

Summary
The papain-like protease (PLpro) of coronaviruses is an attractive antiviral target to inhibit both viral replication and interference of the host immune response. We have identified and characterized three novel classes of small molecules, thiophene, cyanofuran, and triazoloquinazoline, as PLpro inhibitors. Thiophene inhibited the PLpro of two major coronaviruses, Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV) including SARS-CoV-2, while cyanofuran and triazoloquinazoline more selectively inhibited MERS-CoV PLpro. Unlike GRL0617, a known PLpro inhibitor, all three compounds contain no naphthyl group but like GRL0617 were predicted to fit on the cleft near the BL2 loop. Docking studies further revealed that the location and direction of the binding determined their specificity to different coronaviruses. Together, our work demonstrates that the BL2 loop and nearby regions are outstanding druggable targets, and our three inhibitors can be applicable to the development of therapeutics for coronavirus infection.
Subject areas: Biochemistry, Virology, Structural biology
Graphical abstract
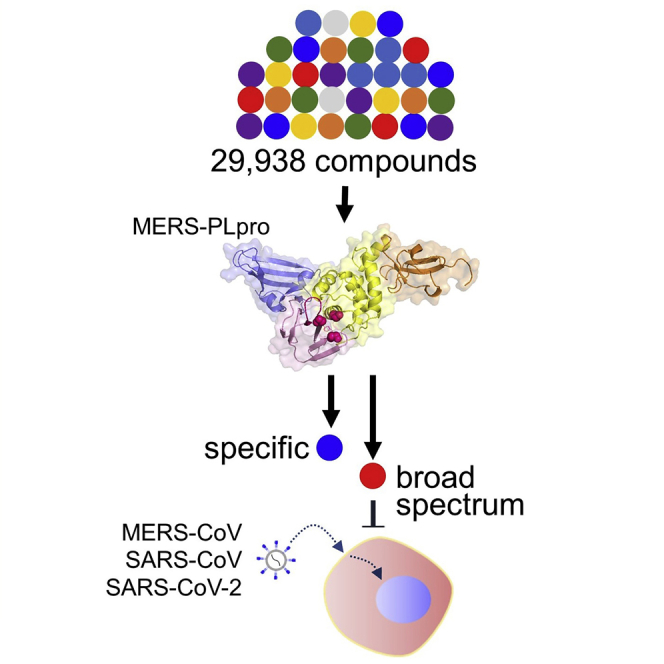
Highlights
-
•
Novel inhibitors were predicted to bind to regions near the BL2 loop of PLpro
-
•
Regions near the BL2 loop contribute to the selectivity of different PLpros
-
•
Identified thiophenes show a broad-spectrum antiviral potency
Biochemistry; Virology; Structural biology
Introduction
Coronaviruses have caused diseases in humans and have led to several epidemic and pandemic crises. To date, seven human coronaviruses (HCoVs) have been reported: HCoV-OC43, Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV), and SARS-CoV-2, which belong to the betacoronavirus genus, as well as HCoV-229E and HCoV-NL63, which belong to the alphacoronavirus genus (Hartenian et al., 2020; van der Hoek et al., 2004). The well-known HCoV-229E and HCoV-OC43 were first reported in 1960 and for decades were considered to be major disease-causing coronaviruses. Identification of SARS-CoV in 2002 (Rota et al., 2003) and MERS-CoV in 2012 (Zaki et al., 2012) proved that these air-borne pathogens can easily transfer between species, spread within the community, and result in an epidemic; and the recently identified SARS-CoV-2 has caused an unprecedented global pandemic and has posed a serious threat to public health and the world economy. By the fall of 2021, over 200 million infection cases and four million deaths by SARS-CoV-2 infection have been reported globally, and casualties are still mounting despite the recent vaccine drive (Grubaugh et al., 2021; Zhou et al., 2020). All these indicate that MERS-CoV and SARS-CoV are highly pathogenic and detrimental to humans. In addition, there is a high likelihood of further retransmission from various zoonotic reservoirs which can result in another serious pandemic. Although several antiviral drugs including neutralizing antibodies (Corti et al., 2021) and molnupiravir (Sheahan et al., 2020; Toots et al., 2019), a nucleoside analog that inhibits viral genome replication, have been developed, additional drugs with novel antiviral mechanisms of action still need to be developed for ready clinical use. This is because the virus can develop resistance mechanisms against nucleoside analogs, and also because a combination of drugs with different mechanisms of action is commonly used in the clinic to improve treatment outcomes.
MERS-CoV and SARS-CoV are single-stranded positive-sense RNA viruses with a genome of ∼30 kb enveloped with spike proteins (de Groot et al., 2013). For entry into host cells, the spike proteins of MERS-CoV and SARS-CoV recognize their respective host cell receptors, dipeptidylpeptidase 4 (DPP4 or CD26) and angiotensin-converting enzyme 2 (ACE2). Upon entry, the viral RNA genome is released from the capsid and undergoes translation to produce viral polyproteins, pp1a and pp1b. The polyproteins are further processed by two cysteine proteases, papain-like protease (PLpro) and a 3-C-like protease (3CLpro), which constitute an essential step for the maturation of 16 non-structural viral proteins (Hartenian et al., 2020; Perlman and Netland, 2009). Therefore, the protease activity of the coronavirus has been implicated as one of the most favorable antiviral drug targets. Especially, PLpro, in addition to being essential for viral proliferation, is involved in other important cellular processes including de-ISGylation and deubiquitination (Lindner et al., 2005; Ratia et al., 2014), thereby repressing host’s innate immunity triggered by interferon regulatory factor 3 (IRF-3) (Bailey-Elkin et al., 2014; Barretto et al., 2005; Clementz et al., 2010; Frieman et al., 2009). Therefore, PLpro is an outstanding antiviral target to inhibit both viral replication and viral interference of the host innate immune response to the viral invasion. To date, several classes of PLpro inhibitors have been identified: the thiopurines 6-mercaptopurine (6-MP) and 6-thioguanine (6-TG) (Chen et al., 2009; Chou et al., 2008); the natural products tanshinones (Park et al., 2012) and geranylated flavonoids (Cho et al., 2013); zinc ion and zinc-conjugated inhibitors (Han et al., 2005); and naphthalene drugs (Ratia et al., 2008). Importantly, a potent naphthalene inhibitor GRL0617 and its naphthalene analogs were reported to specifically bind to the region near the BL2 loop, a region situated near the catalytic cleft, thereby playing an important role in gating the cleft toward the catalytic site of PLpro (Fu et al., 2021; Ghosh et al., 2009; Ratia et al., 2008). Therefore, PLpro, specifically its flexible BL2 loop, is an attractive target for the development of antiviral drugs against HCoVs; and owing to the aforementioned risk of development of viral resistance mechanisms to existing drugs and the effectiveness of combination therapy, uncovering novel classes of compounds as PLpro inhibitors can be an option for therapeutic measures for coronavirus infection.
Here, in our effort to uncover effective antiviral compounds against coronaviruses, we adapted a robust high-throughput compound assay to screen over 29,938 compounds, including FDA-approved drugs. We identified three effective compounds of novel structural classes: thiophene (1), cyanofuran (2), and triazoloquinazoline (3). We found that these three compounds showed differential activities against MERS-CoV PLpro (MERS-PLpro), SARS-CoV PLpro (SARS-PLpro), and SASR-CoV-2 PLpro (SARS-2-PLpro), which was validated with in vitro enzyme assays and immunofluorescence-based cell culture experiments. To further identify the structural properties important for their activity, we explored the series of analogs available, and through our molecular docking study and molecular dynamics simulation (MDS), found that their binding affinity to the cleft near the BL2 loop provides target selectivity against MERS-PLpro and SARS-PLpro. Together, our work identifies novel structural classes of PLpro inhibitors and elucidates their binding properties to the target, which provides essential information for the development of broad-spectrum anti-coronavirus treatments.
