

Abstract
The impact of liver cancer metastasis on protein abundance of 22 drug‐metabolizing enzymes (DMEs) and 25 transporters was investigated using liquid chromatography‐tandem accurate mass spectrometry targeted proteomics. Microsomes were prepared from liver tissue taken from 15 healthy individuals and 18 patients with cancer (2 primary and 16 metastatic). Patient samples included tumors and matching histologically normal tissue. The levels of cytochrome P450 (CYPs 2B6, 2D6, 2E1, 3A4, and 3A5) and uridine 5′‐diphospho‐glucuronosyltransferases (UGTs 1A1, 1A6, 1A9, 2B15, 2B4, and 2B7) were lower in histologically normal tissue from patients relative to healthy controls (up to 6.6‐fold) and decreased further in tumors (up to 21‐fold for CYPs and 58‐fold for UGTs). BSEP and MRPs were also suppressed in histologically normal (up to 3.1‐fold) and tumorous tissue (up to 6.3‐fold) relative to healthy individuals. Abundance of OCT3, OAT2, OAT7, and OATPs followed similar trends (up to 2.9‐fold lower in histologically normal tissue and up to 16‐fold lower in tumors). Abundance of NTCP and OCT1 was also lower (up to 9‐fold). Interestingly, monocarboxylate transporter MCT1 was more abundant (3.3‐fold) in tumors, the only protein target to show this pattern. These perturbations could be attributed to inflammation. Interindividual variability was substantially higher in patients with cancer. Proteomics‐informed physiologically‐based pharmacokinetic (PBPK) models of 50 drugs with different attributes and hepatic extraction ratios (Simcyp) showed substantially lower drug clearance with cancer‐specific parameters compared with default parameters. In conclusion, this study provides values for decreased abundance of DMEs and transporters in liver cancer, which enables using population‐specific abundance for these patients in PBPK modeling.
Study Highlights .
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Predicting the fate of drugs in patients with cancer using physiologically‐based pharmacokinetic (PBPK) models requires knowledge of changes in such populations. Changes in hepatic drug‐metabolizing enzymes (DMEs) and transporters are not fully characterized in metastatic liver cancer.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Protein abundance of DMEs and transporters in healthy controls, histologically normal, and tumorous livers from patients was measured and compared among the groups, and interindividual variability was assessed. Scaled abundance data to tissue levels were used to inform PBPK predictions in cancer.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study provides for the first time population‐specific abundance data of DMEs and transporters for patients with metastatic liver cancer that are necessary for PBPK modeling.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study highlights an overall decreased abundance of DMEs and transporters in liver cancer. These unique data are a valuable resource for PBPK models, which are not fully informed with data on specific populations. This may enable more accurate predictions of pharmacokinetics in patients with cancer.
Colorectal cancer is the second most lethal type of cancer, 1 presenting with metastases to the liver in half of the patients, 2 which leads to poor prognosis. 3 Genetic and epigenetic alterations contribute to its manifestation. 4 Colorectal liver metastasis (CRLM) originates from the colon and metastasizes to the liver through the portal vein, whereas primary liver cancer has hepatic origin. Primary liver cancer is another leading cause of cancer‐related mortality, with its main types being hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma. 1
Whereas liver cancer is ideally treated by surgical resection, this is seldom possible, hence chemotherapy is used instead. 5 , 6 Because the liver is the main site of metabolism, the presence of cancer can affect drug pharmacokinetics (PKs) and pharmacodynamics. Both histologically normal and tumorous tissue contribute to drug metabolism and disposition. Drug‐metabolizing enzymes (DMEs), especially cytochrome P450 (CYP) enzymes and uridine 5′‐diphospho‐glucuronosyltransferases (UGT), in the tumor are responsible for activation and disposition of anticancer prodrugs. 7 However, most anticancer drugs are cleared by DMEs, and their decreased abundance could lead to higher local exposure. Drug transporters are also critical for anticancer drug resistance, which occurs with increased levels of efflux transporters and suppression of uptake transporters. 8
Physiologically‐based pharmacokinetic (PBPK) models are gaining wider acceptance in regulatory decision making in oncology, where recruitment of patients and ethical/safety issues are of great importance. 9 These models require system and drug data for accurate prediction of drug PKs. 10 Cancer populations are very heterogeneous, leading to variable PK profiles (e.g., clearance, exposure, and absorption), owing to changes in various system parameters, including plasma protein, comorbidities, 11 renal function, 12 microsomal protein per gram liver (MPPGL), 13 and abundance of DMEs 14 and transporters. 15 These differences may be driven by inflammation in patients with cancer, where cytokines may lead to downregulation of DMEs. 16 , 17
Protein abundance data in liver cancer are limited, with most studies reporting immunohistochemistry or mRNA data. Yan et al. 14 , 18 have, however, reported quantitative measurement of CYPs and UGTs in patients with HCC using liquid chromatography‐mass spectrometry (LC‐MS) proteomics and highlighted perturbations in cancer. LC‐MS proteomics has also been used to quantify transporters in HCC. 15 Despite the high incidence of CRLM, quantitative data in liver tissue from patients with CRLM is largely unavailable. The qualitative profiles of CYPs, 19 among other DMEs, in histologically normal and cancerous tissues from patients with CRLM 20 highlighted a potential impact of cancer on drug metabolism. Studies on transporter proteins are more scarce, with a focus on mRNA measurement of OATPs 21 or comparisons of abundance between healthy and histologically normal livers from patients with CRLM. 22 A preliminary experiment measured the abundance of DMEs and transporters in CRLM pooled samples. 23 Understanding the range of values in individual patients is necessary for projecting potential clinical consequences of the changes in a probabilistic rather than deterministic PBPK predictions. 24
In this study, we quantified DMEs and transporters in healthy livers from healthy donors and compared with matched histologically normal and cancerous livers from patients with cancer. To our knowledge, this is the most comprehensive report of absolute quantitative measurements of DMEs and transporters using LC‐MS proteomics in CRLM to date. Additionally, the abundance data for individual samples were scaled up to liver tissue content in CRLM using experimentally derived MPPGL in each individual. 13 Finally, we applied scaled data in PBPK simulations to predict the PKs of 50 drugs with varying degrees of metabolism and transporter liability by different enzymes and transporters, different hepatic extraction ratios and attributes (e.g., protein binding in plasma), in order to assess the impact of cancer‐specific abundance of DMEs and transporters in patients with CRLM when conducting PBPK.
