

Abstract
From a designed library of indolyl pyrimidinamines, we identified a highly potent and cell-active chemical probe (17) that inhibits phosphatidylinositol-3-phosphate 5-kinase (PIKfyve). Comprehensive evaluation of inhibitor selectivity confirmed that this PIKfyve probe demonstrates excellent kinome-wide selectivity. A structurally related indolyl pyrimidinamine (30) was characterized as a negative control that lacks PIKfyve inhibitory activity and exhibits exquisite selectivity when profiled broadly. Chemical probe 17 disrupts multiple phases of the lifecycle of β-coronaviruses: viral replication and viral entry. The diverse antiviral roles of PIKfyve have not been previously probed comprehensively in a single study or using the same compound set. Our scaffold is a distinct chemotype that lacks the canonical morpholine hinge-binder of classical lipid kinase inhibitors and has a non-overlapping kinase off-target profile with known PIKfyve inhibitors. Our chemical probe set can be used by the community to further characterize the role of PIKfyve in virology.
Introduction
Phosphatidylinositol-3-phosphate 5-kinase (PIKfyve) is a ubiquitously expressed FYVE finger-containing phosphoinositide kinase that is classified as a lipid kinase. PIKfyve plays diverse roles in membrane trafficking, endosomal transport, GLUT4 translocation, retroviral budding, Toll-like receptor (TLR) signaling, and lysosomal function among others.1−3 PIKfyve binds to and phosphorylates the D-5 position of phosphatidylinositol-3-phosphate (PI(3)P) via its FYVE domain to yield phosphatidylinositol-3,5-bisphosphate (PI(3,5)P2) and phosphatidylinositol 5-phosphate (PI(5)P).1 The essentiality of PIKfyve is supported by the fact that in multicellular organisms, genetically impaired function of this kinase or its partner proteins that regulate PI(3,5)P2 homeostasis results in severe disorders, including embryonic/perinatal death.1
Several inhibitors of PIKfyve have been published with variable potency in cells and selectivity profiling that have been used to define its cellular roles. PIKfyve has been characterized as a critical player in TLR signaling using apilimod as a tool compound (Figure 1). Kinome-wide profiling of apilimod at 1 μM revealed it to be an exquisitely selective inhibitor of PIKfyve. Furthermore, it was found to have a Kd = 75 pM in the DiscoverX PIKfyve binding assay.2 Based on its potent inhibition of IL-12 and IL-23, apilimod has been evaluated in clinical trials for Crohn’s disease and rheumatoid arthritis.4,5 Apilimod inhibits IL-12 and IL-23 through binding to PIKfyve and blocking its phosphotransferase activity.3 It has also been touted as a treatment for B-cell non-Hodgkin lymphoma (B-NHL) based on its broad anticancer activity in vitro and in vivo across all subtypes of B-NHL without toxicity to normal cells. It was proposed that the B-NHL cytotoxicity elicited by apilimod results from its disruption of lysosomal function due to PIKfyve inhibition. More recently, apilimod was investigated in clinical trials for the treatment of COVID-19. The proposed antiviral mechanism of apilimod is prevention of viral invasion via inhibition of host cell proteases.6 Apilimod was also used to demonstrate that PIKfyve inhibition prevents viral infection by Zaire ebolavirus and SARS-CoV-2 by preventing release of the viral contents from endosomes.7
Figure 1.
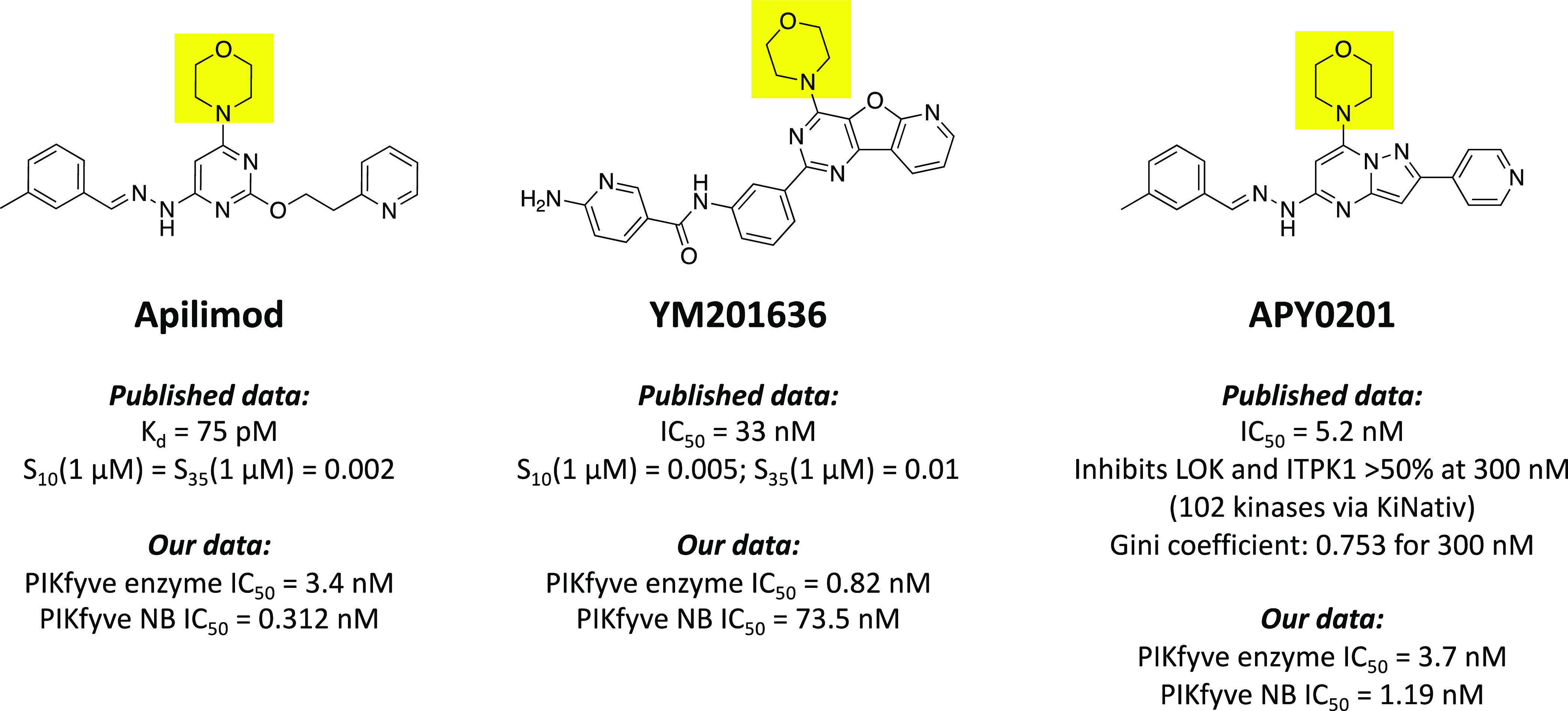
Structures, potency, and selectivity values for published PIKfyve inhibitors. The shared morpholine ring in the three scaffolds is highlighted in yellow.
The ability to inhibit retroviral replication and in insulin activation of GLUT4 surface translocation and glucose influx via targeting PIKfyve was confirmed using YM201636 (Figure 1).8 In addition, YM201636 inhibits the proliferation of liver cancer cells and tumor growth in a murine hepatic cancer model. The proposed mechanism via which it inhibits tumor growth is through promoting EGFR expression, which induces autophagy.9 Dysregulation of autophagy by YM201636 and resultant cell death have been observed in other cell types as well.10 Finally, YM201636 reversibly impairs endosomal trafficking in NIH3T3 murine cells.11 YM201636 is a potent inhibitor of PIKfyve (IC50 = 33 nM) and the recently disclosed kinome-wide selectivity of YM201636 (Figure 1) confirmed it as a very selective inhibitor of PIKfyve.12
Finally, APY0201 is a PIKfyve inhibitor that has demonstrated broad antimultiple myeloma activity.13 APY0201 upregulated genes in the lysosomal pathway, increased cellular vacuolization, and activated the transcription factor EB, a master regulator of lysosomal biogenesis and autophagy.13 Like apilimod, APY0201 was used to confirm the function of PIKfyve in the IL-12/23 production pathway and in inflammatory disease pathologies driven by these interleukins.14 In situ native kinase profiling (KiNativ) using an ATP-competitive probe and Jurkat cell lysate was used to determine the selectivity of APY0201 for 24 human lipid kinases and 83 human protein kinases. This selectivity screening at 300 nM revealed >50% inhibition of LOK and ITPK1 and allowed calculation of a Gini coefficient of 0.753. APY0201 was also profiled versus apilimod against a panel of 137 G protein-coupled receptors (GPCRs), enzymes, ion channels, and transporters and found to demonstrate negligible inhibition relative to its IC50 value in the PIKfyve kinase assay (5.2 nM). Apilimod, by comparison, demonstrated inhibitory activity on some GPCRs.14
ATP-competitive phosphatidylinositol-3-kinase (PI3K) and phosphatidylinositol-3-kinase-related kinase (PIKK) inhibitors have historically possessed a critical morpholine that forms a hydrogen bond with the kinase hinge region.15 Apilimod, YM201636, and APY0201 bear this morpholine (boxed in Figure 1).14 For YM201636, one of the off-target kinases that potently binds (percent of control (PoC) = 9.8 at 1 μM) is a catalytic subunit of PI3Ks, PIK3CB.12 Structural studies around PIKfyve have been limited. All Protein Data Bank entries related to PIKfyve were generated using medium–low resolution Cryo-EM (5.1–6.6 Å). These structures reveal that PIKfyve exists as a complex with five copies of a scaffolding protein known as Vac14. Furthermore, associated models suggest that while this heteroprotein complex interacts with the membrane, the PIKfyve active site is rotated such that it cannot access membrane-incorporated phosphoinositides.16
Here, we present a potent and selective PIKfyve inhibitor that lacks the hinge-binding morpholine of other published inhibitors. Without the presence of morpholine, our off-target kinase profile does not contain other lipid kinases and is non-overlapping with that of known PIKfyve inhibitors. Medicinal chemistry optimization of the indolyl pyrimidinamine scaffold was enabled by a PIKfyve cellular target engagement assay in tandem with kinome-wide selectivity screening. Our chemical probe (17) was evaluated alongside its structurally related negative control (30) and published PIKfyve inhibitors to confirm the role of PIKfyve in mediating several pathways that viruses depend upon for entry, replication, and transmission.
Results and Discussion
Rationale for the Selection of the Parent Inhibitor
To enable illumination of the function of understudied kinases, we identify and synthesize analogues of published scaffolds that inhibit poorly characterized kinases. AMG28 (Figure 2) was selected based on data provided for it in the MRC Kinase Profiling Inhibitor Database. One method by which we define a kinase as understudied is via publication count. If we set a threshold of 100 publications on the human form of a particular kinase, nearly 65% of the kinases targeted by AMG28 with <20% activity remaining at the 1 μM screening concentration meet this criterion. While the MRC profiling data provided insight into the activity of this compound, the panel is only comprised of 140 wild-type (WT) human kinases. Motivated by its inhibition profile and with a desire to screen it in a more comprehensive kinase panel, we first remade AMG28.
Figure 2.

Structures of analogues 1–3 and AMG28.
Once synthesized, AMG28 was screened against the DiscoverX scanMAX platform at 1 μM. This panel includes 403 WT human kinases and thus greatly expands the data generated for AMG28 versus that from MRC. AMG28 demonstrated PoC < 10 for 20 WT human kinases, translating to an adjusted selectivity score of S10(1 μM) = 0.05. When screened at the same concentration (1 μM), several kinases showed low activity remaining in both kinase panels (Figure 3A). Beyond these kinases in common, PIKfyve was identified as a kinase of interest based on its PoC < 1. Enzymatic follow-up in dose–response using commercially available assays was pursued for selected understudied kinases and family members identified in the two large panel screening efforts (MRC and DiscoverX). Part of this effort involved working with SignalChem to establish a PIKfyve enzyme inhibition assay using their commercially available protein. As shown in Figure 3B, AMG28 was validated as an efficacious inhibitor of most of the kinases selected and, notably, the PIKfyve PoC value was confirmed to translate to potent inhibition of PIKfyve enzymatic activity (IC50 = 2.2 nM). The narrow kinome-wide selectivity of AMG28 against 403 WT human kinases coupled with validated inhibition of understudied kinases in an orthogonal enzymatic format encouraged us to pursue structural diversification via analogue synthesis.
Figure 3.
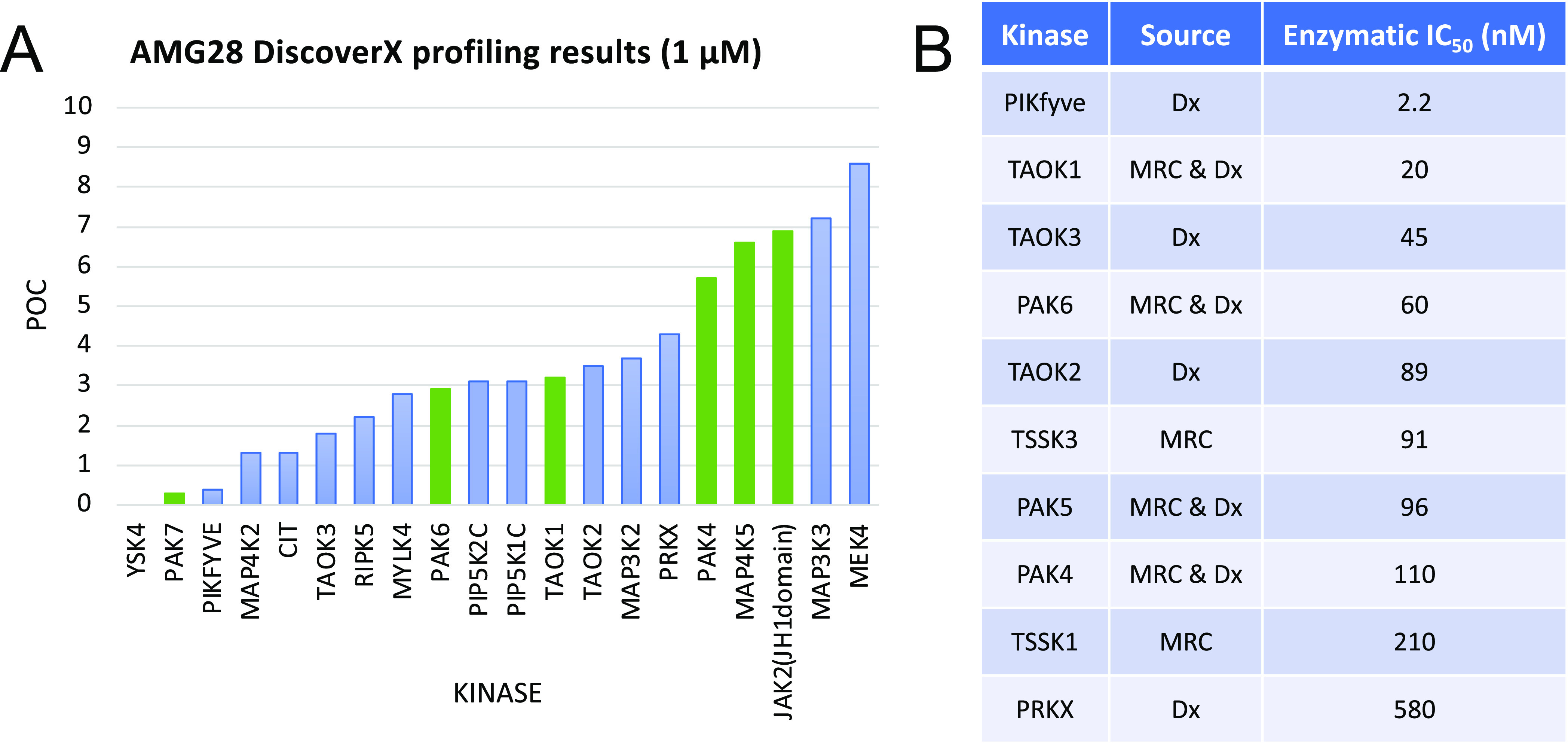
AMG28 is a kinase inhibitor with narrow kinome-wide selectivity. (A) AMG28 DiscoverX screening results for kinases with <10 percent of control (POC) remaining when screened at 1 μM. Kinases common to the MRC panel are represented as green bars. (B) Selective enzymatic follow-up IC50 values for understudied kinases and family members identified via the DiscoverX (Dx) and/or MRC screening campaigns.
Exploring the Role of the Seven-Membered Ring
We began our campaign with a subset of indolyl pyrimidinamine analogues that probed the importance of the seven-membered ring system in AMG28. There are no high-resolution or cocrystal structures of PIKfyve. Our own kinase inhibitor design experience and work of others in the field have confirmed the aminopyrimidine as a common hinge-binding motif.17−20 Although they involve an unrelated human protein kinase that shares only 21% sequence identity with PIKfyve, recently solved cocrystal structures of AMG28 bound to TTBK1 (PDB: 7JXX and 7ZHN) demonstrate that the aminopyrimidine ring in AMG28 makes essential hydrogen bonds with the hinge of TTBK1.21,22 These studies suggest that aminopyrimidine could also bind the hinge of PIKfyve. It was suggested by the authors of the Biogen TTBK1 paper that the three methylene groups of the seven-membered ring help maintain the planarity of the molecule rather than making key contacts with the protein.21 To explore and support these hypotheses as they relate to PIKfyve, we both augmented and decreased the number of methylene groups in the saturated ring system to yield the corresponding six- and eight-membered analogues of AMG28 (1 and 2, respectively, in Figure 2). Furthermore, we made the ring-opened analogue of AMG28 (3, Figure 2). We sent these analogues to DiscoverX for kinome-wide profiling to determine how their PIKfyve PoC values and selectivity compared with AMG28. The DiscoverX scanMAX results for these compounds and AMG28 are summarized in Table 1. We confirmed the PIKfyve PoC values using the orthogonal PIKfyve enzymatic assay at SignalChem. Analogues from Table 1 plus the published PIKfyve inhibitors included in Figure 1 were evaluated. Once the PoC values were validated via the enzymatic assay, we established a PIKfyve cellular target engagement (NanoBRET, NB) assay to support medicinal chemistry optimization of PIKfyve inhibitors. We and others have found that using the NanoBRET assay to drive the analogue design provides a useful and relevant measure of cell affinity for the kinase of interest that also takes into account the ability of compounds to enter cells. All compounds in Table 1 as well as the published PIKfyve inhibitors in Figure 1 were tested in the PIKfyve NanoBRET assay. This first compound set demonstrated that opening the ring to yield analogue 3 did not result in improved PIKfyve potency versus parent AMG28 and caused a loss in kinome-wide selectivity. Analogue 1 maintained the kinome-wide selectivity and potency of AMG28, indicating that a six-membered ring is tolerated by PIKfyve. Finally, expansion to the eight-membered ring produced compound 2 that did not bind with high affinity to PIKfyve or many other kinases in the larger kinome. This small compound set showed that compounds bearing six- and seven-membered rings were effective inhibitors of PIKfyve but require improvement of kinome-wide selectivity to be useful tool compounds. In contrast, incorporation of an eight-membered ring was viewed as a mechanism via which to access highly selective compounds that lack PIKfyve activity.
Table 1. PIKfyve and Selectivity Data for Ring Modification Library and Published Inhibitors2,12.
| compound | PIKfyve PoCa | PIKfyve enzymatic IC50 (nM) | PIKfyve NB IC50 (nM) | S10(1 μM)b | # scanMAX kinases with PoC < 10c |
|---|---|---|---|---|---|
| 1 | 0 | NTd | 220 | 0.05 | 20 |
| AMG28 | 0.4 | 2.2 | 14.1 | 0.052 | 21 |
| 2 | 3.4 | 61 | 2450 | 0.007 | 3 |
| 3 | 0 | 6.1 | 21.1 | 0.084 | 34 |
| Apilimod | NT | 3.4 | 0.312 | 0.002 | 1 |
| YM201636 | 0 | 0.82 | 73.5 | 0.005 | 2 |
| APY0201 | NT | 3.7 | 1.19 | NT | NT |
Percent of control (PoC) values determined at 1 μM via DiscoverX scanMAX profiling.
S10(1 μM): percentage of screened kinases with PoC < 10 at 1 μM.
Number of kinases with PoC < 10 at 1 μM.
NT: not tested.
Probing the Importance and Nature of the Alkyne
Next, we prepared several compounds to solidify our understanding of the importance of ring size, while simultaneously determining the tolerance of PIKfyve to modification of the alkyne portion of AMG28. First, we synthesized matched sets of analogues bearing six-, seven-, and eight-membered rings with the alkyne portion of modified AMG28. The first subset of these compounds (4–6) incorporated modest changes to the terminal alkyne. Through modifying the substituted propargylic alcohol in AMG28 to an alkyne bearing a pyridine, we probed the space in the PIKfyve binding pocket typically occupied by the alkyne. We hypothesized that fewer kinases would tolerate additional bulk on the terminus of the alkyne. Next, we made a matched set of analogues (7–9) in which the alkyne was removed and a para-fluorobenzene ring directly attached in its place. These analogues were designed to determine the importance of the alkyne as well as the affinity of PIKfyve for this biaryl system with a very different shape. Analogues 7–9 will fill a substantially different volume in the kinase ATP site versus those bearing an alkyne.
