

Abstract
Deciphering the relationship between human proteins (genes) and phenotypes is one of the fundamental tasks in phenomics research. The Human Phenotype Ontology (HPO) builds upon a standardized logical vocabulary to describe the abnormal phenotypes encountered in human diseases and paves the way towards the computational analysis of their genetic causes. To date, many computational methods have been proposed to predict the HPO annotations of proteins. In this paper, we conduct a comprehensive review of the existing approaches to predicting HPO annotations of novel proteins, identifying missing HPO annotations, and prioritizing candidate proteins with respect to a certain HPO term. For each topic, we first give the formalized description of the problem, and then systematically revisit the published literatures highlighting their advantages and disadvantages, followed by the discussion on the challenges and promising future directions. In addition, we point out several potential topics to be worthy of exploration including the selection of negative HPO annotations and detecting HPO misannotations. We believe that this review will provide insight to the researchers in the field of computational phenotype analyses in terms of comprehending and developing novel prediction algorithms.
Keywords: Human Phenotype Ontology (HPO), Human protein-phenotype association, HPO annotation, Machine learning, Deep learning
Introduction
In the context of clinical medicine, a phenotype refers to any observable trait or characteristic of a disease, such as morphology, development, biochemical or physiological properties, or behavior (Scheuermann et al. 2009; Robinson 2012). The analysis of phenotype plays a fundamental role in biomedical research and deepens the understanding of the phenotypic spectrum of human disorders (Deans et al. 2015; Yu and Zhang 2015). The use of phenotype information can help empower the discovery of phenotypic mutations and enrich the understanding of disease pathogenesis (Son et al. 2018). To promote the computational analysis of phenotypic data, Robinson et al. (Robinson et al. 2008) developed Human Phenotype Ontology (HPO) to provide a standardized controlled vocabulary describing the phenotypic abnormalities and clinical features related to human diseases. The combination of features is often used for clinical diagnosis in genetics research. With the advent of precision medicine, it is essential to determine the exact genetic causes for a patient and provide individualized diagnosis with a definite molecular profile (Yu and Zhang 2015; Akhmetov and Bubnov 2015). Therefore, deciphering the associations between human proteins and phenotypes can be of great help in the prevention, diagnosis, and therapy of rare diseases (Groza et al. 2015).
It has been estimated that the genetic cause of only about half of the currently named rare diseases (about 7000) has been elucidated, and many more genetic disorders have yet to be recognized (Boycott et al. 2013; Chong et al. 2015). The genetics community believes that considerable quantities of additional Mendelian disease genes remain to be elucidated. So far, only about 4000 human proteins have been annotated with HPO terms. Nonetheless, manual curation is a time-, labor-, and money-consuming process. Hence, developing an accurate and efficient tool to annotate novel human proteins with HPO terms computationally is imperative. The second Critical Assessment of protein Function Annotation algorithms (CAFA2) challenge launched a special track on automatic HPO annotation prediction. The assessment results show the undesirable performance of participants: no methods could significantly outperform the Naive method, a baseline that simply uses the frequency of HPO terms in the HPO annotations as the prediction scores, in the protein-centric mode (Jiang et al. 2016), which highlighted the difficulty of this problem.
Due to the limitation of biomedical technique and the biased research interest of biologists, the current HPO annotations are in fact incomplete. From 2018 to 2020, the average number of annotations per protein has increased by 25%. The missing protein-phenotype associations have a side effect on computational tools (Liu et al. 2020) and mislead the phenotypic analysis of genetic diseases. Identifying missing HPO annotations is needed to ensure the data quality and improve the accuracy of prediction algorithms.
Typically, molecular biologists and physicians are more interested in knowing the set of proteins associated with a certain HPO term. The recent explosion in high-throughput experimental techniques has significantly improved the efficiency in genome-wide association studies (GWAS) (Bush and Moore 2012), which highly promotes the identification of disease-associated mutations (Bromberg 2013). But unfortunately, despite the limited knowledge of phenotype-causative variants, there still exists the sheer quantity of yet experimentally unverified proteins. Computational methods for recognizing probable phenotype-related proteins among the large pool of candidate proteins are therefore necessary to decrease manpower of in vitro verification.
The above tasks can be categorized according to the completeness of annotations: (1) Determining the HPO annotations of no-knowledge proteins, which currently have no experimentally-verified annotations. We can further distinguish based on the optimization target: (a) protein-centric, produces all HPO terms associated with a given protein; (b) term-centric, prioritizes the candidate proteins concerning a certain HPO term. (2) Identifying missing HPO annotations of limited-knowledge proteins, which have been experimentally annotated with some HPO terms but incompletely.
Although many computational methods have been proposed in this area, there are no systematic literature reviews till now. In this paper, we systematically summarized existing works about the above three tasks and attempt to help readers have acquaintance with the current developments and forthcoming applications in this field. The paper is organized as follows. In “Human Phenotype Ontology”, we briefly introduce the basic concepts and characteristics of HPO and HPO annotations. Then, we review the computational methods for predicting HPO annotations of novel proteins (“Methods for Predicting HPO Annotations of Novel Protein”), identifying missing HPO annotations (“Methods for Identifying Missing HPO Annotations”), and phenotype protein prioritization (“Methods for Phenotype Protein Prioritization”). In each section, we first provide the problem formulation, followed by the systematic overview of the existing methods highlighting their advantages and disadvantages, and discuss about promising directions for further improvement. “Potential Topics” outlines the potential topics and future guidelines. Finally, the last section concludes this paper.
Human Phenotype Ontology
Ontology is a technical term denoting a structural and abstract representation of domain knowledge (Smith 2003). An ontology provides a definitive and exhaustive classification of entities. Each entity corresponds to one or more terms in the ontology, and there is a specified semantic relationship between the terms. Therefore, ontology is usually utilized as a standard controlled vocabulary in biomedical research.
Recent years have witnessed an overwhelming number of ontologies in biomedical sciences, such as Gene Ontology (GO) (Ashburner et al. 2000), Mammalian Phenotype Ontology (MPO) (Smith et al. 2005), and Disease Ontology (DO) (Schriml et al. 2012). Inspired by the upsurge of research interest in bio-ontologies and great success achieved by GO, in 2008, Robinson et al. (Robinson et al. 2008) developed Human Phenotype Ontology (HPO), aiming at providing a standard categorization of the abnormal phenotypes encountered in human diseases and of their semantic relationships. Figure 1 presents the partial structure of the current HPO (by October 2020). At present, the HPO contains 15,371 terms organized hierarchically in a Directed Acyclic Graph (DAG), that is, a term can have multiple child nodes and multiple parent nodes. Each HPO term denotes the symptoms or phenotypic abnormalities that characterize a disease. The directed edge between two terms represents the “is-a” (subclass-of) relationship, which implies that the child node is a specialization of its ancestor(s). As a result, the HPO term at the upper level is more general and refers to a broad category of abnormal phenotype, while the HPO term at the bottom level is more detailed, specifically referring to a certain phenotype. Accordingly, the “true-path-rule” can be derived that: whenever a protein is annotated with a given term, then the annotation is propagated to all the ancestors in a recursive way.
Fig. 1.

A fraction of the HPO released by October 2020, where each oval denotes an internal term and each rectangle denotes a leaf term
At present, the HPO consists of seven sub-ontologies under the root term All (HP:0000001) as shown in Fig. 1: Phenotypic Abnormality (HP:0000118), Clinical Modifier (HP:0012823), Clinical Course (HP:0031797), Mode of Inheritance (HP:0000005), Past medical history (HP:0032443), Blood group (HP:0032223), and Frequency (HP:0040279). The description of sub-ontologies is summarized in Table 1. Phenotypic Abnormality (PA) is the largest and core sub-ontology in HPO.
Table 1.
