

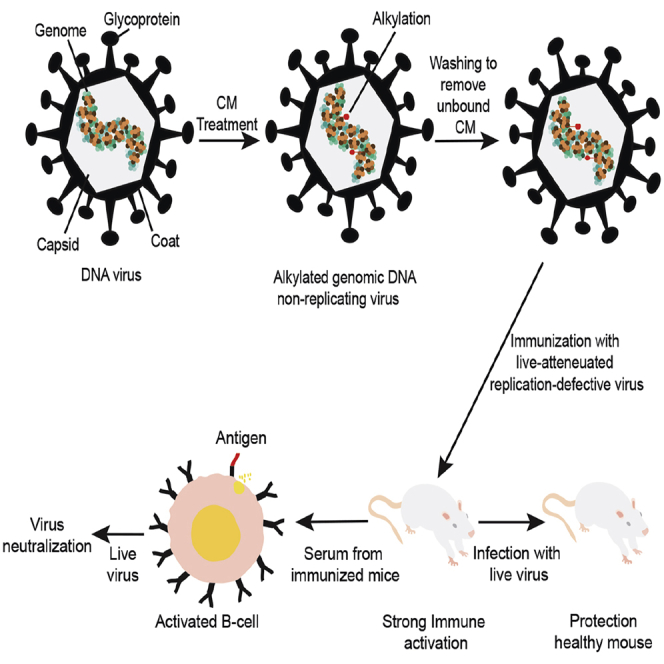
Summary
The development of a chemically attenuated, replication-incompetent virus vaccine can provide protection against diseases caused by DNA viruses. In this study, we have developed a method to produce live-attenuated, replication-defective viruses using centanamycin (CM), a chemical compound that alkylates the A-T-rich minor groove of the DNA and thereby blocks DNA replication. We tested the efficacy of CM to produce live-attenuated, replication-defective human cytomegalovirus, mouse cytomegalovirus, and herpes simplex virus-2 (HSV-2), suggesting a broad application for generating live-attenuated, replication-defective DNA viruses. Mass spectrometry analysis showed that CM alkylate viral DNA at the adenine-N3 position. Moreover, mice immunization with CM-attenuated mouse cytomegalovirus (MCMV) produced a robust immune response and reduced the viral load in immunized animals against challenges with live, wild-type MCMV. Our study offers a unifying and attractive therapeutic opportunity that chemically attenuated live DNA viruses can be readily developed as new frontline vaccines.
Keywords: DNA viruses, cytomegalovirus, herpes simplex virus, vaccines, centanamycin, CM, live-attenuated viruses, inactivated viruses
Graphical abstract
Highlights
-
•
Centanamycin (CM) treatment creates live-attenuated, replication-defective DNA viruses
-
•
Immunization with CM-treated DNA virus induces strong neutralizing antibody response
-
•
Mice immunized with CM-treated mouse CMV are protected against live virus challenges
-
•
A large pool of unique viral antigens is present in CM-treated DNA viruses
Motivation
DNA viruses include major human pathogens such as herpesviruses, adenoviruses, papillomaviruses, enteroviruses, and others. As of now, there is not an established method that can generate live-attenuated, replication-defective viruses for vaccine development. A common technology that can be applied to all DNA viruses presents a feasible solution to this problem. Our proposed technology solution is the use of centanamycin (CM) to develop whole-virion, chemically attenuated, replication-defective live DNA viruses as potential vaccines. We found that CM potently inhibits replication of human cytomegalovirus (HCMV), mouse CMV (MCMV), and herpes simplex virus-2 (HSV-2) in vitro. Our technology can produce live-attenuated, DNA replication-defective viruses in a few hours. It is easy, quickly scalable, adaptable to any type of DNA virus, requires less labor, and is inexpensive. Importantly, we demonstrate that CM-attenuated whole virion MCMV elicits a robust serum neutralizing antibody response in mice and that the vaccinated animals were completely protected from live-virus challenge.
Jaijyan et al. describe an efficient method to generate chemically live-attenuated, replication-defective DNA viruses for vaccine development. CM alkylates the genome of DNA viruses. Upon immunization, CM-treated viruses induce strong immune activation and provide protection against live virus challenges. The method developed here can be applied to all DNA viruses.
Introduction
Viruses are one of the most lethal microorganisms that pose a devastating health risk to human life. At least 31 families of DNA viruses are present that can infect a wide range of hosts, including invertebrates, vertebrates, plants, marine organisms, and others. Many of these viruses are responsible for thousands of infected cases and deaths around the world. There are ample shreds of evidence that have shown that DNA viruses that belong to families like papillomaviridae, hepadnaviridae herpesviridae, and adenoviridae are associated with tumor formation in humans and animals. More specifically, DNA viruses such as Epstein-Barr virus (EBV) (Klein et al., 2007) (Burkitt lymphoma), human papillomavirus (Klein et al., 2007; Parkin, 2006; Schiffman et al., 2007) (vaginal, cervix, head, and neck cancer) (Alhamlan et al., 2021), Kaposi’s sarcoma-associated virus (Chang et al., 1994) (skin cancer), hepatitis B virus (Wong and Goh, 2006) (hepatocarcinoma), and Merkel cell polyomavirus (Feng et al., 2008) (Merkel cell carcinoma) are classified as tumor viruses. Furthermore, DNA viruses are also the causative agents of several diseases, including genital herpes (Sauerbrei, 2016), chickenpox (Ayoade and Kumar, 2020), cytomegalovirus (CMV) retinitis (O'Donnell et al., 1987), roseola, conjunctivitis, pneumonia, cancers, congenital diseases, and many others. Although the antiviral therapies for the human simplex virus, CMV, and varicella-zoster virus reduce the cases significantly, other viruses such as human papillomavirus (HPV), adenovirus, and bovine PV (BPV), where antiviral therapy is limited, continue to cause morbidities and mortalities (Zerr et al., 2012).
The use of replication-defective, live-attenuated DNA viruses is a promising viral intervening strategy to combat many viral diseases that affect humans. A replication-defective virus is incapable of replicating its genome, unable to produce an infectious progeny virus in infected cells, and thus restricted primarily to the site of inoculation. Such a vaccine has many advantages over the classical inactivated viral vaccine as it is safe and inactive, infects host cells, and expresses antigens that can be recognized by the host immune system (Adler et al., 2019; Bloom et al., 2018; Hoshino et al., 2005; Liu et al., 2019; Mocarski et al., 1996; Wang et al., 2016). Studies have shown that the replication-defective viral vaccine can induce sufficient memory T cells to provide complete protection against infection compared with a subunit vaccine (Adler et al., 2019; Wang et al., 2016). Replication-defective viral vaccines have been tested on many viruses. Subcutaneous immunization of mice with replication-defective human simplex virus-1 protects the immunized mice from infection of the eye and ganglion after challenge with a live virus (Morrison and Knipe, 1996). In another study, replication-defective human simplex virus-2 immunization in guinea pigs protects the animal from virus challenge and reduces the replication of the challenged virus in the genital tract (Da Costa et al., 1997). It was found that immunization with replication-defective vaccinia virus induces robust long-term immunity in mice and provides complete protection (Coulibaly et al., 2005). Immunization of mice with replication-defective influenza virus-like particles protects 94% of mice challenged with influenza virus adapted for mice (Watanabe et al., 2002). Recently, Merck & Co. has been testing a replication-defective virus vaccine for CMV in phase 2 clinical trial (Adler et al., 2019). Altogether, these studies indicate the therapeutic potential of developing a novel method that can expedite the production of replication-defective, live-attenuated viruses and will accelerate vaccine development efforts for many untreated viral infections in humans, animals, and plants.
