

Abstract
Background:
Cell-free DNA (cfDNA) is a non-invasive marker of cellular injury. However, its significance in pulmonary arterial hypertension (PAH) is unknown.
Methods:
Plasma cfDNA was measured in two PAH cohorts (A, n=48; B, n=161) and controls (n=48). Data were collected for Registry to Evaluate Early And Long-term PAH Disease Management (REVEAL 2.0) scores and outcome determinations. Patients were divided into REVEAL risk groups: low (≤6), medium (7–8), and high (≥9). Total cfDNA concentrations were compared amongst controls and PAH risk groups by one-way ANOVA. Log-rank tests compared survival between cfDNA tertiles and REVEAL risk groups. Area under the receiver operating characteristics curves (AUC) were estimated from logistic regression models. A sample subset from Cohort B (n=96) and controls (n=16) underwent bisulfite sequencing followed by a deconvolution algorithm to map cell-specific cfDNA methylation patterns with concentrations compared using t-tests.
Results:
In Cohort A, median (interquartile range (IQR)) age was 62 years (47–71), with 75% female and median (IQR) REVEAL 2.0 was 6 (4–9). In Cohort B, median (IQR) age was 59 years (49–71), with 69% female and median (IQR) REVEAL 2.0 was 7 (6–9). In both cohorts, cfDNA concentrations differed amongst PAH patients of varying REVEAL risk and controls (ANOVA P≤0.002) and were greater in the high compared with the low risk category (P≤0.002). In Cohort B, death or lung transplant occurred in 14 of 54, 23 of 53, and 35 of 54 patients in the lowest, middle, and highest cfDNA tertiles, respectively. cfDNA levels stratified as tertiles (log-rank: P=0.0001) and REVEAL risk groups (log-rank: P<0.0001) each predicted transplant-free survival. The addition of cfDNA to REVEAL improved discrimination (AUC 0.72 to 0.78; P=0.02). Compared with controls, methylation analysis in PAH patients revealed increased cfDNA originating from erythrocyte progenitors, neutrophils, monocytes, adipocytes, natural killer cells, vascular endothelium and cardiac myocytes (Bonferroni adjusted P<0.05). Erythrocyte progenitor cell-, cardiac myocyte-, and vascular endothelium-derived cfDNA concentrations were greater in PAH patients with high versus low risk REVEAL scores (P≤0.02).
Conclusions:
Circulating cfDNA is elevated in PAH patients, correlates with disease severity and predicts worse survival. Results from cfDNA methylation analyses in PAH patients are consistent with prevailing paradigms of disease pathogenesis.
Keywords: Cell-free Nucleic Acids, Myocytes, Cardiac, Pulmonary Arterial Hypertension, Endothelium, Vascular, Methylation, Monocytes, Risk Assessment, Biomarkers
Introduction:
Risk prediction in pulmonary arterial hypertension (PAH) remains a major challenge.1, 2 Disease-specific surrogate biomarkers are lacking, leading to continued reliance on subjective functional assessments and invasive hemodynamic measurements. Therefore, novel, non-invasive biomarkers of PAH disease progression remain an unmet need, particularly if such a biomarker is plausibly related to and can inform on the extent of PAH pathogenesis.
Several serum biomarkers have been studied in PAH.3–12 However, wide-spread clinical acceptance has been limited, perhaps because of their lack of specificity and unclear link to PAH pathogenesis. Indeed, only B-type natriuretic peptide (BNP) and its precursor N-terminal Pro-BNP (NT-proBNP) are used in current PAH risk scores.1, 11, 12 Though BNP and NT-proBNP are related to right-ventricular decompensation and volume overload, they are not representative of the contemporary paradigms of PAH pathobiology, including genetic and epigenetic contributions, endothelial damage and disruption, metabolic derangements with a hyperproliferative, anti-apoptotic cellular phenotype, and both systemically and locally dysregulated inflammation.13–16
Cell-free DNA (cfDNA) are circulating short DNA fragments (≈165 base pairs), predominantly from mononucleosomes, that represent cell injury and/or cell-turnover. Elevated total cfDNA concentration has been associated with worse prognoses in heterogeneous conditions such as sepsis, trauma, and malignancy.17–19 In addition, cfDNA has become clinically relevant as a non-invasive marker of solid-organ transplant rejection as well as a tool for genotyping and surveillance in oncology.20–23 Advances in high-throughput methylation sequencing have also enabled the differentiation of cfDNA subsets on the basis of cell origin, facilitating the detection of cell or tissue-specific injury.24–27
Given parallels in the pathogenesis of PAH to diseases characterized by increased cell proliferation and turnover such as cancer and inflammatory-mediated tissue injury such as allograft rejection, this study sought to determine if plasma cfDNA concentrations were elevated in PAH patients, would correlate with PAH disease severity, and predict outcomes. Further, cfDNA methylation analysis was leveraged to identify the cellular origins of cfDNA in PAH, seeking to detect a unique, pathobiologically relevant injury pattern.
Materials and Methods:
Data availability:
The data for the analyses that support the findings of this study are held at the National Institutes of Health (NIH) intramural program. The corresponding author may be contacted for requests with regard to the sharing of data, methods, and materials specific to these analyses.
Patient selection and study design:
World Health Organization (WHO) pulmonary hypertension (PH) Group 1 patients with available plasma were identified at Allegheny General Hospital (Cohort A, n=48) and Tufts Medical Center (Cohort B, n=161) for inclusion in the study. Both centers provided samples from patients in ongoing, institutional review board-approved, prospective studies, with collection of plasma at the time of enrollment right heart catheterization. The Office of Human Subjects Research Protections at the NIH waived institutional review board approval for the use of de-identified plasma samples provided by Allegheny General Hospital and Tufts Medical Center, as all participants provided written informed consent at the time of study enrollment. Allegheny General Hospital and Tufts Medical Center patients were enrolled consecutively from April, 2018 to August, 2019 and July, 2001 to August, 2019 respectively. For patients with multiple hemodynamic studies and plasma samples, only the first sample and associated clinical data were analyzed.
Healthy controls were identified and plasma samples collected at the time of volunteer blood donation at the NIH Clinical Center as part of a Department of Transfusion Medicine NIH institutional review board-approved protocol (99-CC-0168; Collection and Distribution of Blood Components from Healthy Donors for In Vitro Research Use; ClinicalTrials.gov NCT00001846). All healthy controls provided written informed consent. The NIH Blood Bank provided plasma samples from consecutively enrolled healthy adult donors (n=48; mean age 57 years; 32% female). The majority of healthy controls (n=41) were enrolled from September 19, 2019 to November 13, 2019. To have an equal number of healthy controls to compare to PAH patients in Cohort A, 7 additional blood bank healthy control samples were included. Of the total 48 healthy control samples, 16 were randomly selected to undergo additional methylation sequencing analysis, as this was the number of samples that could be run in the flow cell without compromising the depth of sequencing. Healthy controls were not matched to PAH patients for demographics.
Total cell-free DNA analysis:
cfDNA was isolated from plasma (QIAamp Circulating Nucleic Acid Kit, Qiagen) using a validated automated platform (QIASymphony, Quiagen).21 Samples were spiked with a known concentration of lambda DNA to assure adequate cfDNA recovery from plasma. cfDNA was measured using quantitative real-time PCR (Swift Biosciences, Ann Arbor, MI) with final concentrations expressed in ng/mL of plasma. cfDNA quality was confirmed by integrity score (QIAamp Circulating Nucleic Acid Kit, Qiagen) and high-sensitivity gel electrophoresis (Agilent). cfDNA samples with a high integrity score (≥ 0.70), suggesting the presence of longer DNA fragments characteristic of genomic origin, were excluded from analysis.
Cell-free DNA methylation analysis:
The protocol for digital droplet PCR (ddPCR) cfDNA quantification, bisulfite conversion, and methylation sequencing has been previously described.27 This approach relies on the principle that epigenetic fingerprints, such as DNA methylation and chromosomal foot printing have tissue-specificity that is maintained on cfDNA. In brief, cfDNA was extracted from plasma as above and quantified using a ddPCR platform (QX200, Bio-Rad), with primers targeting a nuclear transcription factor gene (EIF2C1). Bisulfite conversion was then performed with the EZ DNA methylation-gold kit (Zymo Research) prior to Methyl-Seq DNA library preparation (Accel-NGS Methyl-Seq DNA Library Kit with Unique Dual Indexing, Swift Biosciences). DNA libraries were pooled in equimolar concentrations and subjected to 2 × 100 bp paired-end DNA sequencing on the Illumina NovaSeq 6000 platform. Sequence reads were mapped to the human reference sequence and underwent quality control and trimming. The Bismark methylation extractor routine was performed, extracting all CpGs for individual samples and bsseq tools were used for methylome visualization and analysis.28, 29 The meth_atlas algorithm was used to identify the cell type of origin for cfDNA and deconvolution of the cfDNA methylome by sample. The algorithm used non-negative least squares methodology26 and a reference of DNA methylation signatures that includes 25 of the major human cell types involved in common diseases. The deconvolution algorithm approximates the plasma cfDNA methylation profile as a linear combination of the methylation profiles of cell types in the reference atlas and has been shown to effectively identify cfDNA cell-of-origin using a small number of loci (~4,000 CpGs). Only CpGs with at least 5× coverage per sample were included in the deconvolution analysis, which was performed with R software version 3.6.3. To obtain the concentrations of cfDNA according to tissue or cell type of origin, the relative estimated proportions of cfDNA were multiplied by the total concentration (or copies per mL) of cfDNA in the same plasma samples. The final analysis was limited to those 11 tissue and cell types with quantifiable cfDNA concentrations in the majority of patients. A heat map was created using the ComplexHeatmap package in R in order to visualize cfDNA concentrations from specific cell types.30 An individual patient on the heatmap is represented as a column. The columns were clustered based on the absolute cfDNA concentration using an unsupervised hierarchical clustering approach that is based on Euclidean distance. A typical cfDNA cell of origin pattern was selected from each of the 4 groups (healthy control, low-risk, medium-risk, and high-risk REVEAL 2.0 [Registry to Evaluate Early and Long-Term PAH Disease Management] score) for visual representation. First the mean cfDNA concentration (log10 ng/mL) for the healthy control, low, medium, and high-risk groups was calculated for each of the 7 cell types that distinguished between healthy controls and PAH patients (erythrocyte progenitors, neutrophils, monocytes, adipocytes, natural killer (NK) cells, vascular endothelium, and cardiac myocytes). Then the sum of the squared differences between the cell-specific cfDNA concentration of each subject and the group mean across the 7 cell types was calculated. Three PAH patients and one healthy control were selected for illustration based on a sum of the squared differences from the mean of < 0.10.