Summary of sub-ontologies in HPO
| Sub-ontology | HPO term ID | #Terms | Description |
|---|---|---|---|
| Phenotypic abnormality | HP:0000118 | 15,150 | Main sub-ontology of the HPO and contain descriptions of clinical abnormalities |
| Clinical modifier | HP:0012823 | 107 | Characterize and specify the phenotypic abnormalities defined in the Phenotypic abnormality sub-ontology, with respect to severity, laterality, age of onset, and other aspects |
| Clinical course | HP:0031797 | 49 | Describe the course a disease typically takes from its onset, progression in time, and eventual resolution or death of the affected individual |
| Mode of inheritance | HP:0000005 | 32 | Describe the pattern in which a particular genetic trait or disorder is passed from one generation to the next |
| Past medical history | HP:0032443 | 16 | Record information about the patient’s medical, personal and family history that might be relevant to the presenting illness or to provide optimal clinical management |
| Blood group | HP:0032223 | 9 | Describe the blood group systems |
| Frequency | HP:0040279 | 7 | Frequency of phenotypic abnormalities within a patient cohort |
The HPO annotations have the following characteristics:
The HPO annotation rigidly follows the hierarchical structure. The HPO annotations of a protein are arranged as a consistent subgraph of HPO, which means that if a vertex is in the subgraph, all the ancestors of this vertex must also be in the subgraph. Thus, the loss-of-function of a protein may lead to multiple abnormal phenotypes simultaneously, and their corresponding HPO terms are not mutually independent but organized hierarchically.
The distribution of HPO annotations presents a serious imbalance. As shown in Fig. 2, the distribution of HPO annotations is unbalanced drastically, which follows a scale-free-like distribution. More than 35.6% of HPO terms are annotated with no more than three proteins, and only 43.8% of HPO terms are associated with over ten proteins.
Current HPO annotations are incomplete. Limited by the development of biomedical technologies, it is difficult to fully understand the genetic causes of human hereditary disorders and observe all the abnormal phenotypes in the disease progress. Therefore, the current HPO annotations are still incomplete. We selected 3717 proteins in the HPO annotations released on 2018-03-09 and tracked the changes in their average number of annotations (that is, how many HPO terms each protein is annotated after applying the true-path-rule) in subsequent releases. As shown in Fig. 3, the average number has increased rapidly from 114.8 to 144.2 in 2 years with an increase of more than 25%.
Fig. 2.
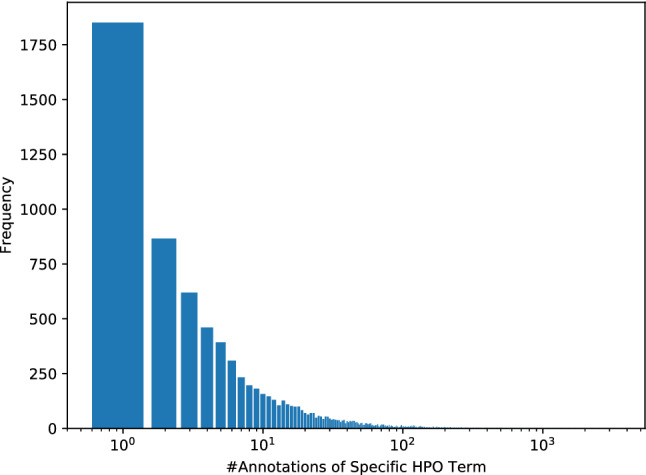
The number of annotated proteins for each HPO term follows a scale-free-like distribution. The x-axis is given on a logarithmic scale
Fig. 3.
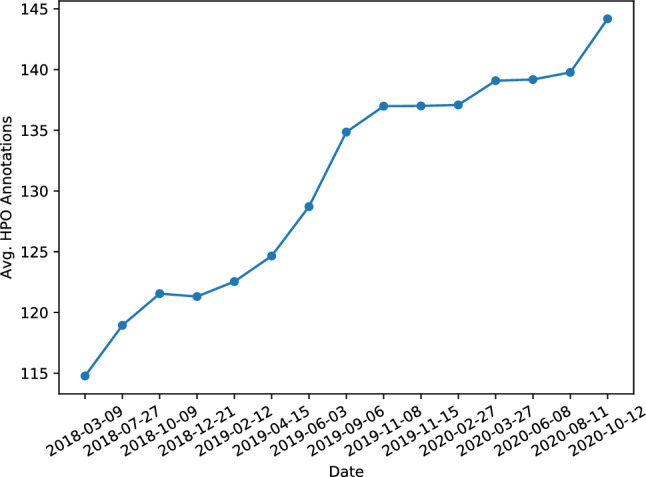
For proteins that already existed in the HPO annotations released on 2018-03-09, the average number of annotations per protein increased over time
Methods for Predicting HPO Annotations of Novel Protein
Problem Formulation
Let be a labeled dataset, where m is the size of the dataset, denotes the i-th protein, and denotes the experimentally-validated annotations of protein from HPO . Here, “” is used to describe consistent subgraph of HPO. We can formulate the HPO annotations prediction of novel protein as follows: given a labeled dataset , and a novel protein p without any known annotation, find a consistent subgraph , such that is closest to the experimental annotation T of p, or more formally, to seek
| 1 |
where is the posterior probability of consistent subgraph given protein p. It is illustrated in Fig. 4. Due to the hierarchical structure, this problem can be modeled as a hierarchical multi-label classification (HMC) problem.
Fig. 4.
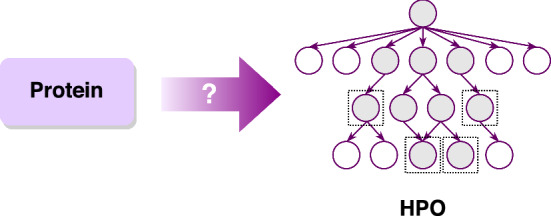
Illustration of predicting HPO annotations of novel proteins. The dark nodes on the right side represent the predicted terms associated with the input proteins. Four leaf nodes are highlighted by dashed line boxes
Existing Methods
The basic idea behind existing methods consists of these aspects: (1) which data sources should be utilized; (2) how should these data sources be integrated; (3) whether the prediction results meet the biological consistency. Table 2 summarizes existing methods from different aspects. Specifically, we say the prediction scores for n HPO terms are consistent if they obey the true-path-rule:
| 2 |
where denotes the parents of term i. In what follows, we review the existing approaches according to the prediction consistency.
Table 2.
Summary of computational methods for predicting HPO annotations of novel proteins
| Method | Data source(s) | Integration strategy | Hierarchically consistent |
|---|---|---|---|
| PhenoPPIOrth (Wang et al. 2013) | PPI, orthology | Weighting, normalization | ✗ |
| HPO2GO (Doğan 2018) | GO annotation | – | ✗ |
| HPOLabeler (Liu et al. 2020) | PPI, GO annotation, sequence, domain | Learning to rank | ✗ |
| PHENOstruct (Kahanda et al. 2015) | PPI, GO annotation, literature, variants | SSVM | ✓ |
| HTD-DAG (Notaro et al. 2017b) | – | – | ✓ |
| TPR-DAG (Notaro et al. 2017b) | |||
| DESCENS (Notaro et al. 2017a) | |||
| DeepPheno (Kulmanov and Hoehndorf 2020) | PPI, gene expression | Hierarchical multi-class multi-label neural network | ✓ |
Inconsistent Methods
Previous studies have revealed that two proteins with strong interactions were likely to be associated with the same phenotype (Oti et al. 2006; Goh et al. 2007). Moreover, researchers have also found that orthologs tended to retain the functions from ancestors (Koonin 2005; Dolinski and Botstein 2007). Based on such findings, Wang et al. (Wang et al. 2013) developed PhenoPPIOrth that utilized protein-protein interactions (PPI) and orthology information to infer protein-phenotype associations. They designed a PPI path considering the interactive partners of the query protein, and an orthology path considering all proteins connected to the given phenotype in the cross-species phenotype network PhenomeNET (Hoehndorf et al. 2011) and the orthologous proteins of the query protein. A potential phenotype could be inferred from the PPI path and orthology path according to the normalized weighted scores which were derived from the above two paths. In fact, PhenoPPIOrth only predicted the relationship between proteins and OMIM rare diseases (Hamosh et al. 2002), and the protein-HPO term associations were produced through the HPO annotations of diseases. However, the HPO integrated Orphanet (Pavan et al. 2017) and DECIPHER (Firth et al. 2009) as well. In consequence, a lot of protein-phenotype pairs were missing and hence hindered the performance.