The main objective of this study is to develop and investigate chemically attenuated replication-defective DNA viruses for their potential as vaccines. CM, also known as ML-970 or AS-I-145, was selected to be the chemical agent for attenuating the viruses because it is a potent anticancer agent that derives its activity from selectively binding and alkylating adenine-N3 atoms in the minor groove of A-T-rich DNA (Kiakos et al., 2018). Studies have shown that CM (Sato et al., 2005a) and its relative CC-1065 (Weiland and Dooley, 1991) bind covalently to the motif (A/T)3A in genomic DNA. CM’s interaction with these A/T motifs can lead to genomic instability, DNA damage, and disruption of essential processes such as DNA replication and gene transcription (Yanow et al., 2008). CM is an indolecarboxamide achiral compound with less toxicity than its parent duocarmycin SA. Its chemical composition includes aromatic amine, aromatic ether, and organochloride. Its complete name is N-[4-amino-1-(2-chloroethyl)-2-naphthyl]-5,6,7-trimethoxy-1H-indole-2-carboxamide. CM and a related analog, tafuramycin A, have also been found to effectively attenuate the malaria parasite Plasmodium sp., which has led to the development of powerful vaccines (Purcell et al., 2008; Stanisic et al., 2018; Yanow et al., 2008). Currently, PlasProtecT, a tafuramycin A-attenuated live Plasmodium vaccine, is undergoing a phase 1b clinical trial in Australia (Stanisic et al., 2018). One unique feature of PlasProtecT is its ability to strongly induce memory T cells to eliminate subsequent Plasmodium infection.
In this study, we tested the efficacy of CM in attenuating human CMV (AD169 and Toledo), herpes simplex virus-2 (HSV-2), and mouse CMV (MCMV). In addition, we also tested the efficacy of CM in generating a replication-defective virus vaccine in an in vivo study whereby the inoculated animals were rechallenged with wild-type MCMV. Lastly, the ability of the potential vaccine to induce the production of specific neutralizing antibodies in inoculated animals was also investigated.
Results
Generation of CM-attenuated live CMV
CMV is a ubiquitous betaherpesvirus that causes asymptomatic infection in healthy individuals. However, human CMV (HCMV) infection can have severe consequences in immunocompromised and organ transplant patients. Moreover, congenital HCMV infection induces fetal malformation and is the leading cause of birth defects in newborns.
We first tested the effects of CM on the growth of HCMV using Toledo-Luc, which has been widely used in the virus growth curve analysis (Dula et al., 2009). The growth curve analysis of Toledo-Luc was studied and compared by both the convention plaque assay and the luciferase assay (Dula et al., 2009). Toledo-Luc did not exhibit any growth defects and grew similar to wild type (WT). Toledo-Luc has been analyzed in vivo using the SCID-hu mouse model (Spiess et al., 2015). In our study, Toledo-Luc was treated with different concentrations of CM for 2 h at room temperature (RT), followed by washing with media and 20% sucrose using ultracentrifugation to remove unbound CM, and then allowed to grow in MRC-5 cells. Toledo-Luc did not replicate when treated with the 100, 10, and 1 μM CM (Figure 1A). At 100, 10, and 1 μM CM, Toledo-Luc neither replicated nor infected the cells, leading to complete inactivation of the virus (Figure 1A). This suggested that 100, 10, and 1 μM CM were capable of inactivating the virus in 2 h of incubation at RT, possibly from the damage of the viral DNA (Figure 1A). These results demonstrated that 100, 10, and 1 μM CM are most potent in preventing HCMV infection and replication. Interestingly, when Toledo-Luc was treated with the 0.1 μM CM for 2 h at RT, we observed luciferase activity in the infected cells, suggesting that virus could infect the cells but did not replicate. These results indicated that treatment of Toledo-Luc with 0.1 μM CM allowed the virus to infect the cells, and the virus got damaged (DNA alkylation) to an extent that did not allow the virus to replicate. Indeed, the previous study has shown that excessive alkylation damages the DNA and prevents its replication and transcription (Xu et al., 2017). This is a novel opportunity for the design of an attenuated vaccine where a chemical treatment renders the virus infected but replication incompetent in infected cells. Furthermore, results confirmed that treatment with 0.01–0.001 μM CM allowed the virus to grow and replicate in the infected cells at a reduced rate, suggesting that the Toledo-Luc guide DNA (gDNA) was alkylated at a low level at these CM concentrations. A decrease in the luciferase activity or less viral titer at low CM concentration depends on the extent of damage (DNA alkylation) to the virus by CM, not by cell lysis. The CM binds to the gDNA of the virus randomly in A-T-rich regions. Therefore, it depends on the CM-mediated alkylation of essential or non-essential genes to determine the growth of the virus after CM treatment at low concentrations.
Figure 1.

Centanamycin treatment generates attenuated replication-defective human cytomegalovirus
(A) In vitro growth curve analysis of Toledo-Luc treated with different concentrations of centanamycin (CM). Toledo-Luc was added to MRC-5 cells after treatment with CM for 2 h at RT, followed by washing. Luciferase activity was measured on the indicated days using IVIS-50. Growth curve lines for 10 and 100 μM CM are hidden under a 1 μM growth curve. The virus was not able to grow in 100, 10, and 1 μM concentrations. At 0.1 μM CM, Toledo-Luc could infect the cells but was unable to replicate, as suggested by the luciferase activity of infected cells. At low concentrations such as 0.01 μM CM, the virus grows initially but did not reach its peak, which may be due to damage to viral DNA caused by CM. Random alkylation of viral gDNA can lead to defective replication of the virus. RLU, relative light units. Data are presented as mean ± SEM. Each data point represents the average triplicate value.
(B) Fluorescence image of ARPE-19 cells infected with CM-treated AD169-GFP. The representative images of infected cells were acquired on the 8th day post-infection. GFP represents the AD169-GFP-infected cells, DIC represents the cells in the bright phase, and merge represents the merge of both images. At 10 μM CM, AD169-GFP was not able to infect the cell, and the virus was inactive. AD169-GFP treated with 1 μM CM was able to infect the cells but unable to replicate. Due to defective replication, the GFP signal of the virus could not spread to adjacent cells and was limited to only 1 or 2 infected cells. These results confirm replication-defective viruses that can infect cells but not replicate. At 0.01 μM CM, AD169-GFP was able to infect and spread to adjacent cells but at a reduced rate.
To understand the pathology of CM-treated viruses, we used AD169-GFP to microscopically examine the treated virus. AD169-GFP used in this study had a pentameric complex repaired that allowed the virus to infect ARPE-19 cells (epithelial cells). The representative fluorescence images of infected ARPE-19 cells were acquired on the 8th day post-infection. The results indicate that at 10 μM, the CM-treated virus was not detected in the infected cells when observed by the fluorescence microscope, strongly suggesting that the virus was not able to infect cells, possibly because of complete inactivation of the treated virus. Similarly, as suggested by very few GFP-positive cells (Figure 1B), AD169-GFP treated with 1 μM CM was not able to replicate in the cells. The viruses that entered in cells could not grow or replicate. In striking contrast, at 0.1 μM CM, the treated virus was detected in the infected cells but did not replicate, as suggested by the GFP signal confined to a few cells, compared with the untreated virus, which replicated efficiently. These results suggest that 1 and 0.1 μM CM treatment caused the virus to mutate to such an extent that allowed the virus to infect the cells but did not let the virus replicate due to damaged DNA (Figure 1B). The GFP signal of the virus was confined to only one or two cells. Exposure of the virus to 0.01 μM CM allowed them to infect the cells, albeit with slow growth due to the CM-mediated alkylation effect of viral gDNA (Figure 1B). The presence of GFP-positive cells upon infection confirmed that the virus could not replicate while potentially infecting the cells at 0.1 and 1 μM CM, suggesting that the virus was not completely inactive. The GFP signal of the virus treated with 0.1 and 1 μM CM did not spread to other cells after 1 week of infection, further confirming the replication-defective nature of the virus. The results obtained from Toledo-Luc and AD169-GFP treatments strongly suggest that CM is an effective chemical agent to generate chemically attenuated CMV under in vitro conditions. Plasmodium parasite treatment with CM generates live-attenuated, replication-defective parasites (Purcell et al., 2008; Stanisic et al., 2018; Yanow et al., 2008). Studies have been performed on malaria parasites, including P. falciparum and P. berghei, that generate chemically attenuated parasites for vaccine development. CM generates DNA alkylation of parasite gDNA and produces replication-defective parasites that are immunogenic. Viruses are also intracellular parasites. We found similar effects of CM treatment in DNA viruses, suggesting the broad application of this therapeutic compound.
