

Abstract
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of leukocytes that are important for tumorigenesis and tumor immunotherapy. They comprise up to 10% of leukocytes in the blood of tumor patients and their depletion may be required for successful tumor immunotherapy. However, the identity of MDSCs remains obscure, primarily due to their heterogeneity and lack of a known lineage-specific transcription factor specifying their differentiation. Using single-cell transcriptomics and gene knockout approaches, we now describe a subset of murine and human myeloid suppressor cells, named rel-dependent monocytes (rMos), which are programmed by the transcription factor c-Rel of the NF-κB family. Unlike MDSCs described previously, the c-Rel-dependent monocytes expressed a high amount of the proinflammatory cytokine IL-1β together with a low level of suppressive molecule arginase 1. Both in vitro and in tumor-bearing mice, these c-Rel+IL-1βhiArg1− monocytes promoted tumor growth by potently suppressing T cell function and showed a strong migratory phenotype, all of which were impaired by c-Rel deficiency or inhibition. Mechanistic studies revealed that c-Rel controlled the expression of monocyte signature genes through a unique transcriptional complex called the c-Rel enhanceosome, and IL-1β:CCL2 crosstalk between tumor cells and the rel-dependent monocytes maintained the suppressive tumor microenvironment. Thus, c-Rel specifies the development of a suppressive monocyte population and could be selectively targeted for treating cancer.
Keywords: myelopoiesis, c-Rel-dependent monocytes, tumor microenvironment, Rel/NF-κB, tumor immunotherapy
INTRODUCTION
Immunotherapy that targets lymphocyte checkpoints to restore anti-cancer immunity has revolutionized cancer therapy1. For most cancer patients, however, little clinical efficacy has been observed2, 3, 4. A significant roadblock to successful immunotherapy is the profoundly immunosuppressive tumor microenvironment (TME)3, 5. High levels of immunosuppressive factors secreted by immunosuppressive cells that are induced and recruited by tumors directly impede anti-tumor immunity leading to poor clinical outcomes. We and several others have described myeloid-derived suppressor cell (MDSC) populations within the TME as preeminent mediators of immunosuppression, which potently inhibit anti-tumor immunity and promote tumorigenesis3, 5, 6, 7.
MDSCs are a heterogeneous population of immature leukocytes that arise in the bone marrow as a result of sustained pathological inflammation8. Myeloid differentiation is a tightly regulated process that controls and limits inflammatory responses. Under normal physiological conditions, myeloid progenitors differentiate into effector myeloid cells including monocytes, macrophages, dendritic cells, and granulocytes9, 10, 11. Under pathological conditions such as cancer, sustained inflammation induced by tumor-secreted molecules, subverts normal myelopoietic processes, allowing the accumulation of aberrant myeloid cell types including granulocytic and monocytic MDSCs (G-MDSCs and M-MDSCs, respectively). Although present in low numbers in healthy individuals, MDSCs accumulate in patients with a variety of solid and hematologic cancers (comprising up to 10% of blood and splenic leukocytes)12, 13, 14. Expansion of MDSC populations within circulating and tumor-infiltrating leukocytes is observed in patients who fail to respond to lymphoid checkpoint blockade5. MDSCs represent a heterogeneous population of immature myeloid cells capable of modulating immune responses15, 16, 17. Few specific markers have been described that identify individual MDSC populations, and the underlying mechanisms of how MDSCs suppress T cells remain to be explored. As such, dissecting the nature of MDSCs is paramount for identifying new clinical targets and devising effective combinatorial therapeutic approaches.
Emerging work suggests that the TME reprograms transcription factor (TF) activity within myeloid precursors to generate and expand suppressive myeloid cells15, 18. c-Rel is a member of the Rel/nuclear factor-κB (NF-κB) family that is implicated in human lymphoid malignancies19, 20, 21. We recently showed that c-Rel is a myeloid checkpoint of anti-tumor immunity that specifies the differentiation of myeloid precursors into pro-tumoral suppressor cells6. The specific MDSC subpopulations induced and regulated by c-Rel, however, are not known.
Here using single-cell transcriptomic techniques, we delineate the landscape of intra-tumoral leukocytes at single-cell resolution, and in doing so, we identify a subset of c-Rel dependent monocytes termed “rel-dependent monocytes” (rMos). These rMos are likely induced in the bone marrow by tumor-secreted cytokines and traffic to tumor sites via an IL-1β:Ccl2 axis. In culture, rMos markedly suppress T-cell proliferation and function with greater potency than the Arg1-expressing myeloid suppressor cells. Overall, these results describe a new subset of MDSCs, regulated by c-Rel, which can be targeted to abrogate immunosuppression and restore anti-tumor immunity.
RESULTS
Identification of a c-Rel-dependent intra-tumoral MDSC subset at single-cell resolution.
We previously showed that c-Rel deficiency significantly reduced the frequency of MDSCs, and markedly inhibited tumor growth in mice6. To understand the effect of c-Rel in MDSC development in the TME, we injected wild-type (WT) (LysM-cre × Rel+/+) and myeloid conditional Rel knockout mice (LysM-cre × RelloxP/loxP) with B16F10 melanoma tumor cells. At 2 weeks post tumor cells injection, viable CD45+ cells from similarly-sized tumors were collected for single-cell RNA sequencing (Fig. 1a). The cell clusters were characterized based on the expression of specific markers (e.g., Mc1r for cancer cells) (Supplementary Fig. 1a) and by using the SingleR package in R (Supplementary Fig. 1a,b). The analysis of the myeloid population (CD45.2+ CD11b+) identified a F4/80+ Arg1hi macrophage population and a CD11b+CCR2− monocyte population. Importantly, we also identified a CD11b+CCR2+IL-1βhiArg1− myeloid population that expressed the highest amounts of c-Rel and whose percentage dropped by 2-fold in LysM-cre × RelloxP/loxP mice, which we refer to as “rel-dependent monocytes (rMos)” (Fig. 1b–d). The rMo subset expressed specific combinations of markers (Fig. 1d and Supplementary Fig. 2a) and when compared to tumor-associated macrophages (TAMs), which are a key component of the immunosuppressive TME, or the CCR2− monocytes, rMos preferentially expressed genes that are highly enriched for immune and inflammatory responses, chemokines, and cytokine signaling pathways (Supplementary Fig. 2b,c). This suggests that rMos are a unique activated myeloid subset that may modulate innate immunity in cancer.
Fig. 1. Identification of c-Rel-dependent monocytes by single-cell RNA sequencing.
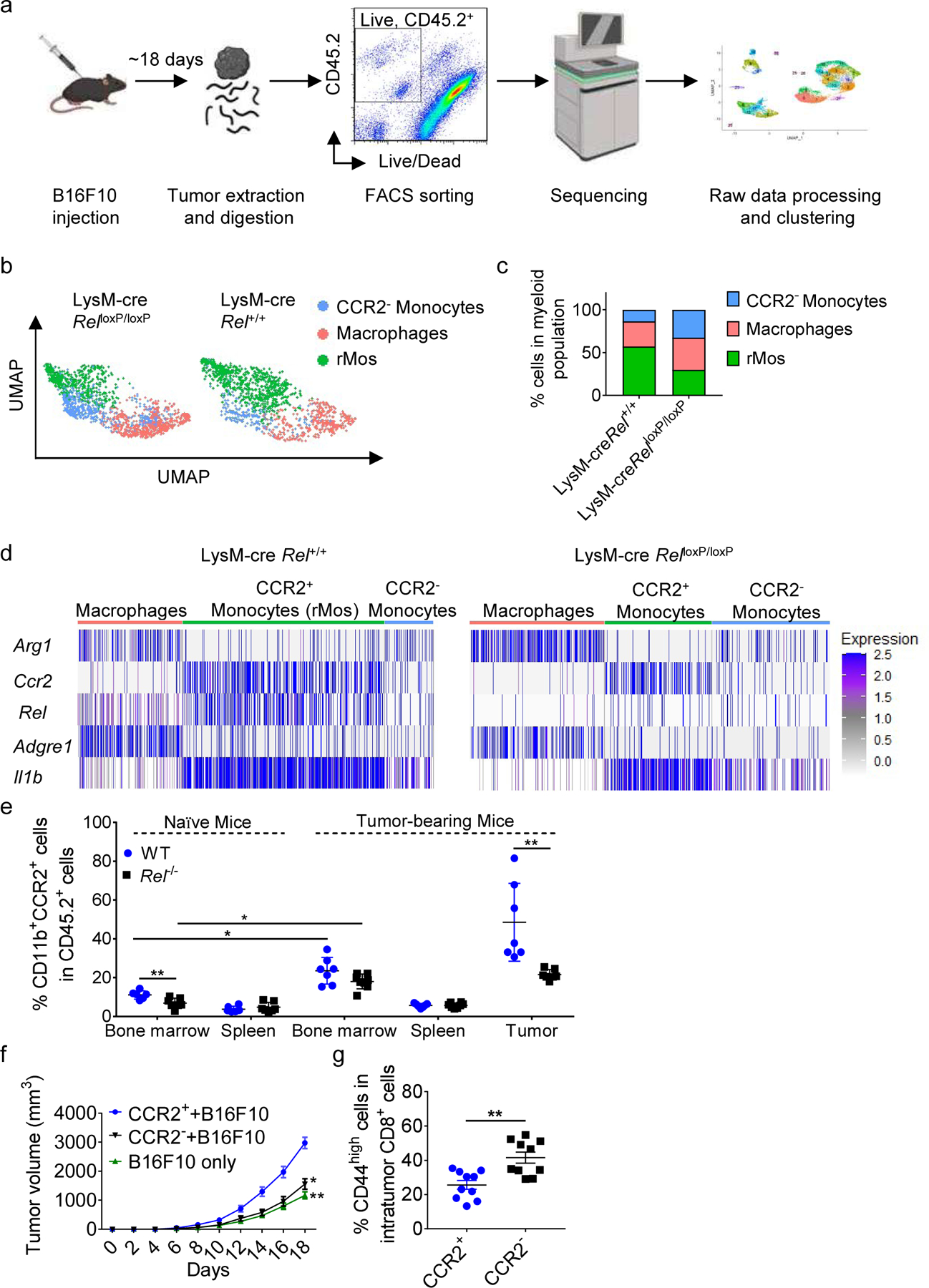
a, Workflow for scRNA-seq of viable CD45.2+ cells isolated by FACS from LysM-cre × Rel+/+ and LysM-cre × RelloxP/loxP mice harboring similarly sized B16F10 tumors (n=5 mice/group).
