

Abstract
The human brain and central nervous system (CNS) present unique challenges to drug development for neurological diseases. One major obstacle is the blood-brain barrier (BBB), which hampers the effective delivery of therapeutic molecules into the brain while protecting it from blood-born neurotoxic substances and maintaining CNS homeostasis. For BBB research, traditional in vitro models rely upon Petri dishes or Transwell systems. However, these static models lack essential microenvironmental factors such as shear stress and proper cell-cell interactions. To this end, organ-on-a-chip (OoC) technology has emerged as a new in vitro modeling approach to better recapitulate the highly dynamic in vivo human brain microenvironment so-called the neural vascular unit (NVU). Such BBB-on-a-chip models have made substantial progress over the last decade, and concurrently there has been increasing interest in modeling various neurological diseases such as Alzheimer’s disease and Parkinson’s disease using OoC technology. In addition, with recent advances in other scientific technologies, we have seen several new opportunities to improve the BBB-on-a-chip platform via multidisciplinary approaches. In this review, we provide an overview of the NVU and OoC technology, discuss recent progress and applications of BBB-on-a-chip for personalized medicine and drug discovery, and delineate current challenges and future directions.
Keywords: organ-on-a-chip, blood-brain barrier, disease modeling, drug discovery, personalized medicine
Graphical Abstract
Organ-on-a-chip technology has emerged as a new in vitro modeling approach to recapitulate the human blood-brain barrier (BBB). BBB-on-a-chip has made substantial progress over the last decade and been used to model various neurological diseases. In this review, we provide an overview of the BBB-on-a-chip models, discuss recent progress and key applications, present current challenges, and propose future directions.
1. Introduction
The human brain is a highly vascularized organ containing vessels of approximately 664-km long with capillaries making up as much as 85% of the vasculature.[1] The neural vascular unit (NVU) refers to a multicellular unit in the brain including cells of the cerebral vasculature and brain parenchyma.[2] As part of the NVU, the blood-brain barrier (BBB) acts as a physiological barrier at the interface between peripheral blood circulation and the central nervous system (CNS). The BBB is indispensable for the proper CNS function and regulation of CNS homeostasis; it controls the transport of substances between the CNS and the blood via various transport mechanisms.[3] The BBB also protects the brain from neurotoxic plasma components, certain chemicals, and pathogens. Functional and structural changes of the BBB are implicated in several neurological diseases and disorders, which have severely impacted individuals worldwide. For example, there are currently estimated to be about 5.7 million people living with Alzheimer’s disease (AD), and the number is projected to reach 13.8 million by 2050.[4] Stroke is the second leading cause of death and the third most common cause of disability.[5] Glioblastoma multiforme (GBM), which accounts for almost 50% of all brain tumors (i.e., glioma), is relatively rare with a global incidence rate of 3.19 per 100,000 people; however, it has a devastatingly poor prognosis with a typical survival rate of 14–15 months rendering it a critical public health problem.[6]
Drug discovery and clinical translation of therapeutics targeting the CNS have been exceedingly difficult for two primary reasons: (1) existing preclinical models’ lack of ability to predict human drug response; and (2) incomplete understanding of the CNS under healthy and pathological conditions.[7–9] For CNS-targeting drugs, it is critically important to account for the highly selective nature of the BBB as the ability to penetrate the barrier effectively is one of the critical determinants of the therapeutic efficacy of such drugs. For this reason, years of research have been devoted to understanding the functional and structural properties of the BBB in healthy or pathological states to develop effective strategies for the delivery of therapeutic molecules into the brain with high efficacy. For BBB research, traditional in vitro BBB models have substantially contributed to the understanding of drug permeability across the BBB. However, it has become apparent that critical factors (i.e., shear stress and proper intercellular interactions) to precisely mimic the highly dynamic three-dimensional (3D) microenvironment of the human BBB are absent from current models. In this regard, in vivo studies using animal models may be preferred; however, they also suffer from interspecies differences in brain physiology leading to the lack of ability to predict human response and poor clinical translation.[10] Additionally, there are long-standing ethical concerns surrounding using animals for research[11] necessitating continuing efforts to minimize the number of animal testing. The aforementioned concerns call for another in vitro platform that allows more accurate recapitulation of the in vivo microenvironment and better predicts drug efficacy and safety in humans.
To that end, microphysiological system (MPS) or organ-on-a-chip (OoC) technology has gained momentum as an alternative in vitro modeling approach to better recapitulate the microenvironment of the human BBB.[12] Since the introduction in 2010 of one of the first microfluidic OoC models, a lung-on-a-chip,[13] OoCs have progressively demonstrated their capability to mimic the in vivo counterparts both functionally and physiologically and potentially serve as new preclinical models to address the high attrition rates in clinical studies by closing the gap between existing preclinical models and humans.[14–17] Termed “BBB-on-a-chip”, the OoC systems can effectively mimic the functional unit of the human brain. Owing to the integration of advanced microfluidic technology, it is now possible to design perfusable OoC devices with multi-compartmentalized chambers and co-culture multiple types of cells while incorporating a flow system that imitates blood circulation. As such, compared to conventional 2D in vitro models, BBB-on-a-chip can better emulate the highly dynamic microenvironment of the brain with physiomechanical cues that are vital for the formation and maintenance of the NVU.[18] Moreover, in contrast to animal models, BBB-on-a-chip can readily be made human-based with well-defined and highly controllable microenvironments, making it possible to isolate and ascertain the roles of specific factors.[12, 19–21]
With recent advances in relevant technologies such as induced pluripotent stem cell (iPSC) technology, artificial intelligence (AI), and biosensors in addition to OoC technology itself, new opportunities have emerged to further improve the functionalities of current BBB-on-a-chip models. These include: (1) the use of iPSCs or iPSC-derived cells to build personalized models;[22] (2) the development of multiorgan-on-a-chip (MOOC) by linking a BBB-on-a-chip unit with other organ models toward the realization of human-body-on-a-chips;[23] (3) integration of in-line sensors for real-time, non-invasive monitoring of functional status;[24] and (4) utilization of AI and robotics/advanced electronics for automation of device operation, data collection, and subsequent data analysis.[25] Therefore, although certain limitations remain to be addressed in the coming years, BBB-on-a-chip has tremendous potential with many untapped opportunities. They may shed further light on the drug transport mechanisms for efficient drug delivery or unveil new pathological alterations associated with different CNS diseases, which is impossible with conventional static models. Moreover, these MPS models can potentially serve the pharmaceutical and healthcare industries as new preclinical models for drug development or as patient-specific clinical models to guide personalized therapy, respectively.
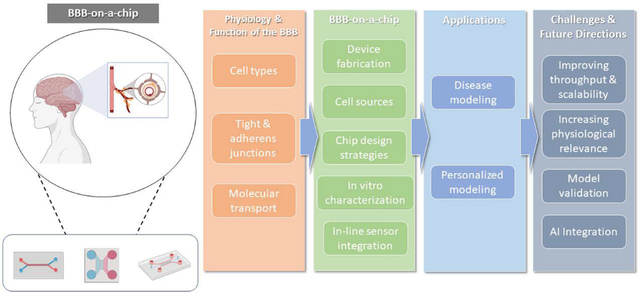
In this review, we provide an overview of the BBB and discuss BBB-on-a-chip technology including recent progress in the field and their use in disease modeling for potential applications in personalized medicine and drug development. Finally, we highlight current challenges and potential future directions.
2. Physiology and Function of the NVU
2.1. The Microenvironment of NVU
2.1.1. Cell Types
In the healthy brain microenvironment, the NVU comprises endothelial cells (ECs), pericytes, and parenchymal cells, which include glial cells (microglia and astrocytes) and adjacent neurons (Figure 1).[26] While sometimes used interchangeably, the BBB is defined as the NVU without the microglial and neuronal components.[27] While ECs are the primary cell type that forms the barrier, the supporting cells such as pericytes and astrocytes are indispensable to achieving a functional BBB as described in the sections that follow. In support of this, co-cultures of human brain microvascular endothelial cells (hBMECs) with pericytes and astrocytes consistently showed improved barrier properties in comparison with monocultures of hBMECs in BBB-on-a-chip models.[28–30]
Figure 1.

Schematic representation of the brain microenvironment. ECs form the luminal side of the BBB by creating a monolayer with TJ proteins to segregate blood from the CNS. The abluminal layer is formed by pericytes and astrocytes. Astrocytes form linkages between ECs and neurons at their end feet. Neurons and microglial cells constitute the rest of the parenchymal cell population. The BBB includes brain ECs, astrocytes, and pericytes whereas the NVU refers to the BBB with neurons and microglia.[27] The inset shows a cross-sectional image of a brain microvessel.
Endothelial cells
BMECs are a major component of the BBB and characterized by high volumes of mitochondria,[31] presence of particular transporter proteins,[32] and low rates of transcytosis.[33] BMECs form capillaries in the CNS with clefts in which adjacent BMECs are held tightly together via the tight junctions (TJs) and adherens junctions (AJs).[34] By forming a cohesive layer at the blood-brain interface, BMECs protect the brain from direct exposure to harmful substances in the blood and regulate the flow of nutrients and metabolites through the BBB to maintain CNS homeostasis. On the other hand, BMECs only allow highly selective uptake of small therapeutic molecules by the brain. This creates a significant challenge for neuropharmacological development aiming to deliver therapeutic substances to the brain to treat CNS diseases.[35]
Pericytes
Pericytes are another critical component of the BBB embedded in the basement membrane (BM).[36, 37] In the brain, there is a higher density of pericytes than in other tissues. They play a pivotal role in BBB formation and stabilization, regulation of TJ protein expression, angiogenesis, regulation of cerebral blood flow, and transcytosis of fluid-filled vesicles across the BBB. Previously, co-cultures of pericytes and BMECs were shown to induce capillary formation and expression of transforming growth factor-β whereas BMEC-astrocyte co-cultures failed to form the capillaries without pericytes and exhibited apoptotic phenotype.[38] It is believed that depending on the differentiation state of pericytes, they have different effects on the BBB.[39] In the resting state, pericytes express a low level of α-smooth muscle actin and stabilize BBB integrity. However, once in the contractile state, they undergo morphological changes and secrete higher levels of permeability factors such as vascular endothelial growth factor (VEGF), matrix metalloproteinase (MMP)-2, and MMP-9 that impair BBB properties.
Astrocytes
Astrocytes are one of the glial cells and regulate BBB formation and maintenance.[40, 41] Astrocytes are a principal source of the extracellular matrix (ECM) and have end-feet serving as the linkage between neurons and blood vessels. They express a water channel, aquaporin 4 and a potassium channel, Kir4 with which they maintain water and ionic homeostasis at the BBB, respectively. Astrocyte-secreted factors such as sonic hedgehog, angiotensin I&II, and apolipoprotein E modulate the expression of TJ proteins like occludin and claudin-5 and polarized localization of transporters such as glucose transporter 1 (GLUT-1).[42, 43] Moreover, astrocytes produce antioxidant molecules (e.g., glutathione peroxidase and superoxide dismutase) that protect the brain from oxidative stress-induced damages by clearing out free radicals produced by neurons.[44, 45]
Neurons
An accumulating body of evidence has suggested the importance of neurons in regulating the BBB properties in vivo.[2] High neural activity characterized by increased levels of glutamate has been implicated in increased BBB permeability.[46] Glutamate can directly modulate BBB permeability by binding glutamate receptors on BMECs[47] although gene expression analysis did not confirm the expression of these receptors in ECs making it an open research question.[2] Also, it was recently discovered that glutamatergic neurons regulate gene expression of a BBB efflux transporter (i.e., p-gp) and EC circadian genes.[48] Neurons are not directly in contact with BMECs; however, astrocytes have receptors for neurotransmitters through which the barrier properties can potentially be modulated.[49] Moreover, in the early developmental stage of the brain, neurons are present and participate in BBB formation.[2]
Microglia
Microglial cells are the only macrophage population in the CNS parenchyma representing 10 to 15% of the total brain cells.[50] Recently, microglia were found to have a dual role in maintaining BBB integrity; under normal conditions, they promote the expression of claudin-5 and are in physical contact with BMECs, but during inflammation, microglia phagocytose astrocytic end-feet and BMECs thereby compromising BBB function.[51] In addition, they are capable of sensing their microenvironments and responsible for the immune response of the CNS and maintenance of CNS homeostasis. Microglia express C1q and complement receptor 3 (CR3) and CR5, all of which are essential components of the classical complement system, an immune mechanism that helps to protect the brain from pathogens and infections and clear cellular debris.[52] Microglial activation occurs in certain diseases through interactions with molecules secreted by other cells (e.g., astrocytes and neurons) via membrane-bound pattern recognition receptors.[53] In pathological settings, circulating inflammatory monocytes in the blood can infiltrate into the brain upon BBB disruption and come in contact with the resident microglia leading to neuroinflammation.[54, 55]
2.1.2. Extracellular matrix
In the brain, the ECM constitutes about 10 – 20 % of the brain volume. There are three distinct types of ECM: the interstitial ECM, perineuronal nets (PNN), and BM.[56] The BM plays a pivotal role in the formation and maintenance of the BBB by providing structural and functional support for pericytes and ECs.[57] The BM consists of the endothelial BM and parenchymal BM, which are separated by pericytes. Major constituents of the BM include collagen IV, laminin, nidogen, and heparin sulfate proteoglycans deposited by BMECs, pericytes, and astrocytes. These ECM proteins contribute significantly to the maintenance of BBB integrity; several studies showed dysfunction of any of the proteins causes BBB disruption to different degrees. The interstitial ECM is primarily synthesized and deposited by glial cells and neurons and reciprocally supports the growth of these cells in the brain parenchyma.[58] It has a highly unique composition compared to non-CNS ECMs, with the major components being proteoglycans, hyaluronan (i.e., hyaluronic acid (HA)), laminin, and tenascins. Another notable characteristic of the brain ECM is its low stiffness, which is estimated to be <1kPa.[59] The soft ECM environment fosters glial migration and neuronal projections essential for neuronal development and maintenance. Neurological diseases are known to be associated with altered ECM compositions and mechanical characteristics leading to a dysregulation of activities and functions of the parenchymal cells.[60] The ECM network surrounding neurons is called a PNN.[61] It consists of hyaluronan and proteoglycans such as aggrecan deposited by neurons and astrocytes. Studies have demonstrated their critical roles in modulating neuronal physiology, in which loss of aggrecan led to structural changes of PNN and promotion of neuronal plasticity.[62]
2.1.3. Shear stress
In the brain microvessels, the shear stress ranges from 4 to 30 dyne/cm2.[63] Shear stress is particularly important for the formation of a functional BBB in vitro as it has considerable effects on BBB properties. Particularly, shear stress upregulates the expression of TJ proteins such as occludin and ZO-1, resulting in significantly reduced permeability and higher trans-endothelial/epithelial electrical resistance (TEER).[28, 64, 65] Unlike ECs from larger vessels, hBMECs, in general, do not change their morphology with no observable cytoskeletal remodeling in response to shear stress.[66] Interestingly, it was recently reported that iPSC-derived BMECs may respond less drastically to shear stress in terms of cell morphology and TJ expression presumably due to the superior ability of iPSC-derived cells to form TJs under static conditions.[67]
2.2. Barrier function and transport pathways
2.2.1. Tight junction and adherens junction proteins
TJs formed between adjacent ECs at the BBB regulate the paracellular transport of molecules contributing to the selective nature and ensuring the proper function of the BBB (Figure 2).[34, 64] Adherens junctions (AJs), on the other hand, hold ECs together and support and maintain BBB integrity.[64] The TJs consist of four major categories of proteins with three transmembrane proteins including claudins, tight junction-associated marvel proteins (TAMPs; e.g., occludin), and junction adhesion molecules (JAMs), and one intracellular protein, zona occludens (ZOs). Vascular endothelial cadherin (VE-cadherin) is a major AJ protein at the BBB.
Figure 2.

TJs and AJs at the BBB. TJs and AJs are formed in between adjacent BMECs. [73, 74] The four major TJ proteins include JAMs, occludin, claudin, and ZO whereas VE-cadherin is a major AJ protein and binds catenins in the cytoplasm. Both TJ and AJ proteins contribute to the structural integrity of ECs through their interactions with actin filaments.
The claudin proteins have extracellular loops (ECLs), which undergo dimerization with those on adjacent cells.[68] The charge and size selectivity of claudins are attributed to their extracellular domains. Most claudins have a PDZ binding motif whose interactions with the other TJ proteins and actin cytoskeleton are instrumental for TJ formation.[34] Claudins are expressed in a tissue type-specific manner with claudin-5 being the primary claudin expressed by BMECs followed by claudin-3. Occludin is one of the first TAMPs discovered. It has two ECLs, which contribute to the selective permeability of the BBB via dimerization.[69] The cytoplasmic domains of occludin play a crucial role in the maintenance of TJ and paracellular permeability. They interact with the ZO proteins, which facilitate the trafficking of occludins to the site of TJs. Both claudins and occludins are rich in phosphorylation sites for various kinases and phosphatases and phosphorylation status affects the permeability and TJ integrity. JAMs are transmembrane proteins with an extracellular domain resembling immunoglobulin G’s structure.[70] The pairing of JAMs bridges between cells at the TJs via homo- or hetero-dimerization. While their expression is not required for TJ formation, they are involved in the trafficking of occludin to the membrane to maintain TJ integrity. As with occludins and claudins, there are multiple phosphorylation sites and a PDZ binding motif in the cytoplasmic domain with which ZO-1 and other PDZ-containing proteins interact. The ZO proteins are intracellular proteins at the TJs and are part of the membrane-associated granulated kinase family.[71] The ZO family includes ZO-1, ZO-2, and ZO-3, and shares similar structures among the family members but have distinct C-terminal sequences, which is thought to be responsible for their functional differences. The central function of ZO proteins is to bind to transmembrane TJ proteins and tether them to actin cytoskeletons. While ZO-1 and ZO-2 are needed for TJ formation, the specific role of ZO-3 remains to be investigated. The ZO proteins interact with claudins via PDZ1 while interactions between the ZO proteins are mediated by PDZ2. The Src homology 3 domain of the ZO proteins is indispensable for the binding of actin cytoskeletons to signaling molecules. The interactions between ZO-1 and actin cytoskeleton filaments occur at an actin-binding region at the C-terminus of the ZO-1 protein. These cytoplasmic interactions are vital for the maintenance of TJ integrity.
VE-cadherin is a major constituent of the AJs serving a critical role in the adhesion of ECs at the BBB.[64] In the cytoplasm, VE-cadherin binds catenins (i.e., p120 and α-, β-, and γ-catenins) through which the actin cytoskeletons interact with the AJs for the maintenance of overall structural integrity.[72] VE-cadherin induces expression of claudin-5 by downregulating FoxO1, which is an inhibitory molecule of the claudin-5 gene and suppressing β-catenin activity, which is essential for FoxO1 function.[64] Therefore, while the AJs do not directly contribute to the paracellular permeability of the BBB, VE-cadherin participates in the formation and maintenance of the TJs.
2.2.2. Transport pathways
Three primary transport systems that control the influx and efflux of molecules exist at the BBB interface.[32, 75] The first is the selective transport of nutrients and other metabolites from the blood to the brain through the BBB. The second is active efflux transporters, which prevent xenobiotics and drugs at the interface from entering the brain by pumping them back into the blood. The third mode of transport is the brain-to-blood efflux transport which eliminates metabolites and neurotoxic compounds from the brain interstitial fluid. Six major pathways associated with the three transport systems are described in Figure 3.[76] While the highly regulated nature of the transport processes is critically important for the proper function and maintenance of the CNS, it also constitutes one of the major obstacles to the effective delivery of therapeutics to the brain. Hence, recapitulation of the transport pathways represents an important aspect of modeling a functional BBB in an OoC system. As discussed later in this review, BBB-on-a-chip has enabled the assessment of specific transport mechanisms especially efflux transporters, transcytosis, and paracellular transport.[28, 65, 77]
Figure 3.

