

Abstract
At-risk alcohol use is associated with multisystemic effects and end-organ injury, and significantly contributes to global health burden. Several alcohol-mediated mechanisms have been identified, with bioenergetic maladaptation gaining credence as an underlying pathophysiological mechanism contributing to cellular injury. This evidence-based review focuses on the current knowledge of alcohol-induced bioenergetic adaptations in metabolically active tissues: liver, cardiac and skeletal muscle, pancreas, and brain. Alcohol metabolism itself significantly interferes with bioenergetic pathways in tissues, particularly the liver. Alcohol decreases states of respiration in the electron transport chain, and activity and expression of respiratory complexes, with a net effect to decrease ATP content. In addition, alcohol dysregulates major metabolic pathways, including glycolysis, the tricarboxylic acid cycle, and fatty acid oxidation. These bioenergetic alterations are influenced by alcohol-mediated changes in mitochondrial morphology, biogenesis, and dynamics. The review highlights similarities and differences in bioenergetic adaptations according to tissue type, pattern of (acute vs. chronic) alcohol use, and energy substrate availability. The compromised bioenergetics synergizes with other critical pathophysiological mechanisms, including increased oxidative stress and accelerates cellular dysfunction, promoting senescence, programmed cell death, and end-organ injury.
Keywords: alcohol, bioenergetics, oxidative phosphorylation, glycolysis, metabolically active tissues
Introduction
At-risk alcohol use is the costliest form of substance use, with the global economic cost estimated to be about 1300 International dollars/adult, of which 38% are direct costs, and 60% due to loss of productivity.1 At-risk alcohol use significantly decreases life expectancy, is linked to more than 2 million years of potential life lost, and is a factor attributed to 5.3% of all annual deaths.2 Alcohol-associated pathophysiology is complex, ranging from the risk of injuries and poisoning resulting from acute intoxication to cumulative organ and tissue injury resulting from chronic at-risk alcohol use, and significant impact on mental health in individuals with alcohol use disorder (AUD).3–5 However, the severity and prognosis of alcohol-induced tissue injury varies between individuals and depends on factors such as genetic makeup, metabolism, age, gender, ethnicity, environment, and lifestyle.4
At-risk alcohol drinking is defined as consumption of more than 3 or 4 drinks on a given day or more than 7 or 14 drinks a week for women and men, respectively.6 Binge drinking, a pattern of alcohol consumption that elevates blood alcohol concentrations to equal or greater than 0.08% (80 mg/dL), generally resulting from consumption of 4–5 alcohol drinks over 2 h is also categorized as high risk.6 The percentage of the adult population that engages in at-risk alcohol drinking is not trivial, with approximately 18% of adults worldwide reporting heavy episodic drinking in the past month, and 6.6% of adults in the United States reporting at-risk alcohol use.7
Several mechanisms are implicated in alcohol-induced multiorgan injury, including alcohol metabolism, oxidative stress, gut dysbiosis and immune activation, inflammation, necrosis and programmed cell death, extracellular matrix remodeling, and epigenomic adaptations.8–22 Recent evidence supports the importance of bioenergetic adaptation as an underlying pathophysiological mechanism shared across tissues affected by alcohol that culminates in metabolic instability and end-organ injury. This evidence-based review focuses exclusively on the current knowledge of alcohol-induced bioenergetic adaptations in metabolically active tissues. For reviews on mechanisms of alcohol-induced tissue injury, please refer to Boyd et al. (2022), Massey et al. (2015), McMahan et al. (2020), Molina et al. (2014), Mandrekar and Szabo (2009), Osna et al. (2022), Simon et al. (2021), and Simon et al. (2022) 23–30 For reviews on alcohol-mediated mitochondrial adaptations, please refer to Hoek et al. (2002), Bailey (2003), Garcia-Ruiz et al. (2013), Nassir and Ibdah (2014), Han et al. (2017), Steiner and Lang (2017), Shang et al. (2020), and Hao et al. (2021).31–38
Relevance of Altered Bioenergetics to Alcohol-induced Tissue Injury
Bioenergetics in the strict sense is described as the oxidation reactions that occur together with the passage of electrons through mitochondrial membrane protein complexes and coenzymes and is coupled to ATP synthesis. This chemiosmotic hypothesis of oxidative phosphorylation (OXPHOS) was proposed by Peter Mitchell in 1961.39 Bioenergetic and metabolism research has since exponentially grown with advances in state-of-the-art techniques to conduct bioenergetic measurements, and their integration with metabolomic, proteomic, and epigenomic data. This approach has significantly improved our understanding of how bioenergetics mechanistically modulate health and disease. At the core of cellular bioenergetics is the mitochondrion, the cell's powerhouse.
Mammalian cell adaptations to energy resources and demands allowed for the evolution of mitochondria with mitochondrial DNA (mtDNA) retaining core genes controlling energy production. Depending on the cellular demands, each cell has a few hundred to thousands of mtDNA copies.40 The cellular energy demands are met through intricate communication between the mitochondria, nucleus, and cytoplasm based on substrate availability, reducing equivalents, and reactive oxygen species (ROS) production.41
Cellular metabolism of energy substrates (glucose, fats, and amino acids) generates ATP needed to meet energy demands and regulates cellular redox state. Glucose is oxidized to pyruvate and in mitochondria containing cells forms acetyl CoA. Fatty acids, on the other hand, are oxidized via the mitochondrial β oxidation pathway to generate acetyl-CoA. Glucogenic amino acids can be converted into glucose, pyruvate or a tricarboxylic acid (TCA) cycle intermediate, and ketogenic amino acids to fat, acetyl CoA or acetoacetyl CoA. The acetyl CoA generated enters the TCA cycle producing Nicotinamide adenine dinucleotide + hydrogen (NADH). The NADH enters the electron transport chain (ETC) and is oxidized by complex I (CI) by transferring electrons (e−) to flavin mononucleotide, iron–sulfur clusters (Fe–S), and coenzyme Q, respectively. Succinate, another intermediate of the TCA cycle is oxidized by CII transferring e− to flavin adenine dinucleotide (FAD), Fe–S and coenzyme Q. Cytochrome c reductase (CIII), on the other hand, accepts e− in 2 steps where ubiquinone (CoQ) and ubiquinol (CoQH2) binds to CIII, and this cycle is repeated one more time (Q cycle). The e− are finally transferred to oxygen by CIV (cytochrome c oxidase).42 Together with the release of energy, as the e− pass through the redox proton pumps (CI, III, and IV), there is also transport of protons (H+) across the mitochondrial inner membrane to generate a trans-inner membrane electrochemical potential. When ATP is low, H+ enters the matrix through ATP synthase to generate ATP, which is then exported to the cytoplasm by adenine nucleotide translocators (ANTs).43 Because this process of ADP phosphorylation resulting in ATP production requires oxygen, it is known as OXPHOS (Figure 1). This process of mitochondrial ATP synthesis is critically dependent on Ca2+ uptake by the mitochondrial calcium uptake complex.44–48 Additionally, elegant studies in the Sollott laboratory demonstrated that ATP synthase also serves as the mitochondrial K+ transporter and proposed that the transport of both K+ and H+ is responsible for ATP synthesis.49,50 The efficiency with which ETC generates ATP is termed coupling efficiency. In a tightly coupled ETC system, there is maximal ATP production for each calorie used. On the contrary, in a less coupled system, there has to be higher caloric intake for the same amount of ATP to be produced.41
Figure 1.

Bioenergetic mechanisms in the cell. Energy substrates are metabolized via glycolysis, TCA cycle and β-oxidation pathways during which electrons (e−) are transferred to mitochondrial NAD+ and FAD through complex I and II, initiating the ETC. Energy released as e− pass through complexes I, III, and IV generate a trans-inner membrane electrochemical potential. Protons enter the matrix through ATP synthase to generate ATP. OCR can be measured in cells using EFA (Seahorse technology) providing measures of OXPHOS and proton leak. ECAR is measured using EFA indicating lactate produced via anaerobic glycolysis.
Chance and Williams described 5 states of mitochondrial respiration using the oxygraphy protocol (Figure 2).
Figure 2.
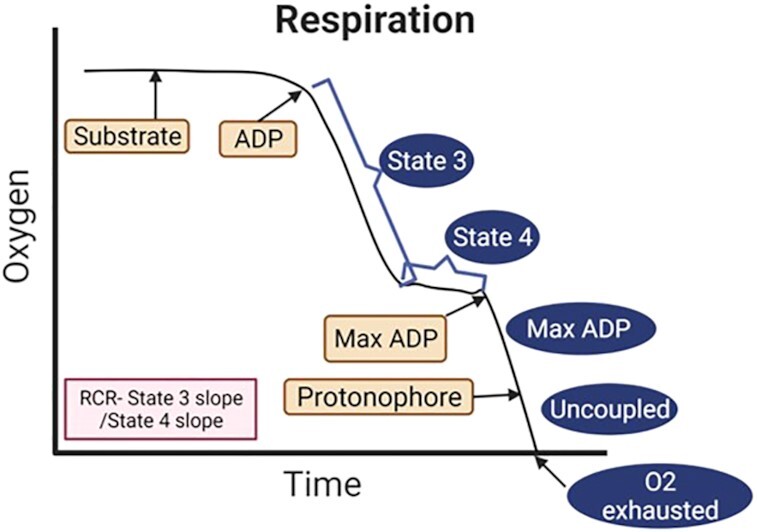
Bioenergetics measured with Clarke electrodes on permeabilized cells or isolated mitochondria. Oxygen consumption is measured in the presence of mitochondrial substrates with or without ADP. State 3 respiration is the maximally activated state and is respiration in the presence of substrates and ADP. State 4 respiration is a resting coupled state and is respiration measured in the presence of substrates alone. Maximal ADP respiration (Max ADP) can be measured if a maximal dose of ADP is added. Uncoupled respiration can be measured after adding a protonophore. RCR is the ratio of respiration state 3/state 4.
State 1 is obtained when the ADP and substrate levels are low, and the respiration rate is slow.
State 2 is obtained when a high concentration of ADP is added, but the respiration rate is low as substrate levels are low.
State 3 is induced when substrates are added, and together with the high ADP concentration already available there is low ATP/ADP ratio, and high oxygen consumption (JO2).51
State 4 has high ATP/ADP ratio and low ADP levels. The JO2 levels are low as oxygen consumption is exclusively from proton leak.52
State 5 indicates ROX or anoxia53 . An increase in state 4 respiration or decreased respiratory coupling ratio (ie, state 3 JO2/state 4 JO2) suggests decreased coupling efficiency.54
The redox state is a balance between ROS produced during cellular metabolism, and the antioxidant system that scavenges ROS. Mitochondrial ROS contain unpaired e− and partial reduction of this molecular oxygen produces superoxide (O2•−) and hydrogen peroxide (H2O2).55,56 O2•− and H2O2 can react with transition metal ions promoting further radical generation, including the highly reactive hydroxyl radical (•OH).41,57 Moreover, a decrease in the trans-inner membrane electrochemical potential induces mitochondrial permeability transition pore (mtPTP), a self-destructing mechanism, that is activated by increases in mitochondrial matrix Ca2+ levels or ROS.41 Several antioxidant mechanisms are in place to quench excess ROS generation. Mitochondrial NADPH + H+ reduces oxidized glutathione (GSSG) to glutathione (2GSH) using glutathione peroxidases.41 Similarly, oxidized thioredoxin-S2 is reduced by NADPH and thioredoxin reductase.58 The matrix Mn superoxide dismutase (MnSOD) and Cu/ZnSOD (also present in the inner mitochondrial membrane), convert O2•− to H2O2. In addition, mitochondrial thioredoxin-2 interacts with mitochondrial peroxiredoxin-3 to decrease ROS.59 Thus, as the oxidative/antioxidative balance is tipped from cell's antioxidant defenses to ROS production, damage of lipids, proteins, and nucleic acids lead to oxidative stress, cell injury, and eventual cell death.34
The overall cellular bioenergetic capacity relies on mitochondrial abundance and quality (Figure 3). Mitochondrial biogenesis allows for increase in size and number of mitochondria and is regulated by peroxisome proliferator-activated receptor (PPAR) γ coactivator 1-/alpha and beta (PGC-1α/b), cotranscriptional factors and its interactions with transcription factors/proteins such as nuclear respiratory factors (NRF-1 and NRF-2), and mitochondrial transcription factor A (TFAM). Together, uncoupling proteins (UCP2), PPARs, thyroid hormone, glucocorticoid, and estrogen and estrogen-related receptors α and γ modulate mitochondrial biogenesis.6061 Studies indicate that bioenergetics is also intricately linked to mitochondrial dynamics involving fusion and fission events.62–64 Optic atrophy protein 1 (OPA1) and mitofusins 1 and 2 (MFN1 and MFN2) are involved in mitochondrial fusion.65–67 Mitochondrial fission is regulated by GTPase dynamin related protein 1 (DRP168), mitochondria fission factor (MFF),69 and fission 1 protein (FIS1).70 In addition, MFN2 is critical for OXPHOS,71–73 and decreased MFN1 and OPA1 affect mitochondrial membrane potential, respiration, and reduce the stability of supercomplexes.74–76 As discussed, mitochondria are susceptible to ROS, and mitochondrial repair processes including mitophagy are critical in preventing propagation of oxidative stress. Alcohol-mediated effects on mitochondrial biogenesis, dynamics, and mitophagy are reviewed elsewhere.30,31,34,77–81
Figure 3.

