

Summary
Whole-genome sequencing (WGS) is the gold standard for fully characterizing genetic variation but is still prohibitively expensive for large samples. To reduce costs, many studies sequence only a subset of individuals or genomic regions, and genotype imputation is used to infer genotypes for the remaining individuals or regions without sequencing data. However, not all variants can be well imputed, and the current state-of-the-art imputation quality metric, denoted as standard Rsq, is poorly calibrated for lower-frequency variants. Here, we propose MagicalRsq, a machine-learning-based method that integrates variant-level imputation and population genetics statistics, to provide a better calibrated imputation quality metric. Leveraging WGS data from the Cystic Fibrosis Genome Project (CFGP), and whole-exome sequence data from UK BioBank (UKB), we performed comprehensive experiments to evaluate the performance of MagicalRsq compared to standard Rsq for partially sequenced studies. We found that MagicalRsq aligns better with true R2 than standard Rsq in almost every situation evaluated, for both European and African ancestry samples. For example, when applying models trained from 1,992 CFGP sequenced samples to an independent 3,103 samples with no sequencing but TOPMed imputation from array genotypes, MagicalRsq, compared to standard Rsq, achieved net gains of 1.4 million rare, 117k low-frequency, and 18k common variants, where net gains were gained numbers of correctly distinguished variants by MagicalRsq over standard Rsq. MagicalRsq can serve as an improved post-imputation quality metric and will benefit downstream analysis by better distinguishing well-imputed variants from those poorly imputed. MagicalRsq is freely available on GitHub.
Keywords: genotype imputation, imputation quality, post-imputation quality control, machine learning, XGBoost
Graphical abstract
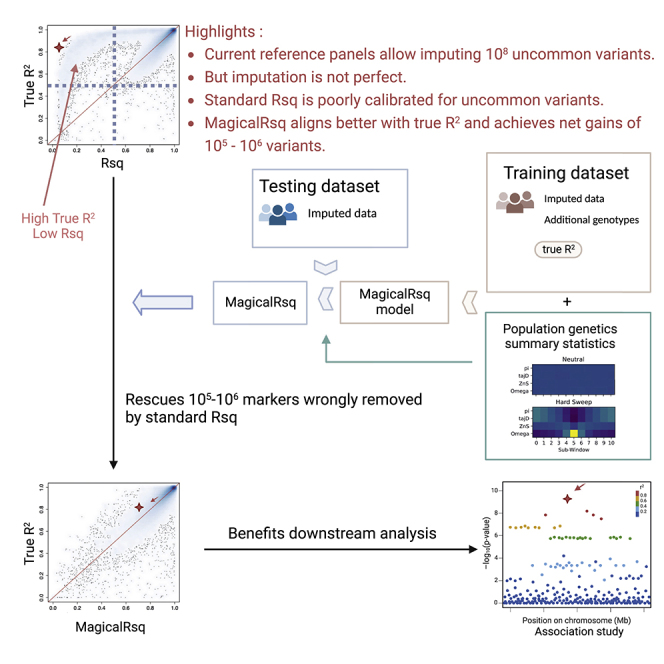
Ever-growing reference panels allow imputation of huge number (∼108) of lower-frequency variants. However, not all variants can be well imputed and standard imputation quality metric poorly reflects true imputation quality, particularly for uncommon variants. We present MagicalRsq, a machine-learning-based and better calibrated post-imputation quality metric, that can rescue 105–106 variants.
Introduction
Genotype imputation is a process of estimating missing genotypes with the aid of reference panel(s). It can effectively boost power for detecting associated variants in genome-wide association studies (GWASs) and narrow down the most strongly associated variants within a genomic region. The latest TOPMed freeze 8 reference panel1 encompasses >300 million variants. However, not all variants that are available in a reference panel can be well imputed in a target cohort.2,3,4 Therefore, post-imputation quality control (QC) is indispensable and critically important to distinguish well-imputed variants from poorly imputed ones. In current standard practice,5,6,7 variant-level imputation quality metrics such as IMPUTE’s INFO,8 minimac’s Rsq,9 or Beagle’s DR210 are adopted to perform such post-imputation quality filtering. Briefly, minimac’s Rsq and IMPUTE’s INFO estimate imputation quality by comparing observed variation in imputed data to expected conditional on allele, genotype, or haplotype frequencies, under the rationale that poorly imputed markers tend to have less than expected variation because lack of information will drag estimated genotype probabilities across individuals toward population average. Beagle calculates the squared Pearson correlation between allele dosages and best-guess genotypes to estimate true imputation quality. These metrics, though slightly different, are highly correlated.11 Hereafter we refer to these standard quality metrics directly from imputation software as Rsq (or standard Rsq). These standard metrics have been proven to be effective discriminators of imputation quality for common variants (minor allele frequency [MAF] > 5%) but are less well calibrated for uncommon (MAF ≤ 5%) variants,12,13,14 which are increasingly prevalent in continuously expanding reference panels.
Realizing that standard Rsq is less accurate for low-frequency (MAF in [0.5%, 5%]) and rare (MAF < 0.5%) variants,12,13,14,15 researchers have explored different strategies to deal with this issue. For example, Liu et al.13 proposed a simple solution: adoption of more stringent thresholds of standard Rsq for variants with lower MAF. Similarly, Coleman et al.16 proposed a procedure to assign the standard Rsq threshold taking MAF into account by using different inflection points for variants within different MAF bins. Both attempted to utilize different thresholds of standard Rsq rather than seeking alternative imputation quality measures. Lin et al.15 address the well-known effect of allele frequency on imputation quality by proposing a new statistic named the imputation quality score (IQS). This score adjusts the MAF-dependent expected concordance between imputed and true genotypes. However, the computation of IQS requires true genotypes for the SNPs of interest, which is impractical in most situations.
In this paper, we propose MagicalRsq, a machine-learning-based genotype imputation quality calibration, by using eXtreme Gradient Boosted trees (XGBoost)17 to effectively incorporate information from various variant-level summary statistics. MagicalRsq requires true R2 information for a subset of individuals and/or a subset of markers (we hereafter refer to both as additional genotypes) to train models that can be applied to all target individuals and all markers. We note that it is rather common that investigators sequence only a subset of markers (genome regions) or individuals due to cost considerations. For example, the NHLBI GO Exome Sequencing Project18 and Exome Aggregation Consortium (ExAC)19 have generated only whole-exome sequencing (WES) data thus far. In addition, some WGS efforts, such as the TOPMed project, have sequenced only subsets of individuals in the constituent cohorts and array-genotyped the remainder.3 With the availability of additional genotype data and our MagicalRsq framework, we can leverage the extra information to improve the calibration of imputation quality.
