

Abstract
With the identification of activating mutations in BRAF across a wide variety of malignancies, substantial effort was placed in designing safe and effective therapeutic strategies to target BRAF. These efforts have led to the development and regulatory approval of three BRAF inhibitors as well as five combinations of a BRAF inhibitor plus an additional agent(s) to manage cancer such as melanoma, non-small cell lung cancer, anaplastic thyroid cancer, and colorectal cancer. To date, each regimen is effective only in patients with tumors harboring BRAFV600 mutations and the duration of benefit is often short-lived. Further limitations preventing optimal management of BRAF mutant malignancies are that treatments of non-V600 BRAF mutations have been less profound and combination therapy is likely necessary to overcome resistance mechanisms, but multi-drug regimens are often too toxic. With the emergence of a deeper understanding of how BRAF mutations signal through the RAS/mitogen activated protein kinase (MAPK) pathway, newer RAF inhibitors are being developed that may be more effective and potentially safer and more rational combination therapies are being tested in the clinic. In this review, we identify the mechanics of RAF signaling through the RAS/MAPK pathway, present existing data on single-agent and combination RAF targeting efforts, describe emerging combinations, summarize the toxicity of the various agents in clinical testing, and speculate as to where the field may be headed.
Introduction/Background
In 2002, the first report of oncogenic mutations in the BRAF gene detailed that point mutations at the V600 position (initially incorrectly described as being at the V599 position) were common in melanoma but also present in other cancers such as colorectal cancer (CRC) and non-small cell lung cancer (NSCLC) (1). These mutations constitutively activated the mutant protein and, as a result, the RAS/mitogen activated protein kinase (MAPK) pathway. Soon after, mutations at other codons in BRAF were identified in many types of malignancies (Figure 1) and shown to variably activate the kinase and, as a result, the RAS/MAPK pathway (2). These discoveries were made in an era when the first small molecule inhibitors targeting oncogenic mutations were being tested, leading to great excitement that effective therapies would be developed for patients with solid tumor malignancies harboring BRAF mutations.
Figure 1: Frequency of BRAF alterations across cancers.
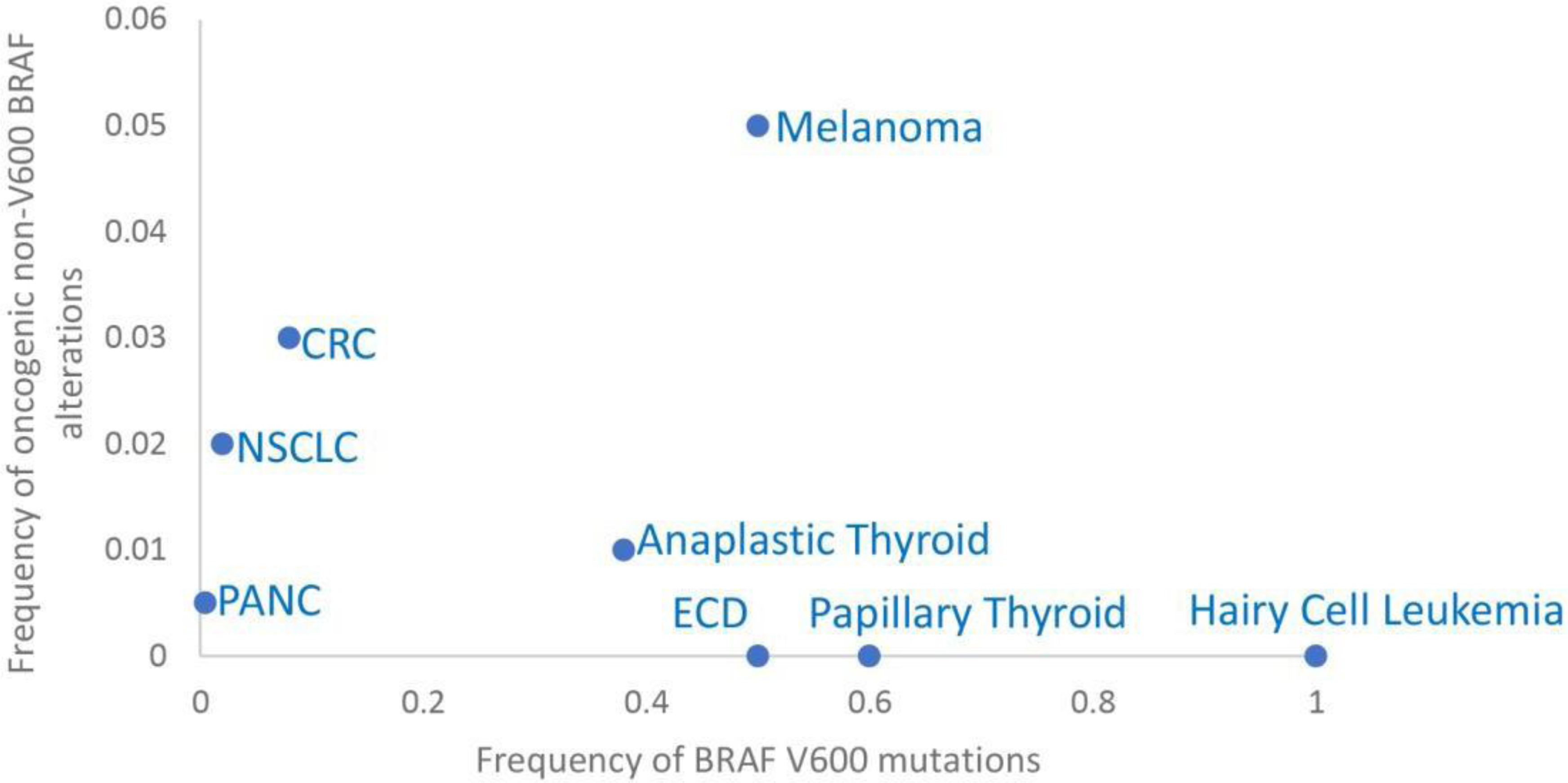
Graph showing portion of V600 BRAF and oncogenic non-V600 BRAF alterations across cancers. CRC: colorectal cancer. ECD: Erdheim Chester Disease. NSCLC: non-small cell lung cancer. PANC: pancreatic cancer.
Indeed, over the course of the subsequent decade, the clinical development of the first potent and specific inhibitor of oncogenic BRAFV600 mutations, vemurafenib, successfully led to approval by the FDA in 2011 to treat patients with advanced, BRAFV600 mutant melanoma (3–5). Over the next ten years, two additional BRAF inhibitors, dabrafenib and encorafenib, have also been approved to treat melanoma, as have each of these BRAF inhibitors in combination with MEK inhibitors for this same indication, and the combination of dabrafenib and trametinib in the adjuvant setting for patients with Stage III, resected, BRAFV600 mutant melanoma (6–10). Moving beyond melanoma, dabrafenib and trametinib have been approved to treat advanced, BRAFV600 NSCLC and locally advanced or metastatic anaplastic thyroid cancer, and encorafenib has been approved in combination with the anti-epidermal growth factor receptor (EGFR) monoclonal antibody, cetuximab, to treat advanced, BRAFV600 mutant CRC (Table 1) (11–13).
Table 1.
Categories of RAF Inhibitors and clinical compounds.
| αC-OUT/DFG-IN (CODI) | αC-IN/DFG-OUT (CIDO) | ||
|---|---|---|---|
| RAF monomer-selective | RAF monomer selective / Paradox Breakers | Equipotent for RAF dimers and monomers | RAF dimer-selective |
| Vemurafenib (Roche/Genentech) |
PLX8394 (Fore Biotherapeutics) |
LY 3009120 (Eli Lilly) |
Belvarafenib (GDC-5573, HM95573 or RO7223619) (Roche/Genentech) |
| Dabrafenib (Novartis) |
PLX7904 | AZ628 (Astrazeneca) |
LXH254 (Novartis) |
| Encorafenib (Pfizer) |
*Lifirafenib (BGB-283) (Beigene) |
Regorafenib (Bayer) |
|
| Sorafenib (Bayer/Onyx) |
|||
| *KIN-2787 (Kinnate) | |||
| *DAY101 (formerly TAK-580, MLN2480) (Day One Pharmaceuticals) | |||
| *BGB-3245 (Beigene) | |||
| *BDTX-BRAF (Black Diamond Therapeutics) | |||
No structural information available. Classification inferred from in vitro activity data.