To evaluate these analogues versus those in Table 1, we first sent them for kinome-wide profiling in the DiscoverX scanMAX panel. All analogues in Table 2 demonstrated improved selectivity scores versus AMG28. In the case of analogues 4 and 6, this reduced the binding affinity exhibited for many kinases across the kinome, echoed by a drastic decrease in PIKfyve activity. Seven-membered analogue 5, in contrast, was confirmed by three orthogonal assays to bind to PIKfyve and result in the inhibition of the enzyme, but it was significantly less active than AMG28. Overall, the pyridyl-bearing alkyne did not result in an improvement of PIKfyve inhibition, which suggested that an aryl ring on the alkyne terminus was not well tolerated by this kinase. Analogues 7–9 also had improved kinome-wide selectivity (all S10(1 μM) values < 0.01) and for compounds 7 and 8, this was not coupled with substantial losses in PIKfyve affinity. One interesting difference between analogues 7 and 8 was observed when comparing their PIKfyve enzymatic and NanoBRET assay IC50 values (Table 2). Despite their structural similarity, for analogue 7, there was a more significant drop-off in potency when moving from the PIKfyve enzymatic assay to the PIKfyve NanoBRET assay.
Table 2. PIKfyve and Selectivity Data for Alkyne Modification Library.

| compound | n | R | PIKfyve PoCa | PIKfyve enzymatic IC50 (nM) | PIKfyve NB IC50 (nM) | S10(1 μM)b | # scanMAX kinases with PoC < 10c |
|---|---|---|---|---|---|---|---|
| 4 | 1 | A | 69 | NTd | 2980 | 0.002 | 1 |
| 5 | 2 | A | 12 | 22 | 885 | 0.012 | 5 |
| 6 | 3 | A | 100 | 580 | 5290 | 0 | 0 |
| 7 | 1 | B | 0.3 | 8.2 | 380 | 0.007 | 3 |
| 8 | 2 | B | 0 | 9.3 | 11.4 | 0.005 | 2 |
| 9 | 3 | B | 37 | NT | 2560 | 0.005 | 2 |
| 10 | 1 | C | 0.9 | NT | 1170 | 0.027 | 11 |
| 11 | 2 | C | 0 | NT | 45.7 | 0.016 | 7 |
| 12 | 1 | D | 2.3 | 34 | 4970 | 0.017 | 7 |
| 13 | 2 | D | 2.2 | 26 | 1560 | 0.02 | 8 |
Percent of control (PoC) values determined at 1 μM via DiscoverX scanMAX profiling.
S10(1 μM): percentage of screened kinases with PoC < 10 at 1 μM.
Number of kinases with PoC < 10 at 1 μM.
NT: not tested.
PIKfyve will tolerate alkyne removal. Once again, the seven-membered analogue (8) was the most potent PIKfyve inhibitor from this subset. Based on its potency, which was confirmed in the PIKfyve enzyme assay, and selectivity (Figure S1B), compound 8 was considered a PIKfyve chemical probe candidate in need of additional characterization. Considering these initial matched sets (4–9) together, the seven-membered ring represents a sweet spot in terms of providing potent PIKfyve inhibition, while the six-membered ring still maintains some potency. These two ring sizes provide analogues with similar selectivity scores when profiled broadly. In contrast, the eight-membered ring is suboptimal for PIKfyve potency but does improve kinome-wide selectivity, suggesting that few kinases tolerate its inclusion.
Another two matched pairs of analogues bearing six- and seven-membered rings were prepared via modifying the alkyne portion of AMG28. These compounds were designed to incorporate the more favorable ring sizes (six and seven) and explore the impact of a more subtle change to the substituted propargylic alcohol of AMG28 (10 and 11) or to the biaryl system of analogues 7–9 (12 and 13). Analogues 10 and 11 were designed to modify the steric bulk directly attached to the carbon bearing the propargylic alcohol. Analogues 12 and 13 were designed to incorporate a smaller 1-methylpyrazole ring system with a different electronic character in place of the para-fluorobenzene. Based on our other analogues, we hypothesized that these changes would be tolerated by PIKfyve and result in potent inhibitors but with unknown kinome-wide selectivity.
Evaluation of analogues 10–13 in the DiscoverX scanMAX panel revealed them to have promising selectivity against the larger kinome. These compounds had selectivity scores that fall between their six- and seven-membered comparators in Tables 1 (1 and AMG28) and 2 (4, 5, 7, and 8). Comparing the modified propargylic alcohols (1, AMG28, 10, and 11) with the pyridine-bearing alkynes highlighted that these smaller groups at the terminus of the alkyne are better tolerated by PIKfyve. In comparing the biaryl systems, those analogues bearing a para-fluorobenzene (7 and 8) were more potent PIKfyve inhibitors than those with a 1-methylpyrazole (12 and 13). Once again, the seven-membered analogues (11 and 13) outperformed their six-membered congeners (10 and 12) in terms of PIKfyve potency. This larger data set (Tables 1 and 2) allowed us to consider the correlations of PIKfyve PoC, enzymatic, and NanoBRET data. Good agreement was found between the two biochemical methods, while some variation was observed in moving into the cell-based assay. Generally, a loss in potency was noted in the NanoBRET assay, a common finding when one moves from a biochemical assay to a cellular context. This is one reason we value the use of this cellular target engagement assay to support optimization, as it identifies compounds with most promise for robust cellular activity.
Diversifying the Terminal Alkyne
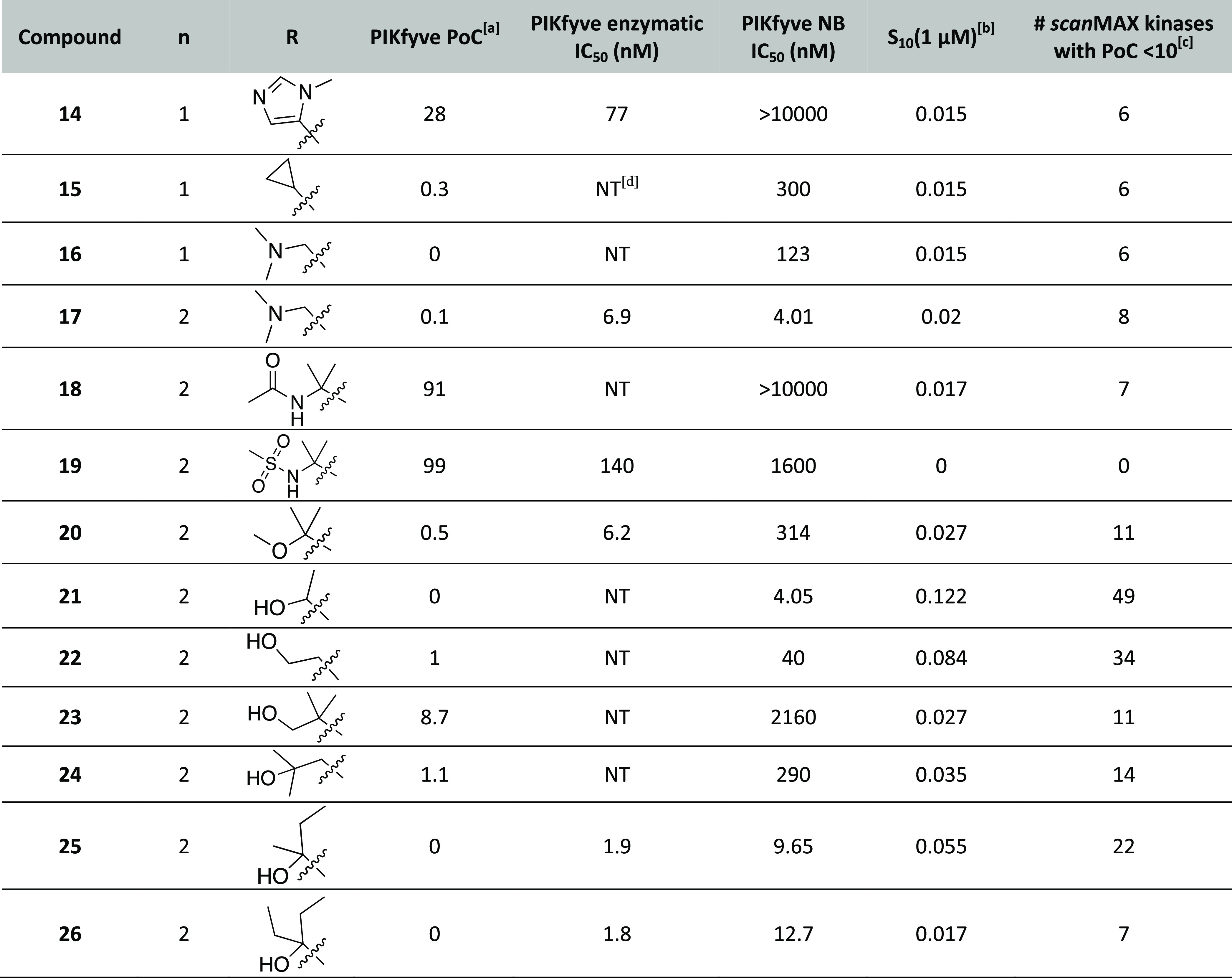
Next, we synthesized several indolyl pyrimidinamines to probe the tolerance of PIKfyve to modification of the alkyne terminus for analogues bearing six- or seven-membered rings. Based on the analogues in Tables 1 and 2, we made a small set of six-membered derivatives bearing an alkyne capped with diverse groups. We designed these to include a five-membered 1-methylimidazole (14), a cyclopropyl ring (15), or a dimethylamino group (16). These substituents represent diversity in size and electronic nature, exploring the size of a ring system tolerated within this portion of the PIKfyve binding pocket as well as nitrogen atoms projected in various directions. They were designed to expand on what we learned from pyridine-bearing analogue 4 and propargylic alcohol analogue 10: smaller groups at the alkyne terminus are better tolerated by PIKfyve. These new analogues aimed to probe whether the hydroxyl group is essential for potent PIKfyve inhibition. We hypothesized that a smaller group on the alkyne terminus would be better tolerated versus the appended aryl system.
When analogues 14–16 were broadly profiled via the DiscoverX scanMAX panel, they demonstrated good selectivity scores and all three bound potently to only 6 of 403 WT human kinases at 1 μM. This places them in the same selectivity range as other six-membered analogues from Tables 1 and 2, which bind 1–20 WT human kinases in the same panel. The PIKfyve potency of these compounds was found to be poor for 1-methylimidazole-bearing analogue 14 but improves when this ring system was replaced with the smaller cyclopropyl (15) or dimethylamino (16) groups. The DiscoverX PIKfyve PoC data was found to correlate well with the PIKfyve NanoBRET assay IC50 values for these analogues. Since they did not improve on the potency and selectivity of probe candidate 8, enzymatic confirmation for analogues 15 and 16 was not pursued. The inactivity of analogue 14 in the PIKfyve NanoBRET assay prompted its evaluation in the PIKfyve enzymatic assay. Its IC50 value in the PIKfyve enzymatic assay suggests that this scaffold, like pyridyl-bearing analogues 6 and 7, suffers from a 40–70-fold loss in potency when moving from the biochemical to the cell-based assay system. All data for 14–16 are summarized in Table 3.
Table 3. PIKfyve and Selectivity Data for Alkyne Optimization Library.
Percent of control (PoC) values determined at 1 μM via DiscoverX scanMAX profiling.
S10(1 μM): percentage of screened kinases with PoC < 10 at 1 μM.
Number of kinases with PoC < 10 at 1 μM.
NT: not tested.
Since moving from the six- to seven-membered system was accompanied by gains in PIKfyve potency (1 versus AMG28, 4 versus 5, 7 versus 8, 10 versus 11, and 12 versus 13), the attachment of the dimethylamino group to the seven-membered core as well as further exploration of the tolerance of PIKfyve to amine-bearing substitution was planned. The seven-membered equivalent of 16 was prepared (17) as well as analogues of AMG28 with the hydroxyl group replaced with an acyl amine (18) or a sulfonamide (19).
Furthermore, we designed additional propargylic alcohol analogues since their potency was still optimal versus other alkyne-bearing derivatives (1, AMG28, 10, and 11 versus 4, 5, and 14–16). Compounds were designed to cap the propargylic alcohol of AMG28 with a methyl group (20) and reduce the bulk on the carbon to which the alcohol is attached (21). Next, three analogues that project the alcohol further into the PIKfyve binding pocket via insertion of a methylene group were selected without bulk (22), with maintained substitution of AMG28 (23), and with a sterically hindered alcohol (24). Finally, two derivatives with increased bulk around the carbon bearing the propargylic alcohol were prepared, one with methyl, ethyl substitution (25) and another with diethyl substitution (26). We hypothesized that reducing the bulk around the propargylic alcohol (21 and 22) would result in analogues that were better tolerated by many kinases and thus reduce their kinome-wide selectivity. Analogues bearing larger groups on the terminus of the alkyne (4 and 5, for example) suggest that the pocket accommodates bulk. Thus, the propargylic alcohol analogues with a methylene unit insertion between the alkyne terminus and alcohol (22–24) or those sterically encumbered around the alcohol (25 and 26) were predicted to be well tolerated by PIKfyve. Finally, the dimethylamino analogue (16) indicated that the hydroxyl group is dispensable and thus analogue 20 was pursued.
We evaluated each of these analogues (17–26) using the DiscoverX scanMAX panel and in the PIKfyve NanoBRET assay. The nitrogen-bearing analogues (17–19) demonstrated good kinome-wide selectivity, binding with PoC < 10 to nine or fewer WT human kinases at 1 μM. The PIKfyve potency of these compounds, however, ranged in the DiscoverX and NanoBRET assays. Only analogue 17 was active and, given its single-digit potency coupled with kinome-wide selectivity (Figure S1A), it was selected for additional characterization as a PIKfyve probe candidate. The enhanced steric bulk (or alternative functionality) on the alkyne terminus in 18 and 19 was not well tolerated by PIKfyve.
Moving to the propargylic alcohol analogues (20–26), the kinome-wide selectivity of these analogues varied widely, and they displayed PoC < 10 for between 7 and 49 kinases at 1 μM. As predicted, the less sterically encumbered analogues 21 and 22 bound the most kinases in the scanMAX panel and are more promiscuous compounds than AMG28. For those analogues where steric bulk was added to the carbon center bearing the alcohol, improved selectivity was only observed with the diethyl analogue (26). In comparison, analogue 25 (methyl, ethyl derivative) and AMG28 demonstrated comparable S10(1 μM) scores. A thorough analysis of analogue 26 was driven by interest in its inhibition of off-target kinases, revealing that analogue 26 lacks the requisite selectivity to be considered as a probe candidate.22 Finally, analogues 20, 23, and 24 exhibited intermediate selectivity (11–14 of 403 WT human kinases with PoC < 10 when profiled at 1 μM). All analogues (20–26) demonstrated PoC < 10 in the PIKfyve assay included in the DiscoverX scanMAX panel. This potency translated to several analogues (21, 25, and 26) with IC50 values <20 nM in the PIKfyve NanoBRET assay.
Follow-up using the PIKfyve enzyme assay was carried out for several compounds that were potent (IC50 < 350 nM) in the PIKfyve NanoBRET assay. It is worth noting that for analogue 20 there was a larger drop-off in potency when moving from the PIKfyve enzymatic assay to the PIKfyve NanoBRET assay when compared to structurally similar analogues. Despite their orthogonally validated PIKfyve potency, the lack of selectivity of these compounds for PIKfyve when profiled broadly precluded their consideration as PIKfyve chemical probes. In pursuit of a negative control compound to be used alongside a PIKfyve chemical probe, analogue 19 was sent for enzymatic follow-up. A suitable negative control compound should bear structural similarity to the chosen chemical probe but lack potency in the PIKfyve NanoBRET and enzymatic assays and demonstrate favorable kinome-wide selectivity. Despite its impressive S10(1 μM) score (Table 3), analogue 19 retained PIKfyve potency and thus was removed from consideration as a negative control compound, highlighting a need for additional analogues. All data for analogues 17–26 are included in Table 3.
Designing a Negative Control
Since a suitable negative control compound had not been identified, we designed analogues that, based on our structure–activity relationships (SAR), would bind few kinases when profiled broadly and lack PIKfyve activity. We wanted to make at least one analogue of our two chemical probe candidates: 8 and 17. Thus, we designed some analogues bearing an alkyne and one with a biaryl system. Our studies included in Tables 1 and 2 narrowed in on the eight-membered ring as demonstrating both reduced affinity for PIKfyve and for the larger kinome, making it the ideal ring system to use in preparation of our negative control candidates. Given the promising selectivity score of analogues 14–16 (S10(1 μM) = 0.015) and the structural similarity of 16 to probe candidate 17, we first designed the eight-membered analogues of these three compounds. We hypothesized that ring expansion of these analogues would not be tolerated by PIKfyve and that a similar potency trend, albeit reduced, would be maintained as was seen for analogues 14–16. To complement the structure of probe candidate 8, we opted to replace the para-fluorobenzene with a smaller cyclopropyl ring and expanded the seven-membered ring to an eight-membered system. We anticipated that moving to the smaller cyclopropyl ring in this position would remove the favorable interactions that the para-fluorobenzene group makes with the PIKfyve binding site. Furthermore, direct attachment of a ring system in analogues 7–9 and 12–13 resulted in favorable kinome-wide selectivity, suggesting that this approach could result in a negative control compound with the requisite selectivity.
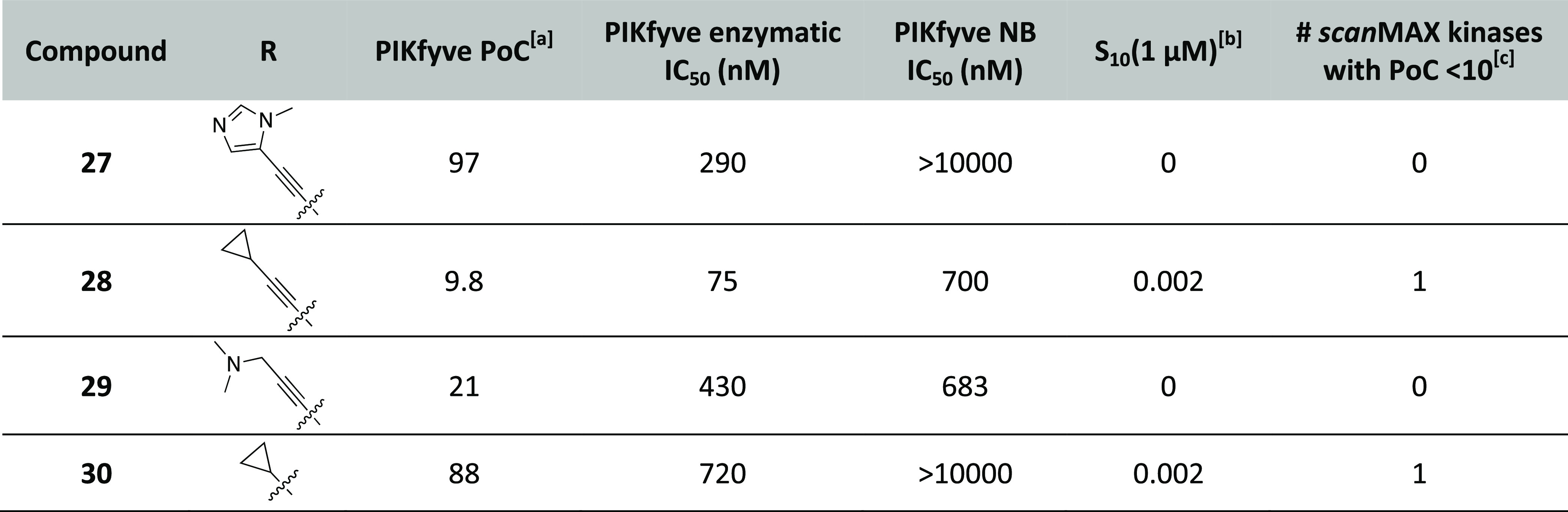
Kinome-wide profiling at 1 μM was carried out for the four analogues (27–30) via the DiscoverX scanMAX platform. All analogues displayed impressive selectivity scores with PoC < 10 for only 2 out of 403 WT human kinases. More variable data were observed when these analogues were tested in the PIKfyve NanoBRET and enzymatic assays. Analogues 28 and 29 maintained potency in the PIKfyve NanoBRET assay, a finding that was confirmed in orthogonal follow-up using the PIKfyve enzyme assay. In comparison, when N-methylimidazole was added to the alkyne terminus to produce analogue 27, PIKfyve potency in the NanoBRET assay was completely lost. The maintenance of some activity in the PIKfyve enzymatic assay but not in the PIKfyve NanoBRET assay suggests that cell entry could contribute to this loss in activity. Analogue 27 was excluded from consideration as a negative control due to its maintenance of PIKfyve enzymatic inhibition (IC50 < 300 nM) and potential cell penetrance issues. Moving to analogue 30, PIKfyve NanoBRET potency was lost and this loss in potency was echoed in the PIKfyve enzymatic assay (IC50 > 700 nM). This compound demonstrated the weakest potency of all compounds tested in the PIKfyve enzymatic assay. When we examined the S35(1 μM) DiscoverX scanMAX fraction for analogue 30 (Figure S1C), MAST1 was the only WT human kinase included (PoC = 32). Given its excellent kinome-wide selectivity and lack of PIKfyve potency, compound 30 is a good candidate for our negative control compound.
