

Abstract
Background
Replication stress response is crucial for the maintenance of a stable genome. POLDIP3 (DNA polymerase delta interacting protein 3) was initially identified as one of the DNA polymerase δ (Pol δ) interacting proteins almost 20 years ago. Using a variety of in vitro biochemical assays, we previously established that POLDIP3 is a key regulator of the enzymatic activity of Pol δ. However, the in vivo function of POLDIP3 in DNA replication and DNA damage response has been elusive.
Methods
We first generated POLDIP3 knockout (KO) cells using the CRISPR/Cas9 technology. We then investigated its biological functions in vivo using a variety of biochemical and cell biology assays.
Results
We showed that although the POLDIP3‐KO cells manifest no pronounced defect in global DNA synthesis under nonstress conditions, they are sensitive to a variety of replication fork blockers. Intriguingly, we found that POLDIP3 plays a crucial role in the activation and maintenance of the DNA damage checkpoint in response to exogenous as well as endogenous replication stress.
Conclusion
Our results indicate that when the DNA replication fork is blocked, POLDIP3 can be recruited to the stalled replication fork and functions to bridge the early DNA damage checkpoint response and the later replication fork repair/restart.
Keywords: DNA damage checkpoint, DNA polymerase delta, POLDIP3, replication stress
DNA replication stress is a major cause of genome instability and tumorigenesis. In this article, we show that a previously identified DNA polymerase δ interacting protein, POLDIP3, functions to bridge the early DNA replication stress response event (DNA damage checkpoint activation) and the late DNA replication stress response events (DNA polymerase δ, or Pol δ4, dependent fork repair/restart).

1. INTRODUCTION
DNA replication is one of the most important cellular processes, the fidelity of which is critical for the fitness of the organism. For example, in humans, errors resulting from DNA replication of the stem cells may account for two‐thirds of cancer incidence. 1 , 2 Therefore, organisms, from yeast to humans, have developed elaborate pathways to cope with replication stress and to ensure the accurate and complete replication of DNA during each cell cycle. 3 , 4 , 5 , 6
In eukaryotes, three DNA polymerases are involved in DNA replication during S phase: DNA polymerase alpha (α), DNA polymerase delta (δ), and DNA polymerase epsilon (ε). 6 DNA polymerase α (Pol α) initiates DNA synthesis by synthesizing a short RNA primer. DNA polymerase ε (Pol ε) is mainly responsible for the synthesis of the leading strand, whereas DNA polymerase δ (Pol δ) is mainly responsible for the synthesis of the lagging strand.
To identify novel Pol δ interacting proteins, we previously performed a two‐hybrid screening using the second subunit of human Pol δ, PolD2/p50, as the bait. 7 Two of the PolD2/p50 interacting proteins identified were novel with no known functions. We thus named them Polymerase δ interacting protein 38 (PDIP38) and polymerase δ interacting protein 46 (PDIP46) based on their sizes. PDIP46 was later renamed as DNA polymerase delta i nteracting protein 3 (POLDIP3). Subsequently, Richardson and colleagues reported the identification of SKAR, which is the mouse homologue of POLDIP3, as a binding partner and substrate of S6 kinase 1 (S6K1). 8 It was reported later that the human enhancer of rudimentary is also a binding partner of POLDIP3. 9 Using a variety of in vitro biochemical assays, we have established that POLDIP3 is a robust activator of Pol δ. 10 , 11 Using purified recombinant proteins, we showed that POLDIP3 directly binds PCNA and PolD2/p50 in vitro. Using a variety of in vitro functional assays, we further demonstrated that POLDIP3 stimulates the enzymatic activity of Pol δ in the primer extension and strand displacement assays as well as its ability to overcome a hairpin secondary structure during DNA synthesis. However, the in vivo function(s) of POLDIP3 in DNA replication and DNA damage response (DDR) remains largely unclear.
To elucidate the function of POLDIP3 in DNA replication and DDR in vivo, we first deleted POLDIP3 gene in a variety of human cancer cell lines using CRISPR/Cas9 technology. We found that the POLDIP3 knockout (KO) cells are sensitive to a variety of DNA replication blockers, including cisplatin [cis‐diamminedichloroplatinum (II), CDDP] and mitomycin C (MMC). We also found that POLDIP3 plays a crucial role in activating and maintaining the DNA damage checkpoint in response to the exogenous as well as the endogenous replication stress. Our results thus indicate that when DNA replication is blocked, POLDIP3 is recruited to the stalled replication fork and functions to bridge the early DNA damage checkpoint response and the later replication fork repair/restart.
2. MATERIALS AND METHODS
2.1. Expression and purification of recombinant proteins
For the pull‐down assays, GST‐POLDIP3 was expressed as previously described. 10 His tagged RPA34 was subcloned into the PET21b vector and expressed in Escherichia coli BL21DE3 (pLys) by induction with 0.1 mM isopropyl thiogalactopyranoside (IPTG) overnight. His‐tagged Tipin was purchased from Addgene and expressed in Sf9 cells. GST‐ or His‐tagged proteins were purified using either glutathione beads (GE Healthcare Life Sciences) or Ni‐NTA agarose (Qiagen).