HSV-2-GFP treatment with CM
To determine whether CM can attenuate other DNA viruses, we tested its effects on HSV-2-expressing GFP (HSV-2-GFP), for which there is no vaccine available as of now. HSV-1 and HSV-2 are human pathogens causing mucosal infections and establishing latent infections in sensory neurons. These viruses reactivate to cause disease manifestation and severe complications. Patients who are Immunocompromised and those with AIDS are at life-threatening risk. The HSV infections and genital herpes pandemic severity in people infected with HIV require an effective vaccine for HSV treatment.
HSV-2-GFP was treated with different concentrations of CM ranging from 100 to 0.01 μM (10-fold serial dilution at each concentration), and growth curves were determined using the plaque assay (Figure 2A). The results obtained from the growth curve analysis of HSV-2-GFP treated with CM confirmed that 100 μM CM completely inhibited virus infection of ARPE-19 cells (Figure 2A). HSV-2-GFP treatment with 10 μM CM led to the virus attenuation, and the virus was not able to spread to adjacent cells (Figure 2A). HSV-2-GFP treatment with 1 μM caused reduced growth compared with the untreated control. The reduced growth of the virus did not attain a peak, as is the case for the untreated virus, suggesting irreversible damage to virus genomic DNA through alkylation. Growth curve analysis showed a dose-dependent growth of the HSV-2-GFP in response to CM (Figure 2A). Consistent with the microscopic results obtained from 36 h post-infection, 100 μM-treated HSV-2-GFP was not detected in infected cells, indicating complete inactivation of the virus (Figure 2B). The virus was able to infect cells but did not replicate when treated with 10 μM CM, suggesting chemical attenuation of the virus (Figure 2B). Treatment of virus with 1 μM CM leads to viral infection of host cells and extremely slow spread to neighboring cells (Figure 2B). These results suggest that CM can attenuate HSV-2, as well as other DNA viruses. At 10 μM CM, the HSV-2-GFP signal was restricted to one or two cells, suggesting that the virus was able to infect cells but could not spread to adjacent cells. This confirms that CM treatment can generate live-attenuated, replication-defective DNA viruses.
Figure 2.

CM treatment generates replication-defective HSV-2
(A) Growth curve analysis of CM-treated HSV-2-GFP in the ARPE-19 cells. HSV-2-GFP was treated with different concentrations of CM for 2 h at RT, followed by washing using 20% sucrose, and added to ARPE-19 cells at an MOI of 0.2. The experiment was performed in triplicates. A standard plaque assay was performed to determine the growth curves of HSV-2-GFP using samples collected at indicated time points. At 100 μM CM, the virus was inactive and unable to infect the cells. HSV-2-GFP treated with 10 μM CM could not grow in infected cells due to defective replication. At 1 μM CM, the virus was able to infect cells and grew at a reduced rate. Data are presented as mean ± SEM.
(B) Fluorescence image of HSV-2-GFP treated with different concentrations of CM. GFP represents the HSV-2-GFP-infected cells, DIC represents the cells in the bright phase, and merge represents the merge of both images. The images were taken at 40× objective post 36 h of virus infection. HSV-2-GFP treated with 10 μM CM was able to infect cells, but the virus cannot further grow due to defective replication produced by CM-mediated alkylation of viral DNA. The untreated virus grows efficiently. HSV-2-GFP treated with 100 μM CM was completely inactive. Treatment of virus with 1 μM CM leads to host cell infection and a slow spread to neighboring cells. The alkylation of viral DNA can lead to defective replication and transcription of genes.
CM attenuates the DNA virus through DNA alkylation
To understand the molecular mechanism underlying CM-mediated attenuation of the virus, we analyzed CM-treated viral DNA using agarose gel electrophoresis, restriction digestion, and heat-labile fragmentation assays. AD169 genomic DNA was treated with 1 and 10 μM CM for 2 h at RT and analyzed in 0.6% agarose gel. Untreated AD169 genomic DNA was used as a control. Results showed that there was no significant difference in the viral DNA bands, mobility, and gel pattern of treated and untreated viral gDNA (Figure 3A). The DNA band was less intense in the CM-treated samples, which could be due to the slow degradation of AD169 gDNA in presence of CM. To further characterize the effect of CM on the virus, we isolated the viral gDNA and treated it with the 10 μM CM for 2 h at RT, followed by digestion with the EcoR1 restriction enzyme. The digestion pattern shows that CM-treated DNA has no major missing bands due to the alkylation (Figure 3B). CMV genomic DNA treated with CM was digested in the presence of EcoR1, indicating that CM did not interfere with the enzyme activity of restriction enzymes. These results also suggested that CM did not interfere with the protein or enzyme activity of the virus and specifically alkylated the DNA only. However, the CM-treated viruses were alkylated efficiently when compared with the control, which was observed by DNA fragmentation at 50°C, while untreated viruses have intact DNA (Figure 3C). The CM-treated viral DNA fragmentation is consistent with the alkylation of N3 in adenine that produces thermolabile adduct (Sato et al., 2005a).
Figure 3.

CM binds with the virus DNA
(A) An agarose gel image of AD169 gDNA treated with and without CM. The gDNA of AD169 was treated with different concentrations of CM for 2 h at RT and loaded into 0.6% agarose gel. AD169 gDNA without CM was used as a control. No significant difference was observed in the gel pattern of viral DNA treated with CM compared with treated without CM.
(B) An agarose gel image of digested AD169 gDNA. AD169 gDNA was treated with 10 μM of CM, followed by digestion with EcoR1-HF. AD169 gDNA without CM was used as a control. Restriction digestion of viral DNA by EcoRI in the presence of CM suggests that CM does not affect protein or enzyme activity. The restriction digestion pattern of CM-treated AD169 gDNA was similar to untreated AD169 gDNA.
(C) AD169 gDNA was treated with 1 and 10 μM CM, followed by incubation at 50°C for 30 min to analyze thermolabile alkylation of viral DNA. The CM-treated gDNA was thermolabile, while untreated gDNA was stable without any degradation. This suggests that CM interacts with viral genomic DNA and produces extensive alkylation. The CM-mediated alkylation of viral genomic DNA was thermolabile at 50°C.
(D) A structure of the nucleotide adduct formed with CM. CM-treated viral gDNA was thermolabile, and the alkylated adenine nucleotide N3 cleaved off from DNA at 50°C. LC-MS analysis of the cleaved nucleotide showed a peak at m/z 552 for the molecular ion of adduct that corresponded to the N3 alkylation structure.
CM exerts its virus attenuation effect through alkylating adenine at the N3 atom
The CM-mediated alkylation of viral gDNA is thermolabile, and the alkylated nucleotide is cleaved off from DNA at 50°C, thus causing DNA fragmentation (Sato et al., 2005a). Therefore, we determined the exact nucleotide affected by CM. CMV gDNA was treated with 10 μM CM for 2 h at RT, followed by incubation at 50ºC. The samples were prepared and analyzed by liquid chromatography-mass spectrometry (LC-MS). The LC-MS results showed a peak at m/z 552 for the molecular ion of the adduct that corresponded to the N3 alkylation structure (Figure 3D). Previous study has also shown that CM exerts its effect on DNA through the alkylation of N3 of adenine, which is a thermolabile (Sato et al., 2005b).
MCMV-Luc growth curve analysis under in vitro conditions
In order to demonstrate the efficacy of CM in vivo, first we analyzed the growth curve of MCMV in vitro when exposed to different concentrations of CM. This would identify the CM concentration required to accurately attenuate the MCMV. In this assay, we used MCMV-Luc, which has a luciferase cassette encoded, and virus growth was measured as photon counts in the in vivo imaging system (IVIS). MCMV-Luc was treated with different concentrations of CM at RT for 2 h, followed by wash with media and 20% sucrose and ultracentrifugation. Thereafter, the treated virus was added to mouse fibroblast 3T3 cells in a 24-well plate in triplicate at an MOI of 0.1. MCMV growth curves were determined by measuring the luciferase activity every day using an IVIS. These results unambiguously demonstrated that 1 μM CM allowed the virus to infect host cells, but the virus cannot replicate (Figure 4A), while at 10 μM CM, MCMV-Luc could not infect the cells, indicating complete inhibition. These results concluded that 1 μM CM alkylated the MCMV genomic DNA to an extent that the virus could infect the cells but not replicate because its DNA was damaged. Accordingly, different concentrations of CM will result in different levels of inhibition of virus replication. Virions after adequate CM treatment will have all the proteins that are required for the infection of a cell. On other hand, these virions cannot replicate further or transcribe new genes as a result of DNA alkylation. We used this important information to perform an in vivo analysis of CM inhibition on MCMV replication in mice.
