

Abstract
Aims:
Sirtuin 1 (SIRT1) is a key player in liver physiology and a therapeutic target against hepatic inflammation. We evaluated the role of SIRT1 in the proinflammatory context and oxidative stress during acetaminophen (APAP)-mediated hepatotoxicity.
Results:
SIRT1 protein levels decreased in human and mouse livers following APAP overdose. SIRT1-Tg mice maintained higher levels of SIRT1 on APAP injection than wild-type mice and were protected against hepatotoxicity by modulation of antioxidant systems and restrained inflammatory responses, with decreased oxidative stress, proinflammatory cytokine messenger RNA levels, nuclear factor kappa B (NFκB) signaling, and cell death. Mouse hepatocytes stimulated with conditioned medium of APAP-treated macrophages (APAP-CM) showed decreased SIRT1 levels; an effect mimicked by interleukin (IL)1β, an activator of NFκB. This negative modulation was abolished by neutralizing IL1β in APAP-CM or silencing p65-NFκB in hepatocytes. APAP-CM of macrophages from SIRT1-Tg mice failed to downregulate SIRT1 protein levels in hepatocytes. In vivo administration of the NFκB inhibitor BAY 11-7082 preserved SIRT1 levels and protected from APAP-mediated hepatotoxicity.
Innovation:
Our work evidenced the unique role of SIRT1 in APAP hepatoprotection by targeting oxidative stress and inflammation.
Conclusion:
SIRT1 protein levels are downregulated by IL1β/NFκB signaling in APAP hepatotoxicity, resulting in inflammation and oxidative stress. Thus, maintenance of SIRT1 during APAP overdose by inhibiting NFκB might be clinically relevant.
Rebound Track:
This work was rejected during standard peer review and rescued by Rebound Peer Review (Antioxid Redox Signal 16: 293–296, 2012) with the following serving as open reviewers: Rafael de Cabo, Joaquim Ros, Kalervo Hiltunen, and Neil Kaplowitz. Antioxid. Redox Signal. 28, 1187–1208.
Keywords: : paracetamol, SIRT1, oxidative stress, antioxidant defense, inflammation, hepatotoxicity, interleukin 1β
Innovation .
Our work has evidenced for the first that sirtuin 1 overexpression confers protection against acetaminophen (APAP)-mediated hepatotoxicity by targeting interleukin 1β/nuclear factor kappa B-mediated inflammation, thereby increasing antioxidant defenses. These results might have clinical implications in the design of novel therapies for APAP intoxication.
Rebound Track .
This work was rejected during standard peer review and rescued by Rebound Peer Review (Antioxid Redox Signal 16: 293–296, 2012) with the following serving as open reviewers: Rafael de Cabo, Joaquim Ros, Kalervo Hiltunen, and Neil Kaplowitz. The comments by these reviewers supporting the rescue are listed below:
Rafael de Cabo (decabora@grc.nia.nih.gov): I am a qualified reviewer (per Antioxid Redox Signal 16: 293–296) and move to rescue this article that was initially rejected during the regular peer review process. After I have carefully reviewed all versions of the article, reviewer comments, and the response to the reviewers provided, I would suggest accepting the article. I concur with the enthusiasm about the significance and quality of work that can be read throughout in some of the reviewers' comments and consider that the major concerns raised during the review process have been entirely satisfied. The authors employed a battery of in vivo and in vitro models that successfully dissect the mechanistic aspects of their initial hypothesis. This study provided novel evidence for the negative modulation of sirtuin 1 (SIRT1) via interleukin (IL)1β/nuclear factor kappa B (NFκB) pathway following acetaminophen (APAP) intoxication. The authors shed new light into the underlying mechanisms that drive these effects on SIRT1 and how the maintenance of SIRT1 levels after an APAP challenge is protective against hepatotoxicity in promoting antioxidant and repressing inflammatory responses. Therefore, in the interests of science, I take full responsibility to rescue this work from rejection.
Joaquim Ros (joaquim.ros@cmb.udl.cat): I am a qualified reviewer (per Antioxid Redox Signal 16: 293–296) and move to rescue this article that was rejected during the regular peer review process after reviewing all versions of the article and detailed blind reviewer's comments. The article entitled “SIRT1 controls acetaminophen hepatotoxicity by modulating inflammation and oxidative stress” by Rada et al. describes key findings concerning the role of SIRT1 in controlling the toxic effects of APAP in the liver and the interplay between macrophages and hepatocytes in this process. The work is well executed and well written, including data from in vitro, in vivo, as well as data obtained from samples of APAP-overdosed patients. Although the exact mechanism by which APAP is downregulating SIRT1 expression is not fully provided, the data connecting the modulation of SIRT1 protein levels with the IL1β/NFκB axis are conclusive and represent a novel and highly relevant contribution to understand the hepatotoxicity caused by APAP. Furthermore, the in vivo experiments using BAY11-7082 are of special interest to reach the conclusions described in the article. Therefore, in the interest of science, I take full responsibility to rescue this work from rejection, recommending its acceptance to the journal. Regarding the anonymous peer review process, although this reviewer could agree with some of the concerns initially raised by blind reviewers, the truth is that the authors have fully answered these concerns very satisfactorily, making a great effort to include new data and amending the article according to the suggestions made by the blind reviewers. Based on these comments, I believe that this article merits publication in Antioxid Redox Signal. Therefore, in the interests of science, I take full responsibility to rescue this work from rejection.
Kalervo Hiltunen (kalervo.hiltunen@oulu.fi): I am a qualified reviewer (per Antioxid Redox Signal 16: 293–296) and move to rescue this article that was rejected during the regular peer review process after reviewing all versions of the article and detailed reviewer comments. APAP/paracetamol (APAP) is a widely used antipyretic and pain-relieving drug and it is generally considered safe. However, at a higher dose, APAP can cause severe acute liver injury. The mechanism of APAP toxicity is a target of intensive research. The current work tackled regulatory events leading to liver toxicity by APAP using the mouse as model organism. The authors approached the set problem from many angles using monitoring and manipulating hepatic cell signaling pathways with inhibitors, genetic modification of target animals, and cell cultures. Western blotting, microscopical inspection of histological specimens, and cell biological cytological approaches were the main methods for readouts of the responses. The article deepens our understanding on the mechanism on APAP-triggered liver injury, the experiments have been conducted with care, the data are clearly presented, the article is logically written, and the drawn conclusions are supported by the given experimental data. Based on these novel findings, I support acceptance of this work for publication in Antioxid Redox Signal. The original article was reviewed by five anonymous peer reviewers. The tones of the reviewers' statements were generally positive, but also a number of comments were raised. This is expectable considering the multicity of used approaches, large numbers of parameters analyzed, and complexity of mechanisms resulting in APAP-induced liver failure. The authors show now additional data obtained through immunoblotting of liver specimen from human and mice intoxicated by APAP, which addressed the previous concerns. In addition, the authors show now supporting data and revised the text with a suggestion that in SIRT1-Tg mice liver infiltrating MoMFs are likely polarized towards M2, protecting APAP-mediated inflammation. This agrees with a previous study showing that deletion of myeloid SIRT1 stimulates the proinflammatory M1-like polarization of macrophages and increases the activation of NFκB. Altogether, the authors have addressed the major criticisms raised by the anonymous reviewers in a satisfactory manner and strengthened the article. Therefore, in the interests of science, I take full responsibility to rescue this work from rejection.
Neil Kaplowitz (kaplowit@usc.edu): I am a qualified reviewer (per Antioxid Redox Signal 16: 293–296) and move to rescue this article that was rejected during the regular peer review process after reviewing all versions of the article and detailed reviewer comments. This is a very carefully conducted study that presents a large body of data in support of an extremely novel hypothesis for the role of dysregulation of hepatocellular SIRT1 by IL-1 produced by macrophage acting on hepatocytes by an NFκB mechanism in response to APAP. Transgenic overexpression of SIRT1 strongly protected, which is itself extremely novel and the presumption is that enhanced antioxidant defense is the mechanism. The article is written in a clear and circumspect manner and uncertainties are acknowledged appropriately. I have considered the five reviewers' comments and am satisfied that most are very satisfactorily addressed. Like all good science, the findings are provocative and will require additional studies, particularly to more definitely link the in vitro and in vivo data. However, the authors present an interesting hypothesis, a variety of different approaches to support their hypothesis, and technically well-executed experiments. I believe this should be accepted and the rest of us in the field can determine if the hypothesis is correct or needs modification. Ultimately, the known mechanistic contributors to promoting or protecting against APAP downstream of covalent binding include sustained c-Jun N-terminal kinase activation, its interplay with mitochondria, mitophagy, and mitochondrial biogenesis. These will need to be addressed in future studies to fit into the authors' hypothesis. Therefore, in the interests of science, I take full responsibility to rescue this work from rejection.
Introduction
The liver is the main organ in charge of drug catabolism and also the major site susceptible to drug injury. Drug-induced liver injury (DILI) is the leading cause of hepatic dysfunction in the United States and Europe. In fact, hepatotoxicity is the single most frequent reason for removing approved medications from the market (41). Therefore, new advances in developing of strategies aimed to reduce DILI are critical to increase the availability of many drugs for the treatment of a wide range of diseases. In this regard, in vivo and in vitro preclinical models of DILI are valuable tools for identifying molecular mechanisms responsible for hepatocyte death and for testing new candidate genes responsible for the susceptibility to DILI as well as therapeutic interventions.
Most of the knowledge about DILI has come from studies with paracetamol, called acetaminophen (APAP) in the United States, since at high doses this drug causes liver injury in both humans and experimental animals (23). At nontoxic doses (up to 4 g per day in adult humans), APAP is eliminated by glucuronide and sulfate conjugation. The initial step in APAP hepatotoxicity is the formation of the reactive metabolite N-acetyl-p-aminobenzoquinone imine (NAPQI) by cytochrome P450 2e1 (CYP2E1). At therapeutic doses, NAPQI is detoxified by glutathione (GSH) and eliminated in urine or bile as APAP-cysteine, APAP-N-acetylcysteine, and APAP–APAP-GSH.
After an overdose, glucuronidation and sulfation routes become saturated, and extensive bioactivation of APAP leads to a rapid depletion of the hepatic GSH pool. Subsequently, NAPQI binds to cysteine groups on cellular proteins forming APAP-protein adducts. NAPQI also binds to mitochondrial proteins, which, in turn, causes oxidative stress that triggers signaling pathways through mitochondrial toxicity, leading to lethal cell injury (8). This massive mitochondrial dysfunction and nuclear DNA damage are the main causes of necrotic cell death of the hepatocytes on APAP treatment.