Despite this great progress, there remain great unmet needs for patients whose tumors harbor BRAF mutations with respect to BRAF inhibition. First and foremost, therapeutic resistance to BRAF inhibition (or existing combinations with RAF inhibitors) is nearly universal. The development of improved treatment strategies, including novel agents and combinations of agents, is critical to optimize the management of patients with tumors harboring these mutations. In this review, we describe the mechanics of RAF biology and signaling, the clinical significance of these mechanics as it pertains to the development of effective RAF inhibitors, the major existing clinical data, and ongoing and/or future strategies investigating combinatorial regimens.
Mechanics
BRAF is a member of the RAF family of serine/threonine kinases (the others are ARAF and CRAF) that are components of the RTK-RAS–RAF–MEK–ERK growth factor signaling pathway (RAS/MAPK signaling) (14). RAF proteins share similar domain organization, with the catalytic domain in the C terminus and regulatory elements in the N-terminus. Inactive RAF proteins exist as monomers in the cytosol. Activation of RAF is complex: it includes recruitment to the membrane, homo and heterodimerization of RAF family members and several phosphorylation and dephosphorylation events (15) (Figure 2).
Figure 2: Combinatorial pharmacologic strategies tailored to BRAF alteration and RAS/MAPK signaling context.
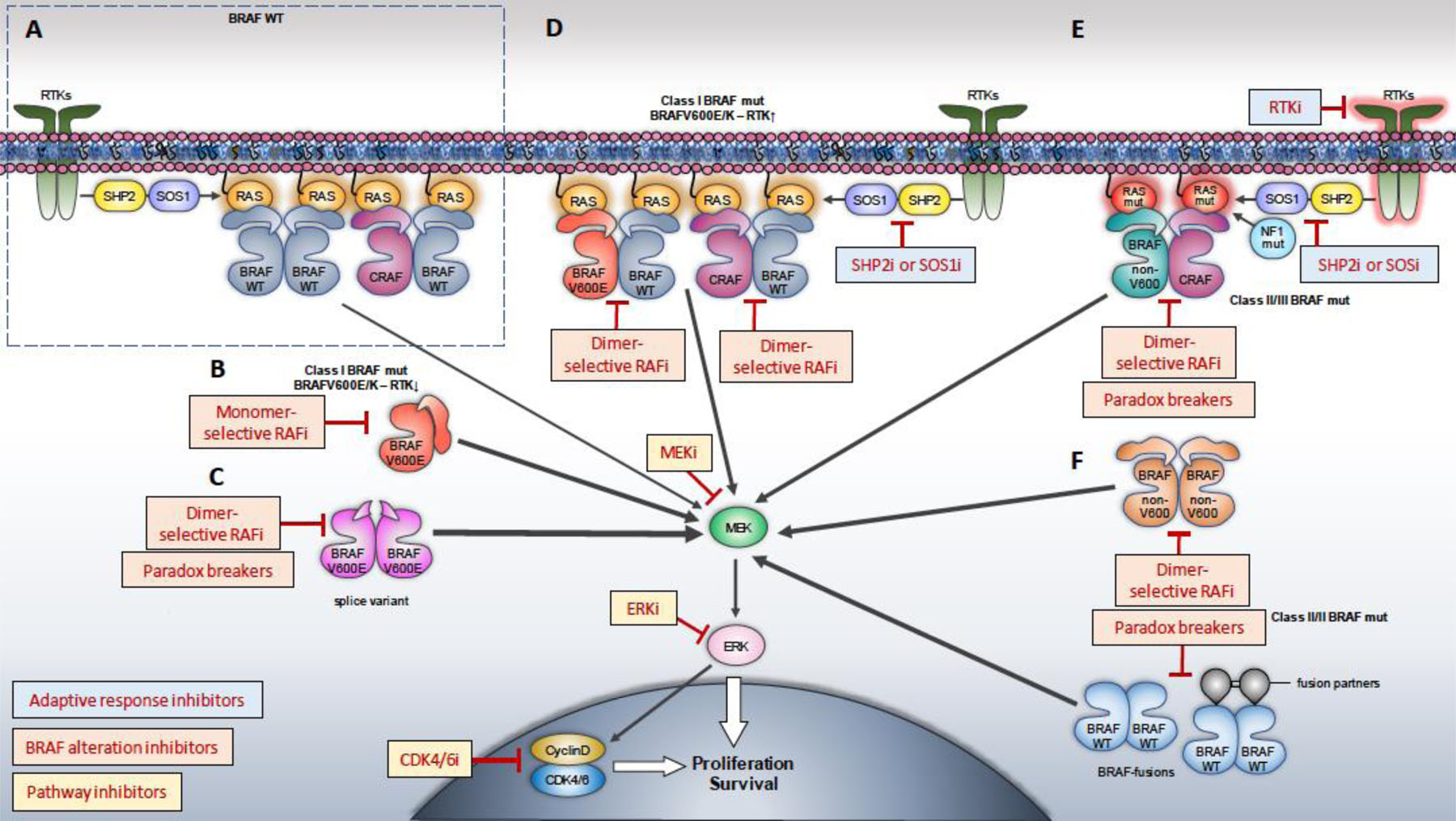
Therapeutic strategies aimed at inhibiting the totality of RAS/MAPK signaling output would require a) a direct “pathway” inhibitor (e.g. MEK, ERK or CDK4/6 inhibitor), b) a BRAF alteration inhibitor (e.g. RAF monomer, RAF dimer, equipotent or Paradox Breaker inhibitor) and c) an “adaptive response” inhibitor (e.g. SHP2, SOS, RTK, or a RAF dimer inhibitor). A. Physiologic (“normal”) RAS/MAPK signaling. B. For tumors expressing Class I (BRAFV600E/K) and low RTK activity (e.g. a portion of melanomas), RAF monomer-selective inhibitors in combination with MEK inhibitors are effective. C. Acquired resistance to RAF monomer inhibitors is most commonly the result of RAF dimerization (expression of dimeric splice variant shown here, as an example), indicating tumor sensitivity to RAF dimer (or equipotent) inhibitors or Paradox Breakers in this context. D. For tumors expressing Class I (BRAFV600E/K) and high RTK activity (e.g. a portion of Colorectal Cancers), RAF monomer-selective inhibitors should be combined with “adaptive response” inhibitors (e.g. EGFR, or RAF dimer inhibitors), in addition to pathway inhibitors. E. Effective targeting of tumors expressing class II/III BRAF altered proteins that are either bound to RAS, or form RAS-independent dimers (F), would require an inhibitor targeting RAF (e.g. RAF dimer (or equipotent) inhibitor, or a Paradox Breaker), a pathway inhibitor (e.g. MEKi, ERKi) and an adaptive response inhibitor (e.g. SHP2i, SOSi, RTKi). Achieving both potent RAS/MAPK inhibition in the tumor and acceptable therapeutic index requires understanding and optimizing the biochemical properties of each inhibitor and their combination.
Image by Christos Adamopoulos.
RAS/MAPK signaling mediates signals from cell surface receptors to the nucleus to promote cell proliferation and survival. Ligand binding results in activation and phosphorylation of Receptor Tyrosine Kinases (RTKs, e.g. EGFR) creating docking sites that promote the recruitment to RTK (and consequently to the membrane and in close proximity to RAS) of a multiprotein complex including among other proteins, SHP2, GAB1, GRB2 and SOS (16,17). Interaction of the guanine nucleotide exchange factor (GEF) SOS with RAS results in guanine exchange, GTP loading, and RAS activation (i.e. transition to the RAS-ON state) (18). Activated RAS recruits RAFs to the membrane by binding their RAS-Binding Domain. Recruitment of RAF to the membrane results in activation by oligomerization and by phosphorylation at activating sites by kinases residing in the membrane, presumably members of the PAK family. Once activated, RAF kinases phosphorylate and activate MEK (MEK1/2), which in turn phosphorylate and activate ERK (ERK1/2). Activated ERK phosphorylates a large number of substrates. A subset of ERK targets drive transcription of genes that drive proliferative and survival signals (such as myc, cyclin D1, AP-1, members of the Fos and ETV families etc). Importantly, homeostatic mechanisms are in place to ensure regulation of the pathway in normal cells: ERK activation also promotes negative feedback suppression of the pathway, by multiple mechanisms: upregulation of members of the DUSP and Sprouty families of phosphatases that dephosphorylate upstream components of the pathway, including ERK1/2, suppression of expression and activity of RTKs, as well as direct phosphorylation of EGFR, BRAF, CRAF and other proteins by ERK1/2 (19). Relief of negative feedback upon treatment with inhibitors of components of RAS/ERK signaling is a major cause of adaptive drug resistance, as it results in RTK upregulation, RAS activation and RAF dimerization promoting recovery of ERK activity (20–23).
