

SUMMARY
Integrins are validated drug targets with six approved therapeutics. However, small molecule inhibitors to three integrins failed in late-stage clinical trials for chronic indications. Such unfavorable outcomes may in part be caused by partial agonism, i.e., stabilization of the high-affinity, extended-open integrin conformation. Here we show that failed, small molecule inhibitors of integrins αIIbβ3 and α4β1 stabilize the high-affinity conformation. Furthermore, we discovered a simple chemical feature present in multiple αIIbβ3 antagonists that stabilizes integrins in their bent-closed conformation. Closing inhibitors contain a polar nitrogen atom that stabilizes, via hydrogen bonds, a water molecule that intervenes between a serine residue and the metal in the metal ion-dependent adhesion site (MIDAS), expulsion of which is requisite for transition to the open conformation. This change in metal coordination is general to integrins, suggesting broad applicability of the drug-design principle to the integrin family, as validated with a distantly related integrin, α4β1.
Keywords: integrins, drug discovery, conformational change, thrombosis, autoimmune disease, membrane receptors
Graphical Abstract
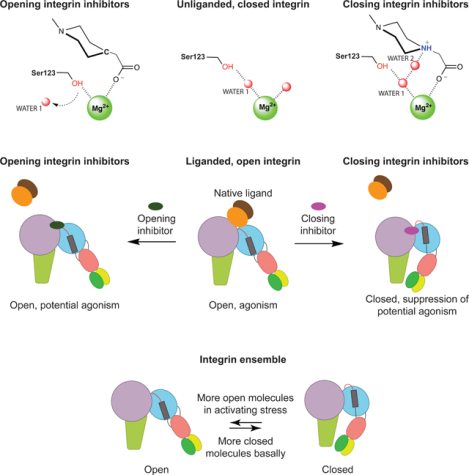
In brief:
The design principle for integrin inhibitors.
INTRODUCTION
In time and with water, everything changes. —Leonardo da Vinci
Integrins are attractive therapeutic targets with central functions in multiple diseases (Cox et al., 2010; Ley et al., 2016; Shimaoka and Springer, 2003). The 24 integrin αβ heterodimers have evolved highly specialized functions. Antibodies to integrin α4β7/α4β1 and specific for α4β7 are approved for multiple sclerosis and ulcerative colitis, respectively. Integrin ligand binding pockets are unusually good binding sites for small molecules. Small molecules to the leukocyte integrin LFA-1 (αLβ2), and the fibrinogen receptor on platelets, integrin αIIbβ3, are approved topically for dry eye disease and acutely to prevent thrombosis, respectively. However, despite emergence of new targets, including integrins αVβ6 and αVβ8 in fibrosis and immuno-oncology, there are no approved chronic (oral) small molecule integrin antagonists.
An intense pharmaceutical effort to develop oral antagonists of αIIbβ3 following the success of parenteral ligand mimetics (Scarborough and Gretler, 2000) was fueled by anticipation of “the dawn of a new era in antithrombotic therapy, the era of αIIbβ3 antagonism” (Topol et al., 1999). However, oral integrin antagonists (Chew et al., 2001; Topol et al., 2003), as well as longer-term dosing of parenteral inhibitors (Théroux et al., 1998), were beset with massive failure. Five phase III trials of oral αIIbβ3 inhibitors in a total of 42,530 patients failed (Chew et al., 2001; Topol et al., 2003). In all trials, mortality was significantly higher in treated patients than in the placebo control, even though the need for urgent revascularization was significantly reduced. Mortality was lessened by concurrent aspirin treatment, suggesting that platelet-mediated events were responsible for the bad outcome. The contrast between the benefit of short-term and harm of long-term αIIbβ3 therapy was striking, and suggested “the presence of clinically relevant ‘toxic’ mechanisms” (Chew et al., 2001).
The reason for these failures appeared to stem, at least in part, from agonism derived from integrin conformational change (Aga et al., 2004; Cox et al., 2000; Murphy et al., 1998; Peter et al., 2001). Most integrins, including αIIbβ3, are present predominantly in a low-affinity, bent-closed (BC) conformation on cell surfaces but change to a high-affinity extended-open (EO) state when bound to their biological ligands (Frelinger et al., 1988; Li et al., 2017; Takagi et al., 2002) (Fig. 1A). A known complication of αIIbβ3 antagonism is pre-existing or drug exposure-stimulated production of antibodies to integrin conformational epitopes or ligand-induced binding sites (LIBS) (Bosco et al., 2005; Bougie et al., 2012; Onitilo, 2006; Topol et al., 1999). After the Phase III failures, αIIbβ3 antagonist research and development was shut down and publications in the field greatly declined.
FIGURE 1. Integrin domains, conformational change, and effect of drugs on the αIIbβ3 headpiece.
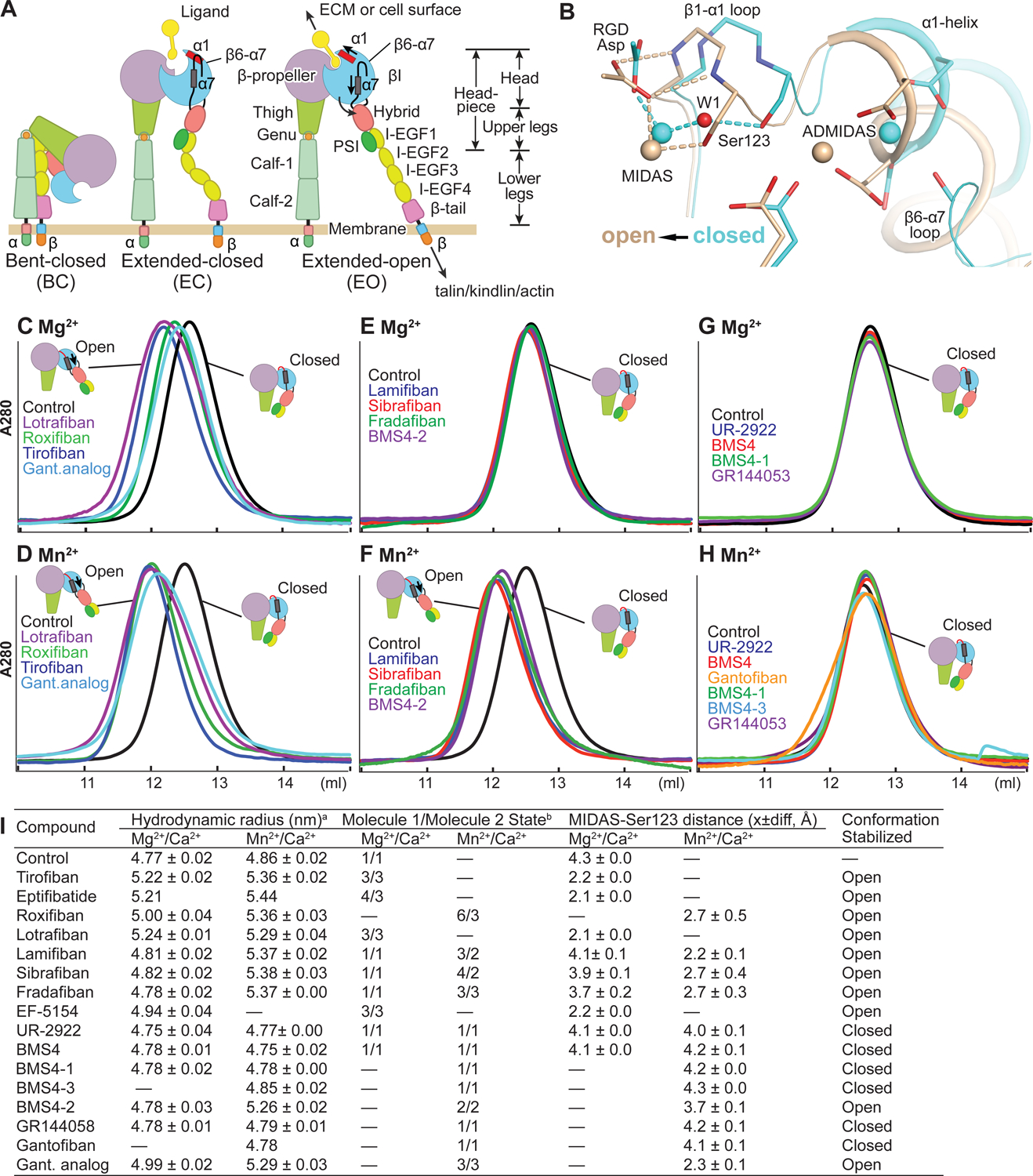
(A) Integrin domain organization and conformational states. In integrin headpiece opening, which increases affinity for biological ligands, pistoning of the α7-helix (purple bar) of the βI domain is linked to α1-helix pistoning (red bar), swing-out of the hybrid domain (curved arrow) and (B) rearrangement of loops at the ligand-binding site. (B) Shows αIIbβ3 bound to RGD in the closed (cyan carbons and metals) and open (wheat carbons and metals) conformations from PDB codes 3ZDY and 3ZE2 chains C+D+J, respectively. Loops and helices are thin and thicker tubes. Key metal coordinating sidechains, backbone and the RGD Asp sidechain are shown in stick. Oxygens are red and nitrogens blue. Metal ions and water 1 are shown as spheres. (C-H) Overlaid Superdex 200 chromatograms in absence (control, black) and presence of 10 μM drug in running buffer with curves in same color as the name of the drug. Running buffer contained 1 mM Mg2+, 1 mM Ca2+ or 2 mM Mn2+ 0.2 mM Ca2+ as indicated. Open and closed headpieces are shown in cartoon in insets (I) Summary of effects of drugs in gel filtration and when soaked into closed αIIbβ3 headpiece crystals. aHydrodynamic radii from Superdex 200 gel filtration. Control and tirofiban measurements are mean and s.d. of ≥ 3 experiments; other measurements are mean and difference from mean of two experiments, except for eptifibatide and gantofiban which were run once. bConformational states (Zhu et al., 2013). MIDAS metal ion - Ser-123 sidechain oxygen atom distances are shown as mean and difference for the two headpiece molecules in the asymmetric unit.
Thus, the story of how a just-emerging class of αIIbβ3 antagonist worked—which did not induce LIBS—was never told. Several inhibitors were published as non-LIBS inducers, but how they were discovered and the chemical properties that differentiated them from LIBS inducers were not described (Aga et al., 2004; Baba et al., 2001; Breth et al., 2005; Dickfeld et al., 2001). Furthermore, development of αIIbβ3 antagonists was stopped prior to their first structures bound to αIIbβ3, which were all with LIBS inducers (Xiao et al., 2004). A subsequently developed LIBS-inducing integrin αVβ3 inhibitor also failed in clinical trials in cancer, and was suggested to be agonistic (Ley et al., 2016; Li et al., 2019; Reynolds et al., 2009). Although non-LIBS inducing αIIbβ3 and αVβ3 inhibitors have been reported by academic investigators, they have not been extended to integrins with other β-subunits (Adair et al., 2020; Kereiakes et al., 2020; Li et al., 2019; Zhu et al., 2012).
A principle for inhibiting integrin conformational change by small molecule antagonists that could be extended to integrins with other β-subunits would be desirable. Consider a dangerous safety signal with firategrast, a dual α4β1/α4β7 integrin antagonist, in a phase 2 trial in multiple sclerosis (Miller et al., 2012). High doses were significantly beneficial, a medium dose had no significant effect, and a low dose was significantly harmful, a profile consistent with action of a partial agonist and induction of the active conformation of the integrin. However, little is known about firategrast, including whether it induces the high affinity state of α4β1.
Here, we hypothesized that the rich chemical matter developed for αIIbβ3 antagonism could be used to uncover a chemical principle by which inhibitors could bind but not induce the high affinity integrin state, and that this principle might be broadly transferable to integrin targets of current clinical interest. We determined high resolution structures of αIIbβ3 bound to opening-stabilizing (LIBS-inducing) and closure-stabilizing (non-LIBS-inducing) compounds. The key chemical principle to emerge, a hydrogen bond to a water molecule of importance in integrin conformational change, is elegant in its simplicity. Drug molecules that stabilize this water not only do not induce LIBS epitope expression, but also suppress LIBS expression by stabilizing the closed, low affinity integrin conformation.
To challenge our understanding of the chemical features required for closing inhibitors, we extrapolated what we learned from αIIbβ3 to the highly dissimilar integrin α4β1. We found that firategrast, which is no longer in pharmaceutical development, stabilizes the open, high-affinity integrin conformation. Searching the chemical literature for α4β1 antagonists, we found a pair of two compounds that were similar but differed in one position by the presence of a nitrogen atom able to hydrogen bond to water or a carbon atom lacking this ability; these compounds stabilized the closed and open states, respectively. This success and the invariance in integrin β-subunits of the metal ion dependent adhesion site (MIDAS) Mg2+ ion and the MIDAS serine sidechain, between which the key water locates, suggest that the closing principle described here is likely extendable to all integrins.
RESULTS
αIIbβ3 inhibitors stabilize distinct integrin conformations.
Integrins on cell surfaces and as soluble ectodomain fragments equilibrate between three conformational states, bent-closed (BC), extended-closed (EC) and extended-open (EO) (Fig. 1A, B). In the absence of ligand binding, the BC state predominates on cell surfaces: >99.8% for α5β1 and >97.7% for α4β1 (Li and Springer, 2018). Integrin αIIbβ3 is also predominately in the BC state in absence of ligand, as shown by lack of reactivity with LIBS antibodies (Frelinger et al., 1988). LIBS antibodies to αIIbβ3 map to regions of the upper and lower legs (Fig. 1A) that become exposed upon extension (Byron et al., 2009; Zhang et al., 2013).
Using multiple assays that extend to the end of Results, we found that inhibitors either stabilized integrin headpiece opening, closure, or were conformationally neutral. Initially, we tested the effect of compounds on the integrin headpiece fragment, which is truncated at the knees (Fig. 1A). The headpiece responds similarly to ligand-mimetic drugs as the ectodomain and intact integrins, but has only two states, closed and open, and thus can directly demonstrate opening in the absence of extension. The integrin headpiece, as demonstrated with α5β1, is more stable in the closed state than the ectodomain, and is thus more difficult to activate (Li and Springer, 2017). In 1 mM Mg2+/1 mM Ca2+ (Mg2+), the clinically tested oral antagonists roxifiban (Pieniaszek et al., 2002) (BMS / DuPont) and lotrafiban (Mould et al., 2001; Topol et al., 2003) (Glaxo), like parenteral tirofiban, caused the headpiece to elute earlier in gel filtration, showing its hydrodynamic radius was increased by opening (Fig. 1C). Drugs studied here are shown in Fig. 2; for convenience we use oral prodrug names for their active forms. Mn2+ increases integrin intrinsic affinity for ligand and also increases the population of the open state (Anderson et al., 2022; Li et al., 2017). As shown by negative stain electron microscopy of detergent soluble, intact αIIbβ3, Mn2+ increases population of the extended-closed and extended-open states, and in presence of a ligand that binds to the open headpiece conformation, enhances population of the extended-open state (Eng et al., 2011).
FIGURE 2. Chemical structures of integrin inhibitors.
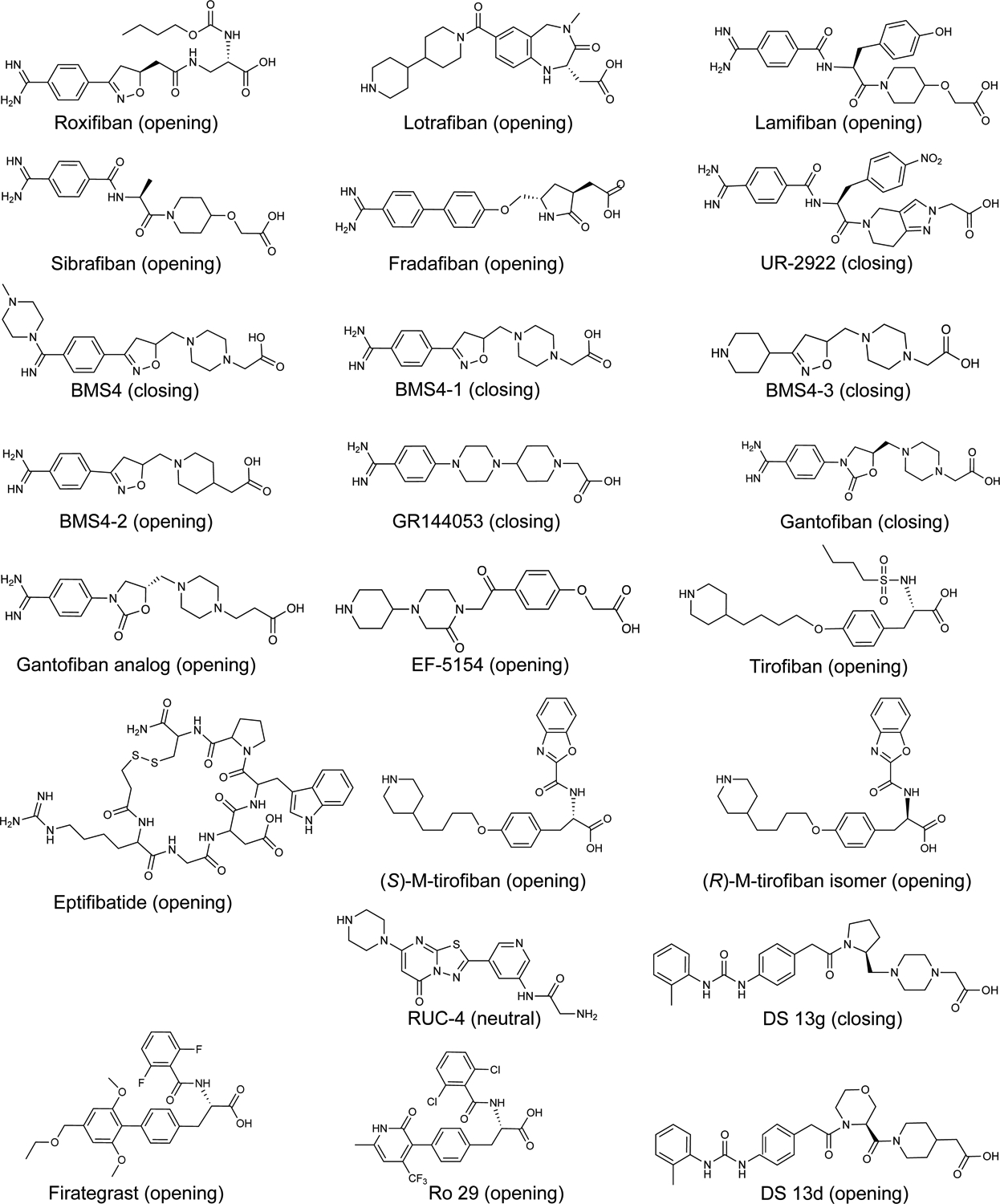
Drawings show the active form used here in the case of prodrugs. Compounds are αIIbβ3 inhibitors, except for four α4β1 inhibitors, shown to the right and below of RUC-4.
In gel filtration of the αIIbβ3 headpiece in absence of compound, 2 mM Mn2+/0.2 mM Ca2+ (Mn2+) increased the hydrodynamic radius slightly, from 4.77 in Mg2+ to 4.88 nm in Mn2+, which suggested a slight increase in population in the ensemble of the open conformation (Fig. 1C&D and Fig. 1I). While clinically tested lotrafiban, roxifiban, and tirofiban varied in the extent to which they shifted headpiece elution in Mg2+, they uniformly increased the hydrodynamic radius to ~5.36 nm in Mn2+. Other clinical stage compounds, lamifiban (Muller et al., 1997; Weller et al., 1996) (Roche, Ro 44–9883), sibrafiban (Weller et al., 1996; Wittke et al., 1999) (Roche, Ro 44–3888), and fradafiban (Muller et al., 1997) (Karl Thomae) did not cause headpiece opening in Mg2+ (Fig. 1E), consistent with reports that lamifiban did not induce exposure of LIBS epitopes on αIIbβ3 on platelets (Murphy et al., 1998). However, in activating Mn2+ buffer, these three compounds induced headpiece opening as completely as tirofiban (Fig. 1F). We term these “opening” inhibitors because they increase the proportion of the open state in conformational ensembles.
Another class of compounds did not induce opening in buffer with Mg2+ or Mn2+. We found UR-2922 (Ube Pharmaceuticals), reported not to induce LIBS epitopes on platelets in Mg2+ (Baba et al., 2001), in a review on integrin antagonists (Cox et al., 2010). We later found BMS4, also reported not to induce LIBS on platelets in Mg2+ (Breth et al., 2005), during a serendipitous search for LIBS antibodies. We found that neither UR-2922 nor BMS4 increased the hydrodynamic radius of the αIIbβ3 headpiece in Mg2+ or Mn2+ (Fig. 1G,H). Furthermore, in Mn2+ these inhibitors shifted the headpiece to a smaller hydrodynamic radius than the control, which was indistinguishable from the control in Mg2+ (Fig. 1G–I). These results are consistent with the suggestion above that a small population of the αIIbβ3 headpiece is in the open conformation in Mn2+ and that these compounds preferentially bind to the closed conformation and increase its population. We term these “closing” inhibitors.
Structural features of opening integrin inhibitors.
In the absence of ligand in Mg2+/Ca2+, αIIbβ3 headpiece–Fab complexes crystallize in the closed state with two molecules per asymmetric unit, giving two independent structural snapshots (Zhu et al., 2013). The key early step in headpiece opening is movement of the loop between the βI domain β1-strand and α1-helix (β1-α1 loop, Fig. 1A,B), which bears three sidechains that coordinate the MIDAS metal ion in an Asp-Xaa-Ser-Xaa-Ser (DXSXS) motif that is conserved in all integrin β-subunits. In previous work, we soaked Arg-Gly-Asp (RGD) peptide at different concentrations with Mg2+or Mn2+ into αIIbβ3 crystals, and trapped αIIbβ3 in closed state 1, intermediate states 2–7, or open state 8 (Zhu et al., 2013). In state 1, the sidechain oxygen of Ser-123 in the MIDAS motif hydrogen bonds to water molecule 1 (W1 in Fig. 3A, B), whereas in states 3–8, movement of the β1-α1 loop repositions the Ser-123 sidechain oxygen to take the place of water 1 (Fig. 1B, Fig. 3C).
FIGURE 3. Crystal structures of the αIIbβ3 headpiece in complex with inhibitors in Mg2+ or Mn2+.
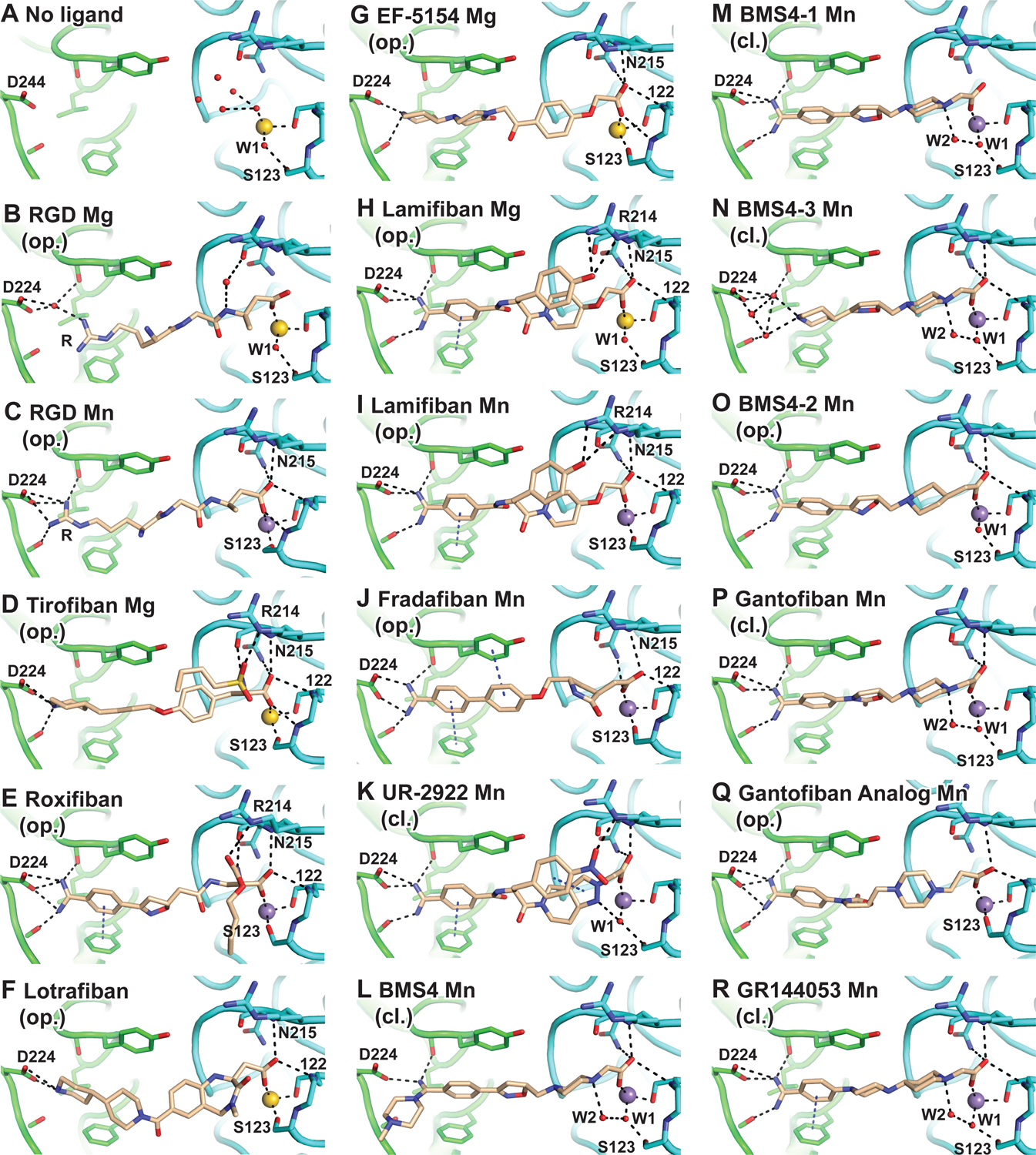
A-R. Structures of molecule 1 in the asymmetric unit (chains A and B) after soaking indicated compounds (0.1–1 mM) (D-R) in 1 or 5 mM Mg2+ and 1 mM Ca2+ or 2 mM Mn2+ and 0.2 mM Ca2+ as indicated into closed αIIbβ3 crystals (Table S1). Opening (op.) or closing (cl.) propensity is noted below each compound name. Comparisons show an unsoaked control (A) PDB ID: 3T3P and 10 mM RGD soaked in presence of 5 mM Mg2+ and 1 mM Ca2+ (B) or 2 mM Mn2+ and 0.1 mM Ca 2+ (C) (chains C, D, and J of PDB IDs 3ZDY and 3ZE2, respectively). Structures are superimposed on the β-propeller and βI domains. αIIb, β3, and compounds or RGD are shown with carbons in green, cyan, and wheat, respectively. MIDAS Mg2+and Mn2+ ions are shown as spheres in yellow and purple, respectively. Water molecule oxygens are shown as smaller red spheres. Backbone is shown as worm trace or sticks for residues that hydrogen bond to ligand. Sidechains that interact with ligand are shown in stick. Black dashes represent metal coordination, hydrogen, and π-π bonds. Relevant residues are labeled. See also Table S1 and Figures S1 and S2.