2.2. Cell culture
All cell lines used in this study were purchased from ATCC. All the cells were grown in Dulbecco's modified Eagle's medium (DMEM, Fisher Scientific) supplemented with 10% of fetal bovine serum (FBS, Atlanta Biologicals). All the cells were grown in a humidified incubator with 5% CO2 at 37°C. 31
2.3. Western blotting, coimmunoprecipitation, and GST pull‐down assays
Western blotting for POLDIP3 and other proteins was performed as described previously. 10 A549 cells were treated with 10 J/m2 ultra violet C (UVC) or without UVC for 4 h and then used to prepare protein lysates. Coimmunoprecipitation was performed using POLDIP3 antibody or a control IgG. The lysates were centrifuged at 14000 rpm for 10 min. POLDIP3 antibody or the control IgG were added to the lysates and the mixtures were incubated at 4°C overnight. The next day, A/G agarose beads (Santa Cruz Biotechnology) were added and the mixtures were incubated at 4°C for another hour. The beads were spun down and washed eight times with radioimmunoprecipitation assay buffer (RIPA buffer) and then suspended in 2X sodium dodecyl sulfate (SDS) loading buffer. The bound proteins were analyzed using sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotted with antibody against RPA34 (Cell Signaling, 2208) or Tipin (Santa Cruz Biotechnology, sc‐135580) or POLDIP3. 10 For GST pull‐down assay, 1 μg of GST‐POLDIP3 or GST was incubated with 1 μg of His‐RPA34 or His‐Tipin in 800 μl of binding buffer (50 mM Tris–HCl, pH 7.8, 1 mM ethylenediaminetetraacetic acid [EDTA], 150 mM NaCl, 0.1% NP‐40, and 0.2 mM phenylmethanesulfonyl fluoride) for 4 h with rotation at 4°C. 15 μl of packed glutathione beads was added and further rotated for another hour. The beads were spun down at 2500 rpm for 5 min, washed eight times with the binding buffer, and suspended in 1X SDS loading buffer. The bound proteins were analyzed by SDS‐PAGE and Western blotted with either antibody against RPA34 (Cell Signaling, 2208) or Tipin (Abcam, ab172792).
2.4. CRISPR/Cas9‐mediated POLDIP3 gene knockout or shRNA‐mediated POLDIP3 knockdown
CRISPR/Cas9‐mediated human POLDIP3 gene knockouts were done according to the manufacturer's instructions (Santa Cruz Biotechnology). Briefly, 1.5 × 105 cells/well were seeded onto six‐well culture plates in 2.5 ml of antibiotic‐free medium per well and allowed to grow to 70% confluency. The cells were then transfected with 2 μg of human POLDIP3 CRISPR/Cas9 KO plasmid (sc‐413603) and POLDIP3 homology‐dependent repair (HDR) plasmid (sc‐413603‐HDR‐2) (Santa Cruz Biotechnology) using 8 μl of UltraCruz transfection reagent (Santa Cruz Biotechnology). Two days after transfection, successful co‐transfection of the CRISPR/Cas9 KO plasmid and the HDR plasmid, which expresses a red fluorescent protein (RFP), was visually confirmed by the detection of RFP via an inverted fluorescence microscope. Stably transfected clones were selected by adding puromycin (1.0 μg/ml) and transfected with Cre vector (sc‐418 923) for the removal of genetic material flanked by the LoxP sites. Control CRISPR/Cas9 plasmid (sc‐418 922) containing a nontargeting 20‐nt scrambled guide RNA was used as the negative control.
Human POLDIP3 shRNA (category no.: RHS4696‐200753940) and a control shRNA (category no.: RHS4743) constructs were purchased from Horizon Discovery Biosciences. The shRNA expression lentivirus were packaged by co‐transfection of the packaging plasmids Δ8.9 and VSVG into HEK293T cells. Forty‐eight hours after transfection, the supernatants were filtered through a sterile 0.45‐μm filter and used for infection of A549 cells. After infection, cells were selected by the addition of 2–3 μg/ml of puromycin and were grown for one more week. Subsequently, cells were maintained in 0.5 μg/ml of puromycin.
2.5. Clonogenic cell survival assays
Clonogenic cell survival assays were performed as previously described. 31 Briefly, A549 cells were first seeded onto 60‐mm cell culture dishes (1200 cells per dish). The attached cells were then treated with different concentrations of CDDP (Sigma‐Aldrich) or MMC (Sigma‐Aldrich) for 3 h. Dimethyl sulfoxide (DMSO) was used as the negative control. After the treatment, the medium was removed, and the cells were washed once with phosphate‐buffered saline (PBS) and replaced with fresh growth medium. The dishes were incubated until the cells in the negative control dishes formed sufficiently large colonies (11–14 days). Subsequently, the media from each dish was removed and then washed with PBS. The colonies were fixed in 5 ml of acetic acid/methanol 1:7 (vol/vol) for 20 min and stained with 5 ml of 0.5% (w/v) crystal violet (Sigma‐Aldrich) in H2O for 4 h. Finally, excess crystal violet was removed using H2O and the dishes were dried. Digital images of the colonies were obtained using a scanning device.
