

Abstract
Genetic testing is increasingly used in the workup and diagnosis of kidney disease and kidney-related disorders of undetermined cause. Out-of-pocket costs for clinical genetic testing have become affordable, and logistical hurdles overcome. The interest in genetic testing may stem from the need to make or confirm a diagnosis, guide management, or the patient’s desire to have a more informed explanation or prognosis. This poses a challenge for providers who do not have formal training in the selection, interpretation, and limitations of genetic tests. In this manuscript, we provide detailed discussion of relevant cases in which clinical genetic testing using a kidney gene panel was applied. The cases demonstrate identification of pathogenic variants for monogenic diseases—contrasting them from genetic risk alleles—and bring up diagnostic limitations and diagnostic utility of these tests in nephrology. This review aims to guide clinicians in formulating pretest conversations with their patients, interpreting genetic variant nomenclature, and considering follow-up investigations. Although providers are gaining experience, there is still risk of testing causing more anxiety than benefit. However, with provider education and support, clinical genetic testing applied to otherwise unexplained kidney-related disorders will increasingly serve as a valuable diagnostic tool with the potential to reshape how we consider and treat many kidney-related diagnoses.
Keywords: genetics, gene panel, genetic testing, genetics, next-generation sequencing
Introduction
Despite standard workup, some patients with kidney disorders may not have a clear diagnosis. Depending on the year, 0.3%–4% of incident ESKD patients have “unknown” listed as their underlying etiology (1), with 20%–27% of patients with CKD/ESKD reporting a positive family history (2–5). Depending on the patient cohort, 10%–65% of patients with a family history may have a genetic cause identified (6–8), whereas up to up to 24% of a more general CKD cohort may have a genetic cause identified (9–11). Implementation of genetic testing has been fruitful, demonstrating notable yield in patients with nephrotic syndrome (12,13), focal segmental glomerulonephritis (14,15), nephrolithiasis (16,17), congenital anomalies of the kidney and urinary tract (18), and cystic kidney disease (19). Genetic sequencing of panels of hundreds of kidney-related genes are now available from multiple commercial or institutional genetic testing services for less than $500 out-of-pocket cost, with sample collection and professional genetic counseling services included. This is affordable to many patients and thus can be more readily offered in a variety of clinical settings (20). Groopman et al. (21) suggest that genetic findings can positively impact clinical care by making a diagnosis or further characterizing an existing clinical diagnosis, and guiding subspecialty referrals. They show that in some—particularly pediatric—cases, a genetic diagnosis may have an important impact on treatment decisions. A genetic diagnosis can also affect screening of living related donors for transplantation (22,23). For some patients, a significant benefit may come simply from providing an explanation for their condition.
Many nephrology providers do not feel comfortable counseling, ordering, or interpretating genetic testing (24). Although a one-time patient referral to a center with genetic kidney disease expertise to make a diagnosis is an option, we expect that the increasing role of genetics in nephrology will push most clinicians to want to over time develop a comfort level with this themselves. This review provides a practical framework of important concepts and nomenclature to utilize in genetic testing of patients with kidney disorders of unclear etiology. For excellent reviews of genetic diagnoses found most often, patient characteristics that predict highest likelihood of genetic diagnosis, and the types of genetic testing useful in kidney disease, we direct the reader to additional references (9,25–27). In this review, we detail the basics of genetic testing in clinical care.
Pretest Preparation and Counseling
Ordering clinicians should provide basic pretest communication and—to the extent of their comfort level—counseling, recognizing that patient education by a certified genetics counselor will be necessary in some circumstances (28). In preparation for pretest discussion, the ordering clinician should know (1) the type of test ordered (2), where to find the list of genes tested and available counseling services, and (3) whether the test will return findings regarding “actionable genes” such as cancer risk or cardiovascular disease that may prompt further testing in addition to genes relevant to kidney-related disease (29). For many testing services, the clinician will be asked to decide in advance whether he or she wishes for variants of unknown significance (VUS) to be included in the results report or withheld. This is of relevance for certain kidney phenotypes and is discussed below.
With the above in mind, we recommend the following be conveyed to the patient as part of pretest counseling (30,31):
The clinician’s expectation of what may or may not be found, and how that would affect management. In other words, define a gestalt pretest probability of making a possible or definitive genetic diagnosis. Also, convey whether cancer risk might be found and reported or will not be considered as part of the test.
Genetic testing may not always provide a definitive result. Sometimes, the results are inconclusive due to either a VUS or the lack of a variant that could be due to the sequencing modality missing it. Genetic testing of additional family members may be a helpful aid in the interpretation of a variant.
A positive test result may have implications for the health of other family members. See discussion of “Is the variant(s) present in all affected family members?” and case 4 below.
A federal law, Genetic Information Nondiscrimination Act of 2008, prohibits health insurance and most employment discrimination based on genetic data, but it does not protect against denial of coverage for other types of coverage such as life and disability insurance (32).
We will discuss the nomenclature used to describe variants (cases 1 and 2) and how the pretest probability of finding a variant will aid in variant interpretation (case 2). Case 3 illustrates interpreting a risk allele in contrast to a pathogenic variant for a monogenic disease. Case 4 illustrates a genetic finding that could have been missed by targeted next-generation sequencing (NGS) and prompts our outlining clinical scenarios where alternative or follow-up genetic testing many be necessary to confirm or detect a relevant variant. Overall, these cases highlight real-life findings and a patient-centered approach. We continue to refine our experience in genetic testing and reach out to certified genetics counselors or genetic specialists to help interpret and manage results when needed.
The Type of Genetic Test
Given the low cost, high sensitivity, and widespread availability of NGS to sequence multiple genes in parallel (also called massively parallel sequencing), this will be the first choice for most situations (21). A kidney gene panel refers to the assessment of multiple genes sequenced by NGS-based methods. Some testing services tailor the list of genes to the specified phenotype (i.e., nephrotic syndrome) (12,33), whereas others have a fixed panel evaluated for any kidney-related indication (8). These lists are often quite thorough; however, we encourage providers to review the panel to ensure the genes of interest are being evaluated. There are many commercially available genetic testing products, and many institutions may also have clinical genetic testing available.
For a NGS panel, DNA obtained from cells in a patient’s blood sample, cheek swab, or saliva is fragmented and barcoded for sequencing. Unless performing the more complete and costly whole genome sequencing, the next step is to capture and amplify the DNA fragments containing exons—the approximately 1% of the genome that encodes proteins. If the exons of all genes are collected and sequenced, this is known as whole exome sequencing (WES), whereas if the exons of only a subset of genes are used, this is known as “targeted” NGS (20,21,34,35). Either way, analysis is limited to the genes listed in the panel. At present, there are more than 3900 genes associated with monogenic disorders (36), with more than 625 of these associated with kidney or urologic disease (37).
How to Know What to Expect
Certain clinical presentations convey a near certainty that a causative genetic variant exists, whereas in others, genetic testing is being used as one of many tools in the workup. In the latter case, a conclusive positive finding is meaningful, but an inconclusive or negative finding may not be. Figure 1 illustrates factors that correlate with increased likelihood of a genetic diagnosis (6,25). Having an estimated gestalt pretest probability of the likelihood of a positive result is useful both for setting patient expectations and preparing to interpret the genetic test results.
Figure 1.

Patient factors that correlate with an increased likelihood of a genetic diagnosis.
Case 1
A 20-year-old male with normal renal function has a renal ultrasound as part of an initial evaluation for hypertension. Multiple large cysts are noted in each kidney, resulting in mild symmetric enlargement. A follow-up abdominal magnetic resonance imaging scan confirms the kidney cysts and notes absence of liver cysts. He has no known family history of autosomal dominant polycystic kidney disease (ADPKD), kidney failure, or kidney transplant.
