

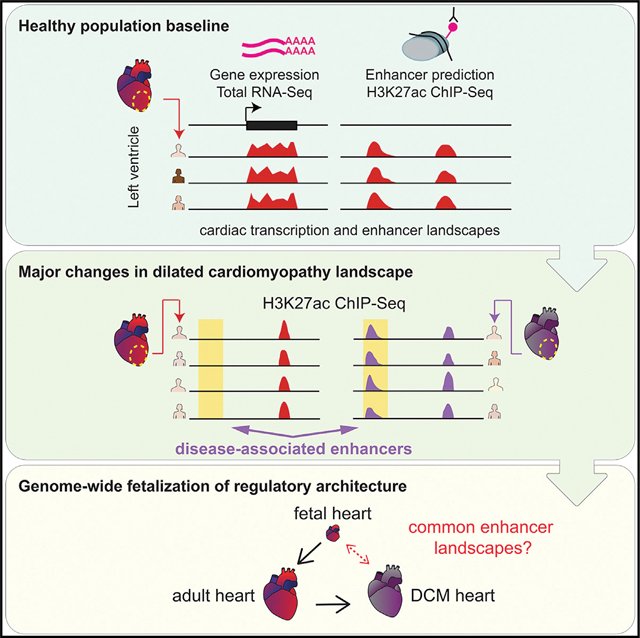
SUMMARY
Heart disease is associated with re-expression of key transcription factors normally active only during prenatal development of the heart. However, the impact of this reactivation on the regulatory landscape in heart disease is unclear. Here, we use RNA-seq and ChIP-seq targeting a histone modification associated with active transcriptional enhancers to generate genome-wide enhancer maps from left ventricle tissue from up to 26 healthy controls, 18 individuals with idiopathic dilated cardiomyopathy (DCM), and five fetal hearts. Healthy individuals have a highly reproducible epigenomic landscape, consisting of more than 33,000 predicted heart enhancers. In contrast, we observe reproducible disease-associated changes in activity at 6,850 predicted heart enhancers. Combined analysis of adult and fetal samples reveals that the heart disease epigenome and transcriptome both acquire fetal-like characteristics, with 3,400 individual enhancers sharing fetal regulatory properties. We also provide a comprehensive data resource (http://heart.lbl.gov) for the mechanistic exploration of DCM etiology.
In brief
Spurrell et al. analyze the transcriptomes and epigenomes of cardiac tissue from healthy individuals and patients with dilated cardiomyopathy, and identify reproducible shifts in the global regulatory landscape. Comparison with fetal heart tissue samples reveals that thousands of predicted enhancer sequences revert to fetal-like chromatin states in heart disease.
Graphical Abstract
INTRODUCTION
Heart failure resulting from cardiomyopathy is a prevailing cause of adult mortality worldwide (World Health Organization, 2022). Idiopathic dilated cardiomyopathy (DCM) is the most common cause of congestive heart failure (Towbin and Bowles, 2002), and is characterized by the dilation and weakening of the left ventricle and decreased ejection fraction in the absence of coronary artery disease or other abnormalities (Hershberger et al., 2010). Approximately one-third of DCM cases are explained by coding mutations in known genes (Hershberger and Siegfried, 2011; Norton et al., 2012). Gene expression studies have been widely used to facilitate the identification of additional causal genetic variation and to understand the transcriptional pathways impacted during heart failure, which may provide new therapeutic targets (Barth et al., 2006; Small and Olson, 2011; Tan et al., 2002). Transcriptional profiling of failing hearts, including those with idiopathic DCM, has identified numerous coding and non-coding genes whose expression is altered in heart disease (Depre et al., 1998; Herrer et al., 2014; Long et al., 2016; Park, 2009; Tan et al., 2002; Yang et al., 2014). These studies have consistently shown that heart failure is associated with the reactivation of a fetal gene program: a set of genes with well-established roles in fetal heart development that are normally repressed in a non-failing adult heart (Dirkx et al., 2013; Oka et al., 2007). Notably, a fetalization mechanism has also been suggested for similar pathways involved in lung disease (Alvira, 2014; Yuan et al., 2018) and tumorigenesis (Saito et al., 2018). However, the full quantitative extent of fetal gene program reactivation in disease processes and the identity of the gene regulatory sequences correlated with these changes remain unknown.
Gene expression is controlled in part by enhancers, a major class of cis-regulatory elements, which can act over large genomic distances to activate gene transcription in specific cell types or developmental stages (Long et al., 2016). Chromatin immunoprecipitation followed by sequencing (ChIP-seq) targeting enhancer-associated chromatin marks (e.g., H3K27ac) can be used to generate accurate genome-wide maps of enhancers active in a tissue of interest (Park, 2009; Won et al., 2008). However, ChIP-seq data sets from human heart samples described to date have not been systematically assessed for reproducibility of the cardiac enhancer landscape in healthy individuals (Bernstein et al., 2010; Dickel et al., 2016; Gillum, 1986; May et al., 2011; Pei et al., 2020; Tan et al., 2020). Recent studies also identified initial, smaller sets of regulatory sequences showing differential epigenomic signatures specific to failing human heart samples (Gilsbach et al., 2018; Tan et al., 2020), but the full extent of disease-associated changes to the cardiac enhancer landscape and its overlap with fetal epigenome signatures have remained unclear.
We performed a comprehensive exploration of human heart enhancers in left ventricle samples from an extensive cohort of control adults and individuals with idiopathic DCM (see study overview in Figure 1). In healthy hearts, we observe a highly reproducible enhancer landscape across individuals. Furthermore, we observe major changes in the regulatory landscape of DCM samples and, through comparison with fetal heart tissue samples, demonstrate that more than 3,400 enhancers adopt fetal-like regulatory activity states in DCM. We also show that these enhancers are enriched in a specific subset of basic helix-loop-helix (bHLH) transcription factor binding sites and are sufficient to drive cardiac expression in vivo in transgenic mouse reporter assays. We demonstrate, through correlation with transcriptome data, that enhancers activated in disease are in close proximity to genes with increased expression in disease. Finally, we identify more than 200 fetal heart genes that are upregulated in adult heart failure. Taken together, our findings indicate that a large number of fetal heart enhancers become consistently reactivated in heart disease, and our results identify the genomic locations and activation states of these enhancers, providing a rich resource for the exploration of mechanistic links between enhancers and clinical cardiac phenotypes.
Figure 1. Overview of this study.