Doğan (Doğan et al. 2018) proposed HPO2GO which was based on an assumption in the light of protein function: if a mutation of a protein resulted in reduced or abolished function X, and it might cause the disease which was characterized as the phenotype Y, then Y could be associated with X. They extracted HPO2GO mappings by calculating the frequency of the HPO-GO co-annotations and filtered unreliable mappings by statistical resampling. Using the generated HPO2GO mappings, we could predict HPO annotations of query protein by taking its existing GO annotations into account. They inferred HPO annotations only from the proteins with similar functions, while ignoring the leverage of PPI. Therefore, the absence of the interaction network, an important and effective information source, degraded the final performance.
Neither PhenoPPIOrth nor HPO2GO was scalable to multiple data sources. To improve the performance by integrating heterogeneous features, Liu et al. (Liu et al. 2020) proposed HPOLabeler which adopted the learning to rank (LTR) (Li 2011) framework. They designed three types of basic models including (1) Logistics Regression models exploiting PPI networks, GO annotations, domains, and sequences; (2) Nearest Neighbor method exploring the local structure of PPI network; (3) Naive method that simply calculated HPO term frequency. Borrowing the idea from information retrieve, they modeled the proteins as a query, and each candidate HPO term as a document, and trained the model to return the optimal rank of those HPO terms with respect to the query protein. Owing to the power of a state-of-the-art LTR algorithm, LambdaMART (Burges 2010), HPOLabeler achieved the best performance. The advantage of HPOLabeler was that it used the “Stacking” strategy (Wolpert 1992) in ensemble learning, which allowed the model to be scalable to multiple data sources.
Consistent Methods
Traditional algorithms for solving hierarchical multi-label classification problems mostly disassembled multi-labels into multiple single labels, and then applied independent binary classifiers to each label. However, such a flat classifier could not learn the inherent relationship between the HPO terms, and it would incur a serious label-imbalance problem especially for the leaf terms. Besides, flat learning usually made the prediction results hierarchically inconsistent, so the interpretability is lost. To address the issues, Kahanda et al. (Kahanda et al. 2015) proposed PHENOstruct to produce hierarchically consistent predictions of the query protein. Specifically, PHENOstruct learned a compatibility function that described the relationship between the given input and the structured output which referred to the possible consistent subgraphs in the HPO hierarchy. In their model, each protein was characterized by sets of features generated from PPI networks, GO, literatures, and variants. The combination of multiple data sources was achieved by joint kernels of structured support vector machine (SSVM). Despite the scalability of heterogeneous information sources, PHENOstruct treated each source equally and did not consider the difference in the contribution to the performance. Therefore, in some cases, the low-quality information sources would degrade the final performance. Furthermore, their method was sensitive to the incomplete annotations and thus it led to the modest performance.
Kulmanov and Hoehndorf (Kulmanov and Hoehndorf 2020) developed DeepPheno to guarantee consistency based on an ontology-aware hierarchical neural network. They employed a two-step procedure in which the model firstly took feature vectors derived from GO annotations and gene expression profiles as input and outputted preliminary predictions and then fixed inconsistency by the proposed hierarchical classification layer. The core idea of this layer is to assign the prediction score of a term with the maximum scores of all its descendants. However, the quality of final predictions was largely influenced by the predictive scores of bottom terms, while the performance of them was possibly undesirable due to the drastic label imbalance. Moreover, their model did not take PPI or other data sources into consideration and was not scalable to heterogeneous input features.
Instead of maintaining the hierarchical structure in the learning process, Notaro et al. (Notaro et al. 2017b) proposed a series of adjustment strategies of the flat learning predictions. The workflow contained two stages: (1) flat learning that trained per-term classifiers and (2) hierarchical combination that modified the predictions to meet the consistency. The first strategy was the hierarchical top-down (HTD-DAG) algorithm, which adjusted the predictions of each base learner from top to bottom, or more precisely, transmitted the predictive score to their children. Another ensemble algorithm was hierarchical true path rule (TPR-DAG) which considered the opposite information flow from bottom to top. Owing to different strategies to select positive children and propagate positive predictions from leaves to root, a lot of variants were derived from the original TPR-DAG algorithm. Additionally, to strengthen the contribution of the bottom HPO terms to the final ensemble result, so that the information would not decay too quickly with the upward transmission, they further extended TPR-DAG to DESCENS (Notaro et al. 2017a) which selected positive descendants rather than direct children. However, the hierarchical combination relied on the quality of flat classifiers, and the final performance might decrease in the worst case.
Discussion
Predicting HPO annotations of novel protein is an extremely challenging task. To date, only a few methods can outperform the Naive method in terms of protein-centric evaluation metrics in the CAFA challenge (Jiang et al. 2016). Jiang et al. (Jiang et al. 2016) imputed it to the large number of HPO terms associated with each protein and ineffective homology information. Recently, Liu et al. (Liu et al. 2020) revealed that the relatively low prediction performance might be attributed to incomplete annotations of new proteins. Further research should be made into this area.
Due to the hierarchical structure of HPO, terms that are far away from the root represent more specific concepts and contain a lot of information. Thus, specific terms are more meaningful in biomedical research. However typically, those terms at the bottom level are annotated with few proteins. The relatively low frequency brings heavy class-imbalance to the machine learning models. As a result, the performance of specific HPO terms shows a decay compared with that of general terms. The computational methods that are able to alleviate the label imbalance are expected to provide predictions for specific terms of high precision.
Despite the efforts in keeping the biological consistency of the prediction, the state-of-the-art performance is still achieved by the inconsistent method, HPOLabeler. The incomplete annotations or the low-quality of specific term predictors have a negative impact on the result. A better hierarchical learning strategy is expected to benefit the consistent methods.
The effectiveness of PPI network in the HPO annotation prediction problem has been recognized (Liu et al. 2020; Kahanda et al. 2015). However, a majority of methods (Kahanda et al. 2015; Kulmanov and Hoehndorf 2020) simply took the adjacency matrix of PPI network as the input feature. Liu et al. (Liu et al. 2020) proposed the Nearest Neighbor method by exploring the direct neighborhood while ignoring the high-order proximity. Additionally, to our best knowledge, DeepPheno (Kulmanov and Hoehndorf 2020) is the only one deploying the deep learning technique. With the emergence of graph neural network (GNN) (Defferrard et al. 2016; Kipf and Welling 2017), numerous GNN-based models have been proposed in many bioinformatics fields, such as disease gene prioritization (Han et al. 2019), polypharmacy side effects identification (Zitnik et al. 2018), and drug repurposing (Wang et al. 2020). The exploitation of GNN will hopefully boost the prediction performance from the more efficient exploration of the PPI network.
Methods for Identifying Missing HPO Annotations
Problem Formulation
Given m proteins and n HPO terms , the known associations between them are represented by a binary matrix . The entry if protein is annotated by HPO term , otherwise . However, does not mean that there must be no relation between them, but only that this link has not been observed yet. Identifying missing HPO annotations is to find the HPO term that but may potentially be related to . It is illustrated in Fig. 5. The problem setting is similar to collaborative filtering with implicit feedback in the recommendation system (Hu et al. 2008).
Fig. 5.
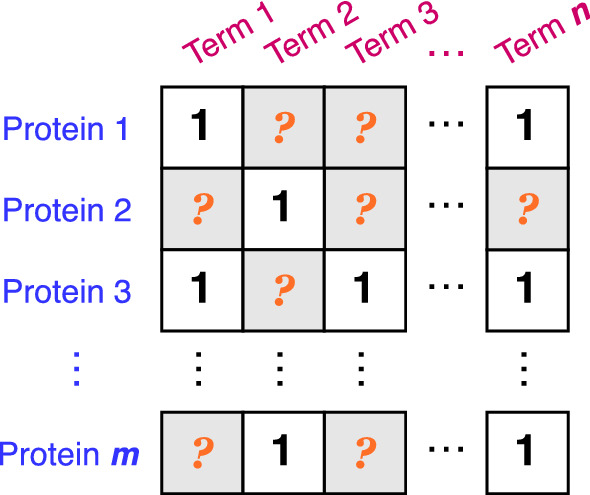
An illustration of the prediction of missing HPO annotations problem. An entry of 1 indicates the association between the corresponding protein and HPO term is known, and an entry filled with question mark means an unobserved relationship. The goal is to figure out which unidentified annotations may be true
Existing Methods
The proposed methods for identifying missing HPO annotations can be categorized into two groups: label propagation-based and matrix completion-based. The basic assumption behind both two categories is that similar proteins tend to be related to the same HPO term. The main difference lies in how to measure the similarity and how to incorporate the similarity into the model. In the following, we review the existing algorithms summarized in Table 3.
