

Abstract
Alternative splicing of RNA occurs frequently in eukaryotic cells and can result in multiple protein isoforms that are nearly identical in amino acid sequence, but have unique biological roles. Moreover, the relative abundance of these unique isoforms can be correlative with diseased states and potentially used as biomarkers or therapeutic targets. However, due to high sequence similarities among isoforms, current proteomic methods are incapable of differentiating native protein isoforms derived from most alternative splicing events. Herein, a strategy employing a nonsynonymous, non-native amino acid (nnAA) pseudo-hapten (i.e. an amino acid or amino acid derivative that is different from the native amino acid at a particular position) as a targeting epitope in splice junction-spanning peptides was successful in directed antibody derivation. After isolating nnAA-specific anti-bodies, directed evolution reduced the antibody’s binding dependence on the nnAA pseudo-hapten and improved binding to the native splice junction epitope. The resulting antibodies demonstrated codependent binding affinity to each exon of the splice junction and thus are splice junction- and isoform-specific. Furthermore, epitope scanning demonstrated that positioning of the nnAA pseudo-hapten within a peptide antigen can be exploited to predetermine the isolated antibody’s specificity at, or near, amino acid resolution. Thus, this nnAA targeting strategy has the potential to robustly derive splice junction- and site-specific antibodies that can be used in a wide variety of research endeavors to unambiguously differentiate native protein isoforms.
Keywords: Alternative splicing, Antibody, Directed evolution, Hapten, Isoform, Splice junction
Introduction
Alternative splicing is a critical RNA processing mechanism that enables a single multi-exon gene to produce more than one unique mRNA. This diversification mechanism has been evolutionarily conserved where events such as exon skipping, usage of alternative donor-acceptor splice sites and intron retention have been reported in species from all major eukaryotic kingdoms [1]. In the human genome, more than 90% of multi-exon genes undergo alternative splicing to generate a highly diverse proteome that far exceeds the number of annotated genes[2,3]. Many alternatively spliced mRNAs are capable of being translated into unique proteins that serve specialized biological roles[3-10]. Moreover, disruptions in the splicing pathway can cause aberrant splicing that produces novel isoforms or increases the production of normally low-level isoforms[11] which can lead to potentially detrimental effects. The impact of aberrant splicing is particularly acute in cancer, where tumors experience up to 30% more alternative splicing events than normal cells and novel exon-exon junctions (EEJs), termed neojunctions, are found unique to tumor samples[11]. Strikingly, it has been estimated that up to one third of all disease-causing mutations affect splicing[2]. However, biological questions about many alternative splicing events at the native protein level in both normal and diseased states have been neglected.
The biological impact of alternative splicing on native protein functions, particularly those derived from exon skipping events, has been largely unexplored in normal and disease states due to a lack of technology available for unambiguously differentiating these highly similar proteins. Current technology including mass spectrometry, indirect tag-based detection and direct antibody-based detection carry undesirable, yet unavoidable, secondary effects such as modifying native protein characteristics and recognizing multiple protein isoforms indiscriminately. Here, this technological gap has been addressed through the implementation of a unique targeted approach to derive splice junction-specific antibodies (Abs) using an alternatively spliced isoform of human centromere protein A (CENP-A) as the target antigen.
CENP-A is a centromere-specific histone protein that is functionally conserved among eukaryotes and is essential for viability due to its pivotal roles in mitotic processes[12-17]. The alternative CENP-A isoform (CENP-A-ΔExon3) is derived from an exon 3 skipping event (Fig. 1A, top), yet its biological role is unresolved. Like most isoforms derived from exon skipping events, currently available Abs to CENP-A are specific to either the N- or C-terminus of the protein and therefore incapable of differentiating the two alternatively spliced isoforms. To achieve the Ab specificity required for unambiguous detection of CEN-P-A-ΔExon3, the unique exon 2/4 splice junction epitope must be targeted with precision to derive Abs that bind to both exon 2 and exon 4 simultaneously. Thus, a two-stage strategy employing a nonsynonymous, non-native amino acid (nnAA) pseudo-hapten (i.e. an amino acid or amino acid derivative that is different from the native amino acid at a particular position) as a targeting epitope in splice junction-spanning peptides was developed for directed Ab derivation. In this in vitro strategy, Abs were first isolated against the predetermined position of the nnAA pseudo-hapten,e.g. phosphoserine, (SEP) and then anti-nnAA Abs were mutagenized to change specificity to the native epitope using directed evolution, resulting in Ab paratopes that simultaneously bind both exons of a splice junction. Application of this nnAA targeting strategy was extended to an in vivo approach, resulting in the successful derivation of additional splice junction-specific Abs. This nnAA targeting strategy can predictably yield splice junction-specific Abs with codependent binding affinity to each exon of any splice junction target.
Fig. 1. Peptide designs and screening results for in vitro derivation of splice junction-specific Abs using a nnAA targeting strategy and directed evolution.

(A, top) Sequence alignment of the canonical human CENP-A and the alternatively spliced human CENP-A-ΔExon3 that lacks exon 3. (A, bottom) Schematics of exon-exon junctions that differ between CENP-A and CENP-A-ΔExon3. The exon 2/4 splice junction specific to CENP-A-ΔExon3 is the target epitope for Ab derivation. (B, left) Five Discovery screening peptides (SEP1–5) spanning the exon 2/4 splice junction of CENP-A-ΔExon3 were synthesized with a single non-native SEP substitution (yellow star) at variable positions across the splice junction. Amino acids are denoted by a circle color coded as per (A). Note that total amino acid length is not shown but the relative position of SEP is depicted. Using two different naïve phage display libraries (AXL40 and AXL41), Discovery screening was performed against the five SEP peptides to select for SEP-specific scFvs. A Discovery scFv schematic is shown with CDRs in purple denoting a SEP-specific paratope. (B, right) Summary of Discovery screening results including all 10 unique screens (i.e., each Discovery library against each SEP peptide). “Positive” hits bind to the SEP peptide in ELISA and are further differentiated based on their ability (“SEP-Independent”) or inability (“SEP-Specific”) to bind to the nonphosphorylated, native sequence peptide. Results for each independent screen are presented in Supplementary Table S2. (C, left) Three SEP-specific scFvs were mutagenized and individual Directed Evolution libraries were generated (DEL6691, DEL6695 and DEL6698). Using each library, Directed Evolution screening was performed against the nonphosphorylated sequence peptides [NAT2 and NAT4, color coded as per (A-B)] to select for scFvs with improved binding strength to the native exon 2/4 splice junction of CENP-A-ΔExon3. Note that NAT2 is identical in sequence and length to SEP3–5 (B, left) except for the SEP residue. Each library was screened under two conditions as per Materials and Methods. An example Directed Evolution scFv schematic is shown with preserved parental CDRs in purple and mutated CDRs in yellow denoting an evolved paratope to the native sequence epitope. Note that mutations can be acquired in any of the six CDRs. (C, right) Summary of Directed Evolution screening results including all six unique screens. “Positive” hits bind to the NAT4 peptide in ELISA and are further differentiated based on their ability (“Improved”) or inability (“No Improvement”) to bind to NAT4 stronger than their SEP-specific parental Discovery clone. Results for each independent screen are presented in Supplementary Table S3B.
