

Abstract

Sphingosine 1-phosphate (S1P) is a pleiotropic signaling molecule that interacts with five native G-protein coupled receptors (S1P1–5) to regulate cell growth, survival, and proliferation. S1P has been implicated in a variety of pathologies including cancer, kidney fibrosis, and multiple sclerosis. As key mediators in the synthesis of S1P, sphingosine kinase (SphK) isoforms 1 and 2 have attracted attention as viable targets for pharmacologic intervention. In this report, we describe the design, synthesis, and biological evaluation of sphingosine kinase 2 (SphK2) inhibitors with a focus on systematically introducing rigid structures in the aliphatic lipid tail present in existing SphK2 inhibitors. Experimental as well as molecular modeling studies suggest that conformationally restricted “lipophilic tail” analogues bearing a bulky terminal moiety or an internal phenyl ring are useful to complement the “J”-shaped sphingosine binding pocket of SphK2. We identified 14c (SLP9101555) as a potent SphK2 inhibitor (Ki = 90 nM) with 200-fold selectivity over SphK1. Molecular docking studies indicated key interactions: the cyclohexyl ring binding in the cleft deep in the pocket, a trifluoromethyl group fitting in a small side cavity, and a hydrogen bond between the guanidino group and Asp308 (amino acid numbering refers to human SphK2 (isoform c) orthologue). In vitro studies using U937 human histiocytic lymphoma cells showed marked decreases in extracellular S1P levels in response to our SphK2 inhibitors. Administration of 14c (dose: 5 mg/kg) to mice resulted in a sustained increase of circulating S1P levels, suggesting target engagement.
Keywords: sphingosine, sphingosine-1-phosphate, sphingosine kinase, conformational rigidity, structure−activity relationships, S1P
Sphingosine 1-phosphate (S1P) is a bioactive lipid that regulates the growth, survival, and migration of several cell types. While the interaction of S1P with a set of G-protein coupled receptors is well established, several intracellular targets of S1P including histone deacetylases,1 tumor-necrosis factor receptor-associated factor 2 (TRAF2),2 and erythrocyte glycolysis3 have been suggested. Cellular synthesis of S1P occurs solely via ATP-dependent phosphorylation of sphingosine (Sph) by sphingosine kinase 1 or 2 (Figure 1).4 Despite being catalytically indistinguishable, the two isoforms differ in cellular localization, which suggests different intracellular roles.5,6 While ablation of all four Sphk alleles in mice indicates that sphingosine kinases are the only source of S1P, some functional redundancy exists as mice with any single functional Sphk allele are viable and fertile.4 Modulating the production of S1P has the potential to affect a wide range of diseases including cancer,7,8 sickle cell disease,9,10 atherosclerosis,11,12 asthma,13,14 diabetes,11 and fibrosis.15 Initial drug discovery efforts have yielded potent SphK1 inhibitors, a process that was aided by the availability of a SphK1 crystal structure.16−19 However, such inhibitors have proven more elusive for SphK2, for which no crystal structure currently exists. Instead, inhibitor design has largely focused on homology modeling and developing ATP-competitive inhibitors.
Figure 1.

S1P pathway.
An SphK2 inhibitor that is often used is ABC294640 (1, Figure 2).20 This inhibitor, which has a Ki value of 10 μM and favorable pharmacokinetic properties, has been deployed in a variety of disease models, including those for ulcerative colitis,21 Crohn’s disease,22 ischemia/reperfusion injury,23 osteoarthritis,22 and colorectal cancer.24 Recently, ABC294640 (opaganib) has progressed into clinical trials for the treatment of cholangiocarcinoma (NCT03414489)25 and COVID-19 (NCT04467840).26 However, opaganib is not without liabilities. In addition to low potency, the molecule has been reported to act as a tamoxifen-mimetic at estrogen receptors.27 Other SphK2-selective inhibitors, namely, (R)-FTY720-OMe28 and K145,29 exhibit optimal selectivity, but are only moderately more potent than ABC294640. Importantly, none of those inhibitors has been documented to affect the pharmacodynamic marker of SphK2 inhibition, which is increased circulating S1P.30−32 In contrast to SphK1, SphK2 deficiency increases circulating S1P markedly.30−32 This seemingly paradoxical effect is due to the role of hepatic SphK2 in clearing S1P from blood.33
Figure 2.

Structures and inhibitory activity of SphK2-selective inhibitors.
Through structure–activity relationship (SAR) studies, our group has developed isoform selective SphK2 inhibitors with increased potency and metabolic durability in mice (Figure 2). SLR080811 (4) has Ki values of 12 and 1.3 μM for SphK1 and SphK2, respectively.30 Exchanging the pyrrolidine ring for an azetidine in SLP120701 (5) led to an improved in vivo half-life of 8 h in mice.31 Using 4 as a lead, subsequent studies led to the discovery of SLM6031434 (6) with a Ki value of 0.4 μM and 50-fold selectivity preference for SphK2 relative to SphK1, and established the importance of the guanyl pyrrolidine and 1,2,4-oxadiazole functionalities.32 Moreover, compound 6 demonstrated significant improvements to both SphK2 potency and selectivity and elicited the in vivo phenotype expected for a SphK2 inhibitor.32 Notable inclusions to the structure of 6 are the additional aryl trifluoromethyl functionality and oxygen heteroatom conjoining the alkyl chain and phenyl ring. This alkoxy substitution has been shown to be beneficial in developing successful SphK2-selective inhibitors with the motif appearing in SLC5091592 (7) as well.34 These analogues have been validated through in vivo testing in mice, where they produced ca. 3-fold increases in blood S1P levels following intraperitoneal injection (5 mg/kg),32 a hallmark of SphK2 inhibition.26−28
To improve potency and selectivity, we initiated an SAR campaign focused on modifying 6. Our design strategy included dividing 6 into three regions: a guanidine-containing pyrrolidine head, a rigid benzo-oxadiazole linker, and an aliphatic tail (Figure 3). In our earlier studies, we established the necessity of the guanidine group and the rigid heteroaromatic 1,2,4-oxadiazole linker for kinase inhibition.31 Recent work published by our lab has shown the availability of a small side cavity within the binding pocket that imparts SphK2 selectivity over SphK1.35 In particular, the trifluoromethyl group clashes with the Ile174 in SphK1, which is exchanged with a smaller Val residue in SphK2. In our current investigation, we focused our studies on the long aliphatic lipid tail present in sphingosine and previous SphK2 inhibitors to determine whether structural rigidification strategies such as the introduction of alkene and aryl motifs can improve the binding affinity and enhance Sph binding pocket occupancy. Conformational constraint has been demonstrated to improve specificity, potency, metabolic stability, and bioavailability.36−42 Our studies suggest that the incorporation of rigid tails can be highly efficacious in increasing both the potency and selectivity of SphK2 inhibitors.
Figure 3.
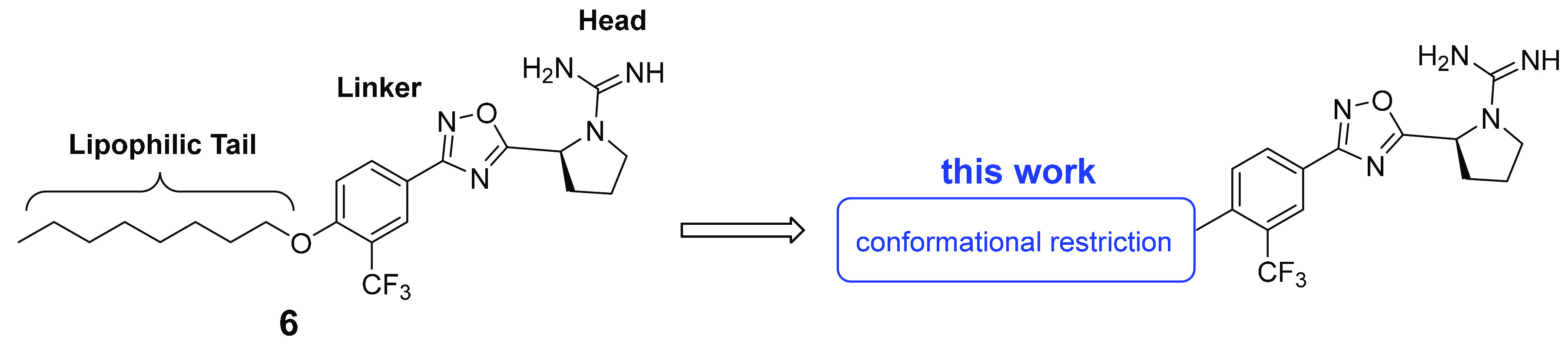
SphK2 pharmacophore.
Results and Discussion
Chemical Synthesis
Initial ω-substitutions were incorporated into the tail region in accordance with Scheme 1. Commercially available aryl bromide 8 was reacted under Sonagashira coupling conditions with terminal alkynes to afford the substituted phenylacetylene intermediates 9a–i. The generated internal alkyne was subjected to complete chemoselective reduction with Lindlar’s catalyst to form the alkyl-substituted benzonitrile intermediates 10a–i. Treatment of 10a–i with hydroxylamine hydrochloride and TEA in refluxing ethanol afforded an isomeric mixture of amidoximes 11a–i in excellent yield. Subsequent HCTU-assisted coupling with N-Boc-l-proline at 110 °C generated the 1,2,4-oxadiazoles 12a–i. Removal of the Boc group was accomplished with gaseous hydrochloric acid to afford the pyrrolidine intermediate as an ammonium salt. Without further purification, this intermediate was converted to the bis-Boc protected guanidines 13a–i via a reaction with N,N′-di-Boc-1H-pyrazole-1-carboxamidine and DIEA. Exposure of 13a–i to hydrogen chloride generated the guanidinium salts 14a–i.
Scheme 1. Synthesis of Derivatives 14a–i.
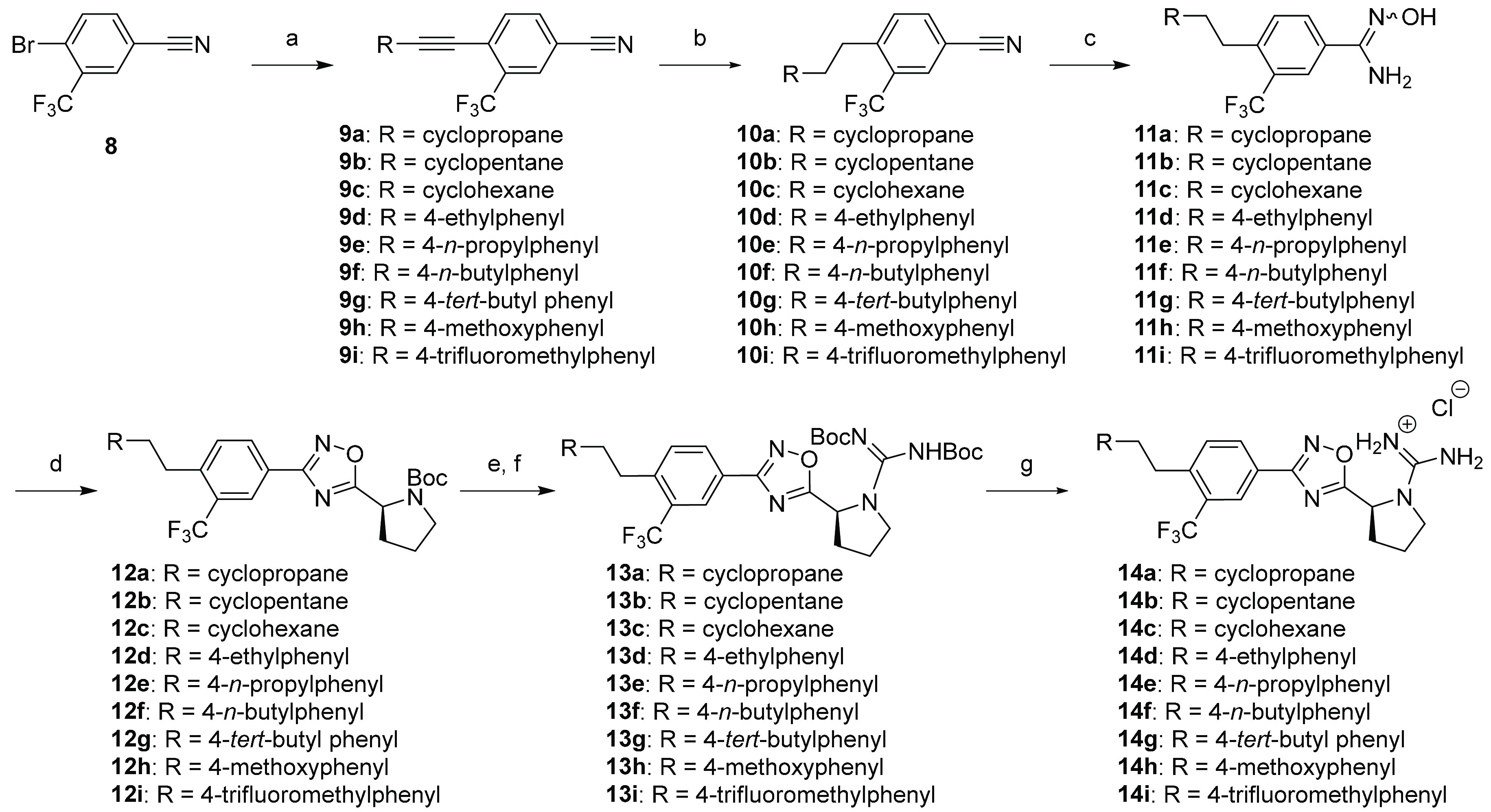
Reaction conditions: (a) Alkyne, Pd(PPh3)2Cl2, CuI, TEA, DMF, 80 °C, 4–12 h; (b) Lindlar’s catalyst, H2, EtOAc, 1–20 h; c) NH2OH·HCl, TEA, EtOH, 80 °C, 6 h; (d) Boc-l-proline, DIEA, HCTU, DMF, 110 °C, 18 h; (e) HCl(g)/MeOH, rt, 4 h; (f) N,N′-di-Boc-1H-pyrazole-1-carboxamidine, DIEA, MeCN, 55 °C, 2–4 h, microwave; (g) 4M HCl/Dioxane, rt, 3 h.
Alkene functionalities were incorporated utilizing the cross-metathesis strategy described in Scheme 2. Benzonitrile 15 was subjected to nucleophilic aromatic substitution with potassium tert-butoxide in THF, producing the O-tert-butyl intermediate 16. Treatment of compound 16 with hydroxylamine hydrochloride and TEA in refluxing ethanol resulted in a mixture of E- and Z-amidoxime 17. Cyclization of 17 with HCTU and N-Boc-l-proline yielded 1,2,4-oxadiazole 18. A one-pot reaction of 18 with gaseous hydrochloric acid and Boc anhydride afforded the N-Boc protected phenol derivative 19. O-Alkylation of 19 with allyl bromide and homoallyl bromide provided the common intermediates 20a and 20b, respectively, with a terminal alkene handle for cross-metathesis. A Grubbs second generation catalyst was employed for cross-metathesis with commercially available terminal alkenes, affording the rigidified ether tail analogues 21a–i. Cross-metathesis with alkenes possessing aliphatic substitution resulted in a mixture of E- and Z-diastereomers (approximately 60:40), while coupling with styrene derivatives generated the E-pyrrolidinium salt intermediate, which was converted to the bis-Boc protected guanidines 22a–iE-isomer almost exclusively. Subsequent treatment with trifluoroacetic acid produced a pyrrolidinium salt intermediate, which was converted to the bis-Boc protected guanidines 22a–i through a one-pot reaction with N,N′-Di-Boc-1H-pyrazole-1-carboxamidine and DIEA. Removal of Boc protecting groups with trifluoroacetic acid afforded compounds 23a–i as gaunidinium salts.
Scheme 2. Synthesis of Ether Tail Derivatives 23a–i.
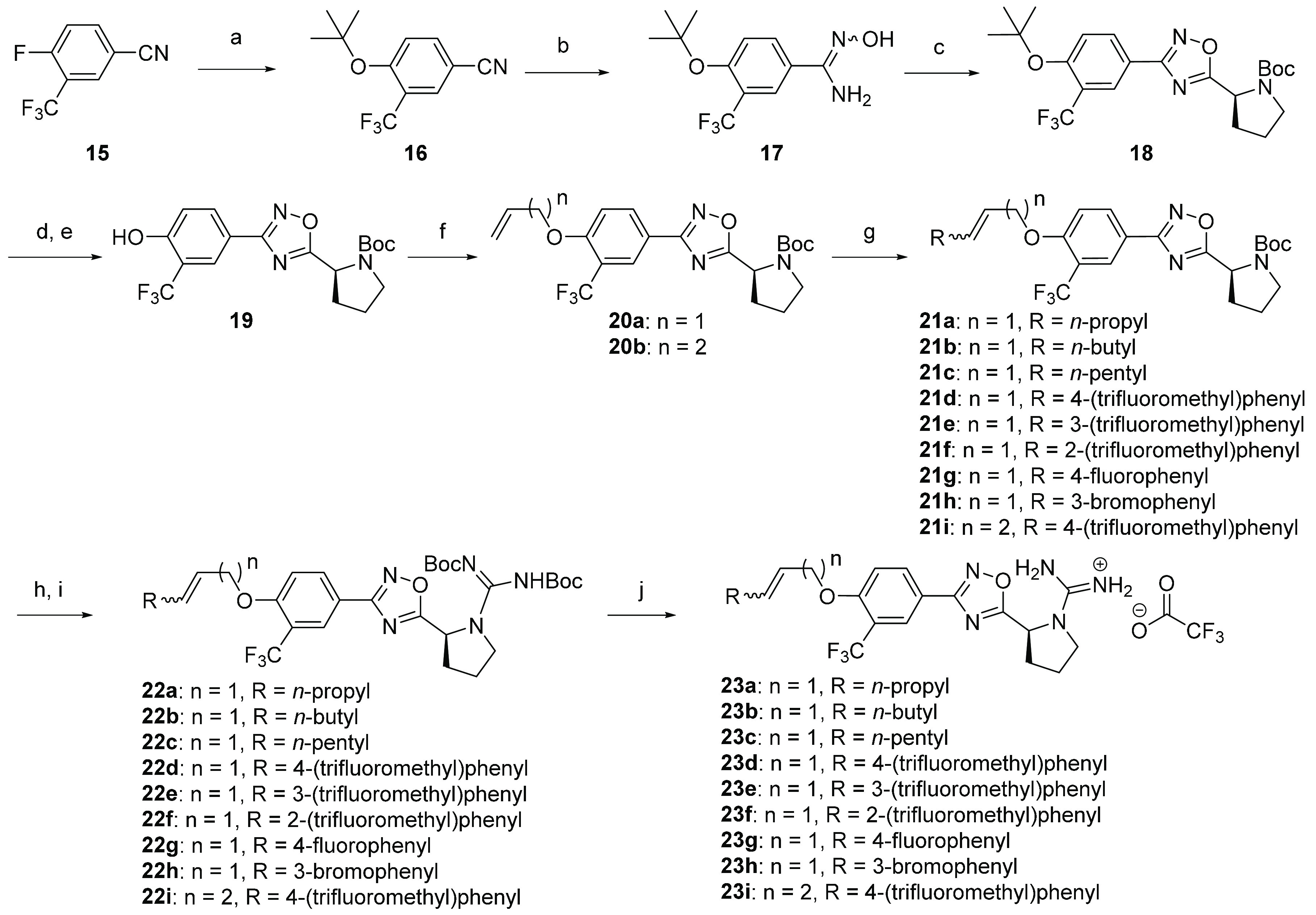
Reaction conditions: (a) KOtBu, THF, 60 °C, 4 h; (b) TEA, NH2OH·HCl, TEA, EtOH, 80 °C, 6 h; (c) Boc-l-proline, DIEA, HCTU, DMF, 110 °C, 18 h; (d) TFA, DCM, 3–6 h, 0 °C to rt; (e) (Boc)2O, TEA, THF, rt, 2–5 h; (f) alkyl bromide, K2CO3, acetone, 90 °C, 2 h, microwave; (g) Grubbs II catalyst, terminal alkenes, DCM, 70 °C, 2–4 h, microwave; (h) TFA, DCM, 0 °C to rt, 3–6 h; (i) N,N′-Di-Boc-1H-pyrazole-1-carboxamidine, DIEA, MeCN, 55 °C, 2–6 h, microwave; (j) TFA, DCM, rt, 4 h.
Aliphatic substituted alkenes generated by Scheme 2 yielded isomeric mixtures of the E- and Z-alkene. To identify the more active isomers, we synthesized E- and Z-alkene analogues in accordance with Scheme 3. Commercially available alkyl bromides containing the requisite alkenes were subjected to SN2 displacement with the N-Boc protected phenol derivative 19. The generated intermediates 24a–e were treated sequentially with trifluoroacetic acid followed by N,N′-Di-Boc-1H-pyrazole-1-carboxamidine and DIEA to form the guanylated intermediates 25a–e. TFA deprotection of both Boc groups yielded compounds 26a–e as trifluoroacetate salts.
Scheme 3. Synthesis of alkyl substituted stereospecific ether tail derivatives 26a–d.
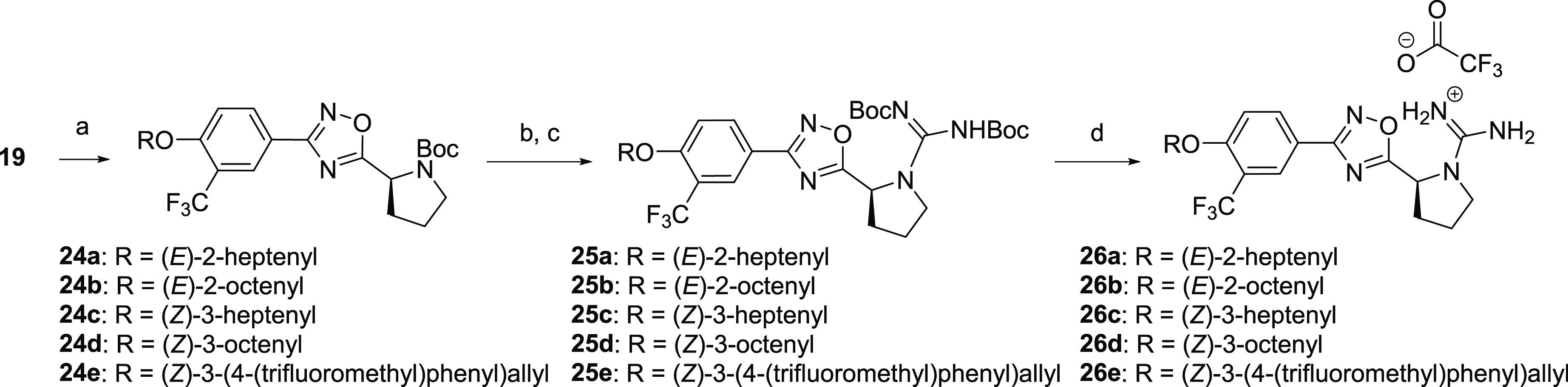
Reaction conditions: (a) Alkyl bromide, K2CO3, I2, acetone, 90 °C, 2 h, microwave; (b) TFA, DCM, 0 °C to rt, 3–6 h; )c) N,N′-Di-Boc-1H-pyrazole-1-carboxamidine, DIEA, MeCN, 55 °C, 2–6 h, microwave; (d) TFA, DCM, rt, 4 h.