Table 3.
Summary of computational methods for identifying missing HPO annotations
| Base | Method | Data source(s) | HPO hierarchy | Optimization |
|---|---|---|---|---|
| Label propagation | LP (Zhu et al. 2003; Zhou et al. 2003) | PPI | – | Closed-form solution |
| DLP (Petegrosso et al. 2017) | PPI | Raw HPO DAG | L-BFGS-B | |
| tlDLP (Petegrosso et al. 2017) | PPI, GO annotation | Raw HPO DAG | L-BFGS-B | |
| Matrix completion | SMC (Lee and Seung 2000) | – | – | ALS |
| AiProAnnotator (Gao et al. 2018) | PPI | Lin method | ALS | |
| HPOAnnotator (Gao et al. 2019) | Multiple PPIs | Lin method | ALS |
Label Propagation-Based Algorithms
Label propagation (LP) (Zhu et al. 2003; Zhou et al. 2003) is a classical semi-supervised learning algorithm that iteratively spread the labels from a few labeled nodes to a large number of unlabeled nodes through the connected graph structure. The rationality of LP lies in the smoothness assumption (Chapelle et al. 2006): if two samples are close in the input space, their labels should be the same. It is consistent with the assumption made on the PPI network. Thus, the model will assign the HPO term of annotated protein to its strongly interacted unannotated partners. The prediction results can be obtained from the closed-form solution.
Petegrosso et al. (Petegrosso et al. 2017) extended vanilla LP to dual label propagation (DLP) by coupling smoothness term imposing smoothness in PPI network and another term imposing smoothness in the HPO hierarchy, which encouraged directly connected phenotypes to be associated with the same protein. Then they further introduced transfer learning to DLP and proposed tlDLP, which utilized GO annotations as an auxiliary data source and let proteins with similar functions be likely to be associated with similar phenotypes. It simultaneously reconstructed protein-HPO term associations matrix and protein-GO term associations matrix, and sought the agreement between predicted phenotype relationships and function relationships through the overlapping proteins. Through L-BFGS-B optimization algorithm (Zhu et al. 1997), DLP and tlDLP achieved better performance than conventional LP.
Matrix Completion-Based Algorithms
Standard matrix completion (SMC) (Lee and Seung 2000) is a typical collaborative filtering algorithm widely used in recommender systems. It decomposes the protein-phenotype associations matrix into the product of two lower dimensionality factor matrices. The missing values are imputed through alternating least square (ALS). Existing matrix completion-based approaches imposed graph Laplacian regularizations in the latent space so as to capture intrinsic relationships between proteins and phenotypes and benefit the representation learning.
AiProAnnotator was proposed by Gao et al. (Gao et al. 2018) which added graph Laplacians on both PPI network and HPO similarity to the objective function of SMC to find a better low-rank approximation solution. It is noteworthy that they adopted the semantic similarity of HPO terms proposed by Lin (Lin 1998) instead of raw HPO DAG. Consequently, the information was enabled to flow between the siblings or ancestor-descendant pairs more than strict parent-child pairs, in order that the model could find similar HPO terms more broadly and deeply. A single PPI network may provide limited information. To integrate multiple PPI networks, Gao et al. (Gao et al. 2019) subsequently proposed HPOAnnotator which extended the graph Laplacian of AiProAnnotator on a single PPI network to multiple networks.
Discussion
The study of identifying missing HPO annotations is still in its infancy. The algorithms reviewed above mostly minimized the difference between the predicted association matrix and the known association matrix through the -norm to implement the approximation process. Besides, they resorted to the graph Laplacian to impose smoothness on the protein side and/or phenotype side, so that similar proteins could be annotated with the same HPO term. Regarding the effectiveness of the PPI network, most algorithms included it as a measure of protein similarity. Some approaches also utilized more information sources, such as GO annotations, to further improve the performance. However, these methods leveraging heterogeneous data sources showed limited performance improvements compared to the methods that used only one auxiliary data source. We expect more studies in exploring the better integration manner of multiple features in the following years.
The above models dwelt in discovering the linear relationships between proteins and HPO terms, and could not capture complex nonlinear relationships. Moreover, these models could only capture low-order topological information from PPI and/or HPO term similarity network(s), and ignored the high-order connectivity. To the best of our knowledge, no researchers have proposed models based on deep learning for this task. Recently, graph convolutional network (GCN) (Kipf and Welling 2017) opens a new paradigm for handling graph-based data in a deep learning fashion, and achieves leading performance in many link prediction tasks in biomedical networks (Han et al. 2019; Zitnik et al. 2018; Wang et al. 2020). The performance improvement would be promising if applying the similar GCN-based architecture to deal with the above issues.
Completing the missing HPO annotations can be seen as a process of extending downward from the leaf nodes of current annotations towards more specific terms, thus the true-path-rule should also be followed. However, neither label propagation-based nor matrix completion-based algorithms could satisfy the consistency. A biological consistent prediction algorithm remains to be explored.
Methods for Phenotype Protein Prioritization
Problem Formulation
Suppose a set of m proteins , where the first l proteins have been annotated with HPO term t. The task is to assign likelihood scores to the remaining unannotated proteins and suggest their potential to be truly associated with the interested phenotype t (Fig. 6). From the perspective of machine learning, we can formulate it as a binary classification problem.
Fig. 6.
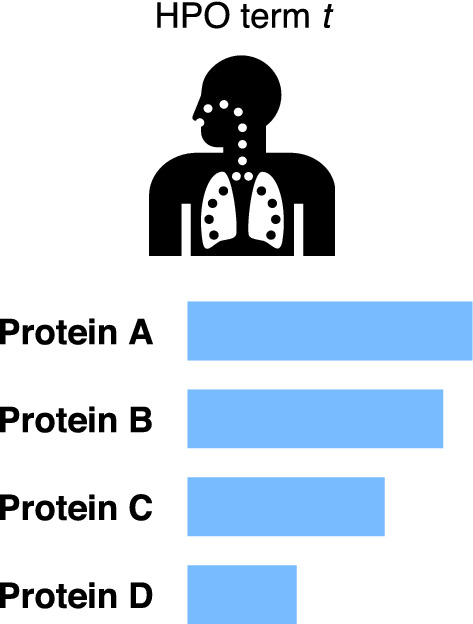
Illustration of phenotype protein prioritization problem. Given a HPO term t, the model assigns likelihood of protein involvement in generating this phenotype and spots the potential proteins which may be truly associated with t
Existing Methods
As far as we know, the phenotype protein prioritization is still a little-tapped problem. Previous efforts mainly focused on ranking candidate proteins with respect to a specific GO term. Existing algorithms can be summarized into two categories according to the way of representation learning on the protein networks: (1) unsupervised learning methods and (2) semi-supervised learning methods. The details are provided in Table 4.
Table 4.
Summary of computational methods for candidate protein prioritization with respect to a particular function
| Method | Mode | Downstream | Network | Technique |
|---|---|---|---|---|
| Mashup (Cho et al. 2016) | Unsupervised | SVM | Multiple | Traditional |
| deepNF (Gligorijevic et al. 2018) | Unsupervised | SVM | Multiple | Deep learning |
| DeepMNE-SVM (Xue et al. 2019) | Unsupervised | SVM | Multiple | Deep learning |
| DeepMNE-CNN (Peng et al. 2021) | Unsupervised | CNN | Multiple | Deep learning |
| BIONIC (Forster et al. 2021) | Unsupervised | LR | Multiple | Deep learning |
| RANKS (Valentini et al. 2016) | Semi-supervised | – | Single | Traditional |
| GeneMANIA (Mostafavi et al. 2008; Mostafavi and Morris 2010) | Semi-supervised | – | Multiple | Traditional |
Unsupervised Methods
The general workflow of unsupervised methods is to first learn the embeddings of the proteins from the input association networks via unsupervised model, and then use the derived embeddings to train a flat classifier for each GO term by a classical supervised learning algorithm, such as support vector machine (SVM).