Leveraging WGS data generated by the Cystic Fibrosis Genome Project (CFGP)2 and WES data from UKB,20 we demonstrate that MagicalRsq substantially outperforms standard Rsq and is a more informative metric for post-imputation QC, particularly for lower-frequency variants. MagicalRsq performs well in the evaluated European and African ancestry cohorts. We carried out comprehensive experiments to mimic different real-life scenarios and showed that MagicalRsq is superior to standard Rsq in almost every scenario. Moreover, as a post-imputation QC metric, MagicalRsq could achieve net gains of thousands or millions of variants that are either well imputed but incorrectly filtered out or poorly imputed but incorrectly retained by standard Rsq. We anticipate MagicalRsq will benefit downstream analysis by better distinguishing well-imputed variants from poorly imputed ones.
Materials and methods
Ethics statement
This research has been conducted using the UK Biobank Resource under Application Number 25953. Furthermore, this study was reviewed by the Cystic Fibrosis Foundation for the use of CF Foundation Patient Registry data and CFGP WGS data. The procedures followed for data collection and processing, DNA sequencing, and analysis were in accordance with the ethical standards of the responsible human rights committees on human experimentation, and proper informed consent was obtained from all individuals.
MagicalRsq model
MagicalRsq adopts the eXtreme Gradient Boosted trees (XGBoost) method17 for better calibration of imputation quality score, particularly for uncommon variants. Tree boosting is a commonly used machine learning approach that has been applied for a wide range of problems.21,22,23,24 MagicalRsq is a supervised learning method where we build models to predict true imputation quality (true R2, squared Pearson correlation between imputed and true genotypes) using a battery of variant-level summary statistics. As a supervised method, MagicalRsq requires true genotypes at a reasonable number of variants (>10k) to derive their true imputation quality scores to train models. For both performance and computational considerations, we build three models separately for common (MAF > 5%), low-frequency (MAF in [0.5%, 5%]) and rare (MAF < 0.5%) variants in our model training process. Note that MAF is estimated based on imputed dosages to better represent realistic settings where we don’t have true WGS genotypes to calculate MAF. As shown in Figure 1, starting from genotype array data in training dataset, minimac imputation is performed to obtain standard Rsq and estimated MAF from imputed dosages. After imputation, using true genotypes at imputed markers not on the initial genotype array, we calculate true imputation quality, i.e., true R2, at these imputed markers. The standard Rsq, estimated MAF, and true R2 will then be carried forward to model training. Note that these statistics are specific for a given training dataset and will vary when using different datasets for model training purposes.
Figure 1.

MagicalRsq workflow
MagicalRsq starts from “training dataset array data” (which are data used for imputation among training individuals) and performs imputation using these data, which gives us standard Rsq and estimated MAF for each marker, in the training dataset. Then we calculate the true R2 by comparing imputed dosages with truth genotypes (established by additional genotype data in the training set). Combining external MAF and alternative allele count (AC), as well as population genetics summary statistics, with the above three metrics (i.e., standard Rsq, estimated MAF, and true R2), we train MagicalRsq models using the XGBoost method where we build supervised models to predict true R2 from all the other features. We then proceed to the testing dataset where we follow the same imputation workflow starting again from array genotype data and obtaining estimated MAF and standard Rsq after imputation. We then calculate MagicalRsq in the testing dataset by plugging in the predictor features into the MagicalRsq models built from the training dataset. Finally, we evaluate the performance of MagicalRsq (and Rsq) by comparing with true R2 in the testing dataset. Yellow highlights represent all the instruments specific for the training dataset, light blue highlights represent the instruments specific for the testing dataset, green highlights represent external information used in both training and testing, and red rectangles represent statistics used during final evaluation and comparison of MagicalRsq and standard Rsq, using true R2 as the gold standard.
Besides the aforementioned imputation summary statistics in the training samples, we also consider multiple variant-level features that are not specific to the training dataset that we use. We first include multiple population genetics features as they are known to impact genotype imputation.9,11,25,26 These population genetics statistics reflect various aspects encompassing haplotype structure, linkage disequilibrium (LD) profile, and the spatial pattern of site frequency spectrum (SFS) structure. Specifically, we consider 11 population genetics summary statistics (Note S1) for six populations (CEU, GWD, JPT, LWK, PEL, and YRI) corresponding to diverse continental ancestry groups including European (EUR), African (AFR), East Asian (EAS), and American (AMR). These 11 population genetics summary statistics, calculated by the positive selection scan program S/HIC,27 are π,28 ,29 Tajima’s D,30 Fay and Wu’s H,29 the maximum frequency of derived mutations (MFDM),31 the number of distinct haplotypes,32 Garud et al.’s32 haplotype homozygosity statistics (H1, H2, H2/H1), Kim and Nielsen’s ,33 and Kelly’s;34 all initially calculated for 100Kb non-overlapping bins.27 We obtain variant-level statistics by application of their corresponding region-level statistics. In addition, considering the known effects of allele frequency on imputation quality reported in previous studies,13,35 we also incorporate external alternative allele counts (AC) per 1,000 samples and MAF in four major ancestral groups (EUR, AFR, EAS, and South Asians [SAS]) derived from TOPMed WGS data in the TOP-LD project,36 and 2nd–4th moments (representing variance, skewness, and kurtosis) for the four ACs. Inclusion of these variant-specific features distinguishes MagicalRsq from standard Rsq and other imputation quality metrics that utilizes only imputation summary statistics.15 We then treat each variant as an observation and build XGBoost models to predict true R2 by leveraging these 79 variant-level features.
Since the true imputation quality (i.e., true R2) is between 0 and 1, we specify the learning task to be tree-based logistic regression and evaluation metric to be root mean square error. To control overfitting, we set early stopping rounds to be 50, i.e., training would stop if the performance did not improve for 50 rounds in an independent validation set.
True imputation quality calculation
We quantify the true imputation quality metric using true R2, which is the squared Pearson correlation between imputed dosages and true genotypes. The true genotypes, coded as 0, 1, and 2, are obtained from the additional genotype data not used for imputation, for example, the WGS data from CFGP or the WES data from UKB. Our evaluation was restricted only to samples with after QCed (QC+) data from both imputed and additional genotype data. Duplicate samples were dropped. Finally, true R2 was calculated for each variant, based on overlapping QC+ samples.
Imputation metric evaluation
We evaluate imputation quality metrics (standard Rsq or MagicalRsq) in testing dataset(s) independent of those used for training. For evaluation, we treat true R2 as the truth and quantify the performance of each quality metric by calculating the squared Pearson correlation, root mean squared error (RMSE), and mean absolute error (MAE) with true R2.
