

Abstract
Resistance to pharmacological treatments is a major public health challenge. Here we introduce Resistor—a structure- and sequence-based algorithm that prospectively predicts resistance mutations for drug design. Resistor computes the Pareto frontier of four resistance-causing criteria: the change in binding affinity (ΔKa) of the (1) drug and (2) endogenous ligand upon a protein’s mutation; (3) the probability a mutation will occur based on empirically derived mutational signatures; and (4) the cardinality of mutations comprising a hotspot. For validation, we applied Resistor to EGFR and BRAF kinase inhibitors treating lung adenocarcinoma and melanoma. Resistor correctly identified eight clinically significant EGFR resistance mutations, including the erlotinib and gefitinib “gatekeeper” T790M mutation and five known osimertinib resistance mutations. Furthermore, Resistor predictions are consistent with BRAF inhibitor sensitivity data from both retrospective and prospective experiments using KinCon biosensors. Resistor is available in the open-source protein design software OSPREY.
1. Introduction
Acquired resistance to therapeutics is a pressing public health challenge that affects maladies from bacterial and viral infections to cancer (Centers for Disease Control and Prevention, 2020; Housman et al., 2014; Zahreddine and Borden, 2013; Assaraf et al., 2019; Gupta et al., 2012; Vasan et al., 2019). There are several different ways cancer cells acquire resistance to treatments, including drug inactivation, drug efflux, DNA damage repair, cell death inhibition, and escape mutations, among others (Housman et al., 2014). Accurate, prospective prediction of resistance mutations could allow for design of drugs that are less susceptible to resistance. While it is unlikely that medicinal chemists will be able to address all of the resistance-conferring mechanisms in cancer cells, progress can be made by the incorporation of increasingly accurate models of the above contributing factors to acquired resistance, leading to the development of more durable therapeutics. To that end, several structure-based computational techniques for therapeutic design and resistance prediction have been proposed.
One such technique is based on the substrate-envelope hypothesis. In short, the substrate-envelope hypothesis states that drugs designed to have the same interactions as the endogenous substrate in the active site will be unlikely to lose efficacy because any mutation that ablates binding to the drug would also ablate binding to the endogenous substrate (Altman et al., 2008). C. Schiffer and B. Tidor’s labs developed the substrate-envelope hypothesis for targeting drug-resistant HIV strains (Prabu-Jeyabalan et al., 2002; King et al., 2004; Altman et al., 2008; Shen et al., 2013). Their design technique has been successfully applied to develop compounds with reduced susceptibility to drug-resistant HIV proteases (Shen et al., 2013).
Another computational technique is to use ensemble-based positive and negative design (Frey et al., 2010; Gainza et al., 2016). There are two specific ways that point mutations can confer resistance to therapeutics: they can decrease binding affinity to the therapeutic or they can increase binding to the endogenous ligand (Frey et al., 2010; Reeve et al., 2015). Protein design with the goal of decreasing binding is known as negative design, and increasing binding is known as positive design. As a concrete example, consider the case of a drug that inhibits the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) to treat lung adenocarcinoma. Here, an active site mutation could sterically prevent the inhibitor from entering the active site (Yan et al., 2017). On the other hand, a different mutation might have no effect on an enzyme’s interactions with the drug but instead increase affinity to its native ligands, resulting in increased phosphorylization of downstream substrates (Yun et al., 2008; Yoshikawa et al., 2013). Because these two distinct pathways to therapeutic resistance exist, it is necessary to predict resistance mutations using both positive and negative design. In other words, predicting resistance can be reduced to predicting a ratio of the change in Ka upon mutation of the protein:endogenous ligand and protein:drug complexes.
Ka is an equilibrium constant measuring the binding and unbinding of a ligand to a receptor. It is defined as:
| (1) |
where kon and koff are the on- and off-rate constants, and [RL], [R], and [L] the equilibrium concentrations of, respectively, the receptor-ligand complex, unbound receptor, and unbound ligand. Ka is the reciprocal of the disassociation constant Kd. K* is an algorithm implemented in the OSPREY computational protein design software that provably approximates Ka (Georgiev et al., 2008; Hallen et al., 2018). It is defined as the quotient of the bound (complex) to unbound (apo protein and apo ligand) partition functions of a protein:ligand system. See the STAR methods for further details on the K* algorithm.
Our lab developed a provable, ensemble-based method using positive and negative K* design to computationally predict and experimentally validate resistance mutations in protein targets (Frey et al., 2010). We then applied this methodology to prospectively predict resistance mutations in dihydrofolate reductase when Staphylococcus aureus was treated with a novel antifolate (Reeve et al., 2015), which we later confirmed in vivo (Reeve et al., 2015, 2016), demonstrating the utility of correctly predicting escape mutations during the drug discovery process.
From these previous works, it is clear that multiple criteria must be combined to decide whether a mutation confers resistance. Often it is the human designers themselves who must choose arbitrary weights for different criteria. Yet multi-objective, or Pareto, optimization techniques would allow designers to combine multiple criteria without choosing arbitrary decision thresholds. Pareto optimization for protein design has been employed by Chris Bailey-Kellogg, Karl Griswold, and co-workers (Parker et al., 2013; Choi et al., 2013; Salvat et al., 2015; Griswold and Bailey-Kellogg, 2016; Choi et al., 2016; Salvat et al., 2017). One such example is PEPFR (Protein Engineering Pareto FRontier), which enumerates the entire Pareto frontier for a set of different criteria such as stability vs. diversity, affinity vs. specificity, and activity vs. immunogenicity (He et al., 2012). Algorithmically, PEPFR combined divide-and-conquer with dynamic or integer programming to achieve an algorithm where the number of divide-and-conquer “divide” steps required for the search over design space is linear only in the number of Pareto optimal designs. Being dependent on multiple criteria, a multi-objective optimization method that ranks solutions, such as Pareto optimization, is particularly suitable for resistance predictions.
Instead of merely finding a single solution optimizing a linear combination of functions, Pareto optimization finds all consistent solutions optimizing multiple objectives such that no solution can be improved for one objective without making another objective worse. Specifically, let Λ be the set of possible solutions to the multi-objective optimization problem, and let λ ∈ Λ. Let F be a set of objective functions and f ∈ F, where is one objective function. A particular solution λ is said to dominate another solution λ′ when
| (2) |
| (3) |
A solution λ is Pareto optimal if it is not dominated. Resistor combines ensemble-based positive and negative design, cancer-specific mutational signature probabilities, and hotspots to identify not only the Pareto frontier, but also the Pareto ranks of all candidate sequences.
The inclusion of mutational signature probabilities in Pareto optimization is possible because distinct mutational processes are operating in different types of cancers (Alexandrov et al., 2013, 2020). Specifically, these mutational processes drive the type and frequency of DNA base substitutions. Alexandrov et al. (2013) postulated each signature to be associated with a biological process (such as ABOPEC activity) or a causative agent (such as tobacco use), although not all associations are definitively known. What is certain is that particular signatures tend to appear in particular types of cancer. For example, 12 single-base substitution signatures, 2 double-base substitution signatures, and 7 indel signatures were found in a large set of melanoma samples, with many of those signatures associated with ultraviolet light exposure (Alexandrov et al., 2020). Building on the work of Alexandrov et al. (2013), Kaserer and Blagg (2018) combined the multiple signatures found in each cancer type to generate overall single-base substitution probabilities. Resistor uses these probabilities to compute the overall probability that mutation events will occur in a gene independent of changes to protein fitness. This amino acid mutational probability is one of the axes we optimize over.
The most computationally complex part of provable, ensemble-based multistate design entails computing the K* scores of the different design states. This is largely because for biological accuracy it is necessary to use K* with continuous sidechain flexibility (Gainza et al., 2012; Qi et al., 2018). Though OSPREY has highly-optimized GPU routines for continuous flexibility (Hallen et al., 2018), energy minimization over a combinatorial number of sequences in a continuous space is, in practice, computationally expensive. Having a method to reduce the number of sequences evaluated would greatly decrease the computational cost. Comets is an empirically sublinear algorithm that provably returns the optimum of an arbitrary combination of multiple sequence states (Hallen and Donald, 2016). Resistor uses Comets to prune sequences whose predicted binding with the drug improves and binding with the endogenous ligand deteriorates. While Comets does not compute the full partition function, it provides a useful method to efficiently prune a combinatorial sequence space, for example when investigating resistant protein targets with more than one resistance mutation. By virtue of pruning using Comets, Resistor inherits the empirical sublinearity characteristics of the Comets sequence search, rendering Resistor sublinear in the size of the sequence space.
The tyrosine kinase EGFR and serine/threonine-protein kinase BRAF are two oncogenes associated with, respectively, lung adenocarcinoma and melanoma. Both kinases are conformationally flexible, but two conformations are particularly determinative to their kinase activity—the “active” and “inactive” conformations. Oncogenic mutations to EGFR include L858R and deletions in exon 19, both of which constitutively activate EGFR (Harrison et al., 2020; Lynch et al., 2004). Likewise, V600E is the most prevalent constitutively activating mutation in BRAF (Davies et al., 2002). Numerous drugs have been developed to treat the EGFR L858R and BRAF V600E mutations. The first generation inhibitors erlotinib and gefitinib competitively inhibit ATP binding in EGFR’s active site, whereas binding by the third generation osimertinib is irreversible (Dowell et al., 2005; Herbst et al., 2004; Soria et al., 2018). For BRAF, the therapeutics dabrafenib, vemurafenib, and encorafenib were designed to target the V600E mutation and are in clinical use, and PLX8394 is in clinical trials (Ballantyne and Garnock-Jones, 2013; Bollag et al., 2012; Shirley, 2018; Janku et al., 2020). Use of Resistor to predict resistance mutations to these drugs would provide strong validation of the efficacy of this approach.
By presenting Resistor, this article makes the following contributions:
A multi-objective optimization algorithm that combines four axes of resistance-causing criteria to rank candidate mutations.
The use of Comets as a provable and empirically sublinear pruning algorithm that removes a combinatorial number of candidate sequences before expensive ensemble evaluation.
A validation of Resistor that correctly predicted eight clinically significant resistance mutations in EGFR, providing explanatory ensemble-bound structural models for acquired resistance.
Prospective predictions with explanatory structural models and experimental validation of resistance mutations for four drugs targeting BRAF mutations in melanoma.