Mitochondrial morphological and functional integrity. Mitochondrial quantity is maintained by biogenesis that is regulated by the major genes, y PPAR γ coactivator 1-/alpha and beta (PGC-1α/b), nuclear respiratory factors (NRF-1 and NRF-2), TFAM. Bioenergetics are closely linked to mitochondrial dynamics that includes fission and fusion events. OPA1 and MFN1 and MFN2 are involved in mitochondrial fusion and mitochondrial fission is regulated by GTPase DRP1, MFF, and fission 1 protein (FIS1). The major mitochondrial repair processes including mitophagy and the major genes implicated are autophagy-related gene (ATG), PINK/PARKIN and microtubule-associated protein 1 light chain 3 (LC3). Some of the major alcohol-mediated changes in mitochondrial morphology include mitochondrial swelling, membrane damage, disoriented cristae, and paracrystalline inclusions.
Experimental Assessment of Cellular Bioenergetics
To better interpret the alcohol-induced alterations in cellular bioenergetics, it is important to have a basic understanding of currently available tools. Metabolic flux analysis provides the highest resolution for bioenergetic measurements through the use of isotopomer labeling patterns to measure internal fluxes.82 In addition, real-time bioenergetic assays using phosphorescent probes measuring external fluxes, and paired with metabolic substrate and inhibitor protocols allow bioenergetic measurements in short time scales. These popular low molecular resolution assays include respirometry and extracellular flux analysis (EFA).83 Respirometry, a potentiometric method that uses platinum and silver electrodes (Clark Oxygraph-2 K; Oroboros) is the most used.84,85 The breakthrough Seahorse technology (Agilent Technologies) measures oxygen consumed, and protons released in a very small volume of media just above the cell monolayer (transient microchamber) using sensor probes (www.seahorsebio.com). This also allows for sequential addition of defined metabolic inhibitors to dynamically measure cellular bioenergetics and mitochondrial function.40,61,86,87 The disadvantage with most of these systems is that they use extreme assay conditions and may not recapitulate the balance of free energies that are available in vivo.88 Here, we briefly discuss the outcome measures using these assays.
Respirometry using Clark electrodes is performed using permeabilized cells or isolated mitochondria. Oxygen consumption is monitored in the presence of mitochondrial substrates (glutamate/malate—complex I substrates; succinate—complex II substrates) with or without ADP to measure respiratory states (Figure 2).89OroborosO2k (Oroboros Instruments) is a 2-chamber respirometer with polarographic oxygen sensors allowing for oxygen calibrations and the results integrated using DatLab software. This allows for high resolution respirometry and has evolved over the years allowing for new standards in bioenergetic research. The disadvantage compared to XF96 is that only 2 samples can be run in parallel, but it allows functional examination of the individual mitochondrial respiratory chain complexes.90,91
XF96 EFA—Intact cells or isolated mitochondria can be used to measure oxygen consumption using the popular Mito stress test. This assay measures (A) basal oxygen consumption rate (OCR): the cell's capacity to meet baseline energy demands and indicates ATP requirement from proton leak. (B) ATP production: this is measured by injecting oligomycin, an ATP synthase inhibitor, and represents basal respiration that drives ATP production. (C) Proton leak: basal respiration that is not coupled to ATP production. An increased proton leak might indicate less efficient mitochondria. (D) Maximal OCR is measured by adding carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone, a potent uncoupler of mitochondrial OXPHOS. Decreases in OCR reflect deficits in mitochondrial biogenesis, damage to mtDNA or the respiration machinery, or limitations in substrate availability or transport. Other measures include (E) reserve capacity: represents the cell's ability to respond to energy demands and (F) nonmitochondrial OCR: measures nonmitochondrial oxygen consumption after the addition of rotenone (CI inhibitor) and antimycin (CIII inhibitor). Generally, this indicates ROS generation or other oxygen-consuming processes, including proinflammatory enzyme activity. Other outcomes that can be measured are respiratory chain complex activities40 and Bioenergetic Health Index (BHI)—log [(reserve capacity × ATP-linked OCR)/nonmitochondrial OCR × proton leak OCR]92 (Figure 4). Though SeaHorse technology has allowed for high throughput screening, is user friendly, and allows for measures in small quantities of samples, Oroboros measures are more sensitive and accurate. Nevertheless, significant improvements are being made in both these technologies to mimic physiological conditions and make them robust systems for bioenergetic measurements.93,94
Figure 4.

EFA using SeaHorse technology. The Mitostress test is commonly used for bioenergetic measures. It measures basal OCR-baseline oxygen consumption. ATP-linked OCR: basal activity linked to ATP production and the level of proton leak (after injecting ATP synthase inhibitor, oligomycin). Proton-leak OCR: mitochondrial OCR that is not oligomycin-sensitive and shows movement of protons, cations, or substrates across the mitochondrial inner membrane. Maximal OCR: the maximal oxygen respiration (after injecting OXPHOS uncoupler). Nonmitochondrial OCR: ROS generation or other oxygen-consuming processes, including proinflammatory enzymes.
Extracellular acidification rate (ECAR), measured either potentiometrically (EFA) or with fluorescent organic cations, is a measure of lactate produced reflecting glycolysis.95,96 Other measures include redox potential of NADH using autofluorescence97,98; enzyme activity assays97; determinations of pH and ion gradients using fluorometric probes99,100; and genetically encoded probes to measure changes of intracellular pH, GSH/GSSG ratio, and NAD(P)H redox state.88
Alcohol and Alcohol Metabolites Contribute to Bioenergetic Adaptations
Seminal studies on alcohol metabolism were performed in the early 1900 s. The 1955 Nobel Prize in Medicine was awarded to Dr. Hugo Theorell for his valuable contributions to unraveling alcohol metabolism, including the development of an enzymatic method of alcohol determination. Virtually all tissues metabolize alcohol, with liver accounting for the greatest percentage of alcohol oxidation, posing a major metabolic burden on this organ.22 Though alcohol has a caloric value of 7 kcal/g, unlike other energy substrates, alcohol is not stored in tissues and remains in the body water until eliminated. In contrast to other macronutrients, alcohol elimination rate is not hormonally regulated.22
Alcohol is metabolized by alcohol dehydrogenase (ADH) mediated conversion to acetaldehyde in the cytoplasm, generating NADH. The reoxidation of the ADH–NADH complex is the rate-limiting step in ethanol oxidation, as the NADH generated must enter mitochondria for oxidation in the ETC.101 Because NADH is impermeable, it uses the malate–aspartate or α-glycerophosphate shuttles to enter the mitochondria. Thus, the cellular redox state during alcohol metabolism depends on both the shuttling capacity and efficiency of the ETC. Acetaldehyde is converted to acetate by acetaldehyde dehydrogenase (ALDH) in the mitochondria, also generating NADH and contributing to a decreased NAD+/NADH ratio. High acetaldehyde concentrations of 0.6–1.5 m m (reported blood levels are in the µm range102,103) are potent inhibitors of the shuttles and the extramitochondrial system that oxidizes NADH is less sensitive to acetaldehyde. Additionally, acetaldehyde inhibits transport of glutamate, phosphate, and citrate that are integral parts of the shuttles, reducing the efficiency of shuttles to transport NADH.104 Thus, the overall acetaldehyde-induced inhibition of the NADH shuttles suggests that acetaldehyde production may be a limiting factor in ethanol metabolism.
Acetate production from ethanol oxidation that is catalyzed by ADH1 occurs primarily in the liver and accounts for most of the hepatic oxygen consumption.105 Acetate can enter metabolic pathways like those used by carbohydrates, fats, and proteins. However, the high NADH levels generated during ethanol oxidation in the liver prevent acetate from entering the TCA cycle because of a reduction of cytoplasmic and intramitochondrial NAD.106 NAD+ is a coenzyme essential for multiple metabolic pathways, including glycolysis, TCA, OXPHOS, and pyruvate dehydrogenase complex. The enzymes in the hydride transfer catalyzes reduction of NAD+ into NADH. Mitochondrial NADH is then utilized by the ETC and is a substrate for ATP production. Thus, an overall decreased NAD+/NADH ratio decreases cellular oxidative metabolism.107 NAD+/NADH ratio also regulates the intracellular redox state, and an increase in NADH generated by alcohol oxidation leads to increased ROS. The decreased NAD+/NADH redox ratio and acetaldehyde formation decreases mitochondrial glutathione (mGSH) levels and decreases the antioxidant reserve of the cell. This alcohol-induced decreased mGSH levels are due partly to the impaired transport of GSH through the inner mitochondrial membrane.108 Additionally, although chronic ethanol exposure does not alter the cytoplasmic redox ratio, the mitochondrial ratio is decreased.109 Ethanol decreases pyruvate levels in the liver110 and the resulting decreased pyruvate carboxylase activity decreases gluconeogenesis.111 All these metabolic alterations ultimately increase hepatic fatty acid synthesis, which together with increased uptake lead to hepatic lipid accumulation.111 Acetate produced by ethanol metabolism can be utilized as energy substrate by tissues, mainly brain, and skeletal and cardiac muscle. Elevated acetate concentrations lead to decreased beta oxidation of long chain fatty acids, the preferred energy substrate in the cardiac and skeletal muscle.112,113 In addition, high acetate levels also increase acetylation of proteins, and affect gene expression by increasing histone acetylation that in turn can alter metabolic and other key functional pathways in specific tissues.111 Thus, the overall decrease in NAD+/NADH ratio and the depleted antioxidant levels inhibit key metabolic pathways, including glycolysis, TCA, fatty acid oxidation (FAO), and gluconeogenesis resulting in decreased ATP production (Figure 5).
Figure 5.

Alcohol metabolism and bioenergetic adaptations. ADH converts alcohol to acetaldehyde in the cytoplasm together with the generation of NADH. NADH is impermeable and uses shuttles to enter the mitochondria. Acetaldehyde is converted to acetate by acetaldehyde dehydrogenase (ALDH), which decreases NAD+/NADH ratio. Acetaldehyde is a potent inhibitor of the shuttles. The acetate generated enters metabolic pathways. But the overall decrease in NAD+/NADH ratio and the depleted antioxidant levels inhibit key metabolic pathways, including glycolysis, TCA, FAO, and gluconeogenesis resulting in decreased ATP production. Chronic heavy alcohol use induces hepatic cytochrome p450 2E1 (CYP2E1) to metabolize alcohol, which produces high amounts of ROS and impairs antioxidant mechanisms. Microsomal enzymes metabolize alcohol and results in ineffective ATP coupling and net energy loss by thermogenesis. Catalase in peroxisomes also metabolizes alcohol. Alcohol can be metabolized by nonoxidative pathways resulting in enzymatic conjugation to fatty acids, sulfate, glucuronic acid, and phospholipids resulting in the generation of FAEEs, phosphatidylethanol, EtS, and EtG and phosphatidylethanol.
Chronic at-risk alcohol use induces additional alcohol metabolic pathways, including hepatic cytochrome p450 2E1 (CYP2E1). Alcohol metabolism through this system produces considerable amounts of ROS and impairs antioxidant mechanisms.114–116 In addition, microsomal enzymes are activated in the liver and other tissues,117,118 which results in ineffective ATP coupling and net energy loss by thermogenesis.119 Some alcohol metabolism occurs by catalase in peroxisomes, though this a minor oxidative pathway. A small fraction of alcohol can be metabolized by nonoxidative pathways resulting in enzymatic conjugation to endogenous metabolites (fatty acids, phospholipids, sulfate, and glucuronic acid) resulting in the generation of fatty acid ethyl esters (FAEEs), phosphatidylethanol, ethyl sulfate (EtS), and ethyl glucuronide (EtG). FAEEs generation is catalyzed by FAEE synthase that is present especially in the brain, heart, liver, and pancreas120 (Figure 5).
