

SUMMARY:
Drug-tolerant persister cells (persisters) evade apoptosis upon targeted and conventional cancer therapies and represent a major non-genetic barrier to effective cancer treatment. Here we show that cells that survive treatment with pro-apoptotic BH3 mimetics display a persister phenotype that includes colonization and metastasis in vivo and increased sensitivity towards ferroptosis by GPX4 inhibition. We found that sublethal mitochondrial outer membrane permeabilization (MOMP) and Holocytochrome c release are key requirements for the generation of the persister phenotype. The generation of persisters is independent of apoptosome formation and caspase activation, but instead, cytosolic Cytochrome c induces activation of Heme-regulated Inhibitor (HRI) kinase and engagement of the integrated stress response (ISR) with consequent synthesis of ATF4, all of which are required for the persister phenotype. Our results reveal that sublethal Cytochrome c release couples sublethal MOMP to caspase-independent initiation of an ATF4-dependent, drug-tolerant persister phenotype.
Keywords: Mitochondrial permeabilization, BCL-2 family, BH3 mimetics, drug-persistence, persister phenotype, cancer, Cytochrome c, ATF4, HRI, EMT, metastasis, drug-resistance, GPX4, ferroptosis, lung adenocarcinoma
Graphical Abstract
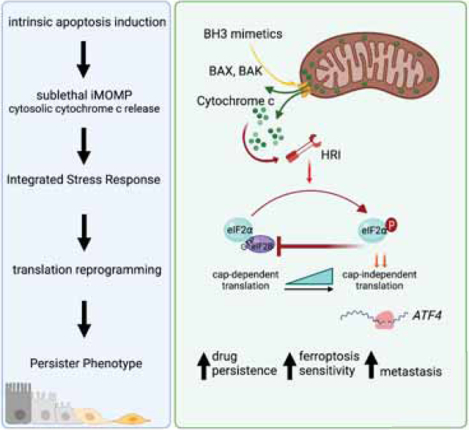
Cancer cells that survive treatment with pro-apoptotic BH3 mimetics do so by undergoing sub-lethal mitochondrial outer membrane permeabilization and induction of a stress response via cytochrome c but independent of caspases. They thus acquire a persister state that enables metastatic colonization in vivo.
INTRODUCTION
In addition to genetic events that mediate resistance to anti-cancer therapies, a major contribution to cancer relapse is the emergence of drug-tolerant cells, a phenomenon called persistence. A key feature of such persister cells is that, in contrast to resistant cells, their drug-unresponsive phenotype is acquired and provides a temporary survival-advantage towards treatment.
Persister cells (PS) can be functionally defined as cells that remain alive following a cell death-inducing treatment, without the selection for resistance mutations. PS from different cancer cell lines treated under different conditions often have a number of features in common that differ from the untreated, parental cells (PT) (Shen et al., 2020). These features, which can serve as a “PS phenotype” include a) transient tolerance of the initial treatment and often other toxic treatments, b) an acquired dependence on the lipid peroxidase, GPX4, c) reduced cell cycle kinetics, and d) similar gene expression profiles, often including, an epithelial to mesenchymal transition (EMT) signature. The ability to adapt to microenvironmental stresses by altering phenotype is often evolutionary conserved and is related to survival strategies in the face of starvation or diapause (García-Jiménez and Goding, 2019; Rehman et al., 2021). Although the features of therapy-tolerant persister cells have been defined, the key molecular events that enable a cancer cell to bypass cell death on exposure to pro-apoptotic cancer therapeutics, especially after initial engagement of the apoptotic machinery, remain poorly understood.
As the best studied and most complex form of regulated cell death, apoptosis can be triggered by a wide range of cellular stresses (e.g. oncogenic, metabolic, genetic) and therapeutics that converge on the intrinsic pathway of apoptosis (Singh et al., 2019). Central to this pathway is the mitochondrion, which harbors in its intermembrane space (IMS) crucial caspase activators such as Holocytochrome c (Kalkavan and Green, 2018).
The BCL-2 family effector proteins BAX, BAK and BOK are responsible for mitochondrial outer membrane permeabilization (MOMP), triggering apoptosis (Bhola and Letai, 2016). Anti-apoptotic proteins bind and hinder effector protein function. The pro-apoptotic BH3-only proteins bind via their BH3 domains to anti-apoptotic proteins and/or directly to BAX and BAK to engage MOMP (Garrido et al., 2006; Tait and Green, 2013). This important function of the BH3 domain has been exploited therapeutically as BH3 mimetics, such as Navitoclax, Venetoclax, and MCL-1 inhibitors (Kalkavan and Green, 2018; Merino et al., 2018).
The initiation of the apoptotic cascade does not inevitably lead to cell death. Ongoing studies have focused on the consequences of incomplete MOMP (iMOMP) and sublethal caspase activity on cell fate, challenging the binary character of MOMP to reveal that cells do not always commit to cell death (Tait et al., 2010). Instead, iMOMP can occur, resulting in a more nuanced caspase activation and cell survival (Ichim and Tait, 2016).
A key and rapid adaptive survival response to a wide range of triggers relies on the phosphorylation of the translation initiation factor eIF2α by one of four kinases, EIF2AK1/HRI, EIF2AK2/PKR, EIF2AK3/PERK and EIF2AK4/GCN2, each responsive to distinct upstream stresses (Pakos-Zebrucka et al., 2016). The resulting translational reprogramming suppresses global cap-dependent translation and enables translation of a subset of mRNAs encoding proteins that function to alleviate the stress and promote survival (Tian et al., 2021). Among these is Activating Transcription Factor 4 (ATF4) that initiates a gene expression program that in the short-term promotes survival by increasing nutrient uptake and autophagy, and suppressing oxidative stress (Costa-Mattioli and Walter, 2020).
Here we reveal that following treatment with pro-apoptotic BH3 mimetics, drug-tolerant persisters arise as a consequence of the activation of mitochondrial-nuclear communication relayed by iMOMP and Cytochrome c-dependent activation of HRI, leading to ATF4 synthesis and function. We found that the persister phenotype, induced by treatment with BH3 mimetics, is dependent on MOMP and the release of Cytochrome c, as well as HRI and ATF4. Further, we define a BAX, BAK, BOK-dependent, ATF4-dependent PS gene signature that can be found in lung cancer patients with residual disease following therapy.
RESULTS
BH3 mimetics induce persister cancer cells
Cancer treatments generally converge on intrinsic apoptosis as their main cell death effector mechanism (Singh et al., 2019). We utilized the highly specific BH3 mimetics ABT-737 and S63845 (S6) (Kotschy et al., 2016), which target BCL-2, BCL-XL, BCL-w (ABT-737) and MCL1 (S6). We first confirmed maximal on-target doses by comparing wildtype (WT) to BAX and BAK double knock-out (DKO) or BAX, BAK and BOK triple knock-out (TKO) cells (Figures 1A, S1A and S1B). WT therapy-persisting cells (persister cells, PS) were released from therapy for 1 day and upon re-exposure to the drugs, proved to be less sensitive to treatment with BH3 mimetics (Figures 1B and S1C). However, a longer drug holiday of 6d restored the drug-sensitivity of the initially drug-tolerant persisters to parental (PT) sensitivity (Figures 1B and S1C), suggesting that the drug-tolerant persister state was transient. We then examined the behavior of PT and BH3 mimetic-induced PS in vivo. We generated PC9 PS by treatment with BH3 mimetics and compared the ability of PS to colonize the lungs to that of the untreated PT cells. PC9 cells were transduced with mNeonGreen or mCherry, and were then used to generate PT and PS, respectively, allowing us to distinguish between each population in vivo. Equal numbers of mNeonGreen-expressing PT and mCherry-expressing PS cells were simultaneously injected i.v. into the tail veins of immunocompromised NSG mice. Nine weeks after injection, lungs were primarily colonized by PS and not by PT (Figure 1C). Similarly, intra-vital assessment of luciferase-labeled cells after tail vein injection showed that mice injected with BH3 mimetic-induced PS from the colon cancer cell line HCT116 had significantly higher tumor burden when compared to mice injected with HCT116 PT cells (Figure 1D and 1E).
Figure 1. Targeting anti-apoptotic Bcl-2 proteins induces persister cells.
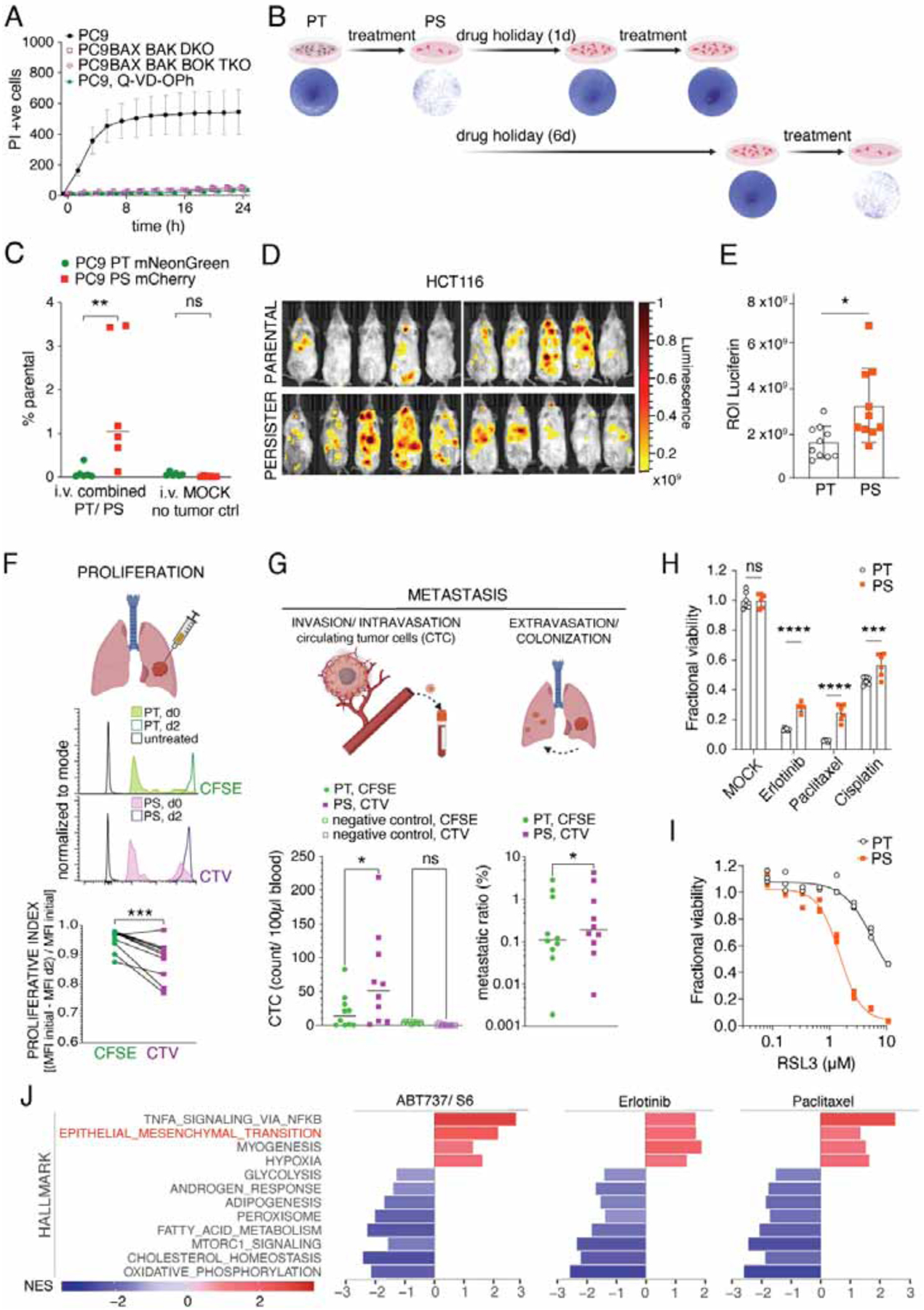
(A) IncuCyte quantification during treatment with 1.5μM ABT737 and 3μM S6 of PC9 WT, PC9 BAX- and BAK-knockout (DKO), PC9 BAX, BAK, BOK TKO cells and PC9 WT cells co-treated with 40μM Q-VD-OPh (mean ± s.d. of n=3 samples/group).
(B) Clonogenic survival of cells treated or not with 1.5 μM ABT737 and 3μM S6 for 6h washed and replated, or treated and then re-seeded again after 1d or 6d and then retreated, washed, and released to growth. Representative of 3 independent experiments, n=3/group.
(C) Flow analysis of mNeonGreen positive PT or mCherry positive PS, from lungs of NSG mice 3 months after combined tail vein injection; n=6 lungs/group. MOCK, lungs without tumor injection.
(D) Intravital images and (E) ROI quantification of luciferase activity 3 weeks after injection of either PT or PS firefly-luciferase-expressing HCT116 cells (n=10 mice, mean ± s.d.). Representative of 2 independent experiments.
(F) Flow analysis of CFSE-labelled PT and CTV-labelled PS. Representative histograms on day 0 before injection and 48h after injection into left apical lungs of NSG mice. Proliferative index calculated by CFSE or CTV MFI signal loss from initial measurement to d2 (n=10 lungs/group, data from 2 pooled experiments).
(G) Flow analysis of CFSE-labelled PT and CTV-labelled PS from RAG1−/− mice (n=10). Left: Quantification of circulating tumor cells from right heart ventricle blood 48h after injection. Right: Metastatic ratio representing the ratio between cells in contralateral lung and primary injection site for PT or PS cells, respectively.
(H) Fractional viability of PC9 PT and PS treated with MOCK, Erlotinib 5μM, Paclitaxel 1μM or Cisplatin 10μM for 3d. N=6 samples/group.
(I) Fractional viability of PC9 PT and PS treated with RSL3 for 3d. N=3 samples/group.
(J) Hallmark pathways enriched across treatment regimens. Enriched pathways were defined as false discovery rate (FDR) <0.05. Normalized enrichment scores (NES) are shown. PS were generated by treatment with ABT737 1.5μM and S6 3μM for 6h, or Erlotinib 5μM for 3d or Paclitaxel 0.5μM for 3d. Statistical analysis by 2-way ANOVA (C, G left, H), unpaired student’s t-test (E), paired student’s t-test (F and G, right) respectively.
*P <0.05; **P <0.01; ***P <0.001; ****P <0.0001. See also Figure S1.
Since these colonization assays mimic only the late phase of the metastatic cascade after intravasation, we exploited an in vivo orthotopic lung tumor model where we simultaneously injected PC9 PS and PT labelled with either Cell Trace Violet (CTV) or carboxyfluorescein succinimidyl ester (CFSE), respectively, into the left lungs of recipient animals. An advantage of these dyes is the strong and equal labeling of cells (Figure 1F). In line with previous studies in vitro, our PS displayed a slower proliferative rate than PT on day 2 after orthotopic injection (Figure 1F) (Chen et al., 2012; Oren et al., 2021; Shen et al., 2020). To test the invasiveness and intravasation ability of the implanted lung tumor cells, we measured circulating-tumor cells (CTC) in blood collected from the right heart ventricle. Interestingly, we found significantly more circulating PS than PT at day 2 (Figure 1G). Next, we measured the metastatic ratio of the injected cells, which we defined as the cancer cell percentage in the contralateral lung divided by its percentage in the primary-injection site. In line with the CTC result, the metastatic ratio revealed a higher metastatic capacity of PS generated from PC9 or A549 cells (Figure 1G and S1D).
Next, we tested whether PS generated using BH3 mimetics led to cross-resistance towards other cancer therapies. We compared PT and PS derived from PC9 and HT29 cells and treated them with targeted and conventional standard-of-care cancer therapies in vitro (Figures 1H and S1E). In both models, PS exhibited significantly increased viability after exposure to each drug.
Recent discoveries on drug-tolerant persisters revealed acquired dependency on the lipid hydroperoxidase GPX4 for their survival (Hangauer et al., 2017; Yang et al., 2016). Viability analysis of our apoptosis PS showed increased sensitivity to GPX4-inhibition by RSL3 compared to PT in PC9 and HT29 cells (Figures 1I, S1F and S1G). In line with this finding, the membrane-targeted lipid ROS sensor C11-BODIPY (Yang et al., 2014) revealed increased lipid ROS levels (Figure S1H). Moreover, the expression of ChaC Glutathione-Specific Gamma-Glutamylcyclotransferase 1 (CHAC1) was upregulated, while the Glutamate-Cysteine Ligase Catalytic Subunit (GCLC) was downregulated in PS when compared to their PT counterpart (Figure S1I), both indicative of decreased glutathione metabolism that may explain the increased dependence on GPX4 (Tang et al., 2021).
To identify common characteristics of PS generated with diverse therapies, we performed RNAseq on therapy-persisting PC9 cells obtained by treatment with conventional chemotherapy, targeted therapy, or BH3 mimetics (Figure 1J). Our analysis revealed common gene expression signatures related to metastasis and invasion, cellular stress responses, upregulated inflammatory signaling and EMT, and downregulated fatty acid metabolism, oxidative phosphorylation and mTORC1 signaling (Figures 1J and S1J). Collectively, these data indicate that PS generated using BH3 mimetics have the gene expression and phenotypic characteristics of drug-tolerant persister cells generated under different conditions as described by others.
The persister phenotype depends largely on Bcl-2 effectors
By binding to the BH3-groove, BH3 mimetics enable the dissociation of the effector proteins BAX and BAK from the antiapoptotic proteins and cause MOMP. However, antiapoptotic, proapoptotic and effector BCL-2 proteins are involved in diverse cell functions, and the BH3 domain is also necessary for the interaction between BH3-only proteins and anti-apoptotic proteins (Kale et al., 2018; Popgeorgiev et al., 2018). To determine whether the presence of BCL-2 family effector proteins is necessary for our persister phenotype we tested PC9 cells deficient in BAX, BAK, and BOK (TKO). We treated these TKO cells with our BH3 mimetics and compared them to the PT cells. Lung colonization capacity of PS (Figure 2A) and their metastatic potential (Figure 2B) were dependent on the BCL2 effector proteins. Moreover, the sensitivity to RSL3-induced ferroptosis was similar between untreated and BH3 mimetic-pretreated TKO cells (Figure 2C and S2A). In line with this finding, C11-BODIPY staining as well as CHAC1 and GCLC transcripts were not changed in BH3 mimetic-pretreated TKO cells when compared to PT (Figures S2B and S2C).
Figure 2. The persister phenotype depends largely on Bcl-2 effectors.
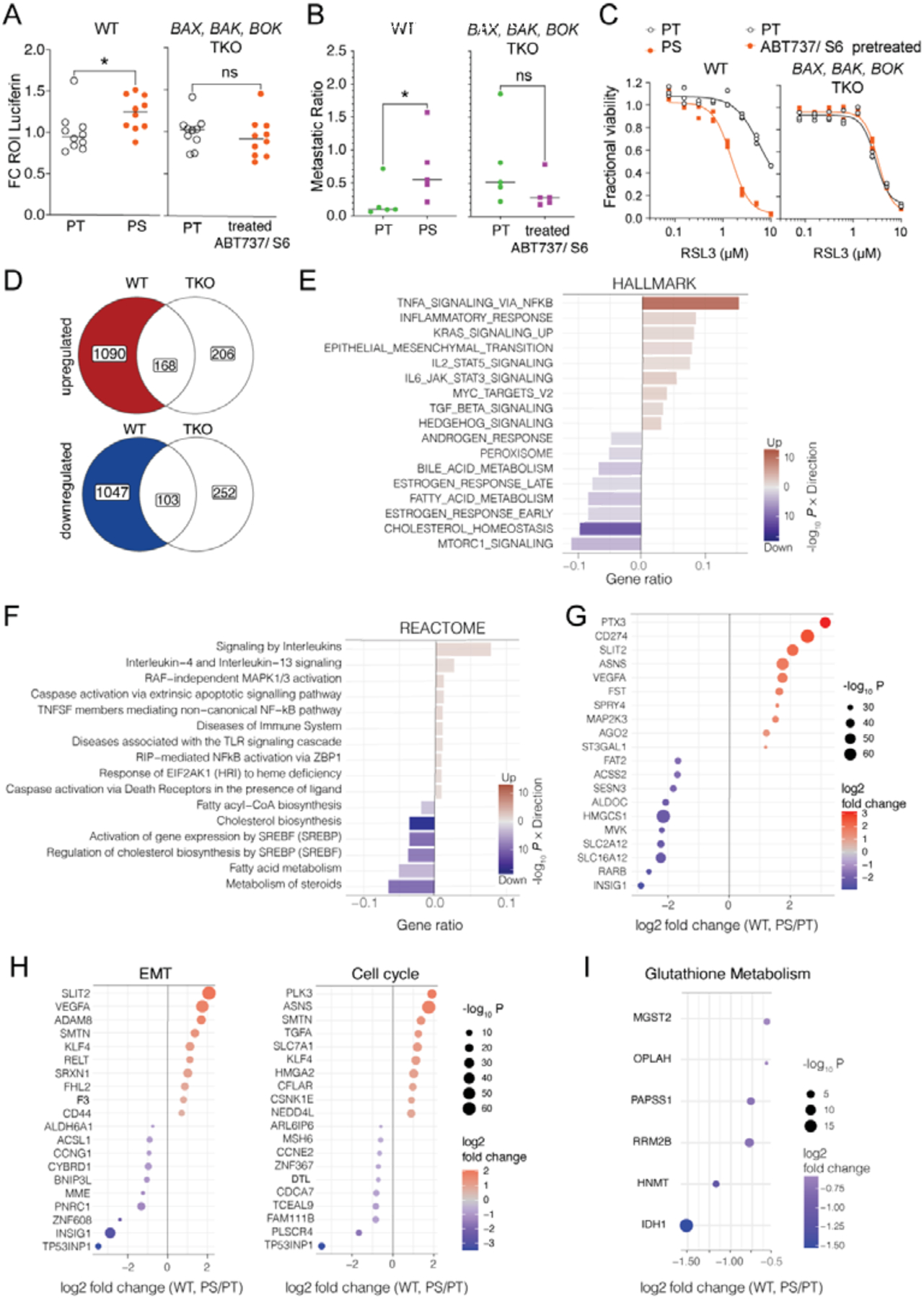
(A) Luciferase activity 2d after injection of firefly luciferase-expressing PT versus PS WT or BAX, BAK, BOK TKO PC9 cells (n=10 mice/group, pooled from 2 independent experiments). All values are standardized to parental values within each experiment =1.