b, UMAP plots of 2830 intratumor myeloid cells that were selected for subsequent analyses. Each dot of the UMAP plot represents a single cell. Cell clusters are color-coded by associated cell types. c, Percentages of myeloid cell types inferred by Panel b.
d, Heatmap showing indicated gene expression across myeloid clusters.
e, Percentages of CD11b+CCR2+ cells in the indicated tissues of naïve and B16F10 tumor-bearing WT and Rel−/− mice, as determined by flow cytometry; for naïve mice: WT, n=6 mice, Rel−/−, n=7 mice; for tumor-bearing mice: WT, n=7 mice, Rel−/−, n=8 mice.
f, Tumor growth in WT mice injected s.c. with B16F10 tumor cells plus CCR2+(rMo)/CCR2− monocytes (n = 10 mice/group).
g, Percentages of CD44high cells in intratumor CD8+ cells in f, as determined by flow cytometry. Statistical significance was determined by a two-tailed unpaired t-test. Data are presented as means ± s.e.m. (e-f), and pooled from two independent experiments (e) (*, P<0.05; **, P<0.01).
To further validate these findings, we profiled and isolated CD11b+CCR2+ cells from naïve and tumor-bearing mice. Our data show that rMos mostly accumulate in the tumor microenvironment of tumor-bearing mice, with a significantly higher percentage in WT mice when compared with Rel−/− mice that are wholly deficient in c-Rel (Fig. 1e and Supplementary Fig. 2d,e). In addition, the presence of CD11b+CCR2+ cells in naïve mice was still significantly higher in WT compared to c-Rel-deficient mice (Fig. 1e). Our findings were also confirmed in a lung cancer model (Supplementary Fig. 2f,g). These data indicate that the generation and development of rMos are c-Rel dependent.
Monocytes similar to rMos could also be generated from bone marrow cultures in vitro in the presence of tumor-related cytokines such as GM-CSF and IL-6. Thus, sorted CCR2+ (rMo) and CCR2− monocytes were s.c. co-injected with B16F10 cells into WT recipient mice. Mice injected with CCR2− monocytes and B16F10 cells had smaller tumors compared with the CCR2+ group (Fig. 1f and Supplementary Fig. 2h). Consistent with this finding, the percentages of activated CD8+ effector T cells in the tumor were significantly increased in the group that was injected with CCR2− monocytes, while the percentages of total CD8+ T cells in the tumor were unchanged (Fig. 1g and Supplementary Fig. 2i).
To test rMos functions in vitro, we isolated them from either tumor or bone marrow cultures and added them to T cell cultures. Compared to tumor-associated macrophages (TAMs) and CCR2− monocytes, rMos displayed stronger immunosuppressive activities in the T cell proliferation assay, which was markedly weaker in the absence of c-Rel (Fig. 2a,b and Supplementary Fig. 3a–d). The level of reactive oxygen species (ROS) and IFNγ were consistent with this observation (Supplementary Fig. 3e,f). Interestingly, Rel deficiency also decreased the suppressive ability of TAMs and CCR2− monocytes (Fig. 2a,b). These results held true for both CD4+ and CD8+ T cells (Fig. 2a,b and Supplementary Fig. 3c,d).
Fig. 2. rMos are potent suppressors of T cells.
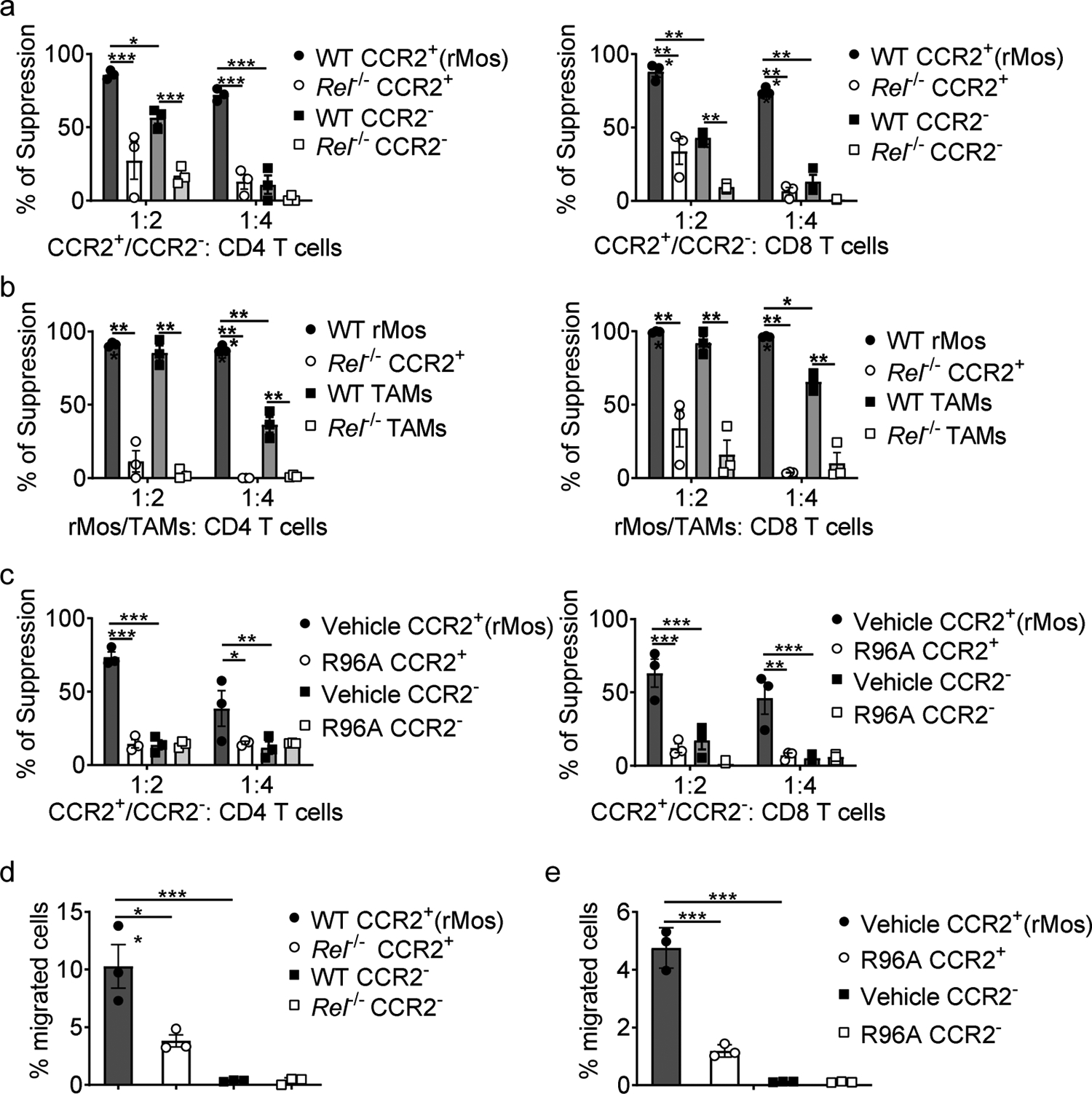
a-c, The degree of suppression, at the indicated effector:T cell ratio, of CD8 (left) and CD4 (right) T cell proliferation by purified WT or Rel−/− CD11b+CCR2+ cells (rMos) and CD11b+CCR2− cells from i) cytokine-treated bone marrow cultures of naïve mice (a, n=3 /group); ii) mice that had similarly sized B16F10 tumors (b, n=3 /group); or iii) cytokine-treated bone marrow cultures of naïve mice, with or without R96A (c, n=3 /group), (*, P<0.05; **, P<0.01; ***, P<0.001).
d, e, Transmigration of CD11b+CCR2+ and CD11b+CCR2− cells, isolated as described in a (d) and c (e), (*, P<0.05; **, P<0.01).
Statistical significance was determined by two-way ANOVA with Tukey post-hoc test (a-c) and one-way ANOVA with Tukey post-hoc test (d,e). For all panels, technical replicates of pooled samples (5 mice per group) were tested; data are presented as means ± s.e.m., and experiments were repeated 2 times independently with similar results; data from one representative experiment are shown.
To further validate the c-Rel-driven phenotype described above, we tested the effect of R96A, a small molecule c-Rel inhibitor compound6, 22. Consistent with the results from the Rel KO system, R96A treatment effectively blocked the function of bone marrow-derived rMos (Fig. 2c and Supplementary Fig. 3g).
We also tested the migratory ability of rMos. Compared to CCR2− cells, an increased percentage of rMos migrated, which was significantly decreased by c-Rel deficiency or pharmaceutical inhibition of c-Rel (Fig. 2d,e). Taken together, our data suggest that rMos are a unique immune population that potently suppresses T cells and actively migrates through the CCR2:CCL2 axis; both of these activities are significantly reduced when c-Rel is absent or inhibited.
c-Rel enhanceosome programs the gene signature of rMos.
The bone marrow is the predominant site for aberrant myelopoiesis during tumor development18. We observed an accumulation of rMos in the bone marrow of tumor-bearing mice when compared to naïve mice (Fig. 1e). Similarly, bone marrow cells cultured with cytokines (GM-CSF and IL-6) expressed rMo signature genes such as Ccr2 and Il1b, which were significantly reduced in the Rel−/− group (2.5-fold and 6-fold reductions, respectively) by Day 5 (Fig. 3a,b). However, by Day 7, Rel−/− cells showed increased expression of Il1b as compared to the WT group, suggesting that non-rMos cells prevailed at later time points of the Rel−/− culture, which is in line with our previous report6.
Fig. 3. Regulation of rMo signature genes by the c-Rel enhanceosome.
