

Abstract
Microorganisms resident in our bodies participate in a variety of regulatory and pathogenic processes. Here, we describe how etiological pathways implicated in Alzheimer’s disease (AD) may be regulated or disturbed by symbiotic microbial activity. Furthermore, the composition of symbiotic microbes has changed dramatically across human history alongside the rise of agriculturalism, industrialization, and globalization. We postulate that each of these lifestyle transitions engendered progressive depletion of microbial diversity and enhancement of virulence, thereby enhancing AD risk pathways. It is likely that the human life span extended into the eighth decade tens of thousands of years ago, yet little is known about premodern geriatric epidemiology. We propose that microbiota of the gut, oral cavity, nasal cavity, and brain may modulate AD pathogenesis, and that changes in the microbial composition of these body regions across history suggest escalation of AD risk. Dysbiosis may promote immunoregulatory dysfunction due to inadequate education of the immune system, chronic inflammation, and epithelial barrier permeability. Subsequently, proinflammatory agents—and occasionally microbes—may infiltrate the brain and promote AD pathogenic processes. APOE genotypes appear to moderate the effect of dysbiosis on AD risk. Elucidating the effect of symbiotic microbiota on AD pathogenesis could contribute to basic and translational research.
Keywords: Alzheimer’s disease, dementia, microbiome, evolutionary medicine, immunoregulation
Introduction
Chronic inflammatory diseases are increasingly recognized as attributable (at least partially) to immunodysregulation resulting from inadequate or adverse exposure to microorganisms.1 In order to appreciate the complex etiology of these diseases, it is necessary to consider the critical role of symbiotic microorganisms in healthy development and function of the human immune system. In addition, understanding how microbiota affect risk and etiology of a specific disease can help us trace human vulnerability to that disease across the evolutionary history of our changing symbiotic microbiota.
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that involves both peripheral and central immunodysregulation.2 Chronic inflammation and impairment of immunoregulatory function precedes cognitive decline by decades,3 suggesting that immunodysregulation may be an early hallmark of AD progression.
Given the association of immunodysregulation and AD pathogenesis, evaluation of a possible relationship between AD and symbiotic microbiota should be investigated. We previously described global epidemiological patterns suggestive of a negative correlation between environmental microbial diversity and age-adjusted AD rates;4 and another group conducted a pilot study suggesting that AD patients, compared with healthy controls, had lower intestinal microbial diversity.5 The relationship between AD and symbiotic microbiota has, otherwise, not been explored, and the evolutionary origins of AD are essentially unknown.6
We focus on the sporadic form of AD, which afflicts individuals most often from age 65 onward. Anthropological evidence suggests that the human life span likely increased into the eighth decade tens of thousands of years ago.7 Although life expectancy at birth is, today, markedly increased compared to earlier epochs of history, this metric is heavily influenced by early life mortality rates (e.g., during infancy and childhood). Evidence suggests that adult-specific life expectancy (i.e., expected years of life for those who already survived to adulthood) has exhibited only a small shift in the recent era,8 although a more dramatic change occurred for a minority of the global population (~15% of nations).9 Long life span is a hallmark feature of the human species, and natural selection may have favored this trait because of human reliance on intelligence and functional competence during later life phases (further discussion of this issue is beyond the scope of our review here).10 Yet, almost nothing is known about when AD emerged alongside the history of humans living into old age.
We adopt an evolutionary medicine approach to advance an argument that recent changes in human microbiota have enhanced AD risk. Evolutionary medicine is a multidisciplinary academic area that applies the concepts of evolutionary biology to the study of human health and disease.11 This approach highlights human coevolution with symbiotic microorganisms and how recent changes in human environment and lifestyle—such as agriculture or industrialization—have altered the composition of symbiotic microbiota.12 These alterations—which include severe reduction or loss of certain microorganisms, adoption of new pathogenic microorganisms, alterations in relative abundances, and overall reduction of microbial diversity—are implicated in chronic inflammatory disease etiology.13 Human14 and murine15 studies have shown that low microbial diversity is associated with disease states. Indeed, the major changes in the human microbiome likely occurred alongside the major epidemiological transitions in human history.16
We briefly review how the microbiome changed across history of major lifestyle transitions, and then review how residential microorganisms of the human gut, oral cavity, nasal cavity, and brain may influence AD pathogenesis. Specifically, we review epidemiological evidence linking the composition of microbes in each body region to AD risk and etiology, the mechanisms by which this influence may occur, and how changes in the composition of microbes in each body region across human history may have enhanced AD risk. We use the term “microbiota” to describe symbiotic microorganisms, and “microbiome” to refer to their genetic composition. We focus on bacteria because they account for the majority of symbionts in the human body, though we acknowledge that other microbes—viruses, fungi, and archaea—may also play a role.17 We limit our discussion to microbiota of the gut, oral, and nasal cavities, and pathogenic microbial infiltration of the brain, while acknowledging that other body regions host microbes that could potentially be involved in modulating AD risk.
Microbial transitions in human history
The human species has experienced at least three major lifestyle shifts over the last 15 thousand (k) years consisting of changes in demography, environments, subsistence, and epidemiology—which, we surmise, coincidentally enhanced AD rates. The shifts were gradual, and different regions of the world experienced these shifts at different rates.18 We highlight below how each transition involved exogenous (e.g., demographic, environmental, dietary, and sanitation) changes that led to endogenous alterations to the composition of microbial communities (Fig. 1). We then speculate that each of these changes in microbial composition likely led to increased risk of immune dysregulation and chronic inflammation (and in some cases, increased risk of microbial translocation to the brain), and thereby progressively to enhanced risk of AD immunopathology. We use the term “industrialized” to refer to any population that experienced an industrial transition, inclusive of what economists may refer to as “postindustrial.”
Figure 1.
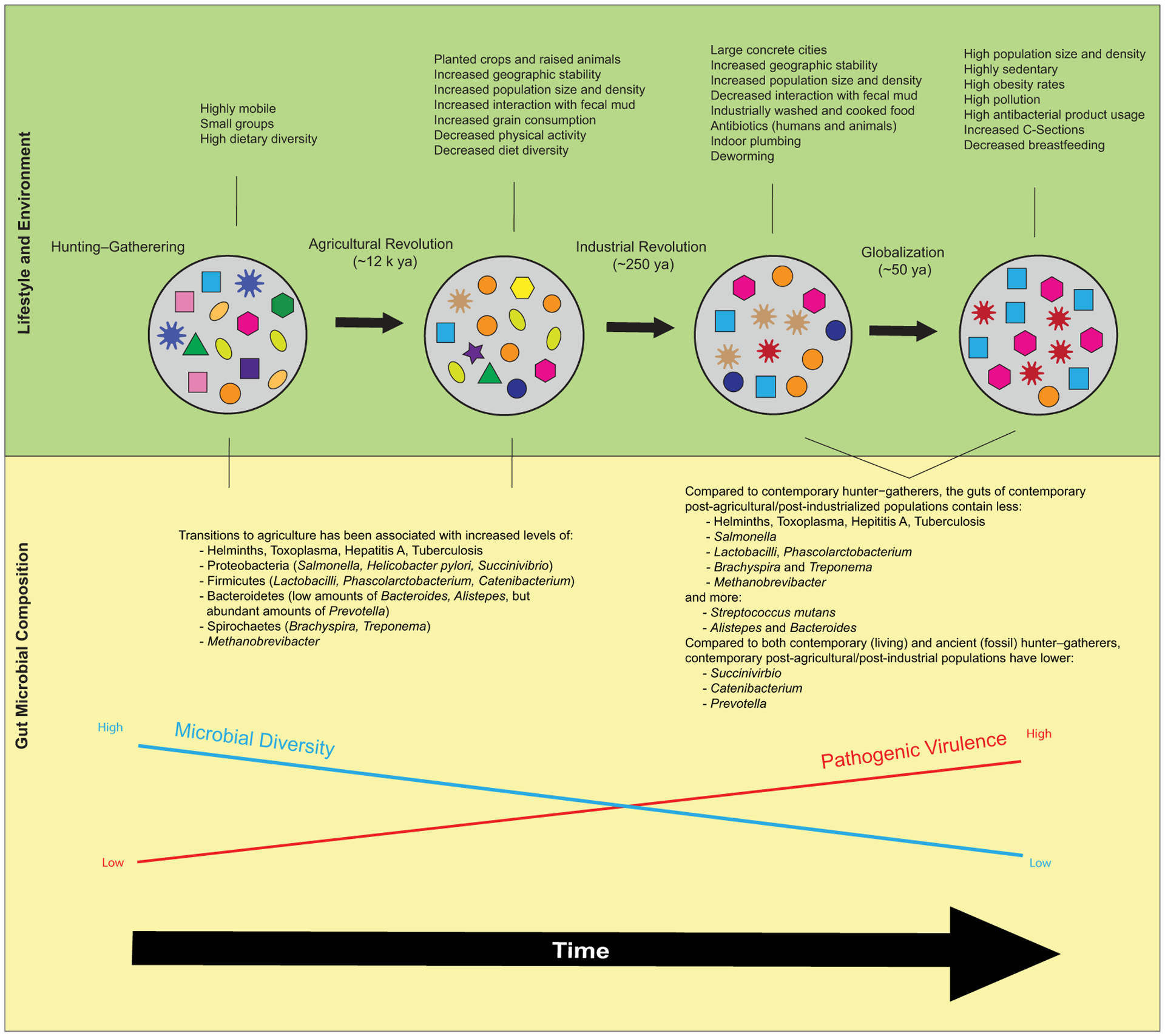
Over the past 15,000 years of human history, there have been a series of major lifestyle transitions that comprised changes in demography, environments, subsistence, and epidemiology. These transitions also likely brought about major changes in the composition of symbiotic microbiota, including generally reduced diversity and enhanced pathogenic virulence. Suppositions regarding pre-Agricultural Revolution gut flora are based on analyses comparing the gut microbial compositions of contemporary people who practice hunting–gathering subsistence strategies in nonindustrialized communities in Tanzania (Hadza), Peru (Matses),97 Malawi, and Venezuela,232 and fossil assemblages of Neanderthals (compilation from Spain, Croatia, Germany, and Russia), Denisovans (from Siberia233), and other early hominins (from Spain234) with those of contemporary industrialized populations in Europe, North America, Asia, and Oceania.97 We note that contemporary hunter–gatherers offer an imperfect proxy for premodern gut microbiomes.235
Agricultural revolution
The advent of agriculture, initially in the Fertile Crescent ~12k years ago (ya) and subsequently spreading to most parts of the world by ~4k ya,19 increased the geographic stability of human habitats, leading to increased population size and density. Pathogenic virulence likely increased due to the evolutionarily selective opportunity of larger numbers of potential human hosts living in closer proximity.20 As for microbial diversity, theoretically, increase to host population density could impose any number of changes upon host microbiota.21 However, experimental22 and correlational23 evidence suggests that increasing symbiont–host encounters when host microbial diversity is low results in greater pathogen transmission between hosts. And the reduced likelihood of dead-end hosts can lead to increased microbial uniformity across hosts.21
With the introduction of agriculture, dietary, social, and demographic changes altered the composition of the human microbiome.24 People living in stable camps planted crops and raised animals, which increased human interaction with fecal mud (both humans and animals).18 Diet diversity decreased and there was a marked increase in grain consumption.20 There was likely an increase in Helminth colonization and orofecal transmission of bacteria, which would have brought novel pathogens into the symbiotic communities of the human body.18
Industrial revolution
The advent of factories and industry further increased the geographic stability of human habitats, giving rise to large, concrete cities with higher population sizes and densities. Initially occurring in 1760s Great Britain and rapidly expanding thereafter, much of the world was industrialized by 1830.25 By living in cities, people experienced less contact with animals and fecal-contaminated mud than before.26 Pathogens likely benefitted from even more advantageous opportunity to infect human hosts due to shared indoor environments and higher population density, and were thereby likely selected to become even more virulent.20 Air pollution became increasingly prevalent as industrial manufacturing and motorized transportation proliferated.27 Given contemporary evidence that indoor, outdoor,28 and dust storm air29 can contain significant microbial load, it is plausible that industrial pollution altered the composition of airborne microbiota.