We further evaluate the performance of MagicalRsq in comparison to standard Rsq in terms of their effectiveness to distinguish between well- and poorly imputed variants. Specifically, using a commonly used 0.8 cutoff,2,3,13,37,38 we compare the numbers of (1) well-imputed variants saved by MagicalRsq, defined as true R2 ≥ 0.8, Rsq < 0.8, and MagicalRsq ≥ 0.8; (2) well-imputed variants missed by MagicalRsq, defined as true R2 ≥ 0.8, Rsq ≥ 0.8, and MagicalRsq < 0.8; (3) poorly imputed variants excluded by MagicalRsq, defined as true R2 < 0.8, Rsq ≥ 0.8, and MagicalRsq < 0.8; and (4) poorly imputed variants included by MagicalRsq, defined as true R2 < 0.8, Rsq < 0.8, and MagicalRsq ≥ 0.8. We then calculate the net gains of variants when applying MagicalRsq for post-imputation QC compared to Rsq, defined as (1) − (2) + (3) − (4).
Data description
The Cystic Fibrosis Genome Project (CFGP) aims to identify genetic modifiers of cystic fibrosis (CF) traits by leveraging WGS data and rich phenotypic information collected through the Cystic Fibrosis Foundation Patient Registry (CFFPR).39 CFGP high-coverage (∼30×) WGS data are available for 5,109 samples representing 5,072 unique individuals. The WGS data contain approximately 90m and 11m high-quality single-nucleotide variants and indels, respectively. The resource represents the largest cohort of CF-affected individuals for which WGS data are available. 5,095 samples representing 5,058 unique individuals remained after sample identity check.2
The UK Biobank (UKB),40 recruiting ∼500,000 people aged between 40 and 69 years in 2006–2010, is a prospective biobank study to study risk factors for common diseases such as cancer, heart disease, stroke, diabetes, and dementia. Participants have been followed up through health records from the UK National Health Service. UKB has genotype data on all enrolled participants, as well as extensive baseline questionnaires and physical measures and stored blood and urine samples. Specifically, genotyping array data at ∼800,000 directly genotyped markers are available for ∼500,000 UKB participants, among whom ∼200,000 also have WES data.20
MagicalRsq experiments
To comprehensively evaluate the performance of MagicalRsq, we performed 14 experiments (Table S2) with 9 imputations (Table S1), leveraging CFGP WGS data2 and UKB WES data.41 These experiments can be categorized in two scenarios. In the first scenario, we have additional directly typed markers (other than array genotypes) available in all target individuals, while in the likely more realistic second scenario, we have additional directly typed markers in only a subset of target individuals.
Imputation overview
For CFGP data, our training dataset contains 1,992 CF samples (hereafter denoted as the CF 2k samples) who have both Illumina 610-Quad array data with 567,784 variants after QC2 and WGS data. An independent set of 3,103 CF samples (hereafter denoted as the CF 3k samples) have WGS data but no array genotypes available. To perform imputation for the latter set of 3,103 samples, we first thinned their WGS data to Illumina 660W array by keeping only the 551,819 QC+ variants overlapped with the 660W array. For the UKB samples, we identified 9,354 participants with significant African ancestry (UKB AFR) and array genotype data available following our previous work.4 Among them, 3,960 individuals also had WES data in the 200k WES release.
Genotype imputation was then performed by uploading QC+ array or thinned WGS data to TOPMed imputation server or Michigan imputation server (web resources), using TOPMed freeze 8 or the 1000 Genomes phase 3 (1000G) as the reference panel, respectively. Phasing was performed using Eagle 242 and imputation was performed by Minimac 4.9
Scenario 1: Availability of additional directly typed markers in all target individuals
We first consider the scenario where all target samples have additional genotype data in addition to the genotyping array data used for imputation. Under this scenario, we first imputed CF 2k samples using their genotypes from Illumina 610-Quad array2 and the 1000G reference panel (Table S1, imputation 1). These individuals also have WGS data which we did not use for imputation but rather used only for obtaining true R2. These true R2 values were used to build models in training “samples” and to evaluate performance in testing “samples,” where “samples” here refer to variants. To avoid information leakage, we trained using variants on even-numbered chromosomes and tested on odd-numbered chromosomes (Table S2, experiment 1). We then imputed the same individuals with the TOPMed freeze 8 reference panel (Table S1, imputation 2) and re-built MagicalRsq models (Table S2, experiment 2) following the same procedures as described above.
We further attempted to relax the MagicalRsq model training requirements to either mix-and-match imputation reference panels or variants in a region (in contrast to genome- or chromosome-wide). Using mix-and-match reference panels, we tried to examine whether MagicalRsq models trained using imputed data from one reference panel can be applied to imputed data from a different reference panel. Specifically, we trained MagicalRsq models using 1000G-imputed variants on odd-numbered autosomes and applied to TOPMed-imputed variants on even-numbered autosomes (Table S2, experiment 3) and vice versa from TOPMed training to 1000G testing (Table S2, experiment 4), in the same 2k CF samples.
By using variants in a region, we tried to mimic realistic scenarios where only some regions (e.g., selected gene(s), selected loci near GWAS regions, exonic regions) are sequenced. Specifically, we assumed that our 2k CF samples are only additionally (besides GWAS array) sequenced in the ±10 Mb region around the CFTR (chr7:107480144–127668447, hg38) and explored training models only using variants in this region, from TOPMed imputed data (Table S2, experiment 5). Note that mutations in the CFTR on chromosome 7 provide the molecular basis of CF and thus a region of high importance in this dataset. We trained MagicalRsq models using 183k variants in this 20 Mb region and applied the models to all other chromosomes. We similarly separated the variants into three MAF categories, leaving 32k common variants, 24k low-frequency variants, and 127k rare variants. We also tried training models with variants on another two regions: chr10:80–100 Mb and chr20:20–40 Mb to assess whether MagicalRsq is robust to different region choices. We then performed additional experiments with 1000G imputed variants on the same three regions (Table S2, experiment 6) to evaluate whether MagicalRsq models trained from regional (versus genome- or chromosome-wide) variants are robust to reference panel choices.
Finally, we trained MagicalRsq models using only exonic variants to assess whether exome-trained models could be applied to other genomic regions. We first retained variants from the CF 2k TOPMed imputed data which are also present in the UKB WES data, leaving 89k common variants, 82k low frequency variants and 635k rare variants. We trained models using these WES variants, and applied to other variants from the same TOPMed imputed data in the CF 2k samples (Table S2, experiment 7).
Scenario 2: Availability of additional directly typed markers in a subset of target individuals
We next consider the scenario where we have additional markers directly assayed in only a subset of samples, while the remaining samples still need imputation. We again leveraged CFGP WGS data. As previously mentioned, we have the CF 2k samples with both the Illumina 610-Quad array genotypes and WGS data and an independent CF 3k samples with WGS data only. We thinned the WGS data of the CF 3k samples to the Illumina 660W array density and performed imputation (Table S1, imputation 3), using TOPMed freeze 8 as reference. We then applied the MagicalRsq models trained from the CF 2k samples to the 3k samples (Table S2, experiment 8).