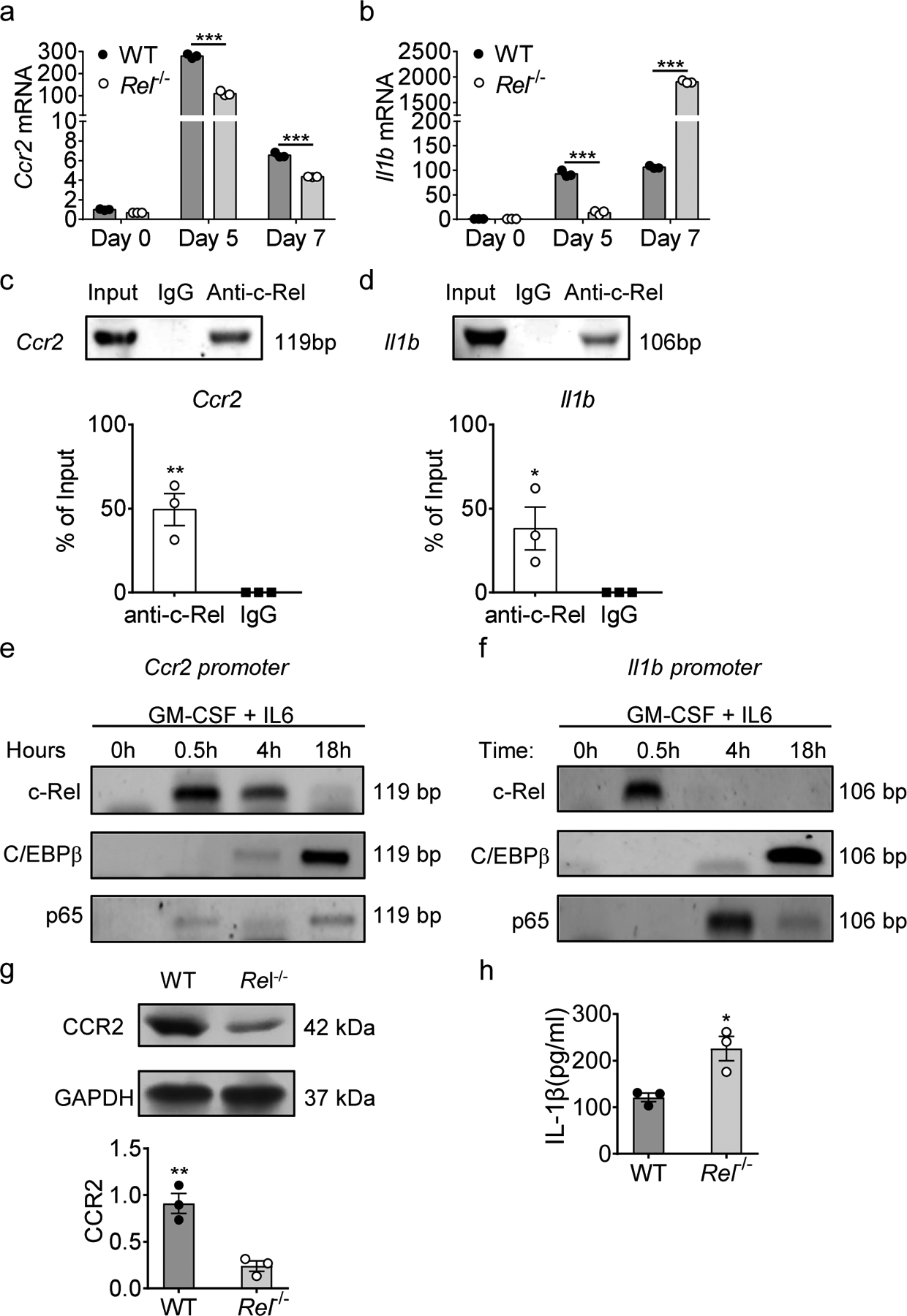
a, b, Relative mRNA levels of Ccr2 (a) and Il1b (b) in bone marrow-derived MDSCs of WT and Rel−/− mice, determined by real-time RT-PCR. (n=3 /group; ***, P<0.001).
c, d, c-Rel binding to the Ccr2 (c) and Il1b (d) gene promoter regions in bone marrow-derived MDSCs as determined by chromatin immunoprecipitation assays (ChIP). Control IgG and input DNA are shown as controls. The bar graph shows the relative c-Rel binding to the Ccr2 gene promoter region compared with input. (*, P<0.05; **, P<0.01)
e, f, The binding kinetics of c-Rel, C/EBPβ, and p65 to the Ccr2 (e) and Il1b (f) gene promoters in cultured bone marrow cells treated with GM-CSF and IL-6 for up to 18 hours as determined by ChIP.
g, CCR2 protein expression in splenic Gr-1+ cells isolated from Rel−/− and WT B16F10 tumor-bearing mice as determined by Western blot (n=3/group) (g). The bar graph shows the relative quantities of the CCR2 protein in the corresponding lane shown left (**, P<0.01;).
h, IL-1β protein in the media of bone marrow-derived MDSCs of WT and Rel−/− mice as determined by ELISA. Statistical significance was determined by two-tailed unpaired t-test (a-d, g,h). Data are presented as means ± s.e.m. (a-d, g,h), and experiments were repeated 2 times independently with similar results; data of one representative experiment are shown.
Next, we investigated how CCR2 and IL-1β gene expression was regulated by c-Rel. Using Chromatin Immunoprecipitation (ChIP), we found that c-Rel binds to the regulatory sites of both Ccr2 and Il1b genes (Fig. 3c,d). We previously described an enhanceosome consisting of c-Rel, C/EBPß, p65, and pSTAT3 in MDSCs6. To investigate whether CCR2 and IL-1β expressions were driven by this c-Rel enhanceosome, we performed a kinetic ChIP analysis for these transcription factors. We found that c-Rel binding to the Ccr2 and Il1b loci was followed by the recruitment of the transcription factors C/EBPβ and p65 after cytokine stimulation (Fig. 3e,f).
To validate our findings that both Ccr2 and Il1b are regulated by the c-Rel enhanceosome factors, we evaluated publicly available ChIP-seq data from myeloid bone-marrow cells (CistromeDB, Accession numbers: p65: 94578, Cebpb: 71767, c-Rel: 39585). Peak visualization in the UCSC browser suggests enrichment of all the enhanceosome factors mentioned above at the enhancer sites within loci of both Ccr2 and Il1b in bone marrow cells ((Supplementary Fig. 4a,b). Corroborating our findings, we observed a decrease in CCR2 protein expression in tumor-infiltrating myeloid Gr1+ cells that were isolated from Rel−/− mice when compared with those of WT mice (Fig. 3g). Moreover, we found significantly increased amounts of IL-1β protein in the culture media of Rel−/− bone marrow-derived MDSCs (BM-MDSC) as compared to the WT group by ELISA (Fig. 3h). Altogether, these findings suggest that c-Rel initiates enhanceosome formation, which functions to induce rMo signature gene expression.
IL-1β−CCL2 crosstalk between rMos and tumor cells sustains immune tolerance.
CCR2-expressing monocytes usually traffic out of the bone marrow in response to CCL2 cues from distant sites23, 24. As described above, rMos preferentially accumulated in the tumor microenvironment of tumor-bearing mice (Fig. 1e). To investigate which cell types in the TME secrete CCL2 to sustain rMo recruitment, we examined Ccl2 expression in our scRNA-seq dataset. Using the VlnPlot function to visualize Ccl2 expression in our expanded data (including all the immune and tumor cells sequenced), we observed high Ccl2 expression in intra-tumoral myeloid cells (rMos, CCR2− MDSCs, and TAMs) (Fig. 4a). Intriguingly, one tumor cell cluster also expressed Ccl2 (Fig. 4a). To independently study this tumor cluster and overall tumor cell heterogeneity, we re-clustered all tumor cells into six distinct cell clusters (Fig. 4b). Interestingly, tumor cluster-6 had high expression of chemokines Cxcl10, Cxcl2, Ccl2, and Ccl8 when compared to the bulk tumor cell population (Fig. 4b). Using the FindAllMarkers function, we identified Cd74, Nfkbia, and Ifitm3 as distinguishing markers for this tumor cell cluster (Fig. 4b). Interestingly, Tumor cluster-6 also expressed IL-1-receptor family genes Il1r1, Il1rap, Il1r2, and Il1rn (Fig. 4c). Il1r1 and Il1rap are the genes expressing the receptor subunits for IL-1β25. Since rMos expressed high levels of IL-1β in the TME, we wondered if IL-1R1+ IL-1Rap+ CD74hi tumor cells could respond to IL-1β signals to promote CCL2 production and sustain rMo recruitment. First, we sorted CD74hi B16F10 tumor cells from B6 tumor-bearing mice. We confirmed increased expression of Ccl2, Il1r1, and Il1rap in CD74hi tumor cells, when compared with the bulk of the tumor cells (Fig. 4d, e). To assess the role of IL-1β in regulating CCL2 expression, we generated IL-1β-rich conditioned media by culturing BM-MDSCs (generated with GM-CSF and IL-6 as in Fig. 2) for 24 hours. By ELISA we confirmed the markedly increased amounts of IL-1β in MDSC-conditioned media when compared with control media (Fig. 4f). We then assessed if the treatment with recombinant IL-1β or IL-1β-rich conditioned media could elicit Ccl2 expression in B16F10 cells. We observed a similar increase in Ccl2 expression following treatment with either recombinant IL-1β or MDSC-conditioned media (Fig. 4g). Co-treatment with the IL-1β-specific inhibitor, Diacerein, abrogated the conditioned media-induced Ccl2 expression, suggesting the effect was IL-1β-mediated (Fig. 4g).
Fig. 4. IL-1β and Ccl2 crosstalk between rMos and tumor cells.