In summary, alcohol metabolism especially in the liver leads to bioenergetic changes that may potentially contribute to alcohol-induced cellular injury as described in the next section. Alcohol metabolism-mediated oxidative stress is also a major mechanism of alcohol-induced tissue injury, as reviewed elsewhere.30,34,36,77,121,122
Alcohol Modulates Cellular Metabolism
Published evidence of alcohol's effects on energy metabolism dates to mid-20th century. In seminal studies by Ammon and Estler using acute and chronic alcohol administration in mice, decreased ATP content was considered the principal cause for functional cellular damage rather than direct toxic effects of alcohol.123 This was later confirmed in preclinical models using chronic ethanol feeding.124–127 The implication for alcohol metabolites was supported by studies showing that acetaldehyde in very high concentrations decreases ATP synthesis in hepatic mitochondria.128,129
Thus, our understanding of alcohol-induced modulation of cellular metabolism is informed by the data from preclinical models coupled with ex-vivo exposure of cells and tissues to alcohol. The most frequently used chronic alcohol feeding models and their salient features are summarized here. Alcohol self-administration of liquid diets, most frequently the Lieber–DeCarli alcohol diet, providing 100% of daily calories is compared to isocalorically matched control diet.130 Other models include a drinking-in-the dark paradigm, where animals have limited periods of access to alcohol in water during the dark cycle for several days or weeks.131 Systemic alcohol injections, generally using intraperitoneal injections,132,133 or intragastric (IG) infusion through surgically implanted intragastric tubes, (eg, Tsukamoto–French model134), or gavages to avoid the influence of taste. In addition, genetically modified mice or rats bred to generate animals with greater or less sensitivity or preference for alcohol have also been used to examine the impact of alcohol on tissue injury.135,136 None of these methods provide a perfect reflection of human pattern, quantity, or frequency of alcohol consumption. However, each model provides insight into differential tissue and organ pathophysiological alterations resulting from the different models of alcohol exposure. Moreover, the integration of results from these in vivo studies with those obtained from ex vivo and in vitro ethanol treatment of cells or tissues allow for interrogation of mechanistic hypotheses.
Alcohol-mediated Bioenergetic Adaptations-evidence From Metabolically Active Tissues Liver
The liver parenchymal cells are rich in mitochondria and are the main sites for alcohol metabolism. Findings from ex vivo, in vitro, preclinical, and clinical studies have demonstrated that alterations in stages of respiration in the ETC, respiratory complexes, glycolysis, ATP content, mitochondrial morphology, and mtDNA mutations initiate and contribute to the progression of alcohol-related liver disease (Figure 6).
Figure 6.

Bioenergetic adaptations in metabolically active tissues. The major alcohol-mediated bioenergetic adaptations in the liver, brain, cardiac and skeletal muscle, and alveolar macrophages.
Alcohol, particularly chronic alcohol, exposure decreases bioenergetic respiratory rates in hepatocytes and isolated hepatic mitochondria. Chronic alcohol feeding decreases both coupled and uncoupled respiratory rates, through direct effects on respiration itself, and the coupling efficiency, with a concomitant decrease in ATP synthesis rate.137,138 Liver mitochondria isolated from rats administered alcohol in drinking water for 10 wk or fed an alcohol diet had significantly decreased state 4 respiratory rate without significant changes in the proton motive force and OXPHOS at coupling site II. These studies showed that decreased activity of the ETC components, which control coupled but not uncoupled respiration, for example, cytochrome c oxidase activity was affected by alcohol.139,140 Liver mitochondria isolated from Lieber–DeCarli alcohol diet fed rats had decreased state 3 respiration rate when multiple substrates were used, and this was associated with decreased CIII and ATP synthase activities.141,142 Thayer and Rottenberg also showed that alcohol decreased states 3 and 4 respiratory rates and respiratory control ratio (RCR) in liver mitochondria.143 Similarly, liver mitochondria had a significant decrease in state 3 respiration and RCR with increased sensitivity to nitric oxide-dependent inhibition of respiration.144 In baboons that were administered alcohol and progressed to fatty liver or advanced fibrosis stages, state 3 respiration was decreased with a decrease in ADP/O ratio and RCR with all substrates. Concomitantly, there was decreased cytochrome expression and glutamate, NADH and succinate dehydrogenase (SDH) activities.145 Even moderate alcohol use (10% ethanol for 4 wk) decreased states 3 and 4 respiration with both glutamate–malate and succinate as substrates, decreased cytochrome c oxidase activity and cytochrome aa3 content, increased SDH activity, and increased Vmax of ANT activity in liver mitochondria, suggesting that any amount of alcohol consumption increases bioenergetic burden.146 The bioenergetic burden is associated with alcohol-mediated hypoxia and is proposed as a significant factor that contributes to hepatotoxicity and necrosis in both peri-venous and peri-portal hepatocytes. Additionally, hepatocytes from alcohol fed-rats had decreased mitochondrial bioenergetic reserve capacity and greater sensitivity to nitric-oxide dependent inhibition of respiration under both normal and hypoxic conditions providing support to the concept that nitric oxide and alcohol increase susceptibility to hypoxia and exacerbate alcohol-induced hepatic injury.147
In addition to alcohol's direct effects on respiration, effects on enzyme expression and activity also contribute significantly to decreased respiration. Chronic alcohol decreases expression of hepatic cytochromes a and b, and cytochrome c oxidase activity.148,149 Male rats fed a Lieber–DeCarli alcohol diet had decreased expression of hepatic mitochondrial respiratory complexes I, III, IV, and V. This was also associated with decreased expression of key genes that regulate mitochondrial function, including PGC-1α, NRF1, TFAM, and mtDNA.150 In vitro ethanol treatment of hepatocytes for 24 h or isolated mitochondria for 3 h, decreased CI and IV enzyme activity. Ethanol also decreased hepatocyte ADP translocase activity and SDH expression.151 As it is known, mitochondrial ETC are organized in supramolecular complexes called respirosomes152 that not only increase efficient transfer between complexes but also reduce electron leakage. However, cells expressing mitochondrion targeted CYP2E1 have significantly lower levels of respirosomes, and both in vivo and in vitro alcohol exacerbate the depletion of these respirosomes. Though a major portion of CYP2E1 is in the endoplasmic reticulum (ER), it is significantly expressed in other organelles, including the mitochondria.153,154 Mitochondrial CYP2E1 is increased in alcohol-treated animals,155 and together with cytochrome c oxidase plays a critical role in alcohol-mediated mitochondrial respiration. Although alcohol significantly decreases mtDNA and mitochondrial genome-encoded mRNA levels, these are potentially secondary effects. Moreover, near complete recovery of respirosomes and DNA damage by mitochondrion-targeted antioxidants and CYP2E1 inhibitors suggest their potential as therapeutic targets to counter alcohol toxicity and tissue damage.156 Mitochondrial bioenergetic adaptations are also influenced by alcohol-induced ER stress. Integrating data obtained from activating transcription factor 4 (ATF4) KO mice, alcoholic hepatitis subjects, and liver specific TFAM over expression revealed ATF4 activation to be a key mediator of ER stress that impairs mitochondrial biogenesis and respiratory function through TFAM-mediated pathways.38
The route of alcohol administration appears to have subtle differential effects on bioenergetic adaptations of liver mitochondria. For example, rats that self-administer an alcohol liquid diet have decreased glutamate/malate-, acetaldehyde- and succinate-driven state 3 respiration, RCR, and expression of CI, III, IV, and V. However, intragastric (IG) alcohol infusion increased glutamate/malate- and acetaldehyde-driven respiration, NAD+/NADH ratio, and decreased succinate-driven respiration.35 These differences may be due to different alcohol concentrations achieved with intragastric administration vs. consumption of alcohol in a liquid diet. IG feeding was also associated with increased respiration driven by acetaldehyde, despite decreased complex I and III protein levels. This could be attributed to a phenomenon known as the mitochondrial threshold effect (excess and reserve of mRNA, tRNA, and respiratory chain complexes).157 This serves as a protective mechanism especially when there are mtDNA mutations and in the presence of chronic stressors such as alcohol. In addition, differential responses are observed according to species used in experimental alcohol administration. In rats, it appears that there is an excess expression of respiratory proteins, which can account for the discrepancy in the correlation between the levels of respiratory complex proteins and mitochondrial respiration. While in mice, the expression of respiratory proteins is not increased, and there is a strong correlation between mitochondrial respiration and expression of respiratory proteins.35 Both alcohol oral self-administration and intragastric infusion increased mitochondrial respiration when glycerol-3-phosphate (which delivers electrons from cytoplasmic NADH to mitochondria) and octanoate (a substrate for beta-oxidation) are used.35 Similarly, dietary composition may also modulate bioenergetic adaptations. In rats fed a high-fat, adequate-protein diet, chronic alcohol decreased state 3 and 4 respiratory rates with both malate–glutamate and succinate substrates. Interestingly, these changes were not observed in high-protein low-fat diets. RCR was also significantly reduced with NAD-linked substrates with high-fat adequate-protein but not high-protein low-fat diet.158 Finally, substrate availability dictates metabolic function. In the setting of limited pyruvate turnover, alcohol decreases glucose and increases lactate production. However, in the presence of alanine (gluconeogenic precursor), alcohol increases total glucose production in fed animals but decreases production in fasted animals, with a concomitant increase in lactic acid.159
Alcohol also mediates glycolytic adaptations, which together with impaired OXPHOS result in a net reduction of ATP and consequent cellular dysfunction. A single oral gavage of 5 g of alcohol/kg body weight decreased ATP generation by glycolysis and maximally increased oxygen uptake in isolated perfused liver 2.5 h after gavage (swift increase in alcohol metabolism).160 This decrease is partly contributed by alcohol oxidation itself, especially when malate/aspartate shuttle intermediates are depleted. The reducing equivalents generated during ethanol oxidation compete with that of glycolysis for transfer to the mitochondria and thus inhibit aerobic glycolysis. Simultaneously, there is a decrease in cytoplasmic NAD+/NADH ratio that decreases anaerobic glycolysis.161 Chronic alcohol interferes with the glycolytic pathway in both normoxic and hypoxic environments in the step between glucose and glyceraldehyde-3-phosphate.162,163 And as discussed with OXPHOS, in the perivenous region of the liver, decreased oxygen tension because of ethanol oxidation significantly contributes to decreased glycolytic ATP production thus increasing the risk for liver injury.164
Whether mitochondrial morphological changes are the result of bioenergetic adaptations or direct effects of alcohol is not fully understood (Figure 3). Mitochondria are often enlarged, display bizarre shapes, have disoriented cristae, and have paracrystalline inclusions.165–167 However, mitochondrial protein content per mtDNA is unchanged and morphological alterations appear to reflect mitochondrial membrane damage rather than an adaptive hypertrophy.145 It is suggested that alcohol induces mitochondrial swelling by increasing mitochondrial membrane permeabilization in 2 phases. In the first phase, alcohol instantaneously causes dilution of the whole media and mitochondrial permeability transition allows redistribution of water and alcohol in a concentration dependent manner. In the second phase, there was a time dependent mitochondrial swelling with high ethanol concentrations. Together, the eventual disruption of the K+ gradient contributes significantly to mitochondrial swelling.168 It is possible that the alcohol-mediated bioenergetic adaptations are mediated by mitochondrial remodeling. Mitochondrial remodeling includes dynamic changes in fission and fusion; mitophagy and biogenesis; and together with calcium signaling, ROS production, and epigenomic alterations.169,170 In fact, Han et al., using the intragastric and oral feeding alcohol models, proposed that mitochondrial remodeling is critical for hepatic mitochondrial adaptations driving bioenergetic changes in response to alcohol.35 However, though mitochondrial remodeling in the short term can be a compensatory mechanism, the chronic stress on the liver long term can lead to liver injury.
Although alcohol metabolism poses a major metabolic burden on the liver, alcohol-mediated bioenergetic adaptations are also observed in metabolically demanding tissues, including the cardiac and skeletal muscle, pancreas, and brain (Figure 6).
Cardiac and Skeletal Muscle
Cardiac and skeletal muscle have high metabolic demand and are rich in mitochondria. In the cardiac tissue, mitochondria account for ∼35% of the volume171 and at rest generate ∼90% of ATP requirements. In skeletal muscle, mitochondria constitute about 3–8% of the volume,172 which is highly influenced by physical activity. Thus, direct, or indirect effects of alcohol-mediated impaired cellular bioenergetics lead to dysfunctional cardiac and skeletal muscle mass.
