

Abstract
How locally adapted ecotypes are established and maintained within a species is a long-standing question in evolutionary biology. Using forest and prairie ecotypes of deer mice (Peromyscus maniculatus), we characterized the genetic basis of variation in two defining traits—tail length and coat color—and discovered a 41-megabase chromosomal inversion linked to both. The inversion frequency is 90% in the dark, long-tailed forest ecotype; decreases across a habitat transition; and is absent from the light, short-tailed prairie ecotype. We implicate divergent selection in maintaining the inversion at frequencies observed in the wild, despite high levels of gene flow, and explore fitness benefits that arise from suppressed recombination within the inversion. We uncover a key role for a large, previously uncharacterized inversion in the evolution and maintenance of classic mammalian ecotypes.
Wide-ranging species that occupy diverse habitats often evolve distinct ecotypes—intraspecific forms that differ in heritable traits relevant to their local environments (1). Ecotypes frequently differ in multiple locally adaptive phenotypes (2), and although ecotypes sometimes show partial reproductive isolation (2), many experience substantial intraspecific gene flow (3). This raises an important question: How are differences in multiple traits maintained between ecotypes when migration acts as a homogenizing force?
One explanation is that natural selection keeps each locus associated with locally adaptive trait variation at migration-selection equilibrium (4). However, in cases of high migration, this requires strong selection acting on many independent alleles. Linkage disequilibrium can play an important role by allowing linked loci, each with potentially weaker selective effects, to establish and be maintained together (5), which can lead to concentrated genetic architectures of ecotype-specific traits (6). Characterizing the genetic basis of the full set of ecotypic differences and the role of migration, selection, and recombination in maintaining these differences is thus critical to understanding local adaptation specifically and biological diversification more generally.
One of the most abundant and widespread mammals in North America is the deer mouse (Peromyscus maniculatus), which is continuously distributed across diverse habitats from the Arctic Circle to central Mexico. In the early 1900s, a taxonomic revision of this species described two distinct ecotypes: a forest and a prairie form (7). Several features distinguish the semiarboreal forest mice that occupy dark-soil habitats from their more terrestrial prairie counterparts that occupy light substrates. Most notably, forest mice typically have longer tails and darker coats than those of prairie mice (7–9), with large differences in these traits maintained between ecotypes despite evidence for gene flow (10, 11). This consistent divergence in multiple traits provides an opportunity to test the mechanisms that establish and maintain ecotypes.
Forest and prairie mice differ in multiple traits
To study divergence between the forest and prairie ecotypes, we selected two focal populations—one from a coastal temperate rainforest (P. m. rubidus, referred to hereafter as the forest ecotype) and one from an arid sagebrush steppe habitat (P. m. gambelii, referred to as the prairie ecotype) in the north-western US—separated by ~500 km (Fig. 1A). After establishing laboratory colonies from wild-caught mice, we measured both the wild-caught mice and their laboratory-reared descendants for four traits previously reported to distinguish forest and prairie ecotypes (7–9): tail, hindfoot, and ear lengths as well as coat color (brightness, hue, and saturation across three body regions). We also measured body length and weight. We found that forest mice had longer tails; longer hind feet; and darker, redder coats compared with prairie mice (Fig. 1, B and C; fig. S1; and table S1). These phenotypic differences persisted in laboratory-born mice raised in common conditions (fig. S2 and table S1), which suggests a strong genetic component to these ecotype-defining traits.
Fig. 1. Forest and prairie mice differ in tail length and pigmentation.
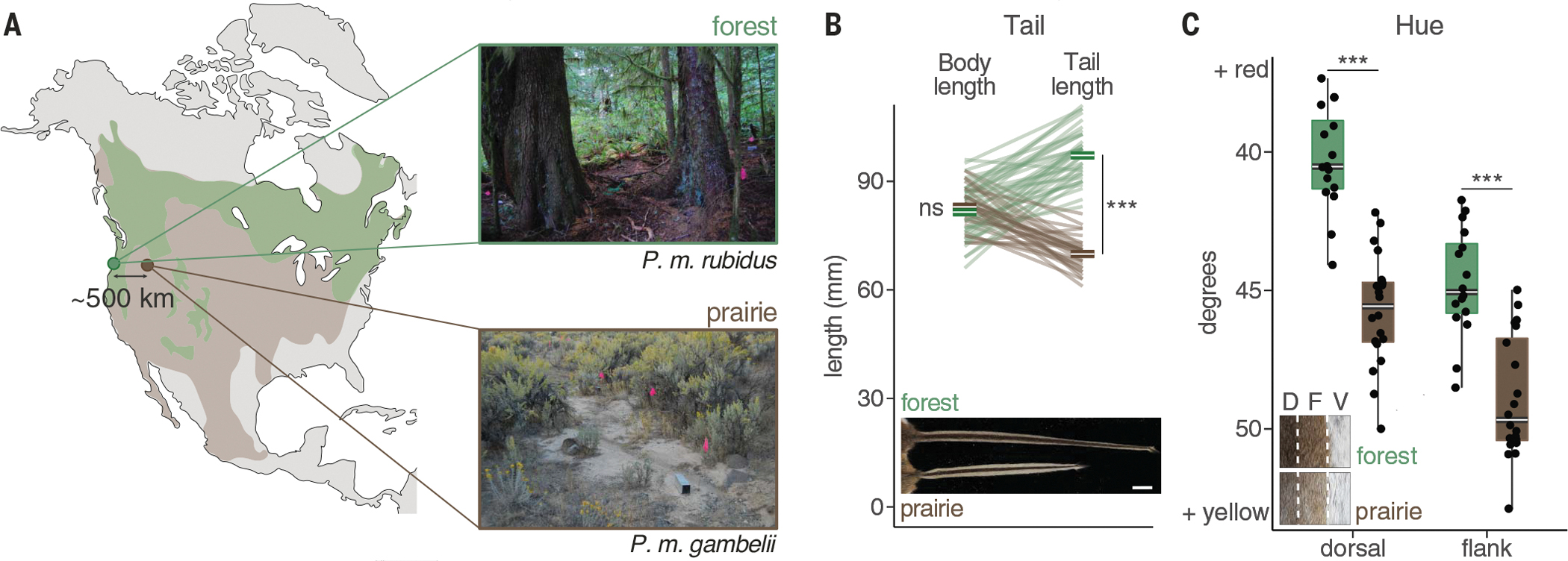
(A) Map shows the approximate range of forest (green) and prairie (brown) deer mouse ecotypes in North America. Collection sites of wild-caught forest (P. m. rubidus, green) and prairie (P. m. gambelii, brown) ecotypes from western and eastern Oregon, USA, respectively, are shown. Photos illustrate representative habitat; pink flags indicate trap lines. (B) Body length (left; not including the tail) and tail length (right) for wild-caught adult mice (n = 38 forest and 32 prairie). Lines connect body and tail measurements for the same individual. Means are shown in bold. (Inset) Image of a representative tail from each ecotype. Scale bar, 1 cm. (C) Coat color (hue) values for the dorsal and flank regions of wild-caught adult mice (n = 16 forest and 20 prairie). Boxplots indicate the median (center white line) and the 25th and 75th percentiles (box extents); whiskers show largest or smallest value within 1.5 times the interquartile range. Black dots show individual data points. (Inset) Dorsal (D), flank (F), and ventral (V) regions from a representative forest and prairie mouse. ns = P > 0.05; ***P < 0.001 (Welch’s t test, two-sided). Original photography in (B) and (C) is copyrighted by the President and Fellows of Harvard College (photo credit: Museum of Comparative Zoology, Harvard University).