Clinical data analysis and REVEAL score calculation:
The REVEAL score, which has been updated as REVEAL 2.0, is a robustly validated, contemporary risk prediction tool.31 Sufficient clinical variables (at least 7) necessary to calculate REVEAL 2.0 scores were provided for each patient in Cohort A. In Cohort B, 11 of the 14 clinical variables necessary for calculating REVEAL 2.0 scores, including age, sex, etiology of PAH, right atrial pressure, pulmonary vascular resistance, baseline heart rate, baseline systolic blood pressure, six-minute walk distance, New York Heart Association (NYHA) functional class, estimated glomerular filtration rate, and BNP/NT-proBNP were available for the majority of patients. REVEAL Lite 2 scores were also calculated for each patient in cohorts A and B.32 Patients were divided into three risk groups based on REVEAL 2.0 score: low (≤ 6), medium (7–8), and high (≥ 9); and REVEAL Lite 2 score: low (≤ 5), medium (6–7), and high (≥ 8), as previously validated.31–33 Transplant-free survival status was available for all patients in Cohort B as of March 9th, 2020. Outcomes in Cohort A were recorded as of May 25th, 2020.
Clinical biomarker analysis:
Concentrations of high-sensitivity cardiac troponin T (hs-cTnT; Roche TnT Gen 5 STAT; Roche, Indianapolis, IN) and high-sensitivity C-reactive protein (hs-CRP; MULTIGENT CRP Vario; Abbott Laboratories Inc., Abbott Park, IL) were determined in a subset of patient samples with sufficient remaining plasma to evaluate the association of cfDNA with additional clinical markers of myocardial injury and inflammation, respectively. The hs-CRP assay reference limit for low risk is < 1.0 mg/L, while the 99th percentile upper reference limit for the hs-cTnT assay is 19 ng/L.
Statistical Analysis:
Cohort A and B demographics, PAH severity, treatment strategies, and medical comorbidities were assessed using Wilcoxon rank sum tests for continuous variables and Pearson’s chi-square or Fisher exact test for categorical variables. Total cfDNA concentrations were log10-transformed and compared amongst healthy control and PAH patient groups of varying risk using one-way ANOVA followed by pairwise comparisons with p-values adjusted using the Tukey-Kramer method. Correlations between cfDNA and clinical variables in Cohorts A and B were assessed using nonparametric Spearman correlation. In Cohort B, cfDNA data were a priori divided into categorical tertiles and treated as a nominal variable for all analyses, an approach that mirrors other risk assessment tools commonly applied to PAH management,1 and is similar to a previously published analysis in lung transplant recipients.34 Differences in demographics and comorbid conditions across cfDNA tertiles were assessed using Pearson’s chi-square or Fisher exact test, and differences in PAH-related clinical variables across tertiles were compared by Kruskal-Wallis test. Additionally, Kaplan-Meier curves were plotted to show transplant-free survival in Cohort B based on cfDNA tertiles, REVEAL 2.0, and REVEAL Lite 2 risk categories. Log-rank tests were used to compare transplant free survival among cfDNA tertiles and REVEAL risk categories, accounting for multiple comparisons by reporting Tukey-Kramer adjusted p-values. After confirming that the proportional hazards assumption was valid,35 Cox proportional hazard models were used to perform adjusted survival analyses accounting for patient age and sex. To compare the predictive ability of REVEAL score and cfDNA for the composite outcome of death or lung transplant, we used logistic regression models to estimate the area under the receiver operating characteristics (ROC) curves for cfDNA tertiles and REVEAL 2.0 risk categorization independently and combined, again adjusting for age and sex. ROC curves were also estimated for BNP and hs-CRP utilizing established risk cut-offs for PAH and cardiovascular disease respectively.2, 36 Areas under the ROC curve (AUC) were compared using generalized U-statistics.37 Finally, log10-transformed cell-specific cfDNA concentrations were compared between PAH patients and healthy controls using t-tests. Specific cell types were selected for further analysis based on a Bonferroni-corrected p-value <0.05. Pairwise comparisons of selected cell-specific cfDNA concentrations were analyzed between PAH risk groups and healthy controls and p-values were adjusted using the Tukey-Kramer method. All analyses were done using SAS version 9.4.
Results:
Cohort demographics
Plasma cfDNA concentrations were quantified in two PAH cohorts (Figure 1). Demographic and clinical data for Cohort A [median (interquartile range (IQR)) age, 62 years (47–71); 75% female; median (IQR) REVEAL 2.0 6 (4–9)] and Cohort B [median (IQR) age, 59 years (49–71); 69% female; median (IQR) REVEAL 2.0 7 (6–9)] are displayed in Table 1.
Figure 1. Pulmonary arterial hypertension (PAH) patient sample flow diagram.
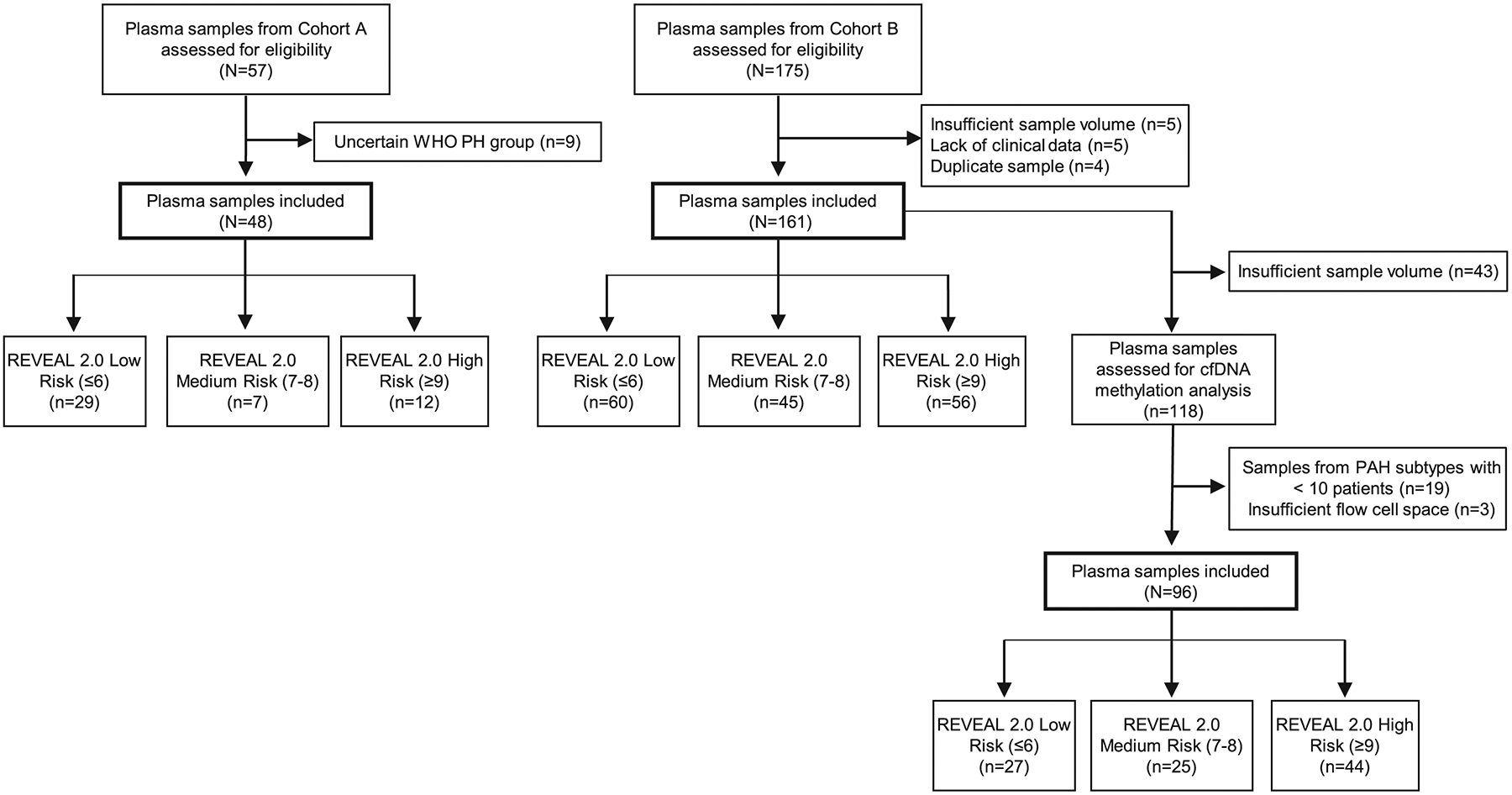
Plasma samples selected for cfDNA methylation analysis (n=96) were obtained from patients with idiopathic (n=42), connective tissue disease-associated (n=35) and portal hypertension-associated PAH (n=19). In addition to PAH patients, plasma samples were obtained in consecutively enrolled healthy controls (n=48; mean age 57 years; 32% female) at the NIH Clinical Center Department of Transfuison Medicine. Of these healthy control samples, 16 were randomly selected to undergo additional methylation sequencing analysis. Abbreviations: WHO, World Health Organization; PH, pulmonary hypertension; cfDNA, cell-free DNA; REVEAL, Registry to Evaluate Early And Long-term PAH Disease Management; IPAH, idiopathic PAH; CTD-PAH, connective tissue disease-associated PAH; PoHTN-PAH, portal hypertension-associated PAH.
Table 1.
