

SUMMARY
Voltage-gated sodium channels (NaV) in nociceptive neurons initiate action potentials required for transmission of aberrant painful stimuli observed in osteoarthritis (OA). Targeting these NaV subtypes with drugs to produce analgesic effects for OA pain management is a developing therapeutic area. Previously, we determined the receptor site for the tamoxifen analog, N-desmethyl tamoxifen (ND-Tam), within a prokaryotic NaV. Here, we report the pharmacology of ND-Tam against eukaryotic NaVs natively expressed in nociceptive neurons. ND-Tam and analogues occupy two conserved intracellular receptor sites in domain II and IV of NaV1.7 to block ion entry using a ‘bind and plug’ mechanism. We find ND-Tam inhibition sodium current is state-dependent— conferring potent frequency- and voltage-dependent block of hyperexcitable nociceptive neuron action potentials implicated in OA pain. When evaluated using an OA pain mouse model, ND-Tam has long lasting efficacy which supports the potential of repurposing ND-Tam analogues as Nav antagonists for OA pain management.
INTRODUCTION
Voltage-gated sodium channels (NaVs) are transmembrane proteins required for electrical signaling in biology. NaVs open their ion-conducting pore and selectively conduct sodium ions in response to membrane depolarization—two features which shape the action potential waveform required for long range signaling in vertebrate nervous systems (Catterall, 2012). There are nine sodium channel subtypes (NaV1.1– 1.9), which are preferentially expressed in excitable cell types of organ systems(Goldin, 2001). In mice and humans, NaV1.1 (SCN1A), NaV1.6 (SCN8A), NaV1.7 (SCN9A), NaV1.8 (SCN10A), and NaV1.9 (SCN11A) are expressed by adult sensory neurons, including dorsal root ganglia (DRG), and play a key role in pain sensation (Toledo-Aral et al., 1997, Akopian et al., 1996, Dib-Hajj et al., 1998, Ramachandra and Elmslie, 2016, Zhang et al., 2017). Aberrant NaV expression patterns in DRG neurons after inflammation contribute to hyperexcitability of sensory neurons observed in chronic pain states, such as osteoarthritis (OA) (Zhu et al., 2020, Miller et al., 2017). An estimated 240 million individuals worldwide have symptomatic OA, with a higher prevalence in women (18%) than in men (10%) age 60 and older (Allen et al., 2022). With OA global incidence rising, pain is one of the most debilitating symptoms, yet few management options exist for patients. Conventional treatment options for OA pain relies mainly on systemic nonsteroidal anti-inflammatory drugs, which are moderately effective but are associated with serious long-term risks (Trelle et al., 2011, Mays, 2001, Kolasinski et al., 2020) . Development of peripheral nerve NaV antagonists as prototypic pain medications has garnered considerable investment since the drug target is not associated with addiction (Emery et al., 2016). However, concerns over drug safety related to poor NaV subtype specificity, and low efficacy has limited their translation for these systemically administered compounds. Intra-articular drug delivery for OA has a number of advantages over systemic administration, including increased local bioavailability, reduced systemic exposure, fewer adverse events and reduced cost (Jones et al., 2019). Currently, intra-articular glucocorticoid injections are strongly recommended as a treatment option for knee OA, albeit with short-term efficacy(Kolasinski et al., 2020), and a number of intra-articular therapeutic candidates for OA are currently in clinical development, including small-molecule therapies (Toyoda et al., 2021). In this manuscript, we investigate drug targeting of recently identified receptor found within NaVs as a method to inhibit nociceptive signaling to attenuate OA-related pain.
X-ray crystallography and cryo-electron microscopy (cryo-EM) methods have led to the structural visualization of prokaryotic and eukaryotic NaVs at high-resolution (as reviewed) (Noreng et al., 2021). These studies have advanced our understanding of biophysical regulation of their ion conductive states, and molecular basis of drug and toxin action (Pan et al., 2019, Yan et al., 2017, Pan et al., 2018, Jiang et al., 2020, Jiang et al., 2021, Shen et al., 2018, Bagneris et al., 2014, Sula et al., 2017, Payandeh et al., 2011). Results from collaboration with the Wallace group structurally identified a unique receptor site for tamoxifen metabolites within NavMs— a prokaryotic sodium channel isolated from Magnetococcus marinus (Sula et al., 2021). Tamoxifen, endoxifen, N-desmethyl tamoxifen (ND-Tam) and 4-hydroxy tamoxifen (4OH-Tam) occupy a binding pocket near the intracellular gate of the NavMs channel, which does not overlap with other known NaV receptor sites (Sula et al., 2021). Prokaryotic sodium channels are related to eukaryotic NaVs but structurally assemble as homotetramers, whereas the eukaryotic α-subunits are a single peptide which form four domains (DI-IV), each containing pore (PM) and voltage sensor modules (VSM) (Ren et al., 2001, Noda et al., 1986). Eukaryotic NaVs likely evolved after gene duplication from a common prokaryotic NaV ancestor (Vien and DeCaen, 2016). Because of their evolutionary relationship, some drug receptor sites in prokaryotic and eukaryotic NaVs are structurally similar— such as the pore fenestration residues that bind to antiepileptic, local anesthetic and antiarrhythmic drugs (Bagneris et al., 2014). However, in other cases, drug receptors sites are divergent among these channels. For example, prokaryotic NaVs are completely insensitive to tetrodotoxin— a marine neurotoxin which blocks eukaryotic NaVs with variable potency among subtypes (Narahashi et al., 1964). Another example of evolutionary divergence in pharmacology is the receptor for the dihydropyridines, which are antihypertensive drugs that bind at distinct sites for eukaryotic Cav1.1 channels and prokaryotic CavAb channels (Tang et al., 2016, Tang et al., 2019, Gao and Yan, 2021). Considering these differences, we set out to functionally determine the conservation of the tamoxifen receptor in sensory neuron NaVs in terms of location and mechanism of action. We evaluate tamoxifen analogues against human NaV subtypes and DRG neuronal action potential firing, leading up to the goal of evaluating their potential as an analgesic in an OA mouse model. To avoid off-target effects associated with tamoxifen pharmacology, we selected ND-Tam for our study since it has the lowest affinity (> 100 times) for the estrogen receptor (ER) among all other tamoxifen metabolites (Katzenellenbogen et al., 1984).
RESULTS
Potency against endogenous and expressed NaVs.
The drug bound structures of NavMs crystallographically identified a unique receptor site for tamoxifen derivative 4OH-Tam and ND-Tam at 2.4 Å and 3.2 Å, respectively (Sula et al., 2021). Because drug occupancy of this receptor caused long lasting inhibition of the NavMs sodium current (INa), we wondered if these properties would translate into effects against sensory neuron NaVs which transmit pain signals. Accordingly, we tested the potency of ND-Tam and 4OH-Tam against endogenously expressed NaVs in cultured DRG neurons isolated from NaV1.8-tdTomato mice by conducting whole cell voltage clamp recordings (Figure 1A, B)(Gautron et al., 2011). Sodium currents were activated by −10 mV depolarization trains (0.2 Hz) from a holding potential of −100 mV, and the extracellularly applied drug effect was evaluated after 5 minutes of application (Figure 1B). The half-maximal potency of INa inhibition (IC50) for ND-Tam and 4OH-Tam was 1.7 μM ± 0.2 and 3.3 μM ± 0.6, respectively (Figure 1C, Table S1). Interestingly, the majority (86 ± 5 %) of INa did not recover after removing ND-Tam from the bath and waiting for 5 minutes (Figure 1B, D), which suggests a slow dissociation of the drug-receptor complex. As discussed in the introduction, the total DRG INa is conducted by several voltage-gated sodium channel subtypes (NaV1.1, NaV1.6, NaV1.7, NaV1.8 and NaV1.9). To determine if there is any subtype specificity, we expressed human orthologues in cell lines (HEK-293 and CHO) co-expressing the human β1 regulatory subunit (hβ1) and conducted voltage-clamp experiments. Cells expressing NaV1.1, NaV1.6, NaV1.7, and NaV1.8 channels produced robust voltage-dependent sodium currents (Figure S1A). However, we did not detect currents from cells expressing NaV1.9, which is possibly due to impaired membrane trafficking with these cells (Lin et al., 2016). We observed nominal differences in potency of 4OH-Tam and ND-Tam among the human NaV subtypes (Figure S1B), where IC50s ranged from 1.9–3.4 μM for 4OH-Tam and 1.7–2.4 μM for ND-Tam (Table S1). Taken together, these results demonstrate that ND-Tam and 4OH-Tam exhibit moderate potency against the DRG sodium current when −100 mV holding potentials are applied, and little selectivity for the NaV channel subtypes expressed in sensory neurons.
Figure 1. Tamoxifen metabolites inhibit endogenous voltage-gated sodium channels expressed in murine DRG neurons.
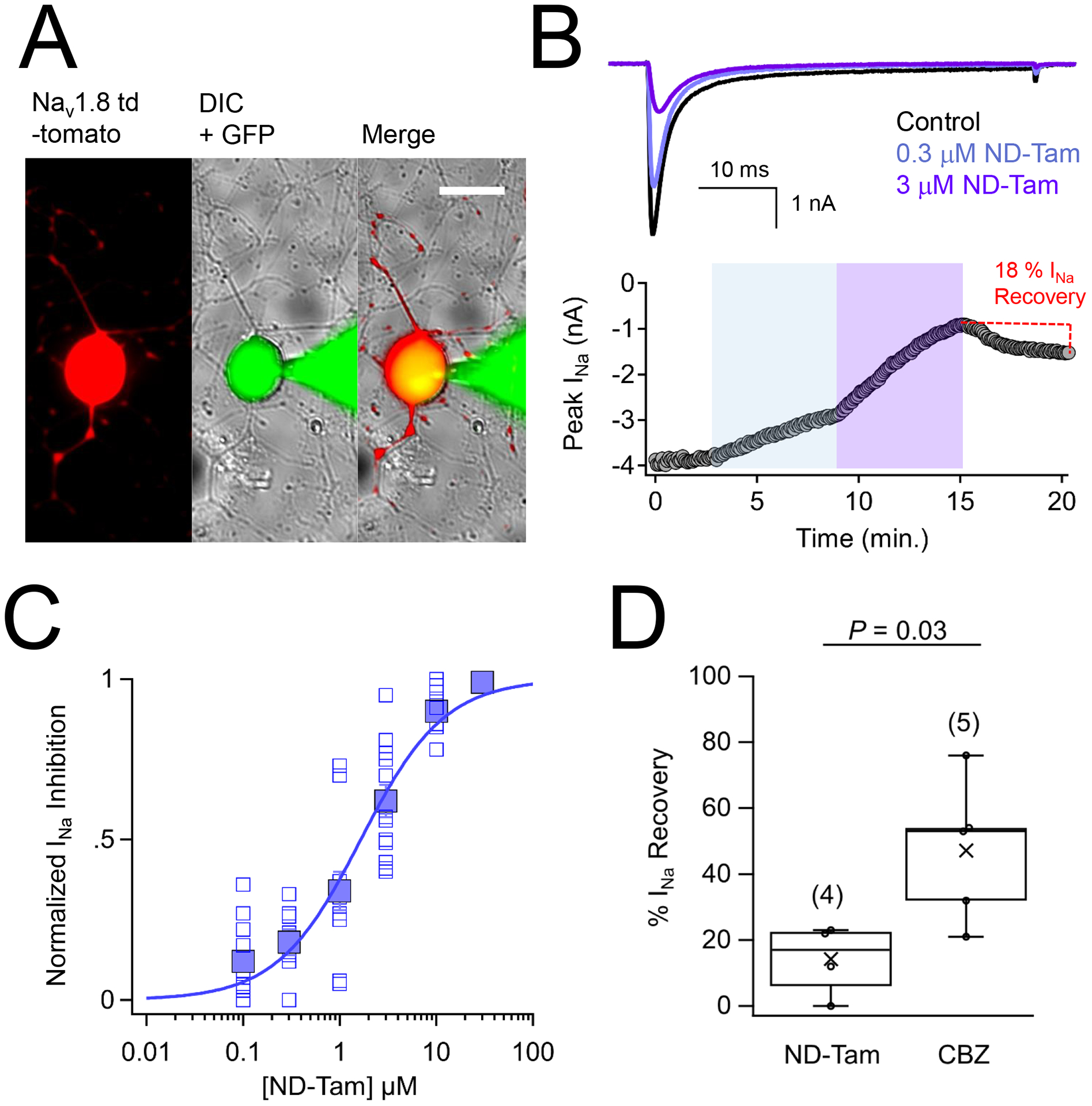
A) DIC and fluorescence images of voltage clamped DRG neuron isolated and cultured from an NaV1.8-tomato mouse. Continuity with the patched neuron is indicted by the GFP (Alexa Fluor 488 dye 1 nM) loaded into the glass patch electrode. Scale bar = 25 μm B) Top, exemplar sodium currents (INa) recorded in control saline and two concentrations of N-desmethyl tamoxifen (ND-Tam) from a voltage-clamped DRG neuron. INa was activated by 0.2 Hz train of 50 ms depolarizations to −10 mV from −100 mV holding potential. Bottom, time course of peak INa inhibition during control conditions and after 5 minutes of extracellular ND-Tam treatment. C) Resulting drug concentration- INa inhibition relationship for ND-Tam fit to the Hill equation. Open symbols represent responses from individual cells and filled symbols represent average response per concentration. Error is equal to S.E.M. (n = 11) D) A comparison of the percent INa recovery after 3–10 μM ND-Tam or 1 mM carbamazepine (CBZ) inhibition. INa recovery was assessed after 5 minutes in control saline post drug exposure. Fewer INa recovered from ND-Tam exposure compared to CBZ exposure, 14.3 ± 5.4 % and 47.2 ± 9.6 %, respectively, and was statistically significant. (P = 0.03). Error is equal to S.E.M. and the number of cells evaluated per treatment group is indicated within the parentheses.