To test whether differences would emerge from comparisons of crystal structures of opening and closing inhibitors bound to αIIbβ3, we determined structures of 16 compounds soaked in the presence of Mg2+ or Mn2+, including 5 compounds soaked in both metal ion conditions. The 21 structures diffracted to 2.52 ± 0.26 Å and were refined to Rfree values of 23.21 ± 1.46 (mean ± s.d.) with excellent electron density for all drug atoms (Fig. 3, Fig. S1, Fig. S2, and Table S1).
Tirofiban, eptifibatide (Xiao et al., 2004), lotrafiban, and EF-5154 in Mg2+ and roxifiban in Mn2+ bound in the αIIbβ3 groove with their Arg-mimetic nitrogen hydrogen-bonded to αIIb residue Asp-224 and their Asp-mimetic carboxyl group coordinated to the β3 MIDAS metal ion (Fig. 3D–G and Supplemental Figs. S1A–E and S2A). Furthermore, β1-α1 loop movement brought the Tyr-122 backbone into position to hydrogen-bond to the drug carboxyl group and Ser-123 displaced water 1 to coordinate directly to the MIDAS metal ion. These opening compounds thus acted similarly to RGD peptides. Three other opening compounds, lamifiban, sibrafiban, and fradafiban, did not cause any conformational change in Mg2+ (Fig. 1I, Fig. 3H, and Figs. S1F,H,J and S2B&D). In contrast, in Mn2+ they induced a 2 Å movement in the β1-α1 loop at the Cα atom of Ser-123 toward ligand, and brought its sidechain oxygen within 2.3 to 2.7 Å of the MIDAS Mn2+ ion, thus displacing and occupying the position of water 1 (Fig. 1I, Fig. 3I,J; Fig. S1G,I,K and Fig. S2C). Moreover, movement also brought the backbone of Tyr-122 into position to form a hydrogen bond to the non-MIDAS coordinating carboxyl oxygen of the drug. 2Fo-Fc density maps at 1.5 σ near the βI domain MIDAS region clearly document displacement of the water by the Ser-123 sidechain (Fig. S1F–K). These results agreed with increased headpiece hydrodynamic radius with lamifiban, sibrafiban, and fradafiban in solution in Mn2+ but not in Mg2+ (Fig. 1I). We could identify no chemical properties or interactions such as hydrogen bonds with αIIbβ3 that uniquely distinguished the compounds that opened the headpiece in Mn2+ and not in Mg2+ (Fig. 3D–J and Fig. S2A–D).
A fundamental chemical principle that defines closing integrin inhibitors.
In contrast to the results with opening integrin inhibitors, soaking with UR-2922 and BMS4 caused no structural perturbation of the closed αIIbβ3 headpiece in Mg2+ or Mn2+ (Fig. 3K,L, Figs. S1L–O and S2E–F). Seeing no unique structural contacts between the integrin and the closing inhibitors, we at first were puzzled by how they stabilized the closed state. During opening, integrin β1-α1 loop movement is linked to sliding of the entire RGD ligand in its binding groove toward Asp-224 in the αIIb subunit (Fig. 1B and Fig. 3B,C) (Zhu et al., 2013). We wondered if closing inhibitors might be longer and more rigid than RGD and block this movement. Pursuing this hypothesis, we synthesized a shorter BMS4 analogue, BMS4–1, and the even shorter BMS4–3 (Fig. 2). Gel filtration showed that BMS4–1 and BMS4–3 were closure stabilizing (Fig. 1G–I). Crystal structures showed that αIIbβ3 remained in the closed state in Mn2+ and bound to BMS4–1 and BMS4–3 essentially identically to BMS4, except that BMS4–3 was so shortened that its piperidine group did not reach αIIb Asp-244 and interacted with it through a network of water molecules (Fig. 3M,N and Fig. S1P,Q). These results falsified the hypothesis that closing propensity of the BMS series was a function of compound length.
The inspirational moment finally came when we noticed that closing compounds either directly hydrogen bonded water 1 (W1), which bridges the MIDAS metal ion to the sidechain of MIDAS residue Ser-123 (UR-2922, Fig. 3K); or indirectly hydrogen bonded to water 1 through water 2 (W2) as in the case of BMS4, BMS4–3, and BMS4–1 (Fig. 3L–N). As water 1 is displaced by the Ser-123 sidechain early in integrin opening and in the final open state (Xiao et al., 2004; Zhu et al., 2013), stabilizing water 1 specifically stabilizes the closed conformation.
UR-2922 has a pyrazole ring with two nitrogens, one of which (N2) hydrogen bonds to water 1 (Fig. 2, Fig. 3K, and Fig. 4A). Neither nitrogen has a hydrogen. Thus, one of the hydrogens of water 1 donates a hydrogen bond to the electron lone pair of N2; in turn, one water 1 oxygen electron lone pair forms a partially covalent (2.1 Å) coordination to the MIDAS metal ion. The UR-2922 complex structures are virtually identical in Mn2+ and Mg2+ (Fig. 3K and Supplemental Fig. S2F) and have strong electron density for water 1 (Fig. 4A and Fig. S1L, M).
FIGURE 4. Stabilization of the water between the MIDAS metal ion and Ser-123 sidechain by closing antagonists.
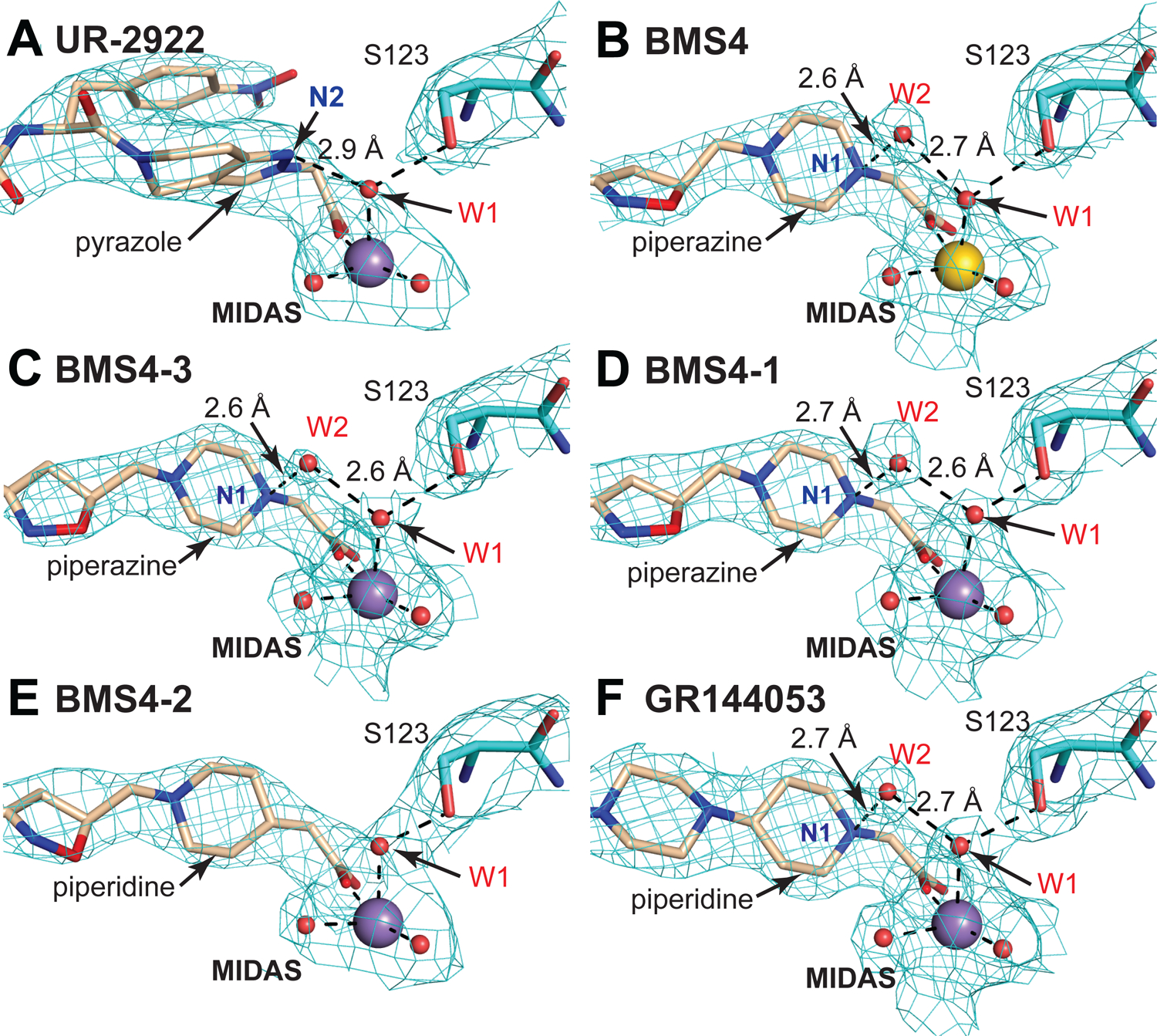
(A-F) The environment around the indicated compound substructures near the MIDAS metal ion, waters, and Ser-123 is shown after superposition on the βI domain. 2Fo-Fc electron density (cyan mesh) is contoured around atoms at 1.5 σ. Colors are as in Fig. 2 including Mg2+ and Mn2+ ions as gold and purple spheres, respectively, and waters as smaller red spheres. Black dashes represent coordination and hydrogen bonds.
The BMS4-bound structures in Mn2+ and Mg2+ and the BMS4–3 and BMS4–1 complexes in Mn2+ all show strong density for both water 1 and water 2 (Fig. 4B–D and Fig. S1N–Q). Density for water in a similar location to water 2 was not observed in any of the complexes with opening-stabilizing inhibitors or in UR-2922 complexes (Fig. 4A, Fig. S1A–K). The piperazine N1 atom is highly basic and is protonated (positively charged) at neutral pH. Thus, water 2 serves as a polarized hydrogen bond acceptor for the protonated piperazine nitrogen N1. Among the BMS4, BMS4–1, and BMS4–3 complex structures, three in Mn2+ and one in Mg2+, each with two independent molecules in the crystal lattice, the strength of the piperazine N–water 2 and water 2–water 1 hydrogen bonds is evidenced by their short heavy atom distances of 2.79±0.18 and 2.64±0.11 (mean±sd, n=6), respectively.
These results define a fundamental chemical principle for designing closing integrin inhibitors: they include a polar atom in the compound that is positioned i) to accept a hydrogen bond from water 1 or ii) to donate a hydrogen bond to water 2, which in turn accepts a hydrogen bond from water 1. The net effect is to stabilize water 1 and prevent its expulsion in integrin opening. These features are specific to closing inhibitors, since no hydrogen bond donors or acceptors, including water, are within 3.5 Å of the position occupied by water 1 in crystal structures of opening inhibitors including RGD (Fig. 3B–J, Figs. S1A–K and S2A–D).
Testing the chemical hypothesis and its ability to identify further closing αIIbβ3 inhibitors.
We challenged the chemical hypothesis by synthesizing BMS4–2, which substituted the piperazine N1 atom in BMS4–1 with a carbon atom (Fig. 2). BMS4–2 soaked into closed αIIbβ3 headpiece crystals in Mn2+ shifted the molecules in the crystal lattice to state 2, in contrast to all closing inhibitors (Fig. 1I). Water 1 remained between the MIDAS and Ser-123 sidechain (Fig. 3O); however, Ser-123 moved closer to the MIDAS Mn2+ ion with a distance of 3.7±0.1 Å, whereas all closing inhibitors showed Ser-123 - Mn2+ distances of 4.2 to 4.4 Å (Fig. 1I). Furthermore, there was no density for water 2 with BMS4–2 (Fig. 4E). In gel filtration, BMS4–2 stabilized headpiece opening in Mn2+ and not in Mg2+ (Fig. 1E,F,I). As BMS4–1 and BMS4–2 differ by a single atom (Fig. 2), these results demonstrated that the piperazine N1 atom is essential for stabilizing the closed integrin state and for positioning water 2 to hydrogen bond to water 1.
Further αIIbβ3 inhibitors with piperazine or piperidine acetic acid moieties were found in review articles and using SciFinder (scifinder.cas.org). Gantofiban (EMD 122347, Merck KGA) is optimized for blocking platelet functions and oral bioavailability in guinea pigs (Gante et al., 1995) (Fig. 2). Coincidentally, we later found that EMD 122347 had been reported to decrease LIBS exposure on platelets (Dickfeld et al., 2001). Although no results were published, gantofiban entered Phase II clinical trials in Europe (Merck KGA) and Japan (Yamanouchi) (https://adisinsight.springer.com). We synthesized gantofiban, which is most active as the (R)-stereoisomer, and its analog with a one carbon longer carboxylic acid, which is most active as the (S)-stereoisomer (Gante et al., 1995) (Fig. 2). Gantofiban does not induce headpiece opening in Mn2+ (Fig. 1H); however, its analog does in both Mg2+ and Mn2+ (Fig. 1C, D). Crystal soaking in Mn2+ showed that gantofiban bound to αIIbβ3 without perturbing its structure and that its piperazine nitrogen hydrogen bonded to water 2 which in turn hydrogen bonded to water 1 (Fig. 3P and Supplementary Fig. S1S). In contrast, the gantofiban analog induced movement of the β1-α1 loop and Ser-123 sidechain oxygen into the position of water 1 and had no equivalent of water 2 (Fig. 3Q and Fig. S1T). Compared to gantofiban, the one-carbon extension of the acid group in its analog displaced the piperazine nitrogen only 1.3 Å further away from the carboxyl group. These results demonstrate the precision with which the hydrogen bond donor for water 2 must be located to stabilize the closed integrin state.
GR144053 (Fig. 2) was selected at Glaxo for potency and long duration of action after oral administration in the cynomolgus monkey (Eldred et al., 1994). Size-exclusion chromatography showed that GR144053 was indeed closing in Mg2+ and Mn2+ (Fig. 1G–I). Crystal structures of αIIbβ3 bound to GR144053 with Mn2+ showed that the headpiece remained in state 1 (Fig. 1I, Fig. 3R). Strong 2Fo-Fc electron density showed the presence of waters 1 and 2 and the piperidine N1 of GR144053 in a strong hydrogen bond network (Fig. 4F and Fig. S1U). Thus, the chemical principles required for closing compounds survived key tests with chemical substitutions and enabled identification of further αIIbβ3 closing compounds.
Transferring the chemical principle of antagonistic inhibitors to a dissimilar integrin.
SciFinder substructure searches for VLA-4 (α4β1) inhibitors found the Daiichi Sankyo compound 13g (DS 13g) (Fig. 2). We synthesized DS 13g and purchased commercially available DS 13d, which was described in the same publication (Chiba et al., 2005) and is similar but contains a carbon atom in place of the nitrogen shown to be essential for stabilizing closure of αIIbβ3 (Fig. 2). We also tested the potent α4β1 antagonist Ro 29 (compound 29 of (Tilley et al., 2013)) and in later experiments firategrast, which had partial agonist-like properties in Phase II clinical trials (Miller et al., 2012) (Fig. 2). Gel filtration in Mn2+ showed that both DS13d and Ro 29 induced opening of the α4β1 headpiece, whereas DS13g did not (Fig. 5A).
FIGURE 5. Closing and opening α4β1 inhibitors and their conformational preferences.
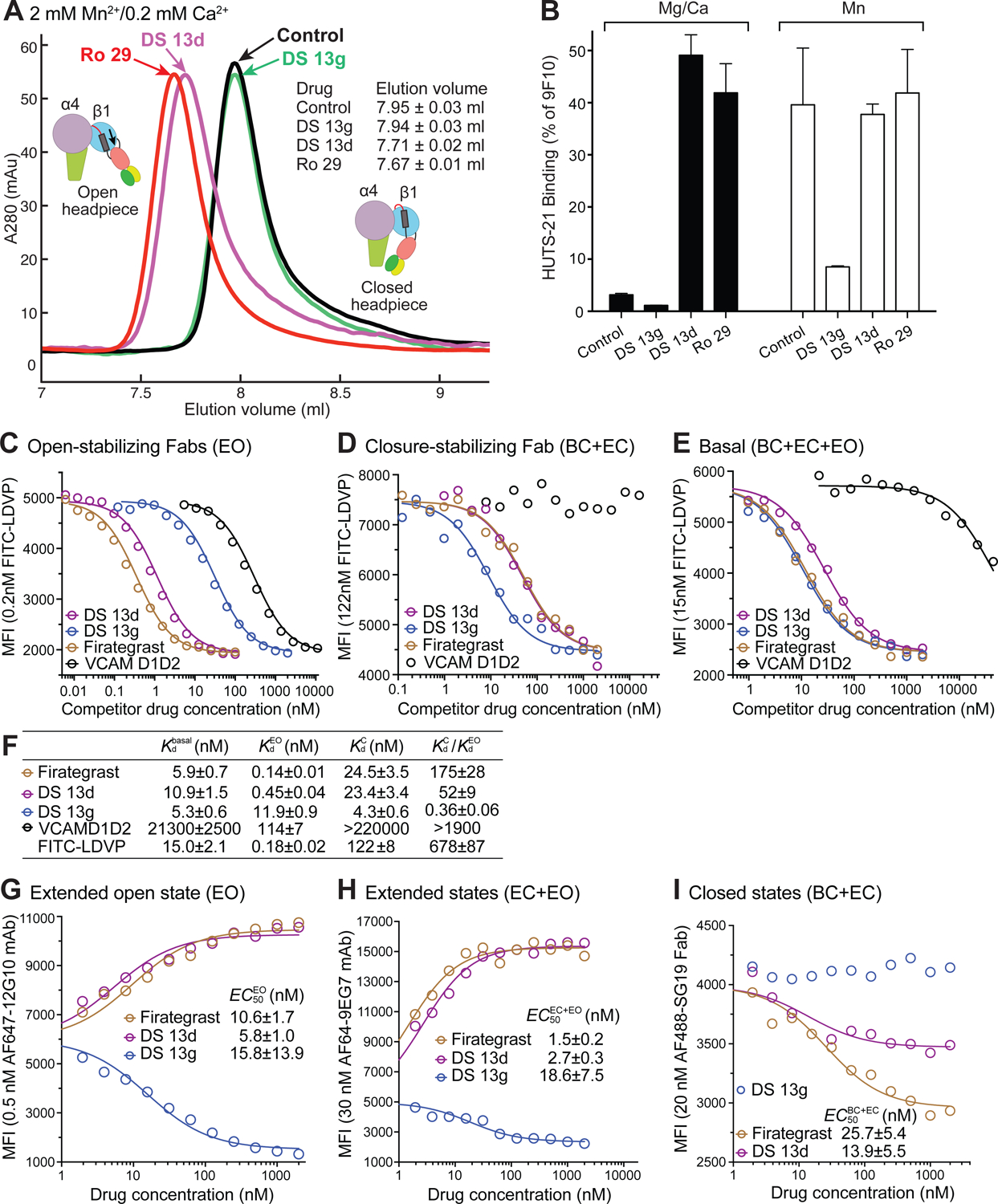
(A) Overlaid size exclusion chromatograms of the α4β1 headpiece in absence (control) and presence of 10 μM drug in running buffer with Mn2+. (B) Drug-induced HUTS-21 LIBS epitope exposure in Jurkat T lymphoma cells in 1 mM Mg2+ and 1 mM Ca2+ (closed bars) or in 1 mM Mn2+ (open bars). Mean fluorescence intensity is shown as % of that with antibody 9F10 to the α4 subunit. (C-E) Competition with FITC-LDVP binding by α4β1 inhibitors and VCAM D1D2 (concentration on the y axis) to Jurkat cells in L15 medium with 1% BSA. Mean fluorescence intensity (MFI) was determined by flow cytometry without washing. (C) EO state stabilized with 4μM 9EG7 Fab and 15nM HUTS4 Fab; (D) Closed states stabilized with 5μM SG19 Fab; (E) basal ensemble. Competition experiments were done multiple times with similar results. (F) Kd and preference for the EO state from C-E and for FITC-LDVP from (Li and Springer, 2018). Kd determined from competitive binding assays with nonlinear least square fits and error propagation (Eq.2, Methods). (G-I) Effect of inhibitors on the binding of conformation-specific antibodies to the β1 subunit. Binding of fluorescently labeled antibodies defined on the y axis was measured by flow cytometry without washing. EC50 values are from fits to Eq.1 with errors from nonlinear least square fits (Methods). No fits are shown when the error was large (one condition each in D and I).
The effect of these α4β1 inhibitors on cell surface α4β1 conformation was examined using HUTS-21 mAb, which binds to the same epitope in the hybrid domain as HUTS-4 (Luque et al., 1996), which is specific for the open conformation (Li et al., 2017). DS 13d and Ro 29 strongly induced HUTS-21 epitope exposure on Jurkat T cells in Mg2+, while DS 13g did not (Fig. 5B). Moreover, exposure of the HUTS-21 epitope by Mn2+ was strongly inhibited by DS 13g, but not by DS 13d or Ro 29.
In the ensemble theory of conformational states (Li et al., 2017), compounds stabilize particular states by having higher affinity for them than alternative states. We therefore measured compound affinities for the closed and open conformations of α4β1 on Jurkat cells. Conformations were stabilized with large excesses of conformation-specific Fab (Li and Springer, 2018). The biological ligand VCAM bound with >1900-fold higher affinity to the open than closed state, while firategrast, DS 13d, and the FITC-diphenylurea LDVP probe (Li and Springer, 2018) bound 175, and 52, and 680-fold better to the open state, respectively (Fig. 5C–F). In contrast, DS 13g bound 2.8-fold better to the closed than the open state.
Conversely, we measured the ability of compounds to shift the basal α4β1 conformational ensemble on Jurkat cells toward the states that they stabilize using fluorescent, conformation-specific antibodies that report the closed, open, and extended conformations (Fig. 5G–I). Firategrast and DS 13d increased the population of the EO state on Jurkat cells while DS 13g decreased its population (Fig. 5G). The compounds had cognate effects on the extended conformations (EC+EO), in agreement with the finding that the open state is always extended (Fig. 5H). In contrast, firategrast and DS 13d decreased the population of the closed states (BC+EC), while DS 13g had no measurable effect (Fig. 5I). The latter result is consistent with the finding that the BC+EC states are 98.9% basally populated on Jurkat cells (Fig. 4D in (Li and Springer, 2018)). The greater decrease of closed states by firategrast than DS 13d (Fig. 5I) is explicated by the greater selectivity of firategrast than DS 13d for the open state, 175-fold versus 52-fold, respectively (Fig. 5F). In summary, a chemical principle for creating closing integrin inhibitors was transferrable from αIIbβ3 to a distant integrin relative and allowed us to identify a closing α4β1 integrin inhibitor, DS 13g, that was “hiding” in plain view in the chemical literature.
Quantifying affinities and conformational preferences of αIIbβ3 antagonists, comparisons to recently described inhibitors, and studies on platelets.