Additionally, technological advances in transportation led to greater volume and distance of population movement, and the resulting greater human mobility also transported microbes to new habitats. For example, Vibrio cholerae was confined to the Indian subcontinent until the 19th century, when intercontinental railroad systems and steamships transported humans harboring it, resulting in cholera pandemics in four continents.30
Agriculture became dominated by single-crop yields,31 which may have further reduced intestinal biodiversity. Food started to be industrially washed and cooked in more sterile environments.26 Eventually, the industrial era brought about indoor plumbing and sewage systems and, beginning in 1945, use of antibiotic medications for humans and livestock that would eventually become widespread.32 Because of these changes in food processing, antibacterial products, and plumbing infrastructure, we speculate that the diversity of the human microbiota decreased.
Globalization era
The past ~50 years have seen exponential acceleration of certain changes that began with industrialization, as well as other novel changes.33 Population density and mobility that began in the era of industrialization dramatically increased. As of 2008 and for the first time in history, the majority of the human population resides in urban rather than rural habitats.34
The composition of air pollution changed, owing to a substantial reduction of coal smoke of the industrial era, for example, in London levels went from 400 mg/m3 in 1922 to <10 μg/m3 today.35 Air pollution today is mostly derived from vehicle emission of airborne particulate matter from combustion of hydrocarbon fuel.36 In addition, commercial passenger aviation became globally available in the 1970s,37 leading to a surge in global mobility that continues today, for example, between 2000 and 2017 saw a 49% increase in international migration.38
Sedentism due to ever-increasing reliance on machinery and technology,39 and diets primarily composed of high-fat, high-sugar, processed foods40 promote exponentially increasing rates of obesity and metabolic disease.
From the 1960s to the 1980s, there was a steady increase in the discovery and development of antibiotic classes,41 with concomitant, prolific use of antibiotic therapies in humans and animals.32 Overuse of antibiotics imposes evolutionary selection for antibiotic resistance42 and pathogenic virulence.43 An expanding catalogue of highly virulent, multidrug-resistant bacteria now causes diseases across the globe, for example, in Europe there are ~400,000 new multidrug-resistant bacterial infections annually.44
Major changes have occurred in perinatal practices, including increases in caesarean (C) section deliveries45 and declines in rates of breastfeeding.46 C-sections and formula feeding can contribute to dysbiosis during critical phases of development.47
Next, we describe how these changes may successively contribute to immunodysregulation and other features of AD pathogenesis, potentially enhancing AD risk across human history.
Gut microbiota
Loss of biodiversity in the human gastrointestinal tract may contribute to AD incidence. The gut hosts the largest microbe reservoir in the body, providing the main source of immune education through sampling of antigens of anaerobic bacteria (99% of the gut microbiome), fungi, protozoa, and archaea.48 Additionally, the gut participates in maintenance of brain homeostasis by producing neurotransmitters, metabolites, and nerve signals that are transmitted along the gut–brain axis.49 Insufficient microbial diversity in the gut can lead to decreased immune efficacy, increased peripheral inflammation, and increased barrier permeability, all of which may perturb brain homeostasis and ultimately contribute to AD pathogenesis (Fig. 2). Lifestyle transitions across human history may be responsible for the depletion of gut microbial diversity that could have enhanced AD risk via this pathway.
Figure 2.
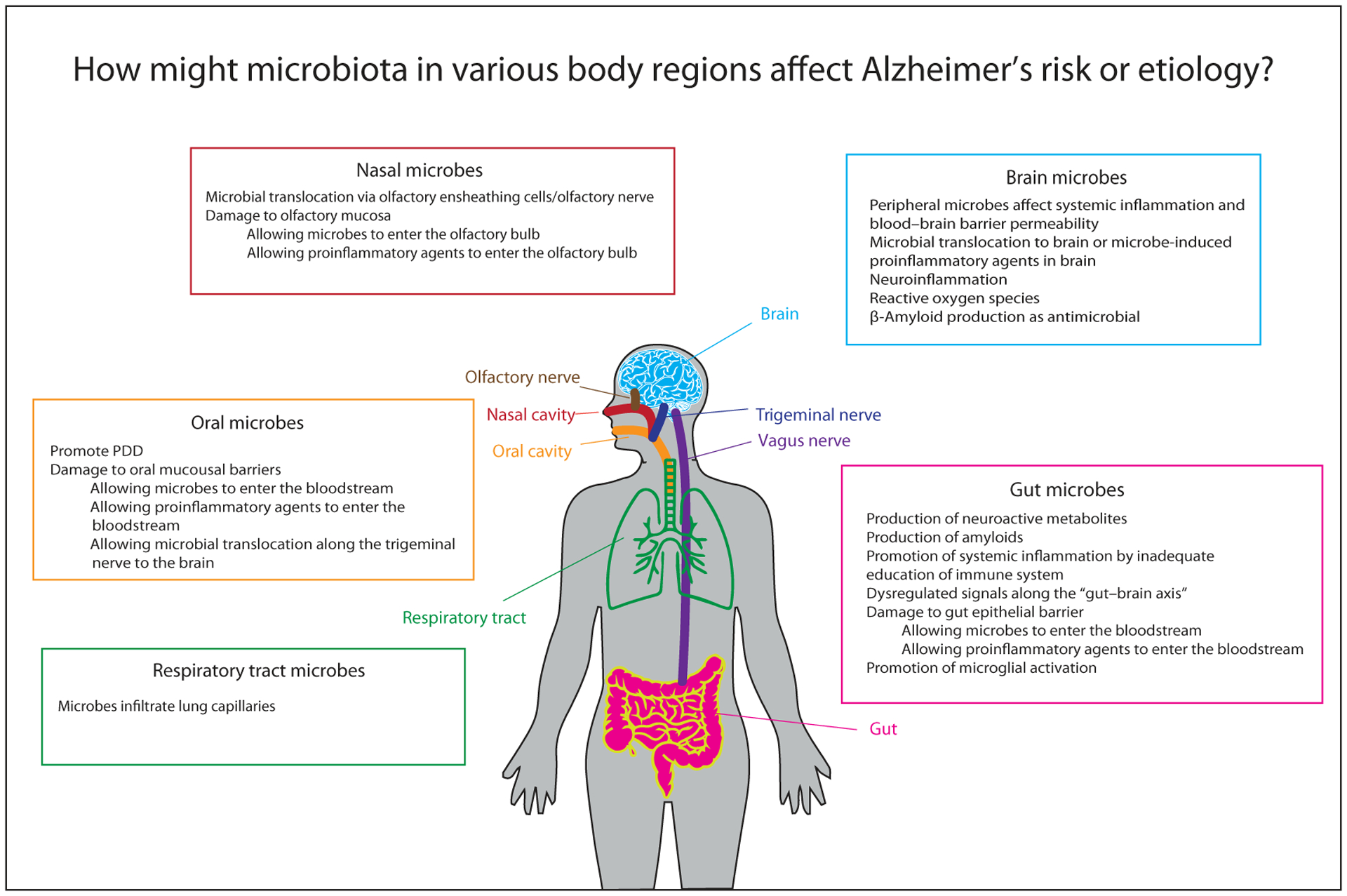
Biomechanisms by which symbiotic microbiota in various regions of the human body may affect Alzheimer’s disease pathogenic processes.
Epidemiological patterns linking gut microbiota to AD
Gut dysbiosis—a high number of pathogenic species or a pathological lack of species diversity in the gut—has been associated with aging, metabolic inflammatory diseases (MIDs), and disorders of the brain, including neurodegenerative disorders. These conditions have overlapping etiologies with AD, and the ways in which gut dysbiosis affects risk (via inflammation and gut epithelial permeability) may also apply to AD.
Aging is the primary AD risk factor. Changes in the gut microbiome across the major lifestyle transitions of human history parallel the age-related changes in the gut microbiome across an individual’s adult life span, such as diminished microbial diversity.50 Geriatric aging is often associated with lower microbial diversity, degradation of gut mucosal barriers,51 and reduced repositories of naive T cells.52 Gut microbial composition is relatively stable over time within an individual.53 However, there is wider interindividual variation among geriatric individuals’ gut microbiomes than among those of younger individuals;14 some geriatric individuals may therefore have more resilience than others to dysbiosis-related immunodysregulation. Additionally, geriatric individuals exhibit lower Bacteroidetes:Firmicutes (B:F) ratios compared with the guts of younger individuals,14 suggesting age-related changes that mirror the difference between contemporary hunter–gatherer (high B:F ratio) and contemporary industrialized populations (low B:F ratio).
Evidence that the gut microbiome can affect MIDs and that MIDs can induce neuronal damage highlights a possible mechanistic pathway linking gut dysbiosis and AD. MIDs, such as obesity,54 hypertension,55 and type-2 diabetes mellitus (T2DM),56 are each independent risk factors for AD. Distinct gut microbiome alterations are associated with MIDs, and the consequent high levels of circulating lipids, glucose imbalance, inflammation, and amplified oxidative stress can lead to neuronal damage.57 AD pathogenesis may exhibit a similar pathological cascade to MIDs, with peripheral inflammation leading to blood–brain barrier (BBB) permeability, neuroinflammation, brain insulin resistance, activated microglia, and neuronal damage.
Obesity may be—at least sometimes or partially—promoted by a dysbiotic gut, which may in turn promote peripheral inflammation, neuroinflammation, and neuronal damage.58 Obesity in human studies has been associated with gut dysbiosis.59 In mice, an obesity phenotype can be transferred through transplant of gut microbiota,60 suggesting gut dysbiosis as a causal factor. Additionally, in obese humans, adipose tissue releases proinflammatory cytokines, adipokines, and chemokines.61 These can contribute to peripheral and central inflammation,62 damage to the hypothalamus,63 decline in white matter integrity,64 and microglial activation.58
T2DM is also an established risk factor for AD,65 a link that may be partially attributable to gut dysbiosis. Lower abundance of butyrate-producing bacteria has been associated with higher T2DM risk in murine and human studies.66 Butyrate and other bacterially produced short-chain fatty acids (SCFAs) may affect host metabolism through modifying ATP67 and glucose68 production. SCFAs may also affect host inflammation by modifying intestinal epithelium integrity69 and differentiation of regulatory T cells (Treg cells).70 These alterations to peripheral metabolism and inflammation may not only affect T2DM risk, but also ultimately may affect central metabolism and inflammation, which are systems whose disturbance is associated with AD.
AD involves neurological dysfunction and can be compared to other psychiatric syndromes that also involve atypical brain circuitry.71 Evidence that gut microbiota can affect the brain in other psychiatric disorders bolsters the theory that gut microbiota could potentially affect AD risk and etiology. Evidence indicates that there are distinct human microbiome profiles for major depressive disorder (MDD),72 autism spectrum disorders (ASDs),73 and schizophrenia.74 Gastrointestinal symptoms are also more prevalent and severe in children with ASD, compared with control children.75 Schizophrenic individuals, compared with controls, have more inflammation and gastrointestinal dysfunction, including bacterial translocation from gut to bloodstream;76 Severance et al. posit that this may lead to overactivated inflammatory responses along the gut–brain axis that lead to psychological changes.
Rodent studies suggest that manipulations of the gut microbiome, such as administering pathogenic bacteria, antibiotics, or germ-free (GF) rearing, can lead to alterations in anxiety-like behaviors.77 Social avoidance behavior in adult mice, used as a model of human anxiety or autism, has been associated with a specific gut microbial profile78 and can be induced by implanting those microbes in the gut.79 Other studies induced depression-like behavior in rodents by fecal transplant from human MDD patients.72,80 Another study observed schizophrenia-like behavior in mice after fecal transplantation from human schizophrenia patients.81
Evidence is emerging that individuals with neurodegenerative diseases have distinct gut microbial profiles. In one study, fecal samples of 25 AD patients contained less microbial richness and diversity compared with 25 control (non-AD) patients.5 Others found the depletion of gut microbes in transgenic mice influenced cerebral β-amyloid deposition, a feature of AD.82,83 Parkinson’s disease (PD) patients have distinctive fecal microbial profiles compared with healthy peers.84 Others have found that when α-synuclein–enriched Lewy bodies (protein aggregates implicated in PD) were injected into the intestinal wall of adult wild-type rats, Lewy bodies were subsequently found in brain tissue.85 These recent studies are among the first to establish patterns between gut dysbiosis and neurodegenerative diseases.
Changes in gut microbiota across human evolutionary history
The major lifestyle transitions in human history were likely each associated with progressively less intestinal biodiversity, which, we argue, can exacerbate AD risk. These historical transitions likely altered gut microbiota through changes in human exposure to animals and soil, food production activities, and diet.