Newly modelled structures of EGFR and BRAF bound to their endogenous ligands and inhibitors in cases where no experimental structures exist.
An implementation of Resistor in our laboratory’s free and open source computational protein design software OSPREY Hallen et al. (2018).
2. Results
2.1. Overview of Resistor
The Pareto optimization in Resistor optimizes four axes: structure-based positive design, structure-based negative design, sequence-based mutational probabilities, and the count of resistance-causing mutations at a given amino acid location. Briefly, we chose these four criteria because they identify mutations that 1) increase affinity to the endogenous ligand in such a way that it outcompetes the inhibitor; 2) decrease the efficacy of the drug by reducing its binding (leading to the same effect); 3) are predicted to occur based on the DNA sequence and excludes those that are unlikely to arise; and, 4) are located at residue positions where many mutations are predicted to confer resistance, thus identifying a position of relative importance. We believe these criteria to be the minimal requirements a cancer clone must fulfill to confer resistance, and we’ve had success predicting retrospective and prospective resistance mutations in a previous study using these four criteria (Kaserer and Blagg, 2018).
In our earlier study, we prioritized potential resistance mutants by first applying four sequence- and structure-based filtering steps and then pruning the remaining predicted resistance mutations by a) chosing the three residue locations with the highest hotspot cardinality (see Section 2.4), and b) ranking the individual amino acids within the hotspots by their mutational probability (Kaserer and Blagg, 2018). In other words, we ranked resistance candidates by two criteria: their hotspot cardinality and mutational probability. With Resistor hotspot cardinality instead becomes one of the Pareto objectives. Our earlier work used the positive and negative design K* scores as a binary resistance filter (Kaserer and Blagg, 2018); here we use them first as a filter and then as two additional Pareto optimization objectives. This allows Resistor to use thermodynamic predictions not only in a binary, qualitative manner (i.e., whether the ratio of K* positive and negative designs indicates resistance) but also in a quantitative manner (i.e. the magnitude of the affinity-driven resistance). Finally, Resistor also transforms mutational probability from the final ranking criteria to one of the four Pareto objectives. In summary, Resistor’s Pareto optimization objective function simultaneously maximizes the ΔKa of the positive design (the protein bound to the endogenous ligand), minimizes the ΔKa of the negative designs (the protein bound to the drug), maximizes the mutational probability, and maximizes the count of resistance-causing mutations per amino acid. Fig. 1 shows an overview how these axes are implemented in our algorithm. It should be mentioned that, as a generalizable method, additional resistance-causing criteria could be trivially added to Resistor for further refinement.
Figure 1. An example Resistor workflow with EGFR.

Resistor finds the Pareto frontier from OSPREY positive and negative designs, mutational probabilities, and resistance hotspots. (A) Two structures are required as input to OSPREY to compute postive and negative design K* scores. The structure for positive design is EGFR (green) bound to its endogenous ligand ATP (blue), for the negative design EGFR is bound to the drug erlotinib (pink). The goal of positive (resp. negative) design is to improve (resp. ablate) binding affinity. A mutation is resistant when its ratio of positive to negative K* scores increases. (B) All residues within 5 Å (purple) of the drug are allowed to mutate to any other amino acid. (C) Comets is used as an efficient, sublinear algorithm to quickly prune infeasible mutations. BWM* is used with a fixed branch width to compute a polynomial-time approximation to the K* score. (D) Candidate mutations that pass the Comets pruning step have their positive and negative K* scores computed in OSPREY. We recommend using the BBK* with MARK* algorithm as it is the fastest for computing K* scores. (E) Candidate resistance mutations are pruned when their ratio of positive to negative K* scores indicates a mutation does not cause resistance or if the target amino acid requires a mutation in all three DNA bases. (F) Resistor computes mutational probabilities using a protein’s coding DNA along with cancer-specific trinucleotide mutational probabilities for lung adenocarcinoma (abbreviated as LuAd), sliding a window (G) over 5′ - and 3′ -flanked codons. (H) Resistor employs a recursive graph algorithm to compute the probability that a particular amino acid will mutate to another amino acid (I). (J) Finally, Resistor uses Pareto optimization on the positive and negative K* scores, the mutational probabilities, and hotspot counts to predict resistance mutants.
2.2. Structure-based Positive and Negative Design
We use the K* algorithm in OSPREY to predict an ε-accurate approximation to the binding affinity (Ka) in four states: 1) the wildtype structure bound to the endogenous ligand; 2) the wildtype structure bound to the therapeutic; 3) the mutated structure bound to the endogenous ligand; and 4) the mutated structure bound to the therapeutic. This ε-accurate approximation is called the K* score (Georgiev et al., 2008; Hallen et al., 2018). In order to calculate the K* score of a protein:ligand complex, it is necessary to have a structural model of the atomic coordinates. Experimentally-determined complexes have been solved for EGFR bound to an analog of its endogenous ligand (PDB id 2itx), to erlotinib (1m17), gefitinib (4wkq), and to osimertinib (4zau) (Yun et al., 2007; Stamos et al., 2002; Yosaatmadja et al., 2014, 2015). Similarly, we used the crystal structure for BRAF bound to dabrafenib (4xv2) and vemurafenib (3og7) (Zhang et al., 2015; Hodis et al., 2012). Experimentally-determined complexes of BRAF bound to encorafenib, PLX-8394, and an ATP analog in an active conformation do not exist, so we instead modelled the ligands into BRAF in its activated conformation (for additional details on model selection and preparation see the STAR methods). We used these predicted complex structures for our resistance predictions.
We added functionality to OSPREY that simplifies the process of performing computational mutational scans. A mutational scan refers to the process of computing the K* score of every possible amino acid mutation within a radius of a ligand. Resistor uses this functionality to create the initial set of candidate mutant sequences by selecting and computing the K* scores for each amino acid within a 5 Å radius of the drug or the endogenous ligand. This generated a search space of 2471 sequences. We then set all residues with sidechains within 3 Å of the mutating residue to be continuously flexible for the Resistor K* designs. Each sequence has an associated conformation space size dependent on the total number of mutable and flexible residues, which one can use as a heuristic to estimate the difficulty of computing a complex’s partition function. The average conformation space size of each sequence was ~5.9 × 1010 conformations, thus computing the partition functions is only possible using OSPREY’s pruning and provable ε-approximation algorithms (Gainza et al., 2012; Hallen et al., 2018; Jou et al., 2020). Empirical runtimes of the positive- and negative-K* designs are shown in the STAR methods. The change in the K* score upon mutation for the endogenous ligand (positive design) and drug (negative design) become two of the four axes of optimization. These two axes also form the basis of a pruning step (described in Section 2.5).
2.3. Computing the Probability of Amino Acid Mutations
To convert the trinucleotide to trinucleotide probabilities into amino acid to amino acid mutational probabilities, Resistor constructs a directed graph with the trinucleotides as nodes and the probability that one trinucleotide mutates into another trinucleotide as directed edges. It then reads the cDNA of the protein in a sliding window of 5′ - and 3′ -flanked codons, since the two DNA bases flanking a codon are necessary to determine the probabilities of either the first or third base of a codon mutating. We designed a recursive algorithm to traverse the graph and find all codons that can be reached within n single-base mutations, where n is an input parameter. The algorithm then translates the target codons into amino acids and, as a final step, sums the different probabilities on each path to an amino acid into a single amino acid mutational probability (see Fig. 1F-I). One can either (a) precompute a cancer-specific codon-to-codon lookup table consisting of every 5′ - and 3′ -flanked codon to its corresponding amino acid mutational probabilities, or (b) read in a sequence’s cDNA and compute the mutational probabilities on the fly. The benefit of (a) is it only needs to be done once per cancer type and can be used on an arbitrary number of sequences. On the other hand, when assigning mutational probabilities to proteins that have strictly fewer than 45 amino acids, it is faster to compute the amino-acid specific mutational signature on the fly. In both cases, the algorithm is strictly polynomial and bounded by O(kn9), where k is the number of codons with flanking base pairs (upper-bounded by 45) and n is the number of mutational steps allowed, which in the case of Resistor is 2. An implementation of this algorithm is included in the free and open source OSPREY repository on GitHub (Hallen et al., 2018).
2.4. Identifying Mutational Hotspots
After calculating the positive and negative change in affinity ΔKa and determining the mutational probability of each amino acid, Resistor prunes the set of candidate mutations (see section 2.5). Post-pruning, it counts the number of mutations at each amino acid location. This count is necessary to determine whether a residue location is likely to become a “mutational hotspot”, namely a residue location where many mutations are predicted to confer resistance. Correctly identifying mutational hotspots is vital because they indicate that a drug is dependent on the wildtype identity of the amino acid at that location, and it is likely that many mutations away from that amino acid will cause resistance. Consequently, the fourth axis used in Resistor’s Pareto optimization is the count of predicted resistance-conferring mutations per residue location, termed hotspot cardinality.
2.5. Reducing the Positive Prediction Space
Prior to carrying out the multi-objective optimization to identify predicted resistance mutations, we prune the set of candidates. First, we introduce a cut-off based on the ratio of K* scores of positive and negative designs, an adaption from Kaserer and Blagg (2018). We determine the average of the K* scores for the drug and endogenous ligand across all of the wildtype designs for the same protein. The cut-off c is:
| (4) |
where c0 is a user-specified constant, is the average of the K* scores for the wildtype protein bound to the endogenous ligand, and is the average of the K* score for the wildtype protein bound to the drug. We recommend in practice to set c0 to be greater than the range () of wildtype K* scores—we set it to 100 for the tyrosine kinase inhibitor (TKI) predictions.2 A mutation m is predicted to be resistant when:
| (5) |
where is the K* score of the endogenous ligand bound to the mutant, and is the K* score of the drug bound to the mutant.
We also prune mutations predicted to completely ablate endogenous ligand binding, i.e., the predicted K* score of the protein and endogenous ligand is 0, because such a mutation renders a critical protein non-functional. This is particularly detrimental to a cancer cell, which relies heavily on the activity of a protein. We lastly prune the predicted resistance mutation candidates by removing all mutations that cannot arise within two DNA base substitutions. Whether an amino acid can be reached within two DNA base substitutions is determined by the algorithm described in section 2.3, and if it cannot, then that particular mutation is assigned a mutational probability of 0 and pruned.