Chemical Probe Selection
To determine whether 8 and 17 meet the chemical probe criteria, we carried out additional enzymatic validation studies to confirm their selectivity criteria.23 These studies were designed to supplement the DiscoverX scanMAX data through determining whether WT human kinases with PoC < 35 in this panel also displayed potent inhibition when evaluated in the corresponding enzyme or NanoBRET assay (where available). All enzyme assays were executed at the Km value for ATP for the specific kinase. Based on the data collected, which is included in Table S1, we found that, in addition to PIKfyve, three of the six kinases in the S35(1 μM) fraction from the DiscoverX scanMAX panel were potently inhibited or engaged by analogue 8: MYLK4, PIP5K1C, and PI5P4Kγ. These three kinases are within 30-fold of the PIKfyve enzymatic and NanoBRET IC50 values. Analogue 8 demonstrates >415-fold selectivity over all other kinases evaluated. As an orthogonal screen, a small panel of understudied kinases was selected based on the inhibition data observed for AMG28 in the MRC panel (Figure 3A). No kinase in this panel was inhibited >55% when analogue 8 was screened at 1 μM in these enzymatic assays (Table S2), so no further follow-up was carried out.
Moving to analogue 17, enzymatic and/or NanoBRET assays were run for 18 of 20 kinases in the S35(1 μM) fraction from the DiscoverX scanMAX panel (Table S3). In addition, the same orthogonal screen of understudied kinases that was executed for analogue 8 was carried out for compound 17 (Table S2). The potent inhibition (PoC = 14) of MAP4K5 at 1 μM, which was also targeted by AMG28 (Figure 3A), warranted additional enzymatic follow-up (Table S3). Finally, the potency of analogue 8 on PI5P4Kγ motivated our exploration of the activity of analogue 17 in the PI5P4Kγ NanoBRET assay. Of the 20 kinases for which we executed orthogonal follow-up, only two kinases in addition to PIKfyve were potently inhibited by analogue 17: MYLK4 and MAP4K5. These two kinases are within 30-fold of the PIKfyve enzymatic and NanoBRET IC50 values. Analogue 17 demonstrates >42-fold selectivity over all other kinases evaluated. The weak binding of compound 17 to MAP4K5 in the DiscoverX scanMAX assay highlights the utility of orthogonal assay formats to confirm the results.
To determine whether the biochemical selectivity profile of analogue 17 was preserved when moving into cells, we ran the MYLK and MAP4K5 NanoBRET assays. The generated NanoBRET IC50 values of 265 nM for MYLK and >10 000 nM for MAP4K5 (Table S3) exposed that analogue 17 demonstrates improved selectivity in cells. A 10-fold biochemical selectivity window for PIKfyve versus MYLK4 became a 66-fold window in cells, while the compound was not found to bind to MAP4K5 in cells (Figure S2). Thus, compound 17 appears to be even more selective for PIKfyve when employed in cell-based studies.
The potency of analogue 17 coupled with its selectivity profile makes it the optimal choice as a PIKfyve chemical probe. Analogue 8, however, is also a high-quality, potent, and selective PIKfyve inhibitor that can be used in parallel, especially given its off-target inhibition profile differs from that of analogue 17. Before launching into cell-based phenotypic assays, we analyzed the aqueous kinetic solubility and mouse liver microsome stability of compound 17. While this small molecule was found to have excellent aqueous solubility (109.1 μM), its microsomal stability is suboptimal. After 30 min incubation with mouse liver microsomes, only 12.6% of the compound remained. While this suggests that our chemical probe is not suitable for use in vivo, it does not preclude its in vitro utility. Studies carried out by Biogen that employed AMG28 as a starting point have led to compounds that are stable in vivo.21,24 The structural modifications that Biogen made offer suggestions to how additional stability could be imparted into our scaffold.
SAR Observed for Indolyl Pyrimidinamines
In summary, we prepared 30 indolyl pyrimidinamines and remade AMG28. Many of these compounds demonstrate potent affinity for PIKfyve: 22 compounds with PoC < 21 in DiscoverX scanMAX assay when screened at 1 μM. 16 of these 22 demonstrated an IC50 < 400 nM in the PIKfyve NanoBRET assay, confirming that this biochemical binding data translated to binding affinity for PIKfyve in cells. Furthermore, the binding affinity of these compounds was validated in the PIKfyve enzymatic assay, solidifying that these are inhibitors of the PIKfyve function. A correlation between PIKfyve NanoBRET and enzyme data was observed for the indolyl pyrimidinamines such that an enzymatic IC50 value of <10 nM was associated with a NanoBRET IC50 value of <25 nM. With two exceptions (7 and 20), an enzymatic IC50 value of >22 nM resulted in a NanoBRET IC50 value of >680 nM. In addition to PIKfyve potency, several compounds display excellent kinome-wide selectivity. We narrowed in on six- and seven-membered analogues as the most efficacious PIKfyve inhibitors in terms of potency and selectivity and determined that the alkyne is dispensable since a biaryl analogue (8) retained probe-like qualities. In all cases where a matched pair of six- and seven-membered analogues was synthesized (1 and AMG28, 4 and 5, 7 and 8, 10 and 11, 12 and 13, and 16 and 17), the seven-membered analogue demonstrated enhanced (3–33-fold) PIKfyve NanoBRET affinity.
PIKfyve Inhibition Prevents β-Coronavirus Replication
We next confirmed that the binding affinity and enzymatic inhibition data of our chemical probe translated to a cellular phenotype through evaluation of the activity of our compound series in relevant cellular assays based on ascribed antiviral activities of published PIKfyve inhibitors. Apilimod, a published PIKfyve inhibitor, prevents infection by SARS-CoV-2 and other viruses. Dose-dependent inhibition of infection was observed in an infectivity assay utilizing SARS-CoV-2 strain 2019-nCoV/USA-WA1/2020. The proposed mechanism via which apilimod elicits this activity is through preventing release of viral contents from endosomes.7 This activity of apilimod coupled with its good tolerance as a drug when used for rheumatoid arthritis or Crohn’s disease as well as a putative ability to block antiviral immune responses motivated its advancement to Phase 2 clinical trials for COVID-19 treatment.4−6
We made use of a previously developed β-coronavirus assay to explore the activity of our compounds versus published PIKfyve inhibitors on β-coronavirus replication.25 This assay employs a fusion of mouse hepatitis virus (MHV) with nanoLuciferase (nLuc) to yield MHV-nLuc virus. MHV belongs to the β-coronavirae genus along with SARS-CoV-2. While MHV is a common mouse pathogen, it is not infectious to humans and it has been widely used as a model to study the virulence of SARS-CoV-2.25,26 Furthermore, there is 94% identity between human and mouse PIKfyve. The optimal titer and time point to analyze viral replication were determined via inoculation of mouse derived-from-brain-tumor (DBT) cells by MHV-NLuc.25 Accordingly, DBT cells were inoculated by MHV-nLuc with an MOI = 0.1 and luciferase measured at 10 h post-infection.
Several of the more selective compounds from Tables 1–4 and the published PIKfyve inhibitors in Figure 1 were evaluated in the optimized MHV-nLuc assay in DBT cells. All of the published inhibitors and many of our PIKfyve inhibitors, including chemical probe candidates 8 and 17, inhibited viral replication with no significant effect on cell viability up to 10 μM (Table 5 and Figures 4A, S3, and S4). Negative control compound 30, which lacks PIKfyve potency, failed to inhibit viral replication but was also nontoxic up to 10 μM (Figures 4A, S3, and S4). Importantly, anti-β-coronavirus activity correlated well with potency in the PIKfyve NanoBRET assay (Table 5 and Figure 4B). The most potent PIKfyve inhibitors were also most efficacious in the antiviral assay, while those devoid of PIKfyve inhibitory potential were unable to block viral replication. The relationship was maintained for indolyl pyrimidinamines of variable potency as well as when the published PIKfyve inhibitors from Figure 1 were added. Figure 4B shows this correlation and the linear regression fit with an R2 = 0.921. That addition of these published inhibitors, with different chemotypes than our series, slightly improves the correlation (R2 = 0.916 for indolyl pyrimidinamines alone) further confirms the relationship between PIKfyve inhibition and anti-β-coronavirus activity.
Table 4. PIKfyve and Selectivity Data for Compounds Designed as Potential Negative Control Options.
Percent of control (PoC) values determined at 1 μM via DiscoverX scanMAX profiling.
S10(1 μM): percentage of screened kinases with PoC < 10 at 1 μM.
Number of kinases with PoC < 10 at 1 μM.
Table 5. PIKfyve NanoBRET versus Viral Replication Data.
| compound | PIKfyve NB IC50 (nM) | PIKfyve NB pIC50 | MHV replication IC50 (nM) | MHV replication pIC50 | SARS-CoV-2 replication IC50 (nM) |
|---|---|---|---|---|---|
| 2 | 2540 | 5.61 | 8470 | 5.07 | NTa |
| 5 | 885 | 6.05 | 485 | 6.31 | NT |
| 7 | 380 | 6.42 | 713 | 6.15 | NT |
| 8 | 11.4 | 7.94 | 71.0 | 7.15 | 73.3 |
| 10 | 1170 | 5.93 | 1920 | 5.72 | NT |
| 12 | 4970 | 5.30 | 5350 | 5.27 | NT |
| 13 | 1560 | 5.81 | 666 | 6.18 | NT |
| 14 | >10 000 | 5.00 | 8050 | 5.09 | NT |
| 17 | 4.01 | 8.40 | 23.5 | 7.63 | 19.5 |
| 26 | 12.7 | 7.90 | 74.0 | 7.13 | 23.9 |
| 28 | 700 | 6.15 | 1800 | 5.75 | NT |
| 30 | >10 000 | 5.00 | >10 000 | 5.00 | >10 000 |
| apilimod | 0.312 | 9.51 | 15.7 | 7.80 | NT |
| YM201636 | 73.5 | 7.13 | 250 | 6.60 | NT |
| APY0201 | 1.19 | 8.92 | 40.4 | 7.39 | NT |
| remdesivir | NT | NT | NT | NT | 86.4 |
NT: not tested.
Figure 4.
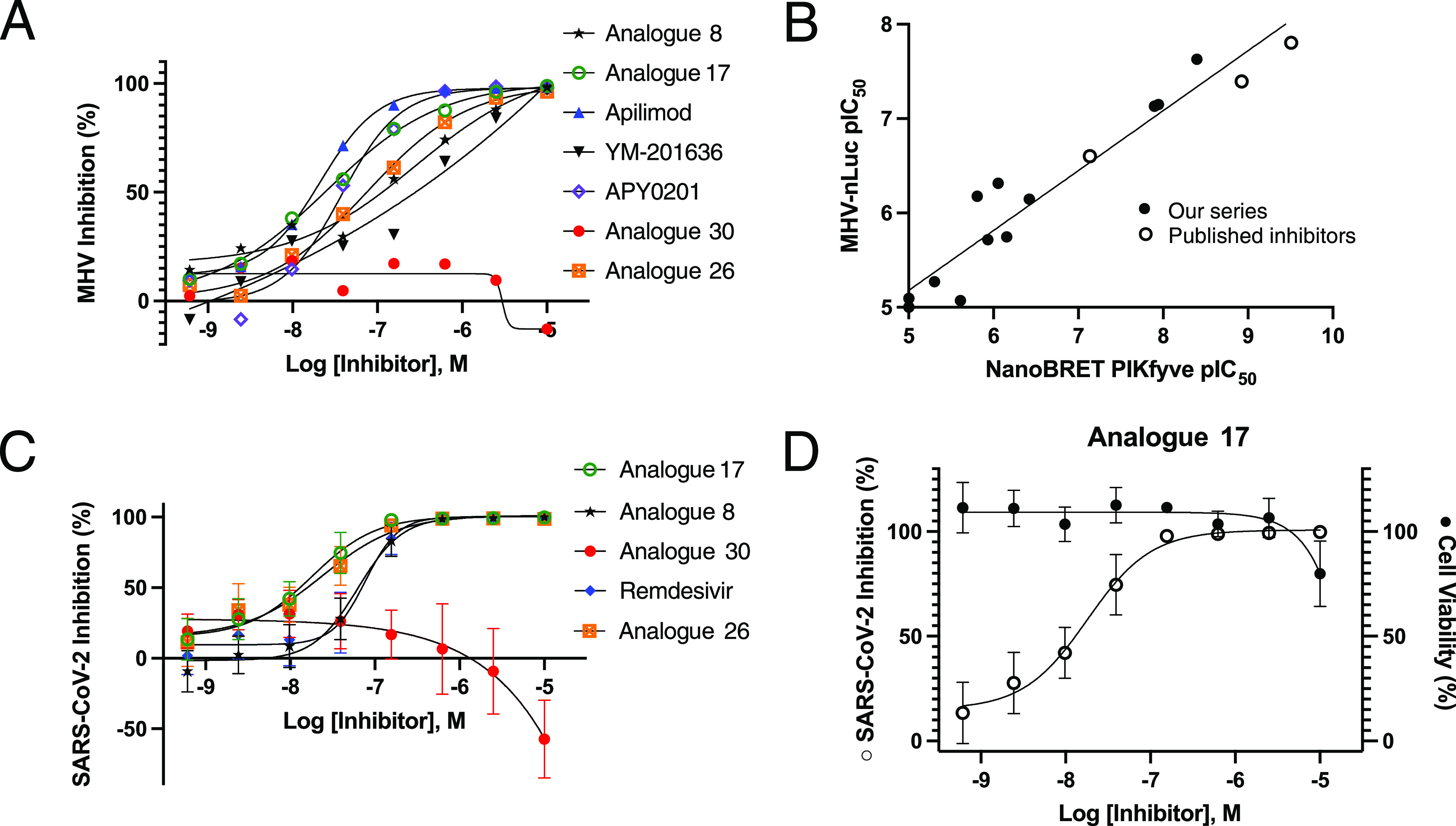
β-Coronavirus replication assay results. (A) Effect of PIKfyve inhibitors on replication of MHV-nLuc in DBT cells. Plot of average value for compounds that exhibit IC50 < 100 nM (n = 3). (B) Correlation of potency for PIKfyve target engagement with inhibition of β-coronavirus replication across our series of indolyl pyrimidinamines and published PIKfyve inhibitors. The analysis uses the pIC50 of the PIKfyve NanoBRET values and the pIC50 of the MHV-nLuc assay values from Table 5 for every analogue tested. The linear regression drawn demonstrates an R2 = 0.921. (C) Effect of PIKfyve inhibitors on viral replication in A549-ACE2 cells infected with SARS-CoV-2-nLuc (n = 4). Error bars represent standard error of the mean (SEM). (D) Effect of probe candidate 17 on viral replication (open circles) in A549-ACE2 cells infected with SARS-CoV-2-nLuc (n = 4). Cell viability determined by LDH assay (closed circles) in A549-ACE2 cells (n = 2). Error bars represent standard error of the mean (SEM).
The three most efficacious inhibitors of viral replication in the MHV-nLuc assay (8, 17, and 26) and the negative control compound (30) from the indolyl pyrimidinamine series were evaluated in a SARS-CoV-2-nLuc assay in human epithelial A549-ACE2 cells (Figures 4C,D, and S5).27 Remdesivir, an antiviral drug FDA-approved for use against SARS-CoV-2,28 was included as a positive control compound (IC50 = 86.4 nM). Probe compound 17 was found to be the most potent inhibitor of SARS-CoV-2 replication (IC50 = 19.5 nM) and outperformed remdesivir in this assay. Analogues 8 and 26 were also potent inhibitors of SARS-CoV-2 replication with IC50 values that were slightly better than remdesivir (Table 5). Importantly, the PIKfyve inhibitors were generally nontoxic to the A549-ACE2 cells and only exhibited toxicity at the highest dose (Figures 4D and S5). Negative control 30 did not inhibit SARS-CoV-2 replication or elicit any toxicity.
PIKfyve Inhibitors Disrupt Multiple Mediators in the Virus Lifecycle
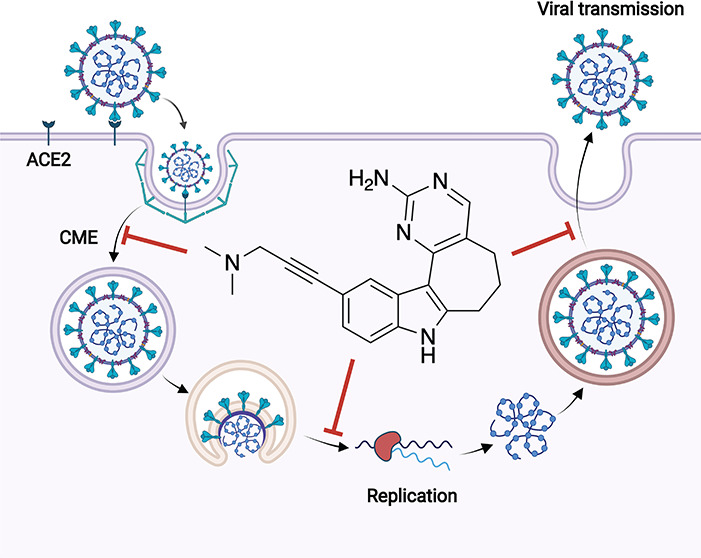
Since phosphatidylinositol-3-phosphate plays roles in membrane dynamics and trafficking as well as endosome processing and lysosomal function,29,30 PIKfyve inhibition is proposed to interrupt multiple processes that viruses hijack for entry and transmission. Reported mechanisms via which PIKfyve inhibition reduces viral infection in host cells include suppression of entry and/or through causing defective viral trafficking prior to entry, prevention of release of viral contents from endosomes, prevention of viral invasion via inhibiting host cell proteases, and/or antagonism of viral replication.6,7,31,32 These roles are consistent with the finding that, among other functions, PIKfyve resides predominately in early endosomes and plays an essential role in maintaining endomembrane homeostasis.32 While engagement of the host cell receptor ACE2 is an important step in viral entry of SARS-CoV-2, subsequent entry steps can vary and be dependent on the cell type. This virus can enter host cells via both clathrin (endosomal) and nonclathrin (not reliant on endosomes) pathways.33 The endosomal system used by SARS-CoV-2 is mediated by phosphoinositides. Endosomal PI(3,5)P2, which is produced via the enzymatic activity of PIKfyve, is implicated as playing a particularly essential role in endosomal homeostasis and regulation of early-to-late endosome dynamics.34,35
Apilimod was previously found to inhibit viral entry and replication during entry of several viruses, including SARS-CoV-2.32,34,35 YM201636 also prevents entry of SARS-CoV-2 S pseudovirions into HEK293/hACE2 cells, suggesting that this reduction of viral entry may be a general feature of PIKfyve inhibition.35 Given these findings, we explored the impact of our PIKfyve inhibitors on viral entry. Since most human coronaviruses are endocytosed prior to infection, we hypothesized that PIKfyve inhibition could specifically impact the clathrin-mediated route of SARS-CoV-2 internalization.36 Clathrin-mediated endocytosis (CME) is the primary pathway of SARS-CoV-2 cell entry.37 A purified spike glycoprotein uptake assay developed by the McPherson lab enabled us to probe whether PIKfyve is required for viral entry of the SARS-CoV-2 spike protein trimer. Furthermore, since this assay relies on CME, its use probes whether this is a mechanism that is interrupted by PIKfyve inhibitors.37
His6-tagged spike protein was incubated with and internalized by HEK293T-ACE2 cells in the presence of either dimethyl sulfoxide (DMSO) (vehicle control), 1 μM SGC-CK2-1 (positive control), or 1 μM PIKfyve inhibitor (Figure 5). When intercellular spike protein was visualized and quantified, vehicle-treated cells showed efficient uptake of the His6-tagged spike protein. In accordance with previous studies, treatment of the cells with 1 μM SGC-CK2-1 efficiently decreased spike protein uptake.25 Similarly, treatment of the cells with 1 μM PIKfyve inhibitors from our series (8 and 17) or apilimod also significantly reduced spike protein uptake versus the vehicle-treated control cells. Notably, 1 μM negative control analogue 30 demonstrated no effect on spike protein uptake in HEK293T-ACE2 cells. Next, since high levels of surface protease expression can promote an alternative route of viral entry via membrane fusion, the uptake of His6-tagged spike protein in Calu-3 and Caco-2 cells was imaged and quantified (Figure 6).38 These more physiologically relevant cell lines were chosen for their high ACE2 expression (Calu-3 and Caco-2) and high transmembrane serine protease (TMPRSS2) expression (Calu-3 only).39,40 As we observed in HEK293T-ACE2 cells, PIKfyve inhibitors (analogue 17 or apilimod) efficiently reduced spike protein uptake versus cells treated with DMSO and/or negative control analogue 30. In both cell lines, PIKfyve inhibitors produced >75% decrease in spike protein uptake, with the most robust reduction (>91%) observed in Caco-2 cells. The fact that uptake was significantly reduced in different cell lines despite their variable expression levels of TMPRSS2 supports that endocytosis is the primary pathway for SARS-CoV-2 uptake into cells. Our chemical series of PIKfyve inhibitors effectively prevents CME-mediated cell entry of the SARS-CoV-2 spike protein.