During necrosis, released damage-associated molecular patterns (DAMPs) activate Toll-like receptors (TLRs) on macrophages and induce proinflammatory signaling pathways leading to cytokine production. These inflammatory mediators also activate other immune cells such as neutrophils and monocytes leading to the recruitment of these leukocytes into the liver to remove cell debris and to prepare the tissue for repair (13, 27). Among these proinflammatory cytokines, both interleukin (IL)1α and IL1β have been recently implicated as critical mediators of APAP hepatotoxicity (3, 14). However, recently controversial evidences based on studies in experimental models and humans have suggested a contrasting, deleterious or beneficial, role of the inflammatory response in the regeneration of the liver after APAP-mediated injury (1, 54).
Sirtuin 1 (SIRT1), an NAD+-dependent histone deacetylase, has been implicated in hepatic metabolism by its ability to deacetylate key metabolic players such as the peroxisome proliferator-activated receptor (PPAR) gamma coactivator 1 alpha (PGC1α), a master regulator of oxidative metabolism and inducer of oxidative stress protection systems such as manganese superoxide dismutase (MnSOD) and catalase (25, 38). SIRT1 is also involved in the control of inflammatory responses, and in this context, an important target for SIRT1 is the nuclear factor kappa B (NFκB) (60), a key mediator of proinflammatory signaling pathways triggered by cytokines, both directly by deacetylation and indirectly through the induction of antioxidant systems that prevent reactive oxygen species (ROS)-induced NFκB nuclear translocation.
It has been reported that SIRT1 activity can contribute to hepatoprotection against some hepatotoxic insults, in particular, to the protection elicited by resveratrol against D-galactosamine/lipopolysaccharide-induced acute liver failure. In addition, a recent study has involved SIRT1/nuclear factor (erythroid-derived 2)-like 2 (Nrf2)/tumor necrosis factor alpha (TNFα) signaling pathway in the effect of luteolin in attenuating acute mercuric chloride exposure-induced hepatotoxicity (59). SIRT1 activity has also been reported to protect against chronic hepatotoxic injures, for example, in the context of alcohol-induced liver injury, and the expression of SIRT1 in hepatic stellate cells is necessary to confer protection against fibrosis, particularly during aging (43). However, to the best of our knowledge, there are no mechanistic studies on the role of SIRT1 in the inflammatory process related to oxidative stress-mediated APAP hepatotoxicity.
In this study, we found that protein levels of SIRT1 are decreased in the liver of humans and mice after acute APAP treatment, and that moderate SIRT1 overexpression in mice protects against oxidative stress and inflammation during APAP hepatotoxicity. We also provide new mechanistic insights on the involvement of IL1β/NFκB pathway in the cross talk between macrophages and hepatocytes that leads to reduction of SIRT1 protein levels in hepatocytes on APAP treatment.
Results
SIRT1 levels are decreased in the liver of humans and mice on acute APAP treatment
To investigate if SIRT1 protein levels are modulated by APAP treatment in human livers, we performed immunohistochemistry analysis in samples from five individuals with severe APAP intoxication who required liver transplantation. Demographic and biochemical characteristics of patients with APAP hepatotoxicity were previously reported (33). As shown in Figure 1A, SIRT1 immunostaining was detected in hepatocytes from individuals with normal liver. Following an acute APAP overdose, SIRT1 immunostaining could be visualized only in the areas of surviving hepatocytes, whereas it was totally absent in the areas of severe liver damage.
FIG. 1.

SIRT1 levels are decreased in the liver of humans and mice on acute APAP treatment. (A) Representative anti-SIRT1 immunostaining in human liver sections from five individuals with severe APAP intoxication that required liver transplantation and from a healthy individual as control. Scale bars = 100 μm. (B) SIRT1 protein levels in liver extracts from wild-type mice i.p. injected 300 mg/kg APAP or saline for 3 or 6 h. Graph depicts SIRT1 densitometric quantification. After quantification of all blots, results are expressed as fold change relative to the saline-injected condition and are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. *p < 0.05 versus saline-injected mice. n = 5–6 mice per group. (C) Overnight fasted mice were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP. After 6 h, mice were sacrificed. Liver sections were stained against SIRT1 protein. Scale bars = 100 μm. ANOVA, analysis of variance; APAP, acetaminophen; i.p., intraperitoneal; SEM, standard error of the mean; SIRT1, sirtuin 1. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
We next evaluated the overall decrease of SIRT1 protein in the hepatic tissue from mice injected with APAP (300 mg/kg) for 3 and 6 h. As depicted in the Western blot analysis of Figure 1B (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars), no differences were found in SIRT1 protein levels 3 h after APAP intoxication, but at 6 h, the decrease in SIRT1 protein levels was statistically significant compared with mice injected with vehicle. In mouse livers, SIRT1 immunostaining was mainly located in the areas surrounding the central veins and also decreased at 6 h after APAP treatment (Fig. 1C).
Mice with moderate overexpression of SIRT1 are more resistant to liver damage during acute APAP treatment
Two previous studies have reported the protection against metabolic damage in the liver of transgenic mice with moderate overexpression of SIRT1 (SIRT1-Tg) in the context of cancer and obesity (12, 40). This fact together with the observed decrease in SIRT1 levels in livers of mice on APAP treatment (Fig. 1B, C) could be indicative of a role of SIRT1 downregulation in APAP toxicity. To test this hypothesis, wild-type and SIRT1-Tg mice were dosed with APAP (300 mg/kg).
As shown in Figure 2A (left panel) and Supplementary Figure S2, in the livers of SIRT1-Tg mice, SIRT1 protein levels were increased by 3.5-fold compared with livers of wild-type mice and, importantly, SIRT1 protein levels were also higher at 6 h post-APAP injection (Fig. 2A, right panel and Supplementary Fig. S2), the time at which reduction of SIRT1 was detected in wild-type mice (Fig. 1B). At this time period, the histological analysis revealed less centrilobular necrotic lesions in SIRT1-Tg mice compared with those of the wild-type mice (Fig. 2B). Moreover, the marked increase in alanine transaminase (ALT) levels found in wild-type mice at 3, 6, and 24 h following APAP injection was attenuated in SIRT1-Tg mice (Fig. 2C) in agreement with liver histology (Fig. 2B).
FIG. 2.

Mice with moderate overexpression of SIRT1 are more resistant to liver damage during acute APAP treatment. Overnight fasted wild-type (SIRT1-WT) and mice overexpressing SIRT1 (SIRT1-Tg) were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP. Mice were sacrificed after 3, 6, or 24 h, as indicated, and livers and serum were collected. (A) Left panels, SIRT1 protein levels detected in liver extracts derived from SIRT1-WT or SIRT1-Tg mice sacrificed before APAP or vehicle injection. Blots were quantified and results are expressed as fold change relative to SIRT1-WT mice. Values are mean ± SEM. ***p < 0.001 versus SIRT1-WT mice according to Student's t-test. n = 5 mice per genotype. Right panels, Immunoblots showing SIRT1 protein levels in liver extracts from SIRT1-WT and SIRT1-Tg mice treated with APAP or saline for 6 h. α-Tubulin was used as loading control. Densitometric quantification depicts SIRT1 protein levels in each condition. After quantification of all blots, results are expressed as fold change relative to the vehicle-injected condition and are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. ***p < 0.001 versus each vehicle-injected control. ###p < 0.001 versus APAP-injected SIRTI-WT mice. n = 6–8 mice per group. (B) Images of hematoxylin and eosin staining of liver sections from wild-type and SIRT1-Tg mice treated with APAP or saline for the indicated times. Scale bars = 200 μm. n = 5 mice per group. (C) Plasma ALT levels measured in the same experimental conditions. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. ***p < 0.001 versus control (vehicle) condition. ##p < 0.01, ###p < 0.001 versus SIRT1-WT mice. n = 6–8 mice per group. (D) Images of TUNEL staining and quantification of TUNEL-positive cells analyzed 24 h after APAP injection. Scale bars: 100 μm. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. *p < 0.05, ***p < 0.001 versus control (vehicle) condition. ###p < 0.001 versus SIRT1-WT. n = 5 mice per group. ALT, alanine transaminase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
APAP-induced DNA fragmentation is regarded as a consequence of mitochondrial dysfunction (6). When TUNEL assay was used to visualize nuclear DNA fragmentation in liver samples, we found that wild-type mice treated with APAP for 24 h had a substantial number of TUNEL-positive cells in the necrotic centrilobular regions (Fig. 2D), while TUNEL-positive cells were less prevalent in the livers of SIRT1-Tg mice.
Taking into account that APAP toxicity is generally associated to oxidative stress due to a rapid GSH depletion, we next determined GSH/GSSG ratio in wild-type and SIRT1-Tg mice following APAP administration. As shown in Figure 3A, basal GSH/GSSG ratio was lower in SIRT1-Tg livers compared to values of the wild-type controls. Moreover, GSH/GSSG ratio was significantly reduced 1 h after APAP intoxication in wild-type mice and remained very low at 6 h. By contrast, in SIRT1-Tg mice, the GSH/GSSG ratio did not decrease 1 h after APAP intoxication and a minor decrease was detected at 6 h.
FIG. 3.

Moderate overexpression of SIRT1 contributes to reduction in oxidative stress by improving the antioxidant capacity during APAP treatment. Overnight fasted SIRT1-WT and SIRT1-Tg mice were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP and sacrificed after the indicated times. (A) GSH/GSSG ratio, protein carbonyl content, and APAP-Cys adducts were analyzed in livers from SIRT1-WT and SIRT1-Tg mice treated with APAP for 6 and 24 h. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. **p < 0.01, ***p < 0.001 versus each control (vehicle) condition. ##p < 0.01, ###p < 0.001 versus SIRT1-WT mice. n = 6–8 mice per group. (B) Immunoblot analysis of catalase and MnSOD levels in total liver extracts from SIRT1-WT and SIRT1-Tg mice 6 h after APAP injection. p85α was used as a loading control. After quantification of all blots, results are expressed as fold change relative to the saline-injected condition and are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. **p < 0.01, ***p < 0.001 versus vehicle-injected mice. #p < 0.05, ##p < 0.01 versus SIRT1-WT mice. n = 6–8 mice per group. (C) Nuclear levels of Sp1 are increased by APAP in wild-type mice and this effect was attenuated in SIRT1-Tg mice. Representative immunoblots showing nuclear Sp1 and Lamin B as a loading control. After quantification of all blots, results are expressed as fold change relative to vehicle condition and are mean ± SEM. Statistical analysis was performed by two-way ANOVA followed by Bonferroni post hoc test. **p < 0.01, ***p < 0.001 versus control (vehicle) condition. ##p < 0.01, ###p < 0.001 versus SIRT1-WT mice. n = 5–6 mice per group. GSH, glutathione; MnSOD, manganese superoxide dismutase; Sp1, specific protein 1.