Six major pathways that regulate the transport of molecules across the BBB.[73, 76] (a) RMT is mediated by receptors on the cell surface to transport target molecules. (b) Adsorptive transcytosis occurs when positively charged molecules come in close proximity to the negatively charged membrane. (c) Efflux transporters such as the ABC function through ATP hydrolysis or ATP binding (e.g. P-gp). (d) In carrier-mediated transport, molecules are loaded onto carriers with high specificity at the cell membrane and transported across the EC layer. (e) Passive diffusion is defined as the non-specific transport of small molecules. (f) Unlike the transcellular pathways, paracellular transport involves hydrophilic molecules passing through the space in between adjacent ECs.
Transcytosis is a mode of molecular transport via either a receptor- or adsorption-mediated mechanism.[33] As mentioned earlier, BMECs have lower transcytotic activity compared to non-neurovascular ECs. Receptor-mediated transcytosis (RMT) is the primary pathway to transport hormones, growth factors, and large molecules such as clathrin-coated pits. The exploitation of the RMT mechanism by linking therapeutic molecules with transport vectors presents a potential opportunity to overcome the BBB for effective drug delivery.[78] Adsorptive transcytosis is a charge-dependent phenomenon in which endocytosis occurs when positively charged molecules such as cationic polymer or lipids come in close proximity to the negatively charged membrane.[79] Plasma-membrane vesicles such as caveolae, loaded with their cargo components, are then transferred from the apical to the basal side of the endothelium.[80] The affinity of adsorption-mediated transcytosis is very low but allows the transport of a large number of molecules compared to RMT.[33] Previously, adsorption-mediated transcytosis was assessed in BBB-on-a-chip by measuring the permeability to albumin.[65, 81] In BBB, this mode of transport is highly inactive due to the expression of the major facilitator superfamily domain containing 2a (Mfsd2a) that establishes a unique lipid environment inhibiting caveolae formation. [82] Efflux transporters serve a critical role in CNS homeostasis and are responsible for the clearance or transport of unwanted substances generated by the brain into the blood circulation.[83] Adenosine triphosphate (ATP) binding cassette (ABC) is one of the major efflux transporters comprised of various efflux pumps that are involved in the excretion of toxic molecules from the brain. These efflux transporters function through ATP hydrolysis or ATP binding. P-glycoprotein (P-gp) is a key ABC efflux transporter of the BBB and is responsible for the removal of hydrophobic drugs. Several neurodegenerative diseases characterized by the accumulation of aggregated peptides are associated with impaired P-gp.[84] Carrier-mediated transport is a special form of transcytosis, in which molecules are loaded onto carriers at the cell membrane and transported across the EC layer.[63] These carriers that mediate the entry have high specificity towards their substrates such as insulin, glucose, amino acids, organic anions, and cations. Specific examples of the carriers include but are not limited to GLUT1 for glucose, MCT1 for lactate and pyruvate, and CrT for creatine[63] with GLUT1 being one of the most common BMEC markers used in studies with BBB-on-a-chip.[22, 28, 65, 77] Passive diffusion is defined as the non-specific transport of small molecules such as ethanol and caffeine.[85] The molecules need to meet certain criteria to passively diffuse through the BBB including sufficient hydrophilicity and lipophilicity.
Paracellular transport is a passive transport pathway in which hydrophilic molecular complexes pass through the space in between adjacent ECs across the BBB.[86] This paracellular transport pathway depends upon the local concentration or gradient of the substance and the molecular size. Temporary disruption of the TJs is one of the techniques widely employed in medicine to facilitate the diffusion of small molecular weight drugs through the paracellular transport pathway. The rest of the five pathways are categorized as transcellular transport.
3. BBB-on-a-chip
BBB-on-a-chip aims to recreate a functional human BBB by having proper 3D spatial arrangements of cells, intercellular communications, and organ-specific mechanical and biochemical gradients.[87] The present section discusses critical aspects of BBB-on-a-chip models including the fabrication methods, cell sources, chip design strategies, and in vitro characterization techniques to study some of the key transport mechanisms described in the previous section.
3.1. Device fabrication
There is a wide range of materials that can be used for OoC development. Synthetic polymers like PDMS, PMMA, and PCL are commonly used to fabricate microfluidic chambers whereas natural materials such as hyaluronic acid (HA), gelatin, chitosan, collagen, and alginate are often employed to reconstitute the ECM.[88] The selection of material needs to be done with careful consideration to meet pre-defined system requirements based upon a multifaceted approach considering: physiological relevance, biocompatibility, physical and mechanical properties, design complexity, cost, scalability, and compatibility with the available chip fabrication method(s). Likewise, chip fabrication techniques should be selected based on a set of selection criteria such as chip material, cost, scalability, and availability of equipment/facility and expertise.
3.1.1. Fabrication of microfluidic chips
PDMS is one of the most widely-used materials for microfluidic-based OoC devices for its biocompatibility, optical transparency, elasticity, and permeability to gas[89] with many of the BBB-on-a-chip reported to date being PDMS-based.[19, 29, 81, 90] Moreover, PDMS has surface chemistry that is highly adjustable via small molecules, nanoparticles, or proteins with tunable stiffness to meet various specifications for specific applications.[91–93] For prototyping, OoC devices are often fabricated using soft lithography techniques, in which one creates replicated structures using a stamp master fabricated traditionally via photolithography.[94] This microfabrication methodology has a high resolution making it possible to fabricate sub-micro and nano-scale structures but suffers from relatively low throughput. Following replica molding, microchannel formation is generally achieved by PDMS-PDMS and/or PDMS-glass bonding using plasma treatment enabling the fabrication of complex 3D geometries and multiple molded layers often required for the construction of multi-compartmental BBB-on-a-chip. However, the use of PDMS comes with a drawback – its tendency to adsorb certain proteins and small hydrophobic molecules and hence potentially confound results by inadvertently reducing the availability of molecules.[95] This could be particularly concerning for preclinical pharmacological and toxicological studies where the accurate assessment of drug effects is of utmost importance. Also, PDMS-based platforms are not apt for large-scale production for a couple of reasons. One is due to the low-throughput nature of the photolithographic process; therefore, other techniques such as laser cutting,[96, 97] injection molding,[98] hot embossing,[99, 100] and 3D printing[101, 102] may be more preferred for large-scale manufacturing of master molds. However, they too come with their own caveats such as high technical demands and complex operational procedures.[103] Secondly, the cost of PDMS is generally higher than that of most thermoplastic polymer materials.[104] Among several options available, PMMA is one of the thermoplastic polymers previously used to construct BBB-on-a-chip models.[105, 106] Similar to PDMS, PMMA is biocompatible and optically transparent with a modifiable surface but has less small-molecule adsorption. However, it is more challenging to fabricate complex microstructures as thermoplastics are rigid materials.[107] Several fabrication methods such as laser cutting, injection molding, and hot embossing can be used to create microchannels with thermoplastics. Particularly, injection molding and hot embossing are two fabrication techniques that can be implemented to facilitate the mass production of thermoplastic-based chips. Also, there are a number of bonding methods available for PMMA to achieve robust bonding between multiple layers such as thermal bonding, solvent bonding, and adhesive bonding. Additional surface treatment is recommended for engraved channels on PMMA to smooth out the surface and avoid unfavorable consequences such as inadvertent disturbance of laminar flow.[108]
3.1.2. Use of bioprinting in creating BBB-on-a-chip
Over the last few years, 3D bioprinting has been increasingly used to construct 3D BBB-on-a-chip models.[109–112] Bioprinters can employ natural or synthetic biomaterials or both to form scaffolds that are biocompatible with organ-specific properties. Natural biomaterials such as HA and collagen IV are naturally found in the brain parenchyma and hence have high physiological relevance and excellent biocompatibility. These natural biomaterials and other polymers such as gelatin and its derivatives (e.g., gelatin methacrylated gel) are among the most popularly-used hydrogels for encapsulation of brain cells to create cell-laden gels. Similarly, synthetic materials such as thiol–ene–epoxy and silicone have also been used to 3D print a BBB-on-a-chip model.[113] Though being artificial, the biochemical and mechanical properties of synthetic materials can be readily modified by mixing with other biomaterials and/or functionalizing with an ECM protein.[110] This allows cell adhesion and ultimately increases their biomimicking ability. Regardless of the biomaterial type, it is essential to ensure good printability of the bioinks, which depends upon several factors such as material viscosity, thermosensitivity, cross-linking capability, and bioprinting modality. A wide variety of printing modalities are available today such as inkjet-based, extrusion-based, and light-assisted printers.[114] For further discussions on applications of bioprinting technology for OoCs, excellent reviews are available elsewhere.[110, 114–116]
3.2. In vitro barrier characterization techniques
BBB characterization is a critical part of developing functional BBB-on-a-chip as barrier properties have long been used as measures to gauge the physiological relevance of constructed BBB-on-a-chip.[117, 118] Traditionally, several techniques such as TEER and permeability assays, and immunostaining methods have been employed, and are commonly used in conjunction with each other.[29]
3.2.1. Trans-endothelial electrical resistance
TEER stands for trans-epithelial/endothelial electrical resistance and is a commonly-used electrophysiological characterization method to measure the integrity of the epithelial and endothelial monolayers that are often used in OoC systems.[117] TEER values quantify the paracellular permeability of a cellular monolayer before the modeled BBB can be used to study the delivery and transport of drugs and chemicals. TEER is calculated as:
where R is the resistance due to the endothelial barrier and A is the area shared by the top and bottom channels within the region between the working and reference TEER electrodes.[119] One advantage of TEER is that it can take real-time measurements by measuring either the ohmic resistance or impedance of the cellular monolayer at different frequencies without damaging cells. Srinivasan et al. extensively review TEER measurement methods and values for several in vitro BBB-on-a-chip models.[120] TEER measurements for rat BBB in vivo can be as high as 5,900 Ω.cm2,[121] which is significantly higher than TEER values reported in the literature for animal and human in vitro BBB models. However, recently iPSCs-derived BMECS were reported to yield a TEER value of higher than 1,000 Ω.cm2, substantially higher than primary or immortalized BMECs.[65] Several factors such as chip design, co-culture versus monoculture, shear stress, as well as ECM coating can play a significant role in observed TEER values. For example, Wang et al. reported that TEER for BMEC monocultures and astrocyte monocultures in their BBB-on-a-chip design peaked at 370 Ω.cm2 and 15 Ω.cm2 respectively, whereas the co-culture reached TEER values as high as 4,400 Ω.cm2 at day 3 and was able to maintain TEER values above 2,000 Ω.cm2 up until day 10.[122] On the other hand, using fibronectin-coated and Matrigel-coated devices, Jeong et al. were able to increase their TEER value 2.5 and 5.5 times, respectively compared to uncoated devices.[123]
3.2.2. Permeability assay
While TEER measures the resistance or impedance of the cellular monolayer, permeability assays provide a different method to assess barrier integrity by measuring the ease with which tracer molecules can cross the BBB cellular monolayer. These experiments are usually performed using molecules of different types, charges, and sizes, as these are key factors that determine barrier permeability.[124] Apparent permeability (i.e., Papp) can be computed as follows:
where Cal and Cl are the concentration of tracer molecules in the brain and vascular channels respectively, Val is the volume in the brain channel, A is the contact area between two compartments, and t is the duration of perfusion.[19] As would be expected, a lower permeability coefficient equates to superior barrier properties. In addition to charge and molecular weight, care should be taken to pick tracer molecules that do not act as ligands for receptors on the cellular monolayer nor as substrates for enzymes as this would interfere with the permeability experiments[124]. FITC-labeled dextran with different molecular weights is one of the most used tracer molecules for permeability assays[30, 65, 125, 126]. As dextrans are transported primarily through the paracellular pathway, their molecular weight has a large effect on permeability assessment.[127] Other tracer molecules used in permeability studies include FITC-labeled albumin[128] and 13C-labeled sucrose and mannitol[129] primarily for RMT and paracellular transport, respectively. Additionally, researchers also use permeability experiments to determine the paracellular transport of antibodies[30, 77] as well as drugs[77, 122] across their modeled BBBs. Nozohouri et al. summarize several iPSC-derived BBB-on-a-chip models along with their TEER and permeability measurement values in their recent review.[130]
3.2.3. BMEC marker analysis
Expression analysis of TJ/AJ proteins such as occludin, claudins, cadherins, and the ZO proteins via immunostaining is another common method for visually and quantitatively characterizing the barrier properties in BBB-on-a-chip models. Specifically, researchers commonly stain for ZO-1 and transmembrane proteins such as claudin-5, and VE-cadherin together with cell markers including but not limited to actin and CD31.[28, 65, 77] In addition, expression levels of P-gp and GLUT-1 have been examined in several studies to investigate the functional phenotype of modeled BBB.[77, 131] The assessment of BMEC markers has been critical in studying the BBB using BBB-on-a-chip to reveal the major advantages of the microfluidic system such as the inclusion of shear stress and proper cell-cell interactions.[65, 81]
3.3. Cell sources
For BBB-on-a-chip models, the source of cells can have a profound impact on the functional properties of modeled BBBs;[132–134] therefore, careful considerations must be given during the selection of cell sources. Due to distinct phenotypes observed in animal BBBs compared to the human BBB,[135, 136] human cells are generally favored for the accurate representation of human physiology and enhanced clinical relevance. In general, the cell sources for in vitro modeling are available in the form of (1) immortalized cell lines, (2) primary cells, or (3) iPSCs or iPSC-derived cells.
3.3.1. Cell lines
Most immortalized cell lines are genetically modified via simian virus 40, which gives them the ability to indefinitely proliferate and hence provide a consistent research sample of interest without any ethical concerns.[137] However, due to the genetic mutations, cell lines may not display primary cell phenotypes and may therefore respond to stimuli differently.[138] As an example, functional and phenotypic differences were previously reported for astrocyte cell lines versus primary cells.[133] It is also worth noting that even among different BMEC lines, they display differential levels of TEER, permeability, and TJ protein expression.[134] Currently, several cell lines are available for use to study astrocytes,[133] neurons,[132] pericytes,[139] and BMECs.[140]
3.3.2. Primary cells
Primary cells are extracted directly from the donors and made available for downstream applications without any gene modifications.[141] Therefore, these cells retain more of the native physiological information compared to genetically-modified cell lines. However, primary cells are limited in their proliferative capacity and possess a tendency to de-differentiate in culture resulting in lower cost performance, especially during preliminary phases of development. For instance, after seven passages, primary astrocytes can undergo drastic phenotypical changes compared with passage one.[142] Likewise, primary neurons stay electrophysiologically active after isolation from the donors but do not proliferate in culture and eventually experience cell death even under proper culturing conditions.[143] Isolation of highly pure populations of primary hBMECs with sufficient yield is also technically and logistically difficult to achieve.[144] Human umbilical vascular endothelial cells (HUVECs) are a type of primary human ECs widely used for the in vitro studies of human vasculatures,[145] including the BBB.[146, 147] Despite this fact, HUVECs are known to possess distinct barrier phenotypes compared to hBMECs including the reduced expression of TJ proteins and higher permeability.[148] Therefore, the use of an appropriate type of ECs is vital for modeling the BBB in vitro.
3.3.3. iPSCs
IPSCs and iPSC-derived cells have progressively become commercially available and used in many studies.[149] These stem cells are derived from human somatic cells and have the ability to differentiate into any type of cells.[150] Their additional advantages over the other cell types include: (1) no ethical issues unlike embryonic stem cells; (2) the less invasive procedure involved in the procurement of iPSCs compared to primary cells; (3) the ability to provide an ample supply of cells from the same donor; and (4) patient-specific genetic composition that enables the personalized modeling of disease and drug response. Over the last decade, studies have proposed robust differentiation protocols to obtain iPSC-derived hBMECs that have phenotypes similar to primary hBMECs with the confirmed expression of TJ proteins and endothelial markers.[151, 152] Pioneering works from Lippmann et al. were the first successful protocols that derived BMECs from iPSC by culturing them onto an appropriate matrix and in media supplemented with retinoic acid (RA). [153, 154] A later study from Qian et al. showed that differentiation of iPSC-BMECs can be archived chemically via sequential Wnt and RA pathway activation that allows iPSCs to progress through an intermediate mesoderm phase and VEGFR2+ endothelial progenitors to CD31+ mature BMEC phenotypes.[151] Shortly afterward, fully-defined media components with specific chemical inhibitors for iPSC-BMECs differentiation were developed.[152, 155] Moreover, in these studies, iPSC-derived BMECs demonstrated more physiologically relevant barrier properties including remarkably lower permeability and higher TEER compared to previously reported values for primary or immortalized hBMECs. In addition to BMECs, the supporting cells in the NVU have been successfully derived from iPSCs including astrocytes,[156] pericytes,[157] neurons,[158, 159] and microglia.[160] To further facilitate iPSC technology for in vitro modeling, certain obstacles still need to be overcome such as the technical complexity, batch-to-batch variation, lack of maturity of differentiated cells, and time-intensive and costly processes associated with the production and differentiation of iPSCs.[161] However, studies on the in vitro modeling of the BBB using iPSC-derived cells have already started to show promising results.[149, 162] Moreover, fueled by recent advances in iPSC technology, there has been increasing interest in using patient-derived cells to improve the clinical relevance of BBB-on-a-chip models.[22, 65, 149] Essentially, patient-specific models enable the studies of patient heterogeneity due to the genetic variations and characteristics linked to the etiology and pathophysiology of various brain diseases. Ultimately, integrating iPSCs and their derivatives with BBB-on-a-chip could be an important step to realizing personalized medicine through patient-specific recapitulation of human physiology.
3.4. BBB-on-a-chip design strategies
3.4.1. Cell culture platforms
The culture platform design for BBB-on-a-chip has evolved from the simple planar petri dish and cell-free synthetic membrane models to sophisticated multi-channel microfluidic systems to recreate the human brain microphysiology more precisely (Figure 4a).[19] Earlier models heavily relied upon acellular methods[163] or planar cultures in Petri dishes[164] without essential microenvironmental factors such as proper intercellular communications and shear stress.[18, 165] On the other hand, in Transwell-based models, multiple cell types can be co-cultured fostering intercellular interactions via paracrine signaling through a porous membrane in-between an EC layer and other cells (e.g., glial cells and neurons).[166] However, as with Petri dish models, Transwell systems are static and do not permit the incorporation of a perfusion system to provide the cells with an adequate level of shear stress, which substantially affects the functional properties of BMECs and BBB formation.[18] As such, the last decade has seen increasing efforts to develop perfusable microdevices to more precisely recapitulate the dynamic BBB microenvironment (Figure 4b–d).
Figure 4.

