

Abstract
Medication (drug) use in human pregnancy is prevalent. Determining fetal safety and efficacy of drugs is logistically challenging. However, predicting (not measuring) fetal drug exposure (systemic and tissue) throughout pregnancy is possible through maternal-fetal (m-f) physiologically-based pharmacokinetic (PBPK) modeling and simulation (M&S). Such prediction can inform fetal drug safety and efficacy. Fetal drug exposure can be quantified in two complementary ways. First, the ratio of the steady-state unbound plasma concentration in the fetal plasma (or AUC) to the corresponding maternal plasma concentration (i.e. Kp,uu). Second, the maximum unbound peak (Cu,max,ss,f) and trough (Cu,min,ss,f) fetal steady-state plasma concentrations. We (and others) have developed a m-f PBPK model that can successfully predict maternal drug exposure. To predict fetal drug exposure, the model needs to be populated with drug specific parameters, of which transplacental clearances (active and/or passive) and placental/fetal metabolism of the drug are critical. Herein, we describe in vitro studies in cells/tissue fractions or the perfused human placenta that can be used to determine these drug-specific parameters. In addition, we provide examples whereby this approach has successfully predicted systemic fetal exposure to drugs that passively or actively cross the placenta. Apart from m-f PBPK models, animal studies also have the potential to estimate fetal drug exposure by allometric scaling. Whether such scaling will be successful is yet to be determined. Here, we review the above approaches to predict fetal drug exposure, outline gaps in our knowledge to make such predictions, and map out future research directions that could fill these gaps.
Keywords: Predicting fetal drug exposure, in vitro to in vivo extrapolation, perfused placenta, maternal-fetal PBPK model, transfected and placental cell lines, transporters and enzymes
Introduction
Pregnant women often take medication (drugs) to treat pre-existing conditions (e.g., epilepsy, depression, hypertension, bacterial or viral infections) or health complications produced by pregnancy (e.g., preeclampsia, nausea, gestational diabetes). In some cases, pregnant women are prescribed medication to treat the maternal-fetal dyad (e.g., progression/prevention of HIV infection) or just the fetus (e.g., antenatal corticosteroids to prevent neonatal morbidity/mortality from preterm labor, digoxin to treat fetal tachycardia). Consequently, 80% of pregnant women take medication throughout their pregnancy.1, 2 Despite prevalent use of medication in pregnancy, more than 90% of clinically approved drugs prescribed to pregnant women are used ‘off-label’ and lack appropriate information on their pharmacokinetics (PK), safety and efficacy in pregnancy.2 In fact, 98% of the industry-sponsored clinical trials in the USA actively excluded pregnant subjects due to legal and practical considerations, and only 1% were specifically designed for pregnant women.3 The above problem is compounded by the fact that determining the safety (especially long-term) and efficacy of drugs in pregnancy is logistically challenging. For example, the use of diethylstilbestrol in pregnancy was found to result in increased risk of breast cancer in mothers and in development of genital tract abnormalities, breast cancer and melanoma in the offspring.4 Administration of thalidomide for pregnancy-induced nausea caused severe limb deformities (phocomelia) in thousands of offsprings.5 For the purposes of this review, the word drug refers to a small molecule and not a large molecule, such as a protein.
Since measuring fetal safety and efficacy of a drug is logistically challenging, measuring a surrogate marker of these endpoints, namely fetal drug exposure, can inform assessment of fetal drug safety and efficacy. While systemic fetal drug concentration can be determined at birth by sampling umbilical vein (UV)/artery (UA) blood, such sampling is impossible earlier in pregnancy. And, as we have detailed before6, a single UV sample at the time of birth does not provide an estimate of fetal drug exposure. To assess such exposure, one must determine the plasma/blood fetal steady-state drug concentration or the area under the plasma concentration-time curve (AUC) (at steady-state or after a single dose). Because fetal drug concentrations are driven by maternal drug concentrations, to take into account the inter-individual variability in the latter, the fetal steady-state plasma concentration (or AUC) is best expressed relative to the corresponding maternal value (i.e., Kp). And, since it is the unbound drug that produces the pharmacological effect of a drug, it is the average fetal steady-state unbound plasma drug concentration (Cu,av,ss,f) relative to the average maternal steady-state unbound plasma drug concentration (Cu,av,ss,m), namely, Kp,uu, that should be estimated. In addition, where the plasma drug concentrations can fluctuate significantly during a dosing interval, it is also important to determine the unbound peak (Cu,max,ss,f) and trough (Cu,min,ss,f) fetal steady-state plasma drug concentrations. Ideally, one should measure fetal tissue drug concentrations where the drug’s efficacy and toxicity is likely to manifest. In practice, neither this nor repeated blood sampling of the fetal circulation (to estimate systemic fetal drug exposure) is possible. Therefore, the only recourse to estimate fetal drug exposure (systemic or tissue) is by maternal-fetal (m-f) physiologically-based pharmacokinetic (PBPK) modeling and simulation (M&S).
PBPK models are physiologically relevant mathematical models wherein individual or grouped organs are depicted as compartments interconnected by relevant blood flow. These models are mechanistic in nature and incorporate intrinsic or system dependent physiological parameters (such as blood flows, tissue volumes, enzyme/transporter abundance), and associated population variability which allows prediction of disposition of any drug in the population of interest.7 The mechanistic nature of PBPK models allows incorporation of the fetus and fetal tissues into the model as well as gestational-dependent changes in both maternal and fetal physiology. Thus, such models can be used to predict maternal-fetal disposition, throughout pregnancy, of any drug administered to the mother provided the models are populated with extrinsic, drug-specific parameters (physicochemical and absorption, distribution, metabolism, elimination (ADME) properties). We8–11, and others12–16 have developed a m-f PBPK model that can successfully predict maternal drug exposure. However, to predict fetal drug exposure, the model needs to be populated with drug specific transplacental clearances (active and/or passive) and placental/fetal metabolism, along with fetal and placental parameters that can affect the fetal drug exposure such as fetal fu, pH of blood, placental binding, ion trapping in the placenta.
In this review, first we describe factors that determine fetal drug exposure and define PK parameters to assess fetal drug exposure. Second, we describe in vitro studies in cells/tissue fractions or in the perfused human placenta that can be used to determine drug-specific disposition parameters in the placenta and the fetal liver. Third, we provide examples whereby these studies, combined with m-f PBPK M&S, have successfully predicted systemic fetal exposure to drugs that passively or actively cross the placenta. Apart from m-f PBPK models, animal studies also have the potential to estimate human fetal drug exposure by allometric scaling. Therefore, we discuss the potential for such scaling to be successful. Finally, we close by highlighting gaps in our knowledge to predict fetal drug exposure and suggest future directions to fill these gaps.
Placental physiology, abundance and location of placental drug transporters and metabolizing enzymes
Any drug administered to the mother will reach the fetus through multiple routes (Fig. 1a). Among these, transplacental transfer is the most important once blood flow to the placenta has been established (late first trimester)17–19. The placenta is of fetal origin and separates the maternal and fetal blood. Maternal blood from the uterine artery floods the intervillous space which acts like a “bath-tub” and bathes the fetal villi lined with the multinucleated syncytiotrophoblast formed by fusion of mononuclear cytotrophoblastic stem cells. This cellular arrangement does not allow paracellular diffusion of drugs across the syncytiotrophoblast layer (Fig. 1b).20 Therefore, placental drug transfer can occur only by transporter-mediated active uptake/efflux and/or passive diffusion. Lipophilic drugs that are unionized at physiological pH generally have high passive diffusion clearance across the placenta provided they are not substrates of efflux transporters. Many drugs taken by pregnant women are substrates of the two most highly expressed efflux transporters in the human placenta, namely P-glycoprotein (P-gp), and Breast Cancer Resistance Protein (BCRP).21–24 These ATP-binding cassette transporters serve to prevent entry of drugs into the placenta (and therefore the fetal compartment) by acting as gatekeepers. They serve this function by effluxing drugs as soon as the drugs attempt to cross the lipid bilayer of the syncytiotrophoblast. These transporters can also efflux drugs from the intracellular compartment. P-gp transports mostly lipophilic, unionized cationic drugs (e.g. HIV protease inhibitors) and some neutral drugs (e.g. digoxin), while BCRP can transport anionic (e.g. nitrofurantoin), cationic (e.g. cimetidine) lipophilic and hydrophilic compounds and their conjugates.25 A substantial number of drugs available in market have some affinity for P-gp. Additionally, both P-gp and BCRP have overlapping substrate selectivity. Therefore, both these efflux transporters, act either alone or in tandem, to reduce fetal exposure to drugs. This has been demonstrated in in vitro, perfused placenta as well as in in vivo (animals) studies. For example, P-gp-mediated transport of calcein-AM and vinblastine was inhibited by P-gp inhibitors in Transwell® assays with human placental choriocarcinoma derived BeWo cells.26 In the dually perfused placenta, the transplacental clearance of cyclosporin A (P-gp substrate) was significantly increased when co-administered with the P-gp inhibitor quinidine or chlorpromazine.27 In an in vivo mouse study, the fetal/maternal plasma concentration ratios of P-gp substrates digoxin, saquinavir, and paclitaxel was significantly greater in Mdr1a/1b(−/−) fetuses than in the Mdr1a/1b(+/+) fetuses of the same dam.28 Furthermore, the P-gp inhibitor cyclosporin A was found to significantly increase the fetal liver distribution of [11C]-verapamil in a PET imaging study in macaques.29 Similarly, in membrane vesicles prepared from human term placenta, BCRP was shown to mediate transport of mitoxantrone30 and glyburide31. BCRP was observed to significantly limit the maternal-to-fetal transport of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)32 and glyburide33 in the perfused human placenta. Additionally, increased fetal exposure of topotecan34, nitrofurantoin35 and glyburide36 was observed in Bcrp1(−/−) mice as compared to the wild-type pregnant mice. Apart from P-gp and BCRP, multidrug resistance proteins 1–3 and 5 (MRP1–3 and 5) are also important in transplacental transport, but to a lesser extent.23, 25 The placenta also expresses other efflux transporters as well as influx transporters (see Table 1). However, studies to demonstrate their functional role in vivo in determining fetal drug exposure, through in vitro or perfused human placenta studies, are limited. The placental transporters show gestational age-dependent abundance/expression e.g. the abundance/expression of P-gp, BCRP, Organic Anion Transporting Polypeptide 1A2 (OATP1A2), OATP2B1, OATP1B1, Organic Cation/Carnitine Transporter 2 (OCTN2) decreases from first trimester to term21, 22, 37–45 while that of Multidrug Resistance Protein 2 (MRP2), Organic Anion Transporter 4 (OAT4) and Organic Cation Transporter 3 (OCT3) increases as gestation progresses.22, 46–49 The abundance/expression of OATP4A1, MRP1, MRP3 and Multidrug And Toxin Extrusion 1 (MATE1) shows no change throughout pregnancy.37, 43, 46, 48, 49 It is important to note that where the abundance of transporters per gram of placental tissue decreases (e.g. P-gp), the overall abundance of the transporters in the whole placenta increases with gestational age because the placenta significantly increases in size with gestational age. Unfortunately, literature reports are controversial regarding the ontogeny of several placental transporters (e.g., P-gp, MRP1, OATP1B1, OCT3)21, 37, 46, 48–50 and additional studies are needed to clarify their ontogeny.
