

Abstract
The SARS-CoV-2 envelope (E) protein is a viroporin associated with the acute respiratory symptoms of COVID-19. E forms cation-selective ion channels that assemble in the lipid membrane of the endoplasmic reticulum Golgi intermediate compartment (ERGIC). The channel activity of E is linked to the inflammatory response of the host cell to the virus. Like many viroporins, E is thought to oligomerize with a well-defined stoichiometry. However, attempts to determine the E stoichiometry have led to inconclusive results, and suggested mixtures of oligomers whose exact nature might vary with the detergent used. Here we employ 19F solid-state NMR and the centerband-only detection of exchange (CODEX) technique to determine the oligomeric number of E's transmembrane domain (ETM) in lipid bilayers. The CODEX equilibrium value, which corresponds to the inverse of the oligomeric number, indicates that ETM associates into pentamers in lipid bilayers, without any detectable fraction of low molecular-weight oligomers. Unexpectedly, at high peptide concentrations and in the presence of the lipid phosphatidylinositol, the CODEX data indicate that more than five 19F spins are spatially close, suggesting that the ETM pentamers cluster in the lipid bilayer. Monte Carlo simulations that take into account peptide-peptide and peptide-lipid interactions yielded pentamer clusters that reproduced the CODEX data. This supramolecular organization of E is likely important for E-mediated virus assembly and budding and for the channel function of the protein.
Keywords: COVID-19, viroporins, 19F solid-state NMR, CODEX, membrane protein, clustering
Graphical Abstract
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the COVID-19 pandemic, encodes three membrane proteins: the spike protein (S), the matrix protein (M), and the envelope protein (E) 1. The vaccine-targeted spike protein mediates entry of the virus into the host cell, while the matrix protein organizes the assembly of new virus particles in the endoplasmic reticulum Golgi intermediate compartment (ERGIC) of the cell. The envelope protein, the least understood among the three proteins, assists in the assembly and budding of the virus and functions as a cation channel 2, 3. The channel activity of E in SARS-CoV-1 is not essential for the virus localization to the ERGIC compartment or the production of new virus particles in cell cultures. However, lack of E channel activity reduced viral fitness and deletion of E gave rise to attenuated viruses in animal models 4. Transient expression of the E protein of SARS-CoV-2 is sufficient to induce in vitro death in human bronchial and alveolar basal epithelial cell lines 5. Moreover, injection of E alone into mice provokes strong immune responses and causes acute respiratory distress symptoms that damage the lung and spleen, similar to that seen in human COVID-19 patients. In mice infected with SARS-CoV-2 the administration of E inhibitors significantly attenuated inflammation and production of viral particles 5. Therefore, E is a potential antiviral drug target to ameliorate the respiratory symptoms of COVID-19 6.
To develop E inhibitors as antiviral drugs, atomic-resolution structural information of the protein is essential 7. We recently determined the high-resolution structure of the E transmembrane domain (ETM) (Fig. 1a) in lipid bilayers using solid-state NMR (ssNMR) spectroscopy 8. In lipid mixtures that mimic the ERGIC membrane and under ionic conditions that produce the closed state of the channel, ETM exhibits well-resolved ssNMR spectra. Measured chemical shifts and interhelical distance constraints allowed determination of the conformation and interhelical packing of the protein 8. In solving this closed-state structure, we assumed that ETM assembles into five-helix bundles in the lipid bilayer. This assumption was based on previous gel electrophoresis and analytical ultracentrifugation (AUC) data of the SARS-CoV-1 E protein 9-11. However, the stoichiometry of the ETM oligomer is so far still inconclusive. Most gel electrophoresis data show multiple bands, many of which are not well resolved. The largest oligomer number observed in these gels depends on the choice of the detergent, the E protein construct length, and the presence or absence of cysteines and reducing agents. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) promotes low molecular weight (MW) oligomers such as dimers and trimers 9, 11, while neutral detergents such as dodecyphosphocholine (DPC), perfluorooctanoic acid (PFO), and C14 betaine promote larger oligomers. These biophysical assays used three construct lengths of the SARS-CoV-1 E protein – ETM (8-38) 10, ETR (8-65) 9, and full-length E (EFL). ETR includes the TM domain and a portion of the cytoplasmic domain and is thus an intermediate construct between ETM and EFL 11. The TM amino acid sequences of SARS-CoV-1 and the original isolate of SARS-CoV-2 E proteins are identical, while the cytoplasmic domains of the two proteins have only four mutations: T55S, V56F, E69R, and G69Δ. Among the three constructs, ETM yielded the best resolved bands in gel electrophoresis data, but still showed a distribution of oligomeric states that varies among different detergents 10, 12. AUC experiments similarly suggest variable oligomeric states. The radial distribution profile of DPC-bound ETM is best fit to a monomer-pentamer equilibrium, while SDS/DPC bound ETR exhibits a mixture of tetramers and pentamers. Compared to ETM, full-length E exhibits a pentameric state only under reducing conditions.
Figure 1.

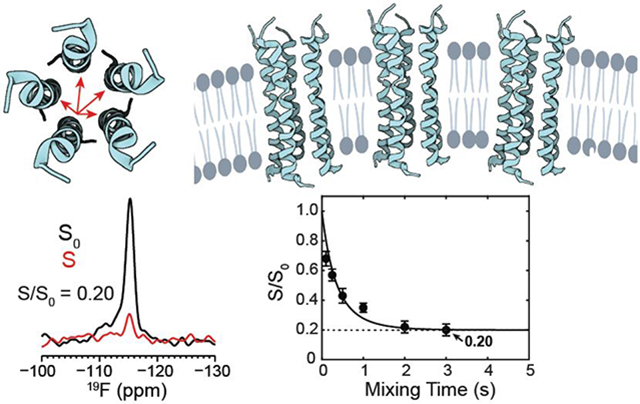
The 19F solid-state NMR approach for determining the stoichiometry of ETM assembly in lipid bilayers. (a) Amino acid sequence diagram of the SARS-CoV-2 E protein. The transmembrane (TM) domain is preceded by a short N-terminal domain (NTD) and is followed by a C-terminal domain (CTD). The amino acid sequences of singly fluorinated ETM peptides used in this study are shown. (b) Structures of the 4-19F-phenylalanine and 19F-Cγ-leucine that are synthetically incorporated into the ETM peptide. (c) Pulse sequence of the 19F CODEX experiment. Filled and open rectangles designate 90° and 180° pulses, respectively. CP: cross-polarization; CW continuous-wave heteronuclear decoupling; TPPM, two-phase modulation heteronuclear decoupling. 19F spin diffusion during the mixing time tm reduces the intensity of a 19F spin echo. The CODEX dephased (S) experiment has a long tm and a short tz, while the CODEX control (S0) experiment has a short tm and a long tz. The sum of the two mixing times is the same between S and S0. (d) Schematic of different stoichiometries of the ETM oligomer in the lipid membrane, with corresponding relative intensities of the CODEX S0 and S spectra.
In addition to SARS-CoV-1 E, the oligomeric structure of the E protein of two other coronaviruses has been investigated experimentally. The Middle East respiratory symptom (MERS)-CoV full-length E was studied in DPC, 1,2-diheptanoylglycerophosphocholine (DHPC) and lyso-myristoylphosphatidylcholine (LMPG) detergents. In analogy to SARS-CoV-1 E, gel electrophoresis data showed a continuum of bands that suggest, but do not definitively determine, a pentameric state for MERS-CoV E 13. AUC data of the protein in C14-betaine micelles provide clearer indications of a pentameric state. The avian infectious bronchitis virus (IBV) E protein was investigated using sucrose gradient analysis and other biochemical experiments 14. The data show that IBV E exists in a low MW pool and a high MW pool in both transfected and infected cells. The low MW component is either monomeric or dimeric while the high MW component is pentameric or hexameric. Importantly, the majority of virion-associated IBV E protein is found in the high MW pool, and production of virus-like particles (VLPs) required the high MW E oligomers. Mutations of key TM residues in the IBV E shifted the populations of the two MW components, affected VLP production, altered the secretory pathway function of the protein, and changed the membrane topology of the protein 15. These results suggest that different oligomeric states of the IBV E may carry out different functions. The potential coexistence of multiple oligomeric states of the E proteins of SARS-CoV-1 and IBV was also suggested by early global search molecular dynamics simulations of E proteins from 13 coronaviruses 12. These in vacuo simulations searched for common low-energy helix orientation, helical bundle handedness and oligomeric sizes. The results suggested that ETM of SARS-CoV-1 and other coronaviruses may adopt a mixture of dimers, trimers, and pentamers.
This survey shows that the oligomeric state of coronavirus E proteins has not been convincingly established, despite efforts in the last two decades. Furthermore, none of the biophysical measurements to date have been conducted in lipid bilayers. Protein stoichiometry in lipid bilayers is a key aspect of the structure and function of membrane proteins. Most membrane proteins oligomerize with well-defined stoichiometries. Mammalian ion channels are dominated by tetrameric helical bundles while viral fusion proteins are predominantly trimeric. Among viroporins, the influenza M2 proton channel is tetrameric, as established by channel activity data 16, biophysical analytical ultracentrifugation data 17, 18, 19F solid-state NMR data in lipid bilayers 19, 20, and X-ray crystal structures in detergents 21 and lipid cubic phases 22. In comparison, the HIV viral protein U (Vpu) assembles into a pentameric cation channel 23-26.