Figure 5.

PIKfyve inhibitors reduce uptake of His6-SARS-CoV-2 spike protein by HEK293T-ACE2 cells. (A) Images of cells treated with 1 μM PIKfyve inhibitors (analogues 8, 17, or apilimod), negative control (analogue 30), CK2 inhibitor (SGC-CK2-1, positive control), or DMSO (vehicle control). Cell nuclei are stained with DAPI, and the spike protein was detected using a His6 antibody. Scale bar (shown in white in the bottom right corner of the images) = 20 μm. (B) Quantification of the His6-SARS-CoV-2 spike protein uptake. The data is from three independent experiments with n = 6 for each condition. AU, arbitrary units; ****p-value < 0.0001. P-values were generated using one-way analysis of variance (ANOVA) with a post-hoc Tukey’s test to compare the means with one another.
Figure 6.

PIKfyve inhibitors reduce uptake of His6-SARS-CoV-2 spike protein by Calu-3 (A, B) and Caco-2 (C, D) cells. (A) Images of Calu-3 cells treated with 1 μM PIKfyve inhibitors (analogue 17 or apilimod), PIKfyve negative control (analogue 30), or DMSO (vehicle control). Scale bar, shown in white in the bottom right corner of the images, is 20 μm for the left and middle panels and 10 μm for the inset. (B) Quantification of the His6-SARS-CoV-2 spike protein uptake in Calu-3 cells. (C) Images of Caco-2 cells treated with 1 μM PIKfyve inhibitors (analogue 17 or apilimod) or PIKfyve negative control (analogue 30). Scale bar, shown in white in the bottom right corner of the images, is 40 μm for the left and middle panels and 20 μm for the inset. (D) Quantification of the His6-SARS-CoV-2 spike protein uptake in Caco-2 cells. The data is from three independent experiments for each cell line with n = 6 for each condition. AU, arbitrary units; ****p-value < 0.0001. For panels (A) and (C), cell nuclei are stained with DAPI, F-actin stained with phalloidin to define cell boundaries, and spike protein detected using a His6 antibody. For panels (B) and (D), p-values were generated using one-way ANOVA, with a post-hoc Tukey’s test to compare the means with one another.
To confirm that the inhibition of CME observed for the SARS-CoV-2 spike protein was also observed for the corresponding pseudovirus, we explored uptake of the lentivirus pseudotyped with SARS-CoV-2 spike glycoprotein by Calu-3 cells. We observed inhibition of pseudoviral entry versus DMSO-treated cells when cells were treated with PIKfyve inhibitors, including analogues 8 and 17 as well as apilimod. Treatment of cells with negative control compound 30 did not inhibit pseudoviral entry (Figure 7). In parallel, bald lentivirus containing the eGFP gene was introduced to Calu-3 cells. This lentiviral pseudovirion was selected as a negative control for studying the viral entry mediated by CME. When treated with the same compounds (apilimod and analogues 8, 17, and 30) or DMSO, entry of bald lentivirus was not impacted (Figure S6). Our PIKfyve inhibitors do not affect viral entry in general. Rather, since PIKfyve inhibitors reduced entry of lentivirus pseudotyped with SARS-CoV-2 spike glycoprotein but not entry of bald lentivirus, the effect of our compounds is specific to viral particles being internalized via the spike protein interacting with ACE2 receptors, a process that utilizes CME.
Figure 7.

Uptake of lentivirus pseudotyped with the SARS-CoV-2 spike glycoprotein is inhibited by PIKfyve inhibitors. Calu-3 cells were incubated with 1 μM of compound (or DMSO) for 1 h followed by the addition of the lentivirus pseudotyped with the SARS-CoV-2 spike glycoprotein for 12 h. Cells were fixed and stained with Hoechst to label the nuclei. Cells were also imaged for spike expression using spike antibody and visualized via Alexa Fluor 488, following transduction of the pseudovirus. Scale bars (shown in white in the bottom right corner of the images) = 100 μm. Bar graph shows the quantification of SARS-CoV-2 pseudovirus infection from experiments: n = 9 from three independent experiments, mean ± standard deviation (SD); one-way ANOVA followed Tukey’s test. ****p < 0.0001.
Studies in multiple cell types have demonstrated that the PI(3,5)P2 depletion following PIKfyve inhibition or deletion leads to significant swelling of endolysosomes.3,41−43 Using apilimod as a tool, inhibition of PIKfyve was linked with disruption of lysosomal function and endolysosomal swelling (vacuolization). The shape and size of endosomes and lysosomes were altered due to apilimod treatment. This activity contributes to the cytotoxicity exerted by apilimod in B-NHL.2 Most viruses, including hepatitis C, dengue, and West Nile, exit cells via the biosynthetic secretory pathway and transmit throughout the body.44,45 However, it was recently reported that β-coronaviruses hijack lysosomes to exit cells and spread systemically. To be able to exist in and exit via lysosomes of infected cells, SARS-CoV-2 deacidify lysosomes and weaken their ability to destroy the virus.46 Thus, targeting lysosomes has been suggested as a method to stop transmission of these viruses. To probe whether our series also disrupts lysosomal homeostasis, we treated SH-SY5Y neuroblastoma cells with DMSO or 5 μM analogues 8, 17, 30, or apilimod for 24 h, stained with LysoTracker, and imaged them. Fluorescence in these images was also quantified (Figure S7). As shown in Figure 8A–E, vacuoles are present in cells treated with 8, 17, or apilimod, but not in those treated with DMSO or 30. Furthermore, there is overlap between the localization of LysoTracker stain and vacuoles in Figure 8B,C,E, which is consistent with most of them being lysosomes. Induced lysosomal dysfunction by PIKfyve inhibitors is a potential mechanism that could be used to slow the spread of β-coronaviruses like SARS-CoV-2.
Figure 8.
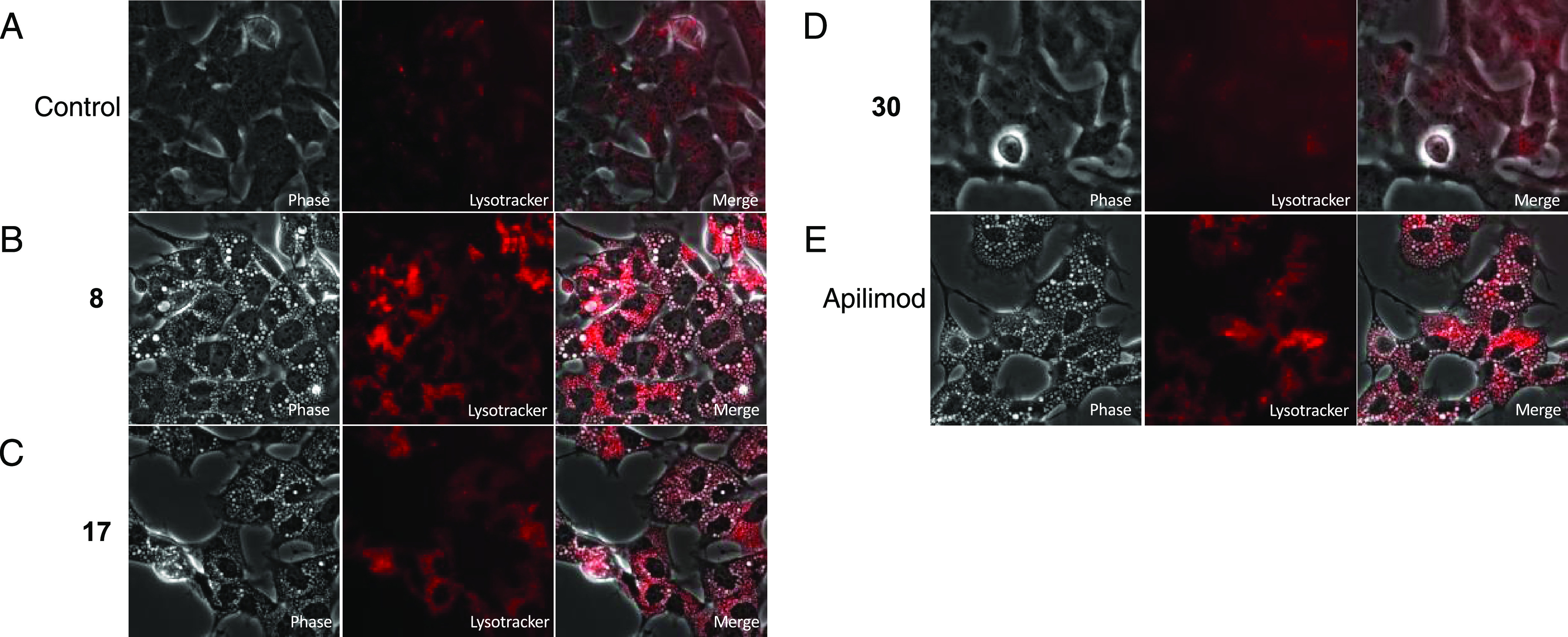
PIKfyve inhibitors impact lysosomal homeostasis. Panels (A–E) show that 50 nM LysoTracker was added 24 h after treatment with 0.05% DMSO (A), 5 μM 8 (B), 5 μM 17 (C), 5 μM 30 (D), or 5 μM apilimod (E) and imaged at 40×.
Chemistry
Similar routes were employed to prepare all intermediates and final compounds. Preparation of AMG28 began with Fischer indole synthesis to produce 34. Indole 34 was also used to prepare analogues 5, 8, 11, 13, and 17–26. Synthesis of analogues 1, 4, 7, 10, 12, and 14–16 commenced with commercially available indole 33, while commercially available indole 35 was used to prepare analogues 2, 6, 9, and 27–30. Intermediates 36–38 were obtained from indoles 33–35 via selective oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). Next, intermediates 39–41 were prepared from 36–38 by refluxing with Bredereck’s reagent and then cyclized to the corresponding aminopyrimidines 42–44 via reacting with a guanidinium species. Final PIKfyve inhibitors 1, 2, 4–6, 10–11, 14–29, and AMG28 were synthesized from 42–44 under Sonogashira coupling conditions (Scheme 1). Similarly, final compound 3 was obtained using commercially available indole 45 and Sonogashira coupling conditions (Scheme 2). Finally, aminopyrimidines 42–44 were used to access PIKfyve inhibitors 7–9, 12, 13, and 30 via the Suzuki cross-coupling methodology (Scheme 3).
Scheme 1. Synthesis of Analogues 1, 2, 4–6, 10–11, 14–29, and AMG28.
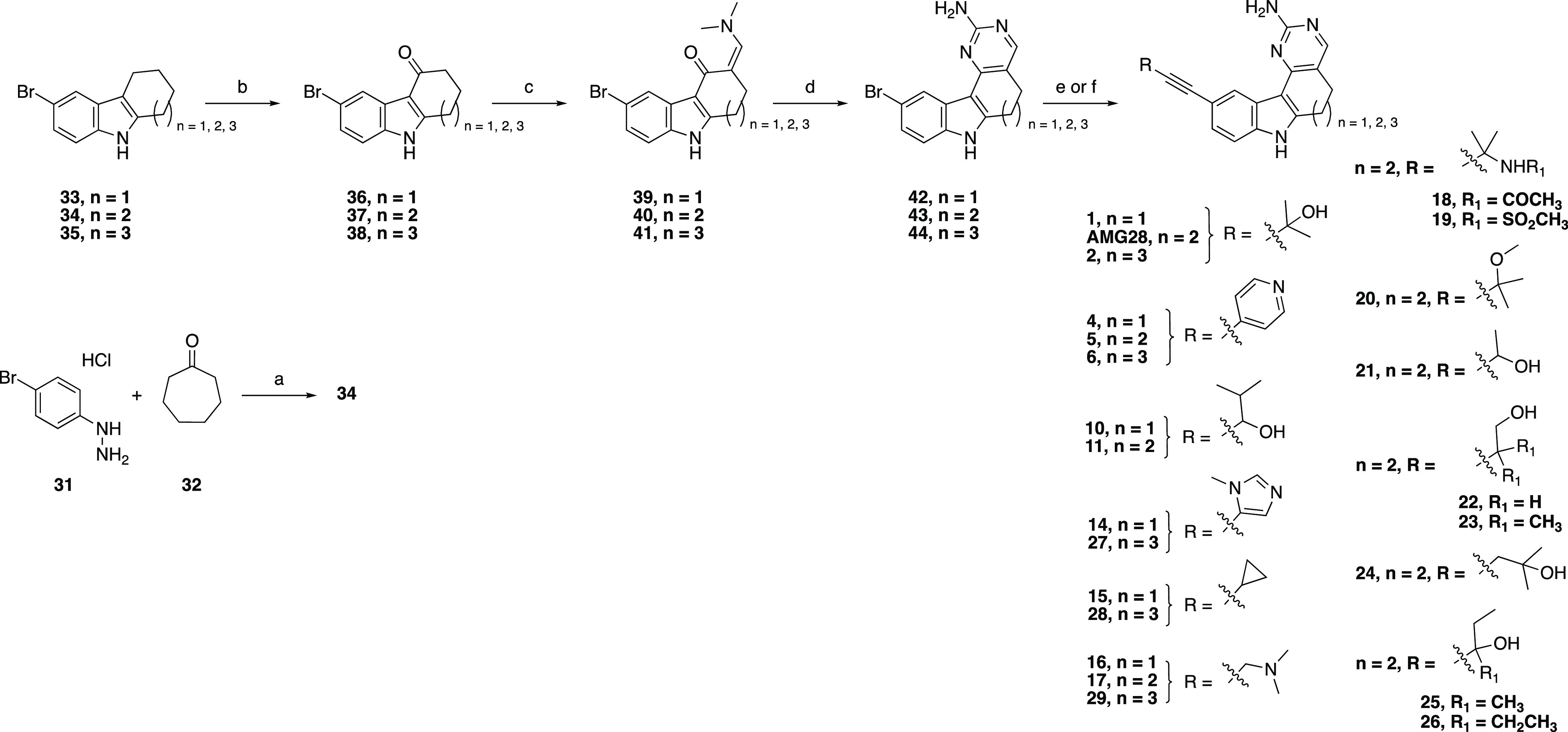
Reagents and conditions: (a) AcOH, 50–120 °C, 3 h, 70%; (b) DDQ, tetrahydrofuran (THF)/water, 0 °C, 45 min, 53–87%; (c) Bredereck’s reagent, toluene, or neat, 110 °C, 3–12 h; (d) NaOMe, guanidine hydrochloride, iPrOH, 100 °C, 12 h, 12–38% over two steps; (e) For 1, 2, 4–6, 10, 11, 14–17, 27–29, and AMG28: Alkyne R, triethylamine (TEA) or N,N-diisopropylamine (DIPA), Pd(PPh3)4, CuI, N,N-dimethylformamide (DMF), or N,N-dimethylacetamide (DMA), 100 °C, 12 h, 1.5–22%; (f) For 18–26: Alkyne R, DIPA, PdCl2(PPh3)2, CuI, propanol, 100 °C, 12 h, 2.4–15%.
Scheme 2. Synthesis of Analogue 3.

Reagents and conditions: (a) 2-methylbut-3-yn-2-ol, TEA, Pd(PPh3)4, CuI, DMA, 100 °C, 12 h, 4.4%.
Scheme 3. Synthesis of Analogues 7–9, 12, 13, and 30.

Reagents and conditions: (a) Boronic acid R, K2CO3, or K3PO4, Pd(PPh3)4, or Pd(dppf)Cl2, dioxane, and/or water, 80–95 °C, 12 h, 4.0–44%.
Conclusions
We suggest analogue 17 as the optimal PIKfyve chemical probe from our indolyl pyrimidinamine series. Analogue 17 meets SGC probe criteria23 based on its potency in the PIKfyve enzymatic (IC50 < 100 nM) and NanoBRET assays (IC50 < 1 μM) and selectivity across the kinome (S10(1 μM) = 0.02), with only two kinases (MAP4K5 and MYLK4) inhibited within 30-fold of its activity in the PIKfyve enzymatic assay. Furthermore, analogue 30 is a suitable negative control to be distributed and utilized alongside the PIKfyve chemical probe due to its >100 × lower potency in the PIKfyve enzymatic and NanoBRET assays coupled with its narrow kinome-wide selectivity (S10(1 μM) = 0). Multiple PIKfyve active inhibitors from our series potently inhibited β-coronavirus replication in MHV-NLuc reporter and SARS-CoV-2-nLuc assays. A deeper dive into the mechanisms via which PIKfyve inhibition disrupts the viral lifecycle demonstrated that analogues 8 and 17 impact both viral entry, specifically mediated by CME, and a potential route of systemic viral spread. In comparing our data with that generated using published PIKfyve inhibitors, we provide an orthogonal scaffold capable of PIKfyve inhibition in cells as a chemical probe set (positive and negative control) to be used by the scientific community to interrogate PIKfyve biology in cells.
Experimental Section
Chemistry: General Information
Reagents were obtained from verified commercial suppliers and used without further characterization or purification. Temperatures are reported in degree celsius (°C); the solvent was removed via a rotary evaporator under reduced pressure; and thin layer chromatography was used to monitor the progress of reactions that were executed under a blanket of nitrogen unless otherwise noted. The following abbreviations are used in schemes and/or experimental procedures: mmol (millimoles), μmol (micromoles), mg (milligrams), min (minutes), equiv (equivalent(s)), r.t. (room temperature), sec (seconds), and h (hours). 1H NMR and/or additional microanalytical data was collected for intermediates and final compounds to confirm their identity and assess their purity. 1H and 13C NMR spectra were obtained in DMSO-d6, CDCl3, or CD3OD-d4 and recorded using Varian or Bruker spectrometers. Magnet strength is indicated in each corresponding line listing. Peak positions are listed in parts per million (ppm) and calibrated versus the shift of the indicated deuterated solvent; coupling constants (J values) are reported in hertz (Hz); and multiplicities are included as follows: singlet (s), doublet (d), doublet of doublets/triplets/quartets (dd/dt/dq), triplet (t), triplet of doublets/triplets (td/tt), quartet (q), quartet of doublets (qd), pentent (p), and multiplet (m). Purity was determined using high-performance liquid chromatography (HPLC). The synthesis of final products was performed by ChemSpace LLC. Identity and purity of these compounds was confirmed upon receipt via NMR and additional microanalytical methods. All final compounds are >95% pure by HPLC analysis. Confirmatory HPLC traces are included in the Supporting Information.
2-Bromo-5,6,7,8,9,10-hexahydrocyclohepta[b]indole (34)
A suspension of 31 (2.0 g, 10.7 mmol) in AcOH (10 mL) was heated at 50 °C for 30 min, then 32 (2.4 g, 21.4 mmol) was added in one portion and the reaction mixture was refluxed for 3 h. After cooling to r.t., AcOH was removed under reduced pressure and the residue was dissolved in EtOAc. The organic phase was washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified with column chromatography (SiO2, 0–40% EtOAc in hexane) to yield 34 (1.14 g, 40% yield). 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.59 (d, J = 1.9 Hz, 1H), 7.17 (dd, J = 8.5, 1.9 Hz, 1H), 7.10 (d, J = 8.5 Hz, 1H), 2.84–2.79 (m, 2H), 2.78–2.73 (m, 2H), 1.93–1.86 (m, 2H), 1.81–1.71 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 139.04, 132.95, 131.20, 123.37, 120.47, 113.70, 112.45, 111.69, 31.79, 29.68, 28.70, 27.49, 24.71. HPLC purity: >95%. HRMS calculated for C13H15BrN [M + H]+: 264.0388. Found: 264.0382.
General Procedure for the Synthesis of Compounds 36–38: Procedure A
A solution of 33–35 (1 equiv) in THF (0.1 M) and H2O (1.0 M) was cooled to 0 °C and treated with a solution of DDQ (2 equiv) in THF (0.5 M) at such a rate that the reaction mixture did not exceed 4 °C. The resulting solution was allowed to stir for an additional 45 min at 0 °C. The solution was concentrated to dryness under reduced pressure. The resulting solids were suspended in EtOAc and saturated NaHCO3 solution. The solids were collected by filtration and washed with additional saturated NaHCO3 solution, followed by water and dried in vacuo to give the title compounds that were used in the next step without further purification.