Figure 4.

CM-treated MCMV immunization provides protection in the mice
(A) Growth curve analysis of CM-treated MCMV-Luc in 3T3 cells. MCMV-Luc was treated with different concentrations of CM for 2 h at RT, followed by washing to remove unbound CM, and added to 3T3 cells in a 24-well plate at 0.1 MOI. Luciferase activity was measured as a relative light unit (RLU) using IVIS-50 on the indicated days. At 10 μM CM, MCMV-Luc was completely inactive. At 1 μM CM, MCMV-Luc can infect the cells but did not replicate. At lower CM concentrations, the virus was infectious and grew at a reduced rate. The experiment was performed in triplicate. Data are represented as mean ± SEM.
(B) In vivo analysis of CM-treated MCMV-Luc. BALB/C mice were injected with untreated and CM-treated MCMV-Luc. MCMV-Luc was treated with 0.1, 1, and 10 μM CM for 2 h at RT, followed by washing to remove unbound CM. Uninfected represents the mice without virus infection, and MCMV-Luc represents the mice injected with the virus. Mice injected with MCMV-Luc treated with 0.1, 1, and 10 μM CM are indicated. The representative images show the growth of treated and untreated MCMV-Luc in mice. MCMV-Luc was completely inactive after treatment with 10 μM CM. At 1 and 0.1 μM CM, MCMV-Luc was limited to the injected site and could not replicate. Three mice were used in each group.
(C) A pictorial representation of the DNA virus attenuation using CM and administration into the mouse. The injected mouse will produce antibodies specific to the attenuated DNA virus. The replication-defective virus carries all the antigens, infects cells, and does not form progeny virus because of alkylated DNA.
(D) A graphical representation of the timeline and strategy for mouse immunizations with CM-treated MCMV. The priming, first booster, and second booster were performed on the indicated days. Immunized mice were challenged with the live virus after 15 days from the last immunization.
(E) Antibody titers determination in the CM-treated MCMV immunized and control mice. Sera samples obtained from immunized and control mice were tested for antibody titration using ELISA. Five mice in each group were used for this assay. ELISA was performed in triplicate. p <0.05 compared with the control mice. Data are represented as mean ± SEM.
(F) An antibody neutralization assay for MCMV. Polyclonal serum samples obtained from immunized and control groups of mice were tested for neutralization titers against MCMV. CM-treated MCMV immunizations in mice generated high neutralization titers. Serially diluted serum (2-fold) samples were added to 1,000 PFUs of MCMV-Luc, followed by incubation for 1 h at RT, and added to 3T3 cells. The viral load was determined in the infected cells by IVIS. The neutralizing titer (NT50) was determined by serial dilution that inhibits a 50% reduction (half maximum) of virus entry into 3T3 cells. p <0.05 compared with control samples.
(G) Determination of viral titer in immunized mice challenged with live wild-type MCMV. Salivary glands of challenged mice were obtained after 10 days of virus administration, and tissue homogenates samples were tested for viral titer analysis using plaque assay. Immunized mice did not show any viral titers, suggesting complete protection. Five mice per group were used for this experiment. p <0.05 compared with the control mice. Data are represented as mean ± SEM.
(H) Adoptive transfer of serum from immunized mice. A passive serum transfer from immunized to unimmunized mice protects against virus challenges. Five naive BALB/C mice were injected intravenously with 150 μL pooled passive serum obtained from CM-treated MCMV immunized mice. After 24 h, the immunized animals were challenged with 10,000 PFU of MCMV. Mice injected with serum obtained from unimmunized animals were used as control. The virus titer was determined in the liver and spleen of control and immunized mice on day 7 post-challenge. Five naive mice were used in each group for this assay. p <0.05 compared with the control group of mice. Data are represented as mean ± SEM.
MCMV-Luc growth analysis under in vivo conditions
MCMV-Luc was purified using a sucrose gradient and titrated in 3T3 cells. We treated 5 × 105 plaque-forming units (PFUs) of the virus with 0.1, 1, and 10 μM CM for 2 h at RT, followed by washing with media using 20% sucrose and ultracentrifugation. Thereafter, 1 × 105 PFUs were injected intraperitoneally into three mice for each concentration. Interestingly, the mice injected with 10 μM CM did not show any infection, suggesting a complete inactivation of the virus. However, the mice injected with 0.1 and 1 μM CM treated MCMV-Luc showed infection, but viruses could not replicate further from the site of infection (Figure 4B). It is conceivable that, during this attenuated infection, the mouse’s immune system might have enough time to recognize the virus as a foreign antigen and generate antibodies specific to the virus. CM-treated MCMV has defective replication and presents an opportunity to be potentially recognized by the host and activate the immune system. MCMV-Luc without CM treatment grew and replicated rapidly (Figure 4B).
Mice challenge assay
To determine the potential vaccine efficacy of CM-attenuated MCMV, we immunized five mice in a group with 1 μM CM-treated 1 × 106 PFUs/mouse (Figures 4C and 4D). A timeline of immunization and the strategy are shown in Figures 4C and 4D. We used 1× PBS to immunize mice in the control group. We did three immunizations at an interval of 15 days, as shown in Figure 4D, and blood was collected 15 days after each inoculation to determine the virus-specific antibody titer using ELISA. The ELISA results showed that immunized mice had high antibody titers after the second immunization and that the titer further increased after the third immunization but at a lower rate (Figure 4E). The serum samples collected from the immunized mice were used to determine neutralizing antibody titers. Results demonstrated that immunization with CM-treated MCMV generated a specific neutralization titer (Figure 4F). After 2 weeks of the last immunization, all animals were healthy because we did not see a difference in the body weight of the animals in the experimental and control groups, thus demonstrating that the CM-attenuated MCMV inoculum was safe and not toxic. The mice were then challenged with the MCMV WT (10,000 PFUs/mouse). Control mice include the animal immunized with 1× PBS along with the adjuvant. To see the virus replication and infection in the mice, salivary glands were isolated after 10 days of post-challenge from the immunized and control groups. At this point, the blood was collected, and the serum was isolated. The serum samples were stored at −80°C until use. The virus titer was determined in salivary glands using a plaque-forming assay. The control mice had a high viral titer value, while the immunized group did not show any viral titer, in tissue homogenate on day 10 post-challenge with WT MCMV (Figure 4G). These results indicate that immunized mice had sufficient neutralizing antibodies that suppressed and eliminated virus infection (Figure 4G). Prior to the end of the experiment, when the mice were sacrificed, none of the animals showed any sign of discomfort or toxicity from the inoculation.
Passive serum immunization protects mice from virus infection
We determined CM-attenuated MCMV vaccine efficacy using pooled passive serum transfer to naive mice followed by the challenge of animals with MCMV WT. The polyclonal serum obtained from CM-attenuated, MCMV immunized animals was injected into five mice via the tail vein. Similarly, the polyclonal serum obtained from unimmunized mice was injected into the control group mice via the tail vein. We used five mice in each group, and each mouse was injected with 150 μL of respective serum. All mice in each group were challenged with 10,000 PFUs of MCMV via the intraperitoneal route after 24 h of serum injection. The mice administered with serum obtained from immunized mice did not show MCMV titer in the spleen and liver, suggesting 100% protection (Figure 4H). In contrast, the mice in the control had high MCMV titers in the spleen and liver. These results demonstrated that CM-attenuated, MCMV immunized mice had sufficient amounts of antibodies that protected mice from the virus challenge. Thus, passive immunization from CM-attenuated MCMV sera could be used for the prevention and treatment of CMV infection. The histology results show that the liver had a well-defined cell shape, membranes, nucleus, and other structures, suggesting no toxicity or other side effects (Figures 5A and 5B). None of the animals showed any sign of discomfort or toxicity from the inoculation. We determined the cytotoxicity of CM-treated viruses using an MTT assay. Cytotoxicity of cells was measured by MTT assay as per manufacturer instructions. The untreated cells were used as a control and compared for the percentage of cells’ survival. The immunogen AD169, Toledo-Luc, and MCMV-Luc treated with different CM concentrations for 2 h at RT followed by washing using 20% sucrose did not show significant cytotoxicity to cells (Figure 5C). It was reported that CM toxicity is below the allowed limit of genotoxic compound (Rayburn et al., 2012).