MATERIALS AND METHODS
Detailed methods are described in Supplementary Methods .
Human liver samples
Matched cancerous and histologically normal liver tissue from adult patients with cancer undergoing hepatectomy (n = 18; HCC primary cancer (n = 1), intrahepatic cholangiocarcinoma primary cancer (n = 1), and CRLM (n = 16)) were sourced from Manchester University NHS Foundation Trust (MFT) Biobank, Manchester, UK. Ethics were covered by MFT Biobank generic ethics approvals (NRES 14/NW/1260 and 19/NW/0644). Healthy human liver microsomes (tumor‐free) from 15 subjects were collected postmortem and provided by Pfizer as microsomes (Groton, CT). These samples were supplied by Vitron (Tucson, AZ) and BD Gentest (San Jose, CA). Ethical approval was covered by the suppliers. Tables S1 and S2 present donor demographic and clinical details.
QconCAT (MetCAT and TransCAT) standards
Two stable isotopes labeled QconCAT standards were used in this study, as previously described 25 : the MetCAT and a modified TransCAT 23 for the quantification of DMEs and transporters, respectively.
Preparation of samples for proteomics
Liver tissue samples were fractionated to microsomes. 13 , 23 Each sample was spiked with known amounts of QconCATs, and prepared using filter‐aided sample preparation. 26 , 27 Unlabeled peptide standards were added to quantify the QconCATs. A pool of healthy samples was prepared for quality control.
Liquid chromatography‐tandem mass spectrometry
Sample peptides were analyzed by an UltiMate 3000 Rapid Separation LC system (Dionex Corporation, Sunnyvale, CA) coupled to a Q Exactive HF Hybrid Quadrupole‐Orbitrap MS (Thermo Fisher Scientific, Waltham, MA).
Analysis and annotation of proteomic data
Proteomic data were processed using MaxQuant 1.6.7.0 (Max Planck Institute, Martinsried, Germany), and searched against a customized database, comprising a human UniprotKB database (74,788 sequences) and QconCAT sequences. For targeted analysis, accurate mass and retention time methodology was used, based on light‐to‐heavy intensity ratios and QconCAT concentrations to calculate protein amounts. 27 , 28 Peptides selected for the quantification are presented in Tables S3 – S8 .
Statistical data analysis
Statistical data analysis was performed using GraphPad Prism 8.1.2 (La Jolla, CA), Microsoft Excel 2016, and R 3.6.3. Nonparametric statistics were used because data did not follow normal distribution. Differences in absolute abundances between the groups were assessed using Mann–Whitney U test. The P value cutoff for statistical significance was set at 0.05. Principal components analysis (PCA) was performed for proteome‐level similarity based on percentage identical peptide and percentage identical protein. 29
Physiologically‐based pharmacokinetic simulations
The effect of applying the experimentally determined DMEs and transporters abundance levels from this study in combination with previously determined scaling factors 13 was assessed using PBPK modeling with Simcyp V20 Release 1 (Certara, Sheffield, UK). Fifty substrates with different attributes and hepatic extraction ratios were used (Table S9 ). The compound files were available within the Simcyp simulator. PBPK simulations were performed using default system parameters available in Simcyp for healthy (“Sim‐Healthy Volunteers”) and cancer (“Sim‐Cancer”) virtual populations, without or with changing MPPGL, 13 abundances of CYPs, UGTs, transporters measured here, and previously measured flavin‐containing monooxygenases and carboxylesterases (pooled samples). 23 Here, we use targeted quantification and flavin‐containing monooxygenase and carboxylesterase that were not measured. Differences in abundance and changes in coefficients of variation, between healthy and histologically normal or cancerous samples were incorporated into the models. The effects of abundance changes on drug exposure following oral administration were assessed using previously described models 13 :
Model 1 (Healthy): default MPPGL and abundance data for healthy population (Simcyp).
Model 2 (Cancer‐D): default MPPGL and abundance data for cancer population (Simcyp).
Model 3 (New Cancer‐ALN): Scaling based on MPPGL from histologically normal tissue (39.0 mg/g) 13 and abundance of DMEs and transporters (relative difference between normal and healthy tissue) were used for the cancer population, assuming the whole liver is histologically normal (maximum metabolic capacity of microsomal enzymes).
Model 4 (New Cancer‐ALC): Scaling based on MPPGL in cancerous tissue (24.8 mg/g) 13 and abundance of DMEs and transporters (relative difference between tumors and healthy tissue) were used for the cancer population, assuming the whole liver is cancerous and liver mass unchanged.
The relative ratios of the clearance (CL) were compared: CL in healthy/CL in new cancer‐ALN or ALC, CL in cancer‐D/CL in new cancer‐ALN or ALC, and CL in new cancer‐ALN/CL in new cancer‐ALC.
RESULTS
Comparison of absolute abundance of CYPs, UGTs, and transporters in healthy, histologically normal and tumor samples
Abundance of CYP2B6, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 (Figure 1a ) in histologically normal samples from patients with cancer was significantly lower than in healthy livers (Mann–Whitney U test, P < 0.05). Abundance of most CYPs in tumors was massively lower relative to healthy (P < 0.05) and histologically normal tissues (P < 0.05).
Figure 1.
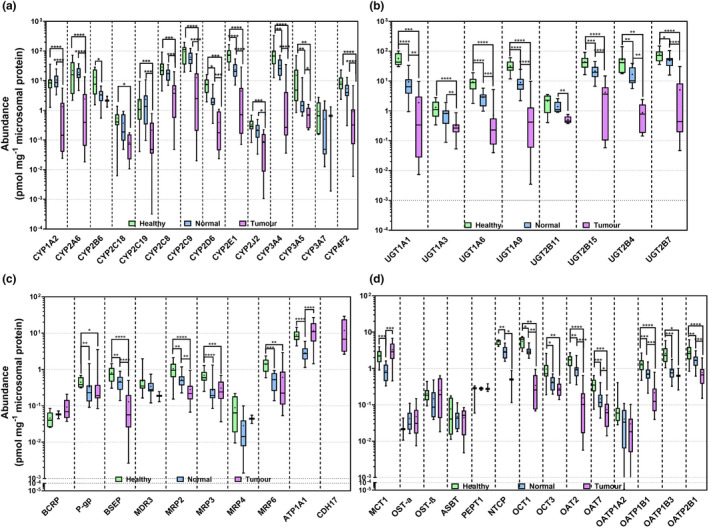
Absolute abundance of cytochrome P450 enzymes (CYPs) (a), UDP‐glucuronosyltransferases (UGTs) (b), ATP‐binding cassette (ABC) transporters, ATPase subunit alpha‐1 (Na+/K+‐ATPase; ATP1A1) and cadherin‐17 (CDH17) (c), and solute carriers (SLCs) (d) in HLM from healthy (n = 15), histologically normal (n = 18) and tumorous (n = 18) tissues. Abundances are represented as box and whiskers plots with the whiskers reflecting the minimum and maximum values, the boxes showing the 25th and 75th percentiles, the lines showing the medians, and the + signs showing the means. Mann‐Whitney test was used to assess differences between healthy and histologically normal, between healthy and tumorous livers and between histologically normal and tumorous livers for each enzyme. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Similarly, most UGTs were significantly decreased in histologically normal tissues compared with healthy controls and in tumors compared with healthy controls and histologically normal tissues (P < 0.05; Figure 1b ).