Materials and methods
Bacterial strains, mammalian cell lines, plasmids, and peptides
Strains, cell lines and plasmids are shown in Supplementary Table S5. All peptide sequences are found in Supplementary Table S6.
Construction of phage display libraries
All libraries employed single-chain variable fragment (scFv)-display strategies[18-20] (Supplementary Tables S1 and S5). AXL40 is a naïve single-framework Discovery library (Abcam)[18,21] with predetermined complementarity determining regions (CDRs) including a 5% molar ratio of four phospho-binding CDR-H2 sequences identified in-house and in [22]. AXL41 is a naïve single-framework Discovery library (Abcam) based on a SEP-specific scFv that has been mutagenized using degenerate nucleotides (i.e., “NNK”) in CDR-L2, CDR-L3, CDR-H2 and CDR-H3. AXL40 and AXL41 have estimated diversities of 1.0 × 1010 and 1.2 × 1010, respectively.
The template phagemid for Directed Evolution phage libraries (DEL6691, DEL6695 and DEL6698) are based on SEP-specific scFv sequences identified herein from phage display biopanning using AXL40 and AXL41. Directed Evolution phage libraries were generated based on methods for AXM mutagenesis[23,24]. DEL6691, DEL6695, and DEL6698 have estimated diversities of 1.0 × 108, 1.7 × 108, and 1.0 × 108, respectively.
The template phagemid for Affinity Maturation libraries (AML6691 Clone 1 and AML6691 Clone 2) are based on evolved scFv sequences identified from phage display biopanning of the Directed Evolution libraries. Affinity Maturation phage libraries were generated as per Directed Evolution libraries, above. AML6691 Clone 1 and Clone 2 libraries have estimated diversities of 7.1 × 107 and 6.3 × 107, respectively.
Isolation of SEP-specific scFvs by phage display biopanning
Ten phage display Discovery screenings using AXL40 and AXL41 libraries against the SEP1–5 peptides were performed as per [18] with optimized conditions for the template phagemid (e.g., growth media and method for phage elution). For rounds 2 and 3 of biopanning, a 10-fold excess of nonbiotinylated, nonphosphorylated peptide competitor was added during positive phage selection against the biotinylated target peptide-coated wells. NAT1 and NAT2 peptides were used as competitors for the SEP1–2 screens and SEP3–5 screens, respectively.
Isolation of Directed Evolution splice junction-specific scFvs by phage display biopanning
Six Directed Evolution and two Affinity Maturation phage display screenings were carried out as per [18] and optimized conditions for the template phagemid (e.g. growth media and method for phage elution). Directed Evolution libraries (DEL6691, DEL6695, DEL6698) were screened under two conditions: (1) three rounds of positive selection against the biotinylated NAT2 peptide, and (2) one round of positive selection against the biotinylated NAT2 peptide followed by two rounds of positive selection against the biotinylated NAT4 peptide. The AML6691 Clone 1 Affinity Maturation library was screened against the biotinylated NAT3 peptide for three rounds of biopanning with successive 10-fold reduction in peptide concentration in rounds 2 and 3. Additionally, 100-fold excess of the nonbiotinylated Exon4 and NAT3 competitor peptide was added during round 2 and round 3, respectively. The AML6691 Clone 2 Affinity Maturation library was screened under the same conditions as the AML6691 Clone 1 library, but using the biotinylated NAT4 peptide for positive phage selection and 10-fold and 100-fold excess of the nonbiotinylated Exon4 competitor peptide in round 2 and round 3, respectively.
ScFv cell supernatant ELISA
Single scFv clones were precultured in 2YT-based media followed by induction with 1 mM IPTG at 30 °C. 96-well Maxisorp plates coated with 10 μg/mL (167 nM) NeutrAvidin (ThermoFisher Scientific, Rockford, IL, USA) were used for enzyme-linked immunosorbent assays (ELISAs) following standard protocols. In brief, plates were washed with phosphate buffered saline (PBS), blocked with 3% bovine serum albumin (BSA)/PBS (American Bioanalytical, Canton, MA) for 1 h at room temperature (RT), washed, and incubated with either 10 μg/mL (3–4 μM, Discovery and Directed Evolution screening), 0.05 μg/mL (15–17 nM, Affinity Maturation screening), or 1 μg/mL (310–35 nM, split-splice junction ELISAs) of the corresponding biotinylated peptide for 1 h at RT. Following a second round of washing-blocking-washing, 100 μL/well of cell supernatant scFv-expressing cultures was incubated for 1 h at RT, washed with PBS/0.1% Tween 20 and incubated with 100 μL/well of goat anti-Myc-HRP polyclonal antibody (1:5000 Abcam, Cambridge, UK; cat, nr. ab1261) for 1 h at RT. Following a PBS/0.1% Tween 20 wash, wells were developed with 1-Step Ultra TMB (100 μL/well; Thermo Scientific, cat. nr. 34028), stopped with 2 M H2SO4 (50 μL/well), and absorbances read at 450 nm.
