

Abstract
Myeloid dendritic cells (DCs) and macrophages are mononuclear phagocytes with key roles in the immune system. As antigen presenting cells, they link innate detection of microbes with programming adaptive immune responses. Myeloid DCs and macrophages also play critical roles in development, promote tissue homeostasis, and direct repair in response to injury and inflammation. As cellular migration and organelle dynamics are intimately connected with these processes, it is necessary to develop tools to track myeloid cell behavior and function. Here, we build on previously established protocols to isolate primary human myeloid cells from peripheral blood and report an optimized method for their genetic modification with lentiviral vectors to study processes related to cell migration, activation, and organelle dynamics. Specifically, we provide a protocol for delivering genetically-encoded fluorescent markers into primary monocyte-derived dendritic cells (MDDCs) and monocyte-derived macrophages (MDMs) to label mitochondria, peroxisomes, and whole cells. We describe the isolation of primary CD14+ monocytes from peripheral blood using positive selection with magnetic beads, and alternatively, isolation based on plastic adherence. Isolated CD14+ cells can be transduced with lentiviral vectors and subsequently cultured in the presence of cytokines to derive MDDCs or MDMs. This protocol is highly adaptable for co-transduction with vectors to knockdown or overexpress genes of interest. These tools enable mechanistic studies of genetically modified myeloid cells through flow cytometry, fluorescence microscopy, and other downstream assays.
Keywords: Gene delivery, viral vectors, innate immunity
INTRODUCTION:
Macrophages and DCs are key cells in the innate immune system, acting to surveil local microenvironments for external and internal threats, and respond when necessary to protect the host (Banchereau & Steinman, 1998; Wynn, Chawla, & Pollard, 2013). In doing so, macrophages and DCs perform a variety of functions. As phagocytic cells, macrophages specialize in clearing pathogens, dead cells, and debris from tissues throughout the body. Similar to macrophages, DCs are phagocytic cells and communicate with other cells of the immune system. Additionally, DCs act as potent antigen presenting cells, helping to polarize effector cells and program adaptive responses (Guilliams et al., 2014). Macrophages and DCs are highly motile, and their proper functioning requires tightly controlled migration, whether to the site of damage as tissue resident cells or to lymphatic tissues to present antigens. To better understand how primary myeloid cells function during the innate immune response, communicate with other cells, and move through their diverse environments, it is important to have good biological tools to study these processes. In this protocol we describe the isolation of CD14+ mononuclear cells from peripheral blood by magnetic bead selection, their genetic modification using newly generated lentiviral vectors that express fluorescent markers, and subsequent differentiation into either MDMs or MDDCs.
Lentiviral vectors are versatile and reliable instruments for stable genetic modification of dividing and non-dividing cells (Naldini, Trono, & Verma, 2016). They have a relatively large packaging capacity (> 10kb) and can be pseudotyped with pan-tropic envelope proteins such as vesicular stomatitis virus G protein (VSV-G), which enables entry into a wide range of target cells (Akkina et al., 1996; Naldini et al., 1996; Reiser et al., 1996). However, primary myeloid cells are notoriously difficult to modify through viral-mediated gene delivery (Gruber, Kan-Mitchell, Kuhen, Mukai, & Wong-Staal, 2000; Tan et al., 2005). In part, this is because myeloid DCs (and, to a lesser degree, macrophages) express an arsenal of restriction factors that make them particularly resistant to viruses and viral vectors (Duggal & Emerman, 2012). One of these restriction factors, SAM and HD domain-containing deoxynucleoside triphosphate triphosphohydrolase 1 (SAMHD1), is responsible for maintaining low levels of dNTPs in the cytosol (Goldstone et al., 2011; Lahouassa et al., 2012), which prevents retroviruses and many lentiviruses from reverse transcribing their genomes upon entry (Hrecka et al., 2011; Laguette et al., 2011).
As part of the escalating evolutionary arms race between viruses and their hosts, some viruses have developed defense mechanisms to counter myeloid cell restriction (Colomer-Lluch, Ruiz, Moris, & Prado, 2018). HIV-2 and various strains of simian immunodeficiency virus (SIV) evolved the lentiviral accessory protein Vpx, which targets SAMHD1 for degradation (Hrecka et al., 2011; Laguette et al., 2011). SAMHD1 degradation leads to a rise in cellular dNTP concentrations and permits efficient reverse transcription of HIV-2 and SIV in myeloid cells and resting T cells (Baldauf et al., 2012; Lahouassa et al., 2012). The discovery that Vpx can be supplied in trans to overcome restriction of HIV-1 and related lentiviruses has dramatically improved our ability to genetically modify MDDCs and MDMs with lentiviral vectors (Goujon et al., 2006). Although myeloid cells can be transduced to some degree with lentiviral vectors in the absence of Vpx, delivering virus-like particles that carry Vpx along with lentiviral vectors improves transduction efficiency by one to two orders of magnitude. This approach and similar strategies (Bobadilla, Sunseri, & Landau, 2013) are now routinely used to genetically modify primary human myeloid cells (Berger et al., 2011; Satoh & Manel, 2013). Here, we provide a detailed description of how lentiviral vectors can be used to modify MDDCs and MDMs, but note that these approaches are not without their caveats (discussed in the Commentary Section).
This protocol can be adapted to study many different biological processes that occur in myeloid cells, including: 1) innate immune stimulation (Johnson et al., 2020), 2) virus infection (such as after exposure to HIV-1 or HIV reporter viruses) (Manel et al., 2010), 3) cell trafficking and migration, 4) organelle dynamics following biological perturbation, and 5) transfer of organelles or cytosolic contents between cells. In this protocol we provide examples of how flow cytometry and fluorescence imaging of live and fixed cells can be employed to track expression of lentiviral reporters. As a resource to accompany this article, we have generated a small catalogue of lentiviral vectors that encode fluorescent transgenes to label mitochondria, peroxisomes, and whole cells (see Materials section of Support Protocol 1). The fluorophores in these vectors are provided in three different colors, with the option to detect green, red, or cyan wavelengths (mEmerald, tagRFP-T, and mCerulean) (Balleza, Kim, & Cluzel, 2018; Rizzo & Piston, 2005; Shaner et al., 2008).
The Basic Protocol in this article describes the process of monocyte isolation, transduction with lentiviral vectors, and subsequent differentiation of MDDCs and MDMs. Alternate Protocol 1 describes a cost-effective method to isolate monocytes through adherence to plastic. Alternate Protocol 2 describes how cells can be co-transduced with vectors to knockdown or overexpress genes of interest in tandem with fluorescent labeling. Support Protocol 1 describes the optimized production and purification of lentiviral vectors for these approaches. Support Protocol 2 details methods for analyzing the expression of surface markers and intracellular proteins through flow cytometry. Lastly, Support Protocol 3 describes the use of both fixed and live cell imaging to visualize expression of fluorescent markers in MDDCs and MDMs.
The procedures involved in this protocol require biosafety level 2 (BSL2) precautions. As donor blood is not typically tested for pathogens before it is received in the laboratory, researchers should work under the assumption that blood-borne pathogens are present and follow the safety guidelines outlined by their institution’s biosafety council. All work should be performed in a biosafety cabinet until samples have been inactivated by fixation. When performing steps in this Protocol, researchers should wear appropriate personal protective equipment including double gloves and a liquid-resistant isolation gown that ties in the back.
CAUTION: Standard lentiviruses are considered a Biosafety Level 2 material. Lentiviruses that alter expression of oncogenes, tumor suppressors, or immune-related genes require BSL2-enhanced safety precautions. Follow all appropriate guidelines and regulations for the use and handling of lentiviral particles.
STRATEGIC PLANNING:
Isolation of primary monocytes from whole blood and their transduction with lentiviral vectors is time and labor-intensive, particularly when using many different transduction conditions in cells from multiple donors. As such, it is important that researchers prepare and plan in advance, giving special consideration to the timing of each step in the protocol. It is advised that researchers prepare and ensure the availability of all reagents, consumables, and equipment prior to starting. In particular, this includes 1) production of recombinant lentiviral vectors and virus-like particles (VLPs) packaging Vpx, 2) validating lots of fetal bovine serum (FBS) for MDDC and MDM culture, 3) arranging delivery or acquisition of leukocytes, 4) scheduling time for blood preparation and transduction, 5) scheduling appropriate cytokine administration and cell culture feedings, and 6) procuring/scheduling equipment for end-point assays.
It should be noted that generation and concentration of lentiviral vectors (Support Protocol 1) should be completed prior to the isolation and collection of monocytes from patient blood. Recombinant lentiviral vectors can be frozen at −80 C without significant loss of titer, so we suggest preparing these at least a week in advance and validating they are functional. We recommend that lentiviral titers be determined on immortalized target cells in culture (such as 293FT cells) before they are used to transduce primary cells. We acknowledge that this is not absolutely necessary for laboratories that prepare lentiviral vectors routinely, but given that several common pitfalls can be avoided by validating high vector titer and transduction efficiency (see Commentary, Troubleshooting Table), it is prudent to confirm whether lentivirus preparations can successfully transduce target cells before beginning the Basic Protocol.
Prior to preparation of peripheral blood mononuclear cells (PBMCs), investigators should decide whether they will use positive selection (Basic Protocol 1) or adherence-based selection (Alternate Protocol 1) to isolate monocytes. As an overview of the most labor-intensive aspects of the process, we include a flow chart to outline the steps of cell isolation and transduction and provide estimates for the amount of time needed to complete the protocol (Figure 1). At every step, consider the self-reflective question posed by John Wooden, “If you don’t have time to do it right, when will you have time to do it again?” Once PBMC recovery has begun, viable MDDCs can be expected within five to six days of monocyte isolation and MDMs expected within seven days of isolation.
Figure 1. Schematic illustration of monocyte isolation, genetic modification, and differentiation of MDDCs and MDMs.
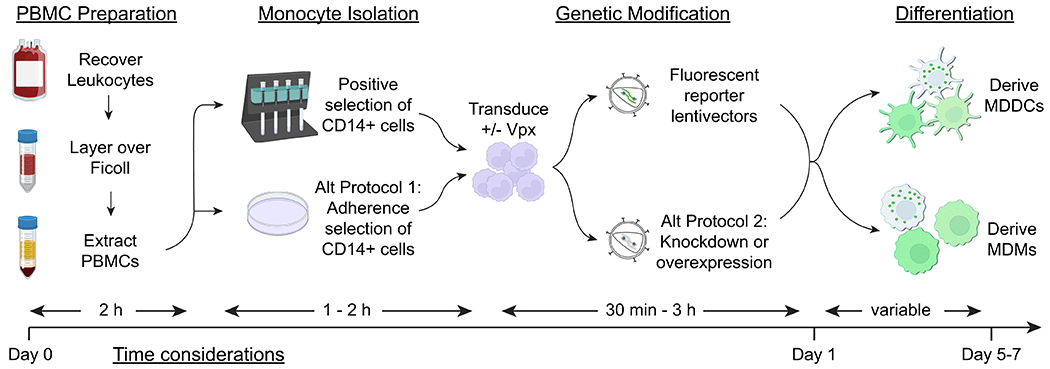
The major steps of the protocols described in this article are shown (PBMC preparation, CD14+ cell isolation, transduction with lentiviral vectors, and differentiation into MDDCs or MDMs), corresponding with estimates of the time investment required at each step.
BASIC PROTOCOL
Basic protocol title: Transduction of MDDCs and MDMs with lentiviral vectors encoding fluorescent markers
Introductory paragraph:
This protocol presents a method to label and genetically modify primary human immune cells for subsequent studies of cell migration, cellular trafficking, and transport of intracellular materials. Here, we describe isolation of CD14+ monocytes from PBMCs through positive selection using magnetic beads, which is the preferred approach when high viability, purity, and yields of monocytes are desired. As a resource to accompany this article, we provide lentiviral vectors to label mitochondria, peroxisomes, and whole cells with three different fluorescent markers. The production of recombinant lentiviral vectors for this protocol is described in Support Protocol 1. Downstream assays for profiling MDDCs and MDMs through flow cytometry and fluorescent imaging are described in Support Protocols 2 and 3.
CAUTION: Take all necessary biosafety precautions for working with blood-borne pathogens. Wear double gloves and a liquid resistant gown that ties in the back.