Conservation of a tamoxifen receptor in DII and DIV of human NaV1.7
Published crystal structures of the prokaryotic NavMs channel complexed with tamoxifen analogs (Tamoxifen, 4OH-tam, endoxifen, ND-Tam) chemically define inner (Sitein) and outer drug receptor sites (Siteout) near the channel gate (Figure 2A)(Sula et al., 2021). While no clear drug-receptor interactions were determined within Siteout, the S6 D220 side chain carboxylate forms hydrogen bond interactions with the ether and amine moieties of the ND-Tam molecules found at Sitein (Figure S2A). Sequence alignment of NavMs with human sensory neuron NaVs suggest that drug interactions are potentially conserved in domains two (DII), three (DIII) and four (DIV) (Figure 2B). After structural alignment of the crystalized NavMs-ND-Tam complex with the cryo-EM NaV1.7+β1+β2 channel coordinates (Shen et al., 2019), the side chain alpha carbon distances (Cα−Cα) of S969 (DII) and D1761 (DIV) observed in the human channel are found within 2 Å of D220 in the prokaryotic sodium channel subunits (Figure S2A). However, Cα−Cα between NavMs D220 and NaV1.7 D1458 (DIII) exceeds 5Å and the hydroxyl side chain of S979 (DII) is facing away from the ND-Tam amine indicating that the distance and proton donor orientation required for hydrogen bonding between the drug and channel is not optimal in this structural model (Figure S2A). Nonetheless, ND-Tam and 4OH-tam inhibits NaV1.7 INa with a steep concentration-dependence (Hill slope coefficient = 2.2 and 1.9, respectively), which suggests two drug binding sites within the channel. To determine if any of the proposed receptor sites in DII, DIII and DIV are responsible for ND-Tam NaV1.7 antagonism, we independently neutralized each of the residues with alanine substitutions. We observed reduced potency of INa inhibition (3–7 times) and a reduction in the slope for DII S969A and DIV D1761A sites, whereas no change in potency or slope was observed for D1458A in domain III (Figure 2D, Table S1). ND-Tam potency was further reduced (IC50 = 36 μM) by more than 21-times in S969A:D1761A double mutant channels compared to WT NaV1.7 (IC50 =1.7 μM). To evaluate the specificity of this result, we evaluated ND-Tam potency against local anesthetic binding site mutations (F1748A and Y1755A) located in the DIV S6 transmembrane pore fenestration (Figure 2B) and observed little change (Figure 2D, Table S1). These data suggest that the S6 NavMs-tamoxifen receptor site is conserved in DII and DIV of NaV1.7, which is located near the exit of the ion conducting pathway into the cell. The mechanism by which ND-Tam NaV occupancy results in INa inhibition is assessed in the following section.
Figure 2. The NaVMs tamoxifen receptor site is conserved in domain II and IV of human NaV1.7.
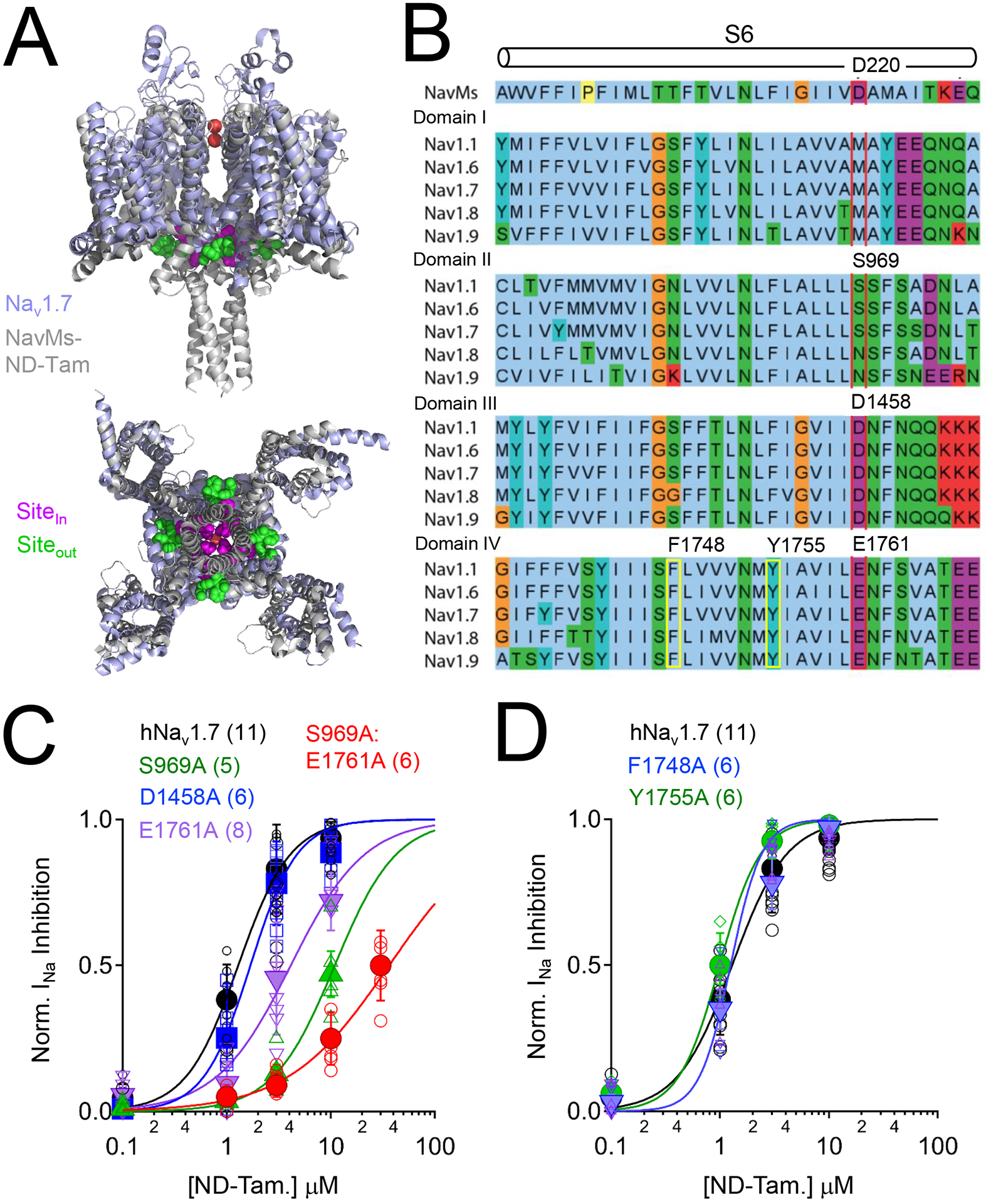
A) Structural alignment of human NaV1.7 (PDB: 6J8J) and the ND-Tam bound prokaryotic NavMs F208L channel (PDB: 6SXG)(Shen et al., 2019, Sula et al., 2021). The NavMs and NaV1.7 channel structures are reported to be in either pre-open or inactivated states. Inner (Sitein) and outer (Siteout) ND-Tam binding sites are indicated. B) Multiple sequence alignment of the sixth transmembrane segment (S6) from NavMs (UnitProt ID: A0L5S6) and each domain (I-IV) of DRG sodium channels NaV1.1 (Q99250), NaV1.6 (Q9UQD0); NaV1.7 (Q15858); NaV1.8 (Q9Y5Y9) and NaV1.9 (Q9UI33) using the standard Clustal color scheme by amino acid character. Proposed location of the ND-Tam receptor is boxed in red. The previously identified local antiesthetic/ anti-arrhythmic receptor site is boxed in yellow. C) Comparative ND-Tam potency for NaV1.7 channels expressing alanine substitutions at proposed receptors for tamoxifens, and D) local anesthetic receptor sites. Drug concentration- INa inhibition relationships for ND-Tam are fit to the Hill equation. Open symbols represent responses from individual cells and filled symbols represent average response per concentration. Error is equal to S.E.M. and the number of cells evaluated per treatment group is indicated within the parentheses.
