

Abstract
The Joubert syndrome (JS), Meckel syndrome (MKS), and nephronophthisis (NPH) ciliopathy spectrum could be the poster child for advances and challenges in Mendelian human genetics over the past half century. Progress in understanding these conditions illustrates many core concepts of human genetics. The JS phenotype alone is caused by pathogenic variants in more than 40 genes; remarkably, all of the associated proteins function in and around the primary cilium. Primary cilia are near-ubiquitous, microtubule-based organelles that play crucial roles in development and homeostasis. Protruding from the cell, these cellular antennae sense diverse signals and mediate Hedgehog and other critical signaling pathways. Ciliary dysfunction causes many human conditions termed ciliopathies, which range from multiple congenital malformations to adult-onset single-organ failure. Research on the genetics of the JS-MKS-NPH spectrum has spurred extensive functional work exploring the broadly important role of primary cilia in health and disease. This functional work promises to illuminate the mechanisms underlying JS-MKS-NPH in humans, identify therapeutic targets across genetic causes, and generate future precision treatments.
Keywords: Joubert, Meckel, nephronophthisis, ciliopathies, genetics
INTRODUCTION
In retrospect, it does not seem terribly surprising that Joubert syndrome (JS), Meckel syndrome (MKS), and nephronophthisis (NPH) represent a spectrum of ciliopathy conditions unified by their overlapping phenotypes, interrelated genetic causes, and shared biological mechanisms linked to primary cilium dysfunction; however, this was not clear when the conditions were first described. In fact, the JS-MKS-NPH spectrum embodies some of the great successes and challenges of understanding Mendelian conditions. In this review, we elaborate on how research on the JS-MKS-NPH spectrum illustrates many fundamental concepts of human genetics—defining diagnostic categories, allelism, pleiotropy, phenotypic expansion, locus (genetic) heterogeneity, lumping and splitting, gene–phenotype correlations, genetic modifiers, and non-Mendelian inheritance. We also cover how the advances in human genetics have been synergistic with research on the biochemical, cellular, and developmental mechanisms underlying these conditions and hold great promise for future precision therapies to improve the health and quality of life of affected individuals. For a broader summary of all ciliopathies, we refer readers to a review by Reiter & Leroux (110).
CLINICAL DESCRIPTIONS
Joubert Syndrome
The first published description of JS was in a family with three children who had developmental delay, ataxia, abnormal breathing patterns, and agenesis of the cerebellar vermis (66). Many families were subsequently described, identifying involvement of the eye, kidney, liver, skeleton, and other body systems (9). It was not until the 1990s that the highly distinctive mid-hindbrain malformation was identified by magnetic resonance imaging (MRI) (88, 129). The molar tooth sign (MTS) appearance on axial MRI results from cerebellar vermis aplasia/hypoplasia, thick and horizontally oriented superior cerebellar peduncles, and often a deep interpeduncular fossa, and is considered pathognomonic for JS (Figure 1). This has allowed a clearer delineation of the associated clinical features in cohorts of patients with the MTS.
Figure 1.
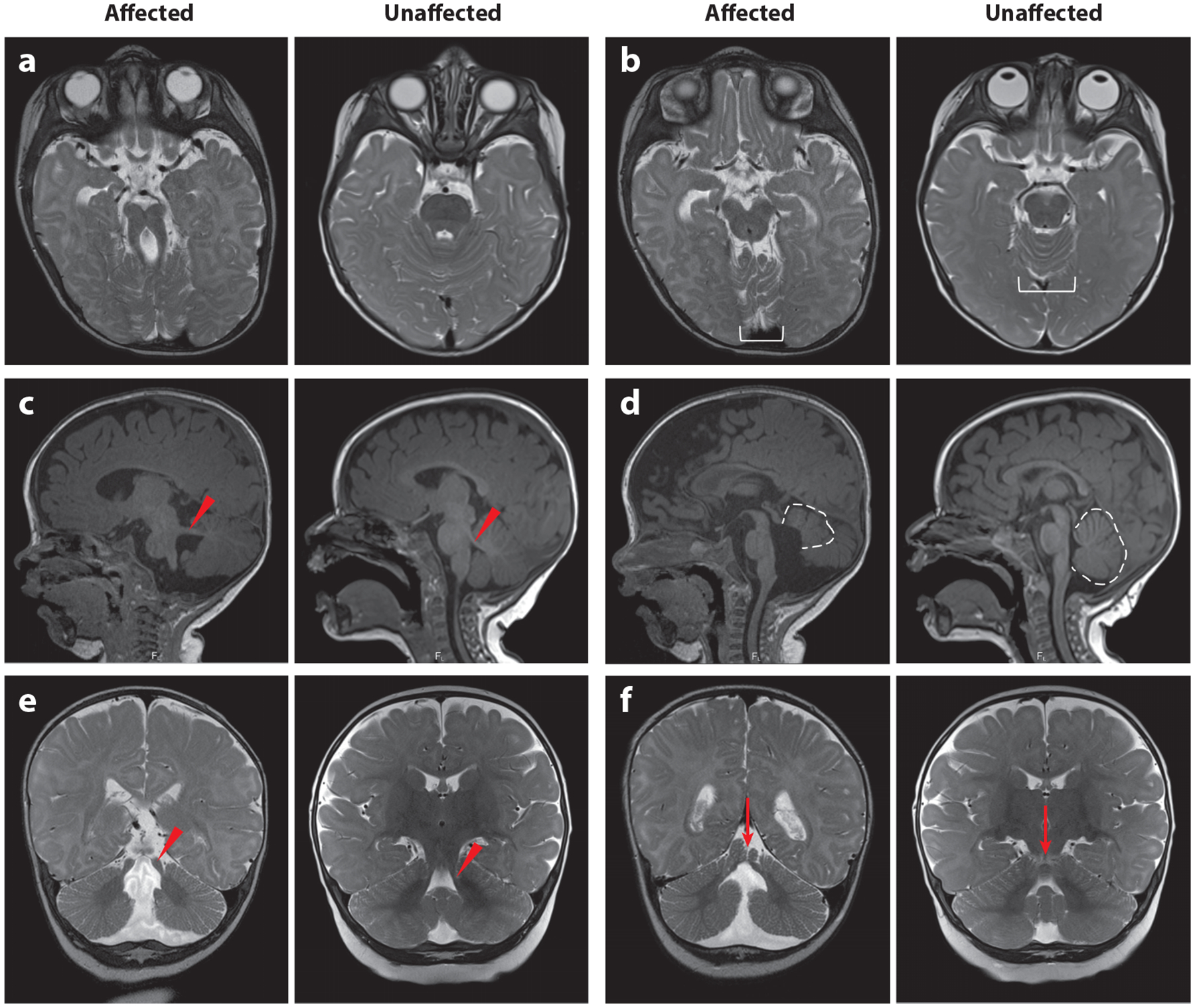
Diagnostic features of JS visualized by MRI. In each pair of images, the left is from an affected individual, and the right is from an unaffected individual of the same age. (a) MTS on a T2-weighted axial image due to the combination of long, thick superior cerebellar peduncles (the roots of the tooth) and a deep interpeduncular fossa (the cutting surface of the tooth). This appearance is not always captured in a single plane, so it is important to evaluate the MRI as a whole rather than relying on a single image. (b) Superior cerebellar foliar dysplasia (white bracket) on an axial T2-weighted image, which is sometimes seen in the absence of MTS in individuals carrying pathogenic variants in the JS-associated genes (note that the folia at this level are usually shaped like a U or V). (c) Horizontally oriented superior cerebellar peduncle (red arrowhead) on a T1-weighted parasagittal image. (d) Vermis hypoplasia (dotted outline), elevated roof of the fourth ventricle, and rostrally displaced fastigium on a T1-weighted sagittal image. (e) Long, thick superior cerebellar peduncles (red arrowhead) on a T2-weighted coronal image. (f) Cerebellar cleft (red arrow, indicating fluid at the cerebellar midline) on a T2-weighted coronal image. Abbreviations: JS, Joubert syndrome; MRI, magnetic resonance imaging; MTS, molar tooth sign. Figure adapted from Reference 9.
Clinically, JS typically presents during infancy with hypotonia, abnormal eye movements, and alternating tachypnea and apnea. Some individuals present as children or adults with mild ataxia and/or cognitive impairment (36, 102, 112). JS is increasingly diagnosed prenatally based on mid-hindbrain imaging abnormalities, often combined with abnormal kidneys, polydactyly, or other features; however, fetuses with JS are commonly assigned to less specific diagnostic categories, such as Dandy–Walker malformation or cerebellar vermis hypoplasia. Additional brain abnormalities seen in some patients include heterotopias (of the cortex, midbrain, and hindbrain, particularly the cervicomedullary junction), polymicrogyria, and agenesis of the corpus callosum (67, 107). Other variable features include polydactyly (~15% of people with JS); coloboma, a congenital eye malformation (~15%); retinal dystrophy (~30%); dysplastic kidneys, including cystic kidney disease and kidney fibrosis (~20%); and liver fibrosis (~15%). JS is not associated with a recognizable facial appearance (19). Until the recent descriptions of an autosomal dominant form of mild JS (119, 121), only recessive and X-linked inheritance had been described. The estimated prevalence in Italy is approximately 0.5/100,000 for adults and 2/100,000 for people under 19 years of age (95).
Meckel Syndrome1
The recognizable combination of occipital encephalocele, polycystic kidneys, and polydactyly was described in two siblings by Johann Friedrich Meckel (90) in 1822. This clinical description has evolved with reports of more examples (14) and the identification of genetic causes. Although evidence-based diagnostic criteria have not been established, the four central characteristics of MKS appear to be occipital encephalocele, large cystic–dysplastic kidneys, congenital hepatic fibrosis (also called ductal plate malformation), and polydactyly. A variety of additional malformations have been described, including additional brain abnormalities (cerebellar hypoplasia, hydrocephalus, holoprosencephaly, and anencephaly); coloboma, microphthalmia, or anophthalmia; cleft lip and palate; oral hamartomas; congenital heart disease; ambiguous genitalia; and skeletal dysplasia. Clinically, MKS is diagnosed during pregnancy based on prenatal imaging findings or at the time of delivery. It is typically associated with pre- or perinatal lethality, although survival beyond infancy has been described. So far, only recessive inheritance has been demonstrated for MKS. The estimated prevalence is approximately 2/100,000 (14).
Nephronophthisis
NPH was first described as a clinical entity by Smith & Graham (132) in 1945, as progressive kidney disease in an 8-year-old girl with severe chronic anemia. Autopsy revealed kidneys with innumerable medullary cysts less than 1 cm in size. NPH is most frequently seen in isolation but is also a feature of syndromic conditions, such as Senior–Løken, Dekaban–Arima, COACH (cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis), and Mainzer–Saldino syndromes, as well as cranioectodermal and short-rib thoracic dysplasias and other conditions (138). It can present through adulthood but typically presents in the early teenage years and progresses to kidney failure. This review focuses mainly on genetic forms of NPH that overlap with JS and MKS. Generally, NPH displays autosomal recessive inheritance, and the estimated prevalence is 1/50,000 to 1/100,000.
DISEASE GENE DISCOVERY
We are in the golden age of Mendelian condition gene discovery, enabled by advances in phenotyping, genomic technologies, and data sharing (12). As with many recessive conditions, early gene discovery in JS-MKS-NPH during the 1990s relied on cytogenetics and laborious mapping strategies to identify loci, followed by further laborious Sanger sequencing of candidate genes in the mapped intervals. In fact, cytogenetics enabled the discovery of a relatively common recurrent deletion encompassing the NPHP1 gene as the first genetic cause of NPH in 1997 (57). While NPH was already established as a feature of JS, the first genetic link came in 2004, when NPHP1 deletions were identified in patients with JS. That same year, linkage mapping and Sanger sequencing identified biallelic AHI1 variants in JS patients (35, 43). The first genetic cause of MKS, dysfunction of MKS1, was identified based on a shared chromosome 17q22 haplotype in Finnish families (70). The advent of next-generation sequencing accelerated gene discovery, ultimately resulting in more than 40 genes associated with JS, more than 15 associated with MKS, and more than 15 associated with NPH (Figure 2; Table 1).
Figure 2.