As antibodies are lacking to stabilize the closed and open conformations of β3 integrins, we measured affinity for WT αIIbβ3 on the surface of Expi293F transfectants, where it is predominantly in the BC state as shown by little LIBS exposure, or with an activating glycan wedge (β3 N305T) mutation (Luo et al., 2003) that increases the proportion of the EO state. After measuring affinity of FITC-echistatin (Fig. 6A), we measured its competition by αIIbβ3 inhibitors (Fig. 6B,C and Fig. S3) and determined inhibitor affinity (Fig. 6D). The affinity of FITC-echistatin for αIIbβ3 was increased approximately 6-fold in the αIIbβ3 N305T ensemble. Similarly, all inhibitors characterized in other assays as opening showed higher affinity for the activated mutant than WT αIIbβ3 (lotrafibin, eptifibatide, tirofiban, roxifiban, gantofiban analog, EF-5154, sibrafiban, lamifiban, fradafiban, and BMS4–2). The concentration of cell surface αIIbβ3 in the assays, which was estimated by the ligand depletion effect, was higher than the KD values of the two highest affinity inhibitors, roxifiban and UR-2922 (Methods), which made the error large for UR-2922. Other closing compounds showed higher affinities for the WT than the activated mutant αIIbβ3 (GR144053, gantofiban, BMS4, and BMS4–1).
FIGURE 6. Binding affinity and conformational preference of αIIbβ3 inhibitors.
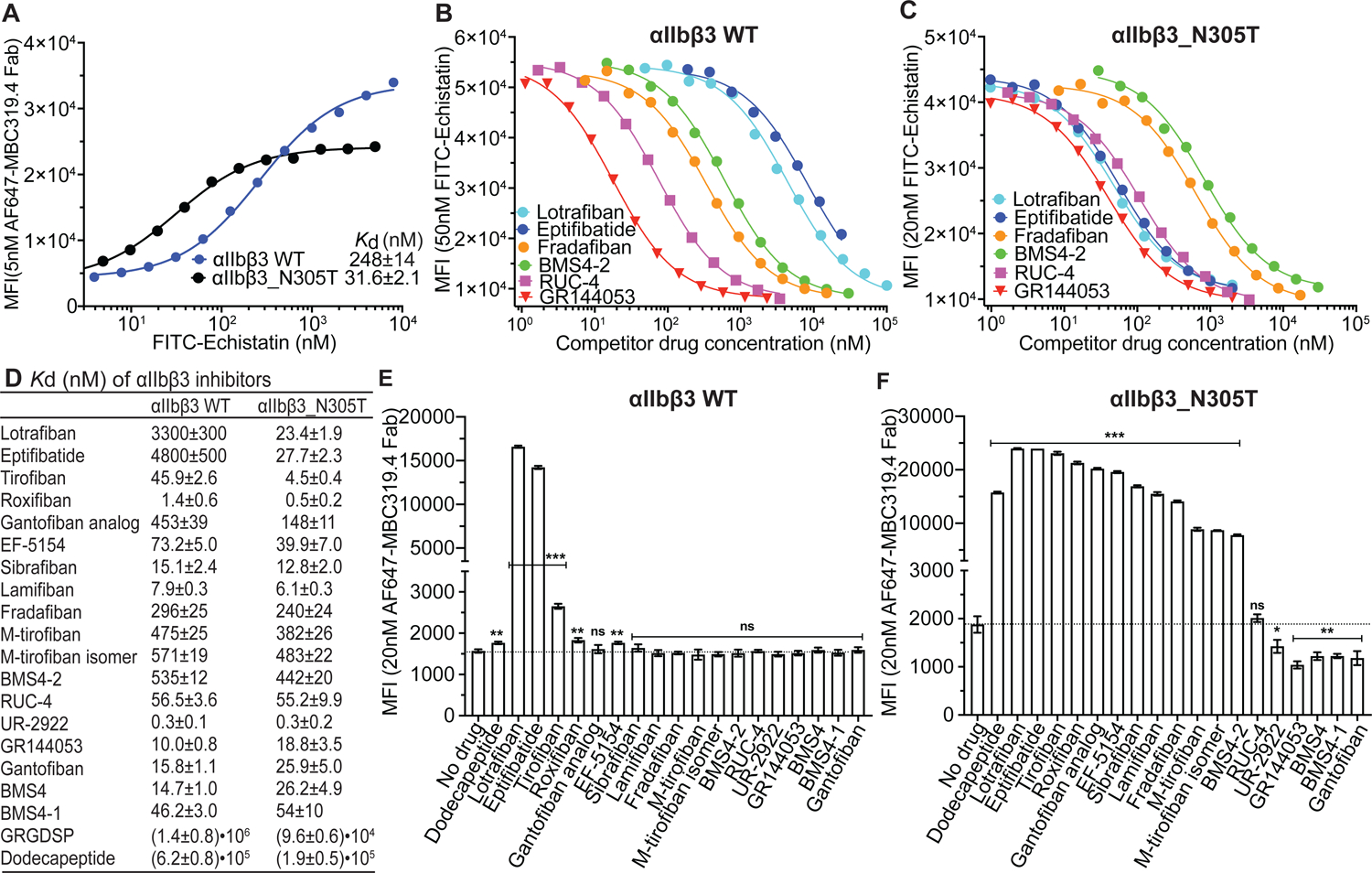
(A) Binding affinity of FITC-echistatin for αIIbβ3 wild type (WT) and αIIbβ3_N305T transfectants determined using 5 nM Alexa647-MBC319.4 Fab in L15 medium with 0.1% BSA. Kd was obtained by fitting to Eq. 3 (Methods). Errors are standard fitting errors from the nonlinear least square fits. (B-C) Competition of representative αIIbβ3 inhibitors with FITC-echistatin (concentration on y axis) for binding to αIIbβ3 WT (B) and to αIIbβ3_N305T (C) on transfectants in L15 medium with 0.1% BSA. Binding assays shown here and in Fig. S3 with other inhibitors were determined multiple times with similar results. Inhibitor concentration range used in the experiment shown was designed based on previous competitive binding curves. (D) Binding affinities determined from competitive binding assays with nonlinear least square fits and error propagation (Eq.4, Methods). (E-F) Effect of αIIbβ3 inhibitors on integrin extension measured with the binding of 20 nM Alexa647-MBC319.4 Fab to αIIbβ3 WT (E) and αIIbβ3_N305T (F) transfectants by flow cytometry without washing in L15 medium with 0.1% BSA. Inhibitors were used at 100x their Kd values for αIIbβ3 WT reported in Panel D. Background MFI of 20 nM Alexa647-MBC319.4 Fab in presence of 2 μM unlabeled MBC319.4 antibody was subtracted from the MFI values of cells treated with or without inhibitors. Binding was measured three times; data show mean and standard deviation. Unpaired two-tailed student’s t-test was between the inhibitor and no drug groups: *: p<0.05; **: p<0.01; ***: p<0.001. See also Figures S3 and S4.
To extend beyond industrial αIIbβ3 antagonists, we examined academic αIIbβ3 antagonists reported not to induce LIBS, RUC-4 (Kereiakes et al., 2020; Li et al., 2014; Zhu et al., 2012) and M-tirofiban (Adair et al., 2020) (Fig. 2). We synthesized both M-tirofiban stereoisomers and both were opening as shown by higher affinity for activated mutant than WT αIIbβ3 ensembles (Fig. 6D). By contrast, RUC-4 had affinities for the WT and mutant ensembles that were indistinguishable.
Conformational preferences of the inhibitors were independently examined by induction of the LIBS epitope recognized by MBC319.4 antibody, specific for the extended states of β3 integrins (Zhang et al., 2013). On WT αIIbβ3 transfectants, lotrafiban and eptifibatide strongly induced LIBS exposure and tirofiban was the next strongest (Fig. 6E). Weaker but significant induction was given by the fibrinogen γ C-terminal dodecapeptide, roxifiban, and EF-5154. High epitope exposure was related to a large increase in affinity for mutationally activated compared to WT αIIbβ3 (Fig. 6E). With the αIIbβ3 N305T ensemble, all compounds that showed significant LIBS induction on the WT αIIbβ3 ensemble gave even higher induction (Fig. 6F). Furthermore, all compounds that were characterized as opening in other assays, as well as M-tirofiban and M-tirofiban isomer, showed significant epitope induction. Conversely, all compounds characterized as closing in other assays, i.e. UR-2922, GR144053, Gantofiban, BMS4, and BMS4–1, significantly suppressed LIBS induction. Closing compounds also suppressed LIBS epitope exposure induced by Mn2+ and mutation of a GFFKR motif in the αIIb-β3 transmembrane domain interface (Fig. S4). RUC-4 had no effect on LIBS epitope expression and thus appears to be conformationally neutral.
We next tested compounds for effect on LIBS epitopes on washed platelets with or without activation by thrombin (Fig. 7A). In the absence of thrombin, lamifiban, sibrafiban, and fradafiban modestly but significantly induced integrin extension. Stronger extension was induced by roxifiban, lotrafiban, eptifibatide, and tirofiban. M-tirofiban and RUC-4 had no significant effect, while M-tirofiban isomer showed modest but significant enhancement of epitope exposure. After activation with thrombin, epitope exposure was increased in the absence of compounds. All opening compounds including M-tirofiban and its isomer enhanced LIBS exposure, with all showing significant induction except for BMS4–2 (p=0.06). LIBS exposure was decreased by all closing compounds (UR-2922, BMS4, BMS4–1 and GR144053), but did not reach significance. RUC-4 showed no effect on integrin extension.
FIGURE 7. Platelet assays and effect of serum on the affinity of αIIbβ3 inhibitors.
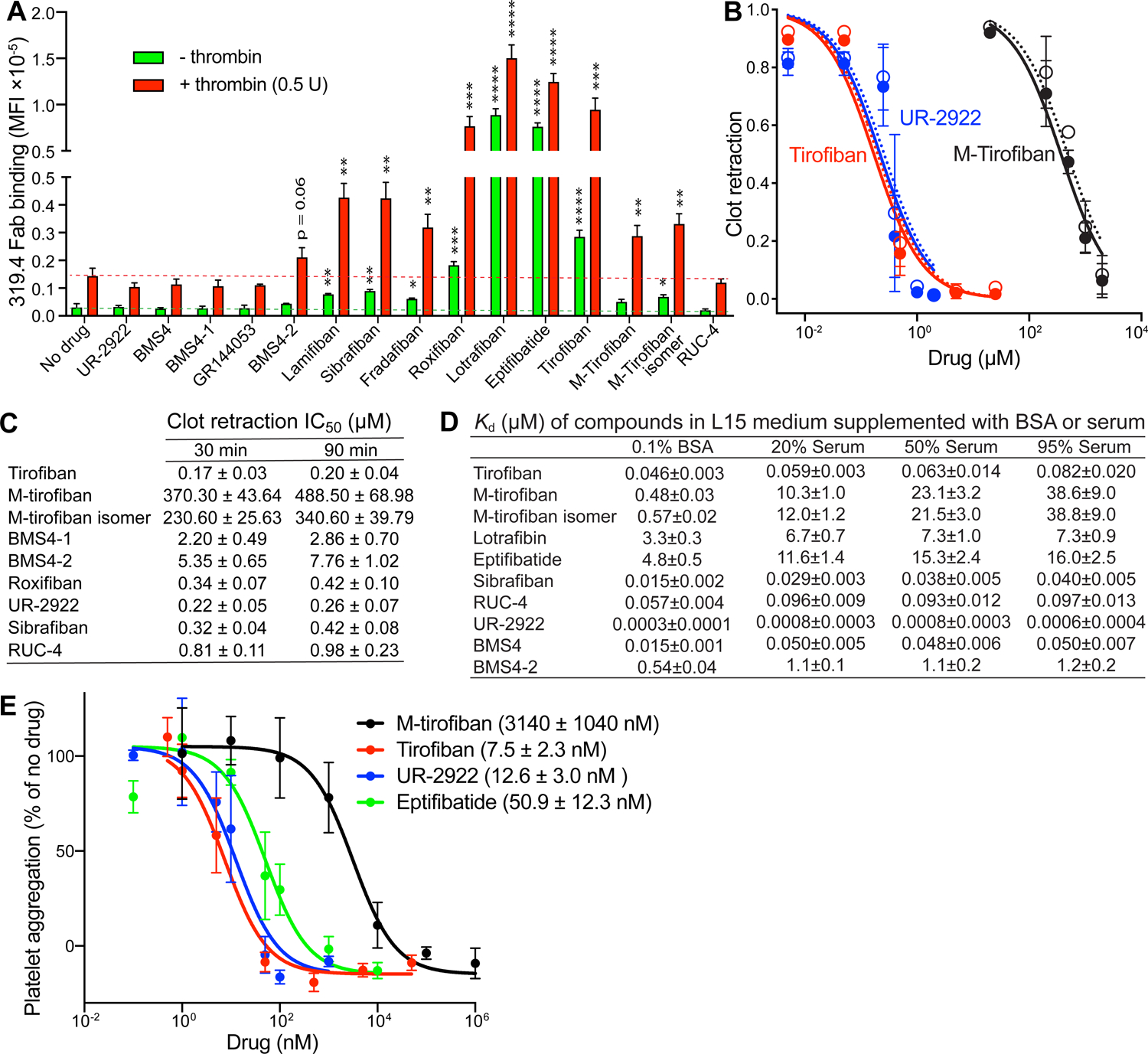
(A) Platelet LIBS exposure. Washed human platelets were pre-incubated with buffer control or αIIbβ3 inhibitors and then treated with or without 0.5 U/ml thrombin and 25 nM Alexa647-conjugated MBC319.4 Fab. Binding was measured by flow cytometry and shown as MFI. Data are mean ± s.d. from three independent experiments. Unpaired two-tailed t test was between the inhibitor and control groups; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. (B) Plots of inhibition of clot retraction (1 – (clot area/whole reaction area)) for three representative αIIbβ3 inhibitors measured at 30 mins (closed circles) and 90 mins (open circles). (C) IC50 values for inhibition of clot retraction by αIIbβ3 inhibitors. (D) Binding affinities of αIIbβ3 inhibitors determined by competing with FITC-echistatin binding to WT αIIbβ3 transfectant in L15 medium supplemented with 0.1% BSA (from Fig. 6) or with different concentrations of serum (Supplementary Fig.S5 B, C). values were determined from nonlinear least square fits (Eq.4, Methods). Errors are standard fitting errors from nonlinear least square fits. Experiments were repeated multiple times with similar results. (E) Inhibition of whole blood platelet aggregation by selected αIIbβ3 inhibitors measured by impedance aggregometry. Data are mean ± s.e.m. (n = 3 different donors). See also Figures S5, S6, and S7.
Clot retraction, which follows conversion of fibrinogen to fibrin during clotting, is largely mediated by binding of platelet αIIbβ3 to fibrin (Jansen and Hartmann, 2021). Representatives of all classes of αIIbβ3 inhibitors inhibited clot retraction (Fig 7B,C and Fig. S5). Both opening inhibitors (tirofiban, BMS4–2, sibrafiban, roxifiban, M-tirofiban, and M-tirofiban isomer) and closing inhibitors (BMS4–1 and UR-2922), as well as conformationally neutral RUC-4, were effective.
An important pharmacologic property of drugs is binding to proteins in the blood stream such as serum albumin. Therefore, we measured the effect of serum on KD values for a representative set of αIIbβ3 inhibitors (Fig. 7D and Fig. S6). In 95% serum M-tirofiban and its isomer showed greatly decreased affinities of 80- and 70-fold, respectively, compared to much smaller decreases by other drugs, such as 1.8-fold by tirofiban. Clot retraction assays are done in the presence of plasma. These results explain the previous failure to find inhibition of clot retraction by M-tirofiban (Adair et al., 2020). We confirmed that M-tirofiban inhibited platelet aggregation in whole blood (Fig. 7E).
DISCUSSION
The previous obscurity of closure-stabilizing integrin inhibitors and the lack of any previous information on their mechanism of action is emphasized by a docking study in a prominent review (Cox et al., 2010) which suggested that the UR-2922 carboxyl group was not in the αIIbβ3 binding pocket and thus could not coordinate with the MIDAS metal ion and induce the active conformation of αIIbβ3. Instead, our crystal structure and structure-function studies led to an unexpected conclusion. We discovered a general chemical mechanism by which closing inhibitors can harness water to stabilize the resting, closed conformation of integrins. Closing compounds must have polar atoms in precisely the correct position to hydrogen bond directly, or indirectly through a water molecule, to a specific MIDAS water molecule that must be expelled in order for the integrin to convert from the closed to the open conformation. Closing inhibitors have no potential for agonism since their binding does not induce integrin conformational change toward the open state. Additionally, and in contrast to both conformationally neutral RUC-4 and opening inhibitors, they stabilize the closed conformation by suppressing conformational change toward the active integrin state.
The relationship between affinity of ligand for integrin conformational states and the ability of ligand binding to alter the populations of these states obeys the laws of classical thermodynamics as previously demonstrated for integrins α4β1 and α5β1 (Li and Springer, 2018; Li et al., 2017). Although the integrin field typically uses only antibodies specific for the extended or open states as reporters for integrin conformational change, we showed that, as predicted by ensemble theory, antibodies to the closed state can also be used as reporters. Several subtleties and implications of integrin ensembles and thermodynamics should be pointed out. Because the open conformation is not stable in the bent integrin conformation, drugs that stabilize opening and closing stabilize the extended and bent states, respectively, as shown explicitly here for integrin α4β1. Thus the studies using Fabs to epitopes in extended αIIbβ3 in cellular assays in transfectants and platelets also reported the ability of compounds to stabilize the open or closed conformations. These Fab-based conformational measurements were in excellent agreement with explicit measurements of effects of compounds on opening of the headpiece by hydrodynamic radius in gel filtration and in crystal structures in Mg2+ and Mn2+. RUC-2, a compound similar to RUC-4, was previously shown not to shift the elution of the αIIbβ3 headpiece in gel filtration or its structure in crystals, in agreement with its lack of effect on LIBS expression as confirmed here (Zhu et al., 2012). As required by thermodynamics, the relative affinities of the compounds for WT and mutant, more open αIIbβ3 ensembles on transfectants also showed an excellent rank order correlation to ability to induce or suppress an extension-reporting epitope on transfectants and platelets. It appears to have been previously unappreciated in the integrin field that opening ligands can vary widely in their ability to induce LIBS, with closing ligands being an extreme example that suppress LIBS and perhaps warrant a change in nomenclature away from LIBS to name epitopes after the type of conformation they report. Equations and their graphical representation in Figure S7 provide guidance on use of reporter antibodies and explain why opening inhibitors with stronger conformational preferences are stronger LIBS inducers. The chemical features that define opening propensity, i.e. affinity for the open relative to the closed conformation, are complex; however, the peptidomimetics eptifibatide, lotrafiban, tirofiban, and roxifiban were among the most strongly opening compounds.
For comparison to small molecules, VCAM, a biological ligand for α4β1 was found to have >1,900 fold higher affinity for the open than the closed conformation. This large difference, and that of 6,000-fold for α5β1 binding to fibronectin (Li et al., 2017), contrast with the lower, but wide ranging, differences in affinity for open and closed states found among opening α4β1 inhibitors. Such comparisons will not be possible for αIIbβ3 inhibitors until methods are developed to obtain pure closed and open αIIbβ3 integrin states. However, the findings on α4β1 already raise the interesting possibility that the large affinity difference for biological ligands is an evolved trait that makes the integrin high and low affinity states on and off-like.
Previously, lamifiban (Ro-44-9883) and sibrafiban (Ro 48–3657) were found not to induce LIBS epitopes on platelets in platelet-rich plasma (Baba et al., 2001; Murphy et al., 1998), which could be consistent with differing assay sensitivities or a more activated αIIbβ3 ensemble on washed platelets than on platelets in platelet-rich plasma. Roche disclosed that it took lamifiban forward in clinical trials because of its potency and low induction of LIBS compared to other inhibitors (Scarborough and Gretler, 2000).
Two academically developed drugs were interesting comparators. M-tirofiban has a large hydrophobic substituent relative to tirofiban that was designed to block movement of the βI domain toward the open state and was reported to not induce opening and to block thrombosis without inhibiting hemostasis or clot retraction (Adair et al., 2020). We found that M-tirofiban showed a lesser preference for activated mutant αIIbβ3 than tirofiban but remained an opening compound, albeit a weak one, and induced statistically significant LIBS exposure both on activated mutant αIIbβ3 transfectants and thrombin-stimulated platelets. The original report showed LIBS exposure by ADP-stimulated platelets that was increased with M-tirofiban but did not reach significance (Adair et al., 2020); thrombin is a stronger platelet stimulator than ADP. The previous report tested only a single concentration of M-tirofiban, and a single, 10-fold lower concentration of tirofiban for inhibition of clot retraction in 20% plasma (Adair et al., 2020). However, we found that in 20% serum the apparent affinity of M-tirofiban is 175-fold lower than tirofiban, and that M-tirofiban completely inhibited clot retraction at the appropriate concentration. The lower potency of M-tirofiban may result from binding of its bulky aromatic modification relative to tirofiban (Figure 2) to hydrophobic binding sites on serum albumin. Our results also call into question the finding that hemostasis was significantly inhibited by tirofiban and not M-tirofiban at 10-fold higher doses in mice with 40% human platelets, since M-tirofiban is 470-fold lower in affinity than tirofiban in 95% serum. All other tested αIIbβ3 inhibitors have been found to inhibit hemostasis as well as thrombosis (Scarborough and Gretler, 2000), and no significant difference in relative potency of inhibition of platelet aggregation versus bleeding time was found between the closing inhibitor UR-2922, sibrafiban (which resembles M-tirofiban in its opening propensity), and xemilofiban, which is a stronger LIBS inducer (Baba et al., 2001). Additionally, we find that M-tirofiban is less efficacious in inhibiting platelet aggregation in whole blood (IC50 of 3140±1040 nM, 420-fold less potent than tirofiban) than originally reported (IC50 of 18±5 nM, 13-fold less potent than tirofiban).
RUC-4 is an academically developed αIIbβ3 inhibitor in clinical trials for first point-of-care treatment of myocardial infarction (Kereiakes et al., 2020; Li et al., 2014). Like the earlier described compound pentamidine (Cox et al., 1996), RUC compounds are specific αIIbβ3 inhibitors that lack a carboxyl group and do not induce LIBS epitopes or integrin headpiece opening (Zhu et al., 2012). RUC-4 occupied a unique intermediate position in our study between opening and closing compounds as a conformationally neutral compound. It had equal affinity for WT and mutant activated αIIbβ3 transfectants and neither enhanced nor suppressed LIBS epitope exposure.
An aspirational goal in the integrin field has been to create small molecule integrin inhibitors that do not induce integrin conformational change or activation. We now have taken this concept one step further, by discovering that closing compounds can suppress integrin opening by shifting the conformational ensemble towards the low affinity closed states. This feature may offer additional clinical benefit. For example, platelets of patients with acute coronary syndrome are significantly more activated than healthy controls even after clinical stabilization (Ault et al., 1999; Davi and Patrono, 2007; Fitzgerald et al., 1986; Trip et al., 1990). Furthermore, treatment of such patients with the αIIbβ3 inhibitor orbofiban increased αIIbβ3 conformational change, antibody-stimulated thromboxane B2 production, and CD63 expression (Cox et al., 2000). Many αIIbβ3 closing compounds with excellent drug-like properties are currently available. These include gantofiban, which entered Phase II clinical trials, and UR-2922. Moreover, GR144053 is commercially available and has been used without knowledge of its closing property in animals with tissue plasminogen activator or losartan after thrombotic injury to suppress neointima formation (Matsuno et al., 1998; Matsuno et al., 1997).
Our results demonstrated that the phase II trial of firategrast in relapsing remitting multiple sclerosis was conducted with an opening inhibitor (Miller et al., 2012). The significant increase in gadolinium-enhanced lesions at the lowest dose, lack of effect at the middle dose and significant decrease at the highest dose are consistent with partial agonism. Drug blood concentrations were measured but not reported. This is unfortunate, because we could have used the data reported here on firategrast binding affinity to cells and opening propensity to estimate at each drug concentration the percentage of drug-bound α4β1 and the percentage of α4β1 that transitioned from bent-closed to extended and open conformations.
It is common for companies and academic investigators alike to measure the potency of integrin inhibitors and biological ligands in Mn2+. Mn2+ increases affinity by two mechanisms: replacing Mg2+ at the MIDAS increases integrin affinity independently of conformation, and replacing Ca2+ at the ADMIDAS stabilizes the open conformation (Anderson et al., 2022). The latter mechanism artefactually inflates the affinity of opening compared to closing inhibitors. Thus, we caution against using Mn2+ to measure drug potency, which might have been one factor that influenced Daiiichi Sanko to put an α4β1 inhibitor into patients that based on its structure is opening (Muro et al., 2009).
Many other closing compound scaffolds can be created based on the essential chemical features that we have established here. Compounds must have carboxyl groups and nearby polar atoms in positions to either accept a hydrogen bond from water 1 or donate a hydrogen bond to water 2. Water 1 in the UR-2922 structure is polarized because one of its hydrogens must orient toward the N atom in UR-2922 and one of its lone electron pairs must orient toward the MIDAS metal ion. Notably, hydrogen bond donation by the protonated piperazine or piperidine nitrogen in the other compound class to relay water 2 may result in the same proposed polarity of water 1 as deduced from first chemical principles from the UR-2922 structure. The importance of donating rather than accepting a hydrogen bond to water 2 is emphasized by comparison to sibrafiban and lamifiban. These opening inhibitors have an oxygen two atoms away from the carboxyl group, in the same position as the nitrogen in closing compounds such as BMS4 (Fig. 2), yet such an ether oxygen cannot donate a hydrogen bond. Not only are sibrafiban and lamifiban not closing but they also do not have a bound water 2, suggesting that proper hydrogen bond polarity is important for forming a water network around the MIDAS metal ion. In contrast to the ether oxygen, it might be possible to create a closing compound with a suitably positioned hydroxyl oxygen. Although the hydrogen bond donor for water 2 is in piperidine or piperazine rings in the structures identified here, linear structures or rings of other size also should be possible.