Figure 1:
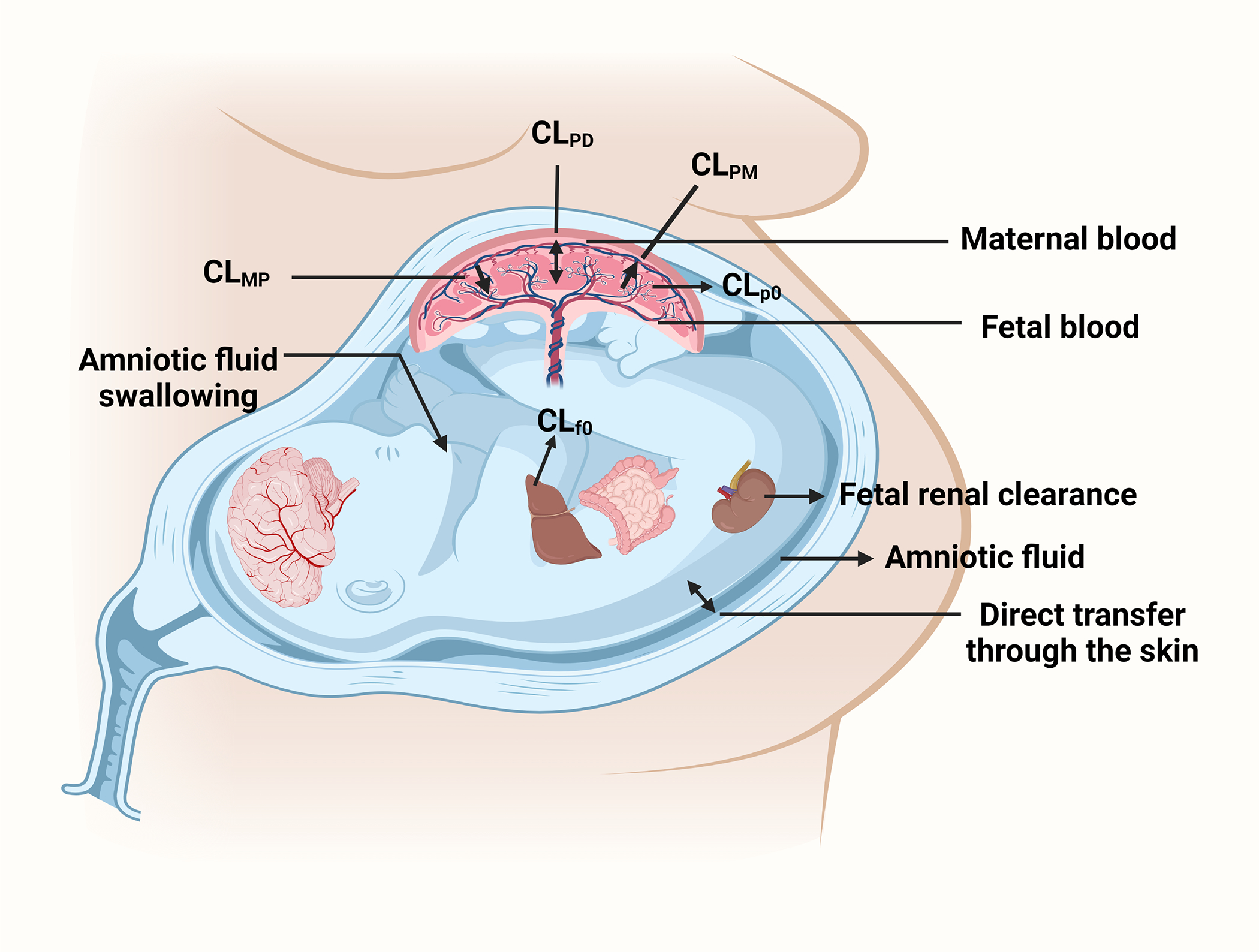
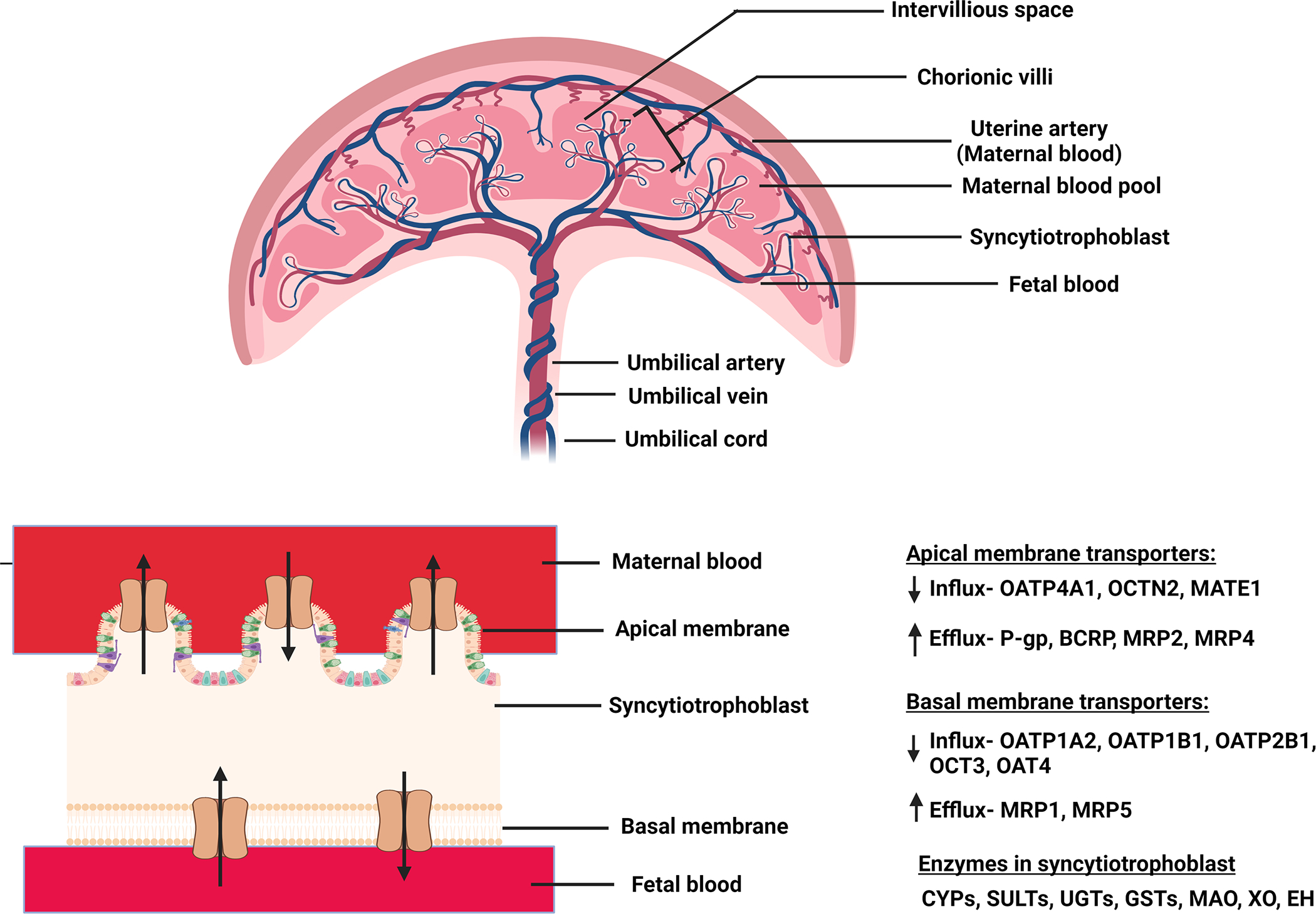
(a) Routes by which drugs distribute into or are cleared from the fetal compartment. (b) Expanded view of the placenta with magnification of the syncytiotrophoblast layer and examples of transporters and enzyme expressed there. Abbreviations: CLPD: passive bi-directional placental clearance; CLMP: transporter-mediated influx clearance; CLPM: transporter-mediated efflux clearance; CLp0: placental metabolic clearance; CLf0: fetal metabolic clearance; CYPs: cytochrome P450s (CYPs); UGTs: UDP-glucuronosyltransferases; SULTs: sulfotransferases, GSTs: glutathione-S-transferases; MAO: monoamine oxidase; XO: xanthine oxidase; EH: epoxide hydrolase. Details of other transporters and enzymes present in the placenta are provided in Table 1.
Table 1:
Drug transporters expressed in the human placenta
| Name of transporter* | Gene/Alternative name | Technique used for detection/Quantification | Localization in the syncytiotrophoblast | Transporter type | Direction of net transport | Potential effect on fetal drug exposure | Reference | ||
|---|---|---|---|---|---|---|---|---|---|
| First trimester | Second trimester | Third trimester/term | |||||||
| P-gp + | ABCB1, MDR1 | R, P | R, P | R, P, A | Apical membrane | Efflux | Fetal to maternal | Reduced | 21, 22, 37–40, 128–131 |
| BCRP + | ABCG2, MXR | R, P | R, P | R, P, A | Apical membrane | Efflux | Fetal to maternal | Reduced | 21, 22, 37, 38, 41, 128–130, 132 |
| MRP1 # | ABCC1 | R | NA | R, P, A | Basal membrane | Efflux | Maternal to fetal | Enhanced | 37, 46, 132–135 |
| MRP2 ^ | ABCC2, CMRP, CMOAT | R, P | R, P | R, P, A | Apical membrane | Efflux | Fetal to maternal | Reduced | 37, 46, 47, 129–131, 135–137 |
| MRP3 # | ABCC3 | R | NA | R, P, A | Apical membrane | Efflux | Fetal to maternal | Reduced | 37, 46, 135–137 |
| MRP4 | ABCC4 | NA | NA | R, P | Apical membrane | Efflux | Fetal to maternal | Reduced | 137 |
| MRP5 | ABCC5 | R | R | R, P | Basal membrane | Efflux | Maternal to fetal | Enhanced | 46, 138 |
| OATP1A2 + | SLCO1A2, SLC21A3, OATP-A | R, P | NA | R, P, A | Basal membrane | Influx | Fetal to maternal | Reduced | 43, 129, 139, 140 |
| OATP1B1+ | SLCO1B1, OATP-C, LST1 | R | NA | NA | Basal membrane | Influx | Fetal to maternal | Reduced | 43, 140 |
| OATP2B1 + | SLC21A9, OATP-B | P | P | R, P, A | Basal membrane | Influx | Fetal to maternal | Reduced | 22, 42, 141 |
| OATP4A1# | SLC21A12, OATP1, OATP-E | R, P | NA | R, P | Apical membrane | Influx | Maternal to fetal | Enhanced | 43, 139 |
| OCT3 ^ | SLC22A3, EMT | R, P | R, P | R, P, A | Basal membrane | Influx | Fetal to maternal | Reduced | 22, 48, 49, 142–144 |
| OCTN2 + | SLC22A5 | NA | NA | R, P, A | Apical membrane | Influx | Maternal to fetal | Enhanced | 44, 45, 145 |
| OAT4^ | SLC22A9, SLC22A11 | R, P | R, P | R, P | Basal membrane | Influx | Fetal to maternal | Reduced | 22, 141, 146, 147 |
| MATE1# | SLC47A1 | R | NA | R | Apical | Influx | Maternal to fetal | Enhanced | 48, 49 |
R = mRNA; P = protein; A = activity measured using the perfused human placenta; NA = not applicable
- bolded transporters are those that are likely to be functionally most important in transplacental transfer of drugs
- no change in expression/abundance with gestational age, per mg protein
- increase in expression/abundance with gestational age, per mg protein
- decrease in expression/abundance with gestational age, per mg protein.
Transporters with no symbol indicate that the data are not available. Reference 22 contains quantitative information for transporters.
In addition to drug transporters, the placenta also expresses a variety of drug metabolizing enzymes including cytochrome P450s (CYPs), UDP-glucuronosyltransferase (UGTs), sulfotransferases (SULTs), glutathione-S-transferases (GSTs) (Table 2). Among them, CYP19A1 (aromatase) is the predominant CYP.51 It is involved in the metabolism of both endogenous compounds (conversion of androgens to estrogens) as well as xenobiotics (such as aflatoxin B152, glyburide53, buprenorphine54, and methadone55). Multiple drugs such as zidovudine56, glyburide57, methadone55, oxcarbazepine58, warfarin59 undergo metabolism in human placental microsomes. Additionally, perfused placental studies have demonstrated the metabolism of bupropion60, oxcarbazepine61, buprenorphine62, tenofovir and emtricitabine63. However, it is important to note that for placental drug metabolism to affect Kp,uu, it must be significant relative to the other clearance pathways in Eq 2 (below). Whether this is the case for the drugs cited above has not been determined. Placental expression of CYP isozymes is generally higher in the first trimester than term.64 In contrast, the expression of UGTs generally remains at about the same levels in the placenta throughout pregnancy.65 It is important to reiterate here that though the abundance of an enzyme per gram of placental tissue may decrease, its total abundance in the whole placenta may increase with gestational age due to the significant gestational age-dependent increase in placental weight. Multiple maternal and environmental factors, such as drug abuse, smoking, alcohol consumption, polluted air, and contaminated food can affect the activity of placental enzymes.66 For example, the activity of UGT and CYP1A is significantly higher in placentae of mothers who smoke and is greatest in women who smoke and consume alcohol.67 In contrast, the catalytic activity of CYP19A1 decreases in mothers who smoke but its protein abundance is not significantly altered.68
Table 2:
Drug metabolizing enzymes expressed in the human placenta and fetal liver
| Enzyme name | Placenta | Fetal liver | ||||||
|---|---|---|---|---|---|---|---|---|
| First trimester | Second trimester | Third trimester/ term | Reference | First trimester | Second trimester | Third trimester/term | Reference | |
| CYP1A1 + | R, P, A | ND | R, P, A | 64, 148–150 | R, P, A | ND | ND | 151 |
| CYP1A2 | R | ND | P | 148, 150 | ND | ND | ND | |
| CYP1B1 | ND | ND | R | 149 | ND | ND | ND | |
| CYP2C8/9 | P, R | ND | P | 148, 152 | ND | R, A | R, A | 153 |
| CYP2D6 | R | ND | R | 148, 149 | ND | R | ND | 154 |
| CYP2E1 | R | ND | R | 64, 148, 149 | ND | ND | ND | |
| CYP2J2 | P | ND | R, P | 149, 152 | ND | ND | R | 149 |
| CYP3A4 | R | ND | R | 64, 148 | ND | R | ND | 154 |
| CYP3A5 | R | ND | R | 64, 148 | R, A | R, A | R, A | 149, 155 |
| CYP3A7 # | R | ND | ND | 148 | P, A | R, P, A | R | 82, 149, 156 |
| CYP19 | A | ND | A | 157, 158 | A | ND | ND | 157 |
| SULT1A1 | A, P | A, P | A, P | 159 | A | R, P, A | A | 71, 72, 160, 161 |
| SULT1A2 | ND | ND | ND | ND | R | ND | 72 | |
| SULT1A3 | A, P | A, P | A, P | 159 | A | R, P, A | A | 71, 72, 160 |
| SULT1B1 | ND | ND | ND | ND | R | ND | ||
| SULT1C2 | ND | ND | ND | ND | R, P | ND | 72, 161 | |
| SULT1C4 | ND | ND | ND | ND | R | ND | 161 | |
| SULT1E1 | A | A | A | 159 | ND | R, P, A | ND | 72, 161 |
| SULT2A1 | A | ND | A | 159 | ND | R, A | ND | 72, 161 |
| SULT2B1 | A | ND | R, A | 159, 162 | ND | R | ND | 161 |
| UGT1A1 | ND | ND | R, P, A | 163 | ND | R | R | 164 |
| UGT1A4 | ND | ND | R, P, A | 163 | ND | R | R | 164 |
| UGT1A6 | ND | ND | R, P, A | 163 | ND | R | R | 164 |
| UGT1A9 | ND | ND | R, P, A | 163 | ND | R | R | 164 |
| UGT2A2 | ND | ND | ND | ND | R | R | 164 | |
| UGT2A3 | ND | ND | ND | ND | R | R | 164 | |
| UGT2B4 | P | ND | R, P, | 65, 67 | R | R | 164 | |
| UGT2B7 | P | ND | R, P | 65, 67 | R | R | R | 164, 165 |
| UGT2B10 | ND | ND | R, A | 65 | ND | R | R | 164 |
| UGT2B11 | ND | ND | R, A | 65 | ND | R | R | 164 |
| UGT2B15 | ND | ND | R, A | 65 | R | R, P | R, P | 164–166 |
| UGT2B17 | ND | ND | ND | R | R | R | 164, 165 | |
| GST | ND | ND | R, A | 167 | ND | A | P | 168, 169 |
| MAO | ND | ND | R, P, A | 170–172 | ND | P, A | ND | 173 |
| X ^ | ND | ND | R, P, A | 174, 175 | A | A | A | 176 |
| PCES1 – 3 | ND | ND | R, A | 177 | ND | ND | ND | |
| CES | ND | ND | ND | ND | A | ND | 178 | |
| Peptidases | P | ND | P | 179 | ND | ND | ND | |
| Epoxide hydrolase | R, P | R, P, A | R, P | 152, 180, 181 | ND | A | ND | 181 |
R = mRNA; P = protein; A = activity was measured in microsomes, S-9, cytosol or homogenates of placental or fetal liver tissue; ND = not determined
- bolded enzymes are those that are likely to be functionally most important in the metabolism of drugs
- no change in activity/expression with gestational age, per mg protein
- increase in activity/expression with gestational age, per mg protein
- decrease in activity/expression with gestational age, per mg protein.
Enzymes with no symbol indicate that the data are not available.