2.6. Immunofluorescent staining
Parental U2OS and U2OS‐POLDIP3‐KO cells (2.0 × 105 cells/well) were first seeded onto six‐well plates. The cells were then transfected twice with different siRNAs and replated on coverslips. The cells were then used for immunostaining 72 h later. Briefly, the cells were first fixed with 4% paraformaldehyde for 15 min, permeabilized at room temperature for 15 min with 0.1% Triton X‐100 in PBS, and then blocked with 2% bovine serum albumin (BSA) in PBS for 1 h. The cells were labeled with different primary antibodies and the appropriate Alexa‐488 (Invitrogen) and Alexa‐546 (Invitrogen) conjugated secondary antibodies. Finally, the cells were washed thrice with PBS for 10 min each time, dried, and mounted using ProLong Antifade reagent with 4′,6‐diamidine‐2′‐phenylindole dihydrochloride (DAPI, Invitrogen). The slides were observed, and the images were obtained using a Zeiss Axiovert 200 M. All the images were obtained at a magnification of 40X. Antibodies used for immunofluorescent staining include Chk1‐pS345 (Cell Signaling, 2348), TRF2 (Millipore, 05–521), γH2AX (Abcam, ab81299), and POLDIP3 as previously reported. 10
2.7. Graphing and statistics
The results were presented as means ± standard errors of the mean. All statistical tests and graphing were performed using GraphPad Prism, version 7.01. Unpaired t‐test was applied for statistical analysis. p‐values ≤0.05 were considered statistically significant.
2.8. DNA fiber assay
Cells (1.0 × 105 cells/well) were seeded onto six‐well plates and grew to 70% confluency. DNA fiber assay was performed as described. 33 Briefly, 5‐iodo‐2′‐deoxyuridine (IdU, Sigma‐Aldrich) was added to the cell culture to a final concentration of 25 μM. The cells were incubated for 30 min. The media was then removed, and the cells were washed thrice with serum‐free media. Growth media containing 100 μM CldU (Sigma‐Aldrich) was added to the plates, and the cells were incubated for another 30 min. After the double labeling, the cells were washed with ice‐cold PBS, harvested, and resuspended in 200–400 cells/μl ice‐cold PBS. 2 μl of the cell suspension was spotted at the end of the microscope slides (Fisherbrand 15–188‐48) and air‐dried for 5 min. Subsequently, 7 μl of lysis buffer (200 mM Tris–HCl, pH 7.5, 50 mM EDTA, and 0.5% SDS) was added to the cell suspension, gently mixed using a pipette tip, and incubated for 2 min. After cell lysis, the slides were tilted to 15 degree to allow the DNA fibers to spread along the slides. Once the spreaded fibers had dried, the slides were fixed in methanol/acetic acid (3:1) solution in a staining jar for 10 min. Subsequently, the slides were washed in distilled water and immersed in 2.5 M HCl for 80 min. After DNA denaturation, the slides were washed thrice with PBS for 5 min each time, and then the blocking solution (5% BSA in PBS) was applied for 20 min. After the blocking buffer was removed, the slides were incubated with monoclonal anti‐IdU antibody (Sigma‐Aldrich, SAB3701448) and monoclonal anti‐BrdU antibody (BU1/75) (Thermo Scientific, MA1‐82088) for 2 h at room temperature. Subsequently, the slides were washed thrice with PBS for 5 min each time. The secondary antibodies (1:500 for goat anti‐mouse Alexa Fluor 594, 1:200 for goat anti‐rat Alexa Fluor 488) were applied on each slide for 1 h. After the second antibodies were removed, the slides were washed thrice with PBS for 5 min each time, and subsequently a drop of mounting medium was spotted onto each slide; coverslips were applied by gently pressing down. The slides were sealed with transparent nail polish, allowed to dry, and stored at −20°C for further analysis. The slides were observed, and images were obtained using a Zeiss Axiovert 200 M with a black and white CCD AxioCam and pseudo‐colored with Axiovision 4.8 software. All the images were obtained at 100× magnification and were analyzed using the ImageJ (http:rsbweb.nih.gov/ij).
3. RESULTS
3.1. POLDIP3‐deficient cells are sensitive to replication stress inducers
POLDIP3 was first identified as a Pol δ interacting protein in a two‐hybrid screening. 7 Our previous studies using purified recombinant POLDIP3 have demonstrated that POLDIP3 can robustly stimulate the various enzymatic activities of Pol δ in vitro. 10 However, the in vivo function of POLDIP3 in DNA replication and DDR remains largely unclear.
To investigate the in vivo function of POLDIP3, we first genetically deleted human POLDIP3 gene using CRISPR/Cas9 technology in multiple cancer and transformed cell lines (A549, DU145, and HEK293T) (Figure 1A and Figure S1). We were able to successfully recover multiple POLDIP3 KO clones from each cell line. The growth rates of the POLDIP3‐KO cells were indistinguishable from their parental cells, indicating that POLDIP3 is dispensable for the viability of these cells. We then performed cell cycle analysis of two independent POLDIP3‐KO clones of the A549 cells and found no pronounced difference in G1, S, and G2 distribution when compared to the wild‐type (WT) A549 cells (Figure 1B), indicating that POLDIP3 is also dispensable for cell cycle regulation under nonstressed conditions.