Biological Evaluation
Retaining both the guanyl pyrrolidine head and 1,2,4-oxadiazole linker found in 6, analogues were designed and synthesized as described in Schemes 1–3. These analogues were screened for their inhibitory activity against each recombinant human SphK enzymes in accordance with a previously established protocol.31 Briefly, SphK1 or SphK2 plasmids were transfected into HEK293T cells. Cell lysates were generated containing the respective recombinant enzymes and were treated with [γ32P]ATP sphingosine at either 10 μM for SphK1 or 5 μM for SphK2, which is the KM for each enzyme. This assay generates radiolabeled S1P, which is separated via thin layer chromatography, isolated, and detected by radiography. Thus, treatment with inhibitor is expected to decrease the quantified amount of [γ32P]-S1P. For our studies, we set a stringent 0.3 μM inhibitor for SphK2 inhibition as these compounds were designed as potent SphK2 inhibitors. Because we are only interested in compounds with minimal SphK1 inhibitory activity, compounds were tested at 1 μM inhibitor to detect SphK1 inhibition that would not be revealed at lower concentrations. Results are reported as a percent of control without inhibitor. The percent inhibitory activity results are listed in Table 1 with 6 serving as the standard. In this assay, 6 exhibited 51% hSphK2 inhibition at 0.3 μM and 6% hSphK1 inhibition at 1.0 μM.
Table 1. SphK1 and SphK2 Inhibitory Activitiesa.
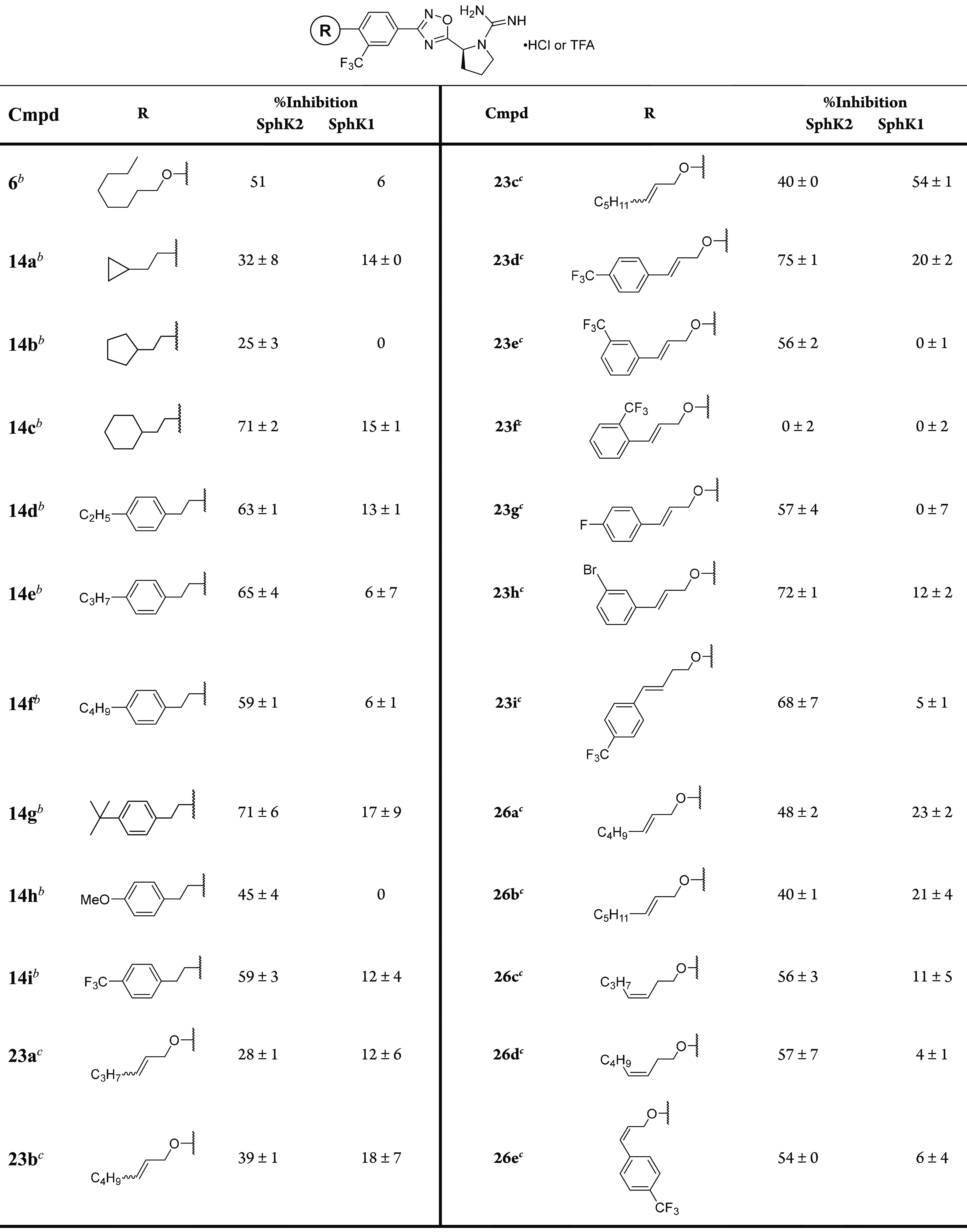
SphK activity is presented as percent of control during which no inhibitor is introduced. Recombinant human SphK1 or SphK2 was isolated from cell lysate, and enzyme activity was measured with either 5 μM (SphK1) or 10 μM (SphK2) sphingosine and 250 μM [γ32P]ATP. Compounds were assayed at 1.0 μM (SphK1) or 0.3 nM (SphK2). All measurements were performed in triplicate.
Compound is a hydrochloride salt.
Compound is a trifluoroacetate salt.
Structure–Activity Relationship Study
The first alterations to 6 consisted of introducing terminal cycloalkane functionalities at the ω-position. Being lipophilic and bulky in nature, these analogues probe the chemical environment and three-dimensional space of the SphK2 binding pocket. The cyclopropyl derivative 14a exhibited modest (32%) SphK2 inhibition and 14% SphK1 inhibition, proving to be less potent and selective than 6. However, replacing the cyclopropyl ring with cyclopentyl 14b and cyclohexyl 14c structures led to significant improvements in SphK2 potency. The cyclohexyl derivative 14c exhibited 71% SphK2 inhibition and 15% SphK1 inhibition. As these results suggest that this region of the binding pocket is large, we replaced the cyclohexyl ring with a phenyl ring and appended ethyl (14d), propyl (14e), butyl (14f), and tert-butyl (14g) groups. Exchanging the tert-butyl group with a more polar electron donating methoxy (14h) or a trifluoromethyl (14i) group had a negative impact on inhibiting SphK2, most likely due to the hydrophobic properties of this region of the binding pocket. Among these analogues, 14g was the most potent inhibitor with similar activity as 14c.
As an alternative strategy to investigate the effect of conformational restriction on the aliphatic tail, we introduced a double bond with increasing pendant alkyl groups in 23a–c, with 23c having the same relative number of carbon chain as 6. As depicted in Scheme 2, cross metathesis with monosubstituted terminal alkenes and a Grubbs second generation catalyst resulted in a 60:40 mixture of diastereomers. Homologating the tail from six to eight carbons (23a, 23b, and 23c, respectively) led to stepwise increases in potency but with a corresponding decrease in selectivity. As these analogues are isomeric mixtures, one orientation might have contributed to the observed loss in enzyme specificity. To identify the more potent diastereomer, we synthesized the pure E- and Z-alkene analogues. Using Scheme 3, we synthesized the E double bond geometry with a heptenyl chain 26a and octenyl chain 26b and found a slightly diminished inhibitory activity and SphK2 selectivity. These analogues exhibited comparable potency to 6 but still possessed poor enzyme selectivity. The corresponding Z isomers, heptenyl 26c and octenyl 26d, however, not only were more potent but also improved SphK2 selectivity. The observed differences in activity between the O-allylic E-alkene structures 26a,b and the O-homoallylic Z-alkene structures 26c,d provide valuable insight into the spatial geometry of the SphK enzyme binding pockets.
To determine a synergistic effect of the phenyl ring (14d–i) and alkene in 26a–d, a series of styrene-containing analogues was synthesized. Placing a para-trifluoromethyl styryl group in 23d resulted in an impressive selectivity and 75% SphK2 inhibition. Moving the trifluoromethyl substituent to the meta (23e) or ortho (23f) positions gave precipitous loss in activity. Likewise, placing a smaller fluorine atom at the 4-position of the ring diminished activity while a bromine on the 3-position restored activity. With this information at hand, we aimed to capitalize on 23d by inserting a methylene unit to generate homoallylic derivative 23i, but this analogue did not provide appreciative improvement in activity. Finally, as the Z isomers of the aliphatic tail series were more potent, we synthesized the corresponding Z isomer (26e) of the most potent compound of the styryl series (23d). Surprisingly, compound 26d is less potent (54% inhibition) than 23d.
With the structure–activity profile of tail analogues in hand, we determined the inhibitory constants (Ki) and selectivity for the most potent analogues (Table 2). Consistent with the results from the initial screen, 14c exhibited marked selectivity for SphK2 (∼200 fold) and a Ki of 90 nM. Replacement of the cyclohexyl motif with phenyl rings with pendant propyl (14e) or trifluoromethyl (14i) resulted in slightly diminished inhibitory activity (Ki = 132 and 165 nM, respectively) as well as slight decreases in SphK2 selectivity. Compared to the planar nature of aromatic rings, the cyclohexyl group has a larger three-dimensional volume, potentially filling the binding pocket more adequately and enabling for additional lipophilic interactions with SphK2. Surprisingly, when 14i was further rigidified with an alkene in 23d, both the potency (Ki = 105 nM) and selectivity (120-fold) toward SphK2 improved. Our studies suggest that the flexibility provided by two methylene carbons is beneficial for selectivity as it allows for compounds to accommodate subtle differences in the binding pockets of the two enzymes. To date, 14c remains the most potent and selective SphK2 inhibitor identified, achieving this activity presumably through its ability to fill the entirety of SphK2 binding pocket and participate in hydrogen bonding with a catalytically important Asp308 residue (Figure 4, vide infra).
Table 2. Ki and cLogP Values for SphK2-Selective Inhibitorsa.
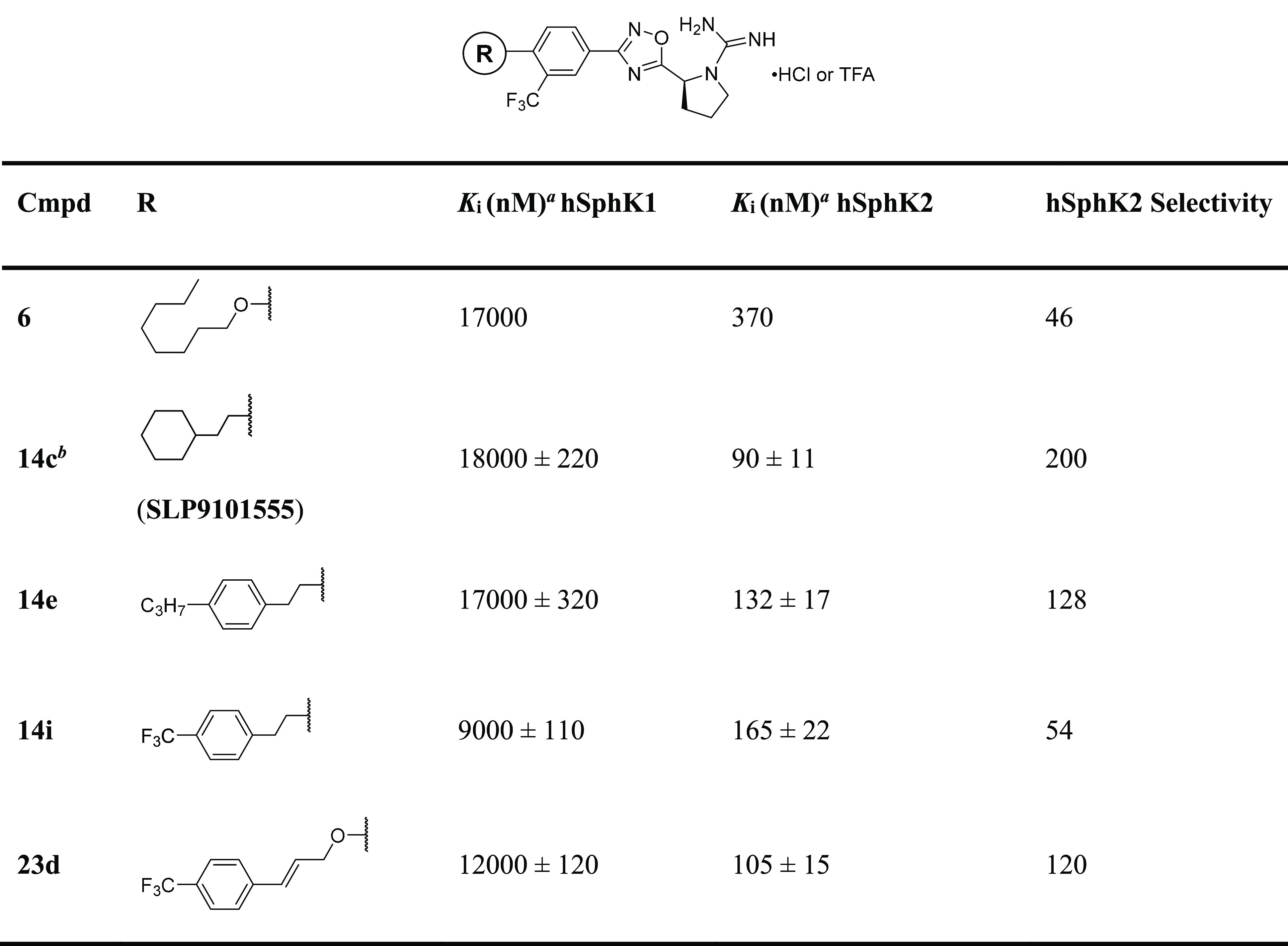
Inhibitory constants for recombinant enzymes were obtained by kinetic analysis of S1P production using variable concentration of sphingosine and a fixed concentration of ATP in the presence or absence of compounds as described previously.30 Selectivity for each compound was determined by dividing the KM for SphK2 by the KM for SphK1. hSphK2 selectivity for each compound was determined by dividing Ki for hSphK1 by the Ki for hSphK2.
Ki for mouse SphK2 is 110 ± 13 nM and mouse SphK1 is >10 000 nM.
Figure 4.
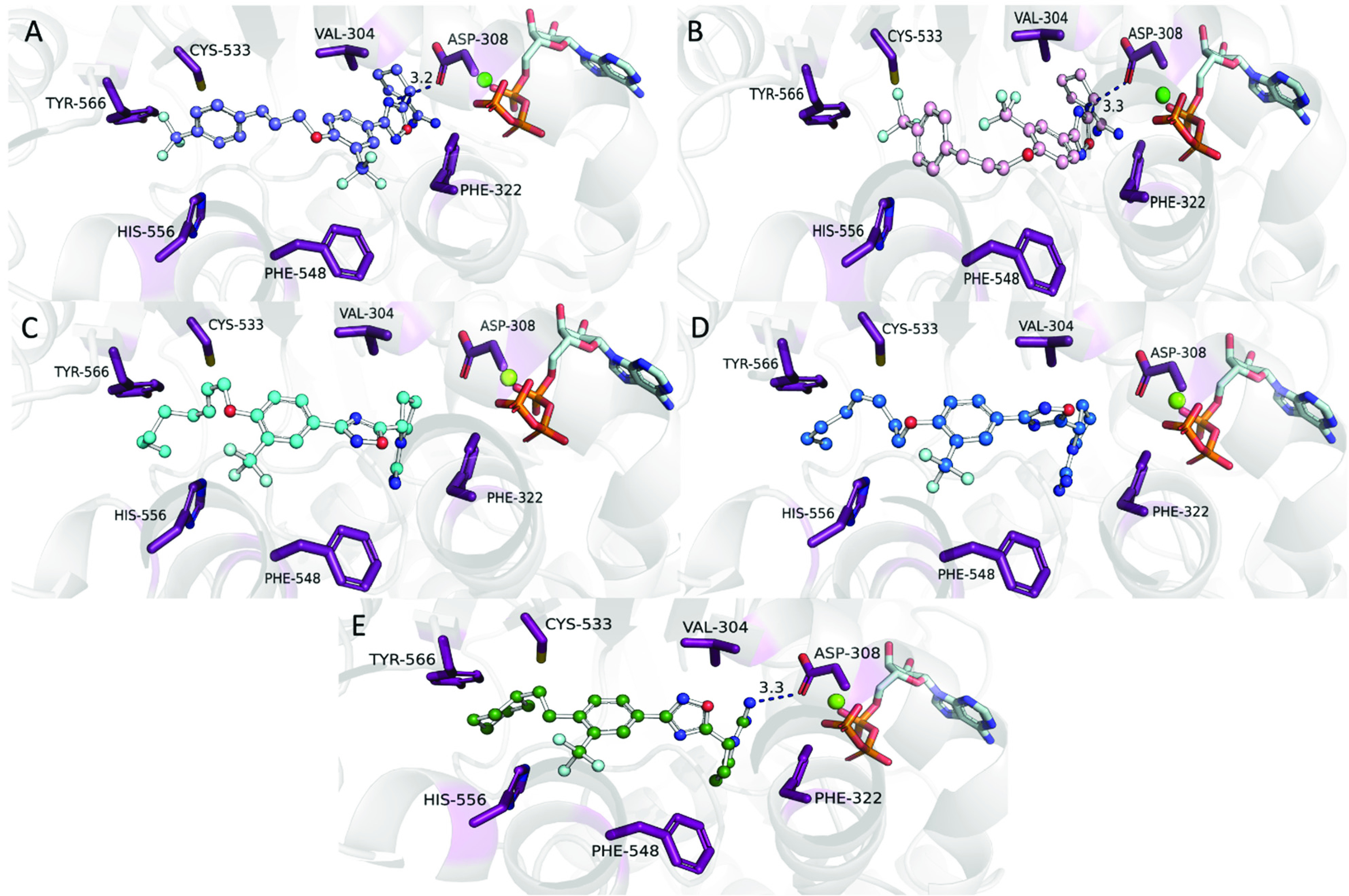
Molecular docking of top compounds indicates positioning of inhibitors in the back of the binding pocket influence binding potency. Molecular docking of 23d (blue, A) and 23e (pink, B) in an hSphK2 homology model position the ligand toward the top of the binding cavity near ATP. The hSphK2 homology model is based on the template of hSphK1 with sphingosine cocrystallized (PDB ID: 3VZB). Analysis of alkene bond geometry in alkyl tail analogues 26d (cyan, C) and 26a (blue, D) through molecular docking in an hSphK2 homology model show utilization of the side pocket near Phe548. Compound 14c (green, E) utilizes the side pocket near Phe548 to position the trifluoromethyl group and participates in electrostatic interactions with Asp308. Hydrogen bonds are shown in blue dashed lines.
Molecular Docking
To understand better the significance of alkene bond geometry in SphK2 inhibition and positioning of ligands in the binding pocket, we used molecular modeling to assess interactions and fit of inhibitors within the binding cavity. Structures were docked in a SphK2 homology model generated from a cocrystal structure of hSphK1 in combination with ADP and Mg2+ (PDB ID: 3VZB).43,44 To more accurately predict the position of inhibitors in the Sph binding cavity, ADP was replaced with ATP and the entire complex then energy minimized to adjust for the additional phosphate presence. Molecular docking with (E)-23d and (Z)-26e showed the that double bond geometry is critical for maximizing interactions in the binding pocket as the E-alkene extends the tail region deeper into the binding pocket (Figure 4A). This allows for stronger interactions between the tail portion of 23d and the amino acid residues Cys-533 and Tyr-566. However, the most notable difference between the two models involves the orientation of the compounds within the SphK2 binding pocket. Changing the bond geometry from the E- to the Z-alkene results in the rotation of the internal phenyl ring bearing the ortho-trifluoromethyl group, which leads to the weakening or complete disappearance of interactions with Phe548 and Val304. Additionally, while still capable of interacting with Phe322, the oxygen heteroatom in the 1,2,4-oxadiazole is too far removed from Asp308 to participate in hydrophilic interactions or hydrogen bonding. The hydrogen bond interactions between the guanyl moiety on the pyrrolidine ring and Asp308 is also weakened as the distance is further increased.
In a sharp deviation from 23d and 26e, the alkene bond geometry in 26a and 26d did not appear to significantly impact the orientation of the internal phenyl ring or headgroup region of either compound. Most of the key interactions between these moieties and exposed amino acid residues were retained in the molecular docking model of both compounds. This trend continued into the tail region as no major differences in amino acid residue interactions were observed across these two analogues. In both cases, the aliphatic chains form a bent “J”-shaped conformation similar to what is observed when sphingosine is bound into the active site.43 This is consistent with several reports that describe the shape of the SphK2 binding pocket and also helps to explain the observed differences in potency and enzyme selectivity between 26a and 26d.27−29,31 The additional methylene unit and the Z-alkene geometry of 26d allow it to more accurately conform to the “J”-shaped binding pocket of SphK2. Docking of the most potent and selective inhibitor 14c in SphK2 reveals several interactions key to the observed activity: (1) the cyclohexyl ring fills the binding cleft formed by Cys533, His556, and Tyr 566, (2) the trifluoromethyl group favorably interacts with the residues (Phe548, Leu544, and Leu547) that form a side cavity within the Sph binding pocket of SphK2,35 and (3) the guanidine moiety is perfectly situated to form a hydrogen bond with Asp308. In contrast, other compounds with high potency (e.g., 23d) adjust their position in the side pocket region near Phe548 but are more constrained in binding pocket due to size and form a weaker hydrogen bond between the oxadiazole ring and Asp308. Molecular docking of these inhibitors in SphK1 was performed to determine whether the affinity for the side cavity near Phe548 (Phe303 in SphK1) would be observed and if this is potential feature that provides SphK2 selectivity over SphK1 (see Figure S1, Supporting Information). These results demonstrate a lack of ability of these compounds to utilize the side pocket near Phe303 (compounds 26a,d and 14c). Hydrogen bonding of Asp178 to the oxadiazole in compounds 23d,e were not observed in SphK1. These combined results indicate a lack of general favorability and fitness in the SphK1 binding cavity as compared to the SphK2 results, supporting our rationale for design and selectivity.