A large inversion is associated with tail length and coat color
Using an unbiased forward-genetic approach, we identified genomic regions linked to ecotype differences in morphology. We intercrossed forest and prairie mice in the laboratory to generate 555 second-generation (F2) hybrids (forest female × prairie male, n = 203 F2s; prairie female × forest male, n = 352 F2s) and performed quantitative trait locus (QTL) mapping for each trait (12) (Fig. 2, fig. S3, and table S2). We identified five regions associated with tail length variation [total percent variance explained (PVE): 27%; individual PVE: 2.6 to 12.1%]. Only one region, on chromosome 15, was strongly and significantly associated with coat color variation (PVE, dorsal hue: 40.0%; PVE, flank hue: 45.6%). Each QTL exhibited incomplete dominance, and the forest allele was always associated with forest traits—longer tails or redder coats. The one significant QTL for coat color overlapped with the largest-effect locus associated with tail length (95% Bayesian credible intervals: dorsal hue = 0.4 to 40.5 Mb; flank hue = 0.4 to 39.4 Mb; tail length = 0.4 to 41.5 Mb). Thus, a single region on chromosome 15 was strongly associated with ecotype differences in both tail length and coat color.
Fig. 2. A region on chromosome 15 is strongly associated with both tail length and coat color.
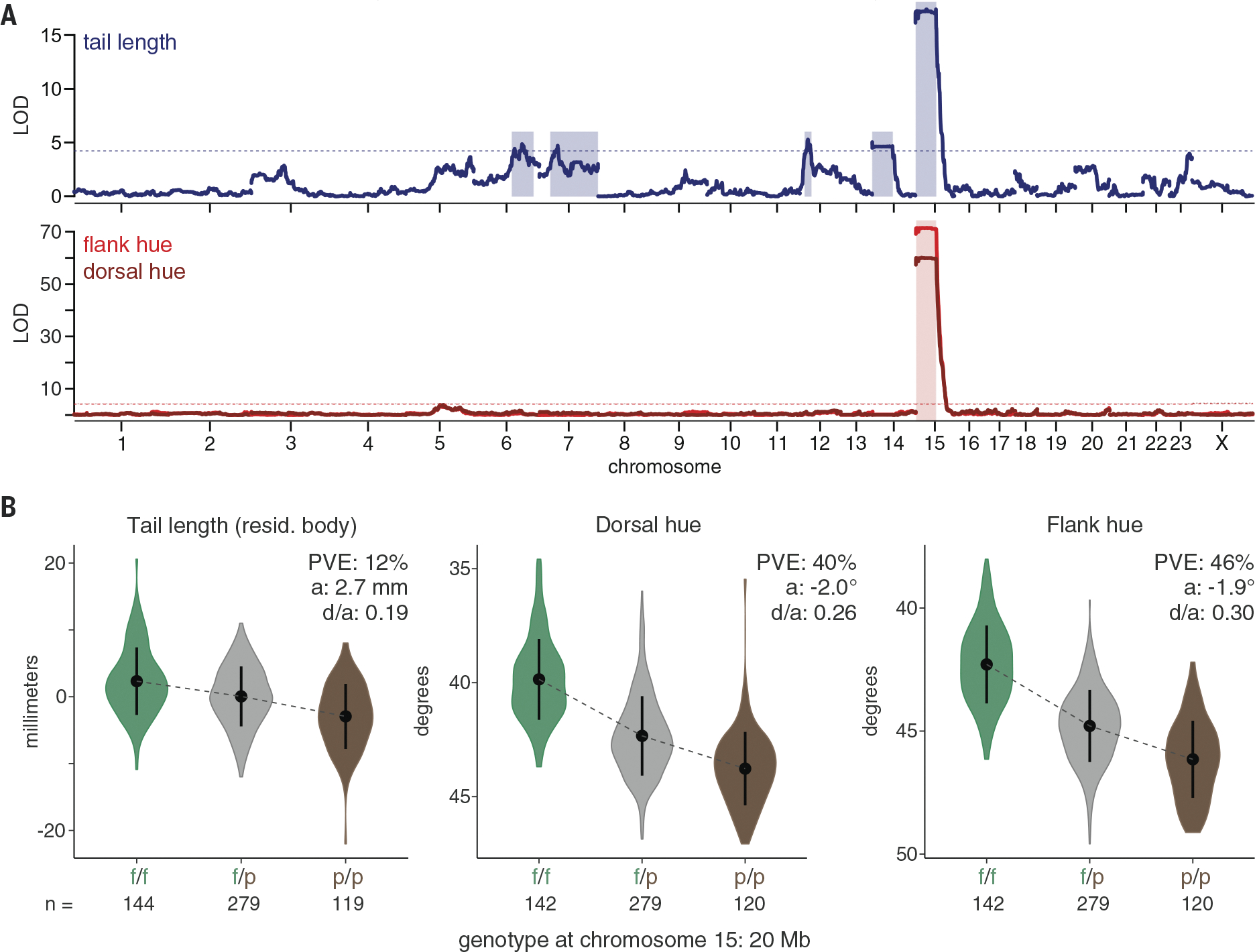
(A) Statistical association [log of the odds (LOD) score] of ancestry with tail length (top; blue) and dorsal and flank hue (bottom; dorsal, dark red; flank, light red) in laboratory-reared F2 hybrids (tail, n = 542; hue, n = 541). Physical distance (in base pairs) is shown on the x axis; axis labels indicate the center of each chromosome. Dotted lines indicate the genome-wide significance threshold (α = 0.05) based on permutation tests, and shaded rectangles indicate the 95% Bayesian credible intervals for all chromosomes with significant QTL peaks. For tail length analysis, body length was included as an additive covariate. (B) Tail length (left; shown after taking the residual against body length in the hybrids), dorsal hue (center), and flank hue (right) of F2 hybrids, binned by genotype at 20 Mb on chromosome 15 (f/f, homozygous forest; f/p, heterozygous; p/p, homozygous prairie) (sample sizes are given below the x axes). Points and error bars show means ± standard deviations. PVE, percent of the variance explained by genotype; a, additive effect of one forest allele; d/a, absolute value of the dominance ratio.