Structure-activity relationship indicates ‘bind and plug’ model of NaV inhibition
To explore the molecular basis of NaV drug antagonism, we compared the direct binding of chemical ND-Tam analogs to NaV1.7 channel, and functional inhibition of sodium currents recorded from DRG neurons (Figure 3A). The parental molecular structure of ND-Tam can be separated into the receptor-binding N-methyl 2 phenoxyethanamine (NM2P) and triphenylene (TPE) fragments, which we call the ‘binder’ and ‘plug’, respectively. These fragments, along with 4OH-Tam and synthesized benzophenone analogs lacking the TPE fragment, 4-hydroxy-N-desmethyl benzophenone (4OH-ND-BP) and 4-hydroxy benzophenone (4OH-BP), were tested for affinity for the human NaV1.7 tamoxifen receptor in a competitive binding assay using a tritium labeled tamoxifen (H3-Tam). All analogs have affinity (Ki = 0.96–2.9 μM) with the channel, with exception of TPE (Figure 3B, Table S2). This was an expected result given that the hydrophobic plug does not have defined hydrogen-bond interactions within the tamoxifen drug bound NavMs channel structures (Figure 3A, Figure S2A). However, none of these compounds, except for 4OH-Tam and ND-Tam, inhibit DRG neuron INa (Figure 3B). This was an unexpected result given the established specific binding of NM2P, 4OH-ND-BP and 4OH-BP to NaV1.7 protein, and because the hydrogen bonds formed between the channel and analogs are expected to be preserved. These data suggest that NaV occupancy at the receptor site alone does not produce sodium current antagonism. By substituting the ND-Tam molecule found in DII and DIV of the NavMs-NaV1.7 aligned structures with the inert drug fragments, we observed that the TPE hydrophobic plug moiety of 4OH-Tam and ND-Tam molecules encroach 7Å into the diameter of the ion-conducting pathway (Figure S2B). By comparison, the remaining analogs (NM2P, 4OH-ND-BP and 4OH-BP) may occupy DII and DIV receptor sites but would leave the pore unobstructed in this model. Based on this limited SAR data set, we propose that the efficacy of the analogues is dependent on the combination of the ‘channel-binding’ (NM2P) and ‘hydrophobic plug’ (TPE) chemical moieties. Features that enhance ND-Tam potency for hyperexcitable sensory neuron sodium channels is explored in the next section.
Figure 3. Structure related activity of analogs at the NaV tamoxifen receptor, as assessed by competitive binding results and DRG sodium current inhibition.
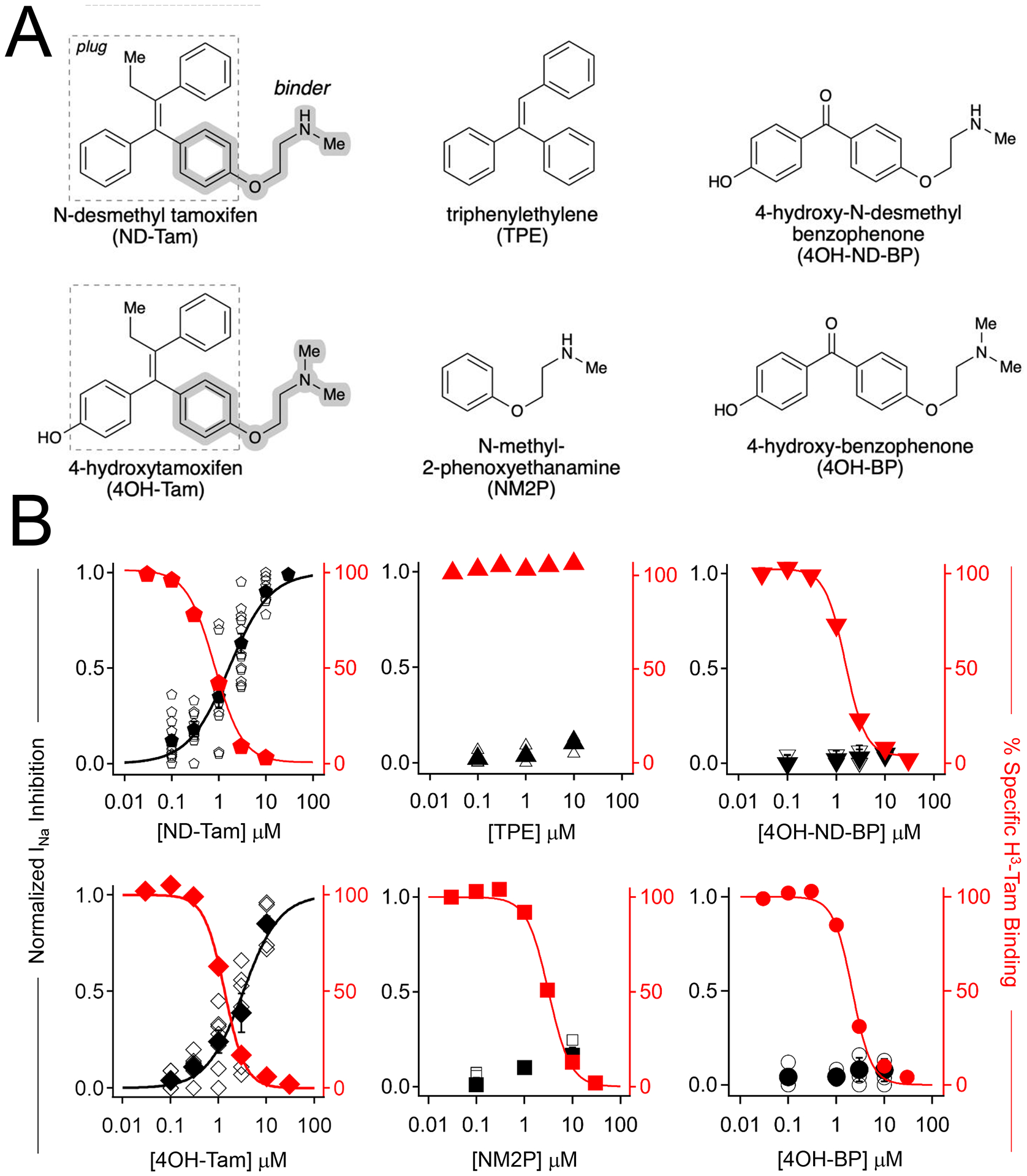
A) Molecular structures of the tested tamoxifen analogues. B) Normalized sodium current inhibition recorded DRG responses (black), and percent specific binding of tritium labeled tamoxifen (red) to membrane preparations expressing NaV1.7, plotted as a function of tamoxifen analog concentration. Open symbols are results recorded from individual neurons. Filled symbols represent the average response at each concentration. Percent specific binding was averaged from four trials. Error is equal to S.E.M.
NaV state-dependent inhibition in sensory neurons.
NaVs undergo changes in structural conformation when neuronal membranes are depolarized. Here, NaVs transition from the closed to open and inactivated states, which initiates and terminates the repetitive depolarizing peak waveforms (i.e., spiking behavior) observed in sensory neuron action potentials. State-dependent accessibility of receptor sites by analgesic and antiepileptic drugs provides a selective block of NaVs of hyperexcitable sensory neuronal circuits— a mechanism which is postulated to contribute to their clinical efficacy(Yang and Kuo, 2005, Castaneda-Castellanos et al., 2002). To test for state-dependent block of NaVs, we compared steady state voltage-dependent conductance and inactivation before after blocking ≈ 45% of the total INa with 1 μM ND-Tam. We observed no change in the half-maximal activation of conductance (GV1/2), which suggests that ND-Tam prevents DRG NaVs from conducting without altering activation (Figure 4A, B). However, we observed a significant (P = 0.015) shift in half-maximal voltage dependence of inactivation (Inact. V½) by −8 mV after ND-Tam treatment, suggesting that drug affinity may be enhanced in the NaV inactivated state. To evaluate the proposed mechanism of action, we assessed ND-Tam antagonism of DRG INa over more depolarized holding potentials (Figure 4C). We observed enhanced potency of inhibition (IC50 = 3.9 nM ± 0.7 and 112 nM ± 15) when −40 mV and −60 mV holding potentials were used, compared to the data sets collected with a −100 mV holding potential (IC50 =1.7 μM ± 0.2, Table S1). As previously reported, the primary mechanism of NavMs inhibition by ND-Tam is stabilization of the non-conducting inactivated channel state, where the recovery rate from this non-conducted state was delayed by ≈10x. However, when assessed against DRG neurons, ND-Tam treatment had nominal impact on the recovery time from inactivation (τrec. = 1.1 ± 0.1 ms) when compared to untreated neurons (τrec. = 1.4 ± 0.1 ms) (Figure 4D). For comparison of ND-Tam effects among clinically used compounds reported to have voltage-dependent shifts in potency against NaVs, we also tested carbamazepine (CBZ), lidocaine (Lid.), and cannabidiol (CBD) (Ghovanloo et al., 2018, Willow et al., 1985). Inhibition by CBD, CBZ and lidocaine was most potent when cell membranes were at held more depolarized potential, −60 mV (Figure S3, Table S1). However, none of these compounds were as potent as ND-Tam, which reached half-maximal inhibition at 112 nM using this holding potential (Figure S3, Table S1). Finally, we assessed the frequency dependence DRG INa inhibition by 300 nM ND-Tam at 0.1, 10 and 20 Hz (Figure S4A, B). When expressed as a fraction of the pulse number, we observed enhanced current inhibition at greater depolarization frequencies. Taken together, our findings indicate ND-Tam exhibits both voltage and frequency dependent blockade, suggesting that this drug preferentially binds to the open and inactivated NaV states. The implication of ND-Tam inhibition of sensory neuronal action potentials and in vivo efficacy are considered in the following sections.
Figure 4. The mechanism of N-desmethyl tamoxifen inhibition of sensory neuron sodium currents.