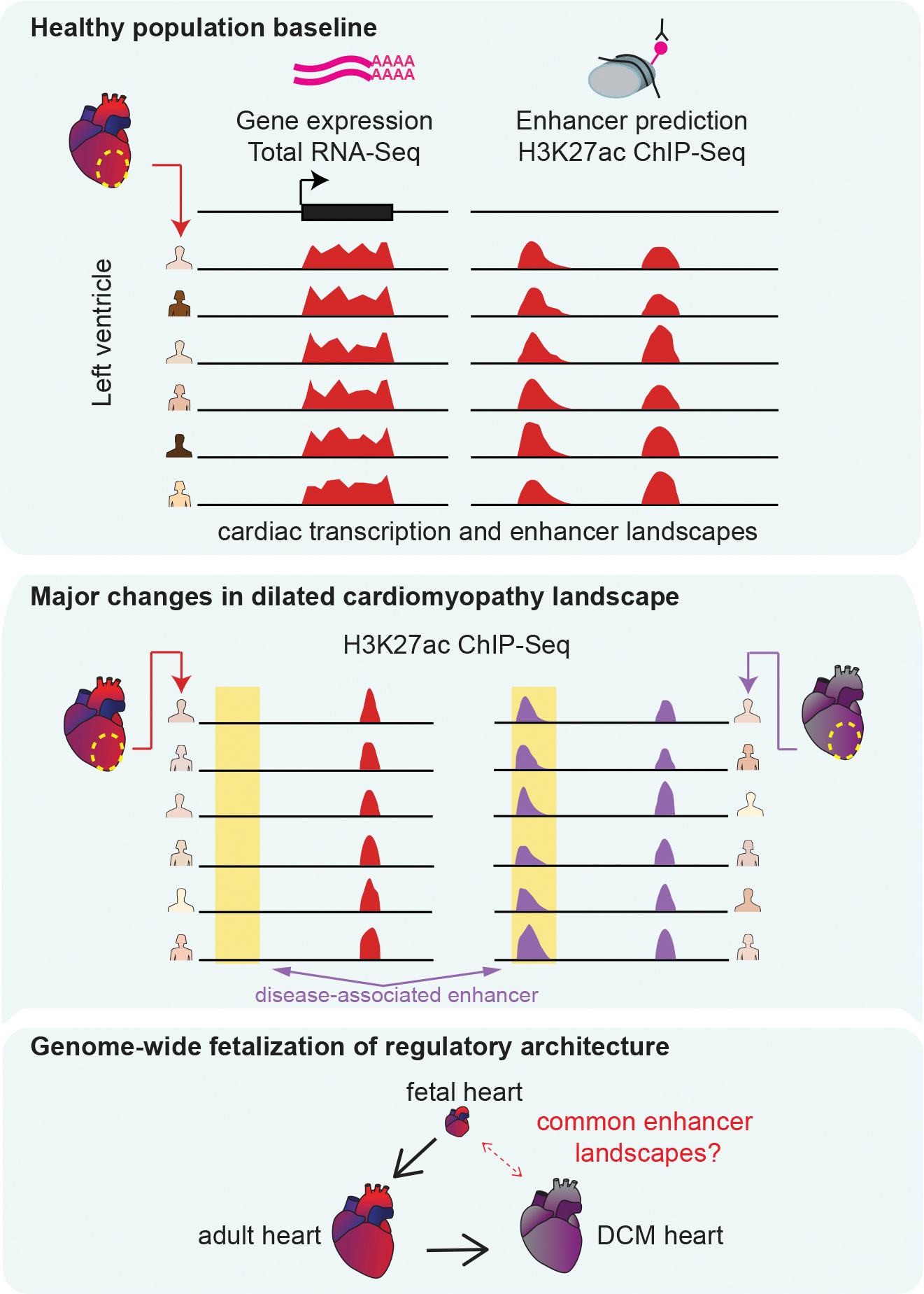
We performed RNA-seq and ChIP-seq on left ventricle samples from 18 healthy donors. We identified reproducible enhancer predictions and gene expression patterns in healthy heart (top). Next, we identified global alterations in enhancer occupancy from 18 hearts with idiopathic dilated cardiomyopathy (DCM, center). Last, we compared adult healthy and disease states with five embryonic or fetal hearts and identified similarities in the enhancer landscapes of DCM and fetal heart (bottom).
RESULTS
Highly reproducible heart enhancer landscape in healthy individuals
To assess the heterogeneity of the gene regulatory landscape in healthy human hearts, we used chromatin profiling to identify putative enhancer regions. We performed ChIP-seq with an antibody to H3K27ac on left ventricle samples from 16 male and 10 female healthy adult donor hearts for which an acceptable transplant recipient could not be found (Figure 1 and Table 1 and Table S1). The healthy cohort included 18 European American and eight African American donors ranging in age from 20 to 65 years old. In parallel, we performed RNA sequencing (RNA-seq) on these samples to enable a comparison of each enhancer profile with its associated transcriptome. We defined enhancers as genomic regions with significant H3K27ac enrichment that are at least 1 kb away from the nearest transcription start site (see STAR Methods). All chromatin and RNA-seq data from these samples, as well as disease samples described below, are available for download and browsing in an accompanying web resource (http://heart.lbl.gov). Across all 26 control samples, we identified a total of 46,826 predicted distal enhancers present in at least one sample, with more than 70% (33,317) found in at least two individuals (Figure S1A and Table S2A). Healthy samples had an average of 14,394 putative distal enhancers (Figure 2A, top). Each sample shared on the average between 57% and 90% of predicted enhancers with any other sample. The observed numbers of peaks shared between samples was significantly larger than that expected by chance (Figure 2A, bottom). Peaks found in a larger number of samples had higher average ChIP-seq enrichment signals (Figure S1B). To determine the functions of genes associated with the cardiac enhancers predicted through this approach, we examined the enrichment of functional ontology terms (McLean et al., 2010) among genes located near candidate regions showing strong H3K27ac enrichment in at least 24 healthy controls (see STAR Methods). Among the most significantly enriched annotations derived from mouse gene expression studies, 11 of the top 20 are cardiovascular-related functions (Table S2B). Our results indicate that the enhancer landscape in healthy human hearts is overall highly reproducible, with quantitatively modest differences between individuals.
Table 1.
Clinical characteristics of adult subjects
| Non-failing controls |
DCM |
||
|---|---|---|---|
| EA | AA | EA | |
|
| |||
| Total subjects | 18 | 8 | 18 |
| Gender: Male/Female | 10/8 | 6/2 | 10/8 |
| Age at surgery (years) | 50 ± 10 | 49 ± 13 | 54 ± 8 |
| BMI (kg/m2) | 29 ± 7 | 26 ± 4 | 27 ± 6 |
| Prior hypertension (%) | 33 | 37.5 | 28 |
| Prior diabetes (%) | 5.5 | 14† | 11 |
| LVEF (%) | 63 ± 6 | 54 ± 8† | 14 ± 3* |
| Heart mass (g) | 373 ± 74 | 424 ± 82 | 525 ± 95* |
Quantitative data provided as mean ± standard deviation.
p < 0.05 significantly different between EA DCM and EA non-failing cohorts by two-tailed t test.
Data unavailable for some subjects.
AA, African American; BMI, body mass index; EA, European American; LVEF, left ventricular ejection fraction. For additional clinical information about subjects see Table S1.
Figure 2. The cardiac enhancer landscape is highly reproducible across individuals.
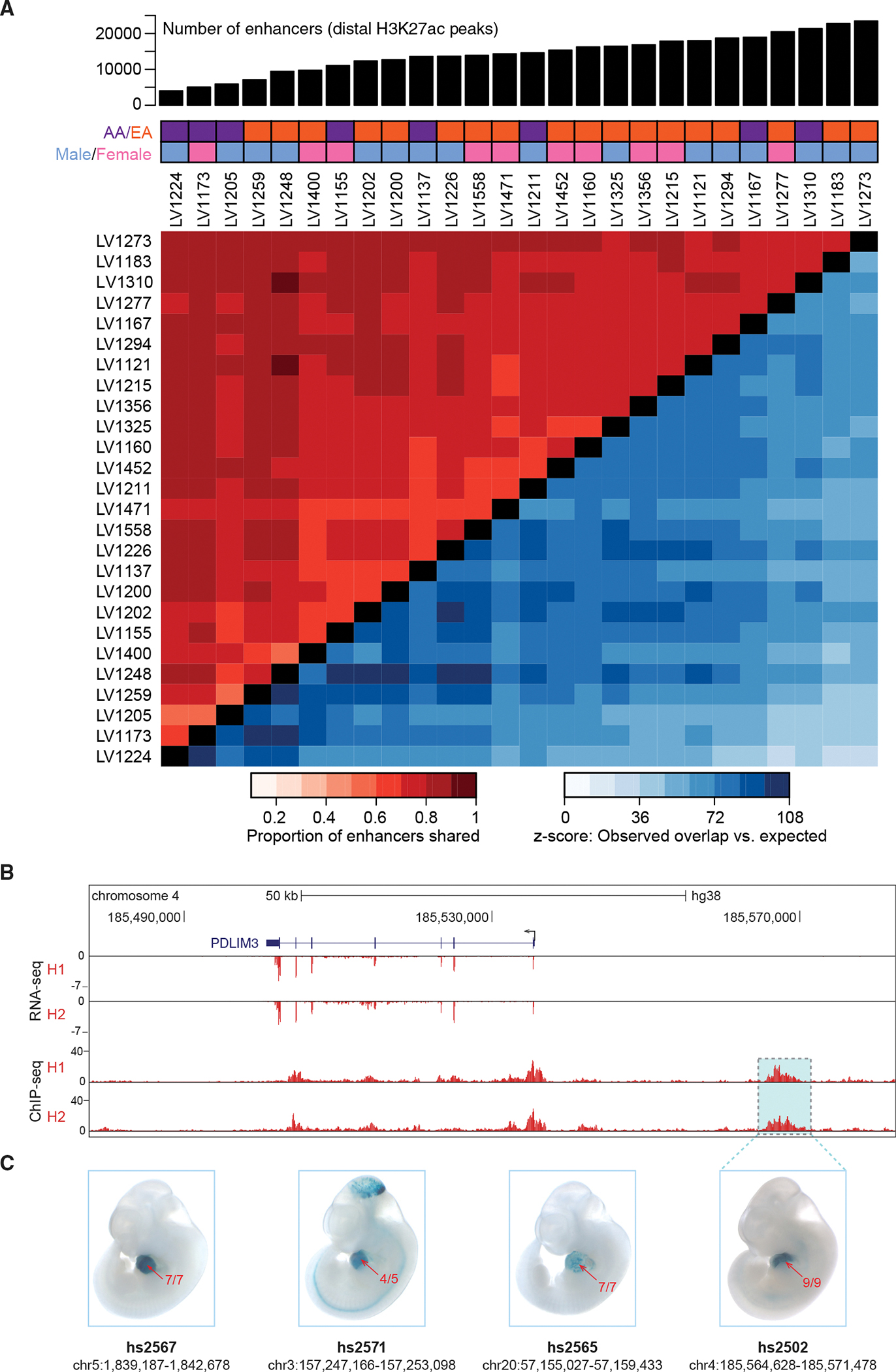
(A) Top: number of predicted enhancers per sample for 26 healthy left ventricle samples. Middle: demographic information for each subject (AA, African American; EA, European American). Bottom: Heatmap showing the proportion of peaks shared between samples (red tones in top left), along with z-scores indicating how many standard deviations the observed number of shared peaks exceeded random expectation (blue tones in bottom right, see STAR Methods).