To elucidate the oligomeric state of the SARS-CoV-2 E protein in lipid bilayers, and to ascertain if the protein might adopt two distinctly different oligomeric states, here we apply 19F solid-state NMR spectroscopy to fluorinated ETM peptides in lipid bilayers. Specifically, we employ the centerband-only detection of exchange (CODEX) technique, which allows counting of the number of 19F spins within a distance of about 2 nm 19, 27-30. We synthesized singly fluorinated ETM peptides and reconstituted them into ERGIC-mimetic lipid bilayers and several other simplified lipid membranes. We show that ETM assembles into pentamers in all lipid compositions studied. Unexpectedly, we discovered that these ETM pentamers cluster in membranes that contain phosphatidylinositol, whose derivatives are involved in intracellular signaling. This suggests a possible mechanism with which interactions between E and viral and host membrane proteins may play a role in the pathogenic activities of the virus.
Materials and Methods
Synthesis of 4-19F-Phe labeled ETM peptides
The TM domain of the SARS-CoV-2 E protein spanning residues 8-38 and containing a 4-19F-Phe23 label was produced by Fmoc solid-phase peptide synthesis. The amino acid sequence is ETG TLIVNSVLLF LAFVVFLLVT LAILTALR-NH2. We used a custom-built fast-flow synthesizer that achieves each coupling in 3 minutes per residue 31. H-Rink amide ChemMatrix resin at 0.10 mmol scale (0.20 g at 0.5 mmol/g loading size) was loaded into the reactor, which was kept at 70°C using a water bath. Each amino acid was coupled in ten-fold excess (1.0 mmol). The Fmoc-amide and side chain protected amino acids were dissolved in hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) solution (2.5 mL per residue, 0.38 M HATU in dimethylformamide (DMF) solution, 9.5 eq). For each amino acid coupling, diisopropylethylamine (DIPEA, 348 μL, 2.0 mmol, 20 eq) was added to the vial, and the amino acids were delivered via a syringe pump (3.9 mL/min, 45 s). The coupling was followed by a wash with DMF (20 mL/min, 55 s) delivered using an HPLC pump. Each residue was deprotected with a DMF solution containing 20% piperidine (20 mL/min, 25 s), followed by a DMF wash (20 mL/min, 65 s) to remove residual piperidine. After the final amino acid coupling, the solvent was flushed from the reactor, and the resin was washed three times with dichloromethane, then dried under room temperature (RT) vacuum overnight. The following day the peptide was deprotected and cleaved from the resin using 5 mL of trifluoroacetic acid / phenol / water / triisopropylsilane solution (88 : 5 : 5 : 2 by volume), shaking for 2 h at RT. The resin was filtered off, and the crude peptide was precipitated from the cleavage solution with cold diethyl ether, then washed twice with cold diethyl ether before being dried under house vacuum (50 mBar, RT) overnight. The resulting crude peptide was dissolved in trifluoroethanol (TFE) and purified by preparative reverse-phase HPLC using a Vydac C4 column (22 mm × 250 mm, 10 μm particle size) and a linear gradient of 80-100% methanol (with the remaining fraction water) over 25 min at a flow rate of 10 mL/min. The peptide was eluted at ~99% methanol. Fractions containing the peptide were assessed for relative purity by MALDI mass spectrometry (Fig. S1) (observed m/z: 3381.0, calculated m/z: 3378.2) and concentrated under reduced pressure on a rotary evaporator, followed by lyophilization. About 20 mg purified peptide was obtained as a white powder from the 0.10 mmol synthesis scale, corresponding to an overall yield of ~5%.
Synthesis of γ-19F-Leu18 labeled ETM
Fmoc-4-fluoro-leucine was synthesized from commercially available 2(S)-amino-4-fluoro-4-methylpentanoic acid using typical Fmoc protection condition 32 for amino acid (FmocOsu, NaHCO3 in H2O-acetone). The C-F group is installed on the Cγ carbon of the amino acid, thus in the rest of the paper we designate this residue as γ-19F-Leu. ETM peptide (residues 8-38) containing γ-19F-Leu at residue 18 (Ac-ETG TLIVNSVLγ-FLF LAFVVFLLVT LAILTALR-NH2) was synthesized on TantaGel S Ram resin (0.25 mmol/g, 0.1 mmol scale) using an automated microwave peptide synthesizer (Biotage Alstra + Initiator). The deprotection step was carried out for 5 min at 70 °C with 20% 4-methylpiperidine in DMF (4.5 mL) and each coupling step was done twice for 5 min at 75 °C with Fmoc-protected amino acids (5 eq), HCTU (4.98 eq), and DIPEA (10 eq) in DMF at a final concentration of 0.125 M amino acids. The N-terminus of the peptide was acetylated using Ac2O (10 eq), and DIPEA (20 eq) in DMF for 1 h at room temperature. The peptide was cleaved from the resin by 3-h treatment with a 95% TFA solution (10 mL) containing 2.5% TIPS and 2.5% water. The crude peptide was obtained by blowing off TFA using nitrogen gas, followed by precipitation with cold diethyl ether. The crude peptide was purified by RP-HPLC (C4 Vydac column, mobile phase A: 0.1% TFA in H2O, B: 0.1% TFA in i-PrOH/CH3CN/H2O (60/30/10), flow rate: 10 mL/min, gradient: 60% to 100% B over 40 minutes). Analytical HPLC and MALDI (observed m/z: 3421.75, calculated m/z: 3421.15 for MH+(C165H274N36O40F+)) confirmed the chemical entity and purity.
Preparation of Proteoliposomes
To reconstitute ETM into lipid bilayers, purified peptide was dissolved in methanol at 1 mg/mL and dry lipids in chloroform at a total concentration of 15 mg/mL. The peptide solution was added to the lipid solution, then the bulk organic solvent was removed with a stream of nitrogen gas (RT, 1 h). Residual organic solvent was further removed under house vacuum (50 mBar, RT, 4 h), followed by lyophilization overnight. The dry proteoliposome film was resuspended in 3 mL pH 7.5 Tris buffer (25 mM tris, 25 mM NaCl, 1 mM EDTA, 0.07 mM NaN3), vortexed and sonicated five times, five seconds each. The proteoliposome solution was incubated for 1 h at RT, with gentle agitation every 10 minutes. The homogenous solution was then frozen in liquid nitrogen until solid (90 s) and thawed in a 42°C water bath until warmed (4 minutes). This freeze-thaw cycle was repeated 8 times total to produce multilamellar vesicles. The homogeneous opaque white proteoliposome solution was ultracentrifuged at 311,000 × g at 10 °C for 4 h to obtain an opaque off-white membrane pellet. The wet pellet was dried in a desiccator at RT until the sample reached a hydration level of ~40% (w/w) of the total mass of protein, lipid and water, . The pellet was packed into a 1.9 mm Bruker MAS rotor at 5,000 × g with a benchtop Beckman Coulter swinging-bucket rotor.
To investigate the effect of the lipid composition on the supramolecular organization of ETM, we reconstituted ETM into four membrane mixtures. The main lipid mixture is an ERGIC mimetic membrane 33-35, composed of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (16:0-18:1 PC; POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (16:0-18:1 PE; POPE), L-α-phosphatidylinositol (Liver, Bovine; PI), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (16:0-18:1 PS; POPS) and cholesterol (chol). All lipids were purchased from Avanti Polar Lipids without further purification. The molar ratios of POPC : POPE : PI : POPS : Chol in this ERGIC membrane are 9 : 4 : 2.6 : 1.4 : 3. Three ERGIC membrane samples with different peptide (monomer) to phospholipid (P : L) molar ratios of 1 : 8.5, 1 : 17, and 1 : 34 were prepared. Cholesterol is not considered in the peptide to phospholipid ratios, and cholesterol concentrations are presented separately. We also prepared two simplified ERGIC-like membranes. The first membrane omits cholesterol, thus has a molar composition of POPC : POPE : PI : POPS = 9 : 4 : 2.6 : 1.4. The P : L molar ratio was 1 : 17. The second membrane omits cholesterol and phosphatidylinositol. The POPC : POPE : POPS molar ratios were 9 : 4 : 1.4 while the P : L molar ratio was 1 : 17. Finally, we prepared a model membrane containing 1,2-dimyristoyl-sn-glycero-3-phosphocholine (14:0 PC, DMPC) and 1,2-dimyristoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (14:0 PG, DMPG) at a molar ratio of 7 : 3. The P : L molar ratio for this sample was 1 : 30.
Solid-State NMR Spectroscopy
19F solid-state NMR experiments were conducted on an Avance III HD 600 MHz 1H (14.1 T) spectrometer using a Bruker 1.9 mm HFX MAS probe. Additional 19F solid-state NMR experiments were conducted on an Avance III HD 400 MHz 1H (9.4 T) spectrometer using a Bruker 4 mm HFX probe for 19F experiments. All 19F externally referenced to 5-19F-trptophan at −122.1 ppm on the CF3Cl scale. Typical radiofrequency (RF) field strengths were 50-80 kHz for 1H and 50-62 kHz for 19F.
19F CODEX experiments were conducted at 18 kHz MAS at thermocouple reported temperatures of 240-244 K. Typical sample heating at 18 kHz MAS results in sample temperature is 2-4 K greater than thermocouple reported temperatures. A 1H excitation pulse of 71.4 kHz was used, followed by cross-polarization (CP) for 750 us at the sideband matching condition of ω1,H (80 kHz) – ω1,F (62 kHz) = ωr (18 kHz). A linear 70–100% ramp was used on the 19F spin-lock pulse. This 1H-19F cross polarization was followed by a short (1 ms) pre-trigger z-filter for rotor synchronization. Two rotor-synchronized 180°-pulse trains recouple the 19F chemical shift anisotropy (CSA) under MAS. Three 180°-pulses were used in each pulse train, giving a CSA recoupling time of 2τr = 0.11 ms per pulse train. The two CSA recoupling periods sandwich the mixing time tm, during which no pulses or decoupling was applied. The mixing time was rotor synchronized with triggering on the second 90° pulse. To correct for spin-lattice relaxation during the mixing time, we added a z-filter (tz) after the second 180°-pulse train. The CODEX experiment was run in pairs. The exchange experiment (S) was run with the desired tm first and a short (10 μs) tz period second, while the control experiment (S0) was run with the short tz period first and the long mixing period second. The normalized integrated intensity, S/S0, of the centerband is reported for each mixing time.