Production and purification of soluble scFvs
Following scFv clone culturing and induction with 1 mM IPTG for 18 h at 30°C, cells were pelleted at 1900 x g for 25 min at RT and incubated at − 40 °C for 20 min. Thawed cell pellets were resuspended in 5 mL lysis buffer [B-PER (Thermo Scientific, cat. nr. 78248) supplemented with 1 mM phenylmethansulfonyl fluoride (PMSF), protease inhibitors (Sigma, cat. nrs. C7268, A1153, L2884, P5318), 1:10,000 Pierce Universal Nuclease (Thermo Scientific, cat. nr. 88702), 4 mM benzamidine (MilliporeSigma, cat. nr. 434760), and 10 mM imidazole], incubated on a platform shaker at RT for 25 min, and then centrifuged at 15,000 x g for 25 min at 4 °C. The lysate supernatant was added to a PBS-equilibrated column containing 200 μL of HisPur Cobalt Resin (Thermo Scientific, cat. nr. 89966), allowed to pass through by gravity flow at 4 °C, and washed twice with 2 mL of PBS/10 mM imidazole. Bound scFvs were eluted with 600 μL PBS/200 mM imidazole/10% glycerol and analyzed by Coomassie Plus assay and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
ScFv protein ELISA
96-well Maxisorp plates were coated with NeutrAvidin and processed as described for scFv cell supernatant ELISAs with the following modifications. For antigen titration ELISAs, plates were coated with peptides serially diluted 1:2 from 2 μg/mL (620–870 nM) to 0.03125 μg/mL (10–14 nM) and scFvs were tested at 1 μg/mL (32 nM). For scFv titration ELISAs, plates were coated with peptides at 10 μg/mL (3–4 μM) and scFvs were serially diluted 1:3 from 2 μg/mL (64 nM) to 0.0001 μg/mL (3 pM) or 1:2 from 3 μg/mL (95 nM) to 0.02344 μg/mL (744 pM).
Immunizations and polyclonal IgG production
Rabbit immunizations and whole blood sample collections were performed by Covance Laboratories (Princeton, NJ, USA) under Institutional Animal Care and Use Committee (IACUC)-approved conditions consistent with the National Institutes of Health (NIH) Animal Use policy. Two immunization strategies were performed: (1) co-immunizations with SEP6 and SEP7 followed by single immunizations with NAT6, and (2) single immunizations with NAT5 followed by single immunizations with NAT6 (for SEP and NAT sequences, see Supplementary Table S6). Whole blood (25 mL) was collected after the final boost in each series.
Monoclonal IgG isolation, production, and purification
Peripheral blood mononuclear cells (PBMCs) were isolated and cultured from whole blood. IgG-containing supernatants were tested in ELISA against the SEP6/SEP7, NAT5 and NAT6 peptides. cDNA was generated from PBMC populations positive against NAT5 and NAT6 and antibody heavy and light chain genes were PCR amplified. PCR products were purified, cloned into antibody expression vectors, and sequenced. Unique antibody sequences were used to produce recombinant monoclonal IgGs and purified by Protein A affinity chromatography.
Production and purification of alanine scan peptides fused to maltose binding protein (MBP)
pMAL-c5X plasmids were transformed into NEBExpress® cells (New England Biolabs, Ipswich, MA, USA) and single colonies were precultured in LB-based media then grown to an optical density 600 nm (OD600) of 0.5 for induction with 0.3 mM IPTG for 1 h at 37 °C. Cells were pelleted, stored at − 20 °C overnight, incubated with 5 mL of lysis buffer (as per soluble scFv purification), and then centrifuged at 20,000 x g for 20 min at 4 °C. Fusion proteins were purified as per soluble scFv purification but with 250 μL HisPur Cobalt Resin and eluting with 1 mL PBS/200 mM imidazole/15% glycerol.
Monoclonal IgG ELISA for epitope scanning
96-well Maxisorp plates were coated with 6 μg/mL (120 nM) of each MBP-fusion protein (wild-type exon 2/4 splice junction sequence fused to MBP, 15 alanine scan sequences independently fused to MBP, and MBP alone) and incubated overnight at 4 °C. Antigen-coated plates were processed as described for scFv cell supernatant ELISAs with the following modifications. Purified monoclonal IgGs were serially diluted 1:3 from 5 μg/mL (33 nM) to 0.1852 μg/mL (1 nM) and detected with goat anti-rabbit-HRP polyclonal antibody (1:5000; Jackson ImmunoResearch, West Grove, PA, USA, cat. nr. 111–035–144). Epitope scanning ELISAs were performed in triplicate. To calculate “% binding activity” against each antigen, the OD450 for MBP alone was subtracted from the OD450 for each MBP-fusion protein at each corresponding IgG concentration (background-corrected OD450). The background-corrected OD450 for each alanine scan MBP-fusion was divided by the background-corrected OD450 for the wild-type splice junction MBP-fusion for each corresponding IgG concentration (“% binding activity”). “% binding activity” was averaged across triplicate results for each IgG concentration (“average % binding activity”). To normalize for monoclonal IgGs with differing affinities, the IgG concentration that gave an average background-corrected OD450 of 1.2–1.6 with the lowest standard deviation (SD) against the wild-type splice junction MBP-fusion was used for the final reported “average % binding activity” against each alanine scan construct.
Generation of CENP-A and CENP-A-ΔExon3 overexpression lysates
Freestyle™ 293-F cells were transfected with the pSNAPf expression vectors using 40 μL of 293fectin™ Reagent and following the manufacturer’s protocol (Gibco, cat. nr. 12347–019) alongside a negative control transfection omitting DNA. At 48 h post-transfection, cells were pelleted at 500 x g for 10 min, resuspended in RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM benzamidine, and protease inhibitors) at a concentration of 1 × 107 cells/mL, and incubated on a nutator at 4 °C for 30 min. Lysates were centrifuged at 12,000 x g for 20 min and supernatants used in western blot analyses.
Western blot
Clarified lysates were mixed 1:1 with 2x Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA, USA, cat. nr. 1610737) supplemented with 0.17 M dithiothreitol (DTT) and incubated at 95 °C for 5 min. Protein standards (Bio-Rad, cat nr. 1610373) and an equivalent number of lysed cells were electrophoresed (Bio-Rad, cat, nr, 5671095) and proteins transferred to nitrocellulose membranes (Bio-Rad, cat, nr. 1704159) using the Bio-Rad Trans-Blot Turbo System. Membranes were blocked with 5% MTBST (5% w/v skim milk powder, TBS, 0.1% Tween 20) for 1 h at RT and incubated with monoclonal IgG at 1 μg/mL (7 nM) overnight at 4 °C. Membranes were washed 3 × 10 min with TBST, incubated with goat anti-rabbit-HRP polyclonal antibody (1:10,000; Jackson ImmunoResearch, cat. nr. 111–035–144) for 1 h at RT, washed, and developed using Enhanced Chemiluminescence (ECL) reagent (Bio-Rad, cat. nr. 1705061).