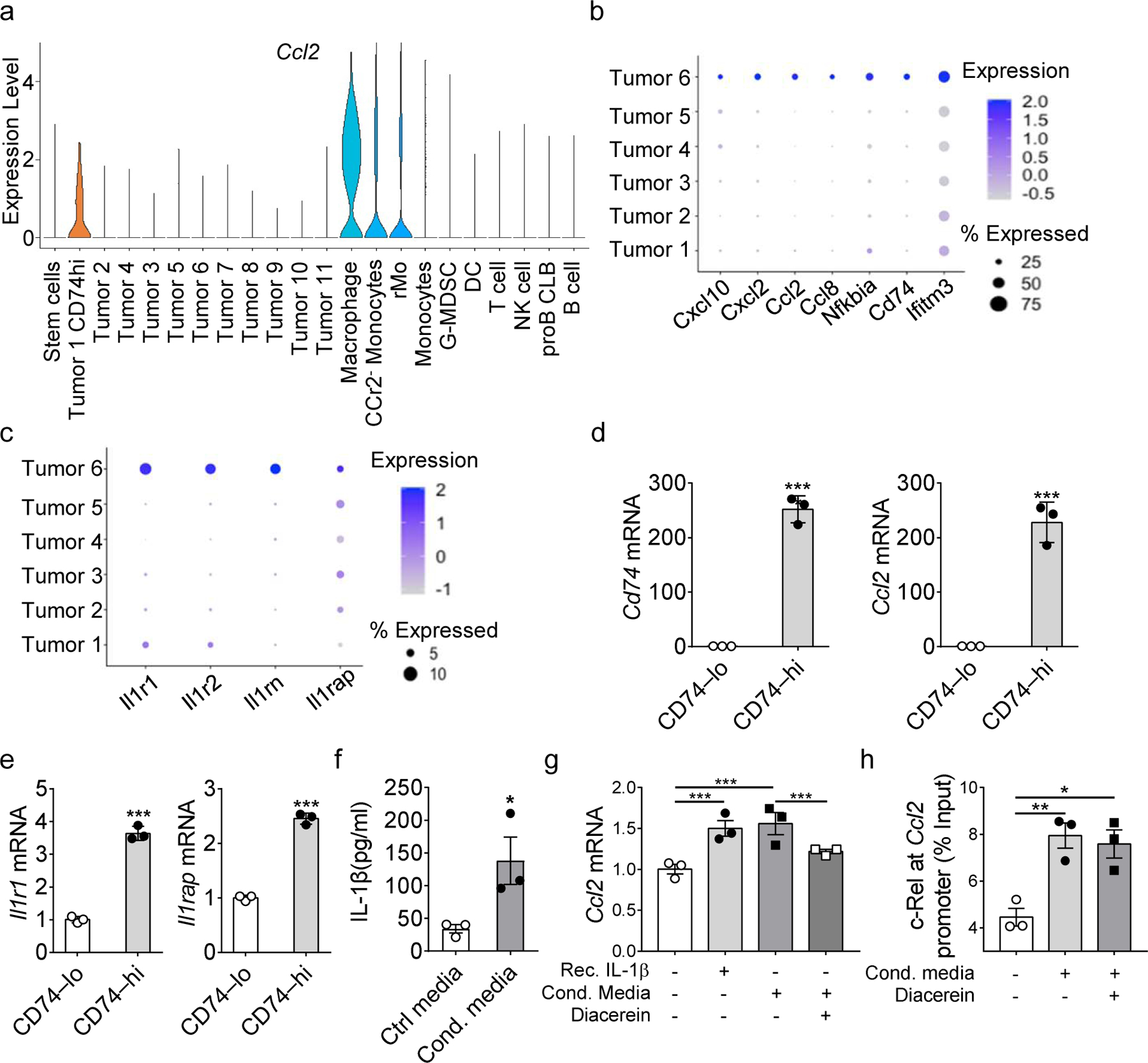
a, Violin plots showing Ccl2 gene expression in tumor clusters and immune clusters.
b, c, Bubble plot showing the expression of indicated chemokines (b) and Il1-receptor family proteins (c) across different tumor clusters.
d, e, Relative mRNA levels of Cd74, Ccl2 (d) and Il1r1, Il1rap (e) between CD74hi and CD74lo tumor cells as determined by real-time RT-PCR.
f, IL-1β secreted in the conditioned media of MDSC cultures compare with control media as determined by ELISA.
g, h, Relative Ccl2 mRNA level of B16F10 cells (g) and Raw264.7 cells (h) with indicated treatments as determined by real-time RT-PCR.
i, j, Bar graphs show the relative c-Rel binding to the Ccl2 gene promoter region in B16F10 cells (i) and Raw264.7 cells (j) with indicated treatments compared with input as determined by ChIP. Statistical significance was determined by two-tailed unpaired t-test (d-f) and one-way ANOVA with Tukey post-hoc test (g-j). For panel d-j, data are presented as means ± s.e.m., and experiments were repeated 2 times independently with similar results; data of one representative experiment are shown.
c-Rel is an important transcriptional regulator for Ccl2 in cancer and immune cells26, 27, 28. Therefore, we performed ChIP analyses of B16F10 cells treated similarly as in Fig. 4g and confirmed increased binding of c-Rel to Ccl2 regulatory sites upon stimulation with conditioned media (Fig. 4h). Collectively, the data reported here suggest that tumor cells secret chemokine CCL2 via c-Rel to recruit rMos to the TME, and that IL-1β secretion by rMos, in turn, enhances the expression of CCL2, forming an immune tolerant microenvironment conducive to tumor growth.
rMos can differentiate into Arg1-expressing macrophages within the TME.
To investigate myeloid maturation and differentiation patterns, we performed a pseudotime analysis on the myeloid clusters, using the Monocle package in R. The analysis suggested that some rMos differentiated into CCR2− monocytes, some of which could then become Arg1+ F4/80+ TAMs (Fig. 5a–c). As shown in the dendrogram, rMos, CCR2− monocytes, and TAMs are clearly separated within the myeloid clusters, suggesting that the CCR2− monocytes represented an intermediate population between rMos and TAMs (Fig. 5d).
Fig. 5. The relationship between rMos and TAMs in TME.
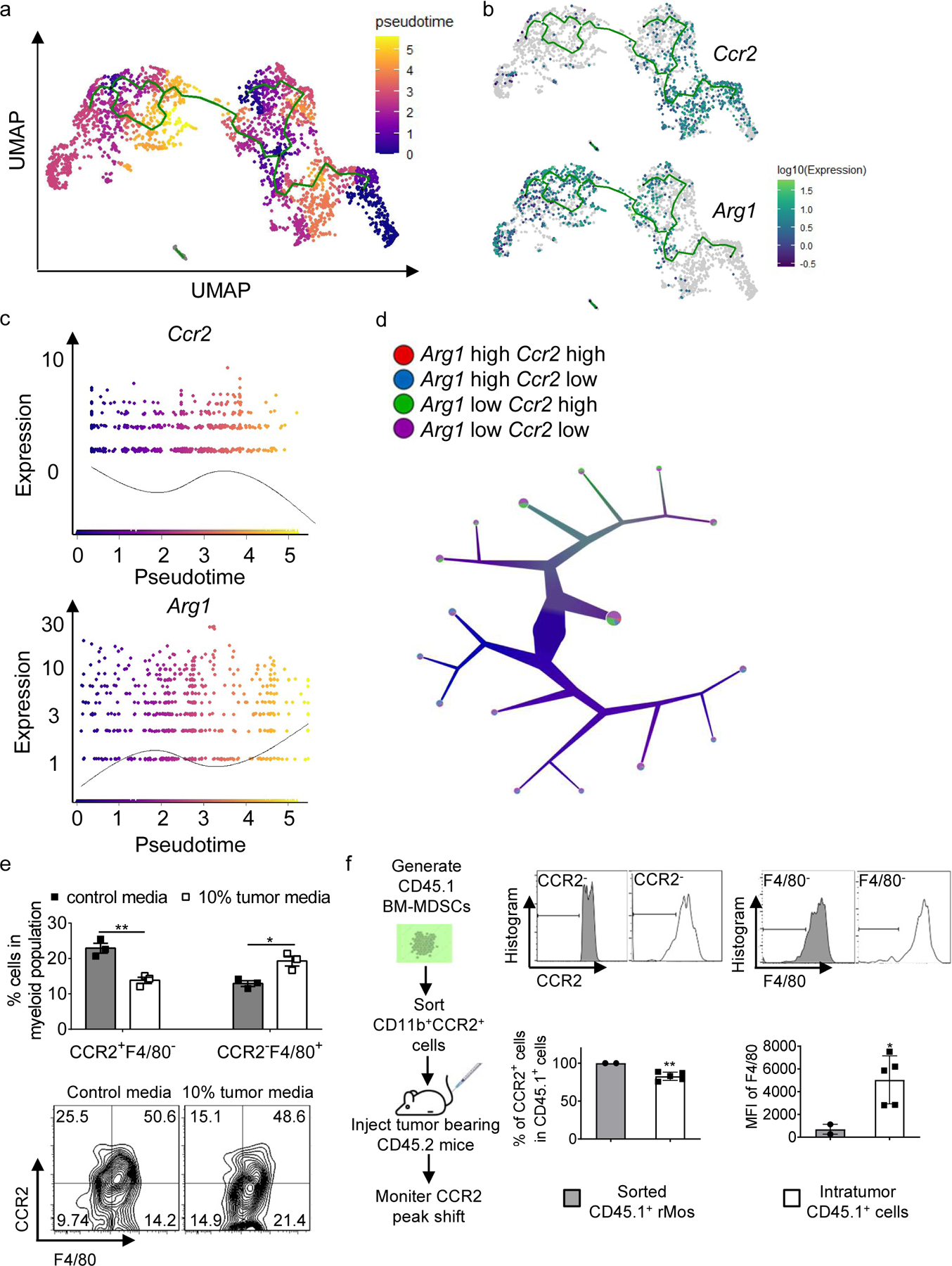
a, Monocle analysis on Myeloid populations resulted in branched trajectory with 6 distinct Monocle states (color code for each state is indicated).
b, Ccr2 and Arg1 expression levels along the branched trajectory.
c, Pseudotime plot illustrating expression of Ccr2 and Arg1 genes over pseudotime.
d, Color-coded tree with the expression level of Ccr2 and Arg1 by TooManyCells analysis.
e, Percentages of indicated cell subsets in BM-MDSCs cultured with control media or 10% tumor cell-conditioned media as determined by flow cytometry (n=3 samples/group).
f, Adoptive transfer experiment flowchart (left), stacked histograms (upper right), and bar graph (lower right) showing CCR2 and F4/80 levels in sorted CD45.1+ cells (pooled from 8 donor mice) and CD45.1+ cells in the tumors of recipient mice (n=5 mice).