Materials:
Reagents, solutions, and biological samples:
Disposable nitrile gloves
Liquid resistant isolation gown that ties in back (Medline Industries, cat. no. NONLV350)
Germicidal Bleach, Concentrated, 121 Ounce Bottle (Staples, cat. no. 30966)
70% ethanol
Disposable bench protector (MedPride cat. no. MPR-90503)
Conical Tubes (50 ml and 15 ml)
Source of leukocytes (whole blood, leukopack, leukofilter, or equivalent (local/regional blood bank))
Female luer x 1/8” hose barb adapter (autoclaved) (Cole Parmer, cat. no. EW-45501-04)
60 ml syringes with luer-lock tips (BD, cat. no. 13-689-8)
Serological pipettes (25 ml, 10 ml, 5 ml)
Ficoll-Paque Premium (GE Healthcare, cat. no. 17-5541-03)
Recovery Buffer (see recipe in Reagents and Solutions)
Phosphate buffered saline (Thermofisher, cat. no. 14190235)
Ammonium-Chloride-Potassium (ACK) Lysing Buffer (Thermofisher, cat. no. A1049201)
3 ml Sterile transfer pipettes (Fisher Scientific, cat. no. 13-711-20)
1.5 ml Eppendorf tubes (Corning, cat. no. MCT-175-c)
Hemocytometer (Millipore Sigma, cat. no. Z359629)
Falcon® 70 μm Cell Strainers (Fisher Scientific, cat. no. 08-771-2)
Magnetic activating cell sorting (MACS) buffer (see recipe in Reagents and Solutions)
BSA, Bovine Serum Albumin Fraction V, heat shock, (Sigma, cat. no. 3116956001)
CD14 MicroBeads, human (Miltenyi Biotec, Inc, cat. no. 130-050-201)
LS Columns (Miltenyi Biotec, Inc, cat. no. 130-042-401)
EDTA (0.5 M) for cell culture, pH 8.0, (VWR, cat. no. 46-034-CI)
Qualified, heat-inactivated FBS (ThermoFisher / Gibco, cat. no. 26140079, Lot #1982147 selected for low activation)
Penicillin/Streptomycin (Invitrogen, cat. no. 15140122)
HEPES 1 M (Invitrogen, cat. no. 15630080)
Gibco™ 2-Mercaptoethanol (BME) for cell culture (ThermoFisher, cat. no. 21985023)
RPMI + L-glutamine (Invitrogen, cat. no. 11875093)
RPMI Complete Media (see recipe in Reagents and Solutions)
Recombinant Human GM-CSF, 100 μg (Peprotech, cat. no. 300-03-100UG)
Recombinant Human IL-4, 500 μg (Peprotech, cat. no. 200-04-500UG)
Recombinant Human M-CSF, 100 μg (Peprotech, cat. no. 300-25-100UG)
Polybrene (Millipore cat. no. TR-1003-G)
SIV-VLPs containing Vpx (see Supporting Protocol 1)
Lentiviral vectors (see Supporting Protocol 1)
Tissue culture-treated plates (GenClone, cat. no. 25-203)
Hardware and Instruments:
Biosafety cabinet (BSL Class 2 or equivalent, ThermoFisher Scientific, cat. no. 1371)
Vacuum line and trap (Vactrap or equivalent, VWR, cat. no. 76207-620)
Pipette aid (Drummond Scientific Company, cat. no. 4-000-101)
Pipettes and tips (20 μL, 200 μL and 1000 μL)
Children’s safety scissors (Fiskars, cat. no. 194160-1064)
Tissue culture microscope (Nikon or equivalent)
Table-top clinical centrifuge (ThermoFisher Scientific, cat. no. 75004539)
QuadroMACS Separator (Miltenyi Biotec, cat. no. 130-090-976)
Protocol steps with step annotations:
Setup:
- Before beginning, check supplies of anti-CD14 magnetic beads, LS columns, cell strainers, room temperature Recovery Buffer, 4°C PBS, and room temp ACK Lysing Buffer. We advise taking note of the protocol schematic and planning experiments according to the time considerations shown in Figure 1.Note: Prepare a biosafety cabinet for experiments. Check waste levels and empty vacuum trap if needed. Prepare a freshly diluted solution of bleach (1:10 v/v) to sterilize any serological pipettes that touch blood. Place an absorbent pad on the work surface of the biosafety cabinet to catch potential drips of fluid.
- Prepare MACS buffer (Reagents and Solutions) and filter buffer with a 0.2 micron filter.In most cases this is prepared on the day of monocyte isolation but can be prepared in advance and stored at 4°C.
- Prepare collection tubes and reagents for each donor:
-
aOne 50 ml conical tube to collect blood flushed from leukofilters
-
bTwo 50 ml conical tubes containing 15 ml Ficoll-Paque
-
cOne luer lock adapter
-
dOne 60 ml syringe
-
a
Part 1. Recover leukocytes from whole blood
Leukocytes can be acquired from many different sources. This protocol describes how to isolate fresh leukocytes from whole blood leukofilters that are used during the processing of routine blood donations. Alternative sources include fresh whole blood from phlebotomy/venipuncture or leukopaks from apheresis collections. In some locations, leukocytes can be procured from blood centers that provide products specifically for research, and in some cases, reverse-flush the contents of leukofilters back into blood collection bags prior to delivery to research laboratories (e.g. New York Blood Center). Since the process of recovering cells from leukofilters is not intuitive, we describe this process below, in case alternative sources of leukocytes are not available. To prevent coagulation, blood should be maintained at room temperature during transportation and through the first part of this protocol until the buffy coat is separated from the bulk of the red blood cells (step #20), at which point cells can be handled at temperatures between 4°C and room temperature.
-
5Clean a pair of children’s safety scissors by spraying generously with 70% EtOH.Note: Sharps are not typically allowed in BSL2 labs, but this is a safe and reliable method to access the filter.
-
6Cut the tubing on the side of the filter that the arrow is pointing toward and snugly attach a luer lock adapter to the exposed end of tubing.The arrow on the filter indicates the direction that the blood was pushed through the filter during processing – you want to flush in the opposite direction of the arrow.
-
7
Remove and save the plunger from a 60 ml syringe and then connect the syringe onto the luer lock adapter.
-
8Cut the tubing on the opposite end of the filter.Be sure to keep the filter flat (i.e. lay on the absorbent pad) otherwise blood will begin to drip out.
-
9
Open a 50 ml conical for receipt of blood (tube from step 3a).
-
10Draw up ~20 ml of recovery buffer using a 25 ml serological pipette. Keeping the pipette filled and held in one hand, pick up the syringe and filter with the opposite hand and hold it above the open 50 ml conical.Take care to keep the filter and tubing relatively horizontal until the open end of the tubing is directly above the 50 ml conical. If the open end of the tubing points downward, gravity will take control, and likely lead to blood dripping all over your work surface instead of into the 50 ml conical.
-
11
Pipette the recovery buffer into the syringe and set down the empty pipette without contaminating the tip.
-
12Attach the plunger to the syringe and forcefully flush the contents of the filter into the 50 ml conical just until you begin to pass air through the filter.You will see air bubbles emerge from the draining end of the tubing once all of the fluid has passed through.
-
13
Lay down the filter and syringe and detach the syringe from the luer lock. Pull off the plunger and then reattach the syringe to the luer lock.
-
14
Determine the total volume of liquid that was ejected from the filter (typically ~40 ml) and calculate how much recovery buffer you need to add to bring the total to ~50 ml.
-
15Flush the filter again with a second push of recovery buffer (similar to step #11) to bring the total volume to ~50 ml.Note: You will typically use around 10 ml of recovery buffer for this second flush. At this stage, it is common for bloody air bubbles to extend over the top of the 50 ml conical used for collection; be careful not to generate so many bubbles that they cascade down the side of the conical.
-
16
Dispose of the filter and syringe into biohazard waste.
-
17Using the same 25 ml pipette, aspirate the extra blood bubbles that are above the rim of the 50 ml conical, and rinse the pipette with a freshly diluted solution of bleach (1:10 v/v).After rinsing with bleach, re-sheath the pipette in its paper packaging to avoid drips and dispose of the pipette in a biohazard sharps trash.
Part 2. Buffy coat spins to isolate peripheral blood mononuclear cells (PBMCs)
-
18Layer the blood over Ficoll-Paque cushions.Invert conicals with blood a few times to mix gently. Split the blood evenly into two 50 ml conicals (tubes from step 3b) that should already have 15ml of Ficoll (without disturbing the Ficoll layer). One way to do this is to tilt a tube on a very flat angle and then very slowly and carefully pipette in ~24-25 ml of blood. Setting the pipette aide eject speed to “S” will help control the flow – make sure to set this before pipetting. As you near the finish you can slowly tilt the conical upright. Do this again for a second tube with the same volume of blood to make sure tubes for each donor are balanced.
-
19Spin red blood cells (RBCs) through the Ficoll layer to isolate white blood cells (the “buffy coat”).Cap tubes and carefully spin at 1080 RCF (2,150 RPM in a TX-1000 rotor) for 30 minutes at room temperature in a table-top clinical centrifuge with low brake.Note: It is important to do this in a well-balanced centrifuge that vibrates minimally when up to speed. It is also important to ensure the centrifuge is at room temperature, as cold spins can lead to coagulation of the blood.
-
20Harvest buffy coats.Red blood cells should now be in the bottom Ficoll layer, with platelets & serum on the top layer and your PBMCs in the middle buffy coat. Use a 3 ml sterile transfer pipette to draw off the PBMC buffy coat layers, pooling the two coats from each 50 ml conical into a new 50 ml conical for each donor. Do not disturb the bottom Ficoll-Paque layer. It’s okay to take as much of the top layer as you need to recover all cells from the buffy coat.
-
21Wash Cells with PBS.Pour in enough cold (4°C) PBS to each PBMC tube to make a total volume of 50 ml. Invert gently to mix. You will likely see dead cell aggregates or cell clumps, so pass all 50 ml through a 70 μm cell strainer into a new 50 ml conical. Spin at 200 RCF (900 RPM in a TX-1000 rotor) for 10 minutes at 4°C to remove platelets and sediment the leukocytes. Then aspirate the supernatant from the pelleted leukocytes, taking care to avoid disturbing the pellet.
Part 3. ACK lysis of red blood cells (RBC)
-
22Lyse RBCs.Lyse the cell pellet in 5 ml ACK Lysing Buffer, pipetting up and down ~10 times to resuspend the pellet completely; incubate for 4-5 minutes at room temperature.
-
23Wash cells with PBS.Within 5 minutes of adding ACK, bring up volume to 50 ml using cold PBS and spin at 330 RCF (1,200 RPM in a TX-1000 rotor) for 4 minutes at 4°C, then aspirate supernatant. If cell clumps are present after ACK lysis, these are likely aggregates of dead cells and debris in the sample and should be strained away (i.e. the sample should be filtered into a new conical using a 70 μm strainer before continuing with the prep). Most donors will need at least one straining either after removal of the buffy coat or at this ACK lysis step.
-
24Resuspend PBMCs in MACS buffer.Resuspend the cell pellet in 10 ml of cold MACS buffer by pipetting up and down 10-12 times, then bring total volume to 50 ml with more MACS buffer. As before, any aggregates or cell clumps in the sample that fail to resuspend should be filtered into a new conical using 70 μm strainers before continuing with the protocol.
-
25Count PBMCs.Dilute 10 μl of cells into 190 μl of PBS and count them using a hemocytometer to determine the total number of PBMCs present (typically around 500 M cells per unit of blood).
-
26Pellet PBMCs.Spin cells at 330 RCF (1,200 RPM in a TX-1000 rotor) for 4 minutes at 4°C and then aspirate the supernatant.
Part 4. Positive selection of CD14+ cells using LS columns
-
27Resuspend the PBMC cell pellet.Divide the total number of cells by 500 million to determine the number of mls of MACS buffer to use for resuspension. For example, if there are 500x106 cells in total, resuspend the cell pellet with 1 ml MACS buffer. If there are fewer than 200 million cells, use 400 μl for resuspension. If there are more than 500 million cells, consider the maximum amount that can be loaded onto the column. We typically do not isolate CD14+ cells from more than 500 million cells per donor. Excess cells can be frozen in liquid nitrogen (typically 10 million cells per ml in 90 % FBS, 10 % DMSO; 0.5 – 1 ml per cryotube) or discarded.
-
28Incubate PBMCs with anti-CD14 beads.Add 100 μl of human anti-CD14 beads for each 100 million cells, swirl gently and incubate for 30 minutes at 4°C in the fridge. Note: New MACS beads may have a different ratio of beads to cells, so always consult the instructions provided with the kit.
-
29Equilibrate LS columns.Near the end of the bead incubation, prepare LS columns for each donor. For each donor, attach a column to the magnet and place a 15 ml conical in the rack to collect flow-through. To equilibrate the column, add 3 ml of degassed MACS buffer (making sure to avoid bubbles) and let it drip into the flow-through tube.
-
30Wash excess beads from cells.When cells have finished incubating with beads, bring up the volume to ~35 ml with cold MACS buffer. Spin at 330 RCF (1,200 RPM in a TX-1000 rotor) for 4 min at 4°C. Resuspend cells in 1 ml of MACS buffer for every 100 million cells in the sample.
-
31Load cells onto LS columns.Pass cells through a 70 μm nylon filter as you apply the cell suspension to the top of the LS column. Hold the filter at an angle over the column and let cells drip through.
-
32Wash LS columns.After all of the cell suspension has passed through the columns by gravity flow, wash the column with 5 ml of cold MACS buffer. CD14-(negative) cells in the flow-through fraction can be banked and used in downstream assays. Otherwise, aspirate the flow-through to make room for more washes. Wash the column three more times, each time using 3 ml MACS buffer. Before each wash, make sure that the previous wash has stopped dripping from the columns.
-
33Elute CD14+ cells from columns.Remove the column from the magnet, place the column into the neck of a new 15 ml conical, add 5 ml of MACS buffer, and then quickly use the LS column plunger to forcefully flush the cells out of the column.
-
34Count CD14+ cells.Mix cells by gently pipetting up and down ~5 times with a P1000. We recommend against mixing by inversion or mixing with a serological pipette. Dilute 10 μl of cells into 190 μl MACS buffer, mix thoroughly, and count 10 ul of this dilution using a hemocytometer. Note: CD14+ cells are incredibly phagocytic and including a viability dye like trypan blue is not helpful for counting as most of the cells will rapidly take up dye. Thus, we prefer manual counting on a hemocytometer to using an automated cell counter. Typically, one unit of blood (i.e. 500 million PBMCs) will yield between 20 million and 50 million CD14+ cells.