FIGURE 1.
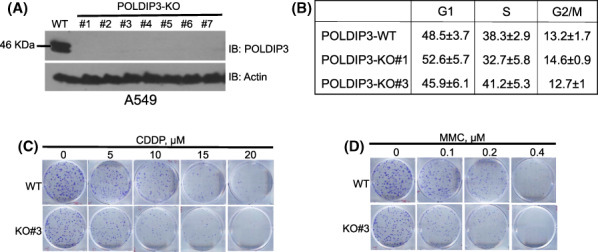
POLDIP3 (DNA polymerase delta interacting protein 3)‐deficient cells are sensitive to replication stress inducers, CDDP (cis‐diamminedichloroplatinum (II)) and MMC (mitomycin C). (A) Cell lysates from the wild‐type (WT) A549 cells and POLDIP3 knockout (KO) A549 clones 1–7 were immunoblotted (IB) with different antibodies, as indicated on the right. (B) Cell cycle analysis of the WT A549 cells (POLDIP3‐WT) and two POLDIP3‐KO clones (#1 and #3). (C, D) Equal number of POLDIP3‐WT and POLDIP3‐KO#3 cells were treated with different concentrations of (C) CDDP and (D) MMC for 3 h and allowed to grow in fresh growth medium for 11 more days. Cells were finally stained with crystal violet.
Next, we tested whether the POLDIP3‐KO cells are sensitive to DNA damaging agents. Interestingly, as shown in Figure 1C,D, POLDIP3‐KO cells are sensitive to CDDP and MMC treatment, both of which are known to form intra‐ and inter‐strand crosslinks with DNA and block the progression of DNA replication forks. These data suggest that POLDIP3 is likely involved in the regulation of DNA replication stress response and/or repair/restart of the stalled replication forks.
3.2. POLDIP3‐deficient cells are defective in the activation and maintenance of DNA damage checkpoint
To further investigate the role of POLDIP3 in DNA replication stress response, we first performed a focused screening of known DDR proteins that may interact with POLDIP3 by immunoprecipitation using A549 cell lysates. Interestingly, we detected strong interactions between POLDIP3 and RPA34 and between POLDIP3 and Tipin (Figure 2A). Furthermore, we also observed strong interactions between POLDIP3 and RPA34 and between POLDIP3 and Tipin using purified recombinant proteins (Figure 2B), suggesting that the in vivo interactions observed in Figure 2A are most likely through direct protein–protein interaction. RPA34 is part of the heterotrimeric complex, called replication protein A (RPA), which selectively binds and protects the single‐stranded DNA. Tipin is part of the so‐called fork protection complex (FPC), which is a key regulator of replisome. In addition to their functions in DNA replication, RPA34 and Tipin are also required for DNA damage checkpoint activation in response to replication stress. 12 , 13
FIGURE 2.
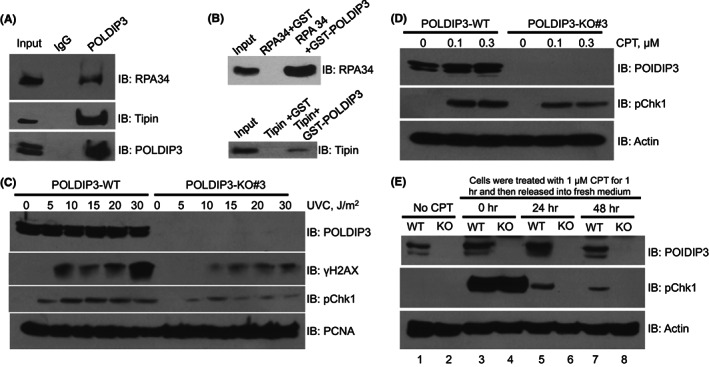
POLDIP3 (DNA polymerase delta interacting protein 3) promotes and sustains the DNA damage checkpoint in response to replication stress. (A) A549 whole cell lysates were used for immunoprecipitation using either normal rabbit IgG or POLDIP3 antibody and then immunoblotted (IB) with different antibodies, as indicated on the right. (B) GST pull‐down assays using purified recombinant proteins, as indicated on the top. (C) Wild‐type (POLDIP3‐WT) and POLDIP3 knockout (POLDIP3‐KO#3) A549 cells were treated with different doses of UVC for 4 h. (D) POLDIP3‐WT and POLDIP3‐KO#3 A549 cells were treated with different doses of CPT (camptothecin) for 1 h. (E) POLDIP3‐WT (WT) and POLDIP3‐KO#3 (KO) A549 cells were first treated with 1 μM CPT for 1 h and then grew in the fresh medium without any CPT for the indicated time. Cell lysates were prepared and immunoblotted (IB) with different antibodies, as indicated on the right.