NMR Spectral Analysis
All MAS NMR spectra were processed using TopSpin 3.6. Typical spectra were processed using Gaussian apodization with LB = −40 Hz and GB = 0.02. For 19F-Phe23 spectra S/S0 values were obtained from integration of the centerband (−100 ppm to −125 ppm). The error bar σ on the S/S0 values was estimated based on the signal-to-noise ratio (SNR) of the spectra using σ = (S/S0)[(SNRS0−2 + SNRS−2)1/2].
Simulation of CODEX data using random distribution and clustered distribution
The CODEX decay curves of multiple oligomers were simulated by first computing the number of oligomers expected in a 100 × 100 nm2 square based on the area of the channel, assuming circular channels of radius , an oligomeric number of , a peptide monomer to lipid molar ratio , and the lipid head group area . All simulations use a lipid head group area based on existing measurements of lipid structural parameters 36, 37:
| (1) |
A random spatial arrangement of oligomers was produced using the random sequential adsorption (RSA) method implemented in MATLAB (Supporting Information) 38. Two pseudorandom numbers (with uniform probability distribution) were generated in the interval [0, 1000]. The center of each oligomer was placed at the x-y coordinate that corresponds to the two randomly generated numbers. If the position of an oligomer overlaps with the previously generated oligomer within the radius of the channel, then the oligomer was rejected. This was repeated times to generate the positions of the oligomers. A channel radius of 12.8 Å was chosen to match the excluded volume of the cylindrical channel in the clustered distribution.
To generate a clustered distribution of oligomers, we started oligomers distributed randomly on a 100 x 100 nm2 square. Oligomers were initially allowed to overlap within their 12.8 Å, as the interaction potential was used to exclude oligomer overlap. The potential of mean force used was constructed based on a pairwise cylinder-cylinder interaction potential for a positive hydrophobic mismatch of 1 nm 39. This mismatch was chosen based on the approximate length of 4 nm for the hydrophobic domain of ETM 8 and the approximate hydrophobic thickness of 3 nm for a POPC bilayer 36. Numerical values for the potential are provided in the Supporting Information. The position of each oligomer was adjusted according to this positive-mismatch potential using an implementation of the Metropolis Monte Carlo method 40. Specifically, a random pore was selected and its total pairwise interaction energy to every other pore, Ei, was calculated under periodic boundary conditions on the 100 x 100 nm2 square. The pore was then randomly moved to a new position and the total pairwise interaction energy of the pore in the new position with all other pores, Ef, was computed. If Ef ≤ Ei, then the new position was accepted. If Ef > Ei, then according to detailed balance the new position was accepted with probability exp (Ei − Ef) where Ei and Ef are in units of kBT. After the new position was either accepted or rejected, a new pore was randomly selected starting from the final state of the previous move. This process was repeated until the average nearest-neighbor distance between oligomers has converged. 50,000 iterations were conducted. The average nearest-neighbor distance was not observed to change significantly after approximately 10,000 iterations.
After the pore positions were generated using either RSA or the Monte Carlo method based on the cylinder-cylinder interaction potential, each oligomer was placed as a regular polygon of vertices with side length . Each polygon was given a random rotation around its center in the bilayer plane. A distance matrix of dimensions was constructed to account for all inter- and intra-oligomer 19F-19F distances. In constructing the distance matrix, boundary effects are assumed to be negligible, and hard-wall boundary conditions are used. Total 19F-19F dipolar coupling converges after summing over approximately a 20 Å radius, including all couplings greater than 12 Hz. Thus, to increase computational efficiency, all distances greater than 50 Å, which correspond to 19F-19F dipolar couplings of less than 0.85 Hz, were neglected. CODEX intensities were then calculated as described previously 19 to produce mixing-time dependent decay curves.
Results
SARS-CoV-2 ETM forms pentamers in lipid bilayers
To investigate the oligomeric state of SARS-CoV-2 E, we synthetized three singly fluorinated ETM peptides (Fig. 1a). 4-19F-Phe23 labeled ETM was the primary construct for the majority of the 19F CODEX experiments presented here, while 4-19F-Phe20 labeled ETM and γ-19F-Leu18 labeled ETM provided additional support (Fig. 1b). Because the protein localizes to the ERGIC membrane during the SARS-CoV-2 virus lifecycle 41, 42, we reconstituted ETM into an ERGIC-mimetic lipid bilayer containing POPC, POPE, POPS, PI and cholesterol (Table 1). To ascertain whether certain lipids in this mixture significantly impact the oligomeric structure, we also prepared simplified membrane mixtures utilizing fewer lipid components.
Table 1.
Membrane compositions, CODEX intensities at the longest mixing times, and best-fit parameters of 419F-Phe23 labeled ETM.
| Lipid and Molar Composition | P : L | 19F T1 (s) | S/S0 | S0/S | r (Å) |
|---|---|---|---|---|---|
| ERGIC: POPC/POPE/PI/POPS/Chol ( 9 : 4 : 2.6 : 1.4 : 3) | 1 : 17 | 4.5 | 0.14 ± 0.03 | 7.1 | 8.8 |
| ERGIC | 1 : 34 | 3.2 | 0.20 ± 0.04 | 5.0 | 8.7 |
| ERGIC | 1 : 8.5 | 4.1 | 0.16 ± 0.03 | 6.3 | 8.6 |
| POPC/POPE/PI/POPS (9 : 4 : 2.6 : 1.4) | 1 : 17 | 4.1 | 0.17 ± 0.02 | 5.9 | 8.4 |
| POPC/POPE/POPS (9 : 4 : 1.4) | 1 : 17 | 0.9 | 0.22 ± 0.06 | 4.2 | 8.4 |
| DMPC/DMPG (7 : 3) | 1 : 30 | 1.5 | 0.21 ± 0.04 | 4.8 | 8.0 |
We use the 19F CODEX technique 19, 28 to determine the oligomeric number of ETM. This technique measures the intensity of a 19F spin echo after spin polarization transfer during a mixing time tm (Fig. 1c). When several 19F spins with different chemical shift tensor orientations are within ~2 nm of each other, polarization transfer among them mediated by 19F-19F dipolar couplings decreases the CODEX echo intensity. This reduced echo intensity results in an intensity ratio of less than 1 between a dephased experiment, S, and a control experiment, S0. The control experiment was conducted to account for T1 relaxation during the mixing time. At sufficiently long mixing times when the magnetization is equilibrated among the spins of an -oligomer, the CODEX intensity decreases to , which corresponds to the fraction of magnetization that resides on the source spin with the initial orientation. Thus, a dimer should result in a CODEX equilibrium S/S0 value of 1/2, a trimer 1/3, a tetramer 1/4, and so on (Fig. 1d).
19F chemical shift is highly sensitive to the local electronic environment of the fluorine atom and is therefore sensitive to molecular structure. One-dimensional (1D) 19F cross-polarization (CP) and direct-polarization (DP) spectra of 4-19F-Phe23-labeled ETM show a dominant peak at − 115 ppm (Fig. 2a), indicating a homogeneous sidechain conformation. Some of the membrane samples also exhibit a minor peak at −111 ppm, whose intensity accounts for 5-25% of the total spectral intensity. The simplicity of these spectra facilitate the determination of the oligomeric state of ETM using CODEX.
Figure 2.

Representative 19F NMR spectra of 4-19F-Phe23 labeled ETM in different lipid membranes. All spectra were measured at a thermocouple reported temperature of 240–244 K and 18 kHz MAS. (a) 19F CP spectra of the six membrane-bound ETM samples. For cholesterol-containing membranes, the peptide : phospholipid : cholesterol ratios are specified. (b-g) CODEX control S0 (black) and dephased S (red) spectra of ETM at selected mixing times. (b) 4 s CODEX spectra of ERGIC-bound ETM at P : L = 1 :17. (c) 3 s CODEX spectra of ERGIC-bound ETM at P : L = 1 : 34. (d) 4 s CODEX spectra of ERGIC-bound ETM at P : L = 1 : 8.5. (e) 4 s CODEX spectra of ETM bound to the cholesterol-free ERGIC membrane at P : L = 1 : 17. (f) 3 s CODEX spectra of ETM bound to the POPC/POPE/POPS membrane at P : L = 1 : 17. The peak doubling indicates the presence of two Phe23 sidechain conformations in this membrane. (g) 2.5 s CODEX spectra of ETM in the DMPC/DMPG membrane at P : L = 1 : 30. The S/S0 values based on the integrated intensities of these CODEX spectra are given in (b-f).
We first measured the CODEX spectra of 4-19F-Phe23 labeled ETM bound to the ERGIC membrane (Fig. 2b). At a P : L of 1 : 17, the T1-corrected CODEX intensities, S/S0, decayed to 0.19±0.02 at a mixing time of 3 s (Fig. 3a, Table 2), suggesting a pentamer. This low S/S0 value excludes the presence of a substantial fraction of small oligomers such as monomers, dimers, or trimers, as they would contribute much higher CODEX intensities of 1, 0.5, and 0.33.
Figure 3.