Demographic and Clinical Data
| Variable | Cohort A (N=48) |
Cohort B (N=161) |
|---|---|---|
| Age, years | 62 (47–71) | 59 (49–71) |
| Female, n (%) | 36 (75) | 111 (69) |
| Body Mass Index, kg/m2 | 26 (23–31) | 27 (22–31)† |
| Etiology, n (%) | ||
| Idiopathic | 16 (33) | 64 (40) |
| Heritable | 4 (8) | 2 (1) |
| Drug/toxin-induced | 3 (6) | 4 (2) |
| Connective tissue disease | 20 (42) | 50 (31) |
| HIV infection | ----- | 3 (2) |
| Portal hypertension | ----- | 26 (16) |
| Congenital heart disease | 5 (11) | 9 (6) |
| PVOD/PCH | ----- | 3 (2) |
| Medications, n (%) | ||
| PDE5 inhibitors | 31 (65) | 64 (40) |
| Endothelin receptor antagonists | 28 (58) | 47 (29) |
| IV/SQ PGI2 | 7 (15) | 26 (16) |
| Inhaled/PO PGI2 or PGI2 receptor agonist | 17 (35) | 6 (4) |
| Soluble guanylate cyclase stimulator | 3 (6) | 2 (1) |
| Calcium channel blockers | 3 (6) | 12 (7) |
| Treatment naïve | 9 (19) | 47 (29) |
| Comorbidities, n (%) | ||
| Hypertension | 21 (44) | 58 (36) |
| Obstructive sleep apnea | 17 (35) | 26 (16) |
| COPD | 6 (13) | 27 (17) |
| Interstitial lung disease | 3 (6) | 9 (6) |
| Cancer | 8 (17) | 15 (9) |
| Diabetes mellitus | 7 (15) | 21 (13) |
| REVEAL 2.0 | 6 (4–9) | 7 (6–9) |
| REVEAL Lite 2 | 5 (4–6) | 7 (5–8) |
| mPAP, mmHg | 36 (25–44) | 44 (34–52) |
| RAP, mmHg | 5 (3–7)* | 10 (7–13) |
| PAWP, mmHg | 9 (6–12)* | 8 (6–13) |
| PVR, WU | 5 (3–8) | 8 (5–10) |
| HR, bpm | 71 (62–84)* | 78 (67–90) |
| SBP, mmHg | 130 (113–141)* | 122 (112–136) |
| DLCO, % predicted | 55 (42–72)* | NR |
| 6MWD, meters | 333 (256–405)* | 358 (158–466)† |
| NYHA class | 2 (2–3)* | 3 (2–3)† |
| NT-proBNP, pg/ml | 288 (78–763)* | NR |
| BNP, pg/ml | NR | 145 (56–418)† |
| Hospitalization within the past 6 months, n (%) | 19 (40) | NR |
Continuous variables are displayed as median (interquartile range).
Indicates less than 48 values available: RAP=47; PAWP=47; HR=37; SBP=46; DLCO=46; 6MWD=45; NYHA=39; NT-proBNP=46
Indicates less than 161 values available: Body mass index=159; NYHA=160; 6MWD=136; BNP=158,
Abbreviations: PAH, pulmonary arterial hypertension; HIV, human immunodeficiency virus; PVOD/PCH, pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis; PDE5, phosphodiesterase 5; PGI2, prostacyclin; COPD, chronic obstructive pulmonary disease; REVEAL, Registry to Evaluate Early and Long-Term PAH Disease Management; mPAP, mean pulmonary artery pressure; RAP, right atrial pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; WU, Wood units; bpm, beats per minute; DLCO, diffusing capacity of the lungs for carbon monoxide; NR, not recorded; 6MWD, six minute walk distance; NYHA, New York Heart Association; NT-proBNP, N-terminal pro brain-type natriuretic peptide; BNP, brain-type natriuretic peptide.
cfDNA correlates with PAH disease severity
In both Cohorts, cfDNA concentrations differed amongst PAH patients of varying REVEAL 2.0 risk and healthy controls (ANOVA P≤0.002; Figure 2) and were greater in the high compared with the low risk REVEAL 2.0 category (P≤0.002; additional pairwise comparisons in Figure 2). In a sensitivity analysis of Cohort B by PAH subtype, cfDNA was also increased in patients with idiopathic, connective tissue disease-associated, and portal hypertension-associated PAH compared with healthy controls (Figure S1). Despite similar age, sex, and comorbidity distributions (Table S1), patients with higher cfDNA levels had more severe PAH based on REVEAL 2.0 scores, invasive hemodynamic measures, 6-minute walk distance, BNP and hs-troponin T concentrations (Table 2). These patients also had higher hs-CRP levels and body mass index. Furthermore, correlation analyses identified similar associations between cfDNA and clinically significant measures of risk and disease severity in both cohorts (Table S2 and Table S3).
Figure 2. Concentrations of cell-free DNA (cfDNA) are elevated in pulmonary arterial hypertension (PAH) patients and correlate with REVEAL 2.0 risk scores in two separate patient cohorts.
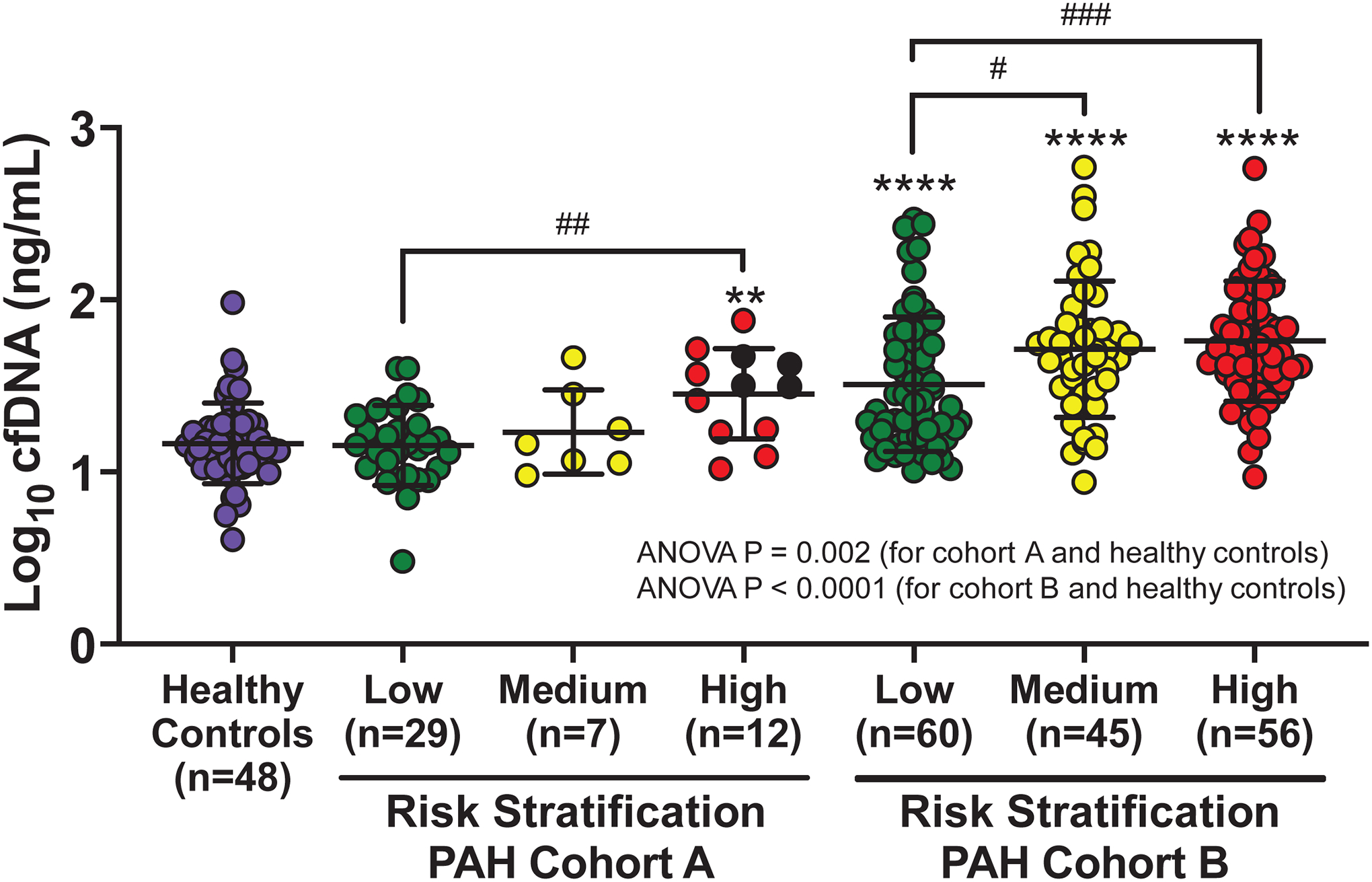
Healthy control and PAH patient cfDNA concentrations are displayed as mean +/− SD of log10-transformed values. Patients were divided into three risk groups based on REVEAL 2.0 score: low (≤ 6), medium (7–8), and high (≥ 9). In Cohort A (n=48), analysis of variance between the healthy control group and the three risk groups of PAH patients was significant (P=0.002) as were pairwise comparisons of high-risk PAH patients with healthy controls and low-risk patients (P=0.002 for both). Four patients in Cohort A died during follow-up (black data points). In Cohort B (n=161), analysis of variance across all 4 groups was also significant (P<0.0001). Pairwise comparisons identified significantly higher cfDNA concentrations in all three PAH risk groups compared with healthy controls (P<0.0001 for all). cfDNA concentration was also significantly greater in medium (P=0.02) and high-risk PAH patients (P=0.0008) compared with low-risk patients. P-values for pairwise comparisons were adjusted for multiple comparisons. ** P<0.01; **** P<0.0001 for PAH risk group versus healthy controls. # P<0.05; ## P<0.01; ### P<0.001 for comparisons between PAH risk groups.
Table 2.