Results
Isolation of SEP-specific scFvs to CENP-A-ΔExon3 nnAA pseudo-haptens using in vitro phage display screening
To specifically target the unique splice junction of CENP-A-ΔExon3 (Fig. 1A), five distinct peptides spanning the exon 2/4 splice junction were synthesized (Supplementary Table S6), each with a single nnAA (SEP) substitution at a different site to ascertain the impact positioning of the SEP substitution relative to the EEJ has on Ab specificity (Fig. 1B, left). Corresponding nonphosphorylated, native sequence peptides (NAT1 and NAT2) were synthesized for counter-selection and counter-screening. Two naïve scFv-displaying phage libraries[18-20] (Discovery AXL40 and AXL41) were independently screened against biotinylated SEP1–5 peptides using three rounds of high-throughput panning and competition in solution to isolate SEP-specific Abs (Discovery screening, Fig. 1B). After the third round of affinity selection, 88 clones from each of the 10 Discovery screens (880 clones in total) were isolated and independently expressed as secretory scFvs (Fig. 1B, right). Each scFv-containing cell supernatant was tested in ELISA against the respective SEP and NAT peptides, and NeutrAvidin alone as a negative control. To classify hits preliminarily as SEP-specific, single clones had to meet three ELISA criteria: (1) < 5-fold signal against the NAT peptide over NeutrAvidin, (2) absorbance reading (OD450) ≥ 0.2 against the SEP peptide with ≥ 2-fold signal over NeutrAvidin, and (3) ≥ 2-fold signal against the SEP peptide over the NAT peptide (Fig. 1B, Supplementary Table S2). In brief, 230 Discovery clones were classified as SEP-specific by meeting all three criteria. Notably, positive hits derived from the Discovery AXL41 screens were more likely to be classified as SEP-specific than those derived from the Discovery AXL40 screens (on average ~80% vs. ~32%, Supplementary Table S2). The performance differences between Discovery libraries can be attributed to unique library designs in both the Ab framework and the diversity of CDRs; Discovery AXL41 was built with a greater inherent propensity to bind phosphorylated serines (see Materials and Methods and Supplementary Table S1). Nevertheless, SEP-specific hits were identified for each of the five SEP peptides.
A subset of SEP-specific hits was sequenced, purified as scFv proteins, and tested in titration ELISAs to validate SEP specificity. A total of 69 unique scFvs were identified (Fig. 1B, right). Four unique scFvs were identified in at least two different SEP screens (Supplementary Table S2), indicating flexible Ab paratopes that rely on SEP for epitope binding but can accommodate variations in the context peptide sequence. The remaining 65 unique scFvs were identified only in their respective Discovery screen. Of the 37 unique scFvs tested in titration ELISAs, 10 scFvs were identified as SEP-specific with ≥ 5-fold background-corrected signal against the SEP peptide over the NAT peptide for at least two antigen or scFv titration points (Fig. 1B, right). Two clonal examples are shown in Fig. 2A-B, left. For scFv titration ELISAs, SEP-specificity was determined under the most stringent conditions using signal comparisons to the NAT peptide with the highest background (NAT3). By testing each scFv under both scFv and antigen titration conditions, and against multiple NAT peptides, SEP-specific scFvs with an expected epitope at the exon 2/4 EEJ of CENP-A-ΔExon3 were confidently identified.
Fig. 2. Successful directed evolution of SEP-specific scFvs to the native exon 2/4 splice junction of CENP-A-ΔExon3.

Unique scFvs from the AXL40 (A, left) and AXL41 (B, left) Discovery screens were purified and tested in antigen and/or scFv titration ELISAs against the SEP and NAT peptides shown in the inset schematic, color coded as per Fig. 1. A total of 10 SEP-specific scFvs were identified (Supplementary Table S2) and two clonal examples are shown here. Note that the scFvs in (A) are unique in both the framework and CDR regions compared to the scFvs in (B), denoted by different shades of grey/purple in the scFv schematics and line graphs. (A and B, right) Unique scFvs from the Directed Evolution screens (DEL6691 and DEL6698) were purified and tested in scFv titration ELISAs against the NAT4 peptide and non-target peptides. Non-target peptides were used as a negative control to assess nonspecific binding events. Three evolved scFvs with 4- to 12-fold improved binding strength to the native exon 2/4 splice junction sequence were identified. Directed Evolution scFv schematics are shown with preserved parental CDRs in purple and mutated CDRs (AA change and position are indicated) in yellow.
In vitro directed evolution of SEP-specific scFvs to the native exon 2/4 splice junction of CENP-A-ΔExon3
To reduce the dependence on the nnAA pseudo-hapten and evolve these SEP-specific scFvs to bind to the nonphosphorylated, native exon 2/4 splice junction of CENP-A-ΔExon3, a combination of Ab mutagenesis and phage display screening was performed. Using a modified AXM mutagenesis[23,24] approach, Directed Evolution libraries (DEL6691, DEL6695 and DEL6698) were generated for three SEP-specific scFvs with a 1.6–1.9% mutation rate throughout the scFv gene (Supplementary Table S3A). SEP-specific scFvs derived from the SEP3–5 Discovery screens and uniquely identified against their target SEP epitope were prioritized for directed evolution. In parallel, each mutagenic library was independently screened (Directed Evolution screening, Fig. 1C) against the NAT peptides using two approaches. After the final round of selection, 44 clones from each of the six screening conditions (264 clones in total) were expressed as secretory scFvs and each cell supernatant was tested in ELISA against the NAT4 peptide and NeutrAvidin alone as a negative control. In parallel, the three Discovery parental clones were tested in the same manner. Directed Evolution clones with improved binding to the native splice junction were identified based on three criteria: (1) OD450 ≥ 0.2 against NAT4, (2) ≥ 2-fold signal against NAT4 over NeutrAvidin, and (3) ≥ 2-fold signal against NAT4 over the parental NAT4 signal. In brief, Directed Evolution screening was successful and yielded 78 hits with improved binding strength to the native splice junction peptide compared to their respective anti-SEP parental clone (Fig. 1C, Supplementary Table S3B). Notably, the vast majority of these hits were derived from the DEL6691 screens. It was expected that some screens would outperform others due to differences in the parental scFv sequences and therefore multiple unique Discovery clones were chosen to proceed to Directed Evolution screening.