6-Bromo-1,2,3,9-tetrahydro-4H-carbazol-4-one (36)
The reaction was carried out according to general procedure A using 33 (8.05 g, 32.1 mmol) in THF (300 mL) and water (35 mL) and DDQ (14.6 g, 64.3 mmol) in THF (130 mL) to afford the title compound (7.45 g, 87% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.07 (s, 1H), 8.05 (d, J = 1.9 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H), 7.29 (dd, J = 8.5, 2.0 Hz, 1H), 2.96 (t, J = 6.2 Hz, 2H), 2.43 (dd, J = 7.1, 5.6 Hz, 2H), 2.11 (p, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 192.92, 153.51, 134.66, 126.23, 124.91, 122.19, 114.13, 113.61, 111.23, 37.61, 23.24, 22.65. HPLC purity: >95%. HRMS calculated for C12H11BrNO [M + H]+: 264.0024. Found: 264.0018.
2-Bromo-6,7,8,9-tetrahydrocyclohepta[b]indol-10(5H)-one (37)
The reaction was carried out according to general procedure A using 34 (16.5 g, 62.6 mmol) in THF (500 mL) and water (70 mL) and DDQ (28.4 g, 125 mmol) in THF (250 mL) to afford the title compound (14.0 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.28 (d, J = 2.0 Hz, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.26 (dd, J = 8.5, 2.0 Hz, 1H), 3.15–3.08 (m, 2H), 2.70–2.63 (m, 2H), 1.97–1.89 (m, 2H), 1.87–1.80 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 196.50, 150.30, 133.90, 129.11, 124.68, 123.01, 114.04, 113.26, 113.03, 42.72, 27.21, 24.21, 21.80. HPLC purity: >95%. HRMS calculated for C13H13BrNO [M + H]+: 278.0181. Found: 278.0174.
2-Bromo-5,6,7,8,9,10-hexahydro-11H-cycloocta[b]indol-11-one (38)
The reaction was carried out according to general procedure A using 35 (10.5 g, 37.6 mmol) in THF (300 mL) and water (35 mL) and DDQ (17.1 g, 75.3 mmol) in THF (150 mL) to afford the title compound (6.43 g, 53% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.40–8.34 (m, 1H), 7.35 (dd, J = 8.5, 0.6 Hz, 1H), 7.27 (dd, J = 8.5, 2.0 Hz, 1H), 3.27 (t, J = 7.1 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H), 1.75–1.70 (m, 4H), 1.41–1.37 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 195.60, 147.78, 133.38, 128.81, 124.54, 123.19, 115.47, 114.24, 113.22, 41.02, 26.84, 24.55, 23.34, 22.89. HPLC purity: >95%. HRMS calculated for C14H15BrNO [M + H]+: 292.0337. Found: 292.0330.
General Procedure for the Synthesis of Compounds 39–41: Procedure B
To a suspension of 36–38 (1 equiv) in toluene (0.3 M, unless otherwise noted) was added Bredereck’s reagent (3 equiv). The resulting mixture was refluxed either for 3 h or overnight. After cooling to r.t., toluene was decanted (unless otherwise noted) and the viscous residue was dried in vacuo to give the crude title compounds that were used in the next step without further purification.
(Z)-6-Bromo-3-((dimethylamino)methylene)-1,2,3,9-tetrahydro-4H-carbazol-4-one (39)
The reaction was carried out according to general procedure B using 36 (7.44 g, 28.2 mmol) in toluene (100 mL) and Bredereck’s reagent (14.7 g, 84.6 mmol) at 110 °C overnight to afford the crude title compound (7.10 g, 74% yield).
(Z)-2-Bromo-9-((dimethylamino)methylene)-6,7,8,9-tetrahydrocyclohepta[b]indol-10(5H)-one (40)
The reaction was carried out according to general procedure B using 37 (14.0 g, 62.6 mmol) in toluene (200 mL) and Bredereck’s reagent (28.4 g, 126 mmol) at 110 °C overnight to afford the crude title compound (13.0 g, 77% yield).
(Z)-2-Bromo-10-((dimethylamino)methylene)-5,6,7,8,9,10-hexahydro-11H-cycloocta[b]indol-11-one (41)
The reaction was carried out according to general procedure B using 38 (6.39 g, 21.9 mmol) and Bredereck’s reagent (7.63 g, 43.8 mmol) at 110 °C for 3 h to afford the crude title compound (5.56 g, 75% yield) after trituration with n-hexane and methyl tert-butyl ether (MTBE), filtration, and drying in vacuo.
General Procedure for the Synthesis of Compounds 42–44: Procedure C
Guanidine hydrochloride (1.5–2 equiv) was added to a hot freshly prepared solution of NaOMe (1.5–3 equiv of Na in 0.4 M methanol), and the mixture was stirred for 10 min, diluted with isopropyl alcohol (IPA, 0.2 M), and filtered. 39–41 (1 equiv) were added to the filtrate, and the mixture was stirred at 100 °C overnight. After completion of the reaction, the mixture was cooled to r.t. and concentrated. The residue was diluted with water and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified either by flash column chromatography on SiO2 with 0–10% MeOH in CH2Cl2 (42, 43) or filtered under vacuum with water (44) to obtain the title compounds.
10-Bromo-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (42)
The reaction was carried out according to general procedure C using NaOMe (1.53 g, 66.6 mmol of Na in 50 mL MeOH), guanidine hydrochloride (3.18 g, 33.3 mmol), IPA (100 mL), and 39 (7.08 g, 22.2 mmol) at 110 °C overnight to afford the title compound (1.10 g, 16% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.84 (s, 1H), 8.39 (d, J = 2.0 Hz, 1H), 7.35 (d, J = 8.5 Hz, 1H), 7.24 (dd, J = 8.5, 2.1 Hz, 1H), 6.26 (s, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.84 (t, J = 7.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.54, 159.75, 153.58, 145.82, 135.18, 126.21, 123.93, 122.70, 113.42, 113.10, 112.95, 108.41, 23.46, 21.56. HPLC purity: >95%. HRMS calculated for C14H12BrN4 [M + H]+: 315.0245. Found: 315.0239.
11-Bromo-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-2-amine (43)
The reaction was carried out according to general procedure C using NaOMe (3.57 g, 77.8 mmol of Na in 100 mL MeOH), guanidine hydrochloride (7.43 g, 77.8 mmol), IPA (200 mL), and 40 (13.0 g, 38.9 mmol) at 110 °C overnight to afford the title compound (4.70 g, 39% yield). 1H NMR (400 MHz, CD3OD-d4) δ 8.81 (dd, J = 1.9, 0.6 Hz, 1H), 7.84 (s, 1H), 7.21 (dd, J = 8.5, 1.9 Hz, 1H), 7.17 (dd, J = 8.5, 0.7 Hz, 1H), 3.16 (t, J = 6.7 Hz, 2H), 2.70–2.62 (m, 2H), 2.06–1.97 (m, 2H). 13C NMR (101 MHz, CD3OD-d4) δ 164.10, 163.01, 156.66, 146.06, 136.50, 130.73, 126.47, 125.86, 122.82, 114.66, 112.74, 111.55, 31.23, 30.72, 25.49. HPLC purity: >95%. HRMS calculated for C15H14BrN4 [M + H]+: 329.0402. Found: 329.0395.
12-Bromo-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (44)
The reaction was carried out according to general procedure C using NaOMe (1.10 g, 24.0 mmol of Na in 50 mL MeOH), guanidine hydrochloride (2.30 g, 24.0 mmol), IPA (100 mL), and 41 (5.50 g, 16.0 mmol) at 110 °C overnight to afford the title compound (2.80 g, 51% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 7.99 (s, 1H), 7.92 (d, J = 1.9 Hz, 1H), 7.18 (d, J = 8.5 Hz, 1H), 7.10 (dd, J = 8.5, 2.0 Hz, 1H), 6.33 (s, 1H), 2.96–2.86 (m, 2H), 2.47–2.37 (m, 2H), 1.72–1.57 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.06, 161.89, 157.82, 142.25, 142.09, 134.27, 134.12, 129.60, 129.56, 123.63, 122.44, 119.62, 112.26, 112.21, 112.00, 108.83, 108.80, 30.76, 27.58, 27.52, 26.65, 20.67. HPLC purity: >95%. HRMS calculated for C16H16BrN4 [M + H]+: 343.0558. Found: 343.0553.
General Procedure for the Synthesis of Compounds 1–6, 10, 11, 14–17, 27–29, and AMG28: Procedure D
A mixture of 42–45 (1 equiv), alkyne (1.5–5 equiv), and TEA or DIPA (1–3 equiv) in DMF or DMA (0.2–0.8 M) was degassed, followed by addition of Pd(PPh3)4 (0.05–0.1 equiv) and CuI (0.05–0.2 equiv). The reaction mixture was heated to 100 °C and stirred overnight. After completion of the reaction, the mixture was cooled to r.t., diluted with EtOAc, and passed through a thin pad of celite. The filtrate was concentrated in vacuo and the crude residue was purified by preparative HPLC (10–100% MeOH in H2O + 0.1% TFA) to give the title compounds.
4-(2-Amino-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-10-yl)-2-methylbut-3-yn-2-ol (1)
The reaction was carried out according to general procedure D using 42 (148 mg, 0.47 mmol), 2-methylbut-3-yn-2-ol (79 mg, 0.94 mmol), TEA (95 mg, 0.94 mmol), Pd(PPh3)4 (55 mg, 47 μmol), and CuI (18 mg, 94 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (28 mg, 19% yield).1H NMR (500 MHz, DMSO-d6) δ 11.79 (s, 1H), 8.28 (d, J = 1.6 Hz, 1H), 7.92 (d, J = 4.1 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.12 (dd, J = 8.4, 1.7 Hz, 1H), 6.20 (s, 2H), 5.40 (s, 1H), 2.97 (t, J = 7.8 Hz, 2H), 2.83 (t, J = 7.8 Hz, 2H), 1.50 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 162.74, 159.85, 153.80, 145.41, 135.85, 124.88, 124.35, 123.77, 114.35, 113.05, 111.69, 108.79, 93.53, 82.01, 63.66, 31.88, 23.56, 21.58. HPLC purity: 95.03%. HRMS (ESI): m/z calculated for C19H19N4O [M + H]+: 319.1559. Found: 319.1555.
4-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-2-methylbut-3-yn-2-ol (AMG28)
The reaction was carried out according to general procedure D using 43 (198 mg, 0.60 mmol), 2-methylbut-3-yn-2-ol (101 mg, 1.2 mmol), TEA (122 mg, 1.2 mmol), Pd(PPh3)4 (70 mg, 60 μmol), and CuI (18 mg, 60 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (3.2 mg, 1.6% yield). The analytical data for AMG28 matches that previously reported.471H NMR (850 MHz, CD3OD-d4) δ 8.74 (s, 1H), 7.88 (s, 1H), 7.26 (d, J = 8.3 Hz, 1H), 7.23–7.17 (m, 1H), 3.23 (t, J = 6.6 Hz, 2H), 2.76–2.70 (m, 2H), 2.12–2.05 (m, 2H), 1.59 (s, 6H). HPLC purity: 96.35%.
4-(2-Amino-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-12-yl)-2-methylbut-3-yn-2-ol (2)
The reaction was carried out according to general procedure D using 44 (198 mg, 0.57 mmol), 2-methylbut-3-yn-2-ol (73 mg, 0.87 mmol), TEA (117 mg, 1.16 mmol), Pd(PPh3)4 (33 mg, 30 μmol), and CuI (6.0 mg, 30 μmol) in DMA (1.0 mL) at 100 °C overnight to afford the title compound (13.2 mg, 6.6% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.08 (s, 1H), 7.87 (d, J = 1.5 Hz, 1H), 7.24 (dd, J = 8.3, 0.7 Hz, 1H), 7.15 (dd, J = 8.3, 1.6 Hz, 1H), 3.06–2.99 (m, 2H), 2.64–2.59 (m, 2H), 1.86–1.79 (m, 4H), 1.56 (s, 6H). 13C NMR (214 MHz, CD3OD-d4) δ 165.02, 163.06, 159.12, 143.29, 137.11, 135.87, 135.82, 131.42, 131.36, 129.08, 126.40, 124.54, 115.64, 111.45, 110.34, 92.31, 84.41, 66.02, 32.26, 31.96, 28.97, 28.23. HPLC purity: 95.56%. HRMS (ESI): m/z calculated for C21H23N4O [M + H]+: 347.1872. Found: 347.1868.
4-(3-(2-Aminopyrimidin-4-yl)-1H-indol-5-yl)-2-methylbut-3-yn-2-ol (3)
The reaction was carried out according to general procedure D using 45 (396 mg, 1.37 mmol), 2-methylbut-3-yn-2-ol (345 mg, 4.11 mmol), TEA (277 mg, 2.74 mmol), Pd(PPh3)4 (159 mg, 0.14 mmol), and CuI (19 mg, 0.14 mmol) in DMA (3.0 mL) at 100 °C overnight to afford the title compound (17.7 mg, 4.4% yield). The analytical data for 3 matches that previously reported.211H NMR (400 MHz, CD3OD-d4) δ 8.59 (s, 1H), 8.17 (s, 2H), 8.07 (d, J = 5.8 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.12 (d, J = 6.0 Hz, 1H), 1.60 (s, 6H). HPLC purity: 100%. HRMS (ESI): m/z calculated for C17H16N4O [M + H]+: 293.1402. Found: 293.1397.
10-(Pyridin-4-ylethynyl)-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (4)
The reaction was carried out according to general procedure D using 42 (140 mg, 0.44 mmol), 4-ethynylpyridine (92 mg, 0.88 mmol), TEA (90 mg, 0.88 mmol), Pd(PPh3)4 (52 mg, 44 μmol), and CuI (17 mg, 88 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (33 mg, 22% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.93 (s, 1H), 8.63 (d, J = 5.1 Hz, 2H), 8.52 (s, 1H), 7.94 (s, 1H), 7.54–7.49 (m, 2H), 7.46 (d, J = 8.3 Hz, 1H), 7.35 (dd, J = 8.3, 1.7 Hz, 1H), 6.24 (s, 2H), 3.00 (t, J = 7.8 Hz, 2H), 2.86 (t, J = 7.8 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.73, 159.63, 153.94, 149.90, 145.80, 136.70, 130.92, 125.14, 125.06, 124.78, 124.47, 113.04, 112.73, 112.09, 109.00, 96.14, 84.82, 23.45, 21.56. HPLC purity: 96.28%. HRMS (ESI): m/z calculated for C21H16N5 [M + H]+: 338.1406. Found: 338.1393.
11-(Pyridin-4-ylethynyl)-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-2-amine (5)
The reaction was carried out according to general procedure D using 43 (187 mg, 0.57 mmol), 4-ethynylpyridine (117 mg, 1.14 mmol), TEA (115 mg, 1.14 mmol), Pd(PPh3)4 (66 mg, 60 μmol), and CuI (13 mg, 60 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (7.5 mg, 3.8% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.95–8.91 (m, 1H), 8.55–8.49 (m, 2H), 7.91 (s, 1H), 7.57–7.53 (m, 2H), 7.39–7.33 (m, 2H), 3.27–3.22 (m, 2H), 2.75–2.72 (m, 2H), 2.12–2.06 (m, 2H). 13C NMR (214 MHz, CD3OD-d4) δ 164.16, 163.18, 156.81, 150.11, 146.25, 138.32, 134.93, 129.07, 128.68, 127.13, 127.01, 123.03, 114.58, 112.22, 111.72, 98.97, 85.10, 31.22, 30.77, 25.66. HPLC purity: 96.99%. HRMS (ESI): m/z calculated for C22H18N5 [M + H]+: 352.1562. Found: 352.1556.
12-(Pyridin-4-ylethynyl)-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (6)
The reaction was carried out according to general procedure D using 44 (188 mg, 0.55 mmol), 4-ethynylpyridine (85 mg, 0.82 mmol), TEA (111 mg, 1.1 mmol), Pd(PPh3)4 (32 mg, 30 μmol), and CuI (6.0 mg, 30 μmol) in DMA (1.0 mL) at 100 °C overnight to afford the title compound (15.9 mg, 8.0% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.50 (d, J = 5.2 Hz, 2H), 8.12–8.08 (m, 2H), 7.50–7.47 (m, 2H), 7.34 (dd, J = 3.8, 1.2 Hz, 2H), 3.08–3.04 (m, 2H), 2.66–2.61 (m, 2H), 1.88–1.79 (m, 4H). 13C NMR (214 MHz, CD3OD-d4) δ 164.80, 163.15, 159.25, 150.11, 143.81, 137.93, 134.80, 129.27, 126.93, 126.69, 126.07, 125.67, 114.16, 111.86, 110.63, 98.63, 85.10, 32.22, 29.00, 28.19. HPLC purity: 97.95%. HRMS (ESI): m/z calculated for C23H20N5 [M + H]+: 366.1719. Found: 366.1711.
1-(2-Amino-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-10-yl)-4-methylpent-1-yn-3-ol (10)
The reaction was carried out according to general procedure D using 42 (142 mg, 0.45 mmol), 4-methylpent-1-yn-3-ol (89 mg, 0.90 mmol), TEA (91 mg, 0.9 mmol), Pd(PPh3)4 (58 mg, 45 μmol), and CuI (17 mg, 90 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (14 mg, 9.0% yield). 1H NMR (400 MHz, CD3OD-d4) δ 8.42 (dd, J = 1.7, 0.8 Hz, 1H), 7.72 (d, J = 1.2 Hz, 1H), 7.39 (dd, J = 8.4, 0.8 Hz, 1H), 7.32 (dd, J = 8.4, 1.6 Hz, 1H), 4.33 (d, J = 5.9 Hz, 1H), 3.14 (td, J = 7.3, 1.2 Hz, 2H), 3.03 (dd, J = 8.3, 6.8 Hz, 2H), 1.93 (dq, J = 13.2, 6.7 Hz, 1H), 1.09 (dd, J = 11.0, 6.7 Hz, 6H). 13C NMR (101 MHz, CD3OD-d4) δ 167.82, 156.71, 153.45, 138.47, 137.89, 128.34, 126.07, 125.92, 118.46, 116.40, 113.08, 110.61, 88.82, 86.81, 69.05, 36.20, 22.69, 22.43, 18.84, 18.27. HPLC purity: 95.34%. HRMS (ESI): m/z calculated for C20H21N5 [M + H]+: 333.1715. Found: 333.1703.
1-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-4-methylpent-1-yn-3-ol (11)
The reaction was carried out according to general procedure D using 43 (190 mg, 0.58 mmol), 4-methylpent-1-yn-3-ol (113 mg, 1.16 mmol), TEA (175 mg, 1.73 mmol), Pd(PPh3)4 (67 mg, 60 μmol), and CuI (15 mg, 60 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (4.0 mg, 2% yield). 1H NMR (400 MHz, CD3OD-d4) δ 8.83 (t, J = 1.1 Hz, 1H), 7.75 (s, 1H), 7.29 (d, J = 0.9 Hz, 2H), 4.35 (d, J = 5.9 Hz, 1H), 3.30–3.25 (m, 2H), 2.83–2.76 (m, 2H), 2.09 (dt, J = 10.3, 6.3 Hz, 2H), 1.92 (dt, J = 13.2, 6.6 Hz, 1H), 1.09 (dd, J = 10.8, 6.7 Hz, 6H). 13C NMR (101 MHz, CD3OD-d4) δ 170.63, 155.39, 151.96, 140.98, 137.68, 128.82, 128.27, 128.17, 122.73, 117.81, 112.15, 111.95, 88.36, 87.53, 69.13, 36.24, 31.37, 31.28, 23.86, 18.82, 18.29. HPLC purity: 95.56%. HRMS (ESI): m/z calculated for C21H23N4O [M + H]+: 347.1872. Found: 347.1867.
10-((1-Methyl-1H-imidazol-5-yl)ethynyl)-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (14)
The reaction was carried out according to general procedure D using 42 (139 mg, 0.44 mmol), 5-ethynyl-1-methyl-1H-imidazole (94 mg, 0.88 mmol), TEA (90 mg, 0.88 mmol), Pd(PPh3)4 (51 mg, 44 μmol), and CuI (17 mg, 88 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (16 mg, 12% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.87 (s, 1H), 8.45 (d, J = 1.6 Hz, 1H), 7.93 (s, 1H), 7.77 (s, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.30 (dd, J = 8.3, 1.7 Hz, 1H), 7.28 (s, 1H), 6.22 (s, 2H), 3.74 (s, 3H), 3.00 (t, J = 7.8 Hz, 2H), 2.85 (t, J = 7.8 Hz, 2H). 13C NMR (214 MHz, CD3OD-d4) δ163.88, 162.89, 153.57, 147.68, 139.69, 138.35, 133.09, 126.41, 126.14, 125.90, 118.56, 115.72, 115.70, 112.64, 110.65, 99.26, 74.99, 32.50, 25.16, 22.93. HPLC purity: 98.86%. HRMS (ESI): m/z calculated for C20H16N6Na [M + Na]+: 363.1334. Found: 363.1347.