Figure 5.

CM-treated MCMV immunization is safe
(A) A histology image of the liver section from control mice. The images were acquired at 10× magnification. Cells, nuclei, and other structures are intact without any cytotoxicity.
(B) A histology image of the liver section from CM-treated MCMV immunized mice. The cells and nuclei can be seen intact without any cell toxicity. The images were acquired at 10× magnification.
(C) Cell viability of ARPE-19, MRC-5, and 3T3 cells incubated with CM-treated viruses after washing of unbound CM. No significant difference was observed in the viability of cells after incubation with CM-treated virus immunogens. After washing, most of the unbound CM is removed, and a minute amount of CM remains bound to the virus genomic DNA. This residual CM is present in small amounts and has many fewer genotoxic effects. p >0.05 compared with uninfected cells.
Discussion
DNA viruses represent a significant threat to global health as well as the economy. In this study, we comprehensively assessed the effect of CM on virus attenuation and the potential to develop whole-virion, chemically (CM)-attenuated, live DNA viruses as potential vaccines. The replication-defective virus carries sufficient, and possibly the largest, antigen pools that infect cells and do not form progeny virus because of its damaged DNA. These live replication-defective viruses represent an excellent opportunity to present antigens and activate immune cells. One of the major advantages of the replication-defective virus vaccine is the safety offered by the robust inhibition of virus replication and the absence of progeny virus production.
In this study, the results showed that CM is an effective antiviral agent that can be used to generate replication-defective, live-attenuated DNA viruses. In fact, the results demonstrated that CM readily entered the virus, alkylated DNA at adenine-N3 in the minor groove of A-T-rich sequences, and effectively produced replication-defective, live-attenuated viruses. Treatment of HCMV with 0.10 μM CM for 2 h at RT produced the replication-defective, live-attenuated virus. However, 10 μM CM was needed to produce replication-defective HSV-2. The viruses treated with higher concentrations of CM (100 μM and above) were non-infectious, non-replicating, and inactive, suggesting an extensive alkylation of viral gDNA. The viruses treated with low concentrations of CM (0.01–0.001 μM) can infect and replicate in the host cells, suggesting a lower level of viral gDNA alkylation, thus demonstrating a dose-dependent relationship that requires the determination of optimal concentration for the specific virus to be attenuated.
Using an MCMV in vivo model, the results presented demonstrate the potential of developing a live, CM-attenuated, replication-defective MCMV for development into a vaccine for subsequent protection against infection from the WT virus. Mice inoculated with the potential vaccine produced a high level of antibody titer sufficient to neutralize live MCMV under in vitro conditions. Moreover, challenging the immunized mice with live MCMV did not produce an infection. Furthermore, the passive serum transfer to unimmunized mice also provided protection against new infections or viral challenges.
We have shown that the underlying mechanism that makes replication-defective viruses after CM treatment is the interaction of CM with the genomic DNA of viruses. Human hepatocytes treated with 80 nmol/L of CM do not have statistically significant chromosomal aberration after 4 or 21 h of exposure (Yanow et al., 2008). CM was not found to be toxic to culture bone marrow cells of mice at a concentration of 8.4 nmol/L (Sato et al., 2005a) and was cleared from murine plasma within 4 h when injected via the intravenous route. The cytotoxicity analysis using the MTT assay confirmed that CM-treated viral immunogen with residual CM was not toxic to the cells. After washing the CM-treated viruses, only a minute residual amount of CM remains with the virus. Furthermore, a much lower amount of CM binds to the virus DNA for attenuation, which is well below the allowed limit of genotoxic compounds (Rayburn et al., 2012). The mechanism and nature of UV-induced DNA damage and DNA damage caused by CM are completely different from one another. UV causes the formation of thymine dimers through [2 + 2] photoaddition reactions, which dramatically deform DNA structures (Knips and Zacharias, 2017), whereas following modeling studies done on similar doucarmycin analogs reported by Boger and Johnson confirmed that CM should bind snugly in the minor groove of A-T-rich sequences, causing little perturbation, and alkylated the N3 position of adenine (Boger and Johnson, 1995).
In summary, results from this study present evidence that chemical attenuation (CM treatment in this case) is an effective method of producing live-attenuated, replication-defective DNA viruses that can be used for immunization to produce broadly neutralizing antibodies. While this study illustrates the ability of CM to attenuate HCMV, MCMV, and HSV and generate live-attenuated, replication-defective viruses, future studies will include its impact on other DNA viruses.
Limitations of the study
This method cannot be applied to generate live-attenuated, replication-defective RNA viruses. CM binds specifically to the A-T-rich sequences of DNA and generates live-attenuated, replication-defective DNA viruses. Viruses that grow at a high rate would require a higher concentration of CM for producing live-attenuated viruses. CM concentration should be identified for a specific DNA virus for attenuation. We recommend performing virus growth curve analysis in the presence of different concentrations of CM and then determining an optimal CM concentration to generate live-attenuated viruses. CM is toxic to cells and viruses, and therefore, we further recommend washing CM-treated viruses after incubation to remove any free and unbound CM and then completing subsequent experiments to establish a growth curve. Future studies will evaluate the efficacy of CM-attenuated CMV to prevent congenital CMV infections and birth defects in newborns. Future work will also establish the feasibility of the method for other DNA viruses of medical importance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-mouse antibody conjugate with AP | Thermofisher | Cat. no. 31320 |
| Bacterial and virus strains | ||
| AD169-GFP with repaired pentamer | Created in the lab | Rutgers University, NJ |
| HSV-2-GFP | Created in the lab | Rutgers University, NJ |
| MCMV-Luc | Dr. Qiyi Tang | Howard University, DC |
| Toledo-Luc | Created in the lab | Rutgers University, NJ |
| Chemicals, peptides, and recombinant proteins | ||
| Centanamycin | Prof. Moses Lee | Georgia State University, USA |
| Ethyl Alcohol | Sigma | Cat. no. E7023-1L |
| Tween-80 | Sigma | Cat. no. P1754-500ML |
| Glucose | Sigma | Cat. no. G8270-1KG |
| Phenol | Sigma | Cat. no. P4557-400ML |
| Critical commercial assays | ||
| MTT assay kit | Sigma | Cat. no. 11465007001 |
| Experimental models: Cell lines | ||
| ARPE-19 | ATCC | CRL-2302™ |
| 3T3 cells | ATCC | CRL-1658™ |
| MRC-5 | ATCC | CCL-171™ |
| Experimental models: Organisms/strains | ||
| BALB/C, Female, 6–8 months | Charles River Laboratories Kingston, NY | https://www.criver.com/products-services/find-model/balbc-mouse?region=3611 |
| Software and algorithms | ||
| Image J, version 1.51 | National Institutes of Health | https://imagej.nih.gov/ij/ download.html |
| Prism, version 8.1.1 | GraphPad | https://www.graphpad.com/ scientific-software/prism/ |
| Photoshop 2020 | Adobe | https://www.adobe.com/products/photoshop.html |
| Image lab | Bio-Rad | https://www.bio-rad.com/ en-us/product/image-lab- software?ID=KRE6P5E8Z |
| Illustrator 2020 | Adobe | https://www.adobe.com/products/illustrator.html |
| Other | ||
| Dulbecco’s Modified Eagle Media (DMEM) | Corning | Cat. no. 50-013-PB |
| Minimum Essential Media | Thermofisher | Cat. no. 11095080 |
| Fetal Bovine Serum, heat-inactivated | Corning | Cat. no. 35-011-CV |
| 0.25% trypsin, 2.25mM EDTA | Corning | Cat. no. 25-053-Cl |
| Syringe filter,.2μm, | Corning | Cat. no. 431219 |
| Syringe | Fisher Scientific | Cat.no. 09-719A |
| Vortex mixer | Fisher Scientific, | Cat. no. 02215365 |
| Water bath | National Appliance Company, | Model 220 |
| Chloroform | Sigma | Cat. no. C2432-1L |
| Sucrose | Sigma | Cat. no. 84097-1KG |
| In Vivo Imaging System-200 (IVIS-200) | PerkinElmer | http://www.perkinelmer.com/Catalog/Product/ID/IVIS200 |
| D-Luciferin Sodium Salt | BD Bioscience | Cat. no. 556876 |
| Ultra-centrifuge | Beckman | https://www.beckman.com/centrifuges/ultracentrifuges |
| Penicillin-streptomycin solution (100X) | Corning | Cat. no. 30-002-Cl |
| RPMI | Corning | Cat. no. 50-020-PB |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dabbu K. Jaijyan, dkj28@njms.rutgers.edu.