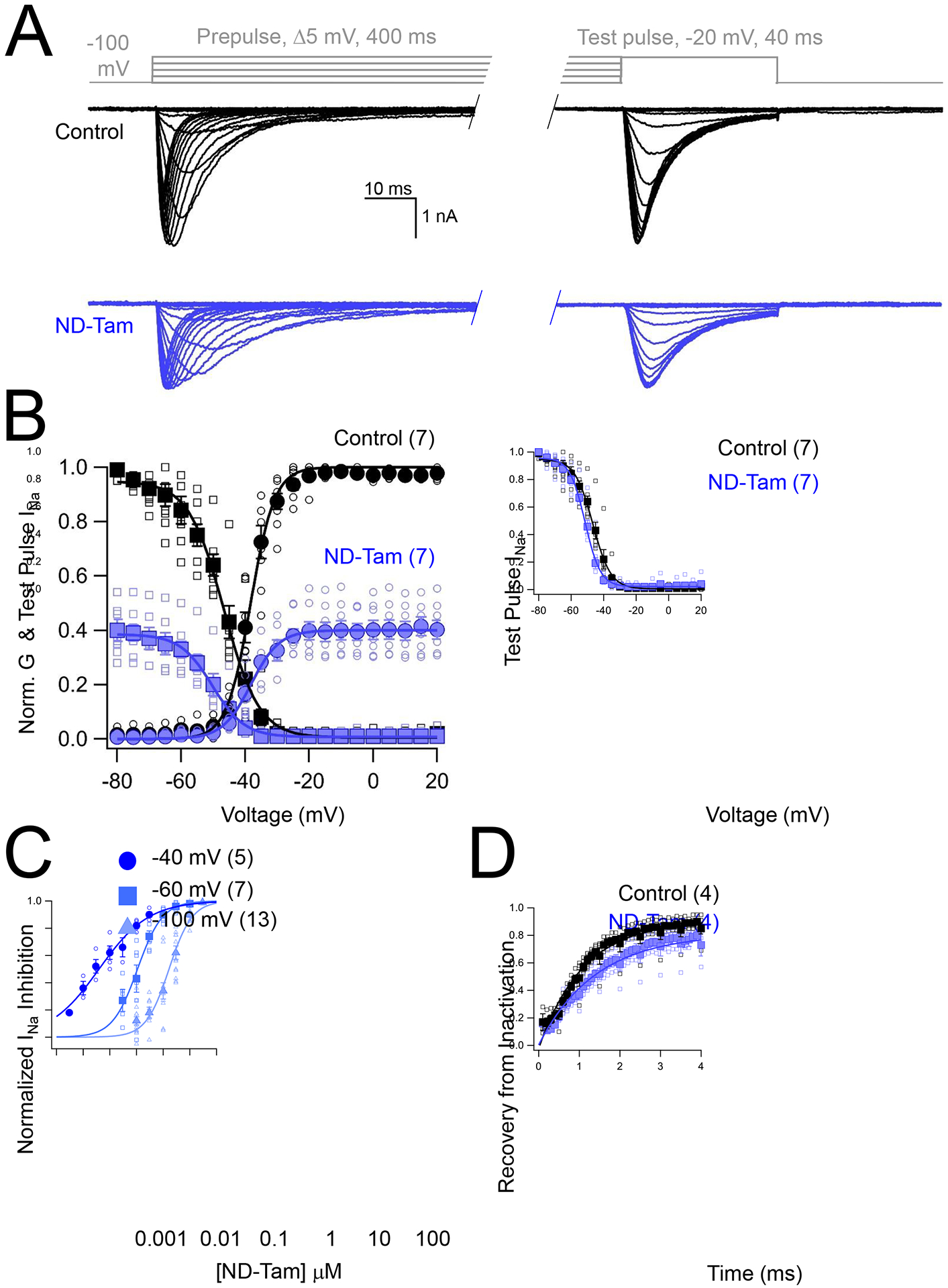
A) Voltage step protocol (gray) and resulting DRG INa traces before and after 1 μM ND-Tam application. B) The impact of ND-Tam on voltage-dependent sodium channel function. Left, conductance (G)-voltage and inactivation-voltage relationships were measured by plotting the average conductance and reduction of test pulse INa, respectively, as a function of pre-pulse potential. Both relationships were fit to a Boltzmann equation. Open symbols represent responses from individual cells and filled symbols represent average responses. The half-maximal conductance-voltage relationship was not different (P = 0.90) when measured in control saline (V1/2 = −38.3 ± 0.3 mV) compared to ND-Tam conditions (V1/2 = −37.7 ± 0.4 mV). Right, the test pulse current from control and drug conditions normalized to the same scale. The voltage-dependence of inactivation in control conditions and after drug treatment were equal to −47.6 ± 0.4 mV and −51.9 ± 0.4 mV, respectively, and were statistically different (P = 0.01), based on results from a paired two-tailed Student’s t-test. C) Voltage-dependent inhibition of DRG INa by ND-Tam. The potency of ND-Tam inhibition of INa was assessed using the voltage protocol described in Figure 1B, while using −40 mV, −60 mV and −100 mV holding potentials. The resulting drug concentration-INa relationships were fit to the Hill equation. The corresponding IC50 for each holding potential is listed in Supplemental Table 1 and the P-value from a two-tailed Student’s t-test comparing −100 mV to −60 mV and −40 mV results are indicated on the graph. Open symbols represent responses from individual cells and filled symbols represent average responses. D) DRG neuron INa recovery from inactivation (τrec.) before and after ND-Tam treatment. Sodium currents were inactivated by a 4 ms pre-pulse to 0 mV followed by an identical test pulse separated by increasing recovery times. The ratio of test pulse and pre-pulse current is plotted as a function of recovery time and fit to a single exponential equation. Recovery of INa from inactivation was delayed after 3 μM ND-Tam exposure (τrec. = 1.1 ± 0.1 ms) compared to control conditions (τrec. = 1.4 ± 0.1 ms) and was statistically significant (P = 0.01). Error is equal to S.E.M. and the number of cells evaluated per treatment group are indicated within the parentheses.
Inhibition of sensory neuron action potentials.
In response to painful stimuli, afferent DRG neurons fire action potentials which result from the opening and closing of ion channels, including NaVs For that reason, we compared the pharmacology of ND-Tam against cultured DRG action potentials recorded using current clamp (Figure 5A). We then compared DRG action potential frequency and amplitude before and after 2–4 minutes of extracellular drug treatment (Figure 5A–C, Table S3). ND-Tam was most potent against the firing frequency (IC50 = 119 nM ± 15) elicited by 80 pA of injected current, which recapitulated potency observed against DRG INa recorded with a −60 mV holding potential and the frequency dependence of drug block reported in our voltage clamp experiments (Figure 5, Figure S3, Figure S4). ND-Tam also inhibited the peak amplitude (IC50 = 0.7 μM ± 0.2) of the rapid depolarizing phase of the action potential carried by INa (Figure 4C). After exchanging the drug for control saline for 2 minutes, ND-Tam inhibition of action potential frequency and peak amplitude was persistent (Figure 5A), which is consistent with the observed slow dissociation of the drug reported in Figure 1D. Consistent with ND-tams frequency-dependent block reported in our voltage clamp experiments, ND-Tam was a more potent inhibitor of DRG action potential parameters than Lidocaine, CBD and CBZ (Figure 5D, E, Table S3). Approximately 10% (11/112) of our cultured neurons exhibited spontaneous action potentials when recorded under neutral current (i.e. no current injected). We excluded this data from the aforementioned analyses and tested a low dose (100 nM) of ND-Tam against these hyper excited neurons (Figure S5). Interestingly, a three-minute extracellular application of 100 nM ND-Tam reduced the frequency of the spontaneous sensory neuron action potentials by 91% (Figure S5A, B), but had insignificant impact on the resting membrane potential (Figure S5C). The drug vehicle (0.001% DMSO) did not reduce spike frequency (9.2%) and amplitude (7.8 %) over the same time course (N=3 neurons), suggesting action potential inhibition in the presence of ND-Tam cannot be attributed to “run down” phenomena or unspecific effects of DMSO. Importantly, action potentials in these neurons can triggered by 40 pA current injection indicating that the cells are still viable after drug treatment despite their electrical quiescence (Figure S5D). Our findings indicate that at low doses, ND-Tam is an effective inhibitor of aberrant firing of hyperexcitable neurons— a feature conferred by the drugs state-dependent block of sodium channels which manifest as voltage- and frequency-dependent inhibition of action potentials. In the next section, we evaluate ND-Tam efficacy in vivo using local administration using a mouse model of OA.
Figure 5. N-desmethyl tamoxifen inhibits DRG action potential amplitude and frequency.
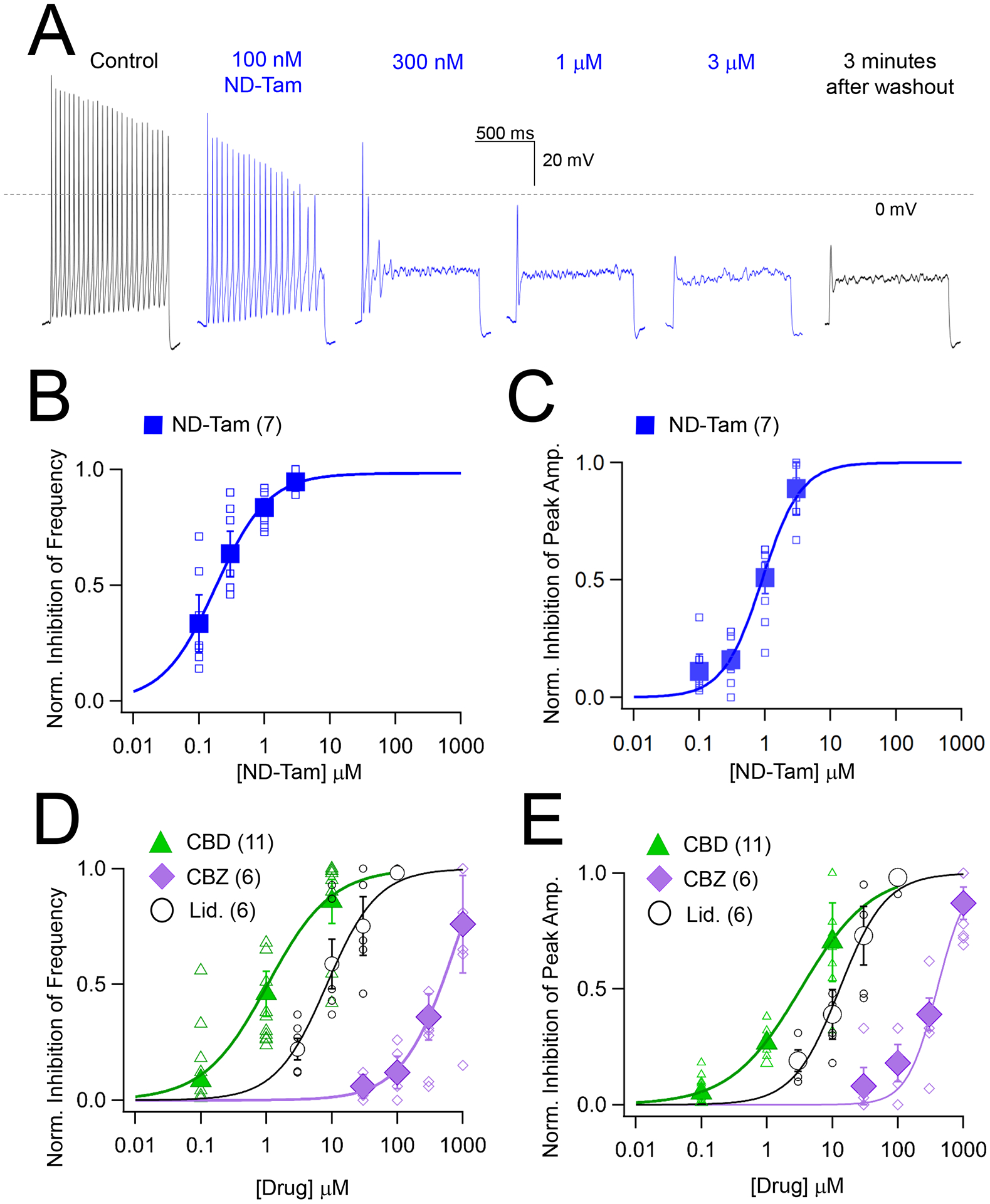
A) Exemplar action potentials recorded from an isolated DRG neuron in control and ND-Tam-treated conditions using a 1 second, 80 pA injection of current. After the experiment was complete, drug was washed out of the bath for five minutes and recovery from inhibition was retested. Consistent with previous reports, the resting membrane potential of the DRG neurons was −59.2 ± 5 mV (Error is equal to S.D., n = 109)(Wang et al., 1994). B, C) The potency of ND-Tam inhibition on DRG action potential frequency and peak amplitude. Open symbols represent responses from individual cells while filled symbols represent average response per concentration. Error is equal to S.E.M. and the number of cells evaluated per treatment group is indicated within the parentheses. D, E) A comparison of the potency of lidocaine (Lid.), cannabidiol (CBD) and carbamazepine (CBZ) inhibition on action potential frequency and peak amplitude. Drug concentration-INa inhibition relationships are fit to the Hill equation. The corresponding half-maximal inhibitory concentrations (IC50) for each drug are listed in Supplemental Table 3. Open symbols represent responses from individual cells and filled symbols represent average response per concentration. Error is equal to S.E.M. and the number of cells evaluated per treatment group are indicated within the parentheses.
Efficacy ameliorating arthritic pain in vivo.