Chronic administration of alcohol in dogs decreased cardiac intramitochondrial isocitrate dehydrogenase, ATP, mitochondrial oxygen consumption, and RCR leading to altered myocardial performance.173,174 Similarly, in rats receiving chronic alcohol there was increased cardiac muscle glucose-6-phosphate dehydrogenase, aldolase, and glyceraldehyde phosphate dehydrogenase activity but decreased isocitrate dehydrogenase activity.175 In the heart, FAEEs are the common alcohol metabolites and myocardial mitochondria incubated with FAEEs decreased RCR and maximal OCR. Furthermore, FAEEs bind to mitochondria, and importantly when incubated with ethanol, 72% of intracellularly synthesized ethyl esters bind to mitochondria. Cleavage of FAEEs and generation of fatty acids results in uncoupling of OXPHOS, thus FAEE may act as a toxic shuttle for fatty acids.176 Acute alcohol also affects cardiac cell bioenergetics. Intraperitoneal alcohol injection results in a significant increase in glycolysis and decreased mitochondrial respiration and RCR within 30 min, which disappear with decreasing blood and myocardium alcohol levels. However, chronic intraperitoneal injection of the same amount of alcohol decreased glycolysis, glycogen content, mitochondrial respiration, and RCR of isolated cardiac mitochondria with decreased ATP and creatine phosphate.177 Thus, it appears that acute alcohol may elicit a compensatory increase in glycolysis to meet the energy demands. In contrast, chronic alcohol administration decreases both glycolytic and mitochondrial respiration potentially leading to significant decreases in ATP production and thus impaired cardiac function. Chronic alcohol also increases cardiac expression of PPARα and decreases PGC-1α indicating impaired cardiac fatty acid metabolism. This was also associated with decreased mRNA, protein, and GAPDH activity indicating impaired glycolytic energy production. In addition, there was an increase in fructose content indicating a compensatory adaptation for activation of alternate glucose metabolism pathways, such as the sorbitol pathway to meet energy demands.178 Contrary to what is observed in the liver, there is a significant increase in mitochondrial enzyme activities (citrate synthase, CI, III, IV, V), and adaptive increase in mtDNA indicative of increased mitochondrial number in the cardiac tissue from alcohol-fed animals. These results suggest tissue-specific bioenergetic adaptations in response to alcohol.179
Functionally, skeletal-muscle mitochondria have a high RCR, ADP/O ratio and a high state-3 respiration rate with different substrates. However, the adverse effects of chronic at-risk alcohol use on skeletal muscle mitochondrial function are unclear. Using unbiased microarray analysis of myotubes treated with 100 m m ethanol, defects in ETC components, endogenous antioxidants, and enzymes regulating TCA cycle were identified to be differentially regulated. In vitro ethanol also impaired cellular respiration, decreased function of complexes I, II, and IV, and reduced OXPHOS. This decreased ATP content and redox ratio dysregulated succinate oxidation in the TCA cycle.180 However, Cardellach et al.181 argue that alcohol-related myopathy is not associated with decreased mitochondrial energy supply. Membrane preparations from liver and skeletal muscle from the same alcohol-fed animals showed that liver membranes developed membrane tolerance while muscle membrane retained normal sensitivity to alcohol effects. The authors conclude that the lack of development of muscle membrane tolerance correlates with lack of chemical changes in phospholipids, normal function of mitochondria and sarcoplasmic reticulum, and that there is not a deficiency in mitochondrial energy supply in the skeletal muscle.181 Using skeletal muscle biopsies from people with at-risk alcohol use, the oxidation rates with different substrates, the activity of respiratory chain complexes, and the cytochrome content were unaffected.182 Similarly, Trounce and co-workers reported normal muscle glycogen, carnitine levels, and normal activities of mitochondrial marker enzymes in people with chronic at-risk alcohol use.183 However, they attributed decreased glycogenolytic and glycolytic enzyme activity to the observed type 2 fiber atrophy that is commonly seen with chronic at-risk alcohol use.182,183 Contrary to this argument, cytochrome c oxidase activity and mitochondrial volumes were lower with higher creatine phosphate content in muscle of people with at-risk alcohol use.184 Additionally, sharing mitochondrial contents by fission–fusion events was decreased by both in vivo genetic perturbations and chronic at-risk alcohol use. A mitofusin 1-dependent pathway was identified, where inhibiting fusion reduced the mitochondrial metabolic reserve and dysregulated calcium oscillations during prolonged stimulation. Thus, prolonged loss of fusion jeopardizes bioenergetics and excitation–contraction coupling, providing a potential mechanism contributing to alcohol-related myopathy.185 Thus, although there are suggestions that at-risk alcohol use may not affect mitochondrial energy supply, alcohol-mediated impairment of bioenergetics may significantly contribute to exercise intolerance, metabolic dyshomeostasis, and impaired regenerative capacity of muscle stem cells.
Work from our group demonstrated that chronic binge alcohol (CBA)-induces skeletal muscle mitochondrial gene dysregulation at end-stage disease of simian immunodeficiency virus (SIV) infection in antiretroviral therapy (ART) naive rhesus macaques. CBA and ART decreased SDH activity in type 1 and type 2b fibers and gene expression of PGC-1β in the skeletal muscle of CBA/SIV macaques compared to uninfected controls. Moreover, the SIV infection-mediated upregulation of mitophagy-related gene expression was prevented by CBA. These findings suggest that SIV infection disrupts mitochondrial homeostasis and combined with CBA, results in differential expression of genes involved in mitochondrial dyshomeostasis.186 Moreover, myoblasts isolated from CBA/SIV/ART+ macaques showed decreased maximal OCR and formoterol, a β adrenergic agonist, partially restored maximal OCR.187 Results from our preclinical studies have now been translated to the study of dysglycemic people living with HIV (PLWH) and at-risk alcohol use. Our data show that a higher Alcohol Use Disorders Identification Test (AUDIT) score was associated with negative indicators of bioenergetic health, including proton leak, nonmitochondrial oxygen consumption, and BHI in skeletal muscle of PLWH. This was also associated with increased mitochondrial volume and decreased expression of genes implicated in mitochondrial function.188 Apart from how bioenergetic changes can regulate skeletal muscle metabolic capacity, we previously published evidence that alcohol-mediated shifts in bioenergetic phenotype underlie impaired differentiation of muscle stem cells. In vitro ethanol treatment of primary macaque myoblasts increased myoblast maximal OCR and decreased glycolytic metabolism (ECAR) at D0 of differentiation, which was associated with ethanol-mediated decreases in fusion index and myotubes per field, indices of myoblast differentiation. Moreover, alcohol impaired myoblast glycolytic metabolism, which may negatively impact the ability of myoblasts to fuse during muscle regeneration in vivo.189
Pancreas and Adipose Tissue
Alcohol-mediated impaired bioenergetic function is seen in alcohol-related pancreatitis. In fact, bioenergetic dysfunction is postulated to be one of the etiopathologies of alcohol-related pancreatitis. Using primary mice and human pancreatic acinar cells or pancreatic acinar AR42J cell line treated with ethanol, acetaldehyde, FAEE, or their combinations, it has been shown that ethanol significantly decreased total ATP production and increased mitochondrial stress.190–193 Acetaldehyde increased ATP from glycolysis but inhibited mitochondrial ATP turnover suggesting a compensatory metabolic adaptation when OXPHOS is impaired. However, FAEE inhibited both glycolytic and mitochondrial ATP production suggesting that alcohol exacerbated the inability of cells to meet cellular energy demands leading to acinar cell dysfunction and apoptosis. In addition, both acetaldehyde and FAEEs decreased spare respiratory capacity and impaired mitochondrial reserve.190 Oxidative ethanol metabolism activating MPTP and mitochondrial failure, is also proposed as a mechanism of decreased ATP production in pancreatic acinar cells.194,191 In particular, the ethanol or FAEE-mediated increased Ca2+ has been identified as a major contributor to the observed decrease in ATP production, increased trypsinogen activation, and cell death associated with acute alcohol-related pancreatitis.191
Clinical and preclinical studies show that alcohol decreases circulating basal insulin levels195,196 and circulating insulin and C-peptide expression in response to glucose.197 Using the Frequently Sampled Intravenous Glucose Tolerance Test, studies from our group have shown that CBA significantly impairs endocrine pancreatic response to a glucose load in SIV-infected macaques.198,199 Moreover, in vitro alcohol exposure decreases glucose-stimulated insulin secretion from human200 and rodent pancreatic islets196,201,202 and increases β-cell apoptosis.203–205 However, the mechanisms responsible for alcohol-mediated impaired pancreatic endocrine function are largely unknown. In type 2 diabetes, there is decreased oxygen consumption, decreased ATP:ADP ratio, and increased oxidative stress in pancreatic β cells. Because mitochondrial function is critical for glucose stimulated insulin release by β cells, and evidence indicate bioenergetic impairments as a pathomechanism in alcohol-induced pancreatitis, studies are warranted to determine whether alcohol-mediated bioenergetic alterations mechanistically contribute to the observed decreased pancreatic insulin secretion. Similarly, alcohol impairs adipocyte metabolic functions206–209 and optimal mitochondrial bioenergetic function is crucial for differentiation, lipogenesis, lipolysis, and secretion of adipokines.210 Preclinical and clinical studies in obesity indicate that mitochondrial oxygen consumption, expression of OXPHOS proteins, and mitochondrial biogenesis are reduced in adipose tissue or isolated adipocytes.211–213 However, whether alcohol impairs adipocyte bioenergetic function is not known. Thus, although these 2 tissues are critical in controlling whole body energy metabolism, there exists a gap in literature on how alcohol-mediated bioenergetic adaptations alter endocrine pancreatic and adipose function.
Brain
The bioenergetic adaptations of the brain to alcohol have been studied in efforts to determine their contribution to addiction and neuronal injury. Studies in zebra fish show that acute alcohol increased brain baseline respiration, CI-mediated OXPHOS, coupling efficiency, bioenergetic efficiency, and residual oxygen consumption to electron transfer system (ROX/ETS) ratio. In contrast, chronic alcohol administration decreased baseline respiration, complex I and II-mediated ETC, and increased ROX state and ROX/ETC ratio.214 In mice administered alcohol (3.8 g/kg) intraperitoneally and sacrificed after 6 h (modeling alcohol hangover), there was a decrease in malate–glutamate state 3 respiration and ATP production rates, and an increase in synaptosome respiration driving proton leak and spare respiratory capacity. This was also associated with decreased synaptosome CI, II, III, and IV activities demonstrating that alcohol-induced bioenergetic adaptations in the brain are potentially due to mitochondrial functional changes at the level of synapses, and that the decreased motor performance could be associated with brain bioenergetic dysregulation.215–217 Similarly, 2 h after an acute intraperitoneal alcohol injection (50 mmol/kg), there was significant inhibition of state 3 respiration in the brain.218 In vitro ethanol (50–200 m m) inhibited depolarization mediated OXPHOS stimulation. Mechanistically, it was demonstrated that ethanol inhibited voltage gated Ca2+ channels thus inhibiting synaptosomal free Ca2+ increase. This was in part by stimulation of the mitochondrial Ca2+/Na+ antiporter, which inhibited free Ca2+ increase in the mitochondrial matrix. The inhibition of the excitation-induced stimulation of synaptic OXPHOS is suggested to contribute to the depressant and narcotic effects of alcohol.219 After 8 h of withdrawal from chronic intermittent alcohol exposure, mitochondria were elongated, and mitochondria-on-a-string were observed in medial prefrontal cortex. This was associated with significant reduction in mitochondrial bioenergetics, including ETC, decreased gene expression of Mfn2, and increased fission.37 In the brain there were no changes in the substrate shuttles and ethanol oxidation, but there was decreased NAD+/NADH ratio and impaired OXPHOS.220 Isolated brain mitochondria from chronic alcohol fed-mice had decreased CI and V activity and decreased carnitine palmitoyl transferase 1 (cPT1) and cPT2 levels that are required of acylation of fatty acids from outer to inner mitochondrial membrane for ATP production. This was also associated with decreased β oxidation of palmitate suggesting that impaired substrate entry step (CI function) can affect ATP production (CV function). There was also increased cytochrome c leakage associated with the decreased cPT1/cPT2, while decreased CI and V paralleled a decrease in depolarization of mitochondrial membrane potential and ATP production.221 Overall, significant evidence supports neuronal bioenergetic adaptations to acute and chronic alcohol exposure.
Changes in mitochondrial bioenergetics are also observed in pups of rats administered alcohol during pregnancy. Cerebella of pups had significantly reduced mRNA levels of mitochondrial genes encoding CII, IV, and V in cerebellar granule cells. In vitro ethanol treatment of cerebellar cells reduced neuronal expression of mitochondrial genes encoding CIV and V, impaired mitochondrial function, and ATP production. These data strongly suggest that mitochondrial dysfunction may contribute to the underlying pathophysiology of fetal alcohol syndrome.222
Several candidate genes are implicated in affecting alcohol-mediated bioenergetic adaptations in the brain. After 1 wk of binge ethanol administration in adolescent rats, it was demonstrated that melanocortin 4 receptor (MC4R) agonist reduces hippocampal oxidative damage by increasing 2 key mitochondrial genes NRF2 and PGC1α, increasing mitochondrial volume, decreasing mitochondrial calcium levels, and increasing respiration complex expression.223–225 Using “drinking in the dark” paradigm, it was shown that alcohol-induced hippocampal oxidative damage and astrocyte activation in adolescent mice were associated with decreased MFN2 expression implicating impaired mitohormetic response and bioenergetic function in alcohol-associated neuroinflammation.226
Thus, published evidence suggest that alcohol-mediated bioenergetic adaptations are shared across metabolically active tissues leading to pathophysiological alterations (Figure 6). How these bioenergetic changes synergistically interplay with other alcohol-mediated pathomechanisms leading to end organ injury remains to be fully understood and is critical in identifying therapeutic targets to ameliorate disease burden.