Genetic testing is pursued because his cyst number and hypertension at this age point to a genetic cause, and diagnosis and prognosis are in question due to lack of family history.
His genetic testing reveals a heterozygous variant in the ALG8 gene and no other pathogenic or likely pathogenic variant in a panel of genes that includes PKD1, PKD2, PRKCSH, SEC63, GANAB, ALG8, ALG9, PKHD1, SEC61B, DNAJB11, IFT140, UMOD, REN, HNF1B (38,39) (Figure 2).
Figure 2.

Abdominal MRI of patient described in Case 1.
Genetic Variant Nomenclature
The Human Genome Variation Society establishes the standards for annotation of genetic variants (40,41). Figure 3 provides a guide to this interpretation. A variant is annotated with reference to the gene and the numerical position(s) in that gene’s coding DNA sequence and protein (chain of amino acids [AA]) sequence. The numbering for the coding DNA starts with nucleotides 1–3, which encode AA number 1, and continues for the length of the protein. Because some genes may have more than one way of splicing its exons together (“alternative transcripts”), which would affect the numbering for the coding DNA and protein sequence, a transcript name such as “NM_024079” may also be provided. Each of the 64 possible three-nucleotide combinations encodes 1 of 20 different AAs or a stop codon. AAs are abbreviated as three letters or a single letter (e.g., “Arg” or “R” for arginine), and the stop codon is abbreviated as “Ter” for termination or “X.” Table 1 illustrates the potential consequences that may result from the substitution, insertion, or deletion of one or a small number of nucleotides.
Figure 3.

Illustration of variant nomenclature for Case 1. Human gene names are capitalized and italicized.
Table 1.
Types of variants resulting from single or small oligonucleotide variants
| Variant Type | Definition | Nucleotide Change | Coding DNA Nomenclature “c. prefix” | Protein Nomenclature “p. prefix” | Comments | Implication |
|---|---|---|---|---|---|---|
| Substitution (synonymous) | Single nucleotide substitution resulting in unchanged AA | c.6A>G | p.Arg2= or p.Arg2Arg |
|
No change in AA; likely benign unless near splice junction | |
| Substitution (missense) | Single nucleotide substitution changing AA | c.5G>T | p.Arg2Ile |
|
AA changed; potentially pathogenic if significant change in important AA | |
| Substitution (nonsense) | Single nucleotide substitution creating a stop codon | c.4A>T | p.Arg2Ter or p.Arg2X |
|
Truncated protein; pathogenic in majority of disease mechanisms | |
| Insertion/deletion (frame shift) | Insertion or deletion of # nucleotides (# ≠ multiple of 3) | c.4delA | p.Arg2fs |
|
Truncated/altered protein; all AAs distal to frame shift are changed; pathogenic in majority of disease mechanisms | |
| Insertion/deletion (nonframeshift) | Insertion or deletion of # nucleotides (# = multiple of 3) | c.4_6delAGA | p.Arg2del |
|
AA deleted; potentially pathogenic if critical AA | |
| Substitution (splice variant) | Single nucleotide substitution | c.21 + 1G>T |
|
Truncated protein expected; splice site abolished if first or second intronic base is modified; pathogenic in majority of disease mechanisms |
AA, amino acid.
Genetic Variant Interpretation
Identified variants affecting a gene of interest are compared with large databases to determine whether they are common or rare in the general population and ethnic subgroups, and whether they have previously been associated with disease. The largest resource for assessing a variant’s frequency in the general population and ethnic subgroups is known as the Genome Aggregation Database (gnomAD) (42). GnomAD provides data from WES and whole genome sequencing for more than 120,000 individuals (more than 240,000 alleles). A minor allele frequency of >1% is considered common. Although a patient’s gene sequence may differ from the reference human genome with common variants, a clinical genetics report will only list variants that are sufficiently rare to be of possible relevance to the rare monogenic disease phenotype in question or that are specific established risk alleles. The American College of Medical Genetics and Genomics and the Association for Molecular Pathology provide criteria for the characterization of a variants as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign,” with “likely” being used when there is a >90% certainty that the variant may be either pathogenic or benign (40).
Truncating versus Nontruncating Variants
An important distinction in variant interpretation is the difference between a variant that results in a shortened (“truncated”) protein versus a variant that still encodes the full-length protein but with a focal modification. The “truncating variant” can be expected to encode a nonfunctional protein fragment that is typically degraded. Examples of truncating variants are (1) nonsense variant (premature termination [“stop”] codon), (2) frameshift “fs” variant (insertion or deletion of nucleotides not a multiple of three), (3) splice variant affecting either of the essential two intronic nucleotides adjacent to a splice junction, or (4) a large deletion of multiple exons (Table 1). In contrast, a “nontruncating” variant encodes a full-length protein that may be fully functional, fully dysfunctional, or partially functional, or alternatively take on a different function—a so-called gain of function. Examples of nontruncating variants include (1) a missense variant (single AA substitution), or (2) small nonframeshifting insertion/deletion of three or six or nine etc. nucleotides (Table 1).
Truncating variants can typically be characterized as likely pathogenic or pathogenic for disorders caused by loss-of-function mechanism even if not previously reported with disease and without family data. In contrast, nontruncating mutations will need to be previously associated with the disorder, found in multiple affected family members, or assessed by biologic assay if they are to be characterized as more than just a VUS.
The ALG8 variant in case 1 is a truncating variant, also known as loss of function variant, because the 179th AA, arginine, is replaced by a stop codon. The ALG8: c.535C>T:p.Arg179Ter variant has been previously reported (43) and can, in the opinion of the authors, be interpreted as pathogenic. The variant classification of “pathogenic” refers to the effect of the variant on the encoded protein but not necessarily whether a single “pathogenic” heterozygous variant can be expected to cause disease as discussed next.
Inheritance and Zygosity
In autosomal dominant (AD) conditions, a single pathogenic variant on one (maternal or paternal) allele—a “heterozygous” variant—results in disease. In autosomal recessive (AR) conditions, both (maternal and paternal) alleles must have a disease-causing variant (i.e., there are no normal copies of the gene). For recessive genotypes, if the two disease-causing alleles are identical, the variant is referred to as “homozygous,” whereas if the two disease-causing alleles are different, the patient’s genotype is referred to as “compound heterozygous.” Patients with a single pathogenic copy of a gene associated with an AR disease are called carriers. They may have either no expression of disease characteristics or, with some diseases, mild or unique attributes (44,45). X-linked variants are on the X chromosome. Males will have greater disease expression of X-linked traits because they only have one X chromosome and thus no normal copy (46,47). De novo mutations spontaneously arose in the gamete or early embryo and are not inherited from either parent.
Considering the Data Linking the Gene to a Phenotype
ALG8 encodes a protein required for the synthesis of sugar molecules known as N-glycans that help protein maturation in the endoplasmic reticulum. ALG8 is essential for appropriate maturation of the ADPKD protein polycystin-1 (43). ALG8 is curated in some but not yet all public databases as an official disease gene for autosomal dominant polycystic kidney and liver phenotypes. The authors and others having growing experience with this genetic diagnosis (38,48). Given what is known about this gene, the lack of variants in the disease genes for typical ADPKD in the case 1 patient, and the phenotype that fits reported cases, we were comfortable providing the patient with reassurance that the ALG8 variant is likely to be the cause of his cystic kidney disease. Based on previously described cases and the mechanism of cyst formation being an incomplete loss of polycystin levels, his course will hopefully be relatively indolent, without renal failure. We expect therefore, based on the non-PKD1 or -PKD2 genetic etiology, that he is at low-risk of progression and thus not a candidate for tolvaptan therapy. In general, genotype alone should not be used to determine those at risk for rapid progression because sometimes even patients with truncating PKD1 mutations may have slowly progressive course (49). For patients with normal renal function and ADPKD, we rely on Mayo imaging classification and/or the finding of average kidney length >16.5 cm in a young patient to determine eligibility for tolvaptan therapy (50).