Given the high potency NaV inhibition demonstrated in cultured DRG sensory neurons, we wanted to investigate whether ND-Tam had analgesic effects in vivo. To do this, we used an established osteoarthritis mouse model where destabilization of the medial meniscus (DMM) induces knee osteoarthritis and associated pain (Miller and Malfait, 2017). As previously shown, NaV1.8-tdTomato positive sensory neurons are present in the synovial joint as visualized by immunohistochemistry (Figure S6) (Ishihara et al., 2021, Obeidat et al., 2019). Previously, we demonstrated that remodeling of NaV1.8+ nerves in the knee joint is a key part of the pathogenesis process of osteoarthritis(Obeidat et al., 2019) and local injection of a relatively high dose of lidocaine (20 mg/kg, ~0.85M) was able to acutely reverse knee hyperalgesia(Miller et al., 2018). Here, we were interested to test whether ND-Tam could also inhibit knee pain, but at a lower dose. Local injection of 50 μM ND-Tam (~0.002 mg/kg) into the knee joint of mice 4 weeks after DMM surgery rapidly inhibited knee hyperalgesia pain compared to injection of vehicle (P = 0.008, 30 minutes after injection) (Figure 6A, Table S4A). Therefore, we were interested to compare the efficacy of this low dose of ND-Tam to similar low doses of CBD and lidocaine. In a direct comparison study, we found that local injection of 50 μM ND-Tam (~0.002 mg/kg), CBD (~0.002 mg/kg), or lidocaine (~0.001 mg/kg) all effectively inhibited knee hyperalgesia 30 minutes after injection (p = 0.002, p < 0.001, and p < 0.001 vs. vehicle, respectively) and remained elevated compared to vehicle control through 3 hours after injection (p = 0.02, p = 0.01, and p = 0.02 vs. vehicle, respectively) (Figure 6B, Table S4B). Area under the curve analysis of the first 3 hours post injection also demonstrated similar efficacy of all 3 drugs (ND-Tam, CBD, lidocaine) compared to vehicle (p < 0.0001, p = 0.0012, p < 0.0001 vs. vehicle, respectively) (Figure 6C, Table S4C). Finally, as a proof of concept of a longer-term local delivery approach, we injected a low dose (0.5 μM) of ND-Tam or lidocaine into the knee joints of mice 4 weeks after DMM surgery each day for 3 days. Following the third injection, mice treated with ND-Tam demonstrated two hours of analgesic relief of knee hyperalgesia compared to only one hour for lidocaine (Figure 6D, Table S4D). Area under the curve analysis of the first 3.5 hours post injection also demonstrated superior efficacy of ND-Tam compared to either lidocaine or vehicle (p = 0.0001, p < 0.0001) (Figure 6E, Table S4E). These results support the potential therapeutic development of ND-Tam for osteoarthritis pain relief.
Figure 6. ND-Tam produces analgesia in mice with knee hyperalgesia 4 weeks post DMM surgery.
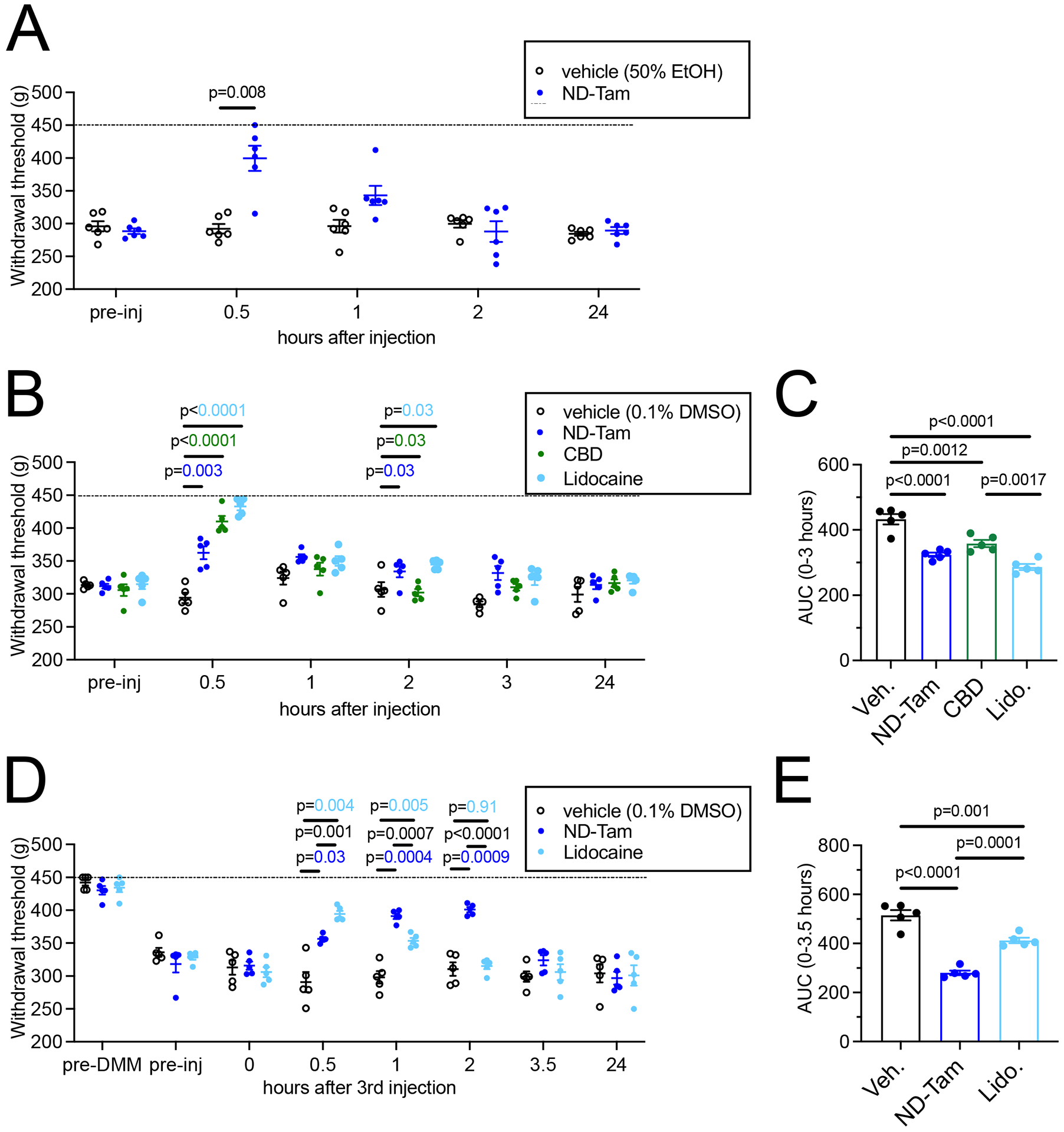
A) Knee withdrawal threshold was measured 4 weeks post DMM surgery and before intra-articular injection of vehicle (50% EtOH) or 50 μM ND-Tam and then at 30 min, 1h, 2h, and 24h post-injection. ND-Tam provided relief from hyperalgesia compared to vehicle 30 minutes after injection (n = 6, P = 0.008). B) Knee withdrawal thresholds assessed 4 weeks post DMM surgery, before and after intra-articular injection of vehicle (0.1% DMSO), 50 μM ND-Tam, 50 μM CBD, or 50 μM lidocaine at 30 min, 1h, 2h, 3h, and 24h time points. P-values are indicated on the graph for time points at which all three drugs are significantly different from vehicle. C) Area under the curve analysis of withdrawal thresholds for vehicle, ND-Tam, CBD, and Lidocaine from 0–3 hours after injection. ND-Tam performed similar to CBD and lidocaine over time (n = 5, P = 0.17, P = 0.13, respectively). D) Knee withdrawal thresholds assessed before DMM surgery (pre-DMM), and 4 weeks post DMM surgery, before (pre-inj) and after 3 days of intra-articular injection of vehicle (0.1% DMSO), 0.5 μM ND-Tam, or 0.5 μM Lidocaine at 0 (day 3 before final injection), 30 min, 1h, 2h, 3.5h, and 24h time points (on day 3 after the final injection). E) Area under the curve analysis of withdrawal thresholds for vehicle, ND-Tam, and Lidocaine from 0–3.5 hours after the 3rd injection. ND-Tam outperforms lidocaine after 3 days of repeated intra-articular injections (n = 5, P = 0.0001). A, B, D) Statistical analysis is stated in the associated data table, Supplemental Table 4; Dashed line indicates maximum of the assay = 450 g.
DISCUSSION
We have reported the pharmacology of tamoxifen analogs against sensory neuron NaVs, both in terms of their molecular receptor interactions and drug chemical properties required for their efficacy. We have focused on the properties of ND-Tam— a first pass metabolic product of tamoxifen that has nominal efficacy against the estrogen receptor. Through direct binding studies, structural alignments, and functional data sets, we establish that ND-Tam antagonizes sodium current by directly blocking the intracellular pore of NaV1.7 at a previously uncharacterized receptor site within domains II and IV. In DRG sensory nerves, ND-Tam is a more potent NaV antagonist of sodium currents and neuronal action potential firing than carbamazepine, lidocaine and cannabidiol— three clinically used drugs. ND-Tam preferentially inhibits sodium channels in the open/inactivated state, a feature observed in anesthetic and anti-epilepsy medications. This property confers voltage- and frequency-dependent drug inhibition of NaVs, which preferentially blocks INa from damaged and/or hyperexcitable neurons, as supported by our DRG current clamp results. We demonstrate ND-Tam efficacy in ameliorating pain in a rodent osteoarthritis arthritis model when injected into the knee joint, supporting its potential for further development and the possibility of repurposing tamoxifen analogues for the treatment of OA associated pain.
In this study, we analyzed existing sodium channel structure coordinates to facilitate our identification of the tamoxifen receptors in DII and DIV of NaV1.7. Clearly, solving high-resolution structures of sensory neuron NaVs (NaV1.7, NaV1.8 and NaV1.9) bound to ND-Tam would further elucidate the chemical interactions and space governing drug-receptor affinity. In addition, future work should determine if drug potency and NaV subtype specificity can be optimized through in silico structure-based design of small molecule ligands for tamoxifen receptor sites. This effort will be challenging given the high degree of conservation of these receptor sites across NaVs. Results from our small-scale SAR study comparing binding and electrophysiology data sets establish critical aspects of NaV-tamoxifen molecular pharmacology. We propose drug efficacy against NaVs is tied to the union of two chemical properties— the affinity of the receptor-binding NM2P moiety and the hydrophobic bulk of TPE pore blocker. A larger analog library could be developed to include greater diversity of ‘binder’ and pore ‘plug’ chemistries, which when tested would elucidate the full potential of this drug-receptor interaction.
To our knowledge, clinical efficacy of tamoxifen analogues for OA pain treatment is untested. Despite the clear need, the time and cost of developing new analgesics is escalating (Pammolli et al., 2011, Chaplan et al., 2010). Currently, bringing a new drug to market requires an average of ten years to develop, and typically costs more than three billion dollars. High attrition rates among new drug candidates and changing regulatory requirements contributes to this trend (Waring et al., 2015). Drug repurposing is the secondary use of already developed drugs for therapeutic uses that are different from those for which they were initially designed. Drug repurposing has advantages over conventional drug discovery approaches, including bypassing Phase 1 clinical trials. Annually, more than 100 million people worldwide are prescribed tamoxifen for the treatment of breast cancer. Orally administered tamoxifen is transformed into ND-Tam and 4OH-Tam by cytochrome P450 enzymes in the liver and its systemic drug safety, pharmacokinetics and pharmacodynamics is reported by several decades of use within a large, heterogenic human population (Crewe et al., 2002, Jordan, 2007). Accordingly, the existing clinical data significantly de-risks tamoxifen metabolites with potentially lower overall development costs. Thus, there are clear benefits to repurposing and developing tamoxifen analogues as prototypic NaV antagonist for the treatment of OA pain.
Limitations of the study.