PAH clinical variables according to cfDNA tertile in Cohort B
| cfDNA Tertile | |||||||
|---|---|---|---|---|---|---|---|
| Low Tertile | Middle Tertile | High Tertile | |||||
| Variable | N | Median (IQR) | N | Median (IQR) | N | Median (IQR) | P-value** |
| Age | 54 | 61 (52–71) | 53 | 59 (48–73) | 54 | 59 (48–71) | 0.99 |
| Body mass index | 52 | 25.5 (21.6–30.2) | 53 | 25.6 (22.0–31.1) | 54 | 29.6 (24.5–32.6) | 0.02 |
| REVEAL 2.0 | 54 | 6 (4–7) | 53 | 8 (7–9) | 54 | 8 (6–9) | <0.0001 |
| REVEAL Lite 2 | 54 | 6 (4–7) | 53 | 7 (6–8) | 54 | 7 (6–8) | 0.0004 |
| mPAP (mmHg) | 54 | 35 (26–50) | 53 | 45 (36–52) | 54 | 48 (42–58) | 0.0001 |
| RAP (mmHg) | 54 | 9 (6–12) | 53 | 9 (6–11) | 54 | 11 (8–15) | 0.007 |
| PAWP (mmHg) | 54 | 7 (5–11) | 53 | 7 (5–12) | 54 | 10 (7–16) | 0.01 |
| PVR (Wood units) | 54 | 5.7 (3.2–8.9) | 53 | 7.0 (4.9–11.1) | 54 | 8.3 (6.0–10.2) | 0.005 |
| Cardiac output (L/min) | 54 | 4.7 (4.0–5.7) | 53 | 4.9 (3.3–6.2) | 54 | 4.5 (3.7–5.5) | 0.58 |
| Heart rate (bpm) | 54 | 76 (67–90) | 53 | 83 (72–95) | 54 | 80 (65–89) | 0.12 |
| SBP (mmHg) | 54 | 119 (111–131) | 53 | 122 (110–139) | 54 | 125 (117–136) | 0.31 |
| 6MWD (meters) | 51 | 450 (269–488) | 46 | 312 (152–413) | 39 | 290 (152–439) | 0.003 |
| NYHA | 54 | 2 (2–3) | 53 | 3 (2–3) | 53 | 3 (2–3) | 0.09 |
| BNP (pg/mL) | 53 | 64 (23–191) | 51 | 143 (62–465) | 54 | 264 (112–734) | <0.0001 |
| hs-Troponin T (ng/L) | 27 | 11.29 (6.65–17.90) | 32 | 13.99 (10.41–20.78) | 22 | 24.89 (13.35–38.18) | 0.002 |
| hs-CRP (mg/L) | 29 | 2.5 (0.8–4.2) | 33 | 6.5 (4.2–15.4) | 23 | 7.3 (4.1–36.0) | <0.0001 |
| Creatinine (mg/dL) | 48 | 0.91 (0.73–1.06) | 51 | 0.93 (0.80–1.21) | 47 | 0.94 (0.79–1.16) | 0.36 |
Subjects were divided into cfDNA tertiles as Low (cfDNA ≤ 28.99 ng/mL), Middle (28.99 < cfDNA ≤ 61.12 ng/mL) or High Tertile (cfDNA > 61.12 ng/mL)
For each variable, differences across cfDNA tertiles were assessed by Kruskal-Wallis test.
Abbreviations: PAH, pulmonary arterial hypertension; cfDNA, cell-free DNA; REVEAL, Registry to Evaluate Early and Long-Term PAH Disease Management; mPAP, mean pulmonary artery pressure; RAP, right atrial pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; SBP, systolic blood pressure; 6MWD, six-minute walk distance; NYHA, New York Heart Association; BNP, brain-type natriuretic peptide; hs, high-sensitivity; CRP, C-reactive protein.
cfDNA predicts transplant-free survival
Available patient follow-up (median 2.7 years) in Cohort B enabled assessment of the relationship between total cfDNA and transplant-free survival. The overall incidence of death or transplant in Cohort B was 45%, with the majority of events being deaths due to progressive PH (64%). Other causes of death included liver/kidney failure (9%), cancer (6%), sepsis (6%), trauma (1%), and unknown (14%). Death or lung transplant occurred in 14 of 54 patients in the lowest cfDNA tertile, 23 of 53 in the middle tertile and 35 of 54 in the highest tertile. A Kaplan-Meier curve, dividing the cohort into three groups based on cfDNA tertiles, demonstrated significantly worse outcomes in patients within higher cfDNA tertiles (log-rank test: P=0.0001; Figure 3). REVEAL 2.0 and REVEAL Lite 2 risk categories also predicted patient outcomes in Cohort B (log-rank test: P<0.0001 for both; Figure S2A and S2B).
Figure 3. Elevated cell-free DNA (cfDNA) concentrations are associated with lower transplant-free survival in Cohort B pulmonary arterial hypertension (PAH) patients.
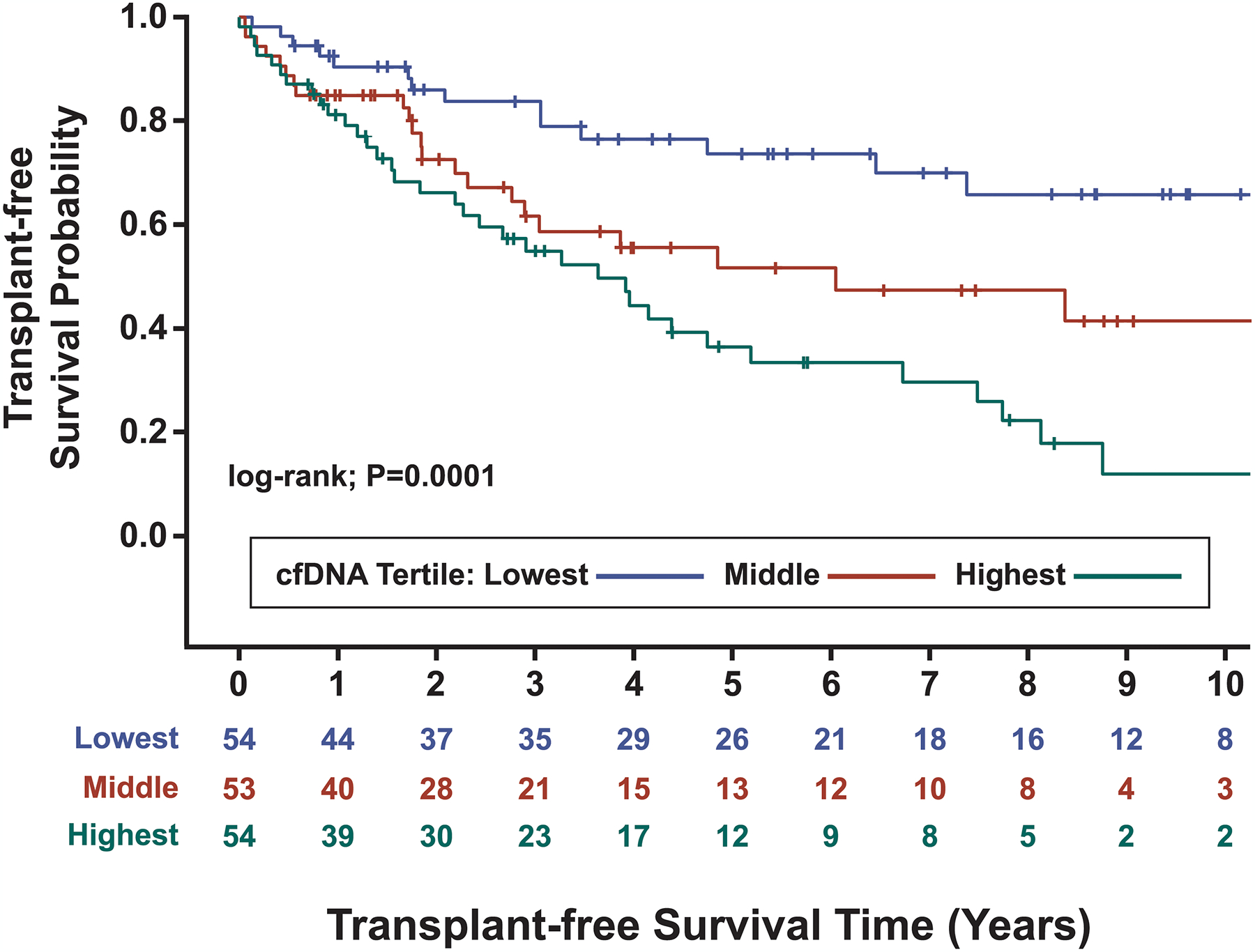
Kaplan-Meier analysis over a median (IQR) follow up time of 2.7 (1.0–5.2) years demonstrated significantly different transplant-free survival amongst cfDNA tertiles (log-rank test: P=0.0001). Corresponding concentrations for each cfDNA tertile are as follows: ≤ 28.99 ng/ml (blue line), 28.99 < cfDNA ≤ 61.12 ng/ml (red line), and > 61.12 ng/ml (green line). Patients were censored at the time of death or transplantation.
The hazard ratio (HR) of death or transplant, accounting for age and sex, was 3.8 (95% CI 2.0–7.0; P<0.001) times higher in the highest cfDNA tertile compared with the lowest and 2.8 (95% CI 1.4–5.6; P=0.003) times higher in the middle tertile compared with the lowest. After accounting for age, sex, and REVEAL 2.0 risk in a Cox proportional hazard model, cfDNA tertiles remained a significant predictor of death or transplant, driven by increased risk in the highest cfDNA tertile compared to the lowest (HR 2.5, 95% CI 1.3–4.9).
Accounting for age and sex, ROC analyses using logistic regression models demonstrated that the AUC did not differ for cfDNA tertiles and REVEAL 2.0 risk (0.75 vs 0.72; P=0.29). Adding cfDNA to REVEAL 2.0 led to a statistically significant AUC improvement (0.72 to 0.78; P=0.02; Figure 4). cfDNA also performed similarly to European Society of Cardiology/European Respiratory Society recommended BNP risk categorization2 (AUC 0.75 vs 0.74; P=0.79). The AUC for BNP improved with the addition of cfDNA (AUC 0.74 to 0.78; P=0.08), and both BNP and cfDNA remained significant independent predictors when included together in a logistic regression model. Finally, neither creatinine nor hs-CRP were statistically significant predictors when added to models including age and sex.
Figure 4. Receiver operating characteristic analyses of REVEAL 2.0 risk and REVEAL 2.0 risk with the addition of cell-free DNA (cfDNA) for predicting death or lung transplant in Cohort B.
