Lysates or glucose media from GSIS experiments were used. Ratios were calculated by dividing proinsulin with insulin for each of the nine replicates. CRP and LBP were measured in serum according to the manufacturer’s instructions (HycultBiotech, NL).
Isolation of flagellin from E. cloacae
E. cloacae was grown overnight in LB media. Bacteria were pelleted at 4000xg for 5 min. Pellet was washed with PBS. The pellet was resuspended in PBS and vigorously shaken for 2 min at 6 m/s (FastPrep). Bacteria were pelleted, and the supernatant was spun at 100.000 xg for 1 h at 4°C. The pellet containing flagellin was resuspended in PBS and heated at 70°C for 20 min to depolymerize the filaments. Protein concentrations were measured with BCA according to the protocol (ThermoFisher, US). TLR5 activity was verified on HEK TLR5 reporter cell line (Invivogen, US).
Monocyte isolation
PBMCs were isolated with Lymphoprep (GE Healthcare) and CD14 MACS beads (Miltenyi) according to manufacturer's instructions.
Macrophage depletion
Pancreatic islet macrophages were depleted with Clodronate-liposome. Islets were isolated and rested for 3 hours. Islets were picked in a small petri dish (40–70 islets per dish) and treated with either clodronate or control liposome for 48 h (1 in 5 diluted in islet media). Islets were washed 3x with 2 mL complete media and picked in fresh media.
Library preparation and sequencing
Library preparation and sequencing was performed at the Wallenberg Laboratory (Sahlgrenska University of Gothenburg, Sweden). Fecal microbiome composition was profiled by sequencing the V4 region of the 16S rRNA gene on an Illumina MiSeq instrument (Illumina RTA v1.17.28; MCS v2.5) with 515 F and 806 R primers designed for dual indexing69 and the V2 Illumina kit (2x250 bp paired-end reads). 16S rRNA genes from each sample were amplified in duplicate reactions in volumes of 25 µL containing 1x Five Prime Hot Master Mix (5 PRIME GmbH), 200 nM of each primer, 0.4 mg/ml BSA, 5% DMSO and 20 ng of genomic DNA. PCR was carried out under the following conditions: initial denaturation for 47 min at 94°C, followed by 25 cycles of denaturation for 45 s at 94°C, annealing for 60 s at 52°C and elongation for 90 s at 72°C and a final elongation step for 10 min at 72°C. Duplicates were combined, purified with the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel) and quantified using the Quant-iT PicoGreen dsDNA kit (Invitrogen). Purified PCR products were diluted to 10 ng/μL and pooled in equal amounts. The pooled amplicons were purified again using Ampure magnetic purification beads (Agencourt) to remove short amplification products; for negative controls, see Deschasaux, Bouter, Prodan, Levin, Groen, Herrema, Tremaroli, Bakker, Attaye, Pinto-Sietsma, van Raalte, Snijder, Nicolaou, Peters, Zwinderman, Backhed and Nieuwdorp.15 Libraries for sequencing were prepared by mixing the pooled amplicons with PhiX control DNA purchased from Illumina. The input DNA had a concentration of 3 pM and contained 15% PhiX and resulted in the generation of about 700 K clusters/mm2 and an overall percentage of bases with quality score higher than 30 (Q30) higher than 70%.
Bioinformatic pipeline
USEARCH (v11.0.667_i86linux64) was used to process the raw sequencing reads. For paired-end merging, we used 30 max. allowed differences in the overlapping region (“maxdiffs”) for the merging step (using the “fastq_mergepairs” command) and max. 1 expected errors (“fastq_maxee”) as a quality filter threshold (using the “fastq_filter” command). Expected error-based read quality filtering is described in detail in Edgar et al. 2015. After merging paired-end reads and quality filtering, remaining contigs were dereplicated and unique sequences were denoised using the UNOISE3 algorithm in order to obtain Amplicon Sequence Variants (ASVs). All merged reads were subsequently mapped against the resulting ASVs to produce an ASV table. ASVs not matching expected amplicon length were filtered out (i.e. ASV sequences longer than 260 bp or shorter than 250 bp). Taxonomy was assigned with the ‘assignTaxonomy’ function from the ‘dada2’ R package (v 1.12.1) and the SILVA (v. 132) reference database. ASV sequences were then aligned using MAFFT (v.7.427) using the auto settings. A phylogenetic tree was constructed from the resulting multiple sequence alignment with FastTree (v.2.1.11 Double Precision) using a generalized time-reversible model (‘-gtr’). The AVS table, taxonomy and tree were integrated using the ‘phyloseq’ R package (v.1.28.0). The ASV table was rarefied to 14,932 counts per sample with vegan v2.5–6. Of 6056 sequenced samples, 24 had insufficient counts (<5000 counts per sample) and were excluded at the rarefaction stage. The final dataset thus contained 6032 samples and 22,532 ASVs. Functional composition was inferred using PICRUSt2 (2.2.0b).