Part 5. Transduction and differentiation into dendritic cells or macrophages
-
35Remove MACS buffer and resuspend cells into an appropriate volume of complete RPMI media with cytokines.Spin down CD14+ cells at 330 RCF (1,200 RPM in a TX-1000 rotor) for 4 minutes at 4°C. Resuspend into complete RPMI media with cytokines and polybrene to a concentration of 1 million CD14+ cells per ml of media.Note: Polybrene should be used at 0.5 μg/ml (final concentration). Regarding cytokines, for MDDCs: GM-CSF should be used at 1:1000 (10 ng/ml final concentration) plus IL-4 used at 1:1000 (50 ng/ml final concentration); for MDMs: M-CSF should be used at 1:500 (20 ng/ml final concentration). Protamine sulfate can be used as an alternative to polybrene.Note: Either TC or non-TC treated plates can be used. If TC treated plates are used, MDMs will take longer to detach when used for experimental procedures. Longer detachment periods can affect the expression of surface proteins and so it is suggested to use non-TC treated plates (Chen, So, Strome, & Zhang, 2015). For MDDCs, differentiation on TC treated plates is associated with higher expression of inflammatory genes and cell surface markers of activation when compared to growth on non-TC treated plates (Sauter, Yi, Li, Roersma, & Appel, 2019). Moreover, the stiffness of the attachment substrate has recently been shown to influence DC activation profiles and metabolic flux (Chakraborty et al., 2021). Since these data indicate that the plate surface and/or attachment tension can impact inflammatory responses in MDDCs (and likely MDMs), the choice of TC treated vs non-TC treated plates is an important parameter to consider.
-
36Inoculate CD14+ cells with SIV-VLPs if desired.Concentrated stocks of SIV-VLPs (prepared as described in Support Protocol 1) should be used at 20-40 μl per 1 million CD14+ cells. Unconcentrated supernatants containing SIV-VLPs should be used at 1-2 ml per 1 million CD14+ cells (amounts should be optimized empirically).Note: The addition of SIV-VLPs can be done before or after plating CD14+ cells. We typically do this “en masse” and add SIV-VLPs to cells in suspension prior to plating.
-
37
Plate into an appropriately sized culture dish (1 million CD14+ cells per ml of media).
-
38Transduce CD14+ cells with lentiviral vectors.If using SIV-VLPs, ensure that at least 30 min has elapsed prior to adding lentivirus. While transduction can occur when adding lentivirus simultaneously with SIV-VLPs, transduction efficiencies will be significantly higher if time is allowed for Vpx to prevent lentiviral restriction (and degrade SAMHD1) prior to inoculation with lentiviral particles.Add lentiviral stocks (prepared as described in Support Protocol 1) to CD14+ cells. Concentrated lentivirus stocks should be used at 10-100 μl per 1 million CD14+ cells (typical stock concentrations are 500 nM p24 as determined by ELISA). Unconcentrated lentiviral supernatants should be used at 0.5-5 ml per 1 million CD14+ cells (optimal amounts should be determined empirically).
-
39
Culture cells in a humidified incubator at 37°C with 5% CO2.
-
40Replenish media on day 1 or day 2 after plating.Simply add more media and cytokines on day 1 or day 2 (add 50% complete media by volume with the same concentrations of cytokines).
-
41Treat immature MDDCs for downstream assays.On day 4, immature MDDCs (in suspension) need to be replated into fresh media with cytokines. Count cells (recovery should be somewhere around 30% of the number of CD14+ cells that were initially plated). Spin down (330 RCF; 1,200 RPM in a TX-1000 rotor for 4 minutes) and resuspend at ~500 k cells per ml in fresh media with cytokines at half strength (1:2000 for GM-CSF and IL-4). It is acceptable to plan experimental end points for DCs to be between day 5 and day 7 after CD14 isolation. While MDDCs may be kept in culture for longer than 7 days, we have observed decreased responses to stimulation in older cultures (see also note for MDMs in Step 41.)
-
42Treat MDMs for downstream assays.By day 4, some cells may have attached. For cells that have not yet attached, collect them by gentle pipetting, spin down (330 RCF; 1,200 RPM in a TX-1000 rotor for 4 minutes) resuspend into fresh media and replate them into the same culture vessel. Before the cells on the plate dry out (i.e. before you begin spinning the cells in suspension) be sure to add back fresh media and cytokines to the plate. Refresh media every 2-3 days as necessary (for example, this can be arranged to accommodate weekday or weekend schedules). Macrophages will attach once differentiated. It is acceptable to plan end points for macrophages to fall between day 7 and day 14 after CD14 isolation. Keep in mind that cellular behavior and responses to stimulation are likely not equivalent for MDMs cultured for 7 days vs. 14 days. For example, inflammatory cytokine output has been reported to decrease in long term cultures of murine bone marrow-derived macrophages and human MDMs (Chamberlain, Holt-Casper, Gonzalez-Juarrero, & Grainger, 2015; Jones et al., 2007). Investigators should be aware of this variable when using in vitro derived cultures and use consistent end points to maintain rigor and reproducibility in their experiments.Please see information about sample data in the Understanding Results section (below).
ALTERNATE PROTOCOL 1
Alternate protocol title: Isolation of monocytes by plastic adhesion
In this protocol, we describe the isolation of CD14+ monocytes using adherence-based selection, which relies on monocyte attachment to tissue culture plates and subsequent washing away of non-adherent cells. Adherence-based selection requires less “hands-on” time in tissue culture than magnetic bead selection and is considerably cheaper. While plastic adhesion results in a less-pure population of cells than positive selection (Nielsen, Andersen, & Moller, 2020), this method represents an attractive alternative for isolating cells when limited by time or financial constraints.
Additional Materials:
Serum free RPMI (ThermoFisher, cat. no. 11875085)
Protocol steps with step annotations:
Process blood to collect leukocytes and lyse RBCs according to steps 1-22 in Basic Protocol 1.
- Count cells following resuspension in PBS post-ACK lysis.Dilute 10 μl of cells into 190 μl of PBS and count them using a hemocytometer to determine the total number of PBMCs present (typically around 500M cells per unit of blood).
- Plate up to 1 x 108 cells on 15 cm cell culture dishes in serum free RPMI and allow monocytes to attach for 2 hours.Note: Either TC coated plates or non-coated plates can be used during this step. If TC coated plates are used, monocytes will adhere faster but be harder to detach when plating for experimental procedures.
- Rinse non-adherent cells from the plate using pre-warmed PBS.After 2 hours, CD14+ cells should be firmly attached to the tissue culture plate. At this point, gently rinse off the non-adherent cells by adding 10 ml of pre-warmed PBS into the plate and pipette up and down ~5 times to remove unattached cells.
Repeat step 4 two more times to flush remaining non-adherent cells.
- Add complete RPMI with cytokines to attached cells.For MDDCs: GM-CSF should be used at 1:1000 (10 ng/ml final concentration) plus IL-4 used at 1:1000 (50 ng/ml final concentration);For MDMs: M-CSF should be used at 1:500 (20 ng/ml final concentration).In both cases, polybrene should be used at 0.5 μg/ml (final concentration) to improve transduction efficiency. Protamine sulfate can be used as an alternative to polybrene.
- Inoculate CD14+ cells with SIV-VLPs (if desired) and transduce with lentiviral vectors according to Basic Protocol 1 or Alternate Protocol 2.It is difficult to determine the precise number of attached CD14+ cells without lifting and counting (which is not recommended). It is reasonable to estimate that 10% of PBMCs will attach using the adherence method and this estimate can be used to determine the amount of SIV-VLPs and lentivirus stocks to use for transduction.Concentrated stocks of SIV-VLPs (prepared as described in Support Protocol 1) should be used at 20-40 μl per 1 million CD14+ cells. Unconcentrated supernatants containing SIV-VLPs should be used at 1-2 ml per 1 million CD14+ cells (amounts should be optimized empirically).If using SIV-VLPs, ensure that at least 30 min has elapsed prior to adding lentivirus and then add lentiviral stocks (prepared and concentrated as described in Support Protocol 1) to CD14+ cells. The total amount of concentrated lentivirus stocks should not exceed 100 μl per 1 million CD14+ cells (typical stock concentrations are 500 nM p24 as determined by ELISA).
- Complete the procedure by culturing cells in a humidified incubator at 37°C with 5% CO2 following steps 38-41 in the Basic Protocol.Investigators should then perform downstream assays to validate vector expression and determine biological effects (examples provided in Support Protocols 2 and 3).
ALTERNATE PROTOCOL 2
Alternate protocol title: Transduction of MDDCs and MDMs with lentiviral vectors to knockdown or overexpress genes of interest
In addition to expressing fluorescent markers in myeloid cells, it is often useful to knockdown or overexpress genes of interest in the same cells that express these markers. One way this can be achieved is through delivering a single transgene that encodes a fluorescent marker along with a cassette for shRNA or cDNA expression (Manel et al., 2010). The lentiviral plasmids that are described herein (pLKO-MCS and related plasmids available through Addgene) include a U6 promoter-driven shRNA cassette should investigators choose to pursue this approach. However, it is sometimes more convenient to deliver fluorescent makers and knockdown/overexpression cassettes using separate vectors, and efficient co-transduction into the same cell can be achieved under optimized conditions. This protocol provides a framework for those conditions and describes a method for co-transduction of vectors that express fluorescent markers together with vectors designed to perturb target genes.
CAUTION: Standard lentiviruses are considered a Biosafety Level 2 material. Lentiviruses that alter expression of oncogenes, tumor suppressors, or immune-related genes require BSL2-enhanced safety precautions. Follow all appropriate guidelines and regulations for the use and handling of lentiviral particles.
Materials:
No additional materials required.
Protocol steps with step annotations:
- Generate shRNA or overexpression lentivectors according to Support Protocol 1.Vector production should be performed prior to preparation of blood. Ideally, vectors should be titered and/or it should be determined whether they are capable of transducing cells at high efficiency before they are used in primary human cells.
Process blood to collect leukocytes and isolate CD14+ cells according to steps 1-33 in Basic Protocol 1.
- Remove MACS buffer and resuspend CD14+ cells into an appropriate volume of complete RPMI media with cytokines.Spin down CD14+ cells at 330 RCF (1,200 RPM in a TX-1000 rotor) for 4 minutes at 4°C. Resuspend into complete RPMI media with cytokines, and polybrene to a concentration of 1 million CD14+ cells per ml of media.Note: Polybrene should be used at 0.5 μg/ml (final concentration). Regarding cytokines, for MDDCs: GM-CSF should be used at 1:1000 (10 ng/ml final concentration) plus IL-4 used at 1:1000 (50 ng/ml final concentration); for macrophages: M-CSF should be used at 1:500 (20 ng/ml final concentration). Protamine sulfate can be used as an alternative to polybrene.Note: Either TC or non-TC coated plates can be used, if TC coated plates are used macrophages will take longer to detach when used for downstream experimental procedures.
- Inoculate CD14+ cells with SIV-VLPs if desired.As SIV-VLPs containing Vpx significantly increase transduction efficiency in MDDCs and MDMs (>1 log order increases in MFI, see Understanding Results Section), they can be particularly helpful when attempting to achieve high efficiency genetic perturbations, especially when co-transducing cells with more than one lentivector, as described here. Concentrated stocks of SIV-VLPs (prepared as described in Support Protocol 1) should be used at 20-40 μl per 1 million CD14+ cells. Unconcentrated supernatants containing SIV-VLPs should be used at 1-2 ml per 1 million CD14+ cells (amounts should be optimized empirically).Note: The addition of SIV-VLPs can be done before or after plating CD14+ cells. We typically do this “en masse” and add SIV-VLPs to cells in suspension prior to plating.
- Plate into an appropriately sized culture dish (1M CD14+ cells per ml of media).We transduce MDDCs and MDMs in a variety of different culture vessels, from 96 well plates to 15 cm dishes and have not observed significant differences in transduction efficiency.
- Transduce CD14+ cells with lentiviral vectors.If using SIV-VLPs, ensure that at least 30 min has elapsed prior to adding lentivirus.Add lentiviral stocks (prepared and concentrated as described in Support Protocol 1) to CD14+ cells. The total amount of concentrated lentivirus stocks should not exceed 100 μl per 1 million CD14+ cells (typical stock concentrations are 500 nM p24 as determined by ELISA).Note: Combining lentiviral vectors will reduce the transduction efficiency of each vector. Thus, to maximize the penetrance of knockdown vectors when combined with fluorescent reporter vectors, we recommend testing ratios between 1:1 and 5:1 (shRNA vector to reporter vector), and optimal amounts should be determined empirically.
- Complete the procedure by culturing cells in a humidified incubator at 37°C with 5% CO2 following steps 38-41 in the Basic Protocol.Investigators should then perform downstream assays to validate vector expression and determine biology effects (examples provided in Support Protocols 2 and 3).
SUPPORT PROTOCOL 1:
Support protocol title: Production and purification of lentiviral vectors for transduction into primary human myeloid cells
This protocol describes the process by which recombinant lentiviral vectors and SIV-VLPs are produced through transfection of HEK293FT cells. To improve the safety of these reagents, viral genes important for transduction are separated into different plasmids, minimizing the risk of recombination and mobilization of infectious agents. In this system, three plasmids are required to produce recombinant lentivirus: 1) a plasmid that encodes genes important for lentiviral capsid structure and packaging (psPax2), 2) a plasmid that encodes an envelope gene (p-CMV-VSV-G (Stewart et al., 2003)), and 3) a plasmid that encodes the transgene cassette to be packaged within the lentivirus (pLKO or equivalent). SIV-VLPs are produced in the same fashion, requiring structural components (pSIV3+ (Mangeot et al., 2000)) and an envelope (p-CMV-VSV-G), but as virus-like particles do not contain a transgene, a third plasmid is not included in transfections to produce SIV-VLPs. To accompany this protocol, we provide new lentiviral reagents to express fluorescent protein markers to tag whole cells, or specifically target the peroxisome, or the mitochondria. Three fluorophores are available: mEmerald, tagRFP-T, and mCerulean. These lentiviral vectors expand our toolkit of reagents that can be used to track cell migration, activation, and organelle dynamics through fluorescence-based assays.