In other cases, if a gene variant is called pathogenic but the implicated gene does not fit the patient’s phenotype, it is always reasonable to consider the level of evidence and experience (i.e., case numbers, pedigrees, biologic mechanism, animal models) that support the implication of a gene with a phenotype because the literature is still a work in progress.
Case 2: A VUS
A 38-year-old male with no past medical history has an elevated creatinine of 1.7 mg/dl. Seven years prior, he had normal renal function. He has no hearing or vision loss and no family history of kidney disease, and he takes no medication. Physical exam and history are unremarkable. Urinalysis consistently demonstrates 2+ blood but no protein. Urine microscopy demonstrates isomorphic red blood cells without casts.
Due to his young age, elevated creatinine, and hematuria of unclear etiology, a renal biopsy is performed. This shows unremarkable light microscopy and immunofluorescence, but diffuse thinning of the glomerular basement membrane on electron microscopy.
This did not explain his renal impairment but suggested thin basement membrane disease which has been associated with mutations in COL4A3, COL4A4, or COL4A5 (51) (Figure 4).
Figure 4.
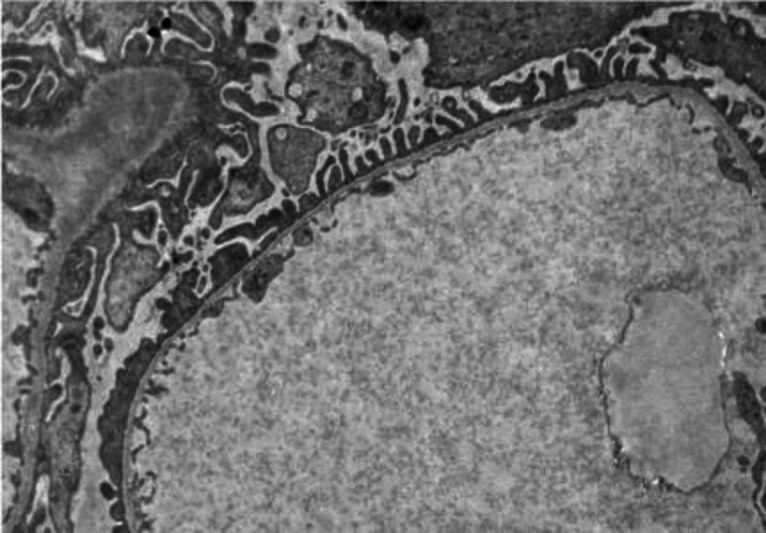
Electron microscopy of renal biopsy in Case 2.
To explore this possibility further and to address patient interest in whether there was a genetic cause, he underwent clinical genetic testing. No positive findings were reported; however, his list of VUSs are shown in Table 2.
Table 2.
Case 2 Variant Report: VUS
| Gene | Associated Disease(s) | Inheritance | Variant | Zygosity | Classification |
|---|---|---|---|---|---|
| COL4A4 | COL4A4-related Alport syndrome | AD and AR | c.816G>A (p. Lys272=) | Heterozygous | Unknown significance |
Provided Interpretation: This synonymous variant is located near an intron-exon boundary located in exon 13 and is predicted by multiple in silico splice predictor algorithms to reduce the splice site activity significantly. This variant has not been reported as associated with a clinical condition in the Human Gene Mutation Database (HGMD) and is absent from the Broad Institute gnomAD dataset.
VUS, variant of uncertain significance; AD, autosomal dominant; AR, autosomal recessive.
VUS
VUSs, as the name implies, are of unknown significance. It is typically advised that the clinician will not base clinical decisions on VUSs. Nonetheless, there are characteristics about a case/gene/VUS that can increase the likelihood that a VUS is indeed pathogenic such that the finding may still provide clinically relevant information that can be interpreted with caution. The following considerations are assessed when determining the potential significance of a VUS (52):
Patient/Family/Phenotype Factors
Does the patient’s phenotype fit with reported phenotype for the disease gene/genotype? Keep in mind that heterozygous VUS will typically not be of relevance for recessive disease unless two variants are found in the same gene.
Is the variant(s) present in all affected family members? This is a very important way to support or exclude the pathogenicity of a variant. Given this, it may be wise to include in pretest counseling that some results may lead to the suggestions that other family members get tested. In general, affected family members will provide more information than unaffected family members—because for some diseases, not all carriers of the pathogenic variant get disease. In contrast, lack of the variant in a family member affected with the same phenotype has the power to rule out that variant as the causative variant in that family.
Gene/Protein/Variant Factors
Does the variant cause a significant alteration in AA characteristics in an important region of the encoded protein? If the specific AA at that position in the protein is conserved (maintained) through evolution of species, this supports its importance. If the affected AA is in a functional domain of the protein, it is more likely to have a consequential effect. If the AA change results in change of size, polarity, or charge of the AA, it is more likely to have an effect than a more subtle change.
Is the variant sufficiently rare in the general population? Rarer is more likely pathogenic in rare monogenic disorders. Often a threshold of <1% in the general population is set to define a variant as being “rare.” This may be a logical threshold for many AR phenotypes because only when two such rare alleles occur in the same person does the phenotype present. However, for AD diseases, the general population frequency for any one pathogenic variant should be expected to be significantly less than the prevalence of the disease, thus certainly <0.1% but typically much rarer than that. For example, the authors consider any variant with frequency in the general cohort or ethnic subpopulations >0.01% as too common to be pathogenic for ADPKD where the 1:1000 disease prevalence is made up of patients who each have any one of many hundreds to thousands of potential unique rare pathogenic alleles. A variant’s frequency in the general population or ethnic subpopulations can be found on the GnomAD resource listed below. Many pathogenic variants will be so rare that they are “novel” or “absent” in GnomAD; the variant was not found in any of the volunteers represented in the GnomAD cohort and thus would not be listed on the site.
Is the variant near a splice junction and predicted by algorithms to effect splicing efficiency?
Is the implicated disease known to be caused by very rare deleterious variants (as opposed to specific gain-of-function mutations)? To use the example of ADPKD again, many disease-causing nontruncating alleles are characterized as VUS because they haven’t been previously reported in association with disease; thus, their consequence cannot be conclusively interpreted. Nonetheless, in a patient with clinically diagnosed ADPKD, finding a VUS in PKD1 that is convincing by the parameters described above is often helpful. For this reason, we suggest requesting VUSs to be reported at least when testing for ADPKD variants.
Clinical geneticists have access to algorithms and prediction scoring systems that incorporate some of this assessment and may innumerate relevant data to the variant interpretation in the report. In addition, assessment will consider whether the specific variant has been reported previously for the provided phenotype. If this information is not provided in the genetic testing report, some details can be found on websites such as ClinGen (53), ClinVar (54), gnomAD (42), and other broad or disease-specific publicly available databases.