Among ATP‐binding cassette (ABC) transporters, P‐gp, BSEP, MRP2, MRP3, and MRP6 decreased in tumorous and histologically normal liver tissue from patients with cancer relative to healthy controls (P < 0.05). A significant decrease in the abundance of BSEP and MRP2 was observed in tumors compared with matched histologically normal livers (P < 0.05).
Similarly, the abundance of most solute carriers (SLC) was lower in livers from patients with cancer. Organic cation transporter (OCT) 1, organic anion transporter (OAT) 2, OAT7, and organic anion transporting polypeptide (OATP) 1B1 and OATP2B1 were significantly downregulated in histologically normal and tumor livers compared with healthy controls and in tumors relative to histologically normal tissues (P < 0.05). The abundance of monocarboxylate transporter 1 (MCT1) was significantly higher in tumors than in normal tissue, whereas Na(+)/NTCP was significantly downregulated (P < 0.05).
Interestingly, the abundance of all DMEs and transporters in our primary tumorous samples was always within the range of the abundance in metastatic tumors (CRLMs). There were, however, only two samples, so the generality of this finding needs verification.
The abundance of DMEs and transporters did not correlate with the age for any of the groups of livers. Our previous study 13 similarly failed to reveal correlations between age and scaling factors for the same normal and tumor samples.
The lower limit of quantification was 0.02 pmol/mg microsomal protein, with high precision and accuracy, based on replicate measurements in a pool of healthy samples (Figure S3 ). The absolute abundance values in individual samples are provided in Tables S3 – S8 . Leukocyte markers were also quantified; CD45 and CD47 were 1.7‐ and 1.2‐fold higher in tumors compared with healthy controls, indicating little difference in infiltration of immune cells.
Proportion of CYPs and UGTs in the three sets of HLMs
The pie charts (Figure 2 ) represent the proportion of DMEs in healthy, histologically normal and tumor samples. CYP2C9 was the most abundant CYP in all groups. In the healthy and histologically normal samples, CYP3A4 was the second most abundant, followed by CYP2E1 and CYP2C8. In tumors, the order was CYP2C8 > CYP2E1, CYP3A4. Among UGTs, UGT2B7 was the most abundant in all groups. The second most abundant UGT was UGT1A1 in healthy and UGT2B15 in histologically normal and tumor samples.
Figure 2.
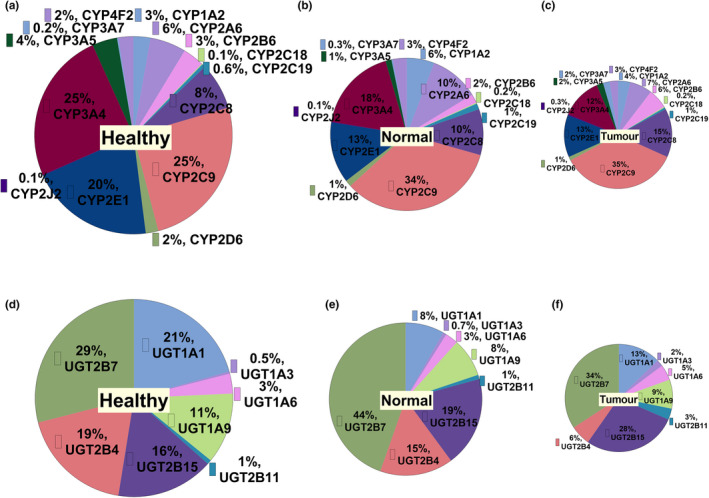
Pie charts representing the proportion of CYPs (a–c), and UGTs (d–f) in healthy, histologically normal and tumorous HLMs, respectively.
Proportion of transporters in the three sets of HLM samples
The most abundant ABC transporter quantified in healthy controls and histologically normal livers was MRP6, followed by MRP2, and BSEP (Figure 3 ). However, the most abundant ABC transporter in liver tumors was P‐gp, followed by MRP6 and MRP3.
Figure 3.
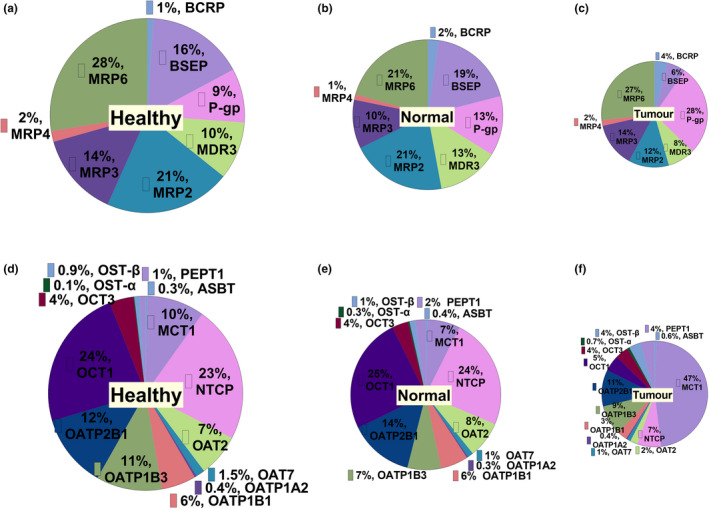
Proportion of ABC transporters (a–c), and SLCs (d–f) in HLM from healthy, histologically normal and tumorous livers, respectively.
The most abundant SLCs quantified in healthy and histologically normal livers were OCT1, NTCP, and OATP2B1. However, MCT1 was most abundant in tumors.