It has been suggested that before the Agricultural Revolution, human gut microbiota comprised a greater variety of species with different relative abundances compared to today. After the Agricultural Revolution, with diets composed of a smaller variety of domesticated species, human gut microbiomes likely exhibited lower diversity86 and relative abundances that reflect grain-dominant diets.87 The transition to agriculturalism also brought an increase in fermentation practices, for example, fermented milks beginning ~10k ya and alcoholic fermentation of barley and grapes beginning ~5k years ago.88 Fermentation converts sugars in an anoxic environment to other products, removing toxic compounds and supplying probiotic bacteria.89
Dietary changes related to the Industrial Revolution likely had significant effects on taxonomic composition and metabolic features of human gut microbiota.90 Postindustrial diets are associated with higher abundance of pathobionts in the gut, which tend to survive on sugar better than other symbiotic species, which survive best on high-fiber diets.91 This transition also marks a distinct decrease in the consumption of fermented foods, likely due to the introduction of refrigeration and shelf-stable processed foods, likely associated with decrease in microbial diversity.
The decrease of dietary and agricultural diversity in the Globalization Era92 likely correlates with lower-than-ever gut biodiversity. The United Nations’ Food and Agricultural Organization described a 75% loss of agricultural biodiversity since the 1900s, with the majority of food today produced by only 12 plant and 5 animal species.93 Gut biodiversity may also be diminished by extensive use of antibiotics in human medicine, antibiotics in domesticated animals, and pharmaceutical-grade pesticides on plants.
Evidence for these historical shifts comes from analyses comparing the gut microbial compositions of contemporary people who practice hunting–gathering subsistence strategies in nonindustrialized communities (Fig. 1). Studies found U.S. individuals’ gut microbiomes have 15–30% fewer species than contemporary hunter–gatherer individuals’ gut microbiomes.94–96 Furthermore, while there is enormous overlap in composition of symbiotic microbes between different industrialized populations, contemporary hunter–gatherer gut microbial compositions were independent both of each other and of the microbiome compositions from industrialized populations.97 The observation that human gut microbial composition is similar across industrialized populations is consistent with the possibility that symbiotic microbial uniformity could partially account for epidemiological similarity across otherwise disparate world regions. Additionally, the guts of dogs parallel human gut diversity because of domestication;98 indeed, the guts of modern dogs of industrialized populations also show dysbiosis and higher incidence of inflammatory bowel disease.99
We contend that there exists a causal pathway from low gut microbial diversity to enhanced AD risk, and that this pathway contributes to the pattern of increasing AD prevalence in today’s industrialized, globalized world.
Biomechanisms potentially linking gut microbiota to AD
Gut microbiota can influence brain function and health in various ways that could be relevant for AD, including producing neuroactive metabolites and amyloids, and promoting systemic inflammation, barrier permeability, and microglial activation. The gut hosts critical immunodevelopmental process by mucosal dendritic cell exposure to microbes, which prompts Treg cell population expansion, thereby establishing tolerance to symbiotic microbiota.48 Treg cell maturation may be specifically induced by particular bacterial products (e.g., SCFAs) as well as certain chloroform-resistant, spore-forming bacteria (e.g., Bacilli, Clostridia, and Firmicutes).100
Neuroactive metabolites.
Gut microbes produce neuroactive molecules and neurotransmitters whose dysfunctions have, elsewhere, been linked to AD.101 For example, gut Bifidobacterium and Lactobacillus produce the neurotransmitter gamma-aminobutyric acid (GABA)102 and may regulate brain GABA-receptor expression by communication along the vagus nerve.103 GABA dysfunction is implicated in AD neuropathy.104 Also, gut microbes may regulate the serotonergic system directly by producing serotonin (in vitro evidence, e.g., Lactobacillus plantarum, Escherichia coli K-12) or by producing (e.g., E. coli) or degrading (e.g., Bacteroides fragilis) serotonin’s precursor molecule, tryptophan.105 Gut-derived serotonin likely has only indirect effects on brain serotonergic function. However, the gut is the only source of tryptophan, derived either from the diet or microbial production.105 Tryptophan crosses the BBB to become available for serotonin synthesis in the brain.106 Dysfunction of peripheral107 and central108 serotonergic systems has been implicated in AD pathophysiology. Additionally, an N-methyl-D-aspartate (NMDA)-targeting neurotoxin that was observed to be elevated in the brains of patients with AD109 may be produced by gut cyanobacteria.110
Amyloids.
Amyloidosis, or the accumulation of amyloid proteins causing damage to the body, is implicated in AD by virtue of the hallmark feature of Aβ accumulation. Amyloid formation is also a widespread, naturally occurring feature of many bacterial clades, including members of Proteobacteria, Bacteroidetes, Chloroflexi, Actinobacteria, and Firmicutes, which utilize amyloids in biofilm development and surface adhesion.111 Further research is needed to clarify whether bacterial amyloids affect host amyloidosis pathogenesis.111
Systemic inflammation.
Gut dysbiosis may induce chronic, systemic inflammation, a condition implicated in AD immunopathogenesis. Typical development of the mammalian immune system requires education by colonizing microbiota in a mutualistic exchange that involves microbes residing within the rich environment of the host’s body, particularly the gut, performing various functions necessary for host survivorship (e.g., nutrient extraction). This careful balance is mediated by the mucus membrane, which minimizes contact between host tissues and microbiota.48 This balance allows the gut to respond appropriately when exposed to pathogenic or mutualistic microbiota.48 When the intestinal ecosystem is out of balance, pathological problems may arise. A dysbiotic gut inadequately educates host T cells, causing exaggerated inflammatory responses and, potentially, chronic inflammation.112 While short-term inflammatory response restores biological equilibrium (e.g., fighting acute infection), chronic inflammation can damage host cells and tissues, causing excessive epithelial permeability. Chronic inflammatory conditions hold open endothelial junctions, which would otherwise only briefly open during acute inflammatory response, to permit inflammatory mediators and immune cells to cross endothelial barriers freely.
Barrier permeability.
Gut dysbiosis may compromise barrier integrity, which may promote AD pathogenesis by allowing translocation of pathogenic agents out of the gut and into the brain. One study demonstrated that butyrate-producing bacteria enhance intestinal epithelial barrier integrity and their absence can contribute to barrier dysfunction.113 Others found more Heliobacter pylori in the sera and gut mucosa of AD patients compared with controls.114 The BBB has been shown to exhibit increased permeability in GF mice compared with pathogen-free conventionally colonized (CC) mice. Transferring microbiota of CC mice to GF mice led to decreased permeability and higher expression of tight junction proteins.115 Pathogen-associated molecular patterns of some microbiota can promote BBB permeability by activating T helper cells to produce type-1 cytokines.116 Barrier permeability can lead to tissue inflammation and bacterial translocation.117
Microglial activation.
Gut microbiota have also been shown to influence microglia. Altered microglial function is implicated in AD pathogenesis.118 One study demonstrated that GF mice (compared with CC mice) expressed global defects of microglia (immature phenotype) causing impaired immune responses.119 Mice with lower microbial diversity had dysfunctional microglia, and recolonization was restorative to microglia. Mice with sufficient SCFA-producing microbes had homeostatic microglia, while mice deficient in SCFA had defective microglia more similar to GF mice.119
Oral cavity
We propose that composition of oral microbiota may be associated with AD via oral infection, leading to microbial translocation to the brain either through the bloodstream or by bypassing the bloodstream via breach of oral mucosal barriers (Fig. 2). Shifts in the composition of oral microbiota over human history may have enhanced AD risk via this pathway. Evidence for this link comes from associations between periodontal disease (PDD) and tooth loss with AD, as well as evidence that oral bacteria are disproportionately prevalent in the trigeminal nerves and brains of AD patients.
Epidemiological patterns linking oral microbiota to AD
Epidemiological evidence suggests that tooth loss may be associated with AD incidence. In a Japanese study, individuals with fewer than half of their teeth by age 50–60 were 2.6 times more likely to later develop AD.120 Similarly, in a Swedish study examining monozygotic twin-pairs discordant for AD, having lost half or more of their teeth by age 35 was strongly associated with AD (odds ratio (OR) = 5.5).121 The authors of these studies speculated that poor oral hygiene and concomitant PDD were the causes of tooth loss, although this was not directly observed. In a longitudinal study of Wisconsin nuns without AD at initial recruitment (ages 75–98), those with fewer teeth (<10) went on to develop AD over the 12-year study period with 2.2 times the risk of peers with more teeth (10+) at the start of the study.122 The temporal order of events in this longitudinal study is consistent with the possibility of causality. A recent study suggests causality in a rodent model: oral infections of Porphyromonas gingivalis in mice led to brain colonization and subsequent brain Aβ production.123 A small-molecule inhibitor targeting P. gingivalis toxic proteases (gingipains) blocked Aβ production, reduced neuroinflammation, rescued neurons in the hippocampus, and reduced bacterial load.123 Altogether, evidence is consistent with the hypothesis that PDD-associated tooth loss contributes to AD risk; further research is needed to establish causality in humans.
Changes in oral microbiota across human evolutionary history
We posit that dietary changes across human history may have enhanced AD risk through changes in microbial composition of the oral cavity. Supportive evidence is beginning to emerge that major lifestyle transitions led to increases in PDD and PDD-associated oral microbiota, and that both have been correlated with AD risk. PDD is considered by archaeologists to be relatively new in human history, emerging around the time of the Agricultural Revolution and then escalating during the Industrial Revolution. Oral pathologies—including tooth decay and PDD—are rare in archaeological assemblages from early hunter–gatherer populations.124 Principal component analysis of microbial beta-diversity comparing the dental calculus of 6 hunter–gatherer European skeletons from the Mesolithic (7550-5450 ya, pre-Agricultural Revolution) and 22 agriculturalist European skeletons from the late Neolithic and Medieval periods (750–400 ya, post-Agricultural Revolution) shows a distinct shift in the composition of oral microbiota.124 Skeletal evidence from other studies suggests a simultaneous increase in PDD incidence.125 This could be due to the decrease in diet diversity during the Agricultural Revolution, relative to the subsistence strategy of hunting–gathering.20 The aforementioned microbial DNA extracted from dental calculus in individuals from agricultural communities in the late Neolithic and Medieval periods shows the presence of PDD-associated microbes, including P. gingivalis and Tannerella, which are absent from the Mesolithic hunter–gatherers.124 Additionally, the dental calculus of individuals from the Neolithic and Medieval farming communities, compared with the Mesolithic hunter–gatherers, had higher concentrations of Treponema,124 which has also been associated with PDD and found at a greater frequency in postmortem brains from AD patients, compared with healthy controls.126 Later, the Industrial Revolution brought an influx of processed and refined sugars to the human diet, which have been implicated in enamel demineralization, subsequent dental caries, and eventual tooth loss, which have been associated with AD risk.120,122
Biomechanisms potentially linking oral microbiota to AD
Oral microbiota may influence AD pathogenesis either indirectly by promoting oral barrier permeability that permits oral microbes or inflammatory mediators to enter the bloodstream, or directly by microbial translocation from oral cavity to brain. Evidence that oral microbes are observed disproportionately in the brains of AD patients supports the plausibility of oral microbial translocation as a contributor to AD pathogenesis.