In Vitro and In Vivo Testing
As 14c was discovered as the most potent and selective SphK2 inhibitor, we investigated its ability to inhibit SphK2 in vitro in the histiocytic lymphoma U937 cell line. Thus, U937 cells were incubated with varying concentrations of 14c and after 2 h, cells were lysed, and levels of S1P were determined by LC-MS/MS. As shown in Figure 5A, administration of 14c evoked a dose-dependent decrease in S1P as expected of SphK2 inhibition.
Figure 5.
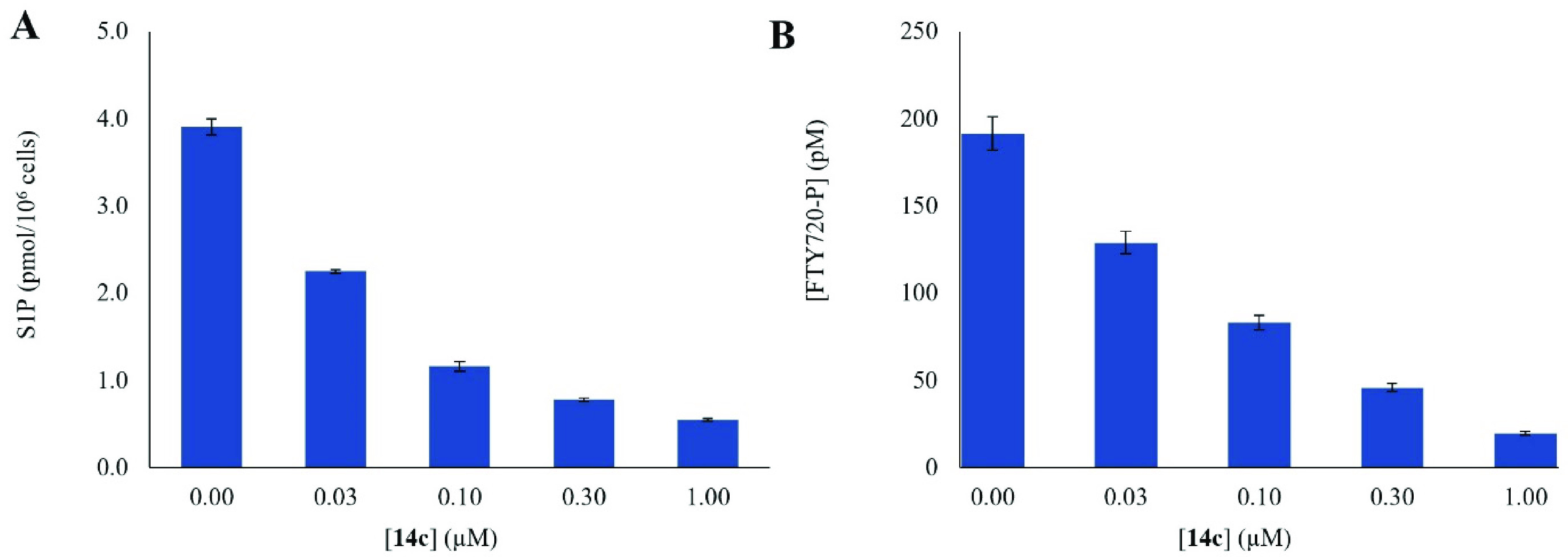
Concentration effect of 14c on intracellular SphK2 inhibition. U937 cells were incubated with 1 μM FTY720 and varying concentrations of 14c. Following incubation, U937 cells were harvested by centrifugation and lysed, and the levels of (A) S1P and (B) FTY720-P were quantified via LC-MS/MS. Each experiment was performed in duplicate.
Because U937 cells express both SphK isoforms, the intracellular phosphorylation state of FTY720 was utilized as a read-out of SphK2 inhibition. As the S1P agonist prodrug FTY720 is primarily phosphorylated by SphK2, the presence of FTY720-phosphate (FTY720-P) correlates with SphK2 activity.45,46 Thus, U937 cells were incubated with FTY720 and 14c at 30–1000 nM. After incubation, FTY720-P levels were determined from cell extracts. As shown in Figure 5B, treatment of U937 cells with 14c resulted in a concentration-dependent decrease in FTY720-P levels. Taken together, these results suggests that 14c is both cell permeable and suppresses the conversion of FTY720 to FTY720-P in whole cells via inhibition of SphK2.
To determine whether 14c inhibits SphK2 in vivo, we dosed C57BL/6 mice with 14c (5 mg/kg, single intraperitoneal injection) and quantified blood S1P using LC-MS/MS. The pharmacodynamic marker of SphK2 deficiency, whether facilitated via genetic means (i.e., germ line ablation of Sphk2)32 or small molecule inhibition,30,31,35 is the increase in levels of blood S1P. Conversely, SphK1 deficiency drives decreases in circulating S1P levels.31,47,48 Dual SphK1/SphK2 inhibitors also show decreases in blood S1P concentrations when there is less than 10-fold selectivity.32 As shown in Figure 6, in vivo administration of 14c resulted in the doubling of S1P levels in blood 4 h post dosing and returning close to baseline after 24 h. The sustained increased levels of S1P, which is similar to the activity of 6, suggests target engagement with SphK2 with once daily dosing.
Figure 6.

S1P levels in the blood of mice injected with 14c. Four wild-type mice were subjected to intraperitoneal (IP) injection with a single dose of 14c (5 mg/kg), and blood was drawn at the indicated time intervals. S1P concentration was quantified via LC-MS/MS, and the standard deviations are shown as error bars.
Conclusions
Given their important role in controlling S1P production and thus potentially in disease progression, SphKs are drug discovery targets. Isoform-selective inhibitors enable perturbation of the natural S1P gradients. In this study, we investigated the effects of conformational restriction on the tail region of SphK2 inhibitor 6. The structure–activity relationship study included rigid motifs such as internal alkenes, cycloalkanes, and substituted aromatics. As sphingosine formed a “J-shaped” structure within the sphingosine kinase substrate pocket, the location of the alkene moiety in the long aliphatic tail was probed. The inclusion of these rigid scaffolds led to the development of several compounds with significantly improved SphK2 potency and selectivity. The most successful of these inhibitors was 14c, which induced SphK2-selective inhibition both in vitro and in vivo. Testing of 14c in U937 cells demonstrated marked SphK2 inhibition that is consistent with a decrease in intracellular phosphorylation of FTY720. To date, 14c remains among the most potent and selective SphK2 inhibitors with 200-fold selectivity (SphK1 Ki = 18 000 nM; SphK2 Ki = 90 nM). Further in vivo testing in mice showed that 14c elicits profound increases in blood S1P, a pharmacodynamic index of target engagement. These studies confirm that 14c functions as a SphK2-selective inhibitor and possesses suitable metabolic stability. In tandem with biological testing, molecular modeling of several synthesized analogues also highlights the necessity of some degree of flexibility between the rigid tail group functionalities and the internal phenyl ring in order to accommodate the “J”-shaped binding pocket of SphK2. Taken together, this study provides valuable insight into the binding interactions of SphK2-selective inhibitors. We anticipate that these results will complement our understanding of the in vivo effects of SphK2 inhibition as well as lead to the development of improved SphK2 inhibitors.
Experimental Section
Sphingosine Kinase Assays
Plasmids encoding SphK1 or SphK2 were used to transfect HEK293T cells, and cleared lysates were prepared after 48 h. SphK activity was measured in kinase assay buffer that consisted of 20 mM Tris-Cl (pH 7.4), 1 mM 2-mercaptoethanol, 1 mM EDTA, 5 mM sodium orthovanadate, 40 mM β-glycerophosphate, 15 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 mM MgCl2, 0.5 mM 4-deoxypyridoxine, 10% glycerol, and 0.01 mg/mL each leupeptin, aprotinin, and soybean trypsin inhibitor. To achieve optimal activity of SphK1 or SphK2, the buffer was supplemented with either 0.5% Triton X-100 or 1 M KCl, respectively. To ascertain any inhibitory effect of compounds, the assay was supplemented with a substrate (d-erythro-sphingosine, 10 μM for SphK1 and 5 μM for SphK2), indicated amount of compounds, 250 μM [γ32P]ATP, and recombinant enzyme (0.02–0.03 mg of total protein). After 20 min at 37 °C, the reaction mixture was extracted with 2 volumes of chloroform/methanol/1 N HCl (100:200:1), and the components in the organic phase were separated by thin layer chromatography using a 1-butanol/glacial acetic acid/water (3:1:1) solvent system. Radiolabeled S1P was detected by autoradiography and identified by migration relative to authentic standards. For quantification, the silica gel containing radiolabeled lipid was scraped into a scintillation vial and measured by liquid scintillation counting. All measurements were taken in triplicate.
SphK Inhibition in Cultured Cells
The effects of 14c on SphK2 inhibition were studied using U937 cells grown in RPMI 1640 media supplemented with l-glutamate, 10% fetal bovine serum, and 1% penicillin/streptomycin at 37 °C in the presence of 5% CO2. At 24 h before adding the inhibitors, the growth media was replaced with media containing 0.5% fetal bovine serum. Cells were then treated for 2 h with 14c in the presence or absence of the SphK2 selective substrate FTY720. Levels of S1P and FTY720 phosphate (FTY720-P) in cells were determined by liquid chromatography mass spectrometry (LC-MS).
Sample Preparation and LC-MS/MS Analysis
Sample preparation protocols were from our previous publication with minor modifications.32 Cell pellets (2–3 × 106 cells), whole blood (10 μL), or plasma (10 μL) was mixed with 2 mL of a methanol/chloroform solution (3:1) and transferred to a sealed glass vial. Suspensions were supplemented with 10 μL of internal standard solution containing 10 pmol of deuterated (d7) S1P and deuterated (d7) sphingosine. The mixture was placed in a bath sonicator for 10 min and incubated at 48 °C for 16 h. The mixture was then cooled to ambient temperature and mixed with 200 μL of 1 M KOH in methanol. The samples were again sonicated and incubated for an additional 2 h at 37 °C. Samples were neutralized by the addition of 20 μL of glacial acetic acid and transferred to 2 mL microcentrifuge tubes. Samples were centrifuged at 12 000g for 12 min at 4 °C. The supernatant fluid was collected in a separate glass vial and evaporated under a stream of nitrogen. Immediately prior to LC-MS analysis, the dried material was dissolved in 0.3 mL of methanol and centrifuged at 12 000g for 12 min at 4 °C. Of the resulting supernatant fluid, 50 μL was analyzed by liquid chromatography-ESI mass spectrometry using a triple quadrupole mass spectrometer (AB-Sciex 4000 Q-trap) coupled to a Shimadzu LC-20Ad LC system. A binary solvent gradient with a flow rate of 1 mL/min was used to separate sphingolipids and drugs by reverse phase chromatography using a Supelco Discovery C18 column (50 mm × 2.1 mm, 5 μm bead size). Mobile phase A consisted of water/methanol/formic acid (79:20:1), and mobile phase B consisted of methanol/formic acid (99:1). The run started with 100% A for 0.5 min. Solvent B was then increased linearly to 100% over the course of 5.1 min and held at 100% for an additional 4.3 min. The column was subsequently re-equilibrated to 100% A for 1 min. Natural sphingolipids were detected using multiple reaction monitoring (MRM) as follows: S1P (380.4 → 264.4); deuterated (d7)C18S1P (387.4 → 271.3), sphingosine (300.5 → 264.4); deuterated (d7) sphingosine (307.5 → 271.3).
Pharmacokinetic Analysis
Mouse studies were conducted using a previously established protocol.31 Samples of 14c (5 mg/kg dissolved in 2% solution of hydroxypropyl-β-cyclodextrin; Cargill Cavitrol 82004) were administered intraperitoneally to groups of four to give C57BL/6J male mice (Jackson Laboratories, Bar Harbor, ME). Control populations received an equal volume injection of the 2% hydroxypropyl-β-cyclodextrin solution vehicle. Blood samples were then collected at the specified time points and analyzed via LC-MS. ASAP time points were collected 1–2 min following drug dosing. Animal protocols were approved prior to experimentation by the University of Virginia’s School of Medicine Animal Care and Use Committee (IACUC approval number 3803).
Molecular Docking
The previously generated SphK2 homology model and the SphK1 crystal structure (PDB ID: 3VZB) were used for docking studies.43,44 The SphK2 homology model coordinates can be found on our Open Science Framework page (https://osf.io/82n73/). Marvin was used for drawing, displaying, and characterizing chemical structures. (Marvin 6.2.2, 2014, ChemAxon; http://www.chemaxon.com). AutoDock Tools49 was used to prepare the protein and ligand files and to define stereochemistry for ligand isomers. AutoDock Vina50 was used to perform the docking. The grid box was set to 20 × 20 × 28 Å3, and a 1.000 Å grid spacing was used. The center of the box (50.0, 62.5, 7.3) was placed at the approximate center of the ligand-binding cavity, with a portion of the ATP binding cavity included. Nine docked poses were predicted for each compound. The lowest energy (kcal/mol) pose for each docked ligand, with the guanidine group oriented toward the head of the binding cavity, was considered as the best pose for the given compound. Free energy of binding scores were cataloged for each docked compound and used as one level of comparison between compounds. The second, more informative aspect of analysis was distance measurements between protein and ligand heavy atoms that would allow for prediction of interactions. Distances were assessed to predict hydrogen bonding (less than 3.5 Å), hydrophobic interactions (3.4–3.9 Å), and distal, weaker interactions (greater than 4.0 Å) that would translate to ligand binding and stability in the binding cavity.
General Material and Synthetic Procedures
Reactions employing microwave radiation were conducted in a Discover SP microwave synthesizer (CEM Corporation). Solvents were dried and purified using a PureSolv solvent purification system prior to use. All chemical reagents were purchased from commercial sources and used without further purification. Thin-layer chromatography (TLC) was performed on aluminum-backed silica gel (200 μm particle diameter and F254 fluorescence backed). Column chromatography was performed on flash grade silica gel (40–63 μm particle diameter) using a Combiflash Rf purification system. 1H NMR spectra were recorded at 400 or 500 MHz, and chemical shifts were reported in ppm with the solvent resonance as an internal standard (CDCl3: 7.26 ppm; CD3OD: 4.87 ppm). The corresponding 13C NMR resonant frequencies were at either 125 or 101 MHz, and chemical shifts were reported in ppm with the solvent resonance as the internal standard (CDCl3: 77.16 ppm; CD3OD: 49.00 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants (Hz), and integration. Rotamers are denoted by an asterisk (*). High-resolution mass spectroscopy (HRMS) was performed on a time-of-flight mass spectrometer with electrospray ionization (ESI). LC-MS analysis was performed with a Thermo Electron TSQ triple quadrupole mass spectrometer equipped with an ESI source. All compounds tested in biological assays had purities greater than or equal to 95% as determined by HPLC analysis (254 nm) and 1H NMR. HPLC traces of lead compounds and NMR spectra (1H and 13C) are provided in the Supporting Information.
General Procedure A: Sonogashira Coupling between Aryl Bromides and Terminal Alkynes
Aryl halide (1.0 equiv) was dissolved in dry DMF and subjected to degassing with a mixture of H2 and N2 three times. PdCl2(PPh3)2 (10 mol %) was added followed by CuI (8 mol %) at room temperature. Terminal alkyne (1.0 equiv) was added followed by addition of TEA. The reaction mixture was stirred and heated to either 90 °C (9a–c) or 50 °C (9d–i). After cooling to room temperature, the resulting solution was concentrated under reduced pressure and partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc (three times). The combined organic layers were washed with brine and dried over anhydrous Na2SO4, and the solvent was removed. The resulting residue was purified by flash chromatography on silica gel to yield the desired product.
General Procedure B: Chemoselective Reduction of Alkynes to Alkanes in the Presence of Nitriles
The internal alkyne intermediate was dissolved in EtOAc (30 mL) followed by addition of Lindlar’s catalyst [(5% Pd-CaCO3 + Pb(OCOCH3)2 + quinoline), 10 mol %]. The reaction mixture was subjected to hydrogenation under a H2 atmosphere for 0.5–2 h until consumption of the starting material was observed via TLC. The reaction mixture was filtered over Celite, and the resulting filtrate was concentrated under reduced pressure and subjected to flash chromatography on silica gel to yield the desired saturated product.
General Procedure C: Conversion of Nitriles to Amidoximes
TEA (3.0 equiv) was added to a solution of the appropriate benzonitrile (1.0 equiv) with hydroxylamine hydrochloride (3.0 equiv) in ethanol (0.5 M solution). The reaction mixture was heated to 80 °C for 6–12 h until the starting material disappeared as monitored via TLC. The organic solvent was removed under reduced pressure, and the residue was purified by flash chromatography to yield the desired product.
General Procedure D: 1,2,4-Oxadiazole Formation through the Coupling of Amidoxime Derivatives with Boc-L-proline
DIEA (1.8 equiv) was added to a solution of the appropriate amidoxime (1.0 equiv) and Boc-l-proline in DMF. HCTU (1.8 equiv) was then added to the resulting mixture at room temperature ,and the mixture was heated to 110 °C for 12–16 h. The reaction progress was followed by TLC. Upon completion, the solution was partitioned between ethyl acetate and aqueous LiBr. The aqueous layer was washed with ethyl acetate three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was subjected to flash chromatography to yield the desired product.
General Procedure E: Removal of Boc Protecting Groups with Gaseous HCl and Subsequent Guanylation of Ammonium Salts
Hydrochloric acid was bubbled through a solution of the N-Boc protected compound (1.0 equiv) in methanol for 2–5 min until complete consumption of the starting material was observed by TLC. The reaction mixture was concentrated under reduced pressure and purified via silica gel column chromatography to yield the corresponding ammonium salt. The residue was then dissolved in a 0.02 M CAN solution and acetonitrile. DIEA (10.0 equiv) and N,N′-Di-Boc-1H-pyrazole-1-carboxamidine (1.1 equiv) were added to the solution. The resulting mixture was placed in a microwave reactor at 50 °C for 2 h. The organic solvent was removed under reduced pressure, and the residue was purified via silica gel column chromatography.
General Procedure F: Removal of Boc Protecting Groups with HCl
To a solution of Boc protected compound (1.0 equiv) was added hydrogen chloride (10.0 equiv; 4 M in dioxane). The reaction mixture was allowed to stir for 2 h or until complete consumption of the starting material was observed by TLC. The reaction mixture was concentrated under reduced pressure and purified via silica gel column chromatography to yield the corresponding ammonium salt.
General Procedure G: O-Alkylation with Alkyl Bromides
The appropriate alkyl halide (1.2 equiv), potassium carbonate (2.0 equiv), and KI (1.5 equiv) were dissolved in acetone in an 8 mL microwave reactor flask. The reaction mixture was heated to 80 °C for 4–12 h in a microwave reactor until TLC indicated the starting material had been fully consumed. The solution was partitioned between EtOAc and water. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified via flash chromatography to yield the desired product.
General Procedure H: Cross Metathesis with Terminal Alkenes Employing a Microwave Reactor
The O-allyl intermediate (1.0 equiv) was dissolved in dry DCM. To the reaction mixture was added sequentially the terminal alkene coupling partner (10.0 equiv) and Grubbs’s II generation catalyst (10 mol %) at room temperature in a microwave reaction capsule. The reaction mixture was subjected to microwave irradiation at 70 °C for 2–4 h until the starting materials had been fully converted. Following complete consumption of the starting material, the solvent was removed under reduced pressure and the residue was subjected to flash column chromatography to afford the desired product.
General Procedure I: Removal of Boc Protecting Groups with TFA and Subsequent Guanylation of Ammonium Salts
To a solution of the N-Boc-protected intermediate (1.0 equiv) in DCM was added a 1 N TFA/DCM solution (20 equiv). The reaction mixture was then stirred at room temperature for 4 h followed by removal of the organic solvent under reduced pressure. The residue was then dissolved in a 0.02 M CAN solution and acetonitrile. DIEA (10.0 equiv) and N,N′-di-Boc-1H-pyrazole-1-carboxamidine (1.1 equiv) were added to the solution. The resulting mixture was placed in a microwave reactor at 50 °C for 2 h. The organic solvent was removed under reduced pressure, and the residue was purified via silica gel column chromatography.
General Procedure J: Removal of Boc Protecting Groups with TFA
To a solution of Boc protected compound (1.0 equiv) in DCM was added TFA (20.0 equiv). The reaction mixture was allowed to stir for 2 h or until complete consumption of the starting material was observed by TLC. The reaction mixture was concentrated under reduced pressure and purified via silica gel column chromatography to yield the corresponding guanidinium salt.
4-(Cyclopropylethynyl)-3-(trifluoromethyl)benzonitrile (9a)
Synthesized according to General Procedure A at 90 °C. Yellow solid (371 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 1.9 Hz, 1H), 7.70 (dd, J = 8.1, 1.7 Hz, 1H), 7.58 (dt, J = 8.1, 0.7 Hz, 1H), 1.51 (tt, J = 8.2, 4.9 Hz, 1H), 1.02–0.92 (m, 2H), 0.94–0.84 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 134.6, 134.6, 132.7 (q, J = 31.4 Hz), 129.6 (q, J = 5.3 Hz), 127.5 (d, J = 2.2 Hz), 122.6 (q, J = 273.8 Hz), 117.5, 110.8, 106.1, 71.2, 9.4, 0.7. HRMS (ESI+): calcd for C13H9F3N [M + H]+ 236.0682, found: 236.0684.
4-(Cyclopentylethynyl)-3-(trifluoromethyl)benzonitrile (9b)
Synthesized according to General Procedure A at 90 °C. Clear oil (202 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.87–7.84 (m, 1H), 7.69 (dd, J = 8.1, 1.7 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 2.93–2.81 (m, 1H), 2.04–1.89 (m, 2H), 1.82–1.66 (m, 4H), 1.67–1.51 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 136.2, 134.5, 134.5, 134.4, 133.1, 132.8, 132.4, 132.1, 129.5, 129.4, 129.3, 127.5, 126.5, 123.8, 121.1, 118.3, 117.3, 110.7, 106.8, 106.8, 76.7, 75.5, 33.5, 33.3, 33.2, 30.9, 29.7, 25.1, 25.0; HRMS (ESI+): calcd for C15H12F3N [M + Na]+ 286.0814, found: 286.2475.
4-(Cyclohexylethynyl)-3-(trifluoromethyl)benzonitrile (9c)
Synthesized according to General Procedure A at 90 °C. Yellow oil (172 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 1.7 Hz, 1H), 7.72 (dd, J = 8.1, 1.7 Hz, 1H), 7.61 (d, J = 8.1 Hz, 1H), 2.68 (tt, J = 8.7, 3.9 Hz, 1H), 1.93–1.82 (m, 2H), 1.82–1.68 (m, 2H), 1.64–1.49 (m, 3H), 1.46–1.30 (m, 3H). This material was taken forward without further characterization.