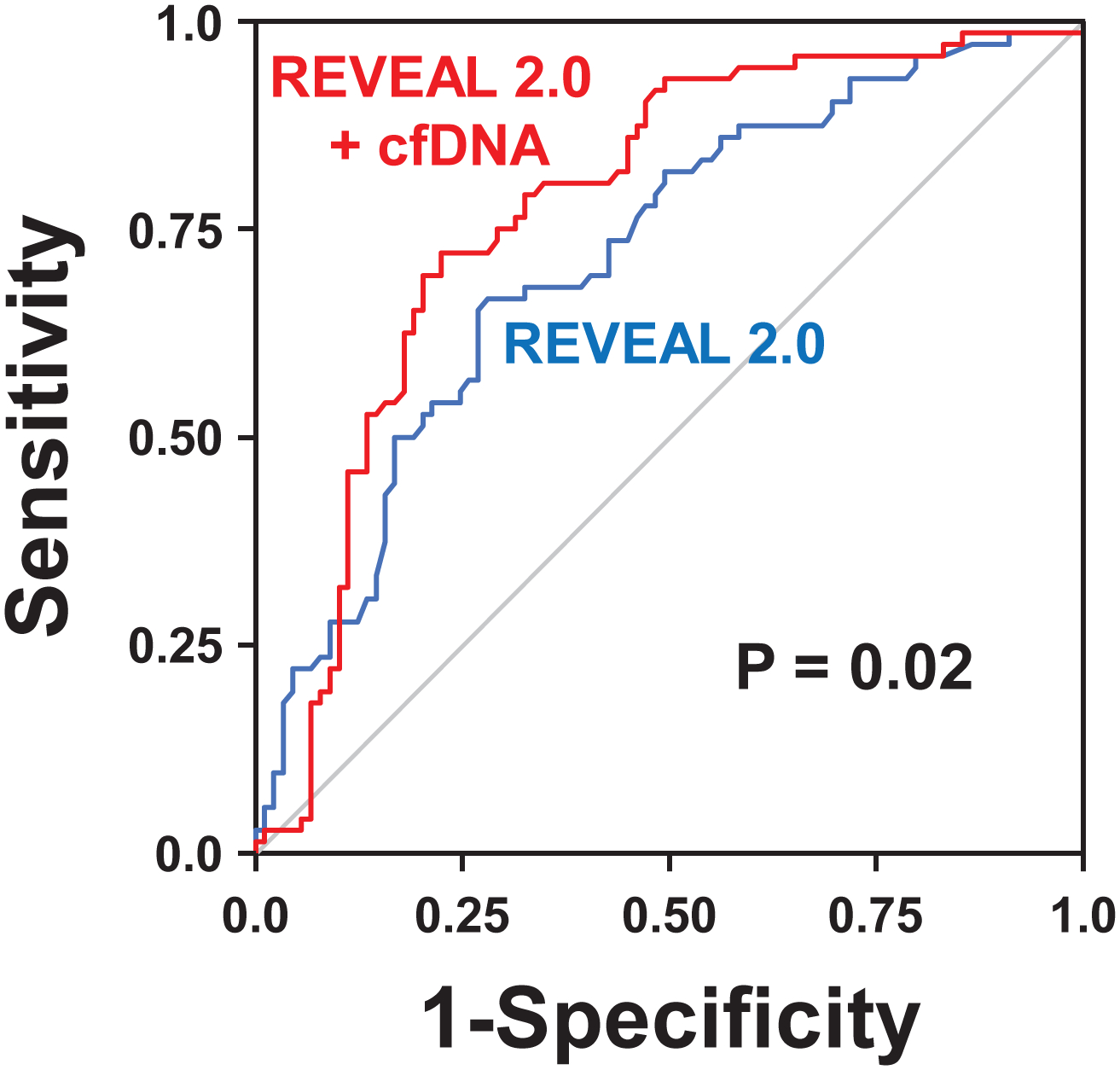
After adjusting for both age and sex, the area under the curve (AUC) of receiver operating characteristics for REVEAL 2.0 risk categories (blue line) was similar to the performance of cfDNA tertiles (line not shown) in Cohort B (AUC of 0.72 versus 0.75; P=0.29). The predictive performance of REVEAL 2.0 risk classification significantly improved with the addition of cfDNA tertiles (red line), with the AUC increasing from 0.72 to 0.78 (P=0.02).
cfDNA methylation sequencing analysis reveals a distinct PAH cellular injury pattern
Samples with sufficient plasma from patients with idiopathic (n=42), connective tissue disease-associated (n=35) and portal hypertension-associated PAH (n=19) in Cohort B (n=96) as well as healthy controls (n=16) underwent cfDNA bisulfite conversion and whole genome sequencing. PAH patients demonstrated a distinct cellular injury pattern compared with healthy controls (Figure 5A), with significantly increased cfDNA arising from hematopoietic cells, including erythrocyte progenitors, neutrophils, monocytes, NK cells, and non-hematopoietic cell types such as cardiac myocytes, vascular endothelium, and adipocytes (Bonferroni adjusted P<0.05 for all). cfDNA concentrations from kidney, bladder, squamous epithelium, and hepatocytes were not significantly different between PAH patients and healthy controls. Pairwise assessments corrected for multiple comparisons revealed that three cell types discriminated between high and low-risk PAH patients (Figure 5B). These included erythrocyte progenitors (P=0.02), vascular endothelium (P=0.004), and cardiac myocytes (P=0.006). Only cfDNA concentrations derived from cardiac myocytes were significantly higher in medium compared with low-risk patients (P=0.04). Levels of cfDNA derived from vascular endothelium also tended to be higher in medium compared with low-risk patients, but this did not reach statistical significance (P=0.08). Representative tissue distributions of cfDNA for a healthy control and three PAH patients of varying REVEAL 2.0 risk (Figure 6) demonstrate that the majority of cfDNA originated from myeloid lineage cells; however, only in PAH patients was there a substantial signal for cfDNA originating from vascular endothelium and cardiac myocytes. The levels of cfDNA from these two cell types implicated in PAH pathobiology increased in accordance with PAH disease severity.
Figure 5. Patients with pulmonary arterial hypertension (PAH) demonstrate a distinct and pathobiologically meaningful cell-free DNA (cfDNA) injury pattern.
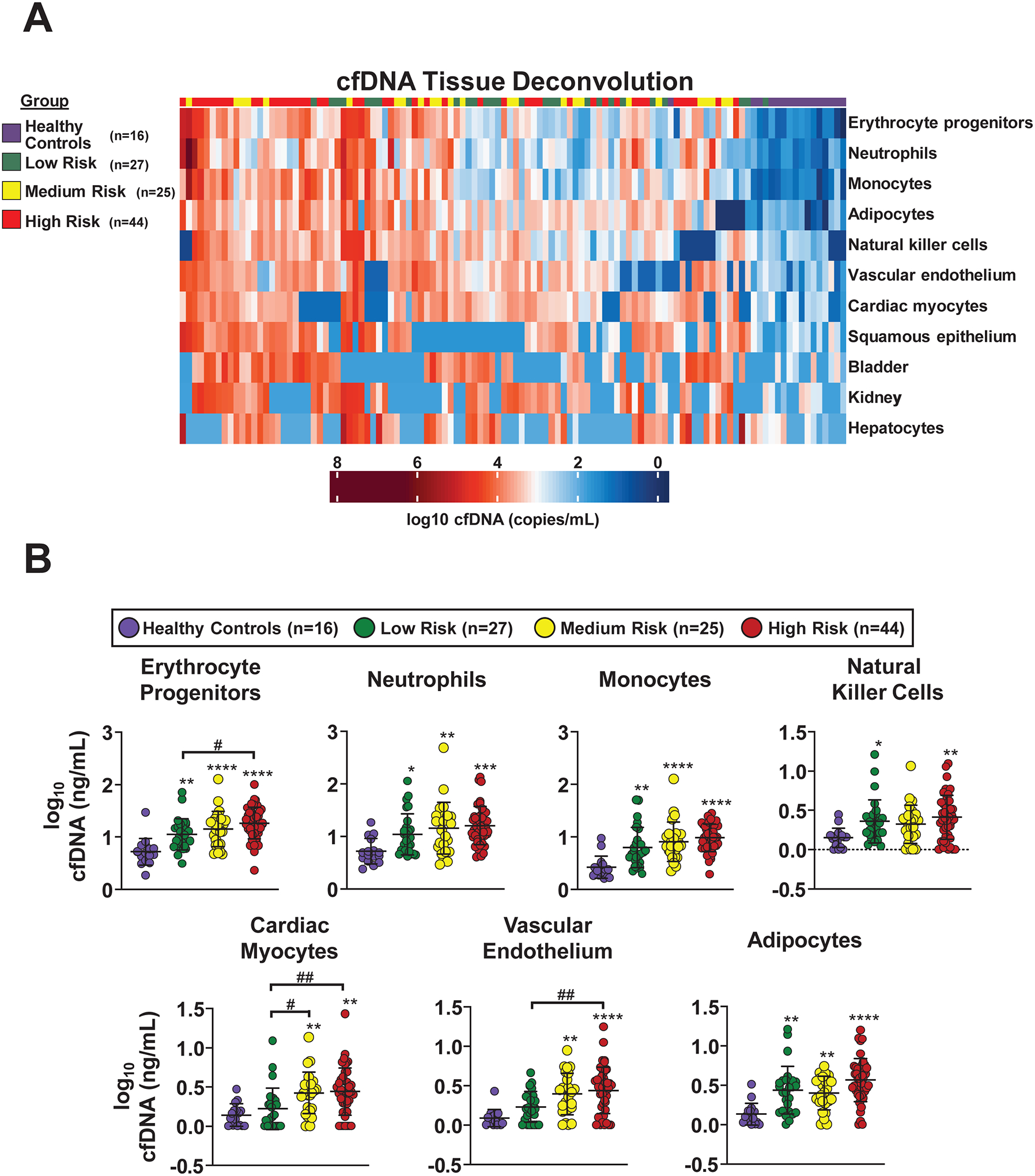
A, Unsupervised clustering of absolute cfDNA concentrations from 11 different cell types in a subset of Cohort B PAH patients (n=96) and healthy controls (n=16; Purple ribbon). After bisulfite sequencing, cfDNA sequence reads were analyzed against a library of 25 cell-specific DNA methylation signatures to deconvolve the cfDNA tissue of origin. The analysis was limited to the 11 cells or tissues that had quantifiable cfDNA present in the majority of patients. Those that were not substantially represented in either PAH patients or healthy controls included B cells, CD4 T cells, CD8 T cells, cortical neurons, thyroid, breast, alveolar epithelium, upper gastrointestinal tract, colon, pancreatic beta cells, pancreatic acinar cells, pancreatic duct cells, prostate, and uterus/cervix. PAH patients were grouped into low (≤ 6, n=27; Green ribbon), medium (7–8, n=25; Yellow ribbon), and high-risk (≥ 9, n=44; Red ribbon) categories based on REVEAL 2.0 risk score. Each column represents an individual patient or healthy control and each row represents a specific cell or tissue type. Therefore, the intersection of each column and row represents an individual’s absolute cfDNA concentration from a specific cell or tissue type. cfDNA is represented in copies/mL of plasma and reported in log scale from lowest (blue) to highest (red) values. B, Cell-specific cfDNA concentrations are displayed as mean +/− SD of log10-transformed values. Patients were divided into three risk groups based on REVEAL 2.0 score: low (≤ 6), medium (7–8), and high (≥ 9). Only the seven cell types that were significantly elevated in PAH patients compared with healthy controls are depicted (Bonferroni adjusted P<0.05 for all). Pairwise comparisons between PAH patients and healthy controls are illustrated with asterisks (* P<0.05; ** P<0.01; *** P<0.001; **** P<0.0001). Pairwise comparisons between PAH patients of varying risk are illustrated with pound signs (# P<0.05; ## P<0.01). Erythrocyte progenitor cells, cardiac myocytes, and vascular endothelium differentiated between PAH patients of varying REVEAL 2.0 risk. All comparisons were corrected for multiple testing.