Materials availability
Any material used in this paper can be requested to lead contact. A material transfer agreement may be executed for transferring a reagent. The USA and worldwide patents have been filed for the material used in this study.
Experimental model and subject details
Female BALB/C mice aged 6–8 months purchased from the Charles River company were used in this study. Animal work was conducted in accordance with all federal regulations in an AAALAC-accredited facility per the standards of the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) at Rutgers University.
Viruses, cell lines, and animals
Viruses used in this study include Toledo-Luc (Dula et al., 2009), AD169-GFP with repaired pentameric complex, MCMV Luciferase (MCMV-Luc, Smith strain), and HSV-2 GFP. Mammalian cell lines used in this work were mouse 3T3 fibroblast cells (ATCC # CRL-1658), human retinal pigment epithelial ARPE-19 (ATCC # CRL-2302), and human lung fibroblast MRC-5 cells (ATCC, # CCL-171). The ARPE-19 and MRC-5 cells were grown in Dulbecco’s modified eagle medium (DMEM) containing 50U/mL penicillin, 50 μg/mL streptomycin (Invitrogen), and 10% fetal bovine serum (FBS, HyClone). The 3T3 cells were grown in minimum essential medium (Gibco) with 50U/mL penicillin, 50 μg/mL streptomycin (Invitrogen), and 10% fetal bovine serum (FBS, HyClone). Viral infections of cells were performed in DMEM containing 5% FBS.
Method details
Centanamycin stock
A 3.3mM stock of CM was prepared in PET (polyethylene glycol 400, absolute ethanol, and Tween-80 in a 6:3:1 ratio)/glucose solution as described previously (Purcell et al., 2008). The working stock of CM for virus treatment was obtained by diluting 3.3mM stock in PET/glucose solution (6:3:1).
Toledo-Luc treatment with CM
A Toledo-Luc containing luciferase expression cassette was used in this study as described previously (Dula et al., 2009). The MRC-5 cells were seeded into 12-well plates. The Toledo-Luc was treated with 100μM, 10μM, 1μM, 0.1μM, 0.01μM, and 0.001μM of CM for 2 hours at room temperature. After 2 hours of treatment with CM, the virus samples were washed with DMEM containing 5% FBS using 20% sucrose and ultracentrifugation and then added to MRC-5 cells at an MOI of 0.1 in triplicate wells. Toledo-Luc treated with the PET vehicle was used as a control. The growth kinetics of Toledo-Luc was determined by a bioluminescence assay. The luciferase activity of the Toledo-Luc virus was measured every 48 hours using an in vivo imaging system (IVIS-50) as described previously (Dula et al., 2009).
Fluorescence microscopy
AD169-GFP with repaired pentamer was incubated with 10μM, 1μM, 0.1μM, and 0.01μM of CM for two hours at room temperature (RT). After treatment, viruses were washed with the DMEM containing 5% FBS using 20% sucrose and ultracentrifugation and added to ARPE-19 cells in triplicate at an MOI of 0.1. Virus growth was analyzed by fluorescence microscopy. In a control experiment, the viruses were treated with the PET vehicle.
Human simplex Virus-2 GFP (HSV-2 GFP) treatment with CM
ARPE-19 (ATCC CRL-2302) cells were grown in 12-well plate in DMEM with 10% FBS and 1% P/S. HSV-2 GFP was treated with different concentrations of CM (100μM, 10μM, 1μM, 0.1μM, and 0.01μM) for two hours at RT. CM-treated viruses were washed with DMEM containing 5% FBS using 20% sucrose and ultracentrifugation to remove unbound CM and added to ARPE-19 cells in triplicate at an MOI of 0.2. The supernatant was collected at 6, 12, 18, 24, 30, 36, and 42 h post-infection. Virus titer was determined using the standard plaque assays on ARPE-19 cells. We also determined the effects of CM on HSV-2 GFP using fluorescence microscopy. Infected live cells were imaged at different time points post-infection.
Analysis of CM and viral gDNA
We analyzed the CM-treated and untreated HCMV gDNA by agarose gel electrophoresis, restriction digestion, and thermal incubation at 50°C. AD169 was grown in ARPE-19 cells and virus particles were purified by 20% sucrose using ultracentrifugation. AD169 gDNA was extracted and the concentration was determined by optical density (OD) at 260/280. Briefly, 90% of confluent ARPE-19 cells were infected at 0.2–0.5 MOI of AD169, and the supernatant was collected after 5–6 days post-infection. Supernatant was then centrifuged at 8,000 RPM for 20 min at 4°C to pellet the cell’s debris. Virus particles were purified using 20% sucrose and ultracentrifugation at 80,000g for one hour at 4°C. AD169 pellet was suspended in two mL of lysis buffer (150mM Tris pH 8.0, 1mM MgCl2 0.2mM EDTA, 200mM NaCl, 1% sodium sarkosyl, 200μg/mL) and lysed. Lysate was then subjected to an equal volume of phenol:chloroform (1:1) followed by chloroform extraction. Approximately, 10μg of AD169 gDNA was incubated with 1μM and 10μM CM at RT for one hour. Control includes 10μg of AD169 gDNA treated with vehicle control in the absence of CM. Treated gDNA was analyzed by agarose gel electrophoresis to see if there was any effect of CM on the HCMV gDNA. In a separate experiment, the CM-treated (1μM and 10μM) AD169 gDNA was incubated at 50°C for 30 minutes to test whether the alkylation of DNA was thermolabile (Sato et al., 2005a). Untreated AD169 gDNA was also treated similarly. In another experiment, 3μg of CM treated (10μM) and untreated AD169 gDNA was digested with EcoRI-HF overnight at 37°C and analyzed by 0.6% agarose gel electrophoresis.
Identification of CM nucleotide adduct in viral DNA
5μg (50μL) of AD169 gDNA was incubated with 10μM of CM for two hours at room temperature followed by a 30-minute incubation at 50°C. Thereafter, DNA samples were adjusted to a volume of 200μL with Tris buffer (pH 9.0). Ethyl acetate (400μL) was added to the DNA samples and vortexed vigorously. The top ethyl acetate layer was collected in a new Eppendorf tube. This extraction process was repeated 4 more times. The combined ethyl acetate extract was allowed to evaporate inside a fume hood, and the residue was dissolved in the HPLC grade acetonitrile and filtered through a 0.2μm filter. LC-MS analysis was performed to identify the CM-nucleobase adduct using Xevo G2-XS QT.
In vivo experiments
First, we determined the growth curve of mouse cytomegalovirus expressing a luciferase gene (MCMV-Luc) under in vitro conditions. MCMV-Luc was treated with 10μM, 1μM, 0.1μM, and 0.01μM CM for two hours at RT followed by a wash using 20% sucrose and ultracentrifugation. After washing, the viruses were added to 3T3 cells in a 24- well plate in triplicate at an MOI of 0.1. The virus growth curves were determined by measuring the luciferase activity every day using an in vivo imaging system (IVIS-50). For the animal experiments, approximately 5 × 105 PFU of MCMV-Luc were treated with 0.1μM, 1μM, and 10μM CM for two hours at RT. Viruses were then washed with RPMI to remove CM using ultracentrifugation and were resuspended in 1mL of RPMI. Thereafter, 200μL of CM-treated MCMV-Luc was injected intraperitoneally into each mouse. We used three mice in each group. The growth of the CM-treated and untreated viruses in injected mice was determined by IVIS. Briefly, the mice were anesthetized using isoflurane and injected with the luciferin substrate (150mg/kg body weight). The injected mice were then incubated for five minutes at RT, and luciferase activity was determined using IVIS (Zhang et al., 2007). In the control experiments, the MCMV-Luc was treated with PET and followed in a similar way as described above.