Recurrent mutations in BRAF are found in about 6% of human cancers (Figure 1) and most commonly result in a substitution of Valine to Glutamic Acid in the activation segment (BRAFV600E). Small molecule inhibitors developed against BRAFV600E (vemurafenib, dabrafenib and encorafenib) showed unique biochemical properties. Early studies on these inhibitors found them to be potent inhibitors of RAS/MAPK in tumors expressing BRAFV600E, but they did not inhibit and instead activated RAS/MAPK signaling in tumors or normal cells with wild-type BRAF (24–26). Further investigation revealed that, in contrast to wild-type RAFs that signal as obligatory dimers (27), BRAFV600 proteins are able to signal as catalytically active monomers, and the biochemical basis of the selective inhibition of BRAFV600E over wild-type BRAF by these RAF inhibitors is their selectivity towards monomeric over dimeric RAF (24,25,28,29). Structurally, these first-generation clinical RAF inhibitors are Type I ½ (αC-helix OUT, DFG-IN, or CODI). The OUT position of the αC-helix of BRAF is not sterically allowed for both promoters in the BRAF dimer resulting in negative allostery for inhibitor binding to the second RAF protomer and providing a structural explanation for their selectivity for monomeric RAF (30). The preference of these inhibitors for binding and inhibiting monomeric RAF is the basis of their increased therapeutic index, but it also predicts that any mechanism that promotes RAF dimerization will confer resistance. An additional property of these inhibitors is a biochemical phenomenon confined so far to RAF inhibitors, the so called “paradoxical activation” of BRAF and downstream signaling promoted by these inhibitors in cells expressing wild-type BRAF. While all kinase inhibitors inhibit their target in all cells, RAF inhibitors paradoxically activate RAF and RAS/MAPK signaling in a RAS-dependent manner in cells expressing wild-type BRAF as a result of recruitment of inactive RAF to active RAS upon binding of inhibitor and subsequent failure of the compound to bind both protomers within the RAF dimer (negative allostery) (30,31).
Thus, RAF dimerization is a major biochemical determinant of tumor response to RAF inhibitors: it is a consequence of relief of negative feedback and wild-type RAS activation in the context of adaptive resistance to RAF inhibitors and it promotes acquired resistance to RAF inhibitors via various mechanisms (RTK upregulation (32,33), acquisition of RAS mutations (34), generation of splice variants that constitutively dimerize (25) or BRAF overexpression (35,36)).
RAF dimerization also limits response of tumors harboring BRAF mutation other than V600 (30,37). Such BRAF alterations include point mutations in the activation segment or the so called glycine-rich loop of BRAF, as well as BRAF fusions containing the catalytic domain of wild-type BRAF fused to various partners. In contrast to BRAFV600 mutants (Class I) that signal as hyperactive monomers, almost all non-V600 BRAF mutants promote RAF activation by dimerization (Class II/III), and they are therefore predicted to be resistant to current clinical RAF inhibitors (30,31,37,38).
The success of RAF inhibitors in a portion of melanomas expressing a potent ERK activator (BRAFV600E) in a cellular context of low RTK activity and absence of other ERK activators has been a fortunate exception to the rule. In the majority of tumors carrying BRAF alterations, mutated BRAF co-exists with other ERK activators (active and/or overexpressed RTKs, RAS mutations, NF1 loss and others) (39). Consequently, in the development of next generation RAF inhibitors and the design of improved therapeutic strategies, targeting of the mutated BRAF oncoprotein should be considered in the context of pharmacologic approaches aimed at potently and durably suppressing the pathway (ERK activity) in the tumor, while minimizing toxicities in normal tissue. For such strategy, the totality of RAF activity (including RAF monomers and all different RAF dimers) in the tumors should be potently inhibited for effective ERK and tumor growth suppression.
Next generation of compounds targeting RAF
On the heels of the clinical success of RAF monomer-selective inhibitors (vemurafenib, dabrafenib and encorafenib), a number of RAF inhibitors with different structural and biochemical properties have been under intense preclinical and clinical development (Table 2). An obvious clinical challenge that needs to be addressed is inhibition of RAF dimers in three contexts: a) to prevent or overcome adaptive or acquired resistance in the BRAFV600E/K setting, b) to target tumors harboring non-V600 BRAF alterations and c) to target dimeric wild-type BRAF in tumors expressing upstream mutations (e.g. mutant RAS, NF1 loss, etc). In terms of biochemical properties, RAF inhibitors in development can be classified in three groups: RAF monomer-selective that do not induce paradoxical activation (“paradox breakers”), equipotent for RAF monomers and dimers, and RAF-dimer selective inhibitors (Table 2). Structurally, paradox breakers (PBs) are CODI, similarly to first generation RAF-monomer selective inhibitors, and RAF dimerization is also predicted to limit their activity. However, in contrast to other CODI inhibitors that also promote recruitment of inactive RAF to RAS, RAF dimerization, and paradoxical activation, PB compounds do not, presumably due to the OUT position of the R506 residue within the αC-helix (30). The therapeutic consequence of this distinction might be important as PB may be able to “break” RAF dimers at clinically achievable concentrations. Further, dimers formed by different RAF family members have been reported to vary in their affinity (40), suggesting that such compounds may confer increased therapeutic index by selectively disrupting RAF dimers that drive tumor growth while spearing RAF dimers that sustain ERK activity in normal tissue.
Table 2.
FDA Approved Indications for RAF Inhibitor Therapies.
| FDA Indication | Treatment | Year of Approval |
|---|---|---|
| Unresectable or metastatic melanoma with BRAF V600E mutation | Vemurafenib Dabrafenib |
2011 2013 |
| Unresectable or metastatic melanoma with BRAF V600E/K mutation | Dabrafenib + Trametinib Vemurafenib + Cobimetinib Encorafenib + Binimetinib |
2014 2015 2018 |
| Erdheim-Chester disease with BRAF V600 mutations | Vemurafenib | 2017 |
| Metastatic non-small cell lung cancer with BRAF V600E mutation | Dabrafenib + Trametinib | 2017 |
| Adjuvant treatment for resected melanoma with BRAF V600E/K mutation with lymph node involvement | Dabrafenib + Trametinib | 2018 |
| Locally advanced or metastatic anaplastic thyroid cancer with BRAF V600E mutation and no satisfactory locoregional treatment options | Dabrafenib + Trametinib | 2018 |
| Metastatic colorectal cancer with BRAF V600E mutation after prior therapy | Encorafenib + Cetuximab | 2020 |
Equipotent and RAF dimer-selective inhibitors stabilize the αC-helix of RAF in the IN position (αC-helix IN, DFG-OUT – CIDO or Type II) and potently bind and inhibit RAF dimers. A number of RAF inhibitors of this class are in preclinical and clinical development and have shown activity in BRAF and RAS-mutant tumors, either as single agents (30,37,41–45) or in combination with MEK inhibitors (46–49). Interestingly, ARAF is not inhibited by one of these inhibitors (LXH254) (50) and activating mutations in ARAF have been reported to confer clinical resistance to Belvarafenib (51), another RAF inhibitor of this class. It is currently unclear whether sparing ARAF is a general property of this class of RAF inhibitors, as well as whether it is another mechanism of resistance or rather confers clinical benefit by increasing therapeutic index when treating tumors driven predominantly by BRAF and CRAF.
Proteolysis Targeted Chimeras (PROTACs) have gained traction recently as an approach to both inhibit and cause proteasome-mediated degradation of a target of interest. Typically, these are bifunctional small molecules, in which a small molecule inhibitor of the target of interest is linked to a chemical moiety that binds an E3 ligase. BRAF-directed PROTACs have been developed and showed preclinical efficacy similar or superior to parent RAF inhibitors in targeting BRAF(V600E) signaling (52,53). As the PROTAC technology requires formation of ternary complexes that may differ across RAF isoforms, it would perhaps enable selective degradation of specific RAF isoforms. Such properties of compounds could be clinically important, as there are reports suggesting a role of CRAF selectively driving growth of RAS-mutant tumors (54,55), or, as aforementioned, of a role of ARAF in escaping inhibition by RAF-dimer inhibitors belvarafenib (51) and LXH254 (50).
Clinical Implications
Treatment with RAF inhibitors ushered in a new era of testing targeted therapies in patients whose tumors express the target of interest. Unlike earlier MEK inhibitor trials that showed limited activity when tested in a genomically-unselected population (56), RAF inhibitors were evaluated in patients whose cancers harbored BRAFV600 mutations. Unexpectedly, the clinical experience with RAF inhibitors revealed that patient response to treatment can vary both by genomic alteration and tissue context as activity differed considerably across cancers.