Because POLDIP3‐KO cells grew normally and manifested no pronounced cell cycle defects (Figure 1B), we speculated that POLDIP3 is not required for global DNA replication. Indeed, when we performed DNA fiber assay, we did not observe any pronounced difference between the POLDIP3‐WT and POLDIP3‐KO cells (Figure S2), indicating that under nonstressed conditions, POLDIP3 is dispensable for genome‐wide DNA synthesis.
To test whether POLDIP3 is involved in DNA damage checkpoint activation, we treated both POLDIP3‐WT and POLDIP3‐KO cells with different doses of UVC, which induces the formation of cyclobutane pyrimidine dimers and other forms of DNA lesions thus impeding the progression of the DNA replication machinery, and examined checkpoint activation by immunoblotting the whole cell lysate with antibodies recognizing γH2AX and the phosphorylated serine‐345 of Chk1 (pChk1) (Figure 2C). Interestingly, we found a profound reduction in both γH2AX and pChk1 in POLDIP3‐KO cells, suggesting that POLDIP3 plays a critical role in the activation of replication stress checkpoint. Consistently, when POLDIP3 was depleted using shRNA, the UVC‐induced DNA damage checkpoint response was also severely compromised compared to the control shRNA infected cells (Figure S3). Similar to the treatment with UVC, when POLDIP3‐KO cells were treated with different doses of camptothecin (CPT), a topoisomerase I inhibitor that also blocks the progression of the replisome, we found that the induction of pChk1 is also attenuated (Figure 2D). To further test if POLDIP3 also plays a role in the maintenance of DNA damage checkpoint, we first treated both POLDIP3‐WT and POLDIP3‐KO cells with a higher dose of CPT (1 μM) for 1 h so that we could monitor the activated DNA damage checkpoint for longer periods. After CPT was removed, the cells were allowed to recover in normal growth medium for 24 or 48 h. Immediately after the higher dose of CPT (1 μM) treatment (Figure 2E, the 0 h time point), we did not observe any difference in pChk1 level between POLDIP3‐WT and POLDIP3‐KO cells, suggesting that the higher dose of CPT also activated the POLDIP3‐independent pathway to activate the DNA damage checkpoint. However, the POLDIP3‐KO cells failed to sustain the DNA damage checkpoint 24 and 48 h after the 1 μM CPT treatment (Figure 2E, lanes 6 and 8), whereas residue pChk1 can still be observed in the POLDIP3‐WT cells (Figure 2E, lanes 5 and 7). This strongly indicates that POLDIP3 is also required for the maintenance of the replication stress checkpoint.
Collectively, our data strongly indicate that although POLDIP3 is dispensable for routine DNA replication under nonstressed conditions, it does play a critical role in the initial activation as well as the maintenance of replication stress checkpoint in vivo.
3.3. In response to endogenous replication stress at ALT telomeres, POLDIP3 is recruited to the stalled replication forks and facilitates the activation of DNA damage checkpoint
We recently reported that depletion of FANCM induces a drastic increase in replication stress at the telomeres in cells that have adopted the alternative lengthening of telomere (ALT) pathway due to the accumulation of the TERRA R‐loops, hybrid molecules formed between the noncoding RNA, TERRA, and telomeric DNA. 14 , 15 We thus referred to this endogenous replication stress model as MR‐SAT (FANCM deficiency–induced replication stress at ALT telomeres). We proposed that the MR‐SAT model can be used to elucidate the molecular mechanism of replication stress responses at a specific endogenous locus. Intriguingly, POLDIP3 was previously identified as one of the ALT telomere‐associated proteins. 16 , 17 However, the exact function of POLDIP3 at the ALT telomeres was not further investigated.
We first examined whether POLDIP3 can be recruited to the ALT telomeres that experience replication stress using the FANCM‐depleted U2OS cells as previously described. 14 , 15 U2OS cells lack telomerase activity and depend on the ALT pathway to maintain their telomeres. We induced robust telomeric replication stress using two different siRNAs targeting FANCM. Cells were then co‐stained with antibodies recognizing POLDIP3 and TRF2, which is a component of the Shelterin complex and is often used as the marker for telomeres. 18 As previously reported, 14 depletion of FANCM in U2OS cells induces a drastic increase in DNA damage and replication stress at the telomeres, as reflected by the dramatically increased appearance of TRF2 foci colocalized with γH2AX foci (Figure 3A) and pChk1 foci (Figure 3B), respectively. Using this MR‐SAT model, we observed a robust recruitment of POLDIP3 to the ALT telomeres experiencing severe replication stress (Figure 3C). Thus, POLDIP3 joins a cohort of DDR, DNA repair, and DNA replication proteins that have been shown to be recruited to these telomeric foci upon FANCM depletion. 14 , 15
FIGURE 3.