Results and discussion
Compounds of novel structural classes were identified as MERS-PLpro inhibitors
We screened for PLpro inhibitors using an in vitro enzymatic assay for which the results were validated by cell-based infection assays (Figure 1A). For this, we codon-optimized and expressed the MERS-PLpro protein in a bacterial strain and adapted a fluorescence-based enzymatic assay where a short peptide-linked AMC (7-amido-4-methylcoumarin) was used as the substrate, as previously described (Kilianski et al., 2013; Ratia et al., 2008). As noted, although MERS-PLpro recognizes K/R-L/I-I/K/V-G-G↓A, we chose a short peptide with the sequence of R-L-R-G-G-AMC instead, because this sequence generated a more robust fluorescence signal (Figure 1B). We tested a series of peptide substrate concentrations (0.8-100 μM) and MERS-PLpro enzyme concentrations (0.3-10 μM) to set up optimal concentrations for the screen at room temperature. Concentrations of 2.5 and 40 μM were selected for MERS-PLpro and the substrate, respectively (Figure S1A). The substrate concentration chosen is lower than the Michaelis constant (KM) which makes it suitable for a screen for competitive inhibitors. The chosen concentrations of MERS-PLpro and the peptide substrate were confirmed using a known PLpro inhibitor, 6-TG (Figure S1C). Using the optimized conditions, we screened a collection of 29,938 compounds including FDA-approved drugs, sourced from ChemDiv, ChemBridge, and Selleckchem, at a single concentration of 10 μM. We performed a high-throughput screen using a 384-well format, and compounds that inhibited the protease activity were selected as positive hits. Our screen was found to be very robust with an average Z′-factor of 0.91 ± 0.01 (Figure S2A). We defined compounds with over 40% inhibition as positive hits and a subset was cherry picked and retested (Figure 1C). Of the 29,938 compounds screened, 257 compounds were identified as primary hits for inhibitors of MERS-PLpro, which is a hit rate of ∼0.86% (Figure 1A). Of the primary hit compounds, we excluded 81 compounds that showed over 20% cytotoxicity (Figure S2B). Then, we further validated the hit compounds by performing the in vitro MERS-PLpro assay at serially diluted compound concentrations, and of the validated hit compounds, three compounds, thiophene (1), cyanofuran (2), and triazoloquinazoline (3) were selected for further assessment (Figures 1D–1F). To further validate the efficacy of these compounds in the host cell, we measured the degree of cellular ubiquitination in the presence of the compounds as MERS-PLpro is known to inhibit cellular ubiquitination. As shown in Figure 2, we have found that all three compounds reversed the inhibition of cellular ubiquitination by MERS-CoV. All these results indicate that the selected compounds prevent the activity of MERS-PLpro in the host cells.
Figure 1.

In vitro screening of drug libraries reveals three novel classes of compounds that effectively inhibit MERS-PLpro
(A) Schematic for the screening workflow. All 29,938 small molecules from the test library, which include FDA-approved drugs, were tested on MERS-PLpro using a fluorescence-based protease assay, and cytotoxicity of the selected 257 primary hit compounds was determined using a commercially available cytotoxicity kit, followed by the final assessment of in vivo efficacy using an immunofluorescence-based MERS-CoV infection assay or viral RNA quantification in virus-infected Vero cells. In addition, the structure-activity relationship (SAR) was examined using compound analogs and molecular docking analysis.
(B) Detailed schematic representation of the high throughput fluorescence assay designed to measure the proteolytic activity of PLpro. AMC: 7-amido-4-methylcoumarin.
(C) Result of the fluorescence-based screen of the 29,938 compounds. Closed red circles represent hits with over 40% inhibition of PLpro, while brown circles indicate the positive controls of each plate. The Z′ factor of the screen was calculated as 0.91 ± 0.01.
(D–F) Dose-response curve determined by the PLpro protease assay of the selected three compounds (D) thiophene (1), (E) cyanofuran (2), and (F) triazoloquinazoline (3). Each data point represents the mean of duplicate assays with ±SEM.
Figure 2.

Identified compounds prevent the reduction of cellular ubiquitination induced by MERS-CoV infection
Virus-infected Huh-7 cells were treated with 50 or 100 μM of the selected compounds 1, 2, and 3 after which cellular ubiquitination was measured by western blot. Ub: ubiquitin.
Thiophene (1) shows a broad-spectrum potency against coronaviruses
As other known competitive inhibitors such as 6-MP and 6-TG show efficacy against both SARS-PLpro and MERS-PLpro, we tested the potency of thiophene (1) against SARS-PLpro in addition to MERS-PLpro using the in vitro enzymatic assay. In the same manner as for MERS-PLpro, a series of peptide substrate concentrations (0.8-100 μM) and SARS-PLpro enzyme concentrations (0.02-0.63 μM) were tested, and 0.02 and 40 μM were selected as optimal concentrations for SARS-PLpro and the substrate, respectively (Figures 1D and S1B) and the chosen concentrations were validated using 6-TG (Figure S1D). As shown in Figure 3B, we found that thiophene (1) has potency against MERS-PLpro as well as SARS-PLpro. The 50% inhibitory concentration (IC50) value of each analog was calculated from the dose-response curve generated from the fluorescence-based PLpro protease assay, and the results indicate that thiophene (1) shows a comparably outstanding potency against MERS-PLpro with an IC50 value of 14.2 μM and a moderate potency against SARS-PLpro with an IC50 value of 156.3 μM (Figure 3B). To identify the structural regions of thiophene important for its affinity to PLpro, we evaluated the available analogs of 1 (Figure 3A). We tested the analogs with the modification of the para-bromo on the phenyl group of 1 with fluorination at different positions of the phenyl ring (-para or -ortho) on the benzamide region. Analog 1a with fluorine at the para position showed a 3-fold decrease in efficacy against MERS-PLpro and a complete loss of efficacy against SARS-PLpro, while 1b with the ortho-fluoro substituent still retained potency against both MERS-PLpro and SARS-PLpro although the potency decreased (Figure 3B). Next, we tested the role of the carboxylic acid at the 3-position on the thiophene scaffold. As shown in Figure 3B, esterification of the carboxylic acid with methyl (1c) or ethyl (1d) moieties resulted in a complete loss of activity against both proteases. This suggests that the carboxylic acid moiety is crucial for the affinity of thiophene (1) to the target protein. Also, we tested analogs with modification on the 4-position of the thiophene scaffold: 1e, 1f, 1g, and 1h. Analog 1e with a tolyl group replacing the thiophene group showed an improved potency on both proteases; 1f with an isopropyl moiety still showed potency, although moderately decreased, on both enzymes; 1g with an introduction of a methyl moiety showed potency only on SARS-PLpro with no activity against MERS-PLpro; 1h with complete removal of the thiophene scaffold showed a 2-fold decrease in potency against MERS-PLpro with no change in potency against SARS-PLpro (Figure 3B).
Figure 3.

Thiophene and its analogs prevent both MERS-CoV and SARS-CoV proliferation
(A) Structural presentation of thiophene (1) and its analogs.
(B) Summary of the potency of each analog. The potency of each thiophene analog is presented as the 50% inhibitory concentration (IC50) value measured using the fluorescence-based MERS-PLpro and SARS-PLpro protease assay. The 50% effective concentration (EC50), 50% cytotoxicity concentration (CC50), and selective index (SI; EC50 divided by CC50) were determined by the immunofluorescence-based infection assay using MERS-CoV-infected Vero cells.
(C) Representative images from the MERS-CoV infection assay. Vero cells were infected with MERS-CoV followed by treatment with compound 1 or the control compound chloroquine (chr) for two days at concentrations ranging from 0.0977 to 50 μM, and images were taken on day 2. Green signals indicate cells infected with MERS-CoV and red signals indicate live Vero cells.
(D and E) RNA quantification of the viral upE gene in MERS-CoV-infected cells after treatment with compound 1 (D) or analog 1a (E). Each data point represents the mean of quadruple assays with ±SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005 compared to untreated control by Student’s t test. untr: untreated control.
(F) Time-of-addition experiment using compound 1. The effect of compound 1 on the early or late stage of infection was determined by adding the compound prior to infection (−1 h), at the time of infection (0), and post-infection (1-6 h) and by subsequently quantifying the upE gene in virus-infected Vero cells. Each data point represents the mean of duplicate assays with ±SD. LPV: lopinavir; CE-5: chloropyridine ester.
(G) Structure of PLpro. PLpro is mainly composed of finger, thumb, palm, and ubiquitin-like (UBL) domains. The BL2 loop and catalytic site important for molecular interaction are included in the palm domain. The BL2 loop and important residues for SARS-PLpro (left) and MERS-PLpro (right) are compared. Crystal structure of SARS-PLpro with its inhibitor (PDB: 3E9S) is adapted from reference (Ratia et al., 2008).