While our results are encouraging, we must acknowledge several limitations in the current study. Although there is strong agreement between our in vitro data (direct binding affinity, potency of INa and action potential firing inhibition) and in vivo OA pain mouse model results, it is difficult determine if ND-Tam analgesic effect is exclusively due to NaV blockade. Indeed, other targets could be involved. Another limitation is set by ND-Tam limited specificity against Nav subtypes— as our results indicate potency is equal among those found in DRG sensory neurons and possibly conserved in other NaVs. Human genetic variants and mouse model studies have implicated NaV1.7, NaV1.8, and NaV1.9 as specific determinants in pain signaling (Dib-Hajj et al., 2005, Faber et al., 2012, Yeomans et al., 2005, Gingras et al., 2014, Nassar et al., 2005). These findings, coupled with known toxicity related to systemic Nav antagonism (i.e., CNS Nav1.2 and cardiac Nav1.5), has set the premise that nociceptive NaV subtypes should be specifically targeted to treat chronic pain. However, despite considerable investment, a subtype-specific drug candidate has yet to emerge. Even without achieving NaV subtype specificity, prototypic tamoxifen analogues may still have therapeutic utility through local drug application. In this study, we evaluated local injection of ND-Tam into the knee synovial capsule, which limits the concentration of ND-Tam systemic exposure and delivers drug directly to the site of injury in OA. This route of administration is analogous to intra-articular therapies used in the clinic (Gerwin et al., 2006). The rate of synovial fluid lymphatic drainage largely depends on the size of the molecule, where drug efficacy is reported 1–32 hours post injection (Gerwin et al., 2006, Habib, 2009). Yet, the rate of ND-Tam drainage from the knee joint is not known. Future work should address this, and safety risks related to non-specific Nav antagonism by ND-Tam drainage from the joint. Our results indicate ND-Tam was more efficacious than lidocaine hours after repeated intra-articular injections, even when using 10,000x (20 mg/kg vs 0.002 mg/kg) lower dose. Further development will focus on developing a slow-release formulation of ND-Tam to extend the duration of effect (Janssen et al., 2016). The effectiveness of this strategy has been demonstrated in a phase 3 trial, where intra-articular injection of a glucocorticoid formulated with microspheres reduced pain 12 weeks post injection (Conaghan et al., 2018). Beyond OA, musculoskeletal pain is frequently highlighted as an unmet medical need and is a leading cause of global burden of disease (Disease et al., 2018). Thus, for diseases like osteoarthritis with peripheral sources of joint pain, targeting the tamoxifen receptor of NaVs might be therapeutically useful.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Paul DeCaen (paul.decaen@northwestern.edu)
Materials Availability
All unique reagents generated in this study are available by request to the lead contact. Expression plasmids will be made available through addgene.com or upon request from the DeCaen lab within 3 months of publication. The synthesized tamoxifen analogs described in this study, including H3-Tam, are available by request pending the compound is in supply.
Data and Code Availability
Imaging, in vivo and electrophysiology data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MDA-MB-231 cells, HEK-293 cells, Parental CHO-K1 and CHO-K1 cell lines expressing NaV beta subunits.
Primary cell cultures
Primary cultured dorsal root ganglion neurons were isolated from 4–6-week-old male C57/B6 mice.
Animal models
Knee sections from 2–4 month old NaV1.8-tdTomato, C57BL/6 mice were used for immunohistochemistry experiments. 10-week old male C57BL/6 mice were used in the DMM pain model.
Animals were housed with food and water ad libitum and kept on 12-h light cycles. Animal procedures were approved by either the Institutional Animal Care and Use Committee at the Feinberg Medical School, Northwestern University or Rush University Medical Center.
METHOD DETAILS
Isolation and primary culture of murine dorsal root ganglia sensory neurons.
Dorsal root ganglia were isolated using a modified version of the previously described protocol. The spinal columns of 4–6-week-old C57/B6 or NaV1.8-Tomato mice (NaV1.8-Td)(Gautron et al., 2011) were removed and DRG neurons from all spinal levels were isolated for primary culture(Ishihara et al., 2021). The collected DRGs, containing both neuronal and non-neuronal cells, were acutely dissociated in DMEM with collagenase IV (2 mg/mL) for 30 minutes, followed by papain (25 U/mL) for 30 minutes. The cells were then triturated, filtered through a 40 μm cell strainer to remove non-dissociated cells, and washed with DMEM. The DRGs were resuspended in 70 μL of pre-warmed growth medium (DMEM F12 + Glutamax supplemented with 0.5% FBS, 0.5% penicillin/streptomycin, 1% N-2 supplement, and β-NGF (5 ng/μL) added on the day of use) and plated on poly-l-lysine (10 mg/mL) and laminin (25 μg/mL) prepared coverslips. The DRGs were incubated at 37°C for 2–4 hours before more growth medium was added to the wells. The cells were then allowed to adhere to the coverslip undisturbed at 37°C for 12–48 hours, until used for electrophysiology recordings.
Electrophysiology of endogenous and heterologously expressed sodium channels.
Plasmids encoding the human version of channels NaV1.1, 1.6, and 1.7 were co-transfected with IRES GFP into HEK cell lines for whole-cell voltage clamp studies (American Type Culture Collection). Transient transfections were performed using lipofectamine 24–48 hours prior to electrophysiology recordings. Chinese hamster ovary (CHO) cells stably expressing human β1 and β2, were transiently transfected with plasmids encoding for either human NaV1.8 or NaV 1.9 plasmids alpha subunits were used to record sodium currents from these channel subtypes. Plasma membrane currents were recorded using borosilicate glass electrodes polished to resistances of 2–4 mΩ. Sodium currents conducted by heterologously expressed NaVs were recorded using the following solutions (in mM). The internal (pipette) solution contained 110 CsF, 30 NaCl, 10 HEPES, and 5 mM EGTA (ethylene glycol- aminoethyl ether-N, N, N0, N0-tetraacetic acid) and the pH was adjusted to 7.3 using CsOH. The extracellular (bath) solution contained 150 NaCl, 10 HEPES, 1.8 CaCl2, and pH was adjusted to 7.4 using NaOH. The osmolality of these solutions was adjusted to 300 mOsm using mannitol. Endogenous DRG sodium currents were recorded using an intracellular solution consisting of 70 CsCl, 70 CsF, 2 EGTA, 5 HEPES, and 5 NaCl; the pH was adjusted to 7.4 with CsOH. The extracellular solution contained 125 NaCl, 25 glucose, 20 TEA-Cl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, and 5 CsCl; the pH was adjusted to 7.4 with TEA-OH. DRG action potentials were measured in current clamp mode using an intracellular solution containing 140 KCl, 10 HEPES, 5 MgCl2, 5 EGTA, 2.5 CaCl2, 4 MgATP, 0.3 GTP, and the pH adjusted to 7.3 using KOH. The extracellular solution contained 140 NaCl, 10 HEPES, 10 glucose, 5.3 KCl, 1 MgCl2, 1.8 mM CaCl2 and the pH adjusted to 7.3 using NaOH. For both voltage and current clamp recordings in DRGs, the intracellular solution osmolality was 280 mOsm and the extracellular solution was adjusted to 325 mOsm with mannitol. Voltage and current clamp data were collected using Multiclamp and Axopatch 200B amplifiers supplied by Molecular Devices. Analog signals were converted to digital signals using a Digidata 1550B and controlled using pClamp 10 software. Currents were digitized at 25 kHz and low pass filtered at 5 kHz. All drug stocks were formulated in DMSO at 10 or 100 mM and stored at −20C° until the day of use. All drug stocks were then diluted into extracellular saline solutions. Drugs were applied using a gravity fed extracellular bath perfusion system with a flow rate of 5–10 ml/minute through a 0.4 ml volume recording chamber.
Current clamp and voltage clamp data were analyzed using Clampfit (Molecular Devices) and IGOR Pro 8.1 (Wavemetrics). Leak current was subtracted using a standard P/−4 protocol. Data from cells whose leak current exceeded −150 pA at −100 mV or whose voltage error exceeded 10 mV were excluded from the final analysis. Series resistance (Rs) was compensated by at least 80% to limit Rs related error to < 3 mV. Rs was monitored periodically throughout the experiment to check for shifts in voltage error. Normalized INa inhibition was determined by taking the ratio of the current at steady state drug block (Idrug) and control current (Icontrol) and expressed as: Normalized INa inhibition = 1 - (Idrug/Icontrol). Normalized INa inhibition was plotted as a function of drug concentration, and the data were fit with the Hill equation: y = base + (max − base) / [1+(IC50/x)^rate], where base and max describe the lower and higher asymptotes, respectively; x is the drug concentration and IC50 is the drug concentration that produces a 50% maximal inhibitory response, and rate is the Hill coefficient. Percent current recovery was calculated by (Irecovery − Idrug) × 100, where Irecovery is the recovered current measured 3–5 min after drug removal. Normalized pre-pulse INa from Figure 3 was converted to normalized conductance (G) using the following equation: G = INa/(Vm-Vrev), where Vm and Vrev is equal to voltage applied across the membrane and reversal potential, respectively. The steady state voltage dependence of activation and inactivation was fit to Boltzmann equation: G/Gmax or I/Imax = 1/(1 + exp((V1/2 − V)/k)), where V1/2 is equal to the half maximal activation (Act. V1/2) or inactivation (Inact. V1/2), and K is equal to the slope. Recovery from inactivation was estimated by fitting the Ipre-pulse/Itest-pulse to the exponential equation: f(x) = B + A•exp[(1/τinact.)x], where x is the time between the pre- and test pulse and τinact. is the half time of total current recovery from inactivation. Gibbs law of free energy was used to calculate the free energy of drug binding: ΔG = −R·T·Ln(Kd), where R = 0.008314 kJ/mol, temperature T = 297 K, and Kd is the apparent association constant estimated by the IC50.
NaV1.7 tritium-labeled tamoxifen competitive binding assay
Membrane from MDA-MB-231 cells, which are the triple negative (estrogen receptor negative) and infected with lenti virus encoding for human NaV1.7 were prepared as follows. Cells were grown to 60–80% confluent and harvested with PBS-based, enzyme-free cell dissociation buffer containing EGTA. Cells were then centrifuged at (14000×g, 4 °C) for 15 min. and homogenized (Tekmar Tissuemizer). The cell homogenate was centrifuged (2000×g, 4 °C) for 10 min. Membrane pellets were suspended in 10 ml of PBS/gram and stored at −80 °C. Total protein concentration was using the Coomassie (Bradford) method. H3-Tamoxifen was synthesized by Tritech AG (Switzerland). On the day of the experiment, suspended membranes expressing NaV1.7 (final protein concentration = 30 μg/well) were added to each of a 96-well plate with 3H-Tam at 30 μM. The plates were incubated at 37 C for 1 hour, aspirated onto filter plates, and rinsed with wash buffer. After addition scintillant (Packard Microscint-20), radioactivity was quantified (Packard Topcount Scintillation Counter). Counts per minute data from binding experiments were converted to percent total specific bound (%TSB) using the following formula: %TSB=[(cpm NSB)/(TB NSB)]100, where TB NSB is the total non-specific binding. Ki values were derived by means of the Cheng and Prusoff equation (Cheng and Prusoff, 1973) Ki = IC50/ 1+[analog]/Kd, using Kd values for [3H-Tam] obtained from saturation assays.
Synthesis and procurement of NaV antagonists.