We additionally leveraged the WES data of UKB AFR participants to assess MagicalRsq’s performance for African-ancestry individuals. We imputed 3,960 UKB AFR samples using both 1000G and TOPMed reference panels (Table S1, imputation 4–7). All these 3,960 individuals also have WES data available. We randomly selected 1,000 samples for MagicalRsq model training and used the remaining for testing, and again trained models separately for common, low-frequency, and rare variants (Table S2, experiments 9 and 10). We also investigated mix-and-match reference panels under this scenario (Table S2, experiments 11 and 12, Note S2). To assess whether other under-represented populations can also benefit from MagicalRsq, we also imputed 4,436 UKB South Asian (SAS)-ancestry individuals using TOPMed freeze 8 reference panel (Table S1, imputation 9). We similarly randomly selected 1,000 samples for building MagicalRsq models and applied the models to the remaining individuals for testing, using genotypes from WES data as truth (Table S2, experiment 13).
We further examined the performance of MagicalRsq when training models using a small subset of variants, instead of all variants available. Specifically, we trained models in the CF 2k samples with different numbers of variants on even-numbered chromosomes and applied such models to the independent 3k samples as described before (Table S2, experiment 14). We randomly selected 10k, 50k, 100k, 200k, 500k, and 1m variants in each of the three MAF categories for model training and repeated five times for more stable inference.
Results
Scenario 1: Availability of additional directly typed markers in all target individuals
We first consider a simple yet realistic scenario where all target samples have additional genotype data in addition to the genotyping array data used for imputation. These additional data could be from a different genotyping array, from WES, or from targeted genotyping or sequencing. We do not consider the scenario where all individuals already have deep WGS data because imputation would no longer be relevant.
Feature importance
We first imputed CF 2k samples using the 1000G reference panel, trained MagicalRsq models using variants on even-numbered chromosomes, and tested on odd-numbered chromosomes (Scenario 1). We performed feature importance analysis to find out major contributors to the training models. We found, as expected, that standard Rsq is by far the most important feature, weighing ∼80% among all the features (Figure S1). European AC is the second most important feature for common variants, while for low frequency and rare variants, the second most important feature is African AC. Because the CF cohorts are predominantly of European ancestry,2 it is not surprising that European AC is influencing the training models. For rarer variants, we suspect the importance of African AC, likely due to the African allele frequency spectrum better capturing rarer variants among individuals with CF.
MagicalRsq outperforms standard Rsq
Compared to standard Rsq, MagicalRsq is more consistent with the true R2 with respect to all three of the evaluation metrics (squared Pearson correlation with true R2, RMSE, and MAE) and for all three MAF categories (Figures 2A and S2). For example, when we compare MagicalRsq to standard Rsq, squared Pearson correlation with true R2 increases by 8.7%–24.1% for common variants, 10.1%–62.7% for low-frequency variants, and 14.3%–17.2% for rare variants (Table S4), across different chromosomes. When using MagicalRsq to replace standard Rsq for post-imputation QC, we have net gains of 33k common variants, 34k low-frequency variants, and 200k rare variants across half of the genome (i.e., even numbered chromosomes) (Table S3 experiment 1). We then imputed the same individuals with the TOPMed freeze 8 reference panel (Table S1, imputation 2), re-built MagicalRsq models (Table S2, experiment 2) following the same procedures, and observed similar improvement (Figures 2B and S3). For example, MagicalRsq increases squared Pearson correlation with true R2 by 1.7%–34.5% for common variants, 37.6%–71.0% for low-frequency variants, and 10.3%–14.3% for rare variants (Table S5), and the net gains of rare variants increase to 411k (Table S3, experiment 2) due to more rare variants in the TOPMed reference panel. Comparing across the three MAF categories, we observe that MagicalRsq shows the most pronounced improvement for low-frequency variants in terms of squared Pearson correlation, regardless of the imputation reference panels.
Figure 2.

Scenario 1, experiments 1–4: Training using even-numbered chromosomes and testing on odd-numbered chromosomes for CF 2k samples
(A and B) Performance comparison between Rsq and MagicalRsq in terms of squared Pearson correlation with true R2 for (A) 1000G-based imputation; (B) TOPMed-based imputation.
(C) Smooth scatterplot showing Rsq or MagicalRsq (x axis) calculated from both matched- (second row) and mis-matched- (third row) models against true R2 (y axis) for both 1000G-based (left) and TOPMed-based (right) imputation, for low-frequency variants on chromosome 13.
Impact of reference panel
We further attempted mix-and-match of the imputation reference panels in training and testing sets to examine whether MagicalRsq trained using imputed data from one reference panel can be applied to imputed data from a different reference panel (Methods). We found that applying 1000G-trained models to TOPMed data performs less well for low-frequency and rare variants, while applying TOPMed-trained models to 1000G data performs less well for common and low-frequency variants (Table S3, experiments 3 and 4). We suspected that reference-panel-specific variants may hinder the prediction accuracy, and then evaluated the mix-and-match performances by restricting to variants shared between the two reference panels. We found that the mix-and-match MagicalRsq were better calibrated than standard Rsq for the shared variants (Note S2), although the improvements are less pronounced than using the matched reference panel (Figure 2C, Table S6).
Number of variants needed for training
In the previous experiments MagicalRsq training models were built on a large subset of markers genome-wide (e.g., all even chromosomes, corresponding to 2.6m, 1.9m, and 12.3m common, low-frequency, and rare variants in TOPMed imputed CF 2k samples). To mimic the scenario where only some regions (e.g., selected gene(s), selected loci near GWAS regions, exonic regions) are sequenced, we trained MagicalRsq models using TOPMed imputed variants on three ∼20 Mb regions: ±10 Mb region around the CFTR (chr7:107480144–127668447, hg38), and two randomly selected regions: chr10:80–100 Mb and chr20:20–40 Mb regions (Table S2, experiment 5). We found that the models performed reasonably well for low-frequency and rare variants, but not for common variants (Figure S6), likely due to the larger fluctuation of Rsq performance for common variants across the genome (Note S2). We then performed additional experiments with 1000G imputed variants on the same regions (Table S2, experiment 6), and the results are highly consistent (Figure S7).
Apply exome-trained models to other genomic regions
As WGS is still not yet available for most studies, many researchers are generating WES data as an alternative.19 We trained our MagicalRsq models using only variants in exomes to test whether such models are generalizable to other genomic regions. Specifically, we trained our models using exonic variants from TOPMed imputed CF 2k cohort (Scenario 1) and applied to variants outside of the exomes (Table S2, experiment 7). The results showed that the WES-trained models improved squared Pearson correlation with true R2 by 3.5%–28.4% for common variants, by 17.8%–68.5% for low-frequency variants, and by 10.1%–13.8% for rare variants (Table S7, Figure S8). We note that the observed improvements are similar to experiment 2 where models were built on half of the genome, indicating that MagicalRsq models perform similarly well when trained with only exonic variants.