4-((4-Ethylphenyl)ethynyl)-3-(trifluoromethyl)benzonitrile (9d)
Synthesized according to General Procedure A at 50 °C. Yellow solid (203 mg, 67%). 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 1.6 Hz, 1H), 7.82–7.72 (m, 2H), 7.49 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 7.9 Hz, 2H), 2.69 (q, J = 7.6 Hz, 2H), 1.25 (t, J = 7.6 Hz, 3H). This material was taken forward without further characterization.
4-((4-Propylphenyl)ethynyl)-3-(trifluoromethyl)benzonitrile (9e)
Synthesized according to General Procedure A at 50 °C. Yellow oil (350 mg, 93%). 1H NMR (500 MHz, CD3OD) δ 8.18–8.11 (m, 1H), 8.02–7.94 (m, 1H), 7.87 (dt, J = 8.1, 0.7 Hz, 1H), 7.47 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.67 (h, J = 7.4 Hz, 2H), 0.96 (t, J = 7.3 Hz, 3H). HRMS (ESI+): calcd for C19H15F3N [M + H]+ 314.1151, found: 314.1164.
4-((4-Butylphenyl)ethynyl)-3-(trifluoromethyl)benzonitrile (9f)
Synthesized according to General Procedure A at 50 °C. Yellow oil (380 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 8.00–7.87 (m, 1H), 7.86–7.68 (m, 2H), 7.50–7.44 (m, 2H), 7.26–7.15 (m, 2H), 2.64 (t, J = 7.7 Hz, 2H), 1.68–1.57 (m, 2H), 1.43–1.29 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 145.4, 135.2, 134.8, 134.4, 132.1, 129.8, 129.2, 128.9, 127.6, 127.4, 117.5, 111.4, 83.9, 35.9, 33.5, 22.4, 14.1. Trifluoromethyl carbon signal not observed. HRMS (ESI+): calcd for C20H17F3N [M + H]+ 328.1308, found: 328.1309.
4-((4-(tert-Butyl)phenyl)ethynyl)-3-(trifluoromethyl)benzonitrile (9g)
Synthesized according to General Procedure A at 50 °C. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
4-((4-Methoxyphenyl)ethynyl)-3-(trifluoromethyl)benzonitrile (9h)
Synthesized according to General Procedure A at 50 °C. Yellow solid (377 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 8.00–7.90 (m, 1H), 7.76 (dd, J = 8.1, 1.7 Hz, 1H), 7.71 (dt, J = 8.1, 0.7 Hz, 1H), 7.50 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 3.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.0, 134.7, 134.1, 133.8, 132.3 (q, J = 31.5 Hz), 129.7 (q, J = 5.3 Hz), 127.0 (q, J = 1.6 Hz), 122.7 (q, J = 273.9 Hz), 117.5, 114.4, 113.8, 111.1, 100.7 (q, J = 0.9 Hz), 83.6, 55.5. HRMS (ESI+): calcd for C17H11F3NO [M + H]+ 302.0787, found: 302.0789.
3-(Trifluoromethyl)-4-((4-(trifluoromethyl)phenyl)ethynyl)benzonitrile (9i)
Synthesized according to General Procedure A at 50 °C. Yellow oil (380 mg, 70%). 1H NMR (500 MHz, CDCl3) δ 8.01–7.95 (m, 1H), 7.84 (dd, J = 8.0, 1.6 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.71–7.63 (m, 4H). HRMS (ESI+): calcd for C17H8F6N [M + H]+ 340.0555, found: 340.0564.
4-(2-Cyclopropylethyl)-3-(trifluoromethyl)benzonitrile (10a)
Synthesized according to General Procedure B. Yellow solid (239 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 1.2 Hz, 1H), 7.74 (dd, J = 8.1, 1.7 Hz, 1H), 7.47 (d, J = 8.1 Hz, 1H), 2.89 (dt, J = 52.0, 7.9 Hz, 2H), 1.67–1.47 (m, 2H), 1.45–1.31 (m, 2H), 0.96–0.85 (m, 2H), 0.50–0.03 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 151.2, 147.7, 135.0, 132.2, 130.0 (q, J = 6.0 Hz), 123.5 (q, J = 274.4 Hz), 117.9, 110.4, 31.9, 22.5, 14.0, 4.8. Trifluoromethyl ipso carbon signal splitting not observed due to overlap. HRMS (ESI+): calcd for C13H13F3N [M + H]+ 240.0995, found: 240.0996.
4-(2-Cyclopentylethyl)-3-(trifluoromethyl)benzonitrile (10b)
Synthesized according to General Procedure B. Clear oil (111 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 1.9 Hz, 1H), 7.79–7.70 (m, 1H), 7.46 (d, J = 8.1 Hz, 1H), 2.89–2.78 (m, 2H), 1.93–1.75 (m, 3H), 1.70–1.47 (m, 6H), 1.21–1.08 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 147.9 (q, J = 1.4 Hz), 135.0 (q, J = 1.1 Hz), 132.2, 129.9 (q, J = 6.0 Hz), 129.8 (q, J = 31.3 Hz), 123.5 (q, J = 274.3 Hz), 117.8, 110.3, 40.3, 38.1, 32.9, 32.7, 25.3.
4-(2-Cyclohexylethyl)-3-(trifluoromethyl)benzonitrile (10c)
Synthesized according to General Procedure B. Yellow oil (125 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 1.7 Hz, 1H), 7.73 (dd, J = 8.1, 1.6 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 2.87–2.78 (m, 2H), 1.89–1.64 (m, 5H), 1.52–1.41 (m, 2H), 1.38–1.12 (m, 4H), 1.03–0.89 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 148.2, 135.0, 132.2, 130.0 (q, J = 5.9 Hz), 129.9 (q, J = 31.1 Hz), 123.5 (q, J = 274.3 Hz), 117.9, 110.2, 39.5, 38.1, 33.3, 30.7 (q, J = 2.0 Hz), 26.7, 26.4. HRMS (ESI+): calcd for C16H19F3N [M + H]+ 282.1464, found: 282.1475.
4-(4-Ethylphenethyl)-3-(trifluoromethyl)benzonitrile (10d)
Synthesized according to General Procedure B. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
4-(4-Propylphenethyl)-3-(trifluoromethyl)benzonitrile (10e)
Synthesized according to General Procedure B. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
4-(4-Butylphenethyl)-3-(trifluoromethyl)benzonitrile (10f)
Synthesized according to General Procedure B. Yellow oil (379 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 1.7 Hz, 1H), 7.72 (dd, J = 8.0, 1.8 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.16–7.05 (m, 4H), 3.17–3.09 (m, 2H), 2.92–2.84 (m, 2H), 2.59 (t, J = 7.7 Hz, 2H), 1.58 (p, J = 6.9 Hz, 2H), 1.36 (h, J = 7.3 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 146.1 (d, J = 1.5 Hz), 141.1, 137.4, 134.9, 132.4, 129.9 (q, J = 5.8 Hz), 129.8 (q, J = 31.4 Hz), 128.6, 128.3, 123.5 (q, J = 274.3 Hz), 117.6, 110.6, 37.1, 35.3, 35.2 (q, J = 1.9 Hz), 33.7, 22.4, 13.9. HRMS (ESI+): calcd for C20H21F3N [M + H]+ 332.1621, found: 332.1635.
4-(4-(tert-Butyl)phenethyl)-3-(trifluoromethyl)benzonitrile (10g)
Synthesized according to General Procedure B. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
4-(4-Methoxyphenethyl)-3-(trifluoromethyl)benzonitrile (10h)
Synthesized according to General Procedure B. Yellow oil (355 mg, 93%). 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 2.1 Hz, 1H), 7.71 (dd, J = 8.0, 1.8 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.08 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 3.80 (s, 3H), 3.16–3.07 (m, 2H), 2.91–2.82 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 158.4, 146.2 (q, J = 1.3 Hz), 135.0, 132.6, 132.4, 130.1 (q, J = 5.8 Hz), 130.0 (q, J = 31.5 Hz), 129.5, 123.5 (q, J = 274.4 Hz), 117.8, 114.2, 110.7, 55.4, 36.7, 35.5 (q, J = 1.9 Hz). HRMS (ESI+): calcd for C17H15F3NO [M + H]+ 306.1100, found: 306.1113.
3-(Trifluoromethyl)-4-(4-(trifluoromethyl)phenethyl)benzonitrile (10i)
Synthesized according to General Procedure B. Yellow oil (377 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 7.96–7.93 (m, 1H), 7.75 (dd, J = 8.0, 0.6 Hz, 1H), 7.59–7.53 (m, 2H), 7.39 (dt, J = 7.7, 0.6 Hz, 1H), 7.33–7.27 (m, 2H), 3.19–3.12 (m, 2H), 3.02–2.94 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 145.42, 144.33, 135.20, 132.42, 130.24 (q, J = 5.8 Hz), 128.90, 125.76 (q, J = 3.8 Hz), 117.61, 111.13, 37.30, 34.85. Trifluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C17H12F6N [M + H]+ 344.0868, found: 344.0869.
4-(2-Cyclopropylethyl)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (11a)
Synthesized according to General Procedure C. White solid (93 mg, 34%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 9.83 (brs, 1H), 7.88 (d, J = 1.6 Hz, 1H), 7.71 (dd, J = 8.0, 2.0 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 5.01 (brs, 2H), 2.83 (dt, J = 50.1, 7.9 Hz, 2H), 1.68–1.47 (m, 2H), 1.45–1.27 (m, 2H), 0.91 (t, J = 6.8 Hz, 2H), 0.47 – −0.03 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 152.1, 152.0*, 143.8 (q, J = 1.6 Hz), 143.4 (q, J = 1.5 Hz)*, 131.7*, 131.5, 130.3*, 130.2, 129.1 (q, J = 1.2 Hz), 129.0 (q, J = 1.2 Hz)*, 128.8 (q, J = 30.1 Hz), 124.3 (q, 272.5 Hz), 123.7 (q, J = 5.8 Hz), 31.9, 22.5, 14.0, 4.6. HRMS (ESI+): calcd for C13H16F3N2O [M + H]+ 273.1209, found: 273.1210.
4-(2-Cyclopentylethyl)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (11b)
Synthesized according to General Procedure C. White solid (90 mg, 72%). 1H NMR (400 MHz, CD3OD) δ 7.92 (d, J = 1.9 Hz, 1H), 7.78 (dd, J = 8.1, 2.0 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 2.85–2.74 (m, 2H), 1.93–1.72 (m, 3H), 1.71–1.45 (m, 6H), 1.27–1.09 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 154.0, 144.5 (q, J = 1.7 Hz), 132.5, 132.2, 130.5 (q, J = 1.2 Hz), 129.2 (q, J = 30.0 Hz), 125.8 (q, J = 273.7 Hz), 124.7 (q, J = 5.7 Hz), 41.5, 39.5, 33.6, 32.9 (q, J = 1.7 Hz), 26.1.
4-(2-Cyclohexylethyl)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (11c)
Synthesized according to General Procedure C. Off-white solid (107 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 1.9 Hz, 1H), 7.89 (dd, J = 8.1, 2.1 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 6.25 (brs, 2H), 2.88–2.74 (m, 2H), 1.89–1.46 (m, 7H), 1.41–1.07 (m, 4H), 1.02–0.79 (m, 2H). HRMS (ESI+): calcd for C16H22F3N2O [M + H]+ 315.1679, found: 315.1695.
4-(4-Ethylphenethyl)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (11d)
Synthesized according to General Procedure C. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
N′-Hydroxy-4-(4-propylphenethyl)-3-(trifluoromethyl)benzimidamide (11e)
Synthesized according to General Procedure C. White solid (350 mg, 89% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 0.9 Hz, 1H), 7.70 (dd, J = 8.1, 1.8 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.14–7.04 (m, 4H), 4.91 (brs, 2H), 3.16–3.07 (m, 2H), 2.91–2.82 (m, 2H), 2.55 (t, J = 7.7 Hz, 2H), 1.62 (h, J = 7.4 Hz, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 146.3, 141.1, 137.5, 135.0, 132.5, 130.1 (q, J = 5.8 Hz), 130.0 (q, J = 30.9 Hz), 128.8, 128.4, 123.5 (q, J = 274.3 Hz), 117.8, 110.7, 37.8, 37.2, 35.3 (q, J = 1.9 Hz), 24.8, 14.0.
4-(4-Butylphenethyl)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (11f)
Synthesized according to General Procedure C. White solid (380 mg, 91%). 1H NMR (400 MHz, CD3OD) δ 8.20 (d, J = 1.9 Hz, 0.4H), 8.00 (dd, J = 8.0, 1.9 Hz, 0.4H), 7.95 (d, J = 1.9 Hz, 0.6H), 7.77 (dd, J = 8.1, 1.9 Hz, 0.6H), 7.48 (d, J = 8.1 Hz, 0.4H), 7.41 (d, J = 8.1 Hz, 0.6H), 3.15–3.02 (m, 2H), 2.92–2.82 (m, 2H), 2.58 (t, J = 7.7 Hz, 2H), 1.58 (t, J = 7.7 Hz, 2H), 1.35 (h, J = 7.3 Hz, 2H), 0.94 (t, J = 7.3 Hz, 3H). HRMS (ESI+): calcd for C20H24F3N2O [M + H]+ 365.1835, found: 365.1843.
4-(4-(tert-Butyl)phenethyl)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (11g)
Synthesized according to General Procedure C. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
N′-Hydroxy-4-(4-methoxyphenethyl)-3-(trifluoromethyl)benzimidamide (11h)
Synthesized according to General Procedure C. White solid (321 mg, 82%). 1H NMR (400 MHz, CD3OD) δ 7.95 (d, J = 2.0 Hz, 1H), 7.77 (dd, J = 8.2, 2.3 Hz, 1H), 7.39 (d, J = 7.3 Hz, 1H), 7.10 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 3.76 (s, 3H), 3.09–3.01 (m, 2H), 2.88–2.80 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 158.2, 152.6, 141.7, 132.9, 131.5, 131.1, 129.0, 129.0, 128.0 (q, J = 29.8 Hz), 127.3 (q, J = 274.0 Hz), 123.3 (q, J = 5.9 Hz), 113.4, 54.2, 36.6, 34.7 (q, J = 1.8 Hz). HRMS (ESI+): calcd for C17H18F3N2O2 [M + H]+ 339.1315, found: 339.1344.
N′-Hydroxy-3-(trifluoromethyl)-4-(4-(trifluoromethyl)phenethyl)benzimidamide (11i)
Synthesized according to General Procedure C. White solid (141 mg, 34%). 1H NMR (500 MHz, CD3OD) δ 7.97 (d, J = 1.9 Hz, 1H), 7.80 (dd, J = 8.2, 1.9 Hz, 1H), 7.61–7.56 (m, 3H), 7.45 (d, J = 8.1 Hz, 1H), 7.40 (d, J = 7.9 Hz, 1H), 3.17–3.09 (m, 2H), 3.05–2.97 (m, 2H).
tert-Butyl (S)-2-(3-(4-(2-Cyclopropylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12a)
Synthesized according to General Procedure D. Yellow oil (61 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.14 (d, J = 7.5 Hz, 1H), 7.44 (t, J = 9.2 Hz, 1H), 5.27–5.03 (m, 1H), 3.77–3.42 (m, 2H), 2.87 (dt, J = 51.2, 8.1 Hz, 2H), 2.48–2.32 (m, 1H), 2.24–1.96 (m, 3H), 1.72–1.51 (m, 2H), 1.48–1.24 (m, 11H), 0.90 (t, J = 6.8 Hz, 2H), 0.52 – −0.00 (m, 1H). HRMS (ESI+): calcd for C23H28F3N3O3Na [M + Na]+ 474.1975, found: 474.1986.
tert-Butyl (S)-2-(3-(4-(2-Cyclopentylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12b)
Synthesized according to General Procedure D. Yellow oil (18 mg, 13%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.50–7.37 (m, 1H), 5.05 (dd, J = 8.1, 3.6 Hz, 1H), 3.74–3.40 (m, 2H), 2.88–2.75 (m, 2H), 2.47–2.27 (m, 1H), 2.21–2.08 (m, 2H), 2.07–1.92 (m, 1H), 1.93–1.72 (m, 3H), 1.68–1.49 (m, 6H), 1.44 (s, 3H)*, 1.28 (s, 6H), 1.19–1.07 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 181.1, 167.6, 153.6, 145.5, 131.9, 130.4, 125.3, 124.6, 80.7, 53.9, 46.5, 40.4, 38.4, 33.2*, 32.7, 32.6, 32.3, 31.7*, 28.5*, 28.3, 25.3, 23.9. Trifluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C25H32F3N3O3Na [M + Na]+ 502.2288, found: 502.2315.
tert-Butyl (S)-2-(3-(4-(2-Cyclohexylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12c)
Synthesized according to General Procedure D. Yellow oil (37 mg, 22%). 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.14 (d, J = 8.3 Hz, 1H), 7.43 (t, J = 9.4 Hz, 1H), 5.23–5.01 (m, 1H), 3.80–3.43 (m, 2H), 2.82 (t, J = 8.6 Hz, 2H), 2.49–2.28 (m, 1H), 2.21–1.95 (m, 3H), 1.83–1.59 (m, 6H), 1.48–1.14 (m, 14H), 1.04–0.93 (m, 2H). HRMS (ESI+): calcd for C26H35F3N3O3 [M + H]+ 494.2625, found: 494.2626.
tert-Butyl (S)-2-(3-(4-(4-Ethylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12d)
Synthesized according to General Procedure D. Yellow oil (56 mg, 16% – 3 steps). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 8.19–8.11 (m, 1H), 7.47–7.36 (m, 1H), 7.18–7.12 (m, 4H), 5.30–5.00 (m, 1H), 3.79–3.64 (m, 1H), 3.64–3.46 (m, 1H), 3.17–3.06 (m, 2H), 2.95–2.86 (m, 2H), 2.64 (q, J = 7.6 Hz, 2H), 2.50–2.33 (m, 1H), 2.24–1.97 (m, 3H), 1.47 (s, 3H)*, 1.31 (s, 6H), 1.24 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 181.2, 180.7*, 167.5, 154.4*, 153.6, 144.0*, 143.8, 142.3, 138.2, 132.1, 132.0*, 130.5, 128.5, 128.1, 128.0, 125.3 (q, J = 5.7 Hz), 125.0, 124.3 (q, J = 273.8 Hz), 80.6, 53.9, 46.8*, 46.5, 37.5, 35.2 (q, J = 1.9 Hz), 32.5, 31.6*, 28.6, 28.5*, 28.3, 24.5*, 23.9, 15.8. HRMS (ESI+): calcd for C28H32F3N3O3Na [M + Na]+ 538.2288, found: 538.2286.
tert-Butyl (S)-2-(3-(4-(4-Propylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12e)
Synthesized according to General Procedure D. Yellow oil (50 mg, 17%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 8.14 (d, J = 7.9 Hz, 1H), 7.47–7.34 (m, 1H), 7.15–7.10 (m, 4H), 5.25–5.03 (m, 1H), 3.79–3.45 (m, 2H), 3.17–3.08 (m, 2H), 2.98–2.86 (m, 2H), 2.57 (t, J = 7.6 Hz, 2H), 2.48–2.32 (m, 1H), 2.25–1.95 (m, 3H), 1.64 (h, J = 7.4 Hz, 2H), 1.47 (s, 3H)*, 1.31 (s, 6H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 181.2, 180.8*, 167.5, 153.6, 144.1, 140.8, 138.2, 132.2, 132.0*, 130.5, 128.7, 128.4, 125.4, 125.1, 124.2 (q, J = 273.7 Hz), 80.7, 80.6*, 54.0, 46.8*, 46.5, 37.8, 37.5, 35.2, 32.6, 31.6*, 28.5*, 28.3, 24.7, 23.9, 14.0. Splitting not observed on trifluoromethyl ipso carbon due to signal overlap. HRMS (ESI+): calcd for C29H34F3N3O3Na [M + Na]+ 552.2444, found: 552.2412.
tert-Butyl (S)-2-(3-(4-(4-Butylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12f)
Synthesized according to General Procedure D. Yellow oil (71 mg, 23%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 8.18–8.10 (m, 1H), 7.45–7.34 (m, 1H), 7.18–7.10 (m, 4H), 5.26–5.03 (m, 1H), 3.78–3.44 (m, 2H), 3.20–3.08 (m, 2H), 2.94–2.86 (m, 2H), 2.59 (t, J = 7.7 Hz, 2H), 2.47–2.33 (m, 1H), 2.22–1.95 (m, 3H), 1.66–1.52 (m, 2H), 1.50–1.23 (m, 11H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 181.2, 180.8*, 167.5, 153.6, 151.3*, 144.1, 143.8*, 141.0, 138.2, 132.1, 132.0*, 130.5, 129.3, 128.7, 128.4, 125.4, 125.1, 124.3 (q, J = 274.4 Hz), 80.6, 53.9, 46.8*, 46.5, 37.5, 35.4, 35.2 (q, J = 1.8 Hz), 33.8, 32.6, 31.6*, 28.5*, 28.3, 24.5*, 23.9, 22.5, 14.1. Splitting not observed on trifluoromethyl ipso carbon due to signal overlap. HRMS (ESI+): calcd for C30H36F3N3O3Na [M + Na]+ 566.2601, found: 566.2602.
tert-Butyl (S)-2-(3-(4-(4-(tert-Butyl)phenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12g)
Synthesized according to General Procedure D. Yellow oil (12 mg, 6% – 4 steps). 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 8.16 (d, J = 7.7 Hz, 1H), 7.51–7.38 (m, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.24–7.14 (m, 2H), 5.24–5.04 (m, 1H), 3.80–3.63 (m, 1H), 3.65–3.42 (m, 1H), 3.20–3.07 (m, 2H), 2.97–2.86 (m, 2H), 2.53–2.29 (m, 1H), 2.26–1.95 (m, 3H), 1.51–1.28 (m, 18H). HRMS (ESI+): calcd for C30H36F3N3O3Na [M + Na]+ 566.2601, found: 566.2712.
tert-Butyl (S)-2-(3-(4-(4-Methoxyphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12h)
Synthesized according to General Procedure D. Yellow oil (158 mg, 32%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (500 MHz, CDCl3) δ 8.36 (d, J = 2.3 Hz, 1H), 8.17–8.09 (m, 1H), 7.37 (dd, J = 25.4, 7.8 Hz, 1H), 7.17–7.08 (m, 2H), 6.88–6.81 (m, 2H), 5.24–5.03 (m, 1H), 3.79 (s, 3H), 3.77–3.65 (m, 1H), 3.62–3.46 (m, 1H), 3.14–3.04 (m, 2H), 2.93–2.82 (m, 2H), 2.49–2.29 (m, 1H), 2.23–2.08 (m, 2H), 2.07–1.96 (m, 1H), 1.47 (s, 3H)*, 1.31 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 181.1, 180.7*, 167.4, 162.5, 158.1, 153.5, 143.8, 143.6*, 133.0, 132.1, 132.0*, 130.4, 129.4, 125.2 (q, J = 5.8 Hz), 125.0, 124.0 (q, J = 274.2 Hz), 113.9, 80.5, 79.4*, 55.3, 53.8, 46.7*, 46.4, 38.6, 35.3 (q, J = 1.9 Hz), 28.6, 28.4*, 28.2, 24.4*, 23.8. Trifluoromethyl ipso carbon signal overlaps with adjacent signals.