Gaps in Knowledge of Alcohol-mediated Bioenergetic Adaptations and Cellular Function
This review focused on describing alcohol-mediated alterations in bioenergetics of tissues central to homeostasis of whole-body energy metabolism. However, it is well established that one of the mechanisms of chronic at-risk alcohol use is immune activation and inflammation, and an inability of the immune system to respond to infections.227–229 Though alcohol-mediated changes in tissue bioenergetics are well characterized, there is a gap in the literature on alcohol-mediated alterations of immunometabolism. Early work from our group showed that alcohol intoxication impairs endotoxin-induced increases in glucose metabolism and attenuates glucose utilization, particularly by tissues rich in immune cells.230,231 Increased metabolic rate by cells of the immune system is a required component of an adequate host defense. More recent evidence of perturbed alcohol-mediated immunometabolism comes from studies in alveolar macrophages. Yeligar and colleagues have demonstrated that ethanol-induced oxidative stress via NOX4 impairs alveolar macrophage phagocytic function. This was attributed to decreased basal respiration, ATP-linked respiration, maximal respiration, and spare capacity of alveolar macrophages, all indicative of bioenergetic adaptations. This was also associated with increased glycolytic capacity, glycolytic reserve, and nonglycolytic acidification, with concomitant increases in hypoxia-induced factor 1α expression and activity, phosphorylation of pyruvate dehydrogenase, and extracellular lactate levels in alveolar macrophages.232 However, immune cells, particularly CD4+ T cells, rely on both glycolysis and OXPHOS for energy requirements for immune responses.233 While naïve CD4+ T cells rely on β oxidation, when activated they rely on aerobic glycolysis.233 The subsequent differentiation depends on distinct metabolic pathways, with Th1, Th2, and Th17 depending on glycolytic pathway, while Treg relies on OXPHOS.234,235 Results from recent studies from our group provide evidence for the first time that ethanol decreased maximal respiration, coupling efficiency, OCR-linked ATP production and decreased BHI with simultaneous increases in OCR-linked proton leak in differentiated CD4+ T cells. This was also associated with increased glycolysis, which when inhibited prevented the ethanol-mediated increase in Th1 cells without affecting Tregs. In addition, ethanol increased Treg mitochondrial volume and altered expression of genes implicated in mitophagy. Overall, the evidence suggests that ethanol impairs CD4+ T cell immunometabolism and mitochondrial repair processes promoting a pro-inflammatory phenotype of CD4+ T cells.236 Whether this immune cell bioenergetic maladaptation functionally contributes to alterations in adaptive immune responses in people with at-risk alcohol is under active investigation.
Summary and Perspectives
Alcohol and its metabolites permeate virtually all tissues eliciting cellular stress. State-of-the-art techniques to determine cellular bioenergetic measurements and their integration with omic and targeted approaches have advanced our understanding of alcohol-induced adaptations to meet the energetic demands in response to cellular stress. In metabolically active tissues, alcohol impairs bioenergetic processes and simultaneously increases oxidative stress, thus decreasing metabolic flexibility. Fundamental to this is dysregulated mitochondrial morphological and functional integrity. This alcohol-mediated vicious cycle of compromised bioenergetics and increased oxidative stress can potentially initiate senescence and programmed cell death ultimately leading to tissue injury. Together, these bioenergetic adaptations synergize with other critical pathophysiological mechanisms exacerbating alcohol-induced end-organ injury. There have been significant strides in elucidating bioenergetic adaptations in metabolically active tissues, but the relevance and significance of alcohol's contribution to altered immunometabolism remains to be fully elucidated. This is especially relevant in lieu of the immune response to an infectious or noninfectious challenge, the ability of the immune system to clear bacterial or viral pathogens, and robust responses to vaccines. Advances in the fundamental knowledge of how alcohol elicits cellular injury will provide insight into novel strategies to ameliorate disease burden.
Funding
This work was supported by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under award number P60 AA009803. LS and PEM have received grant support from the NIH (P60 AA009803).
ACKNOWLEDGEMENTS
All figures were generated using Biorender.com.
Contributor Information
Liz Simon, Department of Physiology and Comprehensive Alcohol-HIV/AIDS Research Center, Louisiana State University Health Sciences Center, 1901 Perdido Street, New Orleans, LA 70112, USA.
Patricia E Molina, Department of Physiology and Comprehensive Alcohol-HIV/AIDS Research Center, Louisiana State University Health Sciences Center, 1901 Perdido Street, New Orleans, LA 70112, USA.
Conflict of Interest
P.E.M. holds the position of Executive Editor for Function and is blinded from reviewing or making decisions for the manuscript.
Data Availability
No new data were generated or analyzed in support of this research.
References
- 1. Manthey J, Hassan SA, Carr S, Kilian C, Kuitunen-Paul S, Rehm J. What are the economic costs to society attributable to alcohol use? A systematic review and modelling study. Pharmacoeconomics. 2021;39(7):809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alcohol. 2018. Available at: https://www.who.int/news-room/fact-sheets/detail/alcohol. Accessed 5 September 2022. [Google Scholar]
- 3. Rehm J, Room R, Monteiro Met al. . Alcohol as a risk factor for global burden of disease. Eur Addict Res. 2003;9(4):157–164. [DOI] [PubMed] [Google Scholar]
- 4. Collaborators GBDA . Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2018;392(10152):1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rehm J, Dawson D, Frick Uet al. . Burden of disease associated with alcohol use disorders in the United States. Alcohol Clin Exp Res. 2014;38(4):1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alcohol Research: Current Reviews Editorial Staff . Drinking patterns and their definitions. Alcohol Res. 2018;39(1):17–18.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/30557143. [PMC free article] [PubMed] [Google Scholar]
- 7. Vladimir Poznyak DR . Global status report on alcohol and health 2018. Switzerland: World Health Organization; 2018. [Google Scholar]
- 8. Fairfield B, Schnabl B. Gut dysbiosis as a driver in alcohol-induced liver injury. JHEP Rep. 2021;3(2):100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qamar N, Castano D, Patt C, Chu T, Cottrell J, Chang SL. Meta-analysis of alcohol induced gut dysbiosis and the resulting behavioral impact. Behav Brain Res. 2019;376:112196. doi: 10.1016/j.bbr.2019.112196. [DOI] [PubMed] [Google Scholar]
- 10. Gukovskaya AS, Mareninova OA, Odinokova IVet al. . Cell death in pancreatitis: effects of alcohol. J Gastroenterol Hepatol. 2006;21(Suppl 3):S10–13. [DOI] [PubMed] [Google Scholar]
- 11. Wang S, Pacher P, De Lisle RC, Huang H, Ding WX. A mechanistic review of cell death in alcohol-induced liver injury. Alcohol Clin Exp Res. 2016;40(6):1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu CY, Wang M, Yu HMet al. . Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci Biotechnol Biochem. 2020;84(8):1621–1628. [DOI] [PubMed] [Google Scholar]
- 13. Miyata T, Nagy LE. Programmed cell death in alcohol-associated liver disease. Clin Mol Hepatol. 2020;26(4):618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol. 2019;70(2):237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adler K, Molina PE, Simon L. Epigenomic mechanisms of alcohol-induced impaired differentiation of skeletal muscle stem cells; role of Class IIA histone deacetylases. Physiol Genomics. 2019;51(9):471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleiber ML, Diehl EJ, Laufer BIet al. . Long-term genomic and epigenomic dysregulation as a consequence of prenatal alcohol exposure: a model for fetal alcohol spectrum disorders. Front Genet. 2014;5:161. doi: 10.3389/fgene.2014.00161. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vore AS, Deak T. Alcohol, inflammation, and blood-brain barrier function in health and disease across development. Int Rev Neurobiol. 2022;161:209–249.. doi: 10.1016/bs.irn.2021.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lotersztajn S, Riva A, Wang S, Dooley S, Chokshi S, Gao B. Inflammation in alcohol-associated liver disease progression. Z Gastroenterol. 2022;60(1):58–66. [DOI] [PubMed] [Google Scholar]
- 19. Lanquetin A, Leclercq S, de Timary Pet al. . Role of inflammation in alcohol-related brain abnormalities: a translational study. Brain Commun. 2021;3(3):fcab154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poole LG, Beier JI, Torres-Gonzales Eet al. . Chronic + binge alcohol exposure promotes inflammation and alters airway mechanics in the lung. Alcohol. 2019;80:53–63.. doi: 10.1016/j.alcohol.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leclercq S, de Timary P, Delzenne NM, Starkel P. The link between inflammation, bugs, the intestine and the brain in alcohol dependence. Transl Psychiatry. 2017;7(2):e1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16(4):667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boyd J, Sexton O, Angus C, Meier P, Purshouse RC, Holmes J. Causal mechanisms proposed for the alcohol harm paradox-a systematic review. Addiction. 2022;117(1):33–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Massey VL, Beier JI, Ritzenthaler JD, Roman J, Arteel GE. Potential role of the gut/liver/lung axis in alcohol-induced tissue pathology. Biomolecules. 2015;5(4):2477–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McMahan RH, Afshar M, Amedee AMet al. . Summary of the 2019 alcohol and immunology research interest group (AIRIG) meeting: alcohol-mediated mechanisms of multiple organ injury. Alcohol. 2020;87:89–95.. doi: 10.1016/j.alcohol.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Molina PE, Gardner JD, Souza-Smith FM, Whitaker AM. Alcohol abuse: critical pathophysiological processes and contribution to disease burden. Physiology (Bethesda). 2014;29(3):203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50(6):1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osna NA, New-Aaron M, Dagur RSet al. . A review of alcohol-pathogen interactions: new insights into combined disease pathomechanisms. Alcohol Clin Exp Res. 2022;46(3):359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Simon L, Edwards S, Molina PE. Pathophysiological consequences of at-risk alcohol use; Implications for comorbidity risk in persons living with human immunodeficiency virus. Front Physiol. 2021;12:758230. doi: 10.3389/fphys.2021.758230. eCollection 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simon L, Souza-Smith FM, Molina PE. Alcohol-associated tissue injury: current views on pathophysiological mechanisms. Annu Rev Physiol. 2022;84:87–112.. doi: 10.1146/annurev-physiol-060821-014008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122(7):2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bailey SM. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic Res. 2003;37(6):585–596. [DOI] [PubMed] [Google Scholar]
- 33. Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Role of mitochondria in alcoholic liver disease. Curr Pathobiol Rep. 2013;1(3):159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2014;20(9):2136–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han D, Johnson HS, Rao MPet al. . Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic Biol Med. 2017;102:100–110.. doi: 10.1016/j.freeradbiomed.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Steiner JL, Lang CH. Etiology of alcoholic cardiomyopathy: mitochondria, oxidative stress and apoptosis. Int J Biochem Cell Biol. 2017;89:125–135.. doi: 10.1016/j.biocel.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shang P, Lindberg D, Starski Pet al. . Chronic alcohol exposure induces aberrant mitochondrial morphology and inhibits respiratory capacity in the medial prefrontal cortex of mice. Front Neurosci. 2020;14:561173. doi: 10.3389/fnins.2020.561173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hao L, Zhong W, Dong Het al. . ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1-TFAM signalling in alcoholic steatohepatitis. Gut. 2021;70(10):1933–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148.. https://doi.org/10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 40. Hill BG, Shiva S, Ballinger S, Zhang J, Darley-Usmar VM. Bioenergetics and translational metabolism: implications for genetics, physiology and precision medicine. Biol Chem. 2019;401(1):3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wallace DC. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man. Proc Natl Acad Sci USA. 2010;107(Suppl 2):8947–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahmad M, Wolberg A, Kahwaji CI. Biochemistry, Electron Transport Chain. Treasure Island (FL): StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 43. Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67(1):101–154. [DOI] [PubMed] [Google Scholar]