(B) Metastatic ratio of CFSE-labelled PT or CTV-labelled PS from PC9 WT or BAX, BAK, BOK TKO PC9 cells. N=5 mice/group (source data for WT from same experiment as Figure 1F).
(C) Fractional viability measured after 3d RSL3 treatment of PT and PS from WT or BAX, BAK, BOK TKO PC9 cells. Representative of 5 independent experiments shown, n=3 samples/group.
(D) Venn-Diagrams illustrating the number of differentially expressed genes (DEGs) in WT (PS/PT) but not in BAX, BAK, BOK TKO (ABT737, S6 treated/MOCK) PC9 cells. The 1090 DEGs that were only upregulated in WT but not in TKO cells (red) and the 1047 DEGs that were only downregulated in WT but not TKO cells (blue) were further analyzed (E to I).
(E) Enrichment analysis of Hallmark and (F) Reactome pathways shown as gene ratios of DEGs in WT (PS/PT) but not in BAX, BAK, BOK TKO (treated/MOCK) PC9 cells. The red bars show the genes up-regulated in PS and the blue down-regulated in PS. Enriched gene sets with adjusted p-value <0.05 are shown.
(G-I) Top 10 up- and downregulated DEGs in WT (PS/PT) but not in BAX, BAK, BOK TKO (treated/MOCK) PC9 cells. (source data as in E and F).
(D-I) DEGs have been defined as FC >1.5 and p-value <0.05. The gene sets were defined as a collection of gene sets in public databases.
Statistical analysis by unpaired student’s t-test (A) or paired student’s t-test (B). *P <0.05. See also Figure S2.
To elucidate the downstream pathways that might be involved in the engagement of the persister phenotype, we conducted RNAseq of WT and TKO cells treated with control (MOCK) or BH3 mimetics. Enrichment analysis of BAX, BAK, BOK-dependent, differentially expressed genes (DEGs) in PS versus PT (Figure 2D) further confirmed the overall dependence on BCL-2 effector proteins for our previously identified pathways, such as the upregulation of NFκB signaling, EMT and the downregulation of mTORC1 signaling and fatty acid metabolism, and revealed additional pathways such as interleukin signaling and “response to EIF2AK1 (HRI) to heme deficiency” (Figures 2E and 2F). Interestingly, many of the DEGs identified have been associated with metastasis, angiogenesis, and cancer immune evasion (Figure 2G and S2D). Moreover, a stratification of the top 10 up- and down regulated DEGs into pathways such as EMT and the cell cycle further identified potential genes implicated in our persister phenotype (Figure 2H).
Previous work has shown that downregulation of glutathione metabolism correlates with sensitivity towards ferroptosis (Oren et al., 2021). Indeed, our data reveals several downregulated DEGs that participate in the glutathione metabolism pathway in WT PS but not in TKO that have been treated with BH3 mimetics (Figure 2I).
Transcriptomic trajectories reveal a transient ISR expression profile in BH3 mimetic persisters
To gain more insight into the mechanisms that underlie the generation of the persister phenotype, we performed single-cell RNAseq on PC9 PT and corresponding PS 1, 3, and 7 days (PS1D, PS3D, and PS7D respectively) after BH3 mimetic treatment. Uniform manifold approximation and projection (UMAP) analysis revealed the distinct transcriptomes of PT and the initial (PS1D) persister phenotype, as well as the trending of late PS (PS3D, PS7D) towards PT (Figure 3A). To dissect the initial events that distinguish PS from PT and late PS, we next performed a trajectory analysis also known as pseudotime, which is a computational method that organizes a population of cells into a progression consistent with patterns of identified, dynamic processes, presumed to represent changes in the cell states over time (Berge et al., 2020; Street et al., 2018). Whereas late PS exhibited comparable pseudotime states to PT, PS1D were enriched for a clearly distinct and earlier state, suggestive of a gradual return to the PT phenotype after BH3 mimetic treatment (Figures 3A). Interestingly, genes from clusters activated relatively early in pseudotime were significantly enriched for the EMT pathway (cluster 1) and the integrated stress response (ISR; cluster 3; Figures 3B, 3C, S2E and S2F). In contrast, clusters corresponding to later pseudotimes (2 and 4) were enriched for genes central to DNA replication and cell cycle progression, both hallmarks for late PS and PT. These cluster enrichments also corresponded to differences in gene set module scores across the PT and PS conditions in real time, independently linking these biological processes with the experimental conditions (Figure 3D). These data also help to indicate specific genes that may underly the PS phenotype with broad involvement in metabolism, inflammation, and cell cycle (Figure S2F). Furthermore, the upregulation of genes involved in the ISR in 1 day PS that gradually waned over the ensuing days corresponded to the transient state of PS (Figures 3D and S2F).
Figure 3. Transcriptomic trajectory analysis reveals transient ISR expression profile in BH3-mimetic persisters.
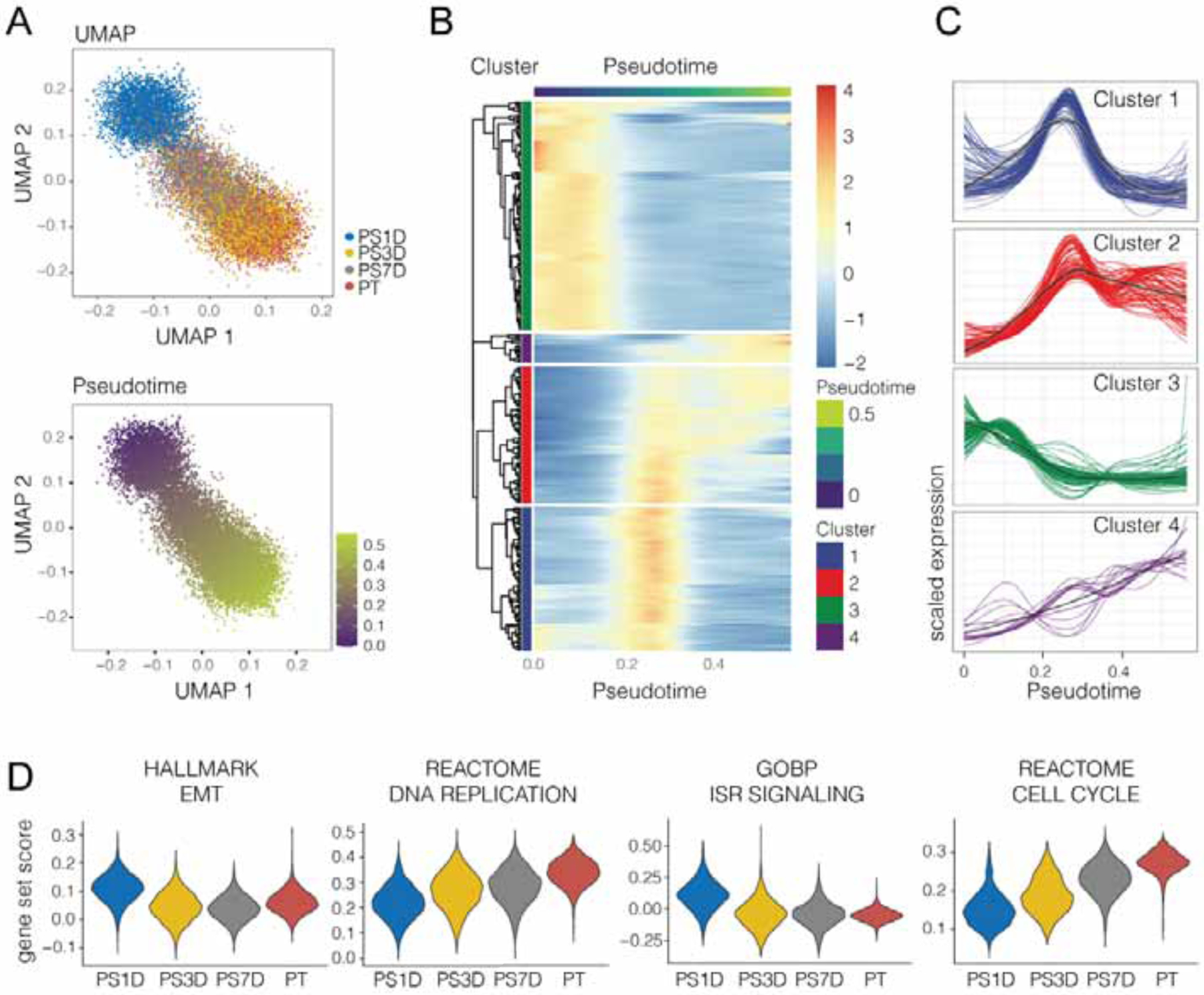
(A) UMAP (upper) and Pseudotime (lower) single cell trajectories of PC9 PT and PS cells harvested after drug holiday of 1 day (PS1D), 3 days (PS3D) or 7 days (PS7D) and subjected to single cell RNAseq. The pseudotime was estimated using the Singleshot R package.
(B) Heatmap of differentially expressed genes along pseudotime (299 genes) identified using the tradeSeq R package. The expression was scaled and centered by gene, and the genes were clustered into four groups using hierarchical clustering.
(C) Scaled expression profiles of gene clusters along pseudotime.
(D) Violin plots visualizing gene set scores of selected gene sets.
See also Figure S2.
BH3 mimetics induce a BCL-2 effector-dependent ISR, independent of caspase activation
We explored if and how the ISR was related to MOMP and the persister phenotype. Consistent with our transcriptomic analysis, we confirmed engagement of the ISR upon BH3 mimetic treatment by detection of the phosphorylation of eIF2α observed within the first hour after treatment and subsequent detection of ATF4 (Figure 4A). Consequently, PS contained elevated levels of nuclear ATF4 compared to PT as assessed by confocal imaging (Figure 4B, 4C and 4D).
Figure 4. BH3 mimetics induce an ISR, dependent on Bcl-2 effectors but independent of caspase activation.
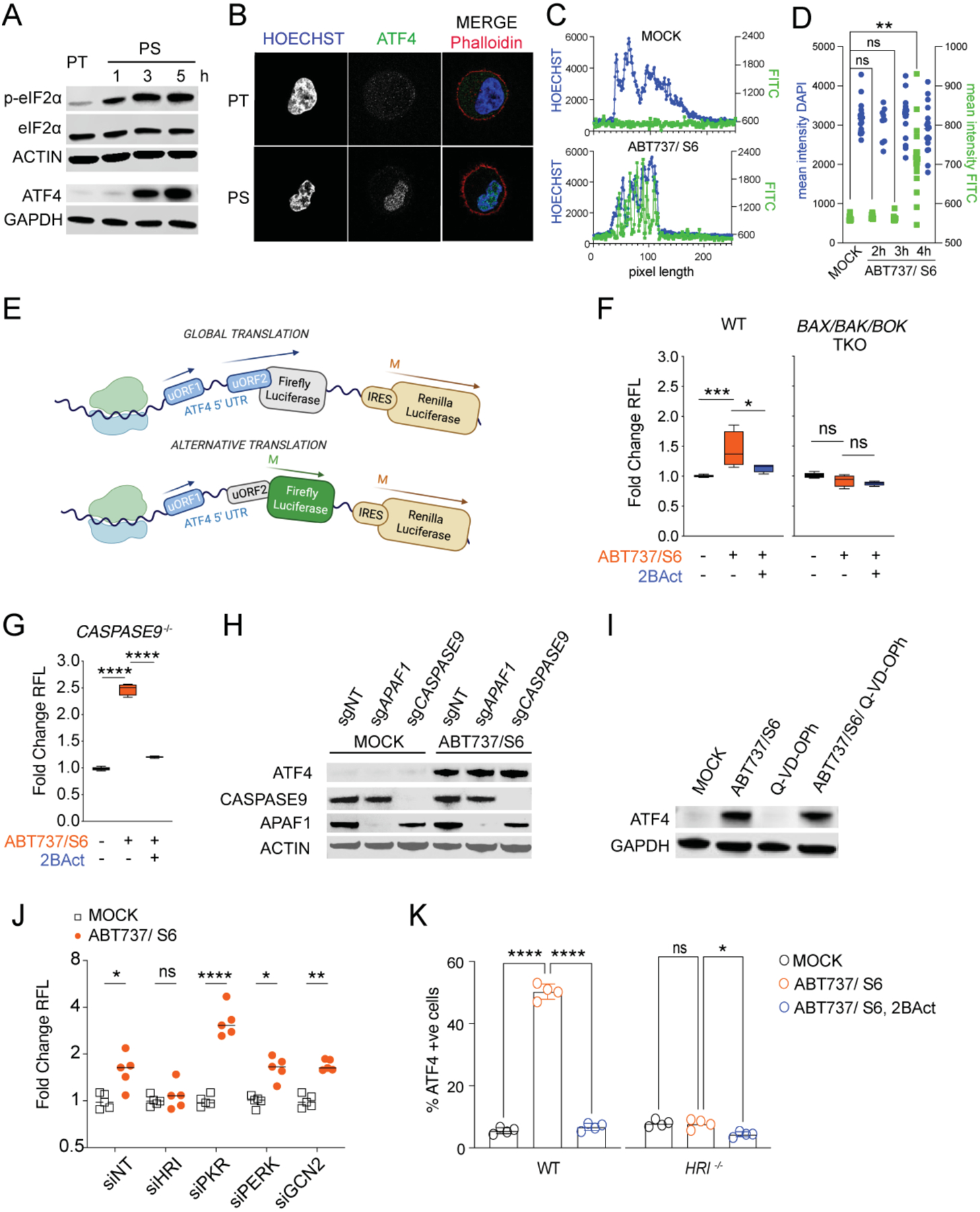
(A) Immunoblot with the indicated antibodies of PC9 PT and PS, generated by treatment with ABT737 1.5μM and S6 3μM for the indicated times.
(B) Confocal microscopy images of PC9 PT and PS after 4h treatment with ABT737 and S6. Cells were stained for Hoechst (blue), ATF4 (green) and Phalloidin (red).
(C) Fluorescence intensity of immunofluorescence from (B) measured in one dimension across the cell through the nucleus. One representative of n=19 PT and n= 18 PS measurements shown.
(D) Mean intensity of immunofluorescence from (B) measured within the nuclear area in PT (n=19) and PS after 2h (n=9), 3h (n=15) or 4h (n=18) treatment. Dots indicate individual cells.
(E) Schematic representation of the luciferase-expressing ATF4 translation reporter construct and its activation under alternative translation conditions.
(F) Luciferase activity assay from (E) showing normalized firefly luciferase activity in PC9 WT and BAX, BAK, BOK TKO cells treated with ABT737 1μM and S6 2μM ± 2Bact 20μM. Data is shown as fold change of RFL compared to MOCK control (mean ± s.d. of n=3 samples/group).
(G) Same as (F) in CASPASE9 KO PC9 cells (mean ± s.d. of n=3 samples/group).
(H) Immunoblot of endogenous ATF4, APAF1 and CASPASE9 in PC9 cells after transient expression of Cas9 and either sgNT, sgAPAF1 or sgCASPASE9. Cells were treated with ABT737 1μM and S6 2μM for 6h.
(I) Immunoblot of endogenous ATF4 in PC9 cells after treatment with ABT737 1μM and S6 2μM and QVD-OPh 10μM.
(J) Luciferase activity assay showing normalized firefly luciferase activity in PC9 cells expressing luciferase ATF4 translation reporter. Cells were silenced with siNT or siRNA for each of the EIF2α- kinases indicated and treated ± ABT737 1μM and S6 2μM for 6h. n=5 samples/group.
(K) Flow analysis of nuclear ATF4 after MOCK or ABT737 1.5μM and S6 3μM ± 2BAct 50μM treatment for 4h in WT or HRI−/− cells. N=4 samples/group.
Statistical analysis by unpaired student’s t-test (D) or 2-way ANOVA (F, G, J and K). *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001. See also Figure S3.
To determine if the translation switch occurs in our BH3 mimetic-induced PS, we engineered an ATF4 translation reporter (Figure 4E). Treatment with BH3 mimetics led to increased reporter activity in WT cells (Figures 4F and S3A). This was inhibited by 2BAct, which prevents phosphorylated eIF2α from reprogramming protein translation (Hetz et al., 2019), and by the ISR inhibitor ISRIB (Figure S3B) (Rabouw et al., 2019). Significantly, BH3 mimetic-treated BAX, BAK, BOK TKO cells failed to activate the luciferase reporter (Figure 4F).
Incomplete MOMP, followed by minimal caspase activity upon apoptosis failure can be associated with cancer cell aggressiveness, mutability, and metastasis (Berthenet et al., 2020; Ichim and Tait, 2016; Oberst et al., 2016). To test whether caspase activation is also necessary for ATF4 activation, we treated Casp9-deficient cells with BH3 mimetics. These cells are unable to activate the caspase cascade (Kuida et al., 1998), but retained the ability to induce the luciferase translation reporter upon BH3 mimetic treatment (Figure 4G). Concordantly, APAF1- and Casp9-deficient cells expressed ATF4 in response to treatment with BH3 mimetics (Figure 4H), as did cells treated with the pan-Caspase inhibitor Q-VD-OPh (Caserta et al., 2003) (Figure 4I).
We then tested which eIF2α kinase is responsible for eIF2α phosphorylation and ATF4 translation. Upon treatment, only cells silenced for HRI (EIF2AK1) displayed decreased luciferase reporter activity compared to cells silenced for the other kinases (Figure 4J). Similarly, treatment with BH3 mimetics of WT but not HRI-deficient cells led to increased nuclear ATF4 protein (Figures 4K and S3C). Therefore, BH3 mimetics induce an ISR via HRI, which is dependent on the BCL-2 effectors but independent of caspase activation.
Engagement of sublethal MOMP is crucial for ISR activation
The dependency on BAX and BAK for the generation of PS suggested that iMOMP in surviving cells might be essential for ISR engagement. We engineered a reporter to reveal mitochondrial permeabilization, using a split-superfoldGFP (sfGFP) system (Figures 5A and B) in which GFP1–10 was fused to mCherry and was localized with a fused IMS-localized tail sequence, while a double GFP11 beta strand was localized to the cytoplasm. We predicted that MOMP would enable the translocation of GFP11 into the IMS by passive diffusion, where it could then associate with GFP1–10 to fluoresce green (Figure 5B). Using this MOMP sensor in A549 PS cells revealed an increased GFP signal (Figure 5C), which was prevented by silencing of BAX and BAK (Figure 5D and S3D). In contrast, cells treated with Raptinal, which induces MOMP in a BAX, BAK, BOK-independent manner (Heimer et al., 2019), showed an increased signal in both siNT and siBAX, siBAK treated cells (Figure 5D). We next tested if the activation of ATF4 protein expression was associated with the extent of MOMP in our PS. Indeed, GFP-high cells that had undergone more extensive sublethal MOMP than GFP-low PS showed higher ATF4 (Figure 5E). Moreover, GFP-high PS displayed increased metastatic potential compared to their GFP-low PS counterparts (Figure 5F). These results support the idea that BCL-2 effector proteins promote ATF4 expression and increased colonization by PS in vivo due to their role in MOMP. Accordingly, WT cells displayed eIF2α phosphorylation and ATF4 expression upon BH3 mimetics or Raptinal treatment (Figure 5G), while BAX, BAK, BOK TKO cells only did so upon treatment with Raptinal (Figures 4F and 5H). Further, Raptinal-treated TKO cells showed an increased metastatic potential in vivo (Figure S3E), which was not observed when such cells were treated with BH3 mimetics (Figure 2B).
Figure 5. Engagement of sublethal MOMP is crucial for ISR activation.