The fetal liver also expresses drug metabolizing enzymes such as CYPs, UGTs, SULTs, xanthine oxidase (Table 2). The most prominent among them is CYP3A7 which accounts for almost 30% of the total fetal CYP content. Its expression increases with gestational age and reaches highest at postnatal days 1 and 7.69 All other CYPs have lower expression in the fetal liver vs. adult liver.70 Among the SULTs, SULT1A1, SULT1A3, SULT2A1, SULT1E1 and SULT1A3 are expressed in the fetal liver.71–74 The SULT enzymes are believed to play a major role in the metabolism of endogenous substances (such as catecholamines, steroids like dehydroepiandrosterone, estrogens) important for fetal development.71–73, 75 The expression of UGTs has also been detected in the fetal liver (Table 2).
The drug reaches the amniotic fluid through urinary excretion by the fetus and by diffusion from the placenta. This drug can be reabsorbed through the intestine when the fetus swallows the amniotic fluid.76 The fraction reabsorbed determines the extent of intestinal reabsorption. This recycling of the drug is likely the highest for hydrophilic drugs that tend to be renally excreted. Irrespective of the extent of drug excretion into the fetal urine, due to this drug recycling, this pathway is akin to distribution into a peripheral fetal compartment rather than irreversible loss from the fetal compartment. Additionally, the fetal skin is also permeable to drugs before keratinization begins at gestational week (GW) 20)77, allowing direct entry of the drug from the amniotic fluid into the fetal circulation (Fig. 1a).
Pharmacokinetic parameters that describe fetal drug exposure
Here we describe four key PK measures that are important to describe fetal drug exposure namely, Kp,uu, the unbound steady-state average (Cu,av,ss,f), peak (Cu,max,ss,f) and trough (Cu,min,ss,f) fetal plasma concentrations. Each is defined in detail below along with factors that determine their value. For the purposes of this discussion, we have assumed that the drug is administered to the mother to steady-state.
Kp,uu and fetal Cu,av,ss plasma concentration
Kp,uu is independent of the drug dose or maternal plasma concentration provided the drug follows linear transplacental and fetal disposition PK (Eq. 1).
| (Eq. 1) |
Where, fu,f and fu,m are unbound drug fractions in fetal and maternal plasma respectively, AUCf and AUCm are area under total plasma concentration-time curves in the fetus and the mother respectively, Cav,ss,f and C av,ss,m are the total plasma steady-state concentrations in the fetus and the mother, respectively.
While it is important to know the absolute Cu,av,ss,f plasma concentration, in vivo, this value is primarily determined by the corresponding maternal concentration. Therefore, the ratio of this concentration and the corresponding maternal concentration, Kp,uu, is usually predicted. Once Kp,uu is predicted, Cu,av,ss,f can be calculated for any corresponding maternal concentration.
Kp,uu is determined by a number of factors (Eq. 2) that determine transplacental and fetal clearance of the drug, namely: i) unbound passive placental diffusion clearance (CLPD); ii) unbound active placental efflux (CLPM), influx (CLMP), or placental metabolic drug clearance (CLp0) and iii) unbound fetal clearance (CLf0) (e.g., fetal hepatic clearance).6
| (Eq. 2) |
As indicated above, the placenta is not well-endowed with enzymes that can extensively metabolize drugs (e.g., CYPs).78 While the placenta expresses influx transporters, to date there is no evidence of drugs that are actively transported into the placenta (and therefore the fetal compartment) via such transporters. Thus, CLMP and CLp0 can usually be considered negligible. But, this is not the case with efflux transporters such as P-gp and BCRP which are highly expressed in the apical membrane of the placental syncytiotrophoblast22 and prevent entry of drugs into the placenta (and therefore the fetal compartment). In the fetus, the drug can be eliminated from the fetal circulation by fetal metabolism in the fetal liver. However, due to its size and selective expression of CYPs (primarily CYP3A7), the fetal liver is usually not a significant determinant of drug metabolism and therefore fetal drug exposure (i.e., CLf0 is usually negligible relative to the transplacental CLPD). Therefore, in general, the fetal Kp,uu is usually dependent on the magnitude of CLPM relative to CLPD (note all clearance values in Eq 2 and 3 are the unbound clearance values) (Eq. 3):
| (Eq. 3) |
When there is negligible placental efflux (CLPM = 0) or placental/fetal elimination, Kp,uu will be 1 because passive transplacental diffusion clearances should be equal in both directions. In this event, fetal unbound AUC will approximate maternal unbound AUC and can be predicted from the latter.79 But, when a drug is actively effluxed (i.e., CLPM > 0), Kp,uu will be less than 1. In that event, the fetal unbound AUC will be less than the maternal unbound AUC and cannot be estimated solely based on maternal drug concentration data. Instead, to predict Kp,uu, one needs the values of all the transplacental clearances or the ratio of the values shown in Eq. 3. Later in this article, we address how these values can be determined through in vitro cell or perfused placenta studies.
Although Kp,uu provides an estimate of fetal exposure at steady-state or over a dosing interval, its major limitation is that it does not provide information about the dynamic variations in fetal unbound plasma concentrations with time. In other words, the value of the peak or trough unbound steady-state fetal plasma drug concentration. However, this limitation can be overcome by incorporating into a m-f PBPK model the exact magnitude of the various transplacental clearances and the volume of distribution of the drug in the fetal compartment. The approach is discussed in detail later in this article.
While Kp,uu is independent of protein binding of the drug, Kp is not. Kp changes with gestational age because the plasma protein concentration in the maternal-fetal compartments change with gestational age.80 For example, fetal α1-acid glycoprotein concentrations increase from almost undetectable level at 12 weeks to 0.3–0.4 g/l at >GW 35, while maternal α1-acid glycoprotein concentrations fluctuate between 0.38 and 1.05 and show no trend with advancing gestational age. These changes lead to an increase in α1-acid glycoprotein fetal/maternal serum concentration ratio from 0.057 at week 12 to 0.37 at >GW 35.80 On the other hand, maternal serum albumin concentration decreases during gestation, while that in the fetus increases with gestational age. As a result, the fetal/maternal serum albumin concentration ratio gradually rises from 0.38 at 12 weeks to 1.20 at term.80 Although Eq. 1 may mathematically suggest that Kp,uu is dependent on the total fetal drug plasma concentration, that would be incorrect. In fact, it is the other way round. To illustrate this point, consider a drug that passively crosses the placenta, is extensively bound to albumin, and is not metabolized in the placenta or the fetal liver. The Kp,uu of this drug will be unity (as only the unbound drug crossed the placenta) irrespective of the gestational age. However, its Kp value will change with gestational age due to changes in albumin concentration in the maternal-fetal compartment.
Fetal and maternal PK parameters that determine fetal unbound peak (Cu,max,ss,f) and trough (Cu,min,ss,f) steady-state plasma concentrations
Fetal unbound peak (Cu,max,ss,f) and trough (Cu,min,ss,f) steady-state plasma concentrations are determined in manner analogous to factors that determine plasma concentrations in a non-pregnant individual after oral absorption of drugs. Thus, when the rates of entry and exit of the drug into and from (back to the mother and by fetal elimination) the fetal compartment are equal, Cu,max,ss,f will be achieved. For this reason, Cu,max,ss,f usually occurs later than the Cu,max,ss,m. However, the greater the transplacental clearance, the faster and higher the Cu,max,ss,f is reached. In addition, the magnitude of Cu,max,ss,f will depend on the volume of distribution of the fetal compartment and fetal clearance (via the placenta or by metabolism). The smaller the fetal volume of distribution, the larger the Cu,max,ss,f and vice-versa. Likewise, the smaller the clearance from the fetal compartment the larger the Cu,max,ss,f. Similarly, Cu,min,ss,f will be determined by the magnitude of fetal drug clearance during the dosing interval. Because maternal concentrations drive fetal concentrations, the higher the maternal concentrations, the higher both Cu,max,ss,f and Cu,min,ss,f will be (see Zhang et al., 20176 for details). Finally, the corresponding total Cmax,ss,f and Cmin,ss,f will be determined by the protein binding of the drug in the fetal compartment.
Predicting fetal systemic and tissue drug concentrations from animal and in vitro studies
As indicated above, measuring fetal (systemic or tissue) exposure to drugs is logistically challenging, especially before term. However, such exposure could be predicted using animal studies or in vitro studies combined with PBPK M&S. Below, we discuss both these approaches.
Animal studies
Several animal models ranging from small rodents such as mice, rats, and guinea pigs to larger animals like sheep and non-human primates have been used to study the transplacental passage and fetal tissue distribution of drugs. The smaller animals (e.g., mice) can be genetically modified to study the role of specific transporters in fetal exposure to drugs. For example, Zhang et al. demonstrated that presence of murine Bcrp significantly limits fetal distribution of nitrofurantoin in pregnant mice while exerting only a minor effect on the systemic clearance of the drug.35 However, small animal studies have some limitations. Due to the small fetal size, fetal blood samples are difficult or impossible to obtain. Thus, both maternal (plasma) and whole fetus and placental tissue drug concentrations are obtained only once (at the time of sacrifice) after drug administration. Consequently, multiple pregnant dams need to be sacrificed and the data pooled to characterize the entire AUC of the drug concentration-time profile. Fortunately, due to inbreeding, inter-animal variability tends to be low and this “naive-pooled” approach to data analysis works relatively well.35, 36, 81, 82 When interpreting the fetal concentrations data, it is important to keep in mind that these concentrations are not in fetal blood or plasma but in the entire fetus. Therefore, for those drugs that bind extensively to fetal tissues, the ratio of the whole fetus/maternal plasma AUC may be greater than unity and should not be interpreted as transport of the drug into the fetal compartment. Also, it is often not possible to determine the drug concentrations in individual fetal tissues. In addition, placental/fetal tissue concentrations of the drug should be corrected for the amount of blood in the organ, especially for drugs that extensively bind to blood components.
In larger animals (sheep and non-human primates), intravascular catheters can be placed both in the dam and the fetus to obtain matched fetal and maternal plasma drug concentration-time profiles over a period sufficient to characterize the corresponding AUCs. The pregnant non-human primates are similar to humans in that they have a discoid placenta with multivillous fetal-maternal interdigitation and a hemo-monochorionic barrier.83 These animals were used to determine the rate and extent of the placental transfer of anti-HIV drugs zidovudine, didanosine, zalcitabine and stavudine.84 These in vivo animal data strongly correlated with those from the perfused human placenta. However, these drugs cross the placenta by passive diffusion and a corresponding correlation for drugs which are actively transported across the placenta or extensively metabolized in the placenta/fetal liver has not been investigated.
The major limitation associated with animal models is the large inter-species differences in (i) placental anatomy, metabolism, and transport, (ii) fetal metabolism and transport (iii) substrate selectivity of drug metabolizing enzymes and transporters and (iv) the abundance of placental/fetal transporters and metabolizing enzymes.83, 85–87 These differences may lead to variations in fetal drug exposure, thus preventing meaningful extrapolation to humans.81, 88 Nevertheless, whether animal data can be used to predict human fetal drug exposure is an open question that has yet to be answered.
In vitro cell and perfused human placenta studies combined with PBPK M&S
Over the years, multiple maternal-fetal (m-f) PBPK models have emerged which show progressive success in predicting maternal and fetal drug exposure.89 Initial m-f PBPK models were focused on estimating variability in maternal drug exposure, with limited emphasis on fetal drug exposure. This was due to lack of curation of gestational age-dependent fetal and placental physiological parameters (e.g., gestational age dependent tissue blood flow) and lack of availability of the abundance of drug transporters and metabolizing enzymes which determine drug CLMP, CLPM, CLp0, and CLf0. However, we6, 22, 79, 90, 91 and others92–98 have begun to fill these gaps in knowledge by generating/curating relevant physiological data (e.g. blood flow, organ sizes, plasma protein concentrations) and describing their gestational-age dependent changes with models that allow interpolation for any gestational age. In addition, we have quantified the abundance of placental transporters at various gestational ages.22 To determine the various parameters which govern fetal exposure, we need to populate the m-f PBPK models with drug-specific CLPD, CLMP, CLPM, CLp0, and CLf0. These parameters can be determined through in vitro cell studies and/or ex vivo perfused human placenta studies. As discussed earlier, there is no evidence of influx transport of drugs across the human placenta (CLMP), and because CLp0 and CLf0 usually contribute minimally to fetal drug exposure, these parameters can be discounted. Therefore, as described below, the remaining two parameters namely CLPD and CLPM, need to be estimated, to populate the m-f PBPK model. However, if CLMP, CLp0 and CLf0 are significant relative to the net transplacental CL, they can be taken into account using the approaches described below, in combination with metabolic depletion studies in placental and fetal liver homogenates or microsomes99, to predict fetal drug exposure.
Two approaches can be utilized to estimate CLPD and CLPM, namely the efflux ratio-relative expression factor (ER-REF) approach using cells (i.e., placental or transporter-transfected cells) or the perfused human placenta (Fig. 2). The former can be used only when the transporter(s) involved are known. While the latter can be used even when the transporter(s) involved are not known. Both these approaches utilize the in vitro to in vivo extrapolation (IVIVE) strategy to scale the magnitude of the unbound bi-directional placental clearance determined in the respective system to that in vivo. The IVIVE scalar for the ER-REF approach is called REF, which accounts for the differences in transporter abundance between the in vitro cell model and in vivo (i.e. the entire placenta). In the case of the perfused placenta, the bi-directional placental clearance determined in a single perfused cotyledon needs to be scaled to the entire placenta (i.e., physiological scaling). This is done by utilizing the average number of cotyledons in the placenta or by scaling the cotyledon weight or volume to the total placental weight or volume. We would like to emphasize here that the data generated by both approaches (cell and perfused placenta studies) can be used to predict Kp,uu, as detailed below, independent of m-f PBPK models provided placental transporter and/or metabolism (and not fetal metabolism) is the major determinant of fetal exposure to drugs. However, the dynamic changes in fetal exposure can be predicted ONLY if these two approaches are combined with the m-f PBPK models.