Statistical significance was determined by two-tailed unpaired t-test (e and f). Data are presented as means ± s.e.m. For panel e, experiments were repeated 2 times independently with similar results, and data of one representative experiment are shown. For panel f, data are pooled from two independent experiments.
To validate these findings experimentally, we applied culture supernatant of B16F10 cells (to mimic the TME) to BM-MDSCs in culture. Flow cytometry results showed a decrease (25.5% to 15.1%) in CCR2+F4/80− cells treated with tumor-conditioned media when compared to non-conditioned media, whereas the F4/80+CCR2− cells increased from 14.2% in the control to 21.4% in the group treated with tumor cell-conditioned media (Fig. 5e). To confirm these results, we performed an adoptive transfer experiment where we generated CD45.1 BM-MDSCs, sorted out CCR2+ rMos, and injected the latter cells intravenously into CD45.2 tumor-bearing mice. We observed decreased CCR2 and increased F4/80 expression in CD45.1 cells in the tumors of recipient mice (Fig. 5f). Altogether, our findings suggest that rMos, CCR2− monocytes, and TAMs are developmentally linked, with some rMos transitioning into Arg1+ TAMs in the TME via an intermediary CCR2− state.
rMos are conserved in human melanoma TME.
To evaluate if rMos are conserved in humans, we explored published single-cell RNA sequencing data of human melanoma samples (GSE123139)29. Li et al. performed single-cell transcriptional profiling of immune cells from tumors of several melanoma patients. We performed clustering of this dataset and identified six distinct immune cell populations (Fig. 6a). Within the monocytic/macrophage-like clusters (CD14+, Clusters 1–3, 5, 6), there were two distinct IL-1β+ populations (Fig. 6b): One CCR2−REL− cluster (Cluster 2) and one CCR2+REL+ cluster (cluster 5) (Fig. 6b). Clusters 1–3 had high expression of the mature macrophage marker CD163 (Fig. 6c). We also assessed the expression of several immunosuppressive factors. NOX2 (CYBB) was highly expressed in all CD14+ clusters except for cluster 6 (Fig. 6c). TGF-ß (TGFB1) was expressed in Clusters 3 and 5, whereas COX2 (PTGS2) was specific to Cluster 5 (Fig. 6d). In fact, Cluster 5 was the only cluster that expressed all immunosuppressive factors examined.
Fig. 6. rMos are present in human melanoma TME.
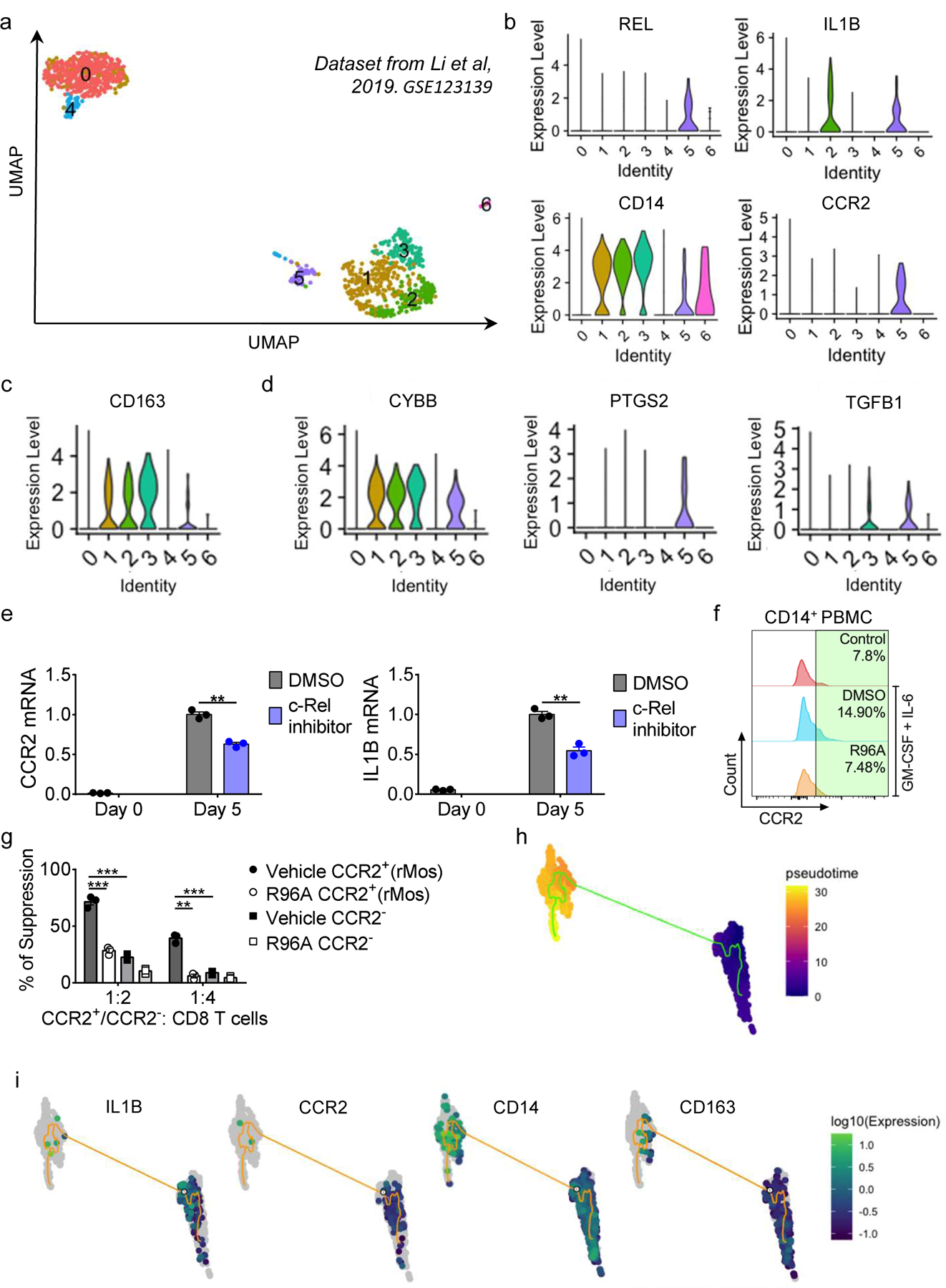
a, UMAP plots of single-cell RNA sequencing data of human melanoma samples (GSE123139).
b-d, Violin plots showing indicated gene expression.
e, Relative mRNA levels of CCR2 and IL1B in human MDSCs as compared to PBMC cells, determined by real-time RT-PCR. (n=3 samples/group; ***, P<0.001)
f, Stacked histograms showing CCR2 levels in human MDSCs with indicated treatment.
g, The degree of suppression, at the indicated effector: T cell ratio, of CD8 T cell proliferation by purified CD11b+CCR2+ cells (rMos) and CD11b+CCR2− cells from cytokine-treated human PBMC cultures with or without R96A. (n = 3 biologically independent cultures per group; **, P<0.01***, P<0.001)
h, Monocle analysis on human myeloid populations resulted in a branched trajectory with 4 distinct Monocle states (color code for each state is indicated).
i, IL1B, CCR2, CD14, and CD163 expression levels along the branched trajectory.
Statistical significance was determined by two-tailed unpaired t-test (e) and two-way ANOVA with Tukey post-hoc test (g). For panels e and f, data are presented as means ± s.e.m., and experiments were repeated 2 times independently with similar results, and data of one representative experiment are shown.
Highly suppressive human MDSCs can be generated by treating PBMCs isolated from healthy donors with GM-CSF and IL-6 for seven days18. To assess if this would induce an rMo subpopulation within MDSCs, we treated PBMCs from a healthy donor with GM-CSF and IL-6, and monitored CCR2 and IL-1β expression. Cytokine treatment markedly increased CCR2 and IL-1β mRNA (Fig. 6e). Co-treatment with the c-Rel inhibitor (R96A)6, inhibited the cytokine-driven increase in CCR2 and IL-1β, suggesting that the effect was partially c-Rel-mediated (Fig. 6e). We also monitored the accumulation of CD14+CCR2+ monocytes by flow cytometry following cytokine treatment with or without the c-Rel inhibitor. Cytokine treatment induced a 2-fold increase in CCR2-expressing CD14+ cells in human PBMCs (Fig. 6f). Co-treatment with R96A abrogated the induction and immune suppression, suggesting that similar to the mouse, a population of Rel-dependent MDSCs is present in humans (Fig. 6f,g). We then performed pseudo time trajectory analysis on the human scRNAseq dataset to evaluate whether rMos (CD14+CCR2+) would transition into mature macrophage subsets (CD14hiCD163hi). We found that IL-1β+ CCR2+ cells transitioned into CD163+ cells (Fig. 6h,i). These results corroborated our findings in Fig. 5 and suggested that human rMos could differentiate into TAMs.
DISCUSSION
Expanded, heterogenous MDSC populations in the TME are significant impediments to immunotherapy such as checkpoint blockade2, 3, 4, 5. Deconstructing MDSC heterogeneity within different tumor contexts is essential to devising targeted approaches to improve immunotherapy. Here we use single-cell RNA sequencing to dissect intratumoral MDSC heterogeneity and investigate myeloid suppressive populations. We identified a subset of strongly suppressive CCR2+Arg1−IL-1βhi monocytic MDSCs, rMos, which are developmentally and functionally dependent on c-Rel. We show that rMos are a product of tumor-induced inflammation in the bone marrow and are recruited to the TME by a CCR2:CCL2 axis. Crucially, in the TME, they are potent immunosuppressors, inhibiting T-cell proliferation and activity, before transitioning into Arg1-expressing macrophages (Supplementary Fig. 5).