Protein carbonyl levels, an indicator of oxidative damage in the liver, were lower in the APAP-injected SIRT1-Tg mice than in wild-type animals. However, no differences were found in either protein expression of the cytochrome P450 isoform involved in APAP catabolism CYP21E (data not shown) nor in the levels of APAP-cysteine adducts analyzed at 6 and 24 h of APAP treatment (Fig. 3A), indicating similar APAP catabolism in both mouse strains and suggesting that the hepatoprotection conferred by SIRT1 overexpression could be more closely related to the control of ROS levels rather than the formation of APAP adducts.
The evaluation of the protein levels of antioxidant enzymes that are transcriptional targets of SIRT1 and PGC1α in the livers of both genotypes of mice revealed that APAP administration led to a substantial decrease of MnSOD and catalase in wild-type mice; these decreases were less pronounced in the livers from APAP-treated SIRT1-Tg mice (Fig. 3B and Supplementary Fig. S3). In agreement with this, nuclear-specific protein 1 (Sp1), a transcription factor induced by oxidative stress (45), was markedly increased by APAP intoxication in wild-type mice and this effect was reduced in SIRT1-Tg mice (Fig. 3C and Supplementary Fig. S3). These results, together with the evaluation of the GSH/GSSG ratio and carbonyl levels, suggest that SIRT1-dependent induction of antioxidant genes is likely contributing to the reduced oxidative stress in the liver of SIRT1-Tg mice.
Differential effect of APAP in nonparenchymal cell populations in livers of wild-type and SIRT1-Tg mice
APAP-induced liver toxicity is accompanied by an inflammatory response involving activation of resident macrophages (Kupffer cells) and recruitment of neutrophils and mononuclear cells into the liver (26, 31, 53). Since SIRT1 has been shown to modulate hepatic inflammation induced by metabolic and oxidative stress (12, 40, 42, 61), and SIRT1-Tg mice showed reduced oxidative stress and APAP liver toxicity, the effect of SIRT1 overexpression in the early inflammatory responses during APAP intoxication was examined. Mice were injected with APAP or vehicle and sacrificed 3 h later. Immune cells of the liver were isolated, labeled with specific antibodies against cell surface markers, and cell populations analyzed by flow cytometry.
Kupffer cells (CD45+CD11b+F4/80+) were similarly depleted in wild-type and SIRT1-Tg livers after APAP injection (Fig. 4A), although newly recruited monocytes (CD11b+CCR2+Ly6C+) were higher in SIRT1-Tg livers than in wild-type livers (Fig. 4B). The analysis of the recruited leukocytes revealed that on APAP treatment, moderate SIRT1 overexpression increased the percentage of infiltrated neutrophils (CD45+Ly6G+F480−) compared with the increase observed in APAP-treated wild-type mice (Fig. 4C). Recruitment of cytotoxic T lymphocytes (CD45+CD3+CD8+) was increased by APAP in wild-type mice, but not in SIRT1-Tg mice, and in the latter group, noncytotoxic T lymphocytes (CD45+CD3+CD8−) were increased compared with the values found in wild-type mice (Fig. 4D).
FIG. 4.

SIRT1 overexpression modulates immune populations in the early stage of APAP toxicity. Overnight fasted SIRT1-WT and SIRT1-Tg mice were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP. Mice were sacrificed after 3 or 6 h, as indicated, and NPCs were isolated and analyzed by flow cytometry. (A) Analysis of active resident macrophages 3 h after APAP injection (percentage of F4/80+CD11b+ cells pregated on CD45+ cells). Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. **p < 0.01 versus control (vehicle) condition. n = 6 mice per group. (B) Newly recruited monocytes 3 h after APAP injection (percentage of CCR2+Ly6C+ cells pregated on CD11b+ cells). Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. ***p < 0.001 versus control (vehicle) condition. #p < 0.05 versus SIRT1-WT mice. n = 6 mice per group. (C) Analysis of neutrophils (Ly6G+F4/80− cells on CD45+ pregated cells). *p < 0.05, ***p < 0.001 versus control (vehicle) condition. ###p < 0.001 versus SIRT1-WT mice. n = 6 mice per group. (D) Study of cytotoxic and noncytotoxic T lymphocytes (percentage of CD3+CD8+ or CD3+CD8− cells on CD45+ pregated cells, respectively). Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. **p < 0.01, ***p < 0.001 versus control (vehicle) condition. ##p < 0.01, ###p < 0.001 versus SIRT1-WT mice. n = 6–8 mice per group. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The analysis of the natural killer T (NKT) population (CD45+CD3+NK1.1+) revealed a strong increase in APAP-treated SIRT1-Tg mice compared with that of APAP-treated wild-type mice (Supplementary Fig. S4). However, in APAP-treated wild-type mice, 18.14 ± 1.60% of this cell population expressed the cytotoxic surface marker CD8 (CD45+CD3+NK1.1+CD8+), whereas in SIRT1-Tg mice, this population was 30% lower than in the wild type (12.05 ± 0.66% in SIRT1-Tg mice). Thus, our data indicate that moderate overexpression of SIRT1 enhances myeloid cell recruitment; however, cytotoxic T lymphocytes, belonging to the acquired immune system, were mostly recruited into the livers of wild-type mice on APAP treatment.
SIRT1-Tg mice have reduced proinflammatory cytokines, increased anti-inflammatory cytokines, and reduced NFκB signaling in APAP hepatotoxicity
We next investigated the impact of non-parenchymal liver cells (NPCs) resident and/or recruited into the liver after APAP treatment in the levels of circulating and hepatic proinflammatory cytokines, as well as in intrahepatic signaling mediators. As shown in Figure 5A, SIRT1-Tg mice had lower plasma levels of the proinflammatory cytokines IL6 and IL1β 6 h post-APAP treatment compared with wild-type mice. No levels of TNFα were detected. Likewise, the hepatic messenger RNA (mRNA) levels of the proinflammatory cytokines IL1β and IL6 were reduced in SIRT1-Tg mice at 3 and 6 h after APAP injection compared with the values of wild-type mice (Fig. 5B). By contrast, the analysis of the mRNA levels of the anti-inflammatory markers IL10 and arginase 1 revealed an increase in APAP-treated SIRT1-Tg mice compared with wild-type mice.
FIG. 5.

SIRT1-Tg mice were protected against the elevation of circulating and hepatic proinflammatory cytokines and NFκB signaling pathway in APAP toxicity. Overnight fasted SIRT1-WT and SIRT1-Tg mice were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP and sacrificed as indicated. (A) Plasma levels of IL6 and IL1β analyzed 6 h after APAP injection. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. ***p < 0.001 versus control (vehicle) condition. ##p < 0.01 versus SIRT1-WT mice. n = 6–8 mice per group. (B) Il1b, Il6, IL10, and Arg1 mRNA levels determined by qRT-PCR 3 and 6 h after APAP injection. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. *p < 0.05, ***p < 0.001 versus control (vehicle) condition. ##p < 0.01, ###p < 0.001 versus SIRT1-WT mice. n = 6–8 mice per group. (C, D, E) Representative immunoblots showing (C) phospho-JNK, (D) IjBa, and (C, D) p85a-PI3K as a loading control and (E) nuclear p65-NFjB and Lamin B as a loading control. After quantification of all blots, results are expressed as fold change relative to control (vehicle) condition and are mean ± SEM. Statistical analysis was performed by two-way ANOVA followed by Bonferroni post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 versus control (vehicle) condition. ###p < 0.001 versus SIRT1-WT mice. n = 5–6 mice per group. IκBα, inhibitor of kappa B alpha; IL, interleukin; JNK, c-Jun N-terminal kinase; mRNA, messenger RNA; NFκB, nuclear factor kappa B; qRT-PCR, quantitative real-time PCR.
Moreover, we analyzed signaling pathways triggered by the proinflammatory milieu of the liver in response to APAP treatment. APAP induced similar increases in the phosphorylation of c-Jun N-terminal kinase (JNK) in liver extracts from wild-type and SIRT1-Tg mice 6 h after APAP intoxication (Fig. 5C and Supplementary Fig. S5). Likewise, no differences in JNK phosphorylation were detected 1 h after APAP intoxication, the time at which wild-type mice presented a significant depletion of GSH compared with SIRT1-Tg mice (Supplementary Figs. S6 and S7). By contrast, substantial differences between genotypes were detected in the NFκB pathway. As shown in Figure 5D (Supplementary Fig. S5), degradation of the inhibitor of kappa B alpha (IκBα) was observed in livers of wild-type mice 6 h after APAP injection, but this effect was absent in SIRT1-Tg mice, reflecting an anti-inflammatory effect. In agreement with this, p65-NFκB was detected in the nucleus exclusively in livers from APAP-injected wild-type animals (Fig. 5E and Supplementary Fig. S5).
Conditioned media from APAP-treated macrophages decreased SIRT1 protein levels in hepatocytes
We next investigated at the molecular level the contribution of the inflammatory environment induced by APAP to the drop of SIRT1 protein levels in hepatocytes. RAW 264.7 macrophages were treated with 5 mM APAP for 8 h; media were then removed and replaced with fresh media for an additional 16 h. This conditioned medium of APAP-treated macrophages (APAP-CM) was used to treat immortalized mouse hepatocytes generated and validated in our laboratory as described in the Materials and Methods section. Figure 6A (Supplementary Fig. S8) shows that SIRT1 was downregulated in hepatocytes treated for 16 h with APAP-CM as compared with hepatocytes incubated with control (vehicle)-CM. By contrast, direct treatment of hepatocytes with toxic doses of APAP previously reported (33) did not modify SIRT1 protein levels (Fig. 6B and Supplementary Fig. S8).