tert-Butyl (S)-2-(3-(3-(Trifluoromethyl)-4-(4-(trifluoromethyl)phenethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (12i)
Synthesized according to General Procedure D. Yellow oil (58 mg, 28%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 1.7 Hz, 1H), 8.16 (d, J = 7.7 Hz, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.43–7.28 (m, 3H), 5.28–5.03 (m, 1H), 3.84–3.46 (m, 2H), 3.24–3.09 (m, 2H), 3.07–2.90 (m, 2H), 2.54–2.32 (m, 1H), 2.27–1.93 (m, 3H), 1.47 (s, 3H)*, 1.31 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 181.3, 180.9*, 167.4, 153.6, 151.4, 145.0, 143.1, 132.1, 130.6, 129.5 (d, J = 30.2 Hz), 128.9, 128.8 (d, J = 29.8 Hz), 125.6 (q, J = 3.8 Hz), 125.5, 124.3 (q, J = 272.1 Hz), 124.2 (q, J = 273.0 Hz), 80.6, 53.9, 46.8*, 46.5, 37.6, 34.8, 32.6, 31.6*, 28.5*, 28.3, 24.5*, 23.9.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(2-cyclopropylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13a)
Synthesized according to General Procedure E. Yellow oil (34 mg, 42% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.09 (brs, 1H), 8.30 (d, J = 2.2 Hz, 1H), 8.13 (dd, J = 8.0, 2.0 Hz, 1H), 7.43 (d, J = 8.1 Hz, 1H), 5.60 (dd, J = 7.9, 4.5 Hz, 1H), 3.95–3.74 (m, 2H), 2.87 (dt, J = 51.1, 8.0 Hz, 2H), 2.51–2.37 (m, 1H), 2.26–2.11 (m, 2H), 2.10–1.96 (m, 1H), 1.70–1.29 (m, 22H), 0.88 (t, J = 7.0 Hz, 2H), 0.48–0.04 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 179.5, 167.6, 162.0, 153.8, 150.5, 145.3, 131.7, 130.5, 129.2 (q, J = 30.2 Hz), 125.4 (q, J = 5.6 Hz), 124.7, 124.4 (q, J = 274.0 Hz), 82.4, 79.8, 55.4, 49.6, 32.8, 32.0, 31.3, 28.2, 22.6, 14.1, 4.7.
tert-Butyl (S)-(((tert-Butoxycarbonyl)imino)(2-(3-(4-(2-cyclopentylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methyl)carbamate (13b)
Synthesized according to General Procedure E. Yellow oil (14 mg, 60% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.31 (d, J = 1.7 Hz, 1H), 8.14 (dd, J = 8.1, 1.8 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 5.62 (dd, J = 7.8, 4.4 Hz, 1H), 3.96–3.77 (m, 2H), 2.88–2.79 (m, 2H), 2.52–2.41 (m, 1H), 2.26–2.14 (m, 2H), 2.10–1.97 (m, 1H), 1.90–1.77 (m, 3H), 1.58–1.38 (m, 24H), 1.23–1.09 (m, 2H). This material was taken forward without further characterization.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(2-cyclohexylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13c)
Synthesized according to General Procedure E. Clear oil (21 mg, 44% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.11 (brs, 1H), 8.31 (d, J = 1.7 Hz, 1H), 8.14 (dd, J = 8.1, 1.8 Hz, 1H), 7.43 (d, J = 8.1 Hz, 1H), 5.61 (dd, J = 7.8, 4.5 Hz, 1H), 3.96–3.73 (m, 2H), 2.88–2.78 (m, 2H), 2.44 (dt, J = 15.3, 7.5 Hz, 1H), 2.21 (s, 2H), 2.05 (q, J = 7.3 Hz, 1H), 1.83–1.10 (m, 29H), 1.02–0.82 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 179.5, 167.6, 150.5, 145.7, 131.8, 130.5, 129.2 (q, J = 30.2 Hz), 125.4 (q, J = 6.0 Hz), 124.6, 124.2 (q, J = 273.8 Hz), 82.5, 79.9, 55.4, 49.6, 39.6, 38.1, 33.3, 30.5, 29.9, 28.2, 26.8, 26.5. N-Boc carbonyl signals not observed. HRMS (ESI+): calcd for C32H45F3N5O5 [M + H]+ 636.3367, found: 636.3355.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(4-ethylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13d)
Synthesized according to General Procedure E. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
tert-Butyl (S)-(((tert-Butoxycarbonyl)imino)(2-(3-(4-(4-propylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methyl)carbamate (13e)
Synthesized according to General Procedure E. Yellow oil (25 mg, 39% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.11 (brs, 1H), 8.35 (d, J = 1.7 Hz, 1H), 8.13 (dd, J = 8.0, 1.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.12 (s, 4H), 5.62 (dd, J = 7.8, 4.5 Hz, 1H), 4.03–3.71 (m, 2H), 3.12 (dd, J = 10.1, 6.4 Hz, 2H), 2.95–2.86 (m, 2H), 2.61–2.40 (m, 3H), 2.26–1.97 (m, 3H), 1.77–1.57 (m, 2H), 1.45 (s, 18H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 179.6, 167.5, 162.1, 153.9, 150.4, 144.0, 140.8, 138.2, 132.1, 130.5, 129.3 (q, J = 30.5 Hz), 128.7, 128.4, 125.5 (q, J = 5.9 Hz), 125.1, 124.4 (q, J = 273.6 Hz), 82.5, 79.8, 55.5, 49.6, 37.8, 37.5, 35.2 (q, J = 2.0 Hz), 31.4, 28.2, 24.7, 24.2, 14.0. HRMS (ESI+): calcd for C35H45F3N5O5 [M + H]+ 672.3367, found: 672.3368.
tert-Butyl (S,E)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(4-butylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13f)
Synthesized according to General Procedure E. Yellow oil (39 mg, 44% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.33 (d, J = 1.8 Hz, 1H), 8.12 (dd, J = 8.0, 1.8 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.19–7.03 (m, 4H), 5.60 (dd, J = 7.8, 4.5 Hz, 1H), 3.96–3.72 (m, 2H), 3.15–3.06 (m, 2H), 2.92–2.84 (m, 2H), 2.61–2.53 (m, 2H), 2.49–2.37 (m, 1H), 2.26–1.97 (m, 3H), 1.66–1.32 (m, 22H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 179.5, 167.5, 158.4, 153.7, 143.9, 140.9, 138.1, 132.0, 130.5, 129.3 (q, J = 30.4 Hz), 128.6, 128.4, 125.5 (q, J = 5.8 Hz), 125.0, 124.2 (q, J = 275.3 Hz), 81.5, 55.4, 49.6, 37.5, 35.3, 35.2 (q, J = 2.0 Hz), 33.8, 29.8, 28.2, 24.1, 22.5, 14.1. HRMS (ESI+): calcd for C36H47F3N5O5 [M + H]+ 686.3524, found: 686.3522.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(4-(tert-butyl)phenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13g)
Synthesized according to General Procedure E. Yellow oil (8 mg, 53% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 1.8 Hz, 1H), 8.15 (dd, J = 8.0, 1.8 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.2 Hz, 2H), 5.65 (dd, J = 7.8, 4.8 Hz, 1H), 4.00–3.89 (m, 1H), 3.88–3.77 (m, 1H), 3.18–3.08 (m, 2H), 2.94–2.86 (m, 2H), 2.55–2.44 (m, 1H), 2.32–2.16 (m, 2H), 2.12–2.05 (m, 1H), 1.46 (s, 18H), 1.32 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 179.9, 166.7, 161.9, 157.2, 153.9, 150.1, 135.6, 131.5, 131.0 (q, J = 31.8 Hz), 129.4, 126.9 (q, J = 5.5 Hz), 126.3, 123.1, 122.5 (q, J = 273.7 Hz), 82.3, 79.6, 55.3, 49.5, 31.2, 29.7, 28.1, 28.0, 24.1. N-Boc carbonyl peaks not observed.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(4-methoxyphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13h)
Synthesized according to General Procedure E. Yellow oil (141 mg, 70% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.35 (d, J = 1.7 Hz, 1H), 8.13 (dd, J = 8.0, 1.8 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 5.62 (dd, J = 7.8, 4.5 Hz, 1H), 3.97–3.85 (m, 1H), 3.85–3.73 (m, 4H), 3.15–3.04 (m, 2H), 2.92–2.81 (m, 2H), 2.53–2.39 (m, 1H), 2.29–2.13 (m, 2H), 2.13–1.95 (m, 1H), 1.44 (s, 18H). 13C NMR (101 MHz, CD3OD) δ 179.6, 167.5, 162.1, 158.2, 153.9, 150.5, 143.9, 133.1, 132.1, 130.5, 129.5, 129.3 (q, J = 30.4 Hz), 125.5 (q, J = 5.8 Hz), 125.1, 124.3 (q, J = 273.8 Hz), 114.1, 82.5, 79.7, 55.4, 49.6, 37.0, 35.4 (q, J = 1.9 Hz), 31.6, 28.2, 28.1, 24.2.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(3-(trifluoromethyl)-4-(4-(trifluoromethyl)phenethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (13i)
Synthesized according to General Procedure E. Yellow oil (38 mg, 52% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.09 (brs, 1H), 8.35 (d, J = 1.7 Hz, 1H), 8.13 (dd, J = 8.0, 1.8 Hz, 1H), 7.57–7.51 (m, 2H), 7.35 (d, J = 8.1 Hz, 1H), 7.33–7.26 (m, 2H), 5.60 (dd, J = 7.8, 4.6 Hz, 1H), 3.96–3.71 (m, 2H), 3.20–3.04 (m, 2H), 3.04–2.90 (m, 2H), 2.51–2.35 (m, 1H), 2.29–2.10 (m, 2H), 2.10–1.94 (m, 1H), 1.45 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 179.7, 167.4, 162.1, 153.9, 150.5, 145.0, 143.0, 132.0, 130.7, 129.5 (q, J = 30.7 Hz), 129.0, 128.9 (q, J = 32.1 Hz), 125.7, 125.6 (q, J = 3.8 Hz), 125.5, 124.4 (q, J = 273.1 Hz), 124.3 (q, J = 272.9 Hz), 82.5, 79.8, 55.5, 49.6, 37.6, 34.8, 31.6, 28.2, 24.2.
(S)-2-(3-(4-(2-Cyclopropylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14a)
Synthesized according to General Procedure F. White solid (3 mg, 28%). 1H NMR (500 MHz, CD3OD) δ 8.28 (d, J = 1.8 Hz, 1H), 8.22 (dd, J = 8.2, 1.8 Hz, 1H), 7.62 (d, J = 8.1 Hz, 1H), 5.47 (dd, J = 8.1, 1.9 Hz, 1H), 3.79 (td, J = 9.2, 2.5 Hz, 1H), 3.63 (td, J = 9.6, 7.1 Hz, 1H), 2.91 (dt, J = 63.2, 8.1 Hz, 2H), 2.64–2.45 (m, 2H), 2.29–2.19 (m, 1H), 2.16–2.02 (m, 1H), 1.75–1.50 (m, 2H), 1.47–1.33 (m, 2H), 0.94 (t, J = 6.8 Hz, 2H), 0.50–0.06 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 179.4, 168.7, 157.1, 146.8 (q, J = 1.6 Hz), 133.5, 131.7, 130.1 (q, J = 30.2 Hz), 125.7 (q, J = 5.8 Hz), 125.5 (q, J = 273.5 Hz), 111.4, 56.5, 33.0, 32.7, 24.3, 23.4, 14.3, 5.1. HRMS (ESI+): calcd for C19H23F3N5O [M + H]+ 394.1849, found: 394.1843.
(S)-2-(3-(4-(2-Cyclopentylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14b)
Synthesized according to General Procedure F. White solid (7 mg, 64%). 1H NMR (400 MHz, CD3OD) δ 8.33–8.21 (m, 2H), 7.65 (d, J = 8.1 Hz, 1H), 5.54–5.46 (m, 1H), 3.82 (t, J = 8.8 Hz, 1H), 3.75–3.60 (m, 1H), 2.94–2.86 (m, 2H), 2.65–2.51 (m, 2H), 2.33–2.07 (m, 2H), 2.00–1.82 (m, 3H), 1.74–1.55 (m, 6H), 1.27–1.17 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 179.4, 168.7, 159.5, 157.1, 148.5, 133.6, 131.7, 125.8, 61.5, 56.5, 47.9, 41.6, 33.6, 26.1, 20.9, 14.5. Trifluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C21H27F3N5O [M + H]+ 422.2162, found: 422.2163.
(S)-2-(3-(4-(2-Cyclohexylethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14c)
Synthesized according to General Procedure F. White solid (7 mg, 63%). 1H NMR (400 MHz, CD3OD) δ 8.28 (d, J = 1.8 Hz, 1H), 8.25–8.17 (m, 1H), 7.61 (d, J = 8.1 Hz, 1H), 5.46 (dd, J = 7.9, 1.9 Hz, 1H), 3.79 (td, J = 9.2, 2.6 Hz, 1H), 3.62 (td, J = 9.7, 7.2 Hz, 1H), 2.91–2.81 (m, 2H), 2.65–2.44 (m, 2H), 2.30–2.02 (m, 2H), 1.87–1.64 (m, 5H), 1.59–1.48 (m, 2H), 1.46–1.13 (m, 4H), 1.00 (qd, J = 12.1, 3.2 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 179.9, 169.1, 157.5, 147.2, 133.9, 132.1, 130.5 (q, J = 30.7 Hz), 126.2, 126.1 (q, J = 6.3 Hz), 125.9 (q, J = 275.9 Hz), 56.9, 39.2, 34.9, 34.4, 33.2, 30.6, 28.2, 27.9, 24.7. HRMS (ESI+): calcd for C22H29F3N5O [M + H]+ 436.2319, found: 436.2357.
(S)-2-(3-(4-(4-Ethylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14d)
Synthesized according to General Procedure F. White solid (28 mg, 50% – 2 steps). 1H NMR (400 MHz, CD3OD) δ 8.30 (d, J = 1.7 Hz, 1H), 8.18 (dd, J = 8.1, 2.1 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.12 (s, 4H), 5.48 (dd, J = 7.9, 1.8 Hz, 1H), 3.79 (td, J = 9.4, 2.6 Hz, 1H), 3.63 (td, J = 9.6, 7.1 Hz, 1H), 3.12 (dd, J = 10.0, 6.5 Hz, 2H), 2.94–2.85 (m, 2H), 2.66–2.44 (m, 4H), 2.23 (q, J = 6.6 Hz, 1H), 2.18–2.03 (m, 1H), 1.21 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 179.5, 168.6, 157.1, 145.6 (q, J = 1.6 Hz), 143.5, 139.3, 133.9, 131.6, 130.2 (q, J = 30.2 Hz), 129.4, 129.0, 126.0, 125.8 (q, J = 5.9 Hz), 125.5 (q, J = 275.1 Hz), 56.5, 38.4, 36.2 (d, J = 1.8 Hz), 32.8, 29.5, 24.3, 16.3. HRMS (ESI+): calcd for C24H26F3N5O [M + H]+ 458.2162, found: 458.2156.
(S)-2-(3-(4-(4-Propylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14e)
Synthesized according to General Procedure F. White solid (9 mg, 48%). 1H NMR (400 MHz, CD3OD) δ 8.34 (d, J = 1.8 Hz, 1H), 8.22 (dd, J = 8.1, 1.6 Hz, 1H), 7.59 (d, J = 8.1 Hz, 1H), 7.16–7.12 (m, 4H), 5.51 (dd, J = 7.9, 1.9 Hz, 1H), 3.83 (td, J = 9.5, 2.7 Hz, 1H), 3.67 (td, J = 9.6, 7.2 Hz, 1H), 3.21–3.12 (m, 2H), 2.99–2.90 (m, 2H), 2.70–2.47 (m, 4H), 2.34–2.22 (m, 1H), 2.22–2.07 (m, 1H), 1.73–1.60 (m, 2H), 0.97 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ 179.5, 168.6, 157.1, 145.7, 141.8, 139.4, 133.9, 131.6, 130.2 (q, J = 30.2 Hz), 129.6, 129.4, 126.0, 125.8 (q, J = 5.9 Hz), 125.6 (q, J = 274.0 Hz), 56.5, 38.7, 38.4, 36.2, 32.7, 25.8, 24.3, 14.1. HRMS (ESI+): calcd for C25H29F3N5O [M + H]+ 472.2324, found: 472.2323.
(S)-2-(3-(4-(4-Butylphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14f)
Synthesized according to General Procedure F. White solid (4 mg, 37%). 1H NMR (500 MHz, CD3OD) δ 8.20 (s, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.49–7.36 (m, 1H), 7.00 (s, 4H), 5.15 (s, 1H), 3.83–3.67 (m, 2H), 3.06–2.95 (m, 2H), 2.86–2.74 (m, 2H), 2.48 (t, J = 7.6 Hz, 2H), 2.22–2.00 (m, 2H), 1.54–1.38 (m, 2H), 1.33–1.15 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, cd3od) δ 179.5, 168.6, 157.1, 145.6, 141.9, 139.3, 133.9, 131.6, 130.2 (q, J = 30.5 Hz), 129.5, 129.4, 126.0, 125.8 (q, J = 6.2 Hz), 125.4 (q, J = 273.8 Hz), 56.6, 38.3, 36.2, 36.1, 35.0, 32.8, 24.4, 23.3, 14.3. HRMS (ESI+): calcd for C26H31F3N5O [M + H]+ 486.2475, found: 486.2466.
(S)-2-(3-(4-(4-(tert-Butyl)phenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14g)
Synthesized according to General Procedure F. White solid (1 mg, 17%). 1H NMR (400 MHz, CD3OD) δ 8.31 (d, J = 1.8 Hz, 1H), 8.19 (dd, J = 8.1, 1.8 Hz, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.3 Hz, 2H), 5.47 (dd, J = 8.0, 2.0 Hz, 1H), 3.79 (td, J = 9.4, 2.6 Hz, 1H), 3.63 (td, J = 9.5, 7.0 Hz, 1H), 3.13 (t, J = 8.1 Hz, 2H), 2.95–2.86 (m, 2H), 2.66–2.41 (m, 2H), 2.34–2.04 (m, 2H), 1.31 (s, 9H). 13C NMR (101 MHz, CD3OD) δ 179.5, 168.6, 157.1, 150.3, 145.7, 139.1, 133.8, 131.6, 130.2 (q, J = 30.4 Hz), 129.1, 126.4, 126.0, 125.8 (q, J = 5.9 Hz), 125.5 (q, J = 273.9 Hz), 56.5, 38.3, 36.2, 35.2, 32.8, 31.8 24.3. HRMS (ESI+): calcd for C26H31F3N5O [M + H]+: 486.2475, found: 486.2480.
(S)-2-(3-(4-(4-Methoxyphenethyl)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14h)
Synthesized according to General Procedure F. White solid (3 mg, 23%). 1H NMR (400 MHz, CD3OD) δ 8.28 (d, J = 1.6 Hz, 1H), 8.16 (dd, J = 8.1, 1.9 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 5.53–5.46 (m, 1H), 3.84–3.73 (m, 4H), 3.69–3.58 (m, 1H), 3.13–3.04 (m, 2H), 2.90–2.81 (m, 2H), 2.66–2.43 (m, 2H), 2.30–2.19 (m, 1H), 2.16–2.04 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 179.4, 168.5, 159.6, 157.0, 145.5 (q, J = 1.6 Hz), 134.0, 133.8, 131.6, 130.4, 130.1 (q, J = 30.2 Hz), 125.9, 125.7 (q, J = 5.9 Hz), 125.6 (q, J = 273.7 Hz), 114.9, 56.4, 55.6, 37.8, 36.2 (q, J = 1.7 Hz), 32.7, 24.3. HRMS (ESI+): calcd for C23H25F3N5O2 [M + H]+ 460.1955, found: 460.1954.
(S)-2-(3-(3-(Trifluoromethyl)-4-(4-(trifluoromethyl)phenethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide Hydrochloride (14i)
Synthesized according to General Procedure F. White solid (4 mg, 35%). 1H NMR (500 MHz, CD3OD) δ 8.32 (d, J = 1.8 Hz, 1H), 8.21 (dd, J = 8.1, 1.8 Hz, 1H), 7.64–7.56 (m, 3H), 7.41 (d, J = 7.9 Hz, 2H), 5.49 (dd, J = 8.0, 1.8 Hz, 1H), 3.79 (td, J = 9.5, 2.6 Hz, 1H), 3.63 (td, J = 9.7, 7.2 Hz, 1H), 3.22–3.15 (m, 2H), 3.08–3.01 (m, 2H), 2.65–2.45 (m, 2H), 2.29–2.19 (m, 1H), 2.16–2.04 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 179.5, 168.6, 157.1, 146.7 (q, J = 1.5 Hz), 144.9 (q, J = 1.6 Hz), 133.8, 131.8, 130.2, 130.1 (q, J = 61.4 Hz), 129.8 (q, J = 63.2 Hz), 126.4 (q, J = 3.9 Hz), 126.3, 125.9 (q, J = 6.0 Hz), 125.6 (q, J = 274.0 Hz), 125.5 (q, J = 272.6 Hz), 56.5, 49.1, 38.3, 35.5, 32.7, 24.3. HRMS (ESI+): calcd for C23H22F6N5O [M + H]+ 498.1723, found: 498.1719.
4-(tert-Butoxy)-3-(trifluoromethyl)benzonitrile (16)
A solution of 4-fluoro-3-(trifluoromethyl)benzonitrile in THF was cooled to 0 °C and purged under nitrogen. Potassium tert-butoxide (1 M in THF) was then added under cooling. The resulting solution was heated to 75 °C for 7–10 h until TLC indicated that the starting material had been fully consumed. The reaction was cooled to room temperature and subsequently partitioned between ethyl acetate and water. The aqueous layer was the extracted twice with ethyl acetate. The combined organic layers were rinsed with brine, dried over anhydrous Na2SO4, filtered, and purified via flash column chromatography (5% ethyl acetate/hexanes). Clear oil (358 mg, 89%). 1H NMR (500 MHz, CDCl3) δ 7.85 (d, J = 2.2 Hz, 1H), 7.71 (dd, J = 8.7, 2.2 Hz, 1H), 7.25 (d, J = 8.9 Hz, 1H), 1.54 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 158.9 (q, J = 1.8 Hz), 136.5, 131.6 (q, J = 5.3 Hz), 123.4 (q, J = 30.9 Hz), 122.6 (q, J = 272.9 Hz), 119.4, 118.1, 103.9, 82.7, 29.0. HRMS (ESI+): calcd for C12H13F3NO [M + H]+ 244.0944, found: 244.0955. Compound is previously known and characterized.