BBB-on-a-chip designs. (a) Evolution of the in vitro culture platform from a 2D Petri dish model to microfluidic OoC. Examples of recent BBB-on-a-chip models. (b) Sandwiched-channel design (Reproduced with permission.[77] Copyright 2019, Springer Nature). (c) Parallel-channel design (Reproduced with permission.[177] Copyright 2021, Springer Nature). (d) Interlinked BBB and brain-on-a-chips with sandwiched design (Reproduced with permission.[81] Copyright 2018, Springer Nature). (e) 3D printed BBB-on-a-chip with sandwiched-channel design (Reproduced with permission.[113] Copyright 2021, Wiley Online Library). (f) BBB-on-a-chip with a bioprinted brain construct (Reproduced with permission.[178] Copyright 2021, Wiley Online Library).
Based on the designs, microfluidic BBB-on-a-chip models can be classified into four distinct types: parallel-channel, sandwiched-channel, tubular-channel, and vasculogenesis-based designs. Sandwiched-channel design is one of the most widely-used device configurations, in which two channels are stacked on top of each other with a porous membrane in-between to separate the top and bottom compartments. The porous membrane acts as the BM and is typically made of PDMS, polyester, PET, or polycarbonate. Additional ECM-coating (e.g., fibronectin, collagen IV, and/or laminin) is often needed for subsequent cell adhesion and the establishment of a functional BBB. Reconstituting the BM and interstitial ECM with similar mechanical properties and/or compositions found in vivo is essential to accurately model brain physiology as they can significantly impact BBB properties.[167] One advantage of this particular channel arrangement is that it enables relatively seamless integration of sensor electrodes for TEER measurement to characterize the barriers. In fact, most TEER measurement methods available today are only compatible with specific chip designs.[119, 120] Also, the location of the endothelium layer (i.e., vascular channel) relative to the parenchymal cells (i.e., brain channel) has appreciable effects on the modeled BBB function. For example, when hBMECs and astrocytes/pericytes were co-cultured on each side of a porous membrane in the bottom and top chambers respectively, hBMECs would receive more signaling molecules from other cells such as antioxidant molecules (e.g., ascorbate and superoxide dismutase) secreted by astrocytes[168] compared to those in a reversed chip configuration with hBMECs in the top chamber.[19]
In contrast to the sandwiched design, BBB-on-a-chip with parallel channels utilize PDMS micropost arrays in place of a porous membrane to establish the interface between the endothelium and brain compartments.[169–171] As with the sandwiched-channel design, the use of macromolecule-permeable BM-like structures enables intercellular communications via paracrine signaling and a degree of direct contact between two cultures through the openings. As a derivative of the parallel-channel design, a circular central tissue chamber (i.e., brain compartment) encircled by two independent outer vascular channels with PDMS micropost arrays has been developed.[172] One shortcoming of the sandwiched- or parallel-channel design is that the channel geometry has a rectangular shape due to the soft lithographic process. In perfusion cultures, this results in flow motions and wall shear stress patterns that deviate from what is observed in the brain capillaries. As a potential solution to that problem, the fabrication of cylindrical channels can be realized by using needles or wires as sacrificial molds, or viscous fingering that creates a hollow cylindrical hydrogel structure inside a microfluidic channel using a partial wash of a high viscosity hydrogel solution by a lower viscosity solution such as media. [131, 162, 173–175] The diameter of the channels can be tuned by adjusting the fluidic flow and/or size of the mold. Subsequent seeding of cells following the channel formation allows the establishment of an endothelial layer in the hollow structure with the outer compartment filled with a cell-laden or acellular hydrogel mimicking the brain parenchyma. While the aforementioned methods induce BBB formation in a more coordinated fashion, the vasculogenesis-based design seeks to induce de novo formation of the brain’s microvascular networks in hydrogel-filled chambers.[171, 176] This approach allows the formation of brain microvessels with more in vivo-like structures by allowing cells to self-organize into vascular networks in 3D environments in vitro. However, as the vessel formation is entirely self-driven by the cells, the stochastic nature can lead to uncontrollable inter-chip variation. To better illustrate the above-mentioned advantages and disadvantages of the platform design strategies, we have summarized the key features of each design in Table 1.
Table 1.
Chip design comparisons.
| Static model | Dynamic model | |||||
|---|---|---|---|---|---|---|
| Feature | Petri dish | Transwell | Sandwiched-channel | Parallel-channel | Tubular-channel | Vasculogenesis-based |
| Multi-compartmentalized | No | Yes | Yes | Yes | Yes | Yes |
| Basement membrane | None | Porous membrane | Porous membrane | PDMS post arrays + hydrogel | Hydrogel | Hydrogel |
| In vivo-like spatial arrangement and channel geometry | No | No | No | No | Yes | Yes |
| TEER measuring method available | No | Yes | Yes | Yes | No | No |
| Permeability measured with tracer molecules | No | Yes | Yes | Yes | Yes | Yes |
| Chip fabrication difficultya) | Low | Medium | High | High | High | High |
| Chip-to-chip variationb) | Low | Low | Medium | Medium | Medium | High |
| Ref. | [164] | [127, 130] | [28, 65, 77, 119] | [169–172] | [131, 162, 173–175] | [171, 176] |
Assuming expertise in only basic laboratory and cell culture techniques.
Contingent upon the technical complexity involved with chip fabrication and cell culture and the nature of the vessel formation process (stochastic vs. coordinated).
3.4.2. 2D versus 3D culture
Besides the culture platform, the transition from 2D to 3D culture has made a major impact on in vitro modeling. An accumulating body of evidence suggests the advantages of cell encapsulation in hydrogels or the formation of spheroids/organoids in faithfully recapitulating the in vivo physiology at the expense of increased experimental complexity.[179, 180] For example, astrocytes are found naturally in the brain embedded in the surrounding ECM and the cell-ECM interactions play an essential role in astrocyte development and function.[181] Indeed, previous studies confirmed that when cultured in 2D versus 3D, astrocytes have highly distinct morphology in which cells were flatter and larger in 2D and smaller and round in 3D.[28, 182] One way to achieve 3D culture is by encapsulating cells in hydrogels, which can be manually pipetted or bioprinted to form 3D constructs.[183] Studies have utilized bioprinting techniques to fabricate hydrogel-based OoC models with a BBB component enabling fine control over the spatial arrangements of printed layers in a relatively high-throughput and reproducible manner.[110] Apart from encapsulation in hydrogels, multicellular 3D organoid-based models of the BBB have been reported, in which cells self-assemble into an organoid with the core largely consisting of glial cells (e.g., astrocytes) and an outer layer being populated with BMECs and pericytes.[184] This approach permits multicellular 3D culture with decent assay throughput[185] although the stochastic nature of organoid formation may lead to inter-assay variations. In addition, 3D cultures of neural stem cells have been achieved through the formation of cell aggregates known as EZ-spheres derived from hiPSCs. [186] In a recent study, EZ-spheres were successfully seeded and co-cultured with iPSC-derived hBMECs within the brain channel of a BBB-on-a-chip.[65] It has increasingly become evident that 3D cultures improve the physiological relevance of in vitro models; however, functional characterization using the conventional methods, especially TEER measurements is still a challenge for both spheroid- and hydrogel-based models in traditional culture plates. As such, 3D culturing cells using Transwell systems has been suggested as a potential solution,[187] thereby enabling the assessment of barrier functions via TEER and permeability assays. As recently demonstrated by Silvani et al. (Figure 4f),[178] a more advanced approach will be to combine bioprinting techniques with microfluidic OoC platforms to permit the culturing of BMECs with shear stress and the parenchymal cells in 3D environments with reduced chip-to-chip variations.
3.5. Integration of in-line sensors
The incorporation of in-line sensors into BBB-on-a-chip devices has been an active field of research over the last decade. However, only a few types of sensors have successfully been integrated.[24] Advantages of such sensors in monitoring OoC systems over traditional methods (e.g., qPCR and immunostaining) include the ability to enable rapid and real-time measurements of biological properties in a noninvasive and continual manner.[188] A variety of sensor types such as electrochemical biosensors (e.g., antibody- or aptamer-based sensors) tagged with redox-active molecules to measure soluble biomarkers or physiological sensors measuring electrochemical impedimetric resistance have long been hypothesized and shown to synergize well with the OoC platforms.[189–191] In addition, optical sensors such as luminescence-based are well-suited for the real-time detection of oxygen, pH, carbon dioxide, glucose, and temperature within OoC systems.[188, 192]
3.5.1. TEER sensors
One example of integrated sensors for BBB-on-a-chip is TEER sensors.[193, 194] It remains challenging to measure TEER with reliable and reproducible readouts as they can readily be influenced by subtle differences in electrode location, insufficient sensitivity, and even non-uniform cell cultures.[119] One way to address the issue is to use mathematical models;[120, 195–197] however, they can only mitigate these inadvertent variations across measurements to a certain degree. Therefore, recent studies employ fully integrated electrodes to measure TEER in their OoC devices, which were demonstrated to yield more reproducible and practical TEER values (Figure 5).[193, 198–201] Particularly, Henry et al. reported a facile layer-by-layer fabrication protocol to assemble the OoCs with integrated electrodes and validated the utility for characterizing the barrier function.[119] On the other hand, Jeong et al.[123] and Maoz et al.[201] developed multi-electrode array (MEA)-based TEER sensors in two separate studies. In both cases, electrodes were deposited onto a polycarbonate layer, which was subsequently incorporated into respective microfluidic devices. In particular, the system developed by Jeong et al. has the ability to perform automated measurements of TEER in 16 individual chambers enabling the simultaneous assessment of multiple samples and thereby greatly improving experimental efficiency.[123] Platinum (Pt)-wire-based electrodes represent another form of TEER sensors.[202] In such TEER chips, Pt wires are inserted into assembled chips through dedicated electrode channels and secured in position using biocompatible glue. Although this type of TEER sensor was first introduced in 2013 by Griep et al.[202], recently a multiplexed OoC with Pt-wire TEER sensors was proposed by Bossink et al.[203] Pt wire-based sensors enable a cleanroom-free approach to TEER sensor integration into BBB-on-a-chip devices; however, microfabricated planar electrodes have superior sensor performance compared to wire-based electrodes. Furthermore, in either case, larger surface areas result in lower noise and are preferred.
Figure 5.

TEER sensor integration. (a-b) An OoC with four deposited electrodes. Reproduced with permission.[119] Copyright 2017, Royal Society of Chemistry. (c-d) TEER-MEA. Reproduced with permission.[201] Copyright 2017, Royal Society of Chemistry. (e-f) An OoC model with Pt-wire electrodes. Reproduced with permission.[203] Copyright 2021, Royal Society of Chemistry.
Several studies have already shown the utility of the above-mentioned TEER sensors, but these sensors are highly variable in their design, device architecture, and sensor performance. Hence, given the sensitive nature of the TEER sensors,[204, 205] currently it is not possible to compare reported TEER values across studies especially if different types of TEER sensors or OoC platforms are used. Therefore, standardization of the TEER measurement method remains a potential area of future BBB-on-a-chip research.
3.5.2. Other sensors
Apart from the TEER sensors, most of the sensing technologies previously incorporated into OoC models are yet to be built into the BBB-on-a-chip systems with a few exceptions. Dissolved oxygen is a microenvironmental parameter that affects BBB integrity in vivo.[206] Stricker et al. presented an oxygen scavenging material with an integrated oxygen sensor.[207] Using a palladium oxygen indicator dye, PdTPTBFP-coated microparticles were directly incorporated into the BBB-on-a-chip. The luminescence lifetime of the PdTPTBFP dye is correlated to the oxygen content within the system (Figure 6a). They observed continuous monitoring of oxygen content over eight hours. In addition, they incorporated an electrochemical oxygen sensor, where oxygen presence is oxidized at the electrode surface, and both electrochemical and optical oxygen sensors demonstrated similar results over eight hours. Although the focus of this work was on creating oxygen management materials for biochips, the incorporation of oxygen sensors provided an interesting example of an in-line oxygen sensor for the BBB-on-a-chip.
Figure 6.