Melanoma
RAF inhibitors were first tested in melanoma with dramatic responses. Single agent RAF inhibitors lead to response in about 50% of patients with BRAFV600E/K melanoma and improve survival over standard chemotherapy (3,5,57). Based on the identification of RAS/MAPK pathway alterations at resistance to RAF inhibitors (25,34,58,59) and preclinical studies showing rebound of RAS/MAPK signaling causing rapid adaptive resistance (20,60,61), combination RAF plus MEK inhibitors were tested. This combination led to a higher response rate than for RAF inhibitor monotherapy and improved progression-free and overall survival (6,8,62). Combination treatment also led to a lower rate of secondary cutaneous cancers and has supplanted monotherapy as the preferred targeted therapy for BRAFV600 mutant melanoma (9,63).
Other cancers
In the phase 1 trial of vemurafenib, there was also a CRC expansion cohort, where more limited activity (5% response rate, 2.1 month median progression-free survival) was seen compared to melanoma (64). Preclinical studies indicated that feedback release of upstream receptors, predominantly EGFR, with RAF inhibition lead to reactivation of the pathway and generation of drug-resistant RAF dimers in CRC (21,65). In vivo treatment of CRC xenografts with combination RAF and EGFR inhibition results in improved efficacy. These data led to clinical trials of combination therapy for BRAFV600E CRC described below.
The effect of vemurafenib in multiple BRAFV600E mutant cancers was tested with a novel basket trial design (66). Among anaplastic thyroid cancers included in this VE-BASKET trial, responses were seen in 2 of 7 patients (66). Concurrent preclinical work suggested reactivation of HER2/HER3 signaling with RAF inhibition through feedback induction of ligand signaling attenuated the effect of RAF inhibitors on signaling and growth of thyroid cell lines (67). A phase 2 trial of dabrafenib and trametinib showed a higher response rate of 69% in anaplastic thyroid cancer (11), and the phase 2 ROAR (Rare Oncology Agnostic Research) basket trial of dabrafenib plus trametinib reported 56% response rate in this population(68). In contrast, the histiocytosis disorder Erdheim Chester disease (ECD) appeared particularly sensitive to RAF inhibitor monotherapy with high response rate and durable regression (66,69). Many of the patients with ECD required treatment dose reductions and still achieved profound responses involving all sites of disease. In NSCLC, the response rate for vemurafenib was about 40% (66). In a phase 2 trial of dabrafenib and trametinib in 57 BRAFV600E NSCLC patients, response was seen in 36 patients (63%) (70). For gliomas, anecdotal responses to vemurafenib have been reported (71), and the interim analysis of the ROAR trial indicated a 33% response rate in high-grade gliomas and 69% response rate in low-grade gliomas to dabrafenib plus trametinib (72). Finally, about 50% of patients with biliary tract cancer had a response to dabrafenib plus trametinib in the ROAR study (73).
Together, these clinical data suggest varied thresholds for response to RAF inhibition. While many tumors may exhibit RAS/MAPK pathway activation and depend on this pathway for growth, the threshold to pharmacologically inhibit the pathway varies by histology so that some tumors (e.g., ECD) may be “simpler” and completely and durably regress with RAF inhibitor while others (e.g., anaplastic thyroid cancer or CRC) may exhibit complex signaling where RAF inhibition is limited by reactivation of multiple signaling loops that attenuate drug effect. Pharmacodynamic studies in the vemurafenib dose escalation study indicated that profound target inhibition is required for response (4), and the clinical data indicate that the threshold to achieve this degree of pathway inhibition varies across cancer types and that the signaling milieu of mutant BRAF varies in different tissue backgrounds. Thus, BRAF inhibitor trials revealed that the presence and nature of ERK-driven negative feedback events varies in different cancers and largely underlies the variability in clinical response.
Combination Therapy for Colorectal Cancer
Combination therapy is needed for clinically meaningful pathway inhibition in BRAFV600E CRC. Combined RAF and MEK inhibition results in modest activity with responses in about 10–20% of patients (74,75). Adding an EGFR antibody (cetuximab or panitumumab) to vemurafenib or dabrafenib also led to modest activity with responses in about 10–20% of patients (66,76,77). Higher response and ERK inhibition in pharmacodynamic studies suggested that the triplet regimen of dabrafenib, trametinib, and panitumumab may support better pathway inhibition and improved activity, but still lower pathway suppression compared to single agent RAF inhibition in melanoma (median 60% pERK modulation in CRC with dabrafenib + trametinib + panitumumab versus median 84% pERK modulation in melanoma with dabrafenib) (77). The doublet of encorafenib plus cetuximab was tested in phase 1 and 2 studies with a response rate of 22% (78,79); encorafenib was hypothesized to have higher activity in CRC because of its slow dissociation from RAF allowing better target inhibition.
Based on these data, the BEACON phase 3 trial tested the investigational therapies of encorafenib and cetuximab and of encorafenib, binimetinib, and cetuximab compared to standard cetuximab-containing treatment. Both investigational targeted therapies were associated with improved overall and progression-free survival compared to standard therapy with overlapping survival curves; the triplet regimen had a response rate of 26% and the doublet regimen had a response rate of 20% (13,80). These data led to the FDA approval of BRAF targeted therapy with the doublet of encorafenib plus cetuximab for CRC and the first positive predictive marker with an approved matched therapy in CRC.
Response rate and duration of response for encorafenib and cetuximab remain below that of RAF inhibitor monotherapy in melanoma (Figure 3). These data suggest better pathway inhibition in melanoma, and current studies are examining the potential to more broadly block receptor reactivation with SHP2 or SOS2 inhibitors to improve activity in CRC. Preclinical data suggest that SHP2 inhibitors could block receptor-mediated feedback activation in a subset of BRAFV600E mutant CRC, particularly those with EGFR or MET activation, but not in CRC where FGFR primarily driver pathway reactivation (33). The limited benefit of adding a MEK inhibitor to encorafenib and cetuximab raises the possibility that incomplete pathway inhibition may be due to release of downstream regulators of ERK activation. Preclinical studies suggest that triplet therapy with RAF, EGFR, and ERK inhibitors may exhibit increased activity and better suppress outgrowth of resistance clones compared to combinations with RAF, EGFR, and MEK inhibitors (81). Expression and regulation of ERK phosphatases varies across tissue types and may contribute to differential activity of RAF inhibitor therapy in different cancers (82–84).
Figure 3: Response to RAF inhibitors across cancers.