The QTL peak on chromosome 15 exhibited a consistently strong association with both morphological traits across half the chromosome (Fig. 3A). This pattern reflects reduced recombination between forest and prairie alleles in the laboratory cross: Only 2 of 1110 F2 chromosomes were recombinant in this region (Fig. 3B). We also found consistently elevated FST (proportion of the total genetic variance explained by population structure) (Fig. 3C) and high linkage disequilibrium (Fig. 3D) across this genetic region in wild populations relative to the rest of the chromosome (whole-genome resequencing: n = 15 forest, n = 15 prairie). Together, these data are consistent with reduced recombination across half of chromosome 15 in both laboratory and wild populations.
Fig. 3. Chromosomal region associated with tail length and coat color is a large inversion.
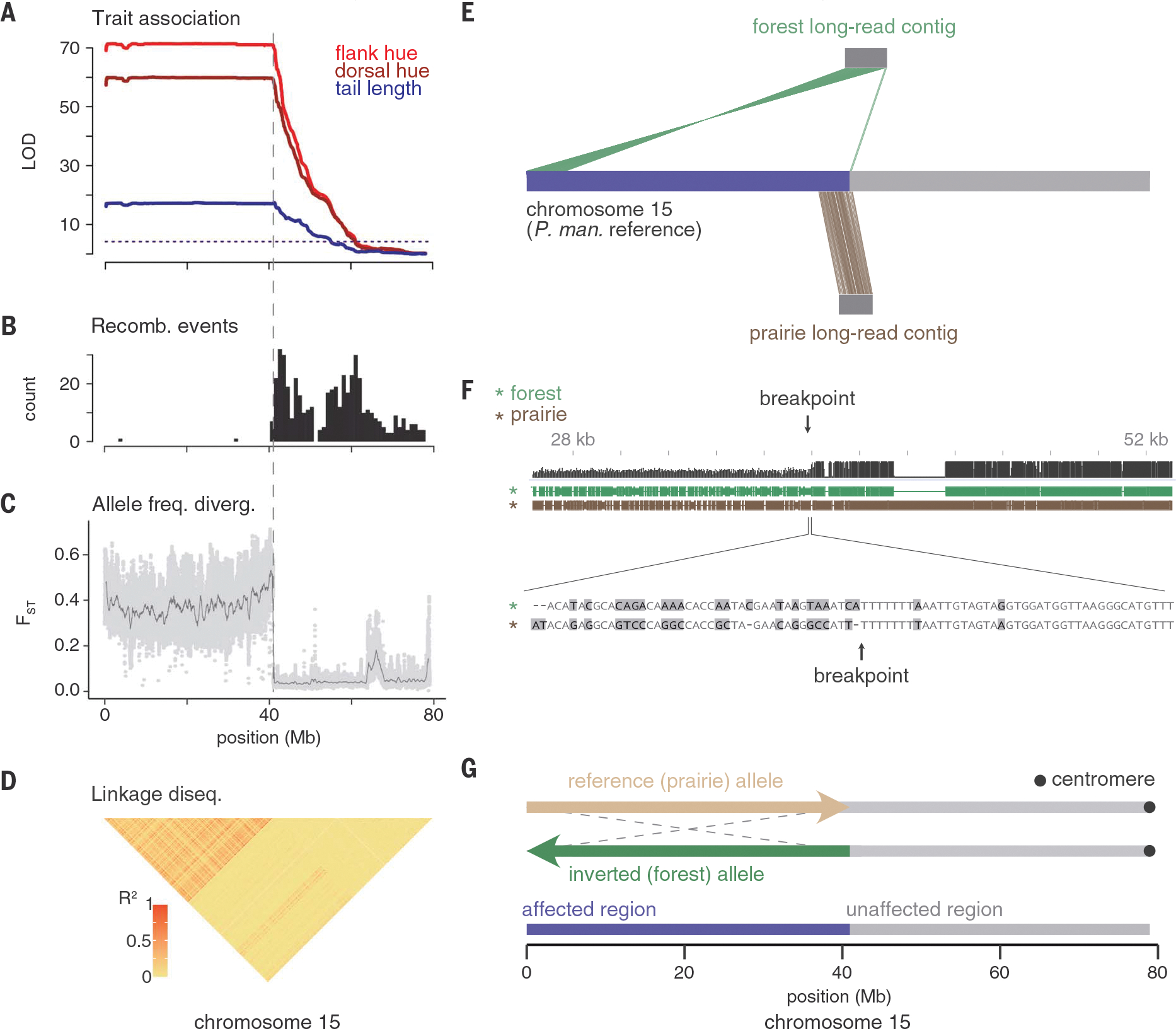
Across chromosome 15, data are from F2 hybrids [(A) and (B)] and wild-caught mice [(C) and (D), (n = 15 forest and 15 prairie)]. (A) LOD score for tail length (blue), dorsal hue (dark red), and flank hue (light red). (B) Number of recombination breakpoint events, binned in 1-Mb windows. (C) FST between forest and prairie mice estimated in 10-kb windows with a step size of 1 kb (light gray dots). Dark gray line shows data smoothed with a moving average over 500 windows. (D) Linkage disequilibrium across forest and prairie mice. Heatmap shows R2 (squared correlation) computed between genotypes at thinned SNPs (12). (E) Contigs assembled from long-read sequencing for one forest (top) and one prairie (bottom) mouse. Only contigs that span the inversion breakpoint are shown. The region of chromosome 15 affected by the inversion is highlighted (purple). (F) (Top) Alignment between regions of the forest and prairie contigs surrounding the breakpoint (black, alignment quality; green, forest contig; brown, prairie contig). Large prairie insertion near the breakpoint is a transposon. (Bottom) Base pair–level alignment around the breakpoint (gray, mismatch). (G) Model of the inverted (green) and reference (tan) alleles. The inversion spans 0 to 40.9 Mb (affected region, purple) and excludes 40.9 to 79 Mb (unaffected region, gray), with predicted centromere location shown in black.