N-desmethyl tamoxifen, 4-hydroxytamoxifen, cannabidiol, carbamazepine, N-methyl 2 phenoxyethanamine and triphenylene and were purchased from Sigma Aldrich. The benzophenone 4-hydroxy-tamoxifen analogs, 4-hydroxy-N-desmethyl benzophenone (4OH-ND-BP) and 4-hydroxy benzophenone (4OH-BP) were prepared by the Scheidt Lab according to literature procedures and all characterization data matched the literature values.(Palermo et al., 2018, Fauq et al., 2010)
In vivo osteoarthritis pain model.
We used a total of n = 47 male C57BL/6 mice. Surgical destabilization of the medial meniscus (DMM) was performed in the right knee of 10-week-old male mice, as previously described(Glasson et al., 2007) (Miller et al., 2012). In experiment 1, a volume of 3 μL of 50 μM ND-Tam formulated in 50% EtOH or vehicle (50% EtOH) was intra-articularly injected into the right knee of mice 4–5 weeks after DMM surgery under isoflurane anesthesia (n = 6 mice/group). Knee hyperalgesia measurement was performed before injection and at 30 mins, 1h, 2h, and 24h time points with a pressure application measurement device (PAM device, Ugo Basile) with the experimenter blinded to the treatment group, as previously described(Miller et al., 2018). In experiment 2, a volume of 3 μL of 50 μM ND-Tam, CBD, or lidocaine formulated in 0.1% DMSO or vehicle (0.1% DMSO) was intra-articularly injected into the right knee of mice 4 weeks after DMM surgery under isoflurane anesthesia (n = 5 mice/group). Knee hyperalgesia measurement was performed before injection and at 30 mins, 1h, 2h, 3h, and 24h time points with a pressure application measurement device (PAM device, Ugo Basile) with the experimenter blinded to the treatment group, as previously described(Miller et al., 2018). In experiment 3, a volume of 3 μL of 0.5 μM ND-Tam or lidocaine formulated in 0.1% DMSO or vehicle (0.1% DMSO) was intra-articularly injected into the right knee of mice each day for 3 consecutive days, 4 weeks after DMM surgery under isoflurane anesthesia (n = 5 mice/group). Knee hyperalgesia measurement was performed before surgery, 4 weeks after surgery before injections were begun, before the third injection, and at 30 mins, 1h, 2h, 3.5h, and 24h post the third injection with a pressure application measurement device (PAM device, Ugo Basile) with the experimenter blinded to the treatment group, as previously described(Miller et al., 2018).
Immunolabeling of neurons in murine knee synovial joints.
NaV1.8-tdTomato mouse knee joints were fixed in 4% paraformaldehyde, decalcified in 10% ethylenediamine tetraacetic acid (EDTA) for 2 weeks, rinsed in PBS and immersed for 72 h in 30% sucrose. The tissue was embedded in OCT compound and sections cut to 20 μm widths. For Tissue sections were rinsed in PBS and incubated for 4 hours at room temperature in primary antibody. Following PBS rinses, the slides were incubated for 2 hours at room temperature in secondary antibody. The slides were rinsed in PBS and treated with Prolong Gold anti-fade reagent. The following dilution of antibodies and were used: anti-sodium channel antibody 1:500x (Sigma, S8809; anti-NeuN antibody 1:1000x (Proteintech, 26975-1-AP); RFP-Booster 1:2000x (Chromotek, rb2AF568-50); anti-ChAT antibody 1:500x (Invitrogen, PA5-29653); DAPI (Invitrogen, D1306) and cell membrane stain 1:1000x (Invitrogen, C10607). The images were obtained using the Nikon A1 confocal microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical methods used to determine significance are described in corresponding figure and table legends. Briefly, electrophysiology data sets were analyzed (GraphPad or Origen) using two tailed paired (equal sample sizes) or unpaired (unequal sample sizes) Student’s t-tests. In vivo data was analyzed using two-way repeated measures ANOVA (Figure 6A, B, D); and one-way ANOVA were performed on each dataset (Figure 6C, E).
Supplementary Material
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-sodium channel antibody | Sigma Aldrich | Cat# S8809 |
| anti-NeuN antibody | ProtienTech | Cat# 26975-1-AP |
| RFP-Booster | Chromotek | Cat# rb2AF568-50 |
| anti-ChAT antibody | Invitrogen | Cat# PA5-29653 |
| Virus strains | ||
| Lentivirus pLVX, human sodium channel beta subunit 1 | Vector Builder, custom | Uniprot ID Q07699 |
| Chemicals | ||
| H3-Tamoxifen | Tritech AG (Switzerland) | CAS# 10540-29-1 |
| 4-hydroxytamoxifen | Sigma Aldrich | Cat# T176 |
| Tamoxifen | Sigma Aldrich | Cat# T5648 |
| Triphenylene | Sigma Aldrich | Cat# T82600 |
| Cannabidiol | Sigma Aldrich | Cat# C7515 |
| Carbamazepine | Sigma Aldrich | Cat# C4024 |
| 4-hydroxy-N-desmethyl benzophenone (4OH-ND-BP) | Scheidt Lab | CAS# 110025-28-0 |
| 4-hydroxy benzophenone (4OH-BP) | Scheidt Lab | CAS# 1137-42-4 |
| Experimental models: Cell lines | ||
| HEK-293 cells | American Type Culture Collection | Cat# CRL-1573 |
| CHO-K1 cells | American Type Culture Collection | Cat# CCL-61 |
| MDA-MB-231 cells, triple negative ER receptor | American Type Culture Collection | Cat# HTB-26 |
| Experimental models: Organisms/strains | ||
| C57/B6 mice | Charles River | Cat# eC57BL/6NCrl |
| C57/B6 mice Nav1.8 td-tomato | John N. Wood University College London, London |
NaV1.8-tdTomato, Gautron et al. |
| Recombinant DNA | ||
| Human NaV1.1, ptracer IRES GFP | Vector Builder | Uniprot ID Q99250 |
| Human NaV1.6, ptracer IRES GFP | Vector Builder | Uniprot ID Q9UQD0 |
| Human NaV1.7, ptracer IRES GFP | Vector Builder | Uniprot ID Q15858 |
| Human NaV1.8, ptracer IRES GFP | Vector Builder | Uniprot ID Q9Y5Y9 |
| Human NaV1.9, ptracer IRES GFP | Vector Builder | Uniprot ID Q9UI33 |
| Software and algorithms | ||
| pCLAMP 11 software, electrophysiology acquisition | Molecular Devices | Version 11 |
| ClampFit software, electrophysiology analysis | Molecular Devices | Version 11 |
| IGOR pro 8.1 software, electrophysiology analysis | Wavemetrics | Version 8.1 |
| OrigenPro, statistical analysis | OrigenLab | 2022b |
| Prism, statistical analysis | GraphPad | Version 9 |
ACKNOWLEDGMENTS
We thank Dr. Christopher H. Thompson and Dr. Al George for their advice analyzing current clamp data sets. We thank Dongjun Ren from the laboratory of Dr. Daniela Menichella for demonstrating the DRG neuron isolation protocol. We thank all members of the DeCaen lab for their useful scientific discussions. The authors acknowledge their respective funding agencies: PGD was supported by NIH NIDDK (1R01 DK123463-01). ML was supported by NU’s Molecular Biophysics Training Program through NIH NIGMS (5T32 GM008382). REM group was supported by NIH NIAMS (R01 AR077019). REM was supported by NIH grants R01AR077019 and P30AR079206. KAS was supported by Northwestern University and LFK was supported by a fellowship from the Sao Paulo Research Foundation (FAPESP - 2019/26414-2).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing or financial interests.
REFERENCES
- AKOPIAN AN, SIVILOTTI L & WOOD JN 1996. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature, 379, 257–62. [DOI] [PubMed] [Google Scholar]
- ALLEN KD, THOMA LM & GOLIGHTLY YM 2022. Epidemiology of osteoarthritis. Osteoarthritis Cartilage, 30, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAGNERIS C, DECAEN PG, NAYLOR CE, PRYDE DC, NOBELI I, CLAPHAM DE & WALLACE BA 2014. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc Natl Acad Sci U S A, 111, 8428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASTANEDA-CASTELLANOS DR, NIKONOROV I, KALLEN RG & RECIO-PINTO E 2002. Lidocaine stabilizes the open state of CNS voltage-dependent sodium channels. Brain Res Mol Brain Res, 99, 102–13. [DOI] [PubMed] [Google Scholar]
- CATTERALL WA 2012. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol, 590, 2577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAPLAN SR, ECKERT IW & CARRUTHERS NI 2010. Drug Discovery and Development for Pain. In: KRUGER L & LIGHT AR (eds.) Translational Pain Research: From Mouse to Man. Boca Raton (FL). [Google Scholar]
- CHENG Y & PRUSOFF WH 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol, 22, 3099–108. [DOI] [PubMed] [Google Scholar]
- CONAGHAN PG, HUNTER DJ, COHEN SB, KRAUS VB, BERENBAUM F, LIEBERMAN JR, JONES DG, SPITZER AI, JEVSEVAR DS, KATZ NP, BURGESS DJ, LUFKIN J, JOHNSON JR, BODICK N & INVESTIGATORS, F. X. P. 2018. Effects of a Single Intra-Articular Injection of a Microsphere Formulation of Triamcinolone Acetonide on Knee Osteoarthritis Pain: A Double-Blinded, Randomized, Placebo-Controlled, Multinational Study. J Bone Joint Surg Am, 100, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CREWE HK, NOTLEY LM, WUNSCH RM, LENNARD MS & GILLAM EM 2002. Metabolism of tamoxifen by recombinant human cytochrome P450 enzymes: formation of the 4-hydroxy, 4’-hydroxy and N-desmethyl metabolites and isomerization of trans-4-hydroxytamoxifen. Drug Metab Dispos, 30, 869–74. [DOI] [PubMed] [Google Scholar]
- DIB-HAJJ SD, RUSH AM, CUMMINS TR, HISAMA FM, NOVELLA S, TYRRELL L, MARSHALL L & WAXMAN SG 2005. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain, 128, 1847–54. [DOI] [PubMed] [Google Scholar]
- DIB-HAJJ SD, TYRRELL L, BLACK JA & WAXMAN SG 1998. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci U S A, 95, 8963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DISEASE, G. B. D., INJURY, I. & PREVALENCE, C. 2018. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet, 392, 1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMERY EC, LUIZ AP & WOOD JN 2016. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets, 20, 975–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FABER CG, LAURIA G, MERKIES IS, CHENG X, HAN C, AHN HS, PERSSON AK, HOEIJMAKERS JG, GERRITS MM, PIERRO T, LOMBARDI R, KAPETIS D, DIB-HAJJ SD & WAXMAN SG 2012. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A, 109, 19444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAUQ AH, MAHARVI GM & SINHA D 2010. A convenient synthesis of (Z)-4-hydroxy-N-desmethyltamoxifen (endoxifen). Bioorganic & Medicinal Chemistry Letters, 20, 3036–3038. [DOI] [PubMed] [Google Scholar]
- GAO S & YAN N 2021. Structural Basis of the Modulation of the Voltage-Gated Calcium Ion Channel Cav 1.1 by Dihydropyridine Compounds*. Angew Chem Int Ed Engl, 60, 3131–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUTRON L, SAKATA I, UDIT S, ZIGMAN JM, WOOD JN & ELMQUIST JK 2011. Genetic tracing of Nav1.8-expressing vagal afferents in the mouse. J Comp Neurol, 519, 3085–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERWIN N, HOPS C & LUCKE A 2006. Intraarticular drug delivery in osteoarthritis. Adv Drug Deliv Rev, 58, 226–42. [DOI] [PubMed] [Google Scholar]