Scenario 2: Availability of additional directly typed markers in a subset of target individuals
The previous scenario assumes that all individuals have additional genotyping (through other genotyping arrays, candidate gene sequencing, WES, etc.). A different scenario where a subset of individuals enjoys higher marker density has become increasingly common.2,3 The remaining samples still need imputation and can benefit from better-calibrated imputation quality metrics. Therefore, we consider scenario 2 where we have additional markers directly assayed in only a subset of individuals. Specifically, we assessed the performance of MagicalRsq assuming no overlap of individuals in the training and testing datasets.
CFGP European samples
Under this scenario, we first applied the MagicalRsq models trained from CF 2k samples using all genome-wide variants to the 3k samples (Scenario 2). We observed similar improvements over standard Rsq for all three MAF categories and every chromosome, again with the most pronounced gains on low-frequency variants (Figure S9, Table S8). Specifically, MagicalRsq improves squared Pearson correlation with true R2 by 2.0%–56.8% for common variants, by 3.0%–6.2% for rare variants, and by 16.3%–91.7% for low-frequency variants, across different chromosomes, compared to standard Rsq (Table S8). MagicalRsq achieves net gains of 18k common variants, 117k low-frequency variants, and 1.4m rare variants (Table S3, experiment 8). MagicalRsq also outperforms Rsq in terms of RMSE or MAE (Figure S9, Table S8), further supporting that MagicalRsq provides better calibrated imputation quality estimation.
UKB African and South Asian samples
We further assessed MagicalRsq’s performance on African-ancestry individuals, leveraging WES data from the UKB. We randomly selected 1,000 individuals as training and the remaining 2,960 as testing (Scenario 2). Consistent with our results in the CF cohort, we found that MagicalRsq is better calibrated than standard Rsq for every variant category for 1000G imputed data (Figure 3A), but interestingly is slightly inferior for TOPMed imputed rare variants (Figure 3B) in terms of squared Pearson correlation with true R2. One explanation is that TOPMed contains more extremely rare variants which are more challenging to impute. Specifically, 62% (664k/1.1m) of TOPMed imputed variants have MAF < 0.1% while only 28% (146k/527k) of 1000G imputed variants have MAF < 0.1%. When restricting to variants with MAF between 0.1% and 0.5% (rare variants with minor allele count [MAC] ≥ 6), MagicalRsq outperforms standard Rsq (Figure 3B). Note that the inferiority is observed only when evaluating using squared Pearson correlation. When comparing MagicalRsq with Rsq as a post-imputation quality filter, we would have a net gain of 12k rare variants. Moreover, though the absolute improvement of squared Pearson correlation is moderate, MagicalRsq is much better aligned with true R2 than Rsq. There was a departure of the 45-degree line when comparing true R2 with Rsq (Figure 3C, top panel), while MagicalRsq correctly rectified such departure (Figure 3C, middle and bottom panels). We again also explored the mix-and-match reference and reached similar conclusions as in scenario 1: MagicalRsq trained using imputed data from a mis-matched reference performs better than Rsq but is inferior to that trained from a matched reference. The performance is substantially better when evaluating only variants shared between the two reference panels, suggesting that some reference-panel-specific variants may hinder the transferability (Note S2, Table S9). We similarly performed experiments on UKB SAS populations to evaluate whether MagicalRsq could benefit other ancestral groups (Scenario 2). We observed that MagicalRsq improved squared Pearson correlation with true R2 by 7.8%–134.4% for common variants, by 13.8%–22.1% for low-frequency variants, and by 14.4%–22.4% for rare variants (Figure S11, Table S10). The results indicate that MagicalRsq is applicable to these under-represented minority populations.
Figure 3.

Scenario 2, experiments 9–12: Training models using 1000 UKB AFR samples and testing on 2,960 independent UKB AFR samples, for all variants with WES available
(A and B) Performance comparison between Rsq and MagicalRsq in terms of squared Pearson correlation with true R2 for (A) 1000G-based imputation; (B) TOPMed-based imputation.
(C) Smooth scatterplot showing Rsq or MagicalRsq (x axis) calculated from both matched (second row) and mis-matched (third row) models against true R2 (y axis) for both 1000G-based (left) and TOPMed-based (right) imputation, for all low-frequency variants with WES available.
Number of variants needed for training
We additionally investigated MagicalRsq’s performance when using a small subset of variants, with randomly selected 10k, 50k, 100k, 200k, 500k, and 1m variants from CF 2k samples and tested on independent CF 3k samples. We repeated this experiment five times for reliability (Scenario 2). We found that even when MagicalRsq models were trained using only 10k variants, they still outperformed Rsq (the red dashed line in Figure 4) for all three MAF categories. When the number of training variants increases, the advantage of MagicalRsq over Rsq is more pronounced (Figures 4 and S12). For example, the average relative increment of squared Pearson correlation with true R2 for MagicalRsq is 2.3% with 10k variants for common variants, which increases to 20.5% with 1m variants contributing to model building (Table S11); the net gains of variants increase from ∼6k to ∼19k for common variants, from ∼94k to ∼123k for low-frequency variants, and from ∼787k to ∼1,085k for rare variants (Table S3, experiment 14). We again noticed that the largest improvement manifests for the low-frequency variant category. For example, when trained with 10k (100K) variants, squared Pearson correlation with true R2 improves by 56.9% (71.0%) for low-frequency variants, substantially more pronounced than the relative increases for common and rare variants, which are 2.3% (13.4%) and 1.9% (9.5%), respectively (Table S11). In addition, we found minimal variation across the five repeats, even when training models with only 10k variants, indicating MagicalRsq is robust to different selected random subsets of variants. Moreover, the computational burden is greatly reduced when using a subset of variants, especially for rare variants. For example, the CPU time was 47 h when training models with all rare variants on even number chromosomes (12.3 million), while it reduced to only 30 min when using randomly selected 50k rare variants (Figure S13, Table S12).
Figure 4.

Scenario 2, experiment 14: Training models using randomly selected subsets of variants
The number of variants used for training varied from 10k to 1m. MagicalRsq models were built based on CF 2k samples and tested on the independent CF 3k samples. We repeated 5 times for each number of variants. Squared Pearson correlation with true R2 was calculated and served as the evaluation metric. The red dashed line denotes the performance of standard Rsq. nvar, number of variants included in model training.
Computation costs
We note that sample size for MagicalRsq models is not the number of individuals but rather the number of variants, which affects the computational costs. We evaluated the CPU time and memory usage, when training from CF 2k samples and applying to CF 3k samples. The CPU time increases exponentially with the number of variants in training, for example, ranging from only 8 min (with 10k variants) to 605 min (with 2.6m variants) for common variants (Figure S13, top panel, Table S12). The memory usage is rather stable with respect to the number of variants in training models, with ∼3 GB for common variants, ∼2 GB for low-frequency variants, and ∼10 GB for rare variants (Figure S13, bottom panel, Table S12).