(B) Paired RNA-seq and ChIP-seq tracks from two samples, H1 and H2, at the PDLIM3 locus.
(C) Transgenic mouse assay validation of four heart enhancers, including one upstream of PDLIM3 (see Figure S2 for results for additional predicted heart enhancers). One representative embryonic day 11.5 embryo is shown for each enhancer, and numbers in red show the reproducibility of heart staining in each transgenic experiment. Information underneath each embryo includes the identification number in the VISTA Enhancer Browser (Visel et al., 2007) (enhancer.lbl. gov) and the human genome coordinates (hg38) of each tested enhancer. Embryos have an average crown-rump length of 6 mm.
To verify that enhancers predicted by this ChIP-seq approach are bona fide cardiac-specific regulatory sequences, we first compared our predicted human heart enhancers with the VISTA Enhancer Browser (https://enhancer.lbl.gov), an existing catalog of in vivo validated enhancers (Visel et al., 2007). This database contains 284 human and mouse loci that have been tested using transgenic mouse assays and have validated activity in the heart. Approximately 51% (n = 146) of these validated heart enhancers overlapped peaks in our adult human heart samples (Table S2A). We additionally performed in vivo validation using transgenic mouse reporter assays (Kothary et al., 1989) at embryonic day 11.5 for several candidate human adult heart enhancers. Examples of reproducible in vivo cardiac reporter activity driven by eight different candidate enhancers, including one near PDLIM3, are shown in Figures 2B and 2C and Figure S2. Together, these results reinforce that predicted adult heart enhancers drive gene expression in heart tissue.
Genome-wide changes in enhancer architecture in DCM
We next evaluated possible global changes in enhancer architecture in heart disease using transcriptome and chromatin profiling on explanted left ventricle samples from individuals with late-stage idiopathic DCM. While less common than DCM associated with ischemia or infarction, idiopathic DCM shares important physiological characteristics with these conditions, including ventricular dilation and clinical signs of heart failure (Berger et al., 1997; Maron et al., 2006). ChIP-seq data were generated from 18 European American DCM cases that were sex-matched and selected to be close in age to the 18 European American healthy control samples (see STAR Methods, Table 1, and Table S1), and we obtained RNA-seq data from 15 of the 18 DCM left ventricle samples. The DCM cohort showed similar inter-individual reproducibility as that seen in the control group (Figure S3A). Healthy and DCM samples clustered separately from each other in principal-component space, both when using gene expression and enhancer activity as inputs (Figures 3B and 3C). Biological sex was not identified as a substantial driver of variance (Figures S3B and S3C). Of the 18,913 genes expressed in at least two individuals, 861 (4.6%) were differentially expressed between disease and health (raw p < 0.01, false discovery rate [FDR]-corrected p < 0.05, and |log2[fold change]| ≥ 1; Figure 3C and Table S3A). From the combined pool of 36 left ventricle samples from European American subjects (healthy controls and DCM), we identified 38,883 predicted enhancers present in at least two individuals (Table S3B) of which 6,850 (18%) were differentially bound between healthy and DCM samples (Ross-Innes et al., 2012) (see STAR Methods, Figure 3C, and Table S3C). These results show that, in addition to the dysregulation of hundreds of cardiac-expressed genes, the genome-wide enhancer architecture is substantially altered in the left ventricle from DCM patients.
Figure 3. Extensive reprogramming of the epigenomic architecture in heart disease.
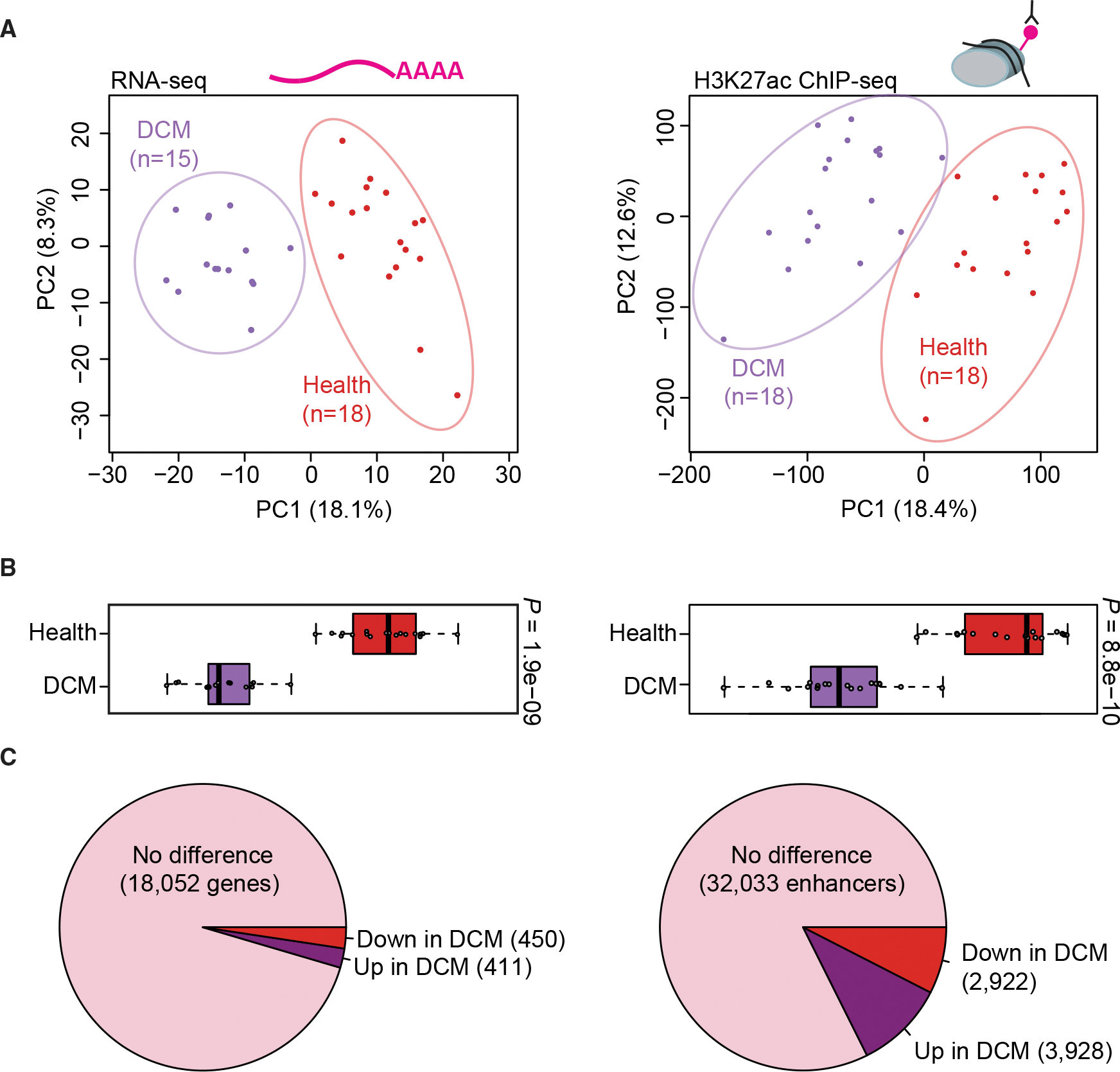
(A) Principal-component analysis showing the first two principal components (PC1 and PC2) for the top 1,000 variably expressed genes from RNA-seq (left) and for distal enhancer peaks from ChIP-seq (right). Each point indicates a unique sample, color coded by cohort.