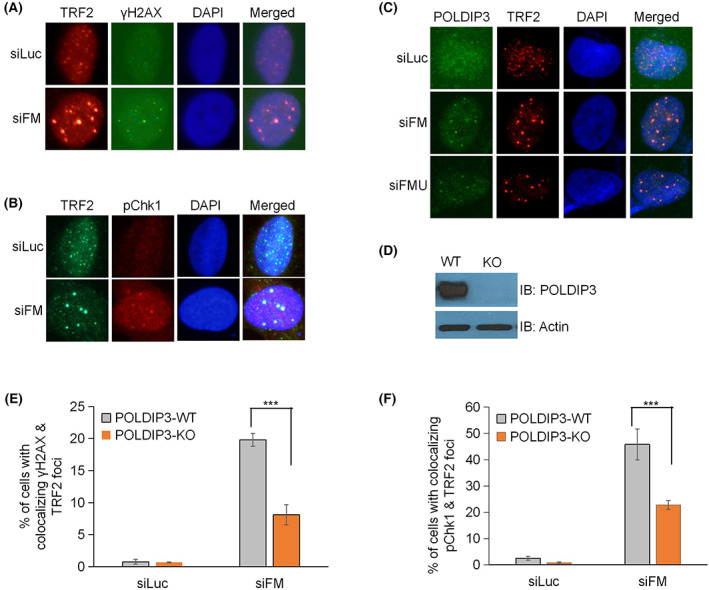
POLDIP3 (DNA polymerase delta interacting protein 3) is recruited to telomeres in FANCM‐deficient ALT (alternative lengthening of telomere) cells and facilities the activation of replication stress–induced DNA damage checkpoint. (A, B) U2OS cells were transfected with siRNA targeting either luciferase (siLuc, negative control) or FANCM (siFM) and then stained with antibodies, as indicated on the top. All the nuclei were stained with DAPI (4′,6‐diamidine‐2′‐phenylindole dihydrochloride) (blue). (C) U2OS cells were transfected with siRNA targeting either luciferase (siLuc) or FANCM (siFM and siFMU) and then stained with antibodies, as indicated on the top. All the nuclei were stained with DAPI (blue). (D) POLDIP3 gene was deleted in U2OS cells using CRISPR/Cas9 technology. Cell lysate from wild‐type (WT) and POLDIP3 knockout (KO) U2OS cells were prepared and immunoblotted (IB) with different antibodies, as indicated on the right. (E, F) POLDIP3‐WT and POLDIP3‐KO U2OS cells were transfected with either siLuc or siFM and then stained with antibodies recognizing TRF2 and γ‐H2AX, or TRF2 and pChk1. All the nuclei were stained with DAPI. Cells with more than three colocalizing TRF2 and γH2AX foci, or TRF2 and pChk foci, were identified. More than 200 POLDIP3‐WT (gray bar) and POLDIP3‐KO (orange bar) cells were counted. All error bars are standard deviation of the mean obtained from three different experiments. Standard two‐tailed Student's t‐test: ***p < 0.001.
To further investigate the potential role of POLDIP3 in the replication stress response at ALT telomeres, we first deleted POLDIP3 gene in U2OS cells using CRISPR/Cas9 technology (Figure 3D) and then examined the replication stress response by further depleting FANCM using siRNA (siFM). Interestingly, the increase in both γH2AX and pChk1 telomeric foci that was usually observed after FANCM depletion was severely suppressed (Figure 3E,F), indicating that POLDIP3 also plays an important role in the activation of the replication stress response at ALT telomeres.
Taken together, these experiments reveal a novel and important additional facet of POLDIP3 function in regulating the replication stress response at ALT telomeres.
4. DISCUSSION
Replication stress response is vital for the accurate and complete replication of eukaryotic genome in every cell cycle. Dysfunction or misregulation of replication stress response will lead to a variety of human diseases, including cancers. 3 Here we report a novel function of POLDIP3 in vivo. Our data demonstrate that POLDIP3 plays an important role in the initial activation as well as the subsequent maintenance of the DNA damage checkpoint induced by either the exogenous or the endogenous replication blockage. These findings are unexpected because POLDIP3 was initially identified as a Pol δ interacting protein. 7 Regarding our new findings, we propose that in cells experiencing replication stress, POLDIP3 is uniquely positioned to bridge the early DDR events, such as the DNA damage checkpoint activation, and the later events, such as the repair/restart of stalled/collapsed replication forks as discussed later (Figure 4).
FIGURE 4.
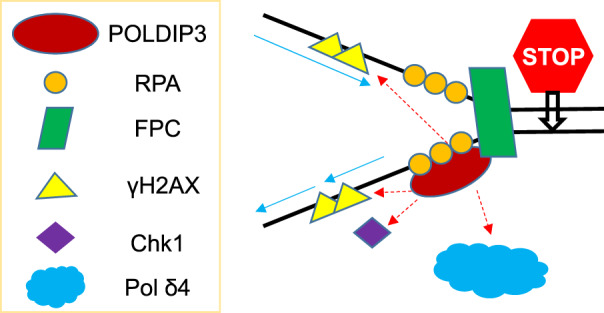
A model of POLDIP3 (DNA polymerase delta interacting protein 3) in replication stress response. In response to replication stress, POLDIP3 functions to bridge the early event (checkpoint activation) with the late events (DNA polymerase δ, or Pol δ4, dependent fork repair/restart).