(a) SEM image of the PtTPTBPF palladium base oxygen sensing dye integrated microparticles and the oxygen sensing integrated into the BBB chip. Reproduced with permission.[207] Copyright 2019, American Chemical Society. (b) Schematic representation of the BBB-on-a-chip integrated with the three-dimensional cell culturing setup, the micro solid-phase extractor, and the final electrospray ion mass spectrometry set-up. Reproduced with permission.[208] Copyright 2016, Elsevier.
One of the significant challenges of integrating sensors into BBB-on-a-chip is the space limitation. One strategy to levitate the space restriction is to attach an external sampler to the chip. An excellent example is the use of this approach demonstrated by Shao et al.[208] As shown in Figure 6b, in their approach, a BBB-on-a-chip was developed with the fluid outflow connected to a C-18 reverse phase micro solid-phase extractor (μSPE). The μSPE is connected to an electrospray ionization mass spectrometer, establishing the microfluidic-based BBB drug permeability analysis setup to quantify the amount of drug substance that has crossed the BBB. This cascade reported a 30-minute analysis time using 5 μL of sample solution, drastically improving analysis efficiency. This technique not only removes the limitation of space for in-line sensor integration into the BBB-on-a-chip but also opens the door to new potential sensors to be integrated into the downstream portion of the cascade.
Significant challenges such as space restriction, leakage, and fouling continue to present themselves as obstacles preventing the rapid development of this field.[204] However, as Liang et al. suggested, many microfabrication techniques and sensor designs have evolved and are becoming available.[24] For instance, Senel et al. recently reported an electrochemical biosensor integrated into a microfluidic device for the detection of dopamine with an excellent limit of detection (0.1 nM) and demonstrated its use to measure dopamine levels in cerebrospinal fluids of mice with PD.[209] Besides dopamine, there are interesting targets of the BBB to be detected via in-line sensors such as vascular endothelial growth factor released by astrocytes and pericytes,[210, 211] von Willebrand factor secreted by BMECs,[212] and any disease-specific markers.[213] In-line measurement of these molecules will provide an efficient means to study the real-time responses of BBBs to external stimuli such as drug candidates. Therefore, integrating sensors into BBB-on-a-chip will remain an interesting research topic with further developments warranted in the years to come.
4. BBB-on-a-chip for disease modeling and development of personalized models
4.1. Disease modeling
Disruption of the CNS and neurovascular abnormalities are implicated in several neurological diseases and disorders such as AD, GBM, PD, amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and stroke.[214, 215] BBB disruption often leads to “a leaky BBB” with endothelial and pericyte degeneration resulting in loss of TJ and AJ.[75] Eventually, the uncontrolled transport of molecules and trafficking of T cells, B cells, and macrophages into the brain ensue. While such vascular pathology is a common phenomenon seen in many neurological diseases, it is often not certain whether it is a preceding or downstream event. It is important to note that the disrupted BBB also has implications for drug delivery. Once BBB breakdown is initiated, structural changes of the BBB occur, which results in the accumulation of cellular and blood-derived debris on the abluminal side.[75] Consequently, the normal interstitial fluid flow is interrupted, preventing therapeutic molecules from successfully reaching their targets. While decades of research have been devoted to understanding neurological diseases, their detailed mechanisms largely remain unknown due to the highly intricate nature of the CNS.[216] In light of this, the in vitro modeling of neuropathology using OoC technology has been of great interest to the research communities to better apprehend disease pathogenesis and progression, identify potential therapeutic targets, or evaluate the therapeutic potential of new drug candidates (Table 2).
Table 2.
BBB-on-a-chip models to study neurological diseases.
| Device characteristics | Barrier characterization methods | Supplementary information | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Target disease | Chip type | Channel design | Culture type and ECM | Cell type(s) | Disease induction method | TEER Sensors | Permeability assessment | Immunostaining | Major on-chip disease phenotypes | Importance/Applications | Ref |
| AD | PDMS-based microfluidic chip (dynamic) | Parallel channels separated by post array | EC: 2D with Matrigel; Brain: 3D with Matrigel and collagen type I gel | EC: hCMEC/D3; Brain: ReN cells | ReN cell line with human familial AD mutations | NA | 3kDa and 40kDa dextrans | Claudin-1, Claudin-5, VE-cadherin | Increased permeability; reduced TJ expression; in vivo like Aβ deposition patterns; Neuronal damage due to neurotoxins | Modeling of AD pathologies in vitro using BBB-on-a-chip model with genetically modified ReN cells | [170] |
| ALS | PDMS-based microfluidic chip (dynamic) | Custom design | EC: 2D with collagen type I gel; Motor unit: 3D with MN spheroids and collagen type I gel/Matrigel | EC: iPSC-ECs; Motor unit: patient-derived hESC- and iPSC-MNs, iPSC-skeletal muscle cells, | Addition of excess glutamic acid; patient-derived cells | NA | 40kDa dextran | ZO-1, occludin, P-glycoprotein | Decreased muscle contraction; muscle function improved by cotreatment with rapamycin and bosutinib through BBB | ALS-on-a-chip using cells from ALS patients and demonstration of compromised muscle function in ALS vs. control model | [225] |
| GBM | PDMS-based microfluidic chip (dynamic) | Central chamber (brain) and outer channel (EC barrier) | EC & Brain: 2D with Matrigel/fibronectin | EC: HUVECs; Brain: CTX-TNA2 rat brain astrocytes, and Met-1 murine metastatic breast cancer cells | Mouse-derived primary cancer cells | NA | 3kDa and 70kDa dextrans; Rho 123; sulforhodamine 101 Acid Chloride | NA | Increased permeability | One of the first BTB-on-a-chip platforms; demonstration of leakier barrier phenotype in BTB vs BBB model | [226] |
| PDMS-based microfluidic + bioprinted chip (dynamic) | Central chamber (brain) and outer channel (EC barrier) | EC: 2D with fibronectin; Brain: 3D in GelMA-alginate and GelMA-fibrin hydrogels | EC: hCMEC/D3 or HUVECs; Brain: human GBM cells | Cancer cell line | NA | 40kDa Texas Red-dextran | ZO-1 | Decreased ZO-1 expression in hCMEC/D3; alteration of cell morphology and decreased expression of vinculin and active Yap1 in GBM cells with microgravity exposure | A dynamic, microfluidic GBM-on-a-chip with bioprinted GBM; elucidation of effects of mechanical cues on BTB and GBM cells | [178] | |
| PDMS-based microfluidic chip (dynamic) | Parallel channels separated by post array | EC & Brain: 3D with fibrinogen | EC: HUVEC; Brain: U-87, patient-derived GBM cells | Cancer cell line; patient-derived cells | NA | 70 kDa dextran | Green fluorescent protein, VE-cadherin, vWF | Colocalization of GBM stem-like cells with perivascular niche; distinct migration trajectory profiles for different GBM cell lines with accompanying changes in gene expression | Recapitulation of 3D microvasculature in GBM; a new potential functional assay to assess the invasiveness of GBM cells for personalized medicine | [227] | |
| PDMS-based microfluidic chip (dynamic) | Central chamber (brain) and outer channel (EC barrier) | EC: 2D with Poly-D-Lysine; Brain: 3D with Matrigel and HA |
EC: hBMECs; Brain: macrophages, microglia, CD8+ T-cells, and patient-derived GBM cells |
Patient-derived cells | NA | NA | NA | Distinct profiles of T cell extravasation, cytotoxic activities, PD-1 expression, and inflammatory cytokine secretion between different GBM subtypes | Development of a patient-specific GBM model that differentiates different GBM subtypes; Enables personalized molecular assessment of effects of anti-PD1 therapy | [228] | |
| HD | PDMS-based microfluidic chip (dynamic) | Sandwiched channels with top (brain) and bottom (vascular) chambers | EC: 2D with collagen IV and fibronectin; Brain: 3D with EZ-spheres and laminin | EC: iPSC-BMECs; Brain: iPSC-EZ spheres | Patient-derived cells | Integrated Au electrodes (AC resistance) | 4, 20 or 70 kDa dextrans; 3,5,3'-triiodothyronine; 2-NBDG; retigabine; levetiracetam; colchicine | GLUT-1, PECAM-1, Caudin-5, Occludin, ZO-1 | Increased permeability with inter-individual variations | Development of BBB-on-a-chip that captures in vivo like complexity of BBB and recapitulates patient heterogeneity in HD phenotype | [65] |
| PD | PDMS-based microfluidic chip (dynamic) | Sandwiched channels with top (brain) and bottom (vascular) chambers | EC & Brain: 2D with collagen type IV, fibronectin, and laminin |
EC: iPSC-BMECs; Brain: primary human brain pericytes, primary human astrocytes, primary human brain microglia, iPSC-neurons |
Treatment with αSyn fibril | NA | 3 kDa dextran, and 0.5 kDa lucifer yellow | Claudin-1, Claudin-5, Occludin, PECAM-1 | Phosphorylation of αSyn129, decreased mitochondrial activity and increase in ROS production, induction of caspase-3 activation and neuroinflammation; increased permeability | Recapitulation of PD pathologies induced by treatment with αSyn fibril in BBB-on-a-chip system | [229] |
| Stroke | PDMS-based microfluidic chip (dynamic) | Parallel channels separated by a smaller middle channel | EC:2D; Brain: 3D with Engelbreth–Holm–Swarm tumor (Cultrex) |
EC: human primary BMECs; Brain: human primary astrocytes, human microglial cell line, human primary vascular pericytes, iPSC-NPCs |
Treatment with low oxygen and deprivation of serum and glucose content | Chopstick silver chloride electrodes (DC resistance) | 4kDa and 70kDa dextrans | PECAM-1, vWF, ZO-1, Claudin-5 | Upregulation of inflammatory genes; increased activity of glutamate and decreased activity of GABA; irregular Ca2+ signaling patterns; increased VEGF; decreased TJ protein expression; pericyte and microglia activation; disruption of AQP4 | Development of stroke-on-a-chip; assessed the neurorestorative effects of several stem cell-based therapies | [177] |
4.1.1. Alzheimer’s disease
AD is one of the most common neurodegenerative diseases among elderly populations.[217, 218] It is the leading cause of dementia, in which one experiences a severe decline in cognitive function. AD pathology is characterized by the deposition of Aβ peptides onto the BBB and the formation of neurofibrillary tangles consisting of clusters of phosphorylated tau proteins. Additionally, AD pathogenesis is often accompanied by thickening and changes in the composition of the BM.[219] Previously, Transwell-based 3D models have been proposed to study the neuropathology.[220, 221] Blanchard et al., for example, co-cultured iPSC-derived BMECs, astrocytes, and mural cells in a Transwell-based system and treated them with a culture medium conditioned with familial AD neurons to model cerebral amyloid angiopathy (CAA), a condition caused by amyloid deposition along the vasculature.[183] Using the AD model, it was revealed that calcineurin/nuclear factor of an activated T cell (NFAT)-signaling and pericytes play pivotal roles in determining the genetic susceptibility of Apolipoprotein E4 (APOE4) versus APOE3 to CAA. In a more recent study, Shin et al. developed a BBB-on-a-chip model with hBMECs and ReNcell VM human neural progenitor cells harboring familial AD mutations (Figure 7).[170] The resulting AD model exhibited significant BBB alterations commonly observed in AD patients, including increased levels of reactive oxygen species, matrix-metalloproteinase-2, and Aβ accumulation, decreased levels of TJ and AJ proteins, and higher BBB permeability. Moreover, the AD model showed the cytotoxic effects of thrombin on neurons and how the restoration of BBB integrity via a pharmacological agent negated the effects of thrombin by preventing its entry into the BBB. Potential limitations of the proposed model include a lack of key BBB components such as astrocytes and pericytes. Astrocytes serve an important role in AD progression; they present altered gene expression patterns in neurodegenerative models and involved in neuroinflammation associated with AD.[221–223] Similarly, pericytes are an imperative player in AD development as evident from a large body of evidence suggesting the loss or degeneration of pericytes being a hallmark of AD pathogenesis.[36, 183, 224]
Figure 7.

Alzheimer’s disease (AD)-on-a-chip. (a) Schematic of the AD-on-a-chip device. (b) Experimental protocol used for the study. (c) Comparisons between wildtype (WT) and AD groups of permeability to tracer molecules of different molecular weights. (d) Claudin-5 expression patterns in WT and AD models. Reproduced with permission.[170] Copyright 2021, Wiley.
4.1.2. Parkinson’s disease
PD is another common neurodegenerative disease among those with age ≥ 65.[230, 231] Degeneration of dopaminergic neurons initially occurs within the ventrolateral substantia nigra and eventually spreads into other brain regions at a later stage. Another pathological characteristic of PD is the accumulation of alpha-synuclein (αSyn) and the subsequent formation of Lewy bodies. In a recent study by Pediaditakis et al.,[229] a BBB-on-a-chip was used to co-culture human iPSC-derived BMECs, pericytes, astrocytes, microglia, and dopaminergic neurons (Figure 8). In the brain channel, the cells were treated with αSyn pre-formed fibrils to induce PD pathogenesis within the chip. Not only did the “substantia nigra brain-chip” recapitulate the pathological events of PD such as the accumulation of pSer129-αSyn, reduced mitochondrial activity, neuroinflammation, and loss of neurons, but it also resulted in BBB disruption as indicated by higher barrier permeability to different tracer molecules.
Figure 8.

Parkinson’s disease (PD) on a chip. (a) Study protocol used for the development of the PD-on-a-chip. (b) Schematic representation of the PD-on-a-chip platform. (c) Expression of phosphorylated αSyn after treatment with αSyn fibrils versus αSyn monomers. (d) Effects of αSyn fibrils on ZO-1 expression and presence of pSer129-αSyn. (e) Permeability of the BBB after treatment with αSyn fibrils or monomers. Reproduced with permission.[229] Copyright 2021, Springer Nature.
4.1.3. Amyotrophic lateral sclerosis
In ALS, loss of motor neurons in the spinal cord and motor cortex is frequently observed, followed by motor paralysis and eventually death.[232] While the pathogenesis of ALS remains largely unknown, past research has identified putative genes that predispose individuals to be familial and/or sporadic ALS, including the superoxide dismutase gene. In a recent study by Osaki et al., an ALS-on-a-chip was developed to test drug candidates and investigate ALS pathogenesis.[225] The device consisted of PDMS chambers with micro-pillars for force measurement. In this chip, iPSC-derived motor neurons, skeletal muscle cells, and ECs were co-cultured. The ALS model exhibited pathological characteristics indicative of ALS, including weaker muscle contraction and increased motor neuron degeneration and apoptosis. When the ALS model was given a cotreatment of rapamycin and bosutinib through the EC layer, it restored the motor functions more effectively compared to a mono-treatment with rapamycin or bosutinib alone. The findings provided further evidence for the importance of the blood-borne drugs’ ability to permeate the BBB to maximize their efficacy to treat ALS and demonstrated the ALS-on-a-chip as a screening tool to effectively assess the therapeutic potential of drug candidates. The ALS model, however, did not have pericytes as part of the BBB. The addition of these cells to the system could help improve the physiological relevance as dysfunctional pericytes are presumably involved in BBB disruption that leads to leakage of red blood cells and subsequent release of neurotoxic molecules that destroy the motor neurons.[233]
4.1.4. Stroke
Ischemic stroke is a leading cause of global mortality and morbidity.[234, 235] It occurs when blood flow to the brain is obstructed due to an embolism, which often results from cardiac diseases and atherosclerosis. Blood occlusion is often followed by a series of catastrophic events due to a lack of oxygen and glucose supply such as abnormal neuronal activities, inflammation, necrosis, and other pathological alterations in the brain microenvironment. Another hallmark of ischemic stroke includes upregulation of proteases within the BBB milieu resulting in degradation of BM proteins and subsequent BBB breakdown.[219] Recently, Lyu et al. investigated the therapeutic potential of stem cell-based therapies to treat ischemic stroke using a BBB-on-a-chip system, which included hBMECs, pericytes, astrocytes, microglia, and neurons (Figure 9).[177] The developed model formed a functional BBB and showed clinically-relevant responses to an ischemic insult. Moreover, gene expression profiling unveiled specific genes responsible for the neurorestorative effects of different stem cells. Importantly, with the stroke-on-a-chip model, it was possible to evaluate the effects of stem cell-based therapies on the host cells and perform mechanistic studies to identify particular genes associated with neurorestoration.
Figure 9.

Stroke-on-a-chip. (a) (i) Schematic diagram of the stroke-on-a-chip device. (ii) Experimental protocol used in the study. (b) Effects of ischemia on cellular morphology and expression of markers by neurons and endothelial cells. (c) Permeability of the modeled BBB in ischemia versus normoxia group. (d) Tracking of hiPSC-derived neural progenitor cells (hNPCs) via (i) GFP, (ii) stem cell markers, and (iii) differentiation markers. Reproduced with permission.[177] Copyright 2021, Springer Nature.
4.1.5. Glioblastoma
Another important application of BBB-on-a-chip technology is the in vitro modeling of GBM. GBM is a rare type of cancer; however, it carries a dismal prognosis with a survival rate of 14–15 months.[6, 236] Currently, little is known about the etiology of the disease, yet research has shed some light on the pathogenesis of GBM. The cancer cells are thought to originate from the subventricular zone in the brain, where cells with neural stem cell (NSC) properties undergo oncogenic transformation as a result of genetic mutations. In the tumor microenvironment (TME), brain tumor stem cells come in direct contact with BMECs. The NVU provides essential growth factors, neurotransmitters, and ECM. GBM cells also interact with tumor-associated macrophages, including the resident microglia and bone marrow-derived macrophages. It is through such interactions that the unique TME supports GBM tumorigenesis, progression, and acquisition of drug resistance.
GBM progression often involves local alterations of the BBB structure and function leading to heterogeneous barrier properties. This BBB disrupted by tumor cells is known as the blood-tumor barrier (BTB).[237] The heterogeneous nature of BTB results in some intact BBB regions with active efflux transporters that prevent the entry of therapeutic agents into the brain. As a way to model and study GBM cell behaviors using patient-derived cells ex vivo, Xiao et al. used a microvasculature-on-a-chip model, in which patient-derived GBM stem-like cells and HUVECs were co-cultured in collagen-filled chambers and characterized.[227] The GBM-on-a-chip effectively showed inter-individual differences in co-localization of GBM cells and HUVECs with distinct gene expression patterns and demonstrated its potential to serve as a clinical model to guide personalized treatment. Cui et al. sought to develop a GBM-on-a-chip for the assessment of immunotherapy by incorporating additional cell types, including macrophages, microglia, and CD8+ T cells (Figure 10).[228] Using the device, the authors were able to show patient-specific epigenetic and immune characteristics that may be responsible for inter-individual differences in response to immunotherapy. Additionally, the GBM model allowed the evaluation of the effects of combination therapy to treat GBM. It is worth noting that astrocytes and pericytes were absent in this model. Astrocytes were recently found to promote tumor cell migration via secretion of C-C motif chemokine ligand 2 (CCL2) [238] and tumor-associated astrocytes are known to promote GBM progression and affect responses to anti-cancer therapies.[239, 240] Likewise, GBM-activated pericytes secrete anti-inflammatory molecules and contribute to the immunosuppressive environment [241] while healthy pericytes may serve to protect the brain from tumor invasion by acting as physical barriers.[242]
Figure 10.

Glioblastoma (GBM)-on-a-chip. (a) Schematic depiction of the GBM-on-a-chip. (b) Experimental protocol followed in the study. (c) Confocal images showing interactions between CD8+ T cells, GBM cells, and EC cells. (d) Proportions of occurrence of apoptosis in GBM cells treated with activated CD8+ T cells in different GBM niches. Reproduced with permission. [228] Copyright 2020, eLife.
4.2. Personalized modeling
BBB-on-a-chip can be personalized using patient-derived cells to model patient-specific characteristics and responses to stimuli of interest. However, obtaining primary patient-derived cells for the BBB has been particularly challenging due to limited access to the human brain tissues and the invasive nature of the isolation process that often results in poor yield and purity.[144] To circumvent this problem, iPSC-derived cells have been investigated as a potential source of patient-derived cells. Several studies support their ability to model inter-individual differences for various neurological diseases[243–246] even when combined with BBB-on-a-chip.[149] In particular, Motallebnejad et al. developed an isogenic BBB-on-a-chip model using iPSC-derived BMECs and astrocytes embedded in a 3D hydrogel.[247] Protein expression analysis of TJ proteins and glial fibrillary acidic protein for BMECs and astrocytes, respectively, confirmed the differentiation of hiPSCs into the respective cell types. Moreover, the BMEC layer yielded a TEER of more than 1,000 Ω.cm2. In another study, Vatine et al. demonstrated a BBB-on-a-chip using hiPSC-derived BMECs and neural progenitor cells, which were differentiated into a mixture of neural progenitors, neurons, and astrocytes on the chip. The model exhibited a physiologically relevant TEER value of approximately 1,500 Ω.cm2 and excellent impermeability to various tracer molecules.[65] In the same study, the authors obtained iPSCs from individuals with and without HD and compared their BBB properties. The results showed higher permeability for the HD group than a healthy control group with the control lines exhibiting inter-individual differences in BBB phenotype. Although the roles of pericytes in HD pathology mostly remain elusive, a recent study demonstrated that pericyte activation may constitute an early step in microvascular alteration seen in HD patients.[248] Therefore, future investigation of pericytes’ roles in HD progression using BBB-on-a-chip will be of interest.
5. Current challenges and future directions
BBB-on-a-chip has made significant progress over the years to provide additional insights into the human BBB under healthy and pathological conditions. However, certain obstacles are yet to be addressed for the platform to become more widely accepted and implemented at a larger scale. Below we summarize the current challenges and how leveraging other technologies such as AI, iPSC, and biosensors may help strategize the approaches to resolve the problems (Figure 11).
Figure 11.