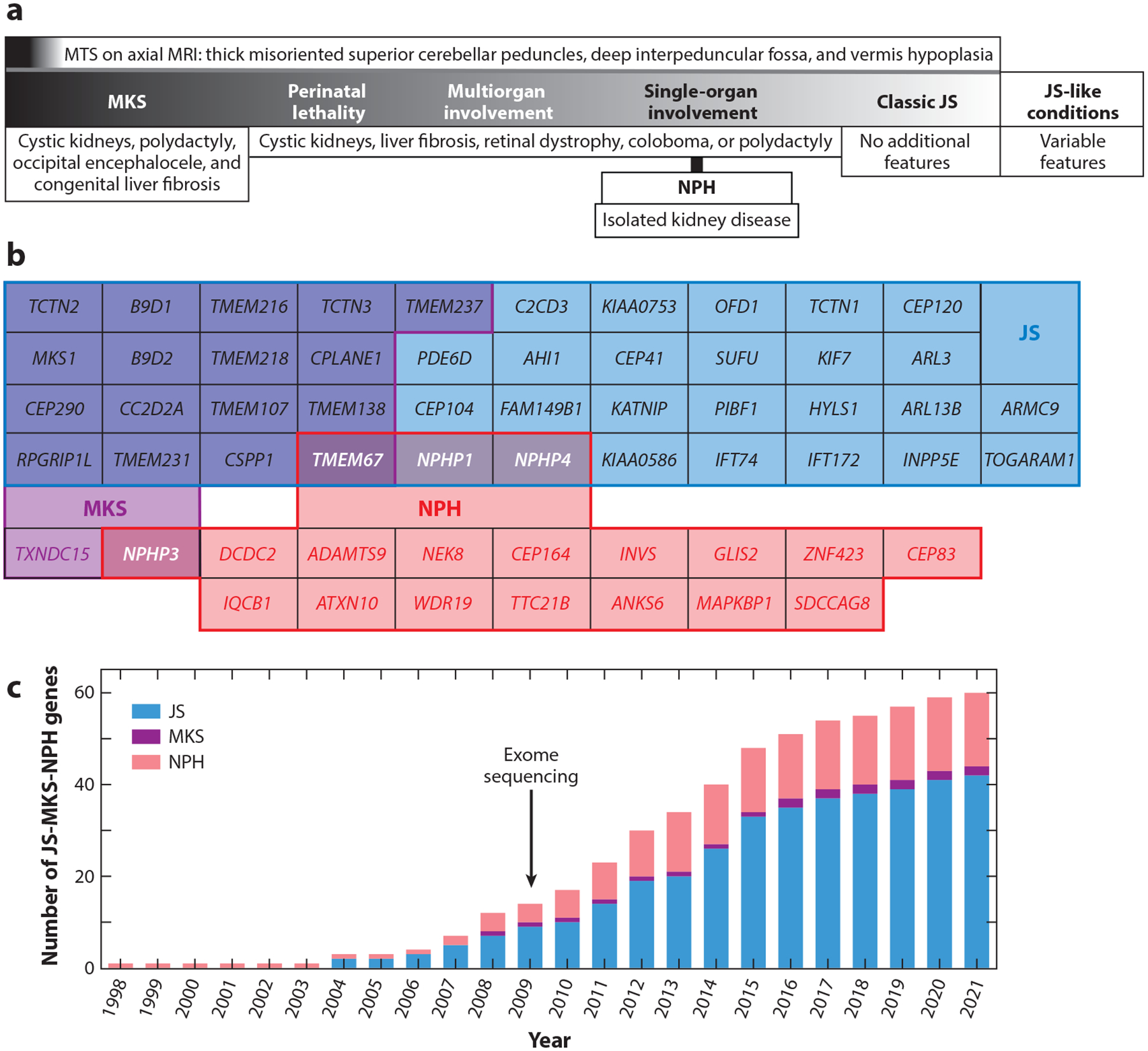
JS-MKS-NPH phenotypic and genetic overlap and gene discovery time line. (a) Coarse phenotypic spectrum, from severe MKS to milder JS. This schematic is based on overlapping genetic etiology and phenotypic manifestations. (b) Overlap of genetic etiology from the JS-MKS-NPH spectrum. Genes associated with JS are in blue, those associated with MKS are in purple, and those associated with isolated NPH are in red, with overlap as indicated by shading. (c) Time line of published JS-MKS-NPH-associated genes. Abbreviations: JS, Joubert syndrome; MKS, Meckel syndrome; MRI, magnetic resonance imaging; MTS, molar tooth sign; NPH, nephronophthisis.
Table 1.
Genes associated with JS, MKS, or isolated NPH
| Human genea | Notation | Alias(es) | Phenotype OMIM | Gene OMIM | Chromosome | First reference(s)b |
|---|---|---|---|---|---|---|
| JS-associated genes | ||||||
| AHI1 | JBTS3 | JOUBERIN | 608629 | 608894 | 6q23.3 | 43 |
| ARL3 | JBTS35 | 618161 | 604695 | 10q24.32 | 5 | |
| ARL13B | JBTS8 | ARL2L1 | 612291 | 608922 | 3q11.2 | 22 |
| ARMC9 | JBTS30 | KIAA1868 | 617622 | 617612 | 2q37.1 | 149 |
| B9D1 | JBTS27 | MKSR1,MKS9 | 617120 | 614144 | 17p11.2 | 111 |
| B9D2 | JBTS34 | MKS10 | 614175 | 611951 | 19q13.2 | 10 |
| C2CD3 | 615944 | 11q13.4 | 141 | |||
| CC2D2A | JBTS9 | KIAA1345,MKS6 | 612285 | 612013 | 4p15.32 | 50 |
| CEP41 | JBTS15 | TSGA14 | 614464 | 610523 | 7q32.2 | 74 |
| CEP104 | JBTS25 | KIAA0562 | 616781 | 616690 | 1p36.32 | 135 |
| CEP120 | JBTS31 | CCDC100 | 617761 | 613446 | 5q23.2 | 126 |
| CEP290 | JBTS5 | NPHP6, MKS4, BBS14 | 610188 | 610142 | 12q21.32 | 118 |
| CPLANE1 | JBTS17 | C5ORF42 | 614615 | 614571 | 5q13.2 | 137 |
| CSPP1 | JBTS21 | 615636 | 611654 | 8q13.1 | 144 | |
| FAM149B1 | JBTS36 | KIAA0974 | 618763 | 618413 | 10q22.2 | 125 |
| HYLS1 | FLJ32915 | 610693 | 11q24.2 | 96 | ||
| IFT74 | JBTS40 | BBS22 | 619582 | 608040 | 9q21.2 | 84 |
| IFT172 | BBS20 | 607386 | 2q23.3 | 53 | ||
| INPP5E | JBTS1 | 213300 | 613037 | 9q34.3 | 18 | |
| KATNIP | JBTS26 | KIAA0556 | 616784 | 616650 | 16p12.1 | 114 |
| KIAA0586 | JBTS23 | TALPID3 | 616490 | 610178 | 14q23.1 | 11 |
| KIAA0753 | JBTS38 | OFIP | 619476 | 617112 | 17p13.1 | 25 |
| KIF7 | JBTS12 | 200990 | 611254 | 15q26.1 | 29 | |
| MKS1 | JBTS28 | BBS13 | 617121 | 609883 | 17q22 | 111 |
| NPHP1 | JBTS4 | 609583 | 607100 | 2q13 | 101 | |
| NPHP4 | KIAA0673 | 607215 | 1p36.31 | 4 | ||
| OFD1 | JBTS10 | CXORF5 | 300804 | 300170 | Xp22.2 | 27 |
| PDE6D | JBTS22 | 615665 | 602676 | 2q37.1 | 143 | |
| PIBF1 | JBTS33 | 617767 | 607532 | 13q21.33 | 151 | |
| RPGRIP1L | JBTS7 | NPHP8,MKS5 | 611560 | 610937 | 16q12.2 | 6 |
| SUFU | JBTS32 | 617757 | 607035 | 10q24.32 | 33,121 | |
| TCTN1 | JBTS13 | 614173 | 609863 | 12q24.11 | 46 | |
| TCTN2 | JBTS24 | MKS8 | 616654 | 613846 | 12q24.31 | 115 |
| TCTN3 | JBTS18 | C10ORF61 | 614815 | 613847 | 10q24.1 | 142 |
| TMEM67 | JBTS6 | MKS3, NPHP11, MECKELIN | 610688 | 609884 | 8q21.1 | 8 |
| TMEM107 | JBTS29 | MKS13 | 617562 | 616183 | 17p13.1 | 71 |
| TMEM138 | JBTS16 | 614465 | 614459 | 11q12.1 | 75 | |
| TMEM216 | JBTS2 | MKS2 | 608091 | 613277 | 11q12.2 | 145 |
| TMEM218 | JBTS39 | 619562 | 619285 | 11q24.2 | 148 | |
| TMEM231 | JBTS20 | MKS11 | 614970 | 614949 | 16q23.1 | 136 |
| TMEM237 | JBTS14 | ALS2CR4 | 614424 | 614423 | 2q33.1 | 62 |
| TOGARAM1 | JBTS37 | FAM19B, KIAA0423 | 619185 | 617618 | 14q21.2 | 73 |
| MKS-associated genes | ||||||
| B9D1 | MKS9 | MKSR1 | 614209 | 614144 | 17p11.2 | 61 |
| B9D2 | MKS10 | 614175 | 611951 | 19q13.2 | 38 | |
| CC2D2A | MKS6 | KIAA1345 | 612284 | 612013 | 4p15.32 | 140 |
| CEP290 | MKS4 | NPHP6, BBS14 | 611134 | 610142 | 12q21.32 | 45 |
| CPLANE1 | C5ORF42 | 614571 | 5q13.2 | 82 | ||
| CSPP1 | 611654 | 8q13.1 | 127 | |||
| MKS1 | MKS1 | BBS13 | 249000 | 609883 | 17q22 | 70 |
| NPHP3 | MKS7 | 267010 | 608002 | 3q22.1 | 131 | |
| RPGRIP1L | MKS5 | NPHP8 | 611561 | 610937 | 16q12.2 | 34 |
| TCTN2 | MKS8 | 613885 | 613846 | 12q24.31 | 124 | |
| TCTN3 | C10ORF61 | 613847 | 10q24.1 | 142 | ||
| TMEM67 | MKS3 | NPHP11, MECKELIN | 607361 | 609884 | 8q22.1 | 133 |
| TMEM107 | MKS13 | 617562 | 616183 | 17p13.1 | 122 | |
| TMEM138 | 614459 | 11q12.1 | 75 | |||
| TMEM216 | MKS2 | 603194 | 613277 | 11q12.2 | 145 | |
| TMEM218 | 619285 | 11q24.2 | 148 | |||
| TMEM231 | MKS11 | 615397 | 614949 | 16q23.1 | 123 | |
| TMEM237 | ALS2CR4 | 614423 | 2q33.1 | 62 | ||
| TXNDC15 | C5ORF14, BUG | 617778 | 5q31.1 | 128 | ||
| Isolated NPH-associated genes | ||||||
| ADAMTS9 | 605421 | 3p14.1 | 26 | |||
| ANKS6 | NPHP16 | PKDR1, SAMCYSTIN | 615382 | 615370 | 9q22.33 | 59 |
| ATXN10 | ATAXIN10 | 611150 | 22q13.31 | 115 | ||
| CEP83 | NPHP18 | CCDC41 | 615862 | 615847 | 12q22 | 42 |
| CEP164 | NPHP15 | KIAA1052 | 614845 | 614848 | 11q23.3 | 23 |
| DCDC2 | NPHP19 | RU2, RU2S | 616217 | 605755 | 6p22.3 | 120 |
| GLLS2 | NPHP7 | 611498 | 608539 | 16p13.3 | 7 | |
| INVS | NPHP2 | 602088 | 243305 | 9q31.1 | 52 | |
| IQCB1 | NPHP5 | KIA0036 | 609254 (SL) | 609237 | 3q13.33 | 100 |
| MAPKBP1 | NPHP20 | JNKBP1 | 617271 | 616786 | 15q15.1 | 85 |
| NEK8 | NPHP9 | 613824 | 609799 | 17q11.2 | 100 | |
| NPHP1 | NPHP1 | 256100 | 607100 | 2q13 | 57 | |
| NPHP3 | NPHP3 | MKS7 | 604387 | 608002 | 3q22.1 | 97 |
| NPHP4 | NPHP4 | KIAA0673 | 606966 | 607215 | 1p36.31 | 92 |
| SDCCAG8 | NPHP10 | CCCAP, BBS16 | 613615 (SL) | 613524 | 1q43 | 98 |
| TMEM67 | NPHP11 | MKS3, MECKELIN | 613550 | 609884 | 8q21.1 | 99 |
| TTC21B | NPHP12 | THM1 | 613820 | 612014 | 2q24.3 | 32 |
| WDR19 | NPHP13 | IFT144 | 614377 | 608151 | 4p14 | 54 |
| ZNF423 | NPHP14 | KIAA0760 | 614844 | 604557 | 16q12.1 | 23 |
Abbreviations: JS, Joubert syndrome; MKS, Meckel syndrome; NPH, nephronophthisis; SL, Senior–Løken syndrome phenotype.
Boldface indicates that the Doherty laboratory led or was involved with the discovery of a gene linked to JS or MKS.
Column lists the first publication(s) to demonstrate an association between the gene and JS, MKS, or isolated NPH.
Gene discovery efforts in JS-MKS-NPH also highlight the need for caution when establishing an association between genes and phenotypes. Several published associations do not meet the highest level of evidence, due to very small numbers of families, inadequate clinical information, and/or stronger support for association with a different phenotype (Supplemental Table 1).
LOCUS HETEROGENEITY AND ALLELISM
The JS-MKS-NPH spectrum represents extreme examples of locus heterogeneity, where the same phenotype is caused by variants at many loci. For instance, the MTS phenotype of JS appeared to be so specific that, initially, it seemed like JS might be associated with only one gene; however, after the first two JS-associated genes accounted for less than 10% of families, it quickly became clear that JS is very genetically heterogeneous. Nonetheless, the fact that all genetic causes of JS affect proteins that function in and around the primary cilium (see the section titled Biochemical and Cellular Mechanisms, below) is a tribute to the specificity of the phenotype. By contrast, the genetic causes of broad diagnostic categories such as autism and intellectual disability implicate many different cellular mechanisms.