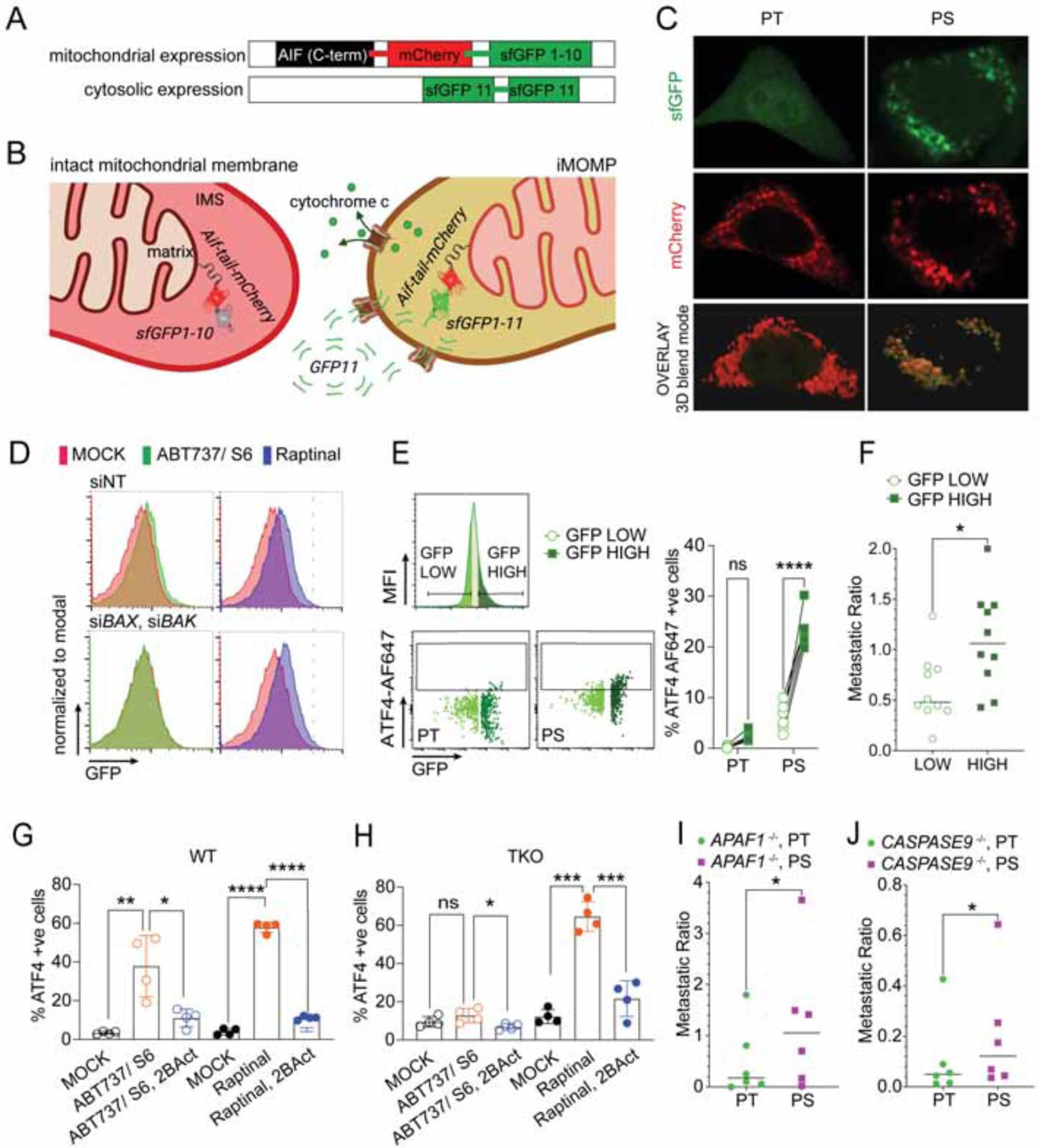
(A) Schematic representation of the mitochondrial and cytosolic expression system for the MOMP sensor and (B) its function upon MOMP.
(C) Confocal microscopy images of A549 PT and PS expressing the MOMP sensor system. PS were generated by 6h treatment with ABT737 1.5μM and S6 3μM and then released overnight before imaging. 3D blend mode image was generated with IMARIS.
(D) Flow analysis of A549 cells expressing the MOMP sensor. Cells were transfected with either non-targeted (siNT) or BAX and BAK targeting siRNA (siBAX, siBAX) and treated with MOCK, ABT737 5μM and S6 10μM or Raptinal 5μM. Representative histograms from n=3 samples/group are shown.
(E) Fow analysis of A549 PT and PS expressing the iMOMP sensor. Left upper panel: representative histogram showing strategy of gating GFP high (25%) and low (25%) cells. Left lower panels: dot plots showing cytosolic ATF4 and GFP in the two stratified groups. Right panel: quantitation of ATF4 positive cells in PT or PS, GFP high and GFP low cells. N=6 samples/group. One representative of 2 independent experiments shown.
(F) Metastatic ratio representing the ratio between cells from the contralateral lung and primary injection site. Analysis of CTV-labelled PS from NSG mice was performed 2d after intrapulmonal injection of either GFP-high or GFP-low sorted PS cells respectively (n=10 mice/group).
(G-H) Flow analysis of nuclear ATF4 after MOCK or ABT737 1.5μM and S6 3μM or Raptinal 5μM ± 2BAct 50μM treatment for 4h in WT (G) or BAX, BAK, BOK TKO cells (H). Mean ± s.d. of n=4 samples/group are shown.
(I, J) Metastatic ratios between cells from the contralateral lung and primary injection site. Analysis of CFSE-labelled PT and CTV-labelled PS from APAF1−/− (I) or CASP9−/− (J) PC9 cells was performed 2d after intrapulmonal combined injection (n=6 mice/group).
Statistical analysis was performed using unpaired student’s t-test (F), 2-way ANOVA (E, G and H) or paired student’s t-test (I and J). *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001. See also Figure S3.
To test the possibility that MOMP, independent of apoptosome and caspase activation, could drive metastatic potential in PS, we also generated PS from APAF1- and Caspase-9-deficient PC9 cells. Indeed, intra-pulmonal injection of PT and PS cells from both lines showed increased metastatic ratio in PS compared to the PT counterpart (Figures 5I and J).
Cytochrome C activates HRI and engages the persister phenotype
Cytochrome c (encoded by CYCS) has a crucial role in the activation of APAF1 and caspases following MOMP (Hao et al., 2005; Li et al., 2000). Strikingly, silencing of CYCS significantly reduced ATF4 activation upon BH3 mimetic treatment (Figures 6A, S4A to C). Additionally, when bovine Cytochrome c was added to cytosolic extracts of WT cells, it led not only to caspase activation as previously described (Liu et al., 1996) but also eIF2α phosphorylation, which depended on the presence of HRI (Figure 6B, 6C and S4D).
Figure 6. Cytochrome c activates HRI and engages the persister phenotype.
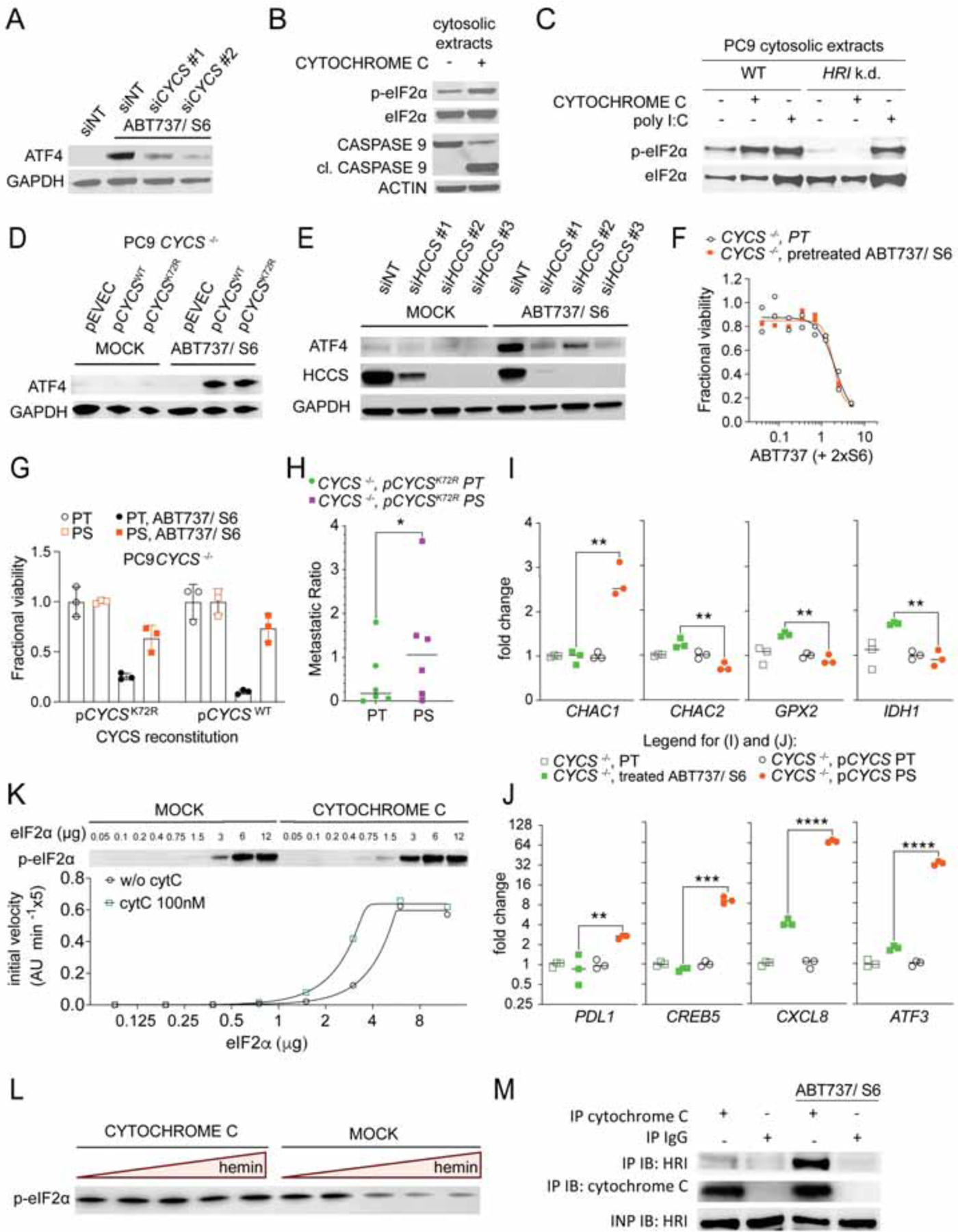
(A) Immunoblot with the indicated antibodies of PC9 cells that had been transfected with non-targeting (siNT) or 2 different CYCS targeting siRNAs, and treated ± ABT737 1.5μM and S6 3μM for 4h. All samples were treated with Q-VD-OPh 20μM.
(B) Immunoblot of cytosolic extracts from PC9 cells treated ± 100nM bovine Cytochrome c for 30min in the presence of 50μM ATP.
(C) Immunoblot of cytosolic extracts from PC9 WT and HRI-silenced cells treated ± 100nM bovine Cytochrome c or 10μg/ml poly I:C for 30 min in the presence of 50μM ATP.
(D) Immunoblot with the indicated antibodies of PC9 CYCS −/− cells, reconstituted ± WT or K72R mutant CYCS and treated ± ABT737 1.5μM and S6 3μM for 4h. All samples were treated with Q-VD-OPh 20μM.
(E) Immunoblot with the indicated antibodies of PC9 cells that had been transfected with non-targeting (siNT) or different HCCS-targeting siRNAs and treated ± ABT737 1.5μM and S6 3μM for 4h. All samples were treated with Q-VD-Oph 20μM.
(F) Fractional viability of PT and BH3-mimetic survivors generated from PC9 CYCS−/− cells treated with a titration of ABT737 and S6. Mean ± s.d. of 3 samples. One representative of n=2 independent experiments shown.
(G) Fractional viability of PT and PS generated from PC9 CYCS−/− cells, reconstituted ± WT or K72R mutant CYCS and treated ± ABT737 1.5μM and S6 3μM. One representative of n=2 independent experiments shown.
(H) Metastatic ratio of PT and PS generated from PC9 CYCS−/− cells that had been reconstituted with CYCS K72R mutant (n=6 mice/group). Statistical analysis by paired student’s t-test. *P <0.05.
(I) and (J) qPCR analysis of transcripts involved in glutathione metabolism (I) or diverse transcripts that have been identified as BCL2 effector protein dependent (J) of PT and PS generated from PC9 CYCS−/− cells, reconstituted ± WT CYCS. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. **P <0.01, ***P <0.001; ****P <0.0001.
(K) Immunoblot and HRI enzyme kinetics in the presence or absence of Cytochrome c 100nm with recombinant proteins. One representative fitting curve from n=3 independent experiments shown.
(L) Immunoblot of in vitro kinase reaction with recombinant eIF2α and HRI, in the presence or absence of Cytochrome c and ± hemin titration (from 2μM max. to 3nM min., 1:5 dilutions in kinase buffer).
(M) Immunoprecipitation of endogenous Cytochrome c or IgG (negative control) from APAF1 KO PC9 cells treated ± ABT737 and S6.
See also Figures S4 and S5.
Next, we asked if the Cytochrome c K72R mutant, which is incapable of activating APAF1 (Hao et al., 2005), can engage the ISR upon treatment with BH3 mimetics (Figures S4E, S4F and S4G). Treatment of cells expressing either WT and or mutant Cytochrome c led to an increase of ATF4, whereas Cytochrome c-deficient cells harboring only an empty vector control (pEVEC) showed no ATF4 protein expression upon treatment (Figure 6D). Similarly, A549 cells that express CYCSK72R, upon silencing of endogenous CYCS with an UTR-targeting siRNA, expressed ATF4 upon treatment with BH3 mimetics, while silencing of both endogenous and ectopically expressed mutant Cytochrome c significantly diminished ATF4 expression (Figure S4H and I). Silencing of Holocytochrome c Synthase (HCCS), required for the generation of mature Cytochrome c, also prevented the synthesis of ATF4 upon BH3 mimetic treatment (Figures 6E and S4C).
We then generated PS from Cytochrome c-deficient cells reconstituted, or not, with WT or K72R mutant Cytochrome c. While BH3 mimetic-pretreated or untreated CYCS-deficient cells showed similar sensitivity towards a variety of drugs tested, PS from either reconstituted cell lines displayed drug-tolerance (Figures 6F, 6G, S5A and B). Additionally, in comparison to PT, PS from cells with reconstituted mutant Cytochrome c displayed enhanced metastatic potential (Figure 6H). While we observed no difference in recovery of BH3 mimetic-pretreated or untreated CYCS-deficient cells, it is likely that such cells do not survive in vivo, and therefore we did not examine their metastatic potential. Therefore, the ability of BH3 mimetics to generate PS is dependent upon Cytochrome c but independent of its ability to induce apoptosis.
We then asked if the genes that had been identified as regulated in PS in a BAX, BAK-dependent manner were also regulated in a CYCS-dependent manner. Genes that are relevant for glutathione metabolism, and hence might account for the RSL3 sensitivity of PS, were up- or downregulated by treatment with BH3 mimetics, dependent upon CYCS expression (Figure 6I). Interestingly, the expression of CHAC1, which is involved in glutathione (GSH) degradation (Crawford et al., 2015), was upregulated only in the presence of Cytochrome c. In contrast, the CHAC cation transport regulator homolog 2 (CHAC2) which competes with CHAC1 to prevent GSH degradation (Wang et al., 2017), was downregulated in a Cytochrome c-dependent manner. Similarly, genes including CREB5, ATF3, PDK1 and PTGS2 showed significantly diminished expression in CYCS-deficient BH3 mimetic treated cells compared to CYCS reconstituted PS (Figure 6J and S5C).
We then performed experiments with recombinant eIF2α and HRI and purified bovine Cytochrome c. We observed increased phosphorylation of eIF2α upon addition of Cytochrome c (Figures 6K and S5D). HRI is constitutively active, and is inhibited by hemin (Berlanga et al., 1998; Igarashi et al., 2008; Yang et al., 1992). We found that the inhibition of HRI by hemin did not occur in the presence of Cytochrome c (Figure 6L). To further investigate if Cytochrome c and HRI physically interact in cells upon treatment with BH3 mimetics, we performed immunoprecipitation of endogenous Cytochrome c and IgG control from PC9 APAF1-deficient cells that had been treated with or without BH3 mimetics, resulting in co-immunoprecipitation of HRI and Cytochrome c (Figure 6M) (A low-level of co-immunoprecipitation of HRI with Cytochrome c in untreated cells was likely due to release of Cytochrome c upon lysis). In another approach, we expressed HRI-mClover or mClover in A549 cells before BH3 mimetic treatment (Figure S5E). Immunoprecipitation of HRI-mClover resulted not only in the co-immunoprecipitation of Cytochrome c, but also of endogenous HRI, indicative of an activation-induced dimerization of HRI, which has been described by others (Bauer et al., 2001). Finally, we generated lysates from HRI-mClover- or mClover-expressing A549 cells and incubated them with equine Cytochrome c. Immunoprecipitation of HRI-mClover resulted not only in the pull-down of Cytochrome c, but also of eIF2α, the substrate of HRI (Figure S5F). Therefore, the engagement of HRI and the ISR, leading to the PS phenotype, is a direct effect of the release of Cytochrome c to the cytosol and its interaction with HRI following iMOMP.
A Cytochrome c – HRI - ATF4 pathway drives the phenotype of persistence
The involvement of ATF4 in various aspects of cancer aggressiveness has been described (Dey et al., 2015). In line with this, cells silenced for ATF4 and treated with BH3 mimetics revealed that the statistically significant upregulation of the EMT pathway is dependent on ATF4 (Figure 7A). Several well-defined transcriptional targets of ATF4 showed increased expression at early pseudotime, characteristic of PS1D in our scRNAseq analysis (Figure S6A). We therefore asked whether ATF4 is the main driver of our observed persister phenotype. Since BH3 mimetic-induced PS also showed an upregulation of other stress-associated modulators, such as NRF2 and HO-1, both of which have been associated with cancer aggressiveness (Bialk et al., 2018; Fox et al., 2020; Inguaggiato et al., 2001; Niture and Jaiswal, 2012), we tested the response to BH3 mimetics upon silencing these regulators. The absence of ATF4 led to significant therapy sensitivity and reduced the occurrence of PS in different cell lines (Figures 7B and S6B to D), while NRF-2 and HO-1 appeared to be dispensable. Early PS displayed an increased sensitivity towards 2BAct (Figure 7C). To test the role of ATF4 in the persister phenotype, we generated ATF4-deficient cells (Figure S6E) and treated them with BH3 mimetics. Surviving cells were exposed to a variety of pro-apoptotic anti-cancer drugs, revealing equal or higher sensitivity to the same drugs as ATF4−/− PT (Figure 7D).
Figure 7. Cytochrome c – HRI – ATF4 axis drives the phenotype of persistence.
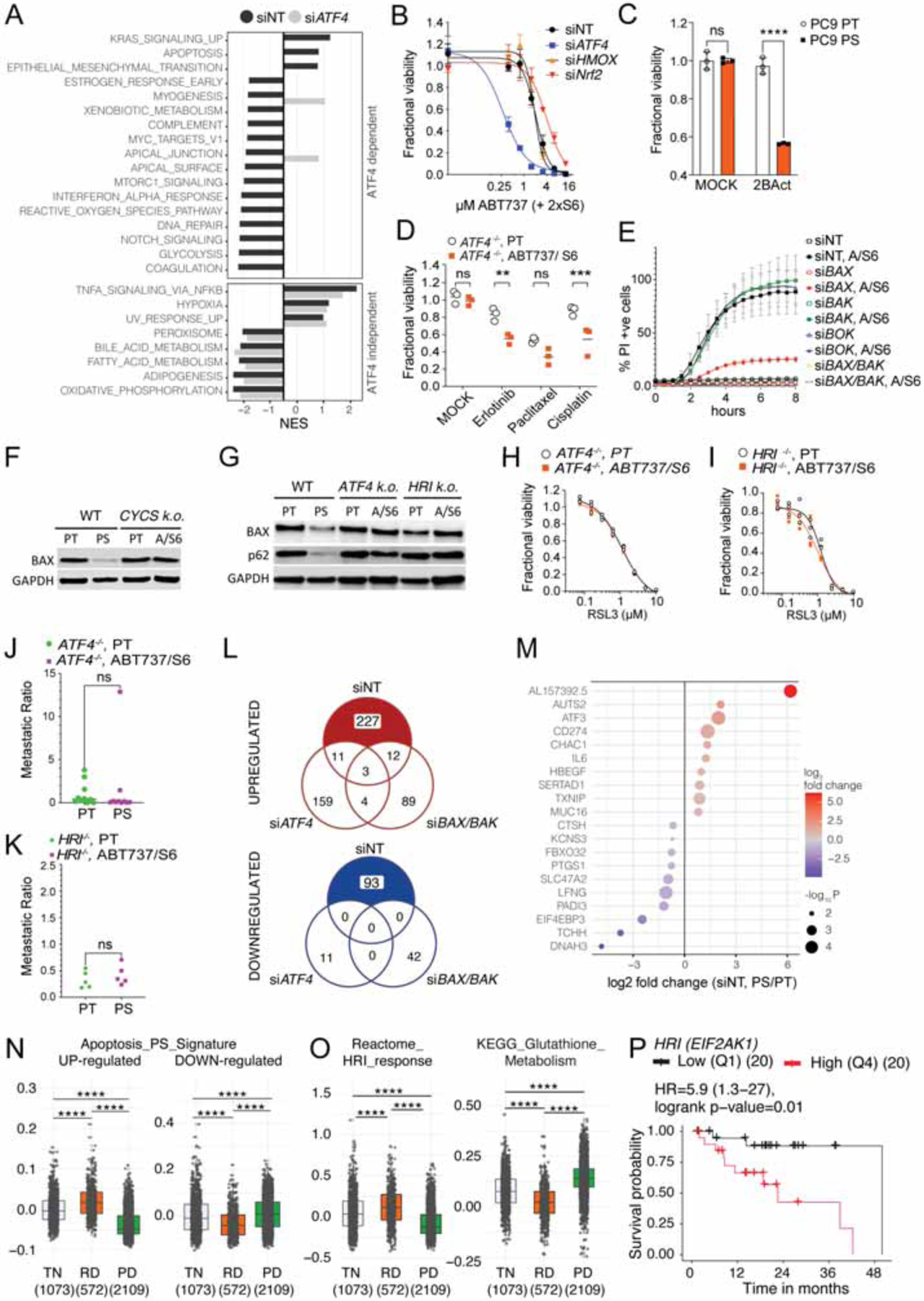
(A) Cancer hallmarks pathway analysis in BH3-mimetic treated versus untreated cells significantly changed in siNT (PS/PT) but not siATF4 (ABT737, S6 treated/MOCK) PC9 cells.