(H) Image of molecular docking of compound 1 into MERS-PLpro (PDB: 4RF1). Docking was performed using the XP Glide docking tool of the Schrodinger platform (v. 2019-2).
Although an in vitro protease assay can provide useful target-specific information, it does not validate the antiviral potency of the compounds in the host cells. Recently, we adapted an immunofluorescence-based assay to monitor MERS-CoV infection in host cells. In this assay, we infected Vero cells with MERS-CoV at a MOI of 0.0625 in the presence of the compounds, then two days after infection, we quantified the viral infection by immunofluorescence using a MERS-CoV spike-specific primary antibody and a fluorescence-conjugated secondary antibody (Figure 1A). As shown in Figure 3C, treatment with both compound 1 and chloroquine, a lysosomotropic compound that prevents endosomal acidification and appears to have antiviral activity (Wang et al., 2020), resulted in the depletion of the MERS-CoV fluorescence signal in the host cells demonstrating antiviral activity. We performed this image-based analysis for 1 and its analogs on MERS-CoV-infected cells and calculated 50% efficient concentration (EC50) values (Figures 3B and S3). Overall, their potencies in the cell-based experiment were similar to the efficacy measured by the in vitro enzymatic assay: 1 was found to have a marked efficacy against MERS-CoV infection (EC50, 2.33 μM), and analogs 1e (EC50, 1.25 μM) and 1f (EC50, 1.06 μM) showed an improved potency. As expected, the analogs 1c and 1d with modification at the carboxylic acid showed no efficacy in cell-based assays. Also, 1h had no activity against MERS-CoV infection. Interestingly, 1b showed an improved IC50 against MERS-PLpro compared to 1a, but 1b had no potency against MERS-CoV infection, which suggests that the modification in 1b may have resulted in poor delivery into the host cells. To further confirm their antiviral activity in the host cells, we also examined the replication of MERS-CoV upon the treatment of 1 or 1e by measuring the copy number of the RNA of the upE gene encoding the E protein of MERS-CoV by real-time quantitative RT-PCR. As shown in Figures 3D and 3E, in vivo potency of 1 and 1e against MERS-CoV was confirmed by an ∼80% decrease in mRNA levels of the upE gene upon treatment with 1 μM of 1 or 1e. As MERS-PLpro is crucial in generating mature proteins including the nsp3 protein which is essential for viral replication (Barretto et al., 2005; Lindner et al., 2005), we reasoned that 1 could be highly effective at an early stage of infection rather than the later stages. To test this hypothesis, we performed a time-of-addition experiment. We treated MERS-CoV infected cells with a compound: prior to infection (−1 h), at the time of infection (0), and post-infection (1-6 h) (Figure 3F). Both 1 and 1e resulted in over 60% protection from the MERS-CoV infection only at an early stage of infection, while drug treatments 2-6 h post-infection had only marginal or negligible effects; and these results were similar to those obtained from two known antiviral compounds, lopinavir (LPV) (Choy et al., 2020) and chloropyridine ester (CE-5) (Ghosh et al., 2008), which are known to prevent the maturation of viral polyproteins.
Next, as we found through the in vitro PLpro assay that 1 and several of its analogs have potency against SARS-PLpro as well, we wanted to validate this activity by analyzing their efficacy in preventing SARS-CoV infection in a cell-based experiment. To this end, we adapted the same immunofluorescence-based infection assay used for the MERS-CoV infection model. We first treated the Vero cells with a compound for 2 h and then infected the cells with SARS-CoV at an MOI of 0.0625. Following a 2-day infection, the cells were subjected to immunofluorescence analysis using a SARS-CoV spike S1 antibody. As shown in Figure S4, 1e was found to have potency against SARS-CoV infection while 1 had only negligible activity. As these results suggested that 1e may have a broad-spectrum inhibitory effect on the PLpro of coronaviruses, we reasoned that 1e could be potent against other coronaviruses including HCoV-OC43 and HCoV-229E. To test this possibility, we treated 1e on HCoV-229E or HCoV-OC43-infected Vero cells and measured viral replication by quantifying the viral RNA. We found that 1e has a marginal potency at 10 μM on both HCoV-OC43 and HCoV-229E while 1 had no antiviral effect (Figure S5). These results suggest that thiophene is capable of inhibiting the PLpro activity of a rather broad spectrum of coronaviruses.
Thiophene may interact with the catalytic cleft near the BL2 loop blocking the access of substrate to the catalytic site
As thiophene does not have a naphthyl group, we wanted to gain insight into the binding properties of 1 on the target protein PLpro. To this end, we explored possible interactions by a computational molecular docking study. The PLpro protein has a right-handed palm-thumb-finger shape and contains an additional ubiquitin-like (UBL) domain at the N-terminus (Figure 3G) (Baez-Santos et al., 2014, 2015; Lei et al., 2014). Although MERS-PLpro and SARS-PLpro share high similarity in their structural architecture, MERS-PLpro recognizes Leu/Ile-X-Gly-Gly-↓-Arg/Glu-X while SARS-PLpro preferentially recognizes Lys over Glu, making its recognition motif Leu-X-Gly-Gly-↓-Arg(or Lys)-X. As reported (Ratia et al., 2008), the PLpro of coronaviruses contains a canonical catalytic triad with Cys-His-Asp in the palm domain and has a flexible BL2 loop near the catalytic cleft which is important for gating the cleft toward the catalytic site. Similarly, our results of docking 1 into the MERS-PLpro crystal structure (PDB: 4RF1 (Bailey-Elkin et al., 2014)) predicted that it would not bind to the catalytic triad (Cys112-H273-D287) (Figures S6A and S6B) but instead would bind to the cleft near the BL2 loop (Figure 3H). We also performed an MDS to further validate the molecular docking, and we found that compound 1 was predicted to not bind to the catalytic site of MERS-PLpro but to bind to the region near the BL2 loop as the docking predicted (Figures S6C and S6D). This suggests that 1 may be a non-competitive inhibitor similar to the known inhibitor of SARS-PLpro, GRL0617. As reported, the BL2 loop plays crucial role in gating access to the catalytic triad, and in PLpro, the BL2 moves toward an open position upon substrate binding (Henderson et al., 2020; Ratia et al., 2008). The cleft region near the BL2 loop of SARS-PLpro binds to a non-competitive inhibitor, GRL0617, through T-shaped stacking (Figure 3G), while interestingly, as shown in Figure 3H, our docking study predicted that 1 binds to a cavity branched from the cleft at the opposite side of the BL2 loop in a “vertically pining shape (VPS).” This cavity is not involved in the interaction of GRL0617 to SARS-PLpro, and notably, MERS-PLpro has different residues constituting the cleft region when compared to SARS-PLpro (Figure 3G). These differences may provide structural restrictions, which might potentially contribute to the unique binding properties of MERS-PLpro and SARS-PLpro. In particular, Y269 of SARS-PLpro is missing in MERS-PLpro; instead, it has T274 which might not be able to form interactions with the naphthyl group of GRL0617 as the phenyl group of the tyrosine does. Also, in place of Q270 of SARS-PLpro which provides a hydrogen bond to GRL0167, MERS-PLpro has A275, which might further contribute to the difference in the binding shape of 1 to MERS-PLpro when compared to the binding shape of GRL0617 to SARS-PLpro. Collectively, our molecular docking study suggested that 1 could be a non-competitive inhibitor, and unlike GRL017, 1 was predicted to bind to the cleft near the BL2 of MERS-PLpro in a different mode of interaction generating a VPS.