This pattern of suppressed recombination could be produced by a large genomic rearrangement (or a set of rearrangements). To determine the nature of any structural variation on chromosome 15, we used PacBio long-read sequencing (n = 1 forest, n = 1 prairie) (12). We generated independent de novo assemblies for each individual and mapped the resulting contigs to the reference genome for P. m. bairdii (12). In the forest individual, one contig mapped near the center of the chromosome (from 41.19 to 40.94 Mb) and then split and mapped in reverse orientation to the beginning of the chromosome (from 0 to 5 Mb). By contrast, in the prairie individual, a single contig mapped continuously to the reference genome in this region (37 to 41.3 Mb) (Fig. 3E). Because we found no other forest-specific rearrangements in this region (fig. S4), we determined that chromosome 15 harbors a simple 41-Mb inversion. Using putative centromere-associated sequences in Peromyscus (12), we determined that the inversion is paracentric, with the centromere located outside of the inversion (Fig. 3G).
Inversions may affect phenotypes directly through the effects of their breakpoints or indirectly by carrying causal mutations (13). Using the long-read sequencing data, we localized the inversion breakpoint to base pair resolution (Fig. 3F and fig. S5). The breakpoint falls within an intron of a long intergenic noncoding RNA (lincRNA), and an additional four annotated genes (two lincRNAs and two protein-coding genes) occur within 200 kb of the breakpoint. Although the breakpoint may disrupt their expression patterns, these genes have no known functions associated with either pigmentation or skeletal phenotypes (table S3). An additional 149 protein-coding genes are located within the inversion, of which 29 contain at least one fixed nonsynonymous mutation between the inversion and reference alleles. Ten of the genes within the inversion (four with nonsynonymous substitutions) are associated with pigmentation phenotypes when disrupted in laboratory mice, and 13 are associated with tail or long-bone length phenotypes in laboratory mice (three with nonsynonymous substitutions and four with associated pigment phenotypes as well; table S4). These 19 genes are thus strong candidates for contributing to tail length and coat color variation.
Inversion frequency and divergence in wild populations
To investigate whether the inversion and associated traits (longer tails and redder coats) may be favored in forested habitats, we collected deer mice across a sharp habitat transition between the focal forest and prairie sites and estimated habitat type and mean soil hue at each capture site (n = 136 mice from 22 sites, supplemented by 12 additional museum specimens from two sites; figs. S6 and S7). We found that much of the transition in both habitat type and soil hue occurs in a narrow region across the Cascade mountain range (Fig. 4, A and B), and the phenotypic clines estimated using either all adult wild-caught individuals or only those from the Cascades region both identified sharp transitions in coat color and tail length that colocalize with this environmental transition (Fig. 4, C and D). Specifically, mean hue changes by 3.2° (63% of the forest-prairie difference), and mean tail length changes by 13 mm (47% of the forest-prairie difference) across the 50-km Cascades region; tail length changes by an additional 4 mm within the next 100 km, coincident with continued changes in forestation (Fig. 4). Together, the strong correlation between phenotype and habitat is consistent with local adaptation.
Fig. 4. Associations between genotype, phenotype, and environment in wild mice.
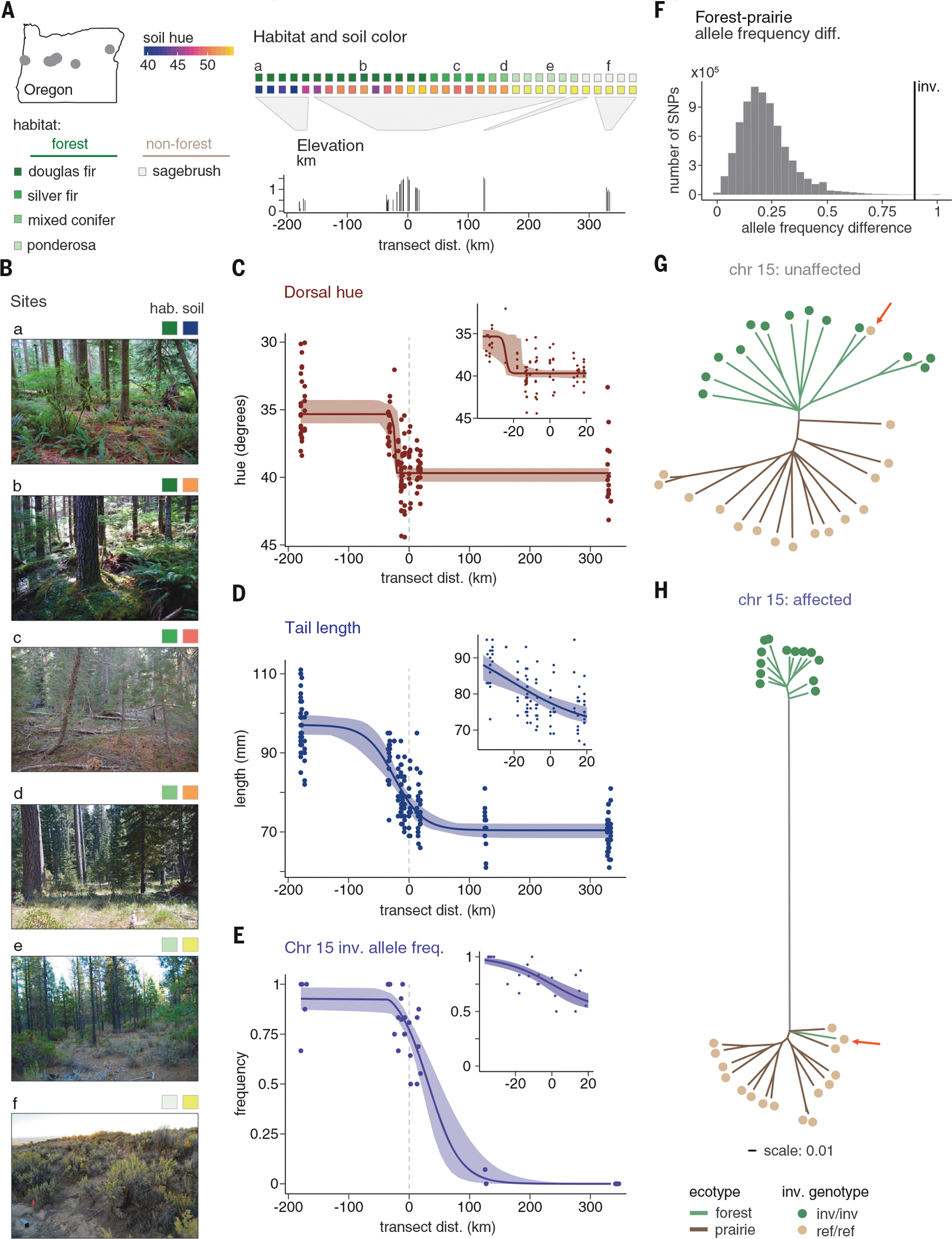