(B) Fractional viability of PC9 cells that had been transfected with siNT, siATF4, siHMOX1 or siNRF2, and treated with a titration of ABT737 and S6. One representative of 2 independent experiments shown.
(C) Fractional viability of PC9 PT and PS that had been treated with 50μM of 2BAct. N=3 samples/group.
(D) Fractional viability of PT and BH3-mimetic survivors that were generated from ATF4−/− cells, and had been treated with MOCK, Erlotinib 5μM, Paclitaxel 1μM or Cisplatin 10μM for 3d. N=3 samples/group.
(E) IncuCyte quantification during treatment with ABT737 (1.5μM) and S6 (3μM) of PC9 cells transfected with siNT, siBAX, siBAK, siBOK or a combination of siBAX and siBAK WT. Data are mean ± s.d. of n=3 samples/group. One representative of 3 independent experiments is shown.
(F) Immunoblot of BAX in WT and CYCS KO cells treated ± ABT737 and S6.
(G) Immunoblot of BAX in WT and ATF4 KO and HRI KO cells treated ± ABT737 and S6.
(H) Fractional viability of PT and BH3-mimetic survivors generated from PC9 ATF4−/− cells treated with a titration of RSL3. One representative of 7 independent experiments shown.
(I) Fractional viability of PT and BH3-mimetic survivors generated from PC9 HRI−/− cells treated with titration of RSL3. One representative of 6 independent experiments shown.
(J) Metastatic ratio of PT and BH3-mimetic survivors generated from PC9 ATF4−/− (n=10 mice/group).
(K) Metastatic ratio of PT and BH3-mimetic survivors generated from PC9 HRI−/− (n=5 mice/group), (same experiment as Figure 1E).
(L) Venn-Diagrams illustrating the number of differentially expressed genes (DEGs) in siNT (PS/PT), siATF4 (ABT737, S6 treated/MOCK) or siBAX/BAK (ABT737, S6 treated/MOCK) PC9 cells. Numbers of up- and down-regulated genes are shown.
(M) DEGs dependent on ATF4 and BAX, BAK. Top 10 genes from 227 up- and 93 down-regulated DEGs that are significantly changed in siNT- but not siATF4- or siBAX/BAK-silenced cells are depicted.
(N) Gene set score for an apoptosis signature generated with DEGs in PS and not PT that are dependent on ATF4 and BAX, BAK (see also M), that are up- or down-regulated in human lung cancers from patients which were treatment-naïve (TN, n=1073 samples), had residual disease (RD, n=572 samples) or had progressive disease (PD, n=2109 samples). The published data set is from Maynard et al., 2020.
(O) Gene set score for Reactome HRI response and KEGG Glutathione metabolism gene set that were up- or down-regulated in human lung cancers from the data set in (N). Statistical analysis was performed using t-test ****P <0.0001.
(P) Kaplan Meier survival curve of patients with EGFR-mutant lung adenocarcinoma expressing high (4th quartile) or low (1st quartile) HRI from TCGA database lung adenocarcinoma (LUAD) cohort. Hazard ratio (HR) and p-value is indicated. N=20/group.
Statistical analysis was performed using 2-way ANOVA (C and D) and paired student’s t-test (J and K). **P <0.01, ***P <0.001; ****P <0.0001. See also Figures S6 and S7.
Since BH3 mimetics induce BCL-2 effector-mediated apoptotic cell death, we wondered if the effector proteins were present in PS. BAX but not the other effector proteins were decreased in PS (Figure S6F). It has been reported that the absence of BAX can lead to chemoresistance (Erler et al., 2004; Haefen et al., 2004). Silencing of BAX, BAK or BOK in PC9 cells before treatment with BH3 mimetics revealed that the absence of BAX alone almost completely abolished sensitivity to the drugs (Figure 7E and S6G) and also decreased sensitivity towards other therapies in PC9 cells (Figure S6H). To evaluate if the decrease in BAX relied on the Cytochrome c-dependent activation of the ISR, we tested CYCS-, ATF4- and HRI-deficient cells treated with BH3 mimetics (Figure 7F and 7G). We observed that BAX was decreased only in WT cells that had survived BH3 mimetic treatment. Transcript levels of BAX were similar in Cytochrome c proficient and deficient cells (Figure S6I), suggesting that post-transcriptional regulation was responsible for the decreased BAX levels in our PS. One possible explanation for the reduction in BAX might be the engagement of autophagy, which can target BAX for degradation (Feng et al., 2018; Guan et al., 2015). ATF4 has been shown to induce autophagy (B’chir et al., 2013; Rzymski et al., 2009) and consistent with this, we observed that the degradation of p62/SQSTM1, a consequence of autophagy, occurred in BH3 mimetic-treated WT but not ATF4- or HRI-deficient cells (Figure 7G).
We examined the sensitivity to GPX4 inhibition and found that the increased sensitivity in PS was abrogated in ATF4- and HRI-deficient BH3 mimetic survivors (Figures 7H, 7I, S6J and S6K), which corresponded to changes in lipid-peroxidation (Figure S6L) and CHAC1 and GCLC expression (Figures S6M and S6N). Next, we tested the metastatic capacity of ATF4- and HRI-deficient cells and their BH3 mimetic treatment-surviving cells in vivo. The increased metastatic ratio of WT PS cells (Figure 1G) was largely abolished in the absence of ATF4 (Figure 7J) or HRI (Figure 7K).
To determine whether ATF4 activation could induce a persister phenotype independent of iMOMP, we treated cells with Tunicamycin (TN), which leads to ATF4 translation independent of BCL-2 effector proteins or Caspase-9 (Figure S6Q). These TN-induced PS revealed decreased sensitivity towards diverse chemotherapies (Figure S6R) and increased sensitivity towards GPX4 inhibition with RSL3 (Figure S6S).
Finally, we performed a stringent comparative analysis of differentially expressed genes in BH3 mimetic-treated WT cells that are absent in treated ATF4- or BAX and BAK-silenced cells (Figure 7L), which revealed several target genes implicated in inflammatory signaling and immune evasion, metabolism, migration and invasion (Figure 7M). We suggest that this gene set represents an “apoptosis persister signature.” To test its clinical relevance, we applied gene set enrichment analysis on published scRNAseq data from lung adenocarcinoma samples (Maynard et al., 2020). Strikingly, gene set module scores for up- or down-regulated apoptosis persister genes were significantly greater among samples from patients with residual disease (RD) compared to those with treatment-naïve (TN) or progressive disease (Figure 7N). Further, the Reactome HRI gene set score was increased in RD, while the KEGG glutathione metabolism gene score was significantly decreased in RD compared to TN (Figure 7O).
Because our experimental results identified HRI as the relevant kinase for translation reprogramming and generation of the persister phenotype in PC9 lung adenocarcinoma (LUAD) cells, we wondered if HRI expression was present in cancer samples. Pathology samples from lung, breast, gastric and ovary cancer showed much stronger expression of HRI compared to their normal tissue counterparts (Figure S7A). HRI gene expression is significantly higher in various tumors when compared to the normal tissues (Figures S7B and C), including LUAD and colon adenocarcinomas. Since RNAseq data revealed a high inter-patient variation in HRI expression (Figure S7C), we asked if HRI (EIF2AK1) expression correlates with patient survival. We interrogated disease free survival and overall survival of patients with HRI high (1st quartile) versus low (4th quartile) tumors. Many HRI-high tumor-bearing patients showed decreased survival and significantly increased hazard ratios compared to patients with HRI-low tumors (Figures 7P and S7D, S7E, S7F and S7G).
DISCUSSION
Our work uncovers a new function for Cytochrome c in promoting survival in the presence of a sublethal incident. We provide evidence of how iMOMP and the release of Cytochrome c drives translation reprogramming in cells that survive the initiation of apoptosis. As a major consequence of eIF2α phosphorylation, ATF4 mRNA translation leads to the initiation of the various downstream pathways that are critical for the persister phenotype, including metabolic reprogramming, cell cycle inhibition, immune evasion, and EMT induction.
Additionally, we found that our persister phenotype includes an increased metastatic potential of PS. While the often-described EMT signature of persister cells is suggestive of an increased metastatic capacity (Nieto, 2011; Shen et al., 2020) and sensitivity towards ferroptosis correlates with both EMT and increased metastasis (Hangauer et al., 2017; Ubellacker et al., 2020; Viswanathan et al., 2017; Yang et al., 2014), a direct connection between a persister phenotype and metastasis has to the best of our knowledge not been previously described.
We identified the heme-regulated inhibitor (HRI) as the critical kinase that leads to eIF2α phosphorylation and translational reprogramming in PC9 cells treated with BH3 mimetic drugs. Our fundamental knowledge about HRI originated in research on erythropoiesis, where iron deficiency leads to the autophosphorylation and dimerization of HRI and thus its activation (Berlanga et al., 1998; Miksanova et al., 2006; Rafie-Kolpin et al., 2003; Yang et al., 1992; Zhang et al., 2019). This evolutionarily conserved pathway appears to be present in non-hematopoetic cells, including cancer cells where we found that high expression of HRI correlates with decreased survival of cancer patients. Our work reveals the crucial impact of HRI on cancer cell survival by linking sublethal Cytochrome c release to HRI activation. It also provides a possible explanation for the increased sensitivity towards ferroptosis in drug-tolerant persisters when GSH metabolism is altered, as a linked trade-off for survival when translational reprogramming takes place. Transcriptomic changes in the metabolome including a downregulation of the mTORC1 pathway that is dependent on sublethal apoptosis initiation and ATF4 activation is in line with findings where mitochondrial dysfunction was accompanied by HRI activation and mTORC1 downregulation (Condon et al., 2021; Zhang et al., 2019).
MOMP can play a role in caspase-independent cell death (CICD), where death is relayed by a loss of mitochondrial function (Lartigue et al., 2009). Previous work on CICD indicated that in the absence of caspase activation cells that had evaded death after the engagement of MOMP and Cytochrome c release were dependent on the activation of the autophagy machinery and intact mitochondria (Colell et al., 2007). Since ATF4 is a well-known activator of autophagy (Sui et al., 2013), our results provide a possible mechanistic explanation for how pro-apoptotic effectors can promote survival. We do not know, however, whether engagement of autophagy is indeed responsible for the transient drug tolerance we observe in PS, or if this applies to PS generated under other conditions.
Previous work has also documented how mitochondrial dysfunction can lead to the cytosolic accumulation of mitochondrial proteins and a mitochondrial unfolded protein response (UPRmt), which in turn activates the expression of ATF4 (Melber and Haynes, 2018). In contrast, our results indicate that unlike the UPRmt, the engagement of ATF4 by the ISR in persister cells does not primarily rely on mitochondrial damage itself but instead is dependent on Cytochrome c release to the cytosol. Recent evidence revealed that release of the IMS protein DELE1 after cleavage by OMA1 can activate the ISR via HRI (Fessler et al., 2020; Guo et al., 2020). However, whether MOMP is necessary for DELE1 release and the biological consequences of DELE1 release in our system remain unclear.
Additionally, iMOMP can lead to sublethal caspase activation (Liu et al., 2015; Miles and Hawkins, 2017; Oberst et al., 2016; Paoli et al., 2013). Although, our data supports a mechanism that is independent of caspases, there is evidence that caspase activation in a drug-persisting cancer cell induces EMT, can activate PKR and increases mutability by the sublethal activation of caspase-activated DNAse (CAD) (Berthenet et al., 2020; Miles and Hawkins, 2017; Saelens et al., 2001). Taken together, it is possible that adaptive responses can be engaged on various levels after sublethal MOMP initiation.
Limitations of the study
Our approach in this study was to investigate – without any other perturbations – the role of sublethal engagement of apoptosis. Thus, we created controlled circumstances to induce PS by the use of BH3 mimetics and in vitro induction of cell death, followed by assessment of the persister phenotype in vitro and in vivo. The extent to which our findings apply to PS induced by other drug treatments and the further application of our findings to patient data from human primary cancers and residual disease will be important. Since other therapeutic treatments can likely engage the ISR via other mechanisms than Cytochrome c-induced HRI activity, the generalizability of our BH3 mimetic persister cells and the mechanism we uncovered requires further investigation. This especially applies to in vivo settings where cells that survive therapeutic insult are more challenging to study.
STAR+METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Douglas R Green (Douglas.Green@stjude.org).
Materials availability
Plasmids generated in this study are obtainable upon request.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO (accession: GSE189639) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cytochrome C – AF 647 | BioLegend | Cat# 612310, RRID:AB_2565241 |
| CD45 – APC | BD | Cat# 559864 RRID:AB_398672 |
| GAPDH (0411) HRP | Santa Cruz | Cat# sc-47724 HRP RRID:AB_627678 |
| HRI (D-12) | Santa Cruz | Cat# sc-365239 RRID:AB_10843794 |
| eIF2alpha | CST | Cat# 9722 RRID:AB_2230924 |
| eIF2 pS51 | Abcam | Cat# ab32157 RRID:AB_732117 |
| ATF-4 | CST | Cat# 11815 RRID:AB_2616025 |
| HRI (D-12) (EIF2AK1) | Proteintech | Cat# 20499-1-AP RRID:AB_10697665 |
| PKR (EIF2AK2) | Santa Cruz | Cat# sc6282 RRID:AB_628150 |
| Perk (EIF2AK3) | CST | Cat# 3192 RRID:AB_2095847 |
| GCN2 (EIF2AK4) | CST | Cat# 40457 RRID:AB_2799177 |
| Nrf2 | Santa Cruz | Cat# sc13032 RRID:AB_2263168 |
| Heme Oxygenase 1 Antibody (A-3) | Santa Cruz | Cat# sc-136960 RRID:AB_2011613 |
| GAPDH-HRP | Santa Cruz | Cat# sc47724-HRP RRID:AB_627678 |
| β-Actin Antibody (C4) HRP | Santa Cruz | Cat# sc-47778 HRP RRID:AB_2714189 |
| Caspase9 | CST | Cat# 9502S RRID:AB_2068621 |
| HCCS | Abcam | Cat# ab234874 |
| APAF-1 | CST | Cat# 5088S RRID:AB_10556958 |
| BAX | CST | Cat# 2772 RRID:AB_10695870 |
| BAK | CST | Cat# 3814 RRID:AB_2290287 |
| BOK | Abcam | Cat# ab186745 RRID:AB_2728737 |
| Cytochrome c (D18C7) Rabbit mAb | CST | Cat# 11940S RRID:AB_2637071 |
| Amersham ECL Rabbit IgG, HRP-linked whole Ab (from donkey) | Amersham | Cat# NA934 RRID:AB_772206 |
| Cytiva’s Amersham ECL Mouse IgG, HRP-linked whole Ab (from sheep) | Amersham | Cat# NA931 RRID:AB_772210 |
| Anti-NRF2 antibody [N2C2] | Genetex | Cat# GTX103322 RRID:AB_1950993 |
| GFP-Booster Alexa Fluor® 488 | Chromotek | Cat# gb2AF488 RRID:AB_2827573 |
| RFP-Booster Alexa Fluor® 568 | Chromotek | Cat# rb2AF568 RRID:AB_2827576 |
| BODIPY™ 581/591 C11 | Thermo Fisher | Cat# D3861 |
| Cytochrome C | BD | Cat# 556432 RRID:AB_396416 |
| GFP-Trap Magnetic Agarose | Chromotek | Cat# gtma-20 RRID:AB_2631358 |
| p62/SQSTM1 | Sigma | Cat# P0067 RRID:AB_1841064 |
| Bacterial and Virus Strains | ||
| NEB®10-beta Competent E.coli (High Efficiency) | NEB | Cat# C3019H |
| NEB 5-alpha Competent E.coli | NEB | Cat# C2987H |
| NEB® Stable Competent E.coli (High Efficiency) | NEB | Cat# C3040H |
| TOPO™ TA Cloning™ Kit for Sequencing, with One Shot™ TOP10 Chemically Competent E. coli | Thermo Fisher | Cat# K4575J10 |
| Biological Samples | ||
| - | ||
| Chemicals, Peptides, a Recombinant Proteins | ||
| ABT-737 | MCE | Cat# HY-50907 |
| S63845 | MCE | Cat# HY-100741 |
| ISRIB | MCE | Cat# HY-12495 |
| 2BAct | AOBIOUS | Cat# 2143542-28-1 |
| Paclitaxel | MCE | Cat# HY-B0015 |
| RSL3 | Selleck Chemicals | Cat# S8155 |
| Irinotecan | MCE | Cat# HY-16562 |
| Erlotinib | Cayman | Cat# 10483 |
| Vemurafenib (PLX4032) | Cayman | Cat# 10618 |
| Raptinal | Millipore Sigma | Cat# SML1745 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| Eif2s1, His tagged human,recombinant | Sigma-Aldrich | Cat# SRP5232 |
| Eif2ak1 (HRI) Human Protein | Thermo Scientific | Cat# PV5856 |
| 3X FLAG® Peptide | Sigma-Aldrich | Cat# F4799 |
| AC DEVD AFC | Enzo Life Sciences | Cat# ALX260032M005 |
| Cytochrome c from Saccharomyces cerevisiae | Sigma-Aldrich | Cat# C2436 |
| Cytochrome C, equine | Sigma-Aldrich | Cat# C2506 |
| Cytochrome C, bovine | Sigma-Aldrich | Cat# C2037 |
| Liberase TM | Sigma-Aldrich | Cat# 5401119001 |
| DNase I, Grade II, from bovine pancreas | Sigma-Aldrich | Cat# 10104159001 |
| Corning® Matrigel® Basement Membrane Matrix, LDEV-Free | Corning | Cat# 356234 |
| Deferoxamine mesylate salt | Sigma-Aldrich | Cat# D9533-1G |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Direct-zol RNA MicroPrep (200 Preps) w/ Zymo-Spin IC Columns (Capped) | Zymo Research | Cat# R2062 |
| Intracellular Fixation & Permeabilization Buffer Set | BD | Cat# 554714 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | Thermo Scientific | Cat# 00-5523-00 |
| Dual-Luciferase Reporter 1000 Assay System | Promega | Cat# E1980 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| CellTrace™ CFSE Cell Proliferation Kit, for flow cytometry | Thermo Scientific | Cat# C34554 |
| Cell Trace Violet | Thermo Scientific | Cat# C34557 |
| Clarity™ Western ECL Substrate, 500 ml, 1705061 | Bio-Rad | Cat# 1705061 |
| Xt MES Running Buffer, 1610789 | Bio-Rad | Cat# 1610789 |
| 20S Proteasome Lysis Buffer | CAYMAN Chemicals | Cat# 10011098 |
| Lipofectamine 3000 Transfection Reagen | Thermo Scientific | Cat# L3000001 |
| Fugene HD Transfection Reagent, Promega, FuGENE HD Transfection | Promega | Cat# E2311 |
| ViaFect Transfection Reagent | Promega | Cat# E4981 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Therno Scientific | Cat# 13778075 |
| Ercc RNA Spike-In Mix | Thermo Scientific | Cat# 4456740 |
| pcDNA3.1/V5-His TOPO TA Expression Kit | Thermo Scientific | Cat# K480001 |
| ANTI-FLAG® M2 Affinity Gel,purified immunoglobulin, buffered aqueous | Sigma-Aldrich | Cat# A2220 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#4309155 |
| Chromium Next GEM Single Cell 3ʹ Library Kit v3.1 16 rxns | 10X Genomics | PN-1000157 |
| Chromium Next GEM Chip G Single Cell Kit, 16 rxns | 10X Genomics | PN-100012 |
| Mycoalert Detection kit | Lonza | Cat# LT07-318 |
| EasySep Dead Cell Removal (Annexin V) kit | STEMCELL technologies | Cat# 17899 |
| Next Seq 500/550 High Output Kit v2 | Illumina | Cat#FC-404-2002 |
| TruSeq Stranded mRNA kits | Illumina | Cat#20020595 |
| Zombie Violet Viability Kit | Biolegend | Cat#423113 |
| Deposited Data | ||
| SuperSeries | GSE189639 | |
| bulk RNA-seq of parental and persister cells generated human cancer cells | GSE189625 | |
| scRNAseq of PC9 parental and persister cells | GSE189638 | |
| Experimental Models: Cell Lines | ||
| H. sapiens: PC9 | ATCC | |
| H. sapiens: HCT116 | Richard Youle | |
| H. sapiens: HT29 | ATCC | |
| H. sapiens: A549 | Richard J. Webby, SJRH | |
| H. sapiens: H226 | ATCC | |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcsscid Il2rgtm1Wjl/SzJ (NSG) | In house | |
| Rag1−/− | In house | |
| Oligonucleotides | ||
| sgRNA see Table S3 | abm (Lopez et al., 2016; Shalem et al., 2014) | N/A |
| siRNA see Table S4 | Dharmacon, IDT | N/A |
| qPCR primer see Table S5 | Origene sequences, (Kim et al., 2010; Spandidos et al., 2008, 2010; Wang et al., 2012) | N/A |
| Recombinant DNA | ||
| Piggybac expression system (inducible) | (Magnúsdóttir et al., 2013) | |
| pPB constitutive CMV | This paper | |
| pPB (puro)-mCherry | This paper | |
| pPB (puro)-mNeonGreen | This paper | |
| pPB (puro)_hUTR(ATF4) luci_IRES RenLuciferase | This paper | |
| pPB (puro) mitoV1 AifTail-mCh-GFP1-10 | This paper | |
| pPB (Blast) | This paper | |
| pPB (Blast) 2xGFP11 (M3) | This paper | |
| pPB (Zeo) | This paper | |
| pPB (Zeo)_CYCS | This paper | |
| pPB (Zeo)_CYCS K72R | This paper | |
| pPB Luciferase-tdTomato | This paper | |
| pcDNA TOPO TA mClover | This paper | |
| pXG290 (HRI-mClover) | (Guo et al., 2020) | Addgene 141284 |
| Px458 pSpCas9(BB)-2A-GFP | (Ran et al., 2013) | Addgene 48138 |
| pLKO5.sgRNA.EFS.PAC | (Heckl et al., 2014) | Addgene 57825 |
| CRISPRv2 GFP | (Walter et al., 2017) | Addgene 82416 |
| pSpCas9(BB)-2A-mCherry | In House | N/A |
| Software and Algorithms | ||
| Trim Galore (v0.5.0) | Babraham Institute | RRID:SCR_011847 |
| Picard (v 2.19.0) | Broad Institute | RRID:SCR_006525 |
| RSEM | Dewey lab | RRID:SCR_013027 |
| DESeq2 (v1.30.1) | Michael Love | RRID:SCR_015687 |
| Cell Ranger (v6.0.1) | 10X Genomics | RRID:SCR_017344 |
| Slingshot (v1.8.0) | Kelly Street | RRID:SCR_017012 |
| STAR (v 2.7.5a) | Alexander Dobin | RRID:SCR_004463 |
| Seurat (v4.0.4) | Satija lab | RRID:SCR_016341 |
| tradeSeq (v1.4.0) | Lieven Clement | RRID:SCR_019238 |
| clusterProfiler (v3.18.1) | Guangchuang Yu | RRID:SCR_016884 |
| R (v 4.0.5) | R project | RRID:SCR_001905 |
| RUVseq (v1.24.0) | Davide Risso | RRID:SCR_006263 |
| FlowJo v10 | FlowJo, LLC | RRID:SCR_008520 |
| Slidebook 6 | Intelligent Imaging Innovations | RRID:SCR_014300 |
| GraphPad Prism 9.0 | GraphPad Software | RRID:SCR_002798 |
| IMARIS 9.8 | Oxford Instruments | RRID:SCR_007370 |
All original code has been deposited at GitHub (https://github.com/markgene/MC015_Persister_public) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHODS
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
RAG1 k.o. (Figure 1E) and NOD.Cg-Prkdcsscid Il2rgtm1Wjl/SzJ (NSG) (all other animal models) mice were used for tumormodels. Age and sex matched cohorts of 5 to 12 weeks old females and males were used in all experiments. The St. Jude Institutional Animal Care and Use Committee approved all procedures in accordance with the Guide for the Care and Use of Animals. All mice were housed in pathogen-free facilities, in a 12-hour light/dark cycle in ventilated cages, with chow and water supply ad libitum.