Relative abundance of CYPs, UGTs, and transporters in HLM from healthy controls and histologically normal and tumor tissues
Figure 4 depicts the relative abundance of proteins (based on mean values) expressed as ratios (fold change). The y‐axis starts at 1 (no change). CYP2B6, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 were more than two‐fold lower in histologically normal livers relative to controls, suggesting downregulation of CYPs in tissue surrounding liver tumors, with more significant decrease in tumors.
Figure 4.
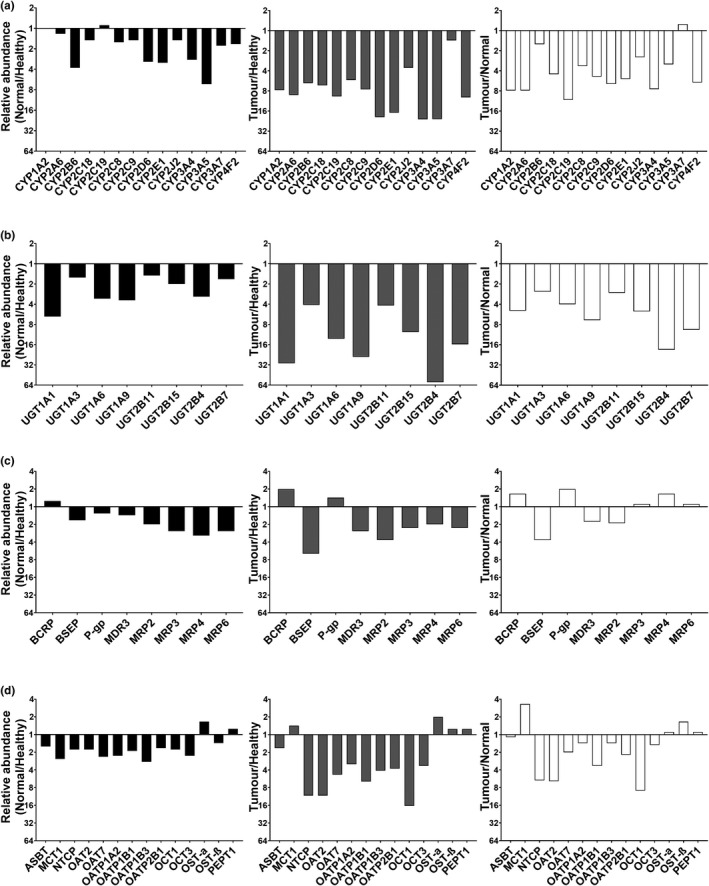
Relative mean abundances of CYPs (a), UGTs (b), ABC (c), and SLC (d) transporters in HLMs. Each panel presents the ratios of mean abundance in histologically normal to healthy controls, tumor to healthy controls, and tumor to histologically normal livers. The y‐axis starts at 1 (no change between the sets). The mean abundances were expressed as pmol of protein per mg of total liver microsomal protein.
Most UGTs were suppressed by more than two‐fold in normal compared with healthy livers, and in tumor relative to healthy liver (up to 58‐fold) and histologically normal specimens (up to 19‐fold).
By contrast, some ABC transporters were more abundant in tumors (mean values). BCRP increased in tumors relative to healthy controls and P‐gp abundance was higher in tumors relative even to histologically normal tissue. However, other efflux transporters (BSEP, MRPs, and MDR3) were suppressed in tumors (up to 6‐fold) relative to healthy controls. MRPs were reduced in histologically normal (up to 3.1‐fold) relative to healthy controls.
Abundance of SLCs in histologically normal tissue relative to healthy controls (Figure 4d ) was lower for MCT1, OAT7, OATP1A2, OATP1B3, and OCT3. Ratios of NTCP, OATs, OATPs, and OCTs in tumors relative to healthy tissue indicated up to 16‐fold decrease in cancer. By contrast, a two‐fold increase was observed for OST‐α in tumors. Last, abundance of NTCP, OATs, OCT1, OATP1B1, and OATP2B1 in tumors compared with histologically normal tissue reflected a decrease of up to 9‐fold in cancer, whereas abundance of MCT1 was 3.3‐fold higher in tumors.
Relative abundance of CYPs, UGTs, and transporters in paired tumor and normal tissue samples
Figure 5 presents the ratios of the abundance in histologically normal relative to matched tumor livers for each individual for CYPs (Figure 5a ), UGTs (Figure 5b ), ABC transporters (Figure 5c ), and SLCs (Figure 5d ). The absence of a data point means the abundance of the target was not determined in one or both of the matched samples. The graphs show that changes in abundance of each target in tumors compared with matched histologically normal tissue were not consistent across samples. The disease did not affect the targets uniformly, and ideally the incorporation of such individual data into models should generate more accurate simulations.
Figure 5.
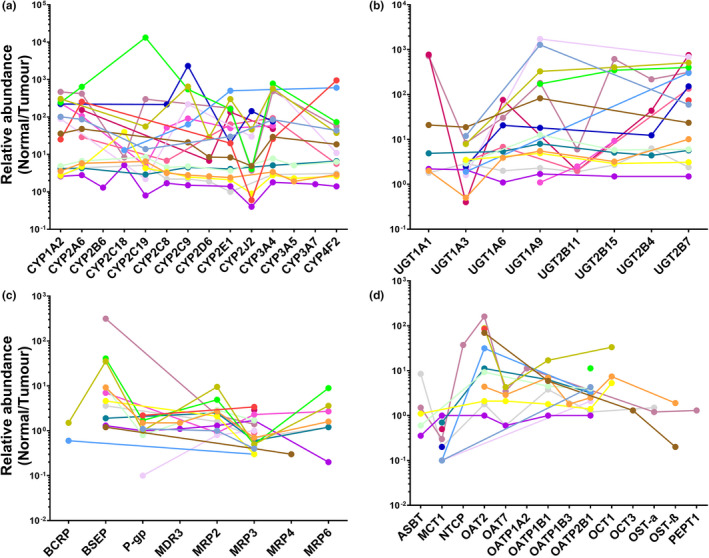
Relative abundance of CYPs (a), UGTs (b), ABC transporters (c), and SLCs (d) in individual HLMs from histologically normal tissue compared to matched tumors (n = 18).