(A) Elevation and habitat characteristics (top row indicates majority habitat category, and bottom row indicates mean soil hue) at sites across an environmental transect. Letters indicate sites shown in (B). Soil hue and habitat category were estimated within 1 km of each site. (Map) Sampled sites across Oregon. Transect distance refers to the east-west distance from the highest-elevation site, and dotted lines in (C), (D), and (E) indicate distance = 0. (B) Photos of capture sites from each habitat type, with habitat and soil classification as in (A). (C to E) Best-fit clines for dorsal hue (C) (n = 143), tail length (D) (n = 180), and inversion genotype (E) (n = 178) fit to the full dataset, with 95% CIs. Insets show best-fit clines using only data from the central Cascades (hue, n = 90; tail, n = 97; genotype, n = 136). (F) Allele frequency differences for the maximally differentiated SNP between forest and prairie mice in 200-bp windows across the genome (12). The inversion forest-prairie allele frequency difference (90%) is shown in black. (G and H) Maximum likelihood trees for unaffected (G) (40.9 to 79 Mb) and affected (H) (0 to 40.9 Mb) regions of chromosome 15, shown on the same scale. Branch colors indicate ecotype (green, forest; brown, prairie), and dots indicate inversion genotype (tan, homozygous reference, n = 15; green, homozygous inversion, n = 14; heterozygous mouse excluded, n = 1). Red arrows highlight the forest mouse homozygous for the reference allele.
The inversion changes substantially in frequency across the habitat transition, from 90% in the forest population to absent in the prairie population (Fig. 4E). This frequency difference of the inversion is extreme: It is greater than the allele frequency difference at the maximally differentiated single-nucleotide polymorphism (SNP) in 99.92% of blocks with similar levels of linkage disequilibrium (12) (Fig. 4F). Moreover, similar to the changes in phenotype, the transition in inversion frequency occurs over only a short distance: Inversion frequency decreases from 100 to 62.5% in the 50-km Cascades region and then drops further within the next 100 km (i.e., inversion frequency drops from 100 to 4% over less than one-third of the total transect distance; Fig. 4E). The sharp change in inversion frequency across the environmental transect, and its extreme forest-prairie allele frequency difference, suggest that the inversion may be favored in forested habitat.
The inversion also strongly contributes to genetic differentiation between the forest and prairie ecotypes by carrying many highly differentiated SNPs. For example, FST between the forest and prairie ecotypes in the inversion region is high compared with the genome-wide average (inversion region: mean FST = 0.376; genome-wide, excluding inversion region: mean FST = 0.071; fig. S8). The strong genetic divergence between the inversion and reference haplotypes is reflected in maximum likelihood–based trees built from the region of chromosome 15 that contains the inversion (affected region: 0 to 40.9 Mb) and the rest of the chromosome (unaffected region: 40.9 to 79 Mb). In the unaffected region, forest and prairie mice cluster by ecotype, with limited divergence between the groups (Fig. 4G). By contrast, in the affected region, mice cluster into two highly distinct groups on the basis of genotypes at the inversion (Fig. 4H). This pattern suggests that the inversion harbors a high density of sites that are divergent between ecotypes.
Evolutionary history of the inversion
To explore the evolutionary history of the inversion, we first estimated a best-fitting demographic model for the forest and prairie populations using neutral sites across the genome to avoid the confounding effects of background selection (12, 14). The data were best fit by a model with a long history of high migration: initial migration rates of 8.3 × 10−7 [prairie-to-forest, 95% confidence interval (CI) = 3.7 × 10−9 to 1.8 × 10−6] and 3.6 × 10−6 (forest-to-prairie, 95% CI = 1.1 × 10−8 to 4.5 × 10−6) after a forest-prairie population split 2.2 million generations ago (95% CI = 1.1 to 5.5 million generations) (Fig. 5A and fig. S9). Because the estimated effective population sizes (Ne) are large (prairie Ne = 1.9 × 106 to 4.3 × 106; forest Ne = 1.8 × 105 to 1.2 × 106), the effective number of migrants per generation (Nem) is consistently high over time: Nem = 3.5 (prairie-to-forest) and Nem = 0.6 (forest-to-prairie), with a recent shift to Nem > 10 in both directions ~30,000 generations ago (Fig. 5A), consistent with high levels of gene flow (15). High migration levels between forest and prairie ecotypes are further supported by genomic data from the Cascades region: We found that the Cascades mice have mixed forest and prairie ancestry genome-wide (fig. S10).
Fig. 5. Evolutionary history of the inversion.