The integrin family may be divided into five subfamilies with distinct ligand specificities. Despite their distant relationship in different subfamilies, we were able to extend the discovery of closing compounds from integrin αIIbβ3 to integrin α4β1. Unlike αIIbβ3, α4β1 inhibitors to the best of our knowledge had not been examined for their ability to induce LIBS epitopes. Our ability to use the chemical principles we had discovered with αIIbβ3 to discover a closing inhibitor of α4β1 validates the generality of these principles. The DSXSX MIDAS motif is invariant in integrin β subunits. Furthermore, the movement of Ser2 in this motif into the position of water 1 has been seen in all structurally characterized integrins, including the β3 integrins αIIbβ3 and αVβ3 (Xiao et al., 2004; Xiong et al., 2002; Zhu et al., 2013), the β1 integrins α5β1 and α6β1 (Arimori et al., 2021; Nagae et al., 2012; Schumacher et al., 2021; Xia and Springer, 2014), the β2 integrin αXβ2 (Sen et al., 2013), the β6 integrin αVβ6 (Dong et al., 2014), the β7 integrin α4β7 (Yu et al., 2012), and even in the atypical β8 integrin αVβ8 (Campbell et al., 2020; Wang et al., 2019). Thus, the general design principles reported here for building closure-stabilizing integrin inhibitors can serve to guide designs of antagonists for other integrins, many of which are currently being pursued in biopharma in challenging diseases including ulcerative colitis, fibrosis, and cancer. Closing integrin inhibitors should not have paradoxical activating effects since they not only potently block ligand binding, but also stabilize the resting integrin conformation against activating stress. This property is distinct from their ability to inhibit ligand binding, which for both closing and opening inhibitors is controlled by their concentrations relative to their IC90 values. The efficacy of closing inhibitors will need to be further tested in animal studies, as well as in clinical trials.
Limitations of the Study.
The molecular mechanisms by which opening integrin inhibitors cause partial agonism remain to be fully defined. Many other factors besides partial agonism may have contributed to clinical failure of parenteral and oral integrin antagonists (Aga et al., 2004). For αIIbβ3, these included the development of antibodies specific for the drug-integrin complex and the difficulty of achieving the desired percentage inhibition of αIIbβ3 in the face of short drug half-lives and causing bleeding at high αIIbβ3 inhibition. Such factors could also lead to the failure of closing antagonists. The ratios of affinities of inhibitors for WT and glycan wedge αIIbβ3 transfectants are for multi-state ensembles and underestimate the difference in affinity between the open and closed states of αIIbβ3. Intact integrins on the cell surface and ectodomain and headpiece fragments differ in free energy difference between open and closed conformations, and therefore differ in sensitivity to the effects of opening and closing inhibitors on their conformations. Although we have established principles for creating closure-stabilizing inhibitors that should be applicable to all integrins, successful oral drugs require many other attributes. These include specificity, adsorption, resistance to metabolism and excretion, and long half-life. The breadth of chemical scaffolds that can hydrogen bond to stabilize water 1 directly or through water 2 is likely to be large and not to require that the hydrogen bonding moiety be in a ring, but remains to be explored, and thus the full scope for adding further desirable chemical features to closing compounds remains unknown.
STAR★METHODS
Resource Availability.
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Timothy Springer (springer@crystal.harvard.edu).
Materials Availability
- Plasmids made in this study have been deposited at AddGene:
- PD2529 CAG αIIb full length: Addgene Plasmid #189796
- PD2529 CAG β3 full length: Addgene Plasmid #189797
- PD2529 CAG β3_N305T full length: Addgene Plasmid #189798
Compounds can be made by following the published synthetic procedures.
Other materials are available from the Lead Contact.
Data and code availability
All structural data has been deposited at the Protein Data Bank.
Diffraction images have been deposited at SBGRID. Datasets for Figures 5C–E and 5G–I, 6A–C and E–F, and Figures S3, S6 and S7 have been deposited at Harvard Dataverse.
Executable Mathematica code for fitting competitive binding data in Fig. 6B–C and Fig. S3, entitled “Data S1-Equation 4.nb” is available in this paper’s supplemental information.
Other data is available on request from the Lead Contact for 3 years.
Experimental Model and Subject Details.
Cell lines were tested for mycoplasma every 60 days and were not authenticated.
Method Details.
Preparation of integrin headpiece samples.
Expression and purification of αIIbβ3 headpiece was as described (Xiao et al., 2004; Zhu et al., 2010). The α4 construct contains the β-propeller and thigh domains, from residue Y1 to R587, with a R558A mutation to eliminate a furin cleavage site in the thigh domain (Yu et al., 2012). The α4 headpiece cDNA fused with a tobacco etch virus (TEV) cleavage site, an ACID coiled-coil, and a StrepII tag was inserted into the pcDNA3.1/Hygro vector. The β1 construct contains PSI, hybrid, βI, and EGF1 domains, from Q1 to E481. The β1 headpiece cDNA fused with a TEV site, a BASE coiled-coil, and a His6 Tag was inserted into the pEF1/Puro vector. The α4β1 headpiece construct was stably expressed in CHO lec 3.2.1.8 cells. Culture supernatant was concentrated and buffer exchanged to TBS buffer (20 mM Tris, 150 mM NaCl, pH 7.4) using a tangential flow concentrator. The α4β1 headpiece was purified using a Ni-NTA column followed by a Strep-Tactin column. The coiled-coils and tags were then removed by TEV digestion as described (Yu et al., 2013), but without the presence of Fab. Finally, the α4β1 headpiece was subjected to Superdex 200 chromatography in TBS buffer with 1 mM Mg2+ and 1 mM Ca2+.
Measuring drug-induced integrin headpiece opening by size-exclusion chromatography.
For size-exclusion chromatography of αIIbβ3 headpiece, a Superdex 200 column was equilibrated with 10 μM of inhibitor in HEPES buffered saline (HBS) plus either 1 mM Mg2+, 1 mM Ca2+ or 2 mM Mn2+, 0.2mM Ca2+. The αIIbβ3 headpiece samples (30 μg) were pre-incubated with 60 μM of drug in corresponding buffers for 30 minutes at room temperature prior to chromatography. Rh values were calculated from peak elution volumes using ovalbumin, BSA, catalase, and aldolase as standards of known Rh (Zhu et al., 2010). Conditions were identical for α4β1 headpiece, except a Zenix-C SEC300 size-exclusion column (Sepax Technologies, Newark, DE) was used, 10 μg of α4β1 headpiece was used, and data was from 3 or more runs.
Crystallization of closed αIIbβ3 headpiece, compound soaking, and structure determination.
Expression and crystallization of αIIbβ3 headpiece-10E5 Fab complex in the closed conformation was as reported (Zhu et al., 2010). Prior to compound soaking, headpiece crystals were stabilized at a higher PEG concentration (15% PEG 8000, 0.2 M ammonium sulfate, 0.1M Tris-HCl, pH 8.9), and glycerol was added in 5% increments to 15%. Compound was added (0.1–1 mM; see Table S1) together with 1 mM MgCl2 and 1 mM CaCl2 or 2 mM MnCl2 and 0.2 mM CaCl2, in the final cryo increment to 20% glycerol. After 4 h crystals were plunge frozen in liquid N2. Diffraction data was collected in GM/CA-CAT at APS. Refinements were with Phenix (Adams et al., 2010). Ligand geometry and protonation states were calculated using Prime (with OPLS2005 force field) and Epik packages from Schrödinger (New York, NY). Geometry-optimized (with hydrogens) ligand coordinates were input to eLBOW (Adams et al., 2010) to generate restraint files for Phenix refinement. Ligands were manually fit to initial Fo-Fc difference maps using Coot and complexes were further refined.
Antibodies.
Hybridomas were AP3 (Newman et al., 1985), AP5 (Kekomaki et al., 1991), LIBS1 (Frelinger et al., 1990), MBC319.4 (Zhang et al., 2013), 12G10 (Mould et al., 1995), 9EG7 (Lenter et al., 1993), SG/19 (Miyake et al., 1992), HUTS4 and HUTS-21 (Luque et al., 1996) and mAb16 (Akiyama et al., 1989). IgG produced from hybridoma was purified by protein G. For Fab fragments, antibodies were digested with papain (500:1 IgG:papain) in PBS with 10 mM EDTA and 10 mM L-Cys at 37°C for 18 h. After buffer exchange with 50 mM Tris (pH 9), the Fab was purified by anion exchange chromatography (HiTrap Q HP, GE Healthcare), after which the fractions containing Fab were concentrated, flash frozen, and stored at −80°C for future use. Antibodies and Fabs were fluorescently labeled with Alexa Fluor 647 NHS Ester or Alexa Fluor 488 NHS Ester (ThermoFisher Scientific) in PBS.
Quantitative fluorescent flow cytometry for α4β1 inhibitor affinity and conformational preference measurements.
Jurkat cells (106 cells/ml in RPMI-1640 medium and 10% FBS) were washed twice with assay medium (Leibovitz’s L-15 medium and 1% BSA) containing 5 mM EDTA, twice with assay medium alone, and resuspended in assay medium. For competitive binding experiments, each 50-μl sample contained 106 cells/ml, the indicated concentration of FITC-LDVP (Tocris Bioscience), the indicated Fabs, and varying concentrations of α4β1 inhibitors or VCAM D1D2 (Li and Springer, 2018) in assay medium. For LIBS antibody epitope exposure assays, each 50-μl sample contained 106 cells/ml, the indicated concentration of fluorescently labeled antibody or Fab, and indicated concentrations of α4β1 inhibitors. Mixtures were allowed to equilibrate at 22°C for 2 h before flow cytometry (BD FAC SCanto II) without washing (Li and Springer, 2018; Li et al., 2017). Binding of FITC-LDVP or fluorescently labeled LIBS antibodies were measured as mean fluorescence intensity (MFIobs) at each indicated inhibitor concentration (Cinhibitor). Competitive binding curves and LIBS antibody exposure curves were both fitted with a 3-parameter dose response curve (Eq.1) with MFI in absence of inhibitor (MFI0), MFI at saturating concentration of inhibitor (MFIsat), and half maximal effective concentration of the inhibitor (EC50), as fitting parameters. Affinity of inhibitors was derived (Eq.2) from the fitted EC50 value in competitive binding curve, the concentration of FITC-LDVP probe (CLDVP), and the binding affinity of the FITC-LDVP probe . Error for was propagated from the errors of EC50 and .
| (Eq.1) |
| (Eq.2) |
Expi293F αV & α5 knock-out cell line.
αV and α5 subunits were sequentially knocked out from Expi293F cells using CRISPR double nicking (Ran et al., 2013). To make the single-guide RNA (sgRNA) expressing plasmids, double stranded DNA oligos, 5’-CACCGGCCCTTCAAGGATTTGAGATGTTT-3’ (αV_guide A), 5’-CACCGCGAAGCATGCCACCAAGCTTGTTT-3’ (αV_guide B), 5’-CACCGGTACAGTGGAGCGCATGCCTGTTT-3’ (α5_guide A), and 5’-CACCGGTACAGTGGAGCGCATGCCTGTTT-3’ (α5_guide B), were cloned into the BbSI site of the pSpCas9n(BB)-2A-puro vector (Addgene: 48141). To create the Expi293F αV knock-out, the pair of sgRNA expressing plasmids targeting αV were transfected into Expi293F cells and knock-out cells were selected by two rounds of panning (Wysocki and Sato, 1978) and one round of FACS sorting, both with αV-specific antibody 17E6 (Mitjans et al., 1995). To create the Expi293F αV & α5 double knock-out, the pair of sgRNA expressing plasmids targeting α5 were transfected into Expi293F αV knock-out cells and the double knock-out cells were selected by two rounds of panning and one round of FACS sorting, both with α5 specific antibody mAb16 (Akiyama et al., 1989).
Quantitative fluorescent flow cytometry for αIIbβ3 inhibitor affinity and conformational preference measurements.
Measurements were done on Expi293F cells with the integrin α5 and αV subunits knocked out (Expi293F α5&αV KO) transiently transfected with αIIb and β3 expression plasmids, or with αIIb and β3_N305T expression plasmids. For transfection, 0.5ug plasmid coding αIIb subunit in PD2529 vector (Atum) and 0.5ug plasmid coding β3 or β3_N305T in PD2529 vector were incubated with 1 μL FectoPro (Polyplus) in 100 uL Opti-MEM (Gibco) for 10 min at room temperature were added per ml of Expi293F α5&αV KO cells (3*106/mL) in Expi293 expression medium (Thermo Fisher Scientific). 24 hrs after transfection, valproic acid and glucose were added to the culture to final concentrations of 3 mM and 0.4%, respectively. 48 hours post transfection, cells were washed twice with assay medium (Leibovitz’s L-15 medium and 0.1% BSA) containing 5 mM EDTA, twice with assay medium alone, and resuspended in assay medium or L15 supplemented with the indicated concentration of serum and 50 mM HEPES, pH7.4. For measuring the affinity of FITC-echistatin probe (United States Biological) to αIIbβ3 WT or αIIbβ3_N305T mutant, each 50-μl sample contained 106 cells/ml, 5 nM Alexa647 labeled MBC319.4 Fab (AF647-MBC319.4), and varying concentrations of FITC-echistatin. For competitive binding experiments, each 50-μl sample contained 106 cells/ml, indicated concentrations of FITC-echistatin, and varying concentrations of αIIbβ3 inhibitors. For LIBS antibody epitope exposure assays, each 50-μl sample contained 106 cells/ml, 20 nM Alexa647 labeled MBC319.4 Fab, and αIIbβ3 inhibitors at concentrations equaling100x their Kd values for αIIbβ3 wild type reported in Fig.5D. The mixture was allowed to equilibrate at 22°C for 2 h before flow cytometry (BD FAC SCanto II) without washing.
FITC-echistatin concentration (Cechistatin) dependent binding of 5nM AF647-MBC319.4 Fab (MFIobs) was fitted to Eq.3 with binding affinity of FITC-echistatin , MFI in absence of FITC-echistatin (MFI0), and MFI at saturating concentration of FITC-echistatin (MFIsat) as fitting parameters.
| (Eq.3) |
The competitive binding curves of the high affinity αIIbβ3 inhibitors, UR-2922 and roxifban), cannot be reliably fitted with the 3-parameter dose response curve (Eq.1) because their Kd values are smaller than the concentration range of integrins in the cell suspension. Under these conditions, binding to cells can significantly deplete the competitor from solution when used at low concentration, and the total concentration of competitor could not be used as an approximation of its free concentration. To solve this problem, we fit the competitive binding curves considering the competitor depletion effect. In detail, αIIbβ3 inhibitor concentration (Cinhibitor) dependent binding of FITC-echistatin (MFIobs), for two inhibitors with high binding affinity, UR-2922 and Roxifiban, was fitted with affinity of inhibitor , MFI in absence of inhibitor (MFI0), MFI at saturating concentration of inhibitor (MFIsat) and the integrin concentration (Cintegrin) expressed on transfectants as fitting parameters, with previously determined FITC-echistatin affinity , inhibitor concentration (Cinhibitor) and FITC-echistatin concentration (Cechistatin) as known parameters (Eq.4). Eq.4 was derived based on the definition of affinity and law of mass conservation detailed in Eqs. 5–10. Function F in Eq. 4 and its derivation are described in the Mathematica file Equation4.nb.
| (Eq.4) |
| (Eq.5) |
| (Eq.6) |
| (Eq.7) |
| (Eq.8) |
| (Eq.9) |
| (Eq.10) |
Global fitting of the competitive binding curves of UR-2922 and roxifiban, with , MFI0, and MFIsat as individual fitting parameters, and Cintegrin as a shared fitting parameter, showed that the effective integrin αIIbβ3 or αIIbβ3_N305T concentration, Cintegrin, at 106 cells/ml of transfectants could be confidently fitted as 15.3 ± 0.6 nM for the WT αIIbβ3 transfectant and 10.2 ± 0.3 nM for the αIIbβ3_N305T transfectant. The competitive binding curves of other αIIbβ3 inhibitors were fitted to Eq.4 in the same way, except that the Cintegrin determined using UR-2922 and roxifiban was used as a known parameter.
α4β1 LIBS antibody binding assays with secondary anti-IgG.
Jurkat cells were incubated with 10 μM compound, together with 10 μg/ml of HUTS-21 IgG or control mAb (mouse IgG) at 4°C in buffer that contained 20 mM HEPES, pH 7.2, 150 mM NaCl and 1% BSA, plus 1 mM MgCl2 and 1 mM CaCl2 or 1 mM MnCl2. After 30 min, cells were washed 3 times in buffers containing the same compound and metal ions, and FITC-conjugated goat anti-mouse IgG (Caltag Laboratories, Burlingame, CA) was used to detect HUTS-21 binding using flow cytometry.
Platelets LIBS exposure assay.
The study with human blood samples was approved by the Institutional Review Board of Medical College of Wisconsin. Blood samples anticoagulated with acid-citrate-dextrose were collected from healthy donors who had not taken any medication known to inhibit platelet function for at least a week. Platelet-rich plasma (PRP) was obtained by spinning the blood samples at 140×g for 12 min at RT. After adding prostaglandin E1 (Sigma, #P5515) to a final concentration of 50 ng/ml, the PRP was centrifuged at 1900×g for 10 min at RT. The platelet pellet was washed in Ringer’s citrate-dextrose buffer (108 mM NaCl, 3.8 mM KCl, 1.7 mM NaHCO3, 21.2 mM sodium citrate, 27.8 mM glucose, 1.1 mM MgCl2, pH 6.5) and resuspended in Tyrode’s buffer (137 mM NaCl, 2.7 mM KCl, 11.9 mM NaHCO3, 0.43 mM NaHPO4, 5.5 mM glucose, 20 mM Hepes, pH 7.4) supplemented with 1% BSA, 1mM CaCl2, and 1 mM MgCl2. 36 μl of washed platelets at 0.2×108/ml were mixed with 4 μl of buffer control or αIIbβ3 inhibitors and incubated for 20 min at RT. Subsequently, 5 μl of thrombin at 5 U/ml and 5 μl of Alexa647-conjugated MBC319.4 Fab at 250 nM were added to 40 μl of treated platelets for 30 min at RT. Final inhibitor concentrations were >70x KD measured in 0.1% BSA with WT αIIbβ3 transfectants. Platelet fluorescence without washing was measured by flow cytometry using LSRII (BD Biosciences). The assay was done on three different days with blood samples from two different donors.
Clot retraction.
261 μl of citrated-PRP was mixed with 29 μl of buffer control or αIIbβ3 inhibitors at different concentrations prepared in Tyrode’s buffer supplemented with 1% BSA for 20 min at RT. Clot retraction was initiated by the addition of 30 μl of 7.5 U/ml thrombin prepared in Tyrode’s buffer supplemented with 1% BSA to a glass tube containing 270 μl inhibitor-treated PRP. The clots were allowed to retract at 37°C. Images were taken at 15-min intervals. The whole reaction and clot areas were calculated with ImageJ software. The clot retraction was presented as 1–(clot area/whole reaction area). The IC50 for 30 and 90 mins of clot retraction was calculated by fitting the data with the function of inhibitor vs response (three parameters) in Prism 8. The assay was done three times with PRP from three donors.
Platelet aggregation.
Whole blood platelet aggregation was measured by impedance aggregometry using a Chrono-log Model 700 aggregometer. Sodium citrate anticoagulated blood was left at RT for 30min before testing and used within 3h. 0.5 ml of whole blood was incubated with 0.5 ml physiologic saline supplemented with different concentrations of αIIbβ3 inhibitors (stock solutions prepared in saline) for 5 min at 37°C without stirring. Aggregation was induced by adding 10 μl ADP (final concentration: 20μM). Platelet aggregation was recorded for 6 min with stirring at 1200rpm at 37°C. Data were presented as the relative aggregation by calculating the background-subtracted impedance (Ohms) as a percentage of impedance in absence of inhibitors. IC50 was calculated by fitting the data with the function of inhibitor vs response (three parameters) in Prism 8. The assay was done three times with whole blood from three different donors.
Integrin inhibitors.
Lamifiban (CAS 144412-49-7), sibrafiban (CAS 144412-18-0), and α4β1/α4β7 inhibitor compound Ro 29 (CAS 1421673-59-7) were generous gifts from Dr. Paul Gillespie (Roche, Nutley, NJ). RUC-4 was a generous gift of Barry Coller (Rockefeller U., NY, NY). EF-5154 (Nomoto et al., 2009) was provided by Meiji Seika Pharma Co. (Yokohama, Japan). Firategrast (CAS 402567-16-2) was purchased from ApexBio Technology (Houston, TX). UR-2922 (CAS 220386-56-1) was synthesized by HDH Pharma (Morrisville, NC). GR144053 (CAS 1215333-48-4) was purchased from Sigma-Aldrich. DS 13d (CAS 317353-73-4) (Chiba et al., 2005) was purchased as TCS 2314 from R&D Systems (Minneapolis, MN). DS 13g (CAS 819078-63-2) was synthesized using the reagents and conditions described in Schemes 1 and 2 and the text in (Chiba et al., 2005). Other compounds were synthesized as described in the protocols below, based on the following references: BMS4 (CAS 270593-59-4), BMS4-1 (CAS 270593-50-5), BMS4-2 (CAS 270088-87-4), and BMS4-3, (Smallheer et al., 2001); fradafiban (CAS 148396-36-5), (Himmelsbach et al., 1996); roxifiban (CAS 170902-52-0), (Zhang Lh et al., 1997); lotrafiban (CAS 171049-14-2), (Ma and Xia, 2001); gantofiban (CAS 167364-04-7) and the gantofiban analog (CAS 167364-14-9) (Gante et al., 1996). The synthetic route to M-tirofiban and its isomer were not published and were as described below. Purity of all compounds was greater than 95%, as confirmed by NMR, LC-MS, and/or elemental analyses.)
Synthetic protocols.
All solvents and reagents obtained from commercial sources were used without further purification. The final products were purified on Agela Cheetah purification system MP200 equipped with Hilic column (4g) with CH3CN/H2O as eluent. The 1H NMR spectra at 400 MHz were performed on a Bruker Avance DRX-400 spectrometer with the chemical shifts (δ in ppm) in the solvent CD3OD referenced at 3.31 ppm and D2O at 4.79 ppm, and coupling constants (J) were given in hertz. The 13C NMR spectra at 101 MHz were performed on the Bruker Avance DRX-400 spectrometer with the chemical shifts (δ in ppm) in the solvent CD3OD referenced at 49.0 ppm. The high resolution mass spectra (HRMS) were measured by Waters Xevo G2 QTof equipped with ESI.
Synthetic scheme of BMS4 (compound 2) (CAS 270593-59-4)
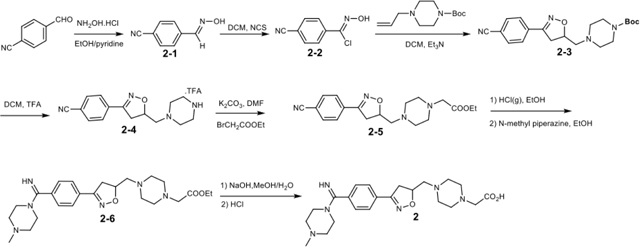
4-Cyanobenzaldoxime (2–1)
To a solution of 4-benzaldehyde (13.13 g, 100 mmol) in EtOH/pyridine (1:1, 200 mL) was added hydroxylamine hydrochloride portion-wise (7.64 g, 110 mmol) under N2 protection. After overnight stirring at room temperature, the solvent was removed half volume and ice water (200 mL) was added to obtain the precipitation, which was washed with ice water and dried to afford the title compound (9.76 g, 83%).
4-Cyanobenzaldoximinochloride (2–2)
To a solution of 4-cyanobenzaldoxime (10.96 g, 75 mmol) in anhydrous DMF (200 mL) was added N-chlorosuccimide (11.93 g, 90 mmol) in portion-wise at room temperature under N2 protection for 16 h. The reaction mixture was poured into ice water (250 mL) and the precipitation was collected by filtration, washed with water and dried in vacuo to afford the title compound (9.88 g, 73%).
tert-Butyl 4-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazine-1-carboxylate (2–3)
Potassium carbonate (18.66 g, 135 mmol) was added to a solution of tert-butyl piperazine-1-carboxylate (10.0 g, 54 mmol) in anhydrous DMF (100 mL) under N2 protection. The mixture was allowed to stir at room temperature for half an hour, followed by addition of allyl bromide (5.0 mL, 59.4 mmol) and 4 h stirring. Saturated sodium bicarbonate solution (50 mL) was then added and the mixture was extracted by ethyl acetate (150 mL×3). The organic layers were combined, washed by brine, dried over MgSO4, and concentrated to offer tert-butyl 4-allylpiperazine-1-carboxylate (10.02 g, 82%).
To a solution of 4-cyanobenzaldoximinochloride (5.42 g, 30 mmol) and tert-butyl 4-allylpiperazine-1-carboxylate (6.79 g, 30 mmol) in dry CH2Cl2 (150 mL) was dropwise added Et3N (3.33 g, 33 mmol) in dry CH2Cl2 (50 mL) in 2 h at room temperature. After stirring at room temperature for 16 h, saturated Na2CO3 solution (100 mL) was added and the organic phase was separated, dried over MgSO4, concentrated under vacuum and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:1) to offer the title product (6.89 g, 62%).