(B) Boxplots showing PC1 for each sample by cohort (left: RNA-seq, right: ChIP-seq). Boxplots indicate median and quartile values for each data set; points indicate individual samples. PC1 separates disease state in both RNA-seq and ChIP-seq data (p values by two-sided Mann-Whitney U test).
(C) Pie charts showing the proportion of unchanged versus differentially expressed genes (left) and differentially bound enhancer peaks (right) relative to each cohort (see STAR Methods for details).
Differential regulation of disease-related genes and pathways
To further assess the regulatory relationship between differentially active enhancers and differentially expressed genes in DCM, we examined correlations between RNA-seq and ChIP-seq data. We observed multiple anecdotal examples of differentially active enhancers located near genes with the same directionality of differential expression. An illustrative example is provided by the SMOC2 locus, where a strong, DCM-specific increase in H3K27ac signal at a site 20 kb upstream of the promoter correlates with a strong increase in SMOC2 expression in DCM (Figure 4A). To examine this effect beyond individual loci, we performed a global correlation analysis, assessing the enrichment of differentially bound candidate enhancers near differentially expressed genes. Differentially bound candidate enhancers with increased binding are indeed enriched near differentially upregulated genes, and candidate enhancers with decreased binding are enriched near downregulated genes, with the most pronounced enrichment observed within the 100-kb window closest to the promoter (Figures 4B and 4C).
Figure 4. Differentially bound distal enhancers associated with heart failure-specific gene expression changes.
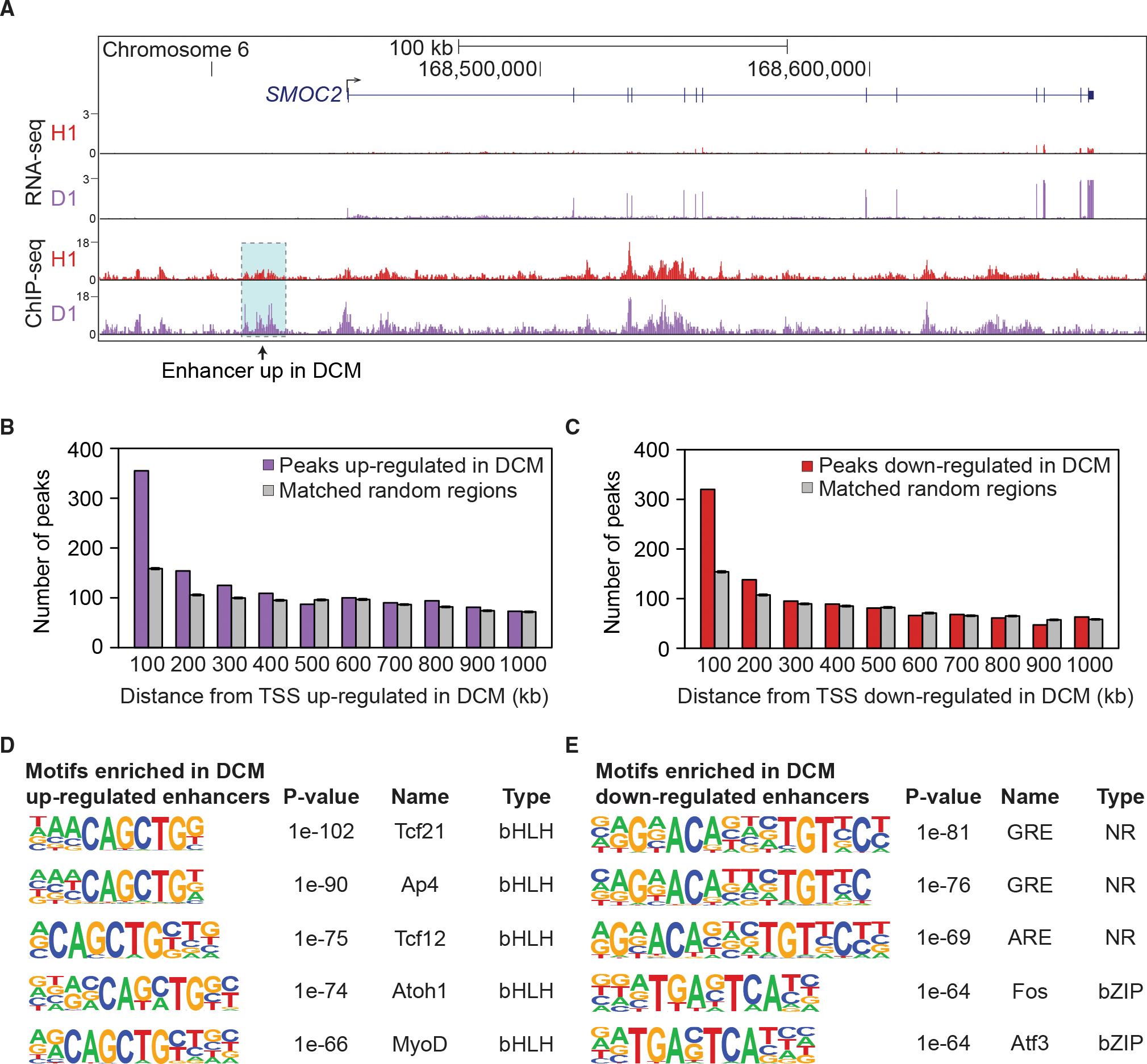
(A) Co-occurrence of increased expression of SMOC2 (by RNA-seq) and increased H3K27ac at a nearby enhancer (by ChIP-seq) in DCM. Shown are data from a representative healthy (H1) and DCM (D1) sample.
(B) Distal ChIP-seq peaks relative to their distance from transcription start sites (TSS) of genes upregulated in DCM, divided into 100-kb bins. Purple: distal peaks upregulated in DCM. Gray: An equal-size, random subset of regions matched to all heart ChIP-seq peaks was assessed. The average across 200 sets of randomized control elements is shown.
(C) Same analysis as in (B), but for genes with decreased expression in DCM and distal peaks downregulated in DCM (red). An equal-size, random subset of regions matched to all heart ChIP-seq peaks was assessed (gray).
(D and E) Transcription factor binding sites enriched in peaks upregulated (D) and downregulated (E) in DCM (Heinz et al., 2010). p values by HOMER.
To assess the biological functions of the differentially expressed genes and differentially active enhancers, we examined their enrichment for biological process ontology terms (McLean et al., 2010). For the 3,928 enhancer peaks with predicted increased activity in DCM, there is enrichment for terms including extracellular matrix remodeling (Table S3D). Similarly, genes upregulated in DCM independently show enrichment for extracellular matrix components and cell adhesion (Table S3E). These results, while not direct evidence of activated pathways in DCM, reinforce our conclusion that DCM-specific enhancers are enriched near genes that are differentially regulated in disease, independently highlighting fibrosis pathways in heart failure (Louzao-Martinez et al., 2016).
To identify candidate transcription factors (TFs) that may be driving altered enhancer activity in DCM, we performed binding site enrichment analysis (Heinz et al., 2010) for distal enhancer peaks with increased or decreased activity in DCM. In peaks with increased activity in DCM, binding sites for bHLH TFs are enriched (Figure 4D). Several of the most significant factors in this class, including AP4 and TCF21, are involved in epithelial-to-mesenchymal transition (Jackstadt et al., 2013; Sazonova et al., 2015), which may contribute to cardiac fibrosis (Zeisberg et al., 2007). In peaks with decreased activity in DCM, there is enrichment in nuclear receptor transcription factor binding sites (Figure 4E). We found particularly pronounced enrichment in glucocorticoid response elements, which are involved in growth and immune response pathways (Ratman et al., 2013). We also found enrichment of bZIP factors that are part of the AP-1 complex, which has been suggested to act as a homeostatic regulator required to maintain a steady state of cell proliferation (Rohini et al., 2018; Shaulian and Karin, 2002). Taken together, these data show that heart disease-associated enhancers are enriched near genes differentially regulated in disease and that known functions of these genes are consistent with roles in heart failure.