Cells
Human lung cancer cell line PC9 was purchased from Millipore Sigma (Cat# 90071810). Human lung cancer cell line A549 was kindly provided by Richard J. Webby (St. Jude Children’s Research Hospital). Human colorectal carcinoma cell line HCT116 was a gift from Richard Youle (Wang and Youle, 2011). Cancer cell line HT29 (human colorectal) and H226 (human Mesothelioma) were purchased from ATCC.
Cells were maintained in complete DMEM media (10% fetal bovine serum (FBS), 2 mM L-glutamine and 100 units/ml penicillin-streptomycin). For culturing CYCS k.o. cells medium was additionally supplemented with 50μg/ml uridine (Sigma) and 1 mM sodium pyruvate (GIBCO). All cells used in this study were cultivated at 37°C with 5% CO2.
All the cell lines used in tumor studies were confirmed as mycoplasma negative using MycoAlert Mycoplasma Detection kit (Lonza #LT07). All cell lines have been authenticated by Hartwell Center (St. Jude Children’s Research Hospital).
Clonogenic survival was assessed by methylene blue staining.
METHOD DETAILS
In vivo colonization and FACS analysis
PC9 stably expressing mNeon Green were used as PT. PC9 cells stably expressing mCherry were treated with ABT737 1.5 μM and S63845 3 μM for 6 h followed by wash and release into complete medium for 2 h, to generate PS. EasySep Dead Cell Removal (Annexin V) kit (Stemcell Technologies) was used to harvest live cells according to the manufacturer’s protocol. PT and PS were injected in combination (0.4 ×106 cells in total per mouse) into the teil vein of NSG mice (200μl in DMEM). When animals were getting symptomatic due to disease progression they have been euthanized in accordance with our existing humane endpoints (9 weeks post injection) to assess organ colonization. For ex vivo processing of lung colonization lungs of animals were resected and kept in PBS. Tissue was minced using scissors and digested in trypsin supplemented with 100 μg/ml Liberase TM (Sigma-Aldrich) and 200 μg/ml Deoxyribonuclease I (Sigma-Aldrich) for 30 min incubation at 37°C 5% CO2. Organs were then resuspended in DMEM containing 5% FCS, pipetted up-and down and passed through a 40 μM strainer. Cells were then washed in FACS Buffer (1g/l sodium azide, 10 g/l PBS powder, 2.5mM EDTA, 1% FCS in MilliQ filtered water). Fixation was performed using 2% formalin in PBS for 10 minutes at room temperature. Cells were washed twice in permeabilization buffer (1mg/ml Saponin in FACS buffer). Cells were then stained with Chromotek GFP- and RFP booster for 30 minutes at 4°C. A final wash in permeabilization buffer was performed before FACS analysis.
In vivo colonization and IVIS Bioluminescence Imaging
PC9 WT or BAX, BAK, BOK TKO cells stably expressing tdTomato and firefly luciferase were used as PT. Another batch of the same cells were treated with ABT737 1.5 μM and S63845 3 μM for 6 h followed by wash and release into complete medium for 2 h, to generate PS. EasySep Dead Cell Removal (Annexin V) kit (Stemcell Technologies) was used to harvest live cells according to the manufacturer’s protocol. PT and PS were injected into separate (0.4 ×106cells per mouse) into the teil vein of NSG mice (200μl in DMEM). Optical imaging of Bioluminescence and/or Fluorescence was performed by using IVIS Spectrum or IVIS-200 imaging systems. 5–10 minutes prior to imaging, animals were injected i.p. with D-Luciferin (15 mg/ml in sterile saline) at a dose of 150 mg/kg (10 μL/g of body weight). After administration, animals were anesthetized using Isoflurane (1.5–2% delivered in 100% O2 at 1 l/min) and maintained via nosecone on heated imaging bed within the system for the duration of the scan. Following imaging, animals were allowed to recover on a heating blanket under observation and supplemented with oxygen as required.
Orthotopic lung tumors (conventional, non-surgical approach) and FACS analysis
Cells were treated with ABT737/S63845 (PC9 1.5/3 μM, A549 5/10 μM) 4–6h followed by wash and release into complete medium for 2 h, to generate PS. EasySep Dead Cell Removal (Annexin V) kit (Stemcell Technologies) was used to harvest live cells according to the manufacturer’s protocol. PT were stained with CFSE and PS with CTV label retention dye (Thermo Scienfic) according to the manufacturer’s protocol, washed twice in complete medium before further processing. Single injection tumor cell suspensions with combinations of PT and PS in Matrigel were prepared in one-milliliter syringes with 30-gauge hypodermic needles for an injection volume of 50 μl (2 ×106 total cells per mouse). Mice were anesthetized with isoflurane and placed in right lateral decubitus position. Local disinfection of the left posterior thorax was performed. Transdermal intrapulmonal injections have been performed at the posterior medial line juxta below the inferior angle of the scapula, through the intercostal space, 5–7 mm into the thorax. After injection mice were turned to the left lateral decubitus position and observed for approximately 15 min. Note: This non-surgical approach is comparable to the published surgical procedure for intrapulmonary injection (Isobe et al., 2013; Okimoto et al., 2017).
For ex vivo processing of lungs to assess metastatic ratio, left (primary injection site) and right (metastatic side) lungs of animals were resected and kept separately in PBS. Tissue was minced using scissors and digested in trypsin supplemented with 100 μg/ml Liberase TM (Sigma-Aldrich) and 200 μg/ml Deoxyribonuclease I (Sigma-Aldrich) for 30 min incubation at 37°C 5% CO2. Organs were then resuspended in DMEM containing 5% FCS, pipetted up-and down and passed through a 40μM strainer.
Cells were stained for surface markers CD45-APC (in initial experiments also CD31-PECy7) for 20 minutes at 4°C. Erythrocyte lysis (NH4Cl2 150mM, KHCO3 10mM, EDTA 0.1mM, pH7.2) was performed at room temperature for 2 minutes. Cells were then washed in FACS Buffer (1g/l sodium azide, 10 g/l PBS powder, 2.5mM EDTA, 1% FCS in MilliQ filtered water) before FACS analysis.
For analysis of circulating tumor cells (CTC) 300 μl blood was collected from the right heart ventricle after mice had been euthanized and collected into vessels containing 30 μl of EDTA (Invitrogen). 100 μl of blood sample per mouse was stained for surface marker CD45-APC for 20 minutes at 4°C. Erythrocyte lysis (NH4Cl2 150mM, KHCO3 10mM, EDTA 0.1mM, pH7.2) was performed at room temperature for 2 minutes. Cells were then washed in FACS Buffer (1g/l sodium azide, 10 g/l PBS powder, 2.5mM EDTA, 1% FCS in MilliQ filtered water) before FACS analysis.
Transient transfection
DNA and siRNA transient transfection was performed over 48 h using Viafect™ (Promega) for HEK, A549 and HT29, FugeneHDR (Promega) for PC9, A549 and HCT116 or Lipofectamine™ 3000 (Invitrogen) for any cell line, and Lipofectamine™ RNAiMAX transfection reagents, respectively. Cells were transfected at 50–75% confluence in serum-free Opti-MEM (Invitrogen) as per the manufacturers’ instructions. The siRNA oligos used were either purchased from Dharmacon as ON-TARGETplus siRNA pools of 4 oligos or from IDT as single siRNAs (see Supplementary Table 4).
Generation of Cell lines
For stable transfections, cells were transfected with a modified Piggybac expression system (Magnúsdóttir et al., 2013) for continuous expression. To remove the inducible expression promoter, we replaced the preexisting 6× Tet-Operators with a CMV promoter and placed the gene of interest under its control. Additionally, another variant of this backbone has been established by replacement of the puromycin resistance gene with zeocin or blasticidin resistance. Stable transfections were then performed by transfection of cells with the expression vector pPB together with a vector expressing the PiggyBac transposase which integrates the expression cassette. Stable transductants were selected after adding 200 μg/mL zeocin™ (Invitrogen), 0.2 μg/mL puromycin (Sigma-Aldrich) or 20 μg/mL blasticidin (Invitrogen) with replacement of the medium every 2 days for max. 14 days or were sorted by flow cytometry for Cerulean-, Venus-, GFP-, or mCherry-positive cells.
Generation of knock out cells was performed by usage of the CRISPR-Cas9 technology. The different lentiviral vectors used are either carrying Cas9 sequence and sgRNA sequence (CRISPRv2 GFP, Addgene, a gift from David Feldser; Px458 pSpCas9(BB)-2A-GFP, Addgene, a gift from Feng Zhang, pSpCas9(BB)-2A-mCherry) or sgRNA carrying vectors (pLKO5.sgRNA.EFS.PAC, Addgene a gift from Benjamin Ebert) were transfected simultaneously with Cas9 expressing vectors mentioned above (Heckl et al., 2014; Ran et al., 2013; Walter et al., 2017). The primer sequences for sgRNAs are listed in Supplementary Table S3 (Lopez et al., 2016; Shalem et al., 2014). Transiently transfected cells were selected after 0.2 μg/mL puromycin (Sigma-Aldrich) or 20 μg/mL blasticidin (Invitrogen) with replacement of the medium every 2 days for max. 10 days or were sorted by flow cytometry for GFP-, or mCherry-positive cells. Single clones were selected and expanded to generate BAX, BAK, BOK triple knock-out cells and CYCS-deficient cells.
Flow Cytometry
For intracellular staining cells were fixed and permeabilized according to the manufacturer’s instructions using Intracellular Fixation & Permeabilization Buffer Set (BD). To monitor Cytochrome c release, cells were incubated with 20μg/ml digitonin in PBS (Sigma-Aldrich) for 10 minutes on ice before fixation and further staining procedure. For nuclear staining of ATF4, the Foxp3 Fixation/Permeabilization (eBioscience) was used according to the manufacturer’s protocol.
Measurement of cellular lipid peroxidation was performed using BODIPY 581/591 C11 (Thermo Fisher). PT and PS have been treated in 12-well plates with 2μM RSL3 overnight in the presence or absence of 40 μM Deferoxamine mesylate salt (DFO). Cells were collected in Hank’s Balanced Salt Solution (HBSS; Gibco) and incubated with 2 μM BODIPY 581/591 C11 for 20 min at 37 °C. Cells were then washed twice in HBSS and analyzed via FACS.
Data acquirement and analysis was performed in a spectral analyzer (CYTEK Aurora 5-laser and 3-Laser) and data was processed using FlowJo software (Tree Star).
IncuCyte Analysis
Cell-death kinetics were monitored by the IncuCyte S3 imaging system (Essen Bioscience). Dead cells were stained with 400 ng/ml propidium iodide (PI) or 25 nM SYTOX Green (Invitrogen). SYTOX Green- or PI-positive cells were quantified by the IncuCyte image analysis software (Essen Bioscience). Data were expressed as positive events per well. Error bars represent the SD from the mean for triplicate samples.
Cytosolic extracts
Cell-free (cytosolic) extracts were generated as previously described (Liu et al., 1996). In our studies we followed the detailed protocol that is provided by the Cold Spring Harbor Protocols (McStay and Green, 2014). Ten 15cm dishes of confluent cells, were washed with ice-cold PBS. Cells were then scraped and harvested into 50ml conical tubes filled with ice-cold PBS. After centrifugation (at 250g for 5min. at 4°C), the supernatant has been removed and the pellet volume has been estimated. An equal volume of homogenization buffer (10mM HEPES pH7, 5mM MgCl2, 0.67 mM DTT, 1 protease inhibitor cocktail tablet (Roche)) has been used to completely resuspend the cells with a 1ml pipette tip. After an incubation of 15 min on ice, the cell suspension has been passed 10 times through a 22-gauge needle using a 3ml syringe. Testing aliquots of the lysate, this procedure has been repeated until more than 80% of nuclei stained blue in Trypan blue solution. Then, the homogenized cell suspension has been centrifuged at 15,000g for 30min 4°C in a 1.5ml microcentrifuge tube, the supernatant has been transferred to a new tube, and the same centrifugation step has been repeated. Next, the supernatant has again been transferred to a new tube and centrifuged at 100,000g for 1h 4°C. Finally, the supernatant has been passed through a 0.22μm centrifugal filter. Protein concentration has been determined by measuring absorbance at 280nm.
HRI eIF2α kinase assay
For kinetic analyses of eIF2α phosphorylation, HRI (25 nM) with or without 100nM Cytochrome c was incubated in kinase buffer (20 mM Tris buffer pH 7.4, 40 mM KCl, 3 mM magnesium acetate and 1 mM DTT) at room temperature for 15 min. A titration of 0.05–12 μg eIF2α was prepared in assay buffer and supplemented with 50 μM ATP. The reaction was initiated by adding the substrate mix to the enzyme mix and then incubated at room temperature for 5 min (initial velocity conditions). The HRI kinase reaction rate using 25 nM HRI is linear for the first 20 min (Figure S5C) as previously published (Guo et al., 2020). The amount of reaction product (phosphorylated eIF2α) formed during the first 5 min was used as the initial velocity. Kinetic constants were determined using least-squares fit with Prism v.9.
qRT-PCR
mRNA for qRT-PCR were extracted using the RNeasy Mini Kit (Qiagen). Reverse transcription reactions were performed with M-MLV reverse transcriptase (Invitrogen) following the manufacturer’s protocol and using random hexamers (IDT). The primer sequences for qPCR are listed in Supplementary Table S5 (Kim et al., 2010; Spandidos et al., 2008, 2010; Wang et al., 2012). Real-time PCR was performed with SYBR™ Green using the QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems).
Western blotting
Cells were either lysed in cell lysis buffer (50mM Tris-Cl pH 7.4, 150 mM NaCl, cOmplete® protease inhibitors cocktail (Roche), and 0.5% Nonidet P-40) or directly in sample buffer (BioRad) containing DTT. To detect phosphorylation, the cell lysis buffer was supplemented with phosSTOP® phosphatase inhibitor cocktail (Roche).
Immunoprecipitation
A549 cells were transiently transfected with HRI-mClover or mClover constructs. 24 h after transfection cells were treated with ABT737 1.5μM, S6 3μM and QvD 20μM for 4 h, washed in ice-cold PBS, trypsinized and again washed in PBS, followed by an incubation in 2% PFA/PBS for 15 minutes at room temperature, turning end-to-end. Then samples were quenched in ice-cold glycine/PBS (0.7M) for 5 minutes at room temperature again turning end-to-end and afterwards washed twice in ice-cold PBS. Immunoprecipitation was performed using GFP-Trap_MA beads (Chromotek, gtma20) according to the manufacturer’s instructions. Lysis was performed in 10mM Tris/Cl pH 7.5, 80mM NaCl, 0.5% NP40 substitute, Phosphatase-inhibitor (Roche) and EDTA-free Protease-inhibitor cocktail (Millipore Sigma). Lysate supernatants were incubated with beads at 4°C for 4h. Proteins captured on the magnetic beads were boiled in 2× SDS loading dye for 10 min before subjecting to SDS–PAGE and western blotting.
For in vitro incubation of HRI-mClover or mClover expressing A549 lysates with equine Cytochrome c, A549 cells were transiently transfected with constructs for 24h, washed in ice-cold PBS, trypsinized and again washed before lysis (same buffer as above). After lysis, supernatants were incubated with 100nm of equine Cytochrome c for 20 minutes at room temperature, followed by GFP-Trap_MA bead incubation at 4°C for 4h. IP was performed as described above.
For Co-immunoprecipitation of endogenous proteins, PC9 APAF1−/− cells were treated with ABT737 1.5 μM and S6 3 μM for 3.5h, then washed in ice-cold PBS, trypsinized and again washed. This was followed by an incubation in 2% PFA/PBS for 15 minutes at room temperature, turning end-to-end. Then samples were quenched in ice-cold glycine/PBS (0.7M) for 5 minutes at room temperature again turning end-to-end and afterwards washed twice in ice-cold PBS. Lysis was performed in 10mM Tris/Cl pH 7.5, 80mM NaCl, 0.5% NP40 substitute, Phosphatase-inhibitor (Roche) and EDTA-free Protease-inhibitor cocktail (Millipore Sigma). Lysate supernatants were incubated with either anti-Cytochrome c antibody (BD #556432, 20 μg) or normal mouse IgG (sc-2025, 20 μg) at 4°C overnight, followed by 2h incubation with Protein A/G PLUS-Agarose (sc-2003, Santa Cruz) at 4°C, and complexes were washed 5 times in cell lysis buffer. Proteins captured were eluted by boiling in 2× SDS loading dye for 10 min before subjecting to SDS–PAGE and western blotting.
Single cell RNA seq
PC9 cells were treated with ABT737 1.5 μM and S63845 3 μM for 6h and then washed and released in complete culture medium overnight (PS1D), 3 days (PS3D) or a week (PS7D) to obtain PS. EasySep Dead Cell Removal (Annexin V) kit (Stemcell Technologies) was used to harvest live cells from PT, PS1D, PS3D and PS7D according to the manufacturer’s protocol. The single cell suspension was resuspended in phosphate-buffered saline containing at the concentration of 1200 cell/ μl. A total of 20000 cells were loaded into each well of a Chromium single cell capture chip (Chromium Single Cell A Chip Kit, 10X Genomics). The captured single cells on droplets were processed for cell lysis, reverse transcription followed by library amplification (Chromium Single Cell 3’ Library and gel Bead Kit v2, 10X Genomics) and indexing as per the manufactures protocol.
RNAseq
For comparison of different treatments of PC9 cells total RNA was extracted from PT or PS which were generated by treatment with ABT737 1.5 μM and S63845 3 μM for 6 h, or Erlotinib 5 μM for 3 d or Paclitaxel 0.5 μM for 3 d (n=3/group).