Cell viability
Cell viability of was measured with CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to manufacturer’s instructions. Ten size-matched islets were used per replicate with five replicates per experiment. Luminescence was read with Promega GLOMAXTM multi detection system.
Statistical analysis
Data were checked for normality with the Shapiro–Wilk test. Paired or unpaired t-test was performed for normal continuous variables and the Wilcoxon signed rank test or Mann-Whitney for other variables. Spearman correlation was used for all correlation analysis. Two-way ANOVA with Šidák multiple comparison was used for glucose and insulin tolerance tests. Statistical analyses were performed using Prism, version 8.3.0 (GraphPad Software, US). Data are provided as mean with SEM . P-values <0.05 were considered statistically significant. All authors had access to the study data and reviewed and approved the final manuscript.
Study approval
The studies were approved by the local Institutional Review Board of the Amsterdam UMC, location AMC in Amsterdam, the Netherlands, and conducted in accordance with the Declaration of Helsinki. Written informed consent was received prior to participation.
Author contributions
T.P.M.S. performed the experiments and prepared the manuscript. M.W. performed experiments. S.R.H., G.S., M.S. and D.D. assisted with animal experiments. S.M., M.dB., A.vdL. and Ö.A. conducted the BARIA cohort. M.B. and M.D. performed bioinformatic analyses. W.M.dV., C.B., H.Y. and C.M. provided bacterial cultures. M.G.B and O.R.B. provided pancreatic biopsies. G.M.D.T., G.J.B. and W.M.V. aided with the writing. B.J.H.B. conducted the HELIUS cohort. B.A.V., M.N., H.H. and C.B.V. supervised the project. D.H.R. developed the theory and supervised the project.
Supplementary Material
Acknowledgments
This study was funded by Diabetes Fonds (Application number: 2015.81) and Marie Skłodowska-Curie Actions (Call H2020-MSCA-IF-2015). CBV was supported by CIHR project grant PJT-165943. M.N. is supported by a personal ZONMW VICI grant 2020 [09150182010020]. B.A.V. is the Children with Intestinal and Liver Disorders (CH.I.L.D) Foundation Chair in Pediatric Gastroenterology. The HELIUS study is conducted by the Amsterdam University Medical Centers, location AMC and the Public Health Service of Amsterdam. Both organizations provided core support for HELIUS. The HELIUS study is also funded by the Dutch Heart Foundation, the Netherlands Organization for Health Research and Development (ZonMw), the European Union (FP-7), and the European Fund for the Integration of non-EU immigrants (EIF). We are most grateful to the participants of the HELIUS study and the management team, research nurses, interviewers, research assistants and other staff who have taken part in gathering the data of this study. The study reported here was additionally supported by (an) additional grant(s) from Dutch Heart Foundation: 2010T084 (K Stronks), ZonMw: 200500003 (K Stronks), European Union (FP-7): 278901 (K Stronks), European Fund for the Integration of non-EU immigrants (EIF): 2013EIF013 (K Stronks). HH is supported by a Senior Fellowship of the Dutch Diabetes Research Foundation (2019.82.004).
Funding Statement
This work was supported by the Diabetes Fonds [2019.82.004]; Diabetes Fonds [2015.81]; Marie Sklodowska-Curie [H2020-MSCA-IF-2015]; Netherlands Organisation for Health Research and Development [09150182010020].
Data availability statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to information that could compromise the privacy of research participants and can only be obtained via a data transfer agreement with the HELIUS board. Further information and requests for resources and reagents should be directed to and will be fulfilled by Torsten P.M. Scheithauer. In vitro experiments and murine data can be found under the following DOI: 10.4121/20484414.