Oral microbes promote PDD, which can damage oral mucosal barriers allowing microbes to enter the bloodstream.127 Observed associations between oral microbial composition and AD may be attributable to damage from trauma, microabrasion, or PDD-caused breaches in mucosal and vascular barriers.128 In PDD, plaque in the area between the gums and tooth is filled with Gram-negative bacteria.129 Damaged barriers in the oral cavity allow for bacteria to enter the bloodstream (transient bacteremia), thereby provoking a proinflammatory response, including cytokines, that may enter the central nervous system (CNS).130 Both PDD127 and AD131 are characterized by type 1 inflammation; type 1–associated cytokines in circulation can traverse the BBB and cause microglial activation, which in AD patients promotes the production of Aβ, tau, cerebrovascular pathology, and neuron death.132
The maxillary and mandibular branches of the trigeminal nerve connect the oral cavity directly to the brain. Studies suggest that microbes may translocate from the oral cavity to the brain directly via the trigeminal nerve, with evidence in AD patients of the PDD-associated bacteria Treponema in the saliva,126 tooth pulp chambers,133 trigeminal ganglia, pons, and, finally, brain structures afflicted in AD.134 Specifically, in postmortem specimens from AD and control subjects, Treponema was detected in the trigeminal ganglia, with more Treponema species in AD compared with control specimens.126 Treponema has been detected in the pons (site where trigeminal ganglia enter brain) as well as in the hippocampus and the frontal lobe cortex at significantly greater concentration and higher number of Treponema species among AD than control brains.126 Six PDD-associated Treponema species have been associated with increased prevalence in AD brains.135
Nasal cavity and respiratory tract
We propose the possibility that air pollution (including microbial) may influence AD risk and shifts in air quality/composition over human history may have increased AD risk. Particulate matter (PM10) in urban environments—but not, for example, natural dust storms—contains high concentrations of Gram-negative bacterial lipopolysaccharides (LPSs),136 suggestive of an industrialization-specific AD risk factor. The olfactory bulb is the part of the brain most exposed to the outside environment. Therefore, this area is particularly vulnerable as a route by which microbes from external environments may affect brain health.137 Additionally, the olfactory bulb is one of the first and hardest hit areas in AD pathogenesis, and olfaction deficit has been implicated in early AD pathology.138 It is plausible that the composition of microbiota in the nasal cavity and respiratory tract could be altered by air pollution in such a way that pathogenic microbes penetrate the olfactory bulb or take other routes to the brain, ultimately inducing AD pathogenic insult (Fig. 2).
Epidemiological patterns linking nasal and respiratory tract microbiota to AD
Air pollution.
Epidemiological evidence links exposure to air pollution to AD incidence and brain insults. In a cross sectional, case–control study in Taiwan, exposure to high concentrations of PM10 and ozone (O3) was positively associated with having AD.139 Jung et al. corroborate these findings in another Taiwanese population in a 9-year longitudinal study by showing that for every 4.34 μg/m3 increase in fine particulate matter (PM2.5) in the local environment, individuals aged 65+ exhibited a 138% increased risk of developing AD, and for every 10.91 parts per billion (ppb) increase in O3, a 211% increased risk.140 A retrospective study among a Canadian cohort aged 55–85 showed higher risk of AD onset for those living <50 miles from a major road, compared with individuals living further away (hazard ratio (HR) = 1.07).141 Longitudinal studies of traffic-related air pollution showed that more long-term exposure to nitrogen oxides was associated with greater risk of AD onset in a Swedish cohort (HR = 1.38),142 and long-term exposure to black carbon was associated with greater risk of having a low mini mental state exam (MMSE) score in a U.S. cohort (OR = 1.30).143
Autopsy studies have demonstrated AD-associated features in the brains of children and young adults in industrial zones of Mexico City—where there are among the highest concentrations in North America of PM10, especially PM2.5, and O3—compared with matched samples from low-air-pollution Mexican cities.144 Mexico City specimens exhibited more Aβ in olfactory ensheathing cells (OECs), more degradation and particulate contamination of nasal and olfactory blub epithelia145 and BBB,146 as well as greater frontal lobe extracellular Aβ and hyperphosporylated tau, as well as frontal lobe upregulated gene expression of pattern recognition receptors that respond to microbial contact and genes indicative of neuroinflammation and oxidative stress.144
Olfactory impairment.
We posit that microbial agents in the nasal cavity may contribute to AD neuropathy in the olfactory bulb, which in turn may explain olfactory impairment in AD. Impaired olfaction (and eventually total loss of the sense of smell, i.e., anosmia) is often an early clinical sign of AD.147,148 AD patients, compared with age-matched controls, exhibit impairment in olfactory detection and identification.149,150 Longitudinal studies of elderly adults without dementia found that olfactory impairment was associated with greater risk of developing mild cognitive impairment (MCI)151 and AD152 over the study periods. A longitudinal study of MCI individuals found that olfactory impairment was associated with greater risk of developing AD.153 Another longitudinal study found that anosmic individuals had 1.9 times higher odds of developing AD compared with normosmic individuals.154
Degree of olfactory impairment among AD patients appears to be correlated with degree of AD neuropathy. Olfactory impairment among AD patients has been correlated with hippocampal volume reduction,155,156 neurofibrillary tangle density in the central olfactory system (entorhinal cortex, CA1-subiculum;157 also among MCI patients151), blood oxygenation level–dependent signal reduction in the primary olfactory cortex,158 and interruptions in the olfactory processing network.159 Another study found that Aβ load was higher among MCI participants who performed poorly on olfactory identification tests compared with healthy controls, but no differences in olfaction between MCI subgroups by Aβ load.160
Olfactory impairment is strongly associated with air pollution, and the relationship may be APOE-genotype conditional.145 The observed correlations between air pollution, anosmia, damage to the olfactory system, AD neuropathy, and AD incidence justify further investigation of the role of nasal cavity microbes in enacting these relationships.
Changes in nasal and respiratory tract microbiota across human evolutionary history
Several observations suggest a relationship between air pollution and changes in nasal and respiratory tract microbial composition. Bacterial or viral infection of the respiratory tract is inherently reflective of alteration to the microbial communities in the respiratory tract. By 1900, the leading cause of death in the United States was pneumonia.161 Bacterial pneumonia has been linked to nitrogen dioxide and PM2.5 exposure.162 Air pollution is also strongly associated with viral infection of the respiratory tract. Longitudinal studies observed dose-dependent relationships between air pollution and croup (typically caused by viral infection) in Germany163 and between air pollution and acute respiratory infection in Kenya164 and Finland.165 Today, microbial infection of the respiratory tract is the singular leading cause of global burden of disease,166 underscoring that the industrialization-related increases in air pollution cause disease primarily through a microbial mechanism. Further research is needed to discern how microbial composition of air pollution affects nasal cavity microbial composition and subsequent health problems.
The Industrial Revolution saw the emergence of manufacturing and motorized transportation, and resultant increase in air pollution.167 For example, sulfur dioxide emissions were negligible before the Industrial Revolution, rose globally across the 19th and 20th centuries,168 and since 1980 have continued to rise in Asia and Africa but have fallen in Europe, South America, and North America.167 We propose that the degree to which air pollution enhances AD risk has mirrored these large-scale patterns, which are consistent with Globalization Era surges in AD rates in developing countries.169
Biomechanisms potentially linking nasal and respiratory tract microbiota to AD
The observed associations between air pollution, nasal and respiratory infection, anosmia, and AD risk may be operating through two routes: first, nasal microbes transmitted along olfactory receptor neurons, and second, respiratory tract microbes transmitted to the brain via lung capillary blood infiltration.
Olfactory mucosa—containing axons directly exposed to the external environment—provides a direct route from the olfactory system to the CNS. Agents, such as bacteria, viruses, prions, nanoparticles, and heavy metals, can damage the olfactory endothelium,170 thereby entering the brain via the olfactory mucosa.171 Air pollution may promote microbial infiltration of the brain either indirectly by residential nasal microbes that take advantage of inorganic-pollution-associated damage to nasal epithelia,172 or directly by airborne microbes that cross from nasal cavity to CNS with microbial barrier breach not relying on barrier damage by inorganic compounds.
Located on the bottom of the brain and separated from the oral cavity by the cribriform plate and olfactory epithelium, the olfactory bulb is particularly vulnerable to exogenous agents. The outer two layers of the olfactory bulb contain OECs. These specialized glial cells ensheathe bundles of non-myelinated olfactory nerve axons.173 OECs provide a channel for olfactory nerve axons to grow from the peripheral nervous system to the CNS by guiding axonal growth of olfactory receptor neurons in olfactory mucosa through the cribriform plate to the olfactory bulb.174 Microbes that have penetrated the olfactory epithelium may be transported along the olfactory or trigeminal nerves to the brain.137 Pathogenic microbes, particularly Chlamydophila pneumoniae, from air pollution can infect the nasal cavity and thereby contribute to AD risk or pathogenesis. C. pneumoniae is one (but not the only) cause of pneumonia and up to 20% of all lower respiratory tract infections.175 Pneumonia is the most common cause of death in patients with AD,176 but evidence suggests that C. pneumoniae infection may participate in AD etiology long after pneumonia symptoms resolve.177 Chronic C. pneumoniae infection may gain entry to the CNS by two routes, either of which may contribute to AD neuropathy. The bacteria may migrate directly from the nasal cavity to infiltrate the brain—indeed, an autopsy study of AD patients found C. pneumoniae in the olfactory bulb epithelium178—or C. pneumoniae may infiltrate lung capillary monocytes that then cross the BBB.179
C. pneumoniae has been observed in the CNS of AD patients more frequently than in non-AD individuals and at CNS locations specifically implicated in AD, suggestive of a role in AD neuropathogenesis. In one study, individuals with AD exhibited C. pneumoniae in their cerebrospinal fluid (CSF) more frequently (44% of AD group) than individuals with vascular dementia (10%) or nondementia controls (1%).180 A postmortem study found C. pneumoniae in AD-affected brain regions among 89% of AD patients and 5% of nondementia controls, unrelated to whether the individuals had pneumonia at the time of death.181 In the brains of AD patients, C. pneumoniae has been found in 20% of neurons, as well as in astrocytes and microglia.182 Astrocytes and microglia react to pathogenic bacteria by producing proinflammatory cytokines and reactive oxygen species (ROS), both associated with AD.182
Herpes simplex virus type 1 (HSV1) may gain access to the brain via the olfactory system, its presence in the brain has been associated with APOE-genotype conditional AD risk, and in vitro studies suggest that HSV1 can cause Aβ deposition and tau phosphorylation.183 HSV1 was shown to migrate from nasal mucosa to the CNS via the olfactory nerve in mice,184 and postmortem study of humans who died of herpes simplex encephalitis detected HSV1 antigen in the olfactory tract, olfactory cortex, and olfactory-connected regions of the limbic system.185 There is evidence supporting the possibility that other microbes may also migrate from the nasal mucosa directly to the CNS bypassing the bloodstream, but the relation of these microbes to AD remains understudied, including viruses such as influenza A, Nipah virus, Sendai virus, equine encephalitis virus, rabies virus, and vesicular stomatitis virus,137 and bacteria such as Neisseria meningitides (which accomplishes this migration by damaging cellular junctions in the olfactory epithelium),186 Burkholderia pseudomallei187 (which can colonize the nasal cavity via inhalation of airborne bacteria),137 and Streptococcus pneumoniae.188 Given that S. pneumoniae is the most common cause of bacterial pneumonia—which is highly comorbid with AD—evidence that nasal S. pneumoniae could directly access the CNS should, in particular, be further investigated in the context of AD.
Brain
The brain connects peripheral dysbiosis to the neurodegenerative processes that characterize AD. Unlike the symbiotic microbiota of other body regions, bacteria in the brain are almost always pathological. Therefore, in this section, we do not focus on epidemiological patterns and lifestyle transitions in human history, but rather directly on how microbial effects in the gut, oral, and nasal cavities coalesce to affect the brain in ways that could influence AD risk or pathogenesis (Fig. 2).
Neuroinflammation
Neuroinflammation may be induced by either peripheral inflammation or microbial translocation in concert with BBB permeability. Systemic inflammation has been shown to increase BBB permeability.189 BBB permeability can expose the brain to cytokines that can lead to neuroinflammation. Extensive exposure to proinflammatory cytokines can impair microglia, decreasing microglia’s ability to clear toxic Aβ, reducing the synaptic remodeling capacity of microglia, leading to irreversible neuronal damage.190 Neuroinflammation is an early feature of AD pathogenesis, preceding Aβ accumulation.191
Reactive oxygen species
The cellular metabolism by-products ROS are involved in redox homeostasis, but excessively high levels of ROS cause oxidative stress, which is implicated in AD etiology. At moderate levels, ROS exhibits antimicrobial properties192 and can be produced by host phagocytes in response to certain pathogenic microbes, such as E. coli.193 In excess, ROS can increase epithelial cell differentiation, proliferation, and apoptosis, which can lead to epithelial injury and inflammation.194 Gut-mediated release of proinflammatory cytokines can also cause oxidative stress.195 ROS may enter the brain via compromised BBB and thereby promote AD-associated processes through destruction of brain tissue,196 increasing the accumulation of Aβ and neurofibrillary tangles197 and Aβ-associated neuronal death.198
Microbial translocation
AD patients exhibit greater abundances of pathogenic microbes, overall bacterial load, and LPS in their brains compared with control patients. In postmortem studies, AD patients had greater abundance of pathogenic microbes in their brains compared with non-AD controls, specifically C. pneumoniae,199 HSV1,183 and Treponema135 (discussed above), as well as Borrelia burgdorferi, which a meta-analysis found was 13 times more frequent in the brains of AD patients than controls.135
AD brains exhibited relatively lower Proteobacteria levels and greater Actinobacteria levels compared with controls, mostly attributed to high levels of Propionibacterium acnes, a species of symbiotic bacteria on skin and in the oral cavity, which has elsewhere been associated with inflammatory disease.200 Additionally, postmortem AD brains were shown to have 5- to 10-fold more bacterial reads than controls.200
Gram-negative bacterial LPS of the human gut are abundantly present in AD-implicated brain regions; specifically, neocortex (7-fold) and hippocampus (21-fold) compared with controls.201 A growing body of evidence suggests that oral microbes can enter human brain tissue specifically in the context of AD. P. gingivalis DNA was observed in CSF and saliva of living people with probable AD.123 Another study found LPS of P. gingivalis in the postmortem brain tissue of 40% of AD patients but absent among controls.202
When BBB permeability increases, more microbes can enter the brain. Microbes may cross the BBB transcellularly (E. coli, S. pneumoniae, Mycobacterium tuberculosis), paracellularly (protozoans, Trypanosoma sp.), or through infected phagocytes (Listeria monocytogenes and M. tuberculosis).203
β-Amyloid
Aβ exhibits antimicrobial properties, and its production may be a feature of the innate immune response to microbes in the brain. We posit that microbial translocation from the periphery would increase Aβ production in the brain.