Ongoing efforts and proposed future directions to enhance the utility of the current BBB-on-a-chip platform. (a) Potential areas of development. (b) An example process flow for the implementation of a potential next-generation BBB-on-a-chip platform.
5.1. Increasing throughput and scalability
The wide implementation of BBB-on-a-chip by the pharmaceutical industry for drug screening has been hindered by the relatively limited throughput of most OoC platforms. Drug screening generally requires the use of multiple well plates (i.e., 384 wells or more). However, most of the BBB-on-a-chip platforms presently do not have such capability. To address this need, a few recent studies have proposed high-throughput BBB-on-a-chip platforms.[30, 249] Wevers et al., for instance, developed a microfluidic BBB-on-a-chip platform with the ability to co-culture hBMECs, pericytes, and astrocytes up to 40 chips at a time and demonstrated its utility to study antibody transport across the BBB.[30]
For industrialization, the traditional chip fabrication processes using soft lithography techniques present a challenge due to their limited scalability. As a potential solution, one can alternatively use a material that is more compatible with industrial manufacturing processes (e.g., hot embossing or injection molding) for mass production such as thermoplastics or resins. In addition, it needs to be realized that increasing the throughput normally results in more labor-intensive and time-consuming processes. Hence, it will be critically important to develop protocols that enable efficient device operation, data collection, and data analysis to streamline the experimental procedures. As an example, one may employ robotics to automate tasks (i.e., chip operation and data collection)[250] and machine learning (ML) to facilitate data analysis.[25, 251]
The throughput of OoCs is determined by not only the number of tests per run but also the number of readouts per test. Most traditional methods for molecular and functional characterization of BBB-on-a-chip such as reverse transcription-polymerase chain reaction and permeability assays via tracer molecules are invasive, not real-time, and require laborious work for sample acquisition. Furthermore, current in-line TEER sensors are not multiplexed, restricting the readout to only TEER measurements. One of the plausible solutions to this problem is to use multiple inline biosensors for the simultaneous detection of multiple soluble markers. The utility of such sensors can be further boosted by exploiting programmable and multiplexable electrochemical instruments (e.g., Potentiostat) to partially, if not fully, automate the data recording and collection processes. Automating experimental procedures using the aforementioned techniques will help improve experimental efficiency by reducing manual labor and human errors, especially for high-throughput screening applications.
5.2. Integration with AI
AI, especially ML and its subfield deep learning (DL), have gained substantial interest in the fields of medicine and life sciences attributed to their ability to learn patterns in voluminous amounts of highly complex data such as medical images and gene expression data.[252–255] Moreover, ML models help reduce inter-operator variability in data analysis.[256] As more data acquired from BBB-on-a-chip systems become available (e.g., immunostaining images or molecular expression data), it will be worthwhile to investigate how best to integrate AI with BBB-on-a-chip platforms. The exploitation of ML will greatly accelerate data analysis and therefore overall experimental efficiency for various BBB-on-a-chip applications including drug screening, in which a large number of samples need to be analyzed.
ML typically requires a large amount of data to be properly trained and validated with the size of data needed being contingent upon the complexity of the data and task at hand.[257] Furthermore, external validation using new data that “have never been seen by the model” is essential for the development of robust ML models with good generalizability and minimal bias.[258] Therefore, the quality and quantity of data available for ML model development are vital for the successful integration of ML with BBB-on-a-chip systems. In addition to model accuracy, model interpretability will also be key to facilitating the adoption of ML, especially DL, which is generally considered a black-box approach.[259] Particularly in the healthcare field, poor model explainability is more concerning than in the other sectors due to a need for high ethical standards and low tolerance towards model errors and uncertainty.
Despite such challenges, the potential applications of ML for BBB-on-a-chip are countless. For example, ML algorithms can be trained with bright-field and corresponding fluorescent images of microtissues cultured on BBB-on-a-chip to perform virtual staining for protein expression analysis.[260] As the AI-assisted approach allows for in silico analysis, it will keep the sample intact, making it possible to continually monitor the on-chip culture with considerably fewer bench experiments. Besides microscopic images, a variety of data types such as omics, soluble biomarker levels, and/or chemical fingerprints of drugs being assessed on BBB-on-a-chip can potentially serve as inputs for ML model development. If adequately trained with such data, ML algorithms may identify and correlate unique molecular signatures with surrogate endpoints (e.g., cell viability or barrier properties) or clinical outcomes if the in vitro data are obtained from patient-specific BBB-on-a-chip models. Hence, the successful integration of AI will be invaluable for not only research but also potential clinical applications of BBB-on-a-chip models.
5.3. Democratizing OoC technology
Compared to conventional in vitro models, OoCs generally require more specialized skills, sound manual dexterity, and training for chip fabrication and device operation. For instance, if a BBB-on-a-chip device with a particular design was not readily available, one would need to be well-versed with microfabrication techniques and require access to facilities to design and fabricate the chip on their own. In addition, the developed device would need to meet all design requirements such as flow dynamics, biocompatibility, and bonding strength with a good seal for multi-layered chips. On-chip culturing of cells can also be challenging for novice users as the protocols for cell seeding and culture are different from those for standard cell culture in traditional culture vessels. Recently, more companies such as Emulate Inc.[94] and Mimetas[261] have started to make OoCs more readily accessible to researchers by offering prefabricated chips and specialized equipment/apparatus that simplify the experimental procedures of OoC research. However, some researchers still resort to fabricating their own OoCs, presumably for budgetary reasons and/or specific design requirements. Therefore, continuing efforts to democratize this technology are essential in the future for BBB-on-a-chip to become broadly accepted by the research communities and industries.
5.4. Improving physiological relevance
Most of the BBB-on-a-chip models are currently fabricated using PDMS, which is known to adsorb small hydrophobic molecules – a longstanding problem with PDMS-based platforms for drug screening applications. As such, an additional treatment of PDMS devices with lipophilic coatings[95] or the use of other non-drug absorbing polymers such as PMMA in place of PDMS will be attractive approaches to circumvent the drug adsorption problem. In addition, most of the current BBB-on-a-chip systems make use of synthetic polymers serving as BM-like structures. As the presence of synthetic materials presents a significant deviation from the native BBB microenvironment, removal of the artificial element without sacrificing the ability to measure barrier properties (e.g., TEER) will substantially enhance the in vitro platform in its utility and physiological relevance.
Apart from the above-mentioned points, most of the existing BBB-on-a-chip systems are missing interactions with other organ units. In humans, cross-talks between multiple organs are critical for the proper functioning of the human body and have considerable influence on the systemic effects of disease or pharmaceutical interventions.[262] Although located distant from each other, the organs can communicate via blood and lymph circulations using various forms of signaling molecules (e.g., cytokines and exosomes). The need for more holistic approaches has fueled the development of MOOC platforms to model inter-organ interactions for the systemic assessment of functional and physiological characteristics in vitro. For example, Lee et al. recently developed a heart-breast-cancer-on-a-chip as a platform to assess the cardiotoxic effects of chemotherapies.[263] The MOOC enables the toxicological studies of chemotherapies in a more clinically-relevant manner compared to conventional in vitro models. Likewise, a BBB-on-a-chip can be incorporated into MOOC systems. By linking with a liver-on-a-chip, one can model the liver-brain interactions to accurately assess the efficacy of CNS-targeting drugs while accounting for drug metabolism by the liver following a similar approach used to develop a heart-liver-on-a-chip by Zhang et al.[191] Additionally, modeling the brain-gut axis using a MOOC approach has recently been proposed to study vesicular transport and elucidate the roles of gut microbiota and their secretome in modulating brain function.[23, 264] It is also worth reiterating the limitations of the current BBB-on-a-chip disease models described in Section 4 of this review due to the microenvironments missing one or more BBB components such as astrocytes and pericytes, which serve critical roles in BBB function and maintenance. Although experimental complexity may increase and throughput may be compromised in exchange for enhanced physiological relevance, the trade-off should carefully be considered at the beginning or early stage of project development.
Further advances in iPSC technology will also be essential for improving the physiological relevance of BBB-on-a-chip models. As discussed in earlier sections, the use of iPSC-derived cells lies at the heart of the development of personalized BBB-on-a-chip models to enable the studies of patient heterogeneity for specific disease applications in vitro. However, iPSC-derived cells tend to be phenotypically immature compared to primary cells, and the generation and differentiation processes remain costly and time-intensive with batch-to-batch variations still being common among different iPSC lines.[161] Therefore, further research is needed in this area to address the remaining challenges and permit a more precise recapitulation of the in vivo physiology using BBB-on-a-chip in a more reproducible and cost-effective manner.
5.5. Validating models following applicable regulatory guidelines
For BBB-on-a-chip to be adopted as a preclinical model and used by pharmaceutical companies, the evaluation of their qualifications will be highly recommended by following guidelines provided by regulatory entities such as the United States Food and Drug Administration (FDA) and the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ) MPS Affiliate. IQ MPS Affiliate was founded at the request of the National Institutes of Health National Center for Advancing Translational Science (NCATS)[265] to provide organ-specific characterization data requirements for OoCs to be implemented for industrial use. Although the guideline is still under development for BBB/CNS models, once published, the comprehensive assessment and validation of BBB-on-a-chip using the established eligibility criteria will bolster its robustness and practical utility facilitating the large-scale implementation of the technology. Similarly, as BBB-on-a-chip continues to show superior physiological relevance and potential to partially, if not fully, replace conventional preclinical models, the development strategies should take into account applicable and evolving FDA regulations around the OoC platforms. This is particularly true if one is seeking to commercialize their BBB-on-a-chip models. In fact, the FDA and major pharmaceutical companies have begun to work with Emulate Inc. to quantitatively validate their OoC products for the safety and efficacy assessment of drugs.[266] As illustrated by this example, close, well-orchestrated collaborations between academic institutions, pharma and healthcare industries, and the FDA are anticipated to become increasingly important in evaluating the BBB-on-a-chip platform as a new in vitro model for drug discovery and personalized medicine.
6. Conclusion
BBB-on-a-chip has revealed new insights into the human brain and offered numerous opportunities to augment the current disease modeling platforms for potential drug development and clinical applications. Nevertheless, the current BBB-on-a-chip platform faces several challenges that hinder its wider adoption such as insufficient throughput and scalability as well as the lack of validation studies confirming their clinical relevance. Prospectively, continuing efforts will be needed to increase the physiological relevance of the on-chip models and assess disease pathologies that remain uninvestigated. It will equally be important to continue to foster the developments in accompanying technologies described herein to augment the BBB-on-a-chip platform. We envision that the multidisciplinary approach presented in this review will allow the development of a next-generation BBB-on-a-chip platform. Ultimately, the future BBB-on-a-chip will have enhanced functionality and facilitate personalized disease modeling with greater potential for large-scale implementation by both academic and commercial institutions.
Acknowledgments
The authors acknowledge funding from the National Institutes of Health (TR003148-01).
Biographies
Author Bibliographies:

Satoru Kawakita is currently a biomedical engineer/data scientist at the Terasaki Institute for Biomedical Innovation (TIBI). He received his M.S. in Biomedical Engineering from the University of Southern California in 2016. His research interests lie in translational research using advanced in vitro models and data science to drive biomedical innovation and improve human health.

Mehmet Remzi Dokmeci received his Ph.D. degree from the University of Michigan, Ann Arbor in Electrical Engineering. Dr. Dokmeci is an Associate Professor at the Terasaki Institute for Biomedical Innovation (TIBI) since July 2020. He was previously an Associate Adjunct Professor in the Radiology Department at University of California-Los Angeles and an Instructor at Brigham and Women’s Hospital, Harvard Medical School. He has been actively involved in research areas such as biomaterials, microfluidic systems for organs-on-a-chip applications, 3D bioprinting, electrical/electrochemical biosensors, and flexible electronics.