Figure 6. Representative cell-free DNA (cfDNA) distributions by origin from a healthy control and pulmonary arterial hypertension (PAH) patients of low, medium, and high risk.
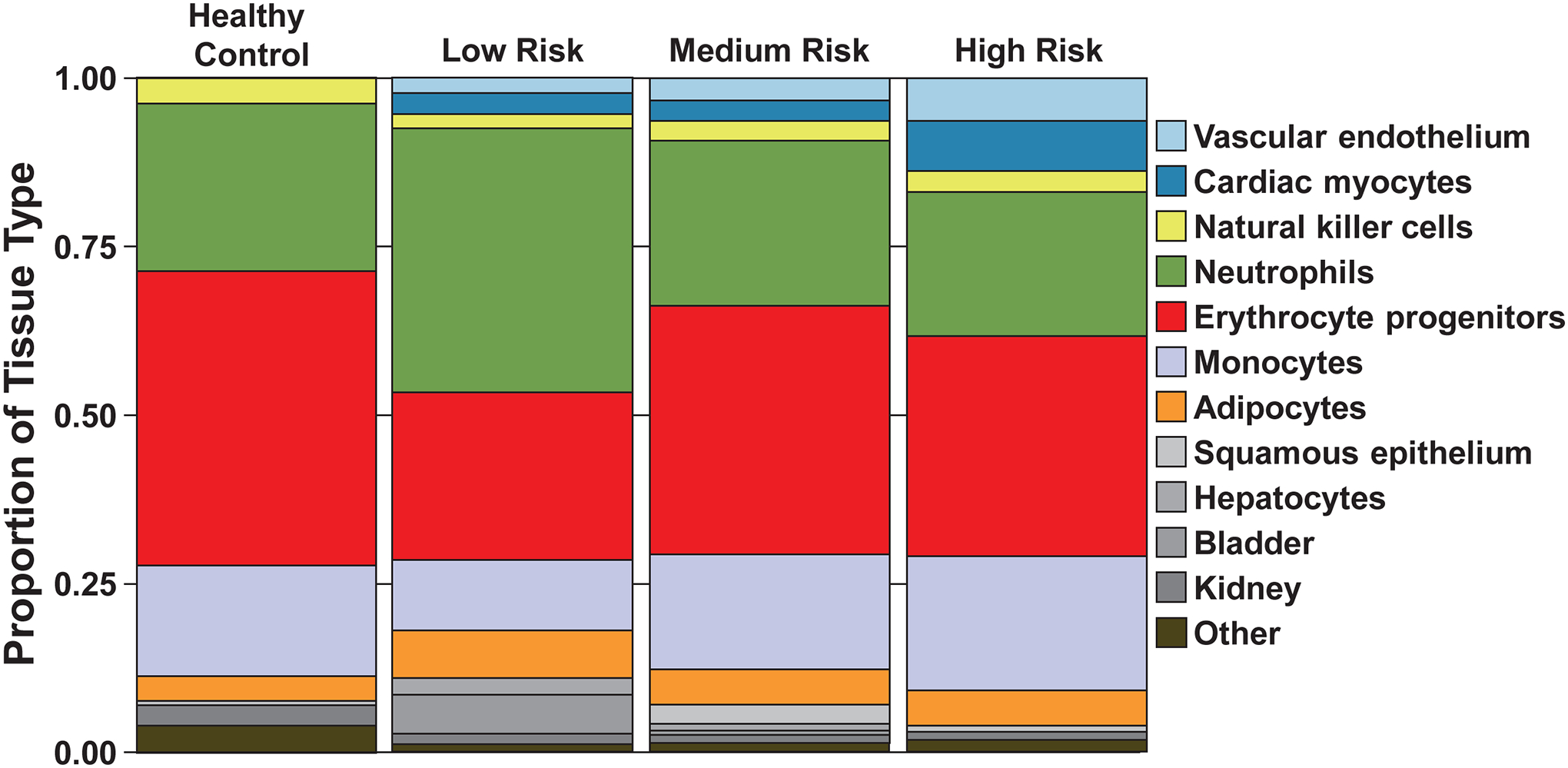
A stacked bar graph displays the proportion of cfDNA (in log10-transformed copies/mL) originating from specific cell or tissue types for a representative healthy control and 3 PAH patients of varying risk. Patients were grouped into low (≤ 6), medium (7–8), and high risk (≥ 9) categories based on REVEAL 2.0 risk score. Each cell or tissue is identified by a specific color, with the length of the stacked segment corresponding to the relative contribution of that source to the total concentration of cfDNA. For healthy control, low, medium, and high risk individuals, the other category consisted of 2, 1, 3, and 4 sources, respectively, that did not have quantifiable cfDNA present in the majority of patients.
Discussion:
This study demonstrates that cfDNA is elevated in PAH patients compared with healthy controls. The results indicate that cfDNA represents a biomarker of disease severity and poor prognosis, with a 3.8 times greater risk of death or transplant identified in the highest tertile of cfDNA compared with the lowest tertile. In the initial analysis (Cohort A), there was a clear relationship between cfDNA concentration and PAH disease severity as determined by increasing REVEAL 2.0 risk score. The second, independent analysis (Cohort B), not only replicated the relationship between cfDNA and PAH disease severity, but also found an association between elevated cfDNA concentration and worse transplant-free survival. Further, cfDNA demonstrated an ability to discriminate that did not differ from the multivariable REVEAL 2.0 model and the addition of cfDNA to REVEAL 2.0 risk improved discrimination. Lastly, the origin of increased cfDNA in PAH was determined, identifying injury from cell types implicated in PAH pathobiology, with increased cfDNA originating from inflammatory cells as well as from vascular endothelial and cardiac myocyte damage. Importantly, a subset of these cell-specific components of cfDNA also differentiated PAH patients by disease severity, implicating their potential role as markers of underlying disease activity.
The distinct PAH cellular injury pattern demonstrated in this study is biologically plausible and in line with the current understanding of PAH pathogenesis, which encompasses dysregulated inflammation, endothelial damage, and myocardial dysfunction. Higher concentrations of cfDNA originating from myeloid-derived inflammatory cells such as monocytes and neutrophils in PAH patients supports an active role for the bone marrow in the pathobiology of pulmonary vascular disease.38–40 Recruitment of circulating monocytes to the lung and the contribution of monocyte-derived interstitial macrophages, in particular, have been demonstrated in hypoxia-induced animal models of PH and PAH patients.41–43 Similarly, neutrophils,44, 45 and neutrophil elastase,46–48 contribute to PAH-associated inflammation.
A predominance of cfDNA derived from erythrocyte progenitors in patients with PAH is in line with an earlier genome-wide expression profiling study that identified an erythroid precursor cell gene signature in PAH patients49 and previous work highlighting the association of hypoxia-inducible factors such as erythropoietin with PAH-associated myeloid abnormalities.50 Notably, factors that prominently influence erythropoiesis intersect with a number of PAH paradigms such as bone morphogenetic protein/transforming growth factor-β (TGF-β) signaling, inflammation, hypoxia-inducible factors and iron homeostasis.51, 52 The link between PAH and abnormal erythropoiesis is further substantiated by recent clinical studies investigating a shared therapeutic approach of targeting pathological SMAD2/3 activation with TGF-β ligand traps.53, 54
Although histopathological studies of explanted lung tissue from PAH patients demonstrate inflammatory infiltrates55–57 consisting of both innate and adaptive immune cells,56, 57 and despite the sizeable proportion of lymphocytes in circulation (normally 20–40% of leukocytes), detectable levels cfDNA derived from B and T lymphocytes were present in fewer than one-third of PAH patients. Thus, while cfDNA concentrations may correlate with the proportion of some circulating leukocyte subsets,27 cfDNA cell of origin analysis is not simply a recapitulation of a peripheral blood cell count and differential. For example, despite the paucity of cfDNA derived from B and T lymphocytes, cfDNA originating from NK cells was increased in PAH patients compared with healthy controls. Interestingly, a reduced number of circulating NK cells as well as alterations in NK cell function have been reported in rodent models of PH and PAH patients.58 Furthermore, in genetic mouse models of NK cell deficiency, animals spontaneously develop pulmonary vascular disease.59 Although not specifically determined in this study, the higher cfDNA concentrations from NK cells may imply an increase in cell turnover and thus offer a potential explanation for lower circulating numbers of functional CD56dimCD16+ NK cells detected in PAH patients.58
This study also identified increased cfDNA from non-hematopoietic cell types associated with PAH pathobiology. For example, increased cfDNA derived from adipocytes observed in PAH patients is consistent with obesity being among the top comorbid conditions in patients with PAH.60 Metabolic syndrome, specifically obesity and insulin resistance, have been implicated as modifiers of pulmonary vascular disease.61, 62 Furthermore, adipose tissue dysfunction is linked to dysregulated inflammation and adipokine imbalance.63
cfDNA derived from vascular endothelium and cardiac myocytes were also among the non-myeloid cells that distinguished between PAH patients in different risk strata. This is consistent with the importance of adverse pulmonary vascular remodeling and right ventricular dysfunction to outcomes in PAH and lends credence to the methylation analysis presented in this study. In aggregate, these findings support the growing body of evidence that PAH is truly a systemic disease.64
If replicated in future studies, the ability to quantify cell injury and/or turnover specific to both the endothelium and myocardium represents a major step forward in PAH biomarker development. Although pulmonary vascular remodeling is the sine qua non of PAH, and endothelial injury is believed to be a critical element that initiates vessel remodeling, there are currently no blood or imaging markers available to specifically detect the extent of lung vascular damage in PAH patients. However, as next generation cfDNA applications continue to mature and the precision of cell- and tissue-specific epigenetic classification improves, the ability to identify cfDNA specific to pulmonary vascular endothelium may soon be possible. Further, chromatin immunoprecipitation-sequencing of cfDNA (cfChIP-seq) targeting specific histone modifications may offer a complementary method for defining injury patterns while concurrently inferring disease-specific transcriptional programs without the need for invasive procedures.65
Despite the ability to elucidate the cellular origins of elevated cfDNA in PAH, significant knowledge gaps in extracellular DNA biology persist, including an unclear primary mechanism of release into circulation and a potential role in innate immune system activation and the development of apoptotic resistance.66–69 Nevertheless, the ability to quantify and visualize patient-specific tissue injury patterns in PAH has the potential to greatly improve the understanding of PAH endovascular phenotypes and potentially develop and choose targeted therapies. Though the present study was not powered to evaluate the prognostic performance of specific cfDNA sub-populations, it remains likely that certain cfDNA concentrations will have more clinical utility than others.