While Aβ may serve an adaptive, antimicrobial function, it may also exacerbate neuronal damage in the context of excessive barrier permeability and microbial translocation. It was first suggested by Soscia et al. that Aβ may be an antimicrobial peptide; they demonstrated that Aβ is active against at least 12 different microorganisms, including Gram-negative and Gram-positive bacteria and the yeast Candida albicans.204 Aβ production has also been shown in response to influenza A,205 herpes simplex virus,206 and pathogenic yeast.207 Exposure of B. burgdorferi spirochetes to rat brain cell cultures induced Aβ production.208 Recent authors, expanding on this evidence (using transgenic Caenorhabditis elegans and murine models), posited that Aβ may be a host response to protect against microbial infections.204,207
APOE
The APOE gene (APOE) and its encoded protein are well-known modifiers of AD risk. APOE allelic variation may play a moderating role in the relationship between dysbiosis and AD risk. APOE isoforms exhibit differential effects at several stages of this pathological cascade: Aβ–microbe complex clearance from the brain, neuroinflammation, and oxidative damage. There are three alleles of APOE (APOE4, −3, and −2), which each translates a unique protein isoform (APOE4, APOE3, and APOE2). APOE4 is also associated with lower peripheral and central concentrations of the APOE protein209 partially due to faster degradation.210 APOE4 is an established risk factor for sporadic AD.211
Evolutionary history of APOE
Evidence suggests that the APOE4 allelic variant associated with AD is the ancestral version of the gene. All other primates have only one allele nearly identical to APOE4.212–214 APOE3 was later formed from a single base mutation ~300 k ya, and another single base mutation formed APOE2 ~200 k ya.212,215,216 It seems unlikely that selection against AD was solely or primarily responsible for the emergence or spread of APOE3 and APOE2. Not only is APOE4 implicated in many different diseases (therefore hard to attribute selection to one over another), but also, nonindustrialized populations exhibit little connection between the APOE4 allele and AD risk.217 We argue that APOE4 might exacerbate some of the deleterious effects of dysbiosis, but in the absence of widespread dysbiosis among premodern human populations, we might suppose that carrying an APOE4 allele may not have independently conferred AD risk.217
APOE and β-amyloid–microbe complex clearance
Recalling the antimicrobial properties of Aβ, it is reasonable to speculate that the ApoE4 isoform prevents Aβ from clearing microbes from the brain more than other isoforms. This speculated higher microbial load in the brains of APOE4 carriers could promote greater neuroinflammatory response. Low-density lipoprotein receptor (LDLR), the main lipoprotein receptor in the brain, mediates the cellular uptake of APOE and Aβ218 and therefore likely also Aβ–microbe complexes. Studies have suggested that APOE and Aβ are in competition for cellular uptake through LDLR.218 Among APOE isoforms, LDLR possesses the highest affinity for the E4 allele (E4 > E3 ≫ E2).219 The higher affinity between LDLR and APOE4 could, conceivably, clear APOE4 at the expense of greater Aβ, and thereby Aβ–microbe complex, retention.
This idea is consistent with observations that APOE4 carriers exhibit reduced Aβ clearance and greater microbial load in the brain. APOE4-expressing mice, compared with other genotypes, exhibited higher levels of Aβ in interstitial fluid and slower rates of Aβ clearance from interstitial fluid.211 A study of postmortem brains of AD patients found those who carried APOE4, compared with noncarriers, were more likely to exhibit C. pneumoniae in AD-afflicted brain regions. The study also found C. pneumoniae in 90% of the brains of APOE4 carriers with AD, but only 5% of APOE4 carriers without AD,181 demonstrating an AD-specific effect. Another study by the same group found that among postmortem AD brains the number of C. pneumoniae–infected cells in AD-afflicted brain regions was higher for APOE4 carriers than other genotypes.220
These studies together demonstrate that APOE4 is associated with reduced Aβ clearance and greater microbial load in the brain specifically in the context of AD, suggesting that AD etiology could, at least sometimes, involve microbial translocation to the brain. While Aβ may typically facilitate microbial clearance from the brain, the APOE4 isoform may be inefficient, compared with other isoforms, at clearing Aβ–microbe complexes from the brain.
APOE and neuroinflammation
APOE is generally an anti-inflammatory molecule. APOE inhibits proinflammatory cytokines, while proinflammatory cytokines downregulate and anti-inflammatory signals upregulate APOE production.221 APOE4 has the weakest anti-inflammatory properties. APOE4 knock-in mice are more susceptible, compared to other genotypes, to LPS- or Aβ-induced inflammation and inflammation-associated damage, such as in traumatic brain injury experiments.222 APOE4 is also more lipid depleted than other isoforms, which may result in less neuronal protection and repair.223 APOE4 is also associated with more AD-specific insult from viruses in the brain, potentially related to inferior capacity to regulate neuroinflammation and consequential neuronal damage. The presence of HSV1 in the brain was associated with AD risk only for APOE4 carriers.183
APOE4 may also exacerbate the effect of oronasal inflammation on AD neuropathy. APOE4 carriers, compared with other genotypes, exhibited more AD-relevant brain pathology144 and AD-associated olfactory dysfunction145 in response to air pollution. Additionally, a longitudinal study found that APOE4 carriers, compared with other genotypes, exhibited stronger relation between olfactory impairment and AD onset.154
APOE and oxidative damage
APOE may also modulate AD risk through its antioxidant qualities,224 with APOE4 exhibiting the weakest protection. APOE4 appears to be less effective against oxidative toxicity225 and more associated with oxidative damage224 than other isoforms. These effects could hold relevance for oxidative stress induced by microbial infection. For example, a study using murine cell culture found that APOE4 cells, compared with APOE3, exhibited greater membrane oxidation and more nitric oxide production in response to Salmonella enteriditis LPS stimulation.226 In a study using synaptosomes isolated from mouse brains, Aβ-induced oxidation caused greater ROS formation among APOE4 specimens compared to other genotypes.227 Others found increased levels of F2-isoprostanes in brains of APOE4 male mice but no genotype differences in female mice.228 Greater ROS production may cause oxidative stress that can lead to a feedback loop of increased Aβ accumulation, neurofibrillary tangles,197 and neuronal damage.198
Conclusion and future directions
Human experiences and exposures to disease risk factors have varied across the course of evolutionary history. As human lifestyles underwent major transitions including the Agricultural Revolution, Industrial Revolution, and globalization, it is likely that major alterations also occurred in the composition of our symbiotic microorganisms. While other diseases have received more attention for their relation to modernization like obesity,229 T2DM,230 and cardiovascular disease231 because links to modernization may seem more obvious, AD should also be considered a disease enhanced by similar pathways during this era.6
Changes across human history to composition of microbial communities in the gut, oral cavity, nasal cavity, and brain may influence immune function and epithelial barrier permeability to modulate various aspects of the AD pathological cascade. The huge scale of changes in human habitats and experiences across history provides a natural experiment to elucidate whether certain disease risk factors or pathogenic processes that appear unavoidable in the globalized world today could be modifiable. As human environments continue to change, this information will aid in forecasting and preparing for disease burden in diverse future habitats.
Improvement in our understanding of the relationship between symbiotic microbes and AD has not only clinical relevance, but also may help us discern what the experiences of aging and the roles of elderly individuals may have been in premodern human society—an issue of interest and debate among anthropologists.6
The interdisciplinary perspective offered by the burgeoning field of evolutionary medicine encourages inquiry into the ultimate origins of diseases,11,20 with potential to contribute to basic and translational research and public health.
Acknowledgment
M.F. was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Award K01 DK105110-01.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Rook GAW 2010. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clin. Exp. Immunol 160: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai KSP, Liu CS, Rau A, et al. 2017. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 88: 876–882. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt R, Schmidt H, Curb JD, et al. 2002. Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia aging study. Ann. Neurol 52: 168–174. [DOI] [PubMed] [Google Scholar]
- 4.Fox M, Knapp LA, Andrews PW, et al. 2013. Hygiene and the world distribution of Alzheimer’s disease. Evol. Med. Public Health 2013: 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogt NM, Kerby RL, Dill-McFarland KA, et al. 2017. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep 7: 13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fox M 2018. ‘Evolutionary medicine’ perspectives on Alzheimer’s disease: review and new directions. Aging Res. Rev 47: 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkes K 2003. Grandmothers and the evolution of human longevity. Am. J. Hum. Biol 15: 380–400. [DOI] [PubMed] [Google Scholar]
- 8.Gurven M & Kaplan H. 2007. Longevity among hunter--gatherers: a cross-cultural examination. Popul. Dev. Rev 33: 321–365. [Google Scholar]
- 9.WHO. 2018. Accessed August 29, 2018. http://www.who.int/nutrition/topics/exclusive_breastfeeding/en/.