FIG. 6.
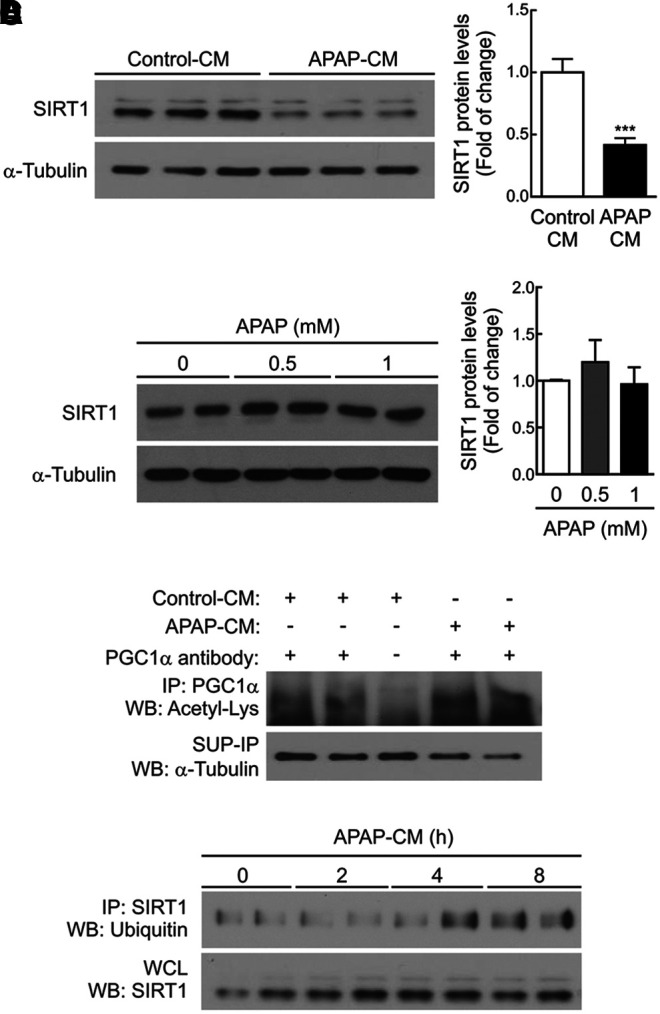
CM from APAP-treated macrophages decreased SIRT1 protein levels in hepatocytes. (A) Hepatocytes were treated 16 h with conditioned media collected from RAW 264.7 cells treated with vehicle or 5 mM APAP for 8 h and then reefed with fresh medium for a further 16 h (control-CM or APAP-CM, respectively). Representative immunoblots showing SIRT1 and α-Tubulin (loading control) protein levels (left). After quantification of all blots, results are expressed as fold change relative to control-CM condition (right). Values are mean ± SEM. ***p < 0.001 versus control-CM according to Student's t-test. (B) Hepatocytes were treated with APAP (0.5 and 1 mM) for 16 h. Representative immunoblot showing SIRT1 and α-Tubulin (loading control) protein levels (left). After quantification of all blots, results are expressed as fold change relative to vehicle condition (right). Values are mean ± SEM. (C) Hepatocytes were treated with control-CM or APAP-CM for 16 h, as indicated (A). PGC1α was immunoprecipitated with anti-PGC1α antibody or protein A agarose as a negative control. Immunoblot against anti-Acetil-Lys represents acetylated PGC1α protein levels. Total α-Tubulin was analyzed in the SUP-IP as a loading protein control. (D) Hepatocytes were treated with APAP-CM for the indicated times in the presence of the proteasome inhibitor MG132 (10 μM) and then the levels of ubiquitinated SIRT1 were analyzed by immunoprecipitation (upper blot). Total SIRT1 protein detected in the WCL is shown in the lower blot. APAP-CM, conditioned medium of APAP-treated macrophages; PGC1α, peroxisome proliferator-activated receptor (PPAR) gamma coactivator 1 alpha; SUP-IP, IP-supernatant; WCL, whole-cell lysate.
As expected, the decrease of SIRT1 by APAP-CM resulted in increased PGC1α acetylation (Fig. 6C and Supplementary Fig. S8). However, similar mRNA levels of SIRT1 were detected in hepatocytes incubated with control-CM and APAP-CM (results not shown), suggesting a post-transcriptional effect of the APAP-CM on SIRT1 expression. These results prompted us to analyze SIRT1 ubiquitination in hepatocytes treated with APAP-CM in the presence of the proteasome inhibitor MG132. As shown in Figure 6D (Supplementary Fig. S8), ubiquitin was detected in anti-SIRT1 immunoprecipitates from hepatocytes incubated for 4–8 h with APAP-CM and, importantly, at these time periods, total SIRT1 protein levels remained unchanged. The data thus suggest that the ubiquitination of SIRT1 in the presence of APAP-CM occurred before its degradation.
Essential role of IL1β in the modulation of SIRT1 protein levels in hepatocytes during APAP treatment
The analysis of mRNA levels of proinflammatory cytokines in RAW 267.4 macrophages treated 8 h with APAP (5 mM) revealed a significant increase in IL1β mRNA (Fig. 7A and Supplementary Fig. S9). Likewise, increased IL1β was found in the CM of RAW 264.7 cells treated with APAP compared to the untreated controls.
FIG. 7.

Role of IL1β in the modulation of SIRT1 expression in hepatocytes treated with APAP. (A) Left panel, Il1b, Il6, and Tnfa mRNA levels were determined by qRT-PCR in RAW 264.7 cells stimulated with vehicle or 5 mM APAP for 8 h. Values are mean ± SEM. ***p < 0.001 versus vehicle according to Student's t-test. Right panel, Media from RAW 264.7 cells treated with vehicle or 5 mM APAP for 8 h were collected, and then, IL1β protein levels were measured. After quantification of all blots, results are expressed as fold change relative to control-CM condition. Values are mean ± SEM. *p < 0.05 versus vehicle according to Student's t-test. (B) Immunoblots showing the active fragment of caspase-1 (p10) detected in RAW 264.7 macrophages stimulated with APAP (5 mM) for 4 h and α-Tubulin showing that similar amounts of protein were loaded in each lane. Representative blots are shown. After quantification of all blots, results are expressed as fold change relative to vehicle condition and are mean ± SEM. **p < 0.01 versus vehicle according to Student's t-test. (C) Upper panels, SIRT1 levels detected in protein extracts from primary mouse (left) or human (right) hepatocytes treated with or without IL1β (20 ng/mL) for 16 h. Densitometric quantifications of protein levels are shown. After quantification of all blots, results are expressed as fold change relative to untreated condition. Values are mean ± SEM. *p < 0.05, ***p < 0.001 versus without IL1β condition according to Student's t-test. Lower panels, Mouse hepatocytes were treated with IL1β (20 ng/mL) for the indicated times, and then, the levels of nuclear p65-NFκB and total IκBα were analyzed by immunoblot. Lamin B and β-Actin were used as nuclear and total protein loading controls, respectively. After quantification of all blots, results are expressed as fold change relative to untreated condition and are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Newman–Keuls multiple comparison test. **p < 0.01, ***p < 0.001 versus without IL1β. (D) Primary mouse hepatocytes were transfected with siRNAs for p65, JNK1/2, or both or with a control scrambled siRNA for 48 h. Then, hepatocytes were treated with control-CM or APAP-CM for 16 h in the presence of siRNAs. Total protein was analyzed by Western blot using the indicated antibodies. Densitometric quantification depicts SIRT1 protein levels in each condition. Values are mean ± SEM. Statistical analysis was performed by two-way ANOVA followed by Bonferroni post hoc test. ***p < 0.001 versus scramble siRNA-transfected hepatocytes treated with control-CM. (E) Control-CM, APAP-CM, and APAP-CM, previously incubated with anti-IL1β neutralizing antibody (0.2 μg/mL) for 1 h, were added to mouse hepatocytes for 16 h, and then, SIRT1 protein levels were analyzed by immunoblot. After quantification of all blots, results are expressed as fold change relative to control-CM condition. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Newman–Keuls multiple comparison test. ***p < 0.001 versus control-CM. #p < 0.05 versus without anti-IL1β neutralizing antibody condition. siRNA, small interfering RNA.
As recent data indicate a role for the inflammasome in APAP hepatotoxicity (14), we next evaluated the effect of APAP on the activation of caspase-1 in RAW 264.7 macrophages. As depicted in Figure 7B (Supplementary Fig. S9), the active fragment of caspase-1 (p10) was detected in RAW 264.7 macrophages exposed to APAP (5 mM) for 4 h. Also, since IL1β was released by macrophages on APAP challenge (Fig. 7A and Supplementary Fig. S9), we tested the possibility of a direct effect of IL1β in the modulation of SIRT1 protein levels in hepatocytes. Both mouse and human hepatocytes directly incubated with IL1β for 16 h had decreased SIRT1 protein expression (Fig. 7C and Supplementary Fig. S9). In contrast, SIRT1 protein levels did not decrease in hepatocytes treated with IL6 or TNFα (Supplementary Figs. S10 and S11).
At the molecular level, IL1β activated the NFκB signaling pathway in mouse hepatocytes as reflected by the time-dependent nuclear accumulation of p65-NFκB in parallel with the degradation of IκBα (Fig. 7C and Supplementary Fig. S9), suggesting that NFκB was likely involved in the effect of IL1β in the modulation of SIRT1 protein levels in hepatocytes by APAP-CM. We tested this hypothesis by two different experimental approaches. First, small interfering RNA (siRNA) was used to decrease p65-NFκB in hepatocytes and then to evaluate the effect of APAP-CM on SIRT1 protein levels. As shown in Figure 7D (Supplementary Fig. S9), silencing of p65-NFκB prevented SIRT1 downregulation in hepatocytes treated with APAP-CM. Of note, silencing of JNK1/2 did not prevent the degradation of SIRT1 in hepatocytes exposed to APAP-CM. In the second approach, APAP-CM was incubated with an anti-IL1β neutralizing antibody for 1 h before the addition to mouse hepatocytes for a further 16 h. Figure 7E (Supplementary Fig. S9) shows that SIRT1 protein levels were preserved in hepatocytes treated with APAP-CM depleted of IL1β.