Ali Khademhosseini is the CEO and distinguished professor at the Terasaki Institute for Biomedical Innovation (TIBI). Formerly, he was a professor and Levi Knight Chair at University of California-Los Angeles (UCLA). He joined UCLA from Harvard University where he was a professor at Harvard Medical School and faculty at the Harvard-MIT’s Division of Health Sciences and Technology, Brigham and Women’s Hospital as well as an associate faculty at Wyss Institute for Biologically Inspired Engineering. His interdisciplinary works mainly include personalized solutions that utilize micro and nanotechnologies to enable a range of therapies for organ failure, cardiovascular diseases, and cancer.
Footnotes
Conflict of Interest
The authors have no conflict of interest.
Contributor Information
Satoru Kawakita, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Kalpana Mandal, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Lei Mou, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA; Department of Clinical Laboratory, Third Affiliated Hospital of Guangzhou Medical University, Guangzhou Medical University, No. 63 Duobao Road, Liwan District, Guangzhou, Guangdong, P. R. China.
Marvin Magan Mecwan, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Yangzhi Zhu, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Shaopei Li, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Saurabh Sharma, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Ana Lopez Hernandez, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Huu Tuan Nguyen, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Surjendu Maity, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Natan Roberto de Barros, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Aya Nakayama, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Praveen Bandaru, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Samad Ahadian, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Han-Jun Kim, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Rondinelli Donizetti Herculano, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA; Department of Bioprocess and Biotechnology Engineering, São Paulo State University (Unesp), School of Pharmaceutical Sciences, Araraquara, SP 14801-902, Brazil.
Eggehard Holler, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Vadim Jucaud, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Mehmet Remzi Dokmeci, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
Ali Khademhosseini, Terasaki Institute for Biomedical Innovation, Los Angeles, California, 90064, USA.
References:
- 1.Sweeney MD; Zhao Z; Montagne A; Nelson AR; Zlokovic BV, Physiological Reviews 2019, 99 (1), 21–78. DOI 10.1152/physrev.00050.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplan L; Chow BW; Gu C, Nature Reviews Neuroscience 2020, 21 (8), 416–432. DOI 10.1038/s41583-020-0322-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segarra M; Aburto MR; Acker-Palmer A, Trends in Neurosciences 2021, 44 (5), 393–405. DOI 10.1016/j.tins.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Association A. s., Alzheimer’s & Dementia 2018, 14 (3), 367–429. DOI 10.1016/j.jalz.2018.02.001. [DOI] [Google Scholar]
- 5.Collaborators GBDS, Lancet Neurol 2019, 18 (5), 439–458. DOI 10.1016/S1474-4422(19)30034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanif F; Muzaffar K; Perveen K; Malhi SM; Simjee SU, Asian Pac J Cancer Prev 2017, 18 (1), 3–9. DOI 10.22034/APJCP.2017.18.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terstappen GC; Meyer AH; Bell RD; Zhang W, Nature Reviews Drug Discovery 2021, 20 (5), 362–383. DOI 10.1038/s41573-021-00139-y. [DOI] [PubMed] [Google Scholar]
- 8.Shlosberg D; Benifla M; Kaufer D; Friedman A, Nat Rev Neurol 2010, 6 (7), 393–403. DOI 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banks WA, Nat Rev Drug Discov 2016, 15 (4), 275–92. DOI 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 10.Aday S; Cecchelli R; Hallier-Vanuxeem D; Dehouck MP; Ferreira L, Trends in Biotechnology 2016, 34 (5), 382–393. DOI 10.1016/j.tibtech.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Baumans V, Gene Therapy 2004, 11 (1), S64–S66. DOI 10.1038/sj.gt.3302371. [DOI] [PubMed] [Google Scholar]
- 12.Ahn SI; Kim Y, Trends Biotechnol 2021, 39 (8), 749–752. DOI 10.1016/j.tibtech.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Huh D; Matthews BD; Mammoto A; Montoya-Zavala M; Hsin HY; Ingber DE, Science 2010, 328 (5986), 1662–1668. DOI doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esch EW; Bahinski A; Huh D, Nature Reviews Drug Discovery 2015, 14 (4), 248–260. DOI 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B; Korolj A; Lai BFL; Radisic M, Nature Reviews Materials 2018, 3 (8), 257–278. DOI 10.1038/s41578-018-0034-7. [DOI] [Google Scholar]
- 16.Low LA; Mummery C; Berridge BR; Austin CP; Tagle DA, Nat Rev Drug Discov 2021, 20 (5), 345–361. DOI 10.1038/s41573-020-0079-3. [DOI] [PubMed] [Google Scholar]
- 17.Ingber DE, Nature Reviews Genetics 2022. DOI 10.1038/s41576-022-00466-9. [DOI] [Google Scholar]
- 18.Cucullo L; Hossain M; Puvenna V; Marchi N; Janigro D, BMC Neurosci 2011, 12, 40-40. DOI 10.1186/1471-2202-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oddo A; Peng B; Tong Z; Wei Y; Tong WY; Thissen H; Voelcker NH, Trends Biotechnol 2019, 37 (12), 1295–1314. DOI 10.1016/j.tibtech.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Lee CS; Leong KW, Curr Opin Biotechnol 2020, 66, 78–87. DOI 10.1016/j.copbio.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown JA; Faley SL; Shi Y; Hillgren KM; Sawada GA; Baker TK; Wikswo JP; Lippmann ES, Fluids Barriers CNS 2020, 17 (1), 38. DOI 10.1186/s12987-020-00200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sances S; Ho R; Vatine G; West D; Laperle A; Meyer A; Godoy M; Kay PS; Mandefro B; Hatata S; Hinojosa C; Wen N; Sareen D; Hamilton GA; Svendsen CN, Stem Cell Reports 2018, 10 (4), 1222–1236. DOI 10.1016/j.stemcr.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raimondi I; Izzo L; Tunesi M; Comar M; Albani D; Giordano C, Frontiers in Bioengineering and Biotechnology 2020, 7 (435). DOI 10.3389/fbioe.2019.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang Y; Yoon J-Y, Sensors and Actuators Reports 2021, 3, 100031. DOI 10.1016/j.snr.2021.100031. [DOI] [Google Scholar]
- 25.Riordon J; Sovilj D; Sanner S; Sinton D; Young EWK, Trends in Biotechnology 2019, 37 (3), 310–324. DOI 10.1016/j.tibtech.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Muoio V; Persson PB; Sendeski MM, Acta Physiologica 2014, 210 (4), 790–798. DOI 10.1111/apha.12250. [DOI] [PubMed] [Google Scholar]
- 27.Cameron T; Bennet T; Rowe EM; Anwer M; Wellington CL; Cheung KC, Micromachines (Basel) 2021, 12 (4). DOI 10.3390/mi12040441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn SI; Sei YJ; Park H-J; Kim J; Ryu Y; Choi JJ; Sung H-J; MacDonald TJ; Levey AI; Kim Y, Nature Communications 2020, 11 (1), 175. DOI 10.1038/s41467-019-13896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Booth R; Kim H, Lab on a Chip 2012, 12 (10), 1784–1792. DOI 10.1039/C2LC40094D. [DOI] [PubMed] [Google Scholar]
- 30.Wevers NR; Kasi DG; Gray T; Wilschut KJ; Smith B; van Vught R; Shimizu F; Sano Y; Kanda T; Marsh G; Trietsch SJ; Vulto P; Lanz HL; Obermeier B, Fluids Barriers CNS 2018, 15 (1), 23. DOI 10.1186/s12987-018-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oldendorf WH; Cornford ME; Brown WJ, Ann Neurol 1977, 1 (5), 409–17. DOI 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 32.Barar J; Rafi MA; Pourseif MM; Omidi Y, Bioimpacts 2016, 6 (4), 225–248. DOI 10.15171/bi.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulgar VM, Frontiers in Neuroscience 2019, 12 (1019). DOI 10.3389/fnins.2018.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zihni C; Mills C; Matter K; Balda MS, Nature Reviews Molecular Cell Biology 2016, 17 (9), 564–580. DOI 10.1038/nrm.2016.80. [DOI] [PubMed] [Google Scholar]
- 35.Pardridge WM, J Cereb Blood Flow Metab 2012, 32 (11), 1959–1972. DOI 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sweeney MD; Ayyadurai S; Zlokovic BV, Nature Neuroscience 2016, 19 (6), 771–783. DOI 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park DY; Lee J; Kim J; Kim K; Hong S; Han S; Kubota Y; Augustin HG; Ding L; Kim JW; Kim H; He Y; Adams RH; Koh GY, Nature Communications 2017, 8 (1), 15296. DOI 10.1038/ncomms15296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsauer M; Krause D; Dermietzel R, The FASEB Journal 2002, 16 (10), 1274–1276. DOI 10.1096/fj.01-0814fje. [DOI] [PubMed] [Google Scholar]
- 39.Thanabalasundaram G; Schneidewind J; Pieper C; Galla H-J, The International Journal of Biochemistry & Cell Biology 2011, 43 (9), 1284–1293. DOI 10.1016/j.biocel.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 40.Kacem K; Lacombe P; Seylaz J; Bonvento G, Glia 1998, 23 (1), 1–10. DOI . [DOI] [PubMed] [Google Scholar]
- 41.Abbott NJ; Rönnbäck L; Hansson E, Nature Reviews Neuroscience 2006, 7 (1), 41–53. DOI 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 42.Haseloff RF; Blasig IE; Bauer HC; Bauer H, Cell Mol Neurobiol 2005, 25 (1), 25–39. DOI 10.1007/s10571-004-1375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seo S; Kim H; Sung JH; Choi N; Lee K; Kim HN, Biomaterials 2020, 232, 119732. DOI 10.1016/j.biomaterials.2019.119732. [DOI] [PubMed] [Google Scholar]
- 44.Cabezas R; El-Bachá RS; González J; Barreto GE, Neurosci Res 2012, 74 (2), 80–90. DOI 10.1016/j.neures.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Ren X; Zou L; Zhang X; Branco V; Wang J; Carvalho C; Holmgren A; Lu J, Antioxid Redox Signal 2017, 27 (13), 989–1010. DOI 10.1089/ars.2016.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vazana U; Veksler R; Pell GS; Prager O; Fassler M; Chassidim Y; Roth Y; Shahar H; Zangen A; Raccah R; Onesti E; Ceccanti M; Colonnese C; Santoro A; Salvati M; D’Elia A; Nucciarelli V; Inghilleri M; Friedman A, J Neurosci 2016, 36 (29), 7727–39. DOI 10.1523/jneurosci.0587-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.András IE; Deli MA; Veszelka S; Hayashi K; Hennig B; Toborek M, J Cereb Blood Flow Metab 2007, 27 (8), 1431–43. DOI 10.1038/sj.jcbfm.9600445. [DOI] [PubMed] [Google Scholar]
- 48.Pulido RS; Munji RN; Chan TC; Quirk CR; Weiner GA; Weger BD; Rossi MJ; Elmsaouri S; Malfavon M; Deng A; Profaci CP; Blanchette M; Qian T; Foreman KL; Shusta EV; Gorman MR; Gachon F; Leutgeb S; Daneman R, Neuron 2020, 108 (5), 937–952.e7. DOI 10.1016/j.neuron.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lalo U; Pankratov Y; Parpura V; Verkhratsky A, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2011, 1813 (5), 992–1002. DOI 10.1016/j.bbamcr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 50.Bachiller S; Jiménez-Ferrer I; Paulus A; Yang Y; Swanberg M; Deierborg T; Boza-Serrano A, Frontiers in Cellular Neuroscience 2018, 12 (488). DOI 10.3389/fncel.2018.00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan J; Ma N; Zhong J; Yu B; Wan J; Zhang W, Molecular Therapy - Nucleic Acids 2021, 26, 970–986. DOI 10.1016/j.omtn.2021.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zabel MK; Kirsch WM, Ageing Res Rev 2013, 12 (3), 749–756. DOI 10.1016/j.arr.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kigerl KA; de Rivero Vaccari JP; Dietrich WD; Popovich PG; Keane RW, Exp Neurol 2014, 258, 5–16. DOI 10.1016/j.expneurol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Q; Barres BA, Nature Reviews Immunology 2018, 18 (4), 225–242. DOI 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- 55.Hickman S; Izzy S; Sen P; Morsett L; El Khoury J, Nature Neuroscience 2018, 21 (10), 1359–1369. DOI 10.1038/s41593-018-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau LW; Cua R; Keough MB; Haylock-Jacobs S; Yong VW, Nature Reviews Neuroscience 2013, 14 (10), 722–729. DOI 10.1038/nrn3550. [DOI] [PubMed] [Google Scholar]
- 57.Xu L; Nirwane A; Yao Y, Stroke and Vascular Neurology 2019, 4 (2), 78–82. DOI 10.1136/svn-2018-000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long KR; Huttner WB, Open Biology 2019, 9 (1), 180216. DOI doi: 10.1098/rsob.180216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnes JM; Przybyla L; Weaver VM, J Cell Sci 2017, 130 (1), 71–82. DOI 10.1242/jcs.191742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonneh-Barkay D; Wiley CA, Brain Pathol 2009, 19 (4), 573–85. DOI 10.1111/j.1750-3639.2008.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fawcett JW; Oohashi T; Pizzorusso T, Nature Reviews Neuroscience 2019, 20 (8), 451–465. DOI 10.1038/s41583-019-0196-3. [DOI] [PubMed] [Google Scholar]
- 62.Rowlands D; Lensjø KK; Dinh T; Yang S; Andrews MR; Hafting T; Fyhn M; Fawcett JW; Dick G, The Journal of Neuroscience 2018, 38 (47), 10102–10113. DOI 10.1523/jneurosci.1122-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wong A; Ye M; Levy A; Rothstein J; Bergles D; Searson P, Frontiers in Neuroengineering 2013, 6 (7). DOI 10.3389/fneng.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luissint A-C; Artus C; Glacial F; Ganeshamoorthy K; Couraud P-O, Fluids and Barriers of the CNS 2012, 9 (1), 23. DOI 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vatine GD; Barrile R; Workman MJ; Sances S; Barriga BK; Rahnama M; Barthakur S; Kasendra M; Lucchesi C; Kerns J; Wen N; Spivia WR; Chen Z; Van Eyk J; Svendsen CN, Cell Stem Cell 2019, 24 (6), 995–1005.e6. DOI 10.1016/j.stem.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 66.Reinitz A; DeStefano J; Ye M; Wong AD; Searson PC, Microvasc Res 2015, 99, 8–18. DOI 10.1016/j.mvr.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeStefano JG; Xu ZS; Williams AJ; Yimam N; Searson PC, Fluids and Barriers of the CNS 2017, 14 (1), 20. DOI 10.1186/s12987-017-0068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greene C; Hanley N; Campbell M, Fluids and Barriers of the CNS 2019, 16 (1), 3. DOI 10.1186/s12987-019-0123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan S; Liu KJ; Qi Z, Brain Circ 2020, 6 (3), 152–162. DOI 10.4103/bc.bc_29_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.JIA W; MARTIN TA; ZHANG G; JIANG WG, Anticancer Research 2013, 33 (6), 2353–2359. [PubMed] [Google Scholar]
- 71.Lochhead JJ; Yang J; Ronaldson PT; Davis TP, Frontiers in Physiology 2020, 11 (914). DOI 10.3389/fphys.2020.00914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sandoval KE; Witt KA, Neurobiology of Disease 2008, 32 (2), 200–219. DOI 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 73.Hajal C; Campisi M; Mattu C; Chiono V; Kamm RD, Biomicrofluidics 2018, 12 (4), 042213. DOI 10.1063/1.5027118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hawkins BT; Davis TP, Pharmacol Rev 2005, 57 (2), 173–85. DOI 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 75.Sweeney MD; Sagare AP; Zlokovic BV, Nat Rev Neurol 2018, 14 (3), 133–150. DOI 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campos-Bedolla P; Walter FR; Veszelka S; Deli MA, Archives of Medical Research 2014, 45 (8), 610–638. DOI 10.1016/j.arcmed.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 77.Park TE; Mustafaoglu N; Herland A; Hasselkus R; Mannix R; FitzGerald EA; Prantil-Baun R; Watters A; Henry O; Benz M; Sanchez H; McCrea HJ; Goumnerova LC; Song HW; Palecek SP; Shusta E; Ingber DE, Nat Commun 2019, 10 (1), 2621. DOI 10.1038/s41467-019-10588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones AR; Shusta EV, Pharm Res 2007, 24 (9), 1759–1771. DOI 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hervé F; Ghinea N; Scherrmann J-M, AAPS J 2008, 10 (3), 455–472. DOI 10.1208/s12248-008-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou M; Shi SX; Liu N; Jiang Y; Karim MS; Vodovoz SJ; Wang X; Zhang B; Dumont AS, Journal of Clinical Medicine 2021, 10 (17). DOI 10.3390/JCM10173795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maoz BM; Herland A; FitzGerald EA; Grevesse T; Vidoudez C; Pacheco AR; Sheehy SP; Park T-E; Dauth S; Mannix R; Budnik N; Shores K; Cho A; Nawroth JC; Segrè D; Budnik B; Ingber DE; Parker KK, Nature Biotechnology 2018, 36 (9), 865–874. DOI 10.1038/nbt.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ben-Zvi A; Lacoste B; Kur E; Andreone BJ; Mayshar Y; Yan H; Gu C, Nature 2014, 509 (7501), 507–511. DOI 10.1038/NATURE13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Golden PL; Pollack GM, J Pharm Sci 2003, 92 (9), 1739–53. DOI 10.1002/jps.10424. [DOI] [PubMed] [Google Scholar]
- 84.Bartels AL, Curr Pharm Des 2011, 17 (26), 2771–7. DOI 10.2174/138161211797440122. [DOI] [PubMed] [Google Scholar]
- 85.Mikitsh JL; Chacko A-M, Perspect Medicin Chem 2014, 6, 11–24. DOI 10.4137/PMC.S13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Keeffe E; Campbell M, Drug Discov Today Technol 2016, 20, 35–39. DOI 10.1016/j.ddtec.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 87.Bhatia SN; Ingber DE, Nature Biotechnology 2014, 32 (8), 760–772. DOI 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 88.Mou L; Jiang X, Advanced Healthcare Materials 2017, 6 (15), 1601403. DOI 10.1002/adhm.201601403. [DOI] [PubMed] [Google Scholar]
- 89.Hassan S; Heinrich M; Cecen B; Prakash J; Zhang YS, 26 - Biomaterials for on-chip organ systems. In Biomaterials for Organ and Tissue Regeneration, Vrana NE; Knopf-Marques H; Barthes J, Eds. Woodhead Publishing: 2020; pp 669–707. [Google Scholar]
- 90.Falanga AP; Pitingolo G; Celentano M; Cosentino A; Melone P; Vecchione R; Guarnieri D; Netti PA, Biotechnology and Bioengineering 2017, 114 (5), 1087–1095. DOI 10.1002/bit.26221. [DOI] [PubMed] [Google Scholar]