Subsequently, 35 Directed Evolution improved clones were sequenced and three unique scFvs were identified (Fig. 1C, Supplementary Table S3B). Although the DEL6691 screen yielded a significantly greater hit rate than DEL6695 or DEL6698 in screening ELISAs, the majority of those hits were identical in sequence. All three evolved clones possessed at least one amino acid mutation in at least one CDR (Fig. 2A-B, right), thus supporting the ELISA data for Ab paratope evolution to the native splice junction epitope. For further binding validation, each scFv was tested in titration ELISAs to identify improved clones with ≥ 2-fold background-corrected signal against NAT4 over the parental NAT4 corrected signal for at least two titration points (Fig. 2A-B, right). Additionally, each scFv was tested against non-target peptide(s) to assess off-target binding events. Since DEL6698 Evolved Clone 1 demonstrated unexpected binding to “Non-target Peptide 1”, it was also tested against “Non-target Peptide 2” (which is nearly identical to “Non-target Peptide 1”) and “Non-target Peptide 3” to reveal no additional off-target binding events. All scFvs demonstrated 4- to 12-fold improved binding strength to the native splice junction peptide.
To further improve the binding affinity of the two DEL6691 evolved clones, another cycle of scFv mutagenesis was performed to generate Affinity Maturation libraries (AML6691 Clone 1 and AML6691 Clone 2; Supplementary Table S4A) and subsequently used in Affinity Maturation phage display screening against the NAT peptides. A combination of antigen reduction and nonbiotinylated peptide competition was used for stringency. In brief, ~58% of ELISA-positive hits demonstrated ≥ 40-fold signal improvement over their parental clone and 21 unique scFvs with CDR mutation(s) were identified (Supplementary Table S4B), thus supporting the ELISA data for affinity maturation of Ab paratopes. An additional 22 unique scFvs with CDR mutation(s) were identified in parallel screens (not discussed herein) for a total of 43 affinity-matured scFvs derived from the AML6691 Clone 1 and Clone 2 libraries. A subset of these scFvs were further tested for specificity to the exon 2/4 splice junction of CENP-A-ΔExon3.
Many Directed Evolution scFvs are specific for the exon 2/4 splice junction of CENP-A-ΔExon3
To determine if the Directed Evolution scFvs from the nnAA targeting strategy met the requirements of splice junction specificity, scFvs were tested in split-splice junction peptide ELISAs. Peptides containing the full exon 2/4 splice junction of CENP-A-ΔExon3 (“NAT3” and “NAT4”), exon 2 in the context of the canonical exon 2/3 splice junction (“Exon2”), and exon 4 in the context of the canonical exon 3/4 splice junction (“Exon4”) were used (Fig. 1A). A total of 29 evolved and affinity-matured scFvs, as well as their corresponding parental clones from Discovery and Directed Evolution screening, were expressed as secretory scFvs and each cell supernatant was tested in comparative ELISAs against the split-splice junction peptides (Fig. 3A, Supplementary Figs. S1, S2). Importantly, 20 scFvs (~69%) demonstrated 5- to 623-fold background-corrected signal against 1 μg/mL of NAT3 and/or NAT4 over the split-splice junction peptides, confirming specificity for the exon 2/4 splice junction.
Fig. 3. Directed Evolution clones demonstrate specificity for the exon 2/4 splice junction of CENP-A-ΔExon3 in split-splice junction ELISAs.
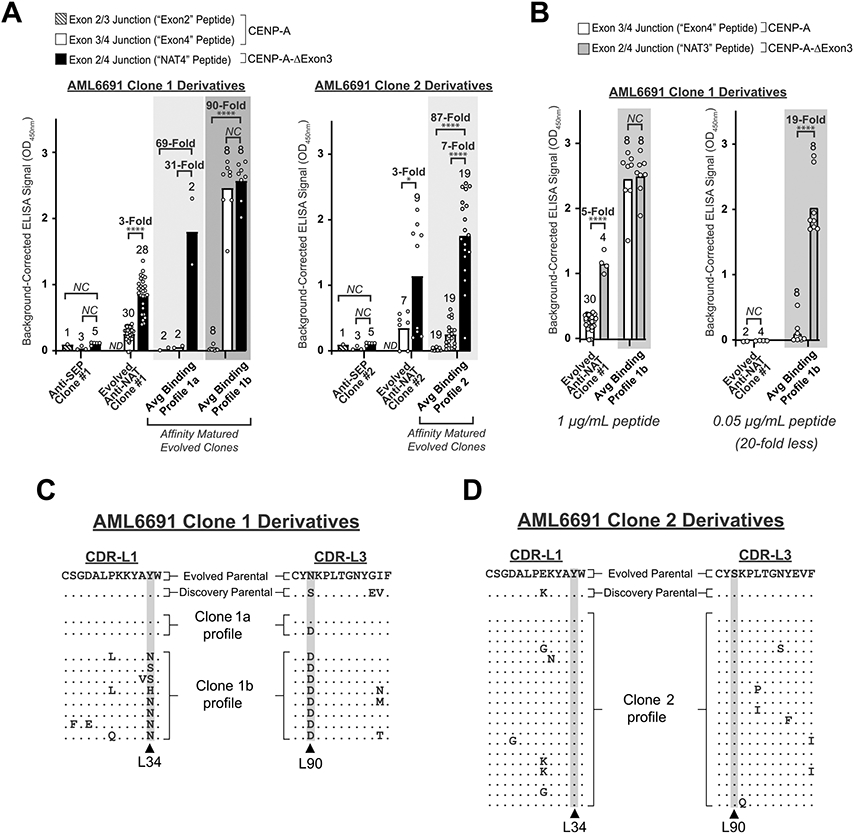
(A) Cell supernatant ELISA graphs comparing splice junction specificity between the Discovery anti-SEP parental scFv (see also Fig. 2A, left), the Directed Evolution anti-NAT parental scFvs (see also Fig. 2A, right), and the affinity matured Directed Evolution anti-NAT scFvs for AML6691 Clone 1 (left) and AML6691 Clone 2 (right). Three CENP-A-ΔExon3 splice junction-spanning peptides were tested: “NAT4” = exon 2/4 splice junction, “Exon2” = exon 2/3 splice junction, and “Exon4” = exon 3/4 splice junction as per Fig. 1A. Each white circle within each bar graph for the Anti-SEP and Evolved Anti-NAT clones represents a biological replicate for scFv expression and ELISA result; bar height represents the average ELISA result of biological replicates. Each white circle within each bar graph for the three “Avg Binding Profile” graphs represents a unique scFv, some of which have biological replicates (Supplementary Figs. S1 and S2) and the average is reported herein; each bar height represents the average ELISA result for the unique scFvs within that group. ND = No Data. NC = No Change. P-values (* = < 0.05, ** = < 0.01, *** = < 0.001, **** = < 0.0001) were calculated using an unpaired t test with Welch’s correction on square root OD450 values in GraphPad Prism on samples with at least two replicates. (B) Cell supernatant ELISA graphs for the AML6691 Clone 1 derivatives against 1 μg/mL (~400 nM) (left) and 0.05 μg/mL (~20 nM) (right) NAT3 (exon 2/4 splice junction) and Exon4 peptides. White circles, bar heights and p-values are defined as in (A). (C and D) CDR-L1 and CDR-L3 protein sequence alignments between the Directed Evolution parental scFv (line 1), the anti-SEP Discovery parental scFv (line 2), and the affinity matured progeny. An enrichment of mutations was identified in the AML6691 Clone 1 affinity matured progeny at Kabat positions L34 and L90 (C), but not in the AML6691 Clone 2 affinity matured progeny (D). No significant enrichment of mutations was identified in any region of the heavy or light chains of the AML6691 Clone 2 progeny.