Disclosure statement
M.N. and W.M.dV. are in the Scientific Advisory Board of Caelus Pharmaceuticals, the Netherlands M.N. is in the SAB of Kaleido, USA, and W.M.dV. is in the SAB of A-Mansia, Belgium. However, none of these are directly relevant to the current paper.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2111951
References
- 1.Weyer C, Bogardus C, Mott DM, Pratley RE.. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104(6):787–23. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Defronzo RABL. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pories WJ, Dohm GL. Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care. 2012;35(12):2438–2442. doi: 10.2337/dc12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, Botezelli J, Asadi A, Hoffman B, Kieffer T, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16(6):723–737. doi: 10.1016/j.cmet.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Erion K, Corkey BE. β-Cell Failure or β-Cell Abuse? Front Endocrinol (Lausanne). 2018;9:532. doi: 10.3389/fendo.2018.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MKJ, Biollaz G, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56(9):2356–2370. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- 7.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 8.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med. 2011;17(11):1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheithauer TPM, Rampanelli E, Nieuwdorp M, Vallance BA, Verchere CB, van Raalte DH, et al. Gut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol. 2020:11. doi: 10.3389/fimmu.2020.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurung M, Li Z, You H, Rodrigues R, Jump DB, Morgun A, Shulzhenko N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. 2020;51:102590. doi: 10.1016/j.ebiom.2019.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrema H, Niess JH. Intestinal microbial metabolites in human metabolism and type 2 diabetes. Diabetologia. 2020;63(12):2533–2547. doi: 10.1007/s00125-020-05268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomes JMG, Costa JA, Alfenas RCG. Metabolic endotoxemia and diabetes mellitus: a systematic review. Metabolism. 2017;68:133–144. doi: 10.1016/j.metabol.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Snijder MB, Galenkamp H, Prins M, Derks EM, Peters RJG, Zwinderman AH, Stronks K. Cohort profile: the Healthy Life in an Urban Setting (HELIUS) study in Amsterdam, The Netherlands. BMJ Open. 2017;7(12):e017873. doi: 10.1136/bmjopen-2017-017873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma S-J, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med. 2018;24(10):1526–1531. doi: 10.1038/s41591-018-0160-1. [DOI] [PubMed] [Google Scholar]
- 16.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 17.Balvers M, Deschasaux M, van den Born B-J, Zwinderman K, Nieuwdorp M, Levin E. Analyzing type 2 diabetes associations with the gut microbiome in individuals from two ethnic backgrounds living in the same geographic area. Nutrients. 2021;13(9):3289. doi: 10.3390/nu13093289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013;7(4):880–884. doi: 10.1038/ismej.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, Chakraborty M, Cheng K, Tao Chan Y, Nøhr MK, et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat Commun. 2019;10(1):3650. doi: 10.1038/s41467-019-11370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran HQ, Ley RE, Gewirtz AT, Chassaing B. Flagellin-elicited adaptive immunity suppresses flagellated microbiota and vaccinates against chronic inflammatory diseases. Nat Commun. 2019;10(1):5650. doi: 10.1038/s41467-019-13538-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji Y, Sun S, Shrestha N, Darragh LB, Shirakawa J, Xing Y, He Y, Carboneau BA, Kim H, An D, et al. Toll-like receptors TLR2 and TLR4 block the replication of pancreatic β cells in diet-induced obesity. Nat Immunol. 2019;20(6):677–686. doi: 10.1038/s41590-019-0396-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(48):20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. 2011;3(9):559–572. doi: 10.1002/emmm.201100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Chekan JR, Dodd D, Hong P-Y, Radlinski L, Revindran V. Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide-degrading enzymes. Proceedings of the National Academy of Sciences, 2014; 111:E3708–E17. doi: 10.1073/pnas.1406156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. doi: 10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 26.Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol. 2004;172(5):3173–3180. doi: 10.4049/jimmunol.172.5.3173. [DOI] [PubMed] [Google Scholar]