Similarly, the JS-MKS-NPH spectrum exemplifies marked allelism, since many different variants in each gene cause each phenotype, in contrast to conditions such as achondroplasia, which is caused almost exclusively by a single variant in a single gene. The distribution of variant types across JS-associated genes illustrates several genetic phenomena, including a copy number variation hot spot (recurrent NPHP1 deletion), a founder variant [TMEM216 c.218G>T (p.R73L) in people of Ashkenazi ancestry], genes sensitive to loss of function (predominantly missense variants in INPP5E), and genes resistant to loss of function (predominantly truncating variants in CEP290) (Figure 3).
Figure 3.
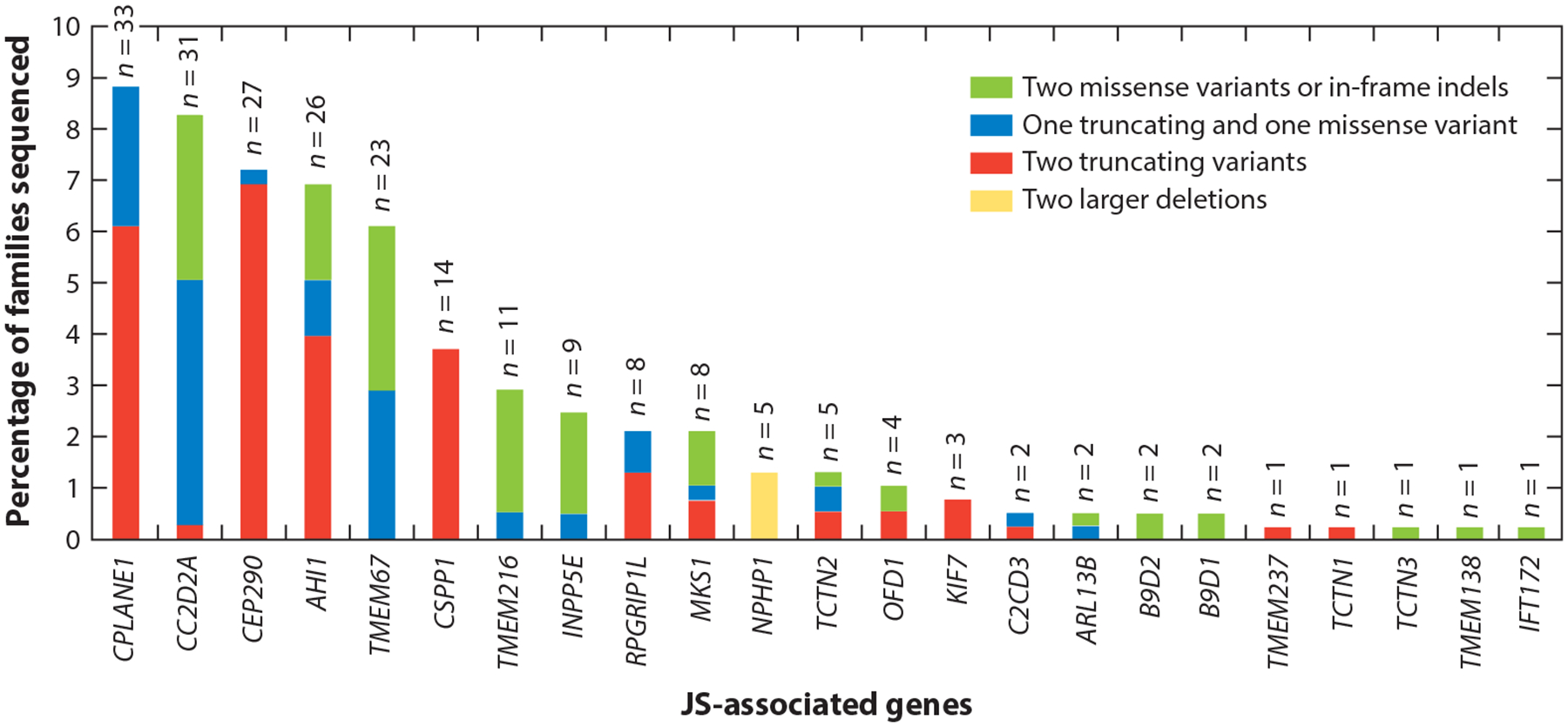
Genetic causes in a large JS cohort, showing the proportion of individuals with JS who carry two rare deleterious variants in each gene. Each bar is broken down to illustrate the relative frequency of the observed variants in each gene: Green indicates two missense variants or small in-frame indels, blue indicates one truncating and one missense variant (including small in-frame indels), red indicates two truncating variants (including nonsense, frameshift, and canonical splice-site variants), and yellow indicates two larger deletions. For each bar, n indicates the total number of people with JS who have variants assessed in this graph. Abbreviations: indel, insertion or deletion; JS, Joubert syndrome. Figure adapted from Reference 10.
CATEGORICAL DIAGNOSES, LUMPING, AND SPLITTING
Categorical clinical diagnoses play a critical role in organizing our thinking about patients. Even in the absence of information about the biological basis, clinical diagnoses of genetic conditions inform prognosis and recurrence risk and guide additional diagnostic evaluations and treatment. For obvious reasons, prior to the 1950s, identifiable DNA differences did not play a role in defining genetic conditions, which was instead based only on clinical information. Thus, MKS and JS were thought to be distinct for decades due to their marked differences in phenotype.
The identification of pathogenic CEP290 variants in both JS (118, 146) and MKS (8) provided the first genetic evidence that these diagnoses should be lumped rather than split. The eventual association of 17 genes with both of these diagnoses indicates that they largely represent the mild and severe ends of the same biological spectrum (Figure 2a). Further observations that JS and MKS can occur in family members with the same causal variants solidified this concept. In parallel, the identification of similar variants in the same genes provided support for the idea that other conditions, such as COACH (37), acrocallosal (29), hydrolethalus (96), Mainzer–Saldino (53) and Varadi–Papp (oral-facial-digital type VI) (82) syndromes, are also part of the JS-MKS-NPH spectrum, although devoted splitters might argue that JS represents more than 40 genetically distinct conditions. When genetic causes appear to impact a defined biological process and result in a coherent recognizable phenotype, we agree with grouping them under an umbrella diagnostic category, such as ciliopathies. This concept has greatly facilitated disease gene discovery and mechanistic understanding by guiding investigators to group patients with similar phenotypes and look for pathogenic variants in functionally related genes (21).
By contrast, the identification of variants in genes for other ciliopathies has split the JS-MKS-NPH spectrum from other ciliopathy diagnoses, such as Bardet–Biedl syndrome (BBS). BBS is a genetically heterogeneous recessive ciliopathy that shares some features with JS (intellectual disability, retinal dystrophy, kidney disease, and polydactyly) but also includes other non-JS features (obesity and hypogonadism) and does not include the defining JS brain malformations. This phenotypic distinction is largely supported by nonoverlapping genetic causes, with the exception of rare patients with possible BBS who carry one or two pathogenic variants in CEP290, MKS1, IFT74, or IFT172 (44). Brain imaging data are not presented for many proposed BBS patients, so it is possible that they have JS with obesity rather than BBS. Similarly, the JS-MKS-NPH spectrum does not genetically overlap with other ciliopathies, such as Alström and Ellis–van Creveld syndromes, cranioectodermal dysplasia, and autosomal dominant polycystic kidney disease. With the rapidly expanding availability of next-generation sequencing and advances in human disease gene discovery, genetic causes now play an important role in defining syndromic conditions.
PLEIOTROPY
As noted above, multiple body systems are commonly impacted in the JS-MKS-NPH spectrum, including the brain (MTS and occipital encephalocele), eye (coloboma and retinal dystrophy), kidney (cystic dysplastic kidneys and NPH), liver (fibrosis), and skeleton (polydactyly and skeletal dysplasia). Less frequent features impact almost all body systems. Identifying the genetic causes explained this remarkable pleiotropy, since primary cilia are present on almost all cells, participate in many signaling pathways, and play widespread roles in the development and function of many body systems.
PHENOTYPIC EXPANSION
Historically, human genetic conditions have been defined by recognizing nonrandom co-occurrence of clinical features in multiple patients, which led to the discovery of the underlying genetic causes. Established genetic causes are then evaluated in phenotypically broader groups of patients who have subsets of the features in the originally identified patients but also have other features, gradually revealing the full spectrum of phenotypes associated with dysfunction of a particular gene. Similar to other Mendelian conditions with known genetic causes, the spectrum of phenotypes associated with many of the JS-MKS-NPH-associated genes has expanded over time. For instance, clearly pathogenic biallelic JS-gene variants have been recently identified in multiple patients who have ocular motor apraxia and other neurological features consistent with JS; however, they do not have the MTS, representing a phenotypic expansion beyond classically defined JS. Similarly, biallelic variants in CEP290, AHI1 (94), and INPP5E (116) can cause isolated retinal dystrophy, and TMEM67 variants can cause isolated NPH (99).
GENE–PHENOTYPE AND GENOTYPE–PHENOTYPE CORRELATIONS
One of the central questions in genetics is how variation in the genome results in phenotypic variation. In Mendelian conditions, high-impact causal variants are major determinants of phenotype, modified by lower-impact variants and environmental factors. In genetically heterogeneous conditions such as the JS-MKS-NPH spectrum, the phenotype is also influenced by the gene in which the causal variants occur. For the purposes of this review, gene–phenotype correlation refers to the relationship between the gene and the phenotype, while genotype–phenotype correlation refers to the relationship between variants within a gene and the phenotype. We define a gene–phenotype correlation as the association between dysfunction of a specific gene and a characteristic constellation of one or more phenotypic features.
The strongest gene–phenotype correlations involve diagnostic categories with relatively little clinical and genetic overlap (e.g., CFTR-related cystic fibrosis versus DMD-related muscular dystrophy). In fact, these gene–phenotype associations help define the diagnostic categories. The situation is more complicated with phenotypically overlapping and genetically heterogeneous conditions; for example, genes associated with autism overlap substantially with those associated with intellectual disability (117). Despite overlapping phenotypes, ciliopathies appear to separate into distinct conditions. For instance, JS and MKS are distinct from other ciliopathies, such as Alström and Ellis–van Creveld syndromes, and from BBS, with very rare possible exceptions (see the section titled Categorical Diagnoses, Lumping, and Splitting, above). Within the JS-MKS-NPH spectrum, each gene has been associated with one or more of the three diagnostic categories (Figure 2; Table 1): 23 genes are associated with JS alone, 1 with MKS alone, and 15 with isolated NPH alone. By contrast, 17 genes are associated with both JS and MKS, 3 are associated with both JS and isolated NPH, 1 is associated with MKS and isolated NPH, and 1 is associated with JS, MKS, and isolated NPH.
Gene–phenotype correlations are more subtle within narrower phenotypes with genetic heterogeneity, such as JS. Early on, it became apparent that some genes were associated with specific variable features of JS. These anecdotal observations have been substantiated by more systematic analyses showing the strongest associations of TMEM67 dysfunction with coloboma and liver fibrosis and CEP290 dysfunction with retinal dystrophy (10). The information on gene–phenotype correlations is limited because these conditions are rare, most of the genetic causes impact only a small proportion of patients, gathering phenotypic data systematically from across the world is challenging, and phenotypic variability is substantial, likely due to genetic modifiers and environmental factors. For a condition like JS, where all genes/proteins function in the same cellular process(es), detecting gene–phenotype correlations appears to be the exception rather than the rule. While gene–phenotype correlation information is limited, the implications are important for informing prognosis and guiding medical monitoring, as well as for focusing treatments on the genetic causes associated with the highest risk of progressive features (9).
Genes associated with more than one phenotypic presentation along the JS-MKS-NPH spectrum provide the opportunity to evaluate genotype–phenotype correlations—that is, whether the predicted deleteriousness of variants within a given gene correlate with the severity of the phenotype, from NPH at the mild end to MKS at the severe end, with JS in the middle. This is analogous to an allelic series in a model system. The initial papers implicating TMEM67 and CC2D2A dysfunction in both MKS and JS found more truncating variants in MKS than in JS (93). TMEM67 dysfunction has been linked to the entire JS-MKS-NPH spectrum. As shown in Figure 4, only biallelic missense variants in TMEM67 are associated with NPH; approximately half of patients with JS carry biallelic missense variants, while the other half carry one missense and one truncating variant; and approximately half of fetuses with MKS carry biallelic truncating variants, while a quarter carry missense and truncating variants and the remaining quarter carry biallelic missense variants. It is likely that other genes display similar genotype–phenotype correlations.
Figure 4.