Depletion of environmental arginine by Arg1 has long been thought to be the predominant mechanism of T-cell suppression in MDSCs30, 31. Our data indicates rMos, which were a more suppressive MDSC subset, suppress T-cell function without high Arg1 expression. These results suggest that Arg1 may not be crucial to the function of this MDSC subset, and Arg1-targeted therapeutic approaches, which have been recently reported32, 33, may not suffice to ablate MDSC-driven immunosuppression. In the absence of arginase expression, we posit ROS as a significant mechanism of immunosuppression for rMos. rMos produced much higher levels of ROS than TAMs.
rMos likely develop in the bone marrow as a result of sustained tumor-induced inflammation. This is mediated by a c-Rel enhanceosome that directly binds and transcribes signature genes such as Ccr2 and Il1b in response to cytokine stimulation. We and several others have proposed that enhanceosomes are key drivers of altered myelopoiesis during pro-tumor immune cell development6, 34, 35, 36, 37. Here we showed that a c-Rel-specific inhibitor, R96A, inhibited rMo differentiation, with no significant toxicity observed6. Therapeutic approaches targeting enhanceosome complexes that specify the development of rMos could thus be crucial to potentiate immunotherapy. We previously showed that the c-Rel inhibitor R96A could block tumor growth in melanoma and lymphoma mouse models6. Crucially, R96A synergized with lymphocyte checkpoint blockade (anti-PD-1 antibodies), presumably by inhibiting rMo development and immunosuppression. Our results suggest that the combination of c-Rel pharmaceutical inhibition with checkpoint blockade would be a viable clinical strategy.
rMos, which express a high level of CCR2, are chemotactically recruited to the tumor-microenvironment via CCL2. We show that CCL2 in the tumor-microenvironment is secreted by myeloid cells (TAMs and CCR2− MDSCs) and CD74hi tumor cells. This tumor cluster had high expression of chemokines Cxcl10, Cxcl2, Ccl2, and Ccl8, which are known to be the regulators of immune cell recruitment38. Melanomas have previously been shown to express a number of chemokines39; in one instance, the CCL21 production by melanomas sustained regulatory T cells and MDSC recruitment to the TME with concomitant immunosuppression40. CCL3, CCL4, and CCL5 are also enriched in melanoma, promoting the recruitment of CCR5+ MDSCs41. Our data suggest that chemokines are not secreted by the bulk of the tumor but rather by a small immunomodulatory tumor subset marked by CD74. Interestingly, CD74 is associated with increased immune cell infiltration and poor clinical outcomes in melanoma42. Our data suggest a hitherto unknown regulatory axis and a potential mechanism for this phenomenon where CD74hi melanoma cells respond to IL-1β secreted by rMos to produce more Ccl2 and sustain rMo recruitment. CD74 could thus represent an intriguing therapeutic candidate to reduce the infiltration of immunosuppressive cells. Inflammasomes are one of the most important mediators of innate immunity and are known to drive various inflammatory disorders via processing the cytokine IL-1β. Nlrp3, an important component of the inflammasome, plays a key role in the processing and secretion of IL-1β. In this study, we found a markedly increased amount of Nlrp3 mRNA in rMos as compared to macrophages and CCR2− monocytes (Supplementary Fig. 2a). These results indicate that there might be an enhanced inflammasome activation to promote IL-1β maturation and secretion in rMos.
In the TME, tumor-derived factors reprogram infiltrating myeloid cells to change their phenotype and immunosuppressive functions43, 44, 45. Specifically, the activation of HIF-1α under hypoxic conditions can drive the differentiation of M-MDSCs into macrophages and induce their expression of Arg146. Here we show that some of the rMos that traffic to the TME through the CCR2:CCL2 axis, lose CCR2 and IL-1β expression and mature into Arg1-expressing TAMs. Interestingly, the TAM population is an abundant source of CCL2 in the TME, suggesting that the trafficking of rMos may also be regulated by a feedback loop through mature Arg1-expressing TAMs.
Whether tissue-resident macrophages, tissue-infiltrating monocytes, or M-MDSCs contribute to the TAM pool is the subject of debate45. Our study shows several distinctive subsets of myeloid cells within the TME. Unlike the CCR2− monocytic population, rMos are characterized by their high expression of genes that regulate immune response and response to cytokines and chemokines, suggesting that they are critical players in the innate immune response. Consistent with this idea, our functional studies show that the suppressive function of rMos exceeds that of the Arg1-expressing TAMs. Consistent with previous studies, our pseudotime analysis shows that some Arg1-expressing TAMs derive from rMos, whereas others derive from other macrophage precursor cells, potentially tissue-resident macrophages46, 47, 48, 49.
In summary, our study identified a suppressive, c-Rel-dependent monocyte (rMo) subset that is induced by tumor-sustained inflammation. rMos are likely generated in the bone marrow and migrate to the TME through a CCR2:CCL2 axis. Defining the phenotypic, developmental, and functional characteristics of rMos as described here is a cornerstone for clarifying MDSC heterogeneity and may open up new avenues for cancer immunotherapy.
METHODS
Mice
Global Rel gene knockout (Rel−/−) C57BL/6 (B6) mice (a gift from Dr. Hsiou-chi Liou, Cornell University) and floxed Rel gene (RelloxP/loxP) B6 mice (a gift from Dr. Sankar Ghosh, Columbia University) were generated as described previously6. LysM-cre (B6.129P2-Lyz2tm1(cre)Ifo/J) and C57BL/6J mice were originally purchased from The Jackson Laboratory. Male and female mice between the ages of 5 and 8 weeks were used in the study. All mice were housed at the University of Pennsylvania animal care facilities under pathogen-free conditions and all procedures were preapproved by the institutional animal care and use committee.
Human samples
Blood samples from human healthy donors were collected by the Human Immunology Core at the University of Pennsylvania with approval from the ethics committee and institutional review board. Written consent was obtained from each healthy donor before blood collection. All experiments involving blood samples from healthy donors were performed in accordance with relevant ethical regulations.
Cells
B16F10 cells were obtained from the ATCC and cultured in Roswell Park Memorial Institute (RPMI) medium supplemented with 0.1mM non-essential amino acids, 1mM sodium pyruvate, 2mM l-glutamine, 25mM HEPES, 55μM 2-mercaptoethanol, 10% FCS and 1% penicillin-streptomycin (100U/ml penicillin, 100μg/ml streptomycin). RAW264.7 cells were obtained from the ATCC and cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 0.1mM nonessential amino acids, 1mM sodium pyruvate, 2mM l-glutamine, 25mM HEPES, 55μM 2-mercaptoethanol, 10% FCS and 1% penicillin-streptomycin (100U/ml penicillin, 100μg/ml streptomycin). All cultures were maintained in a humidified 5% CO2 incubator at 37°C.
Tumor models and tissue collection
To generate subcutaneous tumors, 0.5 × 106 (Fig. 1a)or 0.3 × 106 (Fig. 1f) B16F10 and 0.3 × 106 sorted CCR2+ or CCR2− cells were injected subcutaneously into each mouse. Mice with comparable tumor sizes were sacked and tissues were collected.
Blood was collected from the heart using a 24-gauge needle and syringe. Blood was suspended in 5 ml of red blood cell lysis buffer and incubated at RT for 15 min. Cells were quenched in 5 ml of RPMI with 3% FBS. Cells were then resuspended in FACS buffer and stained for flow cytometry. After the right atrium and left ventricles were punctured, mice were perfused with 15 ml of PBS before the tumor, spleen, and bone marrow were collected.
To obtain tumor-infiltrating cells, tumors were digested in RPMI 1640 (Gibco) supplemented with 0.5 mg/ml collagenase type IV (Gibco) and 0.1 mg/ml DNase I (Sigma-Aldrich) for 20 min at 37°C. Tumors were then treated with Percoll(GE Healthcare Lifesciences, 17089101) and immune cells were isolated for flow cytometry and sorting. Spleens were homogenized and treated with RBC lysis buffer (eBioscience, 00-4300-54) before staining.
Single-cell RNA-sequencing
FACS-isolated CD45+ cells from the 5 pooled tumors from WT (LysM-cre × Rel+/+) or myeloid Rel knockout mice (LysM-cre × RelloxP/loxP) were washed once in PBS (calcium- and magnesium-free) containing 0.04% BSA and resuspended to a concentration of 700–1200 cells/ul. The DNA libraries were prepared using the Chromium Single Cell 3′ Reagents Kits and 10x Genomics Chromium platform for droplet-enabled scRNAseq according to the manufacturer’s instructions. Each library was sequenced on the Illumina HiSeq 4000 platform to achieve an average of 10000 reads per cell. Alignment of 3′ end counting libraries from scRNAseq analyses was completed using 10x Genomics Cell Ranger. The “Cell Ranger Aggr” function was used to normalize the number of confidently mapped reads per cell across the two libraries.
Analysis of single-cell RNA sequencing
Seurat version 3.2.2 was used for cluster identification in scRNAseq datasets50, 51. The raw counts data were read in R and normalized by log transformation. Cells with a mitochondrial ratio above 0.2 were removed. Using Seurat’s “FindIntegrationAnchors” and “IntegrateData” functions, cells from WT and Rel−/− mice were integrated into a single analysis. For analysis of human melanoma data (GSE123139), counts matrix files from samples GSM3496287_AB1891, GSM3496288_AB1892, GSM3496289_AB2093, GSM3496292_AB2096, GSM3496293_AB2097, GSM3496294_AB2098, GSM3496295_AB2099 and GSM3496299_AB2103 were read into Seurat and integrated using both “FindintegrationAnchors” and “IntegrateData” functions. For UMAP (Uniform Manifold Approximation and Projection) dimension reduction and clustering analysis, we used the first 30 principal components. Mc1r and S100a1 were used to identify tumor clusters and SingleR52 was used to identify the cell types for the other clusters. Specific markers were determined using the “FindAllMarkers” function and used in GProfiler53 for enrichment analysis and visualization using the “DoHeatMap” function in Seurat. Violin plots were generated using Seurat’s “VlnPlot” function. Monocle3 was used for pseudotime analysis and to generate the pseudotime expression graphs54, 55. The integrated preprocessed Seurat object was normalized, clustered, and visualized using UMAP for dimensionality reduction. TooManyCells was used to visualize transcriptionally similar cells and generate dendrograms56.