4.1. How does POLDIP3 facilitate the activation of the DNA damage checkpoint?
One of the important questions related to our new findings is, how does POLDIP3 facilitate the activation and maintenance of the DNA damage checkpoint? We showed that POLDIP3 directly interacts with RPA34, which is part of the trimeric RPA complex, as well as Tipin, which is part of the FPC (Figure 2A,B). RPA facilitates the recruitment of ATR‐ATRIP to the vicinity of stalled replication forks and activates the DNA damage checkpoint. 12 Recent cryo‐EM and reconstitution studies using the yeast homologue of FPC suggest that the FPC situates ahead of the CMG helicase complex to modulate the rate of replisome. 19 It is thus conceivable that the binding of POLDIP3 to RPA and Tipin facilitates its recruitment to the stalled replication forks. Our findings that POLDIP3 depletion leads to the attenuation of γH2AX and pChk1 renders these protein–protein interactions functionally significant because it implicates POLDIP3 in the DDR and replication stress checkpoint activation.
The process of assembly and activation of the replication stress response at the stalled replication forks is complex but mainly involves the recruitment and activation of ATR‐ATRIP and subsequently the recruitment and activation of Chk1. 20 , 21 Together they prevent the stalled forks from collapsing and are also involved in the subsequent events of repair and restart of the stalled forks. Our studies reveal here that POLDIP3 deletion greatly affects the activation of Chk1 at the stalled replication forks, indicating that it plays a crucial role in the replication stress checkpoint activation and thus opens up a new perspective of its functions that goes beyond its functions in relation to Pol δ. Clearly further study still needs to be performed. A minimal model of what our findings have demonstrated in terms of its recruitment to stalled replication forks and the implications of its interactions for the DNA damage checkpoint response is shown in Figure 4. Further investigation to understand the multifaceted functions of POLDIP3 in the DNA replication stress response is certainly warranted.
Besides this present work, which implicates POLDIP3 in the replication stress–induced DNA damage checkpoint activation in the context of the ALT telomere model system, that is, the MR‐SAT, other recent studies have also pointed to an involvement of POLDIP3 in replication stress. Most recently, Bjorkman and colleagues reported that POLDIP3 associates with RTEL1, an important DNA helicase, and together they facilitate the disruption of R‐loops ahead of a moving replication fork. 22 Moreover, RTEL1 is also implicated in the abundance and localization of TERRA RNA. 23 We and others have shown that TERRA R‐loops are the major replication barriers at ALT telomeres. 15 , 24 , 25 Here it is also noted that POLDIP3 possesses an RNA recognition motif, which can directly bind RNA. 8 We thus speculate that POLDIP3 may also be involved in the regulation of the biogenesis of TERRA, the localization of TERRA to telomeres, or the formation of TERRA R‐loops by directly binding to TERRA RNA.
4.2. POLDIP3 and the difficult‐to‐replicate regions
It is somewhat surprising to find that POLDIP3 is not required for the viability of the four human cell lines tested here (A549, DU145, HEK293T, and U2OS) given that it plays such an important role in the regulation of Pol δ. 10 , 11 On the other hand, though the three larger subunits of Pol δ4, PolD1/p125, PolD3/p68, and PolD2/p50, are required for the viability of mammalian cells, 26 , 27 , 28 , 29 , 30 the smallest subunit of Pol δ4, PolD4/p12, is not, 31 suggesting that Pol δ3, which consists of PolD1/p125, PolD3/p68, and PolD2/p50, may be sufficient to support the DNA replication under nonstressed conditions and the survival of most, if not all, mammalian cells. The logical question then becomes, what is the in vivo function of Pol δ4? Our recent studies have shed some new light on that. We found that the loss of PolD4/p12 led to a defect in the homologous recombination repair (HDR) pathway as well as sensitization to PARP inhibitors, suggesting that the major in vivo function of Pol δ4 is to facilitate the HDR. 31 It is well established that certain genomic regions are more difficult to replicate (DTR) than others, including centromeres, common fragile sites, rDNA loci, and telomeres. A unique HDR pathway, called break‐induced replication, has been implicated in replicating through many of the DTRs, including the ALT telomeres. 32 We thus speculate that both the checkpoint function of POLDIP3 and its function as the key activator of Pol δ4 will likely be important for the replisome to overcome various replication barriers in the DTRs.
AUTHOR CONTRIBUTIONS
Sufang Zhang, Ernest Y. C. Lee, Marietta Y. W. T. Lee, and Dong Zhang conceived the ideas and designed the experiments. Sufang Zhang and Dong Zhang performed the experiments and analyzed the data. Sufang Zhang, Ernest Y. C. Lee, Marietta Y. W. T. Lee, and Dong Zhang prepared the manuscript.
CONFLICT OF INTEREST
None, Dong Zhang is an Editorial Board member of AMEM and a co‐author of this article. To minimize bias, he was excluded from all editorial decision‐making related to the acceptance of this article for publication.
Funding information
NIEHS, Grant/Award Number: R01 ES014737 USAMRDC, Grant/Award Number: W81XWH‐18‐1‐0353. The research in the D.Z. lab is supported by the research fund from NYIT.
Supporting information
Figure S1
Figure S2
Figure S3
ACKNOWLEDGMENTS
This research was supported in part by R01 ES014737 to Marietta Y. W. T. Lee and a Concept Award from USAMRAA W81XWH‐18‐1‐0353 to Sufang Zhang. Research in Dong Zhang's laboratory was supported by the research fund from NYIT.