19F CODEX of 4-19F-Phe23 labeled ETM in different lipid membranes at various peptide to phospholipid ratios (P : L). Filled circles are the measured CODEX S/S0 values while open circles are the intensity decays of the control spectra S0, normalized to the 100 ms spectral intensity. Solid lines are the best-fit matrix simulations of the CODEX decay using an isolated symmetric oligomer model. Dashed lines are the best exponential fit to the S0 decay to give the 19F T1 relaxation time. (a-c) CODEX decays of ERGIC-bound ETM at varying P : L ratios. (a) P : L = 1 : 17. (b) P : L = 1 : 34. (c) P : L = 1 : 8.5. The best-fit curves use a symmetric pentamer model. (d-f) CODEX intensities shown as inverse S0/S ratios for the same three samples as in (a-c), to better compare the pentamer and hexamer fits to the measured data at long mixing times. The best-fit simulations for a symmetric pentamer model (black) and a symmetric hexamer model (purple) are shown in (d) and (f). (g) CODEX decay of ETM bound to a cholesterol-depleted ERGIC membrane (POPC/POPE/PI/POPS) at P : L = 1 : 17. (h) CODEX decay of the major Phe23 peak in POPC/POPE/POPS membrane-bound ETM at P : L 1 : 17. The S0 decays of the major peak (blue open circles) and minor peak (orange open circles) indicate distinct T1 relaxation times. (i) CODEX decay of DMPC/DMPG bound ETM at a P : L ratio of 1 : 30. The 19F T1 times are longer than 3 s in all membranes except for the major peak of the POPC/POPE/POPS bound sample and the DMPC/DMPG bound ETM.
Table 2.
Measured 19F CODEX S/S0 values of 4-19F-Phe23 labeled ETM in all lipid membranes examined in this study. Some phospholipids are indicated by their last letter for simplicity.
| Mixing Time (s) |
ERGIC P : L = 1 : 17 |
ERGIC P : L = 1 : 34 |
ERGIC P : L = 1 : 8.5 |
POPC/E/I/S P : L = 1 : 17 |
POPC/E/S P : L = 1 : 17 |
DMPC/G P : L = 1 :30 |
|---|---|---|---|---|---|---|
| 0.10 | 0.74 ± 0.02 | 0.68 ± 0.05 | 0.69 ± 0.02 | 0.62 ± 0.02 | 0.70 ± 0.04 | 0.61 ± 0.04 |
| 0.25 | 0.56 ± 0.02 | 0.57 ± 0.04 | 0.64 ± 0.02 | 0.57 ± 0.03 | 0.53 ± 0.04 | 0.58 ± 0.05 |
| 0.50 | 0.49 ± 0.02 | 0.43 ± 0.05 | 0.43 ± 0.02 | 0.45 ± 0.02 | 0.38 ± 0.02 | |
| 1.00 | 0.35 ± 0.02 | 0.35 ± 0.03 | 0.28 ± 0.02 | 0.33 ± 0.02 | 0.28 ± 0.04 | 0.34 ± 0.06 |
| 1.50 | 0.29 ± 0.02 | |||||
| 2.00 | 0.24 ± 0.03 | 0.22 ± 0.04 | 0.19 ± 0.02 | 0.22 ± 0.02 | 0.15 ± 0.08 | 0.24 ± 0.06 |
| 2.50 | 0.21 ± 0.04 | |||||
| 3.00 | 0.19 ± 0.02 | 0.20 ± 0.04 | 0.15 ± 0.01 | 0.18 ± 0.01 | 0.22 ± 0.06 | |
| 4.00 | 0.14 ± 0.03 | 0.16 ± 0.03 | 0.17 ± 0.02 | |||
| Expt. time | 66 hrs | 106 hrs | 50 hrs | 132 hrs | 72 hrs | 181 hrs |
The 3 s CODEX mixing time is the longest documented so far in the literature 19, 20, 27, 43-46. By this mixing time, all viral and bacterial membrane proteins that have been studied so far using this experiment have equilibrated their intensities. However, the intensities of the 1 : 17 ETM sample at 2 s and 3 s are not yet equilibrated. To measure the equilibrium value that would confirm the oligomeric number, we increased the mixing time further to 4 s. This 4 s CODEX experiment was enabled by the unusually long 19F T1 relaxation time of 4.5 s for this ERGIC-bound ETM at this concentration. To our surprise, the 4 s CODEX S/S0 value is even lower, at 0.14±0.03, which would suggest a heptamer (n = 7). However, the continuing drop of the CODEX intensities gives a non-exponential shape to the decay curve, suggesting additional magnetization transfer beyond that of an isolated oligomer. Since the 1 : 17 sample also has a long 19F T1, we hypothesized that multiple ETM oligomers might be clustered in the membrane at this peptide concentration. This clustering both rigidifies the aromatic sidechains, thus slowing down 19F T1 relaxation, and at the same time causes inter-oligomer contacts that accelerate CODEX dephasing at long mixing times. To test this hypothesis, we prepared a two-fold diluted sample with a P : L of 1 : 34. The 19F spectra of this sample (Fig. 2a, c) are unchanged from the 1 : 17 sample, but its CODEX intensities now plateau clearly between 2 s and 3 s to a S/S0 value of 0.20±0.04 (Fig. 3b), indicating a pentamer. This data demonstrates that individual ETM channels are pentameric in ERGIC-mimetic lipid bilayers, and these pentamers cluster at high peptide concentrations. Consistent with the equilibration of the CODEX intensities at the lower peptide concentration, the sample also exhibits a shorter 19F T1 relaxation time of 3.2 s, indicating that the Phe23 sidechain is more dynamic when ETM channels are better separated.
To further verify that multiple ETM pentamers indeed cluster at high peptide concentrations, we prepared a third ERGIC-bound protein sample with a P : L of 1 : 8.5. Compared to the 1 : 17 sample, this highest concentration protein sample displays a similar CODEX intensity of 0.16 ± 0.03 at the longest mixing time of 4 s, but at shorter mixing times the intensities are lower than those of the 1 : 17 sample (Fig. 2d, Fig. 3c). For example, at 2 s and 3 s mixing, the 1 : 8.5 sample exhibits echo intensities of 0.19±0.02 and 0.15±0.01, respectively, which are significantly lower than the intensities of 0.24±0.03 and 0.19±0.02 for the 1 : 17 sample at these times (Table 2). These results confirm that as the peptide concentration increases, inter-pentamer 19F-19F dipolar couplings increasingly suppress the CODEX intensities at long mixing times. This conclusion is consistent with the restored long 19F T1 of 4.1 s for the highly concentrated 1 : 8.5 sample, again indicating that high peptide concentrations immobilize the peptide by clustering of the pentamers.
A plot of the inverse CODEX intensity ratios, S0/S, more clearly differentiates the long-time behavior of the CODEX data and clarifies the concentration-dependent supramolecular organization of ETM in the ERGIC membrane (Fig. 3d-f). The intensities of the 1 : 17 sample fit similarly well to n = 5 or n = 6, but neither matrix calculation fully reproduces both the short and long-time intensities of the data. This is indicative of the presence of two dipolar networks, those within a pentamer and between multiple pentamers (Fig. 3d). In comparison, the inverse intensity ratios of the most diluted sample, at P : L = 1 : 34, is clearly fit with n = 5 (Fig. 3e). At the highest peptide concentration (1 : 8.5), the data are better fit to a hexamer (n = 6) than a pentamer (n = 5), which can be attributed to significant inter-pentamer clustering (vide infra).
The CODEX results obtained from 4-19F-Phe23 labeled peptide are reproduced when fluorine labels are incorporated at two other positions. The 4-19F-Phe20 labeled peptide showed a CODEX intensity of 0.31 ± 0.05 at 500 ms mixing (Fig. 4a). This value is significantly lower than those measured on Phe23 samples at the same mixing time (Table 2) as spin diffusion is more rapid at 9.4 Tesla and 10 kHz MAS than at higher magnetic fields and faster MAS rates. The observed CODEX decay is consistent with a pentamer model while ruling out dimers and trimers. The two 19F peaks for Phe20 have been recently shown to result from different sidechain conformations that correlate with channel closing and opening 47. γ-19F-Leul8 labeled ETM exhibited a CODEX S/S0 intensity of 0.23 ± 0.08 at 2 s mixing (Fig. 4b), consistent with a pentamer model.
Figure 4.

Additional 19F NMR spectra of fluorinated ETM. (a) 500 ms CODEX control S0 (black) and dephased S (red) spectra of 4-19F-Phe20 labeled ETM bound to the ERGIC membrane at P : L = 1 : 13. The spectra were measured on a 400 MHz NMR spectrometer (9.4 T, 19F Larmor frequency 376 MHz) under 10 kHz MAS at a thermocouple reported temperature of 248 K. (b) 2 s 19F CODEX S0 and S spectra of γ-19F-Leu18 labeled ETM, bound to the ERGIC membrane at a P : L ratio of 1 : 17. The spectra were measured on the 600 MHz NMR (14.1 Tesla, 19F Larmor frequency 564 MHz) under 18 kHz MAS at a bearing temperature of 240 K.
To assess whether there is any motional contribution to the CODEX decay, we conducted additional control CODEX experiments at a higher temperature of 250 K. With 1 s mixing, the S/S0 value is 0.36±0.02 (data not shown), which is within the experimental uncertainty of the value at 240 K. With 2 s mixing, the S/S0 value at 250 K is 0.20±0.02, which is still within the uncertainty of the value at 240 K (Table 2). Thus, there is no detectable slow motion of the Phe23 sidechain to cause CODEX decay at the temperature, membrane composition and peptide concentrations used in these experiments. Therefore, the CODEX intensities measured here exclusively reflect 19F spin diffusion.