Scaling of microsomal abundance of CYPs, UGTs, and transporters up to liver tissue content
Using MPPGL data in each individual sample determined previously, 13 individual abundance data for normal and tumor livers were scaled up to tissue levels (Figure S2 ). In Simcyp, population values of enzyme or transporter abundance are entered per mg of microsomal protein or per number of cells and their variabilities. Similarly, MPPGL is created based on the average values in the population and associated variability. However, there is no possibility to define the abundance based on gram of tissue and its variability. Individual MPPGL and corresponding abundance in the same individual give true values of abundance per gram of tissue and its associated variability. The average levels of abundance per gram of tissue and associated variability may vary under linking the individual value compared with the case when values are not linked. For example, the abundance of CYP3A4 in tumor livers is 153 ± 250 pmol of protein/g of liver on a basis of individually linked values whereas without linking (using average MPPGL) these would be 145 ± 243 pmol of protein/g of liver. Nonetheless, the ranking of different liver samples for the abundance of CYP3A4 was hardly affected by applying the individual MPPGL values (Figure 6 ). Similar trends are observed across all the proteins.
Figure 6.
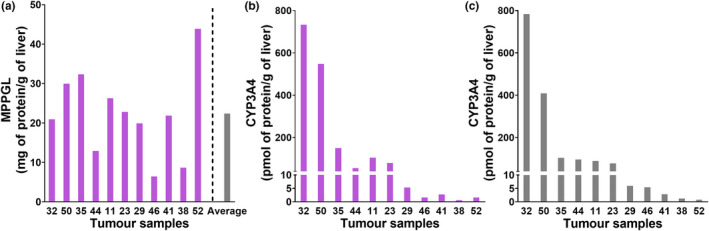
Scaling of protein abundance values to liver tissue levels in cancer (n = 11). Individual microsomal protein per gram of liver tissue (MPPGL) values in tumors were compared to their average MPPGL (a). Individual MPPGL from tumor livers (b) and average MPPGL value (c) were applied to extrapolate the abundance of CYP3A4 to pmol of protein per gram of liver tissue. Eleven cancerous livers were included, for which MPPGL and abundance values were available. Activity of cytochrome P450 reductase (used to correct for microsomal protein loss) was quantified in 12 tumor samples and these were below the limit of quantification in 6 samples. Abundance of CYP3A4 was below the limit of quantification in one sample for which activity of cytochrome P450 reductase was measured.
Principal component analysis
Percentage identical peptides and percentage identical proteins were calculated as previously described, 27 , 29 and the results were analyzed by PCA (Figure S1 ). The healthy and normal liver samples formed two distinct clusters, indicating homogeneity. However, little clustering was observed for the cancer liver samples reflecting the nature of cancer as a phenotypic range rather than one phenotype.
Physiologically‐based pharmacokinetic simulations
Simulations for 50 substrates with different attributes and hepatic extraction ratios were performed. Four models were used (Figure 7 ): model 1 (healthy) based on default MPPGL and abundances of DMEs and transporters (Simcyp) with a healthy population; model 2 (cancer‐D) based on default MPPGL and abundances (Simcyp) with a cancer population; model 3 (new cancer‐ALN) based on MPPGL values and abundances measured here for histologically normal tissue (cancer population); and model 4 (new cancer‐ALC) based on MPPGL 13 and abundances measured here in cancer tissue (cancer population). CL predicted using the model new cancer‐ALC was substantially lower for 66% (up to 22‐fold) and 74% (up to 31‐fold) of the substrates than CL obtained using the cancer‐D and healthy models, respectively, suggesting increase in systemic exposure to several substrates in the case of advanced cancer (most of the liver is cancerous). The most significant change in CL was observed in esomeprazole. The 31‐fold decrease in new cancer‐ALC compared with healthy is explained by the substantial decrease in the abundance of CYP2C19, CYP2C9, CYP3A4, and CYP2A6 that metabolize esomeprazole. Table S10 summarizes the updated parameters specific for cancer that should be used for PBPK modeling.
Figure 7.
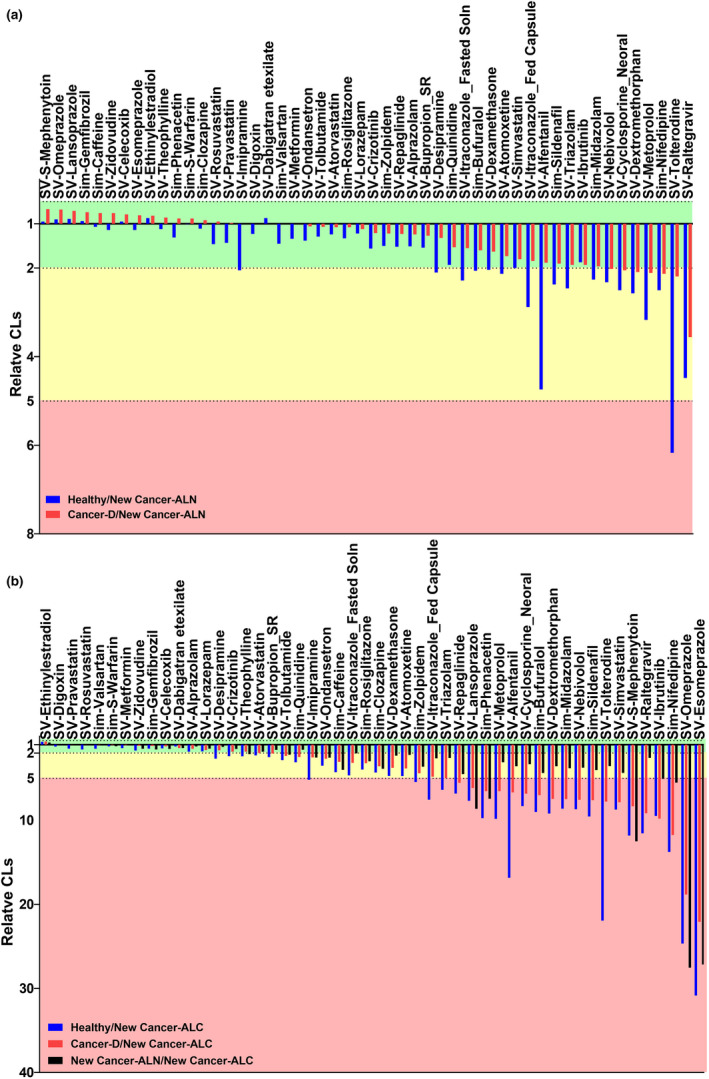
Relative CL of drugs in healthy (blue), and cancer‐D (red) to new cancer‐ALN (a) or new cancer‐ALC (b) populations, and new cancer‐ALN (white) to new cancer‐ALC (b) populations. Healthy: default abundances of enzymes and transporters (Simcyp) with a healthy population. Cancer‐D: default abundances of enzymes and transporters (Simcyp) with a cancer population. New Cancer‐ALN: abundance of enzymes and transporters measured in this study for histologically normal tissue with a cancer population. New Cancer‐ALC: abundances of enzymes and transporters measured in this study for cancer tissue with a cancer population. The green rectangle shows the drugs with less than 2‐fold change in drug clearance (CL) than that obtained using the new cancer‐ALC model, the amber shows 2 to 5‐fold higher CL than that obtained using the new cancer‐ALC model, and the red shows more than 5‐fold higher CL than that obtained using the new cancer‐ALC model.