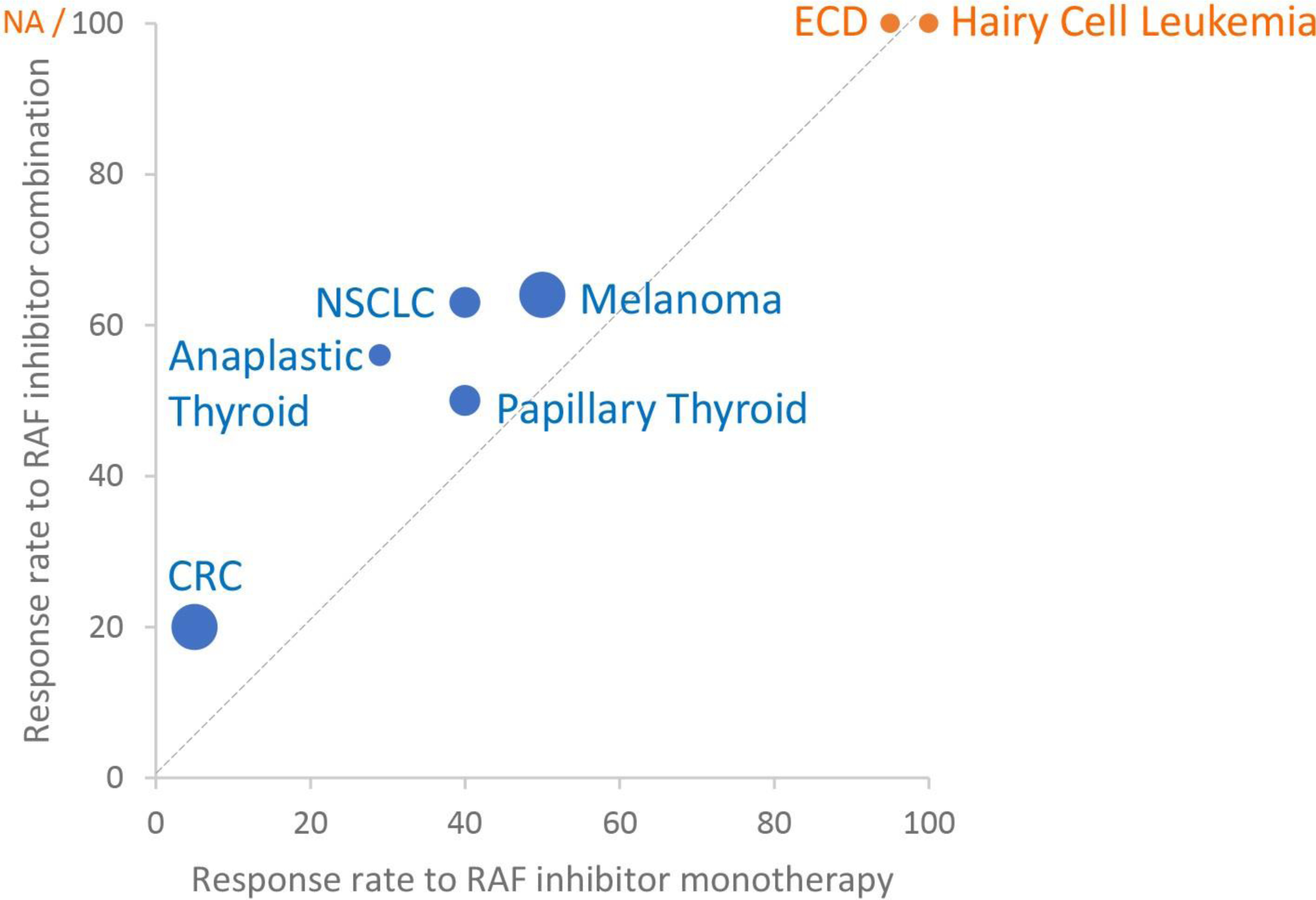
Graph showing response to RAF inhibitor monotherapy or RAF inhibitor combination (RAF and MEK inhibitors or, for colorectal cancer, RAF and EGFR inhibitors). Disease types without reported combination trial data are indicated in orange. Size of data points corresponds to number of trial patients used to estimate response: smallest points (1–50 patients), middle sized points (51–100 patients), large points (>200 patients). Graphed response rate for ECD corresponds to PET response. CRC: colorectal cancer. ECD: Erdheim Chester Disease. NSCLC: non-small cell lung cancer. PANC: pancreatic cancer. NA: not accessed.
Ongoing Trials of First-Generation RAF Inhibitors for BRAFV600 Mutant Tumors
Ongoing trials in several cancer types aim to identify the best targeted therapy or combinations that overcome adaptive resistance. Trials include phase 2 trials of encorafenib and binimetinib in BRAF V600 mutant NSCLC (NCT03915951), BRAF V600E mutant pancreatic cancers (NCT04390243), BRAF V600 mutant hairy cell leukemia (NCT04324112), and BRAF V600 mutant high-grade gliomas (NCT03973918). Combination trials are evaluating the safety and activity of adding SHP2 inhibition to RAF inhibition (NCT04294160, NCT04800822).
RAS/MAPK pathway combinations
Preclinical models, in vitro and in vivo, have demonstrated synergy with blockade of CDK4/6 and MAPK inhibition in melanoma (85–87). To date, however, there has been limited data with CDK4/6 plus MAPK inhibition due to excessive toxicity seen with these combinations. For example, the combination of encorafenib, binimetinib, and the CDK4/6 inhibitor ribociclib was associated with lower response rates than the doublet of encorafenib and binimetinib in the Phase 1 trial, at least in part because the maximum tolerated dose of encorafenib was lower with ribociclib (due to concerns over drug-drug interactions) (88). In trials of MEK and CDK4/6 inhibitors, the toxicity of dual inhibition has been challenging to overcome, even in the face of increased activity in patients with NRAS mutant melanoma with the combination of binimetinib and ribociclib (89,90). Whether there is a path forward for these combinations remains to be seen, but it is possible that alternative approaches to dose escalation that maximize exposure of the MEK and CDK4/6 inhibitors, likely through intermittent dosing schedules, will be a more effective approach (91,92).
Non-RAS/MAPK pathway combinations
With the nearly simultaneous development of BRAF targeted therapy and immune checkpoint inhibitor therapy in melanoma, it seemed logical that combining these two modalities would be tested in the clinic. The first of these combinations was that of vemurafenib with the anti-cytotoxic T lymphocyte associated antigen 4 (CTLA-4) monoclonal antibody ipilimumab in patients with BRAF mutant, advanced melanoma. In this trial, the combination was deemed too toxic due to the development of severe hepatic toxicity in the majority of treated patients (93). However, the preponderance of data supported combined BRAF targeted therapy with inhibition of either the programmed death receptor 1 (PD-1) or its ligand, PD-L1. In particular, MAPK inhibition increases melanocytic antigen expression, T-cell infiltration, and PD-L1 expression in preclinical and clinical analyses (94–96). In the early clinical trials of PD-1/PD-L1 inhibition with BRAF targeted therapy in melanoma, the response rates were promising and the tolerability suitable for further evaluation (97–100). In larger, randomized cohorts, combination BRAF targeted therapy with BRAF/MEK inhibition with either PD-1 or PD-L1 inhibition was consistently associated with improved progression free survival compared to dual BRAF/MEK inhibition, but only statistically significantly so in one of the three randomized cohorts reported (101–103). Thus, only one of these regimens, the combination of vemurafenib, cobimetinib, and atezolizumab has been approved by regulatory authorities for the treatment of unresectable or metastatic, BRAFV600 mutant melanoma, although the optimal use of triplet therapy in this patient population is unknown given the effectiveness of other immunotherapy combinations such as ipilimumab and nivolumab(104). In other BRAF mutant cancers, studies of these combinations are also underway. Specifically, the early results of the combination of dabrafenib, trametinib, and spartalizumab look promising in BRAF mutant, metastatic CRC, although it is unclear if these results will be replicated in larger and multi-institutional datasets (105).
Numerous other combinations with single-agent BRAF or combined BRAF-MEK inhibition have been/are being evaluated in BRAF mutant malignancies. These include targeting specific pathways, such as the PI3K pathway (106), as well as targeting more ethereal cellular processes such as apoptosis (107) (NCT01989585), the molecular chaperone heat shock protein 90 (HSP90) (108,109) (NCT02721459), and autophagy (110). None of these regimens has yet to demonstrate groundbreaking results, however, there are randomized studies completing or being planned that hopefully will shed greater light on these approaches (NCT01989585, NCT04527549).
Intermittent schedules
Preclinical studies suggest that intermittent dosing of RAF inhibitor therapy may provide a means to take advantage of the drug dependence of resistant clones and delay the emergence of resistance. RAF inhibitor-resistant cell lines, including those with BRAFV600E amplification or the BRAF splice variant, grow better in vitro in the presence of drug (111,112). Preclinical modeling with xenograft tumors suggests that a discontinuous dosing strategy (using a schedule of 4 weeks on/2 weeks off treatment in these experiments) substantially delays the emergence of drug resistance (111). Based on these data, a phase 2 clinical trial compared progression-free survival in patients with BRAFV600E metastatic melanoma treated with eight weeks of dabrafenib and trametinib who were then randomized, if they had no progression, to continue with continuous dosing versus to begin intermittent dosing on a schedule of 3 weeks off/5 weeks on (113). The study found a significantly shorter median progression-free survival with the intermittent schedule (5.5 months versus 9 months for intermittent versus continuous dosing). These data indicate the difficulty to clinically translate innovative drug schedules and that intermittent dosing may not forestall clinical resistance. However, it is possible that the treatment lead-in or the schedule of long periods of treatment and breaks may have limited the efficacy of the intermittent dosing strategy.
Non-V600 BRAF Mutants and Novel RAF inhibitors
As described above, oncogenic non-V600 BRAF mutants signal as dimers. Tumors harboring these mutants thus are insensitive to current, monomer-selective RAF inhibitors. Preclinical data and case reports suggest some activity for dabrafenib or the dabrafenib/trametinib combination in non-V600 BRAF mutant tumors (48,114). This activity may be due to the higher potency of dabrafenib compared to other BRAF inhibitors and the potential to hit RAF dimers to some degree at clinical achievable doses.