Mashup (Cho et al. 2016) first analyzed the diffusion state in each network by the random walk with restart (RWR) to characterize the topological context. Low-dimensional embeddings of each node were obtained by jointly minimizing the difference between the observed topological patterns and the parameterized-multinomial logistic distributions for all networks simultaneously. The learned compact representations were then plugged into downstream machine learning models to derive function predictions. However, the integration of heterogeneous networks through multinomial logistic models required high computational complexity, which made it infeasible to large-scale networks.
While Mashup adopted a traditional machine learning technique to learn the low-dimensional embeddings, most previous works obtained representations from the deep auto-encoders. The advantage of deep architecture is that the model can preserve the complex and non-linear network topological structure.
Gligorijevic et al. (Gligorijevic et al. 2018) proposed deepNF which fused multiple interaction networks via multimodal deep auto-encoders. In the first step, they ran RWR and constructed positive pointwise mutual information (PPMI) matrix to convert network structural information into initial high-dimensional node representations. A multimodal auto-encoder was then applied to integrate heterogeneous networks and extract high-level embeddings from the bottleneck layer. The learned compact feature representations were finally fed into off-the-shelf SVM classifiers for function prediction.
The information fusion of multi-networks in deepNF was implemented by dense fully connected layers, ignoring the correlation across different networks. Xue et al. (Xue et al. 2019) revised traditional auto-encoders to incorporate prior constraints. Specifically, the authors assumed that the nodes with must-link constraints were highly similar and closer in the latent space, while the nodes with cannot-link constraints were highly dissimilar and more distant in the low-dimensional space. These constraints were modeled as the penalty term in the loss function of the auto-encoder. DeepMNE chained multiple revised auto-encoders together, where the constraints of each network extracted from the previous auto-encoder were merged and applied to the next auto-encoder as the prior information. The representations derived from the last auto-encoder were used to train the SVM classifiers for protein function prediction. Soon afterward, they proposed a novel convolutional neural network (CNN) as the downstream classifier (Peng et al. 2021), which consisted of three components including convolutional layers, max-pooling layer, and the final fully connected layer. DeepMNE stacked multiple auto-encoders and iteratively extracted constraints one by one, thereby the model was time and memory expensive comparing with deepNF. Moreover, the model was sensitive to the noise because the error in constraints of the first few layers could keep accumulating and misled the node representation learning.
The network structure information was not introduced during the feature extraction but only expressed in the input features generated by RWR. Recently, Forster et al. (Forster et al. 2021) designed a GNN-based auto-encoder, named BINOIC. The network-specific features were first learned from each graph convolutional encoder by aggregating neighbor features and low-dimensional projection. Then various learned features were combined as the integrated representations. The decoder restored the embeddings to the recovered network adjacency matrices, and the model minimized the differences between input and recovered networks to optimize. The integrated embeddings were taken as the input of logistic regression for the downstream task. Owing to the application of GNN, BIONIC was capable of capturing not only low-order proximity but also high-order connectivity. Therefore, the network topology was sufficiently explored when iteratively updating the node representations. But if stacking too many GNN layers, the performance might drop which could partly be attributed to the over-smoothing phenomenon (Li et al. 2018, 2019), thereby hindering the discovery of complex hidden patterns from the graph data.
Semi-supervised Methods
The cornerstone of semi-supervised methods is still the broadly recognized smoothness assumption: proteins with strong interaction possibly share the same function. Therefore, it is reasonable to acquire the label from its annotated neighbors.
RANKS (Valentini et al. 2016) was a general algorithmic scheme for prioritizing candidate nodes with respect to a given property in a biological network, which included a global learning strategy and a local learning strategy. The author proposed to use a kernel (e.g. a random walk kernel) to represent the overall topological structure of the network to accomplish global learning. The generated kernel extended simple connectivity between adjacent nodes to the high-order proximity. Then RANKS adopted a local learning strategy that ranked the candidate nodes with a specific scoring function considering only the annotated direct neighborhood and the edge weights to them in the kernel matrix. For instance, they implemented four types of scoring functions: nearest-neighbors score, k-nearest-neighbors score, average score, and weighted sum with linear decay score. However, RANKS could provide the likelihood between candidate protein and its neighbors with known function annotations only on a single network. If one wanted to exploit multiple interaction networks, it was necessary to integrate them into a composite network in advance. Thus it might lose part of information in the original separate network, making the characterization of the interaction strength biased.
GeneMANIA (Mostafavi et al. 2008) integrated multiple networks based on ridge regression and predicted protein function through label propagation. The first step is to generate a composite functional association network from the weighted sum of individual networks. To obtain the network weights, they proposed a ridge regression model that minimized the difference between the composite network and the target network constructed from the function annotations. Then it predicted GO annotation by applying label propagation on the single composite network. Differing from conventional binary classification problems which only had positive and negative labels, GeneMANIA added the third label for the proteins with unknown functions. In fact, those unlabeled samples were exactly the proteins to be predicted. However, facing the heavy label imbalance in many GO categories, the process of assigning network weights was prone to overfitting. To address the issues, Mostafavi and Morris (Mostafavi and Morris 2010) proposed a novel network combination strategy named SW. In particular, unlike GeneMANIA, which calculated network weights for each GO term separately, SW optimized the network weights on a set of GO terms (e.g. GO hierarchy and term frequency) simultaneously. The following protein function prediction procedure remained the same. Nevertheless, the network weights assignment was independent of the function prediction. We believed that there still existed an improvement room if combining two steps to jointly learn the composite network specifically for functional inference. Although GeneMANIA was available to heterogeneous networks, the prediction process was still carried on a single integrated network generated by adaptive weighted averaging. Therefore, the information loss could also incur when projecting various networks onto a single network representation.
Discussion
Biological networks encode valuable information, but each contains its own individual biases and noise. These issues could be overcomed by proper integration, as multiple networks are complementary to each other. Most unsupervised learning methods are focused on network fusion via auto-encoders, and the produced compact embeddings are too general to possess enough discrimination ability for the specific task. For the semi-supervised learning methods, a lot of them could not make predictions without prior network combinations which might lead to substantial information loss. An end-to-end semi-supervised architecture is expected to make up for the above two shortcomings. Despite the power of GCN (Kipf and Welling 2017) in learning node representation from biological networks, BIONIC still suffers from the intractable over-smoothing problem. Considering the recent progress towards deep GCN model (Li et al. 2019; Chen et al. 2020), more efforts can be made to cope with these issues.
While quite substantial literature has emerged on the function prediction, the phenotype protein prioritization is still under-researched. Although the methodology can be transferred to the phenotype prediction domain in a straightforward manner, it is necessary to design a dedicated algorithm for this task. We believe that more and more algorithms will emerge in this field in the future.
Potential Topics
Selection of Negative HPO Annotations
Negative examples refer to the proteins that are known not to be associated with the given labels. The negative annotations are usually rarely recorded in the proteome databases. For example, in the HPO annotation released in October 2020, only about 1500 negative annotations of diseases were stored, which was far from the scale of positive annotations. This situation arises possibly due to the experimental limitations: experimental assays are usually applied to a single protein, and the protein function may depend on the context, making negative statements/labels quite uncertain, and leading to very few confirmed negative examples. However, for the vast majority of computational annotation prediction tools using machine learning, the selection of negative examples has a significant impact on the final performance: they usually require enough positive and negative examples to train an accurate predictor. Most of the existing methods randomly selected negative examples from unlabeled proteins or treated all unlabeled proteins as negative examples, which could lead to false negatives in the training set. Therefore, a well-designed negative example selection strategy is of great benefit to improving the prediction performance of computational tools.
The current negative example selection algorithms were all in the context of protein function prediction. In fact, similar problems have long been studied in the field of text classification and have been named positive-unlabeled learning (PU learning) (Bekker and Davis 2020). Zhao et al. (Zhao et al. 2008) proposed AGPS based on two-stage PU learning to automatically generate negative examples from unlabeled data in the learning procedure. Youngs et al. (Youngs et al. 2013) proposed ALBias to choose negative examples by a parameterizable Bayesian prior computed from observed annotations, which could be viewed as a generalization of passive two-stage PU learning. However, the ontology structure was overlooked in these methods. To this end, Youngs et al. (Youngs et al. 2014) extended ALBias to SNOB by applying true-path-rule to the annotations to incorporate the hierarchical structure. In addition, they borrowed the idea from the text mining and introduced latent Dirichlet allocation (LDA) to the proposed model NETL (Youngs et al. 2014) to select negative examples according to the learned latent topic distributions. However, there was a pitfall in selecting negative examples based on currently incomplete annotations. Fu et al. (Fu et al. 2016a) proposed NegGOA to mitigate the impact of potentially missing annotations. Then they further considered a small number of available negative annotations and proposed ProPN (Fu et al. 2016b) which modeled the correlations between positive and negative examples as a direct signed hybrid graph. Yu et al. (Yu et al. 2017a) proposed IFDR based on dimensionality reduction with the diffusion component analysis and single value decomposition.