Figure 2:
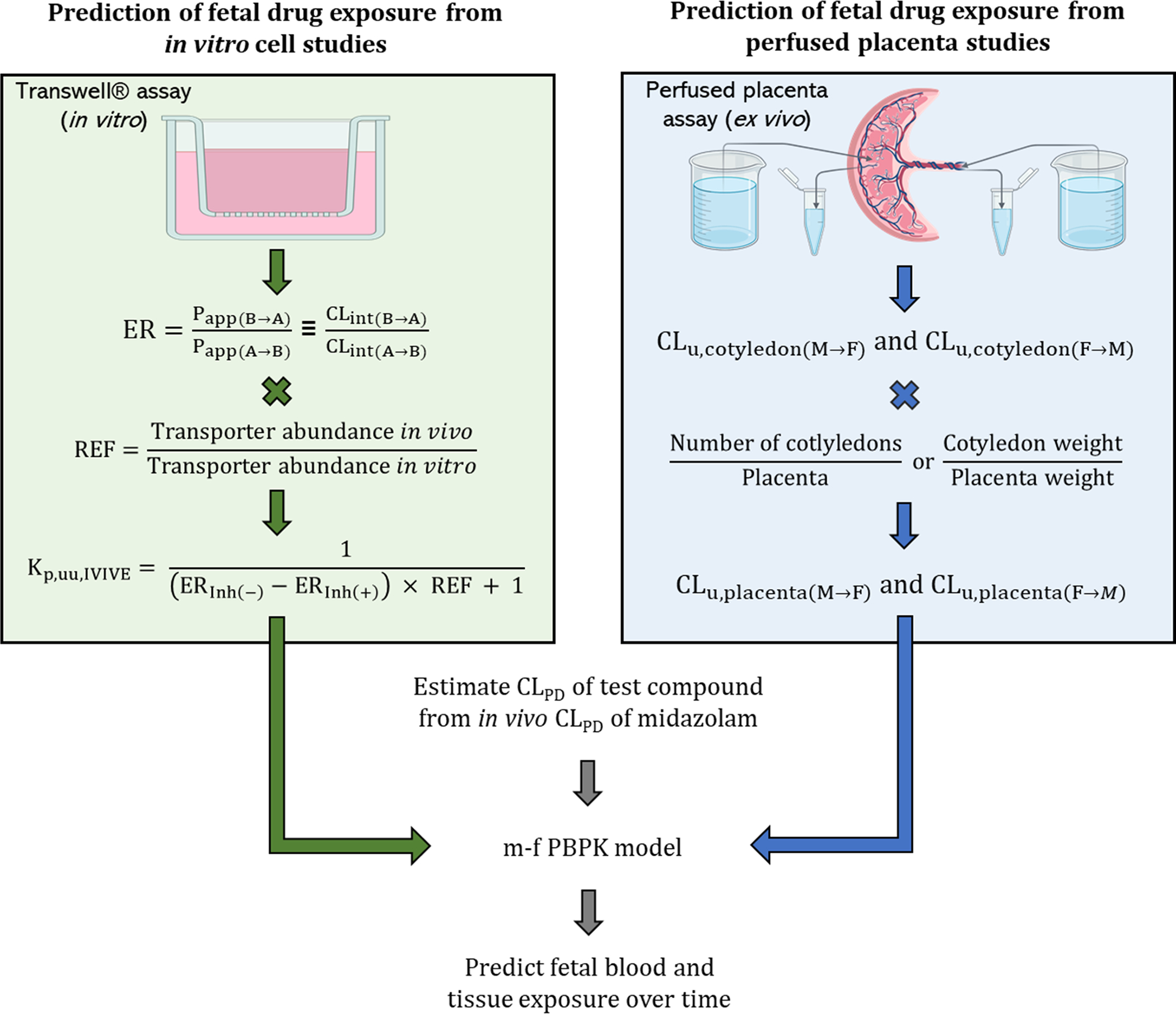
Prediction of fetal drug exposure using the m-f-PBPK approach combined with data from (a) the ER-REF studies (left panel) or (b) the perfused placenta studies (right panel). Although the perfused placenta approach is shown in a single-pass mode, the re-circulation mode could also be used. CLu - unbound clearance; CLint(B→A) and CLint(A→B) - intrinsic drug clearance in the B→A and the A→B direction, respectively; ER - efflux ratio; Papp(B→A) and Papp(A→B) - apparent drug permeability in the B→A and the A→B direction, respectively; F→M - fetal to maternal; M→F - maternal to fetal; REF - relative expression factor.
ER-REF combined with the m-f PBPK approach
This approach requires determination of two parameters, ER and REF (Fig 2, left panel). REF is the ratio of the abundance of the transporter of interest in the placenta and the cell line. The magnitude of drug efflux (or influx) by placental transporters is measured as ER in the Transwell® assay using either placental cell lines or transporter overexpressing cell lines (see below for details). Since the transporter expression in these cell lines is likely to differ from that in the human placenta, the ER measured in these cells needs to be corrected for this difference in abundance using the REF. Unless the abundance on the cell membrane is measured using a method such as biotinylation, combined with quantitative targeted proteomic100, the REF approach assumes that the total abundance of the transporter measured is that on the cell membrane and the transporters that are present there are functional. The advantage of the REF approach (over that of the perfused placenta approach) is that the abundance of various placental transporters in the human placenta of different gestational ages is available. Therefore, the ER-REF approach can be used to predict fetal exposure to drugs once blood flow to the placenta has been established.91
ER determination:
In the Transwell® assay, the polarized cells over-expressing the transporter of interest, are seeded onto a Transwell® insert to create the apical and basal compartments corresponding to the maternal and fetal blood, respectively (Fig. 2, left panel). Then, the drug is added either to the apical or basal chamber (i.e., the donor chamber) and drug concentration in the contralateral receiver chamber is sampled at various times points. The ratio of the apparent drug permeability from the basal-to-apical (B→A) (Papp(B→A)) and apical-to-basal (A→B) (Papp(A→B)) chambers is the ER that represents the magnitude of in vitro transport by the overexpressed transporters across the cell monolayer (Eq. 4).91
| (Eq. 4) |
Assuming that the surface area is the same in both directions, the Papp’s can be set equivalent to CLint(B→A) and CLint(A→B), the intrinsic clearances of drug in the B→A and the A→B directions, respectively; cAA(R) and cAB(R) are cumulative amounts of drug in corresponding receiver compartment; AUCA(D) and AUCB(D) are AUC of the drug in corresponding donor compartment. AUC, rather than initial donor drug concentration, is used to compensate for any depletion of drug in the donor compartment. Proteins are usually not included in the donor chamber. If included, the Papp needs to be determined for the unbound drug.
Then, to predict the in vivo Kp,uu (i.e. Kp,uu,IVIVE), the ER determined in the cell line is scaled (Eq 5)91 by REF as follows:
| (Eq. 5) |
where ERInh(−) and ERInh(+) is the efflux ratio in the absence and presence of complete inhibition of the transporter of interest, respectively. The difference in ER in absence and presence of complete inhibition of transporters provides the magnitude of ER associated with active transport. Alternatively stated, the bi-directional passive diffusion clearance of the drug cancels out when estimating the net active transport ER. In the absence of transport, the ER in the absence and presence of the transporter inhibitor will be identical. Therefore, as predicted, Kp,uu,IVIVE will be 1. For efflux transporter drug substrates, the difference in the ERs will be >1 and Kp,uu,IVIVE, as expected, will be <1. Thus, when Kp,uu,IVIVE is estimated using Eq. 5, it is independent of the absolute magnitude of passive diffusion clearance across the placenta. This is because in this equation only the relative value of the active and passive clearance is taken into consideration. We would like to emphasize again that the Kp,uu,IVIVE can be estimated independent of any m-f PBPK model. However, this value does not provide an estimate of the dynamic exposure of fetal exposure to a drug (i.e. Cu,max,ss,f and Cu,min,ss,f). To do so, one must estimate the absolute value of each bi-directional clearance pathway (active and passive) of the drug, as discussed below under the subheading Transporter-transfected cell lines. Parenthetically, the fraction of a drug transported (e.g., ftP-gp), in vivo, can be estimated as:
| (Eq. 6) |
The choice of the cell line to be used in the Transwell® assay should be one that expresses the transport of interest for the drug in question. Although other transporters are also present in placenta, P-gp and BCRP are the most abundant and many drugs are substrates of one or both these transporters. However, of these two transporters, P-gp plays a more prominent role in governing fetal exposure to drugs that are taken by pregnant women. Hence, the choice of the cells to use in the Transwell® assay could be either a human placental cell line or one that is engineered to overexpress the transporter of interest (e.g., MDCKII cells overexpressing P-gp). These two options are discussed below.
Placental cell lines:
The human placental choriocarcinoma trophoblast cell lines (BeWo, Jar, Jeg-3 and ACH-3P) have been used to address transplacental transport of drugs.101, 102 While BeWo, Jar, and Jeg-3 cells are derived from a trophoblastic tumor, the ACH-3P cell line was derived by fusing primary trophoblast cells from week 12 placenta with human choriocarcinoma cells.103 The BeWo, Jar and Jeg-3 cell lines express relevant efflux drug transporters (P-gp, BCRP, MRP1, etc.) at the mRNA and protein level.46, 104, 105 However, P-gp activity in BeWo cells is low. Jar cells lines have low activity of both P-gp and BCRP.104, 106–108 Of the cell lines, several can form tight junctions109, including a subclone of BeWo cells, a property necessary for use in Transwell® assays to determine drug transport.110 However, the BeWo b30 cell monolayer tight junctions are not easy to establish and maintain for an extended period of time.110 While xenobiotic transport has been shown in these cells, their ability to predict placental transport accurately has not been demonstrated. Therefore, they could be used to identify potential transporters relevant for in vivo transport of drugs.102 However, there are multiple limitations associated with the placental cell lines (Table 3). Due to these limitations, they have not been used for IVIVE of fetal exposure to drugs. Instead, non-placental cell lines, such as Madin-Darby Canine Kidney (MDCKII) cells, transfected with human transporters, have been used as a surrogate.
Table 3:
Merits and limitations of various approaches that can be used to estimate fetal drug exposure
| Merits | Limitations | |
|---|---|---|
|
| ||
| Animal models | ||
|
| ||
| • Allows study of placental transfer and fetal toxicity of drugs at different gestational ages (limited gestational age range for small animals) • Rodents can be genetically modified to study the role of specific transporters in fetal distribution of drugs • Lower interindividual variation due to inbred animals (small animals only); pooling samples from multiple dams/fetuses is feasible • In large animals, both maternal and fetal plasma drug concentration-time profiles can be determined in the same animal |
• Interspecies variability in placental anatomy and transporters, placental and fetal metabolism, substrate selectivity of placental/fetal drug metabolizing enzymes and transporters and abundance of placental/fetal drug metabolizing enzymes and transporters limits extrapolation to humans • Accurate extrapolation from animal models to humans has not been demonstrated • For small animals, due to the small size of the pups, blood samples from pups cannot be obtained. Instead, drug exposure in the entire fetus is determined by pooling multiple pups to generate the AUC of the total fetal drug concentration-time profile. These profiles are then compared with a plasma AUC profile generated in the dam by pooling plasma concentrations obtained by sacrificing the dam at different time points. This is costly and has the potential to increase variability in the data • For small animals, due to size, fetal tissue drug concentration cannot be determined except perhaps near term |
|
|
| ||
| m-f PBPK modeling * | ||
|
| ||
| • Dynamic profiling of fetal drug exposure is possible at term as well as early in gestation (> GW 9) • Prediction of fetal tissue drug exposure is theoretically possible provided the necessary data on fetal tissue enzyme and transporter activity or abundance are available • Can be used to predict fetal exposure to drugs that are effluxed by placental P-gp using the ER-REF or potentially the perfused placenta approach • Can be used to predict fetal exposure to drugs that passively cross the placenta without the need to use either the ER-REF or the perfused placental approach • Can be used to predict the effect of intrinsic (e.g. transporter/metabolic enzyme genetic polymorphisms, disease conditions) and extrinsic factors (e.g. smoking, drug interactions) on fetal drug exposure |
• The limited availability of in vivo data for maternal-fetal exposure to probe drugs that are selectively transported or metabolized by a single transporter or enzyme precludes model validation for such drugs (e.g. those transported solely by BCRP). • As pre-term fetal sampling is not possible, prediction of fetal exposure at gestational ages other than at term cannot be validated • Likewise, prediction of fetal tissue drug exposure cannot be validated due to ethical and logistical constraints in obtaining such data • Lack of data on quantitative contribution and ontogeny of other routes of fetal drug exposure such as direct transfer of drug from the placenta to the amniotic fluid or transfer of drug from the amniotic fluid through the permeable fetal skin may affect model predictions • Lack of mechanistic and quantitative data on major factors that affect maternal drug exposure (and therefore fetal drug exposure) earlier in gestation (such as gestational age dependent changes in transporters, CYPs, UGT and non-CYP metabolizing enzymes) precludes prediction of maternal-fetal exposure to drugs transported or metabolized by these proteins • Lack of data on tissue specific changes in abundance and/or activity in transporters and enzymes in the maternal compartment (e.g. intestine) precludes prediction of maternal-fetal exposure to drugs that are transported or metabolized in these tissues |
|
|
| ||
| ER-REF approach | • Independent of magnitude of passive diffusion clearance in the in vitro system • Not constrained by high degree of technical expertise, limited cell viability and throughput • Approach is validated for P-gp substrates (using MDCKII transfected cell lines) but needs to be validated for other important transporters present in the placenta (e.g. BCRP) • Fraction transport (ft) by a specific transporter can be estimated which can be used to predict the effect of drug-drug interactions, transporter/metabolic enzyme genetic polymorphism and diseases that could modulate fetal drug exposure • Allows dynamic prediction of fetal exposure to drugs at earlier gestational ages by accounting for the ontogeny of placental transporter abundance and change in passive diffusion clearance due to size. To do so, ft, at term, via each transporter must be known |
• Assumes that all quantified transporters are present on the membrane and functional. Otherwise, abundance of transporters should be determined in the cell membrane (e.g. by biotinylation). • Identity of transporter(s) involved in the placental transfer of drug is required • Transporter and metabolizing enzyme abundance in vitro and in vivo should be available • Does not provide absolute measure of passive diffusion clearance (requires calibration with midazolam data) • Approach is validated only for P-gp substrates but not for other transporters |
|
Placental cell lines • More physiologically relevant • Useful to estimate placental transfer (or transport if transport activity is not low) and pharmacologic response to drugs • BeWo b30 cells can form cellular monolayers with tight junctions and hence can be used for flux studies provided transporter functional activity is significant (though it is problematic to establish and maintain the cells) • Can be used as an initial screen of placental transport to identify the transporters potentially relevant in vivo (provided selective inhibitors are available) |
Placental cell lines • Does not represent the complex physiology of the placenta and lacks secondary cell populations (e.g., endothelial cells) where transport can occur • Limited transporter expression decreases the sensitivity of the analysis to identify relevant drug transporter • Quantitative accuracy in prediction of in vivo transplacental clearance of drugs has not been validated • Difficult to predict transplacental transport of drugs that are substrates of multiple transporters unless a selective inhibitor of each transporter is available |
|
|
Transporter overexpressing cell lines (e.g.MDCKII) • Forms tight junctions • Can overexpress the major human placental transporters (P-gp or BCRP), and allows easy detection of transporter activity in vitro • Can be used to predict placental transport of drugs in vivo that are substrates of multiple transporters |
Transporter overexpressing cell lines (e.g.MDCKII) • Does not represent the complex physiology of the placenta and lacks secondary cell populations (e.g., endothelial cells) where transport can occur • Requires studying each transporter separately to estimate the in vivo ft via a given transporter |
|
|
| ||
| Placental perfusion approach | • Most closely resembles the in vivo situation as it incorporates flow and retains placental anatomy • Allows simultaneous study of multiple PK parameters such as passive diffusion, active transport, metabolism, and tissue binding • Inter-individual variability in transplacental clearance can be determined • Does not require prior knowledge about the transporters involved in transplacental transfer • Does not require the assumption that all transporters are on the membrane and functional, as abundance is not quantified • Does not require REF for scaling and hence is free of the assumption that activity is proportional to abundance • Can be used to quantify placental drug accumulation |
• Can determine drug transfer ONLY close to or at term and not at earlier gestational ages (i.e. trimester 1 and 2) • Ability to accurately predict fetal exposure to drugs that are transported by the placenta has not been thoroughly validated • Requires special equipment and technical expertise • Low throughput • The maximum perfusion duration is six hours, after which the tissue is no longer viable, which can pose an issue if insufficient drug has appeared in the receiver compartment within 6 hours • No standardized placenta viability criteria have been established • ft via individual transporters can be determined only if selective inhibitors are available. |
- Merits and limitations of just the m-f PBPK modeling approach are listed here. Those of the ER-REF and the placental perfusion approach are listed later in the table followed by the merits and limitations specific to the placental and transporter overexpressing cell lines
Transporter-transfected cell lines:
MDCKII cells can overcome the major limitations of trophoblast-derived cell lines in that they can form tight junctions and therefore limit paracellular drug transfer which should be absent in the syncytiotrophoblast (a single cell barrier). Additionally, these cells, after transfection, can overexpress the major human placental transporters (e.g., P-gp and BCRP), and thus, allows easy detection of transporter activity in vitro. If a drug is a substrate of both P-gp and BCRP, cells expressing these transporters individually can be used to predict the Kp,uu,IVIVE of such a drug. And, the ft via each transporter can be estimated to allow prediction of possible drug-drug interactions or polymorphisms that could modulate fetal drug exposure. Furthermore, it is also possible to remove any confounding contribution from endogenous transporter by knocking it out (e.g., canine P-gp is knocked out in hMDR1-MDCKcP-gpKO). However, the MDCK cell monolayer does not represent the complex physiology of the placenta and lacks secondary cell populations (e.g., endothelial cells) where transport (though minor contribution) can occur.23
As indicated before, the ER-REF approach considers ONLY the transport-mediated ER or clearance and is independent of the absolute magnitude of CLPD of the drug. Also, it does not provide the absolute value of the maternal-fetal or the fetal-maternal drug clearance. But, to dynamically predict fetal drug exposure, an estimate of the active transport vs. passive clearance is needed. Therefore, we have proposed an approach to estimate the CLPD of any drug by using the in vivo midazolam CLPD as a calibrator (Eq. 7).