Supramolecular organization of ETM is affected by phosphatidylinositol
The observed pentamer clustering, as shown by CODEX intensities below 0.20 at long mixing times, lead to the question of whether certain lipids in the ERGIC mixture are responsible for the clustering of multiple pentamers. To answer this question, we reconstituted 4-19F-Phe23 labeled ETM into three simplified lipid membranes. We first tested a cholesterol-free ERGIC membrane, which contains only the four phospholipids (Table 1). This membrane was chosen based on the recent NMR evidence that cholesterol binds to and mediates the clustering of influenza M2 48, 49, HIV gp41 50, 51 and the amyloid precursor protein 52 in lipid membranes. Contrary to this hypothesis, the cholesterol-free ERGIC-bound ETM at P : L = 1 : 17 gives a similar CODEX equilibrium intensity of 0.17 ± 0.02 at 4 s (Fig 2e, Fig. 3g, Table 2). The cholesterol-free sample also displays a long 19F T1 relaxation time of 4.1 s. Thus, the presence or absence of cholesterol in the ERGIC membrane does not affect pentamer clustering.
We next simplified the membrane further by removing both phosphatidylinositol and cholesterol. Interestingly, this ternary mixture, POPC/POPS/POPE, produced substantial spectral changes. The minor component at −111 ppm increased its intensity relative to the major peak at −115 ppm in the CP spectrum compared to the other samples (Fig. 2a), to about 25% of the total integrated intensity of the two peaks. Moreover, the downfield signal has a similarly long 19F T1 relaxation time (3.6 s) as in the ERGIC membranes while the upfield peak has a much shorter T1 of 0.9 s (Fig. 3h). As a result of these dramatically different T1 relaxation times, the two peaks have similar S0 intensities (60% downfield : 40% upfield) at long CODEX mixing times (Fig. 2f) in qualitative contrast to the other samples. Thus, in the absence of phosphatidylinositol, two populations of the ETM oligomers are present in the membrane, a minor component (~25%) of immobilized peptide with long T1 and a major component of more dynamic peptide. The distinct T1 relaxation behaviors could reflect the heterogeneous dynamics of the entire peptide or the Phe20 sidechain alone. These two populations are not in close contact, as no cross peaks are observed in 2D 19F correlation spectra even after 400 ms of Combined R2nν-Driven (CORD) spin diffusion 53 (Fig. S2). The CODEX intensities of the major component show similarly low S/S0 values as the peptide in other membranes. At 3 s, the intensity is 0.22 ± 0.06, indicating that ETM remains pentameric in this ternary membrane.
Finally, we bound 4-19F-Phe23 labeled ETM to the model membrane DMPC/DMPG at a P : L of 1 : 30. In the absence of cholesterol and phosphatidylinositol and at this relatively low peptide concentration, this membrane sample provides a control for the propensity of ETM to self-assemble in lipid bilayers. We measured a CODEX intensity of 0.21 ± 0.04 at 2.5 s (Fig. 2g, Fig. 3i), again indicating a pentamer. The 19F T1 of the peptide is 1.5 s, suggesting that the ETM pentamers are not clustered under this condition.
To gain insight into the possible arrangements of the ETM pentamers in the clusters and whether they agree with the measured CODEX intensities at long mixing times, we simulated a random distribution and clustered distribution of oligomers. For isolated symmetric pentamers, the CODEX simulation yielded a best-fit pentamer side length of 8.8 Å (Fig. 5a). As shown above, this does not fully reproduce the experimental data at long mixing times. To generate a random distribution of pentamers without inter-oligomer interactions, we implemented the RSA method, whose resulting distribution in a 100 × 100 nm2 square is shown in Fig. 5b. This RSA distribution produces inter-oligomer distances that are, on average, approximately half of those in a regular lattice model. A 100 × 100 nm2 should be occupied by approximately 325 oligomers (Eqn. 1). In a regular lattice, the inter-oligomer spacing is approximately 55 Å. With RSA, the average nearest-neighbor distance from the center of one oligomer to the next decreases to 37 Å. Subtracting twice the channel radius (13 Å) gives an approximate average 19F-19F distance of 11 Å, which is within the detectable range by 19F spin diffusion NMR 54. However, since the rate of 1H-driven spin diffusion depends on r−6, a 11 Å distance on average is only a small correction to the intra-oligomer distance of ~9 Å in the isolated pentamer model. To reproduce the experimentally measured CODEX dephasing, a more specific clustering model is required. We adopted a potential of mean force that accounts for both protein shape and protein-lipid hydrophobic mismatch (Fig. S3) 39 and simulated the resulting distribution using a Metropolis Monte Carlo method (Fig. 5c). The calculated CODEX S/S0 and S0/S curves for these two models are compared with the isolated pentamer simulation in Fig. 5d. The RSA distribution produced only a minor intensity reduction at long mixing times, and thus does not significantly improve the fit over the isolated pentamer fit. The cluster distribution resulted in a noticeable decrease in S/S0 intensity at long mixing times and agreed much better with the experimental CODEX intensities. Therefore, inter-pentamer contacts, which are only significant in the clustered distribution (Fig. 5e), reproduce the observed intensity decays below 0.20 at long mixing times. The improved fit at long mixing times still does not fully reproduce the measured intensities at intermediate mixing times between 1 s and 2 s. Instead, these intensities are well-described by asymmetric pentamers (Fig. 5f). This suggests that the 4-19F label at the Phe23 sidechains may exhibit some rotameric disorder, leading to inequivalent distances between adjacent subunits, as represented by an asymmetric pentamer.
Figure 5.

Alternative ETM oligomerization models and their corresponding CODEX simulations. The experimental data (filled circles) are those of ERGIC-bound 4-19F-Phe23 labeled ETM at P : L = 1 : 17. Simulated CODEX decays are shown in solid lines. (a) Measured CODEX decays together with best-fit simulation using an isolated symmetric pentamer model. The simulation is the same as in Fig. 3a. The nearest-neighbor distance of the best-fit pentamer is 8.8 Å. While the overall agreement between the experiment and simulation is good (), the intensities deviate at intermediate (1–2 s) and long mixing times (4 s). (b) Simulated RSA distribution of pentamers in a 100 x 100 nm2 square. All couplings stronger than 0.85 Hz (50 Å) are considered in the calculation, though convergence occurs around 15 Å. (c) Simulated cluster distribution of pentamers based on a cylinder-cylinder interaction and hydrophobic mismatch model. (d) Measured CODEX decays shown as S/S0 and the inverse S0/S values. These are overlaid with simulated curves for the isolated symmetric pentamer model, the random distribution model, and the clustered distribution model. The clustered pentamer model agrees best with the measured CODEX intensities at long mixing times. (e) Average nearest-neighbor distances from the RSA simulation (blue) and the cluster simulation (red). The distances from the RSA distribution were obtained from 1000 independent simulations. The distances from the interaction potential simulation were obtained from 50,000 Monte Carlo steps to reach a final distribution. (f) Experimental CODEX data with best-fit simulation using an isolated asymmetric pentamer model. Gradient decent fitting yielded better agreement between the experiment and simulations compared to (a), and the nearest-neighbor distances in the model range from 6.5 Å to 10.4 Å.
Calibration of the overlap integral F(0)
The overlap integral Fij(0) is the probability that single-quantum transitions occur at the same frequency for spin i and spin j. It is related to the normalized zero-quantum line shape at zero frequency. As in existing literature 43, 44, 54-57, here we treat F(0) as a parameter to be calibrated from known crystal structures. 19F CODEX of 5-19F-trptophan (Fig. S4a) shows an exponential decay to S/S0 = 0.50 at 400 ms. Fitting to 0.5+0.5e−ct shows excellent agreement with the experimental data (R2 = 0.9935) for the time constant c = 183 s−1 (Fig. S4b). Using the known crystal structure of L-tryptophan hydrochloride 58, we calibrated the F(0) value. L-tryptophan hydrochloride crystallized in the P21 space group, with two orientationally inequivalent molecules in the unit cell. The nearest neighbor H5-H5 distance in the lattice is 4.62 Å. Second moment analysis, summing over additional unit cells until the effective dipolar coupling converges, gives an effective 19F-19F dipolar coupling of 4.99 kHz, which corresponds to an effective distance of 4.16 Å. The rate of 1H-driven spin diffusion driving CODEX is given by kij = 0.5 πωijFij(0) 19, 59. The dipolar coupling (ωij) is given by the dipolar coupling constant, and the powder averaged angular dependence, . Calculating the dipolar coupling based on the converged effective 19F-19F distance allowed the extraction of the F(0) value of 3.4±0.4 μs. A detailed description of the calculation of F(0) is given in the Supporting Information. Performing CODEX matrix calculations at varying F(0) values yields identical results as the exponential fitting does (Fig, S4c-d).
This F(0) value is an order of magnitude smaller than the previously reported value of 37 μs by our group 19. The previous value was measured under 8 kHz MAS on a 9.4 T magnet and should be comparable to the measurements in this work. Recalculation based on the previous data indicates that the previously reported F(0) value incorporated an additional factor of , which was also applied in the matrix calculation scripts for the CODEX simulations. The F(0) values were first obtained from the CODEX decay of samples with known crystal structures, then these F(0) values were used in CODEX calculations with unknown distances. Since the same equation with an error of was used both to calibrate and to determine unknown distances, the previously reported distances are correct, only the numerical value of F(0) was mis-reported. Correcting for this factor, the true F(0) value should be 3.7 μs for 19F and 8.0 μs for 13C. Other literature values for F(0) of 13C spins range from 1.2 to 11.4 μs 55, 57, in agreement with the revised value.
Discussion
SARS-CoV-2 ETM forms pentameric channels that cluster in lipid membranes
The CODEX data obtained here for six ETM samples show that the basic unit of assembly of ETM in lipid bilayers is a pentamer (Fig. 6a). In four membrane mixtures and at three peptide concentrations, the CODEX intensities of 4-19F Phe23 decay to a value between 0.15 – 0.22 at 3 s. At the lowest peptide concentration where inter-oligomer contact is minimal, the CODEX intensities equilibrate to 0.20 (Fig. 3b, e). Thus, when the protein is sufficiently dilute in the membrane so that 19F spin diffusion is restricted to isolated channels, the stoichiometry of the complex can be established to be a pentamer. At higher peptide concentrations, the CODEX intensities decrease below 0.20 in ERGIC-mimetic and phosphatidylinositol-containing membranes, indicating that the pentamers cluster under these conditions (Fig. 6b).