However, some derivative scFvs from AML6691 Clone 1 maturation demonstrated a concentration-dependent specificity profile (“Avg Binding Profile 1b” in Fig. 3A, left). In these cases, significant binding to the Exon4 peptide was observed at higher peptide concentrations but eliminated with 20-fold less peptide, without impacting binding to the full exon 2/4 splice junction peptide (Fig. 3B and Supplementary Fig. S1B). This phenomenon can be attributed to two potential factors: (1) these cell supernatants were not normalized for scFv concentration, and (2) higher affinity scFvs yield saturating ELISAs signals at lower antigen concentrations. Since these splice junction-specific Abs must bind to residues in both exon 2 and exon 4 simultaneously, there is likely some cross-reactivity to either exon alone. By using a greater amount of either Ab or antigen in an ELISA the binding equilibrium shifts and results in saturating ELISA signals for both the NAT and Exon4 peptides, thus necessitating a lower concentration of scFv and/or peptide for these analyses. Altogether, 28 scFvs (~97%) were splice junction-specific with ≥ 5-fold preferential binding to the NAT peptide(s) over the split-splice junction peptides at either antigen concentration (Supplementary Figs. S1 and S2).
The concentration-dependent, splice junction-specific scFvs acquired enriched CDR-L1 and CDR-L3 mutations at Kabat positions L34 and L90, respectively (Fig. 3C). The enriched mutations at L34 appeared to correlate with improved exon 4 binding at higher peptide concentrations. Interestingly, the mutagenic hot spot in CDR-L3 (L90) is one of the same positions previously mutated and selected for during initial directed evolution (Fig. 3C, lines 1 and 2). Furthermore, in the canonical structure for variable light chain domains[25], L34 and L90 are in very close proximity to one another and may have co-evolved to contribute to a distinct paratope for exon 4. These observations indicate that CDR-L1 and/or CDR-L3 may participate in epitope binding directly, or indirectly by altering Ab structure. Derivative scFvs from AML6691 Clone 2 maturation did not acquire enriched mutations in either the light nor heavy chain (CDR-L1 and CDR-L3, Fig. 3D), probably due to sub-optimal screening conditions for this clone. Regardless, this data supports the feasibility of using the nnAA (SEP) targeting strategy and directed evolution to derive splice junction-specific Abs.
Application of the nnAA targeting strategy in vivo
An inherent limitation of this nnAA targeting strategy in vitro is the use of random mutagenesis throughout the Ab heavy and light chain genes to introduce beneficial mutations that would change Ab specificity from the nnAA pseudo-hapten to the native epitope. Capitalizing on the natural immune repertoire of rabbits and in vivo Ab mutagenesis, these mutations are introduced in a CDR-biased manner without manual intervention. Furthermore, the rapid and continuous cycling between Ab mutagenesis and affinity-based selection inherently streamlines the nnAA targeting strategy for directed evolution.
Four sets of peptide immunogens spanning the exon 2/4 splice junction of CENP-A-ΔExon3 were synthesized and two rabbit immunization strategies were performed (Fig. 4A): (1) co-immunizations with SEP6 and SEP7 followed by single immunizations with NAT6, and (2) single immunizations with NAT5 followed by single immunizations with NAT6. Antigen-specific Abs were isolated from each immunization strategy and produced as recombinant monoclonal Abs (mAbs) using B cell cloning. When tested in western blot against whole cell lysates overexpressing SNAP-tag fusion proteins of either the full-length CENP-ΔExon3 or canonical CENP-A, a range of Ab specificity patterns was observed (Fig. 4B) indicating that the isolated mAbs may recognize slightly different epitopes within the same peptide immunogen. Importantly, the majority of isolated mAbs demonstrated splice junction specificity with obvious and large binding differences between the two CENP-A isoforms.
Fig. 4. Application of a nnAA targeting strategy in vivo derived splice junction-specific Abs to unique epitopes when mapped at amino acid level resolution.

Workflow of the nnAA targeting strategy in vivo using SEP-containing peptide immunogens. Zoom boxes to the right reference peptide designs (as per Fig. 1 and Supplementary Table S6) and rabbit immunization strategies (analogous to Fig. 1). Color coded rabbits delineate SEP (purple) and NAT (yellow) immunization series. (B) Western blots using mAbs derived from each immunization strategy against equivalent amounts of mock-transfected FreeStyle™ 293-F whole cell lysates (Lane 1), canonical CENP-A transfected FreeStyle™ 293-F whole cell lysates (Lane 2), and CENP-A-ΔExon3 transfected FreeStyle™ 293-F whole cell lysates (Lane 3). Color coded rabbits reference the corresponding immunization strategy as per (A). All mAbs were tested at 1 μg/mL as purified protein and blots were exposed for five seconds except for mAb32 (*) which was tested as crude supernatant at a 1:10 dilution and exposed for 120 s. Nonspecific bands in the mAb14 and mAb16 blots are noted (•). (C) Summary of epitope scanning results for mAbs derived from each immunization strategy as per (A). Each residue of the exon 2/4 splice junction peptide was serially mutated to alanine or serine (if the native residue is an alanine) and used in ELISAs to determine the binding capabilities of each mAb. A reduction in binding activity (i.e., ELISA signal), as denoted by darker red squares, when a particular residue is mutated to alanine or serine indicates that the native residue at that position is important for mAb binding. Binding activity against mutant peptides is normalized to the native sequence peptide. The dotted line between native sequence positions L6 and A7 denotes the exon-exon junction (EEJ). All mAbs were tested as purified IgGs, except for mAb32 (*) which was tested as crude supernatant, at various dilutions. The nnAA targeting strategy derived mAbs with unique binding profiles (Epitope #1, #2, and #3) that were not observed from a conventional NAT immunization strategy (NAT Epitopes).