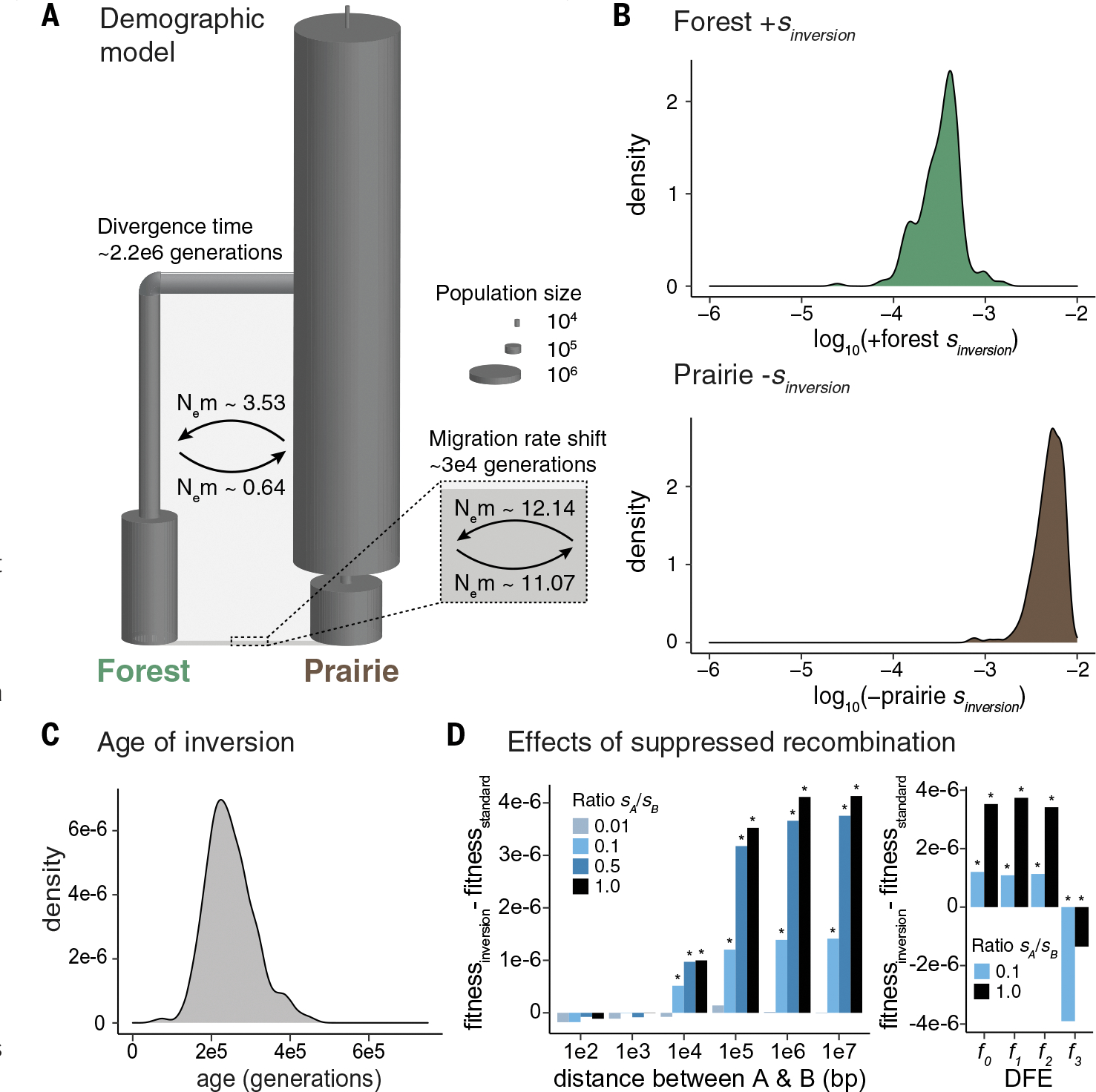
(A) Best-fit demographic model. Ne, effective population size; m, migration rate. (B) Posterior probability distributions for the selection coefficient associated with the inversion in the forest (top, green) and prairie (bottom, brown) populations, when the inversion is introduced 150,000 generations ago (for additional introduction times, see fig. S13). The estimated selection coefficient is positive in forest and negative in prairie. (C) Posterior probability distribution for the age of the inversion. (D) Estimated fitness effects of suppressed recombination within the inversion. Two beneficial loci (A and B) were introduced into the forest population on the inversion or on a standard haplotype, varying the ratio of the selection coefficients for A (sA) and B (sB), with sA + sB kept constant at 3 × 10−4. bp, base pairs. Bar height shows the difference in final mean fitness of the forest population between the inversion and standard haplotype scenarios. Asterisks indicate a significant difference in mean fitness (P < 0.05) computed with permutation tests. (Left) Two beneficial loci at varying distances apart, without deleterious mutations. (Right) Two beneficial loci separated by 100 kb, with deleterious mutations introduced according to distributions of fitness effects (DFE): f0: 100% of mutations neutral (2Ns = 0, where N indicates population size and s indicates selection coefficient); f1: 50% of mutations neutral (2Ns = 0), 50% weakly deleterious (−10 < 2Ns < −1); f2: 33% of mutations neutral (2Ns = 0), 33% weakly deleterious (−10 < 2Ns < −1), 33% moderately deleterious (−100 < 2Ns < −10); f4: 25% of mutations neutral (2Ns = 0), 25% weakly deleterious (−10 < 2Ns < −1), 25% moderately deleterious (−100 < 2Ns < −10), 25% strongly deleterious (−1000 < 2Ns < −100).
These high migration estimates coupled with the large, habitat-associated differences in inversion frequency may indicate a history of natural selection. To test this hypothesis, we simulated the spread of the inversion under our demographic model using SLiM (12). We found that divergent selection was the most likely scenario to explain both the high frequency of the inversion in the forest and its low frequency in the prairie (fig. S11). Using approximate Bayesian computation, we estimated selection coefficients (s) for the inversion of 3.3 × 10−4 (95% CI = 9.2 × 10−5 to 1.6 × 10−3) in the forest population and −4.1 × 10−3 (95% CI = −9.3 × 10−3 to −7.1 × 10−4) in the prairie population (Fig. 5B). These values suggest that the observed distribution of the inversion in the wild is best explained by both positive selection in the forest and negative selection in the prairie, a conclusion robust to the uncertainty in the model parameter estimates (fig. S12) and to variation in the timing of the introduction of the inversion after the forest-prairie split (fig. S13). We also used simulations to assess the minimum age of the inversion required to achieve its divergence from the reference allele (12): We estimated the inversion to be at least 247,000 generations old (95% CI = 149,000 to 384,000 generations or 50,000 to 128,000 years, assuming three generations per year), which suggests that the inversion predates the modern habitat distribution (16) (Fig. 5C). Together, these results suggest that the inversion was most likely established in the forest population under strong divergent selection over the last ~250,000 generations.