Discussion
Genotype imputation has become a standard practice in genomic studies. For post-imputation QC and analysis, the estimated imputation quality metrics (referred to as standard Rsq) provided by the various imputation engines (e.g., Rsq from minimac, INFO from IMPUTE, DR2 from Beagle) are key. In this work, we demonstrate that those estimated quality metrics do not always reflect the true imputation quality, especially for lower-frequency and rare variants. To provide better-calibrated quality metrics for these variants, we propose MagicalRsq, a machine learning enhanced genotype imputation quality estimate, which incorporates multiple variant-level features to improve the calibration of imputation quality estimates. We demonstrate by comprehensive experiments that MagicalRsq performs better than standard Rsq under different circumstances: it not only aligns better with true R2, but can also rescue a substantial number of misclassified variants when replacing standard Rsq as a post-imputation QC metric. We leveraged CFGP WGS data and UKB WES data to show the advantages of MagicalRsq for both European- and African-ancestry individuals. We also performed experiments where MagicalRsq models were built with randomly selected subsets of variants ranging from 10k to 1m variants in each MAF category. Our results showed that MagicalRsq models are robust to different subsets of variants used in model training. We observe slightly better performance with more variants included, but at the cost of exponentially increased computational burden. Considering the tradeoff between computing costs and performance gains, we recommend using 10k to 1m variants in each MAF category when training MagicalRsq models.
We observe that low-frequency variants benefit most from MagicalRsq, which has meaningful implications for downstream analysis. For example, in GWASs, recent publications4,20,43,44,45,46 show multiple examples of GWAS signals from variants in the low-frequency category across diverse populations. Thus, better discerning and including well-imputed low-frequency variants in GWASs could potentially facilitate new discoveries and aid fine-mapping analysis. Our MagicalRsq models can handle rare variants well and we recommend including extremely rare variants down to singletons in model training, especially when the training set is derived from much fewer individuals than the target set. In our experiments, we obtained net gains of 1 million rare variants using a 0.8 cutoff, and the number decreased with a more lenient threshold (Table S14). We note that the squared Pearson correlation is less informative for rare variants due to their extremely low MAF, and thus we recommend applying a more stringent post-imputation QC threshold, which is also consistent with recommendations from existing literature.2,3,13,37,38 Some of the rare variants that are rescued by MagicalRsq have important clinical potentials. For example, a BRCA1 missense variant rs28897686 (chr17:43091783:C:T, hg38, GenBank: NM_007294.4; c.3748G>A [p.Glu1250Lys])47,48,49,50 was well imputed in our UKB AFR target samples with true R2 0.99, but the standard Rsq was only 0.23, which means this variant would have been missed if Rsq was used to perform post-imputation QC. In contrast, the MagicalRsq value of this variant is 0.86, which effectively rescues the variant for further investigations. We also systematically compared the two quality metrics in the UKB AFR experiments by measuring the power of including potential clinically important variants. We downloaded the ClinVar database and checked for large differences between true R2 and Rsq or MagicalRsq for each imputed ClinVar exome variant. In summary, 15 well-imputed variants (true R2 > 0.8) that have large quality differences using Rsq (Rsq < 0.5) are saved by MagicalRsq (MagicalRsq > 0.8). Conversely, there is no well-imputed variant (true R2 > 0.8) that has low MagicalRsq (MagicalRsq < 0.5) and can be saved by Rsq (Rsq > 0.8). These findings show clearly that MagicalRsq is superior to Rsq in association studies and further clinical applications.
The XGBoost method adopted in our models is widely used in both classification and regression problems. It is computationally efficient and requires less tunning procedures than some other machine learning or deep learning methods, such as neural networks. We also tried two deep neural network methods to construct the prediction models, but the performance was inferior to XGBoost-trained models (Note S2, Table S13). Our comparison results are consistent with the literature,51,52,53 showing the advantage of XGBoost in such regression-like problems. We also explored the strategy of directly using true R2 in the training subset with WGS available as a post-imputation quality metric in the target set in our scenario 2. We found that such a strategy works well for common variants, but worse for low-frequency variants and extremely badly for rare variants (Table S15), likely due to poor representation for lower-frequency variants using a smaller subset.
Though we have demonstrated that MagicalRsq performs much better than standard Rsq through comprehensive experiments that mimic real-life scenarios, we do note that there are some limitations and caveats to our study. First, we didn’t include chromosome X for this study mainly due to the complexity of chromosome X coding and imputation and the lack of some key variant-level features (S/HIC’s population genetics summary statistics) that we used for model training. Second, MagicalRsq performance was impaired by some reference-specific variants when applying a model trained with mis-matched reference imputed data, although it nonetheless typically performed better than standard Rsq in this scenario. In practice, we recommend investigators apply MagicalRsq models with matched reference whenever possible for better performance. Third, MagicalRsq performs less well for common variants when training from variants in a particular region, which is likely caused by the fluctuation of Rsq performance across the genome, and such fluctuation was mainly driven by the spanning range of the imputation qualities (Note S2), which impeded the generalizability of such models for common variants.
We note that in our MagicalRsq models, we leverage 79 variant-level features to enhance the imputation quality prediction, but more features could be added into the model. We have released our easily generalizable codes, which allow investigators to incorporate their favorite features into the model and choose whether to include the features we used (i.e., population genetics features summarized by S/HIC or TOP-LD allele-frequency features). We anticipate MagicalRsq to have even better performance when more comprehensive and relevant variant-level features are included. Another future research direction is more general applications and evaluations of MagicalRsq. For the current experiments we performed, rather homogeneous training and target cohorts are used. Even under scenario 2 when training and testing samples are independent with no samples overlap, they still come from the same study (albeit a consortium study involving multiple cohorts in the case of CFGP). Further efforts are also warranted to evaluate the transferability of MagicalRsq models trained on external cohorts, for example, whether MagicalRsq models trained from CFGP data could be applied to UKB cohort.
In summary, MagicalRsq clearly showcases its advantages over standard Rsq in realistic settings. We anticipate that it will benefit the genetic community as a better post-imputation quality metric and will enhance downstream association analysis by rescuing variants.
Acknowledgment
This study is supported by the Cystic Fibrosis Foundation (CUTTIN18XX1, BAMSHA18XX0, KNOWLE18XX0). Y.L. was partially supported by NIH grants U01HG011720, R01HL146500, and R01MH123724. Q.S. was supported by U24AR076730. L.M.R. is additionally supported by KL2TR002490. D.R.S. was supported by U01HG011720 and R35GM138286. C.F. was supported by R01HG009976.
The authors would like to thank the Cystic Fibrosis Foundation for the use of CF Foundation Patient Registry data to conduct this study. Additionally, we would like to thank the patients, care providers, and clinic coordinators at CF centers throughout the United States for their contributions to the CF Foundation Patient Registry.