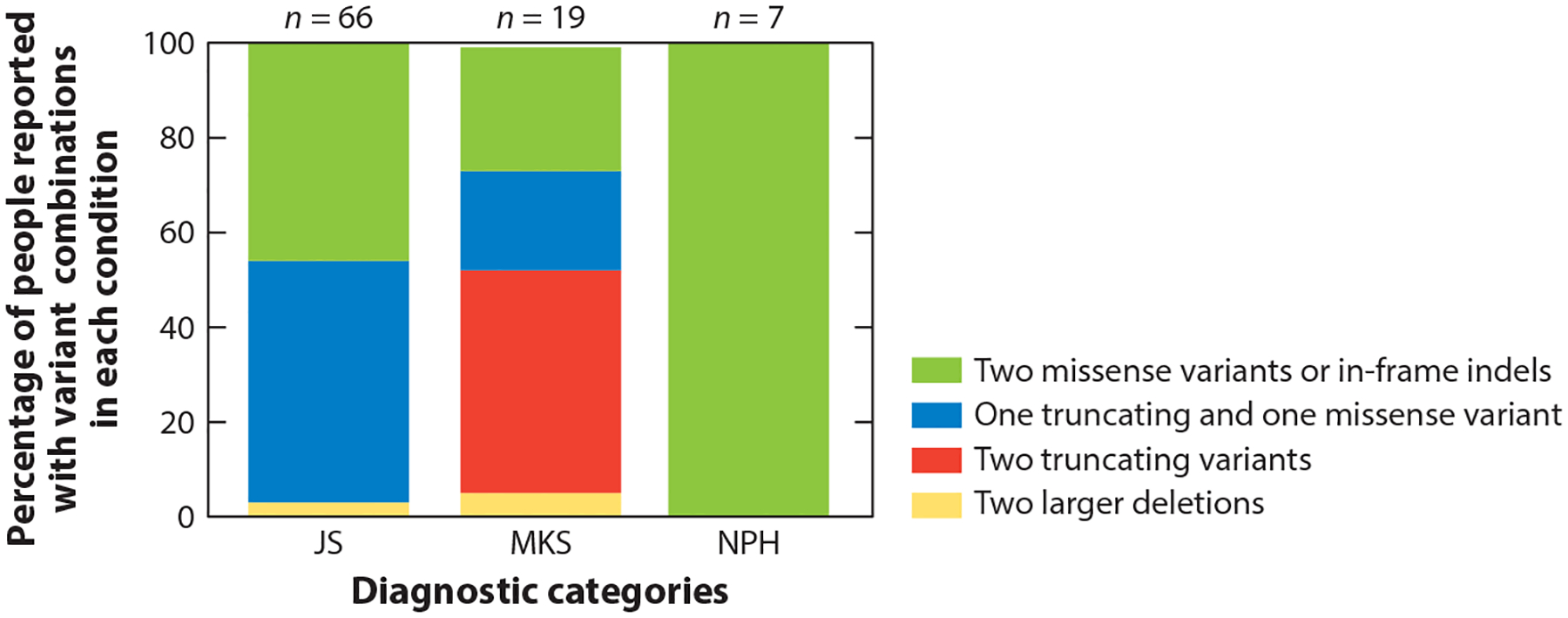
Proportions of individuals with JS, MKS, or isolated NPH who carry two rare deleterious variants in TMEM67. The colors on each bar illustrate the relative frequency of the variants in each diagnostic category: Green indicates two missense variants or small in-frame indels, blue indicates one truncating and one missense variant (including small in-frame indels), red indicates two truncating variants (including nonsense, frameshift and canonical splice-site variants), and yellow indicates two larger deletions. For each bar, n indicates the total number of people found in the literature with each diagnosis who have the variants assessed in this graph. Abbreviations: indel, insertion or deletion; JS, Joubert syndrome; MKS, Meckel syndrome; NPH, nephronophthisis.
In another example exploring allelic series, Shimada et al. (130) found that fibroblasts from people with CEP290-associated JS were different from those from people with Leber congenital amaurosis (LCA). JS-associated fibroblasts did not have detectable CEP290 protein, while this protein was reduced in LCA-associated fibroblasts. Interestingly, ciliogenesis and ciliary length were not affected in LCA-associated fibroblasts, but JS-associated fibroblasts showed impaired ciliogenesis and highly variable ciliary lengths. The JS-associated fibroblasts also showed reduced ciliary ARL13B and ADCY3, whereas these proteins were at near-control levels in LCA-associated fibroblasts.
GENETIC MODIFIERS
As with all Mendelian genetic conditions, the phenotypic expression of the JS-MKS-NPH spectrum in a given person likely depends on the causal gene, the specific effects of the causal variants, genetic modifier variants in the rest of the genome, and environmental factors. The causal gene and variants have the largest impact, while genetic modifiers and environmental factors have smaller effects. Genetic modifiers are well documented in animal systems based on the ability to perform enhancer/suppressor screens in relatively uniform genetic backgrounds. It is more difficult to identify specific genetic modifiers of Mendelian conditions in humans. The best examples come from conditions with uniform genetic causes, such as the identification of DCTN4 variants as modifiers of chronic Pseudomonas infection in cystic fibrosis, often due to the CFTR F508 variant (41).
The situation is more complex for phenotypes in the JS-MKS-NPH spectrum, because these phenotypes can result from hundreds of causal variants across almost 60 published genes. Presumably, these diverse variants have a broad range of functional effects, likely contributing the most variability to the phenotypic presentations, although the functional effects of most variants have not been systematically evaluated. Nonetheless, discordant phenotypes in siblings and individuals with identical causal variants provide strong evidence for the existence of genetic and environmental modifiers in ciliopathies (87). For instance, in the TMEM67 analysis presented above, two siblings homozygous for the p.C615R variant had clear JS with kidney and liver involvement, while several other individuals had kidney and liver involvement with normal brain imaging and no neurological symptoms (99). Similarly, discordant phenotypes in different strains of mice with the same causal variant also support a role for genetic modifiers (3) (see the section titled Genetic Modifiers in Animal Models, below).
Despite the evidence for genetic modifiers of JS-MKS-NPH phenotypes and many anecdotal reports of candidate variants (28, 58), it remains challenging to prove a role for specific variants in humans due to the limited numbers of patients, the diversity of causal variants, the difficulties of quantifying phenotypic severity, and the possibility that multiple variants of small effect rather than single variants of large effect act as modifiers. These factors make it difficult, and perhaps impossible, to prove the impact of most proposed genetic modifiers based on human genetic data alone. To date, the strongest evidence for specific modifiers supports associations of the AHI1 p.R830W and RPGRIP1L p.A229T variants with retinal dystrophy risk in patients with NPH (83) and BBS (69), respectively, although these studies have not been replicated. In one study investigating human genetic modifiers, an RPGRIP1L variant, p.A229T, was detected in people with BBS or NPH with and without retinal degeneration. This variant was found at low frequency in people with retinal degeneration and was absent in people with isolated NPH, suggesting that the p.A229T variant increases the likelihood of retinal involvement (69). Evidence for genetic modifier variants in nonciliopathy genes has not been reported for JS-MKS-NPH phenotypes in humans, and evidence for environmental modifiers in humans is also very limited.
NONMENDELIAN INHERITANCE
Although JS, MKS, and NPH have historically been regarded as almost exclusively recessive conditions, we would be remiss to not consider other modes of inheritance, especially since 5–30% of individuals with JS and 60–70% of individuals with isolated NPH still lack a genetic diagnosis (depending on the cohort and genetic analysis performed) (9, 10, 138).
Dominant Inheritance
Dominant inheritance must be considered in patients with heterozygous variants, particularly when they segregate with a genetic condition in a family or when they occur de novo in single individuals. Dominant inheritance arises from haploinsufficiency, where partial loss of gene function results in a phenotype; dominant negative effects, where variants in one copy of a gene interfere with the function of the other copy; and gain of function, where variants cause increased gene activity or neomorphic effects that differ from the usual gene function. Recently, the first dominant form of JS-MKS-NPH due to inherited and de novo SUFU variants was reported in mildly affected individuals with JS (121).
Oligogenic Inheritance
Oligogenicity most commonly refers to situations in which variants in more than one gene can cause a phenotype. In ciliopathies, one example is triallelic inheritance, in which a combination of biallelic variants in one gene and a heterozygous variant in a second gene has been proposed to cause BBS in some families (68). Additionally, heterozygous variants in two different genes have been proposed to cause NPH in a few families (58). Detailed analysis of one large JS cohort did not reveal substantial evidence for oligogenic inheritance mechanisms, but oligogenicity could not be excluded in a small subset of families (106). In patients with heterozygous variants in more than one JS gene (single hits), unidentified second-hit variants in one of the genes could explain the genetic cause. These patients provide an excellent opportunity to characterize difficult-to-identify variant types, including noncoding variants, structural variants, repeat expansions, and mobile element insertions, among others.
BIOCHEMICAL AND CELLULAR MECHANISMS
Over the past century, remarkable progress has been made in our understanding of ciliopathies such as JS-MKS-NPH and the basic biology of cilium function, in large part due to the synergistic advances in human genetics, biochemical and cell biology techniques, and model organisms.
General Information on Primary Cilia
Primary cilia are micron-scale antenna-like projections found on nearly every cell that sense mechanical, chemical, and light input and mediate signaling pathways. Specialized doublet microtubules polymerize from a modified centriole called the basal body, forming a cylindrical structural core called the axoneme. The distal GTP-capped microtubule tips create a compartment where tip-specific microtubule-associated proteins bind. The proximal ciliary membrane is continuous with the plasma membrane, albeit with a distinct lipid composition. At the base, cilia are partitioned from the cell by the transition zone (TZ), which forms the interface between cellular cytoplasm and cilioplasm; anchors the basal body to the plasma membrane, preventing passive diffusion of large soluble proteins; limits lateral diffusion of membrane-associated proteins and lipids; and facilitates ciliary protein ingress. A devoted intraflagellar transport (IFT) system quickly adjusts protein content. This system uses plus-end-directed kinesin motors to shuttle proteins across the TZ and to the ciliary tip. IFT trains reorganize at the tip and transport proteins bound for ciliary export to the ciliary base using minus-end-directed dynein motors (Figure 5).
Figure 5.
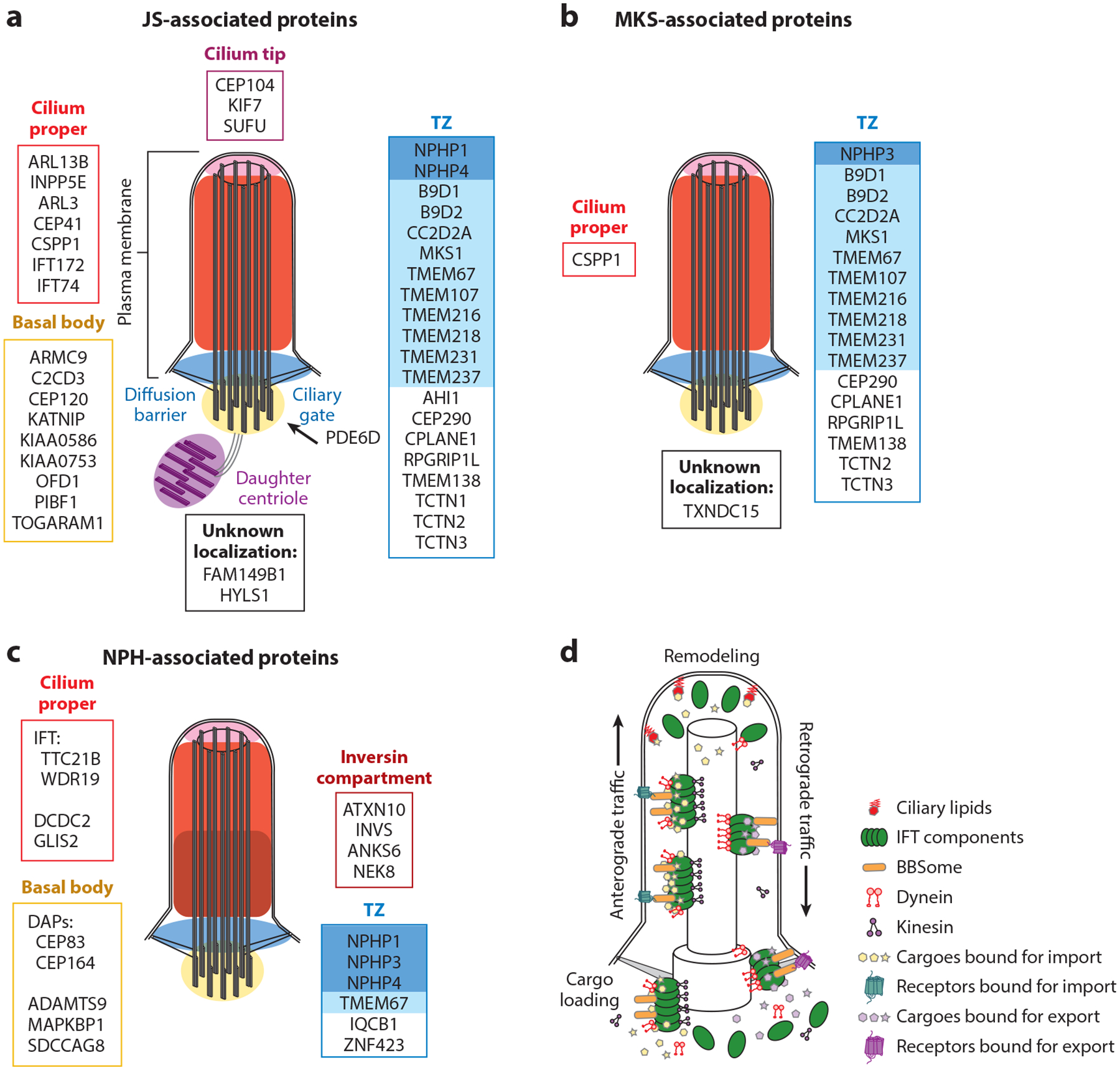
JS-MKS-NPH protein localization and IFT. (a) JS-associated proteins grouped by their localization in steady-state cilia. The TZ is subdivided into the NPHP module (dark blue) and the MKS module (light blue). The ciliary membrane is continuous with the plasma membrane. (b) MKS-associated proteins grouped by their localization in steady-state cilia. (c) NPH-associated proteins grouped by their localization in steady-state cilia. (d) Transport of ciliary proteins by IFT components (green). The BBSome (orange) traffics receptors. Ciliary lipids (red) capture and retain specific proteins. Abbreviations: BBSome, protein complex involved in Bardet–Biedl syndrome; DAP, distal appendage; IFT, intraflagellar transport; JS, Joubert syndrome; MKS, Meckel syndrome; NPH, nephronophthisis; TZ, transition zone.