Our estimates of forest-prairie migration rates and selection on the inversion allowed us to explore possible fitness effects from the inversion’s suppression of recombination. Although it is formally possible that the inversion carries only a single mutation that alone confers a strong enough benefit (s ≥ 3 × 10−4) to explain its current distribution, an alternative hypothesis is that the inversion carries two or more beneficial mutations (e.g., one mutation that contributes to tail length and a second to color variation), each with smaller selection coefficients. In this scenario, theory predicts that the inversion could confer a fitness advantage in the forest beyond the individual mutations it carries by reducing the migration load suffered by each mutation (5, 17, 18). To investigate this possibility, we used our estimates of migration, selection, and recombination to simulate the spread of two beneficial mutations in the forest population either within an inversion or on a freely recombining (standard) haplotype, varying the distance between the mutations (12). We found that if the two mutations are at least 10 kb apart (which is likely, given the inversion size of 41 Mb) and the selection coefficient for the weaker locus is at least 10% of that of the stronger locus [which is possible, given independent evidence for selection acting on coat color and tail length—e.g., (19, 20)], the beneficial mutations are more likely to establish and be maintained at higher frequencies in the forest when carried by the inversion than on the standard haplotype (Fig. 5D and figs. S14 and S15). We also explored possible costs associated with the inversion suppressing recombination (i.e., mutational load accumulation) (21, 22) by introducing deleterious mutations according to four fitness-effect distributions [as described in (14)] into the two–beneficial locus simulations. With weakly or moderately deleterious mutations, the inversion maintained its selective advantage over the standard haplotype in the forest (Fig. 5D and fig. S16). Only when strongly deleterious mutations were introduced did the inversion accumulate a substantial mutational load, which results in the inversion being disadvantageous relative to the standard haplotype in the forest (Fig. 5D and fig. S16). Thus, our results suggest that, under a wide range of conditions, if this inversion carries two or more beneficial mutations, its suppression of recombination likely confers an additional selective advantage in the forest population by linking adaptive alleles in the face of high migration rates.
Discussion
In 1909, Wilfred Osgood described several morphological differences—including tail length and coat color—that distinguish forest and prairie ecotypes of P. maniculatus (7). Long tails are thought to be beneficial for arboreality (8, 9, 23): Long tails have repeatedly evolved in association with forest habitat in deer mice (20) and across mammals (24), and forest mice are better climbers (23), with tail length differences between the ecotypes likely sufficient to affect climbing performance (25). Coat color is subject to pressure from visually hunting predators (19), and many mammals, including deer mice, evolve coats to match local soil color (9, 26). By sampling along an environmental transect, we found evidence that each of these traits is closely associated with habitat (forestation for tail length and soil hue for coat color), which further suggests that these traits are involved in local adaptation.
High migration rates between the forest and prairie ecotypes, as we estimated in this work, makes the strong ecotypic divergence in multiple traits puzzling. By characterizing the genetic architecture of tail length and coat color variation, we help resolve how differences in these traits are maintained between ecotypes: Namely, we discover a previously unknown inversion, involving half a chromosome, that has a large effect on both ecotype-defining traits and in the expected direction (i.e., it is associated with long tails and reddish fur in forest mice). Because recombination between the inversion and the noninverted prairie haplotype is suppressed in heterozygotes, the inversion ensures that longer tail length and redder coat color alleles are coinherited in the forest, despite high levels of gene flow (except in the unlikely scenario that only a single pleiotropic mutation within the inversion affects both traits). The role of this inversion in phenotypically differentiating these ecotypes is consistent with theoretical predictions and empirical examples of concentrated genetic architectures arising under local adaptation with gene flow (6, 27, 28).
Our modeling implicates divergent selection in maintaining the inversion at high frequency in the forest ecotype and absent from the prairie ecotype. The inversion’s selective effects are likely driven by its strong association with tail length and coat color (explaining 12 and 40% of the trait variances, respectively), although it is possible other traits are involved. Although inversions can have phenotypic effects because of their breakpoints disrupting genes or gene expression (13), the inversion’s breakpoint does not occur in or near candidate genes for tail length and coat color variation. Alternatively, inversions may influence phenotypes through the mutations they carry: The inversion is highly differentiated from the reference haplotype, thus harboring many mutations that may influence tail length and/or coat color. We expect that more than one mutation contributes to the inversion’s selective benefit in the forest, given the size of the inversion (41 Mb), its large selection coefficient in the forest (s ≈ 3 × 10−4, or Ns ≈ 120), and its association with two largely developmentally distinct traits. If this is the case, the inversion’s suppression of recombination likely provides an additional benefit (beyond the individual effects of its mutations) in the forest population, as long as strongly deleterious mutations are uncommon. This finding—that recombination suppression is likely beneficial in this system—provides empirical support for the local adaptation hypothesis, which posits that inversions are beneficial in the face of gene flow because they increase linkage disequilibrium between adaptive alleles (5, 17, 18).
One hundred years after Alfred Sturtevant first provided evidence of chromosomal inversions in laboratory stocks of Drosophila (29) and, separately, forest-prairie ecotypes were first described in wild populations of Peromyscus (7), we found that a large chromosomal inversion is key to ecotype divergence in this classic system. Inversions have been identified in association with divergent ecotypes in diverse species, including plants (30–33), invertebrates (34–45), fish (46, 47), and birds (48–52). In mammals, however, evidence for ecotype-defining inversions is limited [(53), but see (54)]. Our results thus underscore the important and perhaps widespread role of inversions in local adaptation, including in mammals, and highlight how selection acting on inversion polymorphisms may maintain intraspecific divergence in multiple traits in the wild.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Lewarch for assistance in the field; S. Niemi, M. Streisfeld, S. Stankowski, G. Binford, S. Bishop, K. Saunders, S. Finch, and T. Schaller for help with field logistics; K. Pritchett-Corning and S. Griggs-Collette for help establishing breeding colonies; Harvard’s Office of Animal Resources for animal care; and M. Omura, J. Chupasko, M. Mullon, and J. Mewherter for help preparing and accessioning museum specimens. G. J. Kenagy, D. S. Yang, and E. Kingsley provided advice at the start of the project; P. Audano provided advice on long-read sequencing and analysis; and N. Edelman, A. Kautt, E. Kingsley, and three anonymous reviewers provided helpful feedback on the manuscript. The University of Washington Burke Museum provided specimens used in this study.