CAUTION: Standard lentiviruses are considered a Biosafety Level 2 material. Lentiviruses that alter expression of oncogenes, tumor suppressors, or immune-related genes require BSL2-enhanced safety precautions. Follow all appropriate guidelines and regulations for the use and handling of lentiviral particles.
Materials:
Polyethylenimine “Max” (PEI Max) (Polysciences, cat. no. 49553-93-7)
Phosphate buffered saline (Thermofisher, cat. no. 14190235)
HCl (Sigma Aldrich, cat. no. 320331)
Sodium Hydroxide (NaOH) (Sigma Aldrich, cat. no. 72068)
Poly-L-lysine (Sigma Aldrich, cat. no. P6282-5MG)
500 ml filter PES 0.22 μm (GenClone, cat. no. 25-227)
15 cm TC coated plates (GenClone, cat. no. 25-203)
Cell culture incubator set to 37°C and 5% CO2 (ThermoFisher Heracell or equivalent)
Pipette aid (Drummond Scientific Company, cat. no. 4-000-101)
Serological pipettes (10 ml, 25 ml)
Sterile water
HEK293FT cells, (Thermofisher, cat. no. R70007)
DMEM complete media (see recipe in Reagent and Solution section)
DMEM (Thermofisher, cat. no. 11995040)
CMV-VSV-G plasmid (a gift from Bob Weinberg: Addgene, 8454)
psPax2 plasmid (a gift from Didier Trono: Addgene, 12260)
pLKO-MCS (Addgene, 185594)
pLKO-cell-mEm (Addgene, 185595)
pLKO-mito-mEm (Addgene, 185596)
pLKO-perox-mEm (Addgene, 185597)
pLKO-cell-tagRFP (Addgene, 185598)
pLKO-mito-tagRFP (Addgene, 185599)
pLKO-perox-tagRFP (Addgene, 185600)
pLKO-cell-mCer (Addgene, 185601)
pLKO-mito-mCer (Addgene, 185602)
pLKO-perox-mCer (Addgene, 185603)
pSIV3+ (Mangeot et al., 2000)
Timer (Traceable, cat. no. 5004CC)
Micropipettes and tips (20 ul, 200 ul, 1000 ul)
Vortex genie (Scientific Industries, Inc, cat. no. SI-0236)
1.5 ml tube (Corning cat. no. MCT-175-c)
Ultracentrifuge tubes (Beckman Coulter, cat. no. 344058)
SW-32 Ti rotor (Beckman Coulter, cat. no. 369650)
Ultracentrifuge (Beckman Optima L-90K Ultracentrifuge or equivalent)
Precision balance (Sartorius, cat. no. BCE822-1S)
Protocol steps with step annotations:
- Prepare PEI.Dissolve 100 mg PEI by adding 50 μl of 12 N HCl followed by 900 μl of PBS in a beaker. Swirl gently while adding ~25ml PBS until the PEI completely goes into solution. Dilute to 1 mg/ml by adding close to 75 ml PBS and adjust final pH to 7.0 using 1 M NaOH dropwise. Filter sterilize through a 0.2 μm filter in hood, aliquot to 5 ml or 10 ml fractions in 15 ml conicals, and store at 4°C. PEI stored at 4°C can be used for at least one year, but re-filtering is recommended every few months.Note: Each batch of PEI needs to be titrated in a test transfection to identify the optimum ratio of PEI to DNA (typically, this is close to 2 ug of PEI for 1 μg of DNA)
- Prepare poly-L-lysine plates.Dissolve 5 mg of poly-L-lysine into 500 ml of deionized water and filter sterilize through a 0.22 μm filter. Pipette 17 ml of poly-L-lysine (0.01 mg/ml) into each 15 cm tissue culture plate needed for transfection and place them in a 37°C incubator for at least 30 min (up to several hours). Remove the poly-L-lysine and rinse each plate 2X with sterile deionized water.Note: Rinse plates quickly so as to not let the poly-L-lysine dry out at high concentration on the plate. After the second wash is removed, poly-L-lysine-coated plates can be stored at room temp or at 4°C in sterile conditions for several days-weeks.Note: Diluted poly-L-lysine can be reused if stored at 4°C in sterile conditions.
- Plate HEK293FT cells for transfection.Approximately 24 h before transfection, plate approximately 6 to 8 million HEK293FT cells in 25 ml media on poly lysine-coated plates, such that at the time of transfection the cells will be 70-80% confluent.Note: If cell density is not ideal, do not proceed as yield will be compromised. Cell number can be adjusted so that transfection can be performed between 16 and 28 h after cell plating without significant reduction in transfection efficiency. Maintain stocks of cells at low passage number and discard older cultures (aged beyond passage #25-30), as lentiviral yields can suffer in high-passage cells.
- Prepare transfection mixtures.For each transfection, prepare 950 μl of DMEM (no antibiotics or supplements) and add the appropriate plasmids (see Table 1 for plasmid amounts). If doing a large number of transfections with the same envelope and packaging construct, you may prepare a master mix (with pCMV-VSV-G and psPax2). Vortex master mix for 6-8, one-second pulses, aliquot 950 μl into 1.5 ml tubes for each transfection. Add transgene plasmids to DMEM containing pCMV-VSV-G and psPax2 (according to amounts in Table 1) or pCMV-VSV-G and pSIV3+ (according to amounts in Table 2).
- Spike in PEI.Set a timer for 10 minutes before spiking in PEI to the first transfection tube. Add PEI (50 μl) to one tube at a time and immediately vortex the tube for ~6, one-second pulses on medium-high. Do not overmix. Spin down briefly to remove contents from lids and incubate for the remainder of the 10 minutes at room temp.Note: Time is critical for this step. PEI-DNA complex incubation times that are too short or too long can reduce transfection efficiency.
- Add transfection complexes to 293FT cells.After the timer goes off for the first tube, slowly add entire contents, dropwise, to 293FT cells (at the appropriate density) in plates, and gently slide the plate back-and-forth and side-to-side to distribute. Place back into humidified 37°C incubator overnight.
- Wash transfection mixtures from cells.The following morning after transfection, aspirate the cell media, gently wash with 4 ml complete media, aspirate, and replenish plates with ~32 ml complete medium.
- Harvest supernatants from transfected cells.Supernatants containing recombinant lentivirus can be harvested between 24 and 48 h after washing cells. We routinely achieve high titers when collecting supernatants between 36 and 48 h after transfection mixtures are washed (step 7). Supernatants should be passed through 0.45 μm syringe filters and collected in 50 ml conicals.Note: This is a potential stopping point. Unconcentrated viral supernatants can be frozen at −80°C prior to concentration.Note: Concentration of viral supernatant improves transduction efficiency and is recommended. However, it is not required and viral supernatant can be added pre-concentration directly to primary myeloid cells.
- Transfer the filtered viral supernatants into ultracentrifuge tubes.Spin down 50 ml conicals with lentiviral supernatants to pellet cell debris that may not have been cleared by the 0.45 μm filter (4 minutes at 330 RCF). Transfer 30 ml of supernatants to clear Beckman ultracentrifuge tubes and balance them to within 0.03 g using an analytical balance set up in the biosafety cabinet.
- Spin down the viral supernatants in an ultracentrifuge.Transfer the balanced ultracentrifuge tubes into swing buckets and load them into an SW-32 Ti rotor. Set the centrifuge for max acceleration and slow deceleration and spin at 115,000 RCF (26,000 RPM) for 2 h at 4°C.
In the hood, remove the supernatant while taking care not to disturb the pellet.
- Resuspend the lentiviral pellet.Add 1 ml of cold, RPMI complete media and incubate on ice for 30 minutes. Resuspend by vigorously pipetting up and down (at least 40-50 times). Avoid introducing bubbles. Store at 4°C for a few hours to overnight.
- Clarify concentrated lentivirus.After the lentivirus has settled for at least a few hours at 4°C, resuspending the contents a second time will enable more particles to go into suspension. Pipette the concentrated stock up and down 40-50 more times (avoiding bubbles). Spin down insoluble particulates (3 minutes at 300 RCF) and carefully transfer the supernatant to a new tube without disturbing the pellet (which may or may not be visible). Aliquot the concentrated supernatant and freeze at −80°C.
Table 1.
Plasmid amounts for transfection into one 15 cm plate to produce recombinant lentivirus
| Plasmid: | μg to be added | Percentage |
|---|---|---|
| pCMV-VSV-G | 3.38 | 15 |
| psPax2 | 8.44 | 37.5 |
| Transgene cassette (e.g. pLKO-cell-mEm) | 10.7 | 47.5 |
| Total: | 22.5 |
Table 2.
Plasmid amounts for transfection into one 15 cm plate to produce SIV-VLPs
| Plasmid: | μg to be added | Percentage |
|---|---|---|
| pCMV-VSV-G | 3.38 | 15 |
| pSIV3+ | 19.12 | 85 |
| Total: | 22.5 |
Please see information about sample data in the Understanding Results section (below).
SUPPORT PROTOCOL 2: Flow cytometry of MDDCs and MDMs
Here, we describe the use of flow cytometry to measure expression levels of fluorescent reporters transduced by lentivectors in concert with expression of host proteins in MDDCs and MDMs. Multicolor flow cytometry is a powerful tool for determining target protein expression, either at the cell surface (through staining unpermeabilized cells) or within the cell (after fixation and subsequent cell permeabilization). Both conventional and spectral flow cytometers are capable of measuring expression levels of dozens of targets at the single cell level simultaneously (advantages and disadvantages of different cytometers are described elsewhere (Nolan & Condello, 2013)). We recommend giving careful consideration to antibody-fluorophore combinations to minimize overlap in emission wavelengths, and to plan for and incorporate single color controls in experimental design. The antibodies suggested in this protocol have minimal overlap and require minimal compensation. Flow cytometry, especially when combined with high-throughput sampling, is a reliable method for quantitatively tracking myeloid cell isolation, differentiation, and innate immune activation. We provide example flow cytometry data for MDDCs in Figure 2A–C and MDMs in Figure 3A–C.
CAUTION: Inactivate all virus dilutions before disposal with a freshly prepared 1:10 dilution of bleach or through fixation with 4% paraformaldehyde. Please take care to not mix bleach with paraformaldehyde (such as following fixation) to avoid generating noxious gas. Prepare a waste beaker with diluted bleach in the biosafety cabinet for ejecting liquid.
Note: Keep all reagents on ice and keep antibodies in the dark.
Note: Work on one plate at a time and do not let cells dry out.
Figure 2. Genetic modification of MDDCs with fluorescent lentiviral reporter vectors.
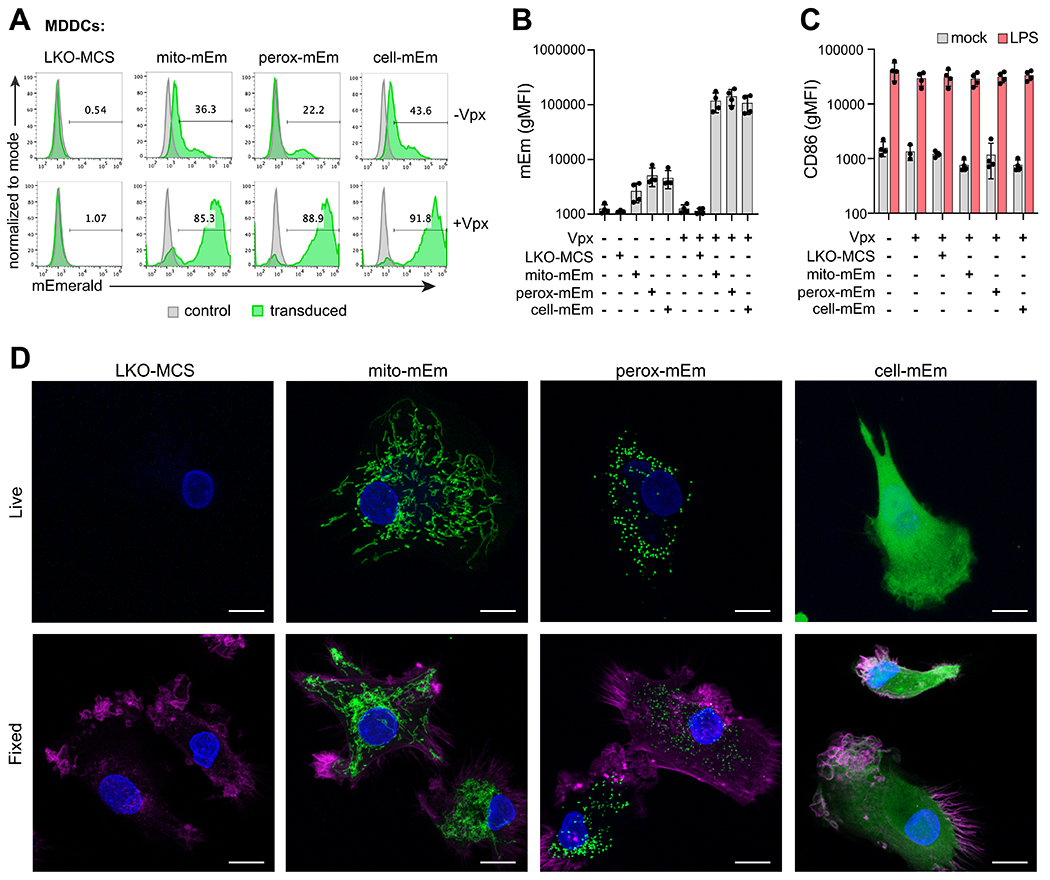
A) Representative flow cytometry histograms of mEmerald expression in MDDCs at day 7 after transduction with LKO-MCS (vector alone), mito-mEm, perox-mEm, or cell-mEm in the presence or absence of SIV-VLPs packaging Vpx. B) Geometric mean fluorescence intensity (gMFI) of mEmerald expression in transduced MDDCs. C) gMFI of CD86 expression in mock-treated or transduced MDDCs that were either unstimulated or stimulated with LPS (100 ng/ml for 24 h). For (C) and (D), n = 4 independent donors. D) Representative z-stack projections (merge of 10-20 planes, 0.5 micron step size) of live and fixed images of transduced MDDCs acquired with an LSM880 Airyscan Fast confocal microscope. Images reveal localization of fluorescent markers (mEmerald expression, green), nuclei (DAPI, blue), and actin (Phalloidin, magenta – only depicted in fixed cell images). Scale bar = 10 μm.