Identifying Central Mechanisms
A central question in the biology of JS-MKS-NPH is how the dysfunction of so many genes results in these phenotypes, as distinct from the other ciliopathies (Figure 6). For instance, some of the JS-MKS-NPH spectrum phenotypes overlap with BBS, which is characterized by intellectual disability, retinal dystrophy, and polydactyly but no structural brain malformation. Despite the overlapping phenotypes, BBS is caused by dysfunction of a cilium-related protein network that is largely distinct from that involved in JS-MKS-NPH. For instance, protein interaction screens using JS-associated proteins as bait pull out other JS-MKS proteins (the Joubertome), while proteins involved in BBS pull out other BBS proteins (the BBSome) with little overlap.
Figure 6.
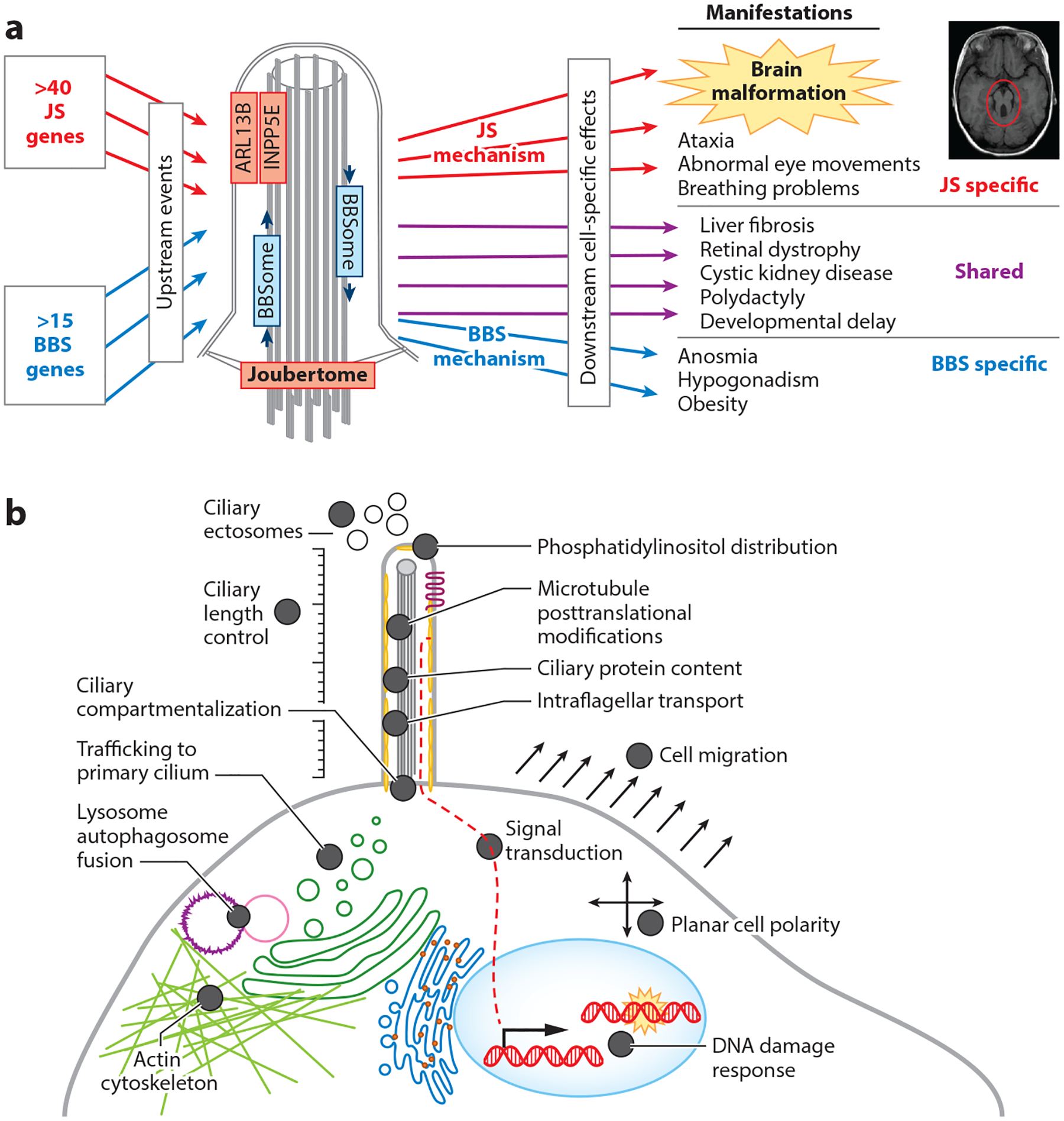
Mechanisms underlying ciliopathies. (a) Unique mechanisms underlying JS and BBS. (b) Diverse cellular dysfunction reported in the literature based on individual JS-MKS protein loss-of-function experiments across model organisms. Abbreviations: BBS, Bardet–Biedl syndrome; JS, Joubert syndrome; MKS, Meckel syndrome.
Extensive work on JS-MKS-NPH-related genes in model organisms with null mutations has revealed a wide variety of defects in cilium number and morphology, multiple signaling pathways, cytoskeletal organization, autophagy, and DNA damage response (Figure 6). Some of these defects have been observed only with dysfunction of one or two genes. For instance, only CEP290, CEP164, and ZNF423 have been implicated in the DNA damage response; therefore, we do not consider this a likely core disease mechanism (23, 30). However, some defects are shared across many genetic causes of the JS-MKS spectrum and are therefore more likely to be central to the pathological mechanisms. Since JS has the tightest clinical phenotype and substantial genetic overlap with MKS, we focus on JS-MKS gene dysfunction here.
Not Just a Transition Zone-opathy
Approximately half of JS-associated proteins localize to the TZ (Figure 5). The other half localize to various ciliary subcompartments, some of them (notably ARL13B and INPP5E) in a TZ-dependent manner. For example, PDE6D shuttles lipid-modified proteins to the cilium but does not localize within the cilium. In addition, dysfunction of the basal body–localizing ARMC9/TOGARAM1 module does not appear to disrupt TZ function, since both TZ components and ciliary ARL13B and INPP5E localize similarly to controls (73). Interestingly, almost all MKS-associated proteins localize to the TZ (Figure 5).
The TZ is often subdivided into protein interaction modules named for the association of their component proteins with certain conditions (i.e., the NPHP and MKS modules, shaded in blue in Figure 5a) (115). This naming convention arose more than a decade ago. Given our evolving knowledge about the genetic spectrum of JS-MKS-NPH, the named modules do not map neatly to the phenotypes; therefore, we discourage referring to protein complexes or modules by phenotypes unless the correlation is consistent, as is the case for the BBSome.
Intraflagellar Transport
IFT dysfunction can interfere with cilium formation, stability, length, and morphology in addition to disrupting signaling pathways. In model organisms, IFT dysfunction phenotypes include abnormal neural tube patterning, skeletal dysplasia, polydactyly, and early lethality. In humans, IFT172 and IFT74 dysfunction causes JS. TTC21B, which encodes IFT139, is associated with isolated NPH and NPH with extrarenal phenotypes (32). Other IFT components and ciliary motors (e.g., the dynein-2 motor complex) are associated with skeletal dysplasia ciliopathies (reviewed in 156) but have not been implicated in JS-MKS-NPH, precluding the involvement of general IFT dysfunction.
Ciliary Microtubule Posttranslational Modifications
Ciliary microtubules undergo a variety of posttranslational modifications, including glutamylation on the outer surface and acetylation in the microtubule lumen. Axonemal posttranslational modifications influence microtubule stability, molecular motor activity, microtubule-associated protein localization, and resistance to mechanical stress. Defects in ciliary posttranslational modifications have been described with dysfunction of multiple JS-associated proteins. In CEP41-associated JS patient fibroblasts, the glutamylating enzyme TTLL6 fails to localize to the cilium, resulting in axonemal hypoglutamylation (74). Similarly, ARL13B promotes ciliary import of the tubulin glutamylating enzymes TTLL5 and TTLL6, and small interfering RNA–mediated ARL13B knockdown results in hypoglutamylation (55). Furthermore, both acetylation and glutamylation are reduced in hypomorphic Kif7L130P mouse embryonic fibroblasts (56). ARMC9- and TOGARAM1-associated JS patient fibroblasts also display reduced axonemal acetylation and polyglutamylation, as confirmed in genome-engineered cell lines and zebrafish (20, 73). Defects in other posttranslational modifications, such as glycylation or detyrosination, have not been identified in the JS disease context. To our knowledge, posttranslational modification defects have not been described in the context of MKS-NPH, but they have been implicated in polycystic kidney disease and other ciliopathies (17, 86).
Ciliary Membrane Phosphatidylinositol Content
The ciliary membrane is a privileged domain, distinct from the rest of the plasma membrane. Part of this distinction is maintained by differences in phosphatidylinositol (PI) content, maintained in part by a single 5′ phosphatase, INPP5E; the role of ciliary PI kinases is less clear. PI(4,5)P2 (the INPP5E substrate) is restricted to the ciliary base, while PI(4)P (the INPP5E product) is the major ciliary PI. In addition, INPP5E may control TZ structure and function by modulating PI(3,4,5)P3 and PI(3,4)P2 (39). INPP5E dysfunction (due to mutation, mislocalization, or insufficient activity) is a common mechanistic defect seen in many genetic causes of JS.
Multiple JS-MKS proteins target and retain INPP5E in cilia. PDE6D shuttles INPP5E to the cilium via its C-terminal farnesylated CaaX motif. Ciliary ARL13B converts ARL3-GDP to ARL3-GTP, a cargo release factor that in turn releases INPP5E from PDE6D in the cilium. Therefore, ciliary localization of INPP5E requires PDE6D (143), ARL3 (5), ARL13B (63), and the INPP5E CaaX motif (108). Notably, TZ dysfunction can also result in reduced ciliary INPP5E due to faulty ciliary ARL13B retention (63).
While INPP5E mislocalization is not an obligate feature of JS based on recent data showing proper ciliary localization in fibroblasts from patients with ARMC9- and TOGARAM1-associated JS (73), whether abnormal ciliary PI localization is present across genetic causes of JS remains an open question (i.e., INPP5E dysfunction due to mutation, mislocalization, or insufficient activity has not been entirely excluded as a unifying mechanism of JS).
Hedgehog Signaling
In vertebrates, the primary cilium plays an obligate role in canonical Hedgehog (Hh) signaling through the coordinated movement of receptors and effectors in and out of the organelle (reviewed in 13). In the absence of Hh ligand, the PTCH receptor localizes to the primary cilium and transports cholesterol from the inner to the outer leaflet of the ciliary membrane, limiting SMO activation. SUFU and KIF7 complex with the transcription factors GLI2 and GLI3 to regulate their ciliary and nuclear localization. In addition, GPR161, a constitutively active orphan G protein–coupled receptor, helps maintain GLI2 and GLI3 in their phosphorylated forms via protein kinase A. Phosphorylated GLI2 and GLI3 are ubiquitinated, resulting in proteolytic cleavage of the activator domain and conversion to transcriptional repressors. KIAA0586 influences both GLI3 activator/repressor ratios and GLI1 expression (31). With stimulation, Hh binds PTCH, which is exported from the cilium, promoting SMO entry and GPR161 exit. With reduced ciliary PTCH, cholesterol accumulates in the inner leaflet and directly activates SMO. GLI2 and GLI3 localization increases at the ciliary tip, and they remain as full-length transcriptional activators that translocate to the nucleus and upregulate targets that include GLI1 and PTCH1. Additionally, noncanonical Hh signaling occurs independently from cilia and involves at least some JS-associated proteins as effectors, notably ARL13B (49).
In addition to the direct involvement of SUFU, KIF7, and KIAA0586 in the Hh signaling pathway (31, 56, 121), aberrant Hh signaling has been observed across most genetic causes of JS-MKS-NPH in patient tissues, patient cell lines, and model systems—specifically, 31 of 42 JS genes, 14 of 19 MKS genes, and 7 of 19 NPH genes (Supplemental Table 2). The most common published cellular indicators of abnormal Hh signaling in JS-MKS-NPH include abnormal protein localization (SMO, GPR161, and GLI2/3), GLI2/3 processing and expression, and target gene expression (GLI1 and PTCH1), while the most common developmental phenotypes are abnormal neural tube patterning and polydactyly. Though not proven, polydactyly and other JS phenotypes (coloboma and potentially kidney disease) are thought to be related to Hh based on similar phenotypes in animal models with disrupted Hh signaling. Despite the extensive links between JS-MKS-NPH and Hh signaling, it remains unclear exactly how the pathway is disrupted across genetic causes. The diverse assays used in a wide variety of models, as well as differences between null and hypomorphic models, make it difficult to discern exactly how Hh is disrupted and whether the disruption is similar across genetic causes.