4-(tert-Butoxy)-N′-hydroxy-3-(trifluoromethyl)benzimidamide (17)
Synthesized according to General Procedure C. White solid (237 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 2.4 Hz, 1H), 7.69 (dd, J = 8.7, 2.4 Hz, 1H), 7.21 (d, J = 8.7 Hz, 1H), 6.70 (brs, 1H), 4.82 (brs, 2H), 1.49 (s, 9H). HRMS (ESI+): calcd for C12H16F3N2O2 [M + H]+ 277.1158, found: 277.1170. Compound is previously known and characterized.
tert-Butyl (S)-2-(3-(4-(tert-Butoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (18)
Synthesized according to General Procedure D. Clear oil (478 mg, 58%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, cdcl3) δ 8.39 (d, J = 2.4 Hz, 1H), 8.29 (dd, J = 8.6, 2.1 Hz, 1H), 7.46–7.35 (m, 1H), 5.25–5.03 (m, 1H), 3.76–3.65 (m, 1H), 3.62–3.45 (m, 1H), 2.49–2.35 (m, 1H), 2.23–1.92 (m, 3H), 1.56 (s, 9H), 1.46 (s, 3H)*, 1.29 (s, 6H). HRMS (ESI+): calcd for C22H29F3N3O4 [M + H]+: 456.2105, found: 456.2113.
tert-Butyl (S)-2-(3-(4-Hydroxy-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (19)
The Boc-protecting group and tert-butoxy functionality were removed according to General Procedure F. The resulting solution was then concentrated under reduced pressure and triturated with diethyl ether. The concentrated material was redissolved in THF. Triethylamine (3.0 equiv) and Boc2O (0.95 equiv) were added. The solution was allowed to stir for 4 h, at which point TLC showed the complete consumption of the starting material. The reaction was concentrated, loaded onto Celite, and subjected to column chromatography (1% methanol/dichloromethane) to afford a pure product. Off-white solid (169 mg, 77%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CD3OD) δ 8.16 (d, J = 2.1 Hz, 1H), 8.08 (d, J = 6.2 Hz, 1H), 7.12–7.03 (m, 1H), 5.17–5.06 (m, 1H), 3.73–3.61 (m, 1H), 3.59–3.48 (m, 1H), 2.56–2.37 (m, 1H), 2.21–1.94 (m, 3H), 1.46 (s, 3H)*, 1.27 (s, 6H). 13C NMR (101 MHz, CD3OD) δ 182.4, 182.0*, 168.6, 160.1, 156.0, 155.4, 133.3, 127.2 (d, J = 5.1 Hz), 124.9 (q, J = 274.1 Hz), 118.5, 118.4 (q, J = 31.8 Hz), 81.9, 81.8*, 55.2, 55.1*, 47.9*, 47.6, 33.2, 32.4*, 28.6*, 28.4, 25.3*, 24.7. HRMS (ESI+): calcd for C18H21F3N3O4 [M + H]+ 400.1479, found: 400.1482.
tert-Butyl (S)-2-(3-(4-(Allyloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (20a)
Synthesized according to General Procedure G. Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 2.2 Hz, 1H), 8.19 (dd, J = 8.7, 1.7 Hz, 1H), 7.10–7.04 (m, 1H), 6.13–5.98 (m, 1H), 5.49 (dq, J = 17.3, 1.7 Hz, 1H), 5.33 (d, J = 10.7 Hz, 1H), 5.22–5.01 (m, 1H), 4.71 (d, J = 4.9 Hz, 2H), 3.80–3.45 (m, 2H), 2.40 (dd, J = 16.1, 8.2 Hz, 1H), 2.21–1.95 (m, 3H), 1.46 (s, 3H)*, 1.30 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 181.0, 180.6*, 167.3, 158.7, 158.6*, 154.4*, 153.6, 132.5, 131.8, 126.8 (q, J = 5.3 Hz), 123.3 (q, J = 272.0 Hz), 119.9 (q, J = 32.0 Hz), 119.1, 118.1, 113.5, 113.4*, 80.6, 80.5*, 69.5, 53.9, 46.8*, 46.5, 32.5, 31.6*, 28.5*, 28.3, 24.5*, 23.8. HRMS (ESI+): calcd for C21H24F3N3O4Na [M + Na]+ 462.1611, found: 462.1609.
tert-Butyl (S)-2-(3-(4-(But-3-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (20b)
Synthesized according to General Procedure G. Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 2.1 Hz, 1H), 8.18 (dd, J = 8.7, 2.0 Hz, 1H), 7.10–7.00 (m, 1H), 5.92 (ddt, J = 17.1, 10.2, 6.8 Hz, 1H), 5.26–5.01 (m, 3H), 4.15 (t, J = 6.5 Hz, 2H), 3.78–3.65 (m, 1H), 3.64–3.44 (m, 1H), 2.60 (q, J = 6.6 Hz, 2H), 2.50–2.29 (m, 1H), 2.23–1.98 (m, 3H), 1.46 (s, 3H)*, 1.29 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 180.9, 180.5*, 167.3, 159.0, 158.9*, 153.6, 133.7, 132.5, 126.7, 123.3 (d, J = 273.3 Hz), 119.6 (d, J = 31.6 Hz), 118.8, 117.6, 113.0, 112.9*, 80.6, 80.5*, 68.5, 53.9, 46.7*, 46.4, 33.4, 32.5, 31.5*, 28.4*, 28.2, 24.5*, 23.8. HRMS (ESI+): calcd for C22H26F3N3O4Na [M + Na]+ 476.1768, found: 476.1778.
tert-Butyl (S)-2-(3-(4-(Hex-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21a)
Synthesized according to General Procedure H. Yellow oil (23 mg, 52%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.17 (dd, J = 8.8, 2.2 Hz, 1H), 7.12–7.03 (m, 1H), 5.87 (dt, J = 14.3, 6.8 Hz, 1H), 5.74–5.61 (m, 1H), 5.23–5.02 (m, 1H), 4.65 (d, J = 5.7 Hz, 2H), 3.78–3.45 (m, 2H), 2.39 (dd, J = 15.5, 7.6 Hz, 1H), 2.22–1.97 (m, 5H), 1.51–1.25 (m, 11H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 180.9, 167.4, 159.0, 153.7, 136.0, 135.9*, 132.4, 126.8, 123.7, 118.8, 113.7, 113.6*, 80.7, 80.6*, 69.8, 53.9, 46.8*, 46.5, 34.5, 32.5, 31.6*, 28.5*, 28.3, 24.5*, 23.9, 22.2, 13.7. Triflluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C24H30F3N3O4Na [M + Na]+ 504.2081, found: 504.2098.
tert-Butyl (S)-2-(3-(4-(Hept-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21b)
Synthesized according to General Procedure H. Yellow oil (28 mg, 60%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.4, 1.5 Hz, 1H), 7.12–7.03 (m, 1H), 5.88 (dt, J = 14.2, 6.8 Hz, 1H), 5.74–5.61 (m, 1H), 5.24–5.01 (m, 1H), 4.82–4.59 (m, 2H), 3.77–3.66 (m, 1H), 3.62–3.45 (m, 1H), 2.48–2.34 (m, 1H), 2.23–1.95 (m, 5H), 1.49–1.27 (m, 13H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 181.0, 180.5*, 167.4, 159.0, 158.8*, 153.6, 136.2, 136.1*, 132.4, 126.8 (q, J = 5.7 Hz), 123.5, 123.1 (q, J = 272.0 Hz), 118.8, 113.7, 113.6*, 110.2, 80.6, 80.5*, 69.8, 53.9, 46.8*, 46.5, 32.5, 32.1, 31.6*, 31.2, 28.5*, 28.3, 24.5*, 23.9, 22.3, 14.0. 13C NMR (126 MHz, CDCl3) δ 179.3, 167.4, 159.0, 136.2, 132.5, 126.9 (d, J = 5.5 Hz), 123.6, 113.7, 69.8, 55.4, 49.6, 32.1, 31.2, 29.9, 28.2, 22.3, 14.0. Trifluoromethyl, ipso, and N-Boc carbonyl carbon signals not observed.
tert-Butyl (S)-2-(3-(4-(Oct-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21c)
Synthesized according to General Procedure H. Yellow oil (25 mg, 72%). 1H NMR (500 MHz, CDCl3) δ 8.29 (d, J = 2.2 Hz, 1H), 8.17 (dd, J = 8.6, 2.5 Hz, 1H), 7.07 (dd, J = 11.0, 8.2 Hz, 1H), 5.87 (dt, J = 15.7, 6.8 Hz, 1H), 5.72–5.60 (m, 1H), 5.21–5.02 (m, 1H), 4.74–4.57 (m, 2H), 3.78–3.45 (m, 2H), 2.47–2.25 (m, 1H), 2.21–1.93 (m, 5H), 1.48–1.14 (m, 15H), 1.00–0.77 (m, 3H). HRMS (ESI+): calcd for C26H34F3N3O4Na [M + Na]+ 532.2394, found: 532.2400.
tert-Butyl (S,E)-2-(3-(3-(Trifluoromethyl)-4-((3-(4-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21d)
Synthesized according to General Procedure H. Clear oil (180 mg, 49%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (500 MHz, CDCl3) δ 8.33 (d, J = 2.7 Hz, 1H), 8.23 (dd, J = 8.4, 1.9 Hz, 1H), 7.59 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.16–7.07 (m, 1H), 6.84 (d, J = 16.1 Hz, 1H), 6.48 (dt, J = 16.1, 5.1 Hz, 1H), 5.22–5.03 (m, 1H), 4.90 (d, J = 5.0 Hz, 2H), 3.76–3.64 (m, 1H), 3.60–3.46 (m, 1H), 2.48–2.31 (m, 1H), 2.20–1.97 (m, 3H), 1.46 (s, 3H)*, 1.30 (s, 6H). HRMS (ESI+): calcd for C28H27F6N3O4Na [M + Na]+ 606.1798, found: 606.1826.
tert-Butyl (S,E)-2-(3-(3-(Trifluoromethyl)-4-((3-(3-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21e)
Synthesized according to General Procedure H. Yellow oil (19 mg, 48%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 2.2 Hz, 1H), 8.20 (dd, J = 8.6, 2.3 Hz, 1H), 7.63 (s, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 6.81 (dt, J = 16.0, 1.7 Hz, 1H), 6.45 (dt, J = 16.0, 5.1 Hz, 1H), 5.20–4.96 (m, 1H), 4.88 (d, J = 3.6 Hz, 2H), 3.69 (dt, J = 13.3, 6.8 Hz, 1H), 3.62–3.40 (m, 1H), 2.37 (tt, J = 14.9, 6.6 Hz, 1H), 2.23–1.91 (m, 3H), 1.45 (s, 3H)*, 1.26 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 181.1, 167.3, 158.2, 137.1, 132.6, 131.7, 129.9, 129.3, 126.8, 125.0, 124.7, 123.4 (q, J = 3.8 Hz), 119.4, 113.5, 80.7, 80.6*, 69.0, 53.9, 46.8*, 46.5, 32.6, 31.6*, 29.9*, 28.3, 24.5*, 23.9. Trifluoromethyl, ipso, and N-Boc carbonyl carbon signals not observed.
tert-Butyl (S,E)-2-(3-(3-(Trifluoromethyl)-4-((3-(2-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21f)
Synthesized according to General Procedure H. Yellow oil (20 mg, 51%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 2.3 Hz, 1H), 8.21 (dd, J = 8.6, 2.3 Hz, 1H), 7.68–7.61 (m, 2H), 7.56–7.47 (m, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.21 (dq, J = 15.9, 2.1 Hz, 1H), 7.13 (t, J = 8.8 Hz, 1H), 6.35 (dt, J = 15.8, 5.1 Hz, 1H), 5.27–5.00 (m, 1H), 4.95–4.86 (m, 2H), 3.78–3.64 (m, 1H), 3.62–3.46 (m, 1H), 2.48–2.34 (m, 1H), 2.23–2.08 (m, 2H), 2.08–1.95 (m, 1H), 1.46 (s, 3H)*, 1.30 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 181.0, 180.5*, 167.4, 159.0, 153.6, 136.2, 132.4, 127.0, 126.8 (q, J = 5.6 Hz), 124.7, 123.5, 122.0, 118.8, 113.6, 110.2, 80.6, 80.5*, 69.8, 53.9, 32.6*, 32.1, 31.2, 28.5*, 28.3, 22.3. Trifluoromethyl, ipso, and N-Boc carbonyl carbon signals not observed. HRMS (ESI+): calcd for C28H28F6N3O4 [M + H]+ 584.1979, found: 584.1985.
tert-Butyl (S,E)-2-(3-(4-((3-(4-Fluorophenyl)allyl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21g)
Synthesized according to General Procedure H. Yellow oil (20 mg, 33%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 2.2 Hz, 1H), 8.20 (d, J = 8.7 Hz, 1H), 7.44–7.34 (m, 2H), 7.13 (t, J = 8.6 Hz, 1H), 7.03 (dd, J = 9.7, 7.6 Hz, 2H), 6.75 (d, J = 16.0 Hz, 1H), 6.31 (dt, J = 16.0, 5.4 Hz, 1H), 5.23–5.01 (m, 1H), 4.86 (d, J = 5.4 Hz, 2H), 3.78–3.44 (m, 2H), 2.43–2.34 (m, 1H), 2.20–2.10 (m, 2H), 2.04–1.97 (m, 1H), 1.46 (s, 3H)*, 1.30 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 181.0, 180.6*, 167.3, 162.7 (d, J = 247.6 Hz), 158.7, 154.4*, 153.6, 132.6, 132.4, 132.3, 128.4 (d, J = 8.0 Hz), 126.9 (d, J = 5.2 Hz), 123.3 (q, J = 270.3 Hz), 122.7, 119.5 (q, J = 31.2 Hz), 119.2, 115.7 (d, J = 21.7 Hz), 113.6, 113.5*, 80.6, 69.4, 53.9, 46.8*, 46.5, 32.5, 31.6*, 28.5*, 28.3, 24.5*, 23.9. HRMS (ESI+): calcd for C27H27F4N3O4Na [M + Na]+ 556.1830, found: 556.1814.
tert-Butyl (S,E)-2-(3-(4-((3-(3-Bromophenyl)allyl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21h)
Synthesized according to General Procedure H. The product was confirmed via HRMS and carried forward as crude to the next step. HRMS (ESI+): calcd for C22H20BrF3N3O2 [M - Boc]+ 494.0686, found: 494.0695.
tert-Butyl (S,E)-2-(3-(3-(Trifluoromethyl)-4-((4-(4-(trifluoromethyl)phenyl)but-3-en-1-yl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (21i)
Synthesized according to General Procedure H. Yellow oil (121 mg, 61%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 2.2 Hz, 1H), 8.24–8.17 (m, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 7.08 (t, J = 9.1 Hz, 1H), 6.58 (d, J = 15.9 Hz, 1H), 6.43 (dt, J = 15.9, 6.9 Hz, 1H), 5.23–5.02 (m, 1H), 4.24 (t, J = 6.1 Hz, 2H), 3.77–3.44 (m, 2H), 2.78 (q, J = 6.4 Hz, 2H), 2.48–2.28 (m, 1H), 2.23–2.06 (m, 2H), 2.06–1.92 (m, 1H), 1.46 (s, 3H)*, 1.29 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 181.0, 180.6*, 167.3, 159.0, 154.4*, 153.6, 140.9, 132.6, 131.7, 129.2 (d, J = 32.2 Hz), 128.6 (d, J = 32.1 Hz), 128.4, 126.9, 126.4, 125.6 (q, J = 3.9 Hz), 124.2 (d, J = 273.3 Hz), 123.3 (d, J = 273.9 Hz), 119.1, 113.1, 80.6, 80.5*, 68.4, 53.9, 46.8*, 46.5, 32.7, 32.5, 31.6*, 28.5*, 28.3, 23.9. Incomplete splitting observed on trifluoromethyl and ipso carbon signals. HRMS (ESI+): calcd for C29H30F6N3O4 [M + H]+ 598.2135, found: 598.2191.
tert-Butyl (S)-(((tert-Butoxycarbonyl)imino)(2-(3-(4-(hex-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methyl)carbamate (22a)
Synthesized according to General Procedure I. Yellow oil (20 mg, 67% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.28 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.7, 2.2 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 5.93–5.81 (m, 1H), 5.74–5.56 (m, 2H), 4.77–4.63 (m, 2H), 3.95–3.74 (m, 2H), 2.51–2.38 (m, 1H), 2.29–1.95 (m, 5H), 1.52–1.41 (m, 20H), 0.90 (t, J = 7.4 Hz, 3H). HRMS (ESI+): calcd for C30H41F3N5O6 [M + H]+ 624.3003, found: 624.3030.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(hept-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22b)
Synthesized according to General Procedure I. Yellow oil (9 mg, 25% – 2 steps). 1H NMR (500 MHz, CDCl3) δ 10.11 (brs, 1H), 8.28 (d, J = 2.2 Hz, 1H), 8.17 (dd, J = 8.7, 2.2 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 5.94–5.82 (m, 1H), 5.70–5.58 (m, 2H), 4.77–4.62 (m, 2H), 3.94–3.75 (m, 2H), 2.44 (p, 7.6 Hz, 1H), 2.28–2.01 (m, 5H), 1.50–1.28 (m, 22H), 0.89 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.5, 167.4, 159.0, 153.8, 136.2, 132.5, 126.9 (d, J = 5.5 Hz), 123.6, 123.2 (q, J = 271.9 Hz), 119.8 (q, J = 30.6 Hz), 118.9, 113.7, 69.8, 55.4, 49.6, 32.1, 31.2, 29.9, 28.2, 28.1, 22.3, 14.0. N-Boc carbonyl and tert-butyoxy carbon signals not observed. HRMS (ESI+): calcd for C31H43F3N5O6 [M + H]+ 638.3160, found: 638.3132.
tert-Butyl (S)-(((tert-Butoxycarbonyl)amino)(2-(3-(4-(oct-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22c)
Synthesized according to General Procedure I. Yellow oil (5 mg, 17% – 2 steps). 1H NMR (500 MHz, CDCl3) δ 10.12 (brs, 1H), 8.28 (d, J = 2.2 Hz, 1H), 8.17 (dd, J = 8.7, 2.2 Hz, 1H), 7.07 (d, J = 8.7 Hz, 1H), 5.92–5.83 (m, 1H), 5.71–5.56 (m, 2H), 4.79–4.62 (m, 2H), 3.96–3.74 (m, 2H), 2.49–2.41 (m, 1H), 2.24–2.00 (m, 5H), 1.53–1.30 (m, 24H), 0.88 (t, J = 7.0 Hz, 3H).
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(3-(trifluoromethyl)-4-(((E)-3-(4-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22d)
Synthesized according to General Procedure I. Yellow oil (100 mg, 45% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.21 (brs, 1H), 8.33 (d, J = 1.7 Hz, 1H), 8.22 (dd, J = 8.7, 2.0 Hz, 1H), 7.59 (d, J = 8.3 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.12 (d, J = 8.8 Hz, 1H), 6.84 (d, J = 16.1 Hz, 1H), 6.48 (dt, J = 16.0, 5.1 Hz, 1H), 5.69–5.60 (m, 1H), 4.90 (d, J = 4.0 Hz, 2H), 3.87 (d, J = 32.7 Hz, 2H), 2.52–2.42 (m, 1H), 2.26–2.16 (m, 2H), 2.09–2.02 (m, 1H), 1.46 (s, 18H).
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(3-(trifluoromethyl)-4-(((E)-3-(3-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22e)
Synthesized according to General Procedure I. Yellow oil (9 mg, 36% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.33 (d, J = 2.2 Hz, 1H), 8.22 (dd, J = 8.7, 2.2 Hz, 1H), 7.65 (s, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.13 (d, J = 8.7 Hz, 1H), 6.88–6.78 (m, 1H), 6.47 (dt, J = 16.0, 5.1 Hz, 1H), 5.61 (dd, J = 7.8, 4.4 Hz, 1H), 4.90 (dd, J = 5.1, 1.7 Hz, 2H), 4.01–3.68 (m, 2H), 2.46 (q, J = 7.1 Hz, 1H), 2.29–2.01 (m, 3H), 1.46 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 179.4, 167.3, 158.5, 137.1, 132.7, 131.7, 131.3 (q, J = 32.2 Hz), 129.9 (q, J = 1.0 Hz), 129.3, 127.1 (q, J = 5.2 Hz), 125.1, 124.7 (q, J = 3.8 Hz), 124.0 (q, J = 273.9 Hz), 123.7 (q, J = 273.2 Hz), 123.4 (q, J = 3.8 Hz), 119.9 (q, J = 31.3 Hz), 119.5, 113.5, 69.1, 55.5, 49.6, 29.9, 28.3, 24.3. N-Boc carbonyl carbon peaks not observed.