| (Eq. 7) |
Where, Papp represents the apparent permeability in MDCKII or another mammalian cell line where the drug is not transported. Once the absolute value of the CLPD has been estimated, the magnitude of the transport clearance, as a fraction of the CLPD, can be obtained from Kp,uu,IVIVE.91
Validation of the ER-REF plus the m-f PBPK model approach:
Any approach or model needs to be validated prior to applying it to prospectively predict fetal drug exposure to a drug for which such data are not available. Therefore, it is logical to ask if this ER-REF approach, combined with the m-f PBPK model, has been validated? It has for P-gp transported drugs91 but needs to be validated for placental transport mediated by BCRP or other transporters. For validation, rich in vivo UV and simultaneously obtained MP concentrations in multiple maternal-fetal dyads that span the AUC profile of the drug are required to assess model prediction performance. But such in vivo data are limited and available for only a limited number of drugs for which the transporters involved (or the lack thereof) have been definitively identified. Using these inclusion criteria, we have successfully validated the ER-REF plus m-f PBPK model approach for four P-gp substate drugs namely dexamethasone DEX, betamethasone BET, darunavir DRV and lopinavir LPV.91 Such validation for BCRP substrates and dual P-gp/BCRP substates is ongoing in our laboratory.
Now that our ER-REF/m-f PBPK model approach has been validated, it can be used to predict fetal drug exposure to other drugs that are P-gp substrates. Moreover, this approach is versatile in that it can be used to predict fetal exposure to P-gp substrate drugs at earlier gestational ages as we have done before.91 The m-f PBPK model purposed by us6, and others (Simcyp version 19 onwards), incorporates multiple fetal organs allowing us to dynamically predict fetal tissue drug concentration such as fetal brain. To do so successfully, one would have to have information (currently lacking) on transporter abundance at the fetal tissue:blood barrier. Plus, the approach would have to be validated for a select number of transporter-selective substrate drugs. The former is possible, but the latter is logistically challenging. Future research should be directed to accomplishing these goals.
Ex-vivo perfused human placenta combined with the m-f PBPK model approach
This model was initially developed by Maurice Panigel and Henning Schneider111, 112 The placental perfusion model allows for the study of multiple pharmacokinetic factors such as passive diffusion, active transport, metabolism, and tissue binding. Unlabored placentas are preferred for this approach since placental cell death and other complications (e.g. infection with exposure to meconium and increased prostaglandin concentrations with inflammation and hypoxia) increase with labor duration.113 In this model, a single cotyledon of the placenta is dually perfused through both the intervillous space and a cannulated fetal artery to represent the maternal and fetal blood flow, respectively. The outflow perfusate is then collected from the intervillous space and the cannulated fetal vein to represent circulating blood returning to the maternal and fetal circulation, respectively. Perfusate buffers are commonly oxygenated with a 95% oxygen and 5% carbon dioxide gas to increase duration of placentae viability. Additionally, proteins (e.g., 2.0 g/L bovine serum albumin) are added to the buffer to allow binding to plasma proteins and to increase drug solubility. In a given cotyledon, the drug is included in the donor perfusate (i.e., either the maternal or the fetal perfusate) and drug concentration is sampled in both the donor and the receiver outflow. The drug perfusion can be set up in two ways: 1) recirculating or 2) single-pass. For the recirculating approach, any outflow from the vein that is not collected for determination of drug concentration is re-circulated. However, for the single-pass approach, the outflow that is not collected for determination of drug concentration goes to waste and the cotyledon is continuously perfused with fresh buffer (with the drug contained in only in the donor perfusate).
Both approaches can be used to evaluate placental drug transfer, but the major difference between them is that the single-pass approach reaches steady-state quicker than the recirculating approach, however the difference in time to steady state is reduced for higher permeability compounds and smaller reservoirs.114 Also, potentially toxic cellular metabolic waste products build up in the re-circulated perfusate while they do not in the singe-pass perfusate. For the re-circulation approach (but not the single-pass approach), the ex vivo fetal/maternal concentration ratio (provided steady-state has been reached) can be directly compared to the in vivo Kp,uu. If steady-state has not been reached or if the single-pass approach is used, the unbound drug clearance ratio (maternal-fetal/fetal-maternal) needs to be estimated to translate to in vivo Kp,uu,IVIVE (see below). Thus, the single-pass method requires both maternal-fetal and fetal-maternal perfusions to estimate in vivo Kp,uu while the re-circulation approach needs perfusion in only one direction, but to steady state. To apply the above approaches to estimate the in vivo Kp,uu, fetal CL would have to be negligible.
Maternal-fetal drug clearance can be estimated when drug is introduced into the maternal perfusate and collected from the fetal venous outflow, and vice versa for the fetal-maternal clearance estimation (i.e. drug is introduced into the fetal perfusate and collected from maternal venous outflow).115 To estimate clearance in both directions, two individual cotyledons must be perfused, one for each clearance estimation. The clearance values estimated from the model can be scaled to the whole placenta, either by scaling the average number of cotyledons in the placenta or by scaling the cotyledon weight or volume to the total placental weight or volume. However, in using this scaling approach, several assumptions are made: 1) the abundance and activity of the transporters and enzymes in the perfused placentae are identical to that in vivo; 2) the entire cotyledon is perfused ex vivo as it is in vivo. The latter assumption is doubtful because in placental perfusion experiments this is usually not the case; part of the cotyledon is not perfused to prevent leakage. Also, because only one fetal artery is cannulated, it is unlikely that the perfused surface area is the same in both directions, maternal-to-fetal and fetal-to-maternal. There will also be inter-cotyledon variability in the surface area perfused. All these factors will likely result in mis-prediction of the in vivo drug placental CL. To determine the contribution of transporters or enzymes in drug transfer, the perfusions can be conducted in the absence and presence of a selective inhibitor of the transporter or enzyme.
It is important to note that perfused placenta can provide an estimate of Kp,uu (provided fetal clearance of drug is negligible) but to predict the dynamic fetal drug exposure, it needs to be combined with a m-f PBPK model which must be populated with the CLMP, CLp0 and CLPM. This combined approach has been employed to dynamically predict fetal exposure to drugs that passively cross the placenta (e.g. acetaminophen116, dolutegravir117, sildenafil118) and are possibly metabolized in the placenta (e.g. tenofovir and emtricitabine)63. This approach has also been used to successfully predict fetal exposure to a drug (darunavir) that is actively transported by the placenta.119 While the authors attributed the higher fetal to maternal clearance vs. maternal to fetal clearance of darunavir to a difference in the physiological blood flows, we believe the difference is due to active P-gp efflux of the drug. As to whether the perfused placenta, combined with PBPK M&S, can predict fetal drug exposure to a wider array of drugs that are transported by the placenta is yet to be determined. It should be noted here that it is not necessary to conduct perfused placenta studies to predict fetal exposure to drugs that passively cross the placenta as such dynamic predictions can be made without conducting these costly and challenging studies (see Zhang et al. 2017)6.
The overall merits and limitations of the above two approaches (ER-REF and perfused placenta) combined with m-f PBPK M&S are provided in Table 3.
Expert opinion on future directions and overall conclusions
There are multiple knowledge gaps in our ability to predict fetal exposure to drugs. First, fetal physiological data for early gestation (<GW9) are incomplete and therefore the present m-f PBPK models cannot be used to predict fetal exposure to drugs prior to GW9. Second, it is impossible to validate fetal drug exposure (systemic or tissue) predictions earlier in gestation because prenatal fetal sampling is not ethical or logistically possible. Third, there is limited information about the ontogeny of non-CYP enzymes in the placenta and the fetal liver. Likewise, for the ontogeny of enzymes and transporters in fetal tissues. Fourth, the ER-REF-PBPK approach is validated for only for P-gp substrates and has yet to be validated for other placental transporters. Fifth, fetal drug exposure estimated by m-f PBPK models at term has been validated for only a limited number of P-gp substate drugs due to paucity of the necessary UV/MP data for such drugs. For the same reasons, fetal tissue drug concentration prediction by the m-f PBPK models cannot be validated. Sixth, the current m-f PBPK models do not incorporate potential absorption of drugs from the amniotic fluid through the skin, which could be important earlier in gestation when the fetal skin is highly permeable.120
It is important to emphasize that, besides factors listed above, fetal drug exposure is also driven by maternal exposure. Therefore, the absolute magnitude of fetal drug exposure cannot be predicted without accurately predicting maternal drug exposure. This can be done using our m-f PBPK model as we have previously shown6, 8–11, 79, 90, 91, if relevant data on transporter and enzyme abundance are available for both healthy pregnant women and those with diseases (e.g. hepatic impairment). In this regard, the impact of pregnancy on the abundance and activity of non-CYP enzymes (such as UGTs, SULTs, carboxylesterases) and hepatic transporters (such as SLC and ABC transporters) are lacking for both healthy pregnant women and those with diseases. Moreover, such data for CYPs are limited to a few isoforms (e.g., CYP1A2, CYP2D6 and CYP3A4) and, where available, they are available mostly for the 3rd trimester (healthy pregnant women only).121, 122 The effect of pregnancy on activity or abundance of metabolic enzymes and transporters in non-hepatic tissues of pregnant women such as intestine, are not available. Such data are particularly important for drugs that are metabolized or transported in the intestine such as CYP3A and BCRP. In contrast, data are available about the effect of pregnancy on renal transporters.123–125 Thus, targeted enzyme and transporter probe drug studies in pregnant women (at all gestational ages) are needed to fill these knowledge gaps to predict both maternal and fetal drug exposure without conducting in vivo studies for every drug administered to pregnant women.
Despite the above gaps, there has been enormous progress in predicting maternal-fetal drug exposure. Mechanistic m-f PBPK models have been developed, populated with much of the relevant physiological data. These model, together with newly devised or refined approaches (e.g. ER-REF), have successfully predicted dynamic fetal exposure to drugs that cross the placenta passively79 and actively90, 91. The next frontier in this area of research is to predict and validate fetal tissue drug exposure. We have already shown that the ER-REF approach is successful in predicting human adult brain and liver drug concentrations where the drugs are actively transported across the tissue:blood barrier.126, 127 Thus, there is every reason to believe that this approach will be successful in predicting fetal tissue drug exposure provided the relevant data on the ontogeny of transporters and enzymes in these tissues are available.
Data Sharing
Since this is a review paper, there are no original data to share.
Funding statement
This work was supported by the National Institutes of Health grant P01DA032507 (JDU), the Bill and Melinda Gates foundation grant INV-006678 (AB, JDU), and the National Institutes of Health grant TL1TR002318 (ARK).
Footnotes
Conflicts of Interest
Authors declare no conflict of interest.