Funding:
This work was partially funded by Putnam Expedition grants from the Museum of Comparative Zoology (MCZ) to E.R.H., a MCZ grant-in-aid of student research to S.T., and the MCZ Chapman Fellowship for the Study of Vertebrate Locomotion to E.R.H. and J.T.G. as well as funding from the American Society of Mammalogists grants-in-aid of research to E.R.H. and O.S.H., the Harvard College Research Program to S.T., and a Society for the Study of Evolution R. C. Lewontin Early Award to O.S.H. E.R.H. was supported by an NIH training grant to Harvard’s Molecules, Cells, and Organisms graduate program (NIH NIGMS T32GM007598) and by the Theodore H. Ashford Fellowship. O.S.H. was supported by a National Science Foundation (NSF) Graduate Research Fellowship, a Harvard Quantitative Biology Student Fellowship (DMS 1764269), and the Molecular Biophysics training grant (NIH NIGMS T32GM008313). J.D.J. was funded by the National Institutes of Health (1R35GM139383–01). H.E.H. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability:
Associated data are available as supplementary files data S1 to S8 and on the NCBI Sequence Read Archive (nos. PRJNA687993, PRJNA688305, and PRJNA816517). Associated scripts are available on Zenodo (55).
REFERENCES AND NOTES
- 1.Turesson G, Hereditas 3, 100–113 (1922). [Google Scholar]
- 2.Lowry DB, Biol. J. Linn. Soc. 106, 241–257 (2012). [Google Scholar]
- 3.Tigano A, Friesen VL, Mol. Ecol. 25, 2144–2164 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Haldane JBS, Math. Proc. Camb. Philos. Soc. 26, 220–230 (1930). [Google Scholar]
- 5.Bürger R, Akerman A, Theor. Popul. Biol. 80, 272–288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeaman S, Whitlock MC, Evolution 65, 1897–1911 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Osgood WH, Am N. Fauna 28, 1–285 (1909). [Google Scholar]
- 8.Blair WF, Evolution 4, 253–275 (1950). [Google Scholar]
- 9.Dice LR, Am. Nat. 74, 212–221 (1940). [Google Scholar]
- 10.Calhoun SW, Greenbaum IF, Fuxa KP, J. Mammal. 69, 34–45 (1988). [Google Scholar]
- 11.Yang DS, Kenagy G, Ecol. Evol. 1, 26–36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Materials and methods are available as supplementary materials.
- 13.Villoutreix R et al. , Mol. Ecol. 30, 2738–2755 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johri P et al. , Mol. Biol. Evol. 38, 2986–3003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright S, Genetics 16, 97–159 (1931). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitt G, Nature 405, 907–913 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Kirkpatrick M, Barton N, Genetics 173, 419–434 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charlesworth B, Barton NH, Genetics 208, 377–382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vignieri SN, Larson JG, Hoekstra HE, Evolution 64, 2153–2158 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Kingsley EP, Kozak KM, Pfeifer SP, Yang DS, Hoekstra HE, Evolution 71, 261–273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berdan EL, Blanckaert A, Butlin RK, Bank C, PLOS Genet. 17, e1009411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charlesworth B, Jensen JD, Annu. Rev. Ecol. Evol. Syst. 52, 177–197 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horner BE, Contrib. Lab. Vertebr. Biol. 61, 1–85 (1954). [Google Scholar]
- 24.Mincer ST, Russo GA, Proc. R. Soc. B. 287, 20192885 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hager ER, Hoekstra HE, Integr. Comp. Biol. 61, 385–397 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrett RDH et al. , Science 363, 499–504 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Faria R, Johannesson K, Butlin RK, Westram AM, Trends Ecol. Evol. 34, 239–248 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Thompson MJ, Jiggins CD, Heredity 113, 1–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sturtevant AH, Proc. Natl. Acad. Sci. U.S.A. 7, 235–237 (1921). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang Z et al. , Genetics 191, 883–894 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang K, Andrew RL, Owens GL, Ostevik KL, Rieseberg LH, Mol. Ecol. 29, 2535–2549 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Lowry DB, Willis JH, PLOS Biol. 8, e1000500 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C-R et al. , Nat. Ecol. Evol. 1, 0119 (2017). [Google Scholar]
- 34.Joron M et al. , Nature 477, 203–206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunte K et al. , Nature 507, 229–232 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Yan Z et al. , Nat. Ecol. Evol. 4, 240–249 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Mérot C et al. , Mol. Biol. Evol. 38, 3953–3971 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng C et al. , Genetics 190, 1417–1432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feder JL, Roethele JB, Filchak K, Niedbalski J, Romero-Severson J, Genetics 163, 939–953 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapun M, Flatt T, Mol. Ecol. 28, 1263–1282 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Koch EL et al. , Evol. Lett. 5, 196–213 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindtke D et al. , Mol. Ecol. 26, 6189–6205 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Dobzhansky T, Evolution 1, 1–16 (1947). [Google Scholar]
- 44.Brelsford A et al. , Curr. Biol. 30, 304–311.e4 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Ayala D, Guerrero RF, Kirkpatrick M, Evolution 67, 946–958 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Jones FC et al. , Nature 484, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matschiner M et al. , Nat. Ecol. Evol. 6, 469–481 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Küpper C et al. , Nat. Genet. 48, 79–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorneycroft HB, Science 154, 1571–1572 (1966). [DOI] [PubMed] [Google Scholar]
- 50.Lamichhaney S et al. , Nat. Genet. 48, 84–88 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Funk ER et al. , Nat. Commun. 12, 6833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanchez-Donoso I et al. , Curr. Biol. 32, 462–469.e6 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Dobigny G, Britton-Davidian J, Robinson TJ, Biol. Rev. 92, 1–21 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Stefansson H et al. , Nat. Genet. 37, 129–137 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Hager ER, Harringmeyer OS, oharring/chr15_inversion: v1.0.0, version 1.0.0, Zenodo; (2022); 10.5281/zenodo.6354918. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Associated data are available as supplementary files data S1 to S8 and on the NCBI Sequence Read Archive (nos. PRJNA687993, PRJNA688305, and PRJNA816517). Associated scripts are available on Zenodo (55).