Cyanofuran (2) and triazoloquinazoline (3) inhibit MERS-CoV PLpro selectively over SARS-CoV PLpro
Unlike thiophene (1), we found cyanofuran (2) and triazoloquinazoline (3) to be highly potent only against MERS-PLpro with no activity against SARS-PLpro (Figures 4 and 5). To identify the structural regions important for the affinity of 2 and 3 to PLpro, we tested the available analogs with important structural modifications (Figures 4A and 5A). For the 3-cyano-furan scaffold (2), we tested analogs with the modification of the chloride on the benzamide group with either fluorine (2b) or iodine (2c) and found that their potency remained unchanged. However, the introduction of trimethoxy (2d) on the benzyl group completely abolished its potency which suggests the benzamide group with substituted halides is essential for its affinity to the target protein. Also, we tested an analog with para-chloride on the phenyl ring at the 5-position of the furan scaffold (2a), and its potency decreased by ∼40% which suggests that the phenyl group without any substituent is crucial for binding (Figure 4B). To validate antiviral efficacy against the authentic virus, we examined the efficacy of 2 in cells infected by MERS-CoV, and it was found to be highly potent (IC50, 0.93 μM) as shown in Figures 4B, 4C, and S7. The in vitro efficacy of 2 was further validated by real-time RT-PCR analysis (Figure 4D). Consistent with the PLpro assay, analogs of 2 showed comparable potency to 2 against MERS-CoV infection while their activity was lower. Interestingly, although 2d had no efficacy in the in vitro PLpro assay, it appeared to retain antiviral efficacy in a cell-based assay (Figure 4B). Although we used a range of substrate concentrations to calculate the IC50 of compound 2d in the in vitro PLpro assay, 2d might have efficacy at a concentration not included in the range tested. Next, we examined whether cyanofuran (2) is as effective at an early stage of infection as thiophene. As shown in Figure 4E, 2 shows a high potency only when added at the beginning of the MERS-CoV infection (−1 h, 0 h) comparable to the effect of chloroquine. Lastly, we performed molecular docking as shown in Figure 4F, and molecular docking of 2 into MERS-PLpro revealed that it was predicted to interact with the key residues of the cleft of MERS-PLpro including Y269 but in a different direction compared to the SARS-PLpro inhibitor, GRL0167. Like compound 1, compound 2 was calculated to bind to the cavity branching from the cleft of MERS-PLpro by interacting more significantly with the cavity region; and similar to compound 1, compound 2 failed to bind to the catalytic site (Figures S8A and S8B). Also, in MDS performed with compound 2 and MERS-PLpro, we found that compound 2, similar to compound 1, was predicted to not bind to the catalytic site of MERS-PLpro although it was predicted to bind to a region upper than the region predicted by molecular docking (Figures S8C and S8D).
Figure 4.

Cyanofuran and its analogs are selectively effective against MERS-CoV infection
(A) Structures of cyanofuran (compound 2) and its analogs.
(B) Measurement of the potency of each analog using the PLpro protease assay. The potency of each analog (IC50) was measured using the MERS-PLpro and SARS-PLpro assay. The EC50, CC50, and SI were determined by the analysis of signal intensities obtained from an immunofluorescence-based infection assay using the MERS-CoV-infected Vero cell model.
(C) Representative images from infection assay using compound 2. Green signals indicate cells infected with MERS-CoV and red signals indicate live Vero cells.
(D) Efficacy of compound 2 evaluated by RNA quantification of the viral upE gene in MERS-CoV-infected Vero cells. Each data point represents the mean of quadruple assays with ±SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005 compared to untreated control by Student’s t test. untr: untreated control.
(E) Time-of-addition experiment using compound 2. Vero cells were treated with a test compound 1 h prior to, at the time of, or 1–6 h post-infection and the viral upE gene was measured by RT-PCR. Each data point represents the mean of duplicate assays with ±SD. chr: chloroquine.
(F) Image obtained by molecular docking of compound 2 into MERS-PLpro using the Schrodinger platform.
Figure 5.

Triazoloquinazoline selectively prevents MERS-CoV proliferation by a similar mode of action as cyanofuran
(A) Representation of the structures of triazoloquinazoline (3) and its analogs.
(B) Summary of the potency of each analog. IC50 values of 3 and its analogs were determined by the dose-response curve obtained from the fluorescence-based protease assay, while the EC50 and CC50 were determined by an infection assay using the Vero cell MERS-CoV infection model.
(C) Representative immunofluorescence images treated with 3 and its analog 3a-1.
(D) RNA quantification of the viral upE gene in MERS-CoV-infected cells treated with compound 3. Each data point represents the mean of quadruple assays with ±SEM. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005 compared to untreated control by Student’s t test.
(E) Molecular docking of triazoloquinazoline into MERS-PLpro (PDB: 4RF1) using Schrodinger XP Glide.
In the case of triazoloquinazoline (3), we tested a total of 15 available analogs as shown in Figure 5A. First, we examined several analogs with modifications in the region containing the benzamide moiety. Replacing this region significantly affected potency: 3a-9 with the modification of a simple carboxylic acid at this site decreased the potency by 3-fold; 3a-4 with an N-isopropyl group resulted in a 4-fold decrease; 3a-2 with a thiophene moiety had further decreased efficacy; and 3a-3 with a substituted cyclohexyl group resulted in a complete loss of potency (Figure 5B). This data suggests that the benzamide group may bind to the limited space of the cleft of MERS-PLpro. Next, we tested several analogs with modifications on the meta-fluorophenyl ring at the 3-position of the triazole scaffold, and none, except for 3c-1, showed efficacy. Finally, we also tested an analog of 3b with a para-methoxy phenyl ring at the 3-position of the triazole scaffold, and no efficacy was found. To examine whether triazoloquinazoline analogs have antiviral activity, we tested all analogs against MERS-CoV infection (Figures 5B, 5C, and S9), and most compounds that showed no efficacy in the PLpro assay showed no efficacy in the cell-based assays, as expected. Interestingly, although 3a-5, 3a-6, and 3a-7 have decreased in vitro potency on MERS-PLpro, their efficacy in the cell-based assay appeared to remain comparable to 3. These results suggest that the region may be important for their delivery into the effective locale in the host cell. In the case of the analog 3a-8 with a meta-methoxy phenyl ring, its potency in the MERS-PLpro assay increased more than 4-fold, however, antiviral efficacy was not detected by the in vivo cell-based assay. Together with the 3a-5, 3a-6, and 3a-7 results, this suggests that the phenyl ring might play crucial roles in the potency of triazoloquinazoline in the host cell. Like 1 and 2, the effectiveness of 3 was confirmed using real-time quantitative RT-PCR (Figure 5D). To obtain molecular insight into possible molecular interactions of 3, molecular docking was performed. As shown in Figure 5E, 3 was predicted to bind through a VPS to an upper region of the cavity further away from the BL2 loop, when compared to 2 and 3, and similar to compounds 1 and 2, compound 3 did not bind to the catalytic site (Figures S10A and S10B). Also, in the MDS analysis of compound 3 and MERS-PLpro, compound 3 was predicted to not bind to the catalytic pocket of MERS-PLpro but to bind to the region near BL2, which is similar to the result generated by the docking analysis (Figures S10C and S10D). Our docking study and MDS suggest that the cavity and its upper regions may contribute to the selectivity of compounds to MERS-PLpro and SARS-PLpro. Importantly, we found that the introduction of mutations in two residues T251 and T310 of MERS-PLpro, specifically substitution with alanine, resulted in a complete loss of expression in the case of the T310A mutant and T310A-T251A double mutant or a significant loss (>50%) of the enzyme activity in the case of the T251A mutant (Figure S11A). When we tested three compounds with T251A mutant, compound 1 showed no potency at a tested concentration (0-50 μM), and the potency of compounds 2 and 3 was decreased significantly (Figures S11B–S11D). These results suggest that T251 plays a crucial role in the interaction of MERS-PLpro with our compounds, especially compound 1 that shows potential as a broad-spectrum PLpro inhibitor. Importantly, these results are in line with those of our molecular docking studies where compound 1 was expected to bind to the cleft most near to the BL2 loop whereas compounds 2 and 3 which were expected to bind to the region further away from the BL2 loop did not show a significant reduction in interaction with MERS-PLpro T251A. In total, our results demonstrate that the residues near the gating of the BL2 loop are crucial for both activity and structural stability of the protease. Therefore, residues near the loop including T251 and T310 can be effective druggable targets.
Inhibitors show potency against SARS-2-PLpro and SARS-CoV-2 infection
Owing to the significance of the recent pandemic caused by SARS-CoV-2, we wanted to test if the selected compounds are efficient in SARS-CoV-2 infection. As shown in Figures 6A–6C, the compounds show a marginal potency against SARS-2-PLpro, and the IC50 of these compounds was barely calculated in the range of tested concentrations. In addition, similar to MERS-CoV and SARS-CoV, we adapted an immunofluorescence-based viral infection assay in cells (Jeon et al., 2020). The assay conditions were validated with the known antiviral drug remdesivir. Treatment with remdesivir resulted in the depletion of the SARS-CoV-2 fluorescence signal in the host cells (Figure S12). Interestingly, when we examined these compounds in a cell-based infection model for SARS-CoV-2, we found that they showed potency at a concentration below their cytotoxicity although their IC50 was over 10-fold lower than those of MERS-CoV infection (Figures 6D–6F). To further validate the potency, we performed a cleavage assay using a 67 kDa protein substrate containing the 10 amino acids at the junction of nsp 2-3 with a cleavage site. Upon PLpro-mediated cleavage at this site, products of two sizes, 25 and 42 kDa, are generated. As shown in Figure 7A, we first confirmed that our recombinant SARS-2-PLpro cleaved the substrate and produced two fragments. When we treated the reaction with the three test compounds, marginal inhibition of SARS-2-PLpro cleavage was observed at the high concentration of 200 μM (Figure 7B).