Wnt Signaling
The role of primary cilia in Wnt signaling is less well understood on a step-by-step level (reviewed in 147). Elimination of cilia using KIF3A short hairpin RNA enhances Wnt pathway activity, indicating that cilia may play an inhibitory role (48). In the absence of Wnt ligand, the axin complex phosphorylates β-catenin, leading to its degradation and reducing its transcriptional activation of target genes. When Wnt binds to the Frizzled class of receptors, phosphorylated DVL/DSH inhibits axin complex formation, which reduces β-catenin phosphorylation. Thus, β-catenin translocates to the nucleus to activate Wnt target genes. While this cascade is not known to proceed within the cilium, the NPH-related ciliary protein INVS can interact with DVL/DSH to prevent its activation, resulting in decreased pathway activity (81). Primary cilia may also balance canonical and noncanonical Wnt signaling, as some effectors, such as Vangl2, have been shown to localize to basal bodies and axonemes (113).
Wnt signaling may well play a role in JS-MKS-NPH. TMEM67 is a Frizzled-like receptor that binds Wnt5a in noncanonical Wnt signaling (1, 2), and Tmem67 knockout mice resemble Wnt5a and Ror2 (encoding a noncanonical Frizzled-like Wnt receptor) knockout mice. TMEM67 interacts with ROR2, and this interaction is required for ROR2 phosphorylation and subsequent Wnt pathway transduction. In Tmem67 mutant mice, cerebella have increased canonical Wnt gene expression, further substantiating the role of cilia to balance the canonical and noncanonical arms of Wnt signaling (1). Conversely, the Ahi1 mutant mouse model shows decreased Wnt signaling and cell proliferation at the cerebellar vermis, a defect that was partially rescued via the Wnt pathway agonist lithium (72).
Protein Composition
For proper function, most cells must make a primary cilium and selectively transport hundreds of proteins in and out of this tiny, privileged compartment (Figure 5). The TZ functions as a ciliary gate to control protein traffic and prevent bidirectional diffusion. IFT moves proteins bound for the cilium across this barrier and works in conjunction with multiple mechanisms for protein retention and selective egress (i.e., the TZ, ciliary lipids, axonemal binding, and the BBSome). The proteins involved in regulating ciliary composition are implicated in a range of ciliopathies, indicating that aberrant ciliary protein content likely contributes to the etiology of these disorders. Specific disease mechanisms could involve abnormal localization of a single key protein or a characteristic set of proteins. Alterations in ciliary protein composition have not been systematically evaluated across genetic causes of JS-MKS-NPH, so this is a major focus of our laboratory.
Crosstalk
Each aspect of ciliary biology discussed here does not occur in isolation; rather, the impacted processes influence each other. For instance, a complex interplay exists among the ciliary proteome, ciliary PIs, and Hh signaling. Hh pathway stimulation promotes a coordinated movement of effectors into and out of primary cilia. The JS-associated protein INPP5E requires an intact TZ for its ciliary localization, and once in the cilium, it controls ciliary PI subtype distribution. Hh pathway activation alters ciliary PI subtype distribution, regulated by INPP5E function, which in turn influences the localization of ciliary proteins (such as the Hh regulators SMO, GPR161, and TULP3) that bind specific PIs (24, 39, 47). Ciliary tubulin-modifying enzymes also impact Hh pathway function; for instance, deglutamylation can reduce SMO and GLI3 ciliary localization (60). Ongoing approaches to understanding the molecular mechanisms underlying JS-MKS-NPH will have to take this complex crosstalk into account.
ANIMAL MODELS
Similar to the advances using biochemical and cell biology techniques, animal model work has illuminated basic ciliary biology, as well as roles for cilia in development and disease. Widely used models such as mice, zebrafish, Caenorhabditis elegans, and Chlamydomonas have played central roles, while less commonly used organisms have also contributed, including unicellular protists (Tetrahymena, Paramecium, Ochromonas, Trypanosoma, and Leishmania), fungi, planarians, sea urchins, Drosophila, and Xenopus. Here, we focus on the most common organisms used to study ciliary biology.
Chlamydomonas
Chlamydomonas may have contributed more to basic ciliary biology than any other model organism, particularly in defining ciliary components and discovering and dissecting mechanisms of intraciliary trafficking (i.e., IFT and the BBSome). In addition, Chlamydomonas was critical in linking the mouse ORPK polycystic kidney disease model to both cilia and IFT (104, 105). This biflagellate algae is inexpensive; fast growing; easy to image (both live and fixed); biochemically tractable, with easily isolated pure cilia; and genetically tractable, with scorable and selectable cilium-related phenotypes (morphology, cilium formation, hatching, swimming, mating, and phototaxing). These attributes make it particularly appropriate for studying ciliary ultrastructure and motility. Conversely, Chlamydomonas lacks organs, many signaling pathways, and other features characteristic of metazoans. Orthologs for approximately a quarter of JS-MKS-related proteins have been identified, mostly TZ and basal body proteins. The new Chlamydomonas Library Project has made more than 60,000 insertional mutants, covering 83% of Chlamydomonas nuclear protein-coding genes, readily available (80). This collection will facilitate analysis of ciliopathy-related proteins in this powerful model for years to come.
Caenorhabditis elegans
Moving from unicellular protozoans to nematodes offers additional advantages. C. elegans is inexpensive, genetically tractable, and easy to grow in large quantities. Unlike in vertebrates, the developmental lineages for all cells have been mapped. C. elegans has more orthologs with functional similarities to human genes than Chlamydomonas has [e.g., TZ function is required for ciliary ARL13B (78) and INPP5E localization]. Genetic screens for mutants with defects in dye uptake (dyf mutants), mechanosensory response (mec mutants), osmotic avoidance (osm mutants), and chemotaxis (che mutants) have efficiently identified ciliary genes. Well-established methods exist for in vivo imaging of tagged ciliary proteins. Disadvantages include a lack of organs and divergent cilium biology; for instance, C. elegans has only highly specialized sensory cilia with unique structural features in a small subset of neurons, which contrasts with the diversity of cilia in other species, and it uses two different plus-end-directed IFT motors instead of the single motor in vertebrates. Nonetheless, C. elegans has yielded important insights into genetic hierarchies of ciliary genes, ciliary-secreted exosomes for intercellular communication, and the links between cilia and polycystic kidney disease (15).
Zebrafish
Zebrafish models have proven useful for validating new ciliopathy-related genes [e.g., CC2D2A (50), ARMC9, and TOGARAM1 (73)], evaluating variant pathogenicity and genetic modifiers (69), and dissecting developmental defects and have potential for small-molecule screens to develop precision treatments. Zebrafish have substantial advantages for the study of human development and disease. They have all of the major organ systems that are most relevant to ciliopathies (brain, retina, kidney, liver, and skeleton); are more cost-effective and reproduce more quickly than mice; develop externally and are transparent, allowing direct visualization of early development; and are genetically tractable, with large panels of mutant lines. Many JS-MKS-NPH zebrafish models have decreased cilia in brain ventricles, kidneys, and/or Kupffer’s vesicles. Similar to humans, zebrafish display pleiotropic manifestations of JS-MKS-NPH gene dysfunction, including retinal degeneration in several models, and pronephric cysts, the zebrafish equivalent of cystic kidneys, present in approximately half of the models (Supplemental Table 3). By contrast, despite close scrutiny, JS-like brain malformations have not been identified. Other phenotypes include a curved body axis, ventriculomegaly or abnormal cerebrospinal fluid flow, laterality defects, craniofacial abnormalities, and microphthalmia. The fact that these phenotypes are seen in multiple JS-MKS-NPH models mirrors the locus heterogeneity seen in humans.
Mice
Mice are the go-to model for human disease. Their genetics, development, and function are much more similar to humans’ than those of other model organisms, except for large animal models and nonhuman primates. Advantages for modeling ciliopathies include the presence of all relevant organs, similar cilia diversity and distribution to those of humans, the availability of sophisticated genetic methods, the availability of cilia-relevant mutants, concordance of signaling pathways and manifestations of disruptions (such as polydactyly for Hh disruption), similar neural tube patterning to that of humans, and the availability of behavioral assays.
These advantages come at a cost, since mice are the most expensive and time intensive of the model organisms discussed here. In addition, they fail to mimic some aspects of JS-MKS-NPH phenotypes. Even more than zebrafish, mouse models recapitulate many of the pleiotropic manifestations of JS-MKS-NPH gene dysfunction in humans. Similar to zebrafish, JS-MKS-NPH mouse models are missing cilia in subsets of tissues (46). Embryonic/perinatal lethality is the most common mouse phenotype, likely explained in part by the null variants in mice compared with the mostly hypomorphic variants seen in humans (Supplemental Table 4). Beyond early lethality, the most common phenotypes are retinal degeneration and microphthalmia/anophthalmia (which likely represents an extreme form of coloboma), confirming the locus heterogeneity seen in humans. Polydactyly is often seen, with up to nine digits in an Ofd1 mouse model. Disrupted Hh signaling is frequently reported, mainly assessed by abnormal neural tube patterning (both ventralized and dorsalized) and Hh target gene expression. Laterality defects are another common feature seen in mice but not humans and may also reflect differences in the pathogenicity of mouse and human variants. Other frequent phenotypes include fibrocystic kidney involvement, cranio-facial malformations, and exencephaly, a more severe form of encephalocele than is typically seen in human fetuses with MKS. Subsets of mouse models have other shared manifestations: skeletal dysplasia, hydrocephalus, ductal plate malformation/congenital liver fibrosis, and curved body axis.
Some gene–phenotype correlations appear to be preserved between human and animal models. For instance, in humans, AHI1 is associated with JS and isolated retinal dystrophy but not with MKS, and mouse models display relatively mild cerebellar, retinal, and kidney phenotypes, without encephalocele or polydactyly (83, 150). CEP290 and AHI1 dysfunction are strongly associated with retinal and renal disease in both humans and mice (10, 83, 109, 150), and AHI1 dysfunction is associated with retinal dystrophy in zebrafish as well (77). By contrast, not all mouse Nphp1 models (65, 79) have kidney involvement, despite NPHP1 dysfunction being the most common cause of human NPH. Similarly, an NPHP4 mouse model also failed to show kidney involvement in mice (153). Furthermore, TMEM67 is associated with high risk of coloboma and liver fibrosis, neither of which have been published for mouse models. This highlights the need for careful evaluation of animal models before using them to dissect mechanisms and develop treatments.
Molar Tooth Sign–Related Joubert Syndrome Brain Phenotypes
Brain imaging and neuropathological data from our group and others indicate that the JS brain malformation is due to defects in fundamental processes required for brain development (103, 107). The cerebellar vermis hypoplasia could be caused by aberrant cell fate specification or decreased proliferation/survival of vermis cell populations. Brain stem heterotopias are likely caused by neuronal migration defects, while decreased midline decussation of axons may be caused by axon guidance defects. Modeling the brain phenotypes of JS in animals has met with limited success. Multiple mouse models display encephalocele, exencephaly, and patterning defects, possibly similar to the severe brain phenotypes observed in fetuses with MKS. Several mouse models recapitulate the cerebellar vermis hypoplasia seen in humans [Ahi1 (72), Cep290 (72), Cep120 (154), and Tmem67 (1)], but only conditional Arl13b and KIAA0586 ortholog 2700049A03Rik knockouts have demonstrated abnormal superior cerebellar peduncle decussation similar to the human MTS (16, 51).
Allelic Series in Mice
Animal models provide a means to directly evaluate the range of phenotypes associated with dysfunction of a particular gene. Interestingly, null Arl13b and Inpp5e models recapitulate some brain phenotypes, but patient alleles engineered into mice do not. Suciu et al. (139) compared neural tube patterning and cerebellar phenotypes in an allelic series, including wild-type controls; a JS-associated allele, Arl13bR79Q/R79Q; a non-cilia-localizing allele, Arl13bV358A/V358A; and an Arl13bΔ/Δ null allele. Only the null allele model displayed abnormal neural tube patterning, reduced cerebellar width, and reduced or absent dorsal contralateral projects of the superior cerebellar peduncles.
Genetic Modifiers in Animal Models
Animal models have historically provided solid evidence for genetic modifiers and oligogenic inheritance, but identifying and characterizing modifiers of JS-MKS-NPH phenotypes in humans has been challenging due to the large number of causal variants and the incredibly diverse genetic background in humans compared with inbred animal models. C. elegans has been particularly useful for identifying interactions between JS-MKS-NPH genes, defining specific genetic hierarchies for ciliogenesis (64, 155). Zebrafish have also highlighted genetic interactions; mild knockdown of Cep290 alone did not show a phenotype, while the same knockdown in cc2d2a mutant fish markedly exacerbated the pronephric cyst phenotype, indicating that CEP290 variants might modify ciliopathy phenotypes in humans (50).
Similar to phenotypic variation in humans with identical causal variants, mouse lines in different genetic backgrounds can sometimes have very different phenotypes. A good example that recapitulates the JS-MKS spectrum is the Tmem67tm1(Dgen/H) knockout mouse. When it was made, two phenotypically distinct lines were isolated, presumably due to differences in the genetic background: A more severe MKS-like line displayed fewer cilia, underactive Hh signaling, highly dysregulated Wnt signaling, and disrupted neural tube patterning, including encephalocele and exencephaly, and a less severe JS-like line displayed cilia with abnormal morphology, overactive Hh signaling, mildly dysregulated Wnt signaling, and mild neural tube patterning defects with cerebellar hypoplasia. Outbreeding Ahi1 and Tmem67 knockout models also reduced early lethality and kidney pathology, respectively (76, 150).