- 27.De Maayer P, Cowan DA. Flashy flagella: flagellin modification is relatively common and highly versatile among the Enterobacteriaceae. BMC Genomics. 2016;17(1):377. doi: 10.1186/s12864-016-2735-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.S-i Y, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, Wilson IA. Structural basis of TLR5-flagellin recognition and signaling. Science. 2012;335(6070):859–864. doi: 10.1126/science.1215584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nackiewicz D, Dan M, Speck M, Chow SZ, Chen Y-C, Pospisilik JA, Verchere CB, Ehses JA. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience. 2020;23(1):100775. doi: 10.1016/j.isci.2019.100775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, Seo JB, Yang B-H, Wollam J, Riopel M, et al. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 2019;29(2):457–74.e5. doi: 10.1016/j.cmet.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nackiewicz D, Dan M, He W, Kim R, Salmi A, Rütti S, Westwell-Roper C, Cunningham A, Speck M, Schuster-Klein C, et al. TLR2/6 and TLR4-activated macrophages contribute to islet inflammation and impair beta cell insulin gene expression via IL-1 and IL-6. Diabetologia. 2014;57(8):1645–1654. doi: 10.1007/s00125-014-3249-1. [DOI] [PubMed] [Google Scholar]
- 32.Hug H, Mohajeri MH, La Fata G. Toll-like receptors: regulators of the immune response in the human gut. Nutrients. 2018;10(2):203. doi: 10.3390/nu10020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 34.Bonifield HR, Hughes KT. Flagellar phase variation in Salmonella enterica is mediated by a posttranscriptional control mechanism. J Bacteriol. 2003;185(12):3567–3574. doi: 10.1128/JB.185.12.3567-3574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50(1):90–97. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Van Olden CC, Van de Laar AW, Meijnikman AS, Aydin O, Van Olst N, Hoozemans JB, De Brauw LM, Bruin SC, Acherman YIZ, Verheij J, et al. A systems biology approach to understand gut microbiota and host metabolism in morbid obesity: design of the BARIA longitudinal cohort study. J Intern Med. 2021;289(3):340–354. doi: 10.1111/joim.13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent L, Bell M, Hay A, Peterson D, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14(5):571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brissova M, Powers AC. Revascularization of transplanted islets: can it be improved? Diabetes. 2008;57(9):2269–2271. doi: 10.2337/db08-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson JD. On the causal relationships between hyperinsulinaemia, insulin resistance, obesity and dysglycaemia in type 2 diabetes. Diabetologia. 2021;64(10):2138–2146. doi: 10.1007/s00125-021-05505-4. [DOI] [PubMed] [Google Scholar]
- 40.Staimez LR, Weber MB, Ranjani H, Ali MK, Echouffo-Tcheugui JB, Phillips LS, Mohan V, Narayan KMV. Evidence of reduced β-Cell function in Asian Indians with mild dysglycemia. Diabetes Care. 2013;36(9):2772–2778. doi: 10.2337/dc12-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen ACH, Loudovaris T, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. 2014;20(12):1417–1426. doi: 10.1038/nm.3705. [DOI] [PubMed] [Google Scholar]
- 42.Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, Zhang E, Almgren P, Ladenvall C, Axelsson AS, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61(7):1726–1733. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, Spiliotis II, Dace W, Mattis KK, Ramracheya R, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun. 2014;5(1):4639. doi: 10.1038/ncomms5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Raalte DH, Verchere CB. Improving glycaemic control in type 2 diabetes: stimulate insulin secretion or provide beta-cell rest? Diabetes Obes Metab. 2017;19(9):1205–1213. doi: 10.1111/dom.12935. [DOI] [PubMed] [Google Scholar]
- 45.Esser N, Utzschneider KM, Kahn SE. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia. 2020;63(10):2007–2021. doi: 10.1007/s00125-020-05245-x. [DOI] [PubMed] [Google Scholar]
- 46.Hajmrle C, Smith N, Spigelman AF, Dai X, Senior L, Bautista A, Ferdaoussi M, MacDonald PE. Interleukin-1 signaling contributes to acute islet compensation. JCI Insight. 2016;1(4):e86055. doi: 10.1172/jci.insight.86055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hudish LI, Reusch JEB, Sussel L. β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest. 2019;129(10):4001–4008. doi: 10.1172/JCI129188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boni-Schnetzler M, Meier DT. Islet inflammation in type 2 diabetes. Semin Immunopathol. 2019;41(4):501–513. doi: 10.1007/s00281-019-00745-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maedler K, Spinas GA, Lehmann R, Sergeev P, Weber M, Fontana A, Kaiser N, Donath MY. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes. 2001;50(8):1683–1690. doi: 10.2337/diabetes.50.8.1683. [DOI] [PubMed] [Google Scholar]