Ethyl 2-(4-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate (2–5)
To a solution of tert-butyl 4-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazine-1-carboxylate (3.70 g, 10 mmol) in CH2Cl2 (10 mL) was added trifluoroacetic acid (10 mL) at 0 °C. After stirring at room temperature for 2 h, the mixture was concentrated and dried. The residue was dissolved in DMF (50 mL), followed by the addition of anhydrous K2CO3 (4.14 g, 30 mmol) and ethyl bromoacetate (1.46 mL, 12 mmol) at room temperature under N2 protection. After stirring overnight, the mixture was diluted with ethyl acetate (100 mL) and successively washed with saturated NaHCO3 solution and brine. The organic phase was dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (ethyl acetate/petroleum ether, 5:1) to offer the title product (2.03g, 57%).
Ethyl 2-(4-((3-(4-(imino(4-methylpiperazin-1-yl)methyl)phenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate (2–6)
Acetyl chloride (3.5 mL) was added dropwise to dry EtOH (6.5 mL) at 0°C under N2 protection to produce 35% HCl/EtOH/EtOAc solution. To this freshly prepared solution was added 2-(4-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate(0.36 g, 1 mmol) at 0°C. After an additional 2 h stirring at 0°C, the mixture was stirred at room temperature for another 48 h, and then concentrated and dried in vacuo. The residue was dissolved in EtOH (5 mL), treated with N-methyl piperazine (1.00 g, 10 mmol) at room temperature under N2 protection for 16 h. The reaction mixture was concentrated, purified by silica gel chromatography (CH2Cl2:MeOH, 50:1 to 10:1) to offer the title product (0.22 g, 48%).
2-(4-((3-(4-(imino(4-methylpiperazin-1-yl)methyl)phenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetic acid (compound 2, BMS4)
Ethyl 2-(4-((3-(4-(imino(4-methylpiperazin-1-yl)methyl)phenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate (91 mg, 0.2 mmol) was hydrolyzed in methanol (1 mL) and H2O (1 mL), followed by the addition of NaOH (24 mg, 0.6 mmol) at room temperature. After stirring for 3 h, the resulting mixture was added 1N HCl (0.6 mL), and then concentrated, purified by hilic column chromatography to offer BMS4 (30 mg, 35%). 1H NMR (400 MHz, D2O) δ (ppm): 7.82(d, J = 7.2 Hz, 2H), 7.61(d, J = 7.2 Hz, 2H), 5.05(m, 1H), 3.93(s, 2H), 3.63(m, 5H), 3.29–2.77(m, 15H), 2.71(s, 3H); 13C NMR (101 MHz, D2O) δ (ppm): 170.1, 165.4, 158.3, 132.8, 129.3, 128.9, 127.7, 78.9, 60.2, 58.3, 52.7, 51.7, 49.7, 47.4, 43.3, 38.5; HRMS (m/z): [M+H]+ calcd. for C22H33N6O3: 429.2614; found 429.2613.
Synthetic scheme of BMS4–1 (compound 3) (CAS 270593-50-5)
![]()
Ethyl 2-(4-((3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate (3–1)
To freshly prepared HCl/EtOH/EtOAc solution (35%, 10 mL) was added 2-(4-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate (2–5) (0.36 g, 1 mmol). After 2 h at 0°C, the mixture was further stirred at room temperature for 48 h, and then concentrated and dried in vacuo. The residue was dissolved in EtOH (5 mL), treated with ammonium carbonate (1.01 g, 10 mmol) at room temperature under N2 protection for 16 h, concentrated, and purified by silica gel chromatography (CH2Cl2:MeOH, 50:1 to 10:1) to give the title product (0.29 g, 78%).
3-(4-((3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetic acid (BMS4–1, compound 3)
Ethyl 2-(4-((3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate (3–1, 0.07 g, 0.2 mmol) was hydrolyzed in MeOH/H2O (1/1, 2 mL) with NaOH (24 mg, 0.6 mmol) at room temperature for 3 h. The reaction was then quenched by 1N HCl (0.6 mL), concentrated and purified by silica column chromatography to offer BMS4–1 (29 mg, 42%). 1H NMR (400 MHz, CD3OD) δ (ppm): 7.94(d, J = 8.4 Hz, 2H), 7.89(d, J = 8.4 Hz, 2H), 5.28(m, 1H), 3.79(s, 2H), 3.72(m, 1H), 3.39–3.31(m, 11H); 13C NMR (101 MHz, CD3OD) δ (ppm):170.6, 167.8, 157.8, 135.7, 130.8, 129.6, 128.7, 78.7, 60.8, 57.4, 52.3, 51.3, 39.6; HRMS (m/z): [M+H]+ calcd. for C17H24N5O3: 346.1879; found 346.1870.
Synthetic scheme of BMS4–2 (CAS 270088-87-4) (compound 5)
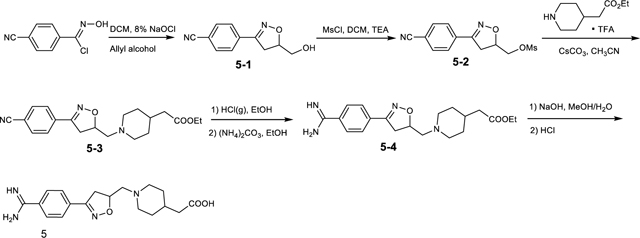
4-(5-(hydroxymethyl)-4,5-dihydroisoxazol-3-yl)benzonitrile (5-1)
To a vigorously stirred solution of 4-cyano-N-hydroxybenzimidoyl chloride (3.6g, 20 mmol) and allyl alcohol (2.32g, 40 mmol) in CH2Cl2 (100 mL) at 0°C was added 8% aqueous solution of NaOCl (37.3 mL, 40 mmol) dropwise, with the temperature below 5°C. The mixture was stirred at room temperature for an additional 15 min. Then the aqueous phase was separated and extracted with CH2Cl2 (100 mL×2). The combined organic layers were washed with brine, dried over MgSO4, and concentrated, and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:2) to offer the title compound (2.75g, 68%).
(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl methanesulfonate (5-2)
To a solution of 4-(5-(hydroxymethyl)-4,5-dihydroisoxazol-3-yl)benzonitrile(1.01 g, 5 mmol) and Et3N (0.84 mL, 6 mmol) in dry CH2Cl2 (20 mL), methanesulfonyl chloride (0.48 mL, 6.00 mmol) was added dropwise at 0°C. The mixture was stirred at room temperature for 2 h, and then quenched with water (10 mL). The aqueous layer was extracted with CH2Cl2 (20 mL×2), and the combined organic phases were successively washed with 1N HCl, saturated NaHCO3 solution, brinedried over MgSO4, and concentrated in vacuo to offer the crude title product (1.21 g, 86%).
Ethyl 2-(1-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperidin-4-yl)acetate (5-3)
To a solution of tert-butyl 4-(2-ethoxy-2-oxoethyl)piperidine-1-carboxylate(1.36 g, 5 mmol) in dry CH2Cl2 (10 mL) was added TFA (10 mL) at 0°C. The resulting mixture was stirred at room temperature for 2 h, and then concentrated and dried in vacuo without further purification. To a solution of (3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl methanesulfonate (1.12 g, 4 mmol) in acetonitrile was added CsCO3 (6.51 g, 20 mmol)and ethyl 2-(piperidin-4-yl)acetate (1.72 g, 5 mmol) at room temperature under N2 protection.The resulting mixture was refluxed for 16 h and then concentrated. The residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:1) to offer the title compound (0.82 g, 46%).
Ethyl 2-(1-((3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperidin-4-yl)acetate (5–4)
Ethyl 2-(1-((3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperidin-4-yl)acetate (0.36 g, 1 mmol) was added to freshly prepared HCl/EtOH/EtOAc solution (35%, 10 mL) at 0°C over 2 h, followed by stirring at room temperature for 48 h. The reaction mixture was then concentrated and dissolved in EtOH (5 mL), followed by the addition of ammonium carbonate (1.01 g, 10 mmol). After stirring at room temperature under N2 protection for 16 h, the mixture was concentrated in vacuo, purified by silica gel chromatography (CH2Cl2/MeOH, 50:1 to 10:1) to offer the title product (0.25 g, 68%).
2-(1-((3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperidin-4-yl)acetic acid (compound 5, BMS 4.2)
To a solution of ethyl 2-(1-((3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)methyl)piperidin-4-yl)acetate(74 mg, 0.2 mmol) in methanol/H2O (1:1, 2 mL) was added NaOH (24 mg, 0.6 mmol). After stirring at room temperature for 3 h, the mixture was quenched by 1N HCl (0.6 mL), concentrated and purified by silica column chromatography to offer BMS4–2 (26 mg, 38%). 1H NMR (400 MHz, CD3OD) δ (ppm): 7.95(d, J = 8.8 Hz, 2H), 7.89(d, J = 8.4 Hz, 2H), 5.34(m, 1H), 3.74(m, 1H), 3.68(m, 2H), 3.44(d, J = 6.2Hz, 2H),3.34(m, 1H), 3.14(t, J = 12.4Hz, 2H), 2.34(d, J = 6.4 Hz, 2H), 2.06(m, 3H), 1.65(m, 2H); 13C NMR (101 MHz, CD3OD) δ (ppm):175.7, 167.9, 157.7, 135.6, 131.0, 129.6, 128.7, 77.3, 60.7, 54.8, 54.0, 39.9, 31.6, 29.8; HRMS (m/z): [M+H]+ calcd. for C18H25N4O3: 345.1927; found 345.1927.
Synthetic scheme of BMS4–3 (compound 10)
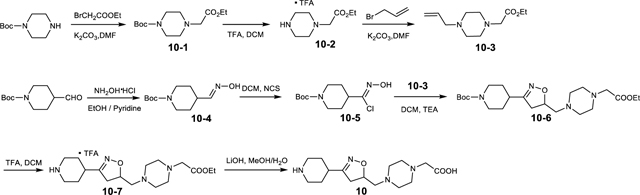
tert-Butyl 4-(2-ethoxy-2-oxoethyl)piperazine-1-carboxylate (10–1)
To a solution of tert-butyl piperazine-1-carboxylate (10.23 g, 55 mmol) in DMF (100 mL) was added K2CO3 (18.66 g, 135 mmol). After stirring at room temperature for 0.5 h, to the mixture was added ethyl bromoacetate (6.6 mL, 59.4mmol). The reaction mixture was stirred at room temperature for 4 h, quenched by saturated NaHCO3 solution (200 mL), and extracted with EtOAc (100 mL×3). The combined organic phases were washed with brine, dried over MgSO4 and concentrated to offer the title product (12.94 g, 88%).
Ethyl 2-(4-allylpiperazin-1-yl)acetate (10–3)
To a solution of tert-butyl 4-(2-ethoxy-2-oxoethyl)piperazine-1-carboxylate (5.74 g, 20 mmol) in CH2Cl2 (20 mL) was added TFA (20 mL) at 0°C. After stirring at room temperature for 2 h, the mixture was concentrated and dried in vacuo. The residue was dissolved in DMF (100 mL), followed by the addition of anhydrous K2CO3 (5.52 g, 40 mmol) and 3-bromopropene (2.03 mL, 24 mmol) at room temperature under N2 protection. After stirring overnight, the mixture was diluted with EtOAc (100 mL) and washed with H2O (100 mL), followed by the addition of saturated NaHCO3 solution and brine. The organic phase was dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (ethyl acetate/petroleum ether, 2:1) to offer the title product (3.31g, 78%).
tert-Butyl (E)-4-((hydroxyimino)methyl)piperidine-1-carboxylate (10–4)
To a solution of tert-butyl 4-formylpiperidine-1-carboxylate (10.67 g, 50 mmol) in 1:1 EtOH/pyridine (100 mL) was added hydroxylamine hydrochloride (3.83 g, 55 mmol) portion-wise under N2 atmosphere. After stirring overnight at room temperature, the solvent was removed half volume and ice water (100 mL) was added to obtain the precipitation, which was washed with ice water and dried to offer the title compound (8.68 g, 76%).
tert-Butyl (Z)-4-(chloro(hydroxyimino)methyl)piperidine-1-carboxylate (10–5)
To a solution of tert-butyl (E)-4-((hydroxyimino)methyl)piperidine-1-carboxylate (6.85 g, 30 mmol) in anhydrous DMF (100 mL) was added N-chlorosuccimide (3.01 g, 36 mmol) in portion-wise at room temperature under N2 protection for 16 h. The reaction mixture was poured into ice water (250 mL) and the precipitation was collected by filtration, washed with water and dried in vacuo to afford the title compound (5.44 g, 69%).
tert-Butyl 4-(5-((4-(2-ethoxy-2-oxoethyl)piperazin-1-yl)methyl)-4,5-dihydroisoxazol-3-yl)piperidine-1-carboxylate (10–6)
To a solution of tert-butyl (Z)-4-(chloro(hydroxyimino)methyl)piperidine-1-carboxylate (2.62 g, 10 mmol) and ethyl 2-(4-allylpiperazin-1-yl)acetate (2.12 g, 10 mmol) in dry CH2Cl2 (100 mL) was added dropwise Et3N (3.33 g, 33 mol) in dry CH2Cl2 (50 mL) in 2 h at room temperature. After stirring at room temperature for 16 h, saturated Na2CO3 solution (150 mL) was added into the mixture and the organic phase was dried over MgSO4, concentrated in vacuo and purified by silica gel chromatography (ethyl acetate/petroleum ether, 5:1) to offer the title product containing 10–3 (1.67 g).
Ethyl 2-(4-((3-(piperidin-4-yl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate trifluoroacetic acid salt (10–7)
To a solution of crude tert-butyl 4-(5-((4-(2-ethoxy-2-oxoethyl)piperazin-1-yl)methyl)-4,5-dihydroisoxazol-3-yl)piperidine-1-carboxylate (0.8 g) in CH2Cl2 (5 mL) was added TFA (5 mL) at 0 °C. After stirring at room temperature for 2 h, the mixture was concentrated and purified by silica column chromatography (CH2Cl2:MeOH, 50:1 to 10:1) to offer the title product (0.36 g).
2-(4-((3-(piperidin-4-yl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetic acid, BMS4–3 (10)
To a solution of ethyl 2-(4-((3-(piperidin-4-yl)-4,5-dihydroisoxazol-5-yl)methyl)piperazin-1-yl)acetate trifluoroacetic acid salt (81 mg, 0.2 mmol) in methanol (1 mL) and H2O (1 mL) was added NaOH (32 mg, 0.8 mmol). After stirring at room temperature for 3 h, to the mixture was added 1N HCl (0.8 mL). The resulting mixture was concentrated and purified by hilic column chromatography to offer BMS4–3 (11 mg, 18%). 1H NMR (400 MHz, D2O) δ (ppm): 4.97 (m, 1H), 3.75 (s, 2H), 3.53 to 3.31 (m, 7H), 3.12 (m, 6H), 2.87 (m, 4H), 2.22(m, 2H), 1.84 (m, 2H); 13C NMR (101 MHz, D2O) δ (ppm):170.2, 163.3, 76.8, 59.9, 51.4, 49.7, 43.3, 39.4, 32.5, 27.6, 25.5; HRMS (m/z): [M+H]+ calcd. for C15H27N4O3: 311.2083; found 311.2076.
Synthetic scheme of Fradafiban (CAS 148396-36-5)
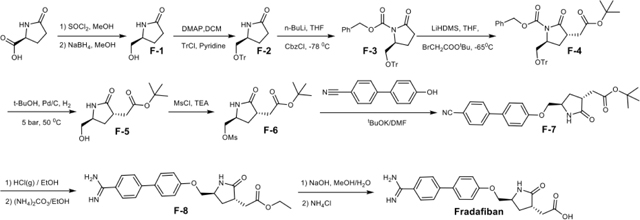
(S)-5-(Hydroxymethyl)-2-pyrrolidone (F-1)
SOCl2 (8.52 mL, 120 mmol) was added dropwise to a solution of (S)-5-oxopyrrolidine-2-carboxylic acid (12.91 g, 100 mmol) in MeOH (100 mL) at 0°C. The resulting mixture was stirred at room temperature for 16 h, concentrated in vacuo, and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:5) to offer intermediate methyl (S)-5-oxopyrrolidine-2-carboxylate (9.88 g, 69%). To this intermediate (7.16 g, 50 mmol) in MeOH (20 mL) was portion-wise added sodium borohydride (2.08 g, 55 mmol) under N2 protection. The mixture was stirred overnight, quenched by saturated NH4Cl (50 mL), and concentrated in vacuo. The residue was then dissolved in water (100 mL) and extracted with CH2Cl2 (100 mL×5). The combined organic phases were washed with brine, dried over MgSO4, and concentrated to offer the crude title compound (3.01 g, 52%).
(5S)-5-Trithloxymethyl-2-pyrrolinone (F-2)
To a solution of (S)-5-hydroxymethyl-2-pyrrolidinone (2.30 g, 20.0 mmol), Et3N (4.2 mL, 30.0 mmol), DMAP (300 mg, 2.67 mmol) in dry CH2Cl2 (200 mL) was added triphenylmethyl chloride (8.40 g, 30.0 mmol) over 5 min at room temperature. After stirring overnight, the mixture was washed with water, dried over MgSO4, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:5) to offer the title product (8.25 g, 75%).
(S)-Benzyl 2-oxo-5-((trityloxy)methyl)pyrrolidine-1-carboxylate (F-3)
To a solution of (5S)-5-trityloxymethyl-2-pyrrolinone (7.55 g, 20 mmol) in dry THF (100 mL) was added a 2.5N solution of n-butyllithium in n-hexane(8.4 mL, 21 mmol) at −78°C. After stirring for 30 min, a solution of benzyl chloroformate (2.86 mL, 21 mmol) was added dropwise. The reaction mixture was stirred at −78 °C for 8 h, then quenched with saturated NH4Cl (20 mL). The mixture was concentrated to remove THF, and extracted with diethyl ether (30 mL×3). The combined organic phases were washed with brine, dried over MgSO4, concentrated in vacuo and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:10) to offer the title compound (4.12 g, 89%).
(3S,5S)-Benzyl 3-(2-(tert-butoxy)-2-oxoethyl)-2-oxo-5-((trityloxy)methyl)pyrrolidine-1-carboxylate (F-4)
To a solution of (S)-benzyl 2-oxo-5-((trityloxy)methyl)pyrrolidine-1-carboxylate (4.92 g, 10 mmol) in THF (50 mL) was added dropwise lithium hexamethyldisilazide in THF (1 M, 10.5 mL) at −65°C under N2 protection. After 20 min, a solution of tert-butyl bromoacetate (1.70 mL, 10.5 mmol) in THF (5 mL) was added dropwise. After stirring at −65°C for 2 h, the mixture was warmed to 0°C, followed by the addition of a saturated NH4Cl solution (20 mL). The mixture was concentrated in vacuo to remove THF, and extracted with EtOAc (20 mL×3). The organic phases were combined and washed with brine, dried over MgSO4, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:4) to afford the title product (5.01 g, 76%).
tert-Butyl 2-((3S,5S)-5-(hydroxymethyl)-2-oxopyrrolidin-3-yl)acetate (F-5)
To a solution of (3S,5S)-benzyl 3-(2-(tert-butoxy)-2-oxoethyl)-2-oxo-5-((trityloxy)methyl)pyrrolidine-1-carboxylate (3.03 g, 5 mmol) in t-butanol (30 mL) was added 10% Pd/C (0.45 g) under a hydrogen pressure of 5 bars. The resulting mixture was stirred for 1.5 days at 50°C. Then the mixture was filtered off and the filtrate was concentrated in vacuo. The residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 5:1) to offer the title product (0.66 g, 58%).
tert-Butyl 2-((3S,5S)-5-(((4’-cyano-[1,1’-biphenyl]-4-yl)oxy)methyl)-2-oxopyrrolidin-3-yl)acetate (F-7)
Methylsulfonyl chloride (0.21 mL, 2.65 mmol) was added dropwise to a solution of tert-butyl 2-((3S,5S)-5-(hydroxymethyl)-2-oxopyrrolidin-3-yl)acetate (0.57 g, 2.5 mmol) and Et3N (0.35 mL, 2.65 mmol) in dry DMF (15 mL) at 0°C. After stirring at room temperature for 8 h, the mixture was quenched with water (20 mL), extracted by CH2Cl2 (20 mL×3), dried over MgSO4, and concentrated to offer crude intermediate F-6 (0.66 g, 86%).
t-BuOK (0.34g, 3 mmol) was added to a solution of 4’-cyano-4-hydroxybiphenyl (0.59g, 3mmol) in DMF (15 mL). After stirring at room temperature for 1 h, F-6 (0.46 g, 1.5 mmol) was added under N2 protection. The resulting mixture was stirred for 2 days at room temperature. The reaction was quenched by water (20 mL) and extracted by CH2Cl2 (20 mL×3). The combined organic phases were washed with brine, dried over MgSO4, concentrated in vacuo and purified by silica gel chromatography (ethyl acetate/petroleum ether, 10:1) to provide the title product (0.35 g, 58%).
Ethyl 2-((3S,5S)-5-(((4’-carbamimidoyl-[1,1’-biphenyl]-4-yl)oxy)methyl)-2-oxopyrrolidin-3-yl)acetate (F-8)
tert-Butyl 2-((3S,5S)-5-(((4’-cyano-[1,1’-biphenyl]-4-yl)oxy)methyl)-2-oxopyrrolidin-3-yl)acetate (0.20 g, 0.5 mmol) was added to freshly prepared HCl/EtOH/EtOAc solution (35%, 5 mL) at 0°C over 2 h, and the resulting mixture was stirred at room temperature for 48 h. The mixture was then concentrated, dried and dissolved in EtOH (5 mL), followed by the addition of ammonium carbonate (0.50 g, 5 mmol). After stirring at room temperature under N2 protection for 16 h, the mixture was concentrated and purified by silica gel chromatography (CH2Cl2/MeOH, 50:1 to 10:1) to offer the title product (0.15 g, 73%).
2-((3S,5S)-5-(((4’-carbamimidoyl-[1,1’-biphenyl]-4-yl)oxy)methyl)-2-oxopyrrolidin-3-yl)acetic acid (Fradafiban)
To a solution of ethyl 2-((3S,5S)-5-(((4’-carbamimidoyl-[1,1’-biphenyl]-4-yl)oxy)methyl)-2-oxopyrrolidin-3-yl)acetate (79 mg, 0.2 mmol) in methanol (1 mL) and H2O (1 mL) was added NaOH (24 mg, 0.6 mmol). After stirring at room temperature for 3 h, to the mixture was added NH4Cl (64 mg, 1.2 mmol), followed by 1 mL of water and stirring for 45 min in an ice/water bath. The precipitate was suction filtered, washed with methanol and water/methanol (2:1), and dried to give Fradifiban (43 mg, 58%). 1H NMR (400 MHz, CD3OD) δ (ppm): 7.87 (dd, J1 = 8.8 Hz, J2 = 8.8 Hz, 4H), 7.69 (d, J = 8.7 Hz, 2H), 7.11 (d, J = 8.8 Hz, 2H), 4.11 (m, 1H), 4.03 (m, 2H), 3.02 (m, 1H), 2.81 (dd, J1 = 16.8 Hz, J2 = 4.1 Hz, 1H), 2.47 (dd, J1 = 16.8 Hz, J2 = 8.9 Hz, 1H), 2.39 (m, 1H), 2.15 (m, 1H); 13C NMR (101 MHz, CD3OD) δ (ppm):175.4, 168.2, 160.9, 147.8, 133.0, 130.8, 129.6, 129.5, 128.2, 127.3, 116.3, 71.9, 53.2, 38.7, 36.3, 31.0; HRMS (m/z): [M+H]+ calcd. for C20H22N3O4: 368.1610; found 368.1603.
Synthetic scheme of Roxifiban (CAS 170902-52-0)
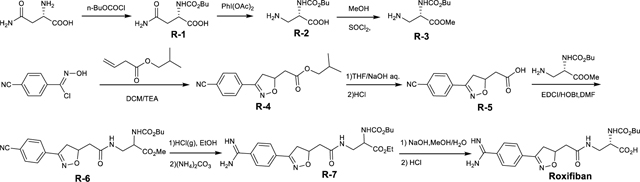
Nα-n-Boc-L-asparagine (R-1)
To a solution of L-asparagine (6.60 g, 50 mmol) in water (100 mL) was added Na2CO3 (6.62 g, 62.5 mol) and THF (50 mL). The mixture was heated to 50–55°C, and a solution of n-butyl chloroformate (8.19 g, 60 mmol) in THF (50 mL) was added over a period of 1 h. The reaction was held at 50–55 °C for an additional 1 h and then cooled to 15°C. The pH of the reaction mixture was adjusted to 2.5–3.5 with 11% aqueous HCl. The crude product was isolated by filtration, and dried in vacuo to offer the crude title product (8.83 g, 76%).
(S)-3-amino-2-((butoxycarbonyl)amino)propanoic acid (R-2)
To a solution of n-propanol (24 mL), EtOAc (24 mL) and water (12 mL) at 5°C was added Nα-n-Boc-L-asparagine (6.97 g, 30 mmol) and iodosobenzene diacetate (11.12 g, 34.5 mmol) under N2 protection. The mixture was warmed to 25°C over 1 h and stirred at the same temperature for an additional 2 h, then slowly heated to 50°C over 90 min and cooled to 5°C. The reaction mixture was held at 5°C for 30 min, filtered, washed with EtOAc and dried in vacuo to offer the crude title product (4.24 g, 69%).