Opposing effects on the modulation of SIRT1 protein levels in hepatocytes treated with APAP-CM from macrophages of SIRT1-Tg or wild-type mice
Since the livers of SIRT1-Tg mice showed reduced IL1β mRNA levels, were insensitive to the activation of NFκB signaling, and preserved SIRT1 protein expression on APAP challenge compared with the effects found on these parameters in wild-type mice (Figs. 2 and 5), we analyzed the contribution of SIRT1 overexpression in macrophages to this protective effect. For this goal, peritoneal macrophages were isolated from wild-type and SIRT1-Tg mice and used to prepare control-CM or APAP-CM and test the differential effects on the modulation of SIRT1 protein levels in hepatocytes.
In mouse hepatocytes treated with APAP-CM from peritoneal macrophages isolated from wild-type mice, SIRT1 protein levels decreased as in hepatocytes treated with APAP-CM from RAW 267.4 macrophages (Figs. 6A, 8A, and Supplementary Fig. S12). In contrast, when mouse hepatocytes were treated with APAP-CM from peritoneal macrophages of SIRT1-Tg mice, the protein levels of SIRT1 remained unchanged.
FIG. 8.

Effect of APAP-CM from peritoneal macrophages from wild-type and SIRT1-Tg mice in the modulation of SIRT1 protein levels in hepatocytes. (A) Peritoneal macrophages were isolated from SIRT1-WT and SIRT1-Tg mice and then used to prepare control-CM or APAP-CM as previously described. Then, mouse hepatocytes were treated with these CM for 16 h. Immunoblots showing SIRT1 protein levels and α-Tubulin as a loading control. Densitometric quantification showing SIRT1 protein levels in each condition. Values are mean ± SEM. Statistical analysis was performed by two-way ANOVA followed by Bonferroni post hoc test. *p < 0.05 versus control-CM. (B) Levels of the active fragment of caspase-1 (p10) detected in peritoneal macrophages derived from SIRT1-WT and SIRT1-Tg mice stimulated with APAP (5 mM) for 3 or 6 h. GAPDH showing similar amounts of protein loaded in each lane. Representative blots are shown. After quantification of all blots, results are expressed as fold change relative to vehicle-treated SIRT1-WT macrophages and are mean ± SEM. Statistical analysis was performed by two-way ANOVA followed by Bonferroni post hoc test. *p < 0.05 versus wild-type macrophages. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To better understand the lack of effect of the APAP-CM from SIRT1-Tg macrophages on the modulation of SIRT1 levels in hepatocytes, we monitored the activation of the inflammasome in response to APAP by measuring the protein levels of active caspase-1. As shown in Figure 8B (Supplementary Fig. S12), a transient increase in the active caspase-1 (p10) fragment was detected in peritoneal macrophages isolated from wild-type mice and treated with APAP (5 mM) for 3 h. Importantly, this effect was not observed in APAP-treated peritoneal macrophages from SIRT1-Tg mice (Fig. 8B and Supplementary Fig. S12).
In vivo administration of the NFκB inhibitor BAY 11-7082 protected from APAP-mediated acute hepatotoxicity
To provide in vivo evidences of the modulation of SIRT1 expression through NFκB signaling during APAP hepatotoxicity, the NFκB inhibitor BAY 11-7082 (5 mg/kg) was injected to wild-type mice 1 h prior APAP intoxication. The efficacy of the NFκB inhibitor was tested by the analysis of nuclear NFκB 6 h after APAP injection.
Nuclear NFκB was detected in mice injected with APAP, but this effect was not observed in mice that received the NFκB inhibitor (Fig. 9A and Supplementary Fig. S13). As shown in Figure 9B and C (Supplementary Fig. S13), liver damage was not visualized in the histological analysis of liver sections from mice injected with BAY 11-7082 before APAP intoxication. Likewise, elevation in ALT was not observed in the group of mice treated with BAY 11-7082. Importantly, in those mice, SIRT1 protein levels were comparable to saline-injected mice (Fig. 9B, D, and Supplementary Fig. S13). Protection against APAP hepatotoxicity and preservation of SIRT1 protein levels were also found in mice that received BAY 11-7082 for 1 h after APAP injection (Fig. 9E–G and Supplementary Fig. S13).
FIG. 9.

In vivo administration of the NFκB inhibitor BAY 11-7082 protected from APAP-mediated acute hepatotoxicity. Overnight fasted wild-type mice were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP or BAY 11-7082 (5 mg/kg) 1 h prior APAP intoxication. Mice were sacrificed after 6 h and livers and serum were collected. (A) Nuclear p65-NFκB and Lamin B as loading control. Blots were quantified and results are expressed as fold change relative to vehicle (saline)-treated mice. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Newman–Keuls test. *p < 0.05 versus control (vehicle)-treated mice, #p < 0.05 versus APAP-treated mice. n = 5 mice per group. (B) Representative images of hematoxylin and eosin staining (upper panels) or anti-SIRT1 immunostaining (lower panels) in liver sections from wild-type mice treated with APAP or injected BAY 11-7082 1 h prior APAP intoxication. Scale bars = 100 μm. (C) Plasma ALT levels measured in the same experimental conditions. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. **p < 0.01 versus control (vehicle)-treated mice, ##p < 0.01 versus APAP-treated mice. n = 5 mice per group. (D) SIRT1 protein levels detected in liver extracts. Blots were quantified and results are expressed as fold change relative to control (vehicle)-treated mice. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. *p < 0.05 versus control (vehicle)-treated mice, ##p < 0.01 versus APAP-treated mice. n = 5 mice per group. (E) Overnight fasted wild-type mice were i.p. injected physiological saline (vehicle) or 300 mg/kg APAP or BAY 11-7082 (5 mg/kg) 1 h after APAP intoxication. Mice were sacrificed after 6 h and livers and serum were collected. Representative images of hematoxylin and eosin staining (upper panels) or anti-SIRT1 immunostaining (lower panels) in liver sections. Scale bars = 100 μm. (F) Plasma ALT levels measured in the same experimental conditions. Values are mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Bonferroni post hoc test. *p < 0.05 versus control (vehicle)-treated mice, #p < 0.05 versus APAP-treated mice. n = 5 mice per group. (G) SIRT1 protein levels detected in liver extracts. Blots were quantified and results are expressed as fold change relative to APAP-treated mice. Values are mean ± SEM. *p < 0.05 versus control (vehicle)-treated mice, ##p < 0.01 versus APAP-treated mice. n = 5 mice per group. H&E, hematoxylin and eosin. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Discussion
APAP-induced hepatotoxicity impacts key molecular processes in hepatocytes such as oxidative stress causing a drop in the GSH/GSSG ratio leading to mitochondrial collapse and ATP depletion (16), unresolved endoplasmic reticulum stress (50), and blockade of the autophagic flux (36), all of which contribute to necroptotic cell death. This work has further shown that APAP also induces a fast drop of antioxidant defenses in parallel to SIRT1 protein degradation in hepatocytes, a process mediated by IL1β/NFκB signaling that plays a key role in ROS-dependent hepatotoxicity.
Recent research has identified a role for nonparenchymal immune cells in the excessive inflammation that occurs during APAP-induced hepatotoxicity, although conflicting data have been generated in different preclinical models (reviewed in 17, 58). Regarding Kupffer cells, one study reported that gadolinium chloride significantly blocked APAP-caused hepatotoxicity in mice, indicating that recruitment of macrophages into the inflammatory sites contributes to APAP-mediated death of hepatocytes (7), whereas when Kupffer cells were depleted with clodronate liposomes, APAP hepatotoxicity was exacerbated due to a reduction in anti-inflammatory cytokines (e.g., IL10), supporting a cytoprotective effect of resident macrophages (21).
Similar controversy has been found with neutrophils. For instance, treatment with neutropenia-inducing antibody administered 24 h prior APAP treatment or deletion of intracellular adhesion molecule (ICAM)-1 in mice decreased the number of FasL-expressing cells and significantly protected mice against APAP-induced liver injury (30) and, on the contrary, Cover et al. (5) did not find protection in ICAM-1-deficient mice. Depletion of both NK and NKT cells decreased interferon gamma (IFNγ) mRNA, neutrophil accumulation, and protected mice from APAP-induced liver injury (29). Likewise, IFNγ-deficiency or administration of an anti-IFNγ neutralizing antibody alleviated APAP-induced liver injury (15) although Masson et al. (32) reported that NKT and NK cells do not play a pathologic role in APAP treatment in C57Bl/6 mice in the absence of dimethylsulfoxide. All of these and other studies (reviewed in 17) failed to confirm that Kupffer cells, neutrophils, or monocytes directly cause liver injury on APAP treatment.
The interplay between hepatocytes and NPCs during APAP hepatotoxicity has been mainly studied in the context of intrahepatic release of DAMPs by dying hepatocytes after APAP toxicity that activated immune liver cells (46). Among DAMPs, DNA activates TLR9, which in turn activates the inflammasome to transcribe pro-IL1β and pro-IL18 and, consequently, TLR9 deficiency or antagonism confers protection against APAP hepatotoxicity (14). Again, controversy has emerged since Williams et al. (53) did not find alterations in APAP hepatotoxicity by modulation of the inflammasome in transgenic mice targeting the inflammasome components.
In the present study, our initial observations of SIRT1 depletion on APAP challenge in both human and mouse livers (Fig. 1), together with the reported role of SIRT1 in the protection against hepatic inflammation linked to metabolic damage (12, 40), or against fibrosis induced by hepatic stellate cell activation in alcohol liver injury (43), prompted us to hypothesize that downregulation of SIRT1 could be a consequence of the enhanced proinflammatory environment that occurs during APAP toxicity due to the prevalent oxidative stress. This hypothesis was tested in mice with moderate overexpression of SIRT1 (SIRT1-Tg) that retained higher SIRT1 protein levels after APAP treatment compared with levels detected in wild-type mice.
Interestingly, in APAP-treated SIRT1-Tg mice, the retained protein levels of SIRT1 in the liver was associated with reduced injury assessed by histology and biomarkers, including ALT, TUNEL, GSH/GSSG ratio, protein carbonylation, and levels of the antioxidant enzymes MnSOD and catalase. Notably, APAP-protein adducts did not differ among the two genotypes of mice, suggesting similar metabolic bioactivation of APAP through CYP2E1. These results challenge the traditional view that APAP-protein adducts are the key drivers in APAP-induced hepatotoxicity. Alternatively, the antioxidant capacity of the cell would be the main component of the resistance to APAP hepatotoxicity in SIRT1-Tg mice since the ensuing oxidative stress per se could be directly responsible for the modulation of the inflammatory responses and, ultimately, for the induction of cell death.