FUTURE DIRECTIONS
For decades, the primary cilium was thought to be a vestigial organelle, and ciliopathies were not recognized as a coherent group of clinically and biologically defined conditions. Advances in medical diagnostics, human genomics, bioinformatics, and basic research on biochemistry, cell biology, and developmental biology have revolutionized our understanding of the JS-MKS-NPH spectrum. Now, we have a nearly complete parts list for the primary cilium, as well as a rapidly evolving understanding of how these parts function together in health and disease. This allows us to provide a specific diagnosis to most families, directly improving prognostic information, guiding medical monitoring and treatment, and enabling carrier testing and prenatal diagnosis.
More complicated than single-gene Mendelian conditions but less byzantine than genetically complex non-Mendelian conditions and traits, the JS-MKS-NPH spectrum now represents a clinically and biologically well-defined system for understanding how variations in the genome result in the range of human phenotypes. Further advances will require new approaches to integrate the impacts of rare, large-effect causal variants with the impacts of more common small-effect variants (genetic background). This will require advanced informatic methods for modeling the effects of variant combinations on biological mechanisms and phenotypes, combined with biological assays and comprehensive clinical data sets to validate the predictions.
Already, improved mechanistic understanding of the progressive retinal, kidney, and liver involvement in ciliopathies is opening the door to precision treatments. Antisense oligonucleotides to induce exon skipping are showing promise in cell-based and animal models. Targeted exon skipping effectively rescued cellular defects in patient fibroblasts and renal epithelial cells (91). Translational read-through drugs have the potential to bypass nonsense variants, resulting in full-length protein production, and have rescued phenotypes in fibroblasts from patients with BBS and Alström syndrome (40). Small molecules to modulate cilium-dependent signaling pathways are also showing promise. Srivastava et al. (134) used an Hh pathway agonist (purmorphamine) or CDK5 antagonist (roscovitine) to correct abnormally long and disorganized cilia in patient renal epithelial cells. Gene therapies are also showing promise. Bbs1 expression using inhaled adenovirus restored olfactory sensory neurons defects and odor responses in Bbs1 mutant mice (152). Finally, the BRILLIANCE clinical trial (ClinicalTrials.gov ID NCT03872479) is the first use of CRISPR-Cas9 to correct a specific disease variant in humans, the recurrent CEP290 variant that causes LCA. This remarkable progress in identifying genetic causes, understanding biological mechanisms, and designing translatable treatments provides hope for people with JS-MKS-NPH conditions and their families in the future.
Supplementary Material
ACKNOWLEDGMENTS
D.D. is supported by National Institutes of Health grant 1R01HD100730; J.C.V. is supported by National Institutes of Health grant 5K99HD100554. We thank members of the Doherty laboratory for reading the manuscript and providing useful feedback.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Meckel syndrome is sometimes referred to as Meckel–Gruber syndrome. Due to the apparent involvement of Dr. Gruber in arguing that the actions of the Nazi movement in Germany were ethically justified, we discourage the use of his name (89).
LITERATURE CITED
- 1.Abdelhamed ZA, Abdelmottaleb DI, El-Asrag ME, Natarajan S, Wheway G, et al. 2019. The ciliary Frizzled-like receptor Tmem67 regulates canonical Wnt/β-catenin signalling in the developing cerebellum via Hoxb5. Sci. Rep 9:5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdelhamed ZA, Natarajan S, Wheway G, Inglehearn CF, Toomes C, et al. 2015. The Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway. Dis. Models Mech 8:527–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, et al. 2013. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet 22:1358–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alazami AM, Alshammari MJ, Baig M, Salih MA, Hassan HH, Alkuraya FS. 2014. NPHP4 mutation is linked to cerebello-oculo-renal syndrome and male infertility. Clin. Genet 85:371–75 [DOI] [PubMed] [Google Scholar]
- 5.Alkanderi S, Molinari E, Shaheen R, Elmaghloob Y, Stephen LA, et al. 2018. ARL3 mutations cause Joubert syndrome by disrupting ciliary protein composition. Am. J. Hum. Genet 103:612–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, et al. 2007. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet 39:882–88 [DOI] [PubMed] [Google Scholar]
- 7.Attanasio M, Uhlenhaut NH, Sousa VH, O’Toole JF, Otto E, et al. 2007. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet 39:1018–24 [DOI] [PubMed] [Google Scholar]
- 8.Baala L, Romano S, Khaddour R, Saunier S, Smith UM, et al. 2007. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet 80:186–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, Chen ML, D’Arrigo S, et al. 2020. Healthcare recommendations for Joubert syndrome. Am. J. Med. Genet. A 182:229–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, et al. 2015. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet 52:514–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bachmann-Gagescu R, Phelps IG, Dempsey JC, Sharma VA, Ishak GE, et al. 2015. KIAA0586 is mutated in Joubert syndrome. Hum. Mutat 36:831–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bamshad MJ, Nickerson DA, Chong JX. 2019. Mendelian gene discovery: fast and furious with no end in sight. Am. J. Hum. Genet 105:448–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bangs F, Anderson KV. 2017. Primary cilia and mammalian Hedgehog signaling. Cold Spring Harb. Perspect. Biol 9:a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barisic I, Boban L, Loane M, Garne E, Wellesley D, et al. 2015. Meckel-Gruber syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur. J. Hum. Genet 23:746–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barr MM, Sternberg PW. 1999. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 401:386–89 [DOI] [PubMed] [Google Scholar]
- 16.Bashford AL, Subramanian V. 2019. Mice with a conditional deletion of Talpid3 (KIAA0586) – a model for Joubert syndrome. J. Pathol 248:396–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berbari NF, Sharma N, Malarkey EB, Pieczynski JN, Boddu R, et al. 2013. Microtubule modifications and stability are altered by cilia perturbation and in cystic kidney disease. Cytoskeleton 70:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, et al. 2009. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet 41:1032–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braddock SR, Henley KM, Maria BL. 2007. The face of Joubert syndrome: a study of dysmorphology and anthropometry. Am. J. Med. Genet. A 143A:3235–42 [DOI] [PubMed] [Google Scholar]
- 20.Breslow DK, Hoogendoorn S, Kopp AR, Morgens DW, Vu BK, et al. 2018. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet 50:460–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunner HG, van Driel MA. 2004. From syndrome families to functional genomics. Nat. Rev. Genet 5:545–51 [DOI] [PubMed] [Google Scholar]
- 22.Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, et al. 2008. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet 83:170–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, et al. 2012. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150:533–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. 2015. Modulation of ciliary phosphoinositide content regulates trafficking and Sonic Hedgehog signaling output. Dev. Cell 34:338–50 [DOI] [PubMed] [Google Scholar]
- 25.Chevrier V, Bruel AL, Van Dam TJ, Franco B, Lo Scalzo M, et al. 2016. OFIP/KIAA0753 forms a complex with OFD1 and FOR20 at pericentriolar satellites and centrosomes and is mutated in one individual with oral-facial-digital syndrome. Hum. Mol. Genet 25:497–513 [DOI] [PubMed] [Google Scholar]
- 26.Choi YJ, Halbritter J, Braun DA, Schueler M, Schapiro D, et al. 2019. Mutations of ADAMTS9 cause nephronophthisis-related ciliopathy. Am. J. Hum. Genet 104:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, et al. 2009. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet 85:465–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coppieters F, Casteels I, Meire F, De Jaegere S, Hooghe S, et al. 2010. Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum. Mutat 31:E1709–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, et al. 2011. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Investig 121:2662–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daly OM, Gaboriau D, Karakaya K, King S, Dantas TJ, et al. 2016. CEP164-null cells generated by genome editing show a ciliation defect with intact DNA repair capacity. J. Cell Sci 129:1769–74 [DOI] [PubMed] [Google Scholar]
- 31.Davey MG, Paton IR, Yin Y, Schmidt M, Bangs FK, et al. 2006. The chicken talpid3 gene encodes a novel protein essential for Hedgehog signaling. Genes Dev. 20:1365–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, et al. 2011. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet 43:189–96. Corrigendum. 2011. Nat. Genet. 43:499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Mori R, Romani M, D’Arrigo S, Zaki MS, Lorefice E, et al. 2017. Hypomorphic recessive variants in SUFU impair the Sonic Hedgehog pathway and cause Joubert syndrome with cranio-facial and skeletal defects. Am. J. Hum. Genet 101:552–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, et al. 2007. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet 39:875–81 [DOI] [PubMed] [Google Scholar]
- 35.Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, et al. 2004. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet 75:979–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doherty D 2009. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol 16:143–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, et al. 2010. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet 47:8–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SG, et al. 2011. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am. J. Hum. Genet 89:94–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dyson JM, Conduit SE, Feeney SJ, Hakim S, DiTommaso T, et al. 2017. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J. Cell Biol 216:247–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eintracht J, Forsythe E, May-Simera H, Moosajee M. 2021. Translational readthrough of ciliopathy genes BBS2 and ALMS1 restores protein, ciliogenesis and function in patient fibroblasts. EBioMedicine 70:103515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, et al. 2012. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet 44:886–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Failler M, Gee HY, Krug P, Joo K, Halbritter J, et al. 2014. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am. J. Hum. Genet 94:905–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, et al. 2004. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet 36:1008–13 [DOI] [PubMed] [Google Scholar]
- 44.Forsyth R, Gunay-Aygun M. 2020. Bardet-Biedl syndrome overview. In GeneReviews, ed. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, et al. Seattle: Univ. Wash. https://www.ncbi.nlm.nih.gov/books/NBK1363 [Google Scholar]
- 45.Frank V, den Hollander AI, Bruchle NO, Zonneveld MN, Nurnberg G, et al. 2008. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum. Mutat 29:45–52 [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, et al. 2011. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet 43:776–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G III, Abedin M, et al. 2015. Phosphoinositides regulate ciliary protein trafficking to modulate Hedgehog signaling. Dev. Cell 34:400–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, et al. 2007. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet 39:1350–60 [DOI] [PubMed] [Google Scholar]
- 49.Gigante ED, Taylor MR, Ivanova AA, Kahn RA, Caspary T. 2020. ARL13B regulates Sonic hedgehog signaling from outside primary cilia. eLife 9:e50434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, et al. 2008. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet 83:559–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo J, Otis JM, Suciu SK, Catalano C, Xing L, et al. 2019. Primary cilia signaling promotes axonal tract development and is disrupted in Joubert syndrome-related disorders models. Dev. Cell 51:759–74.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haider NB, Carmi R, Shalev H, Sheffield VC, Landau D. 1998. A Bedouin kindred with infantile nephronophthisis demonstrates linkage to chromosome 9 by homozygosity mapping. Am. J. Hum. Genet 63:1404–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, et al. 2013. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet 93:915–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, et al. 2013. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet 132:865–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He K, Ma X, Xu T, Li Y, Hodge A, et al. 2018. Axoneme polyglutamylation regulated by Joubert syndrome protein ARL13B controls ciliary targeting of signaling molecules. Nat. Commun 9:3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He M, Subramanian R, Bangs F, Omelchenko T, Liem KF Jr., et al. 2014. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol 16:663–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, et al. 1997. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet 17:149–53 [DOI] [PubMed] [Google Scholar]
- 58.Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, et al. 2007. Evidence of oligogenic inheritance in nephronophthisis. J. Am. Soc. Nephrol 18:2789–95 [DOI] [PubMed] [Google Scholar]
- 59.Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, et al. 2013. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet 45:951–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong SR, Wang CL, Huang YS, Chang YC, Chang YC, et al. 2018. Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and Hedgehog signaling. Nat. Commun 9:1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hopp K, Heyer CM, Hommerding CJ, Henke SA, Sundsbak JL, et al. 2011. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum. Mol. Genet 20:2524–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, et al. 2011. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet 89:713–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, et al. 2012. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. PNAS 109:19691–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jensen VL, Li C, Bowie RV, Clarke L, Mohan S, et al. 2015. Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J. 34:2537–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang ST, Chiou YY, Wang E, Lin HK, Lee SP, et al. 2008. Targeted disruption of Nphp1 causes male infertility due to defects in the later steps of sperm morphogenesis in mice. Hum. Mol. Genet 17:3368–79 [DOI] [PubMed] [Google Scholar]
- 66.Joubert M, Eisenring JJ, Robb JP, Andermann F. 1969. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 9:813–25 [DOI] [PubMed] [Google Scholar]
- 67.Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. 2012. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 123:695–709 [DOI] [PubMed] [Google Scholar]
- 68.Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, et al. 2001. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293:2256–59 [DOI] [PubMed] [Google Scholar]
- 69.Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, et al. 2009. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet 41:739–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, et al. 2006. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet 38:155–57 [DOI] [PubMed] [Google Scholar]