Fetalization of the regulatory landscape in heart disease
Previous studies have reported that some genes that are essential for fetal heart development are downregulated in the adult heart, but become reactivated in failing hearts (Dirkx et al., 2013; Nandi and Mishra, 2015; Tan et al., 2002). However, beyond anecdotal examples, the transcriptome-wide scale of such re-expression of prenatally expressed human genes in DCM has remained unclear. Likewise, it is unknown whether reactivation of these fetal genes is associated with reactivation of fetal regulatory elements. To comprehensively define the fetal gene program reactivated in DCM and to test whether enhancer regulation correlates with the re-expression of fetal genes, we performed ChIP-seq and RNA-seq on five embryonic and fetal human heart samples, covering developmental stages ranging from 8 weeks to 17 weeks post conception (Figure 5A and Tables S4A and S4B; see STAR Methods for sample details). We performed dimensionality reduction by means of principal-component analysis on healthy, DCM, and prenatal samples and found that they show distinct genome-wide transcriptome and enhancer signatures (Figure 5B). Consistent with the notion of reactivation of fetal genes in heart disease, we found that genes differentially regulated during fetal development are enriched among those differentially regulated in DCM relative to healthy adult heart (Figure 5C). To examine if this coincides with enhancers returning to a fetal-like state in heart disease, we focused on 4,547 regions whose activity changed significantly in either fetal or DCM samples (as compared with adult healthy ones). Most of these enhancers (82%, n = 3,471) changed in a concordant fashion, an overlap that was enriched 1.6-fold relative to one expected by chance (Figures 5C and 5D). For the 1,622 enhancers with predicted increased activity in DCM and fetal, there is an enrichment for gene ontology terms associated with fibrosis and extracellular matrix remodeling, along with terms involving transforming growth factor β signaling (Table S4C). Genes in these pathways are upregulated in cardiac development and in heart failure (Azhar et al., 2003; Dobaczewski et al., 2011).
Figure 5. Fetalization is a driver of regulatory change in heart disease.
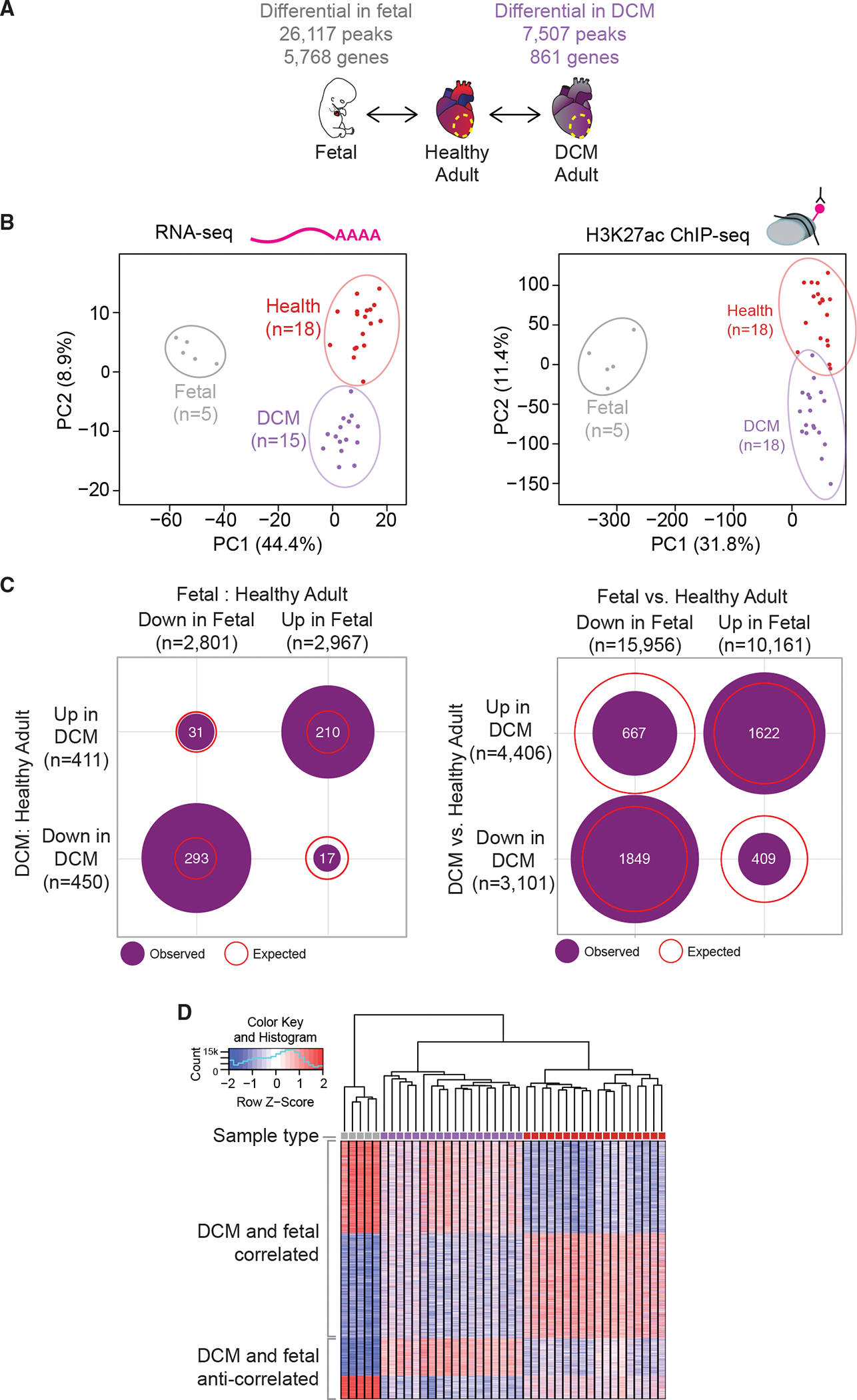
(A) Schematic of experimental comparisons and number of differential genes/peaks.
(B) Principal-component analysis showing the first two principal components (PC1 and PC2) for the top 1,000 variably expressed genes from RNA-seq (left) and for distal enhancer peaks from ChIP-seq (right). Prenatal samples are gray, DCM samples are purple, and healthy samples are red.
(C) Number of enhancer peaks co-regulated in prenatal and DCM states for RNA-seq (left) and ChIP-seq (right). Purple circles show number of peaks overlapping in each quadrant. Red lines show expected overlap between each category.
(D) Heat map showing all differential peaks between 41 samples: 5 prenatal (gray), 18 DCM (purple), and 18 healthy (red).
In addition to enhancers that showed fetal-like regulatory properties in DCM, we also observed populations of enhancers that are either anti-correlated with fetal enhancer activity states (n = 1,076) or that were DCM-specific, i.e., different from both fetal and healthy adult cardiac tissue (n = 2,960). Both of these groups of enhancers indicate that while the genome-wide alterations in enhancer landscape in DCM are dominated by a shift toward fetal-like signatures, there are clearly additional, DCM-specific molecular events, as expected from the pathological context associated with heart disease. To provide additional insight into these gene regulatory processes, we examined enrichment of transcription factor binding site motifs in the enhancers exclusively upregulated in fetal or DCM samples, and ones that have increased activity in both. While fetal-exclusive enhancers tended to be enriched for GATA motifs, both sets of DCM enhancers were enriched for bHLH motifs. However, the shared DCM-fetal group was strongly enriched for TEAD factors, which were also detected in both exclusive groups, but at lower levels of enrichment (Figure S4). Taken together, our results support that the upregulation of fetal genes in DCM is associated with partial reactivation of fetal enhancers. Importantly, the genome-wide expression and enhancer maps generated through this study will enable the targeted follow-up of regulatory events associated with the pervasive reactivation of fetal expression programs, as well as molecular downstream events specifically associated with disease progression.
DISCUSSION
Understanding the mechanisms regulating gene expression in healthy and pathological states of the heart is critical for understanding cardiac biology and disease. By performing enhancer discovery by ChIP-seq on heart tissues from a sizable cohort of individuals, we were able to assess the baseline variation in the heart enhancer landscape across the population. Importantly, we found that the activity of cardiac enhancers is well conserved across individuals. This observation, together with the underlying comprehensive maps of heart enhancers generated through this study, provides an important reference point for assessing the impact of environmental factors, genetic variation, and disease-associated molecular processes on cardiac gene regulation.
Similar to the high reproducibility of the enhancer landscape in the healthy heart, our systematic assessment of enhancers in DCM revealed disease-associated activity changes in thousands of enhancers that were highly reproducible across individuals. This insight implies that pathological disease states of the heart are not associated with a general disruption of coordinated gene expression, but instead show a complex, well-defined shift in genome-wide transcriptional regulation involving a distinct set of distant-acting enhancers.