To establish a rationale link between the protection against APAP hepatotoxicity by preserving SIRT1 protein levels in the liver of SIRT1-Tg mice and the early inflammatory responses, we examined the immune cell populations in the liver 3 h after APAP injection, a time period that precedes SIRT1 degradation. Taking into account that Kupffer cells become depleted in the early course of APAP treatment (9) and that this effect was similar in wild-type and SIRT1-Tg mice, we analyzed the potential renewal of Kupffer cells from newly recruited monocytes that has been shown to be involved in the resolution of liver injury (9, 63). Our data clearly showed a strong elevation of CD11b+CCR2+Ly6C+ cells in the livers of SIRT1-Tg mice as early as 3 h after APAP treatment.
These results are surprising since recently it has been shown that this population plays an important role in APAP hepatotoxicity (34). Such discrepancy suggests that SIRT1 overexpression might be able to prevent the activation of liver infiltrating macrophages thus reducing their contribution to parenchymal damage. This hypothesis is supported by elevations in the anti-inflammatory markers IL10 and arginase-1 in the livers of SIRT1-Tg mice on APAP intoxication compared to the levels of wild-type mice. Likewise, neutrophils were highly elevated in SIRT1-Tg livers and this could be linked to their role in removal of cell debris during regeneration, as suggested by Williams et al. (54), rather than in promoting toxicity.
Also, despite the unclear role of T cells in APAP toxicity (24), our model of SIRT1-Tg mice has revealed that protection against APAP toxicity might be linked to an alteration in the balance between CD8+ cytotoxic T lymphocytes and CD8− noncytotoxic T lymphocytes (T helper) toward an increase in the latter population. In fact, SIRT1 may mediate the differentiation of T helper cells by suppressing STAT3 activity (37). Altogether, these results suggest that the differential immune responses in APAP toxicity in wild-type and SIRT1-Tg mice could be responsible, at least in part, to the reduced hepatic mRNA levels of the proinflammatory cytokines IL6 and IL1β, increased mRNA levels of IL10 and arginase-1, and attenuation of the NFκB proinflammatory signaling in SIRT1-Tg mice.
As previously stated, the relationship between APAP toxicity and the inflammasome is controversial. However, negative regulation of the NLRP3 inflammasome by SIRT1 has been reported in vascular endothelial cells (28). The present work addresses a relationship between IL1β-mediated inflammation by immune cells on APAP challenge and the expression of SIRT1 in hepatocytes. Our initial experiments demonstrated that SIRT1 was ubiquitinated and its protein levels were downregulated by the treatment of hepatocytes with CM of RAW 264.7 macrophages exposed to APAP (APAP-CM), which contained higher levels of IL1β compared with the control-CM. Of note, the effect of APAP on macrophages was likely mediated by the inflammasome since the cleaved caspase-1 fragment (p10) was detected in these immune cells on APAP stimulation for 4 h. Importantly, the effect of APAP-CM in the modulation of SIRT1 protein levels in hepatocytes was mimicked by direct stimulation with IL1β and was abolished by using an anti-IL1β neutralizing antibody or by targeting IL1β downstream signaling with p65-NFκB siRNA. However, in agreement with the similar activation of JNK in the livers of wild-type and SIRT1-Tg mice treated with APAP, silencing of JNK in hepatocytes did not prevent SIRT1 degradation by APAP-CM. These results were somehow unexpected since sustained activation of JNK and its translocation to the mitochondria followed by binding to SH3BP5 or Sab, a protein located on its outer membrane, has been associated with increased mitochondrial ROS production during APAP hepatotoxicity (55, 56).
In this regard, we cannot explain the lack of differences in JNK phosphorylation between wild-type and SIRT1-Tg mice after APAP intoxication. Moreover, in the livers of SIRT1-Tg mice injected APAP for 1 h, the maintenance of considerable GSH levels compared to the depletion detected in wild-type mice (Supplementary Fig. S6) suggests that NAPQI production is inhibited at this early time period. These relevant issues deserve further investigations. In addition to these findings, our data have revealed first that the modulation of SIRT1 protein levels during APAP intoxication was mainly mediated by the IL1β-NFκB signaling pathway and, second, that the preservation of SIRT1 in the livers of SIRT1-Tg mice might be required to enhance the antioxidant defense and protect against APAP toxicity.
These data were assessed by the protective effect of administration of BAY 11-7082, an NFκB inhibitor, to wild-type mice that efficiently protected against the decreased hepatic SIRT1 protein levels and liver damage induced by APAP. Further studies will be necessary to define if additional processes such as mitophagy and mitochondrial biogenesis are linked with SIRT1 during APAP hepatotoxicity.
A variety of factors have been shown to regulate SIRT1 expression at both transcriptional and post-translational levels, including hypoxia, nutrient deprivation, DNA damage, and oxidative stress (20, 62). In the aorta, SIRT1 levels are positively modulated by prostacyclin, an anti-inflammatory prostanoid generated by ciclooxygenase-2 (2). Importantly, in the liver, APAP inhibits prostanoid synthesis (47) and this effect could be linked to the decrease in SIRT1 levels, thereby resulting in the loss of the protective effect of prostacyclin against APAP hepatotoxicity also reported (4, 11). Our data suggest the modulation of SIRT1 protein levels by the proinflammatory milieu during APAP hepatotoxicity and a supporting hypothesis is the existence of a cross talk between macrophages and hepatocytes involving IL1β that acting via NFκB leads to a reduction in SIRT1 protein levels in hepatocytes and these series of events might be relevant in triggering oxidative stress-mediated liver damage (Fig. 10).
FIG. 10.

Model for the interplay between macrophages and hepatocytes in the context of APAP hepatotoxicity. Proposed model for the cross talk between macrophages and hepatocytes modulated by APAP and involving IL1β-mediated NFκB signaling and SIRT1 degradation in hepatocytes. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In addition, we have provided new mechanistic insights to the recent work of Wojnarova et al. (57) who reported decreased SIRT1 activity in rats treated with APAP that was reversed by resveratrol or the SIRT1 activator CAY10591. Wang et al. (52) reported that resveratrol treatment was associated with elevated SIRT1 protein in mouse livers that were protected from APAP hepatotoxicity. In the light of these previous data, our work provides novel aspects by demonstrating first, the mechanism by which SIRT1 is degraded on APAP challenge and, second, the direct effect of SIRT1 overexpression on APAP hepatotoxicity. Regarding this, we have recently reported SIRT1-independent effects of resveratrol in the liver in the context of insulin resistance (10).
Our experiments with APAP-CM from SIRT1-Tg macrophages have demonstrated that SIRT1 overexpression in these immune cells also contributes to the phenotype of SIRT1-Tg mice challenged with APAP. In fact, it has been shown that high glucose significantly downregulates the protein levels of SIRT1 and upregulates IL1β and TNFα mRNAs in RAW 264.7 macrophages (19). Moreover, myeloid-specific deletion of SIRT1 promotes macrophage infiltration into insulin-sensitive organs and aggravates liver inflammation (18, 22). On the contrary, SIRT1 inactivation exacerbates inflammation in human monocytes (48). In the light of these studies, our data suggest the possibility that in SIRT1-Tg mice, the anti-inflammatory effect of SIRT1 in immune cells targeting the inflammasome/IL1β, together with the sustained levels of SIRT1 in hepatocytes, inhibiting the NFκB pathway (60), might act in conjunction, thereby boosting hepatoprotection against oxidative stress during APAP toxicity.
Conclusions
In conclusion and as summarized in the working model proposed in Figure 10, this study provides new insights on the negative modulation of SIRT1 protein levels by the IL1β/NFκB axis in the interplay between immune cells and hepatocytes in the context of oxidative stress during APAP hepatotoxicity, and strongly suggests that targeting this pathway may alleviate ROS-induced liver damage.
Materials and Methods
Reagents and antibodies
Fetal bovine serum (FBS) and culture media were obtained from Invitrogen (Thermo Fisher Scientific, Waltham, MA). Protein A agarose was purchased from Roche Diagnostics (Indianapolis, IN). APAP and MG132 were from Sigma-Aldrich (Saint Louis, MO). The antibodies used in this study were anti-phospho JNK1/2 (Thr183/Tyr185) (No. 4668) and anti-ubiquitin (No. 3936) antibodies from Cell Signaling Technology (Danvers, MA). Anti-PGC1α (sc-13069), anti-JNK (sc-571), anti-IκBα (sc-371), anti-p65 NFκB (sc-372), anti-Sp1 (sc-420), and anti-caspase-1 (p10) (sc-514) antibodies were from Santa Cruz (Palo Alto, CA). Anti-p85α-PI3K (06-195), anti-catalase (219010), anti-Acetyl-Lysine (05-515) and anti-SIRT1 (07-131) antibodies were purchased from Merck Millipore (Billerica, MA). Anti-Lamin B (ab16048) and anti-glyceraldehyde-3-phosphate dehydrogenase (ab8245) antibodies were from Abcam (Cambridge, UK). Anti-MnSOD antibody (ADI-SOD-111-F) was purchased from Enzo Life Sciences (Farmingdale, NY). Anti-α-Tubulin (T5168) and β-Actin (A5441) antibodies were from Sigma-Aldrich. Neutralizing anti-IL1β antibody (AF-401-NA) was from R&D Systems, Inc. (Minneapolis, MN).
Cell culture
Murine RAW 264.7 macrophages, kindly provided by Dr. Tarín (CNIC, Madrid, Spain), were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine at 37°C in a humidified atmosphere with 5% CO2. Elicited peritoneal macrophages were obtained from male mice 4 days after intraperitoneal (i.p.) administration of 2.5 mL of 3% thioglycollate broth as previously described (49). Cells were seeded in six-multiwell plates at a density of 4 × 106 cells/plate and cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS and antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin).
The generation and characterization of immortalized mouse hepatocyte cell line have been previously described (39). Cells were grown in Dulbecco's modified Eagle's medium plus 10% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine.
Human hepatocytes were isolated by the two-step collagenase procedure from nontumor areas of liver biopsies from patients submitted to a surgical resection for liver tumors after obtaining patients' written consent as previously described (39).