(S)-methyl 3-amino-2-((butoxycarbonyl)amino)propanoate hydrochloride (R-3)
To a solution of (S)-3-amino-2-((butoxycarbonyl)amino)propanoic acid (3.06 g, 15 mmol) in methanol (50 mL) was dropwise added thionyl chloride (1.30 mL, 18 mmol) at 0°C. The resulting mixture was stirred at room temperature for 4 h, then concentrated in vacuo. The residue was used without further purification in the subsequent reaction.
Isobutyl 2-(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)acetate (R-4)
To a solution of 4-cyanobenzaldoximinochloride(1.80 g, 10 mmol) and isobutyl vinylacetate (1.42 g, 10 mmol) in CH2Cl2 (50 mL) was added Et3N (1.66 mL, 12 mmol) over a period of 2 h at room temperature. After stirring for 16 h, saturated NaHCO3 solution (50 mL) was added into the mixture. The resulting mixture was extracted by CH2Cl2 (50 mL), washed with water, dried over MgSO4, filtered, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:5) to offer the title compound (1.89 g, 66%).
2-(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)acetic acid (R-5)
To a solution of isobutyl 2-(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)acetate (1.43 g, 5 mmol) in THF (10 mL) and H2O (10 mL) was added NaOH (360 mg, 9 mmol) at room temperature. After stirring for 3 h, to the mixture was added 12 mL of 1N HCl and extracted with EtOAc (15 mL×3). The combined organic phases were washed with brine, dried over MgSO4, filtered, concentrated in vacuo to offer the crude title product (0.99 g, 88%) without further purification.
(2S)-methyl 2-((butoxycarbonyl)amino)-3-(2-(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)acetamido)propanoate (R-6)
The crude product 2-(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)acetic acid (0.92 g, 4 mmol) was dissolved in DMF (15 mL), followed by the addition of EDCI (0.59 g, 4.4 mmol) and HOBt (0.74 g, 4.4 mmol) at 0°C. After stirring for half an hour, to the resulting mixture was added DIEA (1.03 g, 8 mmol) and (S)-methyl 3-amino-2-((butoxycarbonyl)amino)propanoate hydrochloride (1.40 g, 4.4 mmol) at 0°C. After stirring at room temperature for 16 h, water (20 mL) was added, and the mixture was extracted with EtOAc (20 mL × 3). The combined organic phases were washed with brine, dried over MgSO4, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:3) to offer the title compound 0.53 g (28%).
(2S)-ethyl 2-((butoxycarbonyl)amino)-3-(2-(3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)acetamido)propanoate (R-7)
To freshly prepared HCl/EtOH/EtOAc solution (35%, 10 mL) was added (2S)-methyl 2-((butoxycarbonyl)amino)-3-(2-(3-(4-cyanophenyl)-4,5-dihydroisoxazol-5-yl)acetamido)propanoate (0.43 g, 1 mmol) at 0°C for 2 h, and the resulting mixture was stirred at room temperature for 48 h and then concentrated in vacuo. The residue was dried in vacuo, dissolved in EtOH (5 mL), followed by the addition of ammonium carbonate (1.01 g, 10 mmol). After stirring at room temperature under N2 for 16 h, the mixture was concentrated in vacuo, purified by silica gel chromatography (CH2Cl2/MeOH, 50:1 to 10:1) to offer the title product (0.32 g, 70%).
(2S)-2-((butoxycarbonyl)amino)-3-(2-(3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)acetamido)propanoic acid (Roxifiban)
To a solution of (2S)-ethyl 2-((butoxycarbonyl)amino)-3-(2-(3-(4-carbamimidoylphenyl)-4,5-dihydroisoxazol-5-yl)acetamido)propanoate (92 mg, 0.2 mmol) in methanol/H2O (1:1, 2 mL) was added NaOH (24 mg, 0.6 mmol). After stirring at room temperature for 3 h, 1N HCl (0.6 mL) was added to the mixture. The resulting mixture was concentrated in vacuo and purified by hilic column chromatography to offer Roxifiban (31 mg, 38%). 1H NMR (400 MHz, CD3OD) δ (ppm): 7.86(s, 4H), 5.18(m, 1H), 4.36(t, J = 6.5 Hz, 1H), 4.03(m, 2H), 3.67(m, 2H), 3.57–3.50(m, 1H), 3.37–3.30(m, 1H), 2.70(m, 2H), 1.55(m, 2H), 1.31(m, 2H), 0.86(m, 3H); 13C NMR (101 MHz, CD3OD) δ (ppm):172.8, 166.1, 163.1, 162.8, 158.3, 133.6, 129.5, 128.3, 127.5, 135.7, 130.8, 129.6, 128.7, 78.6, 65.8, 53.8, 40.5, 40.0, 39.1, 30.3, 18.4; 12.8. HRMS (m/z): [M+H]+ calcd. for C20H28N5O6: 434.2040; found 434.2040.
Synthetic scheme of Lotrafiban (CAS 171049-14-2)
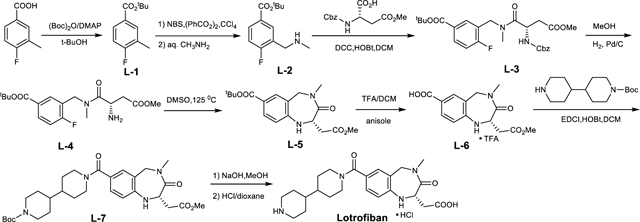
tert-Butyl 4-fluoro-3-methylbenzoate (L-1)
To a solution of 4-fluoro-3-methylbenzoic acid (3.08 g, 20 mmol), di-tert-butyl dicarbonate (8.75 g, 40 mmol) in 50 mL of 2-methylpropan-2-ol was added N,N-dimethylpyridin-4-amine (1.23 g, 10mmol). The resulting mixture was stirred at 25°C for 12 h. Then the mixture was concentrated to give a residue and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:40) to offer the title compound (3.2 g, 76%).
tert-Butyl 4-fluoro-3-methylaminomethylbenzoate (L-2)
To a solution of tert-butyl 4-fluoro-3-methylbenzoate (3.15 g, 15 mmol) in CCl4 (50 mL) was added (PhCO2)2 (1.37 g, 5.7 mmol) and NBS (12.1 g, 18 mmol). The resulting mixture was refluxed for 2 days under N2 protection. After cooling to room temperature, the mixture was washed with water (50 mL), and dried over MgSO4. The solvent was removed and the residue was dissolved in THF (25 mL). A solution of 33% CH3NH2 (4.89 mL, 52.5 mmol) in THF (25 mL) and H2O (12.5 mL) was added dropwise to the mixture. The resulting mixture was stirred for 1 h at room temperature and then concentrated in vacuo. The residue was dissolved by water (50 mL) and extracted with CH2Cl2 (50 mL×3). The combined organic phases were washed with brine, dried over MgSO4, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 5:1) to offer the title compound (1.51 g, 42%).
(S)-tert-butyl 3-((2-(((benzyloxy)carbonyl)amino)-4-methoxy-N-methyl-4-oxobutanamido)methyl)-4-fluorobenzoate (L-3)
To a solution of tert-butyl-4-fluoro-3-methylaminomethylbenzoate (1.20 g, 5 mmol), (S)-2-(((benzyloxy)carbonyl)amino)-4-methoxy-4-oxobutanoic acid (1.41 g, 5 mmol) and HOBt (0.74 g, 5.5 mmol) in dry CH2Cl2 (25 mL) was added a solution of DCC (1.13 g, 5.5 mmol) in CH2Cl2 (10 mL) at room temperature. After stirring for 1 h, the insoluble solid was filtered off, and the filtrate was washed with brine, dried over MgSO4, concentrated in vacuo and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:3) to offer the title compound (1.73 g, 69%).
(S)-tert-butyl 3-((2-amino-4-methoxy-N-methyl-4-oxobutanamido)methyl)-4-fluorobenzoate (L-4)
To a solution of (S)-tert-butyl 3-((2-(((benzyloxy)carbonyl)amino)-4-methoxy-N-methyl-4-oxobutanamido)methyl)-4-fluorobenzoate in methanol (1.51 g, 3 mmol) was added 10% Pd/C (0.15 g) with H2 balloon at room temperature for 4 h. The catalyst was filtered off, and the filtrate was concentrated to offer the crude title product (0.98 g, 89%).
(S)-tert-butyl 2-(2-methoxy-2-oxoethyl)-4-methyl-3-oxo-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine-7-carboxylate (L-5)
A mixture of (S)-tert-butyl 3-((2-amino-4-methoxy-N-methyl-4-oxobutanamido)methyl)-4-fluorobenzoate (0.39 g, 1.06 mmol) in DMSO (10 mL) was stirred at 125°C for 12 h under N2 protection. The resulting mixture was dissolved in water (20 mL) and extracted with CH2Cl2 (20 mL×3). The combined organic phases were washed with brine, dried over MgSO4, filtered, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:1) to offer the title compound (0.19 g, 49%).
(S)-tert-butyl 1’-(2-(2-methoxy-2-oxoethyl)-4-methyl-3-oxo-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine-7-carbonyl)-[4,4’-bipiperidine]-1-carboxylate (L-7)
To a solution of (S)-tert-butyl 2-(2-methoxy-2-oxoethyl)-4-methyl-3-oxo-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine-7-carboxylate(120mg, 0.35mmol) in CH2Cl2 (10 mL) was added TFA (5 mL) and anisole (0.2 mL). After stirring for 3 h, the solvent was evaporated and the residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 5:1) to give the intermediate acid, which was dissolved in dry CH2Cl2 (5 mL). To this solution was added tert-butyl (4,4’-bipiperidine)-1-carboxylate (103 mg, 0.38 mmol), HOBt (52 mg, 0.38 mmol) and EDCI (73 mg, 0.38 mmol). The resulting mixture was stirred for 1 h at room temperature, and diluted with water (10 mL), then extracted with CH2Cl2 (10 mL×3). The combined organic phases were washed with brine, dried over MgSO4, concentrated and purified by silica gel chromatography (ethyl acetate/petroleum ether, 1:3) to offer the title compound (56 mg, 30%).
(S)-2-(7-([4,4’-bipiperidine]-1-carbonyl)-4-methyl-3-oxo-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepin-2-yl)acetic acid (Lotrafiban)
To a solution of (S)-tert-butyl 1’-(2-(2-methoxy-2-oxoethyl)-4-methyl-3-oxo-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine-7-carbonyl)-[4,4’-bipiperidine]-1-carboxylate (33 mg, 0.06 mmol) in methanol (1 mL) and H2O (1 mL) was added NaOH (12 mg, 0.3 mmol). After stirring at room temperature for 8 h, the mixture was concentrated in vacuo. The residue was dissolved in dioxane (2 mL) and then treated with 4 N HCl/dioxane (1 mL) solution for 3 h. The resulting mixture was concentrated in vacuo and purified by hilic column chromatography to offer Lotrafiban (11 mg, 39%). 1H NMR (400 MHz, CD3OD) δ (ppm): 7.11(m, 2H), 6.62(d, J = 8.8 Hz, 1H), 5.58(d, J = 16.4 Hz, 1H), 3.89(d, J = 16.6 Hz, 1H), 3.41(m, 2H), 3.09(s, 3H), 2.95(m, 4H), 2.65(m, 1H), 2.41 – 2.31 (m, 2H), 1.99(m, 2H), 1.80(m, 2H), 1.48(m, 5H), 1.25(m, 3H); 13C NMR (101 MHz, CD3OD) δ (ppm):174.7, 172.6, 172.2, 149.5, 130.7, 129.5, 124.3, 119.5, 116.8, 53.8, 51.9, 45.5, 41.8, 39.7, 36.3, 34.5, 27.2; HRMS (m/z): [M+H]+ calcd. for C23H33N4O4: 429.2502; found 429.2490.
Synthetic scheme of compound 11 (Gantofiban analog) (CAS 167364-14-9)
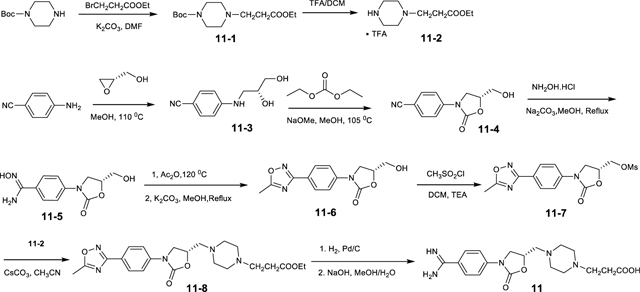
tert-Butyl 4-(3-Ethoxy-3-oxopropyl)-1-piperazinecarboxylate (11–1)
To a solution of tert-butyl-1-piperazine carboxylate (1.86 g, 10 mmol) in DMF (30 ml) was added potassium carbonate (4.86 g, 20 mmol). After 0.5 h, ethyl 3-bromopropanoate (1.29 ml, 10 mmol) was added to the suspension, and the reaction mixture was stirred overnight at room temperature, quenched by saturated NaHCO3 solution (60 mL), extracted with EtOAc (30 mL×3). The combined organic phases were washed with brine, dried over MgSO4 and concentrated to offer the crude title product (2.43 g, 85%).
Ethyl 3-(piperazin-1-yl)propanoate trifluoroacetic acid salt (11–2)
To a solution of tert-Butyl 4-(3-Ethoxy-3-oxopropyl)-1-piperazinecarboxylate (1.43 g, 5 mmol) in CH2Cl2 (10 mL) was added TFA (10 mL) at 0°C. After stirring at room temperature for 2 h, the mixture was concentrated in vacuo, dried in vacuo to offer the crude title compound (1.45 g).
(R)-4-((2,3-dihydroxypropyl)amino)benzonitrile (11–3)
A mixture of 4-aminobenzonitrile (11.8 g, 100 mmol) and (S)-oxiran-2-ylmethanol (3.76 mL, 56 mmol) in MeOH (80 mL) in a sealed tube was heated at 110°C for 3 h. The resulting mixture was concentrated in vacuo, the residue was dissolved in water (300 mL) and extracted with ethyl acetate (300 mL×3). The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (ethyl acetate/petroleum ether, 3:1) to offer the title product (2.86 g, 15%).
(R)-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzonitrile (11–4)
To a mixture of (R)-4-((2,3-dihydroxypropyl)amino)benzonitrile (2.30 g, 12 mmol) in diethyl carbonate (30 mL, 208 mmol) was added 25% sodium methoxide in methanol (1 mL). After the mixture was stirred overnight at 105°C, additional 25% sodium methoxide in methanol (1 mL) was added. After stirring for 5 h, the reaction was quenched by saturated NaHCO3 solution (60 mL), and extracted with EtOAc (60 mL×3). The combined organic phases were washed with brine, dried over MgSO4 and concentrated, purified by silica column chromatography (ethyl acetate/petroleum ether, 1:1) to offer the title product (1.75 g, 67%).
(R)-N’-hydroxy-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzimidamide (11–5)
To a solution of (R)-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzonitrile (1.10 g, 5 mmol) in MeOH (50 mL) was added NH2OH·HCl (1.04 g, 15 mmol) and Na2CO3 (2.12 g, 20 mmol). After stirring for 10 min at room temperature, the reaction mixture was heated to 80 °C and stirred for 6 h. The solvent was removed in vacuo, and the residue was dissolved in water (30 mL), extracted with CH2Cl2 (30 mL×3). The combined organic phases were washed by saturated NaHCO3, dried over MgSO4, and concentrated to offer the crude title compound (0.58 g, 46%).
(R)-5-(hydroxymethyl)-3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)oxazolidin-2-one (11–6)
(R)-N’-hydroxy-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzimidamide (0.50 g, 2 mmol) was dissolved in Ac2O (20 mL), and the mixture was refluxed for 4 h. Then the solvent was removed under the vacuum. The intermediate was dissolved in MeOH (20 mL), to the mixture was added K2CO3 (0.35 g, 2.5 mmol) and refluxed for 6 h. The solvent was removed in vacuo, and the residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 2:1) to obtain the title compound (0.39 g, 73%).
(R)-(3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl methanesulfonate (11–7)
To a solution of (R)-5-(hydroxymethyl)-3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)oxazolidin-2-one (0.33 g, 1.2 mmol) and Et3N (0.20 mL, 1.44 mmol) in dry CH2Cl2 (10 mL), methanesulfonyl chloride (0.12 mL, 1.44 mmol) was added dropwise at 0°C. The mixture was stirred at room temperature for 2 h and then quenched with water (10 mL). The aqueous layer was extracted with CH2Cl2 (10 mL×2), and the combined organic phases were successively washed with 1N HCl, saturated NaHCO3 solution, brine, dried over MgSO4, and concentrated in vacuo to offer the crude title product (0.36 g, 86%).
Ethyl (S)-3-(4-((3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl)piperazin-1-yl)propanoate (11–8)
To a solution of (R)-(3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl methanesulfonate (0.22 g, 0.6 mmol) in acetonitrile was added CsCO3 (0.98 g, 3 mmol) and ethyl 3-(piperazin-1-yl)propanoate trifluoroacetic acid salt (11–2) (0.21 g, 0.75 mmol) at room temperature under N2 protection. The resulting mixture was refluxed for 16 h and then concentrated. The residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 10:1) to offer the title compound (0.16 g, 38%).
(S)-3-(4-((3-(4-carbamimidoylphenyl)-2-oxooxazolidin-5-yl)methyl)piperazin-1-yl)propanoic acid (11, Gantofiban analog)
To a solution of ethyl (S)-3-(4-((3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl)piperazin-1-yl)propanoate (0.088 g, 0.2 mmol) in MeOH (5 mL) was added 10% Pd/C (20% w/w). The suspension was vigorously stirred under H2 atmosphere overnight. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo to obtain the crude intermediate To the intermediate dissolved in methanol/H2O (1:1, 2 mL) was added NaOH (24 mg, 0.6 mmol). After stirring at room temperature for 3 h, the mixture was quenched by 1N HCl (0.6 mL), concentrated and purified by hilic column chromatography to offer the Gantofiban analog (5.6 mg, 6%). 1H NMR (400 MHz, CD3OD) δ (ppm): 7.86(s, 4H), 4.98 – 4.92(m, 1H), 4.25(t, J = 8.0Hz, 1H), 3.98 – 3.88(m, 1H), 3.35 – 3.21(m, 8H), 2.92 – 2.88 (m, 4H), 2.67(t, J = 8.0Hz, 2H).
Synthetic scheme of Gantofiban (CAS 167364-04-7)
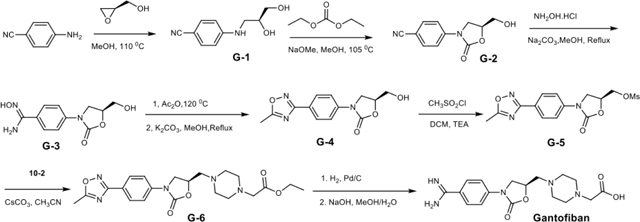
(S)-4-((2,3-dihydroxypropyl)amino)benzonitrile (G-1)
A mixture of 4-aminobenzonitrile (11.8 g, 100 mmol) and (R)-oxiran-2-ylmethanol (3.76 mL, 56 mmol) in MeOH (80 mL) in a sealed tube was heated at 110°C for 3 h. The resulting mixture was concentrated in vacuo, the residue was dissolved in water (300 mL) and extracted with ethyl acetate (300 mL×3). The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (ethyl acetate/petroleum ether, 3:1) to offer the title product (2.38 g, 12%).
(S)-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzonitrile (G-2)
To a mixture of (S)-4-((2,3-dihydroxypropyl)amino)benzonitrile (2.30 g, 12 mmol) in diethyl carbonate (30 mL, 208 mmol) was added 25% sodium methoxide in methanol (1 mL). After the mixture was stirred overnight at 105°C, additional 25% sodium methoxide in methanol (1 mL) was added. After stirring for 5 h, the reaction was quenched by saturated NaHCO3 solution (60 mL), extracted with EtOAc (60 mL×3). The combined organic phases were washed with brine, dried over MgSO4 and concentrated, purified by silica column chromatography (ethyl acetate/petroleum ether, 1:1) to offer the title product (1.85 g, 70%).
(S)-N’-hydroxy-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzimidamide (G-3)
To a solution of (S)-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzonitrile (1.10 g, 5 mmol) in MeOH (50 mL) was added NH2OH·HCl (1.04 g, 15 mmol), Na2CO3 (2.12 g, 20 mmol). After stirring for 10 min at room temperature, the reaction mixture was heated to 80°C and stirred for 6 h. The solvent was removed in vacuo, and the residue was dissolved in water (30 mL) and extracted with CH2Cl2 (30 mL×3). The combined organic phases were washed by saturated NaHCO3, dried over MgSO4, and concentrated to offer the crude title compound (0.61 g, 48%).
(S)-5-(hydroxymethyl)-3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)oxazolidin-2-one (G-4)
(S)-N’-hydroxy-4-(5-(hydroxymethyl)-2-oxooxazolidin-3-yl)benzimidamide (0.50 g, 2 mmol) was dissolved in Ac2O (20 mL), and the mixture was refluxed for 4 h. Then the solvent was removed under the vacuum. The intermediate was dissolved in MeOH (20 mL), to the mixture was added K2CO3 (0.35 g, 2.5 mmol) and refluxed for 6 h. The solvent was removed in vacuo, and the residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 2:1) to obtain the title compound (0.44 g, 78%).
(S)-(3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl methanesulfonate (G-5)
To a solution of (R)-5-(hydroxymethyl)-3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)oxazolidin-2-one (0.33 g, 1.2 mmol) and Et3N (0.20 mL, 1.44 mmol) in dry CH2Cl2 (10 mL), methanesulfonyl chloride (0.12 mL, 1.44 mmol) was added dropwise at 0°C. The mixture was stirred at room temperature for 2 h, and then quenched with water (10 mL). The aqueous layer was extracted with CH2Cl2 (10 mL×2), and the combined organic phases were successively washed with 1N HCl, saturated NaHCO3 solution, brine, dried over MgSO4, and concentrated in vacuo to offer the crude title product (0.35 g, 85%).
Ethyl (R)-3-(4-((3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl)piperazin-1-yl)acetate (G-6)
To a solution of (S)-(3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl methanesulfonate (0.22 g, 0.6 mmol) in acetonitrile was added CsCO3 (0.98 g, 3 mmol) and ethyl 3-(piperazin-1-yl)acetate trifluoroacetic acid salt (10–2) (0.20 g, 0.75 mmol) at room temperature under N2 protection. The resulting mixture was refluxed for 16 h and then concentrated. The residue was purified by silica gel chromatography (ethyl acetate/petroleum ether, 10:1) to offer the title compound (0.17 g, 41%).
(R)-3-(4-((3-(4-carbamimidoylphenyl)-2-oxooxazolidin-5-yl)methyl)piperazin-1-yl)acetic acid (Gantofiban)
To a solution of ethyl (S)-3-(4-((3-(4-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)-2-oxooxazolidin-5-yl)methyl)piperazin-1-yl)acetate (0.085 g, 0.2 mmol) in MeOH (5 mL) was added 10% Pd/C (20% w/w). The suspension was vigorously stirred under H2 atmosphere overnight. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo to obtain the crude intermediate. To the intermediate dissolved in methanol/H2O (1:1, 2 mL) was added NaOH (24 mg, 0.6 mmol). After stirring at room temperature for 3 h, the mixture was quenched by 1N HCl (0.6 mL), concentrated and purified by hilic column chromatography to offer Gantofiban (5.8 mg, 8%). 1H NMR (400 MHz, D2O) δ (ppm): 7.87(d, J = 8.0Hz, 2H), 7.74(d, J = 8.0Hz, 2H), 5.27 – 5.20 (m, 1H), 4.41 (t, J = 8.0Hz, 1H), 3.98 – 3.94 (m, 1H), 3.84 (s, 2H), 3.55 (br, 4H), 3.46 – 3.30 (m, 6H). 13C NMR (101 MHz, D2O) δ (ppm):168.9, 167.5, 155.9, 144.9, 130.0, 123.9, 119.2, 72.6, 60.4, 56.9, 53.1, 51.7, 50.1; HRMS (m/z): [M+H]+ calcd. for C17H24N5O4: 362.1828; found 362.1823.
M-tirofiban and M-tirofiban isomer
Synthesis of M-tirofiban was delayed by lack of publication of its synthetic route (Adair et al., 2020). Our first attempt of stereo-specific synthesis resulted in a major product with a small amount of side product. A second synthesis resulted in M-tirofiban and its stereo-isomer, M-tirofiban isomer. Neither the side-product nor the stereoisomers would be distinguished by the reported mass spectrometry results (Adair et al., 2020).