DISCUSSION
Quantitative proteomics provides useful abundance data for PBPK models in cancer and other disorders. 30 To our knowledge, this is the first comprehensive study on protein abundance of human liver DMEs and transporters in healthy, histologically normal adjacent to tumor and tumorous tissue, and their interindividual variation (as opposed to our previous pilot study in pooled samples). 23 The current analysis confirmed the general trends observed with pooled samples but, importantly, provided a range of protein abundance values and interindividual variability required for population‐based predictions.
CYPs and UGTs are involved in the clearance of more than 90% of drugs, 31 with CYPs metabolizing 80% of clinically used drugs. 32 Although CYPs and UGTs were previously measured in HCC, 14 , 18 to our knowledge, there are no reports on individual abundance data in CRLM. Our data highlight suppression of most CYPs and UGTs in histologically normal compared with healthy controls and in cancerous tissue compared with histologically normal and healthy samples. The extensive downregulation of DMEs in the cancerous tissue implies severe impairment of drug metabolism in these patients, leading to higher exposure to CYP and UGT substrates. The exposure of drugs used in CRLM treatment, such as irinotecan (metabolized by CYP3A4/5 and UGT1A1/9 33 , 34 ) and regorafenib (metabolized by CYP3A4 and UGT1A9 35 ), may increase significantly in these patients, especially when a high proportion of their liver is cancerous. This means that lower drug doses may be required for patients with CRLM to avoid toxicity. Interestingly, a meta‐analysis showed that decreased abundance of CYPs in a virtual cancer population provides better PK predictions for CYP substrates. 36 Our data also highlight that healthy livers from noncancer subjects are better controls. Cancer environment affects abundance in histologically normal livers from patients with cancer, making them less suitable controls.
Drug transporters are important in drug/metabolite disposition and drug–drug interactions 37 and may be involved in chemotherapeutic drug resistance. 8 Transporters involved in drug disposition relevant to CRLM include P‐gp, BCRP, MRP1, MRP2, and OATP1B1 for irinotecan 33 , 34 and regorafenib. 38 Our data revealed lower levels of SLCs in histologically normal tissue relative to healthy controls, and a more significant decrease in cancerous livers. Interestingly, the monocarboxylate transporter MCT1 was the only transporter that increased. Reports suggest that overexpression of MCT1 facilitates lactate efflux, protecting cancer cells, and its inhibition could be useful in drug‐resistant cancers. 39 Most ABC transporters quantified here were either downregulated or unchanged in cancer tissue relative to healthy controls. However, BCRP increased in tumors compared with healthy controls and P‐gp (relative mean abundances) in tumors compared with histologically normal livers from patients with cancer. This may lead to decreased drug accumulation in the tumor cells and could indicate resistance to chemotherapeutic substrates of BCRP and P‐gp. Our data are generally in line with our pilot. 23 Our current study is the first to quantify ABC and SLC transporters in CRLM and report interindividual variability. Overall, our data suggest altered drug disposition in CRLM and recommend MCT1, BCRP, and P‐gp as potential targets for cancer treatment.
The most abundant CYPs and UGTs in all liver sets were similar, with varying percentages of abundance across different groups, consistent with the literature. 26 , 40 , 41 , 42 , 43 The most abundant ABC transporters (in agreement with previous studies) 26 , 44 and SLCs were the same in healthy and normal livers, but different in cancerous livers. Investigating the relative abundance of these proteins is key to understanding differences in drug–drug interactions in patients with CRLM compared with healthy subjects.
The protein profiles, as evidenced by PCA, showed higher heterogeneity in cancer. Abundances of DMEs were more variable in cancerous samples, indicating higher heterogeneity. The maximum interindividual variation across CYPs, UGTs, ABC transporters, and SLCs in healthy samples was 231 (CYP2C19), 11 (UGT1A6), 23 (MRP4), and 16‐fold (ASBT), respectively, which is smaller compared with a previous study using label‐free quantification. 26 However, the maximum fold difference across CYPs, UGTs, ABC transporters, and SLCs in cancer samples was 4063 (CYP2C9), 2784 (UGT1A9), 190 (BSEP), and 179‐fold (OATP1A2), respectively. The ratio of abundance of each protein target in histologically normal to matched tumor livers for each individual was variable across the samples. Considering that normal livers belong to a homogeneous group, the lack of a specific trend in abundance for each individual could be attributed to the heterogeneity in cancerous livers, which may be explained by the fact that individuals differ in cancer cell differentiation, disease severity, type of cancer, previous treatment, and other characteristics.
Absolute abundance was measured in microsomes. Variability in abundance of highly expressed but unrelated HLM proteins can impact the abundance of DMEs and transporters, and abundance levels in tissue may reduce this effect. 45 In our study, we converted abundance into tissue levels using matched MPPGL data measured previously. 13 We did not observe consistency between rank orders of samples between microsomal and tissue abundances, in line with another study 15 demonstrating that normalization with mg of membrane protein or gram of tissue can change the patterns of abundance. Scaling to tissue levels may be more appropriate for PK predictions in PBPK models. Therefore, in our simulations, we used MPPGL and protein abundance to extrapolate to tissue levels.
Based on measured abundance and MPPGL data, we assessed the impact of changes in DME and transporter levels on drug clearance predicted by PBPK simulations of 50 substrates. A substantial difference in drug clearance was observed when using cancer‐specific parameters compared with typical parameters for healthy and default cancer populations, with higher exposure predicted with cancer‐related parameters. This impact on drug clearance increased when the whole liver was assumed to be cancerous. Therefore, abundance data may substantially affect PK profiles, especially when a high proportion of the liver is cancerous (advanced stage). The percentage of cancerous tissue was not known and was not incorporated into the models. Therefore, two extreme cases were used, where the whole liver was considered normal (maximum metabolic capacity) or tumorous. We should highlight that the predicted PK profiles were not compared with clinical data because these are not available for CRLM. Therefore, our simulations did not aim to point the correct method against observations, but to investigate the impact of the abundance of DMEs and transporters on the PK in patients with cancer. Further work should verify our updated models for cancer populations when such clinical data become available.