Figure 3. Genetic modification of MDMs with fluorescent lentiviral reporter vectors.
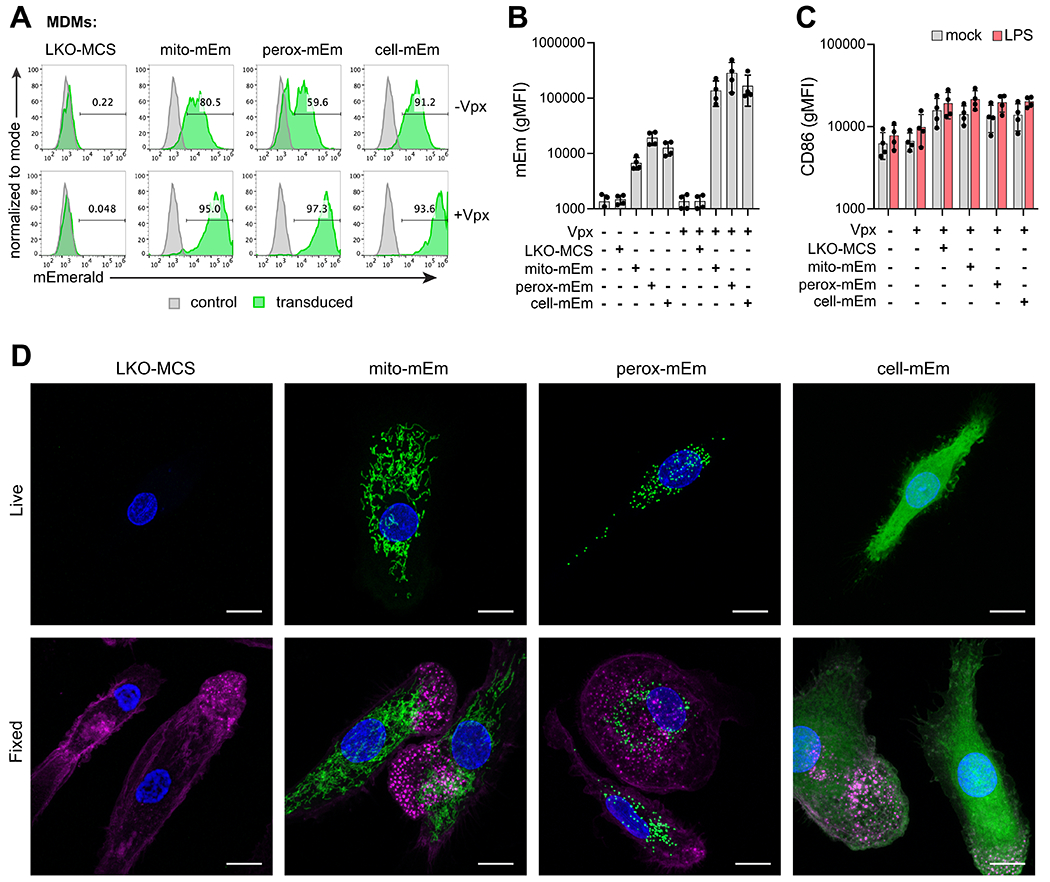
A) Representative flow cytometry histograms of mEmerald expression in MDMs at day 7 after transduction with LKO-MCS (vector alone), mito-mEm, perox-mEm, or cell-mEm in the presence or absence of SIV-VLPs packaging Vpx. B) Geometric mean fluorescence intensity (gMFI) of mEmerald expression in transduced MDMs. C) gMFI of CD86 expression in mock-treated or transduced MDMs that were either unstimulated or stimulated with LPS (100 ng/ml for 24 h). For (C) and (D), n = 4 independent donors. D) Representative z-stack projections (merge of 10-20 planes, 0.5 micron step size) of live and fixed images of transduced MDMs acquired with an LSM880 Airyscan Fast confocal microscope. Images reveal localization of fluorescent markers (mEmerald expression, green), nuclei (DAPI, blue), and actin (Phalloidin, magenta – only depicted in fixed cell images). Scale bar = 10 μm.
Additional Materials:
Reagents, solutions, and biological samples:
Germicidal Bleach, Concentrated, 121 Ounce Bottle (Staples, cat. no. 30966)
RPMI Complete Media (see recipe in Reagents and Solutions)
Phosphate buffered saline (Thermofisher, cat. no. 14190235)
BSA, Bovine Serum Albumin Fraction V, heat shock, (Sigma, cat. no. 3116956001)
Ultrapure LPS from Salmonella minnesota (Invivogen, cat. no. TLRL-SMLPS)
Staining buffer (see recipe in Reagents and Solutions)
Fixation/Permeabilization Solution Kit with BD GolgiPlug (BD, cat. no. 555028)
LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit (ThermoFisher, cat. no. L34955)
Aluminum foil
CD86-PECy5 (ThermoFisher, cat. no. 15-0869-42)
Human Fc receptor block: Human TruStain FcXTM (Biolegend, cat. no.422302)
Accutase (Sigma, cat. no. A6964-100ML)
Hardware and Instruments
Biosafety cabinet (BSL Class 2 or equivalent, ThermoFisher Scientific, cat. no. 1371)
Vacuum line and trap (Vactrap or equivalent, VWR, cat. no. 76207-620)
Pipette aid (Drummond Scientific Company, cat. no. 4-000-101)
Pipettes and tips (20 ul, 200 ul and 1000 ul)
96 well U-bottom plate (non-tissue culture-treated)
Tissue culture microscope (Nikon or equivalent)
Table-top clinical centrifuge (ThermoFisher Scientific, cat. no. 75004539)
Flow cytometer (ThermoFisher, Attune NxT)
Protocol steps with step annotations:
-
1Prepare cells for flow cytometry.Infect or stimulate MDDCs or MDMs according to your needs. In most cases this will be in a 96 well plate format with between 50k and 200k cells per well. As a positive control for MDDC activation, lipopolysaccharide (LPS) can be administered to cells at 100 ng/ml for 16 h prior to staining. If staining for cytokines that are normally secreted (e.g. CXCL10), then ~4-8 h before staining, add GolgiPlug (or GolgiStop) to culture media to prevent secretion and cause intracellular accumulation. Dilute so that final concentration is 1 μl GolgiPlug per ml of culture.
-
2If performing intracellular staining, prepare Perm/Wash and antibody dilutions.Dilute 10X Perm/Wash to 1X with deionized water. Prepare enough Perm/Wash to sufficiently cover three washes and intracellular stains. For example, to stain one 96 well plate, you will need ~40 ml of 1X Perm/Wash buffer. Antibody dilutions for intracellular staining are prepared in Perm/Wash solution. For example, prepare ISG15-PE diluted at 1:100 with enough staining solution for all samples and controls, calculating 50 μl per sample. Keep on ice.
-
3Prepare live/dead stain.For one, 96 well plate, you will need 5.5 ml of diluted live/dead stain. Prepare stocks of live/dead in DMSO as described by the manufacturer and store 5 μl aliquots at −20°C. To prepare diluted live/dead stain, resuspend one 5 μl aliquot of the stock into 5.5 ml of PBS.
-
4Spin down cells (330 RCF, for 4 min) in the 96 well U bottom plate.Using a multichannel, carefully aspirate media without disturbing the cell pellet (by tilting the plate and pipetting along the back wall of each well. Eject liquid into the waste beaker. Visually inspect wells to make sure a pellet is visible after aspiration.
-
5
Using the multichannel, add 100 μl of PBS per well (no need to resuspend).
-
6
Spin cells and aspirate PBS as in step #4.
-
7Add 50 μl Live/Dead stain per well.Gently resuspend with the multichannel by pipetting up and down ~3 times.
-
8
Cover plate with aluminum foil and incubate cells at room temp for 10 min with Live/Dead.
-
9Spin cells and aspirate live/dead stain as in step #4.At this point, if you are working with MDDCs, they should already be in suspension. If you are working with MDMs, you will need to detach them from the plate using Accutase (add 50 μl of Accutase per well to cell pellets, incubate for 5 – 10 min at 37°C, verify that cells have detached, and then continue with the protocol). We prefer using Accutase as it can minimize the enzymatic cleavage of surface proteins when compared to Trypsin while maintaining high viability when compared with physical detachment methods (Chen et al., 2015).
-
10
Wash 1X with 100 μl of staining buffer (SB) – (for intracellular staining)
-or- Wash 1X with 100 μl PBS (for surface marker staining)
-
11
Spin cells and aspirate wash as in step #4.
Surface marker staining (optional)
-
12
Resuspend cells in 50 μl PBS containing 2 μl Fc receptor blocking solution and incubate for 5-10 min at room temperature (covered in foil).
-
13
Spin cells and aspirate Fc block as in step #4
-
14Prepare antibody dilutions in PBS and add 50 μl per well.Ensure that each well in the 96 well plate is aspirated equally. If residual wash buffer is left in some wells this will lead to variability in staining intensity between conditions. Gently resuspend the cell pellets by smoothly pipetting up and down ~6-8 times (resuspension improves staining intensity). As an example stain, CD86-PECy5 should be diluted 1:200 in PBS. Compatible fluorophores can be used simultaneously as long as single-color controls are prepared in parallel.
-
15
Incubate for 15 – 30 min at 4°C (covered in foil to minimize exposure to light).
-
16
Spin cells and aspirate antibody stain as in step #4.
-
17
Wash cells twice with PBS (100 μl each wash).
-
18
If performing intracellular staining, proceed to step #23. Otherwise, resuspend cells in ~75 μl 4% paraformaldehyde (prepared fresh in PBS) and incubate cells at room temp for 10 min (covered in foil).
-
19
Spin cells and aspirate paraformaldehyde as in step #4.
-
20
Wash cells once with PBS (100 μl), spin cells and aspirate PBS as in step #4.
-
21Carefully resuspend cells in 200 μl of PBS.Smoothly pipette cell pellets up and down ~6-8 times. Be sure to avoid introducing bubbles.
-
22
Store plate(s) covered in aluminum foil at 4°C until ready to acquire data by flow cytometry (within 1-2 days).
Intracellular staining (optional, continued from step #18)
-
23Resuspend cells in ~75 μl Fixative (4% paraformaldehyde, provided in kit) and incubate cells at room temp for 10 min (covered in foil).Smoothly pipette up and down ~6-8 times. Avoid introducing bubbles.
-
24
Spin cells and aspirate Fixative as in step #4.
-
25Wash cells twice using 1X Perm/Wash buffer (100 μl each wash).Each time, spin at 330 RCF/1300 RPM for 4 min (no need to resuspend cells during washes).
-
26Aspirate 2nd wash from cells and add 50 μl per well of antibody dilution (prepared in 1X Perm/Wash buffer).Ensure that each well in the 96 well plate is aspirated equally. If residual wash buffer is left in some wells this will lead to variability in staining intensity between conditions. Gently resuspend the cell pellets by smoothly pipetting up and down ~6-8 times (resuspension improves staining intensity).
-
27
Cover the plate with aluminum foil and incubate at room temp for 30 min.
-
28
Spin cells and aspirate antibody stains as in step #4.
-
29
Wash cells 100 μl Perm/Wash.
-
30Aspirate Perm/Wash and carefully resuspend cells in 200 μl of PBS.Smoothly pipette cell pellets up and down ~6-8 times. Be sure to avoid introducing bubbles.
-
31
Store plate(s) covered in aluminum foil at 4°C until ready to acquire data by flow cytometry (within 1-2 days).
Flow Cytometry Analysis
-
32Establish proper voltages for forward scatter, side scatter, and excitation lasers.The main population (by FSC-Area/SSC-Area) should appear near the center of the flow cytometry plot and negative controls for fluorophores should fall within the 2nd and 3rd decades of the x-axis.
-
33
Gate on the main population (FSC-Area/SSC-Area).
-
34
From the main population, gate on single cells determined by FSC-Area/FSC-Height.
-
35
From single cells, gate on live cells by setting an FSC-Area/live-dead 405 gate on the 405-negative population.
-
36Determine expression for fluorescent markers and relevant antibodies according to your experimental design (See Figures 2A–C; 3A–C).Data become more reliable as more events are captured. In general, one should aim to collect between 10 k and 50 k events per sample. However, rare events may require greater cell numbers.
SUPPORT PROTOCOL 3
Support protocol title: Fixed and Live cell imaging of fluorescent markers in MDMs and MDDCs
Visualizing biological processes through microscopy is a powerful way to observe phenotypes and generate new hypotheses. Here, we describe a protocol whereby primary myeloid cells can be visualized through live and fixed cell fluorescence microscopy. This protocol can be adapted for downstream assays to track cell migration and trafficking, responses to viral infection and stimulation, and cell-cell communication. The fluorescent reporters described in this protocol (mEmerald, tagRFP-T, and mCerulean (Balleza et al., 2018; Rizzo & Piston, 2005; Shaner et al., 2008)) have better photostability and pH sensitivity than their fluorescent precursors, and survive fixation, which allows for simultaneous visualization of other cellular markers via dyes and antibody-mediated detection. Representative images of microscopy data generated with these tools are provided for MDDCs (Figure 2D) and MDMs (Figure 3D).