While progress has been made in the field of protein function prediction, the selection of negative HPO annotations is still an untouched area. At present, few negative annotations have been recorded in the HPO database. Identifying irrelevant annotations with aid of ontology structure and auxiliary data sources has the potential for improving the performance of computational tools and should be one of the research spotlights in this area.
Detecting HPO Misannotations
Some HPO annotations are automatically extracted from the OMIM database through a text mining program. As a result, several incorrectly recognized phenotypic abnormalities were introduced into the database, resulting in incorrect annotations. For instance, in HPO annotations released in August 2020, the genes that were associated with Sinusitis (HP:0000246) included ARMC4, BLM, CCDC114, CCDC151, and TTC25. However, these related genes were all removed in the latest release. Existing prediction methods assumed that the known annotations were accurate, and ignored the influence of misannotations on the prediction results. Moreover, the noisy annotations might mislead subsequent research and applications in multiple fields such as clinical diagnosis and treatment. Therefore, detecting HPO misannotations is needed to improve the data quality of the current annotations.
Nevertheless, related work was limited and focused on predicting noisy GO annotations. Some of the methods (Deegan et al. 2010; Wei et al. 2020) analyzed the rationality of function annotation based on the assumption that some protein functions were species-specific. Other methods like NoisyGOA (Lu et al. 2016), NoGOA (Yu et al. 2017b), and NFA (Lu et al. 2018) applied machine learning to infer GO misannotations, considering taxon, ontology structure, and evidence codes.
Despite progress in detecting noisy GO annotations, no effort has been made towards HPO misannotations. In view of the negative impact of inaccurate annotations, exploring an accurate and efficient detecting algorithm will be a direction worthy of in-depth study.
Predicting Protein-Phenotype Associations Under the Specific Mechanisms
Tissue-selective gene expression is an important mechanism by which the same genome can generate differentiated phenotypes among tissues (Bentz et al. 2019). Exploring the pathologic consequences of human genes in specific tissues is typically more essential for insights into disease diagnostics and therapeutics (Barbeira et al. 2018; Hekselman and Yeger-Lotem 2020). The abnormality of the genes in different tissues may incur different diseases and phenotypes. For example, Proprotein Convertase Subtilisin/kexin Type 9 (PCSK9) is a member of the subtilisin family of PCs that encodes a neural apoptosis-regulated convertase 1 (Horton et al. 2007). It specifically enriches in a number of tissues and is involved in lung (Xu et al. 2017), liver (Lee et al. 2019), and brain diseases (Rousselet et al. 2011). One of the key points of predicting tissue-specific pathologic consequences of proteins is the feature representation. Considering the effectiveness of PPI network, it would be important to extend the integrated PPI network on an organismal level to multiple tissue-specific networks (Zitnik and Leskovec 2017; Guan et al. 2012). Elucidating the mechanisms of tissue specificity in human diseases is quite challenging, and still under-researched. The efforts towards this field can be particularly valuable to fill the blank.
Post-translational modification (PTM) is another essential mechanism in protein biosynthesis, whereby the addition, folding, or removal of functional groups leads to drastic alterations in protein function (Mann and Jensen 2003; Seo and Lee 2004). Defects in PTMs have been linked to many human disorders, highlighting the importance of understanding PTMs in the study of disease treatment and prevention (Martin et al. 2011; Anbalagan et al. 2012; Wang et al. 2014). For example, protein caveolin-1 encoded by gene CAV1 is a scaffolding protein within caveolar membranes (Vargas et al. 2002). The role of phosphorylation in the relationships between caveolin-1 and ovarian, breast, rectal, and colon cancers (Wiechen et al. 2001; Joshi et al. 2008) has been gradually revealed. Recently, an integrative database of human PTM-disease associations PTMD was developed which collected thousands of manually curated disease-associated PTM events (Xu et al. 2018). However, considering the complexity of PTM, the role of post-translational protein modifications play in shaping phenotypical traits is still poorly understood. More efforts are welcomed to uncover the relationship between PTMs and phenotypes, and can be helpful for a better appreciation for the disease development, diagnosis, and clinical therapy.
Conclusion
Uncovering the human protein-phenotype associations has long been one of the research hotspots in phenomics study. The emergence of HPO provided a comprehensive logical standard to describe various phenotypic abnormalities encountered in human disorders and facilitated the computational analysis of their genetic causes. In this paper, we reviewed computational methods for three different tasks including predicting HPO annotations of a novel protein, identifying missing HPO annotations, and phenotype protein prioritization.
Despite their promising results, there are several unsolved challenges remained. In summary, we highlight the following key issues:
Specific terms While the specific terms carry more valuable information, the prediction performance of them is typically undesirable, which is partially caused by heavy label imbalance. More efforts are needed to explore an efficient strategy to mitigate the impact of class-imbalance.
Consistency The biological consistency is not preserved in the prediction made by most of the existing methods. Notwithstanding some efforts towards consistent prediction, their performance is still inferior. Developing models that provide consistent predictions without losing performance remains a promising issue.
Data sources A lot of research has demonstrated the effectiveness of PPI networks. However, has the information hidden in the networks been fully exploited? Can we find other fruitful information sources that may boost the prediction performance? How to integrate heterogeneous features more effectively? These questions are left for future research.
Additionally, we pointed out some potential topics including the selection of negative annotations and detecting misannotations. These open problems are understudied in the field of phenotype prediction and will be interesting research directions worth exploring.
Funding
SZ has been supported by National Natural Science Foundation of China (no. 61872094), Shanghai Municipal Science and Technology Major Project (no. 2018SHZDZX01), ZJ Lab, and Shanghai Center for Brain Science and Brain-Inspired Technology. LL has been supported by the 111 Project (no. B18015), Shanghai Municipal Science and Technology Major Project (no. 2017SHZDZX01) and Information Technology Facility, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences.
Data Availability Statement
The datasets analyzed in this review are available on the Human Phenotype Ontology website, “https://hpo.jax.org”.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Code Availability
Not applicable.
Authors’ Contributions
SZ conceived and supervised the work. SZ and LL designed the study. LL drafted the paper. SZ modified the paper. SZ and LL finalized the paper.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publication
Not applicable.