The relationship between total cfDNA concentration and survival as well as the identified biologically plausible unique PAH cellular injury pattern described in this study are clinically relevant and require validation. Total cfDNA is easy to isolate and measure and could improve risk models in the near future. More importantly, the ability to isolate and quantify organ-specific cfDNA provides a unique, non-invasive window into PAH pathogenesis, in a disease where tissue sampling is possible only at the time of transplantation or death. A map of cellular or tissue injury patterns in PAH could allow for the monitoring of specific disease pathogenesis pathways, the institution of personalized “precision medicine,” and the non-invasive monitoring of disease progression. Further, cfDNA monitoring may allow for earlier recognition of pulmonary vascular remodeling and earlier initiation of therapy, prior to the development of right ventricular dysfunction. With this in mind, serial sampling of cfDNA in a prospective fashion is warranted.
Study Limitations:
Limitations of this study include the relatively small PAH cohort sizes, lack of serial samples as well as the retrospective design. Additionally, the ability of cfDNA to predict transplant-free survival in a separate validation cohort was not evaluated. However, confirming the relationship between cfDNA and disease severity in two separate cohorts is encouraging. Further, the identification of such a robust relationship between cfDNA and mortality despite the limited sample size highlights the potential predictive power of cfDNA. Finally, multiple tests were performed to identify the cell-specific cfDNA fractions that were elevated in PAH patients and differentiated between PAH risk groups. A 2-stage adjustment was utilized in an attempt to balance type I and II error rates but likely did not fully control type I error rates. Future larger validation cohorts and prospective studies are needed to confirm these findings.
cfDNA concentrations in Cohort B were substantially higher than in Cohort A. This may be attributable in part to differences in PAH disease severity among the cohorts, including higher REVEAL 2.0 scores, higher New York Heart Association functional class, and greater hemodynamic disturbances in cohort B. However, it is also possible that a component of cfDNA variability between cohorts stemmed from differences in plasma collection, processing, and storage techniques. Even though the quality of cfDNA was confirmed in both cohorts using quantitative PCR and gel electrophoresis, and internal comparisons and risk prediction remain valid, comparisons between archived biorepository samples from independent cohorts must be interpreted with caution. Future prospective trials investigating cfDNA should include standardized collection procedures to mitigate any variability that may be introduced during sample collection, allowing the combination of cohorts for improved statistical power and direct comparisons between patients from different centers.
Conclusion:
Cell-free DNA is elevated in patients with PAH compared with healthy controls and increases with disease severity. Elevated cfDNA predicts worse transplant-free survival and may add prognostic value to currently used risk scores. cfDNA from PAH patients demonstrates a cellular injury pattern primarily originating from myeloid inflammatory cells, but there is also evidence of vascular endothelium as well as cardiac myocyte damage that correlates with disease severity. Validation of these findings in a larger, prospective patient cohort with serial samples will help to better define the role of cfDNA in clinical PAH risk prediction.
Supplementary Material
Clinical Perspective.
What is New?
Circulating cell-free DNA (cfDNA) is elevated in patients with pulmonary arterial hypertension (PAH) compared with healthy controls.
In two independent PAH patient cohorts, cfDNA concentrations increased with severity of disease and predicted transplant-free survival in the larger of the two cohorts.
Methylation patterns revealed increased cfDNA originating from biologically plausible sites including erythrocyte progenitor and myeloid lineage inflammatory cells, vascular endothelium, and cardiac myocytes.
What are the Clinical Implications?
Cell-free DNA concentration could serve as a non-invasive biomarker of underlying disease activity in patients with pulmonary arterial hypertension.
Cell-free DNA measurements may add prognostic value to currently used PAH risk scores.
Cell-free DNA categorized by tissue origin may provide a unique, non-invasive window into PAH pathogenesis.
Acknowledgments:
The authors would like to acknowledge Kelly Byrne for help in formatting and submitting the manuscript.
Sources of Funding:
This research was funded by the National Institutes of Health Clinical Center RASCL award, the National Institutes of Health Distinguished Scholar Award, the Lasker Clinical Research Scholars Program, and the Intramural Research Programs of the National Institutes of Health Clinical Center and the National Heart, Lung, and Blood Institute.
Non-standard Abbreviations and Acronyms
- 6MWD
six minute walk distance
- AUC
area under the curve
- Bpm
beats per minute
- BNP
brain-type natriuretic peptide
- cfChIP-seq
chromatin immunoprecipitation-sequencing
- cfDNA
cell-free DNA
- COPD
chronic obstructive pulmonary disease
- CRP
C-reactive protein
- CTD-PAH
connective tissue disease-associated pulmonary arterial hypertension
- ddPCR
digital droplet PCR
- DLCO
diffusing capacity of the lungs for carbon monoxide
- HR
hazard ratio
- HIV
human immunodeficiency virus
- IPAH
idiopathic pulmonary arterial hypertension
- IQR
interquartile range
- mPAP
mean pulmonary artery pressure
- NIH
National Institutes of Health
- NK
natural killer
- NT-proBNP
N-terminal pro brain-type natriuretic peptide
- NR
not recorded
- NYHA
New York Heart Association
- PAH
pulmonary arterial hypertension
- PAWP
pulmonary artery wedge pressure
- PDE5
phosphodiesterase-5
- PGI2
prostacyclin
- PH
pulmonary hypertension
- PoHTN-PAH
portal hypertension-associated pulmonary arterial hypertension
- PVOD/PCH
pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis
- PVR
pulmonary vascular resistance
- RAP
right atrial pressure
- REVEAL
Registry to Evaluate Early and Long-Term PAH Disease Management
- ROC
receiver operating characteristics
- SBP
systolic blood pressure
- TGF-β
transforming growth factor-β
- WHO
World Health Organization
- WU
Wood units
Footnotes
Disclosures: The authors have no disclosures.
References:
- 1.Galie N, Channick RN, Frantz RP, Grunig E, Jing ZC, Moiseeva O, Preston IR, Pulido T, Safdar Z, Tamura Y, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53: 1801889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119. [DOI] [PubMed] [Google Scholar]
- 3.Nagaya N, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Nakanishi N, Yamagishi M, Kunieda T and Miyatake K. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;160:487–92. [DOI] [PubMed] [Google Scholar]
- 4.Torbicki A, Kurzyna M, Kuca P, Fijalkowska A, Sikora J, Florczyk M, Pruszczyk P, Burakowski J and Wawrzynska L. Detectable serum cardiac troponin T as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation. 2003;108:844–8. [DOI] [PubMed] [Google Scholar]
- 5.Quarck R, Nawrot T, Meyns B and Delcroix M. C-reactive protein: a new predictor of adverse outcome in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;53:1211–8. [DOI] [PubMed] [Google Scholar]
- 6.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–7. [DOI] [PubMed] [Google Scholar]
- 7.Hu EC, He JG, Liu ZH, Ni XH, Zheng YG, Gu Q, Zhao ZH and Xiong CM. High levels of serum lactate dehydrogenase correlate with the severity and mortality of idiopathic pulmonary arterial hypertension. Exp Ther Med. 2015;9:2109–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simpson CE, Damico RL, Hassoun PM, Martin LJ, Yang J, Nies MK, Vaidya RD, Brandal S, Pauciulo MW, Austin ED, et al. Noninvasive Prognostic Biomarkers for Left-Sided Heart Failure as Predictors of Survival in Pulmonary Arterial Hypertension. Chest. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simpson CE, Damico RL, Hummers L, Khair RM, Kolb TM, Hassoun PM and Mathai SC. Serum uric acid as a marker of disease risk, severity, and survival in systemic sclerosis-related pulmonary arterial hypertension. Pulm Circ. 2019;9:2045894019859477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samokhin AO, Hsu S, Yu PB, Waxman AB, Alba GA, Wertheim BM, Hopkins CD, Bowman F, Channick RN, Nikolic I, et al. Circulating NEDD9 is increased in pulmonary arterial hypertension: A multicenter, retrospective analysis. J Heart Lung Transplant. 2020;39:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frantz RP, Farber HW, Badesch DB, Elliott CG, Frost AE, McGoon MD, Zhao C, Mink DR, Selej M and Benza RL. Baseline and Serial Brain Natriuretic Peptide Level Predicts 5-Year Overall Survival in Patients With Pulmonary Arterial Hypertension: Data From the REVEAL Registry. Chest. 2018;154:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin KM, Rubin LJ, Channick R, Di Scala L, Gaine S, Galie N, Ghofrani HA, Hoeper MM, Lang IM, McLaughlin VV, et al. Association of N-Terminal Pro Brain Natriuretic Peptide and Long-Term Outcome in Patients With Pulmonary Arterial Hypertension. Circulation. 2019;139:2440–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SY and Rubin LJ. Metabolic dysfunction in pulmonary hypertension: from basic science to clinical practice. Eur Respir Rev. 2017;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar R and Graham B. How does inflammation contribute to pulmonary hypertension? Eur Respir J. 2018;51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thenappan T, Ormiston ML, Ryan JJ and Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elinoff JM, Mazer AJ, Cai R, Lu M, Graninger G, Harper B, Ferreyra GA, Sun J, Solomon MA and Danner RL. Meta-analysis of blood genome-wide expression profiling studies in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2020;318:L98–L111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dwivedi DJ, Toltl LJ, Swystun LL, Pogue J, Liaw KL, Weitz JI, Cook DJ, Fox-Robichaud AE, Liaw PC and Canadian Critical Care Translational Biology G. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit Care. 2012;16:R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gögenur M, Burcharth J and Gögenur I. The role of total cell-free DNA in predicting outcomes among trauma patients in the intensive care unit: a systematic review. Crit Care. 2017;21:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tissot C, Toffart AC, Villar S, Souquet PJ, Merle P, Moro-Sibilot D, Perol M, Zavadil J, Brambilla C, Olivier M, et al. Circulating free DNA concentration is an independent prognostic biomarker in lung cancer. Eur Respir J. 2015;46:1773–80. [DOI] [PubMed] [Google Scholar]