Additional Materials:
Reagents, solutions, and biological samples:
MDDCs or MDMs after differentiation (Basic Protocol)
Fixation solution (see recipe in Reagents and Solutions)
Sucrose (Millipore Sigma, cat. no. 8550-5KG)
Calcium Chloride (Thermofisher, cat. no. 2082901KG)
Magnesium Chloride (VWR, cat. no. AC41341-5000)
Triton-X100 (Sigma Aldrich, cat. no. X100-5ML)
Bovine Serum Album (Sigma Aldrich, cat. no. A9418-50G)
Alexa Fluor 555 Phalloidin (Thermofisher, cat. no. A30106)
DAPI (Sigma Aldrich, cat. no. D9542-1MG)
Hoechst 33342 (Sigma Aldrich, cat. no. B2261)
Hardware and Instruments
10 mm FluoroDish Cell Culture Dish, (World Precision Instruments, cat. no. FD3510-100)
Zeiss LSM 880 confocal microscope with Zen Black acquisition software (Version 14.0.20.201).
FIJI image processing software (ImageJ Version 1.53c).
Protocol steps with step annotations:
-
1Plate Cells on to a glass cell culture dish appropriate for imaging.Plate 50,000 cells on a glass 10 mm FluoroDish cell culture dish using 500 μl of complete media and cytokines for MDDCs or MDMs and allow to adhere for 30 min (MDDCs) or 24 h (MDMs).Note: If plating MDDCs, pre-coat plates with Poly-L-Lysine to promote cellular attachment. If plating MDMs, no pre-coating is necessary as macrophages will naturally adhere to the tissue culture dish.Note: If performing fixed cell imaging, proceed to step #9.
Live Cell Imaging (optional)
-
2Prepare Hoechst stain working dilution.Prior to the day of imaging, dissolve Hoechst 33342 in sterile water to a concentration of 2.5 μg/ml to make a stock solution. Aliquot and store the stocks at −20°C. When cells are ready to image, thaw a stock Hoechst aliquot and dilute 1 μl of stock solution in 1 ml of complete culture media [final concentration], and warm to 37°C.
-
3
Aspirate culture media and rinse with 300 μl PBS.
-
4
Aspirate PBS and add 200 μl pre-warmed Hoechst in complete media from step 2 to cells for 10 minutes in 37°C incubator.
-
5
Aspirate Hoechst and add 300 μl fresh complete pre-warmed culture media with cytokines.
-
6Image cells on a Zeiss LSM 880 confocal microscope with Zen Black software containing a Plan-Apochromat 63x/1.4 Oil DIC M27 objective lens and both a 405 nm and 488 nm laser line.Using Airyscan Fast mode, cells are imaged for Hoechst staining and mEmerald expression with the 405 nm and 488 nm laser lines, respectively. Adjust the laser powers and gain settings such that the intensity histogram of each emission spectrum is approximately 30% of the maximum. Z-stacks can then be acquired, using 0.5 micron step sizes across the entire z-range of the sample (typically 16-20 z-planes).Many imaging settings and microscope choices should be decided on based on the biological question being asked and the goal of the experiment. We highlight several of these below:The Zeiss LSM Airyscan Confocal microscope offers both Airyscan Fast and conventional Airyscan modes. Airyscan Fast can be used for live imaging to capture images with ideal temporal resolution. Conventional Airyscan can be used when higher spatial resolution is necessary, however the acquisition time with this approach is slower.When acquiring images with multiple z-planes, investigators must determine the axial resolution required for analysis. As an example, if the researcher intends to merge all z-planes into a single image, then fewer z-planes with larger step sizes are required. If the researcher intends to reconstruct a 3-dimensional image, then higher axial resolution is required (a larger number of z-planes with smaller step sizes) (references). These choices will affect the time required for image acquisition and whether to acquire images from a live or fixed sample.To image dynamic processes, live timelapse recordings may be required. Z-stacks or single z-planes can be acquired over time to observe changes in protein localization or processes. If timelapse imaging is required, we suggest using a microscope equipped with an environmental chamber to maintain 37°C and 5% CO2 to ensure cells remain healthy for long-term imaging. It is also advised to reduce the laser power to prevent phototoxicity.
-
7Retrieve high-resolution information from the raw data with Airyscan Processing.After acquiring raw z-stack images, process the raw data with Zen Black software, applying Airyscan processing by selecting the 2D reconstruction algorithm, with the filter strength set to default values.
-
8
Merge individual z plane images into a maximum intensity projection with FIJI software, omitting the top and bottom z-planes as necessary, if they are outside the boundaries of the cell (example maximum intensity projections are provided in Figures 2D; 3D).
Fixed cell Imaging (optional)
-
9
Pre-warm 1 ml fixation solution (see recipe in Reagents and Solutions) to 37°C.
-
10
Aspirate culture media from plate and rinse with 300 μl PBS (1 mM CaCl2 & 1mM MgCl2).
-
11Aspirate PBS and add 200 μl pre-warmed fixation solution from step 2 to cells for 10 minutes in a chemical fume hood.Note: Any solution with formaldehyde should only be used in a chemical hood and disposed of under EH&S protocols. Pipette tips exposed to fixation solution during step 4 and 5 should be disposed of in a sharps’ biohazard container.
-
12
Remove fixation solution from cells with a pipette and store liquid waste in a container approved for safe disposal of biohazardous materials. Rinse three times with 300 μl PBS, aspirating off after each rinse.
-
13
Add 200 μl 0.2% Triton X-100 in PBS for 10 minutes to permeabilize the cells. Aspirate.
-
14
Rinse in 200 μl PBS and then aspirate off PBS.
-
15Add blocking buffer to the sample.Prepare blocking buffer by dissolving 10 mg of bovine serum albumin in 750 μl of PBS, add 10 μl FBS and top up to 1 ml with PBS (1% FBS, 1% BSA in PBS). Add 200 μl blocking buffer to cells for 1 hour at room temperature to block non-specific binding, then aspirate off blocking buffer.
-
16Aspirate blocking buffer and stain cells with 300 μl of 1X Phalloidin stain for one hour at room temperature in the dark.Prepare a 400X stock solution of Alexa Fluor 555 Phalloidin per the manufacturer’s directions. Stock solution is stable for at least one year when stored at −20°C. To prepare the 1X working solution, add 1 μl of phalloidin stock solution to 1 ml of PBS (1:1000) to make Phalloidin stain.
-
17Aspirate Phalloidin stain and add 200 μl of DAPI (2.5 μg / ml) in PBS to cells for 10 minutes at room temp in the dark.Dissolve DAPI in sterile water to make a stock solution at 2.5 mg/ml. This stock can be stored at −20°C for extended periods of time. To prepare working solution, dissolve 1 μl DAPI stock solution in 1 ml of PBS for a final concentration of 2.5 μl/ml.
-
18
Aspirate off DAPI and then rinse 1X with PBS.
-
19
Proceed to imaging or store in PBS overnight at 4°C in the dark until ready to image on a confocal microscope.
-
20To image cells, a Zeiss LSM 880 confocal microscope with Zen Black software containing a Plan-Apochromat 63x/1.4 Oil DIC M27 objective lens and both a 405 nm, 488 nm, and 561 nm laser line.Using Airyscan Fast mode, cells are imaged with a 405 nm, 488 nm, and 561 nm laser lines, for DAPI, mEmerald, and Phalloidin fluorescence respectively. Adjust the laser powers and gain settings such that the intensity histogram of each emission spectrum is approximately 30% of the maximum. Z stacks can then be acquired using 0.5 micron step sizes across the entire z-range of the sample (typically 16-20 Z-planes).See note on decisions for image acquisition parameters from live cell imaging section.
-
21
After acquiring raw z-stack images, apply Airyscan processing with default settings using Zen Black software to retrieve high-resolution information from the images.
-
22
Merge individual z-plane images into a maximum intensity projection with FIJI software, omitting the top and bottom z-planes as necessary, if they are outside the boundaries of the cell (example maximum intensity projections are provided in Figures 2D; 3D).
REAGENTS AND SOLUTIONS:
293FT Complete Media
DMEM, high glucose, pyruvate (Invitrogen, cat. no. 11995065)
Fetal Bovine Serum (10%, v/v)
HEPES (10 mM)
Pen/Strep (50 U/ml)
- MEM non-essential amino acids (100 nM, ThermoFisher, cat. no. 11140050)All components should be added to DMEM and then media should be sterile filtered through 0.2 μm filters.
Recovery Buffer
Dulbecco’s Phosphate Buffered Saline (without calcium and magnesium)
- EDTA (5 mM)Recovery buffer should be used at room temp and may be stored for several months at room temp or at 4°C if kept sterile.
MACS Buffer
Dulbecco′s Phosphate Buffered Saline (without calcium and magnesium)
Bovine Serum Albumin (BSA) – Fraction V (0.5%)
- EDTA (2.5 mM)To prepare MACS buffer, 2.5 g BSA should be completely dissolved in 500 ml DPBS. After adding EDTA to reach 2.5 mM (2.5 ml of 0.5 M stock), MACS buffer should be sterile filtered through a 0.2 μm filter and kept at 4°C. MACS buffer should be prepared the day of isolation and may be stored for up to two weeks if kept sterile.
RPMI Complete Media (for culturing MDDCs and MDMs)
Serum free RPMI (ThermoFisher, cat. no. 11875085)
Fetal Bovine Serum (10%, v/v)
Glutamine (2 mM)
HEPES (10 mM)
Pen/Strep (50 U/ml)
- Beta-Mercaptoethanol (55 nM) (Thermofisher, cat. no. 21985023)All components should be added and then sterile filtered through 0.2 μm and stored in the fridge for no more than 6-8 weeks.
Staining Buffer (SB)
Dulbecco′s Phosphate Buffered Saline (without calcium and magnesium)
Bovine Serum Albumin (BSA) - Fraction V (0.5%)
- Pen/Strep (50 U/ml)To prepare Staining Buffer, 5 g of BSA should be completely dissolved in 500 ml DPBS. Add Pen/Step to reach a final concentration of 50 u/ml. Filter sterilize through a 0.2 μm filter and store at 4°C.
Fixation solution
Pierce 16% Paraformaldehyde (w/v), Methanol-Free (Thermofisher, cat. no. 28906)
Sterile Water
10X PBS
- SucroseTo prepare Fixation Buffer, dissolve 20 μg of sucrose in 500 μl of sterile water. Add 100 μl of 10X PBS and 250 μl of 16% paraformaldehyde. Bring solution up to 1 ml with sterile water for a final 1X PBS, 2% sucrose, 4% paraformaldehyde fixation solution. 4% paraformaldehyde is stable for up to a week at 4°C or stored for longer periods at −20°C but for best results, it should be made fresh on the day of fixation.
Qualified, heat-inactivated FBS
To heat-inactivate FBS, thaw stock bottles of FBS and warm to 22-37°C. Heat-inactivate by placing in 55°C water bath for 30 min, vigorously swirling bottles every 10 min to ensure even temperature distribution and prevent protein aggregation. Make 50 ml aliquots of heat-inactivated FBS and store at −20°C. See notes regarding FBS lot testing in the Commentary: Critical Parameters Section.
COMMENTARY
Background Information:
The field of immunology can trace its origins to the late 19th century. Elie Metchnikoff discovered macrophages in 1882 while studying marine invertebrates. He identified cells that could phagocytose foreign particles and respond to infection through a process similar to that which occurs in higher-order species. For his work on “immunity” Metchnikoff shared the Nobel Prize with Paul Ehrlich, who helped lay the conceptual foundation for humoral adaptive immunity (Kaufmann, 2008). However, it was not until close to a hundred years later, in the early 1970’s, when the father of modern macrophage biology, Zanvil “Zan” Cohn, and a postdoctoral fellow in his laboratory, Ralph Steinman (also a Nobel laureate), discovered a new class of tree-shaped cells that they termed dendritic cells (Steinman & Cohn, 1973, 1974; Steinman, Lustig, & Cohn, 1974). Their studies built on the important observation that adherent cells from the spleen could initiate immune responses in vitro after they were exposed to antigen (Mosier, 1967). The initial discovery of macrophages, and the subsequent identification of dendritic cells as specialized antigen presenting cells, turned out to be watershed moments for science.
We now appreciate that DCs and macrophages are antigen presenting cells with essential roles in immunity, tolerance, and homeostasis (Banchereau & Steinman, 1998; Wynn et al., 2013). During prenatal development, macrophages arise from yolk sac hematopoiesis and fetal liver hematopoiesis and can then self-renew as tissue resident cells. After birth, monocytes, macrophages, and DCs arise from a common myeloid progenitor in the bone marrow. Classical myeloid DCs (cDCs, also termed conventional DCs) can be separated into two main subtypes based on their functionality and the transcription factor requirements for their development (Guilliams et al., 2014). Type I cDCs (cDC1s) are highly capable of cross presenting exogenous, cell-associated antigens, and are potent stimulators of CD8 T cells. Type II cDCs (cDC2s) primarily present soluble antigens to CD4+ T cells and have roles in the cellular and humoral immune response to extracellular pathogens, allergens, and parasites. Separately from conventional cDC1 and cDC2s, plasmacytoid DCs (pDCs) represent an important arm of the innate immune response and are specialized producers of type I and type III interferons. DCs can be further classified into distinct subsets that are reviewed in detail elsewhere (Anderson, Dutertre, Ginhoux, & Murphy, 2021). Notably, mononuclear phagocytes display a significant amount of plasticity and adopt different characteristics and functionality depending on their microenvironment (Vu Manh, Bertho, Hosmalin, Schwartz-Cornil, & Dalod, 2015).