Finally, our comparison with data from fetal heart tissue uncovered that, beyond the limited number of examples described to date (Dirkx et al., 2013; Oka et al., 2007), hundreds of fetal genes and more than 1,600 fetal enhancers become consistently reactivated in adult heart disease. Our study identified the genomic locations and activation states of this intriguing subset of disease-associated enhancers, offering an entry point for targeted experimental exploration for hundreds of genes with disease-associated activity changes. Reactivation of individual fetal pathways has also been observed in other, non-cardiac disease processes (Alvira, 2014; Saito et al., 2018; Yuan et al., 2018). While limited to anecdotal examples, in conjunction with data from the present study, these reports raise the possibility that widespread reactivation of a subset of fetal enhancers is a more generalized phenomenon in disease, and perhaps non-disease processes that involve tissue plasticity and remodeling.
Taken together, our study highlights how the etiology of a disease process is tightly interlinked with, and possibly driven by, reproducible activity changes across thousands of enhancers in the human genome. The identification of substantial overlap between disease-associated and developmentally active cardiac enhancers reinforces the notion that modulation of genes and pathways normally active in the embryonic and fetal heart may affect disease progression (Dirkx et al., 2013), thus providing inspiration for new therapeutic strategies.
Limitations of the study
We included samples from people of different age, sex, and ethnicity in our study. We also aimed to match healthy adults and those with DCM in terms of sex and age. Biological sex was not identified as a substantial driver of variance and we detected no difference between samples from healthy adults with European and African ancestry. However, it is possible that with increased sample size such differences will become apparent. Similarly, our sample size was insufficient for a conclusive assessment of possible effects of age on the epigenomic landscape, or to investigate interactions between variables, such as age-disease status or ethnicity-disease status.
Our study used epigenomic and transcriptomic methods to assess bulk tissue samples, which consist of heterogeneous cell populations. Complementary single-cell analyses could reveal additional information about disease mechanisms and their relationship with fetal states.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Axel Visel (avisel@lbl.gov).
Materials availability
All plasmids and reagents generated in this study, as well as archived surplus LacZ-stained embryos for selected enhancers are available from the authors.
Data and code availability
All ChIP-seq and RNA-seq data are available through GEO: GSE126573. Images of LacZ-stained embryos are available from the VISTA Enhancer Browser (https://enhancer.lbl.gov), and raw images are available on request from the lead authors.
All original code has been deposited at https://gitlab.com/lotard/spurrell_fetalization and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
All aspects of this study involving human tissue samples were reviewed and approved by the Human Subjects Committee at Lawrence Berkeley National Laboratory (Protocol Nos. 00023126 and 00022756). Procurement of adult human myocardial tissue was performed under protocols approved by Institutional Review Boards at the University of Pennsylvania (Protocol No. 802781) and the Gift-of-Life Donor Program (Pennsylvania, USA). DCM hearts were procured at the time of orthotopic heart transplantation at the Hospital of University of Pennsylvania. All of the 18 DCM patients had routine etiologic workup for cardiomyopathy, as outlined in recent guidelines (Japp et al., 2016). Significant coronary artery disease, exposure to cardiotoxic medications, heavy ethanol use, untreated thyroid disease and unremitting tachycardia were excluded in all cases. Most of the patients did not have endomyocardial biopsy. Two subjects reported a first degree relative with a form of heart disease that could suggest a familial genetic etiology (brother of #1262 reported to have dilated cardiomyopathy; father of #1371 reported to have non-ischemic cardiomyopathy), but in both cases the family member was unavailable for further evaluation. None of the subjects included underwent clinical genetic testing prior to transplantation. Healthy hearts were obtained at the time of organ donation from cadaveric donors. In all cases, hearts were arrested in situ using ice-cold cardioplegia solution, transported on wet ice, and flash frozen in liquid nitrogen within 4 hours of explantation. All samples were full-thickness biopsies obtained from the free wall of the left ventricle. Detailed clinical information for adult DCM and control samples is provided in Table S1. Fetal and embryonic human heart samples were obtained from the Human Developmental Biology Resource at Newcastle University (hdbr.org), in compliance with applicable state and federal laws and with fully informed consent. Embryonic and fetal samples included: one post conception week 8 (Carnegie stage 22) whole heart sample, three post conception week 10 whole heart samples, and one post conception week 17 left ventricle sample. All adult and fetal samples were shipped on dry ice and stored at −80°C until processed.
Transgenic mouse assays
All animal work was reviewed and approved by the Lawrence Berkeley National Laboratory Animal Welfare and Research Committee. Transgenic assays were performed in Mus musculus FVB strain mice and included both male and female embryos. Sample sizes were selected based on our previous experience of performing transgenic mouse assays for >3,000 enhancer candidates (Pennacchio et al., 2006; Visel et al., 2007). Mouse embryos were only excluded from further analysis if they did not carry the reporter transgene or if they were not at the correct developmental stage. Randomization and experimenter blinding were unnecessary and not performed for transgenic assays, as all resulting embryos were treated with identical experimental conditions and no direct comparisons were made between groups of mice.
METHOD DETAILS
ChIP-seq
Chromatin immuno-precipitations were performed as previously described (Visel et al., 2009) with some modifications. Briefly, frozen left ventricle tissue was pulverized with a mortar and pestle, resuspended in PBS, and cross-linked with 1% formaldehyde at room temperature for 10 min. Chromatin was sonicated to obtain fragments with an average size ranging between 100–600 bp. Chromatin was incubated for 2h at 4°C with 5 μg of Active Motif H3K27ac antibody (cat# 39133, lot 01613007). Protein A and G Dynabeads (Invitrogen) were then added to this chromatin/antibody mixture for 30 min at 4°C. Immuno-complexes were sequentially washed. The protein/DNA complexes were eluted in an SDS buffer (1% SDS, 50 mM Tris pH 8.0, 10 mM EDTA) at 37°C for one hour. Samples were treated with Proteinase K at 37°C and reverse-crosslinked overnight. Finally, the DNA was purified on Zymo ChIP clean and concentrate columns (Zymo Research), and the quality was assessed on the Agilent bioanalyzer. The ChIP-seq libraries were prepared using the Illumina TruSeq library preparation kit followed by pooling of libraries. Libraries were pooled and sequenced via single end 50 bp reads on a HiSeq 2500 (6 libraries per lane) or HiSeq 4000 (8 libraries per lane) (Illumina).
ChIP-seq data was analyzed using the ENCODE Uniform Processing Pipelines (https://www.encodeproject.org/pipelines/) implemented at DNAnexus (https://www.dnanexus.com). ChIP-seq data was analyzed using the ENCODE histone ChIP-seq Unary Control Unreplicated (GRCh38) – pipeline version 1.2 (code available from https://github.com/ENCODE-DCC/chip-seq-pipeline). Briefly, reads were mapped to the human genome (GRCh38) using bwa aln v 0.7.10 (Li and Durbin, 2009) with settings -q 5 -l 32 -k2. Duplicate reads were identified and removed using PICARD (https://broadinstitute.github.io/picard/). Non-duplicate reads were used as input for peak calling using MACS 2.0 (Zhang et al., 2008), with the experiment-matched input DNA used as a control. To assess reproducibility between samples within the same cohort (e.g., healthy controls), we performed pairwise comparisons between all possible sample pairs and calculated 1) the proportion of enhancer peaks in common between the two samples relative to the sample with fewer overall peaks, and 2) whether the observed number of peaks in common between two samples exceeded that expected by statistical chance if each sample’s peaks were randomly drawn from the total pool of enhancers from that cohort. For the latter, z-scores were calculated to compare the observed value to the expected value (k) using a normal distribution centered at k and with a standard deviation equal to that expected by random chance.
For the violin plots in Figure S1B, for each one of the TSS-distal regions showing a H3K27ac peak in one or more adult control samples, the TMM-normalized enrichment values were calculated using DiffBind, and further normalized for the size of the region (per kb). Each region was then described using three numbers: 1.) The fraction of regions showing a peak (i.e. a number between 1 and 26, depending on the number of samples showing a peak [FDR ≤ 0.05]); 2.) the average enrichment value across those samples showing a peak; and 3.) the average enrichment value across those samples not showing a peak.