Confluent RAW 264.7 or peritoneal macrophages were treated with vehicle (phosphate-buffered saline or PBS) or APAP (5 mM) for several time periods. In other experiments, macrophages were treated with vehicle or APAP for 8 h, after which, the medium was replaced by fresh media that were collected after 16 h. These conditioned media (control-CM or APAP-CM, respectively) were centrifuged to remove dead cells and directly added (without dilution) to mouse hepatocytes for several time periods.
Human liver biopsies
Human liver samples were obtained from the Department of Pathology, University of Edinburgh, United Kingdom, as previously reported (33). Informed written consent was obtained from each patient.
Animal models
Three-month-old male wild-type and mice overexpressing SIRT1 under the own promoter elements (SIRT1-Tg) (10, 40) maintained on the C57Bl/6J × 129Sv/J genetic background were used throughout this study. Animal experimentation was approved by the Ethics Committee at CSIC (Spain) and was conducted according to the accepted guidelines for animal care of Comunidad de Madrid (Spain). Overnight fasted mice were i.p. injected 300 mg/kg APAP dissolved in physiological saline. Mice were sacrificed at 3, 6, and 24 h after APAP injection and livers and blood were collected. The NFκB inhibitor BAY11-7082 [Sigma-Aldrich; dissolved in dimethyl sulfoxide (DMSO) and diluted in saline solution] was i.p. injected (5 mg/kg) 1 h prior or after APAP intoxication and mice were sacrificed after 6 h of APAP injection. In these experiments, all mice received similar amounts of DMSO.
Liver histology and immunohistochemistry
Histological grading of hepatic necrosis was performed by two blinded observers using hematoxylin and eosin-stained sections as follows: 30% of the total area necrotic (1 point); 30–60% of the total area necrotic (2 points); 60% of the total area necrotic (3 points) as described (33). Immunohistochemistry was performed as described (33).
TUNEL analysis
For cell death detection, paraffin-embedded liver biopsy sections were stained using the In Situ cell death detection kit (Roche) according to the manual instructions.
Isolation and analysis of nonparenchymal liver cells by flow cytometry
NPCs were isolated as described with slight modifications (46). Cells (0.3–0.5 × 106 cells/test) were incubated with CD45-FITC (rat IgG; Beckman), CD11b-(Mac1)-PECy7 (rat IgG2bk; eBioscience; Thermo Fisher Scientific), F4/80-APC (rat IgG2ak; eBioscience), Ly6G-PE (rat IgG2ak; Pharmingen, San José, CA), CD3-PECy7 (Hamster IgG; eBioscience), NK1.1-APC (mouse IgG2ak; Pharmingen), CD8a-PE (rat IgG2ak; Cultek, Madrid, Spain), F4/80-PE (rat IgG2ak; eBioscience), Ly6C-FITC (rat IgMk; Pharmingen), and CCR2-APC (rat IgG2B; R&D Systems) or their corresponding isotype controls for 20 min at room temperature. Flow cytometry data were acquired with an FACSCanto II and data analysis was performed using Cytomics FC500 with the CXP program.
Transfection with siRNA
siRNA oligos were synthesized by Dharmacon RNAi Technologies for gene silencing of mouse p65-NFκB and JNK1/2. Immortalized mouse hepatocytes were seeded in 6-cm dishes and incubated at 37°C with 5% CO2 overnight. When 40–50% confluence was reached, cells were transfected with p65-NFκB or JNK1/2 siRNA, or with a scrambled control siRNA at 25 nM concentration following DharmaFECT General Transfection Protocol (Dharmacon, Lafayette, CO). After 36 h, cells were used for experiments.
Homogenization and preparation of tissue extracts
Frozen livers were homogenized in 16 volumes (w/v) of ice-cold lysis buffer containing 50 mM Tris-HCl, 1% Triton X-100, 2 mM EGTA, 10 mM EDTA acid, 100 mM NaF, 1 mM Na4P2O7, 2 mM Na3VO4, 100 μg/mL phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, 1 μg/mL pepstatin A, and 1 μg/mL leupeptin. Livers were homogenized in the same lysis buffer using the Brinkman PT 10/35 Polytron. Extracts were kept ice cold at all times. Liver extracts were cleared by microcentrifugation at 40,000 × g for 20 min at 4°C. The supernatant was aliquoted and stored at −70°C.
Immunoprecipitations and Western blot
After culture and treatments, cells were scraped off in ice-cold PBS, pelleted by centrifugation at 4000 × g for 10 min at 4°C, and resuspended in lysis buffer containing 10 mM Tris-HCl, 5 mM EDTA, 50 mM NaCl, 30 mM disodium pyrophosphate, 50 mM NaF, 100 μM Na3VO4, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, and 10 μg/mL aprotinin pH 7.6. Cellular lysates were clarified by centrifugation at 12,000 × g for 10 min. After protein content determination, equal amounts of protein (600 μg) were immunoprecipitated at 4°C with the corresponding antibodies. The immune complexes were collected on agarose beads and submitted to Western blot analysis. After sodium dodecyl sulfate–polyacrylamide gel electrophoresis, gels were transferred to Immobilon membranes and were blocked using 5% nonfat dried milk or 3% bovine serum albumin (BSA) in 10 mM Tris-HCl and 150 mM NaCl pH 7.5, and incubated overnight with antibodies as indicated in 0.05% Tween-20, 10 mM Tris-HCl, and 150 mM NaCl pH 7.5. Immunoreactive bands were visualized using the ECL Western blotting protocol (Bio-Rad, Hercules, CA).
Protein determination
Protein determination was performed by the Bradford dye method, using the Bio-Rad reagent (Bio-Rad) and BSA as the standard.
Quantitative real-time PCR analysis
Total RNA was extracted with TRIzol® reagent (Invitrogen, Madrid, Spain) and reverse transcribed using a SuperScript™ III First-Strand Synthesis System for quantitative PCR (qPCR) following the manufacturer's indications (Invitrogen). qPCR was performed with an ABI 7900 sequence detector. Primer/probe sets for mouse Sir2, Tnfa, Il6, Il1b, IL10, Arg1, and 18s were purchased as predesigned by TaqMan gene expression assays (Applied Biosystems, Drive Foster City, CA).
Analysis of ALT activity
Blood was collected in heparin and diluted 1/30 with saline (0.9% NaCl). ALT activity was determined by direct measurement with the Reflectron test (Ref. 10745120; Roche Diagnostics).
Serum cytokine detection assay
Serum samples or culture media were collected and immediately frozen at −80°C. TNFα, IL6, and IL1β were determined using Luminex 100 IS (Merck Millipore).
Determination of GSH/GSSG ratio
Total reduced (GSH) and oxidized (GSSG) glutathione levels were measured in the same samples using a colorimetric assay (Assay Designs, Inc., Ann Arbor, MI). The ratio between GSH and GSSG was also calculated as a marker of antioxidant status in liver homogenates (51).
Determination of protein carbonyl content
Protein oxidation of liver homogenates was measured as carbonyl group content according to the method of Richert et al. (44).
Determination of APAP-protein adducts
Analysis of APAP covalently bound to proteins in the liver was measured by initial protease treatment of liver homogenates, followed by high-performance liquid chromatography-electrochemical analysis for APAP-cysteine as previously described (35).
Extraction of nuclear proteins
Livers or hepatocytes were homogenized at 4°C in 10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.2 mM PMSF, 0.75 μg/mL leupeptin, and 0.75 μg/mL aprotinin (Buffer A), allowed to swell on ice for 10 min, and then vortexed for 10 s. Samples were centrifuged and the supernatant containing the cytosolic fraction was stored at −70°C. The pellet was resuspended in cold buffer C (20 mM HEPES-KOH, pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.2 mM PMSF, 0.75 μg/mL leupeptin, and 0.75 μg/mL aprotinin) and incubated on ice for 20 min for high salt extraction. Cellular debris was removed by centrifugation for 2 min at 4°C, and the supernatant fraction was stored at −70°C.
Data analysis
Data are reported as mean ± standard error of the mean. Comparisons between groups were made using Student's t-test. To determine the effects of genotype or APAP injection, one-way or two-way analysis of variance followed by the Bonferroni test, respectively, was carried out. The p-values presented in figures corresponded to the post hoc test. All statistical analyses were performed using the GraphPad Prism 5.0 software (GraphPad Software, Inc., San Diego, CA). Differences were considered statistically significant at p < 0.05.
Supplementary Material
Abbreviations Used
- ALT
alanine transaminase
- ANOVA
analysis of variance
- APAP
acetaminophen
- APAP-CM
conditioned medium of APAP-treated macrophages
- BSA
bovine serum albumin
- CYP2E1
cytochrome P450 2e1
- DAMPs
damage-associated molecular patterns
- DILI
drug-induced liver injury
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSH
glutathione
- H&E
hematoxylin and eosin
- ICAM
intracellular adhesion molecule
- IFNγ
interferon gamma
- IκBα
inhibitor of kappa B alpha
- IL
interleukin
- i.p.