Figure 6.

SARS-CoV-2 ETM forms pentameric α-helical bundles in lipid bilayers. (a) Top view of the ETM pentamer structure, using the solid-state NMR structural model of the closed state of ETM (PDB: 7K3G) 8. Intra-pentamer 19F spin diffusion is indicated by arrows. (b) Model of the clustering of ETM pentamers in ERGIC-mimetic phosphatidylinositol containing lipid bilayers at high peptide concentrations. Spin diffusion between different pentamers is illustrated by arrows.
The low CODEX intensities of less than 0.25 at mixing times longer than 2 s (Table 2) rule out a substantial population of small oligomers such as dimers and trimers. If we assume 20% to be the lower limit of the population of an oligomeric species that can be detected by NMR, then a mixture of 20% dimers and 80% pentamers would give a CODEX equilibrium value of 0.26. This is significantly higher than the measured CODEX equilibrium values of 0.20 for the three ERGIC samples (Table 2). Alternatively, if a dimer is assumed to coexist with a hexamer, then the dimer fraction cannot be larger than 10% in order to agree with the data. Thus, the experimental data rule out the presence of a sizeable fraction of small oligomers.
Furthermore, the 13C, 15N and 19F spectral linewidths of membrane-bound ETM rule out the coexistence of two distinctly different subunit stoichiometries. We found narrow 13C linewidths of ~0.5 ppm and 15N linewidths of ~0.9 ppm for the protein, implying a highly homogenous protein conformation for both the backbone and sidechain. The 19F linewidths of 4-19F Phe23 are ~1.5 ppm, implying that the helix-helix interface of the ETM oligomer is also structurally homogeneous. This high degree of homogeneity coupled with a lack of peak doubling makes the presence of two distinct structural states, a dimer and hexamer, or pentamer and heptamer for example, highly unlikely.
Is it possible that ETM forms a mixture of similarly high molecular-weight oligomers such as tetramers and hexamers, or pentamers and heptamers? Such a mixture cannot be ruled out based on the CODEX data alone. However, we consider this scenario unlikely because even a modest change of the oligomer number will affect the sidechain packing between neighboring helices, which should cause broad 19F peaks. The 19F spectra of 4-19F-Phe23 exhibit a dominant peak at −115 ppm that accounts for 75-95% of the spectral intensity and a minor peak at −111 ppm that represents 5-25% of the spectral intensities. The minor peak became more discernible in the POPC/POPE/POPS membrane due to its long 19F T1 relaxation time compared to the T1 of the major peak (Fig. 2f, Fig. 3h). However, its CODEX intensities are not different from those of the major component within experimental uncertainty. Therefore, for all the lipid membranes examined here, we do not find evidence for a coexistence of multiple oligomeric states.
The stoichiometry of virus membrane proteins can vary in different membrane-mimetic environments. The influenza viroporin, M2, is stably tetrameric in detergents and lipid bilayers, as shown by AUC experiments in detergent micelles 17, 18, solid-state NMR CODEX data in lipid bilayers 19, 20, and X-ray crystal structures in lipid cubic phases 22. Moreover, whole cell current measurements in oocytes that express a mixture of amantadine-sensitive wild-type M2 and amantadine-resistant mutant M2 definitively showed that the tetrameric state is the functionally active state 16. The HIV viroporin Vpu forms pentamers based on gel filtration chromatography data 25, and the pentamer is suggested to be the most thermodynamically stable species by molecular dynamics simulations 24. However, evidence for stoichiometries from tetramers to hexamers has also been reported from crosslinking, gel electrophoresis and AUC data in lipid bilayers 26, 60. The dual-TM-helix hepatitis c virus p7 protein (HCV p7) can adopt either hexamers or heptamers depending on the membrane-mimetic environments 61-64. Compared to these viroporins, SARS-CoV-2 ETM forms stable pentamers in lipid bilayers based on the data shown here. The observation of multiple oligomeric states in various detergents might reflect different structures of the protein formed in these non-native environments and the small differences in thermodynamic stabilities between different oligomeric assemblies.
Increasing experimental evidence has shown that membrane proteins often cluster for function. Ion channels such as KcsA 65 and mammalian K+ and Ca2+ channels 66, 67 have been reported to cluster in the membrane to cause coupled gating. Influenza M2 68, 69 and HIV-1 gp41 50 have been recently shown to cluster in the membrane, mediated by cholesterol 48, 49, 51. This clustering is implicated in membrane curvature generation and may be used by both proteins to achieve their membrane scission and membrane fusion functions, respectively. Electron microscopy studies of the mouse hepatitis virus suggest that its E protein induces membrane curvature 70. SARS-CoV-2 E has a similar role in viral particle formation 71, thus its clustering might help to generate membrane curvature. Fluorescence microscopy data of the subcellular localization of the three membrane proteins (S, M and E) of the SARS-CoV-1 virus found that the E protein forms large membrane clusters that co-localize with endoplasmic reticulum markers 2, 72, but molecular evidence of clustering of E had not been reported until now. The current finding that ETM clusters in the ERGIC-mimetic lipid membrane explains the high rigidity of the peptide in this membrane at ambient temperature 8. Unlike M2 and gp41, clustering of the SARS-CoV-2 E pentamers in lipid membranes is not induced by cholesterol alone. Removal of cholesterol did not raise the CODEX intensities at long mixing times or decrease the 19F T1 (Fig. 3g). Instead, removing both cholesterol and phosphatidylinositol is required to increase the CODEX intensity to about 0.20 (Fig. 3h), which is diagnostic of dispersal of clusters. The isolation of the E pentamers is also manifested by the significantly shortened 19F T1 relaxation time. This result is reproduced in the model membrane DMPC/DMPG, in which ETM exhibits isolated pentamers with a short 19F T1. Thus, supramolecular assembly of ETM pentamers requires the presence of phosphatidylinositol.
The surprising finding that phosphatidylinositol is required for ETM clustering is intriguing. Phosphatidylinositol and its phosphorylated derivatives such as phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-triphosphate (PIP3) are known to self-associate in the membrane in the presence of divalent cations such as calcium 73, and can induce clustering of membrane proteins such as syntaxin-1A to mediate membrane fusion in neurons 74-76. Conversely, some membrane proteins have been reported to associate with and induce clustering of phosphatidylinositol to carry out functions such as signal transduction 77. Moreover, calcium ions are known to cluster anionic phosphatidylserine and phosphatidylinositol 78, 79. Thus, the current finding that the calcium-conducting E channel is clustered by phosphatidylinositol suggests that calcium ions, which are present at much higher concentrations inside the ERGIC than in the cytoplasm, may have the dual action of clustering the E protein via phosphatidylinositol as well as permeating these clustered channels. Future studies should investigate how phosphatidylinositol binds E in the supramolecular assembly in the membrane, and whether and how this interaction facilitates virus assembly and budding.
Structural implications of the interhelical distances between Phe23 sidechains
Simulation of the 19F CODEX decay curves yielded nearest-neighbor intra-pentamer distances of 8-9 Å for the 4-19F-Phe23 labels. This distance is relatively independent of the membrane composition. Previous work by our group solved a 2.1-Å structure of ETM at neutral pH 8. In this structure, Phe23 occupies the heptad position f, interacting with lipids. This configuration would give a nearest-neighbor distance of ~15 Å, which is much longer than observed by the current CODEX data. The interhelical 13C-19F distance constraints that contributed to the structure calculation were measured using recombinant triply fluorinated ETM samples at the three Phe residues. Thus, the sidechain conformations in this closed-state ETM structural model will need to be revised using more extensive distance measurements to accommodate the shorter Phe23-Phe23 distances seen in the CODEX data here. Already, the current 19F spectra of Phe23 indicate that this residue has a single predominant sidechain conformation, which differs from the dual-conformations seen for Phe20 and Phe26, which each manifest two 19F peaks 47. We recently showed that these two sidechain rotamers of Phe20 and Phe26 correspond to a lipid-facing conformer and a more water-accessible conformer at the helix-helix interface. In comparison, the distinct 19F spectra of Phe23 indicate that Phe23 adopts a single rotamer that may point either to the pore or to the helix-helix interface to occlude the channel at neutral pH. In the influenza M2 transmembrane peptide, aromatic gating is accomplished by a pore-facing Trp residue 44, 80-84. The HCV p7 channel also contains three Phe residues at positions 22, 25, and 26. Joint mutations of these three residues to Ala increased the channel activity, suggesting that aromatic interactions within this Phe stack restrict ion current 85. Thus, the HCV p7 channel bears interesting similarities to the SARS-CoV-2 E channel. Future studies to probe how these Phe residues stabilize the five-helix bundle and regulate channel gating will be important for elucidating the structure-function relation of the E protein.
Supplementary Material
Acknowledgement
This work is supported by NIH grant U19AI171110 to M.H. (subaward 13617sc) and W.F.D. The NMR experiments used equipment at the MIT-Harvard Center for Magnetic Resonance, which is supported by the P41 grant GM132079. N.H.S. gratefully acknowledges support by an NSF fellowship 1745302. J.M.-S. gratefully acknowledges the Rubicon Fellowship 452020132 supported by the Dutch Research Council (NWO) and the EMBO Non-Stipendiary Postdoctoral Fellowship ALTF 1056-2020. A.J.D. is supported by an NIH fellowship F31AG069418.