2.6. Resistor Identifies 8 Known Resistance Mutations in EGFR
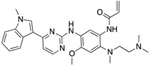
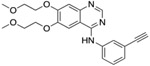
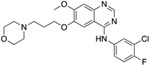
We evaluated a total of 1257 sequences across the three TKIs for EGFR. Among these sequences, the average conformation space size for computing a complex’s partition function was ~1.3 × 107. After we ran the Resistor algorithm on these sequences, a total of 108 mutants were predicted as resistance-conferring candidates for all three inhibitors combined from a purely thermodynamic and probabilistic basis, i.e. these mutations were required to lower affinity of the drug in relation to the endogenous ligand (K* Positive and Negative Design, Fig. 1A-D) and could be formed in patients by less than three base pair exchanges (Calculating Mutational Probabilities, Fig. 1F-I). To further prioritize mutations and identify those that are most likely to be clinically relevant, we then computed the Pareto frontier over the four axes for each drug (Fig. 1J). Out of these 108 candidates, Resistor correctly prioritized eight clinically significant resistance mutants, with 7 of the 8 in the Pareto frontier of the corresponding inhibitor and the remaining mutant in the 2nd Pareto rank (see Table 1). A detailed description of the result for each inhibitor is included in the sections below.
Table 1. Resistor correctly identified 8 resistance mutations in EGFR to erlotinib, gefitinib, and osimertinib.
For osimertinib, G796R, G796S, G796D, and G796C were on the Resistor-identified Pareto frontier. L792H was in the 2nd Pareto rank. For erlotinib, both T790M and G796D were on the Pareto frontier. For gefitinib, T790M was also on the Pareto frontier. Previous studies have documented all of these resistance mutations as occurring in the clinic (Helena et al., 2013; Avizienyte et al., 2008; Chen et al., 2017; Yang et al., 2018; Ou et al., 2017; Fairclough et al., 2019; Li et al., 2021; Yang et al., 2018; Zheng et al., 2017). † indicates that Resistor predicted the mechanism of resistance to be improved binding of the endogenous ligand to the mutant. # indicates that Resistor predicted the mechanism of resistance to be decreased binding of the drug to the mutant. Note that these predicted mechanisms are only attributed here if the predicted change in the log10(ΔK*) ≥ 0.5.
| Resistor Identifies Clinically-Relevant Resistance Mutations in EGFR | ||
|---|---|---|
| Osimertinib | Erlotinib | Gefitinib |
|
|
|
| L792H# | T790M†# | T790M †# |
| G796R†# | G796D# | |
| G796S# | ||
| G796D# | ||
| G796C# | ||
2.6.1. EGFR Treated with Erlotinib and Gefitinib
Of the 462 sequences evaluated for the TKI erlotinib, Resistor identified 50 as candidate resistance mutations. Pareto ranking placed 19 sequences on the frontier, 13 sequences in the second rank, and 11, 6, and 1 sequences in the third, fourth, and fifth ranks, respectively. Resistor correctly identified two clinically significant mutations, T790M and G796D, as being on the Pareto frontier (Helena et al., 2013; Avizienyte et al., 2008). This is concordent with empirical data showing that T790M is, by far, the most prevalent resistance mutation that occurs in lung adenocarcinoma treated with erlotinib (Tate et al., 2019). Similarly, for gefitinib, Resistor evaluated 438 sequences and identified 22 as candidate resistance mutants. The most relevant clinical mutant, T790M, is found on the Pareto frontier.
2.6.2. EGFR and Osimertinib
Resistor evaluated 357 OSPREY-predicted structures of EGFR bound with osimertinib and EGFR bound with its endogenous ligand. Of those, 36 were predicted as resistance candidates. Pareto optimization placed 16 sequences on the frontier, 2 sequences in rank 2, 8 sequences in rank 3, 1 sequence in rank 4, and 5 sequences in rank 5. Resistor correctly identified five clinically significant resistance mutations to osimertinib: L792H, G796R, G796S, G796D, and G796C (Chen et al., 2017; Yang et al., 2018; Ou et al., 2017; Fairclough et al., 2019; Li et al., 2021; Yang et al., 2018; Zheng et al., 2017), and while L792H was in the 2nd Pareto rank, all of the other correctly predicted resistance mutations are on the Pareto frontier.
Two osimertinib resistance mutations in particular stand out: L792H and G796D (see Fig. 2). Both of these mutants have appeared in the clinic (Zheng et al., 2017; Chen et al., 2017; Yang et al., 2018; Ou et al., 2017). OSPREY generated an ensemble of the bound positive and negative complexes upon mutation, providing an explanatory model for how resistance occurs. In both cases, the mutant sidechains are much bulkier than the wildtype sidechain (Fig. 2A and D) and thus are predicted to clash with the original osimertinib binding pose (Fig. 2B and E). Consequently, in both cases the ligand is predicted to translate and rotate to create additional space for the mutant sidechains (Fig. 2C and F). We hypothesize that this movement weakens the other molecular interactions osimertinib makes in the EGFR active site.
Figure 2. Structural models predicted by OSPREY agree with experimental data and explain mechanisms of Osimertinib resistance to EGFR mutations L792H and G796D.

Structural models predicted by OSPREY of EGFR wildtype (blue) and resistance mutations (red) bound to osimertinib (yellow sticks). The histidine (A) and glutamate (D) side chains (red sticks) in the EGFR L792H (A) and G796D (D) mutations are bulkier than the wildtype leucine (A) and glycine (C) residues (blue sticks). They clash with osimertinib in its original binding pose as highlighted by the sphere representation in panels B and E. (C+F) To allow for accommodation of osimertinib in the modelled EGFR mutant structures (red sticks), the inhibitor’s position within the binding pocket moves from the experimentally determined binding pose (yellow sticks). Movements are indicated by black arrows. (F) In case of the G796D mutation, the carboxylate moiety of D796 is predicted to be in close proximity to the osimertinib amide oxygen (highlighted with the dashed circle), thus leading to electrostatic repulsion. This mutation site is adjacent to C797, which reacts with the allyl-moiety of osimertinib to form a covalent bond in the wildtype. Due to the steric and electrostatic properties of the G796D mutant, the allyl group is located further away from C797 in the model, thus preventing covalent bond formation. The movement of the allyl group is indicated by the black arrow.
In the case of G796D, there are additional factors that contribute to acquired resistance. First, the mutation to aspartate introduces a negative charge, which probably leads to electrostatic repulsion with the carbonyl oxygen of the osimertinib amide (Fig. 2F, highlighted with a dashed oval). In addition, the exit vector of the hydrogen bound to the amide nitrogen does not allow a hydrogen bond with the aspartate. Second, the allyl-group of osimertinib must be in close proximity to C797 for covalent bond formation. In fact, C797 is so important to osimertinib’s efficacy that mutations at residue 797 confer resistance (Thress et al., 2015; Arulananda et al., 2017). Even if osimertinib still binds to G796D, the allyl group would have to move away from C797 (Fig. 2F, highlighted with a black arrow). This would prevent covalent bond formation and thus reduce the efficacy of osimertinib considerably. Lastly, it is likely that the mutation away from glycine reduces the conformational flexibility of the loop, incurring an entropic penalty while also plausibly making it more difficult to properly align osimertinib and C797.
2.7. Resistor Predicts Previously Unreported Resistance Mutations in BRAF and Provides Structural Models
In addition to retrospective validation by comparison to existing clinical data for EGFR, we used Resistor to predict how mutations in the BRAF active site could confer resistance. Specifically, we used Resistor to predict which of 1214 BRAF sequences would be resistant to four kinase inhibitors—vemurafenib, dabrafenib, encorafenib, and PLX8394. On the Pareto frontier for vemurafenib are 13 mutations, for dabrafenib 16 mutations, for encorafenib 15 mutations, and for PLX8394 15 mutations. The full sets of predictions are included in the supplementary tables S4-S7. To validate Resistor’s predictions, we compared them with two sources of experimental data: a saturation mutagenesis variant effect assay from Wagenaar et al. (2014) and a cell-based kinase conformation reporter assay termed KinCon (Röck et al., 2019; Mayrhofer et al., 2020). Furthermore, we carried out additional KinCon experiments on a number of Resistor predictions to validate Resistor’s predictive capabilities.
2.7.1. Retrospective and prospective validation of Resistor predictions using the BRAF KinCon biosensor reporter
KinCon, developed by Stefan and colleagues, is an in-cell protein-fragment complementation assay (PCA) that provides a readout of the activity conformation change of full-length BRAF upon mutation or exposure to different inhibitors (Enzler et al., 2020). KinCon’s bioluminescence assay functions by appending parts of a luceriferase enzyme to the N- and C-termini of full-length BRAF and observing the amount of bioluminescence, indicating whether BRAF favors an open, catalytically active or a closed, autoinhibited conformation (see Fig. 3A) (Enzler et al., 2020). Stefan and colleagues have demonstrated that activation of BRAF either via upstream regulators such as EGFR and GTP activated Ras or via tumorigenic mutations cause BRAF to favor an open conformation (Röck et al., 2019; Mayrhofer et al., 2020). The inhibitors bind to BRAF in the ATP binding site and cause BRAF’s N- and C-termini to interact, shifting BRAF back towards a more closed, intermediate state (see Fig. 3A) (Röck et al., 2019; Enzler et al., 2020; Mayrhofer et al., 2020). This implies that for inhibitor binding and BRAF closing to occur, a mutation (or a combination of mutations and/or upstream signaling events) needs first to induce an open conformation. Not all clinically observed BRAF mutations cause opening, even if they activate the MAPK pathway (e.g. L472C) (Mayrhofer et al., 2020; Sen et al., 2012). In the same vein, not all BRAF resistance mutants show increased kinase activity, in fact several are classified as kinase impaired (Mayrhofer et al., 2020; Zheng et al., 2015; Sen et al., 2012). One prominent mutation that shows both increased kinase activity and induces an open conformation is V600E (Fig. 3B). Inhibitor treatment shifts the V600E conformational equilibrium towards a more closed state (Röck et al., 2019; Mayrhofer et al., 2020). In contrast, the gatekeeper mutations T529M and T529I do not confer opening of the kinase conformation and are thus insensitive to inhibitor treatment (Röck et al., 2019). However, in combination with V600E these mutations do confer resistance to BRAF inhibitors to varying degrees. Given that we model a state that is permissive of ligand binding at the outset (i.e., the ligand-bound BRAF complex), our Resistor calculations align very well with the reported KinCon measurements of double mutants (e.g. V600E/T529M and V600E/T529I, see STAR Methods for additional details on modeling).