10-(Cyclopropylethynyl)-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (15)
The reaction was carried out according to general procedure D using 42 (157 mg, 0.50 mmol), ethynylcyclopropane (66 mg, 1.0 mmol), TEA (101 mg, 1.0 mmol), Pd(PPh3)4 (58 mg, 50 μmol), and CuI (19 mg, 100 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (5.0 mg, 3.0% yield). 1H NMR (700 MHz, CD3OD-d4) δ 8.34 (d, J = 1.7 Hz, 1H), 7.74 (s, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.21 (dd, J = 8.4, 1.6 Hz, 1H), 3.10 (t, J = 7.6 Hz, 2H), 2.99 (t, J = 7.6 Hz, 2H), 1.49–1.44 (m, 1H), 0.88 (dt, J = 8.1, 3.3 Hz, 2H), 0.74 (dt, J = 4.7, 3.1 Hz, 2H). 13C NMR (214 MHz, CD3OD-d4) δ 166.59, 158.44, 151.76, 141.63, 137.91, 127.87, 125.99, 125.93, 119.03, 116.19, 112.72, 110.50, 92.23, 77.57, 24.82, 22.54, 8.86. HPLC purity: 96.33%. HRMS (ESI): m/z calculated for C19H17N4 [M + H]+: 301.1453. Found: 301.1445.
10-(3-(Dimethylamino)prop-1-yn-1-yl)-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (16)
The reaction was carried out according to general procedure D using 42 (99 mg, 0.30 mmol), N,N-dimethylprop-2-yn-1-amine (79 mg, 0.9 mmol), DIPA (64 mg, 0.63 mmol), Pd(PPh3)4 (37 mg, 32 μmol), and CuI (13 mg, 64 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (4.7 mg, 4.7% yield). 1H NMR (700 MHz, CD3OD-d4) δ 8.54 (d, J = 2.1 Hz, 1H), 7.77 (d, J = 2.1 Hz, 1H), 7.47 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 4.34 (s, 2H), 3.17 (td, J = 7.6, 2.0 Hz, 2H), 3.07–3.03 (m, 8H). 13C NMR (214 MHz, CD3OD-d4) δ 164.25, 161.84, 149.41, 149.23, 138.55, 127.20, 126.42, 126.02, 115.97, 115.69, 112.78, 110.58, 90.73, 78.97, 43.51, 25.00, 22.76. HPLC purity: 100%. HRMS (ESI): m/z calculated for C19H20N5 [M + H]+: 218.1719. Found: 318.1713.
11-(3-(Dimethylamino)prop-1-yn-1-yl)-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-2-amine (17)
The reaction was carried out according to general procedure D using 43 (199 mg, 0.6 mmol), N,N-dimethylprop-2-yn-1-amine (201 mg, 2.41 mmol), TEA (122 mg, 1.21 mmol), Pd(PPh3)4 (35 mg, 30 μmol), and CuI (7.0 mg, 30 μmol) in DMF (1.5 mL) at 100 °C overnight to afford the title compound (2.9 mg, 1.5% yield). 1H NMR (700 MHz, CD3OD-d4) δ 8.85 (s, 1H), 7.93–7.85 (m, 1H), 7.33–7.24 (m, 2H), 4.00 (s, 2H), 3.24 (t, J = 6.6 Hz, 2H), 2.80 (s, 6H), 2.75–2.72 (m, 2H), 2.09 (dt, J = 10.4, 6.4 Hz, 2H). 13C NMR (126 MHz, CD3OD-d4) δ 164.62, 162.58, 155.68, 146.62, 138.08, 128.93, 128.47, 127.02, 123.11, 114.46, 112.05, 111.61, 91.82, 77.90, 43.38, 31.18, 30.77, 25.54, 18.35. HPLC purity: 100%. HRMS (ESI): m/z calculated for C20H22N5 [M + H]+: 332.1875. Found: 332.1868.
12-((1-Methyl-1H-imidazol-5-yl)ethynyl)-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (27)
The reaction was carried out according to general procedure D using 44 (186 mg, 0.54 mmol), 5-ethynyl-1-methyl-1H-imidazole (86 mg, 0.81 mmol), TEA (110 mg, 1.1 mmol), Pd(PPh3)4 (31 mg, 30 μmol), and CuI (6.0 mg, 30 μmol) in DMA (1.0 mL) at 100 °C overnight to afford the title compound (31 mg, 16% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.06 (d, J = 4.7 Hz, 2H), 7.74 (s, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.24 (dd, J = 7.6, 2.1 Hz, 2H), 6.35 (s, 2H), 3.70 (s, 3H), 3.03–2.98 (m, 2H), 1.75–1.71 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 162.25, 161.87, 158.22, 141.87, 138.98, 135.52, 132.91, 127.76, 124.43, 123.89, 112.25, 110.79, 109.35, 97.95, 31.62, 30.78, 27.60. HPLC purity: 100%. HRMS (ESI): m/z calculated for C22H21N6 [M + Na]+: 369.1828. Found: 369.1822.
12-(Cyclopropylethynyl)-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (28)
The reaction was carried out according to general procedure D using 44 (209 mg, 0.61 mmol), ethynylcyclopropane (81 mg, 1.22 mmol), TEA (136 mg, 1.34 mmol), Pd(PPh3)4 (35 mg, 30 μmol), and CuI (6.0 mg, 30 μmol) in DMA (1.0 mL) at 100 °C overnight to afford the title compound (7.7 mg, 3.9% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.08 (s, 1H), 7.84–7.82 (m, 1H), 7.20 (dd, J = 8.3, 0.7 Hz, 1H), 7.10 (dd, J = 8.3, 1.6 Hz, 1H), 3.06–3.01 (m, 2H), 2.66–2.60 (m, 2H), 1.84 (d, J = 5.7 Hz, 4H), 1.43 (tt, J = 8.2, 5.0 Hz, 1H), 0.85–0.80 (m, 2H), 0.70–0.67 (m, 2H). 13C NMR (214 MHz, CD3OD-d4) δ 165.45, 162.48, 158.29, 143.53, 136.78, 129.03, 126.63, 124.47, 116.76, 113.61, 111.36, 110.10, 91.07, 78.20, 32.25, 28.99, 8.79, 0.82. HPLC purity: 97.53%. HRMS (ESI): m/z calculated for C21H21N4 [M + H]+: 329.1766. Found: 329.1758.
12-(3-(Dimethylamino)prop-1-yn-1-yl)-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (29)
The reaction was carried out according to general procedure D using 44 (99 mg, 0.29 mmol), N,N-dimethylprop-2-yn-1-amine (120 mg, 1.45 mmol), DIPA (59 mg, 0.58 mmol), Pd(PPh3)4 (34 mg, 30 μmol), and CuI (6.0 mg, 30 μmol) in DMA (1.0 mL) at 100 °C overnight to afford the title compound (4.6 mg, 4.6% yield). 1H NMR (700 MHz, CD3OD-d4) δ 8.20 (s, 1H), 8.14 (s, 1H), 7.38–7.32 (m, 2H), 4.30 (s, 2H), 3.19–3.14 (m, 2H), 3.02 (s, 6H), 2.78–2.73 (m, 2H), 1.94–1.88 (m, 4H). 13C NMR (176 MHz, CD3OD-d4) δ 156.06, 148.65, 138.01, 130.77, 128.87, 127.62, 125.97, 122.99, 116.86, 115.25, 114.43, 112.29, 93.01, 75.94, 42.72, 31.84, 29.20, 27.84, 22.03. HPLC purity: 95.61%. HRMS (ESI): m/z calculated for C21H24N5 [M + H]+: 346.2032. Found: 346.2024.
General Procedure for the Synthesis of Compounds 18–26: Procedure E
A mixture of 43 (1 equiv), alkyne (3–3.5 equiv), and DIPA (2–2.5 equiv) in propanol (0.2–0.3 M) was degassed, followed by addition of PdCl2(PPh3)2 (0.1 equiv) and CuI (0.1 equiv). The reaction mixture was heated to 100 °C and stirred overnight. After completion of the reaction, the mixture was cooled to r.t., diluted with EtOAc, and passed through a thin pad of celite. The filtrate was concentrated in vacuo and the crude residue was purified by preparative HPLC (10–100% MeOH in H2O + 0.1% TFA) to give the title compounds.
N-(4-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-2-methylbut-3-yn-2-yl)acetamide (18)
The reaction was carried out according to general procedure D using 43 (264 mg, 0.7 mmol), N-(2-methylbut-3-yn-2-yl)acetamide (302 mg, 2.4 mmol), DIPA (163 mg, 1.61 mmol), Pd(PPh3)2Cl2 (56 mg, 80 μmol), and CuI (13 mg, 80 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (24 mg, 8.0% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.59 (s, 1H), 8.73 (d, J = 1.6 Hz, 1H), 7.97 (s, 1H), 7.92 (s, 1H), 7.25 (d, J = 8.3 Hz, 1H), 7.09 (dd, J = 8.2, 1.6 Hz, 1H), 6.21 (s, 2H), 3.15 (t, J = 6.6 Hz, 2H), 2.64–2.59 (m, 2H), 1.95–1.90 (m, 2H), 1.83 (s, 3H), 1.62 (s, 6H). 13C NMR (214 MHz, CD3OD-d4) δ 172.49, 166.32, 160.72, 147.68, 137.50, 135.99, 128.84, 128.00, 127.48, 116.73, 112.07, 111.40, 91.03, 83.73, 31.29, 30.93, 29.79, 25.12, 23.99, 23.40. HPLC purity: 97.62%. HRMS (ESI): m/z calculated for C22H24N5O [M + H]+: 374.1981. Found: 374.1975.
N-(4-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-2-methylbut-3-yn-2-yl)methanesulfonamide (19)
The reaction was carried out according to general procedure D using 43 (241 mg, 0.73 mmol), N-(2-methylbut-3-yn-2-yl)methanesulfonamide (354 mg, 2.2 mmol), DIPA (148 mg, 1.47 mmol), Pd(PPh3)2Cl2 (51 mg, 70 μmol), and CuI (12 mg, 70 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (36 mg, 12% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.77 (d, J = 1.6 Hz, 1H), 8.16 (s, 1H), 7.90 (d, J = 7.0 Hz, 1H), 7.45 (s, 1H), 7.29 (d, J = 8.2 Hz, 1H), 7.14 (dd, J = 8.2, 1.7 Hz, 1H), 6.18 (s, 2H), 3.18 (td, J = 6.6, 3.5 Hz, 2H), 3.15 (s, 3H), 2.62–2.59 (m, 2H), 1.94 (dt, J = 11.1, 6.1 Hz, 2H), 1.62 (s, 6H). 13C NMR (151 MHz, DMSO-d6) δ 162.05, 161.00, 156.85, 156.78, 144.30, 135.75, 127.34, 126.61, 124.85, 120.33, 110.64, 110.43, 89.68, 84.55, 49.41, 41.99, 40.43, 31.18, 24.05. HPLC purity: 95.31%. HRMS (ESI): m/z calculated for C21H24N5O2S [M + H]+: 410.1651. Found: 410.1645.
11-(3-Methoxy-3-methylbut-1-yn-1-yl)-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-2-amine (20)
The reaction was carried out according to general procedure E using 43 (285 mg, 0.87 mmol), 3-methoxy-3-methylbut-1-yne (255 mg, 2.6 mmol), DIPA (175 mg, 1.73 mmol), PdCl2(PPh3)2 (61 mg, 80 μmol), and CuI (13 mg, 80 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (45 mg, 15% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.76 (s, 1H), 7.26 (d, J = 8.2 Hz, 1H), 7.14 (d, J = 8.3 Hz, 1H), 6.31 (s, 2H), 3.34 (s, 3H), 3.15 (t, J = 6.5 Hz, 2H), 2.63 (d, J = 8.1 Hz, 2H), 1.98–1.90 (m, 2H), 1.50 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 161.09, 144.22, 135.70, 127.24, 126.68, 125.39, 113.21, 110.53, 88.48, 86.04, 70.44, 50.97, 29.75, 29.52, 28.42, 24.13. HPLC purity: 98.7%. HRMS (ESI): m/z calculated for C21H23N4O [M + H]+: 347.1872. Found: 347.1863.
4-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)but-3-yn-2-ol (21)
The reaction was carried out according to general procedure E using 43 (310 mg, 0.94 mmol), but-3-yn-2-ol (198 mg, 2.82 mmol), DIPA (168 mg, 1.67 mmol), PdCl2(PPh3)2 (66 mg, 90 μmol), and CuI (20 mg, 90 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (8.2 mg, 2.7% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.77 (s, 1H), 7.88 (s, 1H), 7.25 (dd, J = 8.3, 0.7 Hz, 1H), 7.20 (dd, J = 8.3, 1.6 Hz, 1H), 4.71 (q, J = 6.6 Hz, 1H), 3.22 (t, J = 6.6 Hz, 2H), 2.73–2.70 (m, 2H), 2.07 (dt, J = 11.0, 6.6 Hz, 2H), 1.51 (d, J = 6.6 Hz, 3H). 13C NMR (214 MHz, CD3OD-d4) δ 164.34, 163.10, 156.62, 145.85, 137.57, 128.90, 127.80, 126.77, 123.01, 115.88, 111.94, 111.32, 89.55, 86.39, 59.30, 31.21, 30.70, 25.71, 25.00. HPLC purity: 100%. HRMS (ESI): m/z calculated for C19H19N4O [M + H]+: 319.1559. Found: 319.1553.
4-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)but-3-yn-1-ol (22)
The reaction was carried out according to general procedure E using 43 (310 mg, 0.94 mmol), but-3-yn-1-ol (198 mg, 2.8 mmol), DIPA (191 mg, 1.89 mmol), PdCl2(PPh3)2 (66 mg, 90 μmol), and CuI (15 mg, 90 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (7.2 mg, 2.4% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.74 (s, 1H), 7.87 (s, 1H), 7.22 (dd, J = 8.2, 0.7 Hz, 1H), 7.18 (dd, J = 8.2, 1.6 Hz, 1H), 3.75 (t, J = 6.9 Hz, 2H), 3.21 (t, J = 6.6 Hz, 2H), 2.73–2.69 (m, 2H), 2.63 (t, J = 6.8 Hz, 2H), 2.07 (dt, J = 10.9, 6.6 Hz, 2H). 13C NMR (214 MHz, CD3OD-d4) δ 164.20, 162.92, 156.40, 145.50, 137.09, 128.68, 127.48, 126.72, 122.76, 116.61, 111.69, 110.99, 84.35, 84.22, 61.87, 31.04, 30.51, 25.54, 24.20. HPLC purity: 100%. HRMS (ESI): m/z calculated for C19H19N4O [M + H]+: 319.1559. Found: 319.1553.
4-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-2,2-dimethylbut-3-yn-1-ol (23)
The reaction was carried out according to general procedure E using 43 (285 mg, 0.87 mmol), 2-methylpent-4-yn-2-ol (255 mg, 2.6 mmol), DIPA (175 mg, 1.73 mmol), PdCl2(PPh3)2 (61 mg, 90 μmol), and CuI (15 mg, 90 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (12.6 mg, 4.0% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.71 (s, 1H), 7.84 (s, 1H), 7.22 (dd, J = 8.2, 0.7 Hz, 1H), 7.19 (dd, J = 8.3, 1.6 Hz, 1H), 3.51 (s, 2H), 3.21 (t, J = 6.5 Hz, 2H), 2.74–2.69 (m, 2H), 2.09–2.04 (m, 2H), 1.30 (s, 6H). 13C NMR (214 MHz, CD3OD-d4) δ 165.55, 161.71, 153.74, 146.75, 137.26, 128.81, 127.50, 127.36, 122.93, 117.19, 111.95, 111.29, 93.36, 83.91, 72.09, 40.44, 35.17, 31.23, 30.76, 26.23, 25.47, 25.35. HPLC purity: 98.28%. HRMS (ESI): m/z calculated for C21H23N4O [M + H]+: 347.1872. Found: 347.1868.
5-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-2-methylpent-4-yn-2-ol (24)
The reaction was carried out according to general procedure E using 43 (285 mg, 0.87 mmol), 2-methylpent-4-yn-2-ol (255 mg, 2.6 mmol), DIPA (175 mg, 1.73 mmol), PdCl2(PPh3)2 (61 mg, 90 μmol), and CuI (15 mg, 90 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (19 mg, 6.3% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.72 (s, 1H), 8.18 (s, 1H), 7.91 (s, 1H), 7.25 (d, J = 8.2 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 6.19 (s, 2H), 3.17 (t, J = 6.7 Hz, 2H), 2.64–2.61 (m, 2H), 1.99–1.93 (m, 2H), 1.27 (s, 6H). 13C NMR (214 MHz, CD3OD-d4) δ 166.19, 160.43, 151.36, 147.46, 137.11, 128.66, 127.56, 127.13, 122.70, 117.32, 111.76, 111.20, 84.95, 84.92, 71.13, 40.24, 35.38, 31.06, 30.71, 28.74, 24.95. HPLC purity: 95.86%. HRMS (ESI): m/z calculated for C21H23N4O [M + H]+: 347.1872. Found: 347.1865.
1-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-3-methylpent-1-yn-3-ol (25)
The reaction was carried out according to general procedure E using 43 (143 mg, 0.43 mmol), 3-methylpent-1-yn-3-ol (127 mg, 1.3 mmol), DIPA (88 mg, 0.87 mmol), PdCl2(PPh3)2 (30 mg, 40 μmol), and CuI (7.0 mg, 40 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (14.6 mg, 9.7% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.69 (d, J = 1.6 Hz, 1H), 8.27 (s, 1H), 7.92 (s, 1H), 7.25 (d, J = 8.3 Hz, 1H), 7.10 (dd, J = 8.3, 1.6 Hz, 1H), 6.17 (s, 2H), 3.15 (t, J = 6.6 Hz, 2H), 2.61 (d, J = 5.0 Hz, 2H), 1.94 (dt, J = 11.3, 6.3 Hz, 2H), 1.72–1.59 (m, J = 7.0, 6.5 Hz, 2H), 1.45 (s, 3H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (214 MHz, CD3OD-d4) δ 166.22, 160.87, 152.09, 147.58, 137.55, 128.85, 127.70, 127.24, 123.00, 116.59, 112.00, 111.51, 91.44, 85.91, 69.78, 37.87, 31.19, 30.84, 29.69, 9.57. HPLC purity: 100%. HRMS (ESI): m/z calculated for C21H23N4O [M + H]+: 347.1872. Found: 347.1860.
1-(2-Amino-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-11-yl)-3-ethylpent-1-yn-3-ol (26)
The reaction was carried out according to general procedure E using 43 (274 mg, 0.83 mmol), 3-ethylpent-1-yn-3-ol (280 mg, 2.5 mmol), DIPA (168 mg, 1.67 mmol), PdCl2(PPh3)2 (58 mg, 80 μmol), and CuI (13 mg, 80 μmol) in propanol (3.0 mL) at 100 °C overnight to afford the title compound (46.2 mg, 15% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.71 (s, 1H), 7.25 (d, J = 8.1 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 6.21 (s, 2H), 3.15 (t, J = 6.6 Hz, 2H), 2.62 (d, J = 6.7 Hz, 2H), 2.00–1.92 (s, 2H), 1.64 (qd, J = 13.7, 6.8 Hz, 4H), 1.01 (t, J = 7.4 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 161.23, 144.13, 135.50, 127.22, 126.36, 125.32, 113.99, 110.49, 90.86, 84.88, 34.18, 29.66, 29.40, 24.26, 8.80. HPLC purity: 100%. HRMS (ESI): m/z calculated for C22H25N4O [M + H]+: 361.2028. Found: 361.2026.
General Procedure for the Synthesis of Compounds 7–9, 12, and 13: Procedure F
A mixture of 42–44 (1 equiv), boronic acid (1.2–1.6 equiv), and K2CO3 (2 equiv) in a mixture of dioxane (0.2–0.3 M) and water (1.0–2.0 M) was degassed, followed by addition of Pd(PPh3)4 (0.05 equiv). The reaction mixture was heated to 95 °C and allowed to stir overnight. After completion of the reaction, the mixture was cooled to r.t. and diluted with EtOAc and water. The organic layer was washed with water and concentrated in vacuo. The crude residue was purified via preparative HPLC (10–100% MeOH in H2O + 0.1% TFA) to afford the title compounds.
10-(4-Fluorophenyl)-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (7)
The reaction was carried out according to general procedure F using 42 (95 mg, 0.30 mmol), 4-fluorophenylboronic acid (64 mg, 0.47 mmol), K2CO3 (64 mg, 0.63 mmol), and Pd(PPh3)4 (18 mg, 15 μmol) in a mixture of dioxane/water (2.0 mL/0.3 mL) at 95 °C overnight to afford the title compound (29 mg, 29% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.50 (s, 1H), 7.91 (s, 1H), 7.71 (dd, J = 8.4, 5.4 Hz, 2H), 7.45 (d, J = 8.4 Hz, 1H), 7.39 (dd, J = 8.3, 1.9 Hz, 1H), 7.27 (t, J = 8.7 Hz, 2H), 6.18 (s, 2H), 2.99 (t, J = 7.7 Hz, 2H), 2.85 (t, J = 7.8 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.64, 160.32, 160.11, 153.53, 145.18, 138.20, 135.94, 131.90, 128.66, 128.59, 125.14, 120.69, 118.76, 115.49, 115.32, 113.00, 111.78, 109.12, 23.62, 21.69. HPLC purity: 100%. HRMS (ESI): m/z calculated for C20H16FN4 [M + H]+: 331.1359. Found: 331.1352.