- 44. De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem. 2016;85:161–192.. doi: 10.1146/annurev-biochem-060614-034216. [DOI] [PubMed] [Google Scholar]
- 45. Pizzo P, Pozzan T. Mitochondrialand: what will come next?. Function (Oxf). 2022;3(1):zqab073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Filadi R, Basso E, Lefkimmiatis K, Pozzan T. Beyond intracellular signaling: the ins and outs of second messengers microdomains. Adv Exp Med Biol. 2017;981:279–322.. https://doi.org/10.1007/978-3-319-55858-5_12. [DOI] [PubMed] [Google Scholar]
- 47. Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. Mitochondrial Ca2+ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013;17(6):965–975. [DOI] [PubMed] [Google Scholar]
- 48. Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: the calcium connection. Biochim Biophys Acta. 2010;1797(6–7):607–618. [DOI] [PubMed] [Google Scholar]
- 49. Juhaszova M, Kobrinsky E, Zorov DBet al. . ATP synthase K+- and H+-fluxes drive ATP synthesis and enable mitochondrial K+-“uniporter” function: II. Ion and ATP synthase flux regulation. Function (Oxf). 2022;3(2):zqac001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Juhaszova M, Kobrinsky E, Zorov DBet al. . ATP synthase K+- and H+-fluxes drive ATP synthesis and enable mitochondrial K+-“uniporter” function: I. Characterization of ion fluxes. Function (Oxf). 2022;3(2):zqab065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217(1):409–427.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/13271404. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 52. Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416(1):15–18. [DOI] [PubMed] [Google Scholar]
- 53. Gnaiger E, Aasander Frostner E, Abdul Karim Net al. . Mitochondrial respiratory states and rates. MitoFit Preprint Arch. 2019. doi: 10.26124/mitofit:190001.v5. [Google Scholar]
- 54. Lark DS, Torres MJ, Lin CT, Ryan TE, Anderson EJ, Neufer PD. Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am J Physiol Cell Physiol. 2016;311(2):C239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Halliwell B. Antioxidant defence mechanisms: from the beginning to the end (of the beginning). Free Radic Res. 1999;31(4):261–272. [DOI] [PubMed] [Google Scholar]
- 57. Halliwell B, Gutteridge JM, Cross CE. Free radicals, antioxidants, and human disease: where are we now?. J Lab Clin Med. 1992;119(6):598–620.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/1593209. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 58. Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271.. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 59. Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys. 2007;465(1):119–126. [DOI] [PubMed] [Google Scholar]
- 60. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders—a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. 2017;1863(5):1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hill BG, Benavides GA, Lancaster JR Jret al. . Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393(12):1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bulthuis EP, Adjobo-Hermans MJW, Willems P, Koopman WJH. Mitochondrial morphofunction in mammalian cells. Antioxid Redox Signal. 2019;30(18):2066–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sauvanet C, Duvezin-Caubet S, di Rago JP, Rojo M. Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin Cell Dev Biol. 2010;21(6):558–565. [DOI] [PubMed] [Google Scholar]
- 64. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212(4):379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee H, Smith SB, Yoon Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem. 2017;292(17):7115–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117(Pt 26):6535–6546. [DOI] [PubMed] [Google Scholar]
- 67. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305(5685):858–862. [DOI] [PubMed] [Google Scholar]
- 68. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein DRP1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12(8):2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Otera H, Wang C, Cleland MMet al. . MFF is an essential factor for mitochondrial recruitment of DRP1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191(6):1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Y, Chan DC. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc Natl Acad Sci USA. 2007;104(47):18526–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bach D, Pich S, Soriano FXet al. . Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278(19):17190–17197. [DOI] [PubMed] [Google Scholar]
- 72. Mourier A, Motori E, Brandt Tet al. . Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol. 2015;208(4):429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pich S, Bach D, Briones Pet al. . The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14(11):1405–1415. [DOI] [PubMed] [Google Scholar]
- 74. Son JM, Sarsour EH, Kakkerla Balaraju Aet al. . Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell. 2017;16(5):1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Z, Wakabayashi N, Wakabayashi Jet al. . The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. 2011;22(13):2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Patten DA, Wong J, Khacho Met al. . OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014;33(22):2676–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mantena SK, King AL, Andringa KK, Landar A, Darley-Usmar V, Bailey SM. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J Gastroenterol. 2007;13(37):4967–4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hajnoczky G, Buzas CJ, Pacher P, Hoek JB, Rubin E. Alcohol and mitochondria in cardiac apoptosis: mechanisms and visualization. Alcohol Clin Exp Res. 2005;29(5):693–701. [DOI] [PubMed] [Google Scholar]
- 79. Basavarajappa BS, Subbanna S. Epigenetic mechanisms in developmental alcohol-induced neurobehavioral deficits. Brain Sci. 2016;6(2):12.doi:10.3390/brainsci6020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Williams JA, Ding WX. Role of autophagy in alcohol and drug-induced liver injury. Food Chem Toxicol. 2020;136:111075. doi: 10.1016/j.fct.2019.111075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Szabo G, Hoek JB, Darley-Usmar Vet al. . RSA 2004: combined basic research satellite symposium—session three: alcohol and mitochondrial metabolism: at the crossroads of life and death. Alcohol Clin Exp Res. 2005;29(9):1749–1752. [DOI] [PubMed] [Google Scholar]
- 82. Antoniewicz MR. A guide to (13)C metabolic flux analysis for the cancer biologist. Exp Mol Med. 2018;50(4):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gerencser AA, Neilson A, Choi SWet al. . Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81(16):6868–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Haller T, Ortner M, Gnaiger E. A respirometer for investigating oxidative cell metabolism: toward optimization of respiratory studies. Anal Biochem. 1994;218(2):338–342. [DOI] [PubMed] [Google Scholar]
- 85. Ripple MO, Kim N, Springett R. Mammalian complex I pumps 4 protons per 2 electrons at high and physiological proton motive force in living cells. J Biol Chem. 2013;288(8):5374–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dranka BP, Benavides GA, Diers ARet al. . Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51(9):1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chacko BK, Smith MR, Johnson MSet al. . Mitochondria in precision medicine; linking bioenergetics and metabolomics in platelets. Redox Biol. 2019;22:101165. doi: 10.1016/j.redox.2019.101165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schmidt CA, Fisher-Wellman KH, Neufer PD. From OCR and ECAR to energy: perspectives on the design and interpretation of bioenergetics studies. J Biol Chem. 2021;297(4):101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lesnefsky EJ, Hoppel CL. Oxidative phosphorylation and aging. Ageing Res Rev. 2006;5(4):402–433. [DOI] [PubMed] [Google Scholar]
- 90. Long Q, Huang L, Huang K, Yang Q. Assessing mitochondrial bioenergetics in isolated mitochondria from mouse heart tissues using oroboros 2k-oxygraph. Methods Mol Biol. 2019;1966:237–246.. doi: 10.1007/978-1-4939-9195-2_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Doerrier C, Gnaiger E. Oxygen flux analysis: DatLab real-time. Available at: https://wiki.oroboros.at/images/1/18/MiPNet19.18E_O2FluxAnalysis.pdf. Accessed 12 March 2022. [Google Scholar]
- 92. Chacko BK, Kramer PA, Ravi Set al. . The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond). 2014;127(6):367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhdanov AV, Ogurtsov VI, Taylor CT, Papkovsky DB. Monitoring of cell oxygenation and responses to metabolic stimulation by intracellular oxygen sensing technique. Integr Biol (Camb). 2010;2(9):443–451. [DOI] [PubMed] [Google Scholar]
- 94. Gu X, Ma Y, Liu Y, Wan Q. Measurement of mitochondrial respiration in adherent cells by Seahorse XF96 Cell Mito Stress Test. STAR Protoc. 2021;2(1):100245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schmidt CA, McLaughlin KL, Boykov INet al. . Aglycemic growth enhances carbohydrate metabolism and induces sensitivity to menadione in cultured tumor-derived cells. Cancer Metab. 2021;9(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fisher-Wellman KH, Lin CT, Ryan TEet al. . Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem J. 2015;467(2):271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med. 2016;100:53–65.. doi: 10.1016/j.freeradbiomed.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fisher-Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, Muoio DM. Mitochondrial diagnostics: a multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Rep. 2018;24(13):3593–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Takahashi A, Zhang Y, Centonze E, Herman B. Measurement of mitochondrial pH in situ. BioTechniques. 2001;30(4):804–808. [DOI] [PubMed] [Google Scholar]
- 100. Roe MW, Lemasters JJ, Herman B. Assessment of Fura-2 for measurements of cytosolic free calcium. Cell Calcium. 1990;11(2-3):63–73. [DOI] [PubMed] [Google Scholar]
- 101. Berry MN. The action of pyruvate on ethanol oxidation by intact isolated liver cells. Biochem J. 1971;123(4):41P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Peters TJ, Ward RJ, Rideout J, Lim CK. Blood acetaldehyde and ethanol levels in alcoholism. Prog Clin Biol Res. 1987;241:215–230.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/2886995. PMID: 2886995. [PubMed] [Google Scholar]
- 103. Takase S, Yasuhara M, Takada A, Ueshima Y. Changes in blood acetaldehyde levels after ethanol administration in alcoholics. Alcohol. 1990;7(1):37–41. [DOI] [PubMed] [Google Scholar]
- 104. Cederbaum AI, Lieber CS, Rubin E. Effect of acetaldehyde on activity of shuttles for the transport of reducing equivalents into the mitochondria. FEBS Lett. 1973;37(1):89–92. [DOI] [PubMed] [Google Scholar]
- 105. Lundquist F, Tygstrup N, Winkler K, Mellemgaard K, Munck-Petersen S. Ethanol metabolism and production of free acetate in the human liver. J Clin Invest. 1962;41(5):955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Forsander OA. Influence of the metabolism of ethanol on the lactate/pyruvate ratio of rat-liver slices. Biochem J. 1966;98(1):244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang Y, Sauve AA. NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016;1864(12):1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lluis JM, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124(3):708–724. [DOI] [PubMed] [Google Scholar]
- 109. Gordon ER. The effect of chronic consumption of ethanol on the redox state of the rat liver. Can J Biochem. 1972;50(9):949–957. [DOI] [PubMed] [Google Scholar]
- 110. Krebs HA, Freedland RA, Hems R, Stubbs M. Inhibition of hepatic gluconeogenesis by ethanol. Biochem J. 1969;112(1):117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wilson DF, Matschinsky FM. Ethanol metabolism: the good, the bad, and the ugly. Med Hypotheses. 2020;140:109638. doi: 10.1016/j.mehy.2020.109638. [DOI] [PubMed] [Google Scholar]
- 112. Bertocci LA, Jones JG, Malloy CR, Victor RG, Thomas GD. Oxidation of lactate and acetate in rat skeletal muscle: analysis by 13C-nuclear magnetic resonance spectroscopy. J Appl Physiol. 1997;83(1):32–39. [DOI] [PubMed] [Google Scholar]
- 113. Putman CT, Spriet LL, Hultman E, Dyck DJ, Heigenhauser GJ. Skeletal muscle pyruvate dehydrogenase activity during acetate infusion in humans. Am J Physiol. 1995;268(5 Pt1):E1007–1017. [DOI] [PubMed] [Google Scholar]
- 114. Cederbaum AI. Introduction-serial review: alcohol, oxidative stress and cell injury. Free Radic Biol Med. 2001;31(12):1524–1526.. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11744324. Accessed 5 September 2022. [DOI] [PubMed] [Google Scholar]
- 115. Jing L, Jin CM, Li SSet al. . Chronic alcohol intake-induced oxidative stress and apoptosis: role of CYP2E1 and calpain-1 in alcoholic cardiomyopathy. Mol Cell Biochem. 2012;359(1-2):283–292. [DOI] [PubMed] [Google Scholar]
- 116. Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83(6):519–548. [DOI] [PubMed] [Google Scholar]