Among the limitations of the study are the high interindividual variability and the small number of patients. Additionally, the systems parameters (albumin, AAG, and hematocrit) used in the default cancer population (Simcyp) are not specific for patients with CRLM but derived from several cancer types. Regarding the experimental method, we tried to reduce the variability. The models in healthy population (Simcyp) are validated and, therefore, we did not change the abundance in this population for our simulations. For the new cancer populations, the relative ratios of the abundance of cancerous or histologically normal to healthy controls were calculated and the default abundances (Simcyp) were modified accordingly. This minimizes the risk of differences in the abundances because of the method (our method vs. methods used for Simcyp default abundances). However, access to the observed data can show the exact impact of these factors on the in vivo clearance.
To conclude, this study provides, for the first time, absolute protein abundance data for a large array of hepatic DMEs and transporters in individual cancer samples with a focus on CRLM. DMEs were substantially downregulated and transporters were also altered in cancer. Interindividual variability was higher in cancer. Abundance data were scaled to tissue levels, highlighting the importance of abundance values with MPPGL data in the same livers. PBPK simulations demonstrated higher drug exposure with cancer population‐specific abundance data relative to a healthy population. Therefore, appropriate abundance values specific for cancer populations may contribute to more accurate PK predictions, and our quantitative data will be valuable in addressing gaps in cancer PBPK models. The values reported here should enable updating systems parameters within existing PBPK platforms in relation to abundance of DMEs and transporters to reflect biological values. Verification with observed clinical PK data creates the basis for what has been coined as “master files” to be used with different new drugs substrates.
FUNDING
This work was supported by Merck KGaA, Frankfurter Str. 250, F130/005, 64293 Darmstadt, Germany.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
A.M.V., Z.M.A., B.A., S.A.P., J.B., and A.R.‐H. wrote the manuscript. A.M.V., S.A.P., J.B., and A.R.‐H. designed the research. A.M.V. performed the research. A.M.V., Z.M.A., and B.A. analyzed the data.
Supporting information
Figure S1
Figure S2
Figure S3
Table S1–S10
Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank Manchester University NHS Foundation Trust (MFT) Biobank, Manchester, UK, and Pfizer (Groton, CT) for providing the samples with demographic and clinical data, and the Biological Mass Spectrometry Core Research Facility (BioMS, RRID: SCR_020987), University of Manchester, for LC‐MS/MS instrumentation and support. The authors also acknowledge Merck KGaA for financial support.
- 1. Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Maher, B. , Ryan, E. , Little, M. , Boardman, P. & Stedman, B. The management of colorectal liver metastases. Clin. Radiol. 72, 617–625 (2017). [DOI] [PubMed] [Google Scholar]
- 3. Siegel, R.L. , Miller, K.D. & Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Nikolouzakis, T. et al. Improving diagnosis, prognosis and prediction by using biomarkers in CRC patients (review). Oncol. Rep. 39, 2455–2472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitchell, D. , Puckett, Y. & Nguyen, Q.N. Literature review of current management of colorectal liver metastasis. Cureus 11, e3940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen, X. , Liu, H.‐P.‐L. , Li, M. & Qiao, L. Advances in non‐surgical management of primary liver cancer. World J. Gastroenterol. 20, 16630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michael, M. & Doherty, M.M. Tumoral drug metabolism: overview and its implications for cancer therapy. J. Clin. Oncol. 23, 205–229 (2005). [DOI] [PubMed] [Google Scholar]
- 8. Akhdar, H. , Legendre, C. , Aninat, C. & More, F. Anticancer Drug Metabolism: Chemotherapy Resistance and New Therapeutic Approaches. (ed. Paxton, J. ) Topics in Drug Metabolism. 137–170 (IntechOpen, London, UK, 2012). [Google Scholar]
- 9. Yoshida, K. , Budha, N. & Jin, J.Y. Impact of physiologically based pharmacokinetic models on regulatory reviews and product labels: Frequent utilization in the field of oncology. Clin. Pharmacol. Ther 101, 597–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rostami‐Hodjegan, A. Physiologically based pharmacokinetics joined with in vitro–in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin. Pharmacol. Ther. 92, 50–61 (2012). [DOI] [PubMed] [Google Scholar]
- 11. Cheeti, S. , Budha, N.R. , Rajan, S. , Dresser, M.J. & Jin, J.Y. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm. Drug Dispos. 34, 141–154 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Suri, A. , Chapel, S. , Lu, C. & Venkatakrishnan, K. Physiologically based and population PK modeling in optimizing drug development: a predict‐learn‐confirm analysis. Clin. Pharmacol. Ther. 98, 336–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vasilogianni, A.‐M. et al. Hepatic scaling factors for in vitro–in vivo extrapolation of metabolic drug clearance in patients with colorectal cancer with liver metastasis. Drug Metab. Dispos. 49, 563–571 (2021). [DOI] [PubMed] [Google Scholar]
- 14. Yan, T. et al. Significantly decreased and more variable expression of major CYPs and UGTs in liver microsomes prepared from HBV‐positive human hepatocellular carcinoma and matched pericarcinomatous tissues determined using an isotope label‐free UPLC‐MS/MS method. Pharm. Res. 32, 1141–1157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Billington, S. et al. Transporter expression in noncancerous and cancerous liver tissue from donors with hepatocellular carcinoma and chronic hepatitis C infection quantified by LC‐MS/MS proteomics. Drug Metab. Dispos. 46, 189–196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shinko, D. , Diakos, C.I. , Clarke, S.J. & Charles, K.A. Cancer‐related systemic inflammation: the challenges and therapeutic opportunities for personalized medicine. Clin. Pharmacol. Ther. 102, 599–610 (2017). [DOI] [PubMed] [Google Scholar]
- 17. Dunvald, A.D. , Järvinen, E. , Mortensen, C. & Stage, T.B. Clinical and molecular perspectives on inflammation‐mediated regulation of drug metabolism and transport. Clin. Pharmacol. Ther. 112, 277–290 (2022). [DOI] [PubMed] [Google Scholar]