RNA quality was assessed by 2100 Bioanalyzer RNA 6000 Nano assay (Agilent). Libraries were prepared using TruSeq Stranded mRNA kits (Illumina) and subjected to 100 cycle paired-end sequencing on the Illumina HiSeq platform.
scRNA-seq data processing
The libraries were sequenced on Illumina HiSeq with 500 million clusters per library. Cell Ranger (10X Genomics) (version 6.0.1) was used to process the raw sequencing data and align the reads to the GRCh38 reference genome. The count matrix was filtered as follows. All mitochondrial and ribosomal protein genes were removed, as were any genes expressed in 0.15% cells or less per library. Genes expressed at high level similar to mitochondrial and ribosomal genes were also removed, including MALAT1, FTL and FTH1. Cells were removed by individual libraries based on the number of genes per cell, proportion of reads mapped to mitochondrial and ribosomal genes (see supplementary table S2 for the threshold). Each library was normalized by dividing the UMI counts of each gene within a cell by the total number of UMIs in the cell and natural-log transformed using log1p function in R base package. A cell cycle score of each cell was calculated using CellCycleScoring function in Seurat package (Stuart et al., 2019) and cell cycle genes (Tirosh et al., 2016). The libraries were down-sampled to the same number of cells (3753 cells), merged and scaled by number of genes, proportion of reads mapped to mitochondrial genes, and cell cycle phases S and G2M scores. Principal component analysis (PCA) was run on the scaled data. The cells were projected onto a 2-dimension space of Uniform Manifold Approximation and Projection (UMAP) using the first 40 principal components of PCA as input and the parameters a=200, b=0.4 (other parameters were defaulted).
Trajectory analysis and differential expression along the pseudotime
Trajectory analysis was performed with Slingshot using UMAP projection as input, starting cells of PS1D sample, and approx_points=150 (Street et al., 2018). The analysis of gene expression along the trajectory was performed with tradeSeq R package followed its official documentation with default parameters (Berge et al., 2020); 299 genes were differentially expressed along the pseudotime testing against 2-fold change (l2fc=1) identified using associationTest function. The genes were clustered using hierarchical clustering method with the modelled gene expression (centered and scaled) and cut into 4 clusters.
Gene set score calculation
The gene set score measures the average gene expression level of given a gene set. It was calculated using AddModuleScore function of Seurat package with default parameters. The gene sets were defined based on bulk RNA-seq in this study (Figure 7N) or from MSigDB database (Liberzon et al., 2015) (Figure 7O).
Bulk RNA-seq data analysis
Paired-end sequencing reads were mapped by the pipeline of St Jude Center for Applied Bioinformatics. Basically, the reads were trimmed with Trim Galore (version 0.5.0) (Krueger et al., 2021) with default parameters. Then reads were aligned to the reference mouse hg38 assembly plus ERCC spike-in sequences using STAR (version 2.7.5a) (Dobin and Gingeras, 2015). The resulting alignments, recorded in BAM file, were sorted, indexed, and marked for duplicates with Picard MarkDuplicates function (version 2.19.0) (Broad-Institute, 2019). Transcript quantification was calculated using RSEM (Li and Dewey, 2011). To examine if there is a global change in gene expression, RUVg function in RUVSeq package was used for normalization followed by DESeq2 with reads mapped ERCC spike-in according to RUVSeq manual (Risso et al., 2014). No global change was found, and differential gene expression analysis was carried out using DESeq2 with standard procedure (normalized by the median-of-ratios method with default parameters (Love et al., 2014).
Enrichment analysis
Gene set enrichment analysis (GSEA) was performed against MSigDB database (Liberzon et al., 2015) with R package clusterProfiler (version 3.18.1) (Yu et al., 2012) (Figure 1J, 7A, S1J). Wald statistic calculated from DESeq2 was used for gene ranking. Over-representation enrichment was performed against MSigDB with R package clusterProfiler (version 3.18.1) (Figures 2E, 2F, S2F).
Gene expression and survival analysis
For HRI gene expression comparison between human normal and cancer samples we utilized RNA sequencing expression data of 9,736 tumors and 8,587 normal samples from the TCGA and the GTEx projects which were analyzed by the Webtool GEPIA2 (Tang et al., 2019). Same datasets and software were used to determine disease free survival and overall survival in dependence of HRI expression in various tumors estimated by Mantel-Cox test (Figure S7D, S7E).
Gene expression, survival and clinical information of TCGA LUAD cohort were downloaded from UCSC Xena (Goldman et al., 2020). EGFR-mutated lung adenocarcinomas were filter by sample_type of “Primary tumor” and egfr_mutation_performed of “YES”. Gene expression RNAseq (IlluminaHiSeq) was used, in which the expression value is log2(x+1) where x is the RSEM value. Survival analysis was performed in R (Figure 7P).
Kaplan Meier Plots have been generated by use of the online webtool Kaplan-Meier Plotter with univariate Cox regression analysis. Patients have been trichotomized into 1st and 4th quartile for HRI (217735_s_at) expression using the GSE51105 dataset for gastric cancer (Brasacchio et al., 2018; Busuttil et al., 2014; Szász et al., 2016), GSE3149 dataset for ovarian cancer (Bild et al., 2006; Győrffy et al., 2012) and TCGA dataset for renal papillary cell cancer (Nagy et al., 2021) (Figure S7F) and E-MTAB-365 dataset for breast cancer relapse-free survival (Figure S7G) (Guedj et al., 2012; Győrffy, 2021; Rème et al., 2013).
Pathology
The antibody used for immunohistochemistry corresponding to human normal and cancer tissue was polyclonal rabbit anti-HRI HPA016496 (Millipore Sigma). Images were obtained from the Human Protein Atlas (https://www.proteinatlas.org/ENSG00000086232-EIF2AK1).
Supplementary Material
Figure S1. Targeting anti-apoptotic Bcl-2 proteins induces persister cells, related to Figure 1 (A) Western Blot of WT and BAX, BAK, BOK TKO PC9 cells with the indicated antibodies.
(B) Western Blot of WT and BAX- and BAK-deficient PC9 cells with the indicated antibodies.
(C) Fractional viability of PC9 PT and PS after either 1d drug holiday (PS d1) or 7d drug holiday (PSd7) treated with ABT737 and S6. n=2 samples/condition. One representative of 2 independent experiments is shown.
(D) Metastatic ratio representing the ratio between cells in contralateral lung and primary injection site for CFSE-labelled A549 PT or CTV-labelled A549 PS cells respectively. N=5 mice. Paired t-test: *P< 0.05
(E) Fractional viability of HT29 PT and PS treated with MOCK, Oxaliplatin 10 μM, Irinotecan 4 μM, Etoposide 4 μM or Vemurafenib 20 μM for 3 d. n=3 samples/group. 2-way ANOVA: ****P< 0.0001; NS, not significant (P >0.05).
(F) Fractional viability of HT29 PT and PS treated with RSL3 for 3d. N=3 samples/group.
(G) IC50 values from fractional viability assays comparing PC9 PT and PS treated with titrations of RSL3 for 3d. n=7 independent experiments. Paired t test: ***P< 0.001.
(H) MFI of C11-Bodipy in PC9 PT and PS treated ± RSL3, in the presence or absence of DFO. n=3 samples/group. 2-way ANOVA: *P< 0.05, ***P< 0.001, ****P< 0.0001
(I) qPCR analysis of CHAC1 or GCLC transcripts representing glutathione metabolism in PC9 PT and PS. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. **P <0.01, ****P <0.0001.
(J) KEGG and Reactome pathways commonly enriched across treatment regimens. Enrichment was calculated by GSEA (see Methods). The commonly enriched pathways were defined as those that have false discovery rate (FDR) < 0.05 and the same direction in different treatments. Normalized enrichment scores (NES) are shown. PS were generated by treatment with ABT737 1.5 μM and S6 3 μM for 6 h, or Erlotinib 5 μM for 3 d or Paclitaxel 0.5 μM for 3 d.
Figure S2. The persister phenotype depends largely on Bcl-2 effectors, related to Figure 2 and 3 (A) IC50 values from fractional viability assays comparing untreated versus ABT737 (1.5 μM) and S6 (3 μM) pretreated BAX, BAK, BOK-deficient PC9 (TKO) treated with titrations of RSL3 for 3d. n=5 independent experiments. Paired t test: NS, not significant (P> 0.05).
(B) MFI of C11-Bodipy in untreated versus ABT737 (1.5 μM) and S6 (3 μM) pretreated BAX, BAK, BOK-deficient PC9 (TKO) treated ± RSL3, in the presence or absence of DFO. Same experiment as Figure S1H.
(C) qPCR analysis of CHAC1 or GCLC transcripts related to glutathione metabolism in untreated versus ABT737 (1.5 μM) and S6 (3 μM) pretreated BAX, BAK, BOK-deficient PC9 (TKO). N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. ns, not significant (P >0.05). Same experiment as Figure S1I.
(D) Heatmap of normalized count of the top 10 up- or down-regulated, differentially expressed genes (DEGs) in WT (PS/PT) but not in BAX, BAK, BOK TKO (ABT737, S6 treated/MOCK) PC9 cells. The normalized counts were scaled with root mean square (RMS) by gene and genotype.
(E) Scaled expression profiles of gene clusters along pseudotime. (same as Figure3)
(F) Enrichment analysis of differentially expressed genes (80 in cluster 1, 76 in cluster 2, 127 in cluster 3, 16 in cluster 4) in Reactome, Hallmark or C2:Chemical and genetic perturbations (C2:CGP) collections. Enrichment analysis was performed using the over-representation approach.
Figure S3. BH3 mimetics induce an ISR, dependent on Bcl-2 effectors and Imomp but independent of caspase activation, related to Figures 4 and 5. (A) Double-luciferase assay of PC9 cells expressing ATF4 translation reporter upon treatment with ABT737 1.5 μM and S6 3 μM in combination ± 2BAct 50 μM for indicated times. N=5 samples per group and timepoint.
(B) Double-luciferase assay of WT or BAX, BAK, BOK TKO PC9 cells expressing ATF4 translation reporter and treated with Tunicamycin 10 μM, ISRIB 100 μM and 2BAct 50 μM overnight. N=8 samples/group.
(C) Immunoblot of PC9 cells transiently transfected with Cas9 and non-targeting or HRI targeting sgRNAs.
(D) Cytofluorimetric analysis of mean fluorescence intensity (MFI) of ATF4 (AF647) in siNT or siBAX and siBAK transfected A549 cells treated ± ABT737 5 μM and S6 10 μM. (n=4/group; 2-way ANOVA: **P <0.01)
(E) Metastatic ratio representing the ratio between cells from the contralateral lung and primary injection site. For quantification cytofluorimetric analysis of CFSE-labelled PT and CTV-labelled PS (generated by treatment with Raptinal) from BAX, BAK, BOK TKO PC9 cells was performed 2 d after intrapulmonal combined injection (n=10 mice/group).
(A and B) Statistical analysis was performed using 2-way ANOVA. *P <0.05; ****P <0.0001; NS, not significant (P> 0.05).
Figure S4. Cytochrome C activates HRI and engages the persister phenotype, related to Figure 6 (A) FACS histograms and (B) fold change quantification of Cytochrome c release PC9 cells treated with indicated siRNAs and ± ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM for 4 h.
(C) Double-luciferase assay of PC9 cells expressing ATF4 translation reporter and silenced with indicated siRNAs before treatment with ABT737 1.5 μM and S6 3 μM for 6.5 h. N=4 samples/group. 2way ANOVA: ****P< 0.0001, ***P< 0.001, *P< 0.05; NS, not significant (P> 0.05).
(D) Left: Immunoblot with the indicated antibodies of cytosolic extracts from PC9 cells transiently transfected with sgNT or sgHRI, treated with serial dilutions of Cytochrome c (3–50 μM). Right: Quantification by fold change of normalized samples. One representative of 2 similar experiments shown.
(E) FACS blots of PC9 cells treated with either sgNT (non-targeted) or sgCYCS to generate stable knock-out cells by use of CRISPR/Cas9 and reconstituted with indicated pCYCS WT or pCYCSK72R vectors. Mitochondrial Cytochrome c was measured after 4 h treatment with MOCK control or a combination of ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM.
(F) Percentage of Cytochrome c positive cells from (E) ± treatment with ABT737 1.5 μM, S6 3 μM and Q-VD-OPh for 6.5 h. n=6 samples/group.
(G) Incucyte analysis of PI positive CYCS−/− cells that had been reconstituted with either pCYCS WT or pCYCSK72R, and treated ± ABT737 1.5 μM and S6 3 μM. n=4 samples/group.
(H) Experimental schematic and (I) western blot of A549 cells expressing the CDS of K72R mutated CYCS, and were treated with or without a combination of ABT737 5 μM, S6 10 μM and Q-VD-OPh 20 μM for 4h.
Figure S5. Cytochrome C activates HRI and engages the persister phenotype, related to Figure 6 (A) PT and PS generated using ABT737 1.5 μM and S6 3 μM of PC9 CYCS−/− or (B) PC9 CYCS−/− reconstituted with WT CYCS. Fractional viability was determined after 3 d treatment with Mock, Erlotinib 5 μM, Paclitaxel 1 μM or Cisplatin 10 μM. N=3 samples/group. 2-way ANOVA: **P <0.01; ****P <0.0001; NS, not significant (P> 0.05).
(C) qPCR analysis of transcripts that have been identified as BAX, BAK-dependent in PT and PS generated from PC9 CYCS−/− cells, reconstituted with or without WT CYCS. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. ***P <0.001; ****P <0.0001.
(D) Immunoblot and densitometric quantitation of hosphor-eIF2α from in vitro kinase reaction with recombinant eIF2α 12 μg and HRI 25 nM, in the presence of ATP 50 μM incubated for indicated times (30 seconds to 20 minutes) showing initial velocity conditions. One representative of 2 independent experiments shown.
(E) Immunoprecipitation of mClover in HRI-mClover- or mClover-transfected A549 cells after treatment with ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM for 4 h.
(F) Immunoprecipitation of HRI-mClover or mClover from transfected A549 cells after in vitro, post lysis incubation with equine cytochrome C (100nm).
Figure S6. Cytochrome c - HRI - ATF4 axis drives the phenotype of persistence, related to Figure 7 (A) Slingshot trajectory showing log-transformed counts and fitted values for indicated genes along pseudotime. P-value was calculated from association test in tradeSeq package. Data from single cell RNAseq of PT and PS PC9 cells (see legend, Figure 3).
(B) AnnexinV staining of HCT116 WT and ATF4−/− cells treated with or without ABT737 10 μM and S6 20 μM. N=4 samples, mean bar ± SD. Significance was calculated using 2-way ANOVA. ***p < 0.001.
(C) Fractional viability of A549 cells that have been transfected with siNT, siATF4, siHMOX1 or siNRF2, and treated with titration of a combination of ABT737 and S6 overnight. One representative of 2 independent experiments shown.
(D) Immunoblot with the indicated antibodies of PC9 cells that have been transfected with siNT, siATF4, siHMOX1 or siNRF2.
(E) Immunoblot with the indicated antibodies of PC9 cells after CRISPR/Cas9 knockout (KO) of ATF4 or using non-targeted sgRNA (sgNT) treated with or without ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM.
(F) Immunoblot with the indicated antibodies of PT and PS generated from PC9 (ABT737 1.5μM, S6 3μM), H226 (ABT737 2.5μM, S6 5μM), A549 (ABT737 5μM, S6 10μM) and HT29 (ABT737 4μM, S6 8μM) overnight.
(G) Immunoblot with the indicated antibodies of PC9 cells that have been transfected with siNT, siBAX, siBAK or siBOK.
(H) PC9 cells were transfected with siNT or siBAX, for 48 h, following which they were treated with the indicated drugs for 48h. Fractional viability (mean ± S.D.) is shown. One representative of 3 independent experiments with n=8 samples/group.
(I) qPCR analysis of BAX transcript in PT and ABT737 (1.5 μM) and S6 (3 μM) treated PC9 CYCS−/− cells, reconstituted with or without WT CYCS. N=3 replicate samples are shown.
(J) IC50 values from fractional viability assays comparing PT versus ABT737 (1.5 μM) and S6 (3 μM) pretreated ATF4 KO or (K) HRI KO PC9 cells, treated with titrations of RSL3 for 3d. n=7 (ATF4 KO), n=6 (HRI KO) independent experiments. Paired t test: ns, not significant (P> 0.05).
(L) MFI of C11-Bodipy in PC9 WT, ATF4 KO or HRI KO PT and ABT737 (1.5 μM) and S6 (3 μM) pretreated cells, treated with or without RSL3. 2-way ANOVA: **P< 0.01, ****P< 0.0001, ns, not significant (P> 0.05). n=3 samples per group.
(M) qPCR analysis of CHAC1 or GCLC transcripts representing glutathione metabolism in PT versus ABT737 (1.5 μM) and S6 (3 μM) treated PC9 ATF4 KO or (N) HRI KO cells. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. ns, not significant (P >0.05).
(O) Fractional viability and (P) IC50 values from fractional viability assays of PC9 cells transfected with siNT or siBAX/BAK or siATF4 or siHRI respectively treated with RSL3 for 3d. n=3 independent experiments. Paired t test: ns, not significant (P> 0.05).
(Q) Double-luciferase assay of PC9 WT or BAX, BAK, BOK TKO or CASPASE9 KO cells expressing ATF-4 translation reporter upon treatment with Tunicamycin in combination with or without 2BAct for 6,5h. N=4 samples per group and timepoint.
(R) Fractional viability of PC9 PT and PS generated by Tunicamycin treatment and then treated with MOCK, Erlotinib 10 μM, Paclitaxel 1 μM or Cisplatin (CPD) 10 μM 3 d. n=4 samples per group. 2-way ANOVA: ***P< 0.001, ****P<0.0001; ns, not significant (P> 0.05).
(S) Fractional viability of PC9 WT or BAX, BAK, BOK TKO PT and PS generated by Tunicamycin treatment and then treated with RSL3 3 d. n=4 samples per group. 2-way ANOVA: ****P< 0.0001; ns, not significant (P> 0.05).
Figure S7. Cytochrome c - HRI - ATF4 axis drives the phenotype of persistence, related to Figure 7 (A) Immunohistochemistry showing HRI expression in representative examples of human normal and associated cancer tissues. Scale bars, 100 μm.
(B) List of cancer name abbreviations used in (C) and (D) and number of subjects analyzed.
(C) HRI expression in various tumors (T, red dots) compared to normal tissue (N, green dots). Red cancer name abbreviations indicate significant changes between normal and tumor (ANOVA).
(D) Dependence of disease-free survival and (E) overall survival on HRI expression in various tumors estimated by Mantel-Cox analysis. Hazard ratio (HR) shown (red = worse outcome), red squares statistically significant p<0.05 (ANOVA).
(F) Kaplan Meier overall survival curve of patients with gastric cancer (left, dataset GSE51105), ovarian cancer (middle, dataset GSE3149) and renal papillary cell cancer (right, dataset TCGA) belonging to high (4th quartile) or low (1st quartile) of HRI expression. Hazard ratio (HR) and p-value are indicated.
(G) Kaplan Meier relapse-free survival curve of patients with HER2+ breast cancer (dataset E-MTAB-365) belonging to high (4th quartile) or low (1st quartile) of HRI expression. Hazard ratio (HR) and p-value are indicated.
SUPPLEMENTARY TABLE S3. sgRNA sequences. Related to the STAR methods section.
SUPPLEMENTARY TABLE S4. siRNA sources. Related to the STAR methods section.
SUPPLEMENTARY TABLE S5. qPCR Primer sequences. Related to the STAR methods section.
Highlights.
Sublethal BH3 mimetic treatment generates persisters in caspase-independent manner
The persister phenotype implements a heightened metastatic potential
Nuanced iMOMP and Cytochrome c release function as rheostat for ATF4 activation
The persister phenotype is orchestrated by the Integrated Stress Response and ATF4
ACKNOWLEDGEMENTS
The authors thank Richard Cross and Greig Lennon (SJCRH) for technical assistance and Aseem Ansari, Paul Thomas, Cliff Guy, Gustavo Palacios, Shelbi Christgen, Diego Rodriguez, Joelle Magne, Emilio Boada Romero (SJCRH), Jane-Jane Chen (MIT), and Cristina Muñoz Pinedo (Idibell, Barcelona) for thoughtful insights and discussions. We also thank the Hartwell Center for RNA sequencing and the Center for In Vivo Imaging and Therapeutics (supported by SJCRH, NCI R50 CA211481, and NCI P30 CA021765) for preclinical imaging (SJCRH). This work was supported by ALSAC (SJCRH), and by the U.S. National Cancer Institute grant R35 CA231620 to D.R.G. and the German Research Foundation (DFG, KA 4830/1-1) to H.K.
Original Piggybac vectors were provided by Kazuhiro Murakami (RIKEN, Kobe, Japan). As indicated, some of the results published here are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests. D.R.G. consults for Ventus Therapeutics, Inzen Therapeutics, and Horizon Therapeutics.
STATISTICAL ANALYSIS
Please refer to the legend of the figures for description of sample sizes and statistical tests performed. Data were plotted and analyzed with GraphPad Prism 9.0 software. Differences were considered statistically significant when the p-value was less than 0.05, and otherwise not significant (ns).
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Douglas Green (douglas.green@stjude.org).