- 117. Cederbaum AI, Dicker E, Lieber CS, Rubin E. Factors contributing to the adaptive increase in ethanol metabolism due to chronic consumption of ethanol. Alcohol Clin Exp Res. 1977;1(1):27–31. [DOI] [PubMed] [Google Scholar]
- 118. Alderman J, Kato S, Lieber CS. The microsomal ethanol oxidizing system mediates metabolic tolerance to ethanol in deermice lacking alcohol dehydrogenase. Arch Biochem Biophys. 1989;271(1):33–39. [DOI] [PubMed] [Google Scholar]
- 119. Pirola RC, Lieber CS. Hypothesis: energy wastage in alcoholism and drug abuse: possible role of hepatic microsomal enzymes. Am J Clin Nutr. 1976;29(1):90–93. [DOI] [PubMed] [Google Scholar]
- 120. Heier C, Xie H, Zimmermann R. Nonoxidative ethanol metabolism in humans-from biomarkers to bioactive lipids. IUBMB Life. 2016;68(12):916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Piano MR, Phillips SA. Alcoholic cardiomyopathy: pathophysiologic insights. Cardiovasc Toxicol. 2014;14(4):291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Song BJ, Akbar M, Abdelmegeed MAet al. . Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014;3:109–123.. doi: 10.1016/j.redox.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ammon HP, Estler CJ. Influence of acute and chronic administration of alcohol on carbohydrate breakdown and energy metabolism in the liver. Nature. 1967;216(5111):158–159. [DOI] [PubMed] [Google Scholar]
- 124. Bottenus RE, Spach PI, Filus S, Cunningham CC. Effect of chronic ethanol consumption of energy-linked processes associated with oxidative phosphorylation: proton translocation and ATP-Pi exchange. Biochem Biophys Res Commun. 1982;105(4):1368–1373. [DOI] [PubMed] [Google Scholar]
- 125. Gordon ER. ATP metabolism in an ethanol induced fatty liver. Biochem Pharmacol. 1977;26(13):1229–1234. [DOI] [PubMed] [Google Scholar]
- 126. Spach PI, Bottenus RE, Cunningham CC. Control of adenine nucleotide metabolism in hepatic mitochondria from rats with ethanol-induced fatty liver. Biochem J. 1982;202(2):445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Walker JE, Gordon ER. Biochemical aspects associated with an ethanol-induced fatty liver. Biochem J. 1970;119(3):511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Cederbaum AI, Lieber CS, Rubin E. Effects of chronic ethanol treatment of mitochondrial functions damage to coupling site I. Arch Biochem Biophys. 1974;165(2):560–569. [DOI] [PubMed] [Google Scholar]
- 129. Mitchell MC, Herlong HF. Alcohol and nutrition: caloric value, bioenergetics, and relationship to liver damage. Annu Rev Nutr. 1986;6:457–474.. doi: 10.1146/annurev.nu.06.070186.002325. [DOI] [PubMed] [Google Scholar]
- 130. Lieber CS, DeCarli LM. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989;24(3):197–211.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/2667528. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 131. Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6 J mice. Physiol Behav. 2005;84(1):53–63. [DOI] [PubMed] [Google Scholar]
- 132. Steiner JL, Lang CH. Alcohol intoxication following muscle contraction in mice decreases muscle protein synthesis but not mTOR signal transduction. Alcohol Clin Exp Res. 2015;39(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Steiner JL, Kimball SR, Lang CH. Acute alcohol-induced decrease in muscle protein synthesis in female mice Is REDD-1 and mTOR-independent. Alcohol Alcohol. 2016;51(3):242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tsukamoto H, Gaal K, French SW. Insights into the pathogenesis of alcoholic liver necrosis and fibrosis: status report. Hepatology. 1990;12(3 Pt 1):599–608. [DOI] [PubMed] [Google Scholar]
- 135. Colombo G, Lobina C, Carai MA, Gessa GL. Phenotypic characterization of genetically selected Sardinian alcohol-preferring (sP) and -non-preferring (sNP) rats. Addict Biol. 2006;11(3-4):324–338. [DOI] [PubMed] [Google Scholar]
- 136. Mayfield J, Arends MA, Harris RA, Blednov YA. Genes and alcohol consumption: studies with mutant mice. Int Rev Neurobiol. 2016;126:293–355.. doi: 10.1016/bs.irn.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Thayer WS, Rubin E. Effects of chronic ethanol intoxication on oxidative phosphorylation in rat liver submitochondrial particles. J Biol Chem. 1979;254(16):7717–7723.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/572826. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 138. Thayer WS. Effects of ethanol on proteins of mitochondrial membranes. Ann NY Acad Sci. 1987;492(1):193–206. [DOI] [PubMed] [Google Scholar]
- 139. Piquet MA, Nogueira V, Devin Aet al. . Chronic ethanol ingestion increases efficiency of oxidative phosphorylation in rat liver mitochondria. FEBS Lett. 2000;468(2-3):239–242. [DOI] [PubMed] [Google Scholar]
- 140. Murphy MP, Tipton KF. Effects of chronic ethanol feeding on rat liver mitochondrial energy metabolism. Biochem Pharmacol. 1992;43(12):2663–2667. [DOI] [PubMed] [Google Scholar]
- 141. Marcinkeviciute A, Mildaziene V, Crumm S, Demin O, Hoek JB, Kholodenko B. Kinetics and control of oxidative phosphorylation in rat liver mitochondria after chronic ethanol feeding. Biochem J. 2000;349(Pt 2):519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Spach PI, Cunningham CC. Control of state 3 respiration in liver mitochondria from rats subjected to chronic ethanol consumption. Biochim Biophys Acta. 1987;894(3):460–467. [DOI] [PubMed] [Google Scholar]
- 143. Thayer WS, Rottenberg H. Comparative effects of chronic ethanol consumption on the properties of mitochondria from rat brain and liver. Alcohol Clin Exp Res. 1992;16(1):1–4. [DOI] [PubMed] [Google Scholar]
- 144. Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar VM. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38(1):141–147. [DOI] [PubMed] [Google Scholar]
- 145. Arai M, Leo MA, Nakano M, Gordon ER, Lieber CS. Biochemical and morphological alterations of baboon hepatic mitochondria after chronic ethanol consumption. Hepatology. 1984;4(2):165–174. [DOI] [PubMed] [Google Scholar]
- 146. Zentella de Pina M, Villalobos-Molina R, Saavedra-Molina A, Riveros-Rosas H, Pina E. Effects of moderate chronic ethanol consumption on rat liver mitochondrial functions. Alcohol. 1989;6(1):3–7. [DOI] [PubMed] [Google Scholar]
- 147. Zelickson BR, Benavides GA, Johnson MSet al. . Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim Biophys Acta. 2011;1807(12):1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Koch OR, Boveris A, Favelukes S, Schwarcz de Tarlovsky M, Stoppani AO. Biochemical lesions of liver mitochondria from rats after chronic alcohol consumption. Exp Mol Pathol. 1977;27(2):213–220. [DOI] [PubMed] [Google Scholar]
- 149. Rubin E, Bacchin P, Gang H, Lieber CS. Induction and inhibition of hepatic microsomal and mitochondrial enzymes by ethanol. Lab Invest. 1970;22(6):569–580.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/5430804. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 150. Sun Q, Zhong W, Zhang W, Zhou Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: role of zinc deficiency. Am J Physiol Gastrointest Liver Physiol. 2016;310(3):G205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Devi BG, Henderson GI, Frosto TA, Schenker S. Effect of acute ethanol exposure on cultured fetal rat hepatocytes: relation to mitochondrial function. Alcohol Clin Exp Res. 1994;18(6):1436–1442. [DOI] [PubMed] [Google Scholar]
- 152. Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199(2):223–231. [DOI] [PubMed] [Google Scholar]
- 153. Neve EP, Ingelman-Sundberg M. A soluble NH(2)-terminally truncated catalytically active form of rat cytochrome P450 2E1 targeted to liver mitochondria(1). FEBS Lett. 1999;460(2):309–314. [DOI] [PubMed] [Google Scholar]
- 154. Robin MA, Anandatheerthavarada HK, Fang JK, Cudic M, Otvos L, Avadhani NG. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N terminus and requires mitochondrial specific electron transfer proteins for activity. J Biol Chem. 2001;276(27):24680–24689. [DOI] [PubMed] [Google Scholar]
- 155. Robin MA, Sauvage I, Grandperret T, Descatoire V, Pessayre D, Fromenty B. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett. 2005;579(30):6895–6902. [DOI] [PubMed] [Google Scholar]
- 156. Bansal S, Srinivasan S, Anandasadagopan Set al. . Additive effects of mitochondrion-targeted cytochrome CYP2E1 and alcohol toxicity on cytochrome c oxidase function and stability of respirosome complexes. J Biol Chem. 2012;287(19):15284–15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370(Pt 3):751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wahid S, Khanna JM, Carmichael FJ, Israel Y. Mitochondrial function following chronic ethanol treatment: effect of diet. Res Commun Chem Pathol Pharmacol. 1980;30(3):477–491.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/7196064. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 159. Freinkel N, Cohen AK, Arky RA, Foster AE. Alcohol hypoglycemia. Ii. A postulated mechanism of action based on experiments with rat liver slices. J Clin Endocrinol Metab. 1965;25(1):76–94. [DOI] [PubMed] [Google Scholar]
- 160. Yuki T, Thurman RG. The swift increase in alcohol metabolism. Time course for the increase in hepatic oxygen uptake and the involvement of glycolysis. Biochem J. 1980;186(1):119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Berry MN, Gregory RB, Grivell AR, Phillips JW, Schon A. The capacity of reducing-equivalent shuttles limits glycolysis during ethanol oxidation. Eur J Biochem. 1994;225(2):557–564. [DOI] [PubMed] [Google Scholar]
- 162. Baio DL, Czyz CN, Van Horn CG, Ivester P, Cunningham CC. Effect of chronic ethanol consumption on respiratory and glycolytic activities of rat periportal and perivenous hepatocytes. Arch Biochem Biophys. 1998;350(2):193–200. [DOI] [PubMed] [Google Scholar]
- 163. Van Horn CG, Cunningham CC. Contributions of dietary carbohydrate and ethanol to alterations in liver glycogen levels and glycolytic activity. Alcohol. 1999;19(2):139–144. [DOI] [PubMed] [Google Scholar]
- 164. Young TA, Bailey SM, Van Horn CG, Cunningham CC. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol. 2006;41(3):254–260. [DOI] [PubMed] [Google Scholar]
- 165. Rubin E, Lieber CS. Early fine structural changes in the human liver induced by alcohol. Gastroenterology. 1967;52(1):1–13.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/6018368. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 166. Rubin E, Lieber CS. Fatty liver, alcoholic hepatitis and cirrhosis produced by alcohol in primates. N Engl J Med. 1974;290(3):128–135. [DOI] [PubMed] [Google Scholar]
- 167. Lieber CS, DeCarli L, Rubin E. Sequential production of fatty liver, hepatitis, and cirrhosis in sub-human primates fed ethanol with adequate diets. Proc Natl Acad Sci USA. 1975;72(2):437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ma L, Dong JX, Wu Cet al. . Spectroscopic, polarographic, and microcalorimetric studies on mitochondrial dysfunction induced by ethanol. J Membr Biol. 2017;250(2):195–204. [DOI] [PubMed] [Google Scholar]
- 169. Santulli G, Monaco G, Parra V, Morciano G. Editorial: mitochondrial remodeling and dynamic inter-organellar contacts in cardiovascular physiopathology. Front Cell Dev Biol. 2021;9:679725. doi: 10.3389/fcell.2021.679725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Gottlieb RA, Bernstein D. Mitochondrial remodeling: rearranging, recycling, and reprogramming. Cell Calcium. 2016;60(2):88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Stride N, Larsen S, Hey-Mogensen Met al. . Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur J Heart Fail. 2013;15(2):150–157. [DOI] [PubMed] [Google Scholar]
- 172. Larsen S, Nielsen J, Hansen CNet al. . Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590(14):3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Pachinger OM, Tillmanns H, Mao JC, Fauvel JM, Bing RJ. The effect of prolonged administration of ethanol on cardiac metabolism and performance in the dog. J Clin Invest. 1973;52(11):2690–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Pachinger O, Mao J, Fauvel JM, Bing RJ. Mitochondrial function and excitation-contraction coupling in the development of alcoholic cardiomyopathy. Recent Adv Stud Cardiac Struct Metab. 1975;5:423–429.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/1237922. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 175. Sardesai VM, Provido HS. The effect of chronic ethanol ingestion on myocardial glucose and energy metabolism. J Nutr. 1978;108(12):1907–1912. [DOI] [PubMed] [Google Scholar]
- 176. Lange LG, Sobel BE. Mitochondrial dysfunction induced by fatty acid ethyl esters, myocardial metabolites of ethanol. J Clin Invest. 1983;72(2):724–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gvozdjak A, Borovic F, Bada V, Kruty F, Niederland TR, Gvozdjak J. Myocardial cell damage due to ethanol. Recent Adv Stud Cardiac Struct Metab. 1975;7:451–457.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/1226455. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 178. Yan X, Wu L, Lin Qet al. . From the cover: alcohol inhibition of the enzymatic activity of glyceraldehyde 3-phosphate dehydrogenase impairs cardiac glucose utilization, contributing to alcoholic cardiomyopathy. Toxicol Sci. 2017;159(2):392–401. [DOI] [PubMed] [Google Scholar]