One clinical approach for non-V600 BRAF mutant tumors is to target downstream of BRAF with MEK or ERK inhibitors. The ERK inhibitor ulixertinib (BVD-523) was tested in several molecularly defined cohorts in a phase 1 dose escalation and expansion (115), and 14 partial responses were observed in 101 patients who received at least the recommended phase 2 dose. Among 27 patients with non-V600 BRAF alterations, five experienced a response, all of whom had tumors harboring class II BRAF mutants (23). The NCI-MATCH study included a subprotocol where non-V600 BRAF mutant tumors were treated with trametinib (116). Thirty-two patients with non-V600 BRAF mutant tumors (1 fusion, 12 class II, 19 class III) received trametinib. Only one patient (3%) achieved a partial response, and this subprotocol did not meet its primary response endpoint. Notably, patients with a gastrointestinal primary made up 25% of study patients, primarily consisting of patients with CRC; five patients in this study had tumors with concurrent RAS mutations (1 HRAS, 2 KRAS, 2 NRAS); and the class III BRAF D594 mutants were the most common alteration (11 cases). In a phase 2 trial, trametinib was evaluated in melanoma with non-V600 BRAF mutants (117). While a small study enrolling nine patients, three patients achieved a partial response. In a recent meta-analysis, 411 patients with a non-V600 BRAF mutation and treated with RAS/MAPK pathway inhibition (including BRAF, MEK, ERK inhibitors as well as combined therapies) were analyzed. Responses were seen in 119 of these 411 patients (29%) and factors associated with better response included patients with tumors harboring a mutation with a higher predicted pERK activation (using the Class II versus Class III designation (38)) and patients with melanoma tumor type, whereas poorer outcomes with associated with patients enrolled to prospective study and patients treated with single-agent RAF inhibitor (118). These trial data also suggest varying thresholds for inhibition with likely lower activity in class III BRAF mutants, which have persistent upstream activation, and certain cancer types (i.e, gastrointestinal or prostate primary).
New RAF inhibitors (Table 1), described above, have the potential to inhibit RAF mutant dimers directly. An ongoing phase 1 trial is evaluating the safety and efficacy of the BRAF PB PLX8394 across cancer types. In this trial, different formulations have been tested to enhance drug dose. As of the last report (119), over 75 patients have been treated. Responses have been reported for some patients with BRAFV600E or non-V600 BRAF alterations, including a complete response reported in a patient with BRAF-fusion melanoma and partial responses in patients with other cancers (gliomas [n=3], ovarian [n=2], and papillary thyroid, small bowel, colorectal, and anaplastic thyroid [all n=1]).
The RAF inhibitors lifirafenib (BGB-283, equipotent) and belvarafenib (RAF dimer-selective) have completed phase 1 trials, enrolling patients with BRAF or RAS mutant tumors (51,120–122). Responses to lifirafenib were seen primarily in patients with BRAFV600 mutant tumors (8/47), including melanoma, papillary thyroid cancer, and low grade serous ovarian cancer patients; a patient with a non-V600 BRAF NSCLC had an unconfirmed partial response. Seventy-two patients were included in the dose escalation of belvarafenib and partial responses were seen in NRAS-mutant melanoma, BRAF-mutant melanoma, KRAS-mutant sarcoma, and BRAF-mutant GIST. At the phase 2 dose of belvarafenib, among 63 patients, responses were seen in patients with NRAS Q61 mutant melanoma (2/9), BRAF V600E mutant melanoma (2/6), BRAF V600E mutant CRC (2/7), as well as in one patient with bladder cancer harboring a KRAS G12D mutation (51). Current clinical trial efforts are exploring combining belvarafenib with the MEK inhibitor cobimetinib. The initial data with this combination shows 7 responders in the 32 patients treated in dose escalation. Five of the responders had NRAS Q61 mutant melanoma, one had BRAF V600E mutant melanoma, and the last had KRAS G12D mutant CRC(122).
Toxicities of BRAF Targeted Therapies
Toxicity has always been a central issue with targeting the MAPK pathway because of the biological importance of this pathway across cell types and the need for profound pathway inhibition within the tumor (4). Treatment of BRAFV600E melanoma with MEK inhibitors and RAF inhibitors leads to tumor regression by inhibiting ERK, but MEK inhibitors, which have a narrow therapeutic index, are associated with a response rate of about 20% (123) compared to a response rate of about 50% for RAF inhibitors. This difference highlights how toxicity directly limits the pathway inhibition that can be achieved and consequently treatment response.
While RAF inhibitors have a broad therapeutic index due to differing effects in BRAF mutant tumor and BRAF wild-type tissues, patients still need to be monitored closely for safety and some combinations have been difficult to administer because of toxicity. The toxicities of RAF inhibitors can be divided into those that are on-target and thus shared across molecules, and those that are off-target which tend to be molecule specific. Each of the first-generation RAF inhibitors cause “on-target” toxicity through paradoxical activation of the MAPK pathway in non-mutant, MAPK signaling-dependent cells. The most common shared toxicities across first generation RAF inhibitors include hyperkeratosis, palmoplantar erythrodysaesthesia syndrome (aka hand-foot syndrome), rash, arthralgias, and cutaneous malignancies (typically keratoacanthoma and/or squamous cell carcinoma). Predictably, each of these adverse effects are reduced with the addition of a MEK inhibitor, which reduce RAS/MAPK signaling related to upstream monomeric as well as dimeric RAF activity. Other common toxicities include nausea, vomiting, fatigue, and diarrhea; each of which is likely more non-specific and not reduced with the combination with a MEK inhibitor.
Other classes of RAF inhibitors have been less extensively studied in the clinic than first-generation RAF inhibitors, but safety data is emerging with these agents. For example, the PB PLX8394 has been tested in an ongoing Phase 1/2 trial with preliminary data in 74 patients demonstrating that the agent, which needs to be given in combination with cobicistat to enhance oral absorption of the drug, causes no significant dermatologic toxicity and no secondary cutaneous malignancies; the most common adverse events seen were elevated transaminases, elevated bilirubin, nausea, vomiting, diarrhea, and fatigue (124). Several equipotent monomer/dimer RAF inhibitors have reported phase 1 data. The most common treatment related adverse events for LY3009120 were fatigue, nausea, reduced appetite, and rash, including maculopapular and acneiform. Most common emergent adverse events with lifirafenib were fatigue and dermatitis acneiform rash; thrombocytopenia was the most common dose-limiting toxicity. For belvarafenib, the most common treatment-emergent adverse events were rash, dermatitis acneiform, and pyrexia and the dose-limited toxicities were skin adverse effects. Finally, LXH254 is tolerated as a single agent and in combination with the ERK inhibitor LTT462, with most common toxicities being rash and fatigue. Of note, there have been reports of an acute neuropathy from LXH254 and its use in combination with LTT462 in a small number of patients (4%)(125). This is not a typical toxicity for inhibitors of this pathway and thus the rate and severity of this toxicity should be closely studied.
Future Directions
It took less than a decade from the discovery of oncogenic BRAF to FDA-approval of a class 1 BRAF inhibitor for the treatment of melanoma. Since, BRAFV600 targeted therapy has been optimized in melanoma, anaplastic thyroid cancer, and NSCLC to involve dual pathway inhibition targeting BRAF and MEK. In CRC, dual BRAF and EGFR targeting has demonstrated significant benefit and become a standard regimen. Still, therapeutic resistance remains the dominant limiting factor for optimal BRAF targeting, and the development of more effective strategies to inhibit BRAF targeting is the most critical unmet need in the field. Fortunately, newer agents are making it into the clinic and combinatorial strategies are emerging. It is anticipated that novel approaches to drug development may be required to build multi-drug regimens to optimize MAPK inhibition while targeting other critical signaling pathways (e.g. PI3K) and cellular processes (e.g. apoptosis, autophagy, etc.); these efforts are just beginning. Additionally, in an era that has seen a paradigm shift from cytotoxic therapy to immunotherapy, there also have been important efforts to better characterize the impact of ERK inhibition on the tumor-immune microenvironment and determine the value of combining MAPK inhibition with anti-PD-1/PD-L1 therapy. The initial results show promise, but it is clear that the optimal use of these regimens and biomarker approaches to select patients most likely to benefit have not yet been determined. Finally, it is critical to note that the recent success in the development of highly effective approaches to target BRAF mutant malignancies has been a partnership between bench researchers, pharmaceutical companies, and academically focused clinical and translational investigators. For the field to move into the next phase, this partnership will need to continue to strengthen so that patients with BRAF mutant cancers will live better and longer lives.