References
- Akhmetov I, Bubnov RV. Assessing value of innovative molecular diagnostic tests in the concept of predictive, preventive, and personalized medicine. EPMA J. 2015;6(1):19. doi: 10.1186/s13167-015-0041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbalagan M, Huderson B, Murphy L, Rowan BG. Post-translational modifications of nuclear receptors and human disease. Nucl Recept Signal. 2012;10(1):nrs-1001. doi: 10.1621/nrs.10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira AN, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9(1):1–20. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker J, Davis J. Learning from positive and unlabeled data: a survey. Mach Learn. 2020;109(4):719–760. doi: 10.1007/s10994-020-05877-5. [DOI] [Google Scholar]
- Bentz AB, Thomas GWC, Rusch DB, Rosvall KA. Tissue-specific expression profiles and positive selection analysis in the tree swallow (Tachycineta bicolor) using a de novo transcriptome assembly. Sci Rep. 2019;9(1):1–12. doi: 10.1038/s41598-019-52312-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14(10):681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- Bromberg Y. Disease gene prioritization. PLoS Comput Biol. 2013;9(4):e1002902. doi: 10.1371/journal.pcbi.1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burges C (2010) From RankNet to LambdaRank to LambdaMART: an overview. Technical report, Microsoft Research
- Bush WS, Moore JH. Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. doi: 10.1371/journal.pcbi.1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapelle O, Schölkopf B, Zien A (eds) (2006) Semi-Supervised Learning. The MIT Press. 10.7551/mitpress/9780262033589.001.0001
- Chen M, Wei Z, Huang Z, Ding B, Li Y (2020) Simple and deep graph convolutional networks. In: Proceedings of the 37th international conference on machine learning, ICML 2020, 13–18 July 2020, virtual event. Proceedings of machine learning research, vol 119, pp 1725–1735. PMLR
- Cho H, Berger B, Peng J. Compact integration of multi-network topology for functional analysis of genes. Cell Syst. 2016;3(6):540–548. doi: 10.1016/j.cels.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JX, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97(2):199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans Andrew R, et al. Finding our way through phenotypes. PLoS Biol. 2015;13(1):e1002033. doi: 10.1371/journal.pbio.1002033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan JI, Dimmer EC, Mungall CJ. Formalization of taxon-based constraints to detect inconsistencies in annotation and ontology development. BMC Bioinform. 2010;11:530. doi: 10.1186/1471-2105-11-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defferrard M, Bresson X, Vandergheynst P (2016) Convolutional neural networks on graphs with fast localized spectral filtering. In: Advances in neural information processing systems 29: annual conference on neural information processing systems 2016, December 5–10, 2016, Barcelona, Spain, pp 3837–3845
- Doğan T. HPO2GO: prediction of human phenotype ontology term associations for proteins using cross ontology annotation co-occurrences. PeerJ. 2018;6:e5298. doi: 10.7717/peerj.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinski K, Botstein D. Orthology and functional conservation in eukaryotes. Annu Rev Genet. 2007;41:465–507. doi: 10.1146/annurev.genet.40.110405.090439. [DOI] [PubMed] [Google Scholar]
- Firth HV, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84(4):524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster DT, Boone C, Bader GD, Wang B (2021) BIONIC: biological network integration using convolutions. bioRxiv. 10.1101/2021.03.15.435515
- Fu G, Wang J, Yang B, Yu G. NegGOA: negative GO annotations selection using ontology structure. Bioinformatics. 2016;32(19):2996–3004. doi: 10.1093/bioinformatics/btw366. [DOI] [PubMed] [Google Scholar]
- Fu G, Yu G, Wang J, Guo M. Protein function prediction using positive and negative examples (in Chinese) J Comput Res Dev. 2016;53(8):1753–1765. doi: 10.7544/issn1000-1239.2016.20160196. [DOI] [Google Scholar]
- Gao J, Yao S, Mamitsuka H, Zhu S (2018) AiProAnnotator: low-rank approximation with network side information for high-performance, large-scale human protein abnormality annotator. In: IEEE international conference on bioinformatics and biomedicine, BIBM 2018, Madrid, Spain, December 3–6, 2018, pp 13–20. IEEE Computer Society. 10.1109/BIBM.2018.8621517
- Gao J, Liu L, Yao S, Mamitsuka H, Zhu S. HPOAnnotator: improving large-scale prediction of HPO annotations by low-rank approximation with HPO semantic similarities and multiple PPI networks. BMC Med Genom. 2019;12(10):187. doi: 10.1186/s12920-019-0625-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic V, Barot M, Bonneau R. deepNF: deep network fusion for protein function prediction. Bioinformatics. 2018;34(22):3873–3881. doi: 10.1093/bioinformatics/bty440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh K, Cusick ME, Valle D, Childs B, Vidal M, Barabási A. The human disease network. Proc Natl Acad Sci USA. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groza T, et al. The human phenotype ontology: semantic unification of common and rare disease. Am J Hum Genet. 2015;97(1):111–124. doi: 10.1016/j.ajhg.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, et al. Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol. 2012;8(9):e1002694. doi: 10.1371/journal.pcbi.1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002;30(1):52–55. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Yang P, Zhao P, Shang S, Liu Y, Zhou J, Gao X, Kalnis P (2019) GCN-MF: disease-gene association identification by graph convolutional networks and matrix factorization. In: Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining, KDD 2019, Anchorage, AK, USA, August 4–8, 2019, pp 705–713. ACM. 10.1145/3292500.3330912
- Hekselman I, Yeger-Lotem E. Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nat Rev Genet. 2020;21(3):137–150. doi: 10.1038/s41576-019-0200-9. [DOI] [PubMed] [Google Scholar]
- Hoehndorf R, Schofield PN, Gkoutos GV. PhenomeNET: a whole-phenome approach to disease gene discovery. Nucleic Acids Res. 2011;39(18):e119. doi: 10.1093/nar/gkr538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton Jay D, Cohen Jonathan C, Hobbs Helen H. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32(2):71–77. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Koren Y, Volinsky C (2008) Collaborative filtering for implicit feedback datasets. In: Proceedings of the 8th IEEE international conference on data mining (ICDM 2008), December 15–19, 2008, Pisa, Italy, pp 263–272. IEEE Computer Society. 10.1109/ICDM.2008.22
- Jiang Y, et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016;17(1):184. doi: 10.1186/s13059-016-1037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B, et al. Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal adhesion dynamics and tumor cell migration and invasion. Cancer Res. 2008;68(20):8210–8220. doi: 10.1158/0008-5472.CAN-08-0343. [DOI] [PubMed] [Google Scholar]
- Kahanda I, Funk C, Verspoor K, Ben-Hur A. PHENOstruct: prediction of human phenotype ontology terms using heterogeneous data sources [version 1; peer review: 2 approved] F1000Research. 2015;4:259. doi: 10.12688/f1000research.6670.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipf TN, Welling M (2017) Semi-supervised classification with graph convolutional networks. In: 5th International conference on learning representations, ICLR 2017, Toulon, France, April 24–26, 2017, conference track proceedings. OpenReview.net
- Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- Kulmanov M, Hoehndorf R. DeepPheno: predicting single gene loss-of-function phenotypes using an ontology-aware hierarchical classifier. PLoS Comput Biol. 2020;16(11):e1008453. doi: 10.1371/journal.pcbi.1008453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DD, Seung HS (2000) Algorithms for non-negative matrix factorization. In: Advances in neural information processing systems 13, Papers from Neural Information Processing Systems (NIPS) 2000, Denver, CO, USA, pp 556–562. MIT Press
- Lee JS, et al. PCSK9 inhibition as a novel therapeutic target for alcoholic liver disease. Sci Rep. 2019;9(1):1–16. doi: 10.1038/s41598-019-53603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A short introduction to learning to rank. IEICE Trans Inf Syst. 2011;94-D(10):1854–1862. doi: 10.1587/TRANSINF.E94.D.1854. [DOI] [Google Scholar]
- Li Q, Han Z, Wu X-M (2018) Deeper insights into graph convolutional networks for semi-supervised learning. In: Proceedings of the thirty-second AAAI conference on artificial intelligence, (AAAI-18), the 30th innovative applications of artificial intelligence (IAAI-18), and the 8th AAAI symposium on educational advances in artificial intelligence (EAAI-18), New Orleans, Louisiana, USA, February 2–7, 2018, pp 3538–3545. AAAI Press
- Li G, Müller M, Thabet AK, Ghanem B (2019) DeepGCNs: can GCNs go as deep as CNNs? In: 2019 IEEE/CVF international conference on computer vision, ICCV 2019, Seoul, Korea (South), October 27–November 2, 2019, pp 9266–9275. IEEE. 10.1109/ICCV.2019.00936
- Lin D (1998) An information-theoretic definition of similarity. In: Shavlik JW (ed) Proceedings of the fifteenth international conference on machine learning (ICML 1998), Madison, Wisconsin, USA, July 24–27. Morgan Kaufmann, pp 296–304