- 20.Agbor-Enoh S, Jackson AM, Tunc I, Berry GJ, Cochrane A, Grimm D, Davis A, Shah P, Brown AW, Wang Y, et al. Late manifestation of alloantibody-associated injury and clinical pulmonary antibody-mediated rejection: Evidence from cell-free DNA analysis. The Journal of Heart and Lung Transplantation. 2018;37:925–932. [DOI] [PubMed] [Google Scholar]
- 21.Agbor-Enoh S, Tunc I, De Vlaminck I, Fideli U, Davis A, Cuttin K, Bhatti K, Marishta A, Solomon MA, Jackson A, et al. Applying rigor and reproducibility standards to assay donor-derived cell-free DNA as a non-invasive method for detection of acute rejection and graft injury after heart transplantation. J Heart Lung Transplant. 2017;36:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corcoran RB and Chabner BA. Application of Cell-free DNA Analysis to Cancer Treatment. N Engl J Med. 2018;379:1754–1765. [DOI] [PubMed] [Google Scholar]
- 23.Agbor-Enoh S, Shah P, Tunc I, Hsu S, Russell S, Feller E, Shah K, Rodrigo ME, Najjar SS, Kong H, et al. Cell-Free DNA to Detect Heart Allograft Acute Rejection. Circulation. 2021;143:1184–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgard B, Blennow K, Zetterberg H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A. 2016;113:E1826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zemmour H, Planer D, Magenheim J, Moss J, Neiman D, Gilon D, Korach A, Glaser B, Shemer R, Landesberg G, et al. Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nat Commun. 2018;9:1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, Samet Y, Maoz M, Druid H, Arner P, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andargie TE, Tsuji N, Seifuddin F, Jang MK, Yuen PS, Kong H, Tunc I, Singh K, Charya A, Wilkins K, et al. Cell-free DNA maps COVID-19 tissue injury and risk of death and can cause tissue injury. JCI Insight. 2021;6: e147610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen KD, Langmead B and Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krueger F and Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu Z, Eils R and Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–9. [DOI] [PubMed] [Google Scholar]
- 31.Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, McGoon MD, Pasta DJ, Selej M, Burger CD, et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest. 2019;156:323–337. [DOI] [PubMed] [Google Scholar]
- 32.Benza RL, Kanwar MK, Raina A, Scott JV, Zhao CL, Selej M, Elliott CG and Farber HW. Development and Validation of an Abridged Version of the REVEAL 2.0 Risk Score Calculator, REVEAL Lite 2, for Use in Patients With Pulmonary Arterial Hypertension. Chest. 2021;159:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson JJ, Lau EM, Lavender M, Benza R, Celermajer DS, Collins N, Corrigan C, Dwyer N, Feenstra J, Horrigan M, et al. Retrospective Validation of the REVEAL 2.0 Risk Score With the Australian and New Zealand Pulmonary Hypertension Registry Cohort. Chest. 2020;157:162–172. [DOI] [PubMed] [Google Scholar]
- 34.Agbor-Enoh S, Wang Y, Tunc I, Jang MK, Davis A, De Vlaminck I, Luikart H, Shah PD, Timofte I, Brown AW, et al. Donor-derived cell-free DNA predicts allograft failure and mortality after lung transplantation. EBioMedicine. 2019;40:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin DY, Wei LJ and Ying Z. Checking the Cox Model with Cumulative Sums of Martingale-Based Residuals. Biometrika. 1993;80:557–572. [Google Scholar]
- 36.Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO 3rd, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. [DOI] [PubMed] [Google Scholar]
- 37.DeLong ER, DeLong DM and Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–45. [PubMed] [Google Scholar]
- 38.Asosingh K, Farha S, Lichtin A, Graham B, George D, Aldred M, Hazen SL, Loyd J, Tuder R and Erzurum SC. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood. 2012;120:1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai Z, Li M, Wharton J, Zhu MM and Zhao YY. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2alpha. Circulation. 2016;133:2447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan L, Chen X, Talati M, Nunley BW, Gladson S, Blackwell T, Cogan J, Austin E, Wheeler F, Loyd J, et al. Bone Marrow-derived Cells Contribute to the Pathogenesis of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2016;193:898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, Zhang Y, Watson A, Sembrat J, Rojas M, et al. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends upon Mobilization of Blood-Borne Monocytes. J Immunol. 2018;200:3612–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu YA, Malakhau Y, Yu CA, Phelan SJ, Cumming RI, Kan MJ, Mao L, Rajagopal S, Piantadosi CA and Gunn MD. Nonclassical Monocytes Sense Hypoxia, Regulate Pulmonary Vascular Remodeling, and Promote Pulmonary Hypertension. J Immunol. 2020;204:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N and Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor S, Dirir O, Zamanian RT, Rabinovitch M and Thompson AAR. The Role of Neutrophils and Neutrophil Elastase in Pulmonary Arterial Hypertension. Front Med (Lausanne). 2018;5:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Florentin J, Zhao J, Tai YY, Vasamsetti SB, O’Neil SP, Kumar R, Arunkumar A, Watson A, Sembrat J, Bullock GC, et al. Interleukin-6 mediates neutrophil mobilization from bone marrow in pulmonary hypertension. Cell Mol Immunol. 2021;18:374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rose F, Hattar K, Gakisch S, Grimminger F, Olschewski H, Seeger W, Tschuschner A, Schermuly RT, Weissmann N, Hanze J, et al. Increased neutrophil mediator release in patients with pulmonary hypertension--suppression by inhaled iloprost. Thromb Haemost. 2003;90:1141–9. [DOI] [PubMed] [Google Scholar]
- 47.Sweatt AJ, Miyagawa K, Rhodes CJ, Taylor S, Del Rosario PA, Hsi A, Haddad F, Spiekerkoetter E, Bental-Roof M, Bland RD, et al. Severe Pulmonary Arterial Hypertension Is Characterized by Increased Neutrophil Elastase and Relative Elafin Deficiency. Chest. 2021;160:1442–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S and Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. [DOI] [PubMed] [Google Scholar]
- 49.Cheadle C, Berger AE, Mathai SC, Grigoryev DN, Watkins TN, Sugawara Y, Barkataki S, Fan J, Boorgula M, Hummers L, et al. Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients. PLoS One. 2012;7:e34951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, Tang WH, Loyd JE, Theil K, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quatredeniers M, Mendes-Ferreira P, Santos-Ribeiro D, Nakhleh MK, Ghigna MR, Cohen-Kaminsky S and Perros F. Iron Deficiency in Pulmonary Arterial Hypertension: A Deep Dive into the Mechanisms. Cells. 2021;10:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, Davies MV, Alexander MJ, Devine M, Loveday KS, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20:408–14. [DOI] [PubMed] [Google Scholar]
- 54.Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR, Souza R, Waxman A, Escribano Subias P, Feldman J, et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2021;384:1204–1215. [DOI] [PubMed] [Google Scholar]
- 55.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908. [DOI] [PubMed] [Google Scholar]
- 57.Perros F, Dorfmuller P, Montani D, Hammad H, Waelput W, Girerd B, Raymond N, Mercier O, Mussot S, Cohen-Kaminsky S, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:311–21. [DOI] [PubMed] [Google Scholar]
- 58.Ormiston ML, Chang C, Long LL, Soon E, Jones D, Machado R, Treacy C, Toshner MR, Campbell K, Riding A, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation. 2012;126:1099–109. [DOI] [PubMed] [Google Scholar]
- 59.Ratsep MT, Moore SD, Jafri S, Mitchell M, Brady HJM, Mandelboim O, Southwood M, Morrell NW, Colucci F and Ormiston ML. Spontaneous pulmonary hypertension in genetic mouse models of natural killer cell deficiency. Am J Physiol Lung Cell Mol Physiol. 2018;315:L977–L990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poms AD, Turner M, Farber HW, Meltzer LA and McGoon MD. Comorbid conditions and outcomes in patients with pulmonary arterial hypertension: a REVEAL registry analysis. Chest. 2013;144:169–176. [DOI] [PubMed] [Google Scholar]
- 61.Frank RC, Min J, Abdelghany M, Paniagua S, Bhattacharya R, Bhambhani V, Pomerantsev E and Ho JE. Obesity Is Associated With Pulmonary Hypertension and Modifies Outcomes. J Am Heart Assoc. 2020;9:e014195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hemnes AR, Luther JM, Rhodes CJ, Burgess JP, Carlson J, Fan R, Fessel JP, Fortune N, Gerszten RE, Halliday SJ, et al. Human PAH is characterized by a pattern of lipid-related insulin resistance. JCI Insight. 2019;4:e123611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mair KM, Gaw R and MacLean MR. Obesity, estrogens and adipose tissue dysfunction - implications for pulmonary arterial hypertension. Pulm Circ. 2020;10:2045894020952019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosenkranz S, Howard LS, Gomberg-Maitland M and Hoeper MM. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation. 2020;141:678–693. [DOI] [PubMed] [Google Scholar]
- 65.Sadeh R, Sharkia I, Fialkoff G, Rahat A, Gutin J, Chappleboim A, Nitzan M, Fox-Fisher I, Neiman D, Meler G, et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotechnol. 2021;39:586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anunobi R, Boone BA, Cheh N, Tang D, Kang R, Loux T, Lotze MT and Zeh HJ. Extracellular DNA promotes colorectal tumor cell survival after cytotoxic chemotherapy. J Surg Res. 2018;226:181–191. [DOI] [PubMed] [Google Scholar]
- 67.Tumburu L, Ghosh-Choudhary S, Barbu EA, Yang S, Harrison Ware LD, Tunc I, Seifuddin FT, Pirooznia M, Zhu J and Thein SL. Cell-Free Mitochondrial DNA Is Elevated in Sickle Cell Disease Patients, and Serve As a Potential Proinflammatory DAMP. Blood. 2018;132:1068–1068. [Google Scholar]
- 68.Aucamp J, Bronkhorst AJ, Badenhorst CPS and Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc. 2018;93:1649–1683. [DOI] [PubMed] [Google Scholar]
- 69.Fernando MR, Jiang C, Krzyzanowski GD and Ryan WL. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS One. 2017;12:e0183915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data for the analyses that support the findings of this study are held at the National Institutes of Health (NIH) intramural program. The corresponding author may be contacted for requests with regard to the sharing of data, methods, and materials specific to these analyses.