Immunization with CM-attenuated replication-defective virus
For the first immunization, 6x106 PFU of MCMV were treated with 1μM CM for two hours at RT. The viruses were purified through 20% sucrose in RPMI and ultracentrifugation to remove unbound CM. Five BALB/C mice per group were immunized subcutaneously with the attenuated MCMV (1 × 106 PFU per mouse) along with alum adjuvant. The second and third immunizations were performed with CM-treated 0.5 × 106 PFU with alum adjuvant per mouse. Subcutaneous immunizations were performed on day 1, day 15, and day 30. Pre-bleed was performed before immunization and serum samples were stored at −80°C until use. Blood samples were collected 15 days after each immunization. Blood samples were centrifuged, and the serum was collected for storage at −80°C until use. The mice administered with PBS/alum adjuvant were used as control and treated in a similar manner.
Virus-specific antibody response in immunized mice
Mice sera samples were analyzed by ELISA to see if MCMV-specific antibodies were present in the immunized mice. MCMV virus particles (2 μg/mL) were coated (50μL/well) into the 96 wells plate at 4ᵒC overnight. The uncoated viruses were removed by washing three times with 1× PBST (0.1% Tween 20) and blocked with 1% BSA in 1× PBST for two hours at 37ᵒC. Serially diluted serum samples were added in triplicates and incubated for 2 hours at 37ᵒC. The plate was washed three times with 1× PBST (0.1% Tween 20). AP-conjugated secondary antibody diluted in 1% BSA (1:1000) was added to each well (50μL/well) and incubated for 1 hour at 37ᵒC. Thereafter, the plate was washed five times to remove the unbound secondary antibodies. Next, alkaline phosphatase substrate was added to each well, and the plate was read at 405 nm in an ELISA reader (Tecan).
Virus neutralization assay
A plaque reduction assay was performed to determine the neutralizing antibodies in the serum obtained from immunized mice. Serum from unimmunized mice was used as a control. Briefly, 3T3 cells were seeded into a 24-well plate. Serially diluted serum samples (2-fold) in MEM were added to the 1,000 PFU of MCMV-Luc and incubated for one hour at RT. After incubation, MCMV-Luc (1,000 PFU/well) was added to the 3T3 cells, and the media was changed after 4 hours of incubation. Virus infection continued for 48 hours, and the viral load was determined in relative light units (RLU value) using the IVIS. The 50% neutralization titer (NT50) was determined by calculating the antibody titer that is required to neutralize 50% of the virus load.
Mice challenge assay
After 15 days of the last immunization, the mice in each group were challenged by intraperitoneal injection of 1 × 104 PFU of MCMV. After 10 days of virus administration, salivary glands were isolated from the challenged animals and the tissue homogenates were prepared to determine MCMV titer using a plaque assay. Unimmunized mice challenged with MCMV were served as control. For the plaque assay, the tissue lysate (0.2mL) was diluted serially and added to the 3T3 cells in a 24-well plate. The virus titer was determined using tissue homogenate prepared from each mouse. The liver tissues were also obtained for histology to analyze any sign of cytotoxicity due to CM-treated MCMV immunization.
Passive sera transfer
To determine the vaccine protective efficacy in vivo, we did a passive serum transfer assay using polyclonal serum obtained from immunized mice. About 150μL of pooled serum/mouse was intravenously injected into five non-immune BALB/C mice (six months old) via the tail vein. Animals in the control groups were administered with 150μL of polyclonal serum obtained from unimmunized mice. After 24 hours of serum administration, the mice were challenged with 10,000 PFU of MCMV-WT via intraperitoneal injection. After seven days of challenge, the spleen and liver were harvested and homogenized in MEM media that contained 10% FBS. About 0.2mL of homogenate was titrated using a standard plaque assay.
The cytotoxicity of each immunogen
The cytotoxicity of each immunogen was measured by the MTT assay kit (Sigma). Briefly, ARPE-19, MRC-5, and 3T3 cells were seeded into the 96 well plate at a density of 8,000 cells per well in 100μL of DMEM (containing 10% FBS). AD169, Toledo-Luc, and MCMV-Luc were treated with different concentrations of CM (100μM to 0.001μM) for two hours at RT followed by a wash to remove unbound CM using 20% sucrose and ultracentrifugation. After the washings, CM-treated viruses were added to the respective cells and incubated for 24 hours. The media was changed with 25μL of MTT (5 mg/mL). After 6 hours of incubation, 100μL of dimethylsulfoxide (DMSO) was added to each well. The cell viability was determined by the absorbance of each well at 570 nm (Spectrophotometer, TECAN) and normalized by comparing with the untreated group.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 8.1.1. Luciferase assays were performed, and luciferase expression was quantified. Mean represents the average of triplicate values. Data are represented as mean ± SD. The significance of the difference was analyzed by analysis of variance (ANOVA). A p value<0.05 was considered significant. The statistical details are provided in the figure legends.
Acknowledgments
We would like to thank Dr. Qiyi Tang (Howard University, Washington, DC, USA) for providing the MCMV-Luc. We would like to say thank you to Dr. Jian Lu for making HSV-2-GFP. We would also like to thank Dr. Shikha Singh for her help in the scientific discussion. We would like to thank Alison Gu for proofreading the manuscript. Research reported in this publication was supported by the Rutgers HealthAdvance program, partially supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under award number U01HL150852.
Author contributions
The idea was conceived by D.K.J., H.Z., and M.L. The experiments were performed by D.K.J. Results were analyzed by D.K.J., H.Z., K.G., and M.L. The manuscript was written by D.K.J., M.L., K.G., and H.Z. The manuscript was revised by D.K.J., M.L., K.G., and H.Z.
Declaration of interests
United States and worldwide patents have been filed based on the research findings in this paper.
Published: September 8, 2022
Contributor Information
Dabbu Kumar Jaijyan, Email: dkj28@njms.rutgers.edu.
Hua Zhu, Email: zhuhu@njms.rutgers.edu.
Data and code availability
-
•
All the data published in this paper will be available from the lead contact upon request.