- 18. Yan, T. et al. Severely impaired and dysregulated cytochrome P450 expression and activities in hepatocellular carcinoma: implications for personalized treatment in patients. Mol. Cancer Ther. 14, 2874–2886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lane, C.S. et al. Identification of cytochrome P450 enzymes in human colorectal metastases and the surrounding liver: a proteomic approach. Eur. J. Cancer 40, 2127–2134 (2004). [DOI] [PubMed] [Google Scholar]
- 20. van Huizen, N.A. et al. Up‐regulation of collagen proteins in colorectal liver metastasis compared with normal liver tissue. J. Biol. Chem. 294, 281–289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wlcek, K. et al. The analysis of organic anion transporting polypeptide (OATP) mRNA and protein patterns in primary and metastatic liver cancer. Cancer Biol. Ther. 11, 801–811 (2011). [DOI] [PubMed] [Google Scholar]
- 22. Kurzawski, M. et al. The reference liver—ABC and SLC drug transporters in healthy donor and metastatic livers. Pharmacol. Rep. 71, 738–745 (2019). [DOI] [PubMed] [Google Scholar]
- 23. Vasilogianni, A.‐M. et al. Proteomics of colorectal cancer liver metastasis: a quantitative focus on drug elimination and pharmacodynamics effects. Br. J. Clin. Pharmacol. 88, 1811–1823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hariparsad, N. et al. Current practices, gap analysis and proposed workflows for PBPK modeling of cytochrome P450 induction: an industry perspective. Clin. Pharmacol. Ther. 10.1002/cpt.2503. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 25. Russell, M.R. et al. Alternative fusion protein strategies to express recalcitrant QconCAT proteins for quantitative proteomics of human drug metabolizing enzymes and transporters. J. Proteome Res. 12, 5934–5942 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Couto, N. et al. Quantification of proteins involved in drug metabolism and disposition in the human liver using label‐free global proteomics. Mol. Pharm. 16, 632–647 (2019). [DOI] [PubMed] [Google Scholar]
- 27. Al‐Majdoub, Z.M. et al. Proteomic quantification of human blood‐brain barrier SLC and ABC transporters in healthy individuals and dementia patients. Mol. Pharm. 16, 1220–1233 (2019). [DOI] [PubMed] [Google Scholar]
- 28. Al‐Majdoub, Z.M. et al. Quantification of proteins involved in intestinal epithelial handling of xenobiotics. Clin. Pharmacol. Ther. 109, 1136–1146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al Feteisi, H. , Al‐Majdoub, Z.M. , Achour, B. , Couto, N. , Rostami‐Hodjegan, A. & Barber, J. Identification and quantification of blood–brain barrier transporters in isolated rat brain microvessels. J. Neurochem. 146, 670–685 (2018). [DOI] [PubMed] [Google Scholar]
- 30. Sharma, S. , Suresh Ahire, D. & Prasad, B. Utility of quantitative proteomics for enhancing the predictive ability of physiologically based pharmacokinetic models across disease states. J. Clin. Pharmacol. 60, S17–S35 (2020). [DOI] [PubMed] [Google Scholar]
- 31. Rowland, A. , Miners, J.O. & Mackenzie, P.I. The UDP‐glucuronosyltransferases: their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 45, 1121–1132 (2013). [DOI] [PubMed] [Google Scholar]
- 32. Zanger, U.M. & Schwab, M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141 (2013). [DOI] [PubMed] [Google Scholar]
- 33. de Man, F.M. , Goey, A.K.L. , van Schaik, R.H.N. , Mathijssen, R.H.J. & Bins, S. Individualization of irinotecan treatment: a review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin. Pharmacokinet. 57, 1229–1254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma, M. & McLeod, H. Lessons learned from the irinotecan metabolic pathway. Curr. Med. Chem. 10, 41–49 (2003). [DOI] [PubMed] [Google Scholar]
- 35. Rey, J.B. , Launay‐Vacher, V. & Tournigand, C. Regorafenib as a single‐agent in the treatment of patients with gastrointestinal tumors: an overview for pharmacists. Target Oncol. 10, 199–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwenger, E. et al. Harnessing meta‐analysis to refine an oncology patient population for physiology‐based pharmacokinetic modeling of drugs. Clin. Pharmacol. Ther. 103, 271–280 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Liang, X. , Staiger, K.M. , Riddle, E. , Hao, J. & Lai, Y. Role of Transporters in Drug Disposition and Drug–Drug Interactions. Identification and Quantification of Drugs, Metabolites, Drug Metabolizing Enzymymes, and Transporters (Second Edition). (eds. Ma, S. & Chowdhury, S.K. ) 311–337 (Elsevier B.V., New York, NY, 2020). [Google Scholar]
- 38. Ohya, H. et al. Regorafenib is transported by the organic anion transporter 1B1 and the multidrug resistance protein 2. Biol. Pharm. Bull. 38, 582–586 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Payen, V.L. , Mina, E. , Van Hée, V.F. , Porporato, P.E. & Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 33, 48–66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohtsuki, S. et al. Simultaneous absolute protein quantification of transporters, cytochrome P450s and UDP‐glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab. Dispos. 40, 83–92 (2011). [DOI] [PubMed] [Google Scholar]
- 41. Achour, B. , Russell, M.M.R. , Barber, J. & Rostami‐Hodjegan, A. Simultaneous quantification of the abundance of several cytochrome p450 and uridine 5′‐diphospho‐glucuronosyltransferase enzymes in human liver microsomes. Drug Metab. Dispos. 42, 500–510 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Zhang, H.‐F. et al. Correlation of cytochrome P450 oxidoreductase expression with the expression of 10 isoforms of cytochrome P450 in human liver. Drug Metab. Dispos. 44, 1193–1200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Achour, B. , Barber, J. & Rostami‐Hodjegan, A. Expression of hepatic drug‐metabolizing cytochrome P450 enzymes and their intercorrelations: a meta‐analysis. Drug Metab. Dispos. 42, 1349–1356 (2014). [DOI] [PubMed] [Google Scholar]
- 44. Vildhede, A. , Wiśniewski, J.R. , Norén, A. , Karlgren, M. & Artursson, P. Comparative proteomic analysis of human liver tissue and isolated hepatocytes with a focus on proteins determining drug exposure. J. Proteome Res. 14, 3305–3314 (2015). [DOI] [PubMed] [Google Scholar]
- 45. Achour, B. , Al Heteisi, H. , Lanucara, F. , Rostami‐Hodjegan, A. & Barber, J. Global proteomic analysis of human liver microsomes: rapid characterization and quantification of hepatic drug‐metabolizing enzymes. Drug Metab. Dispos. 45, 666–675 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Table S1–S10
Supporting Information