intraperitoneal
- JNK
c-Jun N-terminal kinase
- MnSOD
manganese superoxide dismutase
- mRNA
messenger RNA
- NAPQI
N-acetyl-p-aminobenzoquinone imine
- NFκB
nuclear factor kappa B
- NKT
natural killer T
- NPCs
non-parenchymal liver cells
- PBS
phosphate-buffered saline
- PGC1α
peroxisome proliferator-activated receptor (PPAR) gamma coactivator 1 alpha
- qPCR
quantitative PCR
- qRT-PCR
quantitative real-time PCR
- ROS
reactive oxygen species
- SEM
standard error of the mean
- siRNA
small interfering RNA
- SIRT1
sirtuin 1
- Sp1
specific protein 1
- SUP-IP
IP-supernatant
- TLRs
Toll-like receptors
- TNFα
tumor necrosis factor alpha
- WCL
whole-cell lysate
Acknowledgments
This work was funded by SAF2015-65267-R (MINECO/FEDER), CIBERdem (ISCIII, Spain), and INFLAMES (ISCIII PIE14/00045, cofunded by ERDF, “Investing in your future”) to A.M.V.; IJCI-2014-19381 (MINECO/FEDER) to P.R. and A.M.V.; SAF2014-52009-R (MINECO/FEDER) to S.A.; SAF2015-63904-R (MINECO/FEDER) to M.M.; CP14/00181 (ISCIII/FEDER) to A.G.-R.; and PI13/00021 (ISCIII/FEDER) to J.M. CIBERehd (ISCIII) to A.G.-R. and J.M.; Grant 37/371 from Al Jouf University to M.A.M. We also acknowledge H2020 Marie Sklodowska-Curie ITN-TREATMENT (Grant Agreement No. 721236) (European Commission).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R, Possamai LA, Bruce M, McPhail M, Starling C, Wagner B, Barnardo A, Pomplun S, Auzinger G, Bernal W, Heaton N, Vergani D, Thursz MR, and Wendon J. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 56: 735–746, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Barbieri SS, Amadio P, Gianellini S, Tarantino E, Zacchi E, Veglia F, Howe LR, Weksler BB, Mussoni L, and Tremoli E. Cyclooxygenase-2-derived prostacyclin regulates arterial thrombus formation by suppressing tissue factor in a sirtuin-1-dependent-manner. Circulation 126: 1373–1384, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Blazka ME, Elwell MR, Holladay SD, Wilson RE, and Luster MI. Histopathology of acetaminophen-induced liver changes: role of interleukin 1 alpha and tumor necrosis factor alpha. Toxicol Pathol 24: 181–189, 1996. [DOI] [PubMed] [Google Scholar]
- 4.Cavar I, Kelava T, Heinzel R, and Culo F. The role of prostacyclin in modifying acute hepatotoxicity of acetaminophen in mice. Coll Antropol 33(Suppl 2): 25–29, 2009. [PubMed] [Google Scholar]
- 5.Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, and Jaeschke H. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 216: 98–107, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, and Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther 315: 879–887, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Choi DY, Ban JO, Kim SC, and Hong JT. CCR5 knockout mice with C57BL6 background are resistant to acetaminophen-mediated hepatotoxicity due to decreased macrophages migration into the liver. Arch Toxicol 89: 211–220, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A 81: 1327–1331, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dambach DM, Watson LM, Gray KR, Durham SK, and Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology 35: 1093–1103, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Rodriguez A, Santamaria B, Mas-Gutierrez JA, Rada P, Fernandez-Millan E, Pardo V, Alvarez C, Cuadrado A, Ros M, Serrano M, and Valverde AM. Resveratrol treatment restores peripheral insulin sensitivity in diabetic mice in a sirt1-independent manner. Mol Nutr Food Res 59: 1431–1442, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Guarner F, Boughton-Smith NK, Blackwell GJ, and Moncada S. Reduction by prostacyclin of acetaminophen-induced liver toxicity in the mouse. Hepatology 8: 248–253, 1988. [DOI] [PubMed] [Google Scholar]
- 12.Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, and Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun 1: 3, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holt MP, Cheng L, and Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol 84: 1410–1421, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, and Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 119: 305–314, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, and Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J 16: 1227–1236, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Jaeschke H, McGill MR, and Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44: 88–106, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaeschke H, Williams CD, Ramachandran A, and Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int 32: 8–20, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeon BT, Kim KE, Heo RW, Shin HJ, Yi CO, Hah YS, Kim WH, Lee SI, and Roh GS. Myeloid-specific deletion of SIRT1 increases hepatic steatosis and hypothalamic inflammation in mice fed a high-fat diet. Metab Brain Dis 29: 635–643, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Jia Y, Zheng Z, Wang Y, Zhou Q, Cai W, Jia W, Yang L, Dong M, Zhu X, Su L, and Hu D. SIRT1 is a regulator in high glucose-induced inflammatory response in RAW264.7 cells. PLoS One 10: e0120849, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin J, Iakova P, Jiang Y, Medrano EE, and Timchenko NA. The reduction of SIRT1 in livers of old mice leads to impaired body homeostasis and to inhibition of liver proliferation. Hepatology 54: 989–998, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, and Pohl LR. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol 15: 1504–1513, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Ka SO, Song MY, Bae EJ, and Park BH. Myeloid SIRT1 regulates macrophage infiltration and insulin sensitivity in mice fed a high-fat diet. J Endocrinol 224: 109–118, 2015. [DOI] [PubMed] [Google Scholar]
- 23.Kaplowitz N. Acetaminophen hepatoxicity: what do we know, what don't we know, and what do we do next? Hepatology 40: 23–26, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Krenkel O, Mossanen JC, and Tacke F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg Nutr 3: 331–343, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, and Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127: 1109–1122, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem Res Toxicol 22: 1376–1385, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawson JA, Farhood A, Hopper RD, Bajt ML, and Jaeschke H. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol Sci 54: 509–516, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Yang X, He Y, Wang W, Zhang J, Zhang W, Jing T, Wang B, and Lin R. Negative regulation of NLRP3 inflammasome by SIRT1 in vascular endothelial cells. Immunobiology 222: 552–561, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Liu ZX, Govindarajan S, and Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 127: 1760–1774, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Liu ZX, Han D, Gunawan B, and Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 43: 1220–1230, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Liu ZX. and Kaplowitz N. Role of innate immunity in acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol 2: 493–503, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Masson MJ, Carpenter LD, Graf ML, and Pohl LR. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 48: 889–897, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mobasher MA, Gonzalez-Rodriguez A, Santamaria B, Ramos S, Martin MA, Goya L, Rada P, Letzig L, James LP, Cuadrado A, Martin-Perez J, Simpson KJ, Muntane J, and Valverde AM. Protein tyrosine phosphatase 1B modulates GSK3beta/Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity. Cell Death Dis 4: e626, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, Heymann F, Kalthoff S, Lefebvre E, Eulberg D, Luedde T, Marx G, Strassburg CP, Roskams T, Trautwein C, and Tacke F. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 64: 1667–1682, 2016. [DOI] [PubMed] [Google Scholar]
- 35.Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, and Mayeux PR. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos 30: 446–451, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Ni HM, Bockus A, Boggess N, Jaeschke H, and Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55: 222–232, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, and Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol 11: 492–500, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olmos Y, Sanchez-Gomez FJ, Wild B, Garcia-Quintans N, Cabezudo S, Lamas S, and Monsalve M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid Redox Signal 19: 1507–1521, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pardo V, Gonzalez-Rodriguez A, Guijas C, Balsinde J, and Valverde AM. Opposite cross-talk by oleate and palmitate on insulin signaling in hepatocytes through macrophage activation. J Biol Chem 290: 11663–11677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, and Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A 105: 9793–9798, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polson J, Lee WM; and American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 41: 1179–1197, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, and Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9: 327–338, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramirez T, Li YM, Yin S, Xu MJ, Feng D, Zhou Z, Zang M, Mukhopadhyay P, Varga ZV, Pacher P, Gao B, and Wang H. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J Hepatol 66: 601–609, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richert S, Wehr NB, Stadtman ER, and Levine RL. Assessment of skin carbonyl content as a noninvasive measure of biological age. Arch Biochem Biophys 397: 430–432, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, and Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci 23: 3597–3606, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanz-Garcia C, Ferrer-Mayorga G, Gonzalez-Rodriguez A, Valverde AM, Martin-Duce A, Velasco-Martin JP, Regadera J, Fernandez M, and Alemany S. Sterile inflammation in acetaminophen-induced liver injury is mediated by Cot/tpl2. J Biol Chem 288: 15342–15351, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schildknecht S, Daiber A, Ghisla S, Cohen RA, and Bachschmid MM. Acetaminophen inhibits prostanoid synthesis by scavenging the PGHS-activator peroxynitrite. FASEB J 22: 215–224, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Takeda-Watanabe A, Kitada M, Kanasaki K, and Koya D. SIRT1 inactivation induces inflammation through the dysregulation of autophagy in human THP-1 cells. Biochem Biophys Res Commun 427: 191–196, 2012. [DOI] [PubMed] [Google Scholar]
- 49.Traves PG, Pimentel-Santillana M, Rico D, Rodriguez N, Miethke T, Castrillo A, Theodorakis EA, Martin-Sanz P, Palladino MA, and Bosca L. Anti-inflammatory actions of acanthoic acid-related diterpenes involve activation of the PI3K p110gamma/delta subunits and inhibition of NF-kappaB. Chem Biol 21: 955–966, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y, Xavier R, Chung RT, Tirosh B, and Shibolet O. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol 59: 495–503, 2013. [DOI] [PubMed] [Google Scholar]
- 51.Valdecantos MP, Perez-Matute P, Gonzalez-Muniesa P, Prieto-Hontoria PL, Moreno-Aliaga MJ, and Martinez JA. Lipoic acid improves mitochondrial function in nonalcoholic steatosis through the stimulation of sirtuin 1 and sirtuin 3. Obesity (Silver Spring) 20: 1974–1983, 2012. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Jiang Y, Fan X, Tan H, Zeng H, Wang Y, Chen P, Huang M, and Bi H. Hepato-protective effect of resveratrol against acetaminophen-induced liver injury is associated with inhibition of CYP-mediated bioactivation and regulation of SIRT1-p53 signaling pathways. Toxicol Lett 236: 82–89, 2015. [DOI] [PubMed] [Google Scholar]
- 53.Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, Kanneganti TD, Park BK, and Jaeschke H. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol 252: 289–297, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, and Jaeschke H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol 275: 122–133, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Win S, Than TA, Han D, Petrovic LM, and Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 286: 35071–35078, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Win S, Than TA, Min RW, Aghajan M, and Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 63: 1987–2003, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wojnarova L, Kutinova Canova N, Farghali H, and Kucera T. Sirtuin 1 modulation in rat model of acetaminophen-induced hepatotoxicity. Physiol Res 64(Suppl 4): S477–S487, 2015. [DOI] [PubMed] [Google Scholar]
- 58.Woolbright BL. and Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol 66: 836–848, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang D, Tan X, Lv Z, Liu B, Baiyun R, Lu J, and Zhang Z. Regulation of Sirt1/Nrf2/TNF-alpha signaling pathway by luteolin is critical to attenuate acute mercuric chloride exposure induced hepatotoxicity. Sci Rep 6: 37157, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, and Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23: 2369–2380, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, Stevens SM, Jr., and You M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146: 801–811, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan Y, Cruzat VF, Newsholme P, Cheng J, Chen Y, and Lu Y. Regulation of SIRT1 in aging: roles in mitochondrial function and biogenesis. Mech Ageing Dev 155: 10–21, 2016. [DOI] [PubMed] [Google Scholar]
- 63.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, and Varol C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol 193: 344–353, 2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.