Acknowledgments:
P.I.P is supported by the NIH/NCI (R01 CA204314, R01 CA240362, and R01CA238229), the Irma T. Hirschl Trust, the Manhasset Women’s Coalition Against Breast Cancer, the Breast Cancer Alliance, the Melanoma Research Foundation, the Melanoma Research Alliance and Tisch Cancer Institute (TCI) developmental awards. R.J.S. is supported by NIH/NCI (R01 CA193970, R01 CA229851, UM1 CA186709, U54 CA224068). R.Y. is supported by the Memorial Sloan Kettering Cancer Center National Institutes of Health Cancer Center Support Grant P30 CA008748 and R01 C233736.
Disclosure of Potential Conflicts of Interest
P.I.P reports research funding to his institution by Black Diamond Therapeutics and Verastem Oncology.
R.J.S. has received research funding from Merck and has consulted/served on scientific advisory boards for Bristol Myers Squibb, Eisai, Iovance, Merck, Novartis, Pfizer. R.Y. has received research funding from Array BioPharma/Pfizer, Boehringer Ingelheim, and Mirati Therapeutics and has consulted for Array BioPharma/Pfizer, Natera, and Mirati Therapeutics.
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 2.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116:855–67. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010;467:596–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30–9. [DOI] [PubMed] [Google Scholar]
- 8.Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76. [DOI] [PubMed] [Google Scholar]
- 9.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19:603–15. [DOI] [PubMed] [Google Scholar]
- 10.Long GV, Hauschild A, Santinami M, Atkinson V, Mandalà M, Chiarion-Sileni V, et al. Adjuvant dabrafenib plus trametinib in Stage III BRAF-mutated melanoma. N Engl J Med 2017;377:1813–23. [DOI] [PubMed] [Google Scholar]
- 11.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol 2018;36:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 2017;18:1307–16. [DOI] [PubMed] [Google Scholar]
- 13.Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol 2021;39:273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young A, Lyons J, Miller AL, Phan VT, Alarcón IR, McCormick F. Ras signaling and therapies. Adv Cancer Res 2009;102:1–17. [DOI] [PubMed] [Google Scholar]
- 15.Terrell EM, Morrison DK. Ras-mediated activation of the raf family kinases. Cold Spring Harb Perspect Med 2019;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010;141:1117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007;26:3113–21. [DOI] [PubMed] [Google Scholar]
- 18.Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell 2017;170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A 2009;106:4519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012;22:668–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014;13:928–42. [DOI] [PubMed] [Google Scholar]
- 23.Yaeger R, Corcoran RB. Targeting alterations in the RAF-MEK pathway. Cancer Discov 2019;9:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011;480:387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5. [DOI] [PubMed] [Google Scholar]
- 27.Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009;461:542–5. [DOI] [PubMed] [Google Scholar]
- 28.Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 2013;49:751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol 2015;22:37–43. [DOI] [PubMed] [Google Scholar]
- 30.Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell 2016;30:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer 2017;17:676–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010;18:683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep 2019;26:65–78.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaeger R, Yao Z, Hyman DM, Hechtman JF, Vakiani E, Zhao H, et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to Its inhibition. Cancer Res 2017;77:6513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015;28:370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017;548:234–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garman B, Anastopoulos IN, Krepler C, Brafford P, Sproesser K, Jiang Y, et al. Genetic and genomic characterization of 462 melanoma patient-derived xenografts, tumor biopsies, and cell lines. Cell Rep 2017;21:1936–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med 2019;25:284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Z, Yuan X, Du R, Cheung SH, Zhang G, Wei J, et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol Cancer Ther 2015;14:2187–97. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res 2013;73:7043–55. [DOI] [PubMed] [Google Scholar]
- 43.Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov 2016;6:300–15. [DOI] [PubMed] [Google Scholar]
- 44.Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 2015;28:384–98. [DOI] [PubMed] [Google Scholar]
- 45.Botton T, Talevich E, Mishra VK, Zhang T, Shain AH, Berquet C, et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep 2019;29:573–88.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan X, Tang Z, Du R, Yao Z, Cheung SH, Zhang X, et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in K-RAS mutant tumors. Mol Oncol 2020;14:1833–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yen I, Shanahan F, Merchant M, Orr C, Hunsaker T, Durk M, et al. Pharmacological induction of RAS-GTP confers RAF inhibitor sensitivity in KRAS mutant tumors. Cancer Cell 2018;34:611–25.e7. [DOI] [PubMed] [Google Scholar]
- 48.Dankner M, Lajoie M, Moldoveanu D, Nguyen TT, Savage P, Rajkumar S, et al. Dual MAPK inhibition is an effective therapeutic strategy for a subset of class II BRAF mutant melanomas. Clin Cancer Res 2018;24:6483–94. [DOI] [PubMed] [Google Scholar]
- 49.Adamopoulos C, Ahmed TA, Tucker MR, Ung PMU, Xiao M, Karoulia Z, et al. Exploiting allosteric properties of RAF and MEK inhibitors to target therapy-resistant tumors driven by oncogenic BRAF signaling. Cancer Discov 2021;11:1716–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monaco KA, Delach S, Yuan J, Mishina Y, Fordjour P, Labrot E, et al. LXH254, a potent and selective ARAF-sparing inhibitor of BRAF and CRAF for the treatment of MAPK-driven tumors. Clin Cancer Res 2021;27:2061–73. [DOI] [PubMed] [Google Scholar]
- 51.Yen I, Shanahan F, Lee J, Hong YS, Shin SJ, Moore AR, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 2021;594:418–23. [DOI] [PubMed] [Google Scholar]
- 52.Alabi S, Jaime-Figueroa S, Yao Z, Gao Y, Hines J, Samarasinghe KTG, et al. Mutant-selective degradation by BRAF-targeting PROTACs. Nat Commun 2021;12:920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Posternak G, Tang X, Maisonneuve P, Jin T, Lavoie H, Daou S, et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat Chem Biol 2020;16:1170–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blasco MT, Navas C, Martín-Serrano G, Graña-Castro O, Lechuga CG, Martín-Díaz L, et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and C-RAF. Cancer Cell 2019;35:573–87.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blasco RB, Francoz S, Santamaría D, Cañamero M, Dubus P, Charron J, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 2011;19:652–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62. [DOI] [PubMed] [Google Scholar]
- 57.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012;366:707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A 2009;106:20411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer 2010;102:1724–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sturm OE, Orton R, Grindlay J, Birtwistle M, Vyshemirsky V, Gilbert D, et al. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal 2010;3:ra90. [DOI] [PubMed] [Google Scholar]
- 62.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012;367:1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2018;19:1315–27. [DOI] [PubMed] [Google Scholar]
- 64.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 2015;33:4032–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100–3. [DOI] [PubMed] [Google Scholar]
- 66.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov 2013;3:520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH, et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 2020;135:1929–45. [DOI] [PubMed] [Google Scholar]
- 70.Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF Inhibition in BRAF(V600)-Mutant Gliomas: Results From the VE-BASKET Study. J Clin Oncol 2018;36:3477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen PY, Stein A, van den Bent M, De Greve J, Wick A, de Vos F, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol 2022;23:53–64. [DOI] [PubMed] [Google Scholar]
- 73.Subbiah V, Lassen U, Élez E, Italiano A, Curigliano G, Javle M, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 2020;21:1234–43. [DOI] [PubMed] [Google Scholar]
- 74.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol 2015;33:4023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sullivan RJ, Weber J, Patel S, Dummer R, Carlino MS, Tan DSW, et al. A phase Ib/II study of the BRAF inhibitor encorafenib plus the MEK inhibitor binimetinib in patients with BRAF(V600E/K) -mutant solid tumors. Clin Cancer Res 2020;26:5102–12. [DOI] [PubMed] [Google Scholar]