The VUS listed in case 2 was one of six reported VUSs from a panel testing 385 “kidney genes” in this patient. Although it is reasonable to pass over most VUSs, in cases such as this where the pretest probability for finding a COL4A3 or COL4A4 heterozygous variant was considered high, the reported VUS in COL4A4 can be considered with added interest. COL4A4 encodes the α4 collagen protein integral to the α3α4α5 helix that forms the adult glomerular basement membrane. Patients with heterozygous mutations negatively impacting COL4A4 may have AD Alport syndrome or thin basement membrane disease (51,55). Although this VUS is a synonymous variant—no change in the 272nd AA lysine—algorithms identified that this novel (never before reported) variant occurs near an exon-intron boundary and is predicted based on the surrounding sequence to interfere with splicing. Unlike variants in the first or second base of the intron from the exon/intron junction, which will undoubtedly abolish splicing, exonic nucleotides near the splice site may also alter splicing—potentially resulting in exclusion of exon 13 in the case above—but their consequence is less predictable. This case certainly raises the likelihood that indeed this variant has consequence; however, without biologic assessment or more familial data, this suspicion remains unproven.
Case 3: APOL1 Risk Alleles
A 38-year-old male with hypertension and prediabetes presents to clinic for evaluation of proteinuria. His mother had developed ESKD in her late 30s, but the details of her diagnosis are not well known by the family. His physical exam, aside from mild hypertension, is unremarkable. His laboratory data show a serum creatinine of 1.2 mg/dl, a urine protein-to-creatinine ratio of 0.97 mg/mg Cr, and no other abnormalities. His renal biopsy shows focal segmental glomerulosclerosis (FSGS). Due to his family history and young age of disease onset, the patient underwent genetic testing. There were no variants reported for monogenic causes of FSGS; however, positive variants were found, as detailed in Table 3.
Table 3.
Case 3 Variant Report: Two APOL1 risk alleles
| Gene | Condition(s) | Inheritance | Variant(s) | Zygosity | Classification |
|---|---|---|---|---|---|
| APOL1 | Susceptibility to ESKD and focal segmental glomerulosclerosis | Complex | G1 allele | Heterozygous | Risk allele |
| APOL1 | G2 allele | Heterozygous | Risk allele |
This case highlights the discovery of a risk allele—an allele that on its own is not pathogenic but can increase the risk of developing a disease in the setting of other clinical or environmental “second hit” factors. APOL1 risk alleles (termed G1 or G2) were discovered among people with recent West African ancestry who had higher rates of ESKD, even after accounting for traditional risk factors. Further studies demonstrated that individuals homozygous for either allele (G1/G1 or G2/G2) or heterozygous for both alleles (G1/G2) as seen in this case are at significant risk for developing hypertensive ESKD, biopsy-proven FSGS, HIV-associated nephropathy, and possibly coronavirus disease 2019–related glomerular disease (56–60). APOL1 G1 or G2 heterozygosity will be encountered frequently by a clinician performing genetic testing because more than one third of Black individuals carry one high-risk allele—thought to confer no significant added risk—whereas 13% have a high-risk genotype (60,61). The discovery of the high-risk genotype may lead the clinician to consider its associated disease(s) and/or aggressively treat known risk factors that may contribute to the “second hit” such as HIV or other viral infections, hypertension, and obesity.
Use of Genetic Testing in Living Related Kidney Donation
Living kidney donors are at increased risk of developing of CKD/ESKD (62). Although this may be attributed to reduced nephron mass following donation, an alternative explanation could be that some living related donors might carry the genetic cause or genetic risk that already destined them to kidney problems. As such, it may be valuable to attempt to identify a genetic cause of CKD/ESKD in a recipient such that if found, the living related donor can be screened for the same variant (single variant testing would be all that would be necessary). This is particularly of relevance when the donor is younger than the age at which clinical manifestations would become apparent (22,23,63–65).
The presence of risk alleles such as APOL1 is a different situation because <15% of those with the high risk APOL1 genotype will develop ESKD (66). Even though studies indeed report an increased risk of ESKD in donors with the APOL1 high-risk genotype, that is a risk that some living related donors may still wish to take in order to give their loved one a kidney (67). Importantly, attitudes toward genetic testing in potential donors are mixed, with some demonstrating dissatisfaction knowing that genetic testing may preclude them from donating their kidney to a loved one and would be willing to undergo donation despite these potential risks of transplant (68).
Case 4: An Unexpected but Fitting Diagnosis
A 53-year-old female with hypertension and insulin-dependent diabetes mellitus is noted to have chronic hypomagnesemia. Multiple family members have been diagnosed with type 2 diabetes and CKD attributed to diabetes. She does not take any diuretics, proton pump inhibitors, or alcohol. During a prior hospitalization, she rarely achieved normal magnesium levels despite significant intravenous and oral magnesium repletion. Renal ultrasound demonstrates symmetric, echogenic kidneys in addition to multiple right upper pole simple cysts measuring up to 1.7 cm in diameter.
Due to severe renal magnesium wasting, renal cysts, CKD, and positive family history for CKD, there was moderate suspicion for a genetic etiology, although it was unclear to what extent diabetes could explain the familial kidney disease.
Serum creatinine: 1.5 mg/dl (gradual upward trend from a creatinine of 1.0 six years ago).
Serum magnesium: 1.4 mg/dl (normal range 1.7–2.4 mg/dl) despite supplementation with magnesium lactate 336 mg twice daily.
Fractional excretion of magnesium: 25% (expected <3% in patients with hypomagnesemia).
Urinalysis is remarkable for 1+ protein, 2+ glucose.
Findings are detailed in Table 4.
Table 4.
Case 4 Variant Report: Whole gene deletion
| Gene | Associated Disease(s) | Inheritance | Variant | Zygosity | Classification |
|---|---|---|---|---|---|
| HNF1B | Renal cysts and diabetes syndrome | AD | Whole gene deletion | Heterozygous | Pathogenic |
AD, autosomal dominant.
Given the whole gene deletion, the patient underwent clinical chromosomal microarray analysis to confirm the variant and define the extent of the deletion. This confirmed the presence of a large (1.4 Mb) heterozygous deletion at 17q12, which includes the disease gene HNF1B.
When NGS May Not Be Enough
Large deletions such as seen in this case can be characterized as a type of structural variation. Since the heterozygous deletion results in only one remaining allele, these deletions are also referred to as copy number variation (CNV; i.e., only one copy of the gene is present rather than the usual two copies provided by the maternal and paternal allele). CNVs, broadly defined as deletions or duplications of large segments of the genome, can occur de novo or be inherited (69). They are not optimally detected by the short reads of targeted NGS or WES. Although NGS-based sequencing will be sufficient as the genetic testing modality for many patients with kidney disorders and was sufficient to detect the large deletion in this case, Table 5 outlines specific indications where alternative genetic testing modalities are used as the preferred initial test, follow-up to a suspected missed variant, or confirmation of a detected variant, as in case 4 (35,70–73).
Table 5.
Specific indications for alternative sequencing modalities in nephrology
| Clinical Scenario | Alternative Testing Options |
|---|---|
| Large deletions or chromosomal rearrangements suspected (typically pediatric: CAKUT, multi-organ manifestations) or needed as a follow-up assessment to a negative test with suboptimal assessment of copy number variation (e.g., whole gene deletions of NPHP1) or to confirm large deletion found by WES or targeted NGS | SNP or chromosomal microarray Other options: • Comparative genomic hybridization • Whole genome sequencing • Multiplex ligation probe amplification |
| ADTKD-MUC1 suspected (ADTKD phenotype but no mutations found in other ADTKD genes) | Variant-specific testing available without cost from the Broad Institute (contact ableyer@wakehealth.edu) |
| Single variant testing: An established familial pathogenic variant is known, and patient desires testing only for presence/absence of that variant | Sanger sequencing following PCR amplification of a small genomic region containing the variant location |
| Gold standarda for ADPKD | Sanger sequencing of long-range PCR amplicons designed to amplify only PKD1, PKD2, but not the duplicated regions (pseudogenes) homologous to PKD1 |
| High suspicion for genetic etiology but no variant found | Consider testing for large deletions (above), and/or contacting testing facility to ask about sequencing quality for specific genes of interest |
ADTKD, autosomal dominant tubulointerstitial kidney disease; ADPKD, autosomal dominant polycystic kidney disease; CAKUT, congenital anomalies of the kidney and urinary tract; NGS, next-generation sequencing; SNP, single nucleotide polymorphisms; WES, whole exome sequencing.