References
- 1.Mitchell AA, Gilboa SM, Werler MM, et al. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol. 2011;205(1): 51 e51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scaffidi J, Mol BW, Keelan JA. The pregnant women as a drug orphan: a global survey of registered clinical trials of pharmacological interventions in pregnancy. BJOG. 2017;124(1): 132–140. [DOI] [PubMed] [Google Scholar]
- 3.Shields KE, Lyerly AD. Exclusion of pregnant women from industry-sponsored clinical trials. Obstet Gynecol. 2013;122(5): 1077–1081. [DOI] [PubMed] [Google Scholar]
- 4.Carey JL, Nader N, Chai PR, Carreiro S, Griswold MK, Boyle KL. Drugs and Medical Devices: Adverse Events and the Impact on Women’s Health. Clin Ther. 2017;39(1): 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JH, Scialli AR. Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol Sci. 2011;122(1): 1–6. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Imperial MZ, Patilea-Vrana GI, Wedagedera J, Gaohua L, Unadkat JD. Development of a Novel Maternal-Fetal Physiologically Based Pharmacokinetic Model I: Insights into Factors that Determine Fetal Drug Exposure through Simulations and Sensitivity Analyses. Drug Metab Dispos. 2017;45(8): 920–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones H, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos. 2013;41(4): 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. A PBPK Model to Predict Disposition of CYP3A-Metabolized Drugs in Pregnant Women: Verification and Discerning the Site of CYP3A Induction. CPT Pharmacometrics Syst Pharmacol. 2012;1: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br J Clin Pharmacol. 2014;77(3): 554–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patilea-Vrana GI, Unadkat JD. Development and Verification of a Linked Delta (9)-THC/11-OH-THC Physiologically Based Pharmacokinetic Model in Healthy, Nonpregnant Population and Extrapolation to Pregnant Women. Drug Metab Dispos. 2021;49(7): 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol. 2012;74(5): 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia B, Heimbach T, Gollen R, Nanavati C, He H. A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pregnancy. AAPS J. 2013;15(4): 1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szeto KX, Le Merdy M, Dupont B, Bolger MB, Lukacova V. PBPK Modeling Approach to Predict the Behavior of Drugs Cleared by Kidney in Pregnant Subjects and Fetus. AAPS J. 2021;23(4): 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dallmann A, Ince I, Solodenko J, et al. Physiologically Based Pharmacokinetic Modeling of Renally Cleared Drugs in Pregnant Women. Clin Pharmacokinet. 2017;56(12): 1525–1541. [DOI] [PubMed] [Google Scholar]
- 16.Dallmann A, Ince I, Coboeken K, Eissing T, Hempel G. A Physiologically Based Pharmacokinetic Model for Pregnant Women to Predict the Pharmacokinetics of Drugs Metabolized Via Several Enzymatic Pathways. Clin Pharmacokinet. 2018;57(6): 749–768. [DOI] [PubMed] [Google Scholar]
- 17.Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol. 2000;157(6): 2111–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogias KJ, Pederson SM, Leemaqz S, et al. Placental Transcription Profiling in 6–23 Weeks’ Gestation Reveals Differential Transcript Usage in Early Development. Int J Mol Sci. 2022;23(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss G, Sundl M, Glasner A, Huppertz B, Moser G. The trophoblast plug during early pregnancy: a deeper insight. Histochem Cell Biol. 2016;146(6): 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al-Enazy S, Ali S, Albekairi N, El-Tawil M, Rytting E. Placental control of drug delivery. Adv Drug Deliv Rev. 2017;116: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathias AA, Hitti J, Unadkat JD. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am J Physiol Regul Integr Comp Physiol. 2005;289(4): R963–969. [DOI] [PubMed] [Google Scholar]
- 22.Anoshchenko O, Prasad B, Neradugomma NK, Wang J, Mao Q, Unadkat JD. Gestational Age-Dependent Abundance of Human Placental Transporters as Determined by Quantitative Targeted Proteomics. Drug Metab Dispos. 2020;48(9): 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joshi AA, Vaidya SS, St-Pierre MV, et al. Placental ABC Transporters: Biological Impact and Pharmaceutical Significance. Pharm Res. 2016;33(12): 2847–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dallmann A, Liu XI, Burckart GJ, van den Anker J. Drug Transporters Expressed in the Human Placenta and Models for Studying Maternal-Fetal Drug Transfer. J Clin Pharmacol. 2019;59 Suppl 1: S70–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ni Z, Mao Q. ATP-binding cassette efflux transporters in human placenta. Curr Pharm Biotechnol. 2011;12(4): 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ushigome F, Takanaga H, Matsuo H, et al. Human placental transport of vinblastine, vincristine, digoxin and progesterone: contribution of P-glycoprotein. Eur J Pharmacol. 2000;408(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 27.Pavek P, Fendrich Z, Staud F, et al. Influence of P-glycoprotein on the transplacental passage of cyclosporine. J Pharm Sci. 2001;90(10): 1583–1592. [DOI] [PubMed] [Google Scholar]
- 28.Smit JW, Huisman MT, van Tellingen O, Wiltshire HR, Schinkel AH. Absence or pharmacological blocking of placental P-glycoprotein profoundly increases fetal drug exposure. J Clin Invest. 1999;104(10): 1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eyal S, Chung FS, Muzi M, et al. Simultaneous PET imaging of P-glycoprotein inhibition in multiple tissues in the pregnant nonhuman primate. J Nucl Med. 2009;50(5): 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolwankar D, Glover DD, Ware JA, Tracy TS. Expression and function of ABCB1 and ABCG2 in human placental tissue. Drug Metab Dispos. 2005;33(4): 524–529. [DOI] [PubMed] [Google Scholar]
- 31.Gedeon C, Anger G, Piquette-Miller M, Koren G. Breast cancer resistance protein: mediating the trans-placental transfer of glyburide across the human placenta. Placenta. 2008;29(1): 39–43. [DOI] [PubMed] [Google Scholar]
- 32.Myllynen P, Kummu M, Kangas T, et al. ABCG2/BCRP decreases the transfer of a food-born chemical carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in perfused term human placenta. Toxicol Appl Pharmacol. 2008;232(2): 210–217. [DOI] [PubMed] [Google Scholar]
- 33.Pollex E, Lubetsky A, Koren G. The role of placental breast cancer resistance protein in the efflux of glyburide across the human placenta. Placenta. 2008;29(8): 743–747. [DOI] [PubMed] [Google Scholar]
- 34.Enokizono J, Kusuhara H, Sugiyama Y. Effect of breast cancer resistance protein (Bcrp/Abcg2) on the disposition of phytoestrogens. Mol Pharmacol. 2007;72(4): 967–975. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Wang H, Unadkat JD, Mao Q. Breast cancer resistance protein 1 limits fetal distribution of nitrofurantoin in the pregnant mouse. Drug Metab Dispos. 2007;35(12): 2154–2158. [DOI] [PubMed] [Google Scholar]
- 36.Zhou L, Naraharisetti SB, Wang H, Unadkat JD, Hebert MF, Mao Q. The breast cancer resistance protein (Bcrp1/Abcg2) limits fetal distribution of glyburide in the pregnant mouse: an Obstetric-Fetal Pharmacology Research Unit Network and University of Washington Specialized Center of Research Study. Mol Pharmacol. 2008;73(3): 949–959. [DOI] [PubMed] [Google Scholar]
- 37.Mason CW, Buhimschi IA, Buhimschi CS, Dong Y, Weiner CP, Swaan PW. ATP-binding cassette transporter expression in human placenta as a function of pregnancy condition. Drug Metab Dispos. 2011;39(6): 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lye P, Bloise E, Javam M, Gibb W, Lye SJ, Matthews SG. Impact of bacterial and viral challenge on multidrug resistance in first- and third-trimester human placenta. Am J Pathol. 2015;185(6): 1666–1675. [DOI] [PubMed] [Google Scholar]
- 39.Sun M, Kingdom J, Baczyk D, Lye SJ, Matthews SG, Gibb W. Expression of the multidrug resistance P-glycoprotein, (ABCB1 glycoprotein) in the human placenta decreases with advancing gestation. Placenta. 2006;27(6–7): 602–609. [DOI] [PubMed] [Google Scholar]
- 40.Gil S, Saura R, Forestier F, Farinotti R. P-glycoprotein expression of the human placenta during pregnancy. Placenta. 2005;26(2–3): 268–270. [DOI] [PubMed] [Google Scholar]
- 41.Yeboah D, Sun M, Kingdom J, et al. Expression of breast cancer resistance protein (BCRP/ABCG2) in human placenta throughout gestation and at term before and after labor. Can J Physiol Pharmacol. 2006;84(12): 1251–1258. [DOI] [PubMed] [Google Scholar]
- 42.Petrovic V, Kojovic D, Cressman A, Piquette-Miller M. Maternal bacterial infections impact expression of drug transporters in human placenta. Int Immunopharmacol. 2015;26(2): 349–356. [DOI] [PubMed] [Google Scholar]
- 43.Patel P, Weerasekera N, Hitchins M, Boyd CA, Johnston DG, Williamson C. Semi quantitative expression analysis of MDR3, FIC1, BSEP, OATP-A, OATP-C,OATP-D, OATP-E and NTCP gene transcripts in 1st and 3rd trimester human placenta. Placenta. 2003;24(1): 39–44. [DOI] [PubMed] [Google Scholar]
- 44.Grube M, Meyer Zu Schwabedissen H, Draber K, et al. Expression, localization, and function of the carnitine transporter octn2 (slc22a5) in human placenta. Drug Metab Dispos. 2005;33(1): 31–37. [DOI] [PubMed] [Google Scholar]
- 45.Chang TT, Shyu MK, Huang MC, et al. Hypoxia-mediated down-regulation of OCTN2 and PPARalpha expression in human placentas and in BeWo cells. Mol Pharm. 2011;8(1): 117–125. [DOI] [PubMed] [Google Scholar]
- 46.Pascolo L, Fernetti C, Pirulli D, Crovella S, Amoroso A, Tiribelli C. Effects of maturation on RNA transcription and protein expression of four MRP genes in human placenta and in BeWo cells. Biochem Biophys Res Commun. 2003;303(1): 259–265. [DOI] [PubMed] [Google Scholar]
- 47.Meyer zu Schwabedissen HE, Jedlitschky G, Gratz M, et al. Variable expression of MRP2 (ABCC2) in human placenta: influence of gestational age and cellular differentiation. Drug Metab Dispos. 2005;33(7): 896–904. [DOI] [PubMed] [Google Scholar]
- 48.Ahmadimoghaddam D, Zemankova L, Nachtigal P, et al. Organic cation transporter 3 (OCT3/SLC22A3) and multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter in the placenta and fetal tissues: expression profile and fetus protective role at different stages of gestation. Biol Reprod. 2013;88(3): 55. [DOI] [PubMed] [Google Scholar]
- 49.Lee N, Hebert MF, Prasad B, et al. Effect of gestational age on mRNA and protein expression of polyspecific organic cation transporters during pregnancy. Drug Metab Dispos. 2013;41(12): 2225–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hodyl NA, Stark MJ, Butler M, Clifton VL. Placental P-glycoprotein is unaffected by timing of antenatal glucocorticoid therapy but reduced in SGA preterm infants. Placenta. 2013;34(4): 325–330. [DOI] [PubMed] [Google Scholar]
- 51.Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi. 2003;123(5): 369–375. [DOI] [PubMed] [Google Scholar]
- 52.Sawada M, Kitamura R, Norose T, Kitada M, Itahashi K, Kamataki T. Metabolic activation of aflatoxin B1 by human placental microsomes. J Toxicol Sci. 1993;18(2): 129–132. [DOI] [PubMed] [Google Scholar]
- 53.Zharikova OL, Fokina VM, Nanovskaya TN, et al. Identification of the major human hepatic and placental enzymes responsible for the biotransformation of glyburide. Biochem Pharmacol. 2009;78(12): 1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deshmukh SV, Nanovskaya TN, Ahmed MS. Aromatase is the major enzyme metabolizing buprenorphine in human placenta. J Pharmacol Exp Ther. 2003;306(3): 1099–1105. [DOI] [PubMed] [Google Scholar]
- 55.Nanovskaya TN, Deshmukh SV, Nekhayeva IA, Zharikova OL, Hankins GD, Ahmed MS. Methadone metabolism by human placenta. Biochem Pharmacol. 2004;68(3): 583–591. [DOI] [PubMed] [Google Scholar]
- 56.Collier AC, Keelan JA, Van Zijl PE, Paxton JW, Mitchell MD, Tingle MD. Human placental glucuronidation and transport of 3’azido-3’-deoxythymidine and uridine diphosphate glucuronic acid. Drug Metab Dispos. 2004;32(8): 813–820. [DOI] [PubMed] [Google Scholar]
- 57.Ravindran S, Zharikova OL, Hill RA, Nanovskaya TN, Hankins GD, Ahmed MS. Identification of glyburide metabolites formed by hepatic and placental microsomes of humans and baboons. Biochem Pharmacol. 2006;72(12): 1730–1737. [DOI] [PubMed] [Google Scholar]
- 58.Myllynen P, Pienimaki P, Raunio H, Vahakangas K. Microsomal metabolism of carbamazepine and oxcarbazepine in liver and placenta. Hum Exp Toxicol. 1998;17(12): 668–676. [DOI] [PubMed] [Google Scholar]
- 59.Rettie AE, Heimark L, Mayer RT, Burke MD, Trager WF, Juchau MR. Stereoselective and regioselective hydroxylation of warfarin and selective O-dealkylation of phenoxazone ethers in human placenta. Biochem Biophys Res Commun. 1985;126(3): 1013–1021. [DOI] [PubMed] [Google Scholar]
- 60.Earhart AD, Patrikeeva S, Wang X, et al. Transplacental transfer and metabolism of bupropion. J Matern Fetal Neonatal Med. 2010;23(5): 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Myllynen P, Pienimaki P, Jouppila P, Vahakangas K. Transplacental passage of oxcarbazepine and its metabolites in vivo. Epilepsia. 2001;42(11): 1482–1485. [DOI] [PubMed] [Google Scholar]
- 62.Nanovskaya T, Deshmukh S, Brooks M, Ahmed MS. Transplacental transfer and metabolism of buprenorphine. J Pharmacol Exp Ther. 2002;300(1): 26–33. [DOI] [PubMed] [Google Scholar]