- Liu L, Huang X, Mamitsuka H, Zhu S. HPOLabeler: improving prediction of human protein-phenotype associations by learning to rank. Bioinformatics. 2020;36(14):4180–4188. doi: 10.1093/bioinformatics/btaa284. [DOI] [PubMed] [Google Scholar]
- Lu C, Wang J, Zhang Z, Yang P, Yu G. NoisyGOA: noisy GO annotations prediction using taxonomic and semantic similarity. Comput Biol Chem. 2016;65:203–211. doi: 10.1016/j.compbiolchem.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Lu C, Chen X, Wang J, Yu G, Yu Z. Identifying noisy functional annotations of proteins using sparse semantic similarity (in Chinese) Sci Sin Inform. 2018;48(8):1035–1050. doi: 10.1360/N112017-00105. [DOI] [Google Scholar]
- Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21(3):255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int. 2011;58(4):458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Morris Q. Fast integration of heterogeneous data sources for predicting gene function with limited annotation. Bioinformatics. 2010;26(14):1759–1765. doi: 10.1093/bioinformatics/btq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(S1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notaro M, Schubach M, Frasca M, Mesiti M, Robinson PN, Valentini G (2017a) Ensembling descendant term classifiers to improve gene-abnormal phenotype predictions. In: International meeting on computational intelligence methods for bioinformatics and biostatistics, pp 70–80. Springer. 10.1007/978-3-030-14160-8_8
- Notaro M, Schubach M, Robinson PN, Valentini G. Prediction of Human Phenotype Ontology terms by means of hierarchical ensemble methods. BMC Bioinform. 2017;18(1):1–18. doi: 10.1186/s12859-017-1854-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti M, Snel B, Huynen MA, Brunner HG. Predicting disease genes using protein–protein interactions. J Med Genet. 2006;43(8):691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavan S, Rommel K, Marquina MEM, Höhn S, Lanneau V, Rath A. Clinical practice guidelines for rare diseases: the orphanet database. PLoS One. 2017;12(1):e0170365. doi: 10.1371/journal.pone.0170365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Xue H, Wei Z, Tuncali I, Hao J, Xuequn Shang. Integrating multi-network topology for gene function prediction using deep neural networks. Brief Bioinform. 2021;22(2):2096–2105. doi: 10.1093/bib/bbaa036. [DOI] [PubMed] [Google Scholar]
- Petegrosso R, Park S, Hwang TH, Kuang R. Transfer learning across ontologies for phenome–genome association prediction. Bioinformatics. 2017;33(4):529–536. doi: 10.1093/bioinformatics/btw649. [DOI] [PubMed] [Google Scholar]
- Peter RN. Deep phenotyping for precision medicine. Hum Mutat. 2012;33(5):777–780. doi: 10.1002/humu.22080. [DOI] [PubMed] [Google Scholar]
- Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83(5):610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousselet E, Marcinkiewicz J, Kriz J, Zhou A, Hatten ME, Annik Prat, Seidah NG. PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J Lipid Res. 2011;52(7):1383–1391. doi: 10.1194/jlr.M014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann RH, Ceusters W, Smith B. Toward an ontological treatment of disease and diagnosis. Summit Transl Bioinform. 2009;2009:116–120. [PMC free article] [PubMed] [Google Scholar]
- Schriml LM, Arze C, Nadendla S, Wayne Chang Y, Mazaitis M, Felix V, Feng G, Kibbe WA. Disease ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40(D1):D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J-W, Lee K-J. Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. BMB Rep. 2004;37(1):35–44. doi: 10.5483/BMBRep.2004.37.1.035. [DOI] [PubMed] [Google Scholar]
- Smith B. Ontology. In: Floridi L, editor. Blackwell Guide to the Philosophy of Computing and Information, Chapter 11. Oxford: Blackwell; 2003. pp. 155–166. [Google Scholar]
- Smith CL, Goldsmith CW, Eppig JT. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;6(1):R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JH, et al. Deep phenotyping on electronic health records facilitates genetic diagnosis by clinical exomes. Am J Hum Genet. 2018;103(1):58–73. doi: 10.1016/j.ajhg.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini G, Armano G, Frasca M, Lin J, Mesiti M, Matteo Re. RANKS: a flexible tool for node label ranking and classification in biological networks. Bioinformatics. 2016;32(18):2872–2874. doi: 10.1093/bioinformatics/btw235. [DOI] [PubMed] [Google Scholar]
- Vargas L, et al. Functional interaction of caveolin-1 with Bruton’s tyrosine kinase and Bmx. J Biol Chem. 2002;277(11):9351–9357. doi: 10.1074/jbc.M108537200. [DOI] [PubMed] [Google Scholar]
- Wang P, Lai W, Li MJ, Xu F, Yalamanchili HK, Lovell-Badge R, Wang J. Inference of gene-phenotype associations via protein–protein interaction and orthology. PLoS One. 2013;8(10):e77478. doi: 10.1371/journal.pone.0077478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-C, Peterson SE, Loring JF. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24(2):143–160. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhou M, Arnold CW (2020) Toward heterogeneous information fusion: bipartite graph convolutional networks for in silico drug repurposing. Bioinformatics 36(Supplement\_1):i525–i533, 07. 10.1093/bioinformatics/btaa437 [DOI] [PMC free article] [PubMed]
- Wei X, Zhang C, Freddolino PL, Zhang Y, Lu Z. Detecting Gene Ontology misannotations using taxon-specific rate ratio comparisons. Bioinformatics. 2020;36(16):4383–4388. doi: 10.1093/bioinformatics/btaa548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechen K, et al. Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumor suppressor gene. Am J Pathol. 2001;159(5):1635–1643. doi: 10.1016/S0002-9440(10)63010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpert DH. Stacked generalization. Neural Netw. 1992;5(2):241–259. doi: 10.1016/s0893-6080(05)80023-1. [DOI] [Google Scholar]
- Xu X, Cui Y, Cao L, Zhang Y, Yin Y, Hu X. PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Exp Ther Med. 2017;13(5):1993–1999. doi: 10.3892/etm.2017.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wang Y, Lin S, Deng W, Peng D, Cui Q, Yu X. PTMD: a database of human disease-associated post-translational modifications. Genom Proteom Bioinform. 2018;16(4):244–251. doi: 10.1016/j.gpb.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Peng J, Shang X (2019) Towards gene function prediction via multi-networks representation learning. In: The thirty-third AAAI conference on artificial intelligence, AAAI 2019, Honolulu, Hawaii, USA, January 27–February 1, 2019, pp 10069–10070. AAAI Press. 10.1609/aaai.v33i01.330110069
- Youngs N, Penfold-Brown D, Drew K, Shasha DE, Bonneau R. Parametric Bayesian priors and better choice of negative examples improve protein function prediction. Bioinformatics. 2013;29(9):1190–1198. doi: 10.1093/bioinformatics/btt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngs N, Penfold-Brown D, Bonneau R, Shasha DE. Negative example selection for protein function prediction: the NoGO database. PLoS Comput Biol. 2014;10(6):e1003644. doi: 10.1371/journal.pcbi.1003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Zhang VW. Precision medicine for continuing phenotype expansion of human genetic diseases. BioMed Res Int. 2015;2015:745043. doi: 10.1155/2015/745043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Fu G, Wang J, Guo M. Predicting irrelevant functions of proteins based on dimensionality reduction (in Chinese) Sci Sin Inf. 2017;47(10):1349–1368. doi: 10.1360/N112017-00009. [DOI] [Google Scholar]
- Yu G, Lu C, Wang J. NoGOA: predicting noisy GO annotations using evidences and sparse representation. BMC Bioinform. 2017;18(1):350. doi: 10.1186/s12859-017-1764-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X-M, Wang Y, Chen L, Aihara K. Gene function prediction using labeled and unlabeled data. BMC Bioinform. 2008;9:57. doi: 10.1186/1471-2105-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Bousquet O, Lal TN, Weston J, Schölkopf B (2003) Learning with local and global consistency. In: Advances in neural information processing systems 16 [Neural information processing systems, NIPS 2003, December 8–13, 2003, Vancouver and Whistler, British Columbia, Canada]. MIT Press, pp 321–328
- Zhu C, Byrd RH, Lu P, Nocedal J. Algorithm 778: L-BFGS-B: Fortran subroutines for large-scale bound-constrained optimization. ACM Trans Math Softw. 1997;23(4):550–560. doi: 10.1145/279232.279236. [DOI] [Google Scholar]
- Zhu X, Ghahramani Z, Lafferty JD (2003) Semi-supervised learning using Gaussian fields and harmonic functions. In: Machine learning, proceedings of the twentieth international conference (ICML 2003), August 21–24, 2003, Washington, DC, USA. AAAI Press, pp 912–919
- Zitnik M, Leskovec J. Predicting multicellular function through multi-layer tissue networks. Bioinformatics. 2017;33(14):i190–i198. doi: 10.1093/bioinformatics/btx252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitnik M, Agrawal M, Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018;34(13):i457–i466. doi: 10.1093/bioinformatics/bty294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed in this review are available on the Human Phenotype Ontology website, “https://hpo.jax.org”.