- 91.Gokaltun A; Yarmush ML; Asatekin A; Usta OB, TECHNOLOGY 2017, 05 (01), 1–12. DOI 10.1142/s2339547817300013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.da Silveira GAT; Rocha Neto JBM; Kerwald J; Carvalho HF; Beppu MM, MEDICAL DEVICES & SENSORS 2021, 4 (1), e10142. DOI 10.1002/mds3.10142. [DOI] [Google Scholar]
- 93.Peng B; Tong Z; Tong WY; Pasic PJ; Oddo A; Dai Y; Luo M; Frescene J; Welch NG; Easton CD; Thissen H; Voelcker NH, ACS Applied Materials & Interfaces 2020, 12 (51), 56753–56766. DOI 10.1021/acsami.0c17102. [DOI] [PubMed] [Google Scholar]
- 94.Huh D; Kim HJ; Fraser JP; Shea DE; Khan M; Bahinski A; Hamilton GA; Ingber DE, Nature Protocols 2013, 8 (11), 2135–2157. DOI 10.1038/nprot.2013.137. [DOI] [PubMed] [Google Scholar]
- 95.van Meer BJ; de Vries H; Firth KSA; van Weerd J; Tertoolen LGJ; Karperien HBJ; Jonkheijm P; Denning C; Ijzerman AP; Mummery CL, Biochemical and Biophysical Research Communications 2017, 482 (2), 323–328. DOI 10.1016/j.bbrc.2016.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Samuel R; Thacker CM; Maricq AV; Gale BK, Journal of Micromechanics and Microengineering 2014, 24 (10), 105007. DOI 10.1088/0960-1317/24/10/105007. [DOI] [Google Scholar]
- 97.Yang L; Wei J; Ma Z; Song P; Ma J; Zhao Y; Huang Z; Zhang M; Yang F; Wang X, Nanomaterials (Basel) 2019, 9 (12), 1789. DOI 10.3390/nano9121789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Attia UM; Marson S; Alcock JR, Microfluidics and Nanofluidics 2009, 7 (1), 1. DOI 10.1007/s10404-009-0421-x. [DOI] [Google Scholar]
- 99.Jeon JS; Chung S; Kamm RD; Charest JL, Biomed Microdevices 2011, 13 (2), 325–333. DOI 10.1007/s10544-010-9496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peng L; Deng Y; Yi P; Lai X, Journal of Micromechanics and Microengineering 2013, 24 (1), 013001. DOI 10.1088/0960-1317/24/1/013001. [DOI] [Google Scholar]
- 101.de Almeida Monteiro Melo Ferraz M; Nagashima JB; Venzac B; Le Gac S; Songsasen N, Scientific Reports 2020, 10 (1), 994. DOI 10.1038/s41598-020-57816-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fritzler KB; Prinz VY, Physics-Uspekhi 2019, 62 (1), 54–69. DOI 10.3367/ufne.2017.11.038239. [DOI] [Google Scholar]
- 103.Shakeri A; Khan S; Didar TF, Lab on a Chip 2021, 21 (16), 3053–3075. DOI 10.1039/D1LC00288K. [DOI] [PubMed] [Google Scholar]
- 104.Volpatti LR; Yetisen AK, Trends in Biotechnology 2014, 32 (7), 347–350. DOI 10.1016/j.tibtech.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 105.Eilenberger C; Rothbauer M; Selinger F; Gerhartl A; Jordan C; Harasek M; Schädl B; Grillari J; Weghuber J; Neuhaus W; Küpcü S; Ertl P, Advanced Science 2021, 8 (11), 2004856. DOI 10.1002/advs.202004856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wong JF; Mohan MD; Young EWK; Simmons CA, Biosensors and Bioelectronics 2020, 147, 111757. DOI 10.1016/j.bios.2019.111757. [DOI] [PubMed] [Google Scholar]
- 107.Chen Y; Zhang L; Chen G, Electrophoresis 2008, 29 (9), 1801–14. DOI 10.1002/elps.200700552. [DOI] [PubMed] [Google Scholar]
- 108.Matellan C; del Río Hernández AE, Scientific Reports 2018, 8 (1), 6971. DOI 10.1038/s41598-018-25202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Neufeld L; Yeini E; Reisman N; Shtilerman Y; Ben-Shushan D; Pozzi S; Madi A; Tiram G; Eldar-Boock A; Ferber S; Grossman R; Ram Z; Satchi-Fainaro R, Science Advances 2021, 7 (34), eabi9119. DOI doi: 10.1126/sciadv.abi9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tang M; Rich JN; Chen S, Advanced Materials 2021, 33 (5), 2004776. DOI 10.1002/adma.202004776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tang M; Tiwari SK; Agrawal K; Tan M; Dang J; Tam T; Tian J; Wan X; Schimelman J; You S; Xia Q; Rana TM; Chen S, Small 2021, 17 (15), 2006050. DOI 10.1002/smll.202006050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tang M; Xie Q; Gimple RC; Zhong Z; Tam T; Tian J; Kidwell RL; Wu Q; Prager BC; Qiu Z; Yu A; Zhu Z; Mesci P; Jing H; Schimelman J; Wang P; Lee D; Lorenzini MH; Dixit D; Zhao L; Bhargava S; Miller TE; Wan X; Tang J; Sun B; Cravatt BF; Muotri AR; Chen S; Rich JN, Cell Research 2020, 30 (10), 833–853. DOI 10.1038/s41422-020-0338-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matthiesen I; Voulgaris D; Nikolakopoulou P; Winkler TE; Herland A, Small 2021, 17 (32), 2101785. DOI 10.1002/smll.202101785. [DOI] [PubMed] [Google Scholar]
- 114.Park JY; Jang J; Kang H-W, Microelectronic Engineering 2018, 200, 1–11. DOI 10.1016/j.mee.2018.08.004. [DOI] [Google Scholar]
- 115.Ma X; Liu J; Zhu W; Tang M; Lawrence N; Yu C; Gou M; Chen S, Adv Drug Deliv Rev 2018, 132, 235–251. DOI 10.1016/j.addr.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yi H-G; Kim H; Kwon J; Choi Y-J; Jang J; Cho D-W, Signal Transduction and Targeted Therapy 2021, 6 (1), 177. DOI 10.1038/s41392-021-00566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Srinivasan B; Kolli AR; Esch MB; Abaci HE; Shuler ML; Hickman JJ, J Lab Autom 2015, 20 (2), 107–26. DOI 10.1177/2211068214561025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arik YB; van der Helm MW; Odijk M; Segerink LI; Passier R; van den Berg A; van der Meer AD, Biomicrofluidics 2018, 12 (4), 042218–042218. DOI 10.1063/1.5023041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Henry OYF; Villenave R; Cronce MJ; Leineweber WD; Benz MA; Ingber DE, Lab on a chip 2017, 17 (13), 2264–2271. DOI 10.1039/c7lc00155j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Srinivasan B; Kolli AR; Esch MB; Abaci HE; Shuler ML; Hickman JJ, Journal of Laboratory Automation 2015, 20 (2), 107–126. DOI 10.1177/2211068214561025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Butt AM; Jones HC; Abbott NJ, J Physiol 1990, 429, 47–62. DOI 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang YI; Abaci HE; Shuler ML, Biotechnol Bioeng 2017, 114 (1), 184–194. DOI 10.1002/bit.26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jeong S; Kim S; Buonocore J; Park J; Welsh CJ; Li J; Han A, IEEE Trans Biomed Eng 2018, 65 (2), 431–439. DOI 10.1109/tbme.2017.2773463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chin E; Goh E, Chapter 9 - Blood–brain barrier on a chip. In Methods in Cell Biology, Doh J; Fletcher D; Piel M, Eds. Academic Press: 2018; Vol. 146, pp 159–182. [DOI] [PubMed] [Google Scholar]
- 125.Zakharova M; Palma do Carmo MA; van der Helm MW; Le-The H; de Graaf MNS; Orlova V; van den Berg A; van der Meer AD; Broersen K; Segerink LI, Lab Chip 2020, 20 (17), 3132–3143. DOI 10.1039/d0lc00399a. [DOI] [PubMed] [Google Scholar]
- 126.Au - Jagadeesan S; Au - Workman MJ; Au - Herland A; Au - Svendsen CN; Au - Vatine GD, JoVE 2020, (157), e60925. DOI doi: 10.3791/60925. [DOI] [PubMed] [Google Scholar]
- 127.Frost TS; Jiang L; Lynch RM; Zohar Y, Micromachines 2019, 10 (8), 533. DOI 10.3390/mi10080533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shao J; Wu L; Wu J; Zheng Y; Zhao H; Lou X; Jin Q; Zhao J, Biomed Microdevices 2010, 12 (1), 81–8. DOI 10.1007/s10544-009-9362-0. [DOI] [PubMed] [Google Scholar]
- 129.Noorani B; Bhalerao A; Raut S; Nozohouri E; Bickel U; Cucullo L, Pharmaceutics 2021, 13 (9). DOI 10.3390/pharmaceutics13091474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nozohouri S; Noorani B; Al-Ahmad A; Abbruscato TJ, Methods Mol Biol 2021, 2367, 47–72. DOI 10.1007/7651_2020_311. [DOI] [PubMed] [Google Scholar]
- 131.Linville RM; DeStefano JG; Sklar MB; Xu Z; Farrell AM; Bogorad MI; Chu C; Walczak P; Cheng L; Mahairaki V; Whartenby KA; Calabresi PA; Searson PC, Biomaterials 2019, 190–191, 24–37. DOI 10.1016/j.biomaterials.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gordon J; Amini S; White MK, Methods in molecular biology (Clifton, N.J.) 2013, 1078, 1–8. DOI 10.1007/978-1-62703-640-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Galland F; Seady M; Taday J; Smaili SS; Gonçalves CA; Leite MC, Neurochem Int 2019, 131, 104538. DOI 10.1016/j.neuint.2019.104538. [DOI] [PubMed] [Google Scholar]
- 134.Eigenmann DE; Xue G; Kim KS; Moses AV; Hamburger M; Oufir M, Fluids Barriers CNS 2013, 10 (1), 33. DOI 10.1186/2045-8118-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thomsen MS; Humle N; Hede E; Moos T; Burkhart A; Thomsen LB, PLoS One 2021, 16 (3), e0236770. DOI 10.1371/journal.pone.0236770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Song HW; Foreman KL; Gastfriend BD; Kuo JS; Palecek SP; Shusta EV, Scientific Reports 2020, 10 (1), 12358. DOI 10.1038/s41598-020-69096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bryan TM; Reddel RR, Crit Rev Oncog 1994, 5 (4), 331–57. DOI 10.1615/critrevoncog.v5.i4.10. [DOI] [PubMed] [Google Scholar]
- 138.Novak R; Ingram M; Marquez S; Das D; Delahanty A; Herland A; Maoz BM; Jeanty SSF; Somayaji MR; Burt M; Calamari E; Chalkiadaki A; Cho A; Choe Y; Chou DB; Cronce M; Dauth S; Divic T; Fernandez-Alcon J; Ferrante T; Ferrier J; FitzGerald EA; Fleming R; Jalili-Firoozinezhad S; Grevesse T; Goss JA; Hamkins-Indik T; Henry O; Hinojosa C; Huffstater T; Jang KJ; Kujala V; Leng L; Mannix R; Milton Y; Nawroth J; Nestor BA; Ng CF; O’Connor B; Park TE; Sanchez H; Sliz J; Sontheimer-Phelps A; Swenor B; Thompson G 2nd; Touloumes GJ; Tranchemontagne Z; Wen N; Yadid M; Bahinski A; Hamilton GA; Levner D; Levy O; Przekwas A; Prantil-Baun R; Parker KK; Ingber DE, Nat Biomed Eng 2020, 4 (4), 407–420. DOI 10.1038/s41551-019-0497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Umehara K; Sun Y; Hiura S; Hamada K; Itoh M; Kitamura K; Oshima M; Iwama A; Saito K; Anzai N; Chiba K; Akita H; Furihata T, Molecular Neurobiology 2018, 55 (7), 5993–6006. DOI 10.1007/s12035-017-0815-9. [DOI] [PubMed] [Google Scholar]
- 140.Daniels BP; Cruz-Orengo L; Pasieka TJ; Couraud P-O; Romero IA; Weksler B; Cooper JA; Doering TL; Klein RS, J Neurosci Methods 2013, 212 (1), 173–179. DOI 10.1016/j.jneumeth.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Richter M; Piwocka O; Musielak M; Piotrowski I; Suchorska WM; Trzeciak T, Frontiers in Cell and Developmental Biology 2021, 9. DOI 10.3389/fcell.2021.711381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bang M; Gonzales EL; Shin CY; Kwon KJ, Biomol Ther (Seoul) 2021, 29 (2), 144–153. DOI 10.4062/biomolther.2020.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Giordano G; Costa LG, Methods Mol Biol 2011, 758, 13–27. DOI 10.1007/978-1-61779-170-3_2. [DOI] [PubMed] [Google Scholar]
- 144.Jamieson JJ; Searson PC; Gerecht S, J Biol Eng 2017, 11, 37–37. DOI 10.1186/s13036-017-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cao Y; Gong Y; Liu L; Zhou Y; Fang X; Zhang C; Li Y; Li J, J Appl Toxicol 2017, 37 (12), 1359–1369. DOI 10.1002/jat.3470. [DOI] [PubMed] [Google Scholar]
- 146.Yeon JH; Na D; Choi K; Ryu SW; Choi C; Park JK, Biomed Microdevices 2012, 14 (6), 1141–8. DOI 10.1007/s10544-012-9680-5. [DOI] [PubMed] [Google Scholar]
- 147.Vandenhaute E; Drolez A; Sevin E; Gosselet F; Mysiorek C; Dehouck M-P, Laboratory Investigation 2016, 96 (5), 588–598. DOI 10.1038/labinvest.2016.35. [DOI] [PubMed] [Google Scholar]
- 148.Uwamori H; Ono Y; Yamashita T; Arai K; Sudo R, Microvasc Res 2019, 122, 60–70. DOI 10.1016/j.mvr.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Workman MJ; Svendsen CN, Fluids and Barriers of the CNS 2020, 17 (1), 30. DOI 10.1186/s12987-020-00191-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shi Y; Inoue H; Wu JC; Yamanaka S, Nature reviews. Drug discovery 2017, 16 (2), 115–130. DOI 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qian T; Maguire SE; Canfield SG; Bao X; Olson WR; Shusta EV; Palecek SP, Science Advances 2017, 3 (11), e1701679. DOI doi: 10.1126/sciadv.1701679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Neal EH; Marinelli NA; Shi Y; McClatchey PM; Balotin KM; Gullett DR; Hagerla KA; Bowman AB; Ess KC; Wikswo JP; Lippmann ES, Stem cell reports 2019, 12 (6), 1380–1388. DOI 10.1016/j.stemcr.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lippmann ES; Azarin SM; Kay JE; Nessler RA; Wilson HK; Al-Ahmad A; Palecek SP; Shusta EV, Nature Biotechnology 2012 30:8 2012, 30 (8), 783–791. DOI 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lippmann ES; Al-Ahmad A; Azarin SM; Palecek SP; Shusta EV, Scientific Reports 2014 4:1 2014, 4 (1), 1–10. DOI 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hollmann EK; Bailey AK; Potharazu AV; Neely MD; Bowman AB; Lippmann ES, Fluids and Barriers of the CNS 2017, 14 (1), 1–13. DOI 10.1186/S12987-017-0059-0/FIGURES/6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jones VC; Atkinson-Dell R; Verkhratsky A; Mohamet L, Cell Death & Disease 2017, 8 (3), e2696–e2696. DOI 10.1038/cddis.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stebbins MJ; Gastfriend BD; Canfield SG; Lee MS; Richards D; Faubion MG; Li WJ; Daneman R; Palecek SP; Shusta EV, Sci Adv 2019, 5 (3), eaau7375. DOI 10.1126/sciadv.aau7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mahajani S; Raina A; Fokken C; Kügler S; Bähr M, Cell Death & Disease 2019, 10 (12), 898. DOI 10.1038/s41419-019-2133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tian R; Gachechiladze MA; Ludwig CH; Laurie MT; Hong JY; Nathaniel D; Prabhu AV; Fernandopulle MS; Patel R; Abshari M; Ward ME; Kampmann M, Neuron 2019, 104 (2), 239–255.e12. DOI 10.1016/j.neuron.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xu R; Li X; Boreland AJ; Posyton A; Kwan K; Hart RP; Jiang P, Nature Communications 2020, 11 (1), 1577. DOI 10.1038/s41467-020-15411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Doss MX; Sachinidis A, Cells 2019, 8 (5), 403. DOI 10.3390/cells8050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Faley SL; Neal EH; Wang JX; Bosworth AM; Weber CM; Balotin KM; Lippmann ES; Bellan LM, Stem Cell Reports 2019, 12 (3), 474–487. DOI 10.1016/j.stemcr.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Di L; Kerns EH; Fan K; McConnell OJ; Carter GT, Eur J Med Chem 2003, 38 (3), 223–32. DOI 10.1016/s0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 164.Bowman PD; Ennis SR; Rarey KE; Lorris Betz A; Goldstein GW, Annals of Neurology 1983, 14 (4), 396–402. DOI 10.1002/ana.410140403. [DOI] [PubMed] [Google Scholar]
- 165.Obermeier B; Daneman R; Ransohoff RM, Nat Med 2013, 19 (12), 1584–96. DOI 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jamieson JJ; Searson PC; Gerecht S, Journal of Biological Engineering 2017, 11 (1), 37. DOI 10.1186/s13036-017-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Katt ME; Linville RM; Mayo LN; Xu ZS; Searson PC, Fluids and Barriers of the CNS 2018, 15 (1), 7. DOI 10.1186/s12987-018-0092-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cabezas R; Avila M; Gonzalez J; El-Bachá RS; Báez E; García-Segura LM; Jurado Coronel JC; Capani F; Cardona-Gomez GP; Barreto GE, Frontiers in cellular neuroscience 2014, 8, 211–211. DOI 10.3389/fncel.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Brown TD; Nowak M; Bayles AV; Prabhakarpandian B; Karande P; Lahann J; Helgeson ME; Mitragotri S, Bioengineering & Translational Medicine 2019, 4 (2), e10126. DOI 10.1002/btm2.10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shin Y; Choi SH; Kim E; Bylykbashi E; Kim JA; Chung S; Kim DY; Kamm RD; Tanzi RE, Advanced Science 2019, 6 (20), 1900962. DOI 10.1002/advs.201900962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hajal C; Offeddu GS; Shin Y; Zhang S; Morozova O; Hickman D; Knutson CG; Kamm RD, Nature Protocols 2022, 17 (1), 95–128. DOI 10.1038/s41596-021-00635-w. [DOI] [PubMed] [Google Scholar]
- 172.Deosarkar SP; Prabhakarpandian B; Wang B; Sheffield JB; Krynska B; Kiani MF, PloS one 2015, 10 (11), e0142725–e0142725. DOI 10.1371/journal.pone.0142725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim JA; Kim HN; Im S-K; Chung S; Kang JY; Choi N, Biomicrofluidics 2015, 9 (2), 024115–024115. DOI 10.1063/1.4917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Herland A; van der Meer AD; FitzGerald EA; Park T-E; Sleeboom JJF; Ingber DE, PLOS ONE 2016, 11 (3), e0150360. DOI 10.1371/journal.pone.0150360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bischel LL; Young EWK; Mader BR; Beebe DJ, Biomaterials 2013, 34 (5), 1471–1477. DOI 10.1016/J.BIOMATERIALS.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Campisi M; Shin Y; Osaki T; Hajal C; Chiono V; Kamm RD, Biomaterials 2018, 180, 117–129. DOI 10.1016/j.biomaterials.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lyu Z; Park J; Kim K-M; Jin H-J; Wu H; Rajadas J; Kim D-H; Steinberg GK; Lee W, Nature Biomedical Engineering 2021, 5 (8), 847–863. DOI 10.1038/s41551-021-00744-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Silvani G; Basirun C; Wu H; Mehner C; Poole K; Bradbury P; Chou J, Advanced Therapeutics 2021, 4 (11), 2100106. DOI 10.1002/adtp.202100106. [DOI] [Google Scholar]