To further characterize the specificity of the mAbs derived from each immunization strategy, each was tested for binding in independent ELISAs against peptides with an alanine or serine residue replaced serially across the native exon 2/4 splice junction of CENP-A-ΔExon3 (Fig. 4C). An observed reduction in binding activity when a particular residue is mutated to alanine or serine indicates that the native residue at that position is important for mAb binding. MAbs with significant binding to the canonical CENP-A protein isoform (e.g., mAb5, mAb7 and mAb32, Fig. 4B) relied heavily on native residues in only one of the exons of the splice junction peptide (darker red squares, Fig. 4C). MAbs that were more specific for the CENP-A-ΔExon3 protein isoform (e.g., mAb14, mAb16, etc., Fig. 4B) relied heavily on residues in both exons of the splice junction peptide. Importantly, mAbs derived from the nnAA targeting strategy had unique epitope mapping results compared to mAbs derived from the conventional NAT immunization strategy (mAb31 and mAb32), supporting the feasibility of using the nnAA targeting strategy in vivo to derive highly specific Abs to the splice junction epitope at or near an amino acid level resolution.
Discussion
The availability of affinity reagents that can specifically detect alternatively spliced proteins is lacking, despite the observation that more than 90% of multi-exon genes in humans are alternatively spliced [2,3] and exon skipping is the predominant alternative splicing event in mammals[26]. Here, a unique hapten targeting strategy that takes advantage of modular Ab paratopes and Ab degeneracy after affinity maturation was successfully implemented to derive splice junction-specific Abs. A nnAA pseudo-hapten (SEP) was used as a targeting epitope in splice junction-spanning peptide antigens, selecting for Abs with paratopes specific for the SEP-containing region (Fig. 1B and Fig. 2A-B, left). In agreement with previous observations[22], phospho-specific CDRs incorporated into naïve phage display libraries act as “hot spots” for driving initial selection. This design strategy results in highly diverse Ab libraries with inherent propensity to bind phosphorylated residues via a predefined paratope (e.g., AXL41, Supplementary Table S2). With careful choice in the positioning of the nonsynonymous SEP in peptide antigens, SEP-specific Abs can be derived with predetermined specificity for one exon of the splice junction. Through a Directed Evolution screening strategy, the SEP-specific Abs can be evolved to bind to the unmodified, native splice junction epitope (Fig. 1C and Fig. 2A-B, right). This phenomenon of Ab degeneracy has previously been reported when attempting to affinity mature anti-SEP Abs without losing their SEP-specificity[27]. Capitalizing on this feature, here splice junction-specific Abs were derived with modular paratopes – a first region binding the native, nonphosphorylated epitope in one exon and a second region binding the opposite exon.
Although random mutagenesis was successful in driving directed evolution in vitro, restricting Ab mutagenesis only to the CDRs is expected to improve this process significantly. For Discovery clones with a known phospho-specific CDR, mutagenesis of this CDR alone and in combination with others can be used to determine the optimal strategy for paratope evolution. With improved genetic encoding of non-canonical amino acids during protein production[28] or by using non-native canonical amino acids as pseudo-haptens, the nnAA targeting strategy can be expanded beyond peptide antigens to full-length proteins or protein fragments which are more conformationally relevant.
While Ab development using in vivo methods is far less customizable than phage display methods, they offer significant advantages to improve this nnAA targeting strategy. By capitalizing on the natural rabbit immune repertoire and the continual cycling between Ab mutagenesis and affinity-based selection, a multitude of Ab frameworks are simultaneously selected for or against in vivo and the process of directed evolution occurs without any manual intervention. Unlike scFvs derived via phage display, Abs derived in vivo are functionally selected for in the most desired immunoglobulin format (i.e., IgG) and Ab mutagenesis is biased more towards CDRs. More studies are needed both in vitro and in vivo using a variety of splice junction sequences and SEP positions to further reveal patterns in paratope:epitope interactions and determine optimal phospho-peptide designs to robustly derive splice junction-specific Abs.
Conclusions
Abs are important affinity reagents used to specifically detect antigens of interest in a wide variety of biological assays. However, most Abs fail to distinguish between alternatively spliced protein isoforms, particularly those derived from exon-skipping events, due to high sequence similarities among isoforms. Many protein isoforms can be differentiated by unique splice junction epitopes, but require Ab development at near amino acid level precision. Here, splice junction-specific Ab derivation employing a nnAA pseudo-hapten (SEP) as a targeting epitope was successful. Using in vitro mutagenesis and directed evolution strategies, anti-SEP Abs were evolved to bind to the nonphosphorylated, native sequence splice junction. The resultant Abs demonstrated true splice junction specificity in ELISAs, where significant binding was only observed to peptides containing both exons of the splice junction and not to peptides that contained each exon independently. By comparing sequences of anti-SEP Abs and the resulting directed evolution Abs, mutations in the CDRs that support the paratope evolution were identified. This nnAA targeting strategy was also deployed in vivo, resulting in the successful derivation of splice junction-specific Abs. The binding epitopes of these Abs were mapped with an epitope scanning approach to reveal Abs that bind to distinct sets of splice junction residues not observed for those derived from a conventional immunization strategy. Thus, this nnAA targeting strategy can be exploited to predetermine an Ab’s specificity to a splice junction, or any antigen, at or near amino acid level resolution.
Supplementary Material
Acknowledgments
We would like to thank Dr. Margaret Kiss and Dr. Melissa Batonick for their discussions on experimental strategy. We thank Dr. Xiaofeng Li, Mary Ferguson, Meghan Kelly, Amanda Chapman, Holland Driscoll, Kezzia Jones, Shannon McBride, Nicole Pauloski, and Christine McCann for their laboratory support and collaboration on experimental design. We thank Dr. Jay Chaplin and Dr. Gillian Johnson for their reviews and comments while drafting this manuscript.
Funding
This work was supported in part by the Innovative Molecular Analysis Technologies (IMAT) Program from the National Institutes of Health [5R21CA240199]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding source was not involved in the research endeavors or manuscript preparation.