- 10.Kaplan H, Hill K, Lancaster J, et al. 2000. A theory of human life history evolution: diet, intelligence, and longevity. Evol. Anthropol. Issues News Rev 9: 156–185. [Google Scholar]
- 11.Stearns SC, Nesse RM, Govindaraju DR, et al. 2010. Evolutionary perspectives on health and medicine. Proc. Natl. Acad. Sci. USA 107: 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho I & Blaser MJ. 2012. The Human Microbiome: at the interface of health and disease. Nat. Rev. Genet 13: 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proal AD, Albert PJ & Marshall TG. 2013. The human microbiome and autoimmunity. Curr. Opin. Rheumatol 25: 234–240. [DOI] [PubMed] [Google Scholar]
- 14.Claesson MJ, Jeffery IB, Conde S, et al. 2012. Gut microbiota composition correlates with diet and health in the elderly. Nature 488: 178–184. [DOI] [PubMed] [Google Scholar]
- 15.Hildebrand F, Nguyen TLA, Brinkman B, et al. 2013. Inflammation-associated enterotypes, host genotype, cage and interindividual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 14: R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter J & Ley R. 2011. The human gut microbiome: ecology and recent evolutionary changes. Annu. Rev. Microbiol 65: 411–429. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman C, Dollive S, Stephanie G, et al. 2013. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8: e66019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rook GAW 2012. Hygiene hypothesis and autoimmune diseases. Clin. Rev. Allerg. Immunol 42: 5–15. [DOI] [PubMed] [Google Scholar]
- 19.Barker G 2006. The Agricultural Revolution in Prehistory: Why did Foragers become Farmers? Oxford: Oxford University Press. [Google Scholar]
- 20.Stearns S & Medzhitov R. 2016. Evolutionary Medicine. Sinauer Associates. [Google Scholar]
- 21.Keesing F, Belden LK, Daszak P, et al. 2010. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 468: 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson Pieter TJ, Lund Peder J, Hartson Richard B, et al. 2009. Community diversity reduces Schistosoma mansoni transmission, host pathology and human infection risk. Proc. Biol. Sci 276: 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang JY, Antonopoulos DA, Kalra A, et al. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile—associated diarrhea. J. Infect. Dis 197: 435–438. [DOI] [PubMed] [Google Scholar]
- 24.De Filippo C, Cavalieri D, Di Paola M, et al. 2010. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 107: 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landes DS 1969. The Unbound Prometheus: Technological Change and Industrial Development in Western Europe from 1750 to the Present. London: Cambridge University Press. [Google Scholar]
- 26.Rook GAW 2011. Hygiene and other early childhood influences on the subsequent function of the immune system. Dig. Dis 29: 144–153. [DOI] [PubMed] [Google Scholar]
- 27.Brimblecombe P 2012. The Big Smoke (Routledge Revivals): A History of Air Pollution in London since Medieval Times. Routledge. [Google Scholar]
- 28.Prussin AJ & Marr LC. 2015. Sources of airborne microorganisms in the built environment. Microbiome 3: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffin DW, Westphal DL & Gray MA. 2006. Airborne microorganisms in the African desert dust corridor over the mid-Atlantic ridge, Ocean Drilling Program, Leg 209. Aerobiologia 22: 211–226. [Google Scholar]
- 30.Lee K & Dodgson R. 2003. Globalisation and cholera: implications for global governance. In Health Impacts of Globalization—Towards Global Governance. 1st ed. Lee Kelley, Ed.: 123–143. UK: Palgrave Macmillan. [Google Scholar]
- 31.Barlett PF 2013. Industrial agriculture in evolutionary perspective. Cult. Anthropol 2: 137–154. [Google Scholar]
- 32.Adegoke AA, Faleye AC, Singh G, et al. 2016. Antibiotic resistant superbugs: assessment of the interrelationship of occurrence in clinical settings and environmental niches. Molecules 22: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson PR 1992. Globalization: Social Theory and Global Culture. Sage. [Google Scholar]
- 34.UNFPA. 2007. State of world population 2007. UNFPA. [Google Scholar]
- 35.Anderson HR 2009. Air pollution and mortality: a history. Atmos. Environ 43: 142–152. [Google Scholar]
- 36.Brimblecombe P 2006. The Clean Air Act after 50 years. Weather 61: 311–314. [Google Scholar]
- 37.Dowling S 2014. January 30, 2014. Accessed January 3, 2019. http://www.bbc.com/future/story/20140130-how-air-travel-shrunk-the-globe.
- 38.United Nations. 2017. Population facts. United Nations. [Google Scholar]
- 39.Lakdawalla D & Philipson T. 2009. The growth of obesity and technological change. Econ. Hum. Biol 7: 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cordain L, Eaton SB, Sebastian A, et al. 2005. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr 81: 341–354. [DOI] [PubMed] [Google Scholar]
- 41.Ventola CL 2015. The antibiotic resistance crisis. P T 40: 277–283. [PMC free article] [PubMed] [Google Scholar]
- 42.Read AF & Woods RJ. 2014. Antibiotic resistance management. Evol. Med. Public Health 2014: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geisinger E & Isberg RR. 2017. Interplay between antibiotic resistance and virulence during disease promoted by multidrug-resistant bacteria. J. Infect. Dis 215: S9–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prestinaci F, Pezzotti P & Pantosti A. 2015. Antimicrobial resistance: a global multifaceted phenomenon. Pathog. Glob. Health 109: 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Center for Health Statistics. 2017. Health, United States, 2016: with chartbook on long-term trends in health. Hyattsville, MD: National Center for Health Statistics. [PubMed] [Google Scholar]
- 46.WHO. 2017. World Health Organization. August 1, 2017. Accessed April 25, 2018. http://www.who.int/news-room/detail/01-08-2017-babies-and-mothers-worldwide-failed-by-lack-of-investment-in-breastfeeding.
- 47.Yassour M, Vatanen T, Siljander H, et al. 2016. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med 8: 343ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belkaid Y & Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157: 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayer EA, Tillisch K & Gupta A. 2015. Gut/brain axis and the microbiota. J. Clin. Invest 125: 926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salazar N, Valdés-Varela L, González S, et al. 2017. Nutrition and the gut microbiome in the elderly. Gut Microbes 8: 82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collado MC, Derrien M, Isolauri E, et al. 2007. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl. Environ. Microbiol 73: 7767–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Claesson MJ, Cusack S, O’Sullivan O, et al. 2011. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 108: 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S & Jazwinski SM. 2018. The gut microbiota and healthy aging: a mini-review. Gerontology 64: 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu WL, Atti AR, Gatz M, et al. 2011. Midlife overweight and obesity increase late-life dementia risk: a population-based twin study. Neurology 76: 1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skoog I & Gustafson D. 2006. Update on hypertension and Alzheimer’s disease. Neurol. Res 28: 605–611. [DOI] [PubMed] [Google Scholar]
- 56.Maher PA & Schubert DR. 2009. Metabolic links between diabetes and Alzheimer’s disease. Expert Rev. Neurother 9: 617–630. [DOI] [PubMed] [Google Scholar]
- 57.Rojas-Gutierrez E, Muñoz-Arenas G, Treviño S, et al. 2017. Alzheimer’s disease and metabolic syndrome: a link from oxidative stress and inflammation to neurodegeneration. Synapse 71: e21990. [DOI] [PubMed] [Google Scholar]
- 58.Guillemot-Legris O & Muccioli GG. 2017. Obesity-induced neuroinflammation: beyond the hypothalamus. Trends Neurosci. 40: 237–253. [DOI] [PubMed] [Google Scholar]
- 59.Ley RE, Turnbaugh PJ, Klein S, et al. 2006. Human gut microbes associated with obesity. Nature 444: 1022–1023. [DOI] [PubMed] [Google Scholar]
- 60.Ridaura VK, Faith JJ, Rey FE, et al. 2013. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341. 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makki K, Froguel P & Wolowczuk I. 2013. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013. 10.1155/2013/139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferreira ST, Clarke JR, Bomfim TR, et al. 2014. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. 10: S76–S83. [DOI] [PubMed] [Google Scholar]
- 63.Thaler JP, Yi C-X, Schur EA, et al. 2012. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest 122: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreno-Navarrete JM, Blasco G, Puig J, et al. 2017. Neuroinflammation in obesity: circulating lipopolysaccharide-binding protein associates with brain structure and cognitive performance. Int. J. Obes 41: 1627–1635. [DOI] [PubMed] [Google Scholar]
- 65.Vagelatos NT & Eslick GD. 2013. Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev 35: 152–160. [DOI] [PubMed] [Google Scholar]
- 66.Brunkwall L & Orho-Melander M. 2017. The gut microbiome as a target for prevention and treatment of hyperglycaemia in type 2 diabetes: from current human evidence to future possibilities. Diabetologia 60: 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Donohoe DR, Garge N, Zhang X, et al. 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 13: 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Vadder F, Kovatcheva-Datchary P, Goncalves D, et al. 2014. Microbiota-generated metabolites promote metabolic benefits via gut–brain neural circuits. Cell 156: 84–96. [DOI] [PubMed] [Google Scholar]
- 69.Peng L, Li Z-R, Green RS, et al. 2009. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr 139: 1619–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cushing K, Alvarado DM & Ciorba MA. 2015. Butyrate and mucosal inflammation: new scientific evidence supports clinical observation. Clin. Transl. Gastroenterol 6: e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lyketsos CG, Carrillo MC, Ryan JM, et al. 2011. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 7: 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng P, Zeng B, Zhou C, et al. 2016. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 21: 786–796. [DOI] [PubMed] [Google Scholar]
- 73.Krajmalnik-Brown R, Lozupone C, Kang D-W & Adams JB. 2015. Gut bacteria in children with autism spectrum disorders: challenges and promise of studying how a complex community influences a complex disease. Microb. Ecol. Health Dis 26. 10.3402/mehd.v26.26914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen Y, Xu J, Li Z, et al. 2018. Analysis of gut microbiota diversity and auxiliary diagnosis as a biomarker in patients with schizophrenia: a cross-sectional study. Schizophr. Res 197: 470–477. [DOI] [PubMed] [Google Scholar]
- 75.Chaidez V, Hansen RL & Hertz-Picciotto I. 2014. Gastrointestinal problems in children with autism, developmental delays or typical development. J. Autism Dev. Disord 44: 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Severance EG, Gressitt KL, Stallings CR, et al. 2013. Discordant patterns of bacterial translocation markers and implications for innate immune imbalances in schizophrenia. Schizophr. Res 148: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Neufeld K-AM, Kang N, Bienenstock J, et al. 2011. Effects of intestinal microbiota on anxiety-like behavior. Commun. Integr. Biol 4: 492–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arentsen T, Raith H, Qian Y, et al. 2015. Host microbiota modulates development of social preference in mice. Microb. Ecol. Health Dis 26. 10.3402/mehd.v26.29719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gacias M, Gaspari S, Santos P-MG, et al. 2016. Microbiota-driven transcriptional changes in prefrontal cortex override genetic differences in social behavior. eLife 5. 10.7554/eLife.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kelly JR, Borre Y, O’ Brien C, et al. 2016. Transferring the blues: depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res 82: 109–118. [DOI] [PubMed] [Google Scholar]
- 81.Zheng P, Zeng B, Liu M, et al. 2019. The gut microbiome from patients with schizophrenia modulates the glutamate-glutamine-GABA cycle and schizophrenia-relevant behaviors in mice. Sci. Adv 5: eaau8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Minter MR, Zhang C, Leone V, et al. 2016. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep 6: 30028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harach T, Marungruang N, Duthilleul N, et al. 2017. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep 7. 10.1038/srep41802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scheperjans F, Aho V, Pereira PAB, et al. 2015. Gut microbiota are related to Parkinson’s disease and clinical phenotype: gut microbiota in Parkinson’s disease. Mov. Disord 30: 350–358. [DOI] [PubMed] [Google Scholar]
- 85.Holmqvist S, Chutna O, Bousset L, et al. 2014. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128: 805–820. [DOI] [PubMed] [Google Scholar]
- 86.Tasnim N, Abulizi N, Pither J, et al. 2017. Linking the gut microbial ecosystem with the environment: does gut health depend on where we live? Front. Microbiol 8. 10.3389/fmicb.2017.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schnorr SL, Candela M, Rampelli S, et al. 2014. Gut microbiome of the Hadza hunter–gatherers. Nat. Commun 5: 3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Campbell-Platt G 1994. Fermented foods—a world perspective. Food Res. Int 27: 253–257. [Google Scholar]