Final synthetic route (scheme 1)
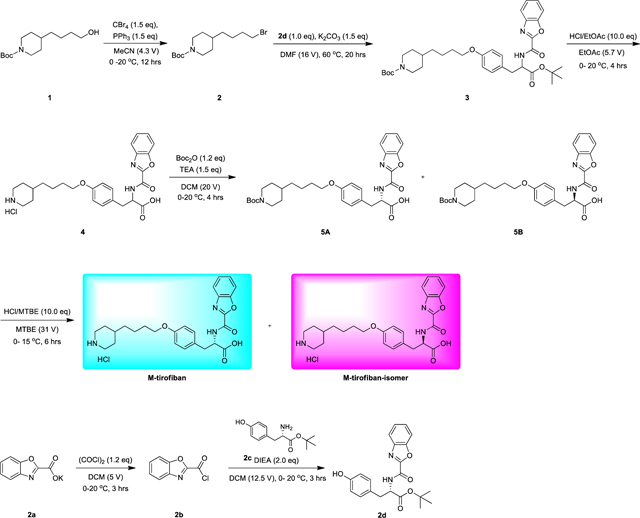
Initial synthetic route: (scheme 2)
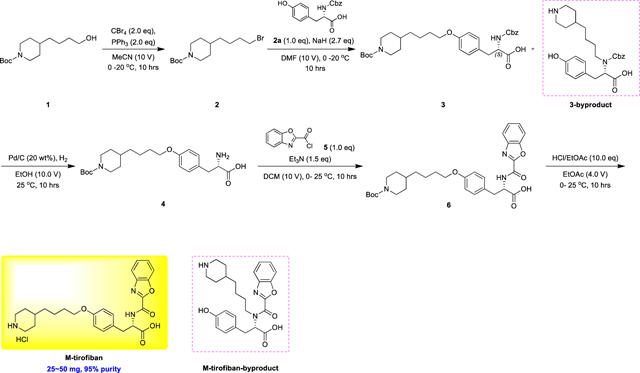
Summary:
In scheme 2, the title compound was obtained after prep-HPLC separation. However, there were two sets of 1H NMR peaks. We analyzed the HNMR and the synthetic route carefully again. The suspected impurity was the M-tirofiban-byproduct which would be difficult to remove by prep-HPLC due to the very close polarity.
The impurity should have been formed during synthesis of compound 3 due to the excess NaH. Thus, a new batch was synthesized using scheme 1.
In scheme 1, two peaks of target compound were detected by chiral HPLC. Racemization would have occurred in the alkylation reaction of step 2. The HCl salt of the final product could not be separated by chiral chromatography i.e., supercritical fluid chromatography (SFC). Therefore, Boc was re-added to the amine and then the isomers were separated by SFC. Finally, M-tirofiban and M-tirofiban isomer were obtained after Boc removal.
Compound 2
Compound 1 (700 mg, 2.72 mmol, 1.00 eq) was added to a round bottom flask charged with ACN (3.0 mL), then PPh3 (1.07 g, 4.08 mmol, 1.50 eq) and CBr4 (1.35 g, 4.08 mmol, 1.50 eq) were added at 0°C. The mixture was stirred at 20°C for 12 hour. TLC (petroleum ether: ethyl acetate = 5:1, Rf = 0.43) indicated compound 1 was consumed completely and one new main spot formed. The reaction was quenched by ice-water (10 mL) at 0°C, and then extracted with MTBE (20 mL and 10 mL). The combined organic layers were concentrated under reduced pressure to give a residue. The crude product was purified by column chromatography (SiO2, petroleum ether/ethyl acetate= 20/1 to 5/1) to give compound 2 (800 mg, 2.50 mmol, 91.8% yield) as a colorless oil. 1H NMR (400 MHz CDCl3) δ 4.03 – 4.07 (m, 2H), 3.39 (t, J = 6.8 Hz, 2H), 2.61 – 2.68 (m, 2H), 1.80 – 1.86 (m, 2H), 1.61 – 1.65 (m, 2H), 1.42 – 1.43 (m, 11H), 1.22 – 1.26 (m, 2H), 1.06 – 1.07 (m, 2H)
Compound 2b
To a solution of compound 2a (400 mg, 1.98 mmol, 1.00 eq) in DCM (2 mL) was added dropwise (COCl)2 (301 mg, 2.37 mmol, 207 uL, 1.20 eq) at 0°C. The resulting mixture was stirred at 20°C for 3 hrs. TLC (petroleum ether: ethyl acetate = 3:1, Rf = 0.43) indicated intermediate (quenched by MeOH) was consumed completely and one new spot formed. The reaction mixture was concentrated under reduced pressure below 40°C to give compound 2b (350 mg, 1.93 mmol, 97.5% yield) as purple solid, which was used in the next step without further purification.
Compound 2d
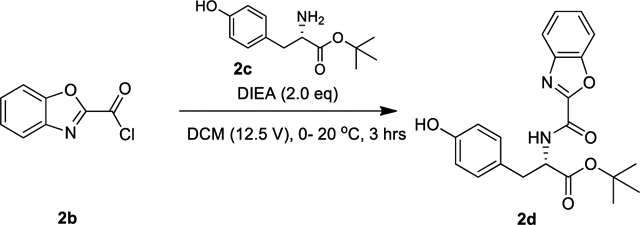
To a solution of compound 2c (400 mg, 1.69 mmol, 1.00 eq) in DCM (5 mL) was added dropwise DIEA (436 mg, 3.37 mmol, 587 uL, 2.00 eq) and compound 2b (306 mg, 1.69 mmol, 1.00 eq) at 0°C. The resulting mixture was stirred at 20 °C for 3 hrs. LCMS showed no compound 2c remained, and one major new peak (Rt = 1.888 min, m/z −55 = 327.3) with desired product was detected. The reaction was quenched by ice-water (10 mL) at 0°C, and then extracted with EtOAc (20 mL, then 10 mL). The combined organic layers were concentrated under reduced pressure to give compound 2d (500 mg, 1.31 mmol, 77.6% yield) as a red oil, which was used in the next step without purification.
Compound 3
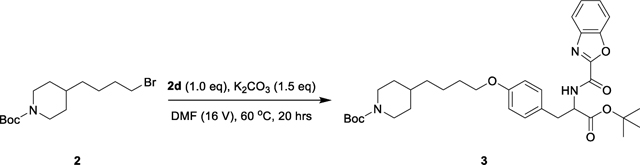
To a solution of compound 2d (500 mg, 1.31 mmol, 1.00 eq) and compound 2 (419 mg, 1.31 mmol, 1.00 eq) in DMF (8 mL) was add K2CO3 (271 mg, 1.96 mmol, 1.50 eq) at 25°C. The resulting mixture was stirred at 60°C for 20 hrs. LCMS showed no compound 2d remained, and one major new peak (Rt = 1.058, m/z = 466.1) with desired product was detected. The reaction was quenched by ice-water (30 mL) at 0°C, and then extracted with EtOAc (20 mL, 10 mL). The combined organic layers were concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 4/1 to 2/1, Rf = 0.43) to give compound 3 (350 mg, 563 μmol, 43.1% yield) as a light-yellow solid. 1H NMR (400 MHz CDCl3) δ 7.85 (d, J = 8.0 Hz, 1H), 7.68 – 7.77 (m, 2H), 7.47 – 7.54 (m, 2H), 7.16 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 4.95 – 5.00 (m, 1H), 4.06 – 4.08 (m, 2H), 4.10 – 4.19 (m, 2H), 3.95 (t, J = 6.4 Hz, 2H), 3.16 – 3.28 (m, 2H), 1.67 – 1.80 (m, 2H), 1.30 – 1.49 (m, 27H), 1.07 – 1.30 (m, 2H)
Compound 4
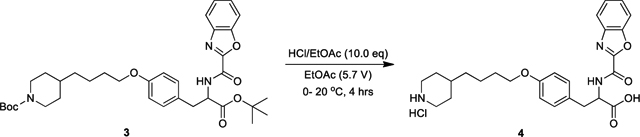
To a solution of compound 3 (350 mg, 563 μmol, 1.00 eq) in EtOAc (2.0 mL) was added dropwise HCl/EtOAc (4 M, 1.41 mL, 10.0 eq) at 0°C. The resulting mixture was stirred at 20°C for 4 hrs. LCMS showed no compound 3 remained, and one major new peak (Rt = 2.047, m/z = 466) with desired mass was detected. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by prep-HPLC (HCl condition). Column: Phenomenex Luna C18 200×40mm×10um; mobile phase: [water (0.04%HCl)-ACN]; B%: 10%–50% ACN, 8 min). The fractions after prep-HPLC were lyophilized directly to give Compound 4 (150 mg, 322 μmol, 57.2% yield) as a white solid. 1H NMR (400 MHz DMSO) δ 13.01 (br s, 1H), 9.34 (d, J = 8.0 Hz, 1H), 8.92 (br s, 1H), 8.66 (br s, 1H), 7.83 – 7.90 (m, 2H), 7.53 – 7.56 (m, 2H), 7.16 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.4 Hz, 2H), 4.58 – 4.64 (m, 1H), 3.85 (t, J = 6.4 Hz, 2H), 3.07 – 3.17 (m, 4H), 2.73 – 2.76 (m, 2H), 1.70 – 1.73 (m, 4H), 1.30 – 1.34 (m, 1H), 1.19 – 1.25 (m, 6H)
Compounds 5A&5B
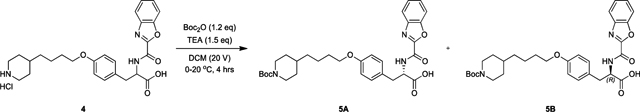
To a solution of compound 4 (150 mg, 299 μmol, 1.00 eq, HCl) in DCM (3 mL) was added Boc2O (78.3 mg, 359 μmol, 82.4 uL, 1.20 eq) and Et3N (45.3 mg, 448 μmol, 62.4 uL, 1.50 eq) at 0°C. The resulting mixture was stirred at 20 °C for 4 hrs. LCMS showed no compound 4 remained, and one major new peak (Rt = 0.873, m/z = 466) with desired product was detected. The reaction mixture was poured into 20% citric acid (~ 5 mL) and extracted with EtOAc (~ 20 mL, 10 mL), the organic layers were washed with brine (10 mL), dried over Na2SO4 and concentrated to give the product. The product was separated further by chiral SFC (Thar SFC 80) using a DAICEL Chiralpak AD column (250mm×30mm, 10um); mobile phase: A: CO2 and isocratic elution with B: 38% 0.1%NH3H2O ETOH for 6 min at 40°C with a system back pressure of 100 bar. The fractions after SFC were concentrated to give compound 5A (65.0 mg, 115 μmol, 76.9% yield) as a light-yellow solid and Compound 5B (70.0 mg, 123. μmol, 82.8% yield) as a light-yellow solid. Comparison of the elution in SFC of the 95% pure, chiral M-tirofiban synthesized in scheme 1 showed that peak 2 in SFC was compound 5A, the desired (S)-M-tirofiban; peak 1 was compound 5B, the undesired (R)-M-tirofiban isomer.
(S)-M-tirofiban
To a solution of compound 5A (65.0 mg, 115 μmol, 1.00 eq) in MTBE (2.0 mL) was added dropwise HCl/MTBE (4.0 M, 287 uL, 10.0 eq) at 0°C. The resulting mixture was stirred at 15°C for 6 hrs. TLC (petroleum ether: ethyl acetate = 0:1, Rf = 0.01) indicated the intermediate was consumed completely and one new spot formed. The reaction mixture was concentrated under reduced pressure below 40°C to give a residue. The crude product was triturated with MTBE (2 mL) at 20°C for 20 min, and then lyophilized to remove the solvent residue. Compound (S)-M-tirofiban (22.0 mg, 43.8 μmol, 38.1% yield, HCl) was obtained as a thick white solid. 1H NMR (400 MHz DMSO) δ 12.98 (br s, 1H), 9.30 (d, J = 8.4 Hz, 1H), 8.92 (br s, 1H), 8.66 (br s, 1H), 7.83 – 7.90 (m, 2H), 7.53 – 7.56 (m, 2H), 7.16 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.4 Hz, 2H), 4.60 – 4.64 (m, 1H), 3.85 (t, J = 6.4 Hz, 2H), 3.07 – 3.17 (m, 4H), 2.73 – 2.76 (m, 2H), 1.70 – 1.73 (m, 4H), 1.19 – 1.35 (m, 7H)
(R)-M-tirofiban isomer
To a solution of compound 5B (70.0 mg, 124 μmol, 1.00 eq) in MTBE (2.0 mL) was added dropwise HCl/MTBE (4.0 M, 309 uL, 10.0 eq) at 0°C. The resulting mixture was stirred at 15°C for 6 hrs. TLC (petroleum ether: ethyl acetate = 0:1, Rf = 0.01) indicated the intermediate was consumed completely and one new spot formed. The reaction mixture was concentrated under reduced pressure below 40°C to give a residue. The crude product was triturated with MTBE (2 mL) at 20°C for 20 min, and then lyophilized to remove the solvent residue. Compound (R)-M-tirofiban-isomer (58.0 mg, 115. μmol, 93.4% yield, HCl) was obtained as a white solid. 1H NMR (400 MHz CDCl3) δ 12.99 (br s, 1H), 9.28 (d, J = 8.0 Hz, 1H), 8.57 (br s, 2H), 7.83 – 7.90 (m, 2H), 7.53 – 7.56 (m, 2H), 7.16 (d, J = 8.8 Hz, 1H), 6.77 (d, J = 8.4 Hz, 2H), 4.59 – 4.60 (m, 1H), 3.85 (t, J = 6.4 Hz, 2H), 3.07 – 3.17 (m, 4H), 2.73 – 2.76 (m, 2H), 1.70 – 1.73 (m, 4H), 1.22 – 1.35 (m, 8H)
Supplementary Material
FIGURE S1. Related to Fig. 3. 2Fo-Fc electron density maps of drugs bound to aIIbβ3 headpiece in the order of their appearance in the main text. Metal conditions (Mg or Mn) for compound soaking are described in Table S1. Electron density (cyan mesh) is contoured around atoms at 1.5 σ. Colors are as in Fig. 2: Mg2+ and Mn2+ ions as gold and purple spheres, respectively, and waters as smaller red spheres. Black dashes represent metal coordination and key hydrogen bonds.
Figure S2. Related to Fig. 3. Crystal structures of the αIIbβ3 headpiece in complex with inhibitors in Mg or Mn. Structures of molecule 1 in the asymmetric unit (chains A and B) after soaking indicated compounds (1 mM) in 1 mM (or 5 mM Mg2+ for UR-2922) and 1 mM Ca2+ or 2 mM Mn2+ and 0.2 mM Ca2+ as indicated into closed αIIbβ3 crystals. Structures are superimposed on the β-propeller and βI domains. αIIb, β3, and compounds or RGD are shown with carbons in green, cyan, and wheat, respectively. MIDAS Mg2+ and Mn2+ ions are shown as spheres in yellow and purple, respectively. Water molecule oxygens are shown as smaller red spheres. Backbone is shown as worm trace or sticks for residues that hydrogen bond to ligand. Sidechains that interact with ligand are shown in stick. Black dashes represent metal coordination, hydrogen, and pi-pi bonds. Relevant residues are labeled.
Figure S3. Related to Fig. 6. Competitive binding to measure the affinity of αIIbβ3 inhibitors. αIIbβ3 inhibitors competed the concentration of FITC-Echistatin shown on the y-axis for binding to αIIbβ3 WT (A) and to αIIbβ3_N305T (B) expressed on transiently transfected Expi293F α5 &αV KO cells. Mean fluorescence intensity (MFI) was determined by flow cytometry without washing in L15 medium with 0.1% BSA. Competitive binding curves were fitted to Eq.4 in Methods.
Figure S4. Related to Fig. 6. Closing αIIbβ3 inhibitors suppress integrin conformational change on cell surface as detected by LIBS epitope exposure using flow cytometry. (A and B) LIBS1 antibody (A) and AP5 antibody (B) binding (as percentage of AP3 fluorescence intensity) in CHO cells stably expressing full-length αIIbβ3 treated with the indicated compounds in the presence of 1 mM Mg2+ and 1 mM Ca2+ (filled bars) or 2 mM Mn2+ and 0.1 mM Ca2+ (open bars). (C) HEK293T cells transfected with hyperactive αIIbβ3 with a GFFKR/GAAKR mutation in the αIIb transmembrane domain were analyzed for LIBS1 expression as in A.
Figure S5. Related to Fig. 7. Inhibition of clot retraction by αIIbβ3 inhibitors. (A) Human PRP was pre-incubated with buffer control or indicated concentrations of αIIbβ3 inhibitors, and then thrombin was added at 0.75 U/ml to stimulate clot retraction. The tubes were imaged at 30 and 90 mins. (B) Clot retraction was quantified with ImageJ and presented as 1– (clot area/whole reaction area). Data were fitted with the function of inhibitor vs response (three parameters) in Prism 8.
Figure S6. Related to Fig. 7. Influence of serum on the binding affinities of selected αIIbβ3 inhibitors. (A) Binding affinity of αIIbβ3 WT expressed on transiently transfected Expi293F integrin α5 and αV knock-out cells for FITC-Echistatin determined by binding of 5 nM Alexa647-MBC319.4 Fab in L15 medium supplemented with varying concentrations of serum. Binding curves were fitted to Eq.3 for the binding affinity of FITC-Echistatin at different serum concentrations. Errors were standard fitting errors from nonlinear least square fits. (B-C) Competitive binding to measure the affinity of αIIbβ3 inhibitors. αIIbβ3 inhibitors at the indicated concentrations competed binding of FITC-Echistatin at the concentration shown on the y axis to αIIb β3 WT expressed on transiently transfected Expi293 α5 &αV KO cells. Mean fluorescence intensity (MFI) was determined by flow cytometry without washing.
Figure S7. Called in Discussion. Related to Figs. 5, 6, and 7. Influence of inhibitors with varying preference for the open state on extension-stabilizing antibody epitope exposure. (A) Equations for calculating the population of extended states of integrin in presence of inhibitors with different binding affinities to the closed and EO states. (B) A scenario to explain why wild type and active mutant integrin αIIbβ3 show different sensitivities in inhibitor-induced LIBS antibody epitope exposure assays. Free energy values used for the BC, EC and EO states of wild type αIIBβ3 are −4, −1.8 and 0 kcal/mol, respectively; free energy values used for the BC, EC and EO states of active mutant are −1.2, −1.0 and 0 kcal/mol, respectively, representing a 2.8 kcal/mol destabilization of the closed states by the mutation.
Any conformation-specific antibody can be a LIBS reporter. However, antibodies must be used at concentrations near their EC50 values to be good LIBS reporters. It appears that many useful, high-affinity, conformation-specific antibodies are not used in the literature because the typical antibody concentrations investigators use are too high for these antibodies to report epitope exposure. All LIBS antibodies must be titrated to find the optimal concentration.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 12G10 | (Mould et al., 1995) Hybridoma from Martin Humphries | Produced from hybridoma in lab |
| 9EG7 | (Lenter et al., 1993) Hybridoma from Dietmar Vestweber | Produced from hybridoma in lab |
| SG/19 | (Miyake et al., 1992) Hybridoma from Jun Takagi | Produced from hybridoma in lab |
| MBC319.4 | (Zhang et al., 2013) Hybridoma from Richard Aster | Produced from hybridoma in lab |
| LIBS1 | (Frelinger et al., 1990) Hybridoma from Mark Ginsberg | Produced from hybridoma in lab |
| AP5 | (Kekomaki et al., 1991) Antibody from Dan Bougie, Versiti | Cat#153747 |
| HUTS4, HUTS-21 | (Luque et al., 1996) Hybridoma from Carlos Cabamas | Produced from hybridoma in lab |
| mAb16 | (Akiyama et al., 1989) Hybridoma from Kenneth Yamada | Produced from hybridoma in lab |
| 17E6 | (Mitjans et al., 1995) Antibody from Simon Goodman, Merck KGA | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Thrombin | Sigma-Aldrich | Cat#10602400001 |
| Prostaglandin E1 | Sigma-Aldrich | Cat#P5515 |
| FITC-echistatin | US Biological | Cat#E0256–01A.1 |
| FITC-LDVP | Tocris Bioscience | Cat#4577 |
| Alexa Fluor™ 647 NHS Ester | Thermo Fisher Scientific | Cat#A20006 |
| Alexa Fluor™ 488 NHS Ester | Thermo Fisher Scientific | Cat#A20000 |
| Dodecapeptide (HHLGGAKQAGDV) | Phoenix Pharmaceuticals | Cat#025–35 |
| Tirofiban | Merck | CAS 144494-65-5 |
| Eptifibatide | Sigma-Aldrich | SML1042; CAS 188627-80-7 |
| Orbofiban (active form) | Pfizer | CAS 199530-77-3 |
| Roxifiban (active form) | This paper | CAS 170725-15-2 |
| Lotrafiban (active form) | This paper | CAS 171049-14-2 |
| Lamifiban | Roche | CAS 144412-49-7 |
| Sibrafiban (active form) | Roche | CAS 144412-18-0 |
| Fradafiban (active form) | This paper | CAS 148396-36-5 |
| EF-5154 | Meiji Seika Pharma | CAS 177276-17-4 |
| UR-2922 | This paper | CAS 220386-56-1 |
| BMS4 | This paper | CAS 270593-59-4 |
| BMS4–1 | This paper | CAS 270088-90-9 |
| BMS4–3 | This paper | N/A |
| BMS4–2 | This paper | CAS 270088-87-4 |
| GR144058 | Sigma-Aldrich | G6418; CAS 150040-23-6 |
| Gantofiban | This paper | CAS 167364-04-7 |
| Gantofiban analog | This paper | CAS 167364-14-9 |
| RUC-4 | Barry Coller | CAS 1448313-27-6 |
| M-tirofiban (both isomers) | This paper | (S)-isomer: CAS 2410607-77-9; (R)-isomer: N/A |
| Firategrast | Apexbio | Cat#A3415 CAS 402567-16-2 |
| Ro 29 | Roche | CAS 1421673-59-7 |
| DS 13d | Tocris Bioscience | Cat#TCS 2314 CAS 317353-73-4 |
| DS 13g | This paper | CAS 819078-63-2 |
| Deposited Data | ||
| Drug-bound αIIbβ3 structures | This paper | PDB accession IDs for structures are in Table S1 |
| Drug-bound αIIbβ3 structure diffraction images | SBGRID accession IDs for diffraction images are in Table S1: https://data.sbgrid.org/data/ | |
| Experimental Models: Cell Lines | ||
| Jurkat | ATCC | Cat#TIB-152 |
| Expi293F | Thermo Fisher Scientific | Cat# A14527 |
| Expi293F α5&αV knock-out cells | This paper | N/A |
| HEK293 expression αllbβ3 with GAAKR mutant | (Luo et al., 2004) | N/A |
| Recombinant DNA | ||
| PD2529 CAG αIIb full length | This paper | Addgene Plasmid #189796 |
| PD2529 CAG β3 full length | This paper | Addgene Plasmid #189797 |
| PD2529 CAG β3_N305T full length | This paper | Addgene Plasmid #189798 |
| pSpCas9n(BB)-2A-puro | (Ran et al., 2013) | Addgene Plasmid #4814 |
| Software and Algorithms | ||
| Mathematica | Wolfram Mathematica version 12 | https://www.wolfram.com/mathematica/ |
| Function F in Eq. 4 and its derivation | This paper | Mathematica file Equation4.nb |
| Phenix | (Adams et al., 2010) | http://www.phenix-online.org |
| XDS | (Kabsch, 2010) | xds.mpimf-heidelberg.mpg.de |
| COOT | (Emsley et al., 2010) | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Maestro, Prime, and Epik | Schrödinger Inc. | https://www.schrodinger.com |
| PyMOL | Schrödinger Inc. | https://pymol.org/2/ |
| SciFinder | https://scifinder-n.cas.org | https://scifinder-n.cas.org |
| Plot2 | Apple Inc. App store | https://plotdoc.micw.org |
| Prism9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Integrin inhibitors in failed clinical trials stabilized the open conformation.
Inhibitor designs were discovered that stabilize the closed integrin conformation.
Inhibitor design principles were transferrable from integrin αIIbβ3 to α4β1.
Closing inhibitors may lack partial agonism seen with opening inhibitors.
Acknowledgements.
We thank Rui Qiu and Christine Hwang for experimental work early in this project. Supported by NIH grants HL131729 (T.A.S.) and HL131836 (Jq.Z.). We thank the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357 for beamline support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests. T.A.S. is a founder, stock owner, and director of Morphic Therapeutic. FY.L. is an employee of Morphic Therapeutic.
Quantification and statistical analysis.
All of the statistical analyses and how significance was defined are described in Figure legends. Equations and software used are described in the Methods.
SUPPLEMENTAL INFORMATION
Data S1. Equation 4, related to Fig. 6, Fig. S3.