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(3-(trifluoromethyl)-4-(((E)-3-(2-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22f)
Synthesized according to General Procedure I. Yellow oil (10 mg, 40% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.07 (brs, 1H), 8.32 (d, J = 2.1 Hz, 1H), 8.21 (dd, J = 8.7, 2.2 Hz, 1H), 7.68–7.61 (m, 2H), 7.52 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.22 (d, J = 16.8 Hz, 1H), 7.13 (d, J = 8.7 Hz, 1H), 6.36 (dt, J = 15.8, 5.1 Hz, 1H), 5.60 (dd, J = 7.8, 4.5 Hz, 1H), 4.92 (dd, J = 5.2, 1.8 Hz, 2H), 3.97–3.75 (m, 2H), 2.50–2.40 (m, 1H), 2.28–2.16 (m, 2H), 2.09–1.97 (m, 1H), 1.45 (s, 18H).
tert-Butyl ((E)-((tert-Butoxycarbonyl)amino)((S)-2-(3-(4-(((E)-3-(4-fluorophenyl)allyl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22g)
Synthesized according to General Procedure I. Yellow oil (12 mg, 47% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.31 (d, J = 2.1 Hz, 1H), 8.20 (dd, J = 8.7, 2.2 Hz, 1H), 7.38 (dd, J = 8.6, 5.5 Hz, 2H), 7.12 (d, J = 8.7 Hz, 1H), 7.02 (t, J = 8.7 Hz, 2H), 6.74 (dd, J = 16.0, 1.5 Hz, 1H), 6.30 (dt, J = 16.0, 5.4 Hz, 1H), 5.63 (dd, J = 7.9, 4.4 Hz, 1H), 4.86 (dd, J = 5.5, 1.6 Hz, 2H), 3.98–3.75 (m, 2H), 2.50–2.41 (m, 1H), 2.28–2.00 (m, 3H), 1.45 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 179.4, 167.3, 162.8 (d, J = 247.6 Hz), 158.7, 153.8, 151.3, 148.8, 132.6, 132.5 (d, J = 3.4 Hz), 132.3, 128.4 (d, J = 8.1 Hz), 127.1 (q, J = 5.4 Hz), 123.3 (d, J = 273.4 Hz), 122.8 (d, J = 2.3 Hz), 119.9 (d, J = 31.3 Hz), 119.3, 115.7 (d, J = 21.6 Hz), 113.6, 83.6, 81.9, 69.5, 55.5, 49.7, 36.8, 31.4, 28.2, 24.8. Incomplete splitting observed on trifluoromethyl and ipso carbon signals.
tert-Butyl ((E)-((S)-2-(3-(4-(((E)-3-(3-Bromophenyl)allyl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)((tert-butoxycarbonyl)amino)methylene)carbamate (22h)
Synthesized according to General Procedure I. Yellow oil (9 mg, 18% – 3 steps). 1H NMR (400 MHz, CDCl3) δ 10.09 (brs, 1H), 8.32 (d, J = 2.2 Hz, 1H), 8.21 (dd, J = 8.6, 2.2 Hz, 1H), 7.56 (t, J = 1.8 Hz, 1H), 7.39 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 7.7 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.11 (d, J = 8.7 Hz, 1H), 6.73 (d, J = 16.0 Hz, 1H), 6.39 (dt, J = 16.0, 5.2 Hz, 1H), 5.60 (dd, J = 7.8, 4.4 Hz, 1H), 4.87 (dd, J = 5.3, 1.7 Hz, 2H), 3.97–3.75 (m, 2H), 2.54–2.39 (m, 1H), 2.29–1.97 (m, 3H), 1.46 (s, 18H). 13C NMR (126 MHz, CDCl3) δ 179.4, 167.3, 158.5, 138.5, 138.5, 132.7, 131.7, 131.1, 130.3, 130.3, 129.6, 127.1, 125.5, 124.6, 123.0, 113.5, 84.0, 82.3, 69.1, 49.6, 29.9, 28.2, 28.1. Trifluoromethyl, ipso, and N-Boc carbonyl carbon signals not observed. HRMS (ESI+): calcd for C33H37BrF3N5O6Na [M + Na]+ 758.1772, found: 758.1772.
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(3-(trifluoromethyl)-4-(((E)-4-(4-(trifluoromethyl)phenyl)but-3-en-1-yl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (22i)
Synthesized according to General Procedure I. The product was confirmed via HRMS and carried forward as crude to the next step without further purification. HRMS (ESI+): calcd for C35H40F6N5O6 [M + H]+ 740.2877, found: 740.2900.
(S)-2-(3-(4-(Hex-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23a)
Synthesized according to General Procedure J. White solid (4 mg, 23%). 1H NMR (400 MHz, cd3od) δ 8.26–8.07 (m, 2H), 7.21 (dd, J = 101.6, 8.6 Hz, 1H), 5.99–5.87 (m, 1H), 5.79–5.64 (m, 1H), 5.47–5.39 (m, 1H), 4.71 (dd, J = 5.6, 1.4 Hz, 2H), 3.78 (td, J = 9.2, 2.6 Hz, 1H), 3.62 (td, J = 9.7, 7.3 Hz, 1H), 2.64–2.43 (m, 2H), 2.30–2.06 (m, 4H), 1.44 (h, J = 7.5 Hz, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, cd3od) δ 179.0, 163.7, 157.1, 137.1, 133.7, 127.4, 125.0, 118.5, 115.5, 70.7, 56.5, 33.0, 32.7, 24.3, 23.1, 14.2. Trifluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C20H25F3N5O2 [M + H]+ 424.1955, found: 424.1955.
(S)-2-(3-(4-(Hept-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23b)
Synthesized according to General Procedure J. White solid (7 mg, 88%). 1H NMR (400 MHz, CD3OD) δ 8.25–8.08 (m, 2H), 7.38–7.03 (m, 1H), 5.97–5.84 (m, 1H), 5.74–5.62 (m, 1H), 5.41–5.38 (m, 1H), 4.71–4.63 (m, 2H), 3.81–3.73 (td, J = 9.2, 2.6 Hz, 1H), 3.59 (td, J = 9.2, 7.1 Hz, 1H), 2.60–2.40 (m, 2H), 2.21–2.00 (m, 4H), 1.44–1.24 (m, 4H), 0.94–0.84 (m, 3H). HRMS (ESI+): calcd for C21H27F3N5O2 [M + H]+ 438.2111, found: 438.2110.
(S)-2-(3-(4-(Oct-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23c)
Synthesized according to General Procedure J. White solid (3 mg, 69%). 1H NMR (400 MHz, CD3OD) δ 8.33–8.15 (m, 2H), 7.34 (d, J = 9.1 Hz, 1H), 5.99–5.87 (m, 1H), 5.79–5.61 (m, 1H), 5.44 (dd, J = 7.9, 2.0 Hz, 1H), 4.79–4.64 (m, 2H), 3.78 (td, J = 9.5, 2.6 Hz, 1H), 3.62 (td, J = 9.6, 7.1 Hz, 1H), 2.66–2.44 (m, 2H), 2.32–1.97 (m, 5H), 1.64–1.16 (m, 5H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ 179.2, 168.6, 160.5, 157.1, 137.2, 133.7, 127.2, 125.0, 119.5, 115.6, 70.7, 56.5, 33.3, 32.7, 32.4, 29.8, 24.3, 23.5, 14.4. Trifluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C22H29F3N5O2 [M + H]+ 452.2268, found: 452.2285.
(S,E)-2-(3-(3-(Trifluoromethyl)-4-((3-(4-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23d)
Synthesized according to General Procedure J. White solid (58 mg, 66%). 1H NMR (400 MHz, CD3OD) δ 8.32–8.24 (m, 2H), 7.66–7.61 (m, 4H), 7.43 (d, J = 8.5 Hz, 1H), 6.91 (dt, J = 16.1, 1.7 Hz, 1H), 6.64 (dt, J = 16.1, 5.3 Hz, 1H), 5.44 (dd, J = 7.9, 1.9 Hz, 1H), 4.98 (dd, J = 5.3, 1.7 Hz, 2H), 3.78 (td, J = 9.2, 2.6 Hz, 1H), 3.62 (td, J = 9.6, 7.2 Hz, 1H), 2.64–2.42 (m, 2H), 2.29–2.02 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 176.3, 168.7, 160.4, 141.6, 137.4, 134.0, 132.5, 130.1 (q, J = 31.0 Hz), 128.1, 127.5, 127.4 (q, J = 5.4 Hz), 126.6 (q, J = 3.9 Hz), 119.5, 115.5, 70.3, 55.6, 47.4, 30.2, 24.5. Both trifluoromethyl and one ipso carbon signal not observed. HRMS (ESI+): calcd for C24H22F6N5O2 [M + H]+ 526.1672, found: 526.1671.
(S,E)-2-(3-(3-(Trifluoromethyl)-4-((3-(3-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23e)
Synthesized according to General Procedure J. White solid (5 mg, 63%). 1H NMR (400 MHz, CD3OD) δ 8.36–8.26 (m, 2H), 7.78–7.71 (m, 2H), 7.65–7.52 (m, 1H), 7.48–7.39 (m, 2H), 6.93 (d, J = 16.1 Hz, 1H), 6.63 (dt, J = 16.0, 5.2 Hz, 1H), 5.47 (d, J = 6.9 Hz, 1H), 5.00 (d, J = 4.7 Hz, 2H), 3.88–3.57 (m, 2H), 2.70–2.48 (m, 2H), 2.31–2.22 (m, 1H), 2.20–2.08 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 177.8, 167.1, 158.8, 155.7, 137.4, 132.5, 131.1, 130.8 (q, J = 32.6 Hz), 129.6, 129.2, 125.8 (q, J = 5.0 Hz), 125.3, 124.0 (q, J = 4.1 Hz), 122.8 (q, J = 3.9 Hz), 119.1 (q, J = 32.0 Hz), 118.5, 114.0, 68.9, 55.0, 31.3, 22.9. Trifluoromethyl carbon signals not observed. HRMS (ESI+): calcd for C24H22F6N5O2 [M + H]+ 526.1672, found: 526.1666.
(S,E)-2-(3-(3-(Trifluoromethyl)-4-((3-(2-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23f)
Synthesized according to General Procedure J. White solid (2 mg, 23%). 1H NMR (400 MHz, CD3OD) δ 8.30–8.23 (m, 2H), 7.77 (d, J = 7.9 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.42 (dd, J = 13.6, 8.1 Hz, 2H), 7.21 (d, J = 15.7 Hz, 1H), 6.49 (dt, J = 15.8, 4.5 Hz, 1H), 5.48–5.41 (m, 1H), 4.99 (dd, J = 4.6, 2.0 Hz, 2H), 3.87–3.56 (m, 2H), 2.60–2.44 (m, 2H), 2.32–2.18 (m, 1H), 2.16–1.99 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 179.2, 168.5, 160.1, 157.1, 149.9, 136.8, 133.9, 133.4, 128.9, 128.9, 128.6, 127.2 (q, J = 5.3 Hz), 126.7 (q, J = 5.6 Hz), 119.9, 115.4, 69.7, 56.5, 32.7, 28.1, 24.3. Trifluoromethyl and ipso carbon signals not observed. HRMS (ESI+): calcd for C24H22F6N5O2 [M + H]+ 526.1672, found: 526.1653.
(S,E)-2-(3-(4-((3-(4-Fluorophenyl)allyl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23g)
Synthesized according to General Procedure J. White solid (7 mg, 67%). 1H NMR (400 MHz, CD3OD) δ 8.32–8.24 (m, 2H), 7.52–7.40 (m, 3H), 7.08 (t, J = 8.2 Hz, 2H), 6.84 (d, J = 15.6 Hz, 1H), 6.44 (d, J = 15.0 Hz, 1H), 5.55–5.44 (m, 1H), 4.98–4.92 (m, 2H), 3.90–3.58 (m, 2H), 2.67–2.03 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 179.2, 168.5, 163.9 (d, J = 246.0 Hz), 160.3, 157.0, 134.1, 133.9, 133.3, 129.4 (d, J = 7.9 Hz), 127.2 (q, J = 5.1 Hz), 124.6 (q, J = 273.9 Hz), 124.2, 120.3 (d, J = 31.2 Hz), 119.7, 116.4 (d, J = 22.1 Hz), 115.6, 70.7, 56.6, 32.9, 28.2, 24.4. HRMS (ESI+): calcd for C23H22F4N5O2 [M + H]+ 476.1704, found: 476.1715.
(S,E)-2-(3-(4-((3-(3-Bromophenyl)allyl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23h)
Synthesized according to General Procedure J. White solid (1 mg, 11%). 1H NMR (400 MHz, CD3OD) δ 8.31–8.22 (m, 2H), 7.61 (t, J = 1.8 Hz, 1H), 7.48–7.38 (m, 3H), 7.26 (t, J = 7.9 Hz, 1H), 6.79 (d, J = 16.0 Hz, 1H), 6.52 (dt, J = 16.0, 5.4 Hz, 1H), 5.44 (dd, J = 7.9, 2.0 Hz, 1H), 4.95 (dd, J = 5.4, 1.6 Hz, 2H), 3.78 (td, J = 9.1, 2.6 Hz, 1H), 3.62 (td, J = 9.6, 7.2 Hz, 1H), 2.64–2.44 (m, 2H), 2.35–2.18 (m, 1H), 2.17–2.00 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 179.2, 168.5, 160.2, 157.1, 140.1, 133.9, 132.7, 131.9, 131.5, 130.4, 127.2 (q, J = 5.1 Hz), 126.4, 126.2, 123.8, 119.9, 115.4, 70.3, 56.5, 32.7, 30.8, 24.3. Trifluoromethyl and ipso carbon peaks not observed. HRMS (ESI+): calcd for C23H22BrF3N5O2 [M + H]+ 536.0903, found: 536.0900.
(S,E)-2-(3-(3-(Trifluoromethyl)-4-((4-(4-(trifluoromethyl)phenyl)but-3-en-1-yl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (23i)
Synthesized according to General Procedure J. White solid (33 mg, 26% – 3 steps). 1H NMR (500 MHz, CD3OD) δ 8.32–8.20 (m, 2H), 7.61–7.51 (m, 4H), 7.43–7.35 (m, 1H), 6.65 (d, J = 15.9 Hz, 1H), 6.53 (dt, J = 15.0, 6.5 Hz, 1H), 5.44 (d, J = 6.7 Hz, 1H), 4.33 (t, J = 6.1 Hz, 2H), 3.82–3.71 (m, 1H), 3.70–3.56 (m, 1H), 2.78 (q, J = 6.1 Hz, 2H), 2.62–2.44 (m, 2H), 2.26–2.20 (m, 1H), 2.11–2.06 (m, 1H). 13C NMR (101 MHz, CD3OD) δ 179.2, 168.5, 160.6, 157.1, 142.7, 134.0, 132.5, 130.0, 129.9 (q, J = 31.8 Hz), 127.5, 127.1 (q, J = 5.2 Hz), 126.4 (q, J = 3.9 Hz), 125.6 (q, J = 273.3 Hz), 124.7 (q, J = 271.8 Hz), 120.3 (q, J = 31.3 Hz), 119.6, 115.0, 69.4, 56.5, 33.6, 32.8, 28.1, 24.4. HRMS (ESI+): calcd for C25H24F6N5O2 [M + H]+ 540.1829, found: 540.1819.
tert-Butyl (S,E)-2-(3-(4-(Hept-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (24a)
Synthesized according to General Procedure G. Yellow oil (40 mg, 72%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 2.2 Hz, 1H), 8.17 (dd, J = 8.7, 2.2 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 5.93–5.81 (m, 1H), 5.72–5.60 (m, 1H), 5.22–5.02 (m, 1H), 4.65 (d, J = 5.2 Hz, 2H), 3.78–3.64 (m, 1H), 3.61–3.44 (m, 1H), 2.48–2.34 (m, 1H), 2.22–1.95 (m, 5H), 1.50–1.26 (m, 13H), 0.89 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 180.9, 180.5*, 167.4, 158.9, 158.8*, 153.6, 136.2, 136.2*, 132.4, 126.8 (q, J = 5.5 Hz), 123.5, 123.2 (q, J = 272.2 Hz), 118.8, 113.7, 113.6*, 110.2, 80.6, 80.6*, 69.8, 53.9, 46.8*, 46.5, 32.5, 32.1, 31.6*, 31.2, 28.5*, 28.3, 24.5*, 23.9, 22.3, 14.0. HRMS (ESI+): calcd for C25H32F3N3O4Na [M + Na]+ 519.2269, found: 519.2276.
tert-Butyl (S,E)-2-(3-(4-(Oct-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (24b)
Synthesized according to General Procedure G. Clear oil (41 mg, 72%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.7, 2.3 Hz, 1H), 7.12–7.02 (m, 1H), 5.94–5.78 (m, 1H), 5.72–5.60 (m, 1H), 5.23–4.95 (m, 1H), 4.65 (d, J = 5.4 Hz, 2H), 3.76–3.61 (m, 1H), 3.61–3.44 (m, 1H), 2.49–2.30 (m, 1H), 2.23–1.95 (m, 5H), 1.51–1.15 (m, 15H), 0.92–0.81 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 181.5, 180.5*, 167.4, 159.0, 158.8*, 153.6, 136.3, 136.2*, 132.4, 126.7 (d, J = 5.8 Hz), 123.5, 123.1 (d, J = 272.2 Hz), 119.4 118.8, 113.7, 113.6*, 80.6, 80.5*, 69.8, 53.9, 46.8*, 46.5, 32.5, 32.4, 31.6*, 31.4, 28.7, 28.5*, 28.3, 24.5*, 23.8, 22.6, 14.2. Incomplete splitting observed on trifluoromethyl ipso carbon signal. HRMS (ESI+): calcd for C26H34F3N3O4Na [M + Na]+ 532.2394, found: 532.2400.
tert-Butyl (S,Z)-2-(3-(4-(Hept-3-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (24c)
Synthesized according to General Procedure G. Yellow oil (45 mg, 62%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 2.1 Hz, 1H), 8.19 (dd, J = 8.7, 2.3 Hz, 1H), 7.09–7.00 (m, 1H), 5.62–5.41 (m, 2H), 5.25–4.98 (m, 1H), 4.11 (t, J = 6.8 Hz, 2H), 3.75–3.42 (m, 2H), 2.61 (q, J = 6.9 Hz, 2H), 2.49–2.30 (m, 1H), 2.20–1.92 (m, 5H), 1.51–1.22 (m, 11H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 181.0, 180.5*, 167.4, 159.2, 159.1*, 153.6, 133.0, 132.5, 126.7 (q, J = 5.2 Hz), 124.2, 123.3 (q, J = 273.0 Hz), 119.7 (q, J = 31.9 Hz), 118.8, 113.1, 80.6, 80.5*, 68.8, 53.9, 46.8*, 46.5, 32.5, 31.6*, 29.5, 28.5*, 28.3, 27.3, 24.5*, 23.8, 22.8, 13.9.
tert-Butyl (S,Z)-2-(3-(4-(Oct-3-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (24d)
Synthesized according to General Procedure G. Yellow oil (40 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.18 (d, J = 8.7 Hz, 1H), 7.11–7.01 (m, 1H), 5.60–5.39 (m, 2H), 5.24–5.01 (m, 1H), 4.11 (t, J = 6.7 Hz, 2H), 3.77–3.65 (m, 1H), 3.62–3.46 (m, 1H), 2.60 (q, J = 6.7 Hz, 2H), 2.48–2.34 (m, 1H), 2.23–1.94 (m, 5H), 1.48–1.26 (m, 13H), 0.90 (t, J = 7.0 Hz, 3H). HRMS (ESI+): calcd for C26H34F3N3O4Na [M + Na]+ 532.2394, found: 532.2400.
tert-Butyl (S,Z)-2-(3-(3-(Trifluoromethyl)-4-((3-(4-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (24e)
Synthesized according to General Procedure G. Yellow oil (30 mg, 82%). Rotamer peaks are denoted with an asterisk (*). 1H NMR (500 MHz, CDCl3) δ 8.31 (d, J = 2.5 Hz, 1H), 8.15 (dd, J = 8.7, 1.7 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 6.98–6.90 (m, 1H), 6.81 (d, J = 11.9 Hz, 1H), 6.11 (dt, J = 11.8, 6.4 Hz, 1H), 5.22–5.03 (m, 1H), 4.91 (d, J = 5.9 Hz, 2H), 3.77–3.44 (m, 2H), 2.47–2.30 (m, 1H), 2.20–2.09 (m, 2H), 2.05–1.98 (m, 1H), 1.46 (s, 3H)*, 1.29 (s, 6H).
tert-Butyl ((E)-((tert-Butoxycarbonyl)amino)((S)-2-(3-(4-(((E)-hept-2-en-1-yl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (25a)
Synthesized according to General Procedure I. Yellow oil (19 mg, 37% – 2 steps). 1H NMR (500 MHz, CDCl3) δ 10.02 (brs, 1H), 8.22 (d, J = 2.1 Hz, 1H), 8.11 (dd, J = 8.7, 2.2 Hz, 1H), 7.00 (d, J = 8.7 Hz, 1H), 5.88–5.75 (m, 1H), 5.65–5.47 (m, 2H), 4.58 (d, J = 5.6 Hz, 2H), 3.88–3.64 (m, 2H), 2.44–2.33 (m, 1H), 2.20–1.90 (m, 5H), 1.51–1.08 (m, 22H), 0.83 (t, J = 7.2 Hz, 3H). HRMS (ESI+): calcd for C31H43F3N5O6 [M + H]+ 638.3160, found: 638.3132.
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(4-(((E)-oct-2-en-1-yl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (25b)
Synthesized according to General Procedure I. Yellow oil (18 mg, 33% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.28 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.7, 2.2 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 5.94–5.81 (m, 1H), 5.72–5.56 (m, 2H), 4.65 (dd, J = 5.7, 1.3 Hz, 2H), 3.95–3.74 (m, 2H), 2.56–2.37 (m, 1H), 2.27–1.97 (m, 5H), 1.54–1.23 (m, 24H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 179.3, 167.4, 162.2, 158.9, 136.3, 132.5, 126.9 (q, J = 5.2 Hz), 123.5, 123.2 (q, J = 275.5 Hz), 119.6 (q, J = 31.5 Hz), 118.8, 113.7, 82.5, 79.8, 69.8, 55.4, 49.6, 32.4, 31.4, 29.9, 28.7, 28.2, 28.1, 22.6, 14.2. N-Boc carbonyl carbon signals not observed. HRMS (ESI+): calcd for C32H45F3N5O6 [M + H]+ 652.3316, found: 652.3287.
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(4-(((Z)-hept-3-en-1-yl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (25c)
Synthesized according to General Procedure I. The crude mixture was dried in vacuo and carried forward to the next reaction without purification.
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(4-(((Z)-oct-3-en-1-yl)oxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (25d)
Synthesized according to General Procedure I. Yellow oil (24 mg, 46%). 1H NMR (400 MHz, CDCl3) δ 10.08 (brs, 1H), 8.28 (d, J = 2.1 Hz, 1H), 8.18 (dd, J = 8.7, 2.2 Hz, 1H), 7.06 (d, J = 8.7 Hz, 1H), 5.66–5.43 (m, 3H), 4.11 (t, J = 6.8 Hz, 2H), 3.95–3.74 (m, 2H), 2.60 (q, J = 6.9 Hz, 2H), 2.50–2.39 (m, 1H), 2.31–2.15 (m, 2H), 2.12–1.99 (m, 3H), 1.54–1.30 (m, 22H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.3, 167.4, 159.2, 154.0, 133.3, 132.6, 127.0 (q, J = 5.2 Hz), 124.0, 123.4 (q, J = 272.0 Hz), 119.6 (q, J = 31.4 Hz), 118.8, 113.0, 82.3, 79.9, 68.8, 55.4, 49.6, 31.9, 31.6, 28.2, 27.3, 27.2, 24.2, 22.5, 14.1. N-Boc carbonyl carbon signals not observed. HRMS (ESI+): calcd for C32H45F3N5O6 [M + H]+ 652.3316, found: 652.3307.
tert-Butyl (((tert-Butoxycarbonyl)amino)((S)-2-(3-(3-(trifluoromethyl)-4-(((Z)-3-(4-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methylene)carbamate (25e)
Synthesized according to General Procedure I. Yellow oil (19 mg, 51% – 2 steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (brs, 1H), 8.30 (d, J = 2.1 Hz, 1H), 8.15 (dd, J = 8.7, 2.2 Hz, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 6.94 (d, J = 8.7 Hz, 1H), 6.80 (d, J = 11.7 Hz, 1H), 6.10 (dt, J = 12.2, 6.4 Hz, 1H), 5.61 (dd, J = 7.8, 4.4 Hz, 1H), 4.91 (dd, J = 6.4, 1.7 Hz, 2H), 3.96–3.74 (m, 2H), 2.52–2.38 (m, 1H), 2.26–2.13 (m, 2H), 2.09–1.97 (m, 1H), 1.45 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 179.4, 167.3, 162.2, 158.3, 153.7, 151.4, 139.6, 132.6, 132.5, 130.0 (q, J = 32.7 Hz), 129.1, 128.1, 127.1 (q, J = 5.3 Hz), 125.7 (q, J = 3.8 Hz), 124.0 (q, J = 273.5 Hz), 123.3 (q, J = 272.9 Hz), 120.0 (q, J = 31.5 Hz), 119.4, 113.4, 65.6, 49.6, 28.3, 28.1, 22.1.