- 89.Bell V, Ferrão J, Pimentel L, et al. 2018. One Health, fermented foods, and gut microbiota. Foods 7: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.David LA, Maurice CF, Carmody RN, et al. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zuckerman KE, Hill AP, Guion K, et al. 2014. Over-weight and obesity: prevalence and correlates in a large clinical sample of children with autism spectrum disorder. J. Autism Dev. Disord 44: 1708–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Heiman ML & Greenway FL. 2016. A healthy gastrointestinal microbiome is dependent on dietary diversity. Mol. Metab 5: 317–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.FAO. 2004. What is agrobiodiversity? Fact sheet. Rome, Italy: Food and Agriculture Organization of the United Nations. [Google Scholar]
- 94.Davenport ER, Sanders JG, Song SJ, et al. 2017. The human microbiome in evolution. BMC Biol. 15: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Obregon-Tito AJ, Tito RY, Metcalf J, et al. 2015. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun 6: 6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yatsunenko T, Rey FE, Manary MJ, et al. 2012. Human gut microbiome viewed across age and geography. Nature 486: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mancabelli L, Milani C, Lugli GA, et al. 2017. Meta-analysis of the human gut microbiome from urbanized and pre-agricultural populations. Environ. Microbiol 19: 1379–1390. [DOI] [PubMed] [Google Scholar]
- 98.Song SJ, Lauber C, Costello EK, et al. 2013. Cohabiting family members share microbiota with one another and with their dogs. eLife 2: e00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suchodolski JS, Dowd SE, Wilke V, et al. 2012. 16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease. PLoS One 7: e39333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cekanaviciute E, Pröbstel A-K, Thomann A, et al. 2018. Multiple sclerosis-associated changes in the composition and immune functions of spore-forming bacteria. mSystems 3. 10.1128/mSystems.00083-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhattacharjee S & Lukiw WJ. 2013. Alzheimer’s disease and the microbiome. Front. Cell. Neurosci 7: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yunes RA, Poluektova EU, Dyachkova MS, et al. 2016. GABA production and structure of gadB/gadC genes in Lactobacillus and Bifidobacterium strains from human microbiota. Anaerobe 42: 197–204. [DOI] [PubMed] [Google Scholar]
- 103.Bravo JA, Forsythe P, Chew MV, et al. 2011. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 108: 16050–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lowe SL, Francis PT, Procter AW, et al. 1988. Gamma-aminobutyric acid concentration in brain tissue at two stages of Alzheimer’s disease. Brain 111: 785–799. [DOI] [PubMed] [Google Scholar]
- 105.O’Mahony SM, Clarke G, Borre YE, et al. 2015. Serotonin, tryptophan metabolism and the brain–gut–microbiome axis. Behav. Brain Res 277: 32–48. [DOI] [PubMed] [Google Scholar]
- 106.Ruddick JP, Evans AK, Nutt DJ, et al. 2006. Tryptophan metabolism in the central nervous system: medical implications. Expert Rev. Mol. Med 8: 1–27. [DOI] [PubMed] [Google Scholar]
- 107.Kumar AM, Sevush S, Kumar M, et al. 1995. Peripheral serotonin in Alzheimer’s disease. Neuropsychobiology 32: 9–12. [DOI] [PubMed] [Google Scholar]
- 108.Kepe V, Barrio JR, Huang S-C, et al. 2006. Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 103: 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pablo J, Banack SA, Cox PA, et al. 2009. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol. Scand 120: 216–225. [DOI] [PubMed] [Google Scholar]
- 110.Brenner SR 2013. Blue-green algae or cyanobacteria in the intestinal micro-flora may produce neurotoxins such as Beta-N-Methylamino-L-Alanine (BMAA) which may be related to development of amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson–Dementia-Complex in humans and Equine Motor Neuron Disease in horses. Med. Hypotheses 80: 103–103. [DOI] [PubMed] [Google Scholar]
- 111.Schwartz K & Boles BR. 2013. Microbial amyloids—functions and interactions within the host. Curr. Opin. Microbiol 16: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rook GAW, Raison CL & Lowry CA. 2014. Microbial ‘old friends’, immunoregulation and socioeconomic status: microbes, immunoregulation and SES. Clin. Exp. Immunol 177: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Geirnaert A, Calatayud M, Grootaert C, et al. 2017. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep 7: 11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kountouras J, Tsolaki M, Gavalas E, et al. 2006. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology 66: 938–940. [DOI] [PubMed] [Google Scholar]
- 115.Braniste V, Al-Asmakh M, Kowal C, et al. 2014. The gut microbiota influences blood–brain barrier permeability in mice. Sci. Transl. Med 6: 263ra158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Daniels BP, Holman DW, Cruz-Orengo L, et al. 2014. Viral pathogen-associated molecular patterns regulate blood–brain barrier integrity via competing innate cytokine signals. mBio 5: e01476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mittal R & Coopersmith CM. 2014. Redefining the gut as the motor of critical illness. Trends Mol. Med 20: 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mosher KI & Wyss-Coray T. 2014. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol 88: 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Erny D, Hrabě de Angelis AL, Jaitin D, et al. 2015. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci 18: 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kondo K, Niino M & Shido K. 1994. A case–control study of Alzheimer’s disease in Japan—significance of lifestyles. Dementia 5: 314–326. [DOI] [PubMed] [Google Scholar]
- 121.Gatz M, Mortimer JA, Fratiglioni L, et al. 2006. Potentially modifiable risk factors for dementia in identical twins. Alzheimers Dement. 2: 110–117. [DOI] [PubMed] [Google Scholar]
- 122.Stein PS, Desrosiers M, Donegan SJ, et al. 2007. Tooth loss, dementia and neuropathology in the Nun study. J. Am. Dent. Assoc 138: 1314–1322; quiz 1381–1382. [DOI] [PubMed] [Google Scholar]
- 123.Dominy SS, Lynch C, Ermini F, et al. 2019. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv 5: eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Adler CJ, Dobney K, Weyrich LS, et al. 2013. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet 45: 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kerr NW 1998. The prevalence and natural history of periodontal disease in Britain from prehistoric to modern times. Br. Dent. J 185: 527–535. [DOI] [PubMed] [Google Scholar]
- 126.Riviere GR, Riviere KH & Smith KS. 2002. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol 17: 113–118. [DOI] [PubMed] [Google Scholar]
- 127.Watts A, Crimmins EM & Gatz M. 2008. Inflammation as a potential mediator for the association between periodontal disease and Alzheimer’s disease. Neuropsychiatr. Dis. Treat 4: 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Forner L, Larsen T, Kilian M, et al. 2006. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol 33: 401–407. [DOI] [PubMed] [Google Scholar]
- 129.Loesche WJ & Lopatin DE. 1998. Interactions between periodontal disease, medical diseases and immunity in the older individual. Periodontology 2000 16: 80–105. [DOI] [PubMed] [Google Scholar]
- 130.Offenbacher S 1996. Periodontal diseases: pathogenesis. Ann. Periodontol 1: 821–878. [DOI] [PubMed] [Google Scholar]
- 131.Heppner FL, Ransohoff RM & Becher B. 2015. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci 16: 358–372. [DOI] [PubMed] [Google Scholar]
- 132.Hauss-wegrzyniak B, Vraniak PD & Wenk GL. 2000. LPS-induced neuroinflammatory effects do not recover with time. Neuroreport 11: 1759–1763. [DOI] [PubMed] [Google Scholar]
- 133.Rupf S, Kannengieber S, Merte K, et al. 2000. Comparison of profiles of key periodontal pathogens in periodontium and endodontium. Endod. Dent. Traumatol 16: 269–275. [DOI] [PubMed] [Google Scholar]
- 134.Shoemark DK & Allen SJ. 2015. The microbiome and disease: reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J. Alzheimers Dis 43: 725–738. [DOI] [PubMed] [Google Scholar]
- 135.Miklossy J 2011. Alzheimer’s disease—a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflammation 8: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Song Y, Ichinose T, He M, et al. 2016. Lipopolysaccharide attached to urban particulate matter 10 suppresses immune responses in splenocytes while particulate matter itself activates NF-κB. Toxicol. Res 5: 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dando SJ, Mackay-Sim A, Norton R, et al. 2014. Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev 27: 691–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kovács T, Cairns NJ & Lantos PL. 2001. Olfactory centres in Alzheimer’s disease: olfactory bulb is involved in early Braak’s stages. Neuroreport 12: 285–288. [DOI] [PubMed] [Google Scholar]
- 139.Wu Y-C, Lin Y-C, Yu H-L, et al. 2015. Association between air pollutants and dementia risk in the elderly. Alzheimers Dement. 1: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jung C-R, Lin Y-T & Hwang B-F. 2015. Ozone, particulate matter, and newly diagnosed Alzheimer’s disease: a population-based cohort study in Taiwan. J. Alzheimers Dis 44: 573–584. [DOI] [PubMed] [Google Scholar]
- 141.Chen H, Kwong JC, Copes R, et al. 2017. Living near major roads and the incidence of dementia, Parkinson’s disease, and multiple sclerosis: a population-based cohort study. Lancet 389: 718–726. [DOI] [PubMed] [Google Scholar]
- 142.Oudin A, Forsberg B, Adolfsson AN, et al. 2016. Traffic-related air pollution and dementia incidence in Northern Sweden: a longitudinal study. Environ. Health Perspect 124: 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Power MC, Weisskopf MG, Alexeeff SE, et al. 2011. Traffic-related air pollution and cognitive function in a cohort of older men. Environ. Health Perspect 119: 682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Calderón-Garcidueñas L, Kavanaugh M, Block M, et al. 2012. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimers Dis 28: 93–107. [DOI] [PubMed] [Google Scholar]
- 145.Calderón-Garcidueñas L, Franco-Lira M, Henríquez-Roldán C, et al. 2010. Olfactory dysfunction, olfactory bulb pathology and urban air pollution. Exp. Toxicol. Pathol 62: 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Calderón-Garcidueñas L, Reynoso-Robles R, Vargas-Martínez J, et al. 2016. Prefrontal white matter pathology in air pollution exposed Mexico City young urbanites and their potential impact on neurovascular unit dysfunction and the development of Alzheimer’s disease. Environ. Res 146: 404–417. [DOI] [PubMed] [Google Scholar]
- 147.Velayudhan L 2015. Smell identification function and Alzheimer’s disease: a selective review. Curr. Opin. Psychiatry 28: 173–179. [DOI] [PubMed] [Google Scholar]
- 148.Christen-Zaech S, Kraftsik R, Pillevuit O, et al. 2003. Early olfactory involvement in Alzheimer’s disease. Can. J. Neurol. Sci 30: 20–25. [DOI] [PubMed] [Google Scholar]
- 149.Chan A, Tam J, Murphy C, et al. 2002. Utility of olfactory identification test for diagnosing Chinese patients with Alzheimer’s disease. J. Clin. Exp. Neuropsychol 24: 251–259. [DOI] [PubMed] [Google Scholar]
- 150.Doty RL, Reyes PF & Gregor T. 1987. Presence of both odor identification and detection deficits in Alzheimer’s disease. Brain Res. Bull 18: 597–600. [DOI] [PubMed] [Google Scholar]
- 151.Wilson RS, Arnold SE, Schneider JA, et al. 2009. Olfactory impairment in presymptomatic Alzheimer’s disease. Ann. N.Y. Acad. Sci 1170: 730–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Devanand DP, Michaels-Marston KS, Liu X, et al. 2000. Olfactory deficits in patients with mild cognitive impairment predict Alzheimer’s disease at follow-up. Am. J. Psychiatry 157: 1399–1405. [DOI] [PubMed] [Google Scholar]
- 153.Conti MZ, Vicini-Chilovi B, Riva M, et al. 2013. Odor identification deficit predicts clinical conversion from mild cognitive impairment to dementia due to Alzheimer’s disease. Arch. Clin. Neuropsychol 28: 391–399. [DOI] [PubMed] [Google Scholar]
- 154.Graves A, Bowen JD, Rajaram L, et al. 1999. Impaired olfaction as a marker for cognitive decline: interaction with apolipoprotein E epsilon4 status. Neurology 53: 1480–1487. [DOI] [PubMed] [Google Scholar]
- 155.Murphy C, Jernigan TL & Fennema-Notestine C. 2003. Left hippocampal volume loss in Alzheimer’s disease is reflected in performance on odor identification: a structural MRI study. J. Int. Neuropsychol. Soc 9: 459–471. [DOI] [PubMed] [Google Scholar]
- 156.Kjelvik G, Saltvedt I, White LR, et al. 2014. The brain structural and cognitive basis of odor identification deficits in mild cognitive impairment and Alzheimer’s disease. BMC Neurol. 14: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wilson R, Arnold S, Tang Y, et al. 2007. The relationship between cerebral Alzheimer’s disease pathology and odour identification in old age. J. Neurol. Neurosurg. Psychiatry 78: 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang J, Eslinger PJ, Doty RL, et al. 2010. Olfactory deficit detected by fMRI in early Alzheimer’s disease. Brain Res 1357: 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Murphy C, Cerf-Ducastel B, Calhoun-Haney R, et al. 2005. ERP, fMRI and functional connectivity studies of brain response to odor in normal aging and Alzheimer’s disease. Chem. Senses 30: i170–i171. [DOI] [PubMed] [Google Scholar]
- 160.Bahar-Fuchs A, Chételat G, Villemagne VL, et al. 2010. Olfactory deficits and amyloid-β burden in Alzheimer’s disease, mild cognitive impairment, and healthy aging: a PiB PET study. J. Alzheimers Dis 22: 1081–1087. [DOI] [PubMed] [Google Scholar]
- 161.CDC. 2019. January 2, 2019 Accessed January 8, 2019. https://www.cdc.gov/mmwr/index.html.