Figure 3. KinCon biosensor results for Resistor-predicted mutants.

(A) Schematic depiction of Renilla luciferase (RLuc; F1: fragment 1, F2: fragment 2) PCA-based BRAF kinase conformation (KinCon) reporter system. Conformational rearrangement of the reporter upon (de)activation of the kinase are indicated. Closed kinase conformation induces complementation of RLuc PCA fragments resulting in increased RLuc-emitted bioluminescence signal. (B) Domain organization of the BRAF-KinCon reporter (top) and basal bioluminescent signals of the BRAF-wt (black), V600E (red), and Resistor-predicted mutant (grey) KinCon biosensors. Bars represent the mean signals, relative to BRAF-wt, in relative light units (RLU) with SD of four independent experiments (nodes). Raw bioluminescence signals were normalized on reporter expression levels, determined through western blotting. Asterisk indicates level of significance versus the wild type BRAF biosensor. (C) BRAF-KinCon biosensor dynamics, induced via treatment with respective BRAFi (1μM for 1h) prior to bioluminescence measurement. BRAF wt and V600E KinCon variants serve as control (left). The Resistor-predicted mutants are shown in a separate bar chart (right). Bars represent the mean signals, relative to the DMSO control, in relative light units (RLU) with SEM of four independent experiments (nodes). All experiments were performed in HEK293T cells 48 hours post transfection. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant by t-test.
Specifically, the Resistor predictions of resistance concord with the previous KinCon biosensor results for V600E/T529M and V600E/T529I for three of the four inhibitors: vemurafenib, dabrafenib, and PLX8394 (Röck et al., 2019). In the case of vemurafenib treatment, the proportion of open to closed conformations in the V600E/T529I mutant is not significantly different from the untreated V600E mutant, indicating vemurafenib treatment is not closing the conformational distribution in the double mutant (Röck et al., 2019). These data agree with the Resistor calculation of the ratios of the log10 K* scores, which predict that both double mutants are resistant to vemurafenib, with V600E/T529M more resistant. Treatment of BRAF with PLX8394 follows the same pattern as vemurafenib, namely the V600E/T529I mutant’s closed population increases only 1.2 fold compared to the untreated mutant, and the PLX8394-treated V600E/T529M mutant does not noticeably alter the conformational distribution (Röck et al., 2019). In contrast, the PLX8394-treated V600E mutant’s closed population increases ~3 fold compared to the untreated population, indicating V600E sensitivity to PLX8394 (see Fig. 3C). Resistor correctly predicted the V600E/T529I and V600E/T529M double mutants are resistant to PLX8394, with the change in the ratio of the log10 K* scores of the two mutants suggesting that V600E/T529M confers greater resistance. In the case of dabrafenib, treatment of the V600E/T529I mutant closed the conformational distribution (2.4 fold more closed compared to untreated) more than treatment of the V600E mutation (2 fold more closed compared to untreated), whereas dabrafenib treatment of the V600E/T529M mutant increased the closed conformational population less effectively than the V600E mutant alone (1.4 fold vs. 2 fold). This again agrees with the Resistor predictions, namely that V600E/T529I remains sensitive to dabrafenib but V600E/T529M is resistant. Resistor predicted that the V600E/T529I and V600E/T529M mutants would be resistant to encorafenib, but the KinCon data indicates that these mutants may actually retain sensitivity to encorafenib, as the inhibitor induces BRAF’s closed state.
In addition, all inhibitors except dabrafenib were predicted to be sensitive against the G466V mutation and showed closing of the kinase conformation (Mayrhofer et al., 2020). However, in the case of dabrafenib, the response was comparable to vemurafenib, although vemurafenib was classified as sensitive. Previous KinCon experiments have also shown that G466V (and G466R and G466E (Zheng et al., 2015), see below) impaired kinase function consistent with the reduced endogenous ligand binding predicted by Resistor (see “All BRAF Predictions” supplementary table) (Mayrhofer et al., 2020).
In addition to the above retrospective validation, we chose a few Resistor-predicted mutations and evaluated them using the KinCon reporter. We selected the mutants G466E, G466R, V471F, L505H, and G593D because they were prioritized by Resistor for at least one of the investigated inhibitors and were reported as patient mutations in either the COSMIC (Tate et al., 2019) or cBioPortal (Cerami et al., 2012; Gao et al., 2013) databases, using the curated set of non-redundant studies (see Table 2).
Table 2. Prioritized BRAF mutations selected for experimental testing.
We selected these mutants because they were prioritized by Resistor for at least one of the investigated inhibitors and were reported as patient mutations in either the COSMIC or cBioPortal databases. The numbers in the first four columns indicate the Resistor-predicted Pareto rank with melanoma mutational probabilities. The numbers in the last two columns indicate the number of patient samples containing the mutation reported in the respective database (access date 01/12/2022). Absence of a Pareto rank indicates Resistor predicted the mutant would remain sensitive to the drug.
| Mutation | Vemurafenib | Dabrafenib | Encorafenib | PLX8394 | COSMIC | cBioPortal |
|---|---|---|---|---|---|---|
| G466E | - | 1 | - | - | 49 | 31 |
| G466R | - | 1 | - | - | 17 | 3 |
| V471F | - | - | 2 | 3 | 5 | 2 |
| L505H | - | - | 3 | - | 8 | 10 |
| G593D | 1 | 1 | 1 | 1 | 4 | 0 |
The expression-normalized basal biosensor signal suggests that both G466E and G466R mutants shift the conformation to an opened state, comparable to the highly oncogenic V600E variant and similar to the effect of the common non-small-cell lung cancer mutation G466V (Mayrhofer et al., 2020). The V471F, L505H and G593D mutations, in contrast, did not appear to induce a change in the active conformation (Fig. 3B). When exposed to BRAF inhibitors (Fig. 3C), G466E and G466R mutants showed the highest fold increase of the biosensor signal for all four inhibitors tested. The majority of inhibitors, three out of four, were predicted as sensitive against these mutants. Resistor predicted G466E and G466R to be resistant to dabrafenib, and while Resistor predicted dabrafenib had lower sensitivity compared to encorafenib and PLX8394 (which is consistent with the KinCon results), dabrafenib-treated mutants shifted to a closed comformation at least as much as vemurafenib-treated mutants did. The L505H and G593D KinCon mutants were not affected by any inhibitors, as those mutations do not shift the kinase into an active opened kinase conformation which is required for inhibitor binding. While vemurafenib and dabrafenib do not appear to affect the V471F mutant, encorafenib and PLX8394 did induce a closing of the kinase, suggesting that the structural properties of the inhibitor determine the binding affinity to this mutant. This is particularly intriguing, given that the V471F mutation was selected because we predicted it would confer resistance to encorafenib and PLX8394. While the KinCon results suggest that these two compounds still retain binding to the V471F mutant, the mutant itself did not induce a significant opening of the kinase confirmation required for ligand binding. For the latter three mutations (i.e. L505H, G593D, and V471F), it would therefore be required to induce the open conformation some other way, for example by introducing the V600E mutation similar to T529I and T529M described above, to investigate whether resistance would develop to the inhibitors (Röck et al., 2019).
2.7.2. Retrospective validation of Resistor predictions using BRAF saturation mutagenesis experiments
Wagenaar et al. (2014) examined the effects of BRAF inhibitor binding site mutations on inhibitor efficacy. To do so, they carried out targeted saturation mutagenesis on the BRAF vemurafenib binding site in the A375 human melanoma cell line and challenged the mutants with vemurafenib over a three week period (Wagenaar et al., 2014). They then sequenced the emergent clones and measured the IC50 values of a subset of the mutants. Their work demonstrated a correlation between a mutant’s deep sequencing enrichment, i.e. the increase in the amount of an amino acid sequence in a sample before and after the addition of an inhibitor, and its IC50 value (Wagenaar et al., 2014). We therefore compared their enrichment data to the Resistor predictions and determined Resistor’s vemurafenib resistance prediction specificity to be 91%. There were five Resistor-predicted resistance mutations that had increased enrichment over the three week period: T529M already discussed above (enriched 47.96 fold above the V600E baseline, which was the experiment’s largest change in enrichment), T529L (enriched 18.57 fold above baseline), T529F (enriched 7.87 fold above baseline), G593I (enriched 4.84 fold above baseline), and L514E (enriched 3.73 fold above baseline). Furthermore, Wagenaar et al. determined the relative IC50 values of T529M, T529L, and G593I which were, respectively, 2.05, 2.16, and 3.19 times larger than the IC50 for vemurafenib applied to the V600E mutant. The IC50 of T529F and L514E were not determined.
To further elucidate the molecular mechanisms conferring resistance to the G593I and L514E mutants, we analyzed the OSPREY-predicted structural models. While neither mutant requires a movement of vemurafenib (Fig. 4A) akin to what was observed in the EGFR and osimertinib structures (Fig. 2), the mutations still lead to a loss of favorable interactions and/or the introduction of energetically unfavorable contacts. The residue G593 (Fig. 4B) may facilitate structural adaptions required for BRAF to accommodate the vemurafenib propyl sulfonamide moiety in the rear of the ATP binding site and the G593L mutations may thus constrain the flexibility of this loop region. In addition, the leucine side chain may project near to the fluoro-substituted central phenyl ring and introduce steric clashes (Fig. 4C). The neighboring D594 backbone interacts with the vemurafenib sulphonamide-nitrogen (Fig. 4B), and this interaction would be weakened in the G593L mutant. Furthermore, residue L514 makes a range of hydrophobic contacts with vemurafenib (Fig. 4D), including the central phenyl ring and the propyl-chain, which are lost in the L514E mutant (Fig. 4E).
Figure 4. Structural analysis of BRAF mutations G593I and L514E.

(A) No major movements were required for vemurafenib to bind to the G593I (yellow) and L514E (orange) mutation in comparison to the wild type binding pose (blue). (B) BRAF G593 is located on the N-terminus of the activation loop and may facilitate conformational changes required to accommodate the vemurafenib propyl sulfonamide moiety in the back of the pocket. The backbone of the neighboring D594 residue interacts with the sulfonamide nitrogen of vemurafenib as indicated by black dashed lines. (C) Mutation of G593 to L not only restricts flexibility of the loop, but also puts the leucine side chain in too close proximity to the fluoro-substituted phenyl ring (highlighted with the dashed circle). (D) Residue L514 is involved in a variety of hydrophobic contacts with vemurafenib (indicated by yellow arrows), which are lost in the L514E mutant (E).