REFERENCES
- Adair BD, Alonso JL, van Agthoven J, Hayes V, Ahn HS, Yu IS, Lin SW, Xiong JP, Poncz M, and Arnaout MA (2020). Structure-guided design of pure orthosteric inhibitors of αIIbβ3 that prevent thrombosis but preserve hemostasis. Nat Commun 11, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aga Y, Baba K, Tam S, Nakanishi T, Yoneda K, Kita J, and Ueno H (2004). UR-3216: a new generation oral platelet GPIIb/IIIa antagonist. Curr Pharm Des 10, 1597–1601. [DOI] [PubMed] [Google Scholar]
- Akiyama SK, Yamada SS, Chen WT, and Yamada KM (1989). Analysis of fibronectin receptor function with monoclonal antibodies: Roles in cell adhesion, migration, matrix assembly, and cytoskeletal organization. J Cell Biol 109, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Li J, and Springer TA (2022). Regulation of integrin α5β1 conformational states and intrinsic affinities by metal ions and the ADMIDAS. Mol Biol Cell 33, ar56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimori T, Miyazaki N, Mihara E, Takizawa M, Taniguchi Y, Cabanas C, Sekiguchi K, and Takagi J (2021). Structural mechanism of laminin recognition by integrin. Nat Commun 12, 4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ault KA, Cannon CP, Mitchell J, McCahan J, Tracy RP, Novotny WF, Reimann JD, and Braunwald E (1999). Platelet activation in patients after an acute coronary syndrome: results from the TIMI-12 trial. Thrombolysis in Myocardial Infarction. J Am Coll Cardiol 33, 634–639. [DOI] [PubMed] [Google Scholar]
- Baba K, Aga Y, Nakanishi T, Motoyama T, and Ueno H (2001). UR-3216: a manageable oral GPIIb/IIIa antagonist. Cardiovasc Drug Rev 19, 25–40. [DOI] [PubMed] [Google Scholar]
- Bosco A, Kidson-Gerber G, and Dunkley S (2005). Delayed tirofiban-induced thrombocytopenia: two case reports. J Thromb Haemost 3, 1109–1110. [DOI] [PubMed] [Google Scholar]
- Bougie DW, Rasmussen M, Zhu J, and Aster RH (2012). Antibodies causing thrombocytopenia in patients treated with RGD-mimetic platelet inhibitors recognize ligand-specific conformers of αIIb/β3 integrin. Blood 119, 6317–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breth L, Kochie J, Combs A, Wang S, Smallheer J, Billheimer J, Seiffert D, Hollis G, and O’Neil K (2005). Identification and characterization of antibodies that bind GPIIb/IIIa: antagonist complexes. J Immunol Methods 301, 11–20. [DOI] [PubMed] [Google Scholar]
- Byron A, Humphries J, Askari JA, Craig SE, Mould AP, and Humphries M (2009). Anti-integrin monoclonal antibodies. J Cell Science 122, 4009–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MG, Cormier A, Ito S, Seed RI, Bondesson AJ, Lou J, Marks JD, Baron JL, Cheng Y, and Nishimura SL (2020). Cryo-EM reveals integrin-mediated TGF-β activation without release from latent TGF-β. Cell 180, 490–501 e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew DP, Bhatt DL, Sapp S, and Topol EJ (2001). Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: a meta-analysis of phase III multicenter randomized trials. Circulation 103, 201–206. [DOI] [PubMed] [Google Scholar]
- Chiba J, Machinaga N, Takashi T, Ejima A, Takayama G, Yokoyama M, Nakayama A, Baldwin JJ, McDonald E, Moriarty KJ, et al. (2005). Identified a morpholinyl-4-piperidinylacetic acid derivative as a potent oral active VLA-4 antagonist. Bioorg Med Chem Lett 15, 41–45. [DOI] [PubMed] [Google Scholar]
- Cox D, Aoki T, Seki J, Motoyama Y, and Yoshida K (1996). Pentamidine is a specific, non-peptide, GPIIb/IIIa antagonist. Thromb Haemost 75, 503–509. [PubMed] [Google Scholar]
- Cox D, Brennan M, and Moran N (2010). Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov 9, 804–820. [DOI] [PubMed] [Google Scholar]
- Cox D, Smith R, Quinn M, Theroux P, Crean P, and Fitzgerald DJ (2000). Evidence of platelet activation during treatment with a GPIIb/IIIa antagonist in patients presenting with acute coronary syndromes. J Am Coll Cardiol 36, 1514–1519. [DOI] [PubMed] [Google Scholar]
- Davi G, and Patrono C (2007). Platelet activation and atherothrombosis. N Engl J Med 357, 2482–2494. [DOI] [PubMed] [Google Scholar]
- Dickfeld T, Ruf A, Pogatsa-Murray G, Muller I, Engelmann B, Taubitz W, Fischer J, Meier O, and Gawaz M (2001). Differential antiplatelet effects of various glycoprotein IIb-IIIa antagonists. Thromb Res 101, 53–64. [DOI] [PubMed] [Google Scholar]
- Dong X, Hudson NE, Lu C, and Springer TA (2014). Structural determinants of integrin β-subunit specificity for latent TGF-β. Nat Struct Mol Biol 21, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldred CD, Evans B, Hindley S, Judkins BD, Kelly HA, Kitchin J, Lumley P, Porter B, Ross BC, Smith KJ, et al. (1994). Orally active non-peptide fibrinogen receptor (GpIIb/IIIa) antagonists: identification of 4-[4-[4-(aminoiminomethyl)phenyl]-1-piperazinyl]-1-piperidineacetic acid as a long-acting, broad-spectrum antithrombotic agent. J Med Chem 37, 3882–3885. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng E, Smagghe B, Walz T, and Springer TA (2011). Intact αIIbβ3 extends after activation measured by solution X-ray scattering and electron microscopy. J Biol Chem 286, 35218–35226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DJ, Roy L, Catella F, and FitzGerald GA (1986). Platelet activation in unstable coronary disease. N Engl J Med 315, 983–989. [DOI] [PubMed] [Google Scholar]
- Frelinger AL, Cohen I, Plow EF, Smith MA, Roberts J, Lam SCT, and Ginsberg MH (1990). Selective inhibition of integrin function by antibodies specific for ligand-occupied receptor conformers. J Biol Chem 265, 6346–6352. [PubMed] [Google Scholar]
- Frelinger AL, Lam SCT, Plow EF, Smith MA, Loftus JC, and Ginsberg MH (1988). Occupancy of an adhesive glycoprotein receptor modulates expression of an antigenic site involved in cell adhesion. J Biol Chem 263, 12397–12402. [PubMed] [Google Scholar]
- Gante J, Juraszyk H, Raddatz P, Wurziger H, Bernotat-Danielowski S, Melzer G, and Rippmann F (1995). New peptidomimetics in the chemistry of fibrinogen receptor antagonists. Letters in Peptide Science 2, 135–140. [Google Scholar]
- Gante J, Juraszyk H, Raddatz P, Wurziger H, Bernotat-Danielowski S, Melzer G, and Rippmann F (1996). New antithrombotic RGD-mimetics with high bioavailability. Bioorganic & Medicinal Chemistry Letters 6, 2425–2430. [Google Scholar]
- Himmelsbach F, Volkhard A, Pieper H, Linz G, Weisenberger J, and Mueller T (1996). Cyclic imino derivatives, processes for preparing them and pharmaceutical compositions containing these compounds filed Nov. 19, and published
- Jansen EE, and Hartmann M (2021). Clot retraction: Cellular mechanisms and inhibitors, measuring methods, and clinical implications. Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekomaki R, Dawson B, McFarland J, and Kunicki TJ (1991). Localization of human platelet autoantigens to the cysteine-rich region of glycoprotein IIIa. J Clin Invest 88, 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kereiakes DJ, Henry TD, DeMaria AN, Bentur O, Carlson M, Seng Yue C, Martin LH, Midkiff J, Mueller M, Meek T, et al. (2020). First human use of RUC-4: A nonactivating second-generation small-molecule platelet glycoprotein IIb/iIIa (integrin αiibβ3) inhibitor designed for subcutaneous point-of-care treatment of ST-segment-elevation myocardial infarction. J Am Heart Assoc 9, e016552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenter M, Uhlig H, Hamann A, Jenö P, Imhof B, and Vestweber D (1993). A monoclonal antibody against an activation epitope on mouse integrin chain β1 blocks adhesion of lymphocytes to the endothelial integrin α6β1. Proc Natl Acad Sci USA 90, 9051–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Rivera-Nieves J, Sandborn WJ, and Shattil S (2016). Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discov 15, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Fukase Y, Shang Y, Zou W, Munoz-Felix JM, Buitrago L, van Agthoven J, Zhang Y, Hara R, Tanaka Y, et al. (2019). Novel pure αVβ3 integrin antagonists that do not induce receptor extension, prime the receptor, or enhance angiogenesis at low concentrations. ACS Pharmacol Transl Sci 2, 387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Springer TA (2017). Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc Natl Acad Sci U S A 114, 4685–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Springer TA (2018). Energy landscape differences among integrins establish the framework for understanding activation. J Cell Biol 217, 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Su Y, Xia W, Qin Y, Humphries MJ, Vestweber D, Cabanas C, Lu C, and Springer TA (2017). Conformational equilibria and intrinsic affinities define integrin activation. EMBO J 36, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Vootukuri S, Shang Y, Negri A, Jiang JK, Nedelman M, Diacovo TG, Filizola M, Thomas CJ, and Coller BS (2014). RUC-4: a novel αIIbβ3 antagonist for prehospital therapy of myocardial infarction. Arterioscler Thromb Vasc Biol 34, 2321–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B-H, Springer TA, and Takagi J (2003). Stabilizing the open conformation of the integrin headpiece with a glycan wedge increases affinity for ligand. Proc Natl Acad Sci U S A 100, 2403–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Springer TA, and Takagi J (2004). A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol 2, e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque A, Gomez M, Puzon W, Takada Y, Sanchez-Madrid F, and Cabanas C (1996). Activated conformations of very late activation integrins detected by a group of antibodies (HUTS) specific for a novel regulatory region (355–425) of the common β1 chain. J Biol Chem 271, 11067–11075. [DOI] [PubMed] [Google Scholar]
- Ma D, and Xia C (2001). CuI-catalyzed coupling reaction of beta-amino acids or esters with aryl halides at temperature lower than that employed in the normal Ullmann reaction. Facile synthesis of SB-214857. Org Lett 3, 2583–2586. [DOI] [PubMed] [Google Scholar]
- Matsuno H, Kozawa O, Niwa M, Ito T, Tanabe K, Nishida M, Hayashi H, and Uematsu T (1998). Effect of GR144053, a fibrinogen-receptor antagonist, on thrombus formation and vascular patency after thrombolysis by tPA in the injured carotid artery of the hamster. J Cardiovasc Pharmacol 32, 191–197. [DOI] [PubMed] [Google Scholar]
- Matsuno H, Kozawa O, Niwa M, Kaida T, Hayashi H, and Uematsu T (1997). GR144053, a fibrinogen receptor antagonist, enhances the suppression of neointima formation by losartan, an angiotensin II receptor antagonist, in the injured carotid artery of hamster. Br J Pharmacol 122, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DH, Weber T, Grove R, Wardell C, Horrigan J, Graff O, Atkinson G, Dua P, Yousry T, Macmanus D, et al. (2012). Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 11, 131–139. [DOI] [PubMed] [Google Scholar]
- Mitjans F, Sander D, Adan J, Sutter A, Martinez JM, Jaggle CS, Moyano JM, Kreysch HG, Piulats J, and Goodman SL (1995). An anti-α v-integrin antibody that blocks integrin function inhibits the development of a human melanoma in nude mice. J Cell Sci 108 (Pt 8), 2825–2838. [DOI] [PubMed] [Google Scholar]
- Miyake K, Hasunuma Y, Yagita H, and Kimoto M (1992). Requirement for VLA-4 and VLA-5 integrins in lymphoma cells binding to and migration beneath stromal cells in culture. J Cell Biol 119, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould AP, Garratt AN, Askari JA, Akiyama SK, and Humphries MJ (1995). Identification of a novel anti-integrin monoclonal antibody that recognises a ligand-induced binding site epitope on the β1 subunit. FEBS Lett 363, 118–122. [DOI] [PubMed] [Google Scholar]
- Mould D, Chapelsky M, Aluri J, Swagzdis J, Samuels R, and Granett J (2001). A population pharmacokinetic-pharmacodynamic and logistic regression analysis of lotrafiban in patients. Clin Pharmacol Ther 69, 210–222. [DOI] [PubMed] [Google Scholar]
- Muller TH, Weisenberger H, Brickl R, Narjes H, Himmelsbach F, and Krause J (1997). Profound and sustained inhibition of platelet aggregation by Fradafiban, a nonpeptide platelet glycoprotein IIb/IIIa antagonist, and its orally active prodrug, Lefradafiban, in men. Circulation 96, 1130–1138. [DOI] [PubMed] [Google Scholar]
- Muro F, Iimura S, Sugimoto Y, Yoneda Y, Chiba J, Watanabe T, Setoguchi M, Iigou Y, Matsumoto K, Satoh A, et al. (2009). Discovery of trans-4-[1-[[2,5-Dichloro-4-(1-methyl-3-indolylcarboxamido)phenyl]acetyl]-(4S)-methoxy-(2S)-pyrrolidinylmethoxy]cyclohexanecarboxylic acid: an orally active, selective very late antigen-4 antagonist. J Med Chem 52, 7974–7992. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Pratico D, and Fitzgerald DJ (1998). Functional relevance of the expression of ligand-induced binding sites in the response to platelet GP IIb/IIIa antagonists in vivo. J Pharmacol Exp Ther 286, 945–951. [PubMed] [Google Scholar]
- Nagae M, Re S, Mihara E, Nogi T, Sugita Y, and Takagi J (2012). Crystal structure of α5β1 integrin ectodomain: Atomic details of the fibronectin receptor. J Cell Biol 197, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman PJ, Allen RW, Kahn RA, and Kunicki TJ (1985). Quantitation of membrane glycoprotein IIIa on intact human platelets using the monoclonal antibody, AP-3. Blood 65, 227–232. [PubMed] [Google Scholar]
- Nomoto M, Tatebayashi T, Morita J, Suzuki H, Aizawa K, Kurosawa T, and Komiya I (2009). Physiological models are good tools to predict rat bioavailability of EF5154 prodrugs from in vitro intestinal parameters. J Pharm Sci 98, 1532–1544. [DOI] [PubMed] [Google Scholar]
- Onitilo AA (2006). Delayed profound thrombocytopenia associated with eptifibatide. Am J Hematol 81, 984. [DOI] [PubMed] [Google Scholar]
- Peter K, Schwarz M, Nordt T, and Bode C (2001). Intrinsic activating properties of GP IIb/IIIa blockers. Thromb Res 103 Suppl 1, S21–S27. [DOI] [PubMed] [Google Scholar]
- Pieniaszek HJ Jr., Sy SK, Ebling W, Fossler MJ, Cain VA, Mondick JT, Ma S, and Kornhauser DM (2002). Safety, tolerability, pharmacokinetics, and time course of pharmacologic response of the active metabolite of roxifiban, XV459, a glycoprotein IIb/IIIa antagonist, following oral administration in healthy volunteers. J Clin Pharmacol 42, 738–753. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AR, Hart IR, Watson AR, Welti JC, Silva RG, Robinson SD, Da Violante G, Gourlaouen M, Salih M, Jones MC, et al. (2009). Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat Med 15, 392–400. [DOI] [PubMed] [Google Scholar]
- Scarborough RM, and Gretler DD (2000). Platelet glycoprotein IIb-IIIa antagonists as prototypical integrin blockers: novel parenteral and potential oral antithrombotic agents. J Med Chem 43, 3453–3473. [DOI] [PubMed] [Google Scholar]
- Schumacher S, Dedden D, Nunez RV, Matoba K, Takagi J, Biertumpfel C, and Mizuno N (2021). Structural insights into integrin α5β1 opening by fibronectin ligand. Sci Adv 7, eabe9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen M, Yuki K, and Springer TA (2013). An internal ligand-bound, metastable state of a leukocyte integrin, αXβ2. J Cell Biol 203, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka M, and Springer TA (2003). Therapeutic antagonists and the conformational regulation of integrin structure and function. Nat Rev Drug Disc 2, 703–716. [DOI] [PubMed] [Google Scholar]
- Smallheer JM, Wang S, and Jadhav PK (2001). Isoxazoline fibrinogen receptor antagonists. filed Nov. 5, 2002, and published
- Takagi J, Petre BM, Walz T, and Springer TA (2002). Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–611. [DOI] [PubMed] [Google Scholar]
- Théroux P, Catella-Lawson F, Armstrong P, DeCani J, Hirsh J, Pepine C, Ryan TJ, Pelletier G, Davies R, Flather M, et al. (1998). Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. N Engl J Med 338, 1488–1497. [DOI] [PubMed] [Google Scholar]
- Tilley JW, Sidduri A, Lou J, Kaplan G, Tare N, Cavallo G, Frank K, Pamidimukkala A, Choi DS, Gerber L, et al. (2013). Identification of N-acyl 4-(3-pyridonyl)phenylalanine derivatives and their orally active prodrug esters as dual acting α4β1 and α4β7 receptor antagonists. Bioorg Med Chem Lett 23, 1036–1040. [DOI] [PubMed] [Google Scholar]
- Topol EJ, Byzova TV, and Plow EF (1999). Platelet GPIIb-IIIa blockers. Lancet 353, 227–231. [DOI] [PubMed] [Google Scholar]
- Topol EJ, Easton D, Harrington RA, Amarenco P, Califf RM, Graffagnino C, Davis S, Diener HC, Ferguson J, Fitzgerald D, et al. (2003). Randomized, double-blind, placebo-controlled, international trial of the oral IIb/IIIa antagonist lotrafiban in coronary and cerebrovascular disease. Circulation 108, 399–406. [DOI] [PubMed] [Google Scholar]
- Trip MD, Cats VM, van Capelle FJ, and Vreeken J (1990). Platelet hyperreactivity and prognosis in survivors of myocardial infarction. N Engl J Med 322, 1549–1554. [DOI] [PubMed] [Google Scholar]
- Wang J, Su Y, Iacob RE, Engen JR, and Springer TA (2019). General structural features that regulate integrin affinity revealed by atypical αVβ8. Nat Commun 10, 5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller T, Alig L, Beresini M, Blackburn B, Bunting S, Hadvary P, Muller MH, Knopp D, Levet-Trafit B, Lipari MT, et al. (1996). Orally active fibrinogen receptor antagonists. 2. Amidoximes as prodrugs of amidines. J Med Chem 39, 3139–3147. [DOI] [PubMed] [Google Scholar]
- Wittke B, Mackie IJ, Machin SJ, Timm U, Zell M, and Goggin T (1999). Pharmacokinetics and pharmacodynamics of Ro 44–3888 after single ascending oral doses of sibrafiban, an oral platelet aggregation inhibitor, in healthy male volunteers. J Clin Pharmacol 47, 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki LJ, and Sato VL (1978). “Panning” for lymphocytes: A method for cell selection. Proc Natl Acad Sci USA 75, 2844–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, and Springer TA (2014). Metal ion and ligand binding of integrin α5β1. Proc Natl Acad Sci U S A 111, 17863–17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Takagi J, Wang J-H, Coller BS, and Springer TA (2004). Structural basis for allostery in integrins and binding of fibrinogen-mimetic therapeutics. Nature 432, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, and Arnaout MA (2002). Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science 296, 151–155. [DOI] [PubMed] [Google Scholar]
- Yu Y, Schurpf T, and Springer TA (2013). How natalizumab binds and antagonizes α4 integrins. J Biol Chem 288, 32314–32325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhu J, Mi LZ, Walz T, Sun H, Chen J-F, and Springer TA (2012). Structural specializations of α4β7 an integrin that mediates rolling adhesion. J Cell Biol 196, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Jiang X, Haydar N, Zhang C, Shan H, and Zhu J (2013). Modulation of integrin activation and signaling by α1/α1’-helix unbending at the junction. J Cell Sci 126, 5735–5747. [DOI] [PubMed] [Google Scholar]
- Zhang Lh LH, Chung JC, Costello TD, Valvis I, Ma P, Kauffman S, and Ward R (1997). The Enantiospecific Synthesis of an Isoxazoline. A RGD Mimic Platelet GPIIb/IIIa Antagonist. J Org Chem 62, 2466–2470. [DOI] [PubMed] [Google Scholar]
- Zhu J, Choi W-S, McCoy JG, Negri A, Zhu J, Naini S, Li J, Shen M, Huang W, Bougie D, et al. (2012). Structure-guided design of a high affinity platelet integrin αIIbβ3 receptor antagonist that disrupts Mg2+ binding to the MIDAS. Sci Transl Med 4, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Negri A, Provasi D, Filizola M, Coller BS, and Springer TA (2010). The closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood 116, 5050–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhu J, and Springer TA (2013). Complete integrin headpiece opening in eight steps. J Cell Biol 201, 1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Related to Fig. 3. 2Fo-Fc electron density maps of drugs bound to aIIbβ3 headpiece in the order of their appearance in the main text. Metal conditions (Mg or Mn) for compound soaking are described in Table S1. Electron density (cyan mesh) is contoured around atoms at 1.5 σ. Colors are as in Fig. 2: Mg2+ and Mn2+ ions as gold and purple spheres, respectively, and waters as smaller red spheres. Black dashes represent metal coordination and key hydrogen bonds.
Figure S2. Related to Fig. 3. Crystal structures of the αIIbβ3 headpiece in complex with inhibitors in Mg or Mn. Structures of molecule 1 in the asymmetric unit (chains A and B) after soaking indicated compounds (1 mM) in 1 mM (or 5 mM Mg2+ for UR-2922) and 1 mM Ca2+ or 2 mM Mn2+ and 0.2 mM Ca2+ as indicated into closed αIIbβ3 crystals. Structures are superimposed on the β-propeller and βI domains. αIIb, β3, and compounds or RGD are shown with carbons in green, cyan, and wheat, respectively. MIDAS Mg2+ and Mn2+ ions are shown as spheres in yellow and purple, respectively. Water molecule oxygens are shown as smaller red spheres. Backbone is shown as worm trace or sticks for residues that hydrogen bond to ligand. Sidechains that interact with ligand are shown in stick. Black dashes represent metal coordination, hydrogen, and pi-pi bonds. Relevant residues are labeled.
Figure S3. Related to Fig. 6. Competitive binding to measure the affinity of αIIbβ3 inhibitors. αIIbβ3 inhibitors competed the concentration of FITC-Echistatin shown on the y-axis for binding to αIIbβ3 WT (A) and to αIIbβ3_N305T (B) expressed on transiently transfected Expi293F α5 &αV KO cells. Mean fluorescence intensity (MFI) was determined by flow cytometry without washing in L15 medium with 0.1% BSA. Competitive binding curves were fitted to Eq.4 in Methods.
Figure S4. Related to Fig. 6. Closing αIIbβ3 inhibitors suppress integrin conformational change on cell surface as detected by LIBS epitope exposure using flow cytometry. (A and B) LIBS1 antibody (A) and AP5 antibody (B) binding (as percentage of AP3 fluorescence intensity) in CHO cells stably expressing full-length αIIbβ3 treated with the indicated compounds in the presence of 1 mM Mg2+ and 1 mM Ca2+ (filled bars) or 2 mM Mn2+ and 0.1 mM Ca2+ (open bars). (C) HEK293T cells transfected with hyperactive αIIbβ3 with a GFFKR/GAAKR mutation in the αIIb transmembrane domain were analyzed for LIBS1 expression as in A.
Figure S5. Related to Fig. 7. Inhibition of clot retraction by αIIbβ3 inhibitors. (A) Human PRP was pre-incubated with buffer control or indicated concentrations of αIIbβ3 inhibitors, and then thrombin was added at 0.75 U/ml to stimulate clot retraction. The tubes were imaged at 30 and 90 mins. (B) Clot retraction was quantified with ImageJ and presented as 1– (clot area/whole reaction area). Data were fitted with the function of inhibitor vs response (three parameters) in Prism 8.
Figure S6. Related to Fig. 7. Influence of serum on the binding affinities of selected αIIbβ3 inhibitors. (A) Binding affinity of αIIbβ3 WT expressed on transiently transfected Expi293F integrin α5 and αV knock-out cells for FITC-Echistatin determined by binding of 5 nM Alexa647-MBC319.4 Fab in L15 medium supplemented with varying concentrations of serum. Binding curves were fitted to Eq.3 for the binding affinity of FITC-Echistatin at different serum concentrations. Errors were standard fitting errors from nonlinear least square fits. (B-C) Competitive binding to measure the affinity of αIIbβ3 inhibitors. αIIbβ3 inhibitors at the indicated concentrations competed binding of FITC-Echistatin at the concentration shown on the y axis to αIIb β3 WT expressed on transiently transfected Expi293 α5 &αV KO cells. Mean fluorescence intensity (MFI) was determined by flow cytometry without washing.
Figure S7. Called in Discussion. Related to Figs. 5, 6, and 7. Influence of inhibitors with varying preference for the open state on extension-stabilizing antibody epitope exposure. (A) Equations for calculating the population of extended states of integrin in presence of inhibitors with different binding affinities to the closed and EO states. (B) A scenario to explain why wild type and active mutant integrin αIIbβ3 show different sensitivities in inhibitor-induced LIBS antibody epitope exposure assays. Free energy values used for the BC, EC and EO states of wild type αIIBβ3 are −4, −1.8 and 0 kcal/mol, respectively; free energy values used for the BC, EC and EO states of active mutant are −1.2, −1.0 and 0 kcal/mol, respectively, representing a 2.8 kcal/mol destabilization of the closed states by the mutation.
Any conformation-specific antibody can be a LIBS reporter. However, antibodies must be used at concentrations near their EC50 values to be good LIBS reporters. It appears that many useful, high-affinity, conformation-specific antibodies are not used in the literature because the typical antibody concentrations investigators use are too high for these antibodies to report epitope exposure. All LIBS antibodies must be titrated to find the optimal concentration.
Data Availability Statement
All structural data has been deposited at the Protein Data Bank.
Diffraction images have been deposited at SBGRID. Datasets for Figures 5C–E and 5G–I, 6A–C and E–F, and Figures S3, S6 and S7 have been deposited at Harvard Dataverse.
Executable Mathematica code for fitting competitive binding data in Fig. 6B–C and Fig. S3, entitled “Data S1-Equation 4.nb” is available in this paper’s supplemental information.
Other data is available on request from the Lead Contact for 3 years.