Generation of murine and human MDSCs
Bone marrow-derived MDSCs were generated from the bone marrow of naïve WT or Rel−/− mice using GM-CSF and IL-6 as previously described6. Briefly, following red blood cell lysis, freshly isolated bone marrow cells were cultured in a complete RPMI medium containing GM-CSF (100 ng/ml) (BioLegend, 576306) and IL-6 (100 ng/ml) (BioLegend, 575706), at a density of 5 × 106 cells/ml for seven days. Human MDSCs were similarly generated by treating PBMCs from healthy donors with GM-CSF (100 ng/ml) and IL-6 (100 ng/ml) in a complete RPMI medium, at a density of 5 × 106 cells per ml, for seven days. The c-Rel inhibitor R96A (5μM final concentration) was added to select cultures once per day.
Flow cytometry and sorting
Single-cell suspensions from isolated tissues and cultured BM-MDSC cells were suspended in FACS buffer and incubated for 20 min with fluorescence-labeled antibodies with the different combinations of the following antibodies: CD45.1 (BioLegend, 110743), CD45.2 (BioLegend, 109828), CD11b (BioLegend, 101208), F4/80 (BioLegend, 123115), CCR2 (BioLegend, 150611), CD8 (BioLegend, 100714), CD4 (Invitrogen, MCD0404-3), Ghost Dye™ UV 450 Viability Dye (Cell Signaling, 80862S) and Ghost Dye™ Red 450 Viability Dye (Cell Signaling, 18452S).
Flow cytometry was conducted using FACS Canto and LSR II flow cytometry systems (BD Biosciences). Sorting was done by the Flow Cytometry and Cell Sorting Resource Laboratory at the University of Pennsylvania using BD FACS Aria and Influx sorters (BD Biosciences). Gating for rMo: live, CD45+, CD11b+, CCR2+, F4/80−; gating for tumor-associated macrophages: live, CD45+, CD11b+, CCR2−, F4/80+. All data were analyzed with the FlowJo software (FlowJo LLC, version 10.0.7).
T-cell suppression assay
Murine T cells labeled with 1 μM carboxyfluorescein succinimidyl ester (CFSE) (Thermofisher Scientific, C34554) were activated with plate-bound anti-mouse CD3 (250 ng/ml) (Invitrogen, 16-0031-85) and soluble anti-mouse CD28 (250 ng/ml) (BioLegend, 102102), and co-cultured at different ratios with murine MDSCs for 72 h. Human PBMCs from healthy donors were labeled with 1 μM CFSE, activated with plate-bound anti-human CD3 (1 μg/ml) and soluble anti-human CD28 (5 μg/ml), and cocultured at different ratios with human MDSCs generated in vitro for 72 h. Cells were washed with FACS buffer and stained for 20 min with preconjugated fluorescence-labeled antibodies for CD3, CD4, and CD8. CD8+ and CD4+ T-cell proliferation was measured by flow cytometry using the decrease in CFSE fluorescence in CD8+ and CD4+ T cells compared with unstimulated T cells. The percentage of T-cell suppression was calculated as follows: ((percentage of T-cell proliferation of cultures with anti-CD3 and anti-CD28 − percentage of T-cell proliferation of cultures with anti-CD3, anti-CD28 and MDSCs) / percentage of T-cell proliferation of cultures with anti-CD3 and anti-CD28) × 100.
Migration assay
Transmigration was assessed using a 5.0-μm pore size 96-well ChemoTx transwell system (Neuro Probe). First, 30μL migration buffer with or without 100 ng/ml MCP-1/CCL2 (Biolegend, 578404) was added to the lower chamber. Then, 5 × 104 sorted cells were seeded in the upper chamber. After 6 hours of incubation at 37 °C, the transmigrated cells were collected and counted.
ROS detection assay
A total of 15×103 cells were incubated with 0.5 μM DCFDA (Thermo Fisher Scientific, D399) dye in Hank’s Balanced Salt Solution for 20 minutes (Thermo Fisher Scientific). Fluorescence intensity was measured using a plate reader (at 485/535 nm) and the signal was subtracted from background fluorescence and normalized to the total number of cells.
Enzyme-linked immunosorbent assay
IFNγ and IL-1β in the culture supernatant were measured by sandwich ELISA (Invitrogen, M421B) according to the manufacturer’s protocol.
Quantitative real-time PCR
RNA was isolated from cells and tissues using Qiagen’s RNeasy kit (Qiagen, Cat. No. 74104) according to the manufacturer’s protocol. cDNA synthesis reaction was performed using the SuperScript™ III First-Strand Synthesis System (Thermo Fisher, 18080051). Real-time quantitative PCR of target sequences was performed using an ABI 7500 Fast Real-Time PCR System (Applied Biosystems). Relative mRNA levels were calculated using the ΔCt method using internal controls (18s ribosomal RNA for mice, GAPDH for humans). For primer information please see Supplementary Table 1.
Chromatin Immunoprecipitation
ChIP was performed using the ChIP-IT Express Enzymatic kit (Active Motif), according to the manufacturer’s protocol. Briefly, following culture and treatment with appropriate treatment media, BM-MDSCs, B16F10, RAW264.7, or bone marrow cells were fixed with 1% formaldehyde, then lysed with ice-cold lysis buffer. Chromatin was sheared by enzymatic digestion, input DNA set aside, and protein/DNA complexes immunoprecipitated overnight at 4°C using 2μg of the following antibodies: c-Rel (SCBT, sc-365720, clone G-7), C/EBPß (SCBT, sc-7962, clone H-7), p65 (SCBT, sc-372, clone c-20), pSTAT3 (Cell Signaling Technologies, 91455, D3A7) or control mouse IgG. Reverse crosslinking was then performed at 95°C followed by proteinase K digestion. Identification of genes of interest in bound chromatin was performed by PCR/qPCR using the same primers listed for other qPCR experiments. Transcription factor occupancy of target genes was computed using the percent input method.
Western blot
Cells were lysed using radioimmunoprecipitation assay (RIPA) buffer in the presence of a protease inhibitor cocktail (Roche). Cell lysates were then subjected to 10% SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories). The membranes were probed overnight at 4 °C with antibodies specific for CCR2 (Cell Signaling, 12199) and GAPDH (Cell Signaling, 2118s) according to the manufacturer protocol, followed by incubation for 1 h at room temperature with secondary antibodies conjugated with peroxidase. Membrane-bound immune complexes were detected with the Super ECL Detection Reagent (Thermo Fisher Scientific) on an Amersham Imager 600.
Supplementary Material
Supplementary Fig. 1 related to Fig. 1. rMo signature gene expression and cluster analysis.
a, Heatmap displaying the scaled expression patterns of rMo top marker genes across each tumor and immune clusters.
b, Heat map of hierarchical cluster analysis of immune clusters
Supplementary Fig. 2 related to Fig. 1. rMo marker gene expression and GO analysis.
a, Heatmap displaying the scaled expression patterns of rMo top marker genes across myeloid clusters.
b, c, Gene Ontology (Go) analysis of rMo marker genes in Supplementary Fig. 2a.
d, e, Total CD11b+ CCR2+ cell count in bone marrow, spleen (d) and tumor (e) of naïve mice and tumor-bearing mice in Fig. 1e.
f, Tumor growth in WT and Rel−/− mice that were injected subcutaneously with LLC cells (n = 10 mice per group).
g, Percentages of CD11b+ CCR2+ cells in intratumor CD45.2+ cells in Supplementary Fig. 2f, as determined by flow cytometry.
h, i, Tumor weight (f) and percentages of CD8+ cells in tumors (g) in Fig. 1f (n=10/group). Statistical significance was determined by two-tailed unpaired t-test. Data are presented as means ± s.e.m., and pooled from two independent experiments (d-g) (*, P<0.05; **, P<0.01).
Supplementary Fig. 3 related to Fig. 2. rMos functional analysis.
a, b, Flow cytometry gating strategy of the sorted subsets for subsequent function assays in Fig. 2a (a) and Fig. 2b (b).
c, d, Representative flow cytometry plots from the function assay for Fig. 2a (c), Fig. 2b (d)
e, ROS production by sorted CD11b+CCR2+ (rMos) and CD11b+ F4/80+ (TAMs) cells from B16F10 tumor bearing mice.
f, IFN-γ secreted in the supernatant of a as determined by Elisa.
f, Representative flow cytometry plots from the function assay for Fig. 2c
Statistical significance was determined by two-tailed unpaired t-test (f), or one-way ANOVA with Tukey post-hoc test (g). Data are presented as means ± s.e.m., technical replicates of pooled samples (5 mice per group) were tested; and experiments were repeated 2 times independently with similar results and data of one representative experiment are shown.
Supplementary Fig. 4 related to Fig. 3. Public ChIP-seq data of bone marrow cells.
a, b, Public ChIP-seq data that show the binding peak of c-Rel, CEBPb, and p65 on Ccr2 (a) and Il1b (b) loci of bone marrow myeloid cells.
Supplementary Fig. 5 A working model of rMos.
rMos are generated in the bone marrow, migrate to TME and differentiate into TAMs in TME. Please see the text for details.
Supplementary Table 1: Primers used for PCR
Acknowledgments:
We thank Drs. Hsiou-Chi Liou (Cornell University) for providing the breeding pair of the Rel−/− mice, and Dr. Sankar Ghosh (Columbia University) for floxed Rel gene (RelloxP/loxP) B6 mice. We thank Dr. Daniel P. Beiting from Penn Vet for technical support. We thank Zienab Etwebi, Chin-Nien Lee, Jiyeon Yu , and Mingyue Li for valuable discussions, technical support, and reagents.
This work was supported in part by grants from the National Institutes of Health (NIH), USA (R01-AI152195, R01-AI143676), and Natural Science Foundation, China (32130040).