RNA-seq
RNA was isolated from homogenized left ventricle using the TRIzol Reagent (Life Technologies). RNA samples were DNase-treated with the TURBO DNA-free Kit (Life Technologies), and RNA quality was then assessed using a 2100 Bioanalyzer (Agilent) with an RNA 6,000 Nano Kit (Agilent). RNA sequencing libraries were made using the TruSeq Stranded Total RNA with Ribo-Zero Human/Mouse/Rat kit (Illumina) according to manufacturer instructions. RNA-seq libraries were subjected to an additional purification to remove remaining high molecular weight products as follows: sample volume was increased to 100 μL by addition of 1XTE buffer or Illumina Resuspension Buffer and then incubated with 60 μL Agencourt AMPure XP beads for 4 min. The beads were pelleted by incubation on a magnet, and the entire supernatant was transferred to a tube containing 50 μL of fresh AMPure XP beads and incubated for 4 min. After pelleting the new beads with a magnet, the supernatant was discarded, the beads washed twice with 80% ethanol and the DNA was eluted in 30 μL Illumina Resuspension buffer. The resulting RNAseq libraries were diluted 10×, and their quality and concentration were assessed using a 2100 Bioanalyzer with the High Sensitivity DNA Kit (Agilent) and a Qubit Fluorometer with the Qubit dsDNA HS Assay Kit (Life Technologies). RNAseq libraries were pooled and sequenced via single end 50 bp reads on a HiSeq 2500 (4 libraries per lane) or HiSeq 4000 (6 libraries per lane) (Illumina). RNA-seq was attempted on all adult samples, with libraries for all 18 healthy and 15/18 DCM samples passing minimum quality metrics for inclusion in the analysis.
Like ChIP-seq, RNA-seq data was analyzed using the DNAnexus instance of the ENCODE Uniform Processing Pipeline. RNA-seq data was analyzed using the ENCODE RNA-Seq (Long) Pipeline – 1 (single-end) replicate pipeline (code available from https://github.com/ENCODE-DCC/rna-seq-pipeline). Briefly, reads were mapped to the human genome (GRCh38) using STAR align (V2.12). Genome-wide coverage plots were generated using bam to signals (v2.2.1). Gene expression counts were generated for gencode v24 gene annotations using RSEM (v1.4.1).
Differential binding analysis
Differential peak analysis between two phenotypes (healthy adult vs DCM adult left ventricle; healthy adult left ventricle vs fetal heart) was performed using DiffBind (Ross-Innes et al., 2012) version 2.10.0. We identified all autosomal differential peaks at least 1,000 bp from a transcription start site using Gencode (Frankish et al., 2019) version 27 annotations. Peaks present in at least two individuals were defined using the dba.count function in DiffBind, and these peaks were used to generate a list of consensus peaks for comparison with downstream differential analysis. We used the DBA_DESEQ2 method to call differential peaks using the multiple testing corrected FDR <0.05, a two-sided raw p value <0.01 and |log2(fold change)| >=1. To reduce the dimensionality of the dataset and visualize the samples in two dimensions, the master dataset generated by DiffBind (namely the normalized signal quantifications in all the regions tested across all samples) were log-2 transformed and scaled (z-score by region), and then subject to Principal Component Analysis (PCA) using the prcomp package in R.
Differential gene expression
Differential gene expression analysis (autosomal genes only) between two phenotypes (healthy adult vs DCM adult left ventricle; healthy adult left ventricle vs fetal heart) was performed using edgeR (Robinson et al., 2010) version 3.20.9. First, genes whose expression was very low in most samples were discarded from further analyses (only those genes showing counts per million mapped reads [CPM)] >= 1 in three or more samples were retained for further analyses). Next, estimateCommonDisp and estimate-TagwiseDisp (prior.df = 10) were run to properly handle over-dispersion at the global and single gene level. Across samples, normalization was then performed using TMM, via the function calcNormFactors. Differentially expressed genes were determined using the exactTest function, as those showing the multiple testing corrected FDR <0.05, a two-sided raw p value <0.01 and |log2(fold change)| >=1. The total number of expressed genes includes all genes present in at least two samples with normalized CPM (counts per million) of at least 1. PCA was performed as described in the previous paragraph, using the log2-transformed, TMM-normalized expression values as input. PCA results were robust to the choice of the most variable genes used as input (range tested: 1,000 to 10,000).
Gene enrichment
To identify gene ontology terms enriched in differential regions we ran GREAT (McLean et al., 2010) version 3.0.0 using default settings, including the association rule: Basal+extension: 5,000 bp upstream, 1,000 bp downstream, 1,000,000 bp max extension, curated regulatory domains included. We used the Lift Genome Annotations tool in the UCSC genome browser (genome.ucsc. edu) to convert hg38 regions to hg19 regions required for GREAT input. We identified the most significantly enriched terms by binomial q value using the GO Biological Process and GO Molecular Function ontologies. To identify gene ontology terms represented in differentially expressed genes, we used PANTHER (Thomas et al., 2003) version 10.0 and ran the PANTHER Overrepresentation Test (release 20181113).
Transcription factor binding site analysis
We ran HOMER (Heinz et al., 2010) v. 4.10.3 using findMotifsGenome.pl with the following parameters: -size −500,500 -len 6,7,8,9,10,12,14. We used a binomial distribution to estimate the p value of the enrichments. Input regions were split into 1-kbp bins prior to analysis.
Closest transcription start site comparison
We calculated the nearest transcription start site upregulated in DCM for all 3,928 peaks with increased binding in DCM. As controls, we ran 200 iterations sampling 3,928 peaks from the consensus set of 38,883 peaks present in at least two adult samples. We calculated the mean and standard error of these data and plotted the 95% confidence interval for these controls. We also ran the corresponding analysis of the nearest transcription start site downregulated in DCM for all 2,922 peaks with less binding in DCM, and calculated a control set with 200 iterations randomly sampling 2,922 peaks from the same set of consensus peaks. We calculated the mean and standard error of these data and plotted the 95% confidence interval for these controls.
In vivo transgenic reporter assays
Enhancer candidate regions were cloned into an Hsp68-promoter-LacZ reporter vector (Pennacchio et al., 2006) using Gibson cloning (Gibson et al., 2009) (New England Biolabs [NEB]). Transgenic mouse embryos were generated by pronuclear injection, and F0 embryos were collected at embryonic day 11.5 and stained for LacZ activity as previously described (Kothary et al., 1989; Pennacchio et al., 2006). Only patterns that were observed in at least three different embryos resulting from independent transgenic integration events of the same construct were considered reproducible. The procedures for generating transgenic and engineered mice were reviewed and approved by the Lawrence Berkeley National Laboratory (LBNL) Animal Welfare and Research Committee.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample numbers, experimental repeats and statistical tests are indicated in figures and figure legends or STAR Methods sections above.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| H3K27ac | Active Motif | cat# 39133, lot 01613007; RRID: AB_2793305 |
|
| ||
| Biological samples | ||
|
| ||
| Fetal and embryonic human heart samples | Human Developmental Biology Resource at Newcastle University (hdbr.org) | N/A |
| Healthy adult and DCM hearts | This paper | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| ChIP clean | Zymo Research | D5205 |
| TruSeq library preparation kit | Illumina | IP-202-1012 |
| TURBO DNA-free Kit | Life Technologies | AM1907 |
| Qubit dsDNA HS Assay Kit | Life Technologies | Q32851 |
| TruSeq Stranded Total RNA with Ribo-Zero Human/Mouse/Rat kit | Illumina | 20020597 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data | This paper | GEO: GSE126573 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: FVB | The Jackson Laboratory | RRID:IMSR_JAX:001800 |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: Hsp68-promoter-LacZ | Pennacchio et al. Nature. 2006 | Addgene Plasmid # 37843 |
|
| ||
| Software and algorithms | ||
|
| ||
| ENCODE Uniform Processing Pipelines | N/A | https://www.encodeproject.org/pipelines |
| MACS 2.0 | Zhang et al. (2008) | N/A |
| DiffBind | Ross-Innes et al. (2012) | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| GREAT | McLean et al. (2010) | http://great.stanford.edu/public/html/ |
| HOMER | Heinz et al. (2010) | http://homer.ucsd.edu/homer/ |
| PANTHER | Thomas et al. (2003) | http://pantherdb.org/ |
|
| ||
| Other | ||
|
| ||
| Resource website for the publication | This paper | heart.lbl.gov |
Highlights.