Zhang S, Lee EYC, Lee MYW, Zhang D. DNA polymerase delta interacting protein 3 facilitates the activation and maintenance of DNA damage checkpoint in response to replication stress. Anim Models Exp Med. 2022;5:461‐469. doi: 10.1002/ame2.12274
Contributor Information
Sufang Zhang, Email: zhangsufang@gmail.com.
Marietta Y. W. T. Lee, Email: marietta_lee@nymc.edu.
Dong Zhang, Email: dzhang12@nyit.edu.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and in Supporting Information.
REFERENCES
- 1. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gaillard H, Garcia‐Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15(5):276‐289. [DOI] [PubMed] [Google Scholar]
- 4. Burgers PMJ, Kunkel TA. Eukaryotic DNA Replication Fork. Annu Rev Biochem. 2017;86:417‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berti M, Cortez D, Lopes M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol. 2020;21(10):633‐651. [DOI] [PubMed] [Google Scholar]
- 6. Johansson E, Dixon N. Replicative DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5(6):a012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu L, Rodriguez‐Belmonte EM, Mazloum N, Xie B, Lee MYWT. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem. 2003;278(12):10041‐10047. [DOI] [PubMed] [Google Scholar]
- 8. Richardson CJ, Bröenstrup M, Fingar DC, et al. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol. 2004;14(17):1540‐1549. [DOI] [PubMed] [Google Scholar]
- 9. Smyk A, Szuminska M, Uniewicz KA, Graves LM, Kozlowski P. Human enhancer of rudimentary is a molecular partner of PDIP46/SKAR, a protein interacting with DNA polymerase delta and S6K1 and regulating cell growth. FEBS J. 2006;273(20):4728‐4741. [DOI] [PubMed] [Google Scholar]
- 10. Wang X, Zhang S, Zheng R, et al. PDIP46 (DNA polymerase delta interacting protein 46) is an activating factor for human DNA polymerase delta. Oncotarget. 2016;7(5):6294‐6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee M, Wang X, Zhang S, Zhang Z, Lee E. Regulation and modulation of human DNA polymerase delta activity and function. Genes (Basel). 2017;8(7):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science. 2003;300(5625):1542‐1548. [DOI] [PubMed] [Google Scholar]
- 13. Chou DM, Elledge SJ. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci U S A. 2006;103(48):18143‐18147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, Zhang D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A. 2017;114(29):E5940‐E5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pan X, Chen Y, Biju B, et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R‐loops. Sci Rep. 2019;9(1):19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia‐Exposito L, Bournique E, Bergoglio V, et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alternative Lengthening of Telomeres. Cell Rep. 2016;17(7):1858‐1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao XD, Tu LC, Mir A, et al. C‐BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9‐APEX2. Nat Methods. 2018;15(6):433‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lim CJ, Cech TR. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat Rev Mol Cell Biol. 2021;22(4):283‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baretic D et al. Cryo‐EM Structure of the Fork Protection Complex Bound to CMG at a Replication Fork. Mol Cell. 2020;78(5):926‐940 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yazinski SA, Zou L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu Rev Genet. 2016;50:155‐173. [DOI] [PubMed] [Google Scholar]
- 21. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bjorkman A et al. Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R‐loop resolution. Genes Dev. 2020;34(15–16):1065‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghisays F, Garzia A, Wang H, et al. RTEL1 influences the abundance and localization of TERRA RNA. Nat Commun. 2021;12(1):3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM. RNaseH1 regulates TERRA‐telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun. 2014;5:5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silva B, Pentz R, Figueira AM, et al. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R‐loops. Nat Commun. 2019;10(1):2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uchimura A, Hidaka Y, Hirabayashi T, Hirabayashi M, Yagi T. DNA polymerase delta is required for early mammalian embryogenesis. PLoS One. 2009;4(1):e4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murga M, Lecona E, Kamileri I, et al. POLD3 Is Haploinsufficient for DNA Replication in Mice. Mol Cell. 2016;63(5):877‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou Z, Wang L, Ge F, et al. Pold3 is required for genomic stability and telomere integrity in embryonic stem cells and meiosis. Nucleic Acids Res. 2018;46(7):3468‐3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blomen VA, Májek P, Jae LT, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350(6264):1092‐1096. [DOI] [PubMed] [Google Scholar]
- 30. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR‐Cas9 system. Science. 2014;343(6166):80‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang S, Chao HH, Wang X, Zhang Z, Lee EYC, Lee MYWT. Loss of the p12 subunit of DNA polymerase delta leads to a defect in HR and sensitization to PARP inhibitors. DNA Repair (Amst). 2019;73:64‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Epum EA, Haber JE. DNA replication: the recombination connection. Trends Cell Biol. 2021;32:45‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nieminuszczy J, Schwab RA, Niedzwiedz W. The DNA fibre technique ‐ tracking helicases at work. Methods. 2016;108:92‐98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Data Availability Statement
All data generated or analyzed during this study are included in this published article and in Supporting Information.