- 76.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res 2015;21:1313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corcoran RB, André T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov 2018;8:428–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Geel R, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, et al. A Phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov 2017;7:610–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tabernero J, Geel RV, Guren TK, Yaeger RD, Spreafico A, Faris JE, et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol 2016;34:3544–44.27573652 [Google Scholar]
- 80.Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med 2019;381:1632–43. [DOI] [PubMed] [Google Scholar]
- 81.Hazar-Rethinam M, Kleyman M, Han GC, Liu D, Ahronian LG, Shahzade HA, et al. Convergent therapeutic strategies to overcome the heterogeneity of acquired resistance in BRAF(V600E) colorectal cancer. Cancer Discov 2018;8:417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. Faseb J 2000;14:6–16. [PubMed] [Google Scholar]
- 83.Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov 2007;6:391–403. [DOI] [PubMed] [Google Scholar]
- 84.Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 2002;297:1018–23. [DOI] [PubMed] [Google Scholar]
- 85.Teh JL, Purwin TJ, Greenawalt EJ, Chervoneva I, Goldberg A, Davies MA, et al. An in vivo reporter to quantitatively and temporally analyze the effects of CDK4/6 inhibitor-based therapies in melanoma. Cancer Res 2016;76:5455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen SH, Gong X, Zhang Y, Van Horn RD, Yin T, Huber L, et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 2018;37:821–32. [DOI] [PubMed] [Google Scholar]
- 87.Nassar KW, Hintzsche JD, Bagby SM, Espinoza V, Langouët-Astrié C, Amato CM, et al. Targeting CDK4/6 represents a therapeutic vulnerability in acquired BRAF/MEK inhibitor-resistant melanoma. Mol Cancer Ther 2021;20:2049–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ascierto PA, Bechter O, Wolter P, Lebbe C, Elez E, Miller WH, et al. A phase Ib/II dose-escalation study evaluating triple combination therapy with a BRAF (encorafenib), MEK (binimetinib), and CDK 4/6 (ribociclib) inhibitor in patients (Pts) with BRAF V600-mutant solid tumors and melanoma. J Clin Oncol 2017;35:9518–18. [Google Scholar]
- 89.Schuler MH, Ascierto PA, Vos FYFLD, Postow MA, Herpen CML-V, Carlino MS, et al. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: Safety, efficacy, and recommended phase 2 dose (RP2D). J Clin Oncol 2017;35:9519–19. [Google Scholar]
- 90.Sullivan RJ, Amaria RN, Lawrence DP, Brennan J, Leister C, Singh R, et al. Abstract PR06: Phase 1b dose-escalation study of trametinib (MEKi) plus palbociclib (CDK4/6i) in patients with advanced solid tumors. Mol Cancer Ther 2015;14:PR06–PR06. [Google Scholar]
- 91.Teh JLF, Cheng PF, Purwin TJ, Nikbakht N, Patel P, Chervoneva I, et al. In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov 2018;8:568–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sullivan RJ. Dual MAPK/CDK targeting in melanoma: new approaches, new challenges. Cancer Discov 2018;8:532–33. [DOI] [PubMed] [Google Scholar]
- 93.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368:1365–6. [DOI] [PubMed] [Google Scholar]
- 94.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med 2015;7:279ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res 2015;21:1639–51. [DOI] [PubMed] [Google Scholar]
- 97.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25:936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ribas A, Algazi A, Ascierto PA, Butler MO, Chandra S, Gordon M, et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat Commun 2020;11:6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dummer R, Lebbé C, Atkinson V, Mandalà M, Nathan PD, Arance A, et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: safety run-in and biomarker cohorts of COMBI-i. Nat Med 2020;26:1557–63. [DOI] [PubMed] [Google Scholar]
- 100.Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019;25:941–46. [DOI] [PubMed] [Google Scholar]
- 101.Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med 2019;25:929–35. [DOI] [PubMed] [Google Scholar]
- 102.Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020;395:1835–44. [DOI] [PubMed] [Google Scholar]
- 103.Nathan P, Dummer R, Long GV, Ascierto PA, Tawbi HA, Robert C, et al. LBA43 Spartalizumab plus dabrafenib and trametinib (Sparta-DabTram) in patients (pts) with previously untreated BRAF V600–mutant unresectable or metastatic melanoma: Results from the randomized part 3 of the phase III COMBI-i trial. Ann Oncol 2020;31:S1172. [Google Scholar]
- 104.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J Clin Oncol 2022;40:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Corcoran R Short Oral-26: Clinical efficacy of combined BRAF, MEK, and PD-1 inhibition in BRAFV600E colorectal cancer patients. 2020; Virtual.
- 106.Yam C, Xu X, Davies MA, Gimotty PA, Morrissette JJD, Tetzlaff MT, et al. A multicenter phase I study evaluating dual PI3K and BRAF inhibition with PX-866 and vemurafenib in patients with advanced BRAF V600-mutant solid tumors. Clin Cancer Res 2018;24:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sullivan RJ, Mehnert J, Tawbi H, Lawrence D, Flaherty K, Chen H, et al. Abstract LB-B30: First in human, dose escalation trial of the combination of dabrafenib, trametinib, and navitoclax in patients with BRAF mutant solid tumors. Mol Cancer Ther 2018;17:LB-B30-LB-B30. [Google Scholar]
- 108.Eroglu Z, Chen YA, Gibney GT, Weber JS, Kudchadkar RR, Khushalani NI, et al. Combined BRAF and HSP90 inhibition in patients with unresectable BRAF (V600E)-mutant melanoma. Clin Cancer Res 2018;24:5516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mooradian M, Cleary JM, Cohen JV, Lawrence DP, Buchbinder EI, Giobbie-Hurder A, et al. CTEP 9557: A dose-escalation trial of combination dabrafenib, trametinib, and AT13387 in patients with BRAF mutant solid tumors. J Clin Oncol 2020;38:3609–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nti AA, Serrano LW, Sandhu HS, Uyhazi KE, Edelstein ID, Zhou EJ, et al. Frequent subclinical macular changes in combined BRAF/MEK inhibition with high-dose hydroxychloqoquine as treatment for advamced metastatic BRAF mutant melanoma: preliminary results from a phase I/II clinical treatment trial. Retina 2019;39:502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015;27:240–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Algazi AP, Othus M, Daud AI, Lo RS, Mehnert JM, Truong TG, et al. Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF-mutated melanoma: a randomized phase 2 trial. Nat Med 2020;26:1564–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Noeparast A, Teugels E, Giron P, Verschelden G, De Brakeleer S, Decoster L, et al. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of trametinib and dabrafenib. Oncotarget 2017;8:60094–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer Discov 2018;8:184–95. [DOI] [PubMed] [Google Scholar]
- 116.Johnson DB, Zhao F, Noel M, Riely GJ, Mitchell EP, Wright JJ, et al. Trametinib activity in patients with solid tumors and lymphomas harboring BRAF non-V600 mutations or fusions: results from NCI-MATCH (EAY131). Clin Cancer Res 2020;26:1812–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nebhan CA, Johnson DB, Sullivan RJ, Amaria RN, Flaherty KT, Sosman JA, et al. Efficacy and safety of trametinib in non-V600 BRAF mutant melanoma: a phase II study. Oncologist 2021;26:731–e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dankner M, Wang Y, Fazelzad R, Spreafico A, Cescon DW, Zogopoulos G, et al. Evaluating clinical activity of MAPK targeted therapies (TT) in cancer patients (pts) with non-V600 BRAF mutations: A systematic scoping review and meta-analysis. J Clin Oncol 2021;39:3089–89.34228507 [Google Scholar]
- 119.Janku F, Sherman E, Yaeger R, Parikh A, Sullivan R, Feun L, et al. Abstract CT212: Expanded phase 1/2a study of PLX8394 a novel next generation BRAF inhibitor in patients with advanced, unresectable solid tumors with alterations in BRAF. Cancer Res 2021;81:CT212–CT12. [Google Scholar]
- 120.Desai J, Gan H, Barrow C, Jameson M, Atkinson V, Haydon A, et al. Phase I, open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors. J Clin Oncol 2020;38:2140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim TW, Lee J, Shin SJ, Kim J-S, Kim YJ, Han HS, et al. Belvarafenib, a novel pan-RAF inhibitor, in solid tumor patients harboring BRAF, KRAS, or NRAS mutations: Phase I study. J Clin Oncol 2019;37:3000–00.31557067 [Google Scholar]
- 122.Shin SJ, Lee J, Kim TM, Kim J-S, Kim YJ, Hong YS, et al. A phase Ib trial of belvarafenib in combination with cobimetinib in patients with advanced solid tumors: Interim results of dose-escalation and patients with NRAS-mutant melanoma of dose-expansion. J Clin Oncol 2021;39:3007–07. [Google Scholar]
- 123.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14. [DOI] [PubMed] [Google Scholar]
- 124.Janku F, Sherman EJ, Parkih AR, Feun LG, Tsai F, Allen E, et al. Abstract 5LBA: Interim results from a phase 1/2 precision medicine study of PLX8394- a next generation BRAF inhibitor. 2020; Virtual.
- 125.Wolf J, Planchard D, Heist RS, Solomon B, Sebastian M, Santoro A, et al. 1387P Phase Ib study of LXH254 + LTT462 in patients with KRAS- or BRAF-mutant NSCLC. Ann Oncol 2020;31:S881–S82. [Google Scholar]