11-(4-Fluorophenyl)-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-2-amine (8)
The reaction was carried out according to general procedure F using 43 (192 mg, 0.58 mmol), 4-fluorophenylboronic acid (122 mg, 0.87 mmol), K2CO3 (161 mg, 1.16 mmol), and Pd(PPh3)4 (34 mg, 29 μmol) in a mixture of dioxane/water (2.0 mL/0.3 mL) at 95 °C overnight to afford the title compound (22.7 mg, 44% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.94 (d, J = 1.8 Hz, 1H), 7.92 (s, 1H), 7.75 (dd, J = 8.5, 5.6 Hz, 2H), 7.41–7.33 (m, 2H), 7.22 (t, J = 8.8 Hz, 2H), 6.22 (s, 2H), 3.18 (t, J = 6.6 Hz, 2H), 2.65–2.60 (m, 2H), 1.96 (dt, J = 11.8, 6.4 Hz, 2H). 13C NMR (214 MHz, CD3OD-d4) δ 164.68, 163.89, 163.12, 162.76, 156.52, 145.59, 140.67, 140.65, 137.43, 134.15, 129.98, 129.94, 129.51, 122.70, 122.18, 116.23, 116.13, 112.19, 111.61, 31.13, 30.60, 26.04. HPLC purity: 100%. HRMS (ESI): m/z calculated for C21H18FN4 [M + H]+: 345.1515. Found: 345.1511.
12-(4-Fluorophenyl)-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (9)
The reaction was carried out according to general procedure F using 44 (144 mg, 0.42 mmol), 4-fluorophenylboronic acid (70 mg, 0.50 mmol), K2CO3 (116 mg, 0.84 mmol), and Pd(PPh3)4 (24 mg, 21 μmol) in a mixture of dioxane/water (2.0 mL/0.3 mL) at 95 °C overnight to afford the title compound (48 mg, 32% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.06 (d, J = 16.4 Hz, 2H), 7.64 (dd, J = 8.7, 5.6 Hz, 2H), 7.32 (q, J = 8.4 Hz, 2H), 7.21 (t, J = 8.8 Hz, 2H), 6.30 (s, 2H), 3.02–2.96 (m, 2H), 2.54–2.49 (m, 2H), 1.74 (s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 162.32, 162.23, 162.11, 160.18, 158.08, 141.28, 138.67, 138.64, 135.10, 130.78, 128.53, 128.46, 128.43, 120.42, 118.59, 115.45, 115.29, 110.67, 109.58, 30.93, 27.66, 26.77. HPLC purity: 100%. HRMS (ESI): m/z calculated for C22H20FN4 [M + H]+: 359.1672. Found: 359.1668.
10-(1-Methyl-1H-pyrazol-4-yl)-6,7-dihydro-5H-pyrimido[5,4-c]carbazol-2-amine (12)
The reaction was carried out according to general procedure F using 42 (100 mg, 0.32 mmol), (1-methyl-1H-pyrazol-4-yl)boronic acid (99 mg, 0.47 mmol), K2CO3 (64 mg, 0.63 mmol), and Pd(PPh3)4 (18 mg, 15 μmol) in a mixture of dioxane/water (2.0 mL/0.3 mL) at 95 °C overnight to afford the title compound (36 mg, 36% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.56 (s, 1H), 8.45 (s, 1H), 8.07 (s, 1H), 7.89 (d, J = 2.3 Hz, 2H), 7.37–7.28 (m, 2H), 6.21 (s, 2H), 3.88 (s, 3H), 2.97 (t, J = 7.7 Hz, 2H), 2.84 (t, J = 7.8 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.64, 160.09, 153.49, 144.56, 135.79, 135.15, 127.01, 125.12, 125.00, 123.28, 119.33, 116.96, 112.88, 111.62, 108.81, 38.58, 23.59, 21.66. HPLC purity: 100%. HRMS (ESI): m/z calculated for C18H17N6 [M + H]+: 317.1515. Found: 317.1522.
11-(1-Methyl-1H-pyrazol-4-yl)-5,6,7,8-tetrahydropyrimido[4′,5′:3,4]cyclohepta[1,2-b]indol-2-amine (13)
The reaction was carried out according to general procedure F using 43 (199 mg, 0.60 mmol), (1-methyl-1H-pyrazol-4-yl)boronic acid (189 mg, 0.90 mmol), K2CO3 (167 mg, 1.21 mmol), and Pd(PPh3)4 (35 mg, 30 μmol) in a mixture of dioxane/water (2.0 mL/0.3 mL) at 95 °C overnight to afford the title compound (29 mg, 15% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.42 (s, 1H), 8.88 (s, 1H), 8.15 (s, 1H), 7.91 (d, J = 11.0 Hz, 2H), 7.31 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 8.3 Hz, 1H), 6.29 (s, 2H), 3.87 (s, 3H), 3.16 (t, J = 6.5 Hz, 2H), 2.65–2.59 (m, 2H), 1.94 (dt, J = 11.8, 6.5 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.04, 161.38, 156.59, 143.50, 135.91, 134.77, 128.05, 127.14, 124.45, 123.60, 119.88, 119.53, 119.40, 110.42, 110.40, 38.53, 30.05, 29.72, 23.96. HPLC purity: 100%. HRMS (ESI): m/z calculated for C19H19N6 [M + H]+: 331.1671. Found: 331.1661.
12-Cyclopropyl-6,7,8,9-tetrahydro-5H-pyrimido[4′,5′:3,4]cycloocta[1,2-b]indol-2-amine (30)
A mixture of 44 (226 mg, 0.66 mmol), cyclopropylboronic acid (395 mg, 4.6 mmol), and K3PO4 (697 mg, 3.3 mmol) in dioxane (10 mL) was degassed, followed by the addition of Pd(dppf)Cl2 (27 mg, 33 μmol). The reaction mixture was heated to 80 °C and allowed to stir overnight. After completion of the reaction, the mixture was cooled to r.t. and diluted with EtOAc and water. The organic layer was washed with water and concentrated in vacuo. The crude residue was purified via preparative HPLC to afford the title compound (7.9 mg, 4.0% yield). 1H NMR (850 MHz, CD3OD-d4) δ 8.08 (s, 1H), 7.56 (d, J = 1.7 Hz, 1H), 7.18 (dd, J = 8.3, 0.6 Hz, 1H), 6.87 (dd, J = 8.3, 1.7 Hz, 1H), 3.04–2.99 (m, 2H), 2.63 (s, 2H), 1.99–1.93 (m, 1H), 1.86–1.81 (m, 4H), 0.90–0.85 (m, 2H), 0.66–0.63 (m, 2H). 13C NMR (214 MHz, CD3OD-d4) δ 165.96, 162.18, 157.95, 142.89, 136.61, 136.04, 129.23, 122.97, 121.39, 117.73, 111.21, 109.69, 32.33, 29.00, 28.29, 22.52, 16.47, 8.95. HPLC purity: 100%. HRMS (ESI): m/z calculated for C19H21N4 [M + H]+: 305.1766. Found: 305.1757.
Biological Evaluation: Kinome Screening
The Eurofins DiscoverX Corporation scanMAX assay platform was employed to assess the selectivity of each final compound at a single concentration of 1 μM. The scanMAX assay platform profiles a compound against 403 wild-type (WT) human kinases and produces percent of control (PoC) values.48 A selectivity score can be calculated using the PoC values. Selectivity scores (S10(1 μM)) are included in Tables 1–4 as well as the number of WT human kinases in the scanMAX panel with PoC < 10. Kinome tree diagrams as well as tables of potently inhibited kinases in this screen for analogues 8, 17, and 30 are included in Figure S1. Kd determination using the DiscoverX PIP4K2C assay was carried out for analogue 17 and included in Table S3.
Enzymatic Assays
SignalChem developed an ADP-Glo assay (Promega) to evaluate the enzymatic inhibition of PIKFYVE by several compounds in Tables 1–4, S1, and S3. The assay was optimized to give a high signal-to-noise ratio, which involved using 25 ng/reaction of PIKFYVE protein prepared by SignalChem (Catalog No. P17-31G), 0.1 μg/μL PI(3)P:PS as the substrate, and 25 μM ATP, as suggested by the manufacturer’s protocol. This concentration of ATP is well above the Km value for ATP for PIKfyve, published as 9.9 μM.49 The assay was run with a 60-min incubation of the components at r.t. Executing the assay in dose–response (10-pt curve) in duplicate allowed for the calculation of IC50 values.
Eurofins enzymatic radiometric assays were run at the Km value for ATP using a single concentration (1 μM) in duplicate to produce PoC values for each kinase listed in Table S2. The same assay format, also executed at the Km value for ATP, was used to generate dose–response (9-pt) curves for several kinases listed in Tables S1 and S3. The substrate used, protein constructs, controls, and assay protocol for these enzymatic radiometric kinase assays can be found on the Eurofins website: https://www.eurofinsdiscoveryservices.com.
Several Reaction Biology Corp. (RBC) radiometric HotSpot kinase assays were carried out at the Km value for ATP in dose–response (10-pt curve), specifically for MYLK4, RIPK5, and YSK4. The corresponding IC50 values are included in Tables S1 and S3. Details related to the substrate used, protein constructs, controls, and assay protocol for these HotSpot kinase assays can be accessed via the RBC website: https://www.reactionbiology.com/list-kinase-targets-us-facility.
NanoBRET Assays
Human embryonic kidney (HEK293) cells (hypotriploid, female, fetal) obtained from ATCC were cultured in Dulbecco’s Modified Eagle’s medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Corning). These cells were incubated in 5% CO2 at 37 °C and passaged every 72 h with trypsin (Gibco), ensuring that they never reached confluency.
Constructs for NanoBRET measurements of PIKfyve (PIKfyve-NLuc), PI5P4Kγ (PI5P4Kγ-NLuc), MAP4K5 (MAP4K5-NLuc), and MYLK4 (MYLK4-NLuc) included in Tables 1–5, S1, and S3 were kindly provided by Promega. The NLuc orientations used in the respective assays are indicated in parentheses after each construct. The NanoBRET assays were executed in dose–response (12-pt curves) as described previously.50,51 Assays were carried out as described by the manufacturer using 0.13 μM tracer K8 for PIKfyve, 0.063 μM tracer K8 for PI5P4Kγ, 1 μM tracer K10 for MAP4K5, and 0.13 μM tracer K10 for MYLK4. Representative curves generated for PIKfyve are included in Figure S1 and for MYLK and MAP4K5 versus compound 17 in Figure S2. The NanoBRET data for TYK2(JH2domain-pseudokinase) included in Table S1 was generated by RBC.
Kinetic Solubility
Analiza, Inc. analyzed the kinetic solubility of a 10 mM DMSO stock solution of compound 17 dissolved in phosphate buffered saline (PBS) solution at pH 7.4 as described previously.17 The reported solubility value has been corrected for background nitrogen present in the media and DMSO.
Microsomal Stability Analysis
Analiza, Inc. analyzed the microsomal stability using a 10 mM DMSO stock solution of compound 17, which was diluted upon arrival to 0.5 mM with DMSO and again to 0.1 mM with acetonitrile (ACN). The resulting solution contained 0.1 mM of compound 17 in 20% DMSO/80% ACN. Microsomes were isolated from male (CD-1) mice and supplied at a 20 mg/mL protein concentration. For the assay, the reaction plate was prepared by treating a prewarmed (37 °C) microsomal solution (0.63 mg/mL protein in 100 mM KPO4 with 1.3 mM ethylenediamine tetraacetic acid (EDTA)) with 0.1 mM compound or reference standard and mixing thoroughly by repeated pipetting. The resulting solution was preincubated for 5 min at 37 °C before preparing the T = 0 plates. The T = 0 plate was prepared to include an aliquot of the reaction plate, diluted with cold (4 °C) MeOH, and mixed thoroughly by repeated pipetting and treated with nicotinamide adenine dinucleotide phosphate (NADPH) regeneration solution with mixing via repeated pipetting. The T = 30 incubation plate was prepared by adding NADPH to the reaction plate, sealing, and incubating at 37 °C for 30 min and then transferring an aliquot to a fresh plate and quenching the reaction with cold (4 °C) MeOH. Prior to analysis, all plates were sealed, vortexed, and centrifuged at 3000 RPM at 4 °C for 15 min, and the supernatants analyzed by liquid chromatography time-of-flight mass spectrometry (LC-TOFMS) (AQUASIL C18 column, eluting with a fast generic gradient program). TOFMS data was acquired using Agilent 6538 Ultra High Accuracy TOFMS in extended dynamic range (m/z 100–1000) using generic MS conditions in positive mode. Following data acquisition, exact mass extraction and peak integration was performed using MassHunter Software (Agilent Technologies). Compound stability was calculated as the percent remaining of the unchanged parent compound at 30 min (T = 30) relative to the peak area at T = 0. On-board reference standards were within the acceptable range for the assay and correlated to reported values, ensuring the quality of the assay.
MHV-nLuc Assay
Murine astrocytoma delayed brain-tumor (DBT) cells were cultured at 37 °C in DMEM (Sigma) supplemented with 10% FBS (Gibco) with penicillin and streptomycin (Sigma) added.
The MHV-nLuc virus was engineered as described previously.25 Next, MHV-nLuc virus stocks were grown on DBT cells and the 50% tissue culture infectious dose (TCID50) assay employed to determine their titers. To run the MHV-nLuc assay, DBT cells were plated in 96-well plates at 80% confluency. PIKfyve inhibitors were diluted in DMEM, preparing 4-fold serial dilutions over a concentration range of 15–0.22 μM. The assay protocol was executed as previously described.25 An LDH assay was run in parallel to assess the viability (Figure S3).25 Three biological replicates each with three technical replicates were analyzed using GraphPad Prism to generate the IC50 values included in Table 5 and graph in Figure 4A. These were then converted to pIC50 values for Figure 4. Individual replicates for Figure 4A are included in Figure S4.
SARS-CoV-2-nLuc Assay
Human epithelial A549-ACE2 cells were cultured at 37 °C with 5% CO2 in DMEM supplemented with 10% heat-inactivated FBS, nonessential amino acids, penicillin, and streptomycin. The ic-SARS-CoV-2-NLuc virus was engineered and prepared as described previously.27 PIKfyve inhibitors and remdesivir (positive control) were diluted in DMEM, preparing 4-fold serial dilutions over a concentration range from 10 to 0.0006 μM. The assay protocol was executed in dose–response (8-point curve) as previously described (n = 4, with two technical replicates per assay).27 Briefly, 1 day prior to infection, A549-ACE2 cells were seeded at 20 000 cells per well in 96-well solid black plates. On the day of infection, cells were pretreated with the compound for 1 h prior to infection. Next, ic-SARS-CoV-2-NLuc virus stocks were used to infect A549-ACE2 cells (85–95% confluent) at MOI = 0.02 for 2 h at 37 °C with 5% CO2 with compound treatment maintained during the infection period. After 2 h, the supernatant was removed, infected monolayers rinsed with PBS, and media-containing compound added to each well. At 46 h post-infection, Nano-Glo reagent (Promega) was added to each well according to the manufacturer’s protocol (Promega) and relative light units (RLUs) were measured using a Promega GloMax plate reader. An LDH assay to assess viability (n = 2, with two technical replicates per assay) was run in parallel using 20 000 uninfected A549-ACE2 cells per well in 96-well clear plates.25 After 46 of treatment with compounds in dose–response, an aliquot of the supernatant from each well was taken, centrifuged to remove cell and debris, and transferred to a clean plate. The clarified supernatant was stored frozen until analysis using the Sigma Tox7 kit according to the manufacturer’s protocol. Data was analyzed using GraphPad Prism to generate the graphs and IC50 values included in Figures 4 and S5 and Table 5, respectively.
Spike Uptake Assay in HEK293T-ACE2 Cells
The protocol employed to measure and quantify the uptake of spike protein was previously described.25,37 Briefly, HEK293T-ACE2 cells were seeded onto poly-l-lysine-treated coverslips and allowed to adhere for 24 h at 37 °C. Next, 1 h prior to the addition of spike protein, cell media was changed to starvation media (lacking serum) supplemented with either 1 μM test compound or DMSO (vehicle control). After addition of His6-SARS-CoV-2 spike protein (5 μg per well) to each coverslip, cells were incubated at 0 °C (on ice) for 30 min. Cells were next washed with PBS and fresh starvation media supplemented with either the same test compound at 1 μM or DMSO, followed by a final incubation for 30 min at 37 °C. Prior to fixation, cells were washed for 60 s and then rinsed with acid to remove any extracellular spike protein. Next, cells were washed with PBS and fixed for 10 min with paraformaldehyde at 4 °C. Cells were then permeabilized in 0.2% Triton X-100 in PBS and blocked with 5% bovine serum albumin (BSA). His-tag antibody (HIS.H8) conjugated with Dylight 550 (ThermoFisher) was used to visualize and quantify spike protein uptake. Imaging of cells was executed using a Leica TCS SP8 confocal microscope. Quantification was carried out using Leica LAS X software, with statistical calculations and graphs produced using Prism GraphPad software. Quantification of fluorescence per cell was not necessary since HEK293T-ACE2 cells were at full confluency, resulting in approximately an equal number of cells per field. Statistics were carried out.
Spike Uptake Assay in Calu-3 and Caco-2 Cells
To measure and quantify the uptake of spike protein, the original protocol employed with HEK293T-ACE2 cells was modified.25,37 Calu-3 and Caco-2 were seeded as single cells on poly-d-lysine-treated coverslips and allowed to adhere for 36 h at 37 °C. Seeding as single cells allowed for reduced clustering and cyst formation of Caco-2 cells and allowed for clear separation between Calu-3 cells. Cells were grown in ATCC-formulated Eagle’s minimum essential medium (Catalog No. 30-2003) supplemented with 20% FBS. Next, 2 h prior to the addition of His6-SARS-CoV-2 spike protein, cell media was changed to starvation media (lacking serum) supplemented with either 1 μM test compound or DMSO (vehicle control). Following addition of spike protein (5 μg per well) to each coverslip, cells were incubated at 0 °C (on ice) for 30 min. Cells were next washed with PBS and fresh starvation media supplemented with either 1 μM test compound or DMSO, followed by a final 1 h incubation at 37 °C. Prior to fixation, cells were rinsed for 90 s with acid wash solution (pH = 3.0) to wash away any residual extracellular spike protein. Next, cells were washed with PBS and fixed for 10 min with paraformaldehyde at 4 °C. Cells were then permeabilized in a solution of 0.2% Triton X-100 in PBS and blocked with 5% BSA. A combination of His-tag primary antibody (HIS.H8, ThermoFisher) with Alexa Fluor 488 secondary antibody (ThermoFisher) was used to visualize and quantify spike protein uptake. Cells were imaged using a Leica TCS SP8 confocal microscope and quantification was carried out using Leica LAS X software. Statistical calculations and graphs were produced using Prism GraphPad software. For these studies, the number of cells was factored into the quantification of fluorescence: fluorescence intensity was divided by the number of nuclei in each image. This method accounted for the variable number of cells.
Lentivirus Uptake Assay
Calu-3 cells were seeded at ∼100 000 cells per well (24-well plate) on poly-l-lysine-treated coverslips. At 96 h after seeding, cells were incubated with 1 μM of each compound (or DMSO) for 1 h. Subsequently, pseudovirus SARS-CoV-2 (Catalog No. 78010, BPS Biosciences Inc., Figure 7) or bald GFP lentivirus (Catalog no. 79987, BPS Biosciences Inc., Figure S6) was added to cells for 12 h. After this incubation, virus-containing media was replaced with fresh DMEM media and incubated further for 48 h. Cells were fixed with 4% paraformaldehyde for 20 min at 4 °C and stained with Hoechst. Cells treated with the pseudovirus were then permeabilized, blocked, and stained using Spike antibody (GTX632604) and conjugated with Alexa Fluor 488 (ThermoFisher). Cells were imaged using a Leica TCS SP8 confocal microscope. To quantify the results, nuclei and GFP/Alexa Fluor fluorescing cells were counted using the “Find Maxima” function in ImageJ. GFP/Alexa Fluor counts were then divided by nuclei count for every image. The percentage of infected cells for each image was calculated and analyzed using one-way ANOVA and post-hoc Tukey test.
Lysosomal Staining Protocol
Human neuroblastoma (SH-SY5Y) cells obtained from ATCC were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F12 medium (DMEM/F12, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Corning). SH-SY5Y cells were incubated in 5% CO2 at 37 °C and passaged every 48 h with trypsin (Gibco).