Footnotes
Accession code
The full-length SARS-CoV-2 E protein sequence can be found at NCBI YP_009724392.1, (residues 1–75).
Supporting Information Available
Additional details about the calculation of the overlap integral F(0), MALDI-MS data of ETM peptides, additional NMR spectra, plots of average nearest-neighbor distances from the RSA and Monte Carlo simulations, 5-19F-Trp F(0) calibration data, pairwise interaction potential used, and MATLAB code for simulating CODEX, RSA, and pore clustering. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- [1].Weiss SR, and Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus, Microbiol. Mol. Biol. Rev 69, 635–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schoeman D, and Fielding BC (2019) Coronavirus envelope protein: current knowledge, Virol. J 16, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nieva JL, Madan V, and Carrasco L (2012) Viroporins: structure and biological functions, Nat. Rev. Microbiol 10, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nieto-Torres JL, DeDiego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Fernandez-Delgado R, Castano-Rodriguez C, Alcaraz A, Torres J, Aguilella VM, and Enjuanes L (2014) Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis, PLoS Pathog. 10, e1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xia B, Shen X, He Y, Pan X, Liu FL, Wang Y, Yang F, Fang S, Wu Y, Duan Z, Zuo X, Xie Z, Jiang X, Xu L, Chi H, Li S, Meng Q, Zhou H, Zhou Y, Cheng X, Xin X, Jin L, Zhang HL, Yu DD, Li MH, Feng XL, Chen J, Jiang H, Xiao G, Zheng YT, Zhang LK, Shen J, Li J, and Gao Z (2021) SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target, Cell Res. 31, 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zumla A, Chan JF, Azhar EI, Hui DS, and Yuen KY (2016) Coronaviruses – drug discovery and therapeutic options, Nat. Rev. Drug. Discov 15, 327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Śledź P, and Caflisch A (2018) Protein structure-based drug design: from docking to molecular dynamics, Curr. Opin. Struct. Biol 48, 93–102. [DOI] [PubMed] [Google Scholar]
- [8].Mandala VS, McKay MJ, Shcherbakov AA, Dregni AJ, Kolocouris A, and Hong M (2020) Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers, Nat. Struct. Mol. Biol 27, 1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li Y, Surya W, Claudine S, and Torres J (2014) Structure of a conserved Golgi complex-targeting signal in coronavirus envelope proteins, J. Biol. Chem 289, 12535–12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Parthasarathy K, Ng L, Lin X, Liu DX, Pervushin K, Gong X, and Torres J (2008) Structural flexibility of the pentameric SARS coronavirus envelope protein ion channel, Biophys. J 95, L39–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Parthasarathy K, Lu H, Surya W, Vararattanavech A, Pervushin K, and Torres J (2012) Expression and purification of coronavirus envelope proteins using a modified β-barrel construct, Protein Expr. Purif 85, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Torres J, Wang J, Parthasarathy K, and Liu DX (2005) The Transmembrane Oligomers of Coronavirus Protein E, Biophys. J 88, 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Surya W, Li Y, Verdià-Bàguena C, Aguilella VM, and Torres J (2015) MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels, Virus Res. 201, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Westerbeck JW, and Machamer CE (2015) A Coronavirus E Protein Is Present in Two Distinct Pools with Different Effects on Assembly and the Secretory Pathway, J. Virol 89, 9313–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ruch TR, and Machamer CE (2012) A Single Polar Residue and Distinct Membrane Topologies Impact the Function of the Infectious Bronchitis Coronavirus E Protein, PLoS Pathog. 8, e1002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sakaguchi T, Tu Q, Pinto LH, and Lamb RA (1997) The active oligomeric state of the minimalistic influenza virus M2 ion channel is a tetramer, Proc. Natl. Acad. Sci. U. S. A 94, 5000–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Salom D, Hill BR, Lear JD, and DeGrado WF (2000) pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus, Biochemistry 39, 14160–14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Howard KP, Lear JD, and DeGrado WF (2002) Sequence determinants of the energetics of folding of a transmembrane four-helix-bundle protein, Proc. Natl. Acad. Sci. U. S. A 99, 8568–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luo W, and Hong M (2006) Determination of the Oligomeric Number and Intermolecular Distances of Membrane Protein Assemblies by Anisotropic 1H-Driven Spin Diffusion NMR Spectroscopy, J. Am. Chem. Soc 128, 7242–7251. [DOI] [PubMed] [Google Scholar]
- [20].Williams JK, Zhang Y, Schmidt-Rohr K, and Hong M (2013) pH-Dependent Conformation, Dynamics, and Aromatic Interaction of the Gating Tryptophan Residue of the Influenza M2 Proton Channel from Solid-State NMR, Biophys. J 104, 1698–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, and DeGrado WF (2008) Structural basis for the function and inhibition of an influenza virus proton channel, Nature 451, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thomaston JL, Woldeyes RA, Nakane T, Yamashita A, Tanaka T, Koiwai K, Brewster AS, Barad BA, Chen Y, Lemmin T, Uervirojnangkoorn M, Arima T, Kobayashi J, Masuda T, Suzuki M, Sugahara M, Sauter NK, Tanaka R, Nureki O, Tono K, Joti Y, Nango E, Iwata S, Yumoto F, Fraser JS, and DeGrado WF (2017) XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction, Proc. Natl. Acad. Sci. U. S. A 114, 13357–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].González ME (2015) Vpu Protein: The Viroporin Encoded by HIV-1, Viruses 7, 4352–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Padhi S, Khan N, Jameel S, and Priyakumar UD (2013) Molecular dynamics simulations reveal the HIV-1 Vpu transmembrane protein to form stable pentamers, PLoS One 8, e79779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hussain A, Das SR, Tanwar C, and Jameel S (2007) Oligomerization of the human immunodeficiency virus type 1 (HIV-1) Vpu protein--a genetic, biochemical and biophysical analysis, Virol. J 4, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sharpe S, Yau WM, and Tycko R (2006) Structure and dynamics of the HIV-1 Vpu transmembrane domain revealed by solid-state NMR with magic-angle spinning, Biochemistry 45, 918–933. [DOI] [PubMed] [Google Scholar]
- [27].Buffy JJ, Waring AJ, and Hong M (2005) Determination of Peptide Oligomerization in Lipid Membranes with Magic-Angle Spinning Spin Diffusion NMR, J. Am. Chem. Soc 127, 4477–4483. [DOI] [PubMed] [Google Scholar]
- [28].deAzevedo ER, Bonagamba TJ, Hu W, and Schmidt-Rohr K (1999) Centerband-only detection of exchange: efficient analysis of dynamics in solids by NMR, J. Am. Chem. Soc 121, 8411–8412. [Google Scholar]
- [29].deAzevedo ER, Bonagamba TJ, Hu W, and Schmidt-Rohr K (2000) Principle of centerband-only detection of the exchange and extension to a four-time CODEX., J. Chem. Phys 112, 8988–9001. [Google Scholar]
- [30].Hong M, and Schmidt-Rohr K (2013) Magic-angle-spinning NMR techniques for measuring long-range distances in biological macromolecules, Acc. Chem. Res 46, 2154–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, Berger T, Policarpo RL, Zhang C, Zou Y, Liao X, Spokoyny AM, Jensen KF, and Pentelute BL (2014) Rapid flow-based peptide synthesis, ChemBiochem 15, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Paquet A. (1982) Introduction of 9-fluorenylmethyloxycarbonyl, trichloroethoxycarbonyl, and benzyloxycarbonyl amine protecting groups into O-unprotected hydroxyamino acids using succinimidyl carbonates, Can. J. Chem 60, 976–980. [Google Scholar]
- [33].Schweizer A, Clausen H, van Meer G, and Hauri HP (1994) Localization of O-glycan initiation, sphingomyelin synthesis, and glucosylceramide synthesis in Vero cells with respect to the endoplasmic reticulum-Golgi intermediate compartment, J. Biol. Chem 269, 4035–4041. [PubMed] [Google Scholar]
- [34].Casares D, Escribá PV, and Rosselló CA (2019) Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues, Int. J. Mol. Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].van Meer G, Voelker DR, and Feigenson GW (2008) Membrane lipids: where they are and how they behave, Nat. Rev. Mol. Cell Biol 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hills RD Jr., and McGlinchey N (2016) Model parameters for simulation of physiological lipids, J. Comp. Chem 37, 1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Leftin A, Molugu TR, Job C, Beyer K, and Brown MF (2014) Area per lipid and cholesterol interactions in membranes from separated local-field 13C NMR spectroscopy, Biophys. J 107, 2274–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Evans JW (1993) Random and cooperative sequential adsorption, Rev. Mod. Phys 65, 1281–1329. [Google Scholar]
- [39].Morozova D, Weiss M, and Guigas G (2012) Shape as a determinant of membrane protein cluster formation, Soft Matter 8, 11905–11910. [Google Scholar]
- [40].Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, and Teller E (1953) Equation of State Calculations by Fast Computing Machines, J. Chem. Phys 21, 1087–1092. [Google Scholar]