Enhancer and transcriptome maps of healthy, disease-affected, and fetal heart tissue
Highly reproducible epigenomic landscape in healthy individuals
Disease-associated changes in activity at 6,850 predicted heart enhancers
3,400 disease-associated enhancers with fetal regulatory properties
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants R24HL123879, R01HL162304, and R01HG003988 (to A.V. and L.A.P.) and R01HL105993 (to K.B.M and T.P.C). Research was conducted at the E.O. Lawrence Berkeley National Laboratory and performed under Department of Energy Contract DE-AC02-05CH11231, University of California. The human embryonic and fetal material was provided by the Joint MRC/Wellcome (MR/R006237/1) Human Developmental Biology Resource (www.hdbr.org). I.B. was funded through an Imperial College Research Fellowship. This project used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, which was supported by NIH Instrumentation Grant S10OD018174.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111400.
REFERENCES
- Alvira CM (2014). Nuclear factor-kappa-B signaling in lung development and disease: one pathway, numerous functions. Birth Defects Res. A Clin. Mol. Teratol. 700, 202–216. 10.1002/bdra.23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar M, Schultz JEJ, Grupp I, Dorn GW, Meneton P, Molin DGM, Gittenberger-de Groot AC, Doetschman T, and Doetschman T (2003). Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 14, 391–407. 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AS, Kuner R, Buness A, Ruschhaupt M, Merk S, Zwermann L, Kääb S, Kreuzer E, Steinbeck G, Mansmann U, et al. (2006). Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J. Am. Coll. Cardiol. 48, 1610–1617. 10.1016/j.jacc.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Berger RD, Kasper EK, Baughman KL, Marban E, Calkins H, and Tomaselli GF (1997). Beat-to-beat QT interval variability: novel evidence for repolarization lability in ischemic and nonischemic dilated cardiomyopathy. Circulation 96, 1557–1565. 10.1161/01.cir.96.5.1557. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. (2010). The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 28, 1045–1048. 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depre C, Shipley GL, Chen W, Han Q, Doenst T, Moore ML, Stepkowski S, Davies PJ, and Taegtmeyer H (1998). Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat. Med. 4, 1269–1275. 10.1038/3253. [DOI] [PubMed] [Google Scholar]
- Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, May D, Spurrell CH, Plajzer-Frick I, Pickle CS, et al. (2016). Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 7, 12923. 10.1038/ncomms12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx E, da Costa Martins PA, and De Windt LJ (2013). Regulation of fetal gene expression in heart failure. Biochim. Biophys. Acta 1832, 2414–2424. [DOI] [PubMed] [Google Scholar]
- Dobaczewski M, Chen W, and Frangogiannis NG (2011). Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 51, 600–606. 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Gillum RF (1986). Idiopathic cardiomyopathy in the United States, 1970–1982. Am. Heart J. 111, 752–755. 10.1016/0002-8703(86)90111-0. [DOI] [PubMed] [Google Scholar]
- Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, et al. (2018). Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 9, 391. 10.1038/s41467-017-02762-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrer I, Roselló-Lletí E, Rivera M, Molina-Navarro MM, Tarazón E, Ortega A, Martínez-Dolz L, Triviño JC, Lago F, González-Juanatey JR, et al. (2014). RNA-sequencing analysis reveals new alterations in cardiomyocyte cytoskeletal genes in patients with heart failure. Lab. Invest. 94, 645–653. 10.1038/labinvest.2014.54. [DOI] [PubMed] [Google Scholar]
- Hershberger RE, and Siegfried JD (2011). Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 57, 1641–1649. 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Morales A, and Siegfried JD (2010). Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet. Med. 12, 655–667. 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, Röh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A, and Hermeking H (2013). AP4 is a mediator of epithelial–mesenchymal transition and metastasis in colorectal cancer. J. Cell Biol. 201, 2017OIA33. 10.1083/jcb.2017oia33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japp AG, Gulati A, Cook SA, Cowie MR, and Prasad SK (2016). The diagnosis and evaluation of dilated cardiomyopathy. J. Am. Coll. Cardiol. 67, 2996–3010. 10.1016/j.jacc.2016.03.590. [DOI] [PubMed] [Google Scholar]
- Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, and Rossant J (1989). Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105, 707–714. 10.1242/dev.105.4.707. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187. 10.1016/j.cell.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louzao-Martinez L, Vink A, Harakalova M, Asselbergs FW, Verhaar MC, and Cheng C (2016). Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy. Int. J. Cardiol. 220, 634–646. 10.1016/j.ijcard.2016.06.253. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart Association, et al. (2006). Contemporary definitions and classification of the cardiomyopathies. Circulation 113, 1807–1816. 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, et al. (2011). Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93. 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi SS, and Mishra PK (2015). Harnessing fetal and adult genetic reprograming for therapy of heart disease. J. Nat. Sci. 1, e71. [PMC free article] [PubMed] [Google Scholar]
- Norton N, Robertson PD, Rieder MJ, Züchner S, Rampersaud E, Martin E, Li D, Nickerson DA, and Hershberger RE; National Heart Lung and Blood Institute GO Exome Sequencing Project (2012). evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ. Cardiovasc. Genet. 5, 167–174. 10.1161/CIRCGENETICS.111.961805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Xu J, and Molkentin JD (2007). Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 18,117–131. 10.1016/j.semcdb.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680. 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Harakalova M, Treibel TA, Lumbers RT, Boukens BJ, Efimov IR, van Dinter JT, González A, López B, El Azzouzi H, et al. (2020). H3K27ac acetylome signatures reveal the epigenomic reorganization in remodeled non-failing human hearts. Clin. Epigenetics 12, 106. 10.1186/s13148-020-00895-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. (2006). In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502. 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, and De Bosscher K (2013). How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol. Cell. Endocrinol. 380, 41–54. 10.1016/j.mce.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohini M, Haritha Menon A, and Selvamurugan N (2018). Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int. J. Biol. Macromol. 120, 310–317. 10.1016/J.IJBIOMAC.2018.08.107. [DOI] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Horie M, and Nagase T (2018). TGF-B signaling in lung health and disease. Int. J. Mol. Sci. 19, 2460. 10.3390/ijms19082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazonova O, Zhao Y, Nürnberg S, Miller C, Pjanic M, Castano VG, Kim JB, Salfati EL, Kundaje AB, Bejerano G, et al. (2015). Characterization of TCF21 downstream target regions identifies a transcriptional network linking multiple independent coronary artery disease loci. PLoS Genet. 11, e1005202. 10.1371/journal.pgen.1005202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, and Karin M (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136. 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Small EM, and Olson EN (2011). Pervasive roles of microRNAs in cardiovascular biology. Nature 469, 336–342. 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan FL, Moravec CS, Li J, Apperson-Hansen C, McCarthy PM, Young JB, and Bond M (2002). The gene expression fingerprint of human heart failure. Proc. Natl. Acad. Sci. USA 99, 11387–11392. 10.1073/pnas.162370099.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WLW, Anene-Nzelu CG, Wong E, Lee CJM, Tan HS, Tang SJ, Perrin A, Wu KX, Zheng W, Ashburn RJ, et al. (2020). Epigenomes of human hearts reveal new genetic variants relevant for cardiac disease and phenotype. Circ. Res. 127, 761–777. 10.1161/CIRCRESAHA.120.317254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, and Narechania A (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin JA, and Bowles NE (2002). The failing heart. Nature 415, 227–233. 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, and Pennacchio LA (2007). VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92. 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won K-J, Chepelev I, Ren B, and Wang W (2008). Prediction of regulatory elements in mammalian genomes using chromatin signatures. BMC Bioinf. 9, 547. 10.1186/1471-2105-9-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, 2022. World health statistics 2022: monitoring health for the SDGs, sustainable development goals. Geneva. Available at https://www.who.int/publications/i/item/9789240051157; ISBN 978–92-4–005114-0
- Yang K-C, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, and Nerbonne JM (2014). Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 129, 1009–1021. 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Volckaert T, Chanda D, Thannickal VJ, and De Langhe SP (2018). Fgf10 signaling in lung development, homeostasis, disease, and repair after injury. Front. Genet. 9, 418. 10.3389/fgene.2018.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961. 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All ChIP-seq and RNA-seq data are available through GEO: GSE126573. Images of LacZ-stained embryos are available from the VISTA Enhancer Browser (https://enhancer.lbl.gov), and raw images are available on request from the lead authors.
All original code has been deposited at https://gitlab.com/lotard/spurrell_fetalization and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.