- 63.De Sousa Mendes M, Hirt D, Vinot C, et al. Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br J Clin Pharmacol. 2016;81(4): 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hakkola J, Pasanen M, Hukkanen J, et al. Expression of xenobiotic-metabolizing cytochrome P450 forms in human full-term placenta. Biochem Pharmacol. 1996;51(4): 403–411. [DOI] [PubMed] [Google Scholar]
- 65.Collier AC, Ganley NA, Tingle MD, et al. UDP-glucuronosyltransferase activity, expression and cellular localization in human placenta at term. Biochem Pharmacol. 2002;63(3): 409–419. [DOI] [PubMed] [Google Scholar]
- 66.Stejskalova L, Pavek P. The function of cytochrome P450 1A1 enzyme (CYP1A1) and aryl hydrocarbon receptor (AhR) in the placenta. Curr Pharm Biotechnol 2011;12(5): 715–730. [DOI] [PubMed] [Google Scholar]
- 67.Collier AC, Tingle MD, Paxton JW, Mitchell MD, Keelan JA. Metabolizing enzyme localization and activities in the first trimester human placenta: the effect of maternal and gestational age, smoking and alcohol consumption. Hum Reprod. 2002;17(10): 2564–2572. [DOI] [PubMed] [Google Scholar]
- 68.Huuskonen P, Amezaga MR, Bellingham M, et al. The human placental proteome is affected by maternal smoking. Reprod Toxicol. 2016;63: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet. 1999;37(6): 485–505. [DOI] [PubMed] [Google Scholar]
- 70.Hines RN. Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol. 2007;21(4): 169–175. [DOI] [PubMed] [Google Scholar]
- 71.Richard K, Hume R, Kaptein E, Stanley EL, Visser TJ, Coughtrie MW. Sulfation of thyroid hormone and dopamine during human development: ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J Clin Endocrinol Metab. 2001;86(6): 2734–2742. [DOI] [PubMed] [Google Scholar]
- 72.Stanley EL, Hume R, Coughtrie MW. Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol Cell Endocrinol. 2005;240(1–2): 32–42. [DOI] [PubMed] [Google Scholar]
- 73.Duanmu Z, Weckle A, Koukouritaki SB, et al. Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre- and postnatal human liver. J Pharmacol Exp Ther. 2006;316(3): 1310–1317. [DOI] [PubMed] [Google Scholar]
- 74.Barker EV, Hume R, Hallas A, Coughtrie WH. Dehydroepiandrosterone sulfotransferase in the developing human fetus: quantitative biochemical and immunological characterization of the hepatic, renal, and adrenal enzymes. Endocrinology. 1994;134(2): 982–989. [DOI] [PubMed] [Google Scholar]
- 75.Hobkirk R Steroid sulfotransferases and steroid sulfate sulfatases: characteristics and biological roles. Can J Biochem Cell Biol. 1985;63(11): 1127–1144. [DOI] [PubMed] [Google Scholar]
- 76.Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet. 2004;43(8): 487–514. [DOI] [PubMed] [Google Scholar]
- 77.Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: not just fetal urine anymore. J Perinatol. 2005;25(5): 341–348. [DOI] [PubMed] [Google Scholar]
- 78.Myllynen P, Immonen E, Kummu M, Vahakangas K. Developmental expression of drug metabolizing enzymes and transporter proteins in human placenta and fetal tissues. Expert Opin Drug Metab Toxicol. 2009;5(12): 1483–1499. [DOI] [PubMed] [Google Scholar]
- 79.Zhang Z, Unadkat JD. Development of a Novel Maternal-Fetal Physiologically Based Pharmacokinetic Model II: Verification of the model for passive placental permeability drugs. Drug Metab Dispos. 2017;45(8): 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krauer B, Dayer P, Anner R. Changes in serum albumin and alpha 1-acid glycoprotein concentrations during pregnancy: an analysis of fetal-maternal pairs. Br J Obstet Gynaecol. 1984;91(9): 875–881. [DOI] [PubMed] [Google Scholar]
- 81.Liao MZ, Gao C, Shireman LM, et al. P-gp/ABCB1 exerts differential impacts on brain and fetal exposure to norbuprenorphine. Pharmacol Res. 2017;119: 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shuster DL, Risler LJ, Liang CK, et al. Maternal-fetal disposition of glyburide in pregnant mice is dependent on gestational age. J Pharmacol Exp Ther. 2014;350(2): 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grigsby PL. Animal Models to Study Placental Development and Function throughout Normal and Dysfunctional Human Pregnancy. Semin Reprod Med. 2016;34(1): 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tuntland T, Odinecs A, Pereira CM, Nosbisch C, Unadkat JD. In vitro models to predict the in vivo mechanism, rate, and extent of placental transfer of dideoxynucleoside drugs against human immunodeficiency virus. Am J Obstet Gynecol. 1999;180(1 Pt 1): 198–206. [DOI] [PubMed] [Google Scholar]
- 85.Carter AM. Animal models of human placentation--a review. Placenta. 2007;28 Suppl A: S41–47. [DOI] [PubMed] [Google Scholar]
- 86.Carter AM. Unique Aspects of Human Placentation. Int J Mol Sci. 2021;22(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Furukawa S, Kuroda Y, Sugiyama A. A comparison of the histological structure of the placenta in experimental animals. J Toxicol Pathol. 2014;27(1): 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fujita A, Noguchi S, Hamada R, et al. Limited Impact of Murine Placental MDR1 on Fetal Exposure of Certain Drugs Explained by Bypass Transfer Between Adjacent Syncytiotrophoblast Layers. Pharm Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dallmann A, Pfister M, van den Anker J, Eissing T. Physiologically Based Pharmacokinetic Modeling in Pregnancy: A Systematic Review of Published Models. Clin Pharmacol Ther. 2018;104(6): 1110–1124. [DOI] [PubMed] [Google Scholar]
- 90.Anoshchenko O, Milad MA, Unadkat JD. Estimating fetal exposure to the P-gp substrates, corticosteroids, by PBPK modeling to inform prevention of neonatal respiratory distress syndrome. CPT Pharmacometrics Syst Pharmacol. 2021;10(9): 1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anoshchenko O, Storelli F, Unadkat JD. Successful Prediction of Human Fetal Exposure to P-Glycoprotein Substrate Drugs Using the Proteomics-Informed Relative Expression Factor Approach and PBPK Modeling and Simulation. Drug Metab Dispos. 2021;49(10): 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51(6): 365–396. [DOI] [PubMed] [Google Scholar]
- 93.Abduljalil K, Jamei M, Johnson TN. Fetal Physiologically Based Pharmacokinetic Models: Systems Information on the Growth and Composition of Fetal Organs. Clin Pharmacokinet. 2019;58(2): 235–262. [DOI] [PubMed] [Google Scholar]
- 94.Abduljalil K, Jamei M, Johnson TN. Fetal Physiologically Based Pharmacokinetic Models: Systems Information on Fetal Blood Components and Binding Proteins. Clin Pharmacokinet. 2020;59(5): 629–642. [DOI] [PubMed] [Google Scholar]
- 95.Abduljalil K, Johnson TN, Rostami-Hodjegan A. Fetal Physiologically-Based Pharmacokinetic Models: Systems Information on Fetal Biometry and Gross Composition. Clin Pharmacokinet. 2018;57(9): 1149–1171. [DOI] [PubMed] [Google Scholar]
- 96.Abduljalil K, Pan X, Clayton R, Johnson TN, Jamei M. Fetal Physiologically Based Pharmacokinetic Models: Systems Information on Fetal Cardiac Output and Its Distribution to Different Organs during Development. Clin Pharmacokinet. 2021;60(6): 741–757. [DOI] [PubMed] [Google Scholar]
- 97.Ezuruike U, Blenkinsop A, Pansari A, Abduljalil K. Quantification of Fetal Renal Function Using Fetal Urine Production Rate and Its Reflection on the Amniotic and Fetal Creatinine Levels During Pregnancy. Front Pediatr. 2022;10: 841495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dallmann A, Ince I, Meyer M, Willmann S, Eissing T, Hempel G. Gestation-Specific Changes in the Anatomy and Physiology of Healthy Pregnant Women: An Extended Repository of Model Parameters for Physiologically Based Pharmacokinetic Modeling in Pregnancy. Clin Pharmacokinet. 2017;56(11): 1303–1330. [DOI] [PubMed] [Google Scholar]
- 99.Kumar AR, Patilea-Vrana GI, Anoshchenko O, Unadkat JD. Characterizing and Quantifying Extrahepatic Metabolism of (−)-Δ9-Tetrahydrocannabinol (THC) and Its Psychoactive Metabolite, 11-OH-THC. Drug Metab Dispos. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kumar V, Salphati L, Hop C, et al. A Comparison of Total and Plasma Membrane Abundance of Transporters in Suspended, Plated, Sandwich-Cultured Human Hepatocytes Versus Human Liver Tissue Using Quantitative Targeted Proteomics and Cell Surface Biotinylation. Drug Metab Dispos. 2019;47(4): 350–357. [DOI] [PubMed] [Google Scholar]
- 101.Ikeda K, Utoguchi N, Tsutsui H, et al. In vitro approaches to evaluate placental drug transport by using differentiating JEG-3 human choriocarcinoma cells. Basic Clin Pharmacol Toxicol. 2011;108(2): 138–145. [DOI] [PubMed] [Google Scholar]
- 102.Poulsen MS, Rytting E, Mose T, Knudsen LE. Modeling placental transport: correlation of in vitro BeWo cell permeability and ex vivo human placental perfusion. Toxicol In Vitro. 2009;23(7): 1380–1386. [DOI] [PubMed] [Google Scholar]
- 103.Hiden U, Wadsack C, Prutsch N, et al. The first trimester human trophoblast cell line ACH-3P: a novel tool to study autocrine/paracrine regulatory loops of human trophoblast subpopulations--TNF-alpha stimulates MMP15 expression. BMC Dev Biol. 2007;7: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Evseenko DA, Paxton JW, Keelan JA. ABC drug transporter expression and functional activity in trophoblast-like cell lines and differentiating primary trophoblast. Am J Physiol Regul Integr Comp Physiol. 2006;290(5): R1357–1365. [DOI] [PubMed] [Google Scholar]
- 105.Serrano MA, Macias RI, Briz O, et al. Expression in human trophoblast and choriocarcinoma cell lines, BeWo, Jeg-3 and JAr of genes involved in the hepatobiliary-like excretory function of the placenta. Placenta. 2007;28(2–3): 107–117. [DOI] [PubMed] [Google Scholar]
- 106.Ceckova M, Libra A, Pavek P, et al. Expression and functional activity of breast cancer resistance protein (BCRP, ABCG2) transporter in the human choriocarcinoma cell line BeWo. Clin Exp Pharmacol Physiol. 2006;33(1–2): 58–65. [DOI] [PubMed] [Google Scholar]
- 107.Mark PJ, Waddell BJ. P-glycoprotein restricts access of cortisol and dexamethasone to the glucocorticoid receptor in placental BeWo cells. Endocrinology. 2006;147(11): 5147–5152. [DOI] [PubMed] [Google Scholar]
- 108.Feinshtein V, Holcberg G, Amash A, et al. Nitrofurantoin transport by placental choriocarcinoma JAr cells: involvement of BCRP, OATP2B1 and other MDR transporters. Arch Gynecol Obstet. 2010;281(6): 1037–1044. [DOI] [PubMed] [Google Scholar]
- 109.Rothbauer M, Patel N, Gondola H, Siwetz M, Huppertz B, Ertl P. A comparative study of five physiological key parameters between four different human trophoblast-derived cell lines. Sci Rep. 2017;7(1): 5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Heaton SJ, Eady JJ, Parker ML, et al. The use of BeWo cells as an in vitro model for placental iron transport. Am J Physiol Cell Physiol. 2008;295(5): C1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Panigel M, Pascaud M, Brun JL. [Radioangiographic study of circulation in the villi and intervillous space of isolated human placental cotyledon kept viable by perfusion]. J Physiol (Paris). 1967;59(1 Suppl): 277. [PubMed] [Google Scholar]
- 112.Schneider H, Brownbill P, Albrecht C. Ex vivo dual perfusion of an isolated cotyledon of human placenta: History and future challenges. Placenta. 2021;107: 8–12. [DOI] [PubMed] [Google Scholar]
- 113.Reed LC, Estrada SM, Walton RB, Napolitano PG, Ieronimakis N. Evaluating maternal hyperglycemic exposure and fetal placental arterial dysfunction in a dual cotyledon, dual perfusion model. Placenta. 2018;69: 109–116. [DOI] [PubMed] [Google Scholar]
- 114.Kurosawa K, Chiba K, Noguchi S, Nishimura T, Tomi M. Development of a Pharmacokinetic Model of Transplacental Transfer of Metformin to Predict In Vivo Fetal Exposure. Drug Metab Dispos. 2020;48(12): 1293–1302. [DOI] [PubMed] [Google Scholar]
- 115.Nekhayeva IA, Nanovskaya TN, Deshmukh SV, Zharikova OL, Hankins GD, Ahmed MS. Bidirectional transfer of methadone across human placenta. Biochem Pharmacol. 2005;69(1): 187–197. [DOI] [PubMed] [Google Scholar]
- 116.Mian P, Allegaert K, Conings S, et al. Integration of Placental Transfer in a Fetal-Maternal Physiologically Based Pharmacokinetic Model to Characterize Acetaminophen Exposure and Metabolic Clearance in the Fetus. Clin Pharmacokinet. 2020;59(7): 911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Freriksen JJM, Schalkwijk S, Colbers AP, et al. Assessment of Maternal and Fetal Dolutegravir Exposure by Integrating Ex Vivo Placental Perfusion Data and Physiologically-Based Pharmacokinetic Modeling. Clin Pharmacol Ther. 2020;107(6): 1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Russo FM, De Bie F, Hodges R, Flake A, Deprest J. Sildenafil for Antenatal Treatment of Congenital Diaphragmatic Hernia: From Bench to Bedside. Curr Pharm Des. 2019;25(5): 601–608. [DOI] [PubMed] [Google Scholar]
- 119.Schalkwijk S, Buaben AO, Freriksen JJM, et al. Prediction of Fetal Darunavir Exposure by Integrating Human Ex-Vivo Placental Transfer and Physiologically Based Pharmacokinetic Modeling. Clin Pharmacokinet. 2018;57(6): 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Polin RA, Fox WW, Abman SH. Fetal and neonatal physiology. 3rd ed. Philadelphia, Pa.: W.B. Saunders Co.; 2004. [Google Scholar]
- 121.Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10): 989–1008. [DOI] [PubMed] [Google Scholar]