2.8. Complexity
There are a number of distinct steps in Resistor, each of which has its own complexity. While there are sublinear K* algorithms, such as BBK* (Ojewole et al., 2018) with MARK* (Jou et al., 2020), these algorithms so far have only been applied to positive and negative design with optimization of specific multiple objectives, such as minimizing/maximizing the bound (respectively unbound) state partition functions and their ratios for computing binding affinity or stability. Comets (Hallen and Donald, 2016) provably does multistate design optimizing arbitrary constrained linear combinations of GMEC energies, but Comets does not model the partition functions required for calculating binding affinity. A provable ensemble-based algorithm analogous to Comets for arbitrary multistate design optimization is yet to be developed. Thus, general multistate K* design remains, unfortunately, a problem linear in the number of sequences and thus exponential in the number of mutable residues.
Computing K* itself, as a ratio of partition functions built from the thermodynamic ensembles of the bound to unbound states, can be expensive (Valiant, 1979; Nisonoff, 2015; Viricel et al., 2016). In order to reduce the number of K* problems to solve, Comets is employed as a pruning mechanism for all sequences in which there are more than one mutation. Without Comets, Resistor would need to compute sN K* scores, where s is the number of states and N is the number of sequences. With Comets, Resistor is able to avoid computing many of these K* scores, as Comets has been shown in practice to reduce the number of required GMEC calculations by over 99% and to reduce N for continuous designs by 96%, yielding an overall speedup of over 5 × 105-fold (Hallen and Donald, 2016). Since in this study we considered only single residue mutations we omitted the Comets pruning step, but in any use of Resistor that considers multiple simultaneously mutable residues we believe Comets’ empirical sublinearity will make the difference between feasible and infeasible searches.
Moreover, by using an approximation containing fixed partition function size and sparse residue interaction graphs, we can use the BWM* algorithm (Jou et al., 2016) to compute the K* scores in time , where w is the branch-width and q the number of rotamers per residue. When we have w = O(1) this is polynomial time. In this study we found that the ε-approximation algorithms using adaptively-sized partition functions, such as BBK* with MARK*, were fast enough. However, for larger problems the sparse approximations allow us to approximate the necessary K* scores for resistance prediction in time exponential only in the branch-width, and thus polynomial time for fixed branch-widths.
3. Discussion
In this work, we report Resistor, a computational algorithm to systematically investigate protein mutations and identify those that have a high likelihood of lowering drug potency in comparison to native substrates. In addition, we analyze the probability that such a mutation is generated in cancer patients and thus likely of clinical importance. Our algorithm applies the power of Pareto optimization to resistance predictions, which provides an objective way of prioritizing the most relevant mutations for experimental testing. In addition, we used computationally predicted input structures of ligand-target complexes whenever experimental data was lacking. This broadens the targets on which Resistor can be used, as we have found that the availability of high-resolution experimental ligand-target structures still can present a major bottleneck in computational protein design.
We have applied Resistor to two case studies, EGFR and BRAF, in a retrospective manner and, in case of BRAF, also included prospective experimental data for validation. In EGFR and BRAF, the algorithm correctly identified resistance mutations. Using the vemurafenib data from Wagenaar et al. (2014), which is the most comprehensive dataset on BRAF mutations and vemurafenib resistance available, we determined RESISTOR’s vemurafenib resistance prediction specificity and sensitivity to be 91% and 31%, respectively. In a data-rich setting such as proteomics (e.g. Lilien et al. (2003)), the sensitivity could be regarded as low. However, the prediction of antineoplastic resistance mutations is a sparse data problem. Comprehensive datasets on drug resistance mutations on specific targets are virtually non-existent. We speculate that the reason for this can be found in the large number of individual mutants that must be generated and tested. For example, in our study we used Resistor to investigate 462, 438, and 357 individual mutants for erlotinib, gefitinib, and osimertinib, respectively. While this is computationally feasible, it far exceeds the testing capacities of most experimental groups. Clinical resistance data is even more limited. Furthermore, even for those mutations that have been confirmed to confer clinical resistance in patients, the underlying molecular mechanisms often remain uninvestigated.
Resistor prioritizes escape mutations causing ablation of inhibitor binding and/or tighter substrate binding (the latter as a proxy for KM). However, mutations affecting the drug target could also mediate resistance via other molecular processes, such as altering the stability of conformational states or affinity of protein-protein interactions (Lyczek et al., 2021; Assaraf et al., 2019). One limitation of this present study is that we modelled BRAF in its active conformational state. As Röck et al. (2019) showed, BRAF inhibitors exhibited differences in specificity and efficacy by shifting BRAF’s conformational probability distribution from an open and active to a closed, inactive state. It is plausible that mutations far from the active site could destabilize the closed, inactive state and shift the conformational probability distribution back towards the open, active state. Modeling of large allosteric destabilization of the inactive conformations has been discussed extensively in our previous work (Chen et al., 2009; Gorczynski et al., 2007), but its integration into Resistor is left for future work.
In addition, clinical resistance is caused by several different mechanisms, of which the relative importance of escape mutations can vary greatly. In some kinases, such as c-Abl, EGFR, and FLT3, active site escape mutations are the main cause of acquired resistance (Sierra et al., 2010). In other kinases, such as BRAF, escape mutations are not the main mechanism of acquired resistance (Rizos et al., 2014). Rather, splice variants, amplification, and mutations in related genes such as N-RAS, MEK1, MEK2, IGF-1R, and AKT comprise the majority of cases of clinical resistance (Rizos et al., 2014). From this perspective, the specificity of Resistor for BRAF and vemurafenib is remarkable and the sensitivity is in line with the fraction of resistance mutations whose aetiology is definitively escape via active site mutation.
We believe that the remaining gap can be closed in future work by modelling additional conformational flexibility, kinetics, and the protein-protein interactions of additional effectors. Yet, despite these limitations, Resistor is able to prioritize mutations that are demonstrated to confer resistance in patients. Specifically, our results show that detailed and combinatorial thermodynamic computations can form the basis for predicting escape mutations to TKIs. In the future, since some resistance mutations exploit kinetic phenomena, kinetics could be incorporated for a more comprehensive model.
4. Conclusions
Resistor contributes to the science of predicting resistance mutations by providing an algorithm to enumerate the entire Pareto frontier of multiple resistance-causing criteria. By categorizing predicted resistance mutations by their Pareto rank, it allows the drug discovery community to prioritize escape mutations on the Pareto frontier. Resistor also provides structural justification for the mechanism of each predicted escape mutation by generating an ensemble of predicted structural models upon mutation. In this study, we have applied Resistor to predict resistance mutations in EGFR and BRAF for a number of different therapeutics. We demonstrate that Resistor can also be applied to computationally generated input structures, although the accuracy of the results may be somewhat diminished compared to experimentally determined structures of target-ligand complexes. However, computationally-derived models can still provide useful insights, especially when considering that the availability of experimental structures appears as major bottleneck. While Resistor as described herein optimizes over 4 objectives, as a general method any number of diverse objectives could be added. Resistor can be applied not only to cancer therapeutics, but also to antimicrobial or antiviral drug design. It is our hope that that the drug discovery community can use Resistor to design drugs that are less prone to resistance.
STAR Methods:
Resource Availability:
Lead contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bruce Donald (brd+cellsys22@cs.duke.edu).
Materials availability:
Materials are available upon request to the Lead Contact.
Data and code availability:
OSPREY design specifications and mutational signature probabilities required to reproduce the predictions in this paper have been deposited at in the Harvard Dataverse and are publicly available as of the date of publication. DOIs are listed in the key resources table.
The version of OSPREY used in this paper has been deposited in the Harvard Dataverse and is publicly available as of the date of publication. DOIs are listed in the key resources table. For new empirical designs, we recommend using the latest version of OSPREY available for free at http://www.cs.duke.edu/donaldlab/osprey.php. All code for the OSPREY software package is also available on GitHub at https://github.com/donaldlab/OSPREY3, and is free and open-source.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE |
SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-BRAF | Santa Cruz | F-7: sc-5284 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Vemurafenib | MedChemExpress | HY-12057 |
| Encorafenib | MedChemExpress | HY-15605 |
| Dabrafenib | Selleckchem | S2807 |
| PLX8394 | MedChemExpress | HY-18972 |
| Benzyl-coelenterazine | Nanolight | 301 |
| Transfectin | BioRad | 1703352 |
| Deposited Data | ||
| K* designs, mutational signatures | This studyAlexandrov et al. 2013 |
https://doi.org/10.7910/DVN/DA0WWK
ftp://ftp.sanger.ac.uk/pub/cancer/AlexandrovEtA |
| Coding sequence for wt targets | COSMIC, Bamford et al. 2004 | http://cancer.sanger.ac.uk/cosmic |
| COSMIC mutation data, version 95 | COSMIC, Bamford et al. 2004 | http://cancer.sanger.ac.uk/cosmic |
| cBioPortal mutation data, version 4.0.3 | Cerami et al. 2012 | https://www.cbioportal.org/ |
| Structures of the target-ligand complexes | The Protein Data Bank This study |
http://www.rcsb.org/pdb/home/home.do
https://doi.org/10.7910/DVN/DA0WWK |
| OSPREY-predicted structures of the kinase:ligand complexes | This study | https://doi.org/10.7910/DVN/DA0WWK |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | N/A |
| Recombinant DNA | ||
| KinCon PCA reporters | This paper Roeck et al, 2019 | N/A |
| Software and Algorithms | ||
| OSPREY 3 | Hallen et al. 2018 | https://doi.org/10.7910/DVN/DA0WWK |
| Maestro Release 2021-1 | Schrödinger | https://www.schrodinger.com/products/maestro |
| AmberTools21 | Case et al. 2021 | https://ambermd.org/GetAmber.php#ambertools |
| GOLD version 5.8.0 | Jones et al. 1997 | https://www.ccdc.cam.ac.uk/solutions/csd-discovery/Components/Gold/ |
| MOE 2015.1001 | Chemical Computing Group | https://www.chemcomp.com/ |
Experimental Model and Subject Details:
Cell Culture and Antibodies:
HEK293T cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transient transfections were performed with Transfectin reagent (Bio-Rad, 1703352). Mouse anti-BRAF (Santa Cruz, F-7: sc-5284) antibody was used to determine biosensor expression levels.