As NGS-based methodologies improve, using paired-end sequencing, improved capture reagents, with or without preceding long-range PCR, the superiority of this approach is less apparent, and thus NGS-based panels are often used recently as first test for genetic diagnosis in ADPKD.
Case 4 Continued: Reframing the Patient
HNF1B encodes the essential transcription factor hepatocyte nuclear factor 1β, which is expressed in the pancreas, liver, and kidney (74,75). Recessive loss is embryonically lethal, whereas a range of phenotypes and severities—which may be termed “variable expressivity”—can result from heterozygous variants or loss of this gene. In the kidney, some patients with an HNF1B mutation manifest with congenital anomalies of the kidney and urinary tract, whereas others have a progressive tubulointerstitial kidney disease often with gout with or without kidney cysts. Other potential manifestations are maturity-onset diabetes of the young (formerly known as “renal cysts and diabetes syndrome”), hypomagnesemia, elevated liver function tests, and hyperparathyroidism out of proportion to CKD (76–79). HNF1B gene deletion can be part of a larger chromosome 17q2 deletion syndrome, in which case there may also be developmental, learning, and psychiatric problems (80,81).
In this case, the genetic finding prompted the reframing of the patient’s diabetes diagnosis and her CKD. She had undergone a liver biopsy 1 year prior for workup of persistent mild liver function test elevation, which had shown no actionable findings and arguably could have been avoided if the genetic diagnosis was known at the time. Given the AD nature of this disease, the patient was counseled by the ordering provider about potential implications for her family members. Specifically, 50% of her offspring would be expected to inherit this variant, and she may wish to discuss screening with them.
Depending on the clinician’s comfort level, such post-test counseling can be performed by the clinician or by a genetics counselor—either via the genetic testing service or via an independent clinical referral. Although establishing a definitive pathogenic variant can create the opportunity to establish a potentially presymptomatic diagnosis in affected family members, proactively offering this should not be taken lightly. First, in diseases such as this case with a disease gene (HNF1B) with variable expressivity, knowing that a family member has the variant does not entirely predict the characteristics or severity of disease to expect. Second, genetic diagnosis in pediatrics is typically discouraged other than for familial disease where pre-emptive therapies are available.
Take-Home Points and Looking Ahead
It is important to note that with increasing use of genetic testing, public databases that document variants and their spectrum of clinical consequences will be updated. With time, this will contribute to more informed interpretations of VUSs discovered in the future (82). A clinician can aid in these efforts by accurately providing the patient phenotype at time of ordering the test—allowing the submitted data (phenotype) to be associated with the variant—and encouraging the sequencing service to submit data to public resources. As resources for variant curation improve and additional disease genes are identified, clinicians may consider contacting the testing company to have a patient’s genetic sequencing data reanalyzed. Sequencing services that use WES as opposed to just targeted sequencing of the genes in the panel may be able not only to reinterpret VUSs but also to analyze genes not previously considered.
This review aimed to provide a foundation for nephrology providers to start to develop experience with the affordable tool of clinical genetic testing to aid in the workup of a wide range of unexplained kidney-related abnormalities. Some take home points are the following:
Ordering a clinical genetic test should be feasible and no longer require extensive logistical coordination by the provider.
Pretest discussion (counseling) to establish expectations is recommended as outlined above.
A clinician can choose whether to refer for genetic counseling, and even whether to refer to a center with genetic kidney disease expertise to make a genetic diagnosis, but will want to understand genetic terminology in kidney disease diagnoses.
Everyone has many genetic variants. Avoid overinterpreting VUS. Many, possibly most, VUS are benign, but if the VUS is in a gene that can cause the phenotype, further consideration may help determine clinical relevance.
Seek advice from a genetics counselor or a colleague with genetics experience when there is uncertainty about the consequence or relevance of a variant.
When used in the proper circumstances, genetic testing will allow us to enrich our understanding and care of patients with kidney disorders.
Disclosures
N.K. Dahl reports being an employee of Yale University School of Medicine; consultancy for Otsuka Pharmaceuticals and Vertex; research funding as the PI for clinical trials sponsored by Allena, Kadmon, Reata, Regulus, and Sanofi; honoraria from AstraZeneca, the National Kidney Foundation, and Otsuka Pharmaceutical; an advisory or leadership role for the Natera Scientific Advisory Board and the PKD Foundation; participation in a speakers’ bureau for Otsuka; and other interests or relationships with the Medical Advisory Board, NKF NE Chapter. All remaining authors have nothing to disclose.
Funding
This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant K08 DK119642 and the Doris Duke Charitable Foundation, Clinical Scientist Development award grant #2021195 to W.B.
Author Contributions
All authors were responsible for the conceptualization, wrote the original draft of the manuscript, and reviewed and edited the manuscript.