- 122.Abduljalil K, Pansari A, Jamei M. Prediction of maternal pharmacokinetics using physiologically based pharmacokinetic models: assessing the impact of the longitudinal changes in the activity of CYP1A2, CYP2D6 and CYP3A4 enzymes during pregnancy. J Pharmacokinet Pharmacodyn. 2020;47(4): 361–383. [DOI] [PubMed] [Google Scholar]
- 123.Peng J, Ladumor MK, Unadkat JD. Prediction of Pregnancy-Induced Changes in Secretory and Total Renal Clearance of Drugs Transported by Organic Anion Transporters. Drug Metab Dispos. 2021;49(10): 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bergagnini-Kolev MC, Hebert MF, Easterling TR, Lin YS. Pregnancy Increases the Renal Secretion of N(1)-methylnicotinamide, an Endogenous Probe for Renal Cation Transporters, in Patients Prescribed Metformin. Drug Metab Dispos. 2017;45(3): 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hebert MF, Easterling TR, Kirby B, et al. Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin Pharmacol Ther. 2008;84(2): 248–253. [DOI] [PubMed] [Google Scholar]
- 126.Storelli F, Anoshchenko O, Unadkat JD. Successful Prediction of Human Steady-State Unbound Brain-to-Plasma Concentration Ratio of P-gp Substrates Using the Proteomics-Informed Relative Expression Factor Approach. Clin Pharmacol Ther. 2021;110(2): 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Storelli F, Li CY, Sachar M, et al. Prediction of Hepatobiliary Clearances and Hepatic Concentrations of Transported Drugs in Humans Using Rosuvastatin as a Model Drug. Clin Pharmacol Ther. 2022. [DOI] [PubMed] [Google Scholar]
- 128.Lye P, Bloise E, Dunk C, et al. Effect of oxygen on multidrug resistance in the first trimester human placenta. Placenta. 2013;34(9): 817–823. [DOI] [PubMed] [Google Scholar]
- 129.Tupova L, Ceckova M, Ambrus C, et al. Interactions between Maraviroc and the ABCB1, ABCG2, and ABCC2 Transporters: An Important Role in Transplacental Pharmacokinetics. Drug Metab Dispos. 2019;47(9): 954–960. [DOI] [PubMed] [Google Scholar]
- 130.Neumanova Z, Cerveny L, Ceckova M, Staud F. Interactions of tenofovir and tenofovir disoproxil fumarate with drug efflux transporters ABCB1, ABCG2, and ABCC2; role in transport across the placenta. AIDS. 2014;28(1): 9–17. [DOI] [PubMed] [Google Scholar]
- 131.May K, Minarikova V, Linnemann K, et al. Role of the multidrug transporter proteins ABCB1 and ABCC2 in the diaplacental transport of talinolol in the term human placenta. Drug Metab Dispos. 2008;36(4): 740–744. [DOI] [PubMed] [Google Scholar]
- 132.Afrouzian M, Al-Lahham R, Patrikeeva S, et al. Role of the efflux transporters BCRP and MRP1 in human placental bio-disposition of pravastatin. Biochem Pharmacol. 2018;156: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nagashige M, Ushigome F, Koyabu N, et al. Basal membrane localization of MRP1 in human placental trophoblast. Placenta. 2003;24(10): 951–958. [DOI] [PubMed] [Google Scholar]
- 134.Atkinson DE, Greenwood SL, Sibley CP, Glazier JD, Fairbairn LJ. Role of MDR1 and MRP1 in trophoblast cells, elucidated using retroviral gene transfer. Am J Physiol Cell Physiol. 2003;285(3): C584–591. [DOI] [PubMed] [Google Scholar]
- 135.Zeng H, Chen ZS, Belinsky MG, Rea PA, Kruh GD. Transport of methotrexate (MTX) and folates by multidrug resistance protein (MRP) 3 and MRP1: effect of polyglutamylation on MTX transport. Cancer Res. 2001;61(19): 7225–7232. [PubMed] [Google Scholar]
- 136.St-Pierre MV, Serrano MA, Macias RI, et al. Expression of members of the multidrug resistance protein family in human term placenta. Am J Physiol Regul Integr Comp Physiol. 2000;279(4): R1495–1503. [DOI] [PubMed] [Google Scholar]
- 137.Azzaroli F, Mennone A, Feletti V, et al. Clinical trial: modulation of human placental multidrug resistance proteins in cholestasis of pregnancy by ursodeoxycholic acid. Aliment Pharmacol Ther. 2007;26(8): 1139–1146. [DOI] [PubMed] [Google Scholar]
- 138.Meyer Zu Schwabedissen HE, Grube M, Heydrich B, et al. Expression, localization, and function of MRP5 (ABCC5), a transporter for cyclic nucleotides, in human placenta and cultured human trophoblasts: effects of gestational age and cellular differentiation. Am J Pathol. 2005;166(1): 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Loubiere LS, Vasilopoulou E, Bulmer JN, et al. Expression of thyroid hormone transporters in the human placenta and changes associated with intrauterine growth restriction. Placenta. 2010;31(4): 295–304. [DOI] [PubMed] [Google Scholar]
- 140.Briz O, Serrano MA, MacIas RI, Gonzalez-Gallego J, Marin JJ. Role of organic anion-transporting polypeptides, OATP-A, OATP-C and OATP-8, in the human placenta-maternal liver tandem excretory pathway for foetal bilirubin. Biochem J. 2003;371(Pt 3): 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ugele B, St-Pierre MV, Pihusch M, Bahn A, Hantschmann P. Characterization and identification of steroid sulfate transporters of human placenta. Am J Physiol Endocrinol Metab. 2003;284(2): E390–398. [DOI] [PubMed] [Google Scholar]
- 142.Lee N, Hebert MF, Wagner DJ, et al. Organic Cation Transporter 3 Facilitates Fetal Exposure to Metformin during Pregnancy. Mol Pharmacol. 2018;94(4): 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sata R, Ohtani H, Tsujimoto M, et al. Functional analysis of organic cation transporter 3 expressed in human placenta. J Pharmacol Exp Ther. 2005;315(2): 888–895. [DOI] [PubMed] [Google Scholar]
- 144.Karahoda R, Horackova H, Kastner P, et al. Serotonin homeostasis in the materno-foetal interface at term: Role of transporters (SERT/SLC6A4 and OCT3/SLC22A3) and monoamine oxidase A (MAO-A) in uptake and degradation of serotonin by human and rat term placenta. Acta Physiol (Oxf). 2020;229(4): e13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Karahoda R, Ceckova M, Staud F. The inhibitory effect of antiretroviral drugs on the L-carnitine uptake in human placenta. Toxicol Appl Pharmacol. 2019;368: 18–25. [DOI] [PubMed] [Google Scholar]
- 146.Cha SH, Sekine T, Kusuhara H, et al. Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J Biol Chem. 2000;275(6): 4507–4512. [DOI] [PubMed] [Google Scholar]
- 147.Noguchi S, Nishimura T, Fujibayashi A, Maruyama T, Tomi M, Nakashima E. Organic Anion Transporter 4-Mediated Transport of Olmesartan at Basal Plasma Membrane of Human Placental Barrier. J Pharm Sci. 2015;104(9): 3128–3135. [DOI] [PubMed] [Google Scholar]
- 148.Hakkola J, Raunio H, Purkunen R, et al. Detection of cytochrome P450 gene expression in human placenta in first trimester of pregnancy. Biochem Pharmacol. 1996;52(2): 379–383. [DOI] [PubMed] [Google Scholar]
- 149.Bieche I, Narjoz C, Asselah T, et al. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogenet Genomics. 2007;17(9): 731–742. [DOI] [PubMed] [Google Scholar]
- 150.Avery ML, Meek CE, Audus KL. The presence of inducible cytochrome P450 types 1A1 and 1A2 in the BeWo cell line. Placenta. 2003;24(1): 45–52. [DOI] [PubMed] [Google Scholar]
- 151.Yang HY, Namkung MJ, Juchau MR. Expression of functional cytochrome P4501A1 in human embryonic hepatic tissues during organogenesis. Biochem Pharmacol. 1995;49(5): 717–726. [DOI] [PubMed] [Google Scholar]
- 152.Cizkova K, Tauber Z. Time-dependent expression pattern of cytochrome P450 epoxygenases and soluble epoxide hydrolase in normal human placenta. Acta Histochem. 2018;120(6): 513–519. [DOI] [PubMed] [Google Scholar]
- 153.Treluyer JM, Gueret G, Cheron G, Sonnier M, Cresteil T. Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: in-vivo/in-vitro correlation and inducibility. Pharmacogenetics. 1997;7(6): 441–452. [DOI] [PubMed] [Google Scholar]
- 154.Hakkola J, Pasanen M, Purkunen R, et al. Expression of xenobiotic-metabolizing cytochrome P450 forms in human adult and fetal liver. Biochem Pharmacol. 1994;48(1): 59–64. [DOI] [PubMed] [Google Scholar]
- 155.Vyhlidal CA, Pearce RE, Gaedigk R, et al. Variability in Expression of CYP3A5 in Human Fetal Liver. Drug Metab Dispos. 2015;43(8): 1286–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Topletz AR, Zhong G, Isoherranen N. Scaling in vitro activity of CYP3A7 suggests human fetal livers do not clear retinoic acid entering from maternal circulation. Sci Rep. 2019;9(1): 4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Doody KJ, Carr BR. Aromatase in human fetal tissues. Am J Obstet Gynecol. 1989;161(6 Pt 1): 1694–1697. [DOI] [PubMed] [Google Scholar]
- 158.Mohammed AM, Huuskonen P, Juvonen R, et al. Activities of metabolizing enzymes in human placenta. Toxicol Lett. 2020;326: 70–77. [DOI] [PubMed] [Google Scholar]
- 159.Stanley EL, Hume R, Visser TJ, Coughtrie MW. Differential expression of sulfotransferase enzymes involved in thyroid hormone metabolism during human placental development. J Clin Endocrinol Metab. 2001;86(12): 5944–5955. [DOI] [PubMed] [Google Scholar]
- 160.Pacifici GM. Sulfation of drugs and hormones in mid-gestation human fetus. Early Hum Dev. 2005;81(7): 573–581. [DOI] [PubMed] [Google Scholar]
- 161.Dubaisi S, Barrett KG, Fang H, et al. Regulation of Cytosolic Sulfotransferases in Models of Human Hepatocyte Development. Drug Metab Dispos. 2018;46(8): 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Meloche CA, Falany CN. Expression and characterization of the human 3 beta-hydroxysteroid sulfotransferases (SULT2B1a and SULT2B1b). J Steroid Biochem Mol Biol. 2001;77(4–5): 261–269. [DOI] [PubMed] [Google Scholar]
- 163.Collier AC, Thevenon AD, Goh W, Hiraoka M, Kendal-Wright CE. Placental profiling of UGT1A enzyme expression and activity and interactions with preeclampsia at term. Eur J Drug Metab Pharmacokinet. 2015;40(4): 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Court MH, Zhang X, Ding X, Yee KK, Hesse LM, Finel M. Quantitative distribution of mRNAs encoding the 19 human UDP-glucuronosyltransferase enzymes in 26 adult and 3 fetal tissues. Xenobiotica. 2012;42(3): 266–277. [DOI] [PubMed] [Google Scholar]
- 165.Ekstrom L, Johansson M, Rane A. Tissue distribution and relative gene expression of UDP-glucuronosyltransferases (2B7, 2B15, 2B17) in the human fetus. Drug Metab Dispos. 2013;41(2): 291–295. [DOI] [PubMed] [Google Scholar]
- 166.Divakaran K, Hines RN, McCarver DG. Human hepatic UGT2B15 developmental expression. Toxicol Sci. 2014;141(1): 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Obolenskaya MY, Teplyuk NM, Divi RL, et al. Human placental glutathione S-transferase activity and polycyclic aromatic hydrocarbon DNA adducts as biomarkers for environmental oxidative stress in placentas from pregnant women living in radioactivity- and chemically-polluted regions. Toxicol Lett. 2010;196(2): 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pacifici GM, Franchi M, Colizzi C, Giuliani L, Rane A. Glutathione S-transferase in humans: development and tissue distribution. Arch Toxicol. 1988;61(4): 265–269. [DOI] [PubMed] [Google Scholar]
- 169.Mera N, Ohmori S, Itahashi K, et al. Immunochemical evidence for the occurrence of Mu class glutathione S-transferase in human fetal livers. J Biochem. 1994;116(2): 315–320. [DOI] [PubMed] [Google Scholar]
- 170.Auda GR, Kirk SH, Billett MA, Billett EE. Localization of monoamine oxidase mRNA in human placenta. J Histochem Cytochem. 1998;46(12): 1393–1400. [DOI] [PubMed] [Google Scholar]
- 171.Barnea ER, MacLusky NJ, DeCherney AH, Naftolin F. Monoamine oxidase activity in the term human placenta. Am J Perinatol. 1986;3(3): 219–224. [DOI] [PubMed] [Google Scholar]
- 172.Sivasubramaniam SD, Finch CC, Billett MA, Baker PN, Billett EE. Monoamine oxidase expression and activity in human placentae from pre-eclamptic and normotensive pregnancies. Placenta. 2002;23(2–3): 163–171. [DOI] [PubMed] [Google Scholar]
- 173.Jones JB, Papadaki L, Hubbard J. Monoamine oxidase activity in the fetal lung and liver. Br J Obstet Gynaecol. 1976;83(6): 464–469. [DOI] [PubMed] [Google Scholar]
- 174.Telfer JF, Thomson AJ, Cameron IT, Greer IA, Norman JE. Expression of superoxide dismutase and xanthine oxidase in myometrium, fetal membranes and placenta during normal human pregnancy and parturition. Hum Reprod. 1997;12(10): 2306–2312. [DOI] [PubMed] [Google Scholar]
- 175.Many A, Westerhausen-Larson A, Kanbour-Shakir A, Roberts JM. Xanthine oxidase/dehydrogenase is present in human placenta. Placenta. 1996;17(5–6): 361–365. [DOI] [PubMed] [Google Scholar]
- 176.Vettenranta K, Raivio KO. Xanthine oxidase during human fetal development. Pediatr Res. 1990;27(3): 286–288. [DOI] [PubMed] [Google Scholar]
- 177.Yan B, Matoney L, Yang D. Human carboxylesterases in term placentae: enzymatic characterization, molecular cloning and evidence for the existence of multiple forms. Placenta. 1999;20(7): 599–607. [DOI] [PubMed] [Google Scholar]
- 178.Lassiter TL, Barone S Jr., Moser VC, Padilla S. Gestational exposure to chlorpyrifos: dose response profiles for cholinesterase and carboxylesterase activity. Toxicol Sci. 1999;52(1): 92–100. [DOI] [PubMed] [Google Scholar]
- 179.Imai K, Kanzaki H, Fujiwara H, et al. Expression and localization of aminopeptidase N, neutral endopeptidase, and dipeptidyl peptidase IV in the human placenta and fetal membranes. Am J Obstet Gynecol. 1994;170(4): 1163–1168. [DOI] [PubMed] [Google Scholar]
- 180.Hung TH, Chen SF, Hsieh TT. Soluble epoxide hydrolase in the human placenta throughout gestation. Taiwan J Obstet Gynecol. 2019;58(6): 840–845. [DOI] [PubMed] [Google Scholar]
- 181.Pacifici GM, Temellini A, Giuliani L, Rane A, Thomas H, Oesch F. Cytosolic epoxide hydrolase in humans: development and tissue distribution. Arch Toxicol. 1988;62(4): 254–257. [DOI] [PubMed] [Google Scholar]