Method Details:
Preparation of Empirical and Docked Structures for K* Predictions:
The crystal structures used for the EGFR predictions were adopted from Kaserer and Blagg (2018). A full description of the PDB entries used can be found in that paper’s section Table S7, and details on how the structures were prepared for OSPREY predictions is in that paper’s section Structure Selection and Preparation.
For BRAF, the crystal structures of vemurafenib (PDB id 3og7, Hodis et al. (2012)) and dabrafenib (PDB id 4xv2, Zhang et al. (2015)) in complex with BRAF V600E were selected as input for Resistor. Both structures have been prepared using the default setting of the Protein Preparation Wizard (Sastry et al., 2013) in Maestro (Schrödinger, LLC, New York, NY). In the case of encorafenib and PLX8394, crystal structures of structurally closely related, but not the identical, molecules were available. These experimental complexes were used to generate encorafenib and PLX8394 models. Encorafenib was docked into PDB id 4xv3 (Zhang et al., 2015) using the default settings of the induced fit docking procedure in Maestro (Farid et al., 2006; Sherman et al., 2006a,b; Schrödinger, LLC, New York, NY). For validation, the co-crystallized ligand PLX7922 was re-docked. The highest scored docking pose of encorafenib was selected for further investigation. We found that the conserved substructures in encorafenib and PLX7922 aligned very well in this docking pose.
For PLX8394, re-docking of the co-crystallized ligand PLX7904 (PDB id 4xv1, Zhang et al. (2015)) failed with the induced fit docking procedure, but was successful using a rigid docking workflow in GOLD version 5.8.0 (Jones et al., 1997). The binding site was defined as 6 Å around the ligand and the water molecule HOH905 was set to toggle and spin. The default settings of all other parameters were used.
An experimental structure of the endogenous ligand ADP was available, however, BRAF adopted in inactive conformation in this complex. Apo BRAF in its active conformation (PDB id 4mne, Haling et al. (2014)) was thus combined with ANP-bound protein kinase c-src (PDB id 2src, Xu et al. (1999)) to generate an active, endogenous ligand-bound BRAF complex. This model was used as template to build a BRAF:ADP homology model in the Molecular Operating Environment (Chemical Computing Group ULC) using the default settings. This included refinement steps to resolve potential steric clashes in the rather crude ANP-BRAF input template.
As we note in these preceding paragraphs, in each case the BRAF structure we modeled was in its active conformation. There are some mutations, such as V600E, that are activating mutations and shift BRAF’s conformational probability distribution to the active state (Röck et al., 2019; Mayrhofer et al., 2020). With use of Resistor for mutational scanning of single point mutations within the active site, we assumed that the mutation is either itself activating or is a secondary mutation following an activating mutation, such as V600E. In our discussion of Resistor predictions of the BRAF double mutants V600E/T529M and V600E/T529I in Section 2.7.1, our assumption was that the V600E mutation is the activating mutation (which the existing drugs are effective against) and T529M/I are the secondary, resistance-causing mutations.
For all complexes, water molecules not involved in mediating interactions between the ligand and the target were deleted and only residues with a 12 Å radius around the ligand were kept in the final input structures.
Evaluation of Ligand Affinity:
The command line interface of OSPREY was used to generate distinct YAML design files for each residue within 5 Å of a ligand. These YAML design files specify the input structures, the mutable residues, the flexible residues, and connectivity templates for OSPREY. To create the forcefield parameters files for the inhibitors and endogenous ligands, we used the Antechamber program in the AmberTools software package (Case et al., 2021). Then, to calculate the K* scores we used OSPREY with the following command input:
osprey affinity –design <YAML design file> –epsilon 0.63 –frcmod <force field modification file> –stability–threshold −1
where <YAML design file> was replaced with the individual YAML design file and <force field modification file> was replaced with the AmberTools-generated file. The YAML design and forcefield modification files used in this study are available in the Harvard Dataverse (see Key Resources Table).
Luciferase PCA analyses:
We transiently overexpressed indicated versions of the Rluc-PCA–based KinCon biosensors in 24-well plate formats. Experiments were performed 48h post transfection. For the luciferase-PCA measurements, the growth medium was carefully removed and the cells were washed with phosphate-buffered saline (PBS). Cell suspensions were transferred to 96-well plates and subjected to luminescence analysis using the PHERAstar FSX (BMG Labtech). Luciferase luminescence signals were integrated for 10 seconds following addition of the Rluc substrate benzyl-coelenterazine (NanoLight, #301). Cell lysates were prepared post RLU measurements. Expression levels of the biosensor were determined via western blot analysis.
The K* algorithm:
K* is an ε-accurate algorithm for computing a provable approximation to the affinity constant Ka. It is implemented in the OSPREY computational protein design software package (Lilien et al., 2005; Hallen et al., 2018). K* is defined as the quotient of the bound to unbound partition functions of a protein:ligand system for a given amino acid sequence. For a proof that K* approximates Ka see Appendix A of Lilien et al. (2005).
K* calculates an ε-accurate partition function for three structures: the bound protein:ligand complex (denoted PL), the unbound protein (denoted P), and the unbound ligand (denoted L). Let X be an arbitrary state, X ∈ {P, L, PL}. The partition function is a summation of the Boltzmann-weighted energies for all of the conformations in the thermodynamic ensemble of X. Let s denote an arbitrary amino acid sequence, then the partition function of s in state X (which we donate as qX(s)) is:
| (6) |
where Qx(s) is the entire conformational ensemble of sequence s in state X, and c is a single conformation from that ensemble. E(c) is the energy of conformation c. R is the ideal gas constant and T is the temperature in absolute Kelvin.
The K* score for a sequence s approximates Ka:
| (7) |
By using an A* search over Qx(s) to generate an ordered, gap-free list of low energy conformations, the K* algorithms generates an ε-approximation of the partition function qx(s) and the ensemble-complete K* value. This approximation is known as the K* score.
Inputs to the K* algorithm include 1) an input structure; 2) a conformation library; 3) an energy function; 4) ε, and; 5) flexibility and mutability choices.
Empirical Resistor runtimes:
The Resistor computation entails three stages: 1) computing the positive and negative K* designs; 2) assigning mutational signature probabilities to each mutation, and; 3) run Pareto optimization over the four axes. Steps 2 and 3 empirically take a negligible amount of time, on the order of seconds. Step 1, however, computes two partition functions for each sequence and can take more time. Figure 5 shows the empirical runtime (in seconds) that it took our computers to run the positive and negative K* designs, where a design mutated a residue to each of the 19 other possible amino acids.
Figure 5. Positive and negative design runtimes.

Box-and-whisker plot showing the minimum, maximum, median, first quartile, and third quartile runtimes per inhibitor:kinase pair. The whiskers extend to points that lie within 1.5x the interquartile range. Each dot represents the number of seconds that Resistor took to compute the positive and negative K* designs for a given mutation location in a kinase:inhibitor complex. In other words, each dot represents the computation of 40 K* scores. The computation times across all the inhibitors range from 813 seconds to 972465 seconds, with the average being 40630 seconds or 1015 seconds per sequence. The designs were run on a 24-core, 48-thread Intel Xeon processor with 4 Nvidia Titan V GPUs.
Quantification and Statistical Analysis:
In Fig. 3, the student’s T-test was used to evaluate whether the mean of the RLU of a mutant was significantly different from that of the relative DMSO control. The SEM was used with n = 4. Significance was defined to three different p-levels, where *p < 0.05, **p < 0.01, and ***p < 0.001.
To compute the specificity and sensitivity values reported in Section 3, we used the dataset in supplementary table S1 from Wagenaar et al. (2014). We then reduced this set to those mutants for which Resistor made a prediction (Resistor made predictions for sequences with a mutated amino acid within 5 Å of the inhibitor or endogenous ligand). If Resistor predicted that a mutation caused resistance and Wagenaar et al. indicated that the mutant increased normalized drug enrichment, then that was considered a true positive. If Resistor predicted that a mutation was benign and Wagenaar et al. did not find increased drug encrichment, then that was considered a true negative. The specificity and sensitivity values were computed using their standard formulas.
Supplementary Material
Data S1
The complete set of Resistor-utilized input data for Pareto ranking the EGFR:erlotinib designs.
Data S2
The complete set of Resistor-utilized input data for Pareto ranking the EGFR:gefitinib designs.
Data S3
The complete set of Resistor-utilized input data for Pareto ranking the EGFR:osimertinib designs.
Data S4
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:dabrafenib designs.
Data S5
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:vemurafenib designs.
Data S6
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:encorafenib designs.
Data S7
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:PLX8394 designs.
Box 1: Progress and Potential.
Targeted cancer drugs developed over the past two decades have been instrumental in treating certain types of cancer and extending patient lifespans. These drugs include kinase inhibitors targeting EGFR and BRAF, two important enzymes of the mitogen-activated protein kinase pathway whose dysregulation can lead to many types of cancer, including melanoma and non-small cell lung cancer. The inhibitors are effective for a period of time but the tumors often develop resistance to the drugs, leading once again to cancer progression. The ability to predict how an enzyme target can develop drug resistance would allow for a proactive, resistance-aware approach to drug design. Here we introduce Resistor, an algorithm that uses structure-based computational design to predict how different mutations in an enzyme will affect a drug’s efficacy. It pairs these predictions with empirical data on how likely a mutation is to occur in a given cancer type, which allows researchers to identify “mutational hotspots,” or particular places where mutations are most likely to cause drug resistance. These predictions provide designers new insights during the drug development process that should allow for the quicker development of more durable and longer-lasting cancer therapeutics.
Acknowledgements
BRD & NG: We thank all members of the Donald lab for helpful discussions and the NIH (grants R01-GM078031, R01-GM118543, and R35-GM144042 to BRD) for funding. TK, ES, & AF were funded in whole, or in part, by the Austrian Science Fund (FWF) P34376, P27606, P30441, P32960, and P35159. For the purpose of open access, the author has applied a CC-BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
BRD is a founder of Ten63 Therapeutics, Inc. ES is a founder of KinCon Biolabs. KinCon reporters are subjects of pending patent applications.