(S,E)-2-(3-(4-(Hept-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (26a)
Synthesized according to General Procedure J. White solid (9 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 8.29–8.19 (m, 2H), 7.34 (d, J = 8.8 Hz, 1H), 5.93 (dt, J = 14.5, 6.9 Hz, 1H), 5.69 (dt, J = 15.4, 5.8 Hz, 1H), 5.44 (dd, J = 7.9, 2.0 Hz, 1H), 4.71 (dd, J = 5.8, 1.4 Hz, 2H), 3.78 (td, J = 9.4, 2.7 Hz, 1H), 3.62 (td, J = 9.6, 7.2 Hz, 1H), 2.62–2.47 (m, 2H), 2.32–2.17 (m, 1H), 2.17–2.04 (m, 3H), 1.46–1.27 (m, 4H), 0.91 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 188.9, 179.2, 168.6, 160.5, 148.7, 137.1, 133.7, 129.6, 127.3, 124.9, 120.8 (d, J = 268.9 Hz), 115.5, 70.7, 56.5, 33.0, 32.7, 32.3, 24.3, 23.1, 14.2. Incomplete splitting observed on trifluoromethyl carbon. HRMS (ESI+): calcd for C21H27F3N5O2 [M + H]+ 438.2111, found: 438.2120.
(S,E)-2-(3-(4-(Oct-2-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (26b)
Synthesized according to General Procedure J. White solid (9 mg, 58%). 1H NMR (400 MHz, CD3OD) δ 8.28–8.19 (m, 2H), 7.34 (d, J = 8.6 Hz, 1H), 5.98–5.86 (m, 1H), 5.75–5.63 (m, 1H), 5.44 (dd, J = 7.9, 1.9 Hz, 1H), 4.71 (dd, J = 5.7, 1.4 Hz, 2H), 3.78 (td, J = 9.5, 2.6 Hz, 1H), 3.62 (td, J = 9.6, 7.2 Hz, 1H), 2.64–2.43 (m, 2H), 2.30–2.20 (m, 1H), 2.17–2.00 (m, 3H), 1.42 (p, J = 7.3 Hz, 2H), 1.35–1.20 (m, 4H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 179.2, 168.6, 160.4, 157.1, 137.2, 133.7, 127.1 (q, J = 5.4 Hz), 125.0, 124.5 (q, J = 275.3 Hz), 120.5 (q, J = 31.2 Hz), 119.5, 115.6, 70.7, 56.5, 33.3, 32.7, 32.4, 29.8, 24.3, 23.5, 14.3. HRMS (ESI+): calcd for C22H29F3N5O2 [M + H]+ 452.2268, found: 452.2285.
(S,Z)-2-(3-(4-(Hept-3-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (26c)
Synthesized according to General Procedure J. White solid (20 mg, 40% – 3 steps). 1H NMR (500 MHz, CD3OD) δ 8.34–8.10 (m, 2H), 7.34 (d, J = 8.7 Hz, 1H), 5.73–5.19 (m, 3H), 4.19 (t, J = 6.5 Hz, 2H), 3.78 (td, J = 8.9, 2.6 Hz, 1H), 3.62 (td, J = 9.7, 7.2 Hz, 1H), 2.64–2.42 (m, 4H), 2.34–2.16 (m, 1H), 2.14–2.03 (m, 3H), 1.40 (h, J = 7.4 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ 179.2, 168.6, 160.8, 157.1, 133.9, 133.4, 127.1 (q, J = 5.4 Hz), 125.8, 124.7 (q, J = 271.7 Hz), 120.4 (q, J = 31.4 Hz), 119.5, 114.9, 70.1, 56.5, 32.7, 30.4, 28.1, 24.3, 23.8, 14.1. HRMS (ESI+): calcd for C21H27F3N5O2 [M + H]+ 438.2111, found: 438.2104.
(S,Z)-2-(3-(4-(Oct-3-en-1-yloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (26d)
Synthesized according to General Procedure J. White solid (11 mg, 53%). 1H NMR (500 MHz, CD3OD) δ 8.30–8.11 (m, 2H), 7.35 (d, J = 8.7 Hz, 1H), 5.99–5.88 (m, 1H), 5.77–5.62 (m, 1H) 5.45 (d, J = 7.9 Hz, 1H), 4.72 (d, J = 7.1 Hz, 2H), 3.79 (t, J = 9.2 Hz, 1H), 3.62 (q, J = 7.2 Hz, 1H), 2.63–2.45 (m, 2H), 2.27–2.06 (m, 4H), 1.46–1.25 (m, 6H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 179.2, 168.6, 160.8, 157.1, 133.9, 133.6, 127.1 (q, J = 5.4 Hz), 125.6, 124.7 (q, J = 272.9 Hz), 120.4 (q, J = 31.4 Hz), 119.5, 114.9, 70.1, 56.5, 32.9, 32.7, 28.1, 28.0, 24.3, 23.4, 14.3. HRMS (ESI+): calcd for C22H29F3N5O2 [M + H]+ 452.2268, found: 452.2260.
(S,Z)-2-(3-(3-(Trifluoromethyl)-4-((3-(4-(trifluoromethyl)phenyl)allyl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide 2,2,2-Trifluoroacetate (26e)
Synthesized according to General Procedure J. White solid (8 mg, 54%). 1H NMR (400 MHz, CD3OD) δ 8.29–8.18 (m, 2H), 7.70 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.6 Hz, 1H), 6.87 (d, J = 11.8 Hz, 1H), 6.12 (dt, J = 12.2, 6.3 Hz, 1H), 5.44 (dd, J = 8.0, 1.9 Hz, 1H), 5.02 (dd, J = 6.4, 1.7 Hz, 2H), 3.78 (td, J = 9.2, 2.6 Hz, 1H), 3.62 (td, J = 9.6, 7.2 Hz, 1H), 2.64–2.43 (m, 2H), 2.29–2.01 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 179.3, 168.5, 160.0, 157.1, 141.3, 133.9, 133.3, 130.4, 129.2, 128.1, 127.2 (q, J = 5.3 Hz), 126.4 (q, J = 3.8 Hz), 125.5 (q, J = 271.9 Hz), 124.7 (q, J = 272.3 Hz), 120.6 (q, J = 31.5 Hz), 120.0, 115.3, 66.9, 56.5, 32.7, 24.3. Incomplete splitting observed on ipso carbon due to signal overlap. HRMS (ESI+): calcd for C24H21F6N5NaO2 [M+ Na]+ 548.1492, found: 548.1506.
Acknowledgments
These studies were funded by research grants from the NIH (R01 GM104366 and R01 GM067958).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00017.
Molecular docking, HRMS analysis, 1H NMR, and 13C NMR spectra of new compounds (PDF)
Databank file for 14c 3VZB (PDB)
Databank file for 14c SphK2 (PDB)
Databank file for 23d 3VZB (PDB)
Databank file for 23d SphK2 (PDB)
Databank file for 23e 3VZB (PDB)
Databank file for 23e SphK2 (PDB)
Databank file for 26a 3VZB (PDB)
Databank file for 26a SphK2 (PDB)
Databank file for 26d 3VZB (PDB)
Databank file for 26d SphK2 (PDB)
Databank file for hSphK1 (PDB)
Databank file for hSphK2 (PDB)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): W.L.S. and K.R.L. are among the co-founders of Flux Therapeutics Inc, which was created to commercialize S1P-related discoveries, including SphK inhibitors, discovered and characterized in their laboratories.
Notes
Accession IDs: Human SphK1: NP_001136074, Human SphK2: NP_ 001191089, Mouse SphK1: NP_035581.1, Mouse SphK2: NP_001166032
Supplementary Material
References
- Hait N. C.; Allegood J.; Maceyka M.; Strub G. M.; Harikumar K. B.; Singh S. K.; Luo C.; Marmorstein R.; Kordula T.; Milstien S.; Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez S. E.; Harikumar K. B.; Hait N. C.; Allegood J.; Strub G. M.; Kim E. Y.; Maceyka M.; Jiang H.; Luo C.; Kordula T.; Milstien S.; Spiegel S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K.; Zhang Y.; D’Alessandro A.; Nemkov T.; Song A.; Wu H.; Liu H.; Adebiyi M.; Huang A.; Wen Y. E.; Bogdanov M. V.; Vila A.; O’Brien J.; Kellems R. E.; Dowhan W.; Subudhi A. W.; Jameson-Van Houten S.; Julian C. G.; Lovering A. T.; Safo M.; Hansen K. C.; Roach R. C.; Xia Y. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat. Commun. 2016, 7, 12086. 10.1038/ncomms12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K.; Yamashita T.; Olivera A.; Miller G. F.; Spiegel S.; Proia R. L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M.; Sankala H.; Hait N. C.; Le Stunff H.; Liu H.; Toman R.; Collier C.; Zhang M.; Satin L. S.; Merrill A. H.; Milstien S.; Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- Neubauer H. A.; Pitson S. M. Roles, regulation and inhibitors of sphingosine kinase 2. FEBS Journal 2013, 280, 5317–5336. 10.1111/febs.12314. [DOI] [PubMed] [Google Scholar]
- Kunkel G. T.; Maceyka M.; Milstien S.; Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discovery 2013, 12, 688–702. 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman J.; García-Barros M.; Obeid L. M.; Hannun Y. A. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids 2014, 1841, 1174–1188. 10.1016/j.bbalip.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire M. WANTED: Natural-born sickler. Sci. Transl. Med. 2014, 6, 240ec101. 10.1126/scitranslmed.3009588. [DOI] [Google Scholar]
- Zhang Y.; Berka V.; Song A.; Sun K.; Wang W.; Zhang W.; Ning C.; Li C.; Zhang Q.; Bogdanov M.; Alexander D. C.; Milburn M. V.; Ahmed M. H.; Lin H.; Idowu M.; Zhang J.; Kato G. J.; Abdulmalik O. Y.; Zhang W.; Dowhan W.; Kellems R. E.; Zhang P.; Jin J.; Safo M.; Tsai A.-L.; Juneja H. S.; Xia Y. Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J. Clin. Investig. 2014, 124, 2750–2761. 10.1172/JCI74604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakumar P.; Kaur T.; Singh M. Potential target sites to modulate vascular endothelial dysfunction: Current perspectives and future directions. Toxicology 2008, 245, 49–64. 10.1016/j.tox.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Machida T.; Hamaya Y.; Izumi S.; Hamaya Y.; Iizuka K.; Igarashi Y.; Minami M.; Levi R.; Hirafuji M. Sphingosine-1-phosphate inhibits nitric oxide production induced by interleukin-1β in rat vascular smooth muscle cells. J. Pharmacol. Exp. Ther. 2008, 325, 200–209. 10.1124/jpet.107.127290. [DOI] [PubMed] [Google Scholar]
- Sorrentino R.; Bertolino A.; Terlizzi M.; Iacono V. M.; Maiolino P.; Cirino G.; Roviezzo F.; Pinto A. B cell depletion increases sphingosine-1-phosphate–dependent airway inflammation in mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 571–583. 10.1165/rcmb.2014-0207OC. [DOI] [PubMed] [Google Scholar]
- Price M. M.; Oskeritzian C. A.; Falanga Y. T.; Harikumar K. B.; Allegood J. C.; Alvarez S. E.; Conrad D.; yan J. J.; Milstien S.; Spiegel S. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J. Allergy Clin. Immunol. 2013, 131, 501–511. 10.1016/j.jaci.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne N. J.; Dubois G.; Pyne S. Role of sphingosine-1-phosphate and lysophosphatidic acid in fibrosis. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids 2013, 1831, 228–238. 10.1016/j.bbalip.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Bayraktar O.; Ozkirimli E.; Ulgen K. Sphingosine kinase 1 (SK1) allosteric inhibitors that target the dimerization site. Comput. Biol. Chem. 2017, 69, 64–76. 10.1016/j.compbiolchem.2017.05.006. [DOI] [PubMed] [Google Scholar]
- Papakyriakou A.; Cencetti F.; Puliti E.; Morelli L.; Tricomi J.; Bruni P.; Compostella F.; Richichi B. Glycans meet sphingolipids: Structure-based design of glycan containing analogues of a sphingosine kinase inhibitor. ACS Med. Chem. Lett. 2020, 11, 913–920. 10.1021/acsmedchemlett.9b00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin D. J.; Li Y.; Brown M. L.; Min X.; Schmitt M. J.; Wanska M.; Wang X.; Connors R.; Johnstone S.; Cardozo M.; Cheng A. C.; Jeffries S.; Franks B.; Li S.; Shen S.; Wong M.; Wesche H.; Xu G.; Carlson T. J.; Plant M.; Morgenstern K.; Rex K.; Schmitt J.; Coxon A.; Walker N.; Kayser F.; Wang Z. Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4608–4616. 10.1016/j.bmcl.2013.06.030. [DOI] [PubMed] [Google Scholar]
- Baek D. J.; Macritchie N.; Pyne N. J.; Pyne S.; Bittman R. Synthesis of selective inhibitors of sphingosine kinase 1. Chem. Commun. 2013, 49, 2136–2138. 10.1039/c3cc00181d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.-H.; Kim H.-Y.; Yoon H.-S.; Park W.-J.; Adams D. R.; Pyne N. J.; Pyne S.; Park J.-W. A Novel Selective Sphingosine Kinase 2 Inhibitor, HWG-35D, Ameliorates the Severity of Imiquimod-Induced Psoriasis Model by Blocking Th17 Differentiation of Naïve CD4 T Lymphocytes. Int. J. Mol. Sci. 2020, 21, 8371. 10.3390/ijms21218371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines L. W.; Fitzpatrick L. R.; French K. J.; Zhuang Y.; Xia Z.; Keller S. N.; Upson J. J.; Smith C. D. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig. Dis. Sci. 2008, 53, 997–1012. 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines L. W.; Fitzpatrick L. R.; Green C. L.; Zhuang Y.; Smith C. D. Efficacy of a novel sphingosine kinase inhibitor in experimental Crohn’s disease. Inflammopharmacology 2010, 18, 73–85. 10.1007/s10787-010-0032-x. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Rehman H.; Ramshesh V. K.; Schwartz J.; Liu Q.; Krishnasamy Y.; Zhang X.; Lemasters J. J.; Smith C. D.; Zhong Z. Sphingosine kinase-2 inhibition improves mitochondrial function and survival after hepatic ischemia-reperfusion. J. Hepatol. 2012, 56, 137–145. 10.1016/j.jhep.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xun C.; Chen M.-B.; Qi L.; Tie-Ning Z.; Peng X.; Ning L.; Zhi-Xiao C.; Li-Wei W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94. 10.1186/s13046-015-0205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten C. D.; Garrett-Mayer E.; Chin S. H.; Shirai K.; Ogretmen B.; Bentz T. A.; Brisendine A.; Anderton K.; Cusack S. L.; Maines L. W.; Zhuang Y.; Smith C. D.; Thomas M. B. A Phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 4642–4650. 10.1158/1078-0432.CCR-16-2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M.Opaganib, a sphingosine kinase 2 (SK2) inhibitor in COVID-19 pneumonia: A randomized, double-blind, placebo-controlled phase 2/3 study, in adult subjects hospitalized with severe SARS-CoV-2 positive pneumonia. ClinicalTrials.gov; ClinicalTrials.gov identifier: NCT04467840. Updated July 20, 2021. Accessed June 6, 2022. https://clinicaltrials.gov/ct2/show/NCT04467840.
- Antoon J. W.; White M. D.; Meacham W. D.; Slaughter E. M.; Muir S. E.; Elliott S.; Rhodes L. V.; Ashe H. B.; Wiese T. E.; Smith C. D.; Burow M. E.; Beckman B. S. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology 2010, 151, 5124–5135. 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. G.; Sun C.; Bittman R.; Pyne N. J.; Pyne S. (R)-FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: Effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF-7 breast cancer cells. Cell. Signal. 2011, 23, 1590–1595. 10.1016/j.cellsig.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Guo T. L.; Hait N. C.; Allegood J.; Parikh H. I.; Xu W.; Kellogg G. E.; Grant S.; Spiegel S.; Zhang S. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One 2013, 8, e56471. 10.1371/journal.pone.0056471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Raje M.; Gao M.; Gellett A. M.; Tomsig J. L.; Lynch K. R.; Santos W. L. Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem. J. 2012, 447, 149–157. 10.1042/BJ20120609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan N. N.; Morris E. A.; Kharel Y.; Raje M. R.; Gao M.; Tomsig J. L.; Lynch K. R.; Santos W. L. Structure-activity relationship studies and in vivo activity of guanidine-based sphingosine kinase inhibitors: Discovery of SphK1- and SphK2-selective inhibitors. J. Med. Chem. 2015, 58, 1879–1899. 10.1021/jm501760d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Morris E. A.; Congdon M. D.; Thorpe S. B.; Tomsig J. L.; Santos W. L.; Lynch K. R. Sphingosine kinase 2 inhibition and blood sphingosine-1-phosphate levels. J. Pharmacol. Exp. Ther. 2015, 355, 23–31. 10.1124/jpet.115.225862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Huang T.; Salamon A.; Harris T. E.; Santos W. L.; Lynch K. R. Mechanism of sphingosine 1-phosphate clearance from blood. Biochem. J. 2020, 477, 925–935. 10.1042/BCJ20190730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon M. D.; Kharel Y.; Brown A. M.; Lewis S. N.; Bevan D. R.; Lynch K. R.; Santos W. L. Structure–activity relationship studies and molecular modeling of naphthalene-based sphingosine kinase 2 inhibitors. ACS Med. Chem. Lett. 2016, 7, 229–234. 10.1021/acsmedchemlett.5b00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley C. D.; Morris E. A.; Kharel Y.; Brown A. M.; Huang T.; Bevan D. R.; Lynch K. R.; Santos W. L. Discovery of a small side cavity in sphingosine kinase 2 that enhances inhibitor potency and selectivity. J. Med. Chem. 2020, 63, 1178–1198. 10.1021/acs.jmedchem.9b01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprathe B. W.; Jaen J. C.; Wise L. D.; Heffner T. G.; Pugsley T. A.; Meltzer L. T.; Parvez M. Dopamine autoreceptor agonists as potential antipsychotics. 3. 6-Propyl-4,5,5a,6,7,8-hexahydrothiazolo[4,5-f]quinolin-2-amine. J. Med. Chem. 1991, 34, 2736–2746. 10.1021/jm00113a010. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Negri M.; Jahn-Hoffmann K.; Zhuang Y.; Olgen S.; Bartels M.; Muller-Vieira U.; Lauterbach T.; Hartmann R. W. Synthesis, biological evaluation, and molecular modeling studies of methylene imidazole substituted biaryls as inhibitors of human 17alpha-hydroxylase-17,20-lyase (CYP17)--part II: Core rigidification and influence of substituents at the methylene bridge. Bioorg. Med. Chem. 2008, 16, 7715–7727. 10.1016/j.bmc.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Lucas S.; Negri M.; Heim R.; Zimmer C.; Hartmann R. W. Fine-tuning the selectivity of aldosterone synthase inhibitors: Structure-activity and structure-selectivity insights from studies of heteroaryl substituted 1,2,5,6-tetrahydropyrrolo[3,2,1-ij]quinolin-4-one derivatives. J. Med. Chem. 2011, 54, 2307–2319. 10.1021/jm101470k. [DOI] [PubMed] [Google Scholar]
- Sander K.; Kottke T.; Tanrikulu Y.; Proschak E.; Weizel L.; Schneider E. H.; Seifert R.; Schneider G.; Stark H. 2,4-Diaminopyrimidines as histamine H4 receptor ligands: Scaffold optimization and pharmacological characterization. Bioorg. Med. Chem. 2009, 17, 7186–7196. 10.1016/j.bmc.2009.08.059. [DOI] [PubMed] [Google Scholar]
- Chowdhury S.; Chafeev M.; Liu S.; Sun J.; Raina V.; Chui R.; Young W.; Kwan R.; Fu J.; Cadieux J. A. Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg. Med. Chem. Lett. 2011, 21, 3676–3681. 10.1016/j.bmcl.2011.04.088. [DOI] [PubMed] [Google Scholar]
- Spadaro A.; Negri M.; Marchais-Oberwinkler S.; Bey E.; Frotscher M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD1). PLoS One 2012, 7, e29252. 10.1371/journal.pone.0029252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam P.; Takrouri K.; Chen T.; Sahoo R.; Papadopoulos E.; Chen L.; Wagner G.; Aktas B. H.; Halperin J. A.; Chorev M. Synthesis of rigidified eIF4E/eIF4G inhibitor-1 (4EGI-1) mimetic and their in vitro characterization as inhibitors of protein-protein interaction. J. Med. Chem. 2014, 57, 5094–5111. 10.1021/jm401733v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Min X.; Xiao S.-H.; Johnstone S.; Romanow W.; Meininger D.; Xu H.; Liu J.; Dai J.; An S.; Thibault S.; Walker N. Molecular Basis of Sphingosine Kinase 1 Substrate Recognition and Catalysis. Structure 2013, 21, 798–809. 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- Worrell B. L.; Brown A. M.; Santos W. L.; Bevan D. R. In Silico Characterization of Structural Distinctions between Isoforms of Human and Mouse Sphingosine Kinases for Accelerating Drug Discovery. J. Chem. Inf. Model. 2019, 59, 2339–2351. 10.1021/acs.jcim.8b00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Lee S.; Snyder A. H.; Sheasley-O’Neill S. L.; Morris M. A.; Setiady Y.; Zhu R.; Zigler M. A.; Burcin T. L.; Ley K.; Tung K. S. K.; Engelhard V. H.; Macdonald T. L.; Pearson-White S.; Lynch K. R. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J. Biol. Chem. 2005, 280, 36865–36872. 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- Billich A.; Bornancin F.; Dévay P.; Mechtcheriakova D.; Urtz N.; Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J. Biol. Chem. 2003, 278, 47408–47415. 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- Allende M. L.; Sasaki T.; Kawai H.; Olivera A.; Mi Y.; Van Echten-Deckert G.; Hajdu R.; Rosenbach M.; Keohane C. A.; Mandala S.; Spiegel S.; Proia R. L. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- Kharel Y.; Mathews T.; Gellett A.; Tomsig J.; Kennedy P.; Moyer M.; Macdonald T.; Lynch K. Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem. J. 2011, 440, 345–353. 10.1042/BJ20110817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.