In vitro differentiation of MDDCs from CD14+ monocytes (in the presence of the cytokines GM-CSF and IL-4) has been common practice for more than 20 years (Randolph, Beaulieu, Lebecque, Steinman, & Muller, 1998). Although we note that cDC1s, cDC2s, and MDDCs are phenotypically distinct cell types, with MDDCs being more similar to cDC2s and less-potent primers of T cells than cDC1s (Tamoutounour et al., 2013). Classical cDC1s and monocyte-derived cells also seem to require different transcriptional programs for their differentiation (Briseno et al., 2016). That being said, derivation of MDDCs and MDMs from monocytes in culture remains an attractive experimental platform, especially since monocytes have been observed to differentiate into DCs in vivo, and share some properties with cDCs such as the capacity to cross-present antigens (Jakubzick, Randolph, & Henson, 2017). Studying how MDDCs and MDMs detect and respond to pathogens, activate innate immune pathways, and communicate with other cells can advance our understanding of host-pathogen interactions and reveal fundamental principles in cell biology.
For many years, it was difficult to perform genetic studies in human MDDCs and MDMs. As these cells are equipped to detect exogenous nucleic acids, delivery of DNA or RNA by transfection or electroporation can lead to cell activation and obscure phenotypes that are the target of study. Similarly, although many different viral vectors have been employed to transduce myeloid cells in different contexts (including vectors from adenovirus, pox virus, herpes virus, adeno-associated virus, and retrovirus (Bulcha, Wang, Ma, Tai, & Gao, 2021; Ribas, 2005)), genetic modification of MDDCs and MDMs in this way is relatively intractable because these cells recognize pathogen-associated molecular patterns present in these microbes. Transduction of myeloid-derived cells with HIV-1-derived lentiviral vectors was first reported more than two decades ago (Gruber et al., 2000), but the efficiency was poor, requiring hundreds of infectious particles per cell to achieve moderate levels of transduction. Interestingly, it was discovered that lentiviral vectors derived from SIV displayed higher transduction efficiencies than HIV-1-derived vectors in myeloid cells (Negre et al., 2000). A single accessory protein, Vpx, was responsible for this increase in transduction and could be provided in trans to improve transduction of HIV-1 derived vectors (Goujon et al., 2006). Vpx is present in HIV-2 and several strains of SIV-2 (Etienne, Hahn, Sharp, Matsen, & Emerman, 2013). In 2011, two groups independently made the landmark discovery that Vpx targets the restriction factor SAMHD1 for degradation, which normally prevents reverse transcription of retroviruses (Hrecka et al., 2011; Laguette et al., 2011). Their findings explained why HIV-2 and SIV-derived vectors (but not HIV-1), could efficiently transduce myeloid cells. With these discoveries and early transduction protocols paving the way (Berger et al., 2011; Satoh & Manel, 2013), robust genetic modification of CD14+ monocytes and monocyte-derived cells with lentiviral vectors is now possible through careful control of lentiviral vectors and delivery of the accessory protein Vpx.
CD14+ monocytes can be isolated from whole blood through several different methods. Three commonly used methods include: 1) positive selection using antibodies conjugated to magnetic beads (Basic Protocol), 2) negative selection using antibodies to deplete non-monocytes and leaving CD14+ cells untouched, and 3) adherence-based selection (Alternate Protocol 1). Before proceeding, it is important to consider the impact that each of these methods may have on the purity, viability, and phenotypic characteristics of the cultured CD14+ cells. Positive selection with magnetic beads provides consistently high monocyte purity, yield, and viability. Negative selection also enables isolation of monocytes with high viability, but these preparations are reported to be contaminated with platelets (Nielsen et al., 2020). Selection using magnetic beads is costly and may not be economically feasible for large numbers of blood preparations. As an alternative to positive or negative selection using magnetic beads, isolation of CD14+ monocytes based on their propensity to attach to plastic may be an attractive option for investigators performing pilot experiments, or who would like a cost-effective method for routine blood preparations.
Critical Parameters:
The most important parameters to consider in this protocol are the isolation and cell culture conditions than enable derivation of healthy MDDCs and MDMs (Basic Protocol) and the factors that lead to production of high-titer lentiviral vectors (Support Protocol 1). Giving special attention to these parameters will enable investigators to perform robust experiments and capture relevant cell biology. Below, we outline the major points in these protocols that can determine whether the end result is a great success or an agonizing failure.
It is critical to have a reliable source of leukocytes that can be procured soon after the time of blood draw. Ideally, the procedure should begin with fresh leukocytes or within a few hours after blood donation. As this is not often possible, it is reasonable to begin PBMC preparation using whole blood that was extracted the day before, as long as the samples are maintained at room temperature and transported in good condition. For many steps in this protocol, the adage, “garbage in, garbage out” is relevant. If the material you begin with is compromised (i.e. the blood was donated more than one day prior to PBMC isolation, viability is low, or cells are activated), it is unlikely that the end result will be satisfying.
Investigators should approach these experiments with the understanding that each donor will have a unique genetic signature, and variability between donors is to be expected. Consequently, this variability should be accounted for in the experimental design. Cells from multiple donors should be compared in every experiment and treated as independent biological replicates. The number of different donors needed for a given experiment depends on the nature of the experimental setup, the donor-to-donor variability between measurements, and the observed signal-to-noise in the data. While donor number should be determined on a case-by-case basis, it is not uncommon to test samples from 4 to 8 donors in aggregate (Johnson et al., 2018).
One of the most important parameters to work out, prior to engaging in “full tilt” transduction experiments, is to screen different lots of FBS on multiple donors to identify batches that consistently lead to immature cells (i.e. low baseline activation of CD86, CD80, CD83, CD40 is observed in the absence of stimulation, and proper induction of these genes is observed following directed stimulation). We have observed significant variability in basal activation of MDDCs associated with different lots of FBS (in unstimulated conditions). Aberrant activation can potentially be attributed to many different impurities in the FBS (e.g. nucleic acids, lipopolysaccharides, or damage-associated molecular patterns). To guard against aberrant activation, we test multiple lots of FBS across primary human myeloid cells from multiple donors to identify batches that do not lead to elevated CD86 in unstimulated conditions (as measured by flow cytometry). Identifying “clean” batches of FBS is particularly important when studying phenotypes related to immune activation. We note that differentiation of MDDCs and MDMs can also be performed using autologous or pooled human serum, but in our experience, this leads to greater experimental variability when compared to cells cultured with validated lots of FBS.
Troubleshooting:
The isolation and genetic modification of myeloid cells using lentiviral vectors is a labor-intensive process fraught with challenges. Here, we highlight some of the more common problems encountered with the procedure, indicate their possible causes, and suggest potential solutions (Table 3).
Table 3.
Troubleshooting Guide for Isolation and Transduction of Primary Monocytes
| Problem | Possible Cause | Solution |
|---|---|---|
| Baseline activation of immune cells (e.g. high expression of CD86 and other costimulatory markers without stimulation) | Contaminants present in FBS | Empirically determine lots of FBS that enable differentiation of “immature” cells. |
| Contaminants present in MACS buffer, media, or additives | Prepare buffers and media fresh on the day of isolation and filter sterilize through 0.22 μm filters. | |
| Contaminants present in lentiviral supernatants | Purify lentiviral stocks by ultracentrifugation through sucrose cushions. Make sure insoluble material is clarified from resuspended lentiviral stocks. | |
| Lentiviral transduction reagent used at too high of a concentration | Titrate amounts of lentiviral vectors (and/or SIV-VLPs) and polybrene to determine optimal concentrations. | |
| Poor transfection efficiency | 293FT cells were not cultured under ideal conditions | Use low passage cells, and ensure that cells are plated to reach the proper density at time of transfection (70-80%). |
| Plasmid DNA is damaged or impure | Verify plasmid integrity on a gel and purify through a column or phenol/chloroform extraction. | |
| Transfection complexes failed to form properly | Titrate PEI stocks to determine the optimal ratio of PEI:DNA. Adhere to proper timing as indicated in the protocol. Do not mix complexes too vigorously. | |
| Poor transduction efficiency | Low viral titers | Optimize transfection efficiency (see section above) |
| Lentiviral transduction reagent used at too low of a concentration | Titrate amounts of lentiviral vectors (and/or SIV-VLPs) and polybrene to determine optimal concentrations. | |
| Low yield of PBMCs and/or CD14+ monocytes | Poor separation of cells in the Ficoll-Paque spin to isolate buffy coats | Avoid disturbing the Ficoll layer when pipetting blood. Ensure that centrifuge is well-balanced, and that brake is set to low or off. |
| Coagulation or aggregation of cells | Maintain blood at room temperature until after RBCs are lysed. Filter cell suspension through a 70-micron filter when aggregates are visible. | |
| Cell viability is compromised | Work quickly using fresh material from recent blood donations. | |
| Few events / cell numbers are low during flow cytometry | Too few cells present or cells were lost during staining | Ensure adequate cell numbers are plated and take care to not aspirate the cell pellet during wash steps. Determine if FSC-A/SSC-A voltages are set up properly on the cytometer. |
| Fluidics not connected properly | Verify that all lines are connected properly and that the system is pressurized. | |
| The system is clogged | Thoroughly resuspend your samples (filter them if necessary). Declog the cytometer. | |
| Low cell numbers for immunofluorescence | Low numbers of cells plated | Plate at a higher density |
| Washes detached fixed cells from the cell culture plate | Ensure that all washes and staining steps are gentle to minimize disruption to fixed cells. |
Understanding Results:
To demonstrate successful genetic modification of MDDCs to express fluorescent reporters, we have transduced CD14+ monocytes with either an empty vector lentivirus (LKO-MCS) or vectors that encode proteins to label mitochondria (mito-mEm), peroxisomes (perox-mEm), and whole cells (cell-mEm) (Figure 2). We determined expression levels by flow cytometry (Support Protocol 2) and observed that a subset of the cell population was transduced in the absence of Vpx, and in contrast, roughly ~90% of the population was transduced when Vpx was present (Figure 2A). Analysis of mEmerald geometric mean fluorescence intensity indicated that Vpx increased expression levels by an order of magnitude (Figure 2B). These data are consistent with the published effects of Vpx on lentiviral-mediated gene delivery in myeloid cells (Goujon et al., 2006). Importantly, under these conditions, transduction with lentiviral vectors did not change MDDC responsiveness to LPS stimulation, as determined by induction of the costimulatory molecule CD86 (Figure 2C). However, we cannot rule out that Vpx influences other aspects of myeloid cell biology unseen here, which may include alterations in interferon signaling, NF-κB activity, and gene silencing (Chougui et al., 2018; Cingoz, Arnow, Puig Torrents, & Bannert, 2021; Fink et al., 2022; Yurkovetskiy et al., 2018). Nevertheless, genetic modification of MDDCs through lentiviral-mediated gene delivery can be a useful tool for testing hypotheses related to cell activation, virus infection, and cell-cell communication.
To visualize the subcellular localization of these fluorescent markers, we performed immunofluorescence experiments in live and fixed MDDCs (Support Protocol 3). Live cell imaging through laser-scanning confocal microscopy revealed segmented localization of mito-mEm in cytosolic structures that we interpret to be mitochondria (Figure 2D). Expression of mito-mEm overlaps significantly with fluorescent dyes that target the mitochondria in live cells and with immunostaining of mitochondrial proteins in fixed cells (data not shown), supporting the notion that mEmerald-labeled structures are indeed mitochondria. Similarly, we detected perox-mEm in punctate structures throughout the cytosol, consistent with the organization and localization of peroxisomes. The untargeted mEmerald expression cassette (cell-mEm) was observed to be diffusely expressed throughout the cytosol and nucleus and serves as a general marker of transduced MDDCs. We note that if investigators would like to image cytoskeletal dynamics in live cells in concert with these fluorescent markers, this protocol may be combined with reagents such as SiR-actin or SiR-tubulin to track actin or tubulin, respectively. As a comparison to live cell imaging, we also captured images of transduced MDDCs after fixation (Figure 2D, bottom panels) together with phalloidin staining to mark the architecture of F-actin.
In parallel, we transduced CD14+ monocytes with LKO-MCS, mito-mEm, perox-mEm, or cell-mEm, derived MDMs, and determined expression levels by flow cytometry (Figure 3A). We observed robust expression of mEmerald in MDMs that were transduced by fluorescent lentivectors both with and without Vpx. Similar to effects in MDDCs, the inclusion of Vpx increased expression levels in MDMs by an order of magnitude (Figure 3B). Under the conditions tested here, MDMs did not exhibit a pronounced response to LPS (100 ng/ml, 16 h stimulation), and transduction with lentiviral vectors +Vpx led to a moderate increase in CD86 (Figure 3C). These data emphasize that while lentiviral vectors are useful instruments that enable robust genetic modification of primary blood-derived myeloid cells, researchers should proceed with caution when interpreting biological effects associated with transduction. It is critical to include experimental controls, including mock-treated cells, untransduced cells, and empty vector controls where appropriate. Similar to results in MDDCs, live cell and fixed imaging of MDMs through laser-scanning confocal microscopy revealed targeted localization of fluorescent markers to mitochondria and peroxisomes (for mito-mEm and perox-mEm, respectively) and generalized whole cell labeling for cell-mEm (Figure 3D). Together, these data highlight the versatility of lentiviral vectors for labeling cellular structures in MDDCs and MDMs.