Abbreviations:
- Ab
antibody
- CDR-H
heavy chain complementarity determining region
- CDR-L
light chain complementarity determining region
- CENP-A
centromere protein A
- CENP-A-ΔExon3
centromere protein A alternative isoform with an exon 3 deletion
- EEJ
exon-exon junction
- ELISA
enzyme-linked immunosorbent assay
- mAb
monoclonal antibody
- MBP
maltose binding protein
- NAT
native sequence
- nnAA
nonsynonymous non-native amino acid
- scFv
single-chain variable fragment
- SEP
phosphoserine
Footnotes
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: EF is an employee of, and owns stock in, Abcam. MW owns stock in Abcam, and IP has been filed on some aspects of the methods described. RO declares no relevant financial or non-financial competing interests.
Statement of compliance
All animal experiments were performed in compliance with ARRIVE guidelines and were carried out in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals and the Animal Welfare Act.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.nbt.2022.06.003.
Data availability
Data will be made available on request.
References
- [1].McGuire AM, Pearson MD, Neafsey DE, Galagan JE. Cross-kingdom patterns of alternative splicing and splice recognition. Genome Biol 2008;9:R50. 10.1186/gb-2008-9-3-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008;40:1413–5. 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- [3].Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008;456:470–6. 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. R74-R74 Genome Biol 2004;5. 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Szafranski K, Fritsch C, Schumann F, Siebel L, Sinha R, Hampe J, et al. Physiological state co-regulates thousands of mammalian mRNA splicing events at tandem splice sites and alternative exons. Nucleic Acids Res 2014;42:8895–904. 10.1093/nar/gku532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, et al. Alternative splicing networks regulated by signaling in human T cells. RNA 2012;18:1029–40. 10.1261/rna.032243.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, Kalsotra A, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. 3603–3603 Nat Commun 2014;5. 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, et al. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci 2013;16:499–506. 10.1038/nn.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ergun A, Doran G, Costello JC, Paik HH, Collins JJ, Mathis D, et al. Differential splicing across immune system lineages. Proc Natl Acad Sci USA 2013;110:14324. 10.1073/pnas.1311839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nelson AM, Carew NT, Smith SM, Milcarek C. RNA Splicing in the Transition from B Cells to Antibody-Secreting Cells: The Influences of ELL2, Small Nuclear RNA, and Endoplasmic Reticulum Stress. J Immunol 2018;201:3073. 10.4049/jimmunol.1800557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kahles A, Lehmann K-V, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. e6 Cancer Cell 2018;34:211–24. 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stoler S, Keith KC, Curnick KE, Fitzgerald-Hayes M. A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev 1995;9: 573–86. 10.1101/gad.9.5.573. [DOI] [PubMed] [Google Scholar]
- [13].Palmer DK, O’Day K, Wener MH, Andrews BS, Margolis RL. A 17-kD centromere protein (CENP-A) copurifies with nucleosome core particles and with histones. J Cell Biol 1987;104:805–15. 10.1083/jcb.104.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Meluh PB, Yang P, Glowczewski L, Koshland D, Smith MM. Cse4p Is a Component of the Core Centromere of Saccharomyces cerevisiae. Cell 1998;94:607–13. 10.1016/S0092-8674(00)81602-5. [DOI] [PubMed] [Google Scholar]
- [15].Buchwitz BJ, Ahmad K, Moore LL, Roth MB, Henikoff S. A histone-H3-like protein in C. elegans. Nature 1999;401:547–8. 10.1038/44062. [DOI] [PubMed] [Google Scholar]
- [16].Howman EV, Fowler KJ, Newson AJ, Redward S, MacDonald AC, Kalitsis P, et al. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci USA 2000;97:1148–53. 10.1073/pnas.97.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Régnier V, Vagnarelli P, Fukagawa T, Zerjal T, Burns E, Trouche D, et al. CENP-A is required for accurate chromosome segregation and sustained kinetochore association of BubR1. Mol Cell Biol 2005;25:3967–81. 10.1128/MCB.25.10.3967-3981.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Batonick M, Holland EG, Busygina V, Alderman D, Kay BK, Weiner MP, et al. Platform for high-throughput antibody selection using synthetically-designed antibody libraries. N Biotechnol 2016;33:565–73. 10.1016/j.nbt.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. Bypassing immunization: Human antibodies from V-gene libraries displayed on phage. J Mol Biol 1991;222:581–97. 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- [20].McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990;348:552–4. 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- [21].Zhao Q, Buhr D, Gunter C, Frenette J, Ferguson M, Sanford E, et al. Rational library design by functional CDR resampling. N Biotechnol 2018;45:89–97. 10.1016/j.nbt.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Koerber JT, Thomsen ND, Hannigan BT, Degrado WF, Wells JA. Nature-inspired design of motif-specific antibody scaffolds. Nat Biotechnol 2013;31:916–21. 10.1038/nbt.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Holland EG, Acca FE, Belanger KM, Bylo ME, Kay BK, Weiner MP, et al. In vivo elimination of parental clones in general and site-directed mutagenesis. J Immunol Methods 2015;417:67–75. 10.1016/j.jim.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Holland EG, Buhr DL, Acca FE, Alderman D, Bovat K, Busygina V, et al. AXM mutagenesis: an efficient means for the production of libraries for directed evolution of proteins. J Immunol Methods 2013;394:55–61. 10.1016/j.jim.2013.05.003. [DOI] [PubMed] [Google Scholar]
- [25].Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987;196:901–17. 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- [26].Wang J, Ye Z, Huang TH, Shi H, Jin VX. Computational methods and correlation of exon-skipping events with splicing, transcription, and epigenetic factors. Methods Mol Biol 2017;1513:163–70. 10.1007/978-1-4939-6539-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li D, Wang L, Maziuk BF, Yao X, Wolozin B, Cho YK. Directed evolution of a picomolar-affinity, high-specificity antibody targeting phosphorylated tau. J Biol Chem 2018;293:12081–94. 10.1074/jbc.RA118.003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol 2015;11:496–503. 10.1038/nchembio.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further reading
- [1].Zakharova MV, Beletskaya IV, Kravetz AN, Pertzev AV, Mayorov SG, Shlyapnikov MG, et al. Cloning and sequence analysis of the plasmid-borne genes encoding the Eco29kI restriction and modification enzymes. Gene 1998;208: 177–82. 10.1016/S0378-1119(97)00637-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.