REFERENCES
- Bauer BN, Rafie-Kolpin M, Lu L, Han A, and Chen J-J (2001). Multiple Autophosphorylation Is Essential for the Formation of the Active and Stable Homodimer of Heme-Regulated eIF2α Kinase †. Biochemistry-Us 40, 11543–11551. 10.1021/bi010983s. [DOI] [PubMed] [Google Scholar]
- B’chir W, Maurin A-C, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, and Bruhat A (2013). The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41, 7683–7699. 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge K.V. den, Bézieux H.R. de, Street K, Saelens W, Cannoodt R, Saeys Y, Dudoit S, and Clement L (2020). Trajectory-based differential expression analysis for single-cell sequencing data. Nat Commun 11, 1201. 10.1038/s41467-020-14766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlanga JJ, Herrero S, and Haro C. de (1998). Characterization of the Hemin-sensitive Eukaryotic Initiation Factor 2α Kinase from Mouse Nonerythroid Cells*. J Biol Chem 273, 32340–32346. 10.1074/jbc.273.48.32340. [DOI] [PubMed] [Google Scholar]
- Berthenet K, Ferrer C, Fanfone D, Popgeorgiev N, Neves D, Bertolino P, Gibert B, Hernandez-Vargas H, and Ichim G (2020). Failed Apoptosis Enhances Melanoma Cancer Cell Aggressiveness. Cell Reports 31, 107731. 10.1016/j.celrep.2020.107731. [DOI] [PubMed] [Google Scholar]
- Bhola P, and Letai A (2016). Mitochondria—Judges and executioners of cell death sentences. Mol. Cell 61, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialk P, Wang Y, Banas K, and Kmiec EB (2018). Functional Gene Knockout of NRF2 Increases Chemosensitivity of Human Lung Cancer A549 cells in vitro and in a Xenograft Mouse Model. Mol Ther - Oncolytics 11, 75–89. 10.1016/j.omto.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi M-B, Harpole D, Lancaster JM, Berchuck A, et al. (2006). Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439, 353–357. 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Brasacchio D, Busuttil RA, Noori T, Johnstone RW, Boussioutas A, and Trapani JA (2018). Down-regulation of a pro-apoptotic pathway regulated by PCAF/ADA3 in early stage gastric cancer. Cell Death Dis 9, 442. 10.1038/s41419-018-0470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad-Institute (2019). Picard toolkit (http://broadinstitute.github.io/picard/).
- Busuttil RA, George J, Tothill RW, Ioculano K, Kowalczyk A, Mitchell C, Lade S, Tan P, Haviv I, and Boussioutas A (2014). A Signature Predicting Poor Prognosis in Gastric and Ovarian Cancer Represents a Coordinated Macrophage and Stromal Response. Clin Cancer Res 20, 2761–2772. 10.1158/1078-0432.ccr-13-3049. [DOI] [PubMed] [Google Scholar]
- Caserta TM, Smith AN, Gultice AD, Reedy MA, and Brown TL (2003). Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 8, 345–352. 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, and Parada LF (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colell A, Ricci J-E, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, et al. (2007). GAPDH and Autophagy Preserve Survival after Apoptotic Cytochrome c Release in the Absence of Caspase Activation. Cell 129, 983–997. 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- Condon KJ, Orozco JM, Adelmann CH, Spinelli JB, Helm P.W. van der, Roberts JM, Kunchok T, and Sabatini DM (2021). Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc Natl Acad Sci USA 118, e2022120118. 10.1073/pnas.2022120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368, eaat5314. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford RR, Prescott ET, Sylvester CF, Higdon AN, Shan J, Kilberg MS, and Mungrue IN (2015). Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element*. J Biol Chem 290, 15878–15891. 10.1074/jbc.m114.635144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, Tuttle SW, Feldman MD, Zhang PJL, Fuchs SY, et al. (2015). ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest 125, 2592–2608. 10.1172/jci78031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, and Gingeras TR (2015). Mapping RNA-seq Reads with STAR. Curr Protoc Bioinform 51, 11.14.1–11.14.19. 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, Miller C, Demonacos C, Stratford IJ, and Dive C (2004). Hypoxia-Mediated Down-Regulation of Bid and Bax in Tumors Occurs via Hypoxia-Inducible Factor 1-Dependent and -Independent Mechanisms and Contributes to Drug Resistance. Mol Cell Biol 24, 2875–2889. 10.1128/mcb.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Amgalan D, Singh R, Wei J, Wen J, Wei TP, McGraw TE, Kitsis RN, and Pessin JE (2018). SNAP23 regulates BAX-dependent adipocyte programmed cell death independently of canonical macroautophagy. J Clin Invest 128, 3941–3956. 10.1172/jci99217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E, Eckl E-M, Schmitt S, Mancilla I, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, and Jae LT (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 1–5. 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DB, Garcia NG, McKinney BJ, Lupo R, Noteware LC, Newcomb R, Liu J, Locasale JW, Hirschey MD, and Alvarez JV (2020). NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat Metabolism 2, 318–334. 10.1038/s42255-020-0191-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Jiménez C, and Goding CR (2019). Starvation and Pseudo-Starvation as Drivers of Cancer Metastasis through Translation Reprogramming. Cell Metab 29, 254–267. 10.1016/j.cmet.2018.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, and Kroemer G (2006). Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 13, 1423–1433. 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38, 675–678. 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J-J, Zhang X-D, Sun W, Qi L, Wu J-C, and Qin Z-H (2015). DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis 6, e1624–e1624. 10.1038/cddis.2014.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj M, Marisa L, Reynies A. de, Orsetti B, Schiappa R, Bibeau F, MacGrogan G, Lerebours F, Finetti P, Longy M, et al. (2012). A refined molecular taxonomy of breast cancer. Oncogene 31, 1196–1206. 10.1038/onc.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin Y-HT, Wiita AP, Xu K, Correia AM, and Kampmann M (2020). Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 579, 427–432. 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Győrffy B (2021). Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnology J 19, 4101–4109. 10.1016/j.csbj.2021.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Győrffy B, Lánczky A, and Szállási Z (2012). Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr-Relat Cancer 19, 197–208. 10.1530/erc-11-0329. [DOI] [PubMed] [Google Scholar]
- Haefen C. von, Gillissen B, Hemmati PG, Wendt J, Güner D, Mrozek A, Belka C, Dörken B, and Daniel PT (2004). Multidomain Bcl-2 homolog Bax but not Bak mediates synergistic induction of apoptosis by TRAIL and 5-FU through the mitochondrial apoptosis pathway. Oncogene 23, 8320–8332. 10.1038/sj.onc.1207971. [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Duncan GS, Chang C-C, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, et al. (2005). Specific Ablation of the Apoptotic Functions of Cytochrome c Reveals a Differential Requirement for Cytochrome c and Apaf-1 in Apoptosis. Cell 121, 579–591. 10.1016/j.cell.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, and Ebert BL (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol 32, 941–946. 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer S, Knoll G, Schulze-Osthoff K, and Ehrenschwender M (2019). Raptinal bypasses BAX, BAK, and BOK for mitochondrial outer membrane permeabilization and intrinsic apoptosis. Cell Death Dis 10, 556. 10.1038/s41419-019-1790-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Axten JM, and Patterson JB (2019). Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol 15, 764–775. 10.1038/s41589-019-0326-2. [DOI] [PubMed] [Google Scholar]
- Ichim G, and Tait SWG (2016). A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 16, 539–548. 10.1038/nrc.2016.58. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Murase M, Iizuka A, Pichierri F, Martinkova M, and Shimizu T (2008). Elucidation of the Heme Binding Site of Heme-regulated Eukaryotic Initiation Factor 2α Kinase and the Role of the Regulatory Motif in Heme S ensing by Spectroscopic and Catalytic Studies of Mutant Proteins. J Biol Chem 283, 18782–18791. 10.1074/jbc.m801400200. [DOI] [PubMed] [Google Scholar]
- Inguaggiato P, Gonzalez-Michaca L, Croatt AJ, Haggard JJ, Alam J, and Nath KA (2001). Cellular overexpression of heme oxygenase-1 up-regulates p21 and confers resistance to apoptosis. Kidney Int 60, 2181–2191. 10.1046/j.1523-1755.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- Isobe T, Onn A, Morgensztern D, Jacoby JJ, Wu W, Shintani T, Itasaka S, Shibuya K, Koo PJ, O’Reilly MS, et al. (2013). Evaluation of Novel Orthotopic Nude Mouse Models for Human Small-Cell Lung Cancer. J Thorac Oncol 8, 140–146. 10.1097/jto.0b013e3182725ff9. [DOI] [PubMed] [Google Scholar]
- Kale J, Osterlund EJ, and Andrews DW (2018). BCL-2 family proteins: changing partners in the dance towards death. Cell Death and Differentiation 25, 65–80. 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkavan H, and Green DR (2018). MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25, 46–55. 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-H, Jeong J-Y, Surh Y-J, and Kim K-W (2010). Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res 38, 48–59. 10.1093/nar/gkp865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Toumelin-Braizat G, Chanrion M, Kelly GL, Gong J-NN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- Krueger F, James F, Ewels P, Afyounian E, and Schuster-Boeckler, & B. (2021). FelixKrueger/TrimGalore: v0.6.7 - DOI via Zenodo (0.6.7). Zenodo. [Google Scholar]
- Kuida K, Haydar TF, Kuan C-Y, Gu Y, Taya C, Karasuyama H, Su MS-S, Rakic P, and Flavell RA (1998). Reduced Apoptosis and Cytochrome c–Mediated Caspase Activation in Mice Lacking Caspase 9. Cell 94, 325–337. 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, and Newmeyer DD (2009). Caspaseindependent Mitochondrial Cell Death Results from Loss of Respiration, Not Cytotoxic Protein Release. Mol Biol Cell 20, 4871–4884. 10.1091/mbc.e09-07-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics 12, 323–323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, and Williams RS (2000). Cytochrome c Deficiency Causes Embryonic Lethality and Attenuates Stress-Induced Apoptosis. Cell 101, 389–399. 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim C, Yang J, Jemmerson R, and Wang X (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. [DOI] [PubMed]
- Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, and Li C-Y (2015). Caspase-3 Promotes Genetic Instability and Carcinogenesis. Mol Cell 58, 284–296. 10.1016/j.molcel.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J, Bessou M, Riley JS, Giampazolias E, Todt F, Rochegüe T, Oberst A, Green DR, Edlich F, Ichim G, et al. (2016). Mito-priming as a method to engineer Bcl-2 addiction. Nat Commun 7, 10538. 10.1038/ncomms10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Biorxiv 002832. 10.1101/002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir E, Dietmann S, Murakami K, Günesdogan U, Tang F, Bao S, Diamanti E, Lao K, Gottgens B, and Surani MA (2013). A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat Cell Biol 15, 905–915. 10.1038/ncb2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, Yu EA, Schenk EL, Tan W, Zee A, et al. (2020). Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 182, 1232–1251.e22. 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay GP, and Green DR (2014). Preparation of Cytosolic Extracts and Activation of Caspases by Cytochrome c. Cold Spring Harb Protoc 2014, pdb.prot080275. 10.1101/pdb.prot080275. [DOI] [PubMed] [Google Scholar]
- Melber A, and Haynes CM (2018). UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28, 281–295. 10.1038/cr.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, and Strasser A (2018). BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 34, 879–891. 10.1016/j.ccell.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Miksanova M, Igarashi J, Minami M, Sagami I, Yamauchi S, Kurokawa H, and Shimizu T (2006). Characterization of Heme-Regulated eIF2α Kinase: Roles of the N-Terminal Domain in the Oligomeric State, Heme Binding, Catalysis, and Inhibition. Biochemistry-Us 45, 9894–9905. 10.1021/bi060556k. [DOI] [PubMed] [Google Scholar]
- Miles MA, and Hawkins CJ (2017). Executioner caspases and CAD are essential for mutagenesis induced by TRAIL or vincristine. Cell Death Dis 8, e3062–e3062. 10.1038/cddis.2017.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Á, Munkácsy G, and Győrffy B (2021). Pancancer survival analysis of cancer hallmark genes. Sci Rep-Uk 11, 6047. 10.1038/s41598-021-84787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA (2011). The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu Rev Cell Dev Bi 27, 347–376. 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- Niture SK, and Jaiswal AK (2012). Nrf2 Protein Up-regulates Antiapoptotic Protein Bcl-2 and Prevents Cellular Apoptosis. Journal of Biological Chemistry 287, 9873–9886. 10.1074/jbc.M111.312694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Ichim G, and Tait SWG (2016). Mitochondria and Cell Death. In Mitochondria and Cell Death, Hockenbery DM, ed. (Humana Press; ), pp. 213–226. [Google Scholar]
- Okimoto RA, Breitenbuecher F, Olivas VR, Wu W, Gini B, Hofree M, Asthana S, Hrustanovic G, Flanagan J, Tulpule A, et al. (2017). Inactivation of Capicua drives cancer metastasis. Nat Genet 49, 87–96. 10.1038/ng.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren Y, Tsabar M, Cuoco MS, Amir-Zilberstein L, Cabanos HF, Hütter J-C, Hu B, Thakore PI, Tabaka M, Fulco CP, et al. (2021). Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582. 10.1038/s41586-021-03796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Reports 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli P, Giannoni E, and Chiarugi P (2013). Anoikis molecular pathways and its role in cancer progression. Biochimica Et Biophysica Acta Bba - Mol Cell Res 1833, 3481–3498. 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- Popgeorgiev N, Jabbour L, and Gillet G (2018). Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Frontiers Cell Dev Biology 6. 10.3389/fcell.2018.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouw HH, Langereis MA, Anand AA, Visser LJ, Groot R.J. de, Walter P, and Kuppeveld F.J.M. van (2019). Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc Natl Acad Sci USA 116, 2097–2102. 10.1073/pnas.1815767116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafie-Kolpin M, Han A-P, and Chen J-J (2003). Autophosphorylation of Threonine 485 in the Activation Loop Is Essential for Attaining eIF2α Kinase Activity of HRI. Biochemistry-Us 42, 6536–6544. 10.1021/bi034005v. [DOI] [PubMed] [Google Scholar]
- Ran AF, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature Protocols 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman SK, Haynes J, Collignon E, Brown KR, Wang Y, Nixon AML, Bruce JP, Wintersinger JA, Mer AS, Lo EBL, et al. (2021). Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 184, 226–242.e21. 10.1016/j.cell.2020.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rème T, Hose D, Theillet C, and Klein B (2013). Modeling risk stratification in human cancer. Bioinformatics 29, 1149–1157. 10.1093/bioinformatics/btt124. [DOI] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902. 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T, Milani M, Sin gleton DC, and Harris AL (2009). Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8, 3838–3847. 10.4161/cc.8.23.10086. [DOI] [PubMed] [Google Scholar]
- Saelens X, Kalai M, and Vandenabeele P (2001). Translation Inhibition in Apoptosis Caspase-dependent PKR Activation and eIF2-α Phosphorylation. J Biol Chem 276, 41620–41628. 10.1074/jbc.m103674200. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Vagner S, and Robert C (2020). Persistent Cancer Cells: The Deadly Survivors. Cell 183, 860–874. 10.1016/j.cell.2020.10.027. [DOI] [PubMed] [Google Scholar]
- Singh R, Letai A, and Sarosiek K (2019). Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Bio 20, 175–193. 10.1038/s41580-018-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, and Seed B (2008). A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance. Bmc Genomics 9, 633. 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, and Seed B (2010). PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 38, D792–D799. 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. Bmc Genomics 19, 477. 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, et al. (2013). Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 4, e838–e838. 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szász AM, Lánczky A, Nagy Á, Förster S, Hark K, Green JE, Boussioutas A, Busuttil R, Szabó A, and Győrffy B (2016). Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333. 10.18632/oncotarget.10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, and Green DR (2013). Mitochondrial Regulation of Cell Death. CSH Perspect Biol 5, a008706. 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait S, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Muñoz-Pinedo C, and Green DR (2010). Resistance to Caspase-Independent Cell Death Requires Persistence of Intact Mitochondria. Developmental Cell 18, 802–813. 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Chen X, Kang R, and Kroemer G (2021). Ferroptosis: molecular mechanisms and health implications. Cell Res 31, 107–125. 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Kang B, Li C, Chen T, and Zhang Z (2019). GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 47, W556–W560. 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Zhang S, Zhou L, Seyhan AA, Borrero LH, Zhang Y, and El-Deiry WS (2021). Targeting the Integrated Stress Response in Cancer Therapy. Front Pharmacol 12, 747837. 10.3389/fphar.2021.747837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan, and Wadsworth M (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, Gu Z, McCormick ML, Durham AB, Spitz DR, et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118. 10.1038/s41586-020-2623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter DM, Venancio OS, Buza EL, Tobias JW, Deshpande C, Gudiel AA, Kim-Kiselak C, Cicchini M, Yates TJ, and Feldser DM (2017). Systematic In Vivo Inactivation of Chromatin-Regulating Enzymes Identifies Setd2 as a Potent Tumor Suppressor in Lung Adenocarcinoma. Cancer Res 77, 1719–1729. 10.1158/0008-5472.can-16-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, and Youle R (2011). Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene 31, onc2011497. 10.1038/onc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-K, Yang S-C, Hsu S-C, Chang F-P, Lin Y-T, Chen S-F, Cheng C-L, Hsiao M, Lu FL, and Lu J (2017). CHAC2 is essential for self-renewal and glutathione maintenance in human embryonic stem cells. Free Radical Bio Med 113, 439–451. 10.1016/j.freeradbiomed.2017.10.345. [DOI] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, and Seed B (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40, D1144–D1149. 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JM, London IM, and Chen JJ (1992). Effects of hemin and porphyrin compounds on intersubunit disulfide formation of heme-regulated eIF-2 alpha kinase and the regulation of protein synthesis in reticulocyte lysates. J Biol Chem 267, 20519–20524. 10.1016/s0021-9258(19)88733-1. [DOI] [PubMed] [Google Scholar]
- Yang T, Xu F, Sheng Y, Zhang W, and Chen Y (2016). A targeted proteomics approach to the quantitative analysis of ERK/Bcl-2-mediated anti-apoptosis and multi-drug resistance in breast cancer. Anal Bioanal Chem 408, 7491–7503. 10.1007/s00216-016-9847-7. [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omics J Integr Biology 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Macias-Garcia A, Ulirsch JC, Velazquez J, Butty VL, Levine SS, Sankaran VG, and Chen J-J (2019). HRI coordinates translation necessary for protein homeostasis and mitochondrial function in erythropoiesis. Elife 8, e46976. 10.7554/elife.46976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Targeting anti-apoptotic Bcl-2 proteins induces persister cells, related to Figure 1 (A) Western Blot of WT and BAX, BAK, BOK TKO PC9 cells with the indicated antibodies.
(B) Western Blot of WT and BAX- and BAK-deficient PC9 cells with the indicated antibodies.
(C) Fractional viability of PC9 PT and PS after either 1d drug holiday (PS d1) or 7d drug holiday (PSd7) treated with ABT737 and S6. n=2 samples/condition. One representative of 2 independent experiments is shown.
(D) Metastatic ratio representing the ratio between cells in contralateral lung and primary injection site for CFSE-labelled A549 PT or CTV-labelled A549 PS cells respectively. N=5 mice. Paired t-test: *P< 0.05
(E) Fractional viability of HT29 PT and PS treated with MOCK, Oxaliplatin 10 μM, Irinotecan 4 μM, Etoposide 4 μM or Vemurafenib 20 μM for 3 d. n=3 samples/group. 2-way ANOVA: ****P< 0.0001; NS, not significant (P >0.05).
(F) Fractional viability of HT29 PT and PS treated with RSL3 for 3d. N=3 samples/group.
(G) IC50 values from fractional viability assays comparing PC9 PT and PS treated with titrations of RSL3 for 3d. n=7 independent experiments. Paired t test: ***P< 0.001.
(H) MFI of C11-Bodipy in PC9 PT and PS treated ± RSL3, in the presence or absence of DFO. n=3 samples/group. 2-way ANOVA: *P< 0.05, ***P< 0.001, ****P< 0.0001
(I) qPCR analysis of CHAC1 or GCLC transcripts representing glutathione metabolism in PC9 PT and PS. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. **P <0.01, ****P <0.0001.
(J) KEGG and Reactome pathways commonly enriched across treatment regimens. Enrichment was calculated by GSEA (see Methods). The commonly enriched pathways were defined as those that have false discovery rate (FDR) < 0.05 and the same direction in different treatments. Normalized enrichment scores (NES) are shown. PS were generated by treatment with ABT737 1.5 μM and S6 3 μM for 6 h, or Erlotinib 5 μM for 3 d or Paclitaxel 0.5 μM for 3 d.
Figure S2. The persister phenotype depends largely on Bcl-2 effectors, related to Figure 2 and 3 (A) IC50 values from fractional viability assays comparing untreated versus ABT737 (1.5 μM) and S6 (3 μM) pretreated BAX, BAK, BOK-deficient PC9 (TKO) treated with titrations of RSL3 for 3d. n=5 independent experiments. Paired t test: NS, not significant (P> 0.05).
(B) MFI of C11-Bodipy in untreated versus ABT737 (1.5 μM) and S6 (3 μM) pretreated BAX, BAK, BOK-deficient PC9 (TKO) treated ± RSL3, in the presence or absence of DFO. Same experiment as Figure S1H.
(C) qPCR analysis of CHAC1 or GCLC transcripts related to glutathione metabolism in untreated versus ABT737 (1.5 μM) and S6 (3 μM) pretreated BAX, BAK, BOK-deficient PC9 (TKO). N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. ns, not significant (P >0.05). Same experiment as Figure S1I.
(D) Heatmap of normalized count of the top 10 up- or down-regulated, differentially expressed genes (DEGs) in WT (PS/PT) but not in BAX, BAK, BOK TKO (ABT737, S6 treated/MOCK) PC9 cells. The normalized counts were scaled with root mean square (RMS) by gene and genotype.
(E) Scaled expression profiles of gene clusters along pseudotime. (same as Figure3)
(F) Enrichment analysis of differentially expressed genes (80 in cluster 1, 76 in cluster 2, 127 in cluster 3, 16 in cluster 4) in Reactome, Hallmark or C2:Chemical and genetic perturbations (C2:CGP) collections. Enrichment analysis was performed using the over-representation approach.