Time Considerations:
This protocol can be separated into two main parts: 1) generation of recombinant lentiviral vectors and virus-like particles (Support Protocol 1), and 2) isolation, transduction, and differentiation of MDDCs and MDMs (Basic Protocol). Generation of recombinant lentivectors will take approximately one week to complete, with minimal labor invested each day, assuming all plasmids are prepared in advance and a sufficient quantity of cells are available in culture. Splitting cells for transfection will take 1-2 h. Transfection on the following day will take less than 1 h. Washing transfections and replenishing media on the following day will take 30 min. The most time-consuming steps are on the final day of vector preparation and include harvesting lentiviral supernatants (30 min – 1 h), ultracentrifugation to concentrate supernatants (2 – 3 h), and resuspension, clarification, and aliquoting vector preps (1-2 h of hands-on time, not including several hours to overnight incubation of vector resuspensions). We recommend that lentiviral preparations are tested on immortalized cells in culture prior to beginning work in primary cells (time investment and target cells should be determined by the user, but typically this can take 2-3 days if lentiviral vector preparations are tested in 239FT cells).
Once vectors are prepared, the second part of the protocol will take approximately one week to complete (Figure 1). Preparation of PBMCs will take approximately 2 h once whole blood is procured. Isolation of CD14 positive cells will take 1 – 2 h, depending on whether the Basic Protocol or the adhesion-based selection in Alternate Protocol 1 is used. If adhesion-based selection is used, the hands-on time required from investigators is reduced, but the overall time it takes to acquire CD14+ monocytes is increased. The most variable step in terms of time requirement is the step involving lentiviral transduction. For simple experimental designs, this can be performed in as little as 30 min, but more complicated transductions that involve dozens of conditions and experimental design matrices may take several hours.
After cells are differentiated, which occurs after 5 days of culture for MDDCs and 7 days for MDMs, the downstream assays suggested here (flow cytometry and fluorescence imaging) can be completed in less than a day. Cell surface staining of MDDCs and MDMs can take approximately 1 – 2 h. If performing intracellular staining, this time is increased to 3 – 4 h. Data for up to 96 samples can be collected on a flow cytometer in less than 2 h and is made easier if using a high-throughput sampler. Flow cytometry can occur on the same day as the staining, or, as samples are fixed, they can be stored at 4°C in the dark and processed within 1-2 days. For live cell imaging, experimental procedures require approximately 1 h of preparation, whereas for fixed imaging experiments will generally require 2 h of preparation for fixing and staining the cells. The time required for image acquisition is dependent on the user, and it is reasonable to estimate 2 h of imaging time for up to ~four experimental conditions. More imaging time is required for complex experiments with multiple donors.
ACKNOWLEDGEMENTS:
This work was supported in part by grants from the U.S. National Institutes of Health (U19AI100627 and U19AI135976 to AA, R01 CA247994 to MRJ, and P50 AI150464 to JSJ).
Footnotes
All authors declare that they have no conflicts of interest.
LITERATURE CITED:
- Akkina RK, Walton RM, Chen ML, Li QX, Planelles V, & Chen IS (1996). High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J Virol, 70(4), 2581–2585. doi: 10.1128/JVI.70.4.2581-2585.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DA 3rd, Dutertre CA, Ginhoux F, & Murphy KM (2021). Genetic models of human and mouse dendritic cell development and function. Nat Rev Immunol, 21(2), 101–115. doi: 10.1038/s41577-020-00413-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, … Keppler OT (2012). SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med, 18(11), 1682–1687. doi: 10.1038/nm.2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleza E, Kim JM, & Cluzel P (2018). Systematic characterization of maturation time of fluorescent proteins in living cells. Nat Methods, 15(1), 47–51. doi: 10.1038/nmeth.4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, & Steinman RM (1998). Dendritic cells and the control of immunity. Nature, 392(6673), 245–252. doi: 10.1038/32588 [DOI] [PubMed] [Google Scholar]
- Berger G, Durand S, Goujon C, Nguyen XN, Cordeil S, Darlix JL, & Cimarelli A (2011). A simple, versatile and efficient method to genetically modify human monocyte-derived dendritic cells with HIV-1-derived lentiviral vectors. Nat Protoc, 6(6), 806–816. doi: 10.1038/nprot.2011.327 [DOI] [PubMed] [Google Scholar]
- Bobadilla S, Sunseri N, & Landau NR (2013). Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther, 20(5), 514–520. doi: 10.1038/gt.2012.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, … Murphy KM (2016). Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep, 15(11), 2462–2474. doi: 10.1016/j.celrep.2016.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulcha JT, Wang Y, Ma H, Tai PWL, & Gao G (2021). Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther, 6(1), 53. doi: 10.1038/s41392-021-00487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Chu K, Shrestha A, Revelo XS, Zhang X, Gold MJ, … Winer DA (2021). Mechanical Stiffness Controls Dendritic Cell Metabolism and Function. Cell Rep, 34(2), 108609. doi: 10.1016/j.celrep.2020.108609 [DOI] [PubMed] [Google Scholar]
- Chamberlain LM, Holt-Casper D, Gonzalez-Juarrero M, & Grainger DW (2015). Extended culture of macrophages from different sources and maturation results in a common M2 phenotype. J Biomed Mater Res A, 103(9), 2864–2874. doi: 10.1002/jbm.a.35415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, So EC, Strome SE, & Zhang X (2015). Impact of Detachment Methods on M2 Macrophage Phenotype and Function. J Immunol Methods, 426, 56–61. doi: 10.1016/j.jim.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Chougui G, Munir-Matloob S, Matkovic R, Martin MM, Morel M, Lahouassa H, … Margottin-Goguet F (2018). HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat Microbiol, 3(8), 891–897. doi: 10.1038/s41564-018-0179-6 [DOI] [PubMed] [Google Scholar]
- Cingoz O, Arnow ND, Puig Torrents M, & Bannert N (2021). Vpx enhances innate immune responses independently of SAMHD1 during HIV-1 infection. Retrovirology, 18(1), 4. doi: 10.1186/s12977-021-00548-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer-Lluch M, Ruiz A, Moris A, & Prado JG (2018). Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front Immunol, 9, 2876. doi: 10.3389/fimmu.2018.02876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal NK, & Emerman M (2012). Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol, 12(10), 687–695. doi: 10.1038/nri3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne L, Hahn BH, Sharp PM, Matsen FA, & Emerman M (2013). Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe, 14(1), 85–92. doi: 10.1016/j.chom.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink DL, Cai J, Whelan MVX, Monit C, Maluquer de Motes C, Towers GJ, & Sumner RP (2022). HIV-2/SIV Vpx antagonises NF-kappaB activation by targeting p65. Retrovirology, 19(1), 2. doi: 10.1186/s12977-021-00586-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, … Webb M (2011). HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature, 480(7377), 379–382. doi: 10.1038/nature10623 [DOI] [PubMed] [Google Scholar]
- Goujon C, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, & Cimarelli A (2006). With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther, 13(12), 991–994. doi: 10.1038/sj.gt.3302753 [DOI] [PubMed] [Google Scholar]
- Gruber A, Kan-Mitchell J, Kuhen KL, Mukai T, & Wong-Staal F (2000). Dendritic cells transduced by multiply deleted HIV-1 vectors exhibit normal phenotypes and functions and elicit an HIV-specific cytotoxic T-lymphocyte response in vitro. Blood, 96(4), 1327–1333. [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, … Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol, 14(8), 571–578. doi: 10.1038/nri3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, … Skowronski J (2011). Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature, 474(7353), 658–661. doi: 10.1038/nature10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick CV, Randolph GJ, & Henson PM (2017). Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol, 17(6), 349–362. doi: 10.1038/nri.2017.28 [DOI] [PubMed] [Google Scholar]
- Johnson JS, De Veaux N, Rives AW, Lahaye X, Lucas SY, Perot BP, … Menager MM (2020). A Comprehensive Map of the Monocyte-Derived Dendritic Cell Transcriptional Network Engaged upon Innate Sensing of HIV. Cell Rep, 30(3), 914–931 e919. doi: 10.1016/j.celrep.2019.12.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JS, Lucas SY, Amon LM, Skelton S, Nazitto R, Carbonetti S, … Aderem A (2018). Reshaping of the Dendritic Cell Chromatin Landscape and Interferon Pathways during HIV Infection. Cell Host Microbe, 23(3), 366–381 e369. doi: 10.1016/j.chom.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JA, Chang DT, Meyerson H, Colton E, Kwon IK, Matsuda T, & Anderson JM (2007). Proteomic analysis and quantification of cytokines and chemokines from biomaterial surface-adherent macrophages and foreign body giant cells. J Biomed Mater Res A, 83(3), 585–596. doi: 10.1002/jbm.a.31221 [DOI] [PubMed] [Google Scholar]
- Kaufmann SH (2008). Immunology’s foundation: the 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat Immunol, 9(7), 705–712. doi: 10.1038/ni0708-705 [DOI] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, … Benkirane M (2011). SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature, 474(7353), 654–657. doi: 10.1038/nature10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, … Margottin-Goguet F (2012). SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol, 13(3), 223–228. doi: 10.1038/ni.2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, & Littman DR (2010). A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature, 467(7312), 214–217. doi: 10.1038/nature09337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeot PE, Negre D, Dubois B, Winter AJ, Leissner P, Mehtali M, … Darlix JL (2000). Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J Virol, 74(18), 8307–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosier DE (1967). A requirement for two cell types for antibody formation in vitro. Science, 158(3808), 1573–1575. doi: 10.1126/science.158.3808.1573 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, … Trono D (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science, 272(5259), 263–267. doi: 10.1126/science.272.5259.263 [DOI] [PubMed] [Google Scholar]
- Naldini L, Trono D, & Verma IM (2016). Lentiviral vectors, two decades later. Science, 353(6304), 1101–1102. doi: 10.1126/science.aah6192 [DOI] [PubMed] [Google Scholar]
- Negre D, Mangeot PE, Duisit G, Blanchard S, Vidalain PO, Leissner P, … Cosset FL (2000). Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther, 7(19), 1613–1623. doi: 10.1038/sj.gt.3301292 [DOI] [PubMed] [Google Scholar]
- Nielsen MC, Andersen MN, & Moller HJ (2020). Monocyte isolation techniques significantly impact the phenotype of both isolated monocytes and derived macrophages in vitro. Immunology, 159(1), 63–74. doi: 10.1111/imm.13125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan JP, & Condello D (2013). Spectral flow cytometry. Curr Protoc Cytom, Chapter 1, Unit1 27. doi: 10.1002/0471142956.cy0127s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, & Muller WA (1998). Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science, 282(5388), 480–483. doi: 10.1126/science.282.5388.480 [DOI] [PubMed] [Google Scholar]
- Reiser J, Harmison G, Kluepfel-Stahl S, Brady RO, Karlsson S, & Schubert M (1996). Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc Natl Acad Sci U S A, 93(26), 15266–15271. doi: 10.1073/pnas.93.26.15266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A (2005). Genetically modified dendritic cells for cancer immunotherapy. Curr Gene Ther, 5(6), 619–628. doi: 10.2174/156652305774964758 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, & Piston DW (2005). High-contrast imaging of fluorescent protein FRET by fluorescence polarization microscopy. Biophys J, 88(2), L14–16. doi: 10.1529/biophysj.104.055442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, & Manel N (2013). Gene transduction in human monocyte-derived dendritic cells using lentiviral vectors. Methods Mol Biol, 960, 401–409. doi: 10.1007/978-1-62703-218-6_30 [DOI] [PubMed] [Google Scholar]
- Sauter A, Yi DH, Li Y, Roersma S, & Appel S (2019). The Culture Dish Surface Influences the Phenotype and Cytokine Production of Human Monocyte-Derived Dendritic Cells. Front Immunol, 10, 2352. doi: 10.3389/fimmu.2019.02352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, & Tsien RY (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods, 5(6), 545–551. doi: 10.1038/nmeth.1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, & Cohn ZA (1973). Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med, 137(5), 1142–1162. doi: 10.1084/jem.137.5.1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, & Cohn ZA (1974). Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp Med, 139(2), 380–397. doi: 10.1084/jem.139.2.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Lustig DS, & Cohn ZA (1974). Identification of a novel cell type in peripheral lymphoid organs of mice. 3. Functional properties in vivo. J Exp Med, 139(6), 1431–1445. doi: 10.1084/jem.139.6.1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, … Novina CD (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA, 9(4), 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, … Henri S (2013). Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity, 39(5), 925–938. doi: 10.1016/j.immuni.2013.10.004 [DOI] [PubMed] [Google Scholar]
- Tan PH, Beutelspacher SC, Xue SA, Wang YH, Mitchell P, McAlister JC, … George AJ (2005). Modulation of human dendritic-cell function following transduction with viral vectors: implications for gene therapy. Blood, 105(10), 3824–3832. doi: 10.1182/blood-2004-10-3880 [DOI] [PubMed] [Google Scholar]
- Vu Manh TP, Bertho N, Hosmalin A, Schwartz-Cornil I, & Dalod M (2015). Investigating Evolutionary Conservation of Dendritic Cell Subset Identity and Functions. Front Immunol, 6, 260. doi: 10.3389/fimmu.2015.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, & Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature, 496(7446), 445–455. doi: 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkovetskiy L, Guney MH, Kim K, Goh SL, McCauley S, Dauphin A, … Luban J (2018). Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat Microbiol, 3(12), 1354–1361. doi: 10.1038/s41564-018-0256-x [DOI] [PMC free article] [PubMed] [Google Scholar]