References
- 1.United States Renal Data System : 2021 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States, Bethesda, MD, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2021 [Google Scholar]
- 2.Freedman BI, Soucie JM, McClellan WM: Family history of end-stage renal disease among incident dialysis patients. J Am Soc Nephrol 8: 1942–1945, 1997. 10.1681/ASN.V8121942 [DOI] [PubMed] [Google Scholar]
- 3.Freedman BI, Volkova NV, Satko SG, Krisher J, Jurkovitz C, Soucie JM, McClellan WM: Population-based screening for family history of end-stage renal disease among incident dialysis patients. Am J Nephrol 25: 529–535, 2005. 10.1159/000088491 [DOI] [PubMed] [Google Scholar]
- 4.McClellan WM, Satko SG, Gladstone E, Krisher JO, Narva AS, Freedman BI: Individuals with a family history of ESRD are a high-risk population for CKD: Implications for targeted surveillance and intervention activities. Am J Kidney Dis 53: S100–S106, 2009. 10.1053/j.ajkd.2008.07.059 [DOI] [PubMed] [Google Scholar]
- 5.Connaughton DM, Bukhari S, Conlon P, Cassidy E, O’Toole M, Mohamad M, Flanagan J, Butler T, O’Leary A, Wong L, O’Regan J, Moran S, O’Kelly P, Logan V, Griffin B, Griffin M, Lavin P, Little MA, Conlon P: The Irish Kidney Gene Project—Prevalence of family history in patients with kidney disease in Ireland. Nephron 130: 293–301, 2015. 10.1159/000436983 [DOI] [PubMed] [Google Scholar]
- 6.Connaughton DM, Kennedy C, Shril S, Mann N, Murray SL, Williams PA, Conlon E, Nakayama M, van der Ven AT, Ityel H, Kause F, Kolvenbach CM, Dai R, Vivante A, Braun DA, Schneider R, Kitzler TM, Moloney B, Moran CP, Smyth JS, Kennedy A, Benson K, Stapleton C, Denton M, Magee C, O’Seaghdha CM, Plant WD, Griffin MD, Awan A, Sweeney C, Mane SM, Lifton RP, Griffin B, Leavey S, Casserly L, de Freitas DG, Holian J, Dorman A, Doyle B, Lavin PJ, Little MA, Conlon PJ, Hildebrandt F: Monogenic causes of chronic kidney disease in adults. Kidney Int 95: 914–928, 2019. 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mallett A, Fowles LF, McGaughran J, Healy H, Patel C: A multidisciplinary renal genetics clinic improves patient diagnosis. Med J Aust 204: 58–59, 2016. 10.5694/mja15.01157 [DOI] [PubMed] [Google Scholar]
- 8.Mallett AJ, McCarthy HJ, Ho G, Holman K, Farnsworth E, Patel C, Fletcher JT, Mallawaarachchi A, Quinlan C, Bennetts B, Alexander SI: Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int 92: 1493–1506, 2017. 10.1016/j.kint.2017.06.013 [DOI] [PubMed] [Google Scholar]
- 9.Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, Zhang J, Nestor J, Krithivasan P, Lam WY, Mitrotti A, Piva S, Kil BH, Chatterjee D, Reingold R, Bradbury D, DiVecchia M, Snyder H, Mu X, Mehl K, Balderes O, Fasel DA, Weng C, Radhakrishnan J, Canetta P, Appel GB, Bomback AS, Ahn W, Uy NS, Alam S, Cohen DJ, Crew RJ, Dube GK, Rao MK, Kamalakaran S, Copeland B, Ren Z, Bridgers J, Malone CD, Mebane CM, Dagaonkar N, Fellström BC, Haefliger C, Mohan S, Sanna-Cherchi S, Kiryluk K, Fleckner J, March R, Platt A, Goldstein DB, Gharavi AG: Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 380: 142–151, 2019. 10.1056/NEJMoa1806891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lata S, Marasa M, Li Y, Fasel DA, Groopman E, Jobanputra V, Rasouly H, Mitrotti A, Westland R, Verbitsky M, Nestor J, Slater LM, D’Agati V, Zaniew M, Materna-Kiryluk A, Lugani F, Caridi G, Rampoldi L, Mattoo A, Newton CA, Rao MK, Radhakrishnan J, Ahn W, Canetta PA, Bomback AS, Appel GB, Antignac C, Markowitz GS, Garcia CK, Kiryluk K, Sanna-Cherchi S, Gharavi AG: Whole-exome sequencing in adults with chronic kidney disease: A pilot study. Ann Intern Med 168: 100–109, 2018. 10.7326/M17-1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallett A, Patel C, Salisbury A, Wang Z, Healy H, Hoy W: The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis 9: 98, 2014. 10.1186/1750-1172-9-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gribouval O, Boyer O, Hummel A, Dantal J, Martinez F, Sberro-Soussan R, Etienne I, Chauveau D, Delahousse M, Lionet A, Allard J, Pouteil Noble C, Tête MJ, Heidet L, Antignac C, Servais A: Identification of genetic causes for sporadic steroid-resistant nephrotic syndrome in adults. Kidney Int 94: 1013–1022, 2018. 10.1016/j.kint.2018.07.024 [DOI] [PubMed] [Google Scholar]
- 13.Lovric S, Ashraf S, Tan W, Hildebrandt F: Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol Dial Transplant 31: 1802–1813, 2016. 10.1093/ndt/gfv355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, Venkat-Raman G, Ennis S: Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 31: 961–970, 2016. 10.1093/ndt/gfv325 [DOI] [PubMed] [Google Scholar]
- 15.Yao T, Udwan K, John R, Rana A, Haghighi A, Xu L, Hack S, Reich HN, Hladunewich MA, Cattran DC, Paterson AD, Pei Y, Barua M: Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clin J Am Soc Nephrol 14: 213–223, 2019. 10.2215/CJN.08750718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, Jobst-Schwan T, Vivante A, Schapiro D, Tan W, Warejko JK, Widmeier E, Nelson CP, Fathy HM, Gucev Z, Soliman NA, Hashmi S, Halbritter J, Halty M, Kari JA, El-Desoky S, Ferguson MA, Somers MJG, Traum AZ, Stein DR, Daouk GH, Rodig NM, Katz A, Hanna C, Schwaderer AL, Sayer JA, Wassner AJ, Mane S, Lifton RP, Milosevic D, Tasic V, Baum MA, Hildebrandt F: Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int 93: 204–213, 2018. 10.1016/j.kint.2017.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, Fisher B, Spaneas L, Porath JD, Braun DA, Wassner AJ, Nelson CP, Tasic V, Sayer JA, Hildebrandt F: Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol 26: 543–551, 2015. 10.1681/ASN.2014040388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, Mitrotti A, Fasel DA, Batourina E, Sampson MG, Bodria M, Werth M, Kao C, Martino J, Capone VP, Vivante A, Shril S, Kil BH, Marasà M, Zhang JY, Na YJ, Lim TY, Ahram D, Weng PL, Heinzen EL, Carrea A, Piaggio G, Gesualdo L, Manca V, Masnata G, Gigante M, Cusi D, Izzi C, Scolari F, van Wijk JAE, Saraga M, Santoro D, Conti G, Zamboli P, White H, Drozdz D, Zachwieja K, Miklaszewska M, Tkaczyk M, Tomczyk D, Krakowska A, Sikora P, Jarmoliński T, Borszewska-Kornacka MK, Pawluch R, Szczepanska M, Adamczyk P, Mizerska-Wasiak M, Krzemien G, Szmigielska A, Zaniew M, Dobson MG, Darlow JM, Puri P, Barton DE, Furth SL, Warady BA, Gucev Z, Lozanovski VJ, Tasic V, Pisani I, Allegri L, Rodas LM, Campistol JM, Jeanpierre C, Alam S, Casale P, Wong CS, Lin F, Miranda DM, Oliveira EA, Simões-E-Silva AC, Barasch JM, Levy B, Wu N, Hildebrandt F, Ghiggeri GM, Latos-Bielenska A, Materna-Kiryluk A, Zhang F, Hakonarson H, Papaioannou VE, Mendelsohn CL, Gharavi AG, Sanna-Cherchi S: The copy number variation landscape of congenital anomalies of the kidney and urinary tract [published correction appears in Nat Genet 51: 764, 2019 10.1038/s41588-019-0376-0]. Nat Genet 51: 117–127, 2019. 10.1038/s41588-018-0281-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bullich G, Domingo-Gallego A, Vargas I, Ruiz P, Lorente-Grandoso L, Furlano M, Fraga G, Madrid Á, Ariceta G, Borregán M, Piñero-Fernández JA, Rodríguez-Peña L, Ballesta-Martínez MJ, Llano-Rivas I, Meñica MA, Ballarín J, Torrents D, Torra R, Ars E: A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int 94: 363–371, 2018. 10.1016/j.kint.2018.02.027 [DOI] [PubMed] [Google Scholar]