Figure 6.

The three selected classes of compounds show potency against SARS-CoV-2
(A–C) Dose-response curve determined by the SARS-2-PLpro protease assay using the three selected compounds: 1 (A), 2 (B), and 3 (C).
(D–F) Efficacy and cytotoxicity of the three compounds in the SARS-CoV-2 infection cell model. In total, 10 different concentrations of compounds were tested in the immunofluorescence assay for the compounds 1 (D), 2 (E), and 3 (F), and IC50 and cytotoxicity values were calculated through curve fitting analysis using Prism 6. Each data point represents the mean of triplicate assays with ±SEM.
Figure 7.

Compounds inhibit polyprotein substrate cleavage by SARS-2-PLpro
(A) Polyprotein cleavage assay performed with 1 mM protease and 5 μg of substrate protein. Arrows point at cleavage products.
(B) Cleavage assay performed in the presence of 200 μM of compounds 1, 2, and 3. untr: untreated.
Conclusions
We identified novel PLpro inhibitors with no naphthalene group, which is important for the binding of GRL0617 to SARS-PLpro; and we found by molecular docking that each compound has unique binding properties toward the target protein contributing to target selectivity. Like GRL0617, all three inhibitors were predicted to be non-competitive inhibitors, as in our molecular docking study and MDS they do not fit on the catalytic triad but instead fit on the cleft near the BL2 loop. This result suggests that the BL2 loop is an important target for the treatment of coronaviruses. Furthermore, each class of coronaviruses shows variation in key residues constituting the cleft and the region near the BL2 loop; and this variation can provide structural restriction for accessing the BL2 loop thereby resulting in target specificity. Our compound 1 was found to inhibit both MERS-PLpro and SARS-PLpro/SARS-2-PLpro, while 2 and 3 were selective toward MERS-PLpro. We note that the expected binding of 1 to the target can provide information on structurally important scaffolds for the development of broad-spectrum inhibitors for the PLpro of coronaviruses. Unlike 1, our docking study revealed that 2 and 3 were expected to bind to a cavity branching from the cleft away from the BL2 loop of MERS-PLpro. This binding property can therefore provide insight to develop MERS-PLpro-specific inhibitors. Vaccines are prophylactic and are readily available in limited countries. Currently, in addition to neutralizing antibodies, several non-specific medications including the combinatory treatment of lopinavir-ritonavir and interferon-β1b have been used in the clinic. However, their efficacies can be compromised. Therefore, in view of an urgent need to develop therapeutics targeting viral proteins, our compounds may be promising candidates for drug development.
Limitations of the study
In our present study, we have identified novel structural classes of MERS-PLpro inhibitors, and in our docking analysis and MDS, we have shown that these compounds interact with the region near the BL2 loop of PLpro. Importantly, we found that the mode of binding to the region near the BL2 loop is essential for broad-spectrum potency, which was validated with a cell-based infection model including SARS-CoV-2. However, our study could expand further; although we expect that our compounds could be further engineered as a therapeutic option against SARS-CoV-2, we have no animal data required for pharmaceutical deferment. In addition, performing co-crystallography using our compounds and the PLpro could further validate the mode of action of our inhibitors.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-MERS-CoV spike antibody | Sino Biological | Cat#40069-R723; RRID: AB_2860455 |
| Mouse anti-SARS-CoV spike S1 antibody | Sino Biological | Cat#40150-MM02; RRID: AB_2860459 |
| Rabbit anti-SARS-CoV-2 protein antibody | Sino Biological | Cat#40143-T62; RRID: AB_2892769 |
| Anti-ubiquitin antibody | Cell Signaling Technology | Cat#3936S; RRID: AB_331292 |
| Bacterial and virus strains | ||
| MERS-CoV | Korea Centers for Disease Control and Prevention | GenBank accession#KT029139.1 |
| SARS-CoV | Laboratory of Prof. JSM Peiris (University of Hong Kong) | HK39849 |
| SARS-CoV-2 | National Culture Collection for Pathogens | NCCP43326 |
| Human coronavirus 229E | American Type Culture Collection | Cat#VR-740 |
| Human coronavirus OC43 | American Type Culture Collection | Cat#VR-15558 |
| Chemicals, peptides, and recombinant proteins | ||
| Library of compounds selected on the basis of QED scores | Chembridge library | N/A |
| Antiviral library | ChemDiv | N/A |
| Protease inhibitor library | ChemDiv and Life Chemicals | N/A |
| Natural product standard library | Analyticon and Chemstrong | N/A |
| LOPAC® 1280 library | Sigma-Aldrich | LO1280 |
| FDA-approved drug library | Selleckchem | Cat#L1300 |
| RLRGG-AMC peptides | Bachem | Cat#I-1690.0025 |
| 6-Thioguanine | Sigma-Aldrich | Cat#A4882 |
| Experimental models: Cell lines | ||
| Chlorocebus sabaeus: Vero cells | American Type Culture Collection | Cat#CCL-81; RRID: CVCL_0059 |
| Human: MRC-5 cells | American Type Culture Collection | Cat#CCL-171; RRID: CVCL_0440 |
| Human: Huh7 cells | Korean Cell Line Bank | Cat#10104 |
| Oligonucleotides | ||
| Primer: upE Forward: GCAACGCGCGATTCAGTT Reverse: GCCTCTACACGGGACCCATA |
This paper | N/A |
| Primer: upE-Probe 6-carboxyfluorescein [FAM]-CTCTTCACATAATCGCCCCGAGCTCG-6-carboxy-N,N,N,N′-tetramethylrhodamine [TAMRA] |
This paper | N/A |
| Primer: HCoV-229E Forward: CGCAAGAATTCAGAACCAGAG Reverse: GGCAGTCAGGTTCTTCAACAA |
Esposito et al., 2006 | N/A |
| Primer: HCoV-OC43 Forward: ACTCAAATGAATTTGAAATATGC Reverse: TCACACTTAGGATAATCCCA |
Quiroga et al., 2008 | N/A |
| Software and algorithms | ||
| Image Mining 3.0 plug-in | This paper | http://im.ip-korea.org/ |
| Flare 6.0.1 | Cheeseright et al., 2006 | https://www.cresset-group.com/ |
| Schrodinger (v. 2019-2) | Schrodinger | https://www.schrodinger.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wonsik Lee (wonsik.lee@skku.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Virus strains
Patient-derived MERS-CoV (passage 4, MERS-CoV/KOR/KNIH/002_05_2015; GenBank: KT029139.1) was provided by the Korea Centers for Disease Control and Prevention. The SARS-CoV (strain HK39849) was kindly provided by Prof. JSM Peiris from the University of Hong Kong. SARS-CoV-2 for this study was provided by the National Culture Collection for Pathogens (NCCP43326). Human coronavirus 229E and OC43 (HCoV-229E and HCoV-OC43) were purchased from ATCC. The viral strains are also listed in KRT. All experiments with MERS-CoV, SARS-CoV, and SARS-CoV-2 were performed in BSL-3 facility at Institut Pasteur Korea following the guidelines of the Korea National Institute of Health (KNIH) using the BSL-3 protocol approved by the KCDC.
Cell lines
Vero cells (ATCC, #CCL-81) were cultured in Opti-PRO™ SFM (Gibco, #12309019) supplemented with 4 mM L-glutamine (Gibco) and 1× Antibiotic-Antimycotic (Gibco). MRC-5 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% fetal bovine serum (FBS, Gibco, #16000-044) and 1% penicillin/streptomycin (Sigma-Aldrich, #P4458). Huh7 cells were cultured in Dulbecco`s Modified Eagle Medium (DMEM) supplemented with 2% FBS (Gibco) and 1× Antibiotic-Antimycotic (Gibco). The cell lines are also listed in KRT. All mammalian cells were maintained at 37°C in a humidified 5% CO2 incubator unless otherwise noted.