- 71.Lambacher NJ, Bruel AL, van Dam TJ, Szymanska K, Slaats GG, et al. 2016. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat. Cell Biol 18:122–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, et al. 2011. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med 17:726–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Latour BL, Van De Weghe JC, Rusterholz TD, Letteboer SJ, Gomez A, et al. 2020. Dysfunction of the ciliary ARMC9/TOGARAM1 protein module causes Joubert syndrome. J. Clin. Investig 130:4423–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee JE, Silhavy JL, Zaki MS, Schroth J, Bielas SL, et al. 2012. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet 44:193–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee JH, Silhavy JL, Lee JE, Al-Gazali L, Thomas S, et al. 2012. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science 335:966–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leightner AC, Hommerding CJ, Peng Y, Salisbury JL, Gainullin VG, et al. 2013. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum. Mol. Genet 22:2024–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lessieur EM, Fogerty J, Gaivin RJ, Song P, Perkins BD. 2017. The ciliopathy gene ahi1 is required for zebrafish cone photoreceptor outer segment morphogenesis and survival. Investig. Ophthalmol. Vis. Sci 58:448–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li C, Jensen VL, Park K, Kennedy J, Garcia-Gonzalo FR, et al. 2016. MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLOS Biol. 14:e1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li D, Hu M, Chen H, Wu X, Wei X, et al. 2022. An Nphp1 knockout mouse model targeting exon 2–20 demonstrates characteristic phenotypes of human nephronophthisis. Hum. Mol. Genet 31:232–43 [DOI] [PubMed] [Google Scholar]
- 80.Li X, Zhang R, Patena W, Gang SS, Blum SR, et al. 2016. An indexed, mapped mutant library enables reverse genetics studies of biological processes in Chlamydomonas reinhardtii. Plant Cell 28:367–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lienkamp S, Ganner A, Walz G. 2012. Inversin, Wnt signaling and primary cilia. Differentiation 83:S49–55 [DOI] [PubMed] [Google Scholar]
- 82.Lopez E, Thauvin-Robinet C, Reversade B, Khartoufi NE, Devisme L, et al. 2014. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum. Genet 133:367–77 [DOI] [PubMed] [Google Scholar]
- 83.Louie CM, Caridi G, Lopes VS, Brancati F, Kispert A, et al. 2010. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet 42:175–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luo M, Lin Z, Zhu T, Jin M, Meng D, et al. 2021. Disrupted intraflagellar transport due to IFT74 variants causes Joubert syndrome. Genet. Med 23:1041–49 [DOI] [PubMed] [Google Scholar]
- 85.Macia MS, Halbritter J, Delous M, Bredrup C, Gutter A, et al. 2017. Mutations in MAPKBP1 cause juvenile or late-onset cilia-independent nephronophthisis. Am. J. Hum. Genet 100:323–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Magiera MM, Singh P, Gadadhar S, Janke C. 2018. Tubulin posttranslational modifications and emerging links to human disease. Cell 173:1323–27 [DOI] [PubMed] [Google Scholar]
- 87.Maglic D, Stephen J, Malicdan MC, Guo J, Fischer R, et al. 2016. TMEM231 gene conversion associated with Joubert and Meckel-Gruber syndromes in the same family. Hum. Mutat 37:1144–48 [DOI] [PubMed] [Google Scholar]
- 88.Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, et al. 1997. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol 12:423–30 [DOI] [PubMed] [Google Scholar]
- 89.Mattulat M. 2007. Medizinethik in historischer Perspektive: Zum Wandel arztlicher Moralkonzepte im Werk von Georg Benno Gruber (1884–1977). Stuttgart, Ger.: Steiner [Google Scholar]
- 90.Meckel JF. 1822. Beschreibung zweier durch sehr ähnliche Bildungsabweichungen entstellter Geschwister. Dtsch. Arch. Physiol 7:99–172 [Google Scholar]
- 91.Molinari E, Ramsbottom SA, Srivastava S, Booth P, Alkanderi S, et al. 2019. Targeted exon skipping rescues ciliary protein composition defects in Joubert syndrome patient fibroblasts. Sci. Rep 9:10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mollet G, Salomon R, Gribouval O, Silbermann F, Bacq D, et al. 2002. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat. Genet 32:300–5 [DOI] [PubMed] [Google Scholar]
- 93.Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N, et al. 2009. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum. Mutat 30:1574–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen TT, Hull S, Roepman R, van den Born LI, Oud MM, et al. 2017. Missense mutations in the WD40 domain of AHI1 cause non-syndromic retinitis pigmentosa. J. Med. Genet 54:624–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nuovo S, Bacigalupo I, Ginevrino M, Battini R, Bertini E, et al. 2020. Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology 94:e797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oka M, Shimojima K, Yamamoto T, Hanaoka Y, Sato S, et al. 2016. A novel HYLS1 homozygous mutation in living siblings with Joubert syndrome. Clin. Genet 89:739–43 [DOI] [PubMed] [Google Scholar]
- 97.Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, et al. 2003. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet 34:455–59 [DOI] [PubMed] [Google Scholar]
- 98.Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, et al. 2010. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet 42:840–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, et al. 2009. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J. Med. Genet 46:663–70 [DOI] [PubMed] [Google Scholar]
- 100.Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. 2008. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol 19:587–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, et al. 2004. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet 75:82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parisi MA, Glass I. 2017. Joubert syndrome. In GeneReviews, ed. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, et al. Seattle: Univ. Wash. https://www.ncbi.nlm.nih.gov/books/NBK1325 [PubMed] [Google Scholar]
- 103.Parisi MA, Pinter JD, Glass IA, Field K, Maria BL, et al. 2004. Cerebral and cerebellar motor activation abnormalities in a subject with Joubert syndrome: functional magnetic resonance imaging (MRI) study. J. Child Neurol 19:214–18 [PubMed] [Google Scholar]
- 104.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, et al. 2000. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol 151:709–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. 2002. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol 12:R378–80 [DOI] [PubMed] [Google Scholar]
- 106.Phelps IG, Dempsey JC, Grout ME, Isabella CR, Tully HM, et al. 2018. Interpreting the clinical significance of combined variants in multiple recessive disease genes: systematic investigation of Joubert syndrome yields little support for oligogenicity. Genet. Med 20:223–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Poretti A, Snow J, Summers AC, Tekes A, Huisman T, et al. 2017. Joubert syndrome: neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause. J. Med. Genet 54:521–29 [DOI] [PubMed] [Google Scholar]
- 108.Qiu H, Fujisawa S, Nozaki S, Katoh Y, Nakayama K. 2021. Interaction of INPP5E with ARL13B is essential for its ciliary membrane retention but dispensable for its ciliary entry. Biol. Open 10:bio057653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rachel RA, Yamamoto EA, Dewanjee MK, May-Simera HL, Sergeev YV, et al. 2015. CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet 24:3775–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reiter JF, Leroux MR. 2017. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol 18:533–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, et al. 2014. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J. Rare Dis 9:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Romani M, Micalizzi A, Valente EM. 2013. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 12:894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, et al. 2005. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet 37:1135–40 [DOI] [PubMed] [Google Scholar]
- 114.Sanders AA, de Vrieze E, Alazami AM, Alzahrani F, Malarkey EB, et al. 2015. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 16:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, et al. 2011. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145:513–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sangermano R, Deitch I, Peter VG, Ba-Abbad R, Place EM, et al. 2021. Broadening INPP5E phenotypic spectrum: detection of rare variants in syndromic and non-syndromic IRD. NPJ Genom. Med 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, et al. 2020. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180:568–84.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, et al. 2006. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet 38:674–81 [DOI] [PubMed] [Google Scholar]
- 119.Schroder S, Li Y, Yigit G, Altmuller J, Bader I, et al. 2021. Heterozygous truncating variants in SUFU cause congenital ocular motor apraxia. Genet. Med 23:341–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schueler M, Braun DA, Chandrasekar G, Gee HY, Klasson TD, et al. 2015. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am. J. Hum. Genet 96:81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Serpieri V, D’Abrusco F, Dempsey JC, Cheng YH, Arrigoni F, et al. 2021. SUFU haploinsufficiency causes a recognisable neurodevelopmental phenotype at the mild end of the Joubert syndrome spectrum. J. Med. Genet In press. 10.1136/jmedgenet-2021-108114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shaheen R, Almoisheer A, Faqeih E, Babay Z, Monies D, et al. 2015. Identification of a novel MKS locus defined by TMEM107 mutation. Hum. Mol. Genet 24:5211–18 [DOI] [PubMed] [Google Scholar]
- 123.Shaheen R, Ansari S, Mardawi EA, Alshammari MJ, Alkuraya FS. 2013. Mutations in TMEM231 cause Meckel-Gruber syndrome. J. Med. Genet 50:160–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shaheen R, Faqeih E, Seidahmed MZ, Sunker A, Alali FE, et al. 2011. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum. Mutat 32:573–78 [DOI] [PubMed] [Google Scholar]
- 125.Shaheen R, Jiang N, Alzahrani F, Ewida N, Al-Sheddi T, et al. 2019. Bi-allelic mutations in FAM149B1 cause abnormal primary cilium and a range of ciliopathy phenotypes in humans. Am. J. Hum. Genet 104:731–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, et al. 2015. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum. Mol. Genet 24:1410–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, et al. 2014. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am. J. Hum. Genet 94:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, et al. 2016. Characterizing the morbid genome of ciliopathies. Genome Biol. 17:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen WC, Shian WJ, Chen CC, Chi CS, Lee SK, Lee KR. 1994. MRI of Joubert’s syndrome. Eur. J. Radiol 18:30–33 [DOI] [PubMed] [Google Scholar]
- 130.Shimada H, Lu Q, Insinna-Kettenhofen C, Nagashima K, English MA, et al. 2017. In vitro modeling using ciliopathy-patient-derived cells reveals distinct cilia dysfunctions caused by CEP290 mutations. Cell Rep. 20:384–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Simpson MA, Cross HE, Cross L, Helmuth M, Crosby AH. 2009. Lethal cystic kidney disease in Amish neonates associated with homozygous nonsense mutation of NPHP3. Am. J. Kidney Dis 53:790–95 [DOI] [PubMed] [Google Scholar]
- 132.Smith C, Graham J. 1945. Congenital medullary cysts with severe refractory anemia. Am. J. Dis. Child 69:369–77 [Google Scholar]
- 133.Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, et al. 2006. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet 38:191–96 [DOI] [PubMed] [Google Scholar]
- 134.Srivastava S, Ramsbottom SA, Molinari E, Alkanderi S, Filby A, et al. 2017. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum. Mol. Genet 26:4657–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Srour M, Hamdan FF, McKnight D, Davis E, Mandel H, et al. 2015. Joubert syndrome in French Canadians and identification of mutations in CEP104. Am. J. Hum. Genet 97:744–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Srour M, Hamdan FF, Schwartzentruber JA, Patry L, Ospina LH, et al. 2012. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J. Med. Genet 49:636–41 [DOI] [PubMed] [Google Scholar]
- 137.Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, et al. 2012. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet 90:693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stokman M, Lilien M, Knoers N. 2016. Nephronophthisis. In GeneReviews, ed. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, et al. Seattle: Univ. Wash. https://www.ncbi.nlm.nih.gov/books/NBK368475 [PubMed] [Google Scholar]
- 139.Suciu SK, Long AB, Caspary T. 2021. Smoothened and ARL13B are critical in mouse for superior cerebellar peduncle targeting. Genetics 218:iyab084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila M. 2008. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am. J. Hum. Genet 82:1361–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Perez V, Shida T, et al. 2014. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat. Genet 46:905–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thomas S, Legendre M, Saunier S, Bessieres B, Alby C, et al. 2012. TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet 91:372–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, et al. 2014. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum. Mutat 35:137–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tuz K, Bachmann-Gagescu R, O’Day DR, Hua K, Isabella CR, et al. 2014. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet 94:62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, et al. 2010. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet 42:619–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, et al. 2006. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet 38:623–25 [DOI] [PubMed] [Google Scholar]
- 147.van Amerongen R, Nusse R. 2009. Towards an integrated view of Wnt signaling in development. Development 136:3205–14 [DOI] [PubMed] [Google Scholar]
- 148.Van De Weghe JC, Giordano JL, Mathijssen IB, Mojarrad M, Lugtenberg D, et al. 2021. TMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes. HGG Adv. 2:100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Van De Weghe JC, Rusterholz TDS, Latour B, Grout ME, Aldinger KA, et al. 2017. Mutations in ARMC9, which encodes a basal body protein, cause Joubert syndrome in humans and ciliopathy phenotypes in zebrafish. Am. J. Hum. Genet 101:23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, et al. 2010. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J. Neurosci 30:8759–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen TT, et al. 2015. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol 17:1074–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Williams CL, Uytingco CR, Green WW, McIntyre JC, Ukhanov K, et al. 2017. Gene therapeutic reversal of peripheral olfactory impairment in Bardet-Biedl syndrome. Mol. Ther 25:904–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Won J, Marin de Evsikova C, Smith RS, Hicks WL, Edwards MM, et al. 2011. NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum. Mol. Genet 20:482–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wu C, Yang M, Li J, Wang C, Cao T, et al. 2014. Talpid3-binding centrosomal protein Cep120 is required for centriole duplication and proliferation of cerebellar granule neuron progenitors. PLOS ONE 9:e107943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yee LE, Garcia-Gonzalo FR, Bowie RV, Li C, Kennedy JK, et al. 2015. Conserved genetic interactions between ciliopathy complexes cooperatively support ciliogenesis and ciliary signaling. PLOS Genet. 11:e1005627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, et al. 2018. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum. Mutat 39:152–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.