- 20.Goodwin S, McPherson JD, McCombie WR: Coming of age: Ten years of next-generation sequencing technologies. Nat Rev Genet 17: 333–351, 2016. 10.1038/nrg.2016.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groopman EE, Rasouly HM, Gharavi AG: Genomic medicine for kidney disease. Nat Rev Nephrol 14: 83–104, 2018. 10.1038/nrneph.2017.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas CP, Mansilla MA, Sompallae R, Mason SO, Nishimura CJ, Kimble MJ, Campbell CA, Kwitek AE, Darbro BW, Stewart ZA, Smith RJ: Screening of living kidney donors for genetic diseases using a comprehensive genetic testing strategy. Am J Transplant 17: 401–410, 2017. 10.1111/ajt.13970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas CP, Gupta S, Freese ME, Chouhan KK, Dantuma MI, Holanda DG, Katz DA, Darbro BW, Mansilla MA, Smith RJ: Sequential genetic testing of living-related donors for inherited renal disease to promote informed choice and enhance safety of living donation. Transpl Int 34: 2696–2705, 2021. 10.1111/tri.14133 [DOI] [PubMed] [Google Scholar]
- 24.Berns JS: A survey-based evaluation of self-perceived competency after nephrology fellowship training. Clin J Am Soc Nephrol 5: 490–496, 2010. 10.2215/CJN.08461109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cocchi E, Nestor JG, Gharavi AG: Clinical genetic screening in adult patients with kidney disease. Clin J Am Soc Nephrol 15: 1497–1510, 2020. 10.2215/CJN.15141219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connaughton DM, Hildebrandt F: Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol Dial Transplant 35: 390–397, 2020. 10.1093/ndt/gfz028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knoers N, Antignac C, Bergmann C, Dahan K, Giglio S, Heidet L, Lipska-Ziętkiewicz BS, Noris M, Remuzzi G, Vargas-Poussou R, Schaefer F: Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrol Dial Transplant 37: 239–254, 2022. 10.1093/ndt/gfab218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacyna JE, Radecki Breitkopf C, Jenkins SM, Sutton EJ, Horrow C, Kullo IJ, Sharp RR: Should pretest genetic counselling be required for patients pursuing genomic sequencing? Results from a survey of participants in a large genomic implementation study. J Med Genet 56: 317–324, 2019. 10.1136/jmedgenet-2018-105577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, Miller DT: Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics [published correction appears in Genet Med 19: 484, 2017 10.1038/gim.2017.17]. Genet Med 19: 249–255, 2017. 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- 30.Elliott MD, James LC, Simms EL, Sharma P, Girard LP, Cheema K, Elliott MJ, Lauzon JL, Chun J: Mainstreaming genetic testing for adult patients with autosomal dominant polycystic kidney disease. Can J Kidney Health Dis 8: 20543581211055001, 2021. 10.1177/20543581211055001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hays T, Groopman EE, Gharavi AG: Genetic testing for kidney disease of unknown etiology. Kidney Int 98: 590–600, 2020. 10.1016/j.kint.2020.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joly Y, Ngueng Feze I, Simard J: Genetic discrimination and life insurance: A systematic review of the evidence. BMC Med 11: 25, 2013. 10.1186/1741-7015-11-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vivante A, Hildebrandt F: Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12: 133–146, 2016. 10.1038/nrneph.2015.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsanis SH, Katsanis N: Molecular genetic testing and the future of clinical genomics. Nat Rev Genet 14: 415–426, 2013. 10.1038/nrg3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Besse W: Genetic analysis in kidney disease: Advancing clinical diagnosis and research discovery. Kidney360 1: 720–723, 2020. 10.34067/KID.0003632020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amberger JS, Bocchini CA, Scott AF, Hamosh A: OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 47: D1038–D1043, 2019. 10.1093/nar/gky1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasouly HM, Groopman EE, Heyman-Kantor R, Fasel DA, Mitrotti A, Westland R, Bier L, Weng C, Ren Z, Copeland B, Krithivasan P, Chung WK, Sanna-Cherchi S, Goldstein DB, Gharavi AG: The burden of candidate pathogenic variants for kidney and genitourinary disorders emerging from exome sequencing. Ann Intern Med 170: 11–21, 2019. 10.7326/M18-1241 [DOI] [PubMed] [Google Scholar]
- 38.Cornec-Le Gall E, Torres VE, Harris PC: Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol 29: 13–23, 2018. 10.1681/ASN.2017050483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanktree MB, Haghighi A, di Bari I, Song X, Pei Y: Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol 16: 790–799, 2021. 10.2215/CJN.02320220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424, 2015. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE: HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37: 564–569, 2016. 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- 42.Broad Institute : gnomAD. Available at: https://gnomad.broadinstitute.org. Accessed April 21, 2022
- 43.Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, Gallagher AR, Huang EB, Gulati A, Knight J, Mane S, Tahvanainen E, Tahvanainen P, Sanna-Cherchi S, Lifton RP, Watnick T, Pei YP, Torres VE, Somlo S: Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest 127: 1772–1785, 2017. 10.1172/JCI90129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fava C, Montagnana M, Rosberg L, Burri P, Almgren P, Jönsson A, Wanby P, Lippi G, Minuz P, Hulthèn LU, Aurell M, Melander O: Subjects heterozygous for genetic loss of function of the thiazide-sensitive cotransporter have reduced blood pressure. Hum Mol Genet 17: 413–418, 2008. 10.1093/hmg/ddm318 [DOI] [PubMed] [Google Scholar]
- 45.Hsu YJ, Yang SS, Chu NF, Sytwu HK, Cheng CJ, Lin SH: Heterozygous mutations of the sodium chloride cotransporter in Chinese children: Prevalence and association with blood pressure. Nephrol Dial Transplant 24: 1170–1175, 2009. 10.1093/ndt/gfn619 [DOI] [PubMed] [Google Scholar]
- 46.Germain DP: Fabry disease. Orphanet J Rare Dis 5: 30, 2010. 10.1186/1750-1172-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nozu K, Nakanishi K, Abe Y, Udagawa T, Okada S, Okamoto T, Kaito H, Kanemoto K, Kobayashi A, Tanaka E, Tanaka K, Hama T, Fujimaru R, Miwa S, Yamamura T, Yamamura N, Horinouchi T, Minamikawa S, Nagata M, Iijima K: A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol 23: 158–168, 2019. 10.1007/s10157-018-1629-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mantovani V, Bin S, Graziano C, Capelli I, Minardi R, Aiello V, Ambrosini E, Cristalli CP, Mattiaccio A, Pariali M, De Fanti S, Faletra F, Grosso E, Cantone R, Mancini E, Mencarelli F, Pasini A, Wischmeijer A, Sciascia N, Seri M, La Manna G: Gene panel analysis in a large cohort of patients with autosomal dominant polycystic kidney disease allows the identification of 80 potentially causative novel variants and the characterization of a complex genetic architecture in a subset of families. Front Genet 11: 464, 2020. 10.3389/fgene.2020.00464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanktree MB, Guiard E, Akbari P, Pourafkari M, Iliuta IA, Ahmed S, Haghighi A, He N, Song X, Paterson AD, Khalili K, Pei YPC: Patients with protein-truncating PKD1 mutations and mild ADPKD. Clin J Am Soc Nephrol 16: 374–383, 2021. 10.2215/CJN.11100720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akbari P, Nasri F, Deng SX, Khowaja S, Lee SH, Warnica W, Lu H, Rattansingh A, Atri M, Khalili K, York P: Total kidney volume measurements in ADPKD by 3D and ellipsoid ultrasound in comparison with magnetic resonance imaging. Clin J Am Soc Nephrol 17: 827–834, 2022. 10.2215/CJN.14931121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warady BA, Agarwal R, Bangalore S, Chapman A, Levin A, Stenvinkel P, Toto RD, Chertow GM: Alport syndrome classification and management. Kidney Med 2: 639–649, 2020. 10.1016/j.xkme.2020.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sullivan KE: The scary world of variants of uncertain significance (VUS): A hitchhiker’s guide to interpretation. J Allergy Clin Immunol 147: 492–494, 2021. 10.1016/j.jaci.2020.06.011 [DOI] [PubMed] [Google Scholar]