Method details
Chemical compounds and analogs
A 29,938-compound library originated from an in-house library was used for the high-throughput screen. The library consists of 16,960 compounds selected from the Chembridge library on the basis of quantitative estimates of drug-likeness (QED) scores, 3,200 compounds from the antiviral library (ChemDiv), 4,480 from the protease inhibitor library (ChemDiv or Life Chemicals), 3,040 from the natural product standard library (Analyticon or Chemstrong), 1,280 from the LOPAC library (Sigma-Aldrich), and 978 from the FDA-approved library (Selleckchem). The synthetic compounds were at a stock concentration of 10 mM in DMSO and the natural products were at that of 2 mg/mL in DMSO. RLRGG-AMC peptides were purchased from Bachem. Selected compounds for the analog study were purchased as described in Figure S13.
PLpro protease expression and purification
The coding sequences of MERS-CoV PLpro (MERS-PLpro, aa 1,484–1,802) and SARS-CoV PLpro (SARS-PLpro, aa 1,541–1,854) and SARS-CoV-2 PLpro (SARS-2-PLpro, aa 746–1,060) were obtained from the polyprotein sequences (GenBank: AAY60792.1, KT029139.1, and YP_009725299.1, respectively). Codon-optimized genes (IDT) were cloned into the NdeI/XhoI multicloning sites of the plasmid pET-21a and DNA sequences were verified. The mutations (T251A, T310A, T251A-T310A) were introduced into the wild-type MERS-PLpro by PCR, and the mutants of MERS-PLpro were cloned as above. The introduced mutations were verified by sequencing. Then, the plasmids were transformed into the T7 express lysY/Iq competent E. coli strain (NEB). Ampicillin-resistant colonies were grown at 37°C and induced by the addition of 0.5 mM isopropyl b-D-1-thiogalactopyranoside (IPTG) at OD ∼0.7. After additional culture for 14 h at 20°C, cells were harvested by centrifugation (5,000 g, 10 min) and stored at −70°C. Cell pellets were resuspended in binding buffer (50 mM Tris-Cl, pH 8.0, 500 mM NaCl, 5% glycerol, 20 mM imidazole, and 5 mM β-mercaptoethanol) and lysed by sonication using a Sonic Dismembrator (Fisher Scientific). The cleared lysate was subjected to affinity chromatography using Ni Sepharose® 6 Fast Flow beads (Cytiva). Stepwise gradient elution was carried out using the elution buffer with increasing concentrations of imidazole (50–500 mM). Fractions containing pure C-terminal hexahistidine-tagged PLpro proteins (>95%) were pooled and dialyzed against the storage buffer containing 20 mM Tris-Cl, pH 8.0, 10% glycerol, 100 mM NaCl and 5 mM β-mercaptoethanol, followed by concentration using Amicon® Ultra-15 centrifugal filter units (Merck). The purity and concentration of proteins were determined by SDS-PAGE and a NanoDrop spectrophotometer, respectively.
Fluorescence-based PLpro protease assay
The general procedures were performed as described (Lee et al., 2015; Ratia et al., 2008). To optimize the MERS-PLpro protease assay conditions, varying concentrations of Z-RLRGG-AMC substrate (0.8–100 μM) diluted in assay buffer containing 50 mM HEPES, pH 7.5, 0.01% Triton X-100, 0.1 mg/mL BSA and 5 mM dithiothreitol (DTT) were mixed with varying concentrations of purified MERS-PLpro (0.3–10 μM) diluted in the reaction buffer. Reaction mixtures with a total volume of 40 μL were added to each well of a 384-well non-binding black plate (Greiner Bio-one). Following 1 h incubation at room temperature, fluorescence intensity was monitored at 360 nm (excitation) and 450 nm (emission) using a FlexStation® 3 microplate reader (Molecular Device). The data were plotted using a Prism® 6 analysis tool (GraphPad Software). For optimization of the SARS-PLpro protease assay, all the procedures were the same except for the enzyme concentration range (0.02–0.63 μM) and buffer composition (2 mM reduced L-glutathione instead of 5 mM DTT). The 50% inhibitory concentration (IC50) of a reference compound (6-TG) was determined in 40 μL reaction mixtures containing 40 μM Z-RLRGG-AMC substrate, 2.5 μM MERS-PLpro or 0.02 μM SARS-PLpro/SARS-2-PLpro and varying concentrations of 6-TG compound (1.953–1,000 μM), which were subjected to the same measurement and analysis procedures mentioned above. For determination of efficacy of test compounds on MERS-PLpro T251A, 0–50 μM of compounds and final concentrations of 2.5 μM and 3.125 μM were used for the enzyme and AMC, respectively. For determination of efficacy of test compounds on SARS-2-PLpro, 0–200 μM of compounds and final concentrations of 0.625 μM and 3.125 μM were used for the enzyme and AMC, respectively. The assay was performed in the same assay buffer as for MERS-PLpro.
Primary high-throughput screening
The 29,938-compound library was screened against MERS-PLpro. The screen was carried out in monoplicate. Twenty microliters of MERS-PLpro enzyme (2 μM final concentration) diluted in the assay buffer was added to the wells to which 30 μL of the compounds were dispensed using a JANUS Automated Workstation (Perkin-Elmer). Then, 10 μL of Z-RLRGG-AMC (40 μM final concentration) diluted in the assay buffer was added to initiate the enzyme reaction, after which the assay plates were incubated at room temperature for 1 h. Fluorescence intensity was measured in the same manner as mentioned above. The screening data were processed by the Gyeonggi Bio-center Assay Management System (GAMS) derived from the Pipeline Pilot protocols (Accelrys). The average Z′ factor (Z' = 1–3(σp+σn)/|μp-μn|, where μ and σ are the average and standard deviation, respectively, of the signals of positive (p) and negative (n) control) of the assay plates was calculated to be 0.91.
Cytotoxicity assay
Cell cytotoxicity measurement was performed using the CellTiter-Glo™ assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, Huh7 cells were seeded in a white bottom 384-well plates at a density of 4×103 cells per well. After 24 h, cells were treated with 20 μM of compounds and incubated for 12 h, after which cells were treated with the CellTiter-Glo™ assay reagent. The luminescence was measured using an EnVision® microplate reader (Perkin-Elmer).
Cell-based immunofluorescence infection assay
Vero cells were seeded at 1.2×104 cells per well in black 384-well μClear plates (Greiner bio-one) at 24 h prior to experiment. Test compounds were added to each well before virus infection at a final concentration of 10 μM, and the DMSO concentration was kept at 0.5% or lower. For infection, plates were transferred into a biosafety level 3 (BSL-3) containment laboratory and MERS-CoV at an MOI of 0.0625, SARS-CoV at an MOI of 0.05, and SARS-CoV-2 at an MOI of 0.08 was added. Infection was arrested at 24 h post infection (hpi) by adding 4% PFA followed by immunofluorescence staining. MERS-CoV, SARS-CoV, and SARS-CoV-2 infection was detected using a rabbit anti-MERS-CoV spike antibody (Sino Biological), a mouse anti-SARS-CoV spike S1 antibody (Sino Biological), or a rabbit anti-SARS-CoV-2 protein antibody (Sino Biological), respectively (Jeon et al., 2020). For fluorescence staining, Alexa Fluor 488 goat anti-rabbit or mouse IgG (H+L) secondary antibody (Molecular Probes) was used. Cell viability was evaluated with the Hoechst 33342 stain (Molecular Probes). Data were acquired by taking images at 20× magnification using an Operetta high-content imaging system (Perkin-Elmer), and the images were analyzed by an in-house developed software Image Mining 3.0 (IM 3.0) plug-in. To validate the assay, a dose-response curve (DRC) experiment with the two compounds chloroquine diphosphate and lopinavir, with known antiviral activities against MERS-CoV and SARS-CoV, respectively, was performed (de Wilde et al., 2014). To validate the assay against SARS-CoV-2, DRC analyses with three compounds, remdesivir, chloroquine and lopinavir, with known antiviral activities against SARS-CoV-2 were performed. Test compounds were evaluated by duplicate 10-point DRC experiments at a concentration range of 0.0977–50 μM. Percent inhibition (PI) was normalized as follows: PI = [1 – (INtest – μINmock)/(μINvehicle – μINmock)] × 100, where INtest is percent infection of test compound, μINmock is average of mock, and μINvehicle is the average of infection control. Percent viability (PV) was calculated as follows: PV = (CNtest/μCNmock) × 100%, where CNtest and μCNmock are the viable cell numbers in the treatment groups and average viable cell number in the mock group, respectively. After normalization, the 50% efficient concentration (EC50) and the 50% cytotoxicity concentration (CC50) were calculated with nonlinear regression using GraphPad Prism® software. Selective index (SI) was calculated by dividing CC50 by EC50. All experiments with MERS-CoV, SARS-CoV, and SARS-CoV-2 were performed in BSL-3 facility at Institut Pasteur Korea following the guidelines of the Korea National Institute of Health (KNIH) using the BSL-3 protocol approved by the KCDC.