- GHOVANLOO MR, SHUART NG, MEZEYOVA J, DEAN RA, RUBEN PC & GOODCHILD SJ 2018. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem, 293, 16546–16558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GINGRAS J, SMITH S, MATSON DJ, JOHNSON D, NYE K, COUTURE L, FERIC E, YIN R, MOYER BD, PETERSON ML, ROTTMAN JB, BEILER RJ, MALMBERG AB & MCDONOUGH SI 2014. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One, 9, e105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLASSON SS, BLANCHET TJ & MORRIS EA 2007. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage, 15, 1061–9. [DOI] [PubMed] [Google Scholar]
- GOLDIN AL 2001. Resurgence of sodium channel research. Annu Rev Physiol, 63, 871–94. [DOI] [PubMed] [Google Scholar]
- HABIB GS 2009. Systemic effects of intra-articular corticosteroids. Clin Rheumatol, 28, 749–56. [DOI] [PubMed] [Google Scholar]
- ISHIHARA S, OBEIDAT AM, WOKOSIN DL, REN D, MILLER RJ, MALFAIT AM & MILLER RE 2021. The role of intra-articular neuronal CCR2 receptors in knee joint pain associated with experimental osteoarthritis in mice. Arthritis Res Ther, 23, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANSSEN M, TIMUR UT, WOIKE N, WELTING TJ, DRAAISMA G, GIJBELS M, VAN RHIJN LW, MIHOV G, THIES J & EMANS PJ 2016. Celecoxib-loaded PEA microspheres as an auto regulatory drug-delivery system after intra-articular injection. J Control Release, 244, 30–40. [DOI] [PubMed] [Google Scholar]
- JIANG D, SHI H, TONGGU L, GAMAL EL-DIN TM, LENAEUS MJ, ZHAO Y, YOSHIOKA C, ZHENG N & CATTERALL WA 2020. Structure of the Cardiac Sodium Channel. Cell, 180, 122–134 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG D, TONGGU L, GAMAL EL-DIN TM, BANH R, POMES R, ZHENG N & CATTERALL WA 2021. Structural basis for voltage-sensor trapping of the cardiac sodium channel by a deathstalker scorpion toxin. Nat Commun, 12, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES IA, TOGASHI R, WILSON ML, HECKMANN N & VANGSNESS CT JR. 2019. Intra-articular treatment options for knee osteoarthritis. Nat Rev Rheumatol, 15, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORDAN VC 2007. New insights into the metabolism of tamoxifen and its role in the treatment and prevention of breast cancer. Steroids, 72, 829–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATZENELLENBOGEN BS, NORMAN MJ, ECKERT RL, PELTZ SW & MANGEL WF 1984. Bioactivities, estrogen receptor interactions, and plasminogen activator-inducing activities of tamoxifen and hydroxy-tamoxifen isomers in MCF-7 human breast cancer cells. Cancer Res, 44, 112–9. [PubMed] [Google Scholar]
- KOLASINSKI SL, NEOGI T, HOCHBERG MC, OATIS C, GUYATT G, BLOCK J, CALLAHAN L, COPENHAVER C, DODGE C, FELSON D, GELLAR K, HARVEY WF, HAWKER G, HERZIG E, KWOH CK, NELSON AE, SAMUELS J, SCANZELLO C, WHITE D, WISE B, ALTMAN RD, DIRENZO D, FONTANAROSA J, GIRADI G, ISHIMORI M, MISRA D, SHAH AA, SHMAGEL AK, THOMA LM, TURGUNBAEV M, TURNER AS & RESTON J 2020. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Care Res (Hoboken), 72, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN Z, SANTOS S, PADILLA K, PRINTZENHOFF D & CASTLE NA 2016. Biophysical and Pharmacological Characterization of Nav1.9 Voltage Dependent Sodium Channels Stably Expressed in HEK-293 Cells. PLoS One, 11, e0161450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAYS TA 2001. Antidepressants in the management of cancer pain. Curr Pain Headache Rep, 5, 227–36. [DOI] [PubMed] [Google Scholar]
- MILLER RE, ISHIHARA S, BHATTACHARYYA B, DELANEY A, MENICHELLA DM, MILLER RJ & MALFAIT AM 2017. Chemogenetic Inhibition of Pain Neurons in a Mouse Model of Osteoarthritis. Arthritis Rheumatol, 69, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER RE, ISHIHARA S, TRAN PB, GOLUB SB, LAST K, MILLER RJ, FOSANG AJ & MALFAIT AM 2018. An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER RE & MALFAIT AM 2017. Osteoarthritis pain: What are we learning from animal models? Best Pract Res Clin Rheumatol, 31, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER RE, TRAN PB, DAS R, GHOREISHI-HAACK N, REN D, MILLER RJ & MALFAIT AM 2012. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A, 109, 20602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARAHASHI T, MOORE JW & SCOTT WR 1964. Tetrodotoxin Blockage of Sodium Conductance Increase in Lobster Giant Axons. J Gen Physiol, 47, 965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NASSAR MA, LEVATO A, STIRLING LC & WOOD JN 2005. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol Pain, 1, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NODA M, IKEDA T, SUZUKI H, TAKESHIMA H, TAKAHASHI T, KUNO M & NUMA S 1986. Expression of functional sodium channels from cloned cDNA. Nature, 322, 826–8. [DOI] [PubMed] [Google Scholar]
- NORENG S, LI T & PAYANDEH J 2021. Structural Pharmacology of Voltage-Gated Sodium Channels. J Mol Biol, 433, 166967. [DOI] [PubMed] [Google Scholar]
- OBEIDAT AM, MILLER RE, MILLER RJ & MALFAIT AM 2019. The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis Cartilage, 27, 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALERMO AF, DIENNET M, EL EZZY M, WILLIAMS BM, COTNOIR-WHITE D, MADER S & GLEASON JL 2018. Incorporation of histone deacetylase inhibitory activity into the core of tamoxifen – A new hybrid design paradigm. Bioorganic & Medicinal Chemistry, 26, 4428–4440. [DOI] [PubMed] [Google Scholar]
- PAMMOLLI F, MAGAZZINI L & RICCABONI M 2011. The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov, 10, 428–38. [DOI] [PubMed] [Google Scholar]
- PAN X, LI Z, HUANG X, HUANG G, GAO S, SHEN H, LIU L, LEI J & YAN N 2019. Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu-conotoxin KIIIA. Science, 363, 1309–1313. [DOI] [PubMed] [Google Scholar]
- PAN X, LI Z, ZHOU Q, SHEN H, WU K, HUANG X, CHEN J, ZHANG J, ZHU X, LEI J, XIONG W, GONG H, XIAO B & YAN N 2018. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science, 362. [DOI] [PubMed] [Google Scholar]
- PAYANDEH J, SCHEUER T, ZHENG N & CATTERALL WA 2011. The crystal structure of a voltage-gated sodium channel. Nature, 475, 353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMACHANDRA R & ELMSLIE KS 2016. EXPRESS: Voltage-dependent sodium (NaV) channels in group IV sensory afferents. Mol Pain, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REN D, NAVARRO B, XU H, YUE L, SHI Q & CLAPHAM DE 2001. A prokaryotic voltage-gated sodium channel. Science, 294, 2372–5. [DOI] [PubMed] [Google Scholar]
- SHEN H, LI Z, JIANG Y, PAN X, WU J, CRISTOFORI-ARMSTRONG B, SMITH JJ, CHIN YKY, LEI J, ZHOU Q, KING GF & YAN N 2018. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science, 362. [DOI] [PubMed] [Google Scholar]
- SHEN H, LIU D, WU K, LEI J & YAN N 2019. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science, 363, 1303–1308. [DOI] [PubMed] [Google Scholar]
- SULA A, BOOKER J, NG LC, NAYLOR CE, DECAEN PG & WALLACE BA 2017. The complete structure of an activated open sodium channel. Nat Commun, 8, 14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SULA A, HOLLINGWORTH D, NG LCT, LARMORE M, DECAEN PG & WALLACE BA 2021. A tamoxifen receptor within a voltage-gated sodium channel. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG L, GAMAL EL-DIN TM, LENAEUS MJ, ZHENG N & CATTERALL WA 2019. Structural Basis for Diltiazem Block of a Voltage-Gated Ca(2+) Channel. Mol Pharmacol, 96, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG L, GAMAL EL-DIN TM, SWANSON TM, PRYDE DC, SCHEUER T, ZHENG N & CATTERALL WA 2016. Structural basis for inhibition of a voltage-gated Ca(2+) channel by Ca(2+) antagonist drugs. Nature, 537, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOLEDO-ARAL JJ, MOSS BL, HE ZJ, KOSZOWSKI AG, WHISENAND T, LEVINSON SR, WOLF JJ, SILOS-SANTIAGO I, HALEGOUA S & MANDEL G 1997. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A, 94, 1527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOYODA E, MAEHARA M, WATANABE M & SATO M 2021. Candidates for Intra-Articular Administration Therapeutics and Therapies of Osteoarthritis. Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRELLE S, REICHENBACH S, WANDEL S, HILDEBRAND P, TSCHANNEN B, VILLIGER PM, EGGER M & JUNI P 2011. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ, 342, c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIEN TN & DECAEN PG 2016. Biophysical Adaptations of Prokaryotic Voltage-Gated Sodium Channels. Curr Top Membr, 78, 39–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Z, VAN DEN BERG RJ & YPEY DL 1994. Resting membrane potentials and excitability at different regions of rat dorsal root ganglion neurons in culture. Neuroscience, 60, 245–54. [DOI] [PubMed] [Google Scholar]
- WARING MJ, ARROWSMITH J, LEACH AR, LEESON PD, MANDRELL S, OWEN RM, PAIRAUDEAU G, PENNIE WD, PICKETT SD, WANG J, WALLACE O & WEIR A 2015. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov, 14, 475–86. [DOI] [PubMed] [Google Scholar]
- WILLOW M, GONOI T & CATTERALL WA 1985. Voltage clamp analysis of the inhibitory actions of diphenylhydantoin and carbamazepine on voltage-sensitive sodium channels in neuroblastoma cells. Mol Pharmacol, 27, 549–58. [PubMed] [Google Scholar]
- YAN Z, ZHOU Q, WANG L, WU J, ZHAO Y, HUANG G, PENG W, SHEN H, LEI J & YAN N 2017. Structure of the Nav1.4-beta1 Complex from Electric Eel. Cell, 170, 470–482 e11. [DOI] [PubMed] [Google Scholar]
- YANG YC & KUO CC 2005. An inactivation stabilizer of the Na+ channel acts as an opportunistic pore blocker modulated by external Na+. J Gen Physiol, 125, 465–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEOMANS DC, LEVINSON SR, PETERS MC, KOSZOWSKI AG, TZABAZIS AZ, GILLY WF & WILSON SP 2005. Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of Nav1.7 sodium channels in primary afferents. Hum Gene Ther, 16, 271–7. [DOI] [PubMed] [Google Scholar]
- ZHANG X, PRIEST BT, BELFER I & GOLD MS 2017. Voltage-gated Na(+) currents in human dorsal root ganglion neurons. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU J, ZHEN G, AN S, WANG X, WAN M, LI Y, CHEN Z, GUAN Y, DONG X, HU Y & CAO X 2020. Aberrant subchondral osteoblastic metabolism modifies NaV1.8 for osteoarthritis. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Imaging, in vivo and electrophysiology data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.