In the future, c0 could be learned from running Resistor on a resistance mutation dataset for homologous systems and examining the K* scores.
References
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. , 2020. The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. , 2013. Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman MD, Ali A, Kumar Reddy GK, Nalam MN, Anjum SG, Cao H, Chellappan S, Kairys V, Fernandes MX, Gilson MK, Schiffer CA, Rana TM, Tidor B, 2008. Hiv-1 protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. Journal of the American Chemical Society 130, 6099–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arulananda S, Do H, Musafer A, Mitchell P, Dobrovic A, John T, 2017. Combination osimertinib and gefitinib in c797s and t790m egfr-mutated non–small cell lung cancer. Journal of Thoracic Oncology 12, 1728–1732. [DOI] [PubMed] [Google Scholar]
- Assaraf YG, Brozovic A, Goncalves AC, Jurkovicova D, Linē A, Machuqueiro M, Saponara S, Sarmento-Ribeiro AB, Xavier CP, Vasconcelos MH, 2019. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resistance Updates 46, 100645. [DOI] [PubMed] [Google Scholar]
- Avizienyte E, Ward RA, Garner AP, 2008. Comparison of the egfr resistance mutation profiles generated by egfr-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochemical Journal 415, 197–206. [DOI] [PubMed] [Google Scholar]
- Ballantyne AD, Garnock-Jones KP, 2013. Dabrafenib: first global approval. Drugs 73, 1367–1376. [DOI] [PubMed] [Google Scholar]
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P, 2012. Vemurafenib: the first drug approved for braf-mutant cancer. Nature reviews Drug discovery 11, 873–886. [DOI] [PubMed] [Google Scholar]
- Case DA, Belfon K, Ben-Shalom I, Brozell SR, Cerutti D, Cheatham T, Cruzeiro VWD, Darden T, Duke RE, Giambasu G, et al. , 2021. Amber 2021 . [Google Scholar]
- Centers for Disease Control and Prevention, 2020. Antibiotic / antimicrobial resistance. URL: https://www.cdc.gov/drugresistance/index.html.
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. , 2012. The cbio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemical Computing Group ULC, . Molecular operating environment (moe).
- Chen CY, Georgiev I, Anderson AC, Donald BR, 2009. Computational structure-based redesign of enzyme activity. Proceedings of the National Academy of Sciences 106, 3764–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Zhou F, Shen W, Jiang T, Wu X, Tong X, Shao YW, Qin S, Zhou C, 2017. Novel mutations on egfr leu792 potentially correlate to acquired resistance to osimertinib in advanced nsclc. Journal of Thoracic Oncology 12, e65–e68. [DOI] [PubMed] [Google Scholar]
- Choi Y, Griswold KE, Bailey-Kellogg C, 2013. Structure-based redesign of proteins for minimal t-cell epitope content. Journal of computational chemistry 34, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Ndong C, Griswold KE, Bailey-Kellogg C, 2016. Computationally driven antibody engineering enables simultaneous humanization and thermostabilization. Protein Engineering, Design and Selection 29, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. , 2002. Mutations of the braf gene in human cancer. Nature 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Dowell J, Minna JD, Kirkpatrick P, 2005. Erlotinib hydrochloride. Nature Reviews Drug Discovery 4. [DOI] [PubMed] [Google Scholar]
- Enzler F, Tschaikner P, Schneider R, Stefan E, 2020. Kincon: cell-based recording of full-length kinase conformations. IUBMB life 72, 1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairclough SR, Kiedrowski LA, Lin JJ, Zelichov O, Tarcic G, Stinchcombe TE, Odegaard JI, Lanman RB, Shaw AT, Nagy RJ, 2019. Identification of osimertinib-resistant egfr l792 mutations by cfdna sequencing: oncogenic activity assessment and prevalence in large cfdna cohort. Experimental hematology & oncology 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid R, Day T, Friesner RA, Pearlstein RA, 2006. New insights about herg blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorganic & medicinal chemistry 14, 3160–3173. [DOI] [PubMed] [Google Scholar]
- Frey KM, Georgiev I, Donald BR, Anderson AC, 2010. Predicting resistance mutations using protein design algorithms. Proceedings of the National Academy of Sciences 107, 13707–13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainza P, Nisonoff HM, Donald BR, 2016. Algorithms for protein design. Current opinion in structural biology 39, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainza P, Roberts KE, Donald BR, 2012. Protein design using continuous rotamers. PLoS computational biology 8, e1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. , 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Science signaling 6, pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev I, Lilien RH, Donald BR, 2008. The minimized dead-end elimination criterion and its application to protein redesign in a hybrid scoring and search algorithm for computing partition functions over molecular ensembles. Journal of computational chemistry 29, 1527–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczynski MJ, Grembecka J, Zhou Y, Kong Y, Roudaia L, Douvas MG, Newman M, Bielnicka I, Baber G, Corpora T, et al. , 2007. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins runx1 and cbfβ. Chemistry & biology 14, 1186–1197. [DOI] [PubMed] [Google Scholar]
- Griswold KE, Bailey-Kellogg C, 2016. Design and engineering of deimmunized biotherapeutics. Current opinion in structural biology 39, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Jordan MR, Sultan BJ, Hill A, Davis DH, Gregson J, Sawyer AW, Hamers RL, Ndembi N, Pillay D, et al. , 2012. Global trends in antiretroviral resistance in treatment-naive individuals with hiv after rollout of antiretroviral treatment in resource-limited settings: a global collaborative study and meta-regression analysis. The Lancet 380, 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, et al. , 2014. Structure of the braf-mek complex reveals a kinase activity independent role for braf in mapk signaling. Cancer cell 26, 402–413. [DOI] [PubMed] [Google Scholar]
- Hallen MA, Donald BR, 2016. Comets (constrained optimization of multistate energies by tree search): A provable and efficient protein design algorithm to optimize binding affinity and specificity with respect to sequence. Journal of Computational Biology 23, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen MA, Martin JW, Ojewole A, Jou JD, Lowegard AU, Frenkel MS, Gainza P, Nisonoff HM, Mukund A, Wang S, Holt GT, Zhou D, Dowd E, Donald BR, 2018. Osprey 3.0: Open-source protein redesign for you, with powerful new features. Journal of computational chemistry 39, 2494–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PT, Vyse S, Huang PH, 2020. Rare epidermal growth factor receptor (egfr) mutations in non-small cell lung cancer, in: Seminars in cancer biology, Elsevier. pp. 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Friedman AM, Bailey-Kellogg C, 2012. A divide-and-conquer approach to determine the pareto frontier for optimization of protein engineering experiments. Proteins: Structure, Function, and Bioinformatics 80, 790–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helena AY, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ, 2013. Analysis of tumor specimens at the time of acquired resistance to egfr-tki therapy in 155 patients with egfr-mutant lung cancers. Clinical cancer research 19, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Fukuoka M, Baselga J, 2004. Gefitinib—a novel targeted approach to treating cancer. Nature Reviews Cancer 4, 956–965. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. , 2012. A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S, 2014. Drug resistance in cancer: an overview. Cancers 6, 1769–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, Sherman E, Parikh A, Feun L, Tsai F, Allen E, Zhang C, Severson P, Inokuchi K, Walling J, et al. , 2020. Interim results from a phase 1/2 precision medicine study of plx8394-a next generation braf inhibitor. European Journal of Cancer 138, S2–S3. [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R, 1997. Development and validation of a genetic algorithm for flexible docking. Journal of molecular biology 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Jou JD, Holt GT, Lowegard AU, Donald BR, 2020. Minimization-aware recursive k*: A novel, provable algorithm that accelerates ensemble-based protein design and provably approximates the energy landscape. Journal of Computational Biology 27, 550–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou JD, Jain S, Georgiev IS, Donald BR, 2016. Bwm*: A novel, provable, ensemble-based dynamic programming algorithm for sparse approximations of computational protein design. Journal of Computational Biology 23, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaserer T, Blagg J, 2018. Combining mutational signatures, clonal fitness, and drug affinity to define drug-specific resistance mutations in cancer. Cell chemical biology 25, 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, De Béthune MP, Schiffer CA, 2004. Structural and thermodynamic basis for the binding of tmc114, a next-generation human immunodeficiency virus type 1 protease inhibitor. Journal of virology 78, 12012–12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Yang D, Cui S, Pan E, Yang P, Dai Z, 2021. Ngs-based ctdna profiling after the resistance of second-line osimertinib for patient with egfr-mutated pulmonary adenocarcinoma. OncoTargets and therapy 14, 4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilien RH, Farid H, Donald BR, 2003. Probabilistic disease classification of expression-dependent proteomic data from mass spectrometry of human serum. Journal of computational biology 10, 925–946. [DOI] [PubMed] [Google Scholar]
- Lilien RH, Stevens BW, Anderson AC, Donald BR, 2005. A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. Journal of Computational Biology 12, 740–761. [DOI] [PubMed] [Google Scholar]
- Lyczek A, Berger BT, Rangwala AM, Paung Y, Tom J, Philipose H, Guo J, Albanese SK, Robers MB, Knapp S, et al. , 2021. Mutation in abl kinase with altered drug-binding kinetics indicates a novel mechanism of imatinib resistance. Proceedings of the National Academy of Sciences 118, e2111451118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. , 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. New England Journal of Medicine 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Mayrhofer JE, Enzler F, Feichtner A, Röck R, Fleischmann J, Raffeiner A, Tschaikner P, Ogris E, Huber RG, Hartl M, et al. , 2020. Mutation-oriented profiling of autoinhibitory kinase conformations predicts raf inhibitor efficacies. Proceedings of the National Academy of Sciences 117, 31105–31113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisonoff H, 2015. Efficient Partition Function Estimation in Computational Protein Design: Probabalistic Guarantees and Characterization of a Novel Algorithm. Bachelor’s thesis. Duke University. [Google Scholar]