-
•
No original codes were used in this paper.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adler S.P., Lewis N., Conlon A., Christiansen M.P., Al-Ibrahim M., Rupp R., Fu T.M., Bautista O., Tang H., Wang D., et al. Phase 1 clinical trial of a conditionally replication-defective human cytomegalovirus (CMV) vaccine in CMV-seronegative subjects. J. Infect. Dis. 2019;220:411–419. doi: 10.1093/infdis/jiz141. [DOI] [PubMed] [Google Scholar]
- Alhamlan F.S., Alfageeh M.B., Al Mushait M.A., Al-Badawi I.A., Al-Ahdal M.N. Human papillomavirus-associated cancers. Adv. Exp. Med. Biol. 2021;1313:1–14. doi: 10.1007/978-3-030-67452-6_1. [DOI] [PubMed] [Google Scholar]
- Ayoade F., Kumar S. StatPearls (Treasure Island (FL) 2020. Varicella zoster (chickenpox) [Google Scholar]
- Bloom D.C., Tran R.K., Feller J., Voellmy R. Immunization by replication-competent controlled herpesvirus vectors. J. Virol. 2018;92:e00616-18. doi: 10.1128/JVI.00616-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D.L., Johnson D.S. CC-1065 and the duocarmycins: unraveling the keys to a new class of naturally derived DNA alkylating agents. Proc. Natl. Acad. Sci. USA. 1995;92:3642–3649. doi: 10.1073/pnas.92.9.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Cesarman E., Pessin M.S., Lee F., Culpepper J., Knowles D.M., Moore P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Coulibaly S., Brühl P., Mayrhofer J., Schmid K., Gerencer M., Falkner F.G. The nonreplicating smallpox candidate vaccines defective vaccinia Lister (dVV-L) and modified vaccinia Ankara (MVA) elicit robust long-term protection. Virology. 2005;341:91–101. doi: 10.1016/j.virol.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Da Costa X.J., Bourne N., Stanberry L.R., Knipe D.M. Construction and characterization of a replication-defective herpes simplex virus 2 ICP8 mutant strain and its use in immunization studies in a Guinea pig model of genital disease. Virology. 1997;232:1–12. doi: 10.1006/viro.1997.8564. [DOI] [PubMed] [Google Scholar]
- Dulal K., Zhang Z., Zhu H. Development of a gene capture method to rescue a large deletion mutant of human cytomegalovirus. J. Virol. Methods. 2009;157:180–187. doi: 10.1016/j.jviromet.2008.12.021. [DOI] [PubMed] [Google Scholar]
- Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino Y., Dalai S.K., Wang K., Pesnicak L., Lau T.Y., Knipe D.M., Cohen J.I., Straus S.E. Comparative efficacy and immunogenicity of replication-defective, recombinant glycoprotein, and DNA vaccines for herpes simplex virus 2 infections in mice and Guinea pigs. J. Virol. 2005;79:410–418. doi: 10.1128/JVI.79.1.410-418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiakos K., Englinger B., Yanow S.K., Wernitznig D., Jakupec M.A., Berger W., Keppler B.K., Hartley J.A., Lee M., Patil P.C. Design, synthesis, nuclear localization, and biological activity of a fluorescent duocarmycin analog. Bioorg. Med. Chem. Lett. 2018;28:1342–1347. doi: 10.1016/j.bmcl.2018.03.016. [DOI] [PubMed] [Google Scholar]
- Klein E., Kis L.L., Klein G. Epstein-Barr virus infection in humans: from harmless to life endangering virus-lymphocyte interactions. Oncogene. 2007;26:1297–1305. doi: 10.1038/sj.onc.1210240. [DOI] [PubMed] [Google Scholar]
- Knips A., Zacharias M. Both DNA global deformation and repair enzyme contacts mediate flipping of thymine dimer damage. Sci. Rep. 2017;7:41324. doi: 10.1038/srep41324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Freed D.C., Li L., Tang A., Li F., Murray E.M., Adler S.P., McVoy M.A., Rupp R.E., Barrett D., et al. A replication-defective human cytomegalovirus vaccine elicits humoral immune responses analogous to those with natural infection. J. Virol. 2019;93:e00747-19. doi: 10.1128/JVI.00747-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski E.S., Kemble G.W., Lyle J.M., Greaves R.F. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc. Natl. Acad. Sci. USA. 1996;93:11321–11326. doi: 10.1073/pnas.93.21.11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison L.A., Knipe D.M. Mechanisms of immunization with a replication-defective mutant of herpes simplex virus 1. Virology. 1996;220:402–413. doi: 10.1006/viro.1996.0328. [DOI] [PubMed] [Google Scholar]
- O'Donnell J.J., Jacobson M.A., Mills J. Development of cytomegalovirus (CMV) retinitis in a patient with AIDS during ganciclovir therapy for CMV colitis. N. Engl. J. Med. 1987;316:1607–1608. doi: 10.1056/NEJM198706183162516. [DOI] [PubMed] [Google Scholar]
- Parkin D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- Purcell L.A., Yanow S.K., Lee M., Spithill T.W., Rodriguez A. Chemical attenuation of Plasmodium berghei sporozoites induces sterile immunity in mice. Infect. Immun. 2008;76:1193–1199. doi: 10.1128/IAI.01399-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayburn E., Wang W., Li M., Zhang X., Xu H., Li H., Qin J.J., Jia L., Covey J., Lee M., Zhang R. Preclinical pharmacology of novel indolecarboxamide ML-970, an investigative anticancer agent. Cancer Chemother. Pharmacol. 2012;69:1423–1431. doi: 10.1007/s00280-012-1851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A., McNulty L., Cox K., Kim S., Scott A., Daniell K., Summerville K., Price C., Hudson S., Kiakos K., et al. A novel class of in vivo active anticancer agents: achiral seco-amino- and seco-hydroxycyclopropylbenz[e]indolone (seco-CBI) analogues of the duocarmycins and CC-1065. J. Med. Chem. 2005;48:3903–3918. doi: 10.1021/jm050179u. [DOI] [PubMed] [Google Scholar]
- Sato A., McNulty L., Cox K., Kim S., Scott A., Daniell K., Summerville K., Price C., Hudson S., Kiakos K., et al. A novel class of in vivo active anticancer agents: achiral seco-amino- and seco-hydroxycyclopropylbenz[e]indolone (seco-CBI) analogues of the duocarmycins and CC-1065. J. Med. Chem. 2005;48:3903–3918. doi: 10.1021/jm050179u. [DOI] [PubMed] [Google Scholar]
- Sauerbrei A. Herpes genitalis: diagnosis, treatment and prevention. Geburtshilfe Frauenheilkd. 2016;76:1310–1317. doi: 10.1055/s-0042-116494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M., Castle P.E., Jeronimo J., Rodriguez A.C., Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- Spiess K., Jeppesen M.G., Malmgaard-Clausen M., Krzywkowski K., Dulal K., Cheng T., Hjortø G.M., Larsen O., Burg J.S., Jarvis M.A., et al. Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc. Natl. Acad. Sci. USA. 2015;112:8427–8432. doi: 10.1073/pnas.1509392112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanisic D.I., Fink J., Mayer J., Coghill S., Gore L., Liu X.Q., El-Deeb I., Rodriguez I.B., Powell J., Willemsen N.M., et al. Vaccination with chemically attenuated Plasmodium falciparum asexual blood-stage parasites induces parasite-specific cellular immune responses in malaria-naive volunteers: a pilot study. BMC Med. 2018;16:184. doi: 10.1186/s12916-018-1173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Freed D.C., He X., Li F., Tang A., Cox K.S., Dubey S.A., Cole S., Medi M.B., Liu Y., et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016;8:362ra145. doi: 10.1126/scitranslmed.aaf9387. [DOI] [PubMed] [Google Scholar]
- Watanabe T., Watanabe S., Neumann G., Kida H., Kawaoka Y. Immunogenicity and protective efficacy of replication-incompetent influenza virus-like particles. J. Virol. 2002;76:767–773. doi: 10.1128/JVI.76.2.767-773.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland K.L., Dooley T.P. In vitro and in vivo DNA bonding by the CC-1065 analogue U-73975. Biochemistry. 1991;30:7559–7565. doi: 10.1021/bi00244a027. [DOI] [PubMed] [Google Scholar]
- Wong C., Goh K. Chronic hepatitis B infection and liver cancer. Biomed. Imaging Interv. J. 2006;2:e7. doi: 10.2349/biij.2.3.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Wang W., Wu J., Shin J.H., Wang P., Unarta I.C., Chong J., Wang Y., Wang D. Mechanism of DNA alkylation-induced transcriptional stalling, lesion bypass, and mutagenesis. Proc. Natl. Acad. Sci. USA. 2017;114:E7082–E7091. doi: 10.1073/pnas.1708748114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanow S.K., Purcell L.A., Pradel G., Sato A., Rodriguez A., Lee M., Spithill T.W. Potent antimalarial and transmission-blocking activities of centanamycin, a novel DNA-binding agent. J. Infect. Dis. 2008;197:527–534. doi: 10.1086/526788. [DOI] [PubMed] [Google Scholar]
- Zerr D.M., Boeckh M., Delaney C., Martin P.J., Xie H., Adler A.L., Huang M.L., Corey L., Leisenring W.M. HHV-6 reactivation and associated sequelae after hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2012;18:1700–1708. doi: 10.1016/j.bbmt.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Rowe J., Wang W., Sommer M., Arvin A., Moffat J., Zhu H. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J. Virol. 2007;81:9024–9033. doi: 10.1128/JVI.02666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
All the data published in this paper will be available from the lead contact upon request.
-
•
No original codes were used in this paper.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.