- 179.Jensen C; Teng Y, Frontiers in Molecular Biosciences 2020, 7 (33). DOI 10.3389/fmolb.2020.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Langhans SA, Front Pharmacol 2018, 9, 6–6. DOI 10.3389/fphar.2018.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Johnson KM; Milner R; Crocker SJ, Neurosci Lett 2015, 600, 104–109. DOI 10.1016/j.neulet.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Balasubramanian S; Packard JA; Leach JB; Powell EM, Tissue Eng Part A 2016, 22 (11–12), 885–898. DOI 10.1089/ten.TEA.2016.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Blanchard JW; Bula M; Davila-Velderrain J; Akay LA; Zhu L; Frank A; Victor MB; Bonner JM; Mathys H; Lin Y-T; Ko T; Bennett DA; Cam HP; Kellis M; Tsai L-H, Nature medicine 2020, 26 (6), 952–963. DOI 10.1038/s41591-020-0886-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bergmann S; Lawler SE; Qu Y; Fadzen CM; Wolfe JM; Regan MS; Pentelute BL; Agar NYR; Cho C-F, Nature Protocols 2018, 13 (12), 2827–2843. DOI 10.1038/s41596-018-0066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bose R; Banerjee S; Dunbar GL, Frontiers in Cell and Developmental Biology 2021, 9 (786). DOI 10.3389/fcell.2021.640212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ebert AD; Shelley BC; Hurley AM; Onorati M; Castiglioni V; Patitucci TN; Svendsen SP; Mattis VB; McGivern JV; Schwab AJ; Sareen D; Kim HW; Cattaneo E; Svendsen CN, Stem Cell Res 2013, 10 (3), 417–427. DOI 10.1016/j.scr.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Potjewyd G; Kellett KAB; Hooper NM, Neuronal Signal 2021, 5 (4), NS20210027–NS20210027. DOI 10.1042/NS20210027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhu Y; Mandal K; Hernandez AL; Kawakita S; Huang W; Bandaru P; Ahadian S; Kim H-J; Jucaud V; Dokmeci MR; Khademhosseini A, Current Opinion in Biomedical Engineering 2021, 19, 100309. DOI 10.1016/j.cobme.2021.100309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fuchs S; Johansson S; Tjell AØ; Werr G; Mayr T; Tenje M, ACS Biomaterials Science & Engineering 2021, 7 (7), 2926–2948. DOI 10.1021/acsbiomaterials.0c01110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wikswo JP; F. E. Block I; Cliffel DE; Goodwin CR; Marasco CC; Markov DA; McLean DL; McLean JA; McKenzie JR; Reiserer RS; Samson PC; Schaffer DK; Seale KT; Sherrod SD, IEEE Transactions on Biomedical Engineering 2013, 60 (3), 682–690. DOI 10.1109/TBME.2013.2244891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang YS; Aleman J; Shin SR; Kilic T; Kim D; Mousavi Shaegh SA; Massa S; Riahi R; Chae S; Hu N; Avci H; Zhang W; Silvestri A; Sanati Nezhad A; Manbohi A; De Ferrari F; Polini A; Calzone G; Shaikh N; Alerasool P; Budina E; Kang J; Bhise N; Ribas J; Pourmand A; Skardal A; Shupe T; Bishop CE; Dokmeci MR; Atala A; Khademhosseini A, Proceedings of the National Academy of Sciences 2017, 114 (12), E2293–E2302. DOI 10.1073/pnas.1612906114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ferrari E; Palma C; Vesentini S; Occhetta P; Rasponi M, Biosensors (Basel) 2020, 10 (9). DOI 10.3390/bios10090110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Douville NJ; Tung Y-C; Li R; Wang JD; El-Sayed MEH; Takayama S, Analytical Chemistry 2010, 82 (6), 2505–2511. DOI 10.1021/ac9029345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Marsh GA; McAuley AJ; Brown S; Pharo EA; Crameri S; Au GG; Baker ML; Barr J; Bergfeld J; Bruce M; Burkett K; Durr PA; Holmes C; Izzard L; Layton R; Lowther S; Neave MJ; Poole T; Riddell S; Rowe B; Soldani E; Stevens V; Suen W; Sundaramoorthy V; Tachedjian M; Todd S; Trinidad L; Williams SM; Druce JD; Drew TW; Vasan SS, Transboundary and Emerging Diseases n/a (n/a). DOI 10.1111/tbed.13978. [DOI] [PubMed] [Google Scholar]
- 195.Odijk M; van der Meer AD; Levner D; Kim HJ; van der Helm MW; Segerink LI; Frimat JP; Hamilton GA; Ingber DE; van den Berg A, Lab Chip 2015, 15 (3), 745–52. DOI 10.1039/c4lc01219d. [DOI] [PubMed] [Google Scholar]
- 196.Peters MF; Landry T; Pin C; Maratea K; Dick C; Wagoner MP; Choy AL; Barthlow H; Snow D; Stevens Z; Armento A; Scott CW; Ayehunie S, Toxicological Sciences 2019, 168 (1), 3–17. DOI 10.1093/toxsci/kfy268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Maherally Z; Fillmore HL; Tan SL; Tan SF; Jassam SA; Quack FI; Hatherell KE; Pilkington GJ, The FASEB Journal 2018, 32 (1), 168–182. DOI 10.1096/fj.201700162R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Au - van der Helm MW; Au - Odijk M; Au - Frimat J-P; Au - van der Meer AD; Au - Eijkel JCT; Au - van den Berg A; Au - Segerink LI, JoVE 2017, (127), e56334. DOI doi: 10.3791/56334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tu K-H; Yu L-S; Sie Z-H; Hsu H-Y; Al-Jamal KT; Wang JT-W; Chiang Y-Y, Micromachines 2020, 12 (1). DOI 10.3390/mi12010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Soucy JR; Bindas AJ; Koppes AN; Koppes RA, iScience 2019, 21, 521–548. DOI 10.1016/j.isci.2019.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Maoz BM; Herland A; Henry OYF; Leineweber WD; Yadid M; Doyle J; Mannix R; Kujala VJ; FitzGerald EA; Parker KK; Ingber DE, Lab on a Chip 2017, 17 (13), 2294–2302. DOI 10.1039/c7lc00412e. [DOI] [PubMed] [Google Scholar]
- 202.Griep LM; Wolbers F; de Wagenaar B; ter Braak PM; Weksler BB; Romero IA; Couraud PO; Vermes I; van der Meer AD; van den Berg A, Biomed Microdevices 2013, 15 (1), 145–150. DOI 10.1007/s10544-012-9699-7. [DOI] [PubMed] [Google Scholar]
- 203.Bossink EGBM; Zakharova M; de Bruijn DS; Odijk M; Segerink LI, Lab on a Chip 2021, 21 (10), 2040–2049. DOI 10.1039/D0LC01289K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ricci F; Adornetto G; Palleschi G, Electrochimica Acta 2012, 84, 74–83. DOI 10.1016/j.electacta.2012.06.033. [DOI] [Google Scholar]
- 205.Vigh JP; Kincses A; Ozgür B; Walter FR; Santa-Maria AR; Valkai S; Vastag M; Neuhaus W; Brodin B; Dér A; Deli MA, Micromachines (Basel) 2021, 12 (6). DOI 10.3390/mi12060685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Engelhardt S; Patkar S; Ogunshola OO, Br J Pharmacol 2014, 171 (5), 1210–1230. DOI 10.1111/bph.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sticker D; Rothbauer M; Ehgartner J; Steininger C; Liske O; Liska R; Neuhaus W; Mayr T; Haraldsson T; Kutter JP; Ertl P, ACS Applied Materials & Interfaces 2019, 11 (10), 9730–9739. DOI 10.1021/acsami.8b19641. [DOI] [PubMed] [Google Scholar]
- 208.Shao X; Gao D; Chen Y; Jin F; Hu G; Jiang Y; Liu H, Analytica Chimica Acta 2016, 934, 186–193. DOI 10.1016/j.aca.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 209.Senel M; Dervisevic E; Alhassen S; Dervisevic M; Alachkar A; Cadarso VJ; Voelcker NH, Analytical Chemistry 2020, 92 (18), 12347–12355. DOI 10.1021/acs.analchem.0c02032. [DOI] [PubMed] [Google Scholar]
- 210.Verkhratsky A; Matteoli M; Parpura V; Mothet J-P; Zorec R, EMBO J 2016, 35 (3), 239–257. DOI 10.15252/embj.201592705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Brown LS; Foster CG; Courtney J-M; King NE; Howells DW; Sutherland BA, Frontiers in Cellular Neuroscience 2019, 13 (282). DOI 10.3389/fncel.2019.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lip GYH; Blann A, Cardiovascular Research 1997, 34 (2), 255–265. DOI 10.1016/s0008-6363(97)00039-4. [DOI] [PubMed] [Google Scholar]
- 213.Esparza TJ; Wildburger NC; Jiang H; Gangolli M; Cairns NJ; Bateman RJ; Brody DL, Scientific Reports 2016, 6 (1), 38187. DOI 10.1038/srep38187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rosenberg GA, J Cereb Blood Flow Metab 2012, 32 (7), 1139–1151. DOI 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Profaci CP; Munji RN; Pulido RS; Daneman R, Journal of Experimental Medicine 2020, 217 (4). DOI 10.1084/jem.20190062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Szepesi Z; Manouchehrian O; Bachiller S; Deierborg T, Frontiers in Cellular Neuroscience 2018, 12 (323). DOI 10.3389/fncel.2018.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Knopman DS; Amieva H; Petersen RC; Chételat G; Holtzman DM; Hyman BT; Nixon RA; Jones DT, Nature Reviews Disease Primers 2021, 7 (1), 33. DOI 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Long JM; Holtzman DM, Cell 2019, 179 (2), 312–339. DOI 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Thomsen MS; Routhe LJ; Moos T, J Cereb Blood Flow Metab 2017, 37 (10), 3300–3317. DOI 10.1177/0271678x17722436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kim YH; Choi SH; D’Avanzo C; Hebisch M; Sliwinski C; Bylykbashi E; Washicosky KJ; Klee JB; Brüstle O; Tanzi RE; Kim DY, Nature Protocols 2015, 10 (7), 985–1006. DOI 10.1038/nprot.2015.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Park J; Wetzel I; Marriott I; Dréau D; D’Avanzo C; Kim DY; Tanzi RE; Cho H, Nature Neuroscience 2018, 21 (7), 941–951. DOI 10.1038/s41593-018-0175-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Habib N; McCabe C; Medina S; Varshavsky M; Kitsberg D; Dvir-Szternfeld R; Green G; Dionne D; Nguyen L; Marshall JL; Chen F; Zhang F; Kaplan T; Regev A; Schwartz M, Nature Neuroscience 2020, 23 (6), 701–706. DOI 10.1038/s41593-020-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Monterey MD; Wei H; Wu X; Wu JQ, Frontiers in Neurology 2021, 12. DOI 10.3389/fneur.2021.619626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Nortley R; Korte N; Izquierdo P; Hirunpattarasilp C; Mishra A; Jaunmuktane Z; Kyrargyri V; Pfeiffer T; Khennouf L; Madry C; Gong H; Richard-Loendt A; Huang W; Saito T; Saido TC; Brandner S; Sethi H; Attwell D, Science 2019, 365 (6450), eaav9518. DOI doi: 10.1126/science.aav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Osaki T; Uzel SGM; Kamm RD, Sci Adv 2018, 4 (10), eaat5847. DOI 10.1126/sciadv.aat5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Terrell-Hall TB; Ammer AG; Griffith JI; Lockman PR, Fluids Barriers CNS 2017, 14 (1), 3. DOI 10.1186/s12987-017-0050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Xiao Y; Kim D; Dura B; Zhang K; Yan R; Li H; Han E; Ip J; Zou P; Liu J; Chen AT; Vortmeyer AO; Zhou J; Fan R, Adv Sci (Weinh) 2019, 6 (8), 1801531. DOI 10.1002/advs.201801531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Cui X; Ma C; Vasudevaraja V; Serrano J; Tong J; Peng Y; Delorenzo M; Shen G; Frenster J; Morales RT; Qian W; Tsirigos A; Chi AS; Jain R; Kurz SC; Sulman EP; Placantonakis DG; Snuderl M; Chen W, Elife 2020, 9. DOI 10.7554/eLife.52253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pediaditakis I; Kodella KR; Manatakis DV; Le CY; Hinojosa CD; Tien-Street W; Manolakos ES; Vekrellis K; Hamilton GA; Ewart L; Rubin LL; Karalis K, Nature Communications 2021, 12 (1), 5907. DOI 10.1038/s41467-021-26066-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Poewe W; Seppi K; Tanner CM; Halliday GM; Brundin P; Volkmann J; Schrag A-E; Lang AE, Nature Reviews Disease Primers 2017, 3 (1), 17013. DOI 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 231.Johnson ME; Stecher B; Labrie V; Brundin L; Brundin P, Trends in Neurosciences 2019, 42 (1), 4–13. DOI 10.1016/j.tins.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Osaki T; Uzel SGM; Kamm RD, Science Advances 2018, 4 (10), eaat5847. DOI doi: 10.1126/sciadv.aat5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Coatti GC; Cavaçana N; Zatz M, The Role of Pericytes in Amyotrophic Lateral Sclerosis. In Pericyte Biology in Disease, Birbrair A, Ed. Springer International Publishing: Cham, 2019; pp 137–146. [DOI] [PubMed] [Google Scholar]
- 234.Campbell BCV; De Silva DA; Macleod MR; Coutts SB; Schwamm LH; Davis SM; Donnan GA, Nature Reviews Disease Primers 2019, 5 (1), 70. DOI 10.1038/s41572-019-0118-8. [DOI] [PubMed] [Google Scholar]
- 235.Kuriakose D; Xiao Z, Int J Mol Sci 2020, 21 (20), 7609. DOI 10.3390/ijms21207609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Weller M; Wick W; Aldape K; Brada M; Berger M; Pfister SM; Nishikawa R; Rosenthal M; Wen PY; Stupp R; Reifenberger G, Nature Reviews Disease Primers 2015, 1 (1), 15017. DOI 10.1038/nrdp.2015.17. [DOI] [PubMed] [Google Scholar]
- 237.Arvanitis CD; Ferraro GB; Jain RK, Nat Rev Cancer 2020, 20 (1), 26–41. DOI 10.1038/s41568-019-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hajal C; Shin Y; Li L; Serrano JC; Jacks T; Kamm RD, Sci Adv 2021, 7 (26). DOI 10.1126/sciadv.abg8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Brandao M; Simon T; Critchley G; Giamas G, Glia 2019, 67 (5), 779–790. DOI 10.1002/glia.23520. [DOI] [PubMed] [Google Scholar]
- 240.Nieland L; Morsett LM; Broekman MLD; Breakefield XO; Abels ER, Trends in Neurosciences 2021, 44 (3), 215–226. DOI 10.1016/j.tins.2020.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Valdor R; García-Bernal D; Bueno C; Ródenas M; Moraleda JM; Macian F; Martínez S, Oncotarget 2017, 8 (40), 68614–68626. DOI 10.18632/oncotarget.19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sattiraju A; Mintz A, Adv Exp Med Biol 2019, 1147, 65–91. DOI 10.1007/978-3-030-16908-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Katt ME; Mayo LN; Ellis SE; Mahairaki V; Rothstein JD; Cheng L; Searson PC, Fluids and Barriers of the CNS 2019, 16 (1), 20. DOI 10.1186/s12987-019-0139-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bassil R; Shields K; Granger K; Zein I; Ng S; Chih B, Nature Communications 2021, 12 (1), 5220. DOI 10.1038/s41467-021-25344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Penney J; Ralvenius WT; Tsai L-H, Molecular Psychiatry 2020, 25 (1), 148–167. DOI 10.1038/s41380-019-0468-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Csobonyeiova M; Polak S; Danisovic L, Int J Mol Sci 2020, 21 (6), 2239. DOI 10.3390/ijms21062239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Motallebnejad P; Thomas A; Swisher SL; Azarin SM, Biomicrofluidics 2019, 13 (6), 064119–064119. DOI 10.1063/1.5123476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Padel T; Roth M; Gaceb A; Li JY; Björkqvist M; Paul G, Exp Neurol 2018, 305, 139–150. DOI 10.1016/j.expneurol.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 249.Azizgolshani H; Coppeta JR; Vedula EM; Marr EE; Cain BP; Luu RJ; Lech MP; Kann SH; Mulhern TJ; Tandon V; Tan K; Haroutunian NJ; Keegan P; Rogers M; Gard AL; Baldwin KB; de Souza JC; Hoefler BC; Bale SS; Kratchman LB; Zorn A; Patterson A; Kim ES; Petrie TA; Wiellette EL; Williams C; Isenberg BC; Charest JL, Lab on a Chip 2021, 21 (8), 1454–1474. DOI 10.1039/D1LC00067E. [DOI] [PubMed] [Google Scholar]
- 250.Novak R; Ingram M; Marquez S; Das D; Delahanty A; Herland A; Maoz BM; Jeanty SSF; Somayaji MR; Burt M; Calamari E; Chalkiadaki A; Cho A; Choe Y; Chou DB; Cronce M; Dauth S; Divic T; Fernandez-Alcon J; Ferrante T; Ferrier J; FitzGerald EA; Fleming R; Jalili-Firoozinezhad S; Grevesse T; Goss JA; Hamkins-Indik T; Henry O; Hinojosa C; Huffstater T; Jang K-J; Kujala V; Leng L; Mannix R; Milton Y; Nawroth J; Nestor BA; Ng CF; O’Connor B; Park T-E; Sanchez H; Sliz J; Sontheimer-Phelps A; Swenor B; Thompson G; Touloumes GJ; Tranchemontagne Z; Wen N; Yadid M; Bahinski A; Hamilton GA; Levner D; Levy O; Przekwas A; Prantil-Baun R; Parker KK; Ingber DE, Nature Biomedical Engineering 2020, 4 (4), 407–420. DOI 10.1038/s41551-019-0497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Pérez-Aliacar M; Doweidar MH; Doblaré M; Ayensa-Jiménez J, Computers in Biology and Medicine 2021, 135, 104547. DOI 10.1016/j.compbiomed.2021.104547. [DOI] [PubMed] [Google Scholar]
- 252.Jones DT, Nature Reviews Molecular Cell Biology 2019, 20 (11), 659–660. DOI 10.1038/s41580-019-0176-5. [DOI] [PubMed] [Google Scholar]
- 253.Esteva A; Robicquet A; Ramsundar B; Kuleshov V; DePristo M; Chou K; Cui C; Corrado G; Thrun S; Dean J, Nature Medicine 2019, 25 (1), 24–29. DOI 10.1038/s41591-018-0316-z. [DOI] [PubMed] [Google Scholar]
- 254.Rajkomar A; Dean J; Kohane I, N Engl J Med 2019, 380 (14), 1347–1358. DOI 10.1056/NEJMra1814259. [DOI] [PubMed] [Google Scholar]
- 255.Min S; Lee B; Yoon S, Briefings in Bioinformatics 2016, 18 (5), 851–869. DOI 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 256.Chlebus G; Meine H; Thoduka S; Abolmaali N; van Ginneken B; Hahn HK; Schenk A, PloS one 2019, 14 (5), e0217228–e0217228. DOI 10.1371/journal.pone.0217228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cao C; Liu F; Tan H; Song D; Shu W; Li W; Zhou Y; Bo X; Xie Z, Genomics Proteomics Bioinformatics 2018, 16 (1), 17–32. DOI 10.1016/j.gpb.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Ramspek CL; Jager KJ; Dekker FW; Zoccali C; van Diepen M, Clinical Kidney Journal 2020, 14 (1), 49–58. DOI 10.1093/ckj/sfaa188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Murdoch WJ; Singh C; Kumbier K; Abbasi-Asl R; Yu B, Proc Natl Acad Sci U S A 2019, 116 (44), 22071–22080. DOI 10.1073/pnas.1900654116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Rivenson Y; Wang H; Wei Z; de Haan K; Zhang Y; Wu Y; Günaydin H; Zuckerman JE; Chong T; Sisk AE; Westbrook LM; Wallace WD; Ozcan A, Nature Biomedical Engineering 2019, 3 (6), 466–477. DOI 10.1038/s41551-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 261.Trietsch SJ; Naumovska E; Kurek D; Setyawati MC; Vormann MK; Wilschut KJ; Lanz HL; Nicolas A; Ng CP; Joore J; Kustermann S; Roth A; Hankemeier T; Moisan A; Vulto P, Nature Communications 2017, 8 (1), 262. DOI 10.1038/s41467-017-00259-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Picollet-D’hahan N; Zuchowska A; Lemeunier I; Le Gac S, Trends in Biotechnology 2021, 39 (8), 788–810. DOI 10.1016/j.tibtech.2020.11.014. [DOI] [PubMed] [Google Scholar]
- 263.Lee J; Mehrotra S; Zare-Eelanjegh E; Rodrigues RO; Akbarinejad A; Ge D; Amato L; Kiaee K; Fang Y; Rosenkranz A; Keung W; Mandal BB; Li RA; Zhang T; Lee H; Dokmeci MR; Zhang YS; Khademhosseini A; Shin SR, Small 2021, 17 (15), 2004258. DOI 10.1002/smll.202004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kim M-H; Kim D; Sung JH, Journal of Industrial and Engineering Chemistry 2021, 101, 126–134. DOI 10.1016/j.jiec.2021.06.021. [DOI] [Google Scholar]
- 265.Fabre K; Berridge B; Proctor WR; Ralston S; Will Y; Baran SW; Yoder G; Van Vleet TR, Lab on a Chip 2020, 20 (6), 1049–1057. DOI 10.1039/C9LC01168D. [DOI] [PubMed] [Google Scholar]
- 266.Ma C; Peng Y; Li H; Chen W, Trends Pharmacol Sci 2021, 42 (2), 119–133. DOI 10.1016/j.tips.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]