- 179. Marin-Garcia J, Ananthakrishnan R, Goldenthal MJ. Heart mitochondria response to alcohol is different than brain and liver. Alcohol Clin Exp Res. 1995;19(6):1463–1466. [DOI] [PubMed] [Google Scholar]
- 180. Kumar A, Davuluri G, Welch Net al. . Oxidative stress mediates ethanol-induced skeletal muscle mitochondrial dysfunction and dysregulated protein synthesis and autophagy. Free Radic Biol Med. 2019;145:284–299.. doi: 10.1016/j.freeradbiomed.2019.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Cardellach F, Taraschi TF, Ellingson JS, Stubbs CD, Rubin E, Hoek JB. Maintenance of structural and functional characteristics of skeletal-muscle mitochondria and sarcoplasmic-reticular membranes after chronic ethanol treatment. Biochem J. 1991;274(Pt 2):565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Cardellach F, Galofre J, Grau JMet al. . Oxidative metabolism in muscle mitochondria from patients with chronic alcoholism. Ann Neurol. 1992;31(5):515–518. [DOI] [PubMed] [Google Scholar]
- 183. Trounce I, Byrne E, Dennett X, Santamaria J, Doery J, Peppard R. Chronic alcoholic proximal wasting: physiological, morphological and biochemical studies in skeletal muscle. Aust N Z J Med. 1987;17(4):413–419. [DOI] [PubMed] [Google Scholar]
- 184. Kiessling KH, Pilstrom L, Bylund AC, Piehl K, Saltin B. Effects of chronic ethanol abuse on structure and enzyme activities of skeletal muscle in man. Scand J Clin Lab Invest. 1975;35(6):601–607. [DOI] [PubMed] [Google Scholar]
- 185. Eisner V, Lenaers G, Hajnoczky G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J Cell Biol. 2014;205(2):179–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Duplanty AA, Simon L, Molina PE. Chronic binge alcohol-induced dysregulation of mitochondrial-related genes in skeletal muscle of simian immunodeficiency virus-infected rhesus macaques at end-stage disease. Alcohol Alcohol. 2017;52(3):298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Duplanty AA, Siggins RW, Allerton T, Simon L, Molina PE. Myoblast mitochondrial respiration is decreased in chronic binge alcohol administered simian immunodeficiency virus-infected antiretroviral-treated rhesus macaques. Physiol Rep. 2018;6(5): e13625. doi:10.14814/phy2.13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Levitt DE, Ferguson TF, Primeaux SDet al. . Skeletal muscle bioenergetic health and function in people living with HIV: association with glucose tolerance and alcohol use. Am J Physiol Regul Integr Comp Physiol. 2021;321(5):R781–R790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Levitt DE, Chalapati N, Prendergast MJ, Simon L, Molina PE. Ethanol-impaired myogenic differentiation is associated with decreased myoblast glycolytic function. Alcohol Clin Exp Res. 2020;44(11):2166–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Mukherjee R, Mareninova OA, Odinokova IVet al. . Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. 2016;65(8):1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Petersen OH, Gerasimenko JV, Gerasimenko OV, Gryshchenko O, Peng S. The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiol Rev. 2021;101(4):1691–1744. [DOI] [PubMed] [Google Scholar]
- 192. Huang W, Booth DM, Cane MCet al. . Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut. 2014;63(8):1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci USA. 2004;101(29):10738–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Shalbueva N, Mareninova OA, Gerloff Aet al. . Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology. 2013;144(2):437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Bonnet F, Disse E, Laville Met al. . Moderate alcohol consumption is associated with improved insulin sensitivity, reduced basal insulin secretion rate and lower fasting glucagon concentration in healthy women. Diabetologia. 2012;55(12):3228–3237. [DOI] [PubMed] [Google Scholar]
- 196. Rasineni K, Thomes PG, Kubik JL, Harris EN, Kharbanda KK, Casey CA. Chronic alcohol exposure alters circulating insulin and ghrelin levels: role of ghrelin in hepatic steatosis. Am J Physiol Gastrointest Liver Physiol. 2019;316(4):G453–G461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Patto RJ, Russo EK, Borges DR, Neves MM. The enteroinsular axis and endocrine pancreatic function in chronic alcohol consumers: evidence for early beta-cell hypofunction. Mt Sinai J Med. 1993;60(4):317–320.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/8232378. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 198. Ford SM Jr., Simon L, Vande Stouwe Cet al. . Chronic binge alcohol administration impairs glucose-insulin dynamics and decreases adiponectin in asymptomatic simian immunodeficiency virus-infected macaques. Am J Physiol Regul Integr Comp Physiol. 2016;311(5):R888–R897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Simon L, Torres D, Saravia Aet al. . Chronic binge alcohol and ovariectomy-mediated impaired insulin responsiveness in SIV-infected female rhesus macaques. Am J Physiol Regul Integr Comp Physiol. 2021;321(5):R699–R711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Dragan Nikolić DM, Dimitrijević-Srećković V, Kerkez M, Nikolić B. Effect of alcohol on insulin secretion and viability of human pancreatic islets. Srp Arh Celok Lek. 2017;145(3-4):159–164.. Available at: https://pdfs.semanticscholar.org/5891/145054f6bb2c437891e5a0d5e1e2d364aa60.pdf?_ga=2.151630905.1439910538.1566913613-683285603.1540817223. Accessed 5 September 2022. [Google Scholar]
- 201. Wang S, Luo Y, Feng Aet al. . Ethanol induced impairment of glucose metabolism involves alterations of GABAergic signaling in pancreatic beta-cells. Toxicology. 2014;326:44–52.. doi: 10.1016/j.tox.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 202. Nguyen KH, Lee JH, Nyomba BL. Ethanol causes endoplasmic reticulum stress and impairment of insulin secretion in pancreatic beta-cells. Alcohol. 2012;46(1):89–99. [DOI] [PubMed] [Google Scholar]
- 203. Kim JY, Song EH, Lee HJet al. . Chronic ethanol consumption-induced pancreatic {beta}-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J Biol Chem. 2010;285(48):37251–37262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Dembele K, Nguyen KH, Hernandez TA, Nyomba BL. Effects of ethanol on pancreatic beta-cell death: interaction with glucose and fatty acids. Cell Biol Toxicol. 2009;25(2):141–152. [DOI] [PubMed] [Google Scholar]
- 205. Hodge AM, Dowse GK, Collins VR, Zimmet PZ. Abnormal glucose tolerance and alcohol consumption in three populations at high risk of non-insulin-dependent diabetes mellitus. Am J Epidemiol. 1993;137(2):178–189. [DOI] [PubMed] [Google Scholar]
- 206. Ford SM Jr., Simon Peter L, Berner Pet al. . Differential contribution of chronic binge alcohol and antiretroviral therapy to metabolic dysregulation in SIV-infected male macaques. Am J Physiol Endocrinol Metab. 2018;315(5):E892–E903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Gopal T, Ai W, Casey CA, Donohue TM Jr., Saraswathi V. A review of the role of ethanol-induced adipose tissue dysfunction in alcohol-associated liver disease. Alcohol Clin Exp Res. 2021;45(10):1927–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Zhang W, Zhong W, Sun Xet al. . Visceral white adipose tissue is susceptible to alcohol-induced lipodystrophy in rats: role of acetaldehyde. Alcohol Clin Exp Res. 2015;39(3):416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Pravdova E, Fickova M. Alcohol intake modulates hormonal activity of adipose tissue. Endocr Regul. 2006;40(3):91–104.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/17100551. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 210. De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am J Pathol. 2009;175(3):927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Hallgren P, Sjostrom L, Hedlund H, Lundell L, Olbe L. Influence of age, fat cell weight, and obesity on O2 consumption of human adipose tissue. Am J Physiol. 1989;256(4 Pt 1):E467–474. [DOI] [PubMed] [Google Scholar]
- 212. Dahlman I, Forsgren M, Sjogren Aet al. . Downregulation of electron transport chain genes in visceral adipose tissue in type 2 diabetes independent of obesity and possibly involving tumor necrosis factor-alpha. Diabetes. 2006;55(6):1792–1799. [DOI] [PubMed] [Google Scholar]
- 213. Schottl T, Kappler L, Fromme T, Klingenspor M. Limited OXPHOS capacity in white adipocytes is a hallmark of obesity in laboratory mice irrespective of the glucose tolerance status. Mol Metab. 2015;4(9):631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Muller TE, Nunes MEM, Rodrigues NRet al. . Neurochemical mechanisms underlying acute and chronic ethanol-mediated responses in zebrafish: the role of mitochondrial bioenergetics. Neurochem Int. 2019;131:104584. doi: 10.1016/j.neuint.2019.104584. [DOI] [PubMed] [Google Scholar]
- 215. Bustamante J, Karadayian AG, Lores-Arnaiz S, Cutrera RA. Alterations of motor performance and brain cortex mitochondrial function during ethanol hangover. Alcohol. 2012;46(5):473–479. [DOI] [PubMed] [Google Scholar]
- 216. Karadayian AG, Bustamante J, Czerniczyniec A, Lombardi P, Cutrera RA. Lores-Arnaiz S. Alcohol hangover induces mitochondrial dysfunction and free radical production in mouse cerebellum. Neuroscience. 2015;304:47–59.. doi: 10.1016/j.neuroscience.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 217. Karadayian AG, Lombardi P, Bustamante J, Lores-Arnaiz S. Alcohol hangover effects on brain cortex non-synaptic mitochondria and synaptosomes bioenergetics. Alcohol. 2019;77:113–123.. doi: 10.1016/j.alcohol.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 218. Ribiere C, Sabourault D, Saffar C, Nordmann R. Mitochondrial generation of superoxide free radicals during acute ethanol intoxication in the rat. Alcohol Alcohol Suppl. 1987;1:241–244.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/2827693. Accessed 5 September 2022. [PubMed] [Google Scholar]
- 219. Li HL, Wu S, Rottenberg H. Alcohol inhibits the depolarization-induced stimulation of oxidative phosphorylation in synaptosomes. J Neurochem. 1996;66(4):1691–1697. [DOI] [PubMed] [Google Scholar]
- 220. Rawat AK, Kuriyama K, Mose J. Metabolic consequences of ethanol oxidation in brains from mice chronically fed ethanol. J Neurochem. 1973;20(1):23–33. [DOI] [PubMed] [Google Scholar]
- 221. Haorah J, Rump TJ, Xiong H. Reduction of brain mitochondrial beta-oxidation impairs complex I and V in chronic alcohol intake: the underlying mechanism for neurodegeneration. PLoS One. 2013;8(8):e70833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Chu J, Tong M, de la Monte SM. Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol. 2007;113(6):659–673. [DOI] [PubMed] [Google Scholar]
- 223. Torres AK, Tapia-Rojas C, Cerpa W, Quintanilla RA. Stimulation of melanocortin receptor-4 (MC4R) prevents mitochondrial damage induced by binge ethanol protocol in adolescent rat hippocampus. Neuroscience. 2020;438:70–85.. doi: 10.1016/j.neuroscience.2020.05.005. [DOI] [PubMed] [Google Scholar]
- 224. Tapia-Rojas C, Carvajal FJ, Mira RGet al. . Adolescent binge alcohol exposure affects the brain function through mitochondrial impairment. Mol Neurobiol. 2018;55(5):4473–4491. [DOI] [PubMed] [Google Scholar]
- 225. Tapia-Rojas C, Torres AK, Quintanilla RA. Adolescence binge alcohol consumption induces hippocampal mitochondrial impairment that persists during the adulthood. Neuroscience. 2019;406:356–368.. doi: 10.1016/j.neuroscience.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 226. Mira RG, Lira M, Quintanilla RA, Cerpa W. Alcohol consumption during adolescence alters the hippocampal response to traumatic brain injury. Biochem Biophys Res Commun. 2020;528(3):514–519. [DOI] [PubMed] [Google Scholar]
- 227. Happel KI, Rudner X, Quinton LJet al. . Acute alcohol intoxication suppresses the pulmonary ELR-negative CXC chemokine response to lipopolysaccharide. Alcohol. 2007;41(5):325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Molina PE, Happel KI, Zhang P, Kolls JK, Nelson S. Focus on: alcohol and the immune system. Alcohol Res Health. 2010;33(1-2):97–108.. Available at: https://www.ncbi.nlm.nih.gov/pubmed/23579940. Accessed 5 September 2022. [PMC free article] [PubMed] [Google Scholar]
- 229. Zhang P, Bagby GJ, Happel KI, Raasch CE, Nelson S. Alcohol abuse, immunosuppression, and pulmonary infection. Curr Drug Abuse Rev. 2008;1(1):56–67. [DOI] [PubMed] [Google Scholar]
- 230. Molina PE, Lang CH, Bagby GJ, Spitzer JJ. Ethanol attenuates endotoxin-enhanced glucose utilization. Am J Physiol. 1990;258(4 Pt 2):R987–993. [DOI] [PubMed] [Google Scholar]
- 231. Molina PE, Lang CH, Bagby GJ, D'Souza NB, Spitzer JJ. Ethanol administration diminishes the endotoxin-induced increase in glucose metabolism. Alcohol Clin Exp Res. 1989;13(3):407–412. [DOI] [PubMed] [Google Scholar]
- 232. Morris NL, Harris FL, Brown LAS, Yeligar SM. Alcohol induces mitochondrial derangements in alveolar macrophages by upregulating NADPH oxidase 4. Alcohol. 2021;90:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. McTernan PM, Levitt DE, Welsh DA, Simon L, Siggins RW, Molina PE. Alcohol impairs immunometabolism and promotes naive T cell differentiation to pro-inflammatory Th1 CD4+ T cells. Front Immunol. 2022;13:839390. doi: 10.3389/fimmu.2022.839390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.