Figure S3. BH3 mimetics induce an ISR, dependent on Bcl-2 effectors and Imomp but independent of caspase activation, related to Figures 4 and 5. (A) Double-luciferase assay of PC9 cells expressing ATF4 translation reporter upon treatment with ABT737 1.5 μM and S6 3 μM in combination ± 2BAct 50 μM for indicated times. N=5 samples per group and timepoint.
(B) Double-luciferase assay of WT or BAX, BAK, BOK TKO PC9 cells expressing ATF4 translation reporter and treated with Tunicamycin 10 μM, ISRIB 100 μM and 2BAct 50 μM overnight. N=8 samples/group.
(C) Immunoblot of PC9 cells transiently transfected with Cas9 and non-targeting or HRI targeting sgRNAs.
(D) Cytofluorimetric analysis of mean fluorescence intensity (MFI) of ATF4 (AF647) in siNT or siBAX and siBAK transfected A549 cells treated ± ABT737 5 μM and S6 10 μM. (n=4/group; 2-way ANOVA: **P <0.01)
(E) Metastatic ratio representing the ratio between cells from the contralateral lung and primary injection site. For quantification cytofluorimetric analysis of CFSE-labelled PT and CTV-labelled PS (generated by treatment with Raptinal) from BAX, BAK, BOK TKO PC9 cells was performed 2 d after intrapulmonal combined injection (n=10 mice/group).
(A and B) Statistical analysis was performed using 2-way ANOVA. *P <0.05; ****P <0.0001; NS, not significant (P> 0.05).
Figure S4. Cytochrome C activates HRI and engages the persister phenotype, related to Figure 6 (A) FACS histograms and (B) fold change quantification of Cytochrome c release PC9 cells treated with indicated siRNAs and ± ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM for 4 h.
(C) Double-luciferase assay of PC9 cells expressing ATF4 translation reporter and silenced with indicated siRNAs before treatment with ABT737 1.5 μM and S6 3 μM for 6.5 h. N=4 samples/group. 2way ANOVA: ****P< 0.0001, ***P< 0.001, *P< 0.05; NS, not significant (P> 0.05).
(D) Left: Immunoblot with the indicated antibodies of cytosolic extracts from PC9 cells transiently transfected with sgNT or sgHRI, treated with serial dilutions of Cytochrome c (3–50 μM). Right: Quantification by fold change of normalized samples. One representative of 2 similar experiments shown.
(E) FACS blots of PC9 cells treated with either sgNT (non-targeted) or sgCYCS to generate stable knock-out cells by use of CRISPR/Cas9 and reconstituted with indicated pCYCS WT or pCYCSK72R vectors. Mitochondrial Cytochrome c was measured after 4 h treatment with MOCK control or a combination of ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM.
(F) Percentage of Cytochrome c positive cells from (E) ± treatment with ABT737 1.5 μM, S6 3 μM and Q-VD-OPh for 6.5 h. n=6 samples/group.
(G) Incucyte analysis of PI positive CYCS−/− cells that had been reconstituted with either pCYCS WT or pCYCSK72R, and treated ± ABT737 1.5 μM and S6 3 μM. n=4 samples/group.
(H) Experimental schematic and (I) western blot of A549 cells expressing the CDS of K72R mutated CYCS, and were treated with or without a combination of ABT737 5 μM, S6 10 μM and Q-VD-OPh 20 μM for 4h.
Figure S5. Cytochrome C activates HRI and engages the persister phenotype, related to Figure 6 (A) PT and PS generated using ABT737 1.5 μM and S6 3 μM of PC9 CYCS−/− or (B) PC9 CYCS−/− reconstituted with WT CYCS. Fractional viability was determined after 3 d treatment with Mock, Erlotinib 5 μM, Paclitaxel 1 μM or Cisplatin 10 μM. N=3 samples/group. 2-way ANOVA: **P <0.01; ****P <0.0001; NS, not significant (P> 0.05).
(C) qPCR analysis of transcripts that have been identified as BAX, BAK-dependent in PT and PS generated from PC9 CYCS−/− cells, reconstituted with or without WT CYCS. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. ***P <0.001; ****P <0.0001.
(D) Immunoblot and densitometric quantitation of hosphor-eIF2α from in vitro kinase reaction with recombinant eIF2α 12 μg and HRI 25 nM, in the presence of ATP 50 μM incubated for indicated times (30 seconds to 20 minutes) showing initial velocity conditions. One representative of 2 independent experiments shown.
(E) Immunoprecipitation of mClover in HRI-mClover- or mClover-transfected A549 cells after treatment with ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM for 4 h.
(F) Immunoprecipitation of HRI-mClover or mClover from transfected A549 cells after in vitro, post lysis incubation with equine cytochrome C (100nm).
Figure S6. Cytochrome c - HRI - ATF4 axis drives the phenotype of persistence, related to Figure 7 (A) Slingshot trajectory showing log-transformed counts and fitted values for indicated genes along pseudotime. P-value was calculated from association test in tradeSeq package. Data from single cell RNAseq of PT and PS PC9 cells (see legend, Figure 3).
(B) AnnexinV staining of HCT116 WT and ATF4−/− cells treated with or without ABT737 10 μM and S6 20 μM. N=4 samples, mean bar ± SD. Significance was calculated using 2-way ANOVA. ***p < 0.001.
(C) Fractional viability of A549 cells that have been transfected with siNT, siATF4, siHMOX1 or siNRF2, and treated with titration of a combination of ABT737 and S6 overnight. One representative of 2 independent experiments shown.
(D) Immunoblot with the indicated antibodies of PC9 cells that have been transfected with siNT, siATF4, siHMOX1 or siNRF2.
(E) Immunoblot with the indicated antibodies of PC9 cells after CRISPR/Cas9 knockout (KO) of ATF4 or using non-targeted sgRNA (sgNT) treated with or without ABT737 1.5 μM, S6 3 μM and Q-VD-OPh 20 μM.
(F) Immunoblot with the indicated antibodies of PT and PS generated from PC9 (ABT737 1.5μM, S6 3μM), H226 (ABT737 2.5μM, S6 5μM), A549 (ABT737 5μM, S6 10μM) and HT29 (ABT737 4μM, S6 8μM) overnight.
(G) Immunoblot with the indicated antibodies of PC9 cells that have been transfected with siNT, siBAX, siBAK or siBOK.
(H) PC9 cells were transfected with siNT or siBAX, for 48 h, following which they were treated with the indicated drugs for 48h. Fractional viability (mean ± S.D.) is shown. One representative of 3 independent experiments with n=8 samples/group.
(I) qPCR analysis of BAX transcript in PT and ABT737 (1.5 μM) and S6 (3 μM) treated PC9 CYCS−/− cells, reconstituted with or without WT CYCS. N=3 replicate samples are shown.
(J) IC50 values from fractional viability assays comparing PT versus ABT737 (1.5 μM) and S6 (3 μM) pretreated ATF4 KO or (K) HRI KO PC9 cells, treated with titrations of RSL3 for 3d. n=7 (ATF4 KO), n=6 (HRI KO) independent experiments. Paired t test: ns, not significant (P> 0.05).
(L) MFI of C11-Bodipy in PC9 WT, ATF4 KO or HRI KO PT and ABT737 (1.5 μM) and S6 (3 μM) pretreated cells, treated with or without RSL3. 2-way ANOVA: **P< 0.01, ****P< 0.0001, ns, not significant (P> 0.05). n=3 samples per group.
(M) qPCR analysis of CHAC1 or GCLC transcripts representing glutathione metabolism in PT versus ABT737 (1.5 μM) and S6 (3 μM) treated PC9 ATF4 KO or (N) HRI KO cells. N=3 samples are shown. Statistical analysis was performed using unpaired student’s t-test. ns, not significant (P >0.05).
(O) Fractional viability and (P) IC50 values from fractional viability assays of PC9 cells transfected with siNT or siBAX/BAK or siATF4 or siHRI respectively treated with RSL3 for 3d. n=3 independent experiments. Paired t test: ns, not significant (P> 0.05).
(Q) Double-luciferase assay of PC9 WT or BAX, BAK, BOK TKO or CASPASE9 KO cells expressing ATF-4 translation reporter upon treatment with Tunicamycin in combination with or without 2BAct for 6,5h. N=4 samples per group and timepoint.
(R) Fractional viability of PC9 PT and PS generated by Tunicamycin treatment and then treated with MOCK, Erlotinib 10 μM, Paclitaxel 1 μM or Cisplatin (CPD) 10 μM 3 d. n=4 samples per group. 2-way ANOVA: ***P< 0.001, ****P<0.0001; ns, not significant (P> 0.05).
(S) Fractional viability of PC9 WT or BAX, BAK, BOK TKO PT and PS generated by Tunicamycin treatment and then treated with RSL3 3 d. n=4 samples per group. 2-way ANOVA: ****P< 0.0001; ns, not significant (P> 0.05).
Figure S7. Cytochrome c - HRI - ATF4 axis drives the phenotype of persistence, related to Figure 7 (A) Immunohistochemistry showing HRI expression in representative examples of human normal and associated cancer tissues. Scale bars, 100 μm.
(B) List of cancer name abbreviations used in (C) and (D) and number of subjects analyzed.
(C) HRI expression in various tumors (T, red dots) compared to normal tissue (N, green dots). Red cancer name abbreviations indicate significant changes between normal and tumor (ANOVA).
(D) Dependence of disease-free survival and (E) overall survival on HRI expression in various tumors estimated by Mantel-Cox analysis. Hazard ratio (HR) shown (red = worse outcome), red squares statistically significant p<0.05 (ANOVA).
(F) Kaplan Meier overall survival curve of patients with gastric cancer (left, dataset GSE51105), ovarian cancer (middle, dataset GSE3149) and renal papillary cell cancer (right, dataset TCGA) belonging to high (4th quartile) or low (1st quartile) of HRI expression. Hazard ratio (HR) and p-value are indicated.
(G) Kaplan Meier relapse-free survival curve of patients with HER2+ breast cancer (dataset E-MTAB-365) belonging to high (4th quartile) or low (1st quartile) of HRI expression. Hazard ratio (HR) and p-value are indicated.
SUPPLEMENTARY TABLE S3. sgRNA sequences. Related to the STAR methods section.
SUPPLEMENTARY TABLE S4. siRNA sources. Related to the STAR methods section.
SUPPLEMENTARY TABLE S5. qPCR Primer sequences. Related to the STAR methods section.
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO (accession: GSE189639) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cytochrome C – AF 647 | BioLegend | Cat# 612310, RRID:AB_2565241 |
| CD45 – APC | BD | Cat# 559864 RRID:AB_398672 |
| GAPDH (0411) HRP | Santa Cruz | Cat# sc-47724 HRP RRID:AB_627678 |
| HRI (D-12) | Santa Cruz | Cat# sc-365239 RRID:AB_10843794 |
| eIF2alpha | CST | Cat# 9722 RRID:AB_2230924 |
| eIF2 pS51 | Abcam | Cat# ab32157 RRID:AB_732117 |
| ATF-4 | CST | Cat# 11815 RRID:AB_2616025 |
| HRI (D-12) (EIF2AK1) | Proteintech | Cat# 20499-1-AP RRID:AB_10697665 |
| PKR (EIF2AK2) | Santa Cruz | Cat# sc6282 RRID:AB_628150 |
| Perk (EIF2AK3) | CST | Cat# 3192 RRID:AB_2095847 |
| GCN2 (EIF2AK4) | CST | Cat# 40457 RRID:AB_2799177 |
| Nrf2 | Santa Cruz | Cat# sc13032 RRID:AB_2263168 |
| Heme Oxygenase 1 Antibody (A-3) | Santa Cruz | Cat# sc-136960 RRID:AB_2011613 |
| GAPDH-HRP | Santa Cruz | Cat# sc47724-HRP RRID:AB_627678 |
| β-Actin Antibody (C4) HRP | Santa Cruz | Cat# sc-47778 HRP RRID:AB_2714189 |
| Caspase9 | CST | Cat# 9502S RRID:AB_2068621 |
| HCCS | Abcam | Cat# ab234874 |
| APAF-1 | CST | Cat# 5088S RRID:AB_10556958 |
| BAX | CST | Cat# 2772 RRID:AB_10695870 |
| BAK | CST | Cat# 3814 RRID:AB_2290287 |
| BOK | Abcam | Cat# ab186745 RRID:AB_2728737 |
| Cytochrome c (D18C7) Rabbit mAb | CST | Cat# 11940S RRID:AB_2637071 |
| Amersham ECL Rabbit IgG, HRP-linked whole Ab (from donkey) | Amersham | Cat# NA934 RRID:AB_772206 |
| Cytiva’s Amersham ECL Mouse IgG, HRP-linked whole Ab (from sheep) | Amersham | Cat# NA931 RRID:AB_772210 |
| Anti-NRF2 antibody [N2C2] | Genetex | Cat# GTX103322 RRID:AB_1950993 |
| GFP-Booster Alexa Fluor® 488 | Chromotek | Cat# gb2AF488 RRID:AB_2827573 |
| RFP-Booster Alexa Fluor® 568 | Chromotek | Cat# rb2AF568 RRID:AB_2827576 |
| BODIPY™ 581/591 C11 | Thermo Fisher | Cat# D3861 |
| Cytochrome C | BD | Cat# 556432 RRID:AB_396416 |
| GFP-Trap Magnetic Agarose | Chromotek | Cat# gtma-20 RRID:AB_2631358 |
| p62/SQSTM1 | Sigma | Cat# P0067 RRID:AB_1841064 |
| Bacterial and Virus Strains | ||
| NEB®10-beta Competent E.coli (High Efficiency) | NEB | Cat# C3019H |
| NEB 5-alpha Competent E.coli | NEB | Cat# C2987H |
| NEB® Stable Competent E.coli (High Efficiency) | NEB | Cat# C3040H |
| TOPO™ TA Cloning™ Kit for Sequencing, with One Shot™ TOP10 Chemically Competent E. coli | Thermo Fisher | Cat# K4575J10 |
| Biological Samples | ||
| - | ||
| Chemicals, Peptides, a Recombinant Proteins | ||
| ABT-737 | MCE | Cat# HY-50907 |
| S63845 | MCE | Cat# HY-100741 |
| ISRIB | MCE | Cat# HY-12495 |
| 2BAct | AOBIOUS | Cat# 2143542-28-1 |
| Paclitaxel | MCE | Cat# HY-B0015 |
| RSL3 | Selleck Chemicals | Cat# S8155 |
| Irinotecan | MCE | Cat# HY-16562 |
| Erlotinib | Cayman | Cat# 10483 |
| Vemurafenib (PLX4032) | Cayman | Cat# 10618 |
| Raptinal | Millipore Sigma | Cat# SML1745 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| Eif2s1, His tagged human,recombinant | Sigma-Aldrich | Cat# SRP5232 |
| Eif2ak1 (HRI) Human Protein | Thermo Scientific | Cat# PV5856 |
| 3X FLAG® Peptide | Sigma-Aldrich | Cat# F4799 |
| AC DEVD AFC | Enzo Life Sciences | Cat# ALX260032M005 |
| Cytochrome c from Saccharomyces cerevisiae | Sigma-Aldrich | Cat# C2436 |
| Cytochrome C, equine | Sigma-Aldrich | Cat# C2506 |
| Cytochrome C, bovine | Sigma-Aldrich | Cat# C2037 |
| Liberase TM | Sigma-Aldrich | Cat# 5401119001 |
| DNase I, Grade II, from bovine pancreas | Sigma-Aldrich | Cat# 10104159001 |
| Corning® Matrigel® Basement Membrane Matrix, LDEV-Free | Corning | Cat# 356234 |
| Deferoxamine mesylate salt | Sigma-Aldrich | Cat# D9533-1G |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Direct-zol RNA MicroPrep (200 Preps) w/ Zymo-Spin IC Columns (Capped) | Zymo Research | Cat# R2062 |
| Intracellular Fixation & Permeabilization Buffer Set | BD | Cat# 554714 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | Thermo Scientific | Cat# 00-5523-00 |
| Dual-Luciferase Reporter 1000 Assay System | Promega | Cat# E1980 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| CellTrace™ CFSE Cell Proliferation Kit, for flow cytometry | Thermo Scientific | Cat# C34554 |
| Cell Trace Violet | Thermo Scientific | Cat# C34557 |
| Clarity™ Western ECL Substrate, 500 ml, 1705061 | Bio-Rad | Cat# 1705061 |
| Xt MES Running Buffer, 1610789 | Bio-Rad | Cat# 1610789 |
| 20S Proteasome Lysis Buffer | CAYMAN Chemicals | Cat# 10011098 |
| Lipofectamine 3000 Transfection Reagen | Thermo Scientific | Cat# L3000001 |
| Fugene HD Transfection Reagent, Promega, FuGENE HD Transfection | Promega | Cat# E2311 |
| ViaFect Transfection Reagent | Promega | Cat# E4981 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Therno Scientific | Cat# 13778075 |
| Ercc RNA Spike-In Mix | Thermo Scientific | Cat# 4456740 |
| pcDNA3.1/V5-His TOPO TA Expression Kit | Thermo Scientific | Cat# K480001 |
| ANTI-FLAG® M2 Affinity Gel,purified immunoglobulin, buffered aqueous | Sigma-Aldrich | Cat# A2220 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#4309155 |
| Chromium Next GEM Single Cell 3ʹ Library Kit v3.1 16 rxns | 10X Genomics | PN-1000157 |
| Chromium Next GEM Chip G Single Cell Kit, 16 rxns | 10X Genomics | PN-100012 |
| Mycoalert Detection kit | Lonza | Cat# LT07-318 |
| EasySep Dead Cell Removal (Annexin V) kit | STEMCELL technologies | Cat# 17899 |
| Next Seq 500/550 High Output Kit v2 | Illumina | Cat#FC-404-2002 |
| TruSeq Stranded mRNA kits | Illumina | Cat#20020595 |
| Zombie Violet Viability Kit | Biolegend | Cat#423113 |
| Deposited Data | ||
| SuperSeries | GSE189639 | |
| bulk RNA-seq of parental and persister cells generated human cancer cells | GSE189625 | |
| scRNAseq of PC9 parental and persister cells | GSE189638 | |
| Experimental Models: Cell Lines | ||
| H. sapiens: PC9 | ATCC | |
| H. sapiens: HCT116 | Richard Youle | |
| H. sapiens: HT29 | ATCC | |
| H. sapiens: A549 | Richard J. Webby, SJRH | |
| H. sapiens: H226 | ATCC | |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcsscid Il2rgtm1Wjl/SzJ (NSG) | In house | |
| Rag1−/− | In house | |
| Oligonucleotides | ||
| sgRNA see Table S3 | abm (Lopez et al., 2016; Shalem et al., 2014) | N/A |
| siRNA see Table S4 | Dharmacon, IDT | N/A |
| qPCR primer see Table S5 | Origene sequences, (Kim et al., 2010; Spandidos et al., 2008, 2010; Wang et al., 2012) | N/A |
| Recombinant DNA | ||
| Piggybac expression system (inducible) | (Magnúsdóttir et al., 2013) | |
| pPB constitutive CMV | This paper | |
| pPB (puro)-mCherry | This paper | |
| pPB (puro)-mNeonGreen | This paper | |
| pPB (puro)_hUTR(ATF4) luci_IRES RenLuciferase | This paper | |
| pPB (puro) mitoV1 AifTail-mCh-GFP1-10 | This paper | |
| pPB (Blast) | This paper | |
| pPB (Blast) 2xGFP11 (M3) | This paper | |
| pPB (Zeo) | This paper | |
| pPB (Zeo)_CYCS | This paper | |
| pPB (Zeo)_CYCS K72R | This paper | |
| pPB Luciferase-tdTomato | This paper | |
| pcDNA TOPO TA mClover | This paper | |
| pXG290 (HRI-mClover) | (Guo et al., 2020) | Addgene 141284 |
| Px458 pSpCas9(BB)-2A-GFP | (Ran et al., 2013) | Addgene 48138 |
| pLKO5.sgRNA.EFS.PAC | (Heckl et al., 2014) | Addgene 57825 |
| CRISPRv2 GFP | (Walter et al., 2017) | Addgene 82416 |
| pSpCas9(BB)-2A-mCherry | In House | N/A |
| Software and Algorithms | ||
| Trim Galore (v0.5.0) | Babraham Institute | RRID:SCR_011847 |
| Picard (v 2.19.0) | Broad Institute | RRID:SCR_006525 |
| RSEM | Dewey lab | RRID:SCR_013027 |
| DESeq2 (v1.30.1) | Michael Love | RRID:SCR_015687 |
| Cell Ranger (v6.0.1) | 10X Genomics | RRID:SCR_017344 |
| Slingshot (v1.8.0) | Kelly Street | RRID:SCR_017012 |
| STAR (v 2.7.5a) | Alexander Dobin | RRID:SCR_004463 |
| Seurat (v4.0.4) | Satija lab | RRID:SCR_016341 |
| tradeSeq (v1.4.0) | Lieven Clement | RRID:SCR_019238 |
| clusterProfiler (v3.18.1) | Guangchuang Yu | RRID:SCR_016884 |
| R (v 4.0.5) | R project | RRID:SCR_001905 |
| RUVseq (v1.24.0) | Davide Risso | RRID:SCR_006263 |
| FlowJo v10 | FlowJo, LLC | RRID:SCR_008520 |
| Slidebook 6 | Intelligent Imaging Innovations | RRID:SCR_014300 |
| GraphPad Prism 9.0 | GraphPad Software | RRID:SCR_002798 |
| IMARIS 9.8 | Oxford Instruments | RRID:SCR_007370 |
All original code has been deposited at GitHub (https://github.com/markgene/MC015_Persister_public) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.