SH-SY5Y cells were plated in 35 mm glass bottom dishes and treated for 24 h with either 0.05% DMSO (control) or 5 μM of the indicated compound. After 24 h, 50 nM LysoTracker Red DND-99 (ThermoFisher) was added to each dish. The dishes were incubated for 30 min at 37 °C. Live cells were imaged at 40× magnification. Quantification of fluorescence was performed using ImageJ. Mean intensity of the LysoTracker fluorescence was calculated from five regions per image. Mean intensity was normalized to cell number within each region. Bar graph (Figure S7) represents the average LysoTracker fluorescence (n = 2). Error bars represent the standard deviation.
Acknowledgments
Constructs for NanoBRET measurements of PIKfyve and PI5P4Kγ were kindly provided by Promega. The authors thank SignalChem Biotech Inc. for helping to enable and optimize an enzymatic assay for profiling our compounds against PIKfyve. The TREEspot kinase interaction mapping software was employed in preparation of the kinome trees in Figure S1: http://treespot.discoverx.com. The table of contents graphic was created with Biorender.com. The authors also thank ChemSpace for synthetic support. The authors acknowledge the Department of Chemistry Mass Spectrometry Core Laboratory at the University of North Carolina for their assistance with mass spectrometry experiments. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Genentech, Innovative Medicines Initiative (EU/EFPIA), Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome. Research reported in this publication was supported in part by the NC Biotechnology Center Institutional Support Grant 2018-IDG-1030, NIH U24DK116204. This project was supported by the Rapidly Emerging Antiviral Drug Development Initiative at the University of North Carolina at Chapel Hill with funding from the North Carolina Coronavirus State and Local Fiscal Recovery Funds program, appropriated by the North Carolina General Assembly.
Glossary
Abbreviations Used
- ACN
acetonitrile
- ACE2
angiotensin-converting enzyme 2
- AcOH
acetic acid
- BSA
bovine serum albumin
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DIPA
N,N-diisopropylamine
- DMA
N,N-dimethylacetamide
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- HPLC
high-performance liquid chromatography
- IC50
half-maximal inhibitory concentration
- IPA
isopropylamine
- Km
Michaelis constant
- LC-MS
liquid chromatography-mass spectrometry
- MTBE
methyl tert-butyl ether
- NanoBRET
bioluminescence resonance energy transfer using NanoLuciferase
- NaOMe
sodium methoxide
- nLuc
nanoLuciferase
- NMR
nuclear magnetic resonance
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- SAR
structure–activity relationships
- v/v
volume for volume
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00697.
Molecular formula strings (CSV)
Comprehensive profiling of analogues 8 and 17, an additional figure that details kinome-wide selectivity of probe candidates 8 and 17 as well as negative control 30, cell viability data in DBT cells, SARS-CoV-2 replication versus viability graphs, purity chromatograms, and NMR spectra (PDF)
Author Contributions
○ D.H.D. and F.M.P. contributed equally to this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Shisheva A. PIKfyve: Partners, significance, debates and paradoxes. Cell Biol. Int. 2008, 32, 591–604. 10.1016/j.cellbi.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayle S.; Landrette S.; Beeharry N.; Conrad C.; Hernandez M.; Beckett P.; Ferguson S. M.; Mandelkern T.; Zheng M.; Xu T.; Rothberg J.; Lichenstein H. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 2017, 129, 1768–1778. 10.1182/blood-2016-09-736892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.; Xu Y.; Cheung A. K.; Tomlinson R. C.; Alcázar-Román A.; Murphy L.; Billich A.; Zhang B.; Feng Y.; Klumpp M.; Rondeau J.-M.; Fazal A. N.; Wilson C. J.; Myer V.; Joberty G.; Bouwmeester T.; Labow M. A.; Finan P. M.; Porter J. A.; Ploegh H. L.; Baird D.; De Camilli P.; Tallarico J. A.; Huang Q. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem. Biol. 2013, 20, 912–921. 10.1016/j.chembiol.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz S.; Boumans M. J.; Gerlag D. M.; Lufkin J.; van Kuijk A. W.; Bakker A.; de Boer M.; Lodde B. M.; Reedquist K. A.; Jacobson E. W.; O’Meara M.; Tak P. P. Brief report: A phase IIa, randomized, double-blind, placebo-controlled trial of apilimod mesylate, an interleukin-12/interleukin-23 inhibitor, in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1750–1755. 10.1002/art.34339. [DOI] [PubMed] [Google Scholar]
- Sands B. E.; Jacobson E. W.; Sylwestrowicz T.; Younes Z.; Dryden G.; Fedorak R.; Greenbloom S. Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn’s disease. Inflammatory Bowel Dis. 2010, 16, 1209–1218. 10.1002/ibd.21159. [DOI] [PubMed] [Google Scholar]
- Baranov M. V.; Bianchi F.; van den Bogaart G. The PIKfyve inhibitor apilimod: A double-edged sword against COVID-19. Cells 2021, 10, 30. 10.3390/cells10010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.-L.; Chou Y.-y.; Rothlauf P. W.; Liu Z.; Soh T. K.; Cureton D.; Case J. B.; Chen R. E.; Diamond M. S.; Whelan S. P. J.; Kirchhausen T. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 20803–20813. 10.1073/pnas.2007837117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov O. C.; Sbrissa D.; Shisheva A. YM201636, an inhibitor of retroviral budding and PIKfyve-catalyzed PtdIns(3,5)P2 synthesis, halts glucose entry by insulin in adipocytes. Biochem. Biophys. Res. Commun. 2009, 382, 566–570. 10.1016/j.bbrc.2009.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J.-Z.; Xi Z.-Q.; Niu J.; Li W.; Wang X.; Liang C.; Sun H.; Fang D.; Xie S.-Q. Inhibition of PIKfyve using YM201636 suppresses the growth of liver cancer via the induction of autophagy. Oncol. Rep. 2019, 41, 1971–1979. 10.3892/or.2018.6928. [DOI] [PubMed] [Google Scholar]
- Martin S.; Harper C. B.; May L. M.; Coulson E. J.; Meunier F. A.; Osborne S. L. Inhibition of PIKfyve by YM-201636 dysregulates autophagy and leads to apoptosis-independent neuronal cell death. PLoS One 2013, 8, e60152 10.1371/journal.pone.0060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies H. B. J.; Cooke F. T.; Jat P.; Boucheron C.; Koizumi T.; Hayakawa M.; Kaizawa H.; Ohishi T.; Workman P.; Waterfield M. D.; Parker P. J. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008, 9, 164–170. 10.1038/sj.embor.7401155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares A. C.; Ferreira A.; Mariën J.; Delay C.; Lee E.; Trojanowski J. Q.; Moechars D.; Annaert W.; De Muynck L. PIKfyve activity is required for lysosomal trafficking of tau aggregates and tau seeding. J. Biol. Chem. 2021, 296, 100636 10.1016/j.jbc.2021.100636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Campos C. B.; Zhu Y. X.; Sepetov N.; Romanov S.; Bruins L. A.; Shi C. X.; Stein C. K.; Petit J. L.; Polito A. N.; Sharik M. E.; Meermeier E. W.; Ahmann G. J.; Armenta I. D. L.; Kruse J.; Bergsagel P. L.; Chesi M.; Meurice N.; Braggio E.; Stewart A. K. Identification of PIKfyve kinase as a target in multiple myeloma. Haematologica 2020, 105, 1641–1649. 10.3324/haematol.2019.222729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa N.; Noguchi M.; Takeshita S.; Eviryanti A.; Seki Y.; Nishio H.; Yokoyama R.; Noguchi M.; Shuto M.; Shima Y.; Kuribayashi K.; Kageyama S.; Eda H.; Suzuki M.; Hatta T.; Iemura S.-I.; Natsume T.; Tanabe I.; Nakagawa R.; Shiozaki M.; Sakurai K.; Shoji M.; Andou A.; Yamamoto T. Structure-activity relationship study, target identification, and pharmacological characterization of a small molecular IL-12/23 inhibitor, APY0201. Bioorg. Med. Chem. 2014, 22, 3021–3029. 10.1016/j.bmc.2014.03.036. [DOI] [PubMed] [Google Scholar]
- Andrs M.; Korabecny J.; Jun D.; Hodny Z.; Bartek J.; Kuca K. Phosphatidylinositol 3-kinase (PI3K) and phosphatidylinositol 3-kinase-related kinase (PIKK) inhibitors: Importance of the morpholine ring. J. Med. Chem. 2015, 58, 41–71. 10.1021/jm501026z. [DOI] [PubMed] [Google Scholar]
- Lees J. A.; Li P.; Kumar N.; Weisman L. S.; Reinisch K. M. Insights into lysosomal PI(3,5)P(2) homeostasis from a structural-biochemical analysis of the PIKfyve lipid kinase complex. Mol. Cell 2020, 80, 736–743.e734. 10.1016/j.molcel.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry D. H.; Annor-Gyamfi J. K.; Wells C. I.; Pickett J. E.; Dederer V.; Preuss F.; Mathea S.; Axtman A. D. Identification of pyrimidine-based lead compounds for understudied kinases implicated in driving neurodegeneration. J. Med. Chem. 2022, 65, 1313–1328. 10.1021/acs.jmedchem.1c00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eduful B. J.; O’Byrne S. N.; Temme L.; Asquith C. R. M.; Liang Y.; Picado A.; Pilotte J. R.; Hossain M. A.; Wells C. I.; Zuercher W. J.; Catta-Preta C. M. C.; Ramos P. Z.; Santiago A. D. S.; Couñago R. M.; Langendorf C. G.; Nay K.; Oakhill J. S.; Pulliam T. L.; Lin C.; Awad D.; Willson T. M.; Frigo D. E.; Scott J. W.; Drewry D. H. Hinge binder scaffold hopping identifies potent calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) Iinhibitor chemotypes. J. Med. Chem. 2021, 64, 10849–10877. 10.1021/acs.jmedchem.0c02274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Gupta M. Designing of kinase hinge binders: A medicinal chemistry perspective. Chem. Biol. Drug Des. 2022, 14024. 10.1111/cbdd.14024. [DOI] [PubMed] [Google Scholar]
- Jia C.-C.; Chen W.; Feng Z.-L.; Liu Z.-P. Recent developments of RET protein kinase inhibitors with diverse scaffolds as hinge binders. Future Med. Chem. 2021, 13, 45–62. 10.4155/fmc-2020-0170. [DOI] [PubMed] [Google Scholar]
- Halkina T.; Henderson J. L.; Lin E. Y.; Himmelbauer M. K.; Jones J. H.; Nevalainen M.; Feng J.; King K.; Rooney M.; Johnson J. L.; Marcotte D. J.; Chodaparambil J. V.; Kumar P. R.; Patterson T. A.; Murugan P.; Schuman E.; Wong L.; Hesson T.; Lamore S.; Bao C.; Calhoun M.; Certo H.; Amaral B.; Dillon G. M.; Gilfillan R.; de Turiso F. G. Discovery of potent and brain-penetrant tau tubulin kinase 1 (TTBK1) inhibitors that lower tau phosphorylation in vivo. J. Med. Chem. 2021, 64, 6358–6380. 10.1021/acs.jmedchem.1c00382. [DOI] [PubMed] [Google Scholar]
- Potjewyd F. M.; Marquez A. B.; Chaikuad A.; Howell S.; Dunn A. S.; Beltran A. A.; Smith J. L.; Drewry D. H.; Beltran A. S.; Axtman A. D.. Modulation of tau tubulin kinases (TTBK1 and TTBK2) impacts ciliogenesis. bioRxiv 2022, 2022.2005.2006.490937. DOI: 10.1101/2022.05.06.490937. [DOI] [PMC free article] [PubMed]
- Asquith C. R. M.; Berger B.-T.; Wan J.; Bennett J. M.; Capuzzi S. J.; Crona D. J.; Drewry D. H.; East M. P.; Elkins J. M.; Fedorov O.; Godoi P. H.; Hunter D. M.; Knapp S.; Müller S.; Torrice C. D.; Wells C. I.; Earp H. S.; Willson T. M.; Zuercher W. J. SGC-GAK-1: A chemical probe for cyclin G associated kinase (GAK). J. Med. Chem. 2019, 62, 2830–2836. 10.1021/acs.jmedchem.8b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon G. M.; Henderson J. L.; Bao C.; Joyce J. A.; Calhoun M.; Amaral B.; King K. W.; Bajrami B.; Rabah D. Acute inhibition of the CNS-specific kinase TTBK1 significantly lowers tau phosphorylation at several disease relevant sites. PLoS One 2020, 15, e0228771 10.1371/journal.pone.0228771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Dickmander R. J.; Bayati A.; Taft-Benz S. A.; Smith J. L.; Wells C. I.; Madden E. A.; Brown J. W.; Lenarcic E. M.; Yount B. L. Jr; Chang E.; Axtman A. D.; Baric R. S.; Heise M. T.; McPherson P. S.; Moorman N.; Willson T. M. Host kinase CSNK2 is a target for inhibition of pathogenic SARS-like β-coronaviruses. ACS Chem. Biol. 2022, 17, 1937–1950. 10.1021/acschembio.2c00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner R. W.; Majjouti M.; Alcazar M. A. A.; Mahabir E. Of mice and men: The coronavirus MHV and mouse models as a translational approach to understand SAR-CoV-2. Viruses 2020, 12, 880. 10.3390/v12080880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. J.; Okuda K.; Edwards C. E.; Martinez D. R.; Asakura T.; Dinnon K. H.; Kato T.; Lee R. E.; Yount B. L.; Mascenik T. M.; Chen G.; Olivier K. N.; Ghio A.; Tse L. V.; Leist S. R.; Gralinski L. E.; Schäfer A.; Dang H.; Gilmore R.; Nakano S.; Sun L.; Fulcher M. L.; Livraghi-Butrico A.; Nicely N. I.; Cameron M.; Cameron C.; Kelvin D. J.; de Silva A.; Margolis D. M.; Markmann A.; Bartelt L.; Zumwalt R.; Martinez F. J.; Salvatore S. P.; Borczuk A.; Tata P. R.; Sontake V.; Kimple A.; Jaspers I.; O’Neal W. K.; Randell S. H.; Boucher R. C.; Baric R. S. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020, 182, 429–446.e414. 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; de Castilla D. L.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.-D.; Ruiz-Palacios G. M.; Benfield T.; Fätkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C.; Remdesivir for the treatment of Covid-19 - Final report. N. Engl. J. Med. 2020, 383, 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimbeni A. C.; Codogno P.; Morel E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. 10.1111/febs.13987. [DOI] [PubMed] [Google Scholar]
- Marat A. L.; Haucke V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 2016, 35, 561–579. 10.15252/embj.201593564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberger A. J. B.; Sanyal A.; Ojha R.; Pyle J. D.; Vapalahti O.; Balistreri G.; Kirchhausen T.; Gallagher T. Synergistic block of SARS-CoV-2 infection by combined drug inhibition of the host entry factors PIKfyve kinase and TMPRRSS2 protease. J. Virol. 2021, 95, e00975–00921. 10.1128/JVI.00975-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L.; Yuan S.; Yin X.; Martin-Sancho L.; Matsunaga N.; Pache L.; Burgstaller-Muehlbacher S.; De Jesus P. D.; Teriete P.; Hull M. V.; Chang M. W.; Chan J. F.-W.; Cao J.; Poon V. K.-M.; Herbert K. M.; Cheng K.; Nguyen T.-T. H.; Rubanov A.; Pu Y.; Nguyen C.; Choi A.; Rathnasinghe R.; Schotsaert M.; Miorin L.; Dejosez M.; Zwaka T. P.; Sit K.-Y.; Martinez-Sobrido L.; Liu W.-C.; White K. M.; Chapman M. E.; Lendy E. K.; Glynne R. J.; Albrecht R.; Ruppin E.; Mesecar A. D.; Johnson J. R.; Benner C.; Sun R.; Schultz P. G.; Su A. I.; García-Sastre A.; Chatterjee A. K.; Yuen K.-Y.; Chanda S. K. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu S.; Dick A.; Ju H.; Mirzaie S.; Abdi F.; Cocklin S.; Zhan P.; Liu X. Inhibitors of SARS-CoV-2 entry: Current and future opportunities. J. Med. Chem. 2020, 63, 12256–12274. 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribone S. R.; Paz S. A.; Abrams C. F.; Villarreal M. A. Target identification for repurposed drugs active against SARS-CoV-2 via high-throughput inverse docking. J. Comput.-Aided Mol. Des. 2022, 36, 25–37. 10.1007/s10822-021-00432-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X.; Liu Y.; Lei X.; Li P.; Mi D.; Ren L.; Guo L.; Guo R.; Chen T.; Hu J.; Xiang Z.; Mu Z.; Chen X.; Chen J.; Hu K.; Jin Q.; Wang J.; Qian Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C. B.; Farzan M.; Chen B.; Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol 2022, 23, 3–20. 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayati A.; Kumar R.; Francis V.; McPherson P. S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306 10.1016/j.jbc.2021.100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N.-H.; Nitsche A.; Müller M. A.; Drosten C.; Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel E. C.; Huang C.; Narayanan K.; Makino S.; Tesh R. B.; Peters C. J. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. J. Virol. 2005, 79, 3846–3850. 10.1128/jvi.79.6.3846-3850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J.; Uckeley Z. M.; Doldan P.; Stanifer M.; Boulant S.; Lozach P. Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821 10.15252/embj.2021107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X. P.; Shen D.; Wang X.; Dawson T.; Li X.; Zhang Q.; Cheng X.; Zhang Y.; Weisman L. S.; Delling M.; Xu H. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 2010, 1, 38 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov O. C.; Sbrissa D.; Mlak K.; Kanzaki M.; Pessin J.; Shisheva A. Functional dissection of lipid and protein kinase signals of PIKfyve reveals the role of PtdIns 3,5-P2 production for endomembrane integrity. J. Biol. Chem. 2002, 277, 9206–9211. 10.1074/jbc.M108750200. [DOI] [PubMed] [Google Scholar]
- Min S. H.; Suzuki A.; Stalker T. J.; Zhao L.; Wang Y.; McKennan C.; Riese M. J.; Guzman J. F.; Zhang S.; Lian L.; Joshi R.; Meng R.; Seeholzer S. H.; Choi J. K.; Koretzky G.; Marks M. S.; Abrams C. S. Loss of PIKfyve in platelets causes a lysosomal disease leading to inflammation and thrombosis in mice. Nat. Commun. 2014, 5, 4691 10.1038/ncomms5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran M. S.; Bagchi P.; Cunningham C. N.; Tsai B. Opportunistic intruders: How viruses orchestrate er functions to infect cells. Nat. Rev. Microbiol. 2016, 14, 407–420. 10.1038/nrmicro.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.; Schor S.; Barouch-Bentov R.; Einav S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. 10.1007/s00018-018-2882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Dellibovi-Ragheb T. A.; Kerviel A.; Pak E.; Qiu Q.; Fisher M.; Takvorian P. M.; Bleck C.; Hsu V. W.; Fehr A. R.; Perlman S.; Achar S. R.; Straus M. R.; Whittaker G. R.; de Haan C. A. M.; Kehrl J.; Altan-Bonnet G.; Altan-Bonnet N. β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 2020, 183, 1520–1535.e1514. 10.1016/j.cell.2020.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; McGee L. R.; Fisher B.; Sudom A.; Liu J.; Rubenstein S. M.; Anwer M. K.; Cushing T. D.; Shin Y.; Ayres M.; Lee F.; Eksterowicz J.; Faulder P.; Waszkowycz B.; Plotnikova O.; Farrelly E.; Xiao S.-H.; Chen G.; Wang Z. Inhibiting NF-κB-inducing kinase (NIK): Discovery, structure-based design, synthesis, structure–activity relationship, and co-crystal structures. Bioorg. Med. Chem. Lett. 2013, 23, 1238–1244. 10.1016/j.bmcl.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Fogarty K.; Kashem M.; Bauer A.; Bernardino A.; Brennan D.; Cook B.; Farrow N.; Molinaro T.; Nelson R. Development of three orthogonal assays suitable for the identification and qualification of PIKfyve inhibitors. Assay Drug Dev. Technol. 2017, 15, 210–219. 10.1089/adt.2017.790. [DOI] [PubMed] [Google Scholar]
- Wells C.; Couñago R. M.; Limas J. C.; Almeida T. L.; Cook J. G.; Drewry D. H.; Elkins J. M.; Gileadi O.; Kapadia N. R.; Lorente-Macias A.; Pickett J. E.; Riemen A.; Ruela-de-Sousa R. R.; Willson T. M.; Zhang C.; Zuercher W. J.; Zutshi R.; Axtman A. D. SGC-AAK1-1: A chemical probe targeting AAK1 and BMP2K. ACS Med. Chem. Lett. 2020, 11, 340–345. 10.1021/acsmedchemlett.9b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C. I.; Drewry D. H.; Pickett J. E.; Tjaden A.; Krämer A.; Müller S.; Gyenis L.; Menyhart D.; Litchfield D. W.; Knapp S.; Axtman A. D. Development of a potent and selective chemical probe for the pleiotropic kinase CK2. Cell Chem. Biol. 2021, 28, 546–558.e510. 10.1016/j.chembiol.2020.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.