- [41].Verdiá-Báguena C, Nieto-Torres JL, Alcaraz A, DeDiego ML, Torres J, Aguilella VM, and Enjuanes L (2012) Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids, Virology 432, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nieto-Torres JL, Dediego ML, Alvarez E, Jiménez-Guardeño JM, Regla-Nava JA, Llorente M, Kremer L, Shuo S, and Enjuanes L (2011) Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein, Virology 415, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kwon B, Lee M, Waring AJ, and Hong M (2018) Oligomeric Structure and Three-Dimensional Fold of the HIV gp41 Membrane-Proximal External Region and Transmembrane Domain in Phospholipid Bilayers, J. Am. Chem. Soc 140, 8246–8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kwon B, Roos M, Mandala VS, Shcherbakov AA, and Hong M (2019) Elucidating Relayed Proton Transfer through a His-Trp-His Triad of a Transmembrane Proton Channel by Solid-State NMR, J. Mol. Biol 431, 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee M, Yao H, Kwon B, Waring AJ, Ruchala P, Singh C, and Hong M (2018) Conformation and Trimer Association of the Transmembrane Domain of the Parainfluenza Virus Fusion Protein in Lipid Bilayers from Solid-State NMR: Insights into the Sequence Determinants of Trimer Structure and Fusion Activity, J. Mol. Biol 430, 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Salnikov ES, Raya J, De Zotti M, Zaitseva E, Peggion C, Ballano G, Toniolo C, Raap J, and Bechinger B (2016) Alamethicin Supramolecular Organization in Lipid Membranes from (19)F Solid-State NMR, Biophys. J 111, 2450–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Medeiros-Silva J, Somberg NH, Wang HK, McKay MJ, Mandala VS, Dregni AJ, and Hong M (2022) pH- and Calcium-Dependent Aromatic Network in the SARS-CoV-2 Envelope Protein, J. Am. Chem. Soc 144, 6839–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Elkins MR, Williams JK, Gelenter MD, Dai P, Kwon B, Sergeyev IV, Pentelute BL, and Hong M (2017) Cholesterol-binding site of the influenza M2 protein in lipid bilayers from solid-state NMR, Proc. Natl. Acad. Sci. U. S. A 114, 12946–12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Elkins MR, Sergeyev IV, and Hong M (2018) Determining Cholesterol Binding to Membrane Proteins by Cholesterol 13C Labeling in Yeast and Dynamic Nuclear Polarization NMR, J. Am. Chem. Soc 140, 15437–15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tran N, Oh Y, Sutherland M, Cui Q, and Hong M (2022) Cholesterol-Mediated Clustering of the HIV Fusion Protein gp41 in Lipid Bilayers, J. Mol. Biol 434, 167345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kwon B, Mandal T, Elkins MR, Oh Y, Cui Q, and Hong M (2020) Cholesterol Interaction with the Trimeric HIV Fusion Protein gp41 in Lipid Bilayers Investigated by Solid-State NMR Spectroscopy and Molecular Dynamics Simulations, J. Mol. Biol 432, 4705–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, and Sanders CR (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol, Science 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hou G, Yan S, Sun S, Han Y, Byeon IJ, Ahn J, Concel J, Samoson A, Gronenborn AM, and Polenova T (2011) Spin diffusion driven by R-symmetry sequences: applications to homonuclear correlation spectroscopy in MAS NMR of biological and organic solids, J. Am. Chem. Soc 133, 3943–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Roos M, Wang T, Shcherbakov AA, and Hong M (2018) Fast Magic-Angle-Spinning 19F Spin Exchange NMR for Determining Nanometer 19F-19F Distances in Proteins and Pharmaceutical Compounds, J. Phys. Chem. B 122, 2900–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Olender Z, Reichert D, Müller A, Zimmermann H, Poupko R, and Luz Z (1996) Carbon-13 Chemical-Shift Correlation, Spin Diffusion and Self Diffusion in Isotopically Enriched Tropolone, J. Magn. Reson., Ser. A 120, 31–45. [Google Scholar]
- [56].Williams JK, Shcherbakov AA, Wang J, and Hong M (2017) Protonation equilibria and pore-opening structure of the dual-histidine influenza B virus M2 transmembrane proton channel from solid-state NMR, J. Biol. Chem 292, 17876–17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Xue K, Dervisoglu R, Sowa H, and Andreas LB (2020) Centerband-Only Detection of Exchange NMR with Natural-Abundance Correction Reveals an Expanded Unit Cell in Phenylalanine Crystals, ChemPhysChem 21, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Takigawa T, Ashida T, Sasada Y, and Kakudo M (1966) The crystal structures of L-tryptophan hydrochloride and hydrobromide, Bull. Chem. Soc. Jpn 39, 2369–2378. [DOI] [PubMed] [Google Scholar]
- [59].Vanderhart DL (1987) Natural-abundance 13C-13C spin exchange in rigid crystalline organic solids, J. Magn. Reson. (1969-1992) 72, 13–47. [Google Scholar]
- [60].Lu J-X, Sharpe S, Ghirlando R, Yau W-M, and Tycko R (2010) Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers, Protein Sci. 19, 1877–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Clarke D, Griffin S, Beales L, Gelais CS, Burgess S, Harris M, and Rowlands D (2006) Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro, J. Biol. Chem 281, 37057–37068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, and Rowlands DJ (2003) The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine, FEBS Lett. 535, 34–38. [DOI] [PubMed] [Google Scholar]
- [63].Montserret R, Saint N, Vanbelle C, Salvay AG, Simorre JP, Ebel C, Sapay N, Renisio JG, Böckmann A, Steinmann E, Pietschmann T, Dubuisson J, Chipot C, and Penin F (2010) NMR structure and ion channel activity of the p7 protein from hepatitis C virus, J. Biol. Chem 285, 31446–31461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Luik P, Chew C, Aittoniemi J, Chang J, Wentworth P Jr., Dwek RA, Biggin PC, Vénien-Bryan C, and Zitzmann N (2009) The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy, Proc. Natl. Acad. Sci. U. S. A 106, 12712–12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Visscher KM, Medeiros-Silva J, Mance D, Rodrigues J, Daniëls M, Bonvin A, Baldus M, and Weingarth M (2017) Supramolecular Organization and Functional Implications of K+ Channel Clusters in Membranes, Angew. Chem., Int. Ed. Engl 56, 13222–13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, and Santana LF (2010) Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome, Circ. Res 106, 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lim ST, Antonucci DE, Scannevin RH, and Trimmer JS (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons, Neuron 25, 385–397. [DOI] [PubMed] [Google Scholar]
- [68].Sutherland M, Tran N, and Hong M (2022) Clustering of tetrameric influenza M2 peptides in lipid bilayers investigated by 19F solid-state NMR, Biochim. Biophys. Acta, Biomembr 1864, 183909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Paulino J, Pang X, Hung I, Zhou HX, and Cross TA (2019) Influenza A M2 Channel Clustering at High Protein/Lipid Ratios: Viral Budding Implications, Biophys. J 116, 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Raamsman MJ, Locker JK, de Hooge A, de Vries AA, Griffiths G, Vennema H, and Rottier PJ (2000) Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E, J. Virol 74, 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, Lavillette D, and Denolly S (2021) The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles, J. Biol. Chem 296, 100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nal B, Chan C, Kien F, Siu L, Tse J, Chu K, Kam J, Staropoli I, Crescenzo-Chaigne B, Escriou N, van der Werf S, Yuen KY, and Altmeyer R (2005) Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E, J. Gen. Virol 86, 1423–1434. [DOI] [PubMed] [Google Scholar]
- [73].Wang YH, Collins A, Guo L, Smith-Dupont KB, Gai F, Svitkina T, and Janmey PA (2012) Divalent cation-induced cluster formation by polyphosphoinositides in model membranes, J. Am. Chem. Soc 134, 3387–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Khuong TM, Habets RL, Kuenen S, Witkowska A, Kasprowicz J, Swerts J, Jahn R, van den Bogaart G, and Verstreken P (2013) Synaptic PI(3,4,5)P3 is required for Syntaxin1A clustering and neurotransmitter release, Neuron 77, 1097–1108. [DOI] [PubMed] [Google Scholar]
- [75].James DJ, Khodthong C, Kowalchyk JA, and Martin TF (2008) Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion, J. Cell Biol 182, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Müllar S, Diederichsen U, Fasshauer D, Grubmüller H, Hell SW, Eggeling C, Kühnel K, and Jahn R (2013) Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment, Nat. Struct. Mol. Biol 20, 679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Aleshin AE, Yao Y, Iftikhar A, Bobkov AA, Yu J, Cadwell G, Klein MG, Dong C, Bankston LA, Liddington RC, Im W, Powis G, and Marassi FM (2021) Structural basis for the association of PLEKHA7 with membrane-embedded phosphatidylinositol lipids, Structure 29, 1029–1039.e1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Graber ZT, Shi Z, and Baumgart T (2017) Cations induce shape remodeling of negatively charged phospholipid membranes, Phys Chem Chem Phys 19, 15285–15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Boettcher JM, Davis-Harrison RL, Clay MC, Nieuwkoop AJ, Ohkubo YZ, Tajkhorshid E, Morrissey JH, and Rienstra CM (2011) Atomic view of calcium-induced clustering of phosphatidylserine in mixed lipid bilayers, Biochemistry 50, 2264–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mandala VS, Liao SY, Kwon B, and Hong M (2017) Structural Basis for Asymmetric Conductance of the Influenza M2 Proton Channel Investigated by Solid-State NMR Spectroscopy, J. Mol. Biol 429, 2192–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hu F, Luo W, and Hong M (2010) Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR, Science 330, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, and DeGrado WF (1997) A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity, Proc Natl Acad Sci U S A 94, 11301–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tang Y, Zaitseva F, Lamb RA, and Pinto LH (2002) The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue, J. Biol. Chem 277, 39880–39886. [DOI] [PubMed] [Google Scholar]
- [84].Williams JK, Tietze D, Lee M, Wang J, and Hong M (2016) Solid-State NMR Investigation of the Conformation, Proton Conduction, and Hydration of the Influenza B Virus M2 Transmembrane Proton Channel, J. Am. Chem. Soc 138, 8143–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].StGelais C, Foster TL, Verow M, Atkins E, Fishwick CW, Rowlands D, Harris M, and Griffin S (2009) Determinants of hepatitis C virus p7 ion channel function and drug sensitivity identified in vitro, J. Virol 83, 7970–7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.