- 162.Neupane B, Jerrett M, Burnett RT, et al. 2010. Long-term exposure to ambient air pollution and risk of hospitalization with community-acquired pneumonia in older adults. Am. J. Respir. Crit. Care Med 181: 47–53. [DOI] [PubMed] [Google Scholar]
- 163.Schwartz J, Spix C, Wichmann HE, et al. 1991. Air pollution and acute respiratory illness in five German communities. Environ. Res 56: 1–14. [DOI] [PubMed] [Google Scholar]
- 164.Ezzati M & Kammen DM. 2001. Indoor air pollution from biomass combustion and acute respiratory infections in Kenya: an exposure-response study. Lancet 358: 619–624. [DOI] [PubMed] [Google Scholar]
- 165.Jaakkola JJ, Paunio M, Virtanen M, et al. 1991. Low-level air pollution and upper respiratory infections in children. Am. J. Public Health 81: 1060–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chauhan AJ & Johnston SL. 2003. Air pollution and infection in respiratory illness. Br. Med. Bull 68: 95–112. [DOI] [PubMed] [Google Scholar]
- 167.Klimont Z, Smith SJ & Cofala J. 2013. The last decade of global anthropogenic sulfur dioxide: 2000–2011 emissions. Environ. Res. Lett 8: 014003. [Google Scholar]
- 168.van Zanden J 2014. How Was Life?—Global Well-Being since 1820. OECD. [Google Scholar]
- 169.Wimo A, Jönsson L, Bond J, et al. 2013. The worldwide economic impact of dementia 2010. Alzheimers Dement. 9: 1–11.e3. [DOI] [PubMed] [Google Scholar]
- 170.González-Maciel A, Reynoso-Robles R, Torres-Jardón R, et al. 2017. Combustion-derived nanoparticles in key brain target cells and organelles in young urbanites: culprit hidden in plain sight in Alzheimer’s disease development. J Alzheimer’s Dis. 59: 189–208. [DOI] [PubMed] [Google Scholar]
- 171.Radtke C & Kocsis JD. 2014. Olfactory-ensheathing cell transplantation for peripheral nerve repair: update on recent developments. Cells Tissues Organs 200: 48–58. [DOI] [PubMed] [Google Scholar]
- 172.Adar SD, Huffnagle GB & Curtis JL. 2016. The respiratory microbiome: an underappreciated player in the human response to inhaled pollutants? Ann. Epidemiol 26: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bonfanti R, Musumeci T, Russo C, et al. 2017. The protective effect of curcumin in olfactory ensheathing cells exposed to hypoxia. Eur. J. Pharmacol 796: 62–68. [DOI] [PubMed] [Google Scholar]
- 174.Debon R & Doucette R. 1992. Olfactory ensheathing cells myelinate dorsal root ganglion neurites. Brain Res. 589: 175–179. [DOI] [PubMed] [Google Scholar]
- 175.Choroszy-Król I, Frej-Mądrzak M, Hober M, et al. 2014. Infections caused by Chlamydophila pneumoniae. Adv. Clin. Exp. Med 23: 123–126. [DOI] [PubMed] [Google Scholar]
- 176.Alzheimer’s Association. 2016. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12: 459–509. [DOI] [PubMed] [Google Scholar]
- 177.Balin BJ, Little CS, Hammond CJ, et al. 2008. Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J. Alzheimers Dis 13: 371–380. [DOI] [PubMed] [Google Scholar]
- 178.Albert NM 2000. Inflammation and infection in acute coronary syndromes. J. Cardiovasc. Nurs 15: 13–26. [DOI] [PubMed] [Google Scholar]
- 179.Boman J, Söderberg S, Forsberg J, et al. 1998. High prevalence of Chlamydia pneumoniae DNA in peripheral blood mononuclear cells in patients with cardiovascular disease and in middle-aged blood donors. J. Infect. Dis 178: 274–277. [DOI] [PubMed] [Google Scholar]
- 180.Paradowski B, Jaremko M, Dobosz T, et al. 2007. Evaluation of CSF-Chlamydia pneumoniae, CSF-tau, and CSF-Abeta42 in Alzheimer’s disease and vascular dementia. J. Neurol 254: 154–159. [DOI] [PubMed] [Google Scholar]
- 181.Balin BJ, Gérard HC, Arking EJ, et al. 1998. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol 187: 23–42. [DOI] [PubMed] [Google Scholar]
- 182.Gérard HC, Dreses-Werringloer U, Wildt KS, et al. 2006. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol 48: 355–366. [DOI] [PubMed] [Google Scholar]
- 183.Itzhaki RF 2017. Herpes simplex virus type 1 and Alzheimer’s disease: possible mechanisms and signposts. FASEB J 31: 3216–3226. [DOI] [PubMed] [Google Scholar]
- 184.Boggian I, Buzzacaro E, Calistri A, et al. 2000. Asymptomatic herpes simplex type 1 virus infection of the mouse brain. J. Neurovirol 6: 303–313. [DOI] [PubMed] [Google Scholar]
- 185.Esiri MM 1982. Herpes simplex encephalitis: an immunohistological study of the distribution of viral antigen within the brain. J. Neurol. Sci 54: 209–226. [DOI] [PubMed] [Google Scholar]
- 186.Sjölinder H & Jonsson A-B. 2010. Olfactory nerve—a novel invasion route of Neisseria meningitidis to reach the meninges. PLoS One 5: e14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.St. John JA, Ekberg JAK, Dando SJ, et al. 2014. Burkholderia pseudomallei penetrates the brain via destruction of the olfactory and trigeminal nerves: implications for the pathogenesis of neurological melioidosis. mBio 5: e00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Macedo-Ramos H, Campos FSO, Carvalho LA, et al. 2011. Olfactory ensheathing cells as putative host cells for Streptococcus pneumoniae: evidence of bacterial invasion via mannose receptor-mediated endocytosis. Neurosci. Res 69: 308–313. [DOI] [PubMed] [Google Scholar]
- 189.Elwood E, Lim Z, Naveed H, et al. 2017. The effect of systemic inflammation on human brain barrier function. Brain Behav. Immun 62: 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Navarro V, Sanchez-Mejias E, Jimenez S, et al. 2018. Microglia in Alzheimer’s disease: activated, dysfunctional or degenerative. Front. Aging Neurosci 10: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ferretti MT & Cuello AC. 2011. Does a proinflammatory process precede Alzheimer’s disease and mild cognitive impairment? Curr. Alzheimer Res 8: 164–174. [DOI] [PubMed] [Google Scholar]
- 192.Mittal M, Siddiqui MR, Tran K, et al. 2014. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 20: 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dong TG, Dong S, Catalano C, et al. 2015. Generation of reactive oxygen species by lethal attacks from competing microbes. Proc. Natl. Acad. Sci. USA 112: 2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bhattacharyya A, Chattopadhyay R, Mitra S, et al. 2014. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev 94: 329–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sattar N, Gaw A, Scherbakova O, et al. 2003. Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study. Circulation 108: 414–419. [DOI] [PubMed] [Google Scholar]
- 196.Cheignon C, Tomas M, Bonnefont-Rousselot D, et al. 2018. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14: 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Christen Y 2000. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr 71: 621S–629S. [DOI] [PubMed] [Google Scholar]
- 198.Kadowaki H, Nishitoh H, Urano F, et al. 2005. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 12: 19–24. [DOI] [PubMed] [Google Scholar]
- 199.Itzhaki RF, Wozniak MA, Appelt DM, et al. 2004. Infiltration of the brain by pathogens causes Alzheimer’s disease. Neurobiol. Aging 25: 619–627. [DOI] [PubMed] [Google Scholar]
- 200.Emery DC, Shoemark DK, Batstone TE, et al. 2017. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front. Aging Neurosci 9: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhao Y, Cong L & Lukiw WJ. 2017. Lipopolysaccharide (LPS) accumulates in neocortical neurons of Alzheimer’s disease (AD) brain and impairs transcription in human neuronal-glial primary co-cultures. Front. Aging Neurosci 9: 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Poole S, Singhrao SK, Kesavalu L, et al. 2013. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis 36: 665–677. [DOI] [PubMed] [Google Scholar]
- 203.Kim KS 2008. Mechanisms of microbial traversal of the blood–brain barrier. Nat. Rev. Microbiol 6: 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Soscia SJ, Kirby JE, Washicosky KJ, et al. 2010. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.White MR, Kandel R, Tripathi S, et al. 2014. Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One 9: e101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. 2018. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99: 56–63.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kumar DKV, Choi SH, Washicosky KJ, et al. 2016. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med 8: 340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Miklossy J, Kis A, Radenovic A, et al. 2006. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol. Aging 27: 228–236. [DOI] [PubMed] [Google Scholar]
- 209.Cruchaga C, Kauwe JSK, Nowotny P, et al. 2012. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum. Mol. Genet 21: 4558–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Riddell DR, Zhou H, Atchison K, et al. 2008. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci 28: 11445–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Huynh T-PV, Davis AA, Ulrich JD, et al. 2017. Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res 58: 824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ashford JW 2002. ApoE4: is it the absence of good or the presence of bad? J. Alzheimers Dis 4: 141–143. [DOI] [PubMed] [Google Scholar]
- 213.Finch CE & Sapolsky RM. 1999. The evolution of Alzheimer disease, the reproductive schedule, and apoE isoforms. Neurobiol. Aging 20: 407–428. [DOI] [PubMed] [Google Scholar]
- 214.Hanlon C 1995. Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis 112: 85–90. [DOI] [PubMed] [Google Scholar]
- 215.Glass DJ & Arnold SE. 2012. Some evolutionary perspectives on Alzheimer’s disease pathogenesis and pathology. Alzheimers Dement. 8: 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Singh PP, Singh M & Mastana SS. 2006. APOE distribution in world populations with new data from India and the UK. Ann. Hum. Biol 33: 279–308. [DOI] [PubMed] [Google Scholar]
- 217.Fox M 2018. ‘Evolutionary medicine’ perspectives on Alzheimer’s disease: review and new directions. Ageing Res. Rev 47: 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kanekiyo T, Xu H & Bu G. 2014. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81: 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Johnson LA, Olsen RHJ, Merkens LS, et al. 2014. Apolipoprotein E—low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis 64: 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gérard HC, Wildt KL, Whittum-Hudson JA, et al. 2005. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb. Pathog 39: 19–26. [DOI] [PubMed] [Google Scholar]
- 221.Zhang H, Wu L-M & Wu J. 2011. Cross-talk between apolipoprotein E and cytokines. Mediators Inflamm. 2011: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mannix RC, Zhang J, Park J, et al. 2011. Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab 31: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Rebeck GW 2017. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res 58: 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jofre-Monseny L, Minihane A-M & Rimbach G. 2008. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol. Nutr. Food Res 52: 131–145. [DOI] [PubMed] [Google Scholar]
- 225.Dose J, Huebbe P, Nebel A & Rimbach G. 2016. APOE genotype and stress response—a mini review. Lipids Health Dis. 15: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jofre-Monseny L, de Pascual-Teresa S, Plonka E, et al. 2007. Differential effects of apolipoprotein E3 and E4 on markers of oxidative status in macrophages. Br. J. Nutr 97: 864–871. [DOI] [PubMed] [Google Scholar]
- 227.Lauderback CM, Kanski J, Hackett JM, et al. 2002. Apolipoprotein E modulates Alzheimer’s Aβ (1–42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res. 924: 90–97. [DOI] [PubMed] [Google Scholar]
- 228.Yao J, Petanceska SS, Montine TJ, et al. 2004. Aging, gender and APOE isotype modulate metabolism of Alzheimer’s Aβ peptides and F2-isoprostanes in the absence of detectable amyloid deposits. J. Neurochem 90: 1011–1018. [DOI] [PubMed] [Google Scholar]
- 229.Chaput J-P, Doucet É & Tremblay A. 2012. Obesity: a disease or a biological adaptation? An update. Obes. Rev 13: 681–691. [DOI] [PubMed] [Google Scholar]
- 230.Everson SA, Maty SC, Lynch JW, et al. 2002. Epidemiologic evidence for the relation between socioeconomic status and depression, obesity, and diabetes. J. Psychosom. Res 53: 891–895. [DOI] [PubMed] [Google Scholar]
- 231.Omenn GS 2010. Evolution and public health. Proc. Natl. Acad. Sci. USA 107: 1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schnorr SL, Sankaranarayanan K, Lewis CM, et al. 2016. Insights into human evolution from ancient and contemporary microbiome studies. Curr. Opin. Genet. Dev 41: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kelso J & Prüfer K. 2014. Ancient humans and the origin of modern humans. Curr. Opin. Genet. Dev 29: 133–138. [DOI] [PubMed] [Google Scholar]
- 234.Meyer M, Arsuaga J-L, de Filippo C, et al. 2016. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531: 504–507. [DOI] [PubMed] [Google Scholar]
- 235.Price M 2017. Early human gut bacteria may have cycled with the season. Science 10.1126/science.aap7681. [DOI] [Google Scholar]