Furthermore, we acknowledge use of the Trans-Omics in Precision Medicine (TOPMed) program imputation panel (freeze 8 version) supported by the National Heart, Lung, and Blood Institute (NHLBI); see www.nhlbiwgs.org. TOPMed study investigators contributed data to the reference panel, which was accessed through https://imputation.biodatacatalyst.nhlbi.nih.gov. The panel was constructed and implemented by the TOPMed Informatics Research Center at the University of Michigan (3R01HL-117626-02S1; contract HHSN268201800002I). The TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I) provided additional data management, sample identity checks, and overall program coordination and support. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
Finally, the authors thank the Department of Innovation, Research University and Museums of the Autonomous Province of Bozen/Bolzano for covering the Open Access publication costs.
Declaration of interests
The authors declare no competing interests.
Published: October 4, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.09.009.
Contributor Information
Christian Fuchsberger, Email: cfuchsberger@eurac.edu.
Yun Li, Email: yunli@med.unc.edu.
Web resources
MagicalRsq GitHub page, https://github.com/quansun98/MagicalRsq/
Michigan imputation server, https://imputationserver.sph.umich.edu/
TOP-LD, http://topld.genetics.unc.edu
TOPMed imputation server, https://imputation.biodatacatalyst.nhlbi.nih.gov
True R2 and Rsq calculation scripts, https://yunliweb.its.unc.edu/software.html
Supplemental information
Data and code availability
MagicalRsq is freely available from https://github.com/quansun98/MagicalRsq/.
The CFGP WGS data are available for request to the Cystic Fibrosis Foundation at https://www.cff.org/researchers/whole-genome-sequencing-project-data-requests#requesting-data.
UKB genotyping and WES data are available upon request from the UK Biobank https://www.ukbiobank.ac.uk/.
References
- 1.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53, 831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun Q., Liu W., Rosen J.D., Huang L., Pace R.G., Dang H., Gallins P.J., Blue E.E., Ling H., Corvol H., et al. Leveraging TOPMed imputation server and constructing a cohort-specific imputation reference panel to enhance genotype imputation among cystic fibrosis patients. HGG Adv. 2022;3:100090. doi: 10.1016/j.xhgg.2022.100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowalski M.H., Qian H., Hou Z., Rosen J.D., Tapia A.L., Shan Y., Jain D., Argos M., Arnett D.K., Avery C., et al. Use of >100, 000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15:e1008500. doi: 10.1371/journal.pgen.1008500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Q., Graff M., Rowland B., Wen J., Huang L., Miller-Fleming T.W., Haessler J., Preuss M.H., Chai J.-F., Lee M.P., et al. Analyses of biomarker traits in diverse UK biobank participants identify associations missed by European-centric analysis strategies. J. Hum. Genet. 2022;67:87–93. doi: 10.1038/s10038-021-00968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Bakker P.I.W., Ferreira M.A.R., Jia X., Neale B.M., Raychaudhuri S., Voight B.F. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porcu E., Sanna S., Fuchsberger C., Fritsche L.G. Genotype imputation in genome-wide association studies. Curr. Protoc. Hum. Genet. 2013;Chapter 1:Unit1.25. doi: 10.1002/0471142905.hg0125s78. [DOI] [PubMed] [Google Scholar]
- 7.Naj A.C. Genotype Imputation in Genome-Wide Association Studies. Curr. Protoc. Hum. Genet. 2019;102:e84. doi: 10.1002/cphg.84. [DOI] [PubMed] [Google Scholar]
- 8.Howie B., Marchini J., Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011;1:457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Browning B.L., Zhou Y., Browning S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018;103:338–348. doi: 10.1016/j.ajhg.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 12.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu E.Y., Buyske S., Aragaki A.K., Peters U., Boerwinkle E., Carlson C., Carty C., Crawford D.C., Haessler J., Hindorff L.A., et al. Genotype imputation of Metabochip SNPs using a study-specific reference panel of ∼4, 000 haplotypes in African Americans from the Women’s Health Initiative. Genet. Epidemiol. 2012;36:107–117. doi: 10.1002/gepi.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pistis G., Porcu E., Vrieze S.I., Sidore C., Steri M., Danjou F., Busonero F., Mulas A., Zoledziewska M., Maschio A., et al. Rare variant genotype imputation with thousands of study-specific whole-genome sequences: implications for cost-effective study designs. Eur. J. Hum. Genet. 2015;23:975–983. doi: 10.1038/ejhg.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin P., Hartz S.M., Zhang Z., Saccone S.F., Wang J., Tischfield J.A., Edenberg H.J., Kramer J.R., M Goate A., Bierut L.J., et al. A new statistic to evaluate imputation reliability. PLoS One. 2010;5:e9697. doi: 10.1371/journal.pone.0009697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coleman J.R.I., Euesden J., Patel H., Folarin A.A., Newhouse S., Breen G. Quality control, imputation and analysis of genome-wide genotyping data from the Illumina HumanCoreExome microarray. Brief. Funct. Genomics. 2016;15:298–304. doi: 10.1093/bfgp/elv037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T., Guestrin C. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining - KDD ’16. ACM Press; 2016. XGBoost: A Scalable Tree Boosting System; pp. 785–794. [Google Scholar]
- 18.Auer P.L., Johnsen J.M., Johnson A.D., Logsdon B.A., Lange L.A., Nalls M.A., Zhang G., Franceschini N., Fox K., Lange E.M., et al. Imputation of exome sequence variants into population- based samples and blood-cell-trait-associated loci in African Americans: NHLBI GO Exome Sequencing Project. Am. J. Hum. Genet. 2012;91:794–808. doi: 10.1016/j.ajhg.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karczewski K.J., Weisburd B., Thomas B., Solomonson M., Ruderfer D.M., Kavanagh D., Hamamsy T., Lek M., Samocha K.E., Cummings B.B., et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45:D840–D845. doi: 10.1093/nar/gkw971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backman J.D., Li A.H., Marcketta A., Sun D., Mbatchou J., Kessler M.D., Benner C., Liu D., Locke A.E., Balasubramanian S., et al. Exome sequencing and analysis of 454, 787 UK Biobank participants. Nature. 2021;599:628–634. doi: 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hengl T., Mendes de Jesus J., Heuvelink G.B.M., Ruiperez Gonzalez M., Kilibarda M., Blagotić A., Shangguan W., Wright M.N., Geng X., Bauer-Marschallinger B., et al. SoilGrids250m: Global gridded soil information based on machine learning. PLoS One. 2017;12:e0169748. doi: 10.1371/journal.pone.0169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothschild D., Weissbrod O., Barkan E., Kurilshikov A., Korem T., Zeevi D., Costea P.I., Godneva A., Kalka I.N., Bar N., et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 23.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H., Engkvist O., Wang Y., Olivecrona M., Blaschke T. The rise of deep learning in drug discovery. Drug Discov. Today. 2018;23:1241–1250. doi: 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S., Abecasis G.R., Browning B.L. Genotype Imputation from Large Reference Panels. Annu. Rev. Genomics Hum. Genet. 2018;19:73–96. doi: 10.1146/annurev-genom-083117-021602. [DOI] [PubMed] [Google Scholar]