- 50.Igoillo-Esteve M, Marselli L, Cunha DA, Ladrière L, Ortis F, Grieco FA, Dotta F, Weir GC, Marchetti P, Eizirik DL, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia. 2010;53(7):1395–1405. doi: 10.1007/s00125-010-1707-y. [DOI] [PubMed] [Google Scholar]
- 51.Westwell-Roper CY, Ehses JA, Verchere CB. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes. 2014;63(5):1698–1711. doi: 10.2337/db13-0863. [DOI] [PubMed] [Google Scholar]
- 52.MM OD, Harris HM, Lynch DB, Ross RP, O’Toole PW. Lactobacillus ruminis strains cluster according to their mammalian gut source. BMC Microbiol. 2015;15(1):80. doi: 10.1186/s12866-015-0403-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kajikawa A, Midorikawa E, Masuda K, Kondo K, Irisawa T, Igimi S, Okada S. Characterization of flagellins isolated from a highly motile strain of Lactobacillus agilis. BMC Microbiol. 2016;16(1):49. doi: 10.1186/s12866-016-0667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Massier L, Chakaroun R, Tabei S, Crane A, Didt KD, Fallmann J, von Bergen M, Haange S-B, Heyne H, Stumvoll M, et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut. 2020;69(10):1796. doi: 10.1136/gutjnl-2019-320118. [DOI] [PubMed] [Google Scholar]
- 55.Thomas RM, Jobin C. Microbiota in pancreatic health and disease: the next frontier in microbiome research. Nat Rev Gastroenterol Hepatol. 2020;17(1):53–64. doi: 10.1038/s41575-019-0242-7. [DOI] [PubMed] [Google Scholar]
- 56.Rogers MB, Aveson V, Firek B, Yeh A, Brooks B, Brower-Sinning R, Steve J, Banfield JF, Zureikat A, Hogg M, et al. Disturbances of the Perioperative Microbiome Across Multiple Body Sites in Patients Undergoing Pancreaticoduodenectomy. Pancreas. 2017;46(2):260–267. doi: 10.1097/MPA.0000000000000726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019;178(4):795–806.e12. doi: 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574(7777):264–267. doi: 10.1038/s41586-019-1608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anhê FF, Jensen BAH, Varin TV, Servant F, Van Blerk S, Richard D, Marceau S, Surette M, Biertho L, Lelouvier B, et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat Metab. 2020;2(3):233–242. doi: 10.1038/s42255-020-0178-9. [DOI] [PubMed] [Google Scholar]
- 60.de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, Parkhill J, Charnock-Jones DS, Smith GCS. Human placenta has no microbiome but can contain potential pathogens. Nature. 2019;572(7769):329–334. doi: 10.1038/s41586-019-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Citro A, Campo F, Dugnani E, Piemonti L. Innate immunity mediated inflammation and beta cell function: neighbors or enemies? Front Endocrinol (Lausanne). 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gewirtz AT, Simon PO Jr., Schmitt CK, Taylor LJ, Hagedorn CH, O’Brien AD, Neish AS, Madara JL. Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J Clin Invest. 2001;107(1):99–109. doi: 10.1172/JCI10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen AT, Mandard S, Dray C, Deckert V, Valet P, Besnard P, Drucker DJ, Lagrost L, Grober J. Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: involvement of the GLP-1 pathway. Diabetes. 2014;63(2):471–482. doi: 10.2337/db13-0903. [DOI] [PubMed] [Google Scholar]
- 64.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 65.Ting S-Y, Martínez-García E, Huang S, Bertolli SK, Kelly KA, Cutler KJ, Su ED, Zhi H, Tang Q, Radey MC, et al. Targeted depletion of bacteria from mixed populations by programmable adhesion with antagonistic competitor cells. Cell Host Microbe. 2020;28(2):313–21.e6. doi: 10.1016/j.chom.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, Tramontano M, Driessen M, Hercog R, Jung F-E, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069–1076. doi: 10.1038/nbt.3960. [DOI] [PubMed] [Google Scholar]
- 67.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(1):257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 68.Matsuda K, Tsuji H, Asahara T, Kado Y, Nomoto K. Sensitive quantitative detection of commensal bacteria by rRNA-targeted reverse transcription-PCR. Appl Environ Microbiol. 2007;73(1):32–39. doi: 10.1128/AEM.01224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to information that could compromise the privacy of research participants and can only be obtained via a data transfer agreement with the HELIUS board. Further information and requests for resources and reagents should be directed to and will be fulfilled by Torsten P.M. Scheithauer. In vitro experiments and murine data can be found under the following DOI: 10.4121/20484414.