- 53.Clinical Genome Resource : ClinGen. Available at: https://clinicalgenome.org. Accessed April 21, 2022
- 54.National Library of Medicine : ClinVar. Available at: https://www.ncbi.nlm.nih.gov/clinvar/. Accessed April 21, 2022
- 55.Kashtan CE: Alport syndrome: Achieving early diagnosis and treatment. Am J Kidney Dis 77: 272–279, 2021. 10.1053/j.ajkd.2020.03.026 [DOI] [PubMed] [Google Scholar]
- 56.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010. 10.1126/science.1193032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu H, Larsen CP, Hernandez-Arroyo CF, Mohamed MMB, Caza T, Sharshir M, Chughtai A, Xie L, Gimenez JM, Sandow TA, Lusco MA, Yang H, Acheampong E, Rosales IA, Colvin RB, Fogo AB, Velez JCQ: AKI and collapsing glomerulopathy associated with COVID-19 and APOL 1 high-risk genotype. J Am Soc Nephrol 31: 1688–1695, 2020. 10.1681/ASN.2020050558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA: APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22: 2129–2137, 2011. 10.1681/ASN.2011040388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, Birmingham DJ, Hebert LA, Hicks PJ, Segal MS, Edberg JC, Brown EE, Alarcón GS, Costenbader KH, Comeau ME, Criswell LA, Harley JB, James JA, Kamen DL, Lim SS, Merrill JT, Sivils KL, Niewold TB, Patel NM, Petri M, Ramsey-Goldman R, Reveille JD, Salmon JE, Tsao BP, Gibson KL, Byers JR, Vinnikova AK, Lea JP, Julian BA, Kimberly RP; Lupus Nephritis–End‐Stage Renal Disease Consortium : End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol 66: 390–396, 2014. 10.1002/art.38220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR: Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol 22: 2098–2105, 2011. 10.1681/ASN.2011050519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friedman DJ, Pollak MR: APOL1 and kidney disease: From genetics to biology. Annu Rev Physiol 82: 323–342, 2020. 10.1146/annurev-physiol-021119-034345 [DOI] [PubMed] [Google Scholar]
- 62.O’Keeffe LM, Ramond A, Oliver-Williams C, Willeit P, Paige E, Trotter P, Evans J, Wadström J, Nicholson M, Collett D, Di Angelantonio E: Mid- and long-term health risks in living kidney donors: A systematic review and meta-analysis. Ann Intern Med 168: 276–284, 2018. 10.7326/M17-1235 [DOI] [PubMed] [Google Scholar]
- 63.Niaudet P: Living donor kidney transplantation in patients with hereditary nephropathies. Nat Rev Nephrol 6: 736–743, 2010. 10.1038/nrneph.2010.122 [DOI] [PubMed] [Google Scholar]
- 64.Caliskan Y, Lentine KL: Approach to genetic testing to optimize the safety of living donor transplantation in Alport syndrome spectrum [published online ahead of print January 27, 2022]. Pediatr Nephrol 10.1007/s00467-022-05430-7 [DOI] [PubMed] [Google Scholar]
- 65.Simms RJ, Travis DL, Durkie M, Wilson G, Dalton A, Ong AC: Genetic testing in the assessment of living related kidney donors at risk of autosomal dominant polycystic kidney disease. Transplantation 99: 1023–1029, 2015. 10.1097/TP.0000000000000466 [DOI] [PubMed] [Google Scholar]
- 66.Friedman DJ, Pollak MR: APOL1 nephropathy: From genetics to clinical applications. Clin J Am Soc Nephrol 16: 294–303, 2021. 10.2215/CJN.15161219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doshi MD, Ortigosa-Goggins M, Garg AX, Li L, Poggio ED, Winkler CA, Kopp JB: APOL1 genotype and renal function of Black living donors. J Am Soc Nephrol 29: 1309–1316, 2018. 10.1681/ASN.2017060658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nestor JG, Li AJ, King KL, Husain SA, McIntosh TJ, Sawinski D, Iltis AS, Goodman MS, Walsh HA, DuBois JM, Mohan S: Impact of education on APOL1 testing attitudes among prospective living kidney donors. Clin Transplant 36: e14516, 2022. 10.1111/ctr.14516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watson CT, Marques-Bonet T, Sharp AJ, Mefford HC: The genetics of microdeletion and microduplication syndromes: An update. Annu Rev Genomics Hum Genet 15: 215–244, 2014. 10.1146/annurev-genom-091212-153408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xue Y, Ankala A, Wilcox WR, Hegde MR: Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet Med 17: 444–451, 2015. 10.1038/gim.2014.122 [DOI] [PubMed] [Google Scholar]
- 71.Ali H, Al-Mulla F, Hussain N, Naim M, Asbeutah AM, AlSahow A, Abu-Farha M, Abubaker J, Al Madhoun A, Ahmad S, Harris PC: PKD1 duplicated regions limit clinical utility of whole exome sequencing for genetic diagnosis of autosomal dominant polycystic kidney disease. Sci Rep 9: 4141, 2019. 10.1038/s41598-019-40761-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song X, Haghighi A, Iliuta IA, Pei Y: Molecular diagnosis of autosomal dominant polycystic kidney disease. Expert Rev Mol Diagn 17: 885–895, 2017. 10.1080/14737159.2017.1358088 [DOI] [PubMed] [Google Scholar]
- 73.Wilson PC, Love-Gregory L, Corliss M, McNulty S, Heusel JW, Gaut JP: Beyond panel-based testing: Exome analysis increases sensitivity for diagnosis of genetic kidney disease. Kidney360 1: 772–780, 2020. 10.34067/KID.0001342020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shao A, Chan SC, Igarashi P: Role of transcription factor hepatocyte nuclear factor-1β in polycystic kidney disease. Cell Signal 71: 109568, 2020. 10.1016/j.cellsig.2020.109568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barbacci E, Reber M, Ott MO, Breillat C, Huetz F, Cereghini S: Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development 126: 4795–4805, 1999. 10.1242/dev.126.21.4795 [DOI] [PubMed] [Google Scholar]
- 76.Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, Kuroki H, Kasahara T, Iwamoto Y, Bell GI: Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 17: 384–385, 1997. 10.1038/ng1297-384 [DOI] [PubMed] [Google Scholar]
- 77.Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, Woolf AS, Rizzoni G, Novelli G, Nicholls AJ, Hattersley AT: Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet 68: 219–224, 2001. 10.1086/316945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU, Ellard S, Bockenhauer D: HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 20: 1123–1131, 2009. 10.1681/ASN.2008060633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C: HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol 11: 102–112, 2015. 10.1038/nrneph.2014.232 [DOI] [PubMed] [Google Scholar]
- 80.Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, Adam MP, Pakula AT, Eisenhauer NJ, Uhas K, Weik L, Guy L, Care ME, Morel CF, Boni C, Salbert BA, Chandrareddy A, Demmer LA, Chow EW, Surti U, Aradhya S, Pickering DL, Golden DM, Sanger WG, Aston E, Brothman AR, Gliem TJ, Thorland EC, Ackley T, Iyer R, Huang S, Barber JC, Crolla JA, Warren ST, Martin CL, Ledbetter DH;SGENE Consortium; Simons Simplex Collection Genetics Consortium; GeneSTAR : Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet 87: 618–630, 2010. 10.1016/j.ajhg.2010.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, Tommerup N, Eichler EE, Bellanne-Chantelot C: Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81: 1057–1069, 2007. 10.1086/522591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wenger AM, Guturu H, Bernstein JA, Bejerano G: Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genet Med 19: 209–214, 2017. 10.1038/gim.2016.88 [DOI] [PubMed] [Google Scholar]