Viral RNA quantification
Vero cells were seeded at a density of 3×105 cells per well in 24-well plates (Corning) at 24 h prior to experiments. Test compounds were added to each well before MERS-CoV infection. The final concentrations of test compounds were 1, 3, 10, 30 μM, and the DMSO concentration was kept at 0.5% or lower. The plates were moved to the BSL-3 containment laboratory prior to inoculation with MERS-CoV at an MOI of 0.0625. After 24 h, the cells were washed with PBS (Welgene, Korea) and lysed in RLT buffer of the RNeasy® mini Kit (QIAGEN) supplemented with 1% β-mercaptoethanol (Sigma-Aldrich). The cell lysates were moved to the BSL-2 containment laboratory where total RNA was isolated using the RNeasy® mini Kit and eluted in 50 μL of DEPC-treated water. MERS-CoV viral RNA from the infected cells was quantified by multiplex real-time reverse-transcription polymerase chain reaction (RT-PCR) using the TOPscriptTM One-step RT-PCR DryMIX kit (Enzynomics, Korea). Each 25 μL reaction contained 1 μL of RNA, 400 nM of primer sets and probes specific for the upstream E (upE) gene of MERS-CoV and for GAPDH, an internal control. The primers and probes used for the analysis of the upE and GAPDH were as follows:
| upE | F: GCAACGCGCGATTCAGTT R: GCCTCTACACGGGACCCATA |
| upE-Probe | 6-carboxyfluorescein [FAM]-CTCTTCACATAATCGCCCCGAGCTCG-6-carboxy-N,N,N,N′-tetramethylrhodamine [TAMRA] |
| GAPDH | F: GAAGGTGAAGGTCGGAGTCAAC R: CAGAGTTAAAAGCAGCCCTGGT |
| GAPDH-Probe | 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein [JOE]-TTTGGTCGTATTGGGCGCT-6-TAMRA |
Amplification was performed in the ViiA7 Real-Time PCR System (Thermo Scientific) with the following settings: 50°C for 30 min, 95°C for 3 min, and 40 cycles of 95°C for 30 sec and 58°C for 1 min. The Ct values of the multiplex RT-PCR assay were compared with monoplex values to optimize the conditions. The copy numbers of the targeted upE gene were obtained by normalizing with GAPDH and were presented as fold change by the threshold cycle ΔΔCT method (Corman et al., 2012). For the time-of-addition experiment, MERS-CoV infected cells were treated with a compound 1 h prior to infection, at the time of infection, or 1–6 h post-infection and the upE gene was measured using the same PCR procedure as mentioned above.
HCoV-229E/HCoV-OC43 infection assay
To analyze the antiviral effects of the compounds against HCoV-229E and HCoV-OC43, MRC-5 cells were infected with virus in the absence or presence of the compounds (Ziebuhr and Siddell, 1999). After treatment, total RNA was isolated from MRC-5 cells using the TRIzol reagent (Invitrogen) at the designated time points. cDNA synthesis and RT-PCR were carried out using 2× One Step RT-PCR MasterMix with SYBR Green (MGmed, Korea) according to the manufacturer’s instructions. Two hundred nanogram of RNA of each sample was equally added to 20 μL of reaction mixture. RT-PCR was performed using a LightCycler® 480 instrument at the following settings: 50°C for 30 min, 95°C for 10 min, and 40 cycles of 95°C for 5 s and 60°C for 30 s (Lu et al., 2012). The primer sequences of target genes and internal control (GAPDH) were as follows:
| HCoV-229E | F: CGCAAGAATTCAGAACCAGAG R: GGCAGTCAGGTTCTTCAACAA |
| HCoV-OC43 | F: ACTCAAATGAATTTGAAATATGC R: TCACACTTAGGATAATCCCA |
| GAPDH | F: GTCGGAGTCAACGGATT R: AAGCTTCCCGTTCTCAG |
Ubiquitination assay by western blot
Huh7 cells were seeded at a density of 1×106 cells per well in 6-well plates at 24 h prior to experiments. The plates were moved to the BSL-3 containment laboratory where cells were infected with MERS-CoV at an MOI of 0.01 and treated with test compounds at the concentrations of 50 and 100 μM. At 24 h post-infection, cells were washed with DPBS and lysed with cell lysis buffer (Cell Signaling Technology) containing 1% Triton X-100, Laemmli sample buffer (Sigma-Aldrich), protease inhibitor cocktail (Cell Signaling Technology), and phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich) after which cells were scraped. Supernatant was collected by centrifugation and boiled at 95°C for 5 min after which the samples were transported to the BSL-2 facility for further analysis. Protein concentrations in supernatants were determined by the BCA assay (Thermo Fisher Scientific). The samples were electrophoresed on 4–20% gradient gels and transferred to polyvinylidene difluoride (PVDF) membranes using Trans-Blot® TurboTM transfer pack (Bio-Rad). The membranes were incubated with blocking solution (5% non-fat milk in TBS) for 1.5 h at room temperature, followed by incubation with primary antibodies in 2% non-fat milk in Tris-buffered saline containing 0.05% Tween 20, at 4°C overnight. Antibodies used were mouse anti-ubiquitin antibody (Cell Signaling Technology) at 1:1,000 dilution and rabbit anti-B actin antibody (Cell Signaling Technology) at a dilution of 1:5,000 for loading control. Secondary antibodies were HRP-linked horse anti-mouse IgG (Cell Signaling Technology) and goat anti-rabbit IgG (Cell Signaling Technology) antibodies. Immunoreactivity was detected using SuperSignal West Pico PLUS chemiluminescent substrate (Fisher Scientific). Protein bands were visualized by Chemidoc XRS (Bio-Rad) and intensities of protein bands were quantified using ImageJ.
Gel-based polyprotein cleavage assay
The gel-based polyprotein cleavage assay was performed as previously described (Lim et al., 2021). TLKGG//APTKV (nsp2/3 junction) used as a substrate was modified by attaching the GST-tag and MBP-tag to the N- and C-terminus, respectively (Addgene ID 169195) after which the constructs were expressed in BL21(DE3). The protein was purified as described above in PLpro protease expression and purification section. The enzyme was pre-incubated at a concentration of 1 μM with the selected inhibitor compounds for 10 min. Gel-based assay substrates were added at a concentration of 5 μM and incubated at RT for 24 h in an assay buffer (50 mM HEPES, pH 7.6, 2 mM DTT, 10% glycerol, 0.02% Tween-20) before being run through a gel.
Molecular docking study
Protein sequences and their structure information (SARS-PLpro PDB: 3E9S (Ratia et al., 2008), MERS-PLpro PDB: 4RF1 (Bailey-Elkin et al., 2014)) were downloaded from the Protein Data Bank followed by the removal of water molecules and metal ions using the Schrodinger (v. 2019-2) protein preparation wizard. Test compounds were drawn using the Schrodinger 3D builder and further converted to ligands using the ligand preparation wizard. For docking, the grid box was created using the Schrodinger receptor grid generation wizard with the inner box size set to 16 × 16 × 40 and outer box size set to 52 × 52 × 76. Docking was performed using the Schrodinger XP Glide ligand docking tool, and the generated images were further modified to indicate the important residues and structures.
Molecular dynamics simulation
To evaluate the stability of the molecular docking of three compounds with MERS-PLpro, molecular dynamics simulation (MDS) was performed using Cresset’s Flare 6.0.1. The MDS was initiated using the molecular docking poses of each compound. Under the Amber force field (version GAFF2), charges were calculated by the AM1-BCC method (Jakalian et al., 2002). A solvent system of MDS was built by using a truncated octahedron shape box. Explicit SPC/E water and two chloride molecules were used for solvent and neutralization, respectively. For the equilibration, the default values of each parameter were used. Each simulation was performed for 80 ns at the 300K and 1 bar using a graphics processing unit (GPU) platform. To measure the stability of the protein-ligand complex, the root mean square deviation (RMSD) was calculated every 10 ps.