Footnotes
Competing Interests: YHC is an inventor of the following patent that describes the c-Rel inhibitor used in this study: Chen, Y. H., R. Murali, J. Sun: REL INHIBITORS AND METHODS OF USE THEREOF. USA Patent Number US8609730B2, 2013. YHC is a member of the advisory board of Amshenn Co. and Binde Co.
References
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell 2015, 27(4): 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Haas N, de Koning C, Spilgies L, de Vries IJ, Hato SV. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology 2016, 5(7): e1196312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cassetta L, Bruderek K, Skrzeczynska-Moncznik J, Osiecka O, Hu X, Rundgren IM, et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. Journal for immunotherapy of cancer 2020, 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, et al. Cancer treatment and survivorship statistics, 2019. CA: a cancer journal for clinicians 2019, 69(5): 363–385. [DOI] [PubMed] [Google Scholar]
- 5.Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. Journal of immunology 2018, 200(2): 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li T, Li X, Zamani A, Wang W, Lee CN, Li M, et al. c-Rel Is a Myeloid Checkpoint for Cancer Immunotherapy. Nat Cancer 2020, 1(5): 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan D, Wang J, Sun H, Zamani A, Zhang H, Chen W, et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. The Journal of experimental medicine 2020, 217(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nature reviews Cancer 2013, 13(10): 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer cell 2012, 21(6): 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast cancer research and treatment 2010, 123(1): 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(50): 21248–21255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends in immunology 2016, 37(3): 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature communications 2016, 7: 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zilio S, Serafini P. Neutrophils and Granulocytic MDSC: The Janus God of Cancer Immunotherapy. Vaccines 2016, 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fultang N, Li X, Li T, Chen YH. Myeloid-Derived Suppressor Cell Differentiation in Cancer: Transcriptional Regulators and Enhanceosome-Mediated Mechanisms. Frontiers in immunology 2020, 11: 619253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. European journal of immunology 2010, 40(11): 2969–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Science immunology 2020, 5(44). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 32(6): 790–802. [DOI] [PubMed] [Google Scholar]
- 19.Enciso-Mora V, Broderick P, Ma Y, Jarrett RF, Hjalgrim H, Hemminki K, et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nature genetics 2010, 42(12): 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trynka G, Zhernakova A, Romanos J, Franke L, Hunt KA, Turner G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut 2009, 58(8): 1078–1083. [DOI] [PubMed] [Google Scholar]
- 21.Hussman JP, Beecham AH, Schmidt M, Martin ER, McCauley JL, Vance JM, et al. GWAS analysis implicates NF-kappaB-mediated induction of inflammatory T cells in multiple sclerosis. Genes and immunity 2016, 17(5): 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YH MRSJ. Rel inhibitors and methods of use thereof. US patent number: 8609730; PCT/US2009/030325. 2009(PCT/US2009/030325).
- 23.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nature immunology 2006, 7(3): 311–317. [DOI] [PubMed] [Google Scholar]
- 24.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clinical cancer research : an official journal of the American Association for Cancer Research 2013, 19(13): 3404–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunological reviews 2018, 281(1): 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owen JL, Torroella-Kouri M, Handel-Fernandez ME, Iragavarapu-Charyulu V. GM-CSF up-regulates the expression of CCL2 by T lymphocytes in mammary tumor-bearing mice. International journal of molecular medicine 2007, 20(1): 129–136. [PubMed] [Google Scholar]
- 27.Oda T, Ueda A, Shimizu N, Handa H, Kasahara T. Suppression of monocyte chemoattractant protein 1, but not IL-8, by alprazolam: effect of alprazolam on c-Rel/p65 and c-Rel/p50 binding to the monocyte chemoattractant protein 1 promoter region. Journal of immunology 2002, 169(6): 3329–3335. [DOI] [PubMed] [Google Scholar]
- 28.Lindholm PF, Sivapurapu N, Jovanovic B, Kajdacsy-Balla A. Monocyte-Induced Prostate Cancer Cell Invasion is Mediated by Chemokine ligand 2 and Nuclear Factor-kappaB Activity. Journal of clinical & cellular immunology 2015, 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176(4): 775–789 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heuvers ME, Muskens F, Bezemer K, Lambers M, Dingemans AC, Groen HJM, et al. Arginase-1 mRNA expression correlates with myeloid-derived suppressor cell levels in peripheral blood of NSCLC patients. Lung cancer 2013, 81(3): 468–474. [DOI] [PubMed] [Google Scholar]
- 31.Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2007, 13(2 Pt 2): 721s–726s. [DOI] [PubMed] [Google Scholar]
- 32.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. Journal for immunotherapy of cancer 2017, 5(1): 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Secondini C, Coquoz O, Spagnuolo L, Spinetti T, Peyvandi S, Ciarloni L, et al. Arginase inhibition suppresses lung metastasis in the 4T1 breast cancer model independently of the immunomodulatory and anti-metastatic effects of VEGFR-2 blockade. Oncoimmunology 2017, 6(6): e1316437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 2009, 31(6): 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez GJ, Rao A. Immunology. Cooperative transcription factor complexes in control. Science 2012, 338(6109): 891–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masternak K, Reith W. Promoter-specific functions of CIITA and the MHC class II enhanceosome in transcriptional activation. The EMBO journal 2002, 21(6): 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proceedings of the National Academy of Sciences of the United States of America 2019, 116(25): 12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. The FEBS journal 2018, 285(16): 2944–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. The Journal of investigative dermatology 2002, 118(6): 915–922. [DOI] [PubMed] [Google Scholar]
- 40.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328(5979): 749–752. [DOI] [PubMed] [Google Scholar]
- 41.Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer research 2018, 78(1): 157–167. [DOI] [PubMed] [Google Scholar]
- 42.Zeiner PS, Zinke J, Kowalewski DJ, Bernatz S, Tichy J, Ronellenfitsch MW, et al. CD74 regulates complexity of tumor cell HLA class II peptidome in brain metastasis and is a positive prognostic marker for patient survival. Acta neuropathologica communications 2018, 6(1): 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strauss L, Guarneri V, Gennari A, Sica A. Implications of metabolism-driven myeloid dysfunctions in cancer therapy. Cellular & molecular immunology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sica A, Guarneri V, Gennari A. Myelopoiesis, metabolism and therapy: a crucial crossroads in cancer progression. Cell stress 2019, 3(9): 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiss M, Van Gassen S, Movahedi K, Saeys Y, Laoui D. Myeloid cell heterogeneity in cancer: not a single cell alike. Cellular immunology 2018, 330: 188–201. [DOI] [PubMed] [Google Scholar]
- 46.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. The Journal of experimental medicine 2010, 207(11): 2439–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111(8): 4233–4244. [DOI] [PubMed] [Google Scholar]
- 48.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104(8): 2224–2234. [DOI] [PubMed] [Google Scholar]
- 49.Laoui D, Van Overmeire E, Movahedi K, Van den Bossche J, Schouppe E, Mommer C, et al. Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology 2011, 216(11): 1192–1202. [DOI] [PubMed] [Google Scholar]
- 50.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology 2018, 36(5): 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell 2019, 177(7): 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature immunology 2019, 20(2): 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic acids research 2019, 47(W1): W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566(7745): 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nature methods 2017, 14(10): 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwartz GW, Zhou Y, Petrovic J, Fasolino M, Xu L, Shaffer SM, et al. TooManyCells identifies and visualizes relationships of single-cell clades. Nature methods 2020, 17(4): 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 related to Fig. 1. rMo signature gene expression and cluster analysis.
a, Heatmap displaying the scaled expression patterns of rMo top marker genes across each tumor and immune clusters.
b, Heat map of hierarchical cluster analysis of immune clusters
Supplementary Fig. 2 related to Fig. 1. rMo marker gene expression and GO analysis.
a, Heatmap displaying the scaled expression patterns of rMo top marker genes across myeloid clusters.
b, c, Gene Ontology (Go) analysis of rMo marker genes in Supplementary Fig. 2a.
d, e, Total CD11b+ CCR2+ cell count in bone marrow, spleen (d) and tumor (e) of naïve mice and tumor-bearing mice in Fig. 1e.
f, Tumor growth in WT and Rel−/− mice that were injected subcutaneously with LLC cells (n = 10 mice per group).
g, Percentages of CD11b+ CCR2+ cells in intratumor CD45.2+ cells in Supplementary Fig. 2f, as determined by flow cytometry.
h, i, Tumor weight (f) and percentages of CD8+ cells in tumors (g) in Fig. 1f (n=10/group). Statistical significance was determined by two-tailed unpaired t-test. Data are presented as means ± s.e.m., and pooled from two independent experiments (d-g) (*, P<0.05; **, P<0.01).
Supplementary Fig. 3 related to Fig. 2. rMos functional analysis.
a, b, Flow cytometry gating strategy of the sorted subsets for subsequent function assays in Fig. 2a (a) and Fig. 2b (b).
c, d, Representative flow cytometry plots from the function assay for Fig. 2a (c), Fig. 2b (d)
e, ROS production by sorted CD11b+CCR2+ (rMos) and CD11b+ F4/80+ (TAMs) cells from B16F10 tumor bearing mice.
f, IFN-γ secreted in the supernatant of a as determined by Elisa.
f, Representative flow cytometry plots from the function assay for Fig. 2c
Statistical significance was determined by two-tailed unpaired t-test (f), or one-way ANOVA with Tukey post-hoc test (g). Data are presented as means ± s.e.m., technical replicates of pooled samples (5 mice per group) were tested; and experiments were repeated 2 times independently with similar results and data of one representative experiment are shown.
Supplementary Fig. 4 related to Fig. 3. Public ChIP-seq data of bone marrow cells.
a, b, Public ChIP-seq data that show the binding peak of c-Rel, CEBPb, and p65 on Ccr2 (a) and Il1b (b) loci of bone marrow myeloid cells.
Supplementary Fig. 5 A working model of rMos.
rMos are generated in the bone marrow, migrate to TME and differentiate into TAMs in TME. Please see the text for details.
Supplementary Table 1: Primers used for PCR