- Ojewole AA, Jou JD, Fowler VG, Donald BR, 2018. Bbk*(branch and bound over k*): A provable and efficient ensemble-based protein design algorithm to optimize stability and binding affinity over large sequence spaces. Journal of Computational Biology 25, 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SHI, Cui J, Schrock AB, Goldberg ME, Zhu VW, Albacker L, Stephens PJ, Miller VA, Ali SM, 2017. Emergence of novel and dominant acquired egfr solvent-front mutations at gly796 (g796s/r) together with c797s/g and l792f/h mutations in one egfr (l858r/t790m) nsclc patient who progressed on osimertinib. Lung Cancer 108, 228–231. [DOI] [PubMed] [Google Scholar]
- Parker AS, Choi Y, Griswold KE, Bailey-Kellogg C, 2013. Structure-guided deimmunization of therapeutic proteins. Journal of Computational Biology 20, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M, Nalivaika E, Schiffer CA, 2002. Substrate shape determines specificity of recognition for hiv-1 protease: analysis of crystal structures of six substrate complexes. Structure 10, 369–381. [DOI] [PubMed] [Google Scholar]
- Qi Y, Martin JW, Barb AW, Thélot F, Yan AK, Donald BR, Oas TG, 2018. Continuous interdomain orienta-tion distributions reveal components of binding thermodynamics. Journal of molecular biology 430, 3412–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SM, Gainza P, Frey KM, Georgiev I, Donald BR, Anderson AC, 2015. Protein design algorithms predict viable resistance to an experimental antifolate. Proceedings of the National Academy of Sciences 112, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SM, Scocchera EW, Narendran G, Keshipeddy S, Krucinska J, Hajian B, Ferreira J, Nailor M, Aeschlimann J, Wright DL, Anderson AC, 2016. Mrsa isolates from united states hospitals carry dfrg and dfrk resistance genes and succumb to propargyl-linked antifolates. Cell chemical biology 23, 1458–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. , 2014. Braf inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clinical cancer research 20, 1965–1977. [DOI] [PubMed] [Google Scholar]
- Röck R, Mayrhofer JE, Torres-Quesada O, Enzler F, Raffeiner A, Raffeiner P, Feichtner A, Huber RG, Koide S, Taylor SS, et al. , 2019. Braf inhibitors promote intermediate braf (v600e) conformations and binary interactions with activated ras. Science advances 5, eaav8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvat RS, Parker AS, Choi Y, Bailey-Kellogg C, Griswold KE, 2015. Mapping the pareto optimal design space for a functionally deimmunized biotherapeutic candidate. PLoS Comput Biol 11, e1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvat RS, Verma D, Parker AS, Kirsch JR, Brooks SA, Bailey-Kellogg C, Griswold KE, 2017. Computationally optimized deimmunization libraries yield highly mutated enzymes with low immunogenicity and enhanced activity. Proceedings of the National Academy of Sciences 114, E5085–E5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W, 2013. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Schrödinger, LLC, New York, NY, . Schrödinger release 2020-3: Maestro. [Google Scholar]
- Sen B, Peng S, Tang X, Erickson HS, Galindo H, Mazumdar T, Stewart DJ, Wistuba I, Johnson FM, 2012. Kinase-impaired braf mutations in lung cancer confer sensitivity to dasatinib. Science translational medicine 4, 136ra70–136ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Altman MD, Ali A, Nalam MN, Cao H, Rana TM, Schiffer CA, Tidor B, 2013. Testing the substrate-envelope hypothesis with designed pairs of compounds. ACS chemical biology 8, 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W, Beard HS, Farid R, 2006a. Use of an induced fit receptor structure in virtual screening. Chemical biology & drug design 67, 83–84. [DOI] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R, 2006b. Novel procedure for modeling ligand/receptor induced fit effects. Journal of medicinal chemistry 49, 534–553. [DOI] [PubMed] [Google Scholar]
- Shirley M, 2018. Encorafenib and binimetinib: first global approvals. Drugs 78, 1277–1284. [DOI] [PubMed] [Google Scholar]
- Sierra JR, Cepero V, Giordano S, 2010. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Molecular cancer 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, et al. , 2018. Osimertinib in untreated egfr-mutated advanced non–small-cell lung cancer. New England journal of medicine 378, 113–125. [DOI] [PubMed] [Google Scholar]
- Stamos J, Sliwkowski MX, Eigenbrot C, 2002. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry 277, 46265–46272. [DOI] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. , 2019. Cosmic: the catalogue of somatic mutations in cancer. Nucleic acids research 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. , 2015. Acquired egfr c797s mutation mediates resistance to azd9291 in non–small cell lung cancer harboring egfr t790m. Nature medicine 21, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiant LG, 1979. The complexity of computing the permanent. Theoretical computer science 8, 189–201. [Google Scholar]
- Vasan N, Baselga J, Hyman DM, 2019. A view on drug resistance in cancer. Nature 575, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viricel C, Simoncini D, Barbe S, Schiex T, 2016. Guaranteed weighted counting for affinity computation: Beyond determinism and structure, in: International Conference on Principles and Practice of Constraint Programming, Springer. pp. 733–750. [Google Scholar]
- Wagenaar TR, Ma L, Roscoe B, Park SM, Bolon DN, Green MR, 2014. Resistance to vemurafenib resulting from a novel mutation in the brafv 600 e kinase domain. Pigment cell & melanoma research 27, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC, 1999. Crystal structures of c-src reveal features of its autoinhibitory mechanism. Molecular cell 3, 629–638. [DOI] [PubMed] [Google Scholar]
- Yan XE, Zhu SJ, Liang L, Zhao P, Choi HG, Yun CH, 2017. Structural basis of mutant-selectivity and drug-resistance related to co-1686. Oncotarget 8, 53508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, Bao H, Tong X, Wang X, Shao YW, et al. , 2018. Investigating novel resistance mechanisms to third-generation egfr tyrosine kinase inhibitor osimertinib in non–small cell lung cancer patients. Clinical Cancer Research 24, 3097–3107. [DOI] [PubMed] [Google Scholar]
- Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, McKeage MJ, Squire CJ, 2015. Binding mode of the breakthrough inhibitor azd9291 to epidermal growth factor receptor revealed. Journal of structural biology 192, 539–544. [DOI] [PubMed] [Google Scholar]
- Yosaatmadja Y, Squire C, McKeage C, Flanagan M, 2014. 1.85 angstrom structure of egfr kinase domain with gefitinib. To Be Published . [Google Scholar]
- Yoshikawa S, Kukimoto-Niino M, Parker L, Handa N, Terada T, Fujimoto T, Terazawa Y, Wakiyama M, Sato M, Sano S, et al. , 2013. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 32, 27–38. [DOI] [PubMed] [Google Scholar]
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ, 2007. Structures of lung cancer-derived egfr mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer cell 11, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ, 2008. The t790m mutation in egfr kinase causes drug resistance by increasing the affinity for atp. Proceedings of the National Academy of Sciences 105, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahreddine H, Borden K, 2013. Mechanisms and insights into drug resistance in cancer. Frontiers in pharmacology 4, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, et al. , 2015. Raf inhibitors that evade paradoxical mapk pathway activation. Nature 526, 583–586. [DOI] [PubMed] [Google Scholar]
- Zheng D, Hu M, Bai Y, Zhu X, Lu X, Wu C, Wang J, Liu L, Wang Z, Ni J, et al. , 2017. Egfr g796d mutation mediates resistance to osimertinib. Oncotarget 8, 49671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Tseng LH, Chen G, Haley L, Illei P, Gocke CD, Eshleman JR, Lin MT, 2015. Clinical detection and categorization of uncommon and concomitant mutations involving braf. BMC cancer 15, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
The complete set of Resistor-utilized input data for Pareto ranking the EGFR:erlotinib designs.
Data S2
The complete set of Resistor-utilized input data for Pareto ranking the EGFR:gefitinib designs.
Data S3
The complete set of Resistor-utilized input data for Pareto ranking the EGFR:osimertinib designs.
Data S4
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:dabrafenib designs.
Data S5
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:vemurafenib designs.
Data S6
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:encorafenib designs.
Data S7
The complete set of Resistor-utilized input data for Pareto ranking the BRAF:PLX8394 designs.
Data Availability Statement
OSPREY design specifications and mutational signature probabilities required to reproduce the predictions in this paper have been deposited at in the Harvard Dataverse and are publicly available as of the date of publication. DOIs are listed in the key resources table.
The version of OSPREY used in this paper has been deposited in the Harvard Dataverse and is publicly available as of the date of publication. DOIs are listed in the key resources table. For new empirical designs, we recommend using the latest version of OSPREY available for free at http://www.cs.duke.edu/donaldlab/osprey.php. All code for the OSPREY software package is also available on GitHub at https://github.com/donaldlab/OSPREY3, and is free and open-source.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE |
SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-BRAF | Santa Cruz | F-7: sc-5284 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Vemurafenib | MedChemExpress | HY-12057 |
| Encorafenib | MedChemExpress | HY-15605 |
| Dabrafenib | Selleckchem | S2807 |
| PLX8394 | MedChemExpress | HY-18972 |
| Benzyl-coelenterazine | Nanolight | 301 |
| Transfectin | BioRad | 1703352 |
| Deposited Data | ||
| K* designs, mutational signatures | This studyAlexandrov et al. 2013 |
https://doi.org/10.7910/DVN/DA0WWK
ftp://ftp.sanger.ac.uk/pub/cancer/AlexandrovEtA |
| Coding sequence for wt targets | COSMIC, Bamford et al. 2004 | http://cancer.sanger.ac.uk/cosmic |
| COSMIC mutation data, version 95 | COSMIC, Bamford et al. 2004 | http://cancer.sanger.ac.uk/cosmic |
| cBioPortal mutation data, version 4.0.3 | Cerami et al. 2012 | https://www.cbioportal.org/ |
| Structures of the target-ligand complexes | The Protein Data Bank This study |
http://www.rcsb.org/pdb/home/home.do
https://doi.org/10.7910/DVN/DA0WWK |
| OSPREY-predicted structures of the kinase:ligand complexes | This study | https://doi.org/10.7910/DVN/DA0WWK |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | N/A |
| Recombinant DNA | ||
| KinCon PCA reporters | This paper Roeck et al, 2019 | N/A |
| Software and Algorithms | ||
| OSPREY 3 | Hallen et al. 2018 | https://doi.org/10.7910/DVN/DA0WWK |
| Maestro Release 2021-1 | Schrödinger | https://www.schrodinger.com/products/maestro |
| AmberTools21 | Case et al. 2021 | https://ambermd.org/GetAmber.php#ambertools |
| GOLD version 5.8.0 | Jones et al. 1997 | https://www.ccdc.cam.ac.uk/solutions/csd-discovery/Components/Gold/ |
| MOE 2015.1001 | Chemical Computing Group | https://www.chemcomp.com/ |