- 27.Schrider D.R., Kern A.D. S/HIC: robust identification of soft and hard sweeps using machine learning. PLoS Genet. 2016;12:e1005928. doi: 10.1371/journal.pgen.1005928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nei M., Li W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fay J.C., Wu C.I. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H. A new test for detecting recent positive selection that is free from the confounding impacts of demography. Mol. Biol. Evol. 2011;28:365–375. doi: 10.1093/molbev/msq211. [DOI] [PubMed] [Google Scholar]
- 32.Garud N.R., Messer P.W., Buzbas E.O., Petrov D.A. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015;11:e1005004. doi: 10.1371/journal.pgen.1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y., Nielsen R. Linkage disequilibrium as a signature of selective sweeps. Genetics. 2004;167:1513–1524. doi: 10.1534/genetics.103.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly J.K. A test of neutrality based on interlocus associations. Genetics. 1997;146:1197–1206. doi: 10.1093/genetics/146.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schurz H., Müller S.J., van Helden P.D., Tromp G., Hoal E.G., Kinnear C.J., Möller M. Evaluating the Accuracy of Imputation Methods in a Five-Way Admixed Population. Front. Genet. 2019;10:34. doi: 10.3389/fgene.2019.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang L., Rosen J.D., Sun Q., Chen J., Wheeler M.M., Zhou Y., Min Y.-I., Kooperberg C., Conomos M.P., Stilp A.M., et al. TOP-LD: A tool to explore linkage disequilibrium with TOPMed whole-genome sequence data. Am. J. Hum. Genet. 2022;109:1175–1181. doi: 10.1016/j.ajhg.2022.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu W., Sun Q., Huang L., Bhattacharya A., Wang G.W., Tan X., Kuban K.C.K., Joseph R.M., O’Shea T.M., Fry R.C., et al. Innovative computational approaches shed light on genetic mechanisms underlying cognitive impairment among children born extremely preterm. J. Neurodev. Disord. 2022;14:16. doi: 10.1186/s11689-022-09429-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan Q., Liu E.Y., Croteau-Chonka D.C., Mohlke K.L., Li Y. A comprehensive SNP and indel imputability database. Bioinformatics. 2013;29:528–531. doi: 10.1093/bioinformatics/bts724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knapp E.A., Fink A.K., Goss C.H., Sewall A., Ostrenga J., Dowd C., Elbert A., Petren K.M., Marshall B.C. The cystic fibrosis foundation patient registry. design and methods of a national observational disease registry. Ann. Am. Thorac. Soc. 2016;13:1173–1179. doi: 10.1513/AnnalsATS.201511-781OC. [DOI] [PubMed] [Google Scholar]
- 40.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Hout C.V., Tachmazidou I., Backman J.D., Hoffman J.D., Liu D., Pandey A.K., Gonzaga-Jauregui C., Khalid S., Ye B., Banerjee N., et al. Exome sequencing and characterization of 49, 960 individuals in the UK Biobank. Nature. 2020;586:749–756. doi: 10.1038/s41586-020-2853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loh P.-R., Danecek P., Palamara P.F., Fuchsberger C., A Reshef Y., K Finucane H., Schoenherr S., Forer L., McCarthy S., Abecasis G.R., et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vuckovic D., Bao E.L., Akbari P., Lareau C.A., Mousas A., Jiang T., Chen M.-H., Raffield L.M., Tardaguila M., Huffman J.E., et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214–1231.e11. doi: 10.1016/j.cell.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen M.-H., Raffield L.M., Mousas A., Sakaue S., Huffman J.E., Moscati A., Trivedi B., Jiang T., Akbari P., Vuckovic D., et al. Trans-ethnic and Ancestry-Specific Blood-Cell Genetics in 746, 667 Individuals from 5 Global Populations. Cell. 2020;182:1198–1213.e14. doi: 10.1016/j.cell.2020.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahajan A., Spracklen C.N., Zhang W., Ng M.C.Y., Petty L.E., Kitajima H., Yu G.Z., Rüeger S., Speidel L., Kim Y.J., et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 2022;54:560–572. doi: 10.1038/s41588-022-01058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y., Sun Q., Huang L., Broome J.G., Correa A., Reiner A., NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Raffield L.M., Yang Y., Li Y. eSCAN: scan regulatory regions for aggregate association testing using whole-genome sequencing data. Brief. Bioinformatics. 2022;23:bbab497. doi: 10.1093/bib/bbab497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Judkins T., Hendrickson B.C., Deffenbaugh A.M., Eliason K., Leclair B., Norton M.J., Ward B.E., Pruss D., Scholl T. Application of embryonic lethal or other obvious phenotypes to characterize the clinical significance of genetic variants found in trans with known deleterious mutations. Cancer Res. 2005;65:10096–10103. doi: 10.1158/0008-5472.CAN-05-1241. [DOI] [PubMed] [Google Scholar]
- 48.Pavlicek A., Noskov V.N., Kouprina N., Barrett J.C., Jurka J., Larionov V. Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum. Mol. Genet. 2004;13:2737–2751. doi: 10.1093/hmg/ddh301. [DOI] [PubMed] [Google Scholar]
- 49.Lindor N.M., Guidugli L., Wang X., Vallée M.P., Monteiro A.N.A., Tavtigian S., Goldgar D.E., Couch F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS) Hum. Mutat. 2012;33:8–21. doi: 10.1002/humu.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tavtigian S.V., Deffenbaugh A.M., Yin L., Judkins T., Scholl T., Samollow P.B., de Silva D., Zharkikh A., Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Memon N., Patel S.B., Patel D.P. In: Pattern Recognition and Machine Intelligence: 8th International Conference, PReMI 2019, Tezpur, India, December 17-20, 2019, Proceedings, Part I, B. Deka. Maji P., Mitra S., Bhattacharyya D.K., Bora P.K., Pal S.K., editors. Springer International Publishing; 2019. Comparative analysis of artificial neural network and xgboost algorithm for polsar image classification; pp. 452–460. [Google Scholar]
- 52.Giannakas F., Troussas C., Krouska A., Sgouropoulou C., Voyiatzis I. In: Intelligent Tutoring Systems: 17th International Conference, ITS 2021, Virtual Event, June 7–11, 2021, Proceedings. Cristea A.I., Troussas C., editors. Springer International Publishing; 2021. Xgboost and deep neural network comparison: the case of teams’ performance; pp. 343–349. [Google Scholar]
- 53.Chakraborty D., Elzarka H. Advanced machine learning techniques for building performance simulation: a comparative analysis. J. Building Performance Simulation. 2018;12:193–207. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MagicalRsq is freely available from https://github.com/quansun98/MagicalRsq/.
The CFGP WGS data are available for request to the Cystic Fibrosis Foundation at https://www.cff.org/researchers/whole-genome-sequencing-project-data-requests#requesting-data.
UKB genotyping and WES data are available upon request from the UK Biobank https://www.ukbiobank.ac.uk/.