Quantification and statistical analysis
Student’s t-test was performed for statistical analyses between experimental groups using Prism® 6 software. The results were expressed as the mean ±SEM or ±SD for duplicate, triplicate, or quadruple assays. Differences between groups were inferred as significant for p < 0.05. Statistical details of individual experiments can be found in the figure legends.
Acknowledgments
The authors thank all members of Wonsik Lee’s laboratory and the Bio Center of Gyeonggido Business and Science Accelerator for scientific and experimental discussion. This work was supported by a grant from an R&D Support Program of the Gyeonggi provincial government to K.J., Korea Disease Control and Prevention Agency (KCDC 2021-ER1602-00) to W.L., and National Research Foundation of Korea (NRF) funded by the Korean Government (MSIT) (NRF-2017M3A9G6068245 and NRF-2022M3A9J1081343) to S.K.
Author contributions
Conceptualization, K.J., S.K., and W.L.; methodology, K.J., S.K., J.K., J.C., S.H., I.K., S.O., J.L., H.P., and W.L.; formal analysis, K.J., S.K., J.K., J.C., S.H., I.K., S.O., and W.L.; investigation, K.J., S.K., and W.L.; writing – original draft, W.L.; writing – review & editing, K.J., J.C., S.K., and W.L.; visualization, K.J., S.K., J.C., and W.L.; supervision, K.J. and W.L.; project administration, K.J.
Declaration of interests
The views expressed in this publication are those of the authors.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105254.
Contributor Information
Seungtaek Kim, Email: seungtaek.kim@ip-korea.org.
Wonsik Lee, Email: wonsik.lee@skku.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Baez-Santos Y.M., Mielech A.M., Deng X., Baker S., Mesecar A.D. Catalytic function and substrate specificity of the papain-like protease domain of nsp3 from the Middle East respiratory syndrome coronavirus. J. Virol. 2014;88:12511–12527. doi: 10.1128/JVI.01294-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M., St John S.E., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Elkin B.A., Knaap R.C.M., Johnson G.G., Dalebout T.J., Ninaber D.K., van Kasteren P.B., Bredenbeek P.J., Snijder E.J., Kikkert M., Mark B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014;289:34667–34682. doi: 10.1074/jbc.M114.609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseright T., Mackey M., Rose S., Vinter A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- Chen X., Chou C.Y., Chang G.G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 2009;19:151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- Cho J.K., Curtis-Long M.J., Lee K.H., Kim D.W., Ryu H.W., Yuk H.J., Park K.H. Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorg. Med. Chem. 2013;21:3051–3057. doi: 10.1016/j.bmc.2013.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Chien C.H., Han Y.S., Prebanda M.T., Hsieh H.P., Turk B., Chang G.G., Chen X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008;75:1601–1609. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy K.T., Wong A.Y.L., Kaewpreedee P., Sia S.F., Chen D., Hui K.P.Y., Chu D.K.W., Chan M.C.W., Cheung P.P.H., Huang X., et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786. doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Eckerle I., Bleicker T., Zaki A., Landt O., Eschbach-Bludau M., van Boheemen S., Gopal R., Ballhause M., Bestebroer T.M., et al. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Euro Surveill. 2012;17:20285. doi: 10.2807/ese.17.39.20285-en. [DOI] [PubMed] [Google Scholar]
- Corti D., Purcell L.A., Snell G., Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184:4593–4595. doi: 10.1016/j.cell.2021.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A.M., Galiano M., Gorbalenya A.E., Memish Z.A., et al. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., Jochmans D., Posthuma C.C., Zevenhoven-Dobbe J.C., van Nieuwkoop S., Bestebroer T.M., van den Hoogen B.G., Neyts J., Snijder E.J. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014;58:4875–4884. doi: 10.1128/AAC.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito S., Bosis S., Niesters H.G.M., Tremolati E., Begliatti E., Rognoni A., Tagliabue C., Principi N., Osterhaus A.D.M.E. Impact of human coronavirus infections in otherwise healthy children who attended an emergency department. J. Med. Virol. 2006;78:1609–1615. doi: 10.1002/jmv.20745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Gong G., Grum-Tokars V., Mulhearn D.C., Baker S.C., Coughlin M., Prabhakar B.S., Sleeman K., Johnson M.E., Mesecar A.D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008;18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Takayama J., Aubin Y., Ratia K., Chaudhuri R., Baez Y., Sleeman K., Coughlin M., Nichols D.B., Mulhearn D.C., et al. Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome-coronavirus papain-like protease. J. Med. Chem. 2009;52:5228–5240. doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh N.D., Hodcroft E.B., Fauver J.R., Phelan A.L., Cevik M. Public health actions to control new SARS-CoV-2 variants. Cell. 2021;184:1127–1132. doi: 10.1016/j.cell.2021.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.S., Chang G.G., Juo C.G., Lee H.J., Yeh S.H., Hsu J.T.A., Chen X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry. 2005;44:10349–10359. doi: 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- Hartenian E., Nandakumar D., Lari A., Ly M., Tucker J.M., Glaunsinger B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020;295:12910–12934. doi: 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J.A., Verma N., Harris R.C., Liu R., Shen J. Assessment of proton-coupled conformational dynamics of SARS and MERS coronavirus papain-like proteases: implication for designing broad-spectrum antiviral inhibitors. J. Chem. Phys. 2020;153:115101. doi: 10.1063/5.0020458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- Jeon S., Ko M., Lee J., Choi I., Byun S.Y., Park S., Shum D., Kim S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020;64:e00819–e00820. doi: 10.1128/AAC.00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilianski A., Mielech A.M., Deng X., Baker S.C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013;87:11955–11962. doi: 10.1128/JVI.02105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Lei H., Santarsiero B.D., Gatuz J.L., Cao S., Rice A.J., Patel K., Szypulinski M.Z., Ojeda I., Ghosh A.K., Johnson M.E. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 2015;10:1456–1465. doi: 10.1021/cb500917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Mesters J.R., Drosten C., Anemüller S., Ma Q., Hilgenfeld R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 2014;109:72–82. doi: 10.1016/j.antiviral.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C.T., Tan K.W., Wu M., Ulferts R., Armstrong L.A., Ozono E., Drury L.S., Milligan J.C., Zeisner T.U., Zeng J., et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp3 papain-like protease. Biochem. J. 2021;478:2517–2531. doi: 10.1042/BCJ20210244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Ménard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Yu X., Wang W., Duan X., Zhang L., Zhou W., Xu J., Xu L., Hu Q., Lu J., et al. Characterization of human coronavirus etiology in Chinese adults with acute upper respiratory tract infection by real-time RT-PCR assays. PLoS One. 2012;7:e38638. doi: 10.1371/journal.pone.0038638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.Y., Kim J.H., Kim Y.M., Jeong H.J., Kim D.W., Park K.H., Kwon H.J., Park S.J., Lee W.S., Ryu Y.B. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem. 2012;20:5928–5935. doi: 10.1016/j.bmc.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga M.A., Cappuccio J., Piñeyro P., Basso W., Moré G., Kienast M., Schonfeld S., Cáncer J.L., Arauz S., Pintos M.E., et al. Hemagglutinating encephalomyelitis coronavirus infection in pigs, Argentina. Emerg. Infect. Dise. 2008;14:484–486. doi: 10.3201/eid1403.070825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Kilianski A., Baez-Santos Y.M., Baker S.C., Mesecar A. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10:e1004113. doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Peñaranda S., Bankamp B., Maher K., Chen M.H., et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Sheahan T.P., Sims A.C., Zhou S., Graham R.L., Pruijssers A.J., Agostini M.L., Leist S.R., Schäfer A., Dinnon K.H., 3rd, Stevens L.J., et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020;12:eabb5883. doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toots M., Yoon J.J., Cox R.M., Hart M., Sticher Z.M., Makhsous N., Plesker R., Barrena A.H., Reddy P.G., Mitchell D.G., et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci. Transl. Med. 2019;11:eaax5866. doi: 10.1126/scitranslmed.aax5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J.M., Wolthers K.C., Wertheim-van Dillen P.M.E., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Siddell S.G. Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virol. 1999;73:177–185. doi: 10.1128/JVI.73.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.