

Abstract
Defects in over 500 genes underlie a diverse collection of neuromuscular disorders (NMDs). By definition, NMDs are diseases that arise from defects in muscle or nerve. The subset of NMDs that impact skeletal muscle are referred to as “myopathies” and “muscular dystrophies”, and are due to mutations in genes encoding muscle proteins. Many of these genes encode proteins that provide structural stability or bolster membrane integrity, while others are involved in protein turnover, trafficking, and electrical excitability. In this review, the genetic basis and biological function of mutant proteins associated with myopathies will be discussed. In addition, pathomechanisms and treatment strategies for these disorders will be highlighted. Because the causal genetic defects are known in most cases, strategies that target the underlying molecular defect will likely be the most efficacious approach to therapies, and current strategies utilizing this approach will be briefly mentioned. Since the identification of the first gene associated with a neuromuscular disorder in 1987 to the current day, the field has made tremendous progress and has led in advancing gene therapeutics.
Introduction
Skeletal muscle cells are large and multinucleated, containing highly organized contractile proteins that interact with each other to generate force and allow movement of the body. Each multinucleated muscle cell is surrounded by a thin layer of specialized connective tissue called the basal lamina which serves as an intermediary between the cell and the reticular lamina. This connective tissue bolsters membrane integrity, transmits forces of muscle contraction, and communicates endocrine and paracrine signals to the cell. Each muscle cell is innervated from a single synapse of the motor neuron, with the contact point termed the neuromuscular junction, which is the site where muscle contraction is initiated. Neuromuscular junction signals are transduced to the contractile apparatus via a process called excitation contraction coupling (ECC), which is mediated through a specialized structure called the triad. Defects can arise at essentially all points of the contractile process, from the neuromuscular junction, to the triad, to the contractile apparatus itself, and to the specialized matrix-membrane contacts that maintain and preserve membrane integrity. When mutations arise, they can lead to devastating consequences for skeletal muscle, leading to impaired ambulation, compromised breathing, and in the most severe conditions early death. These muscle diseases are collectively referred to as myopathies and/or muscular dystrophies, depending on the underlying genetic cause and the morphological appearance of the abnormal muscle on biopsy. Disorders that primarily impact the neuromuscular junction are termed myasthenic syndromes. While the latter share many similarities with other primary muscle conditions; however, these disorders will not be discussed further in this review, as they have been recently presented in depth1.
The discovery of the first gene linked to a muscle disease demonstrated that mutations in the DMD gene underlie Duchenne muscular dystrophy (DMD)2,3, followed later by the discovery of dystrophin as its protein product4. This initial finding led to an explosion of information about novel muscle proteins linked to skeletal muscle disorders. Many of the discovered genes and their protein products were previously unknown and thus these discoveries lent new insights and understanding about normal muscle cell biology. Over three decades later, at least 500 genetic loci have been identified and linked to neuromuscular disorders, i.e. disorders which originate from defects in motor neurons (neuropathic origin) or skeletal muscles (myopathic origin). Most of these loci encode for proteins; however, a subset is due to repeat expansions that create toxic RNA (e.g. myotonic dystrophy type 1 and 2), while another set of diseases are caused by aberrant transcriptional activity of a key regulator of myogenesis (DUX4, in fascioscapulohumeral dystrophy types 1 and 2). This review will not cover the myotonic dystrophies5 or FSHD6,7.
In this review, we summarize what is known about myopathies and muscular dystrophies caused by mutations in protein coding loci, with a focus on those that are most common, best understood and that provide the most insight into muscle biology. We will discuss the function(s) of these proteins in skeletal muscle and how mutations lead to disease.
Myopathies Linked to Dystrophin (Dystrophinopathies and Dystroglycanopathies)
Introduction to dystrophin and the dystrophin glycoprotein complex (DGC)
The dystrophin glycoprotein complex (DGC) is a multi-protein membrane complex comprised of intracellular, extracellular and transmembrane proteins8,9. The complex can be divided into two main sub-complexes referred to as the sarcoglycans and the dystroglycans, which are linked together by sarcospan10,11 and retained at the membrane by dystrophin9. There are six different sarcoglycan genes and one dystroglycan gene (DAG) that generates two different DAG proteins (αDAG and βDAG) after post-translational processing of a single polypeptide chain12,13. The skeletal muscle sarcoglycan complex is comprised of four sarcoglycans (α,β,γ,δ).14 Dystrobrevin binds to the DGC, acts as a scaffold and provides additional stabilization; however, the DGC’s membrane association is not wholly dependent on dystobrevin.15
The DMD gene is the largest in the genome (2.2 Mb), which encodes a 14kb dystrophin mRNA. Six different promoters drive expression of dystrophin isoforms in skeletal, cardiac, smooth muscle and brain. These isoforms are named Dp426 (the primary skeletal and cardiac muscle isoform), Dp260, Dp116, Dp140, and Dp71. In skeletal muscle, dystrophin is enriched at costameres, which are sites where terminal Z-discs connect to the sarcolemma, and where force transmission occurs across the membrane16 (Figure 1).
Figure 1: Schematic of the costamere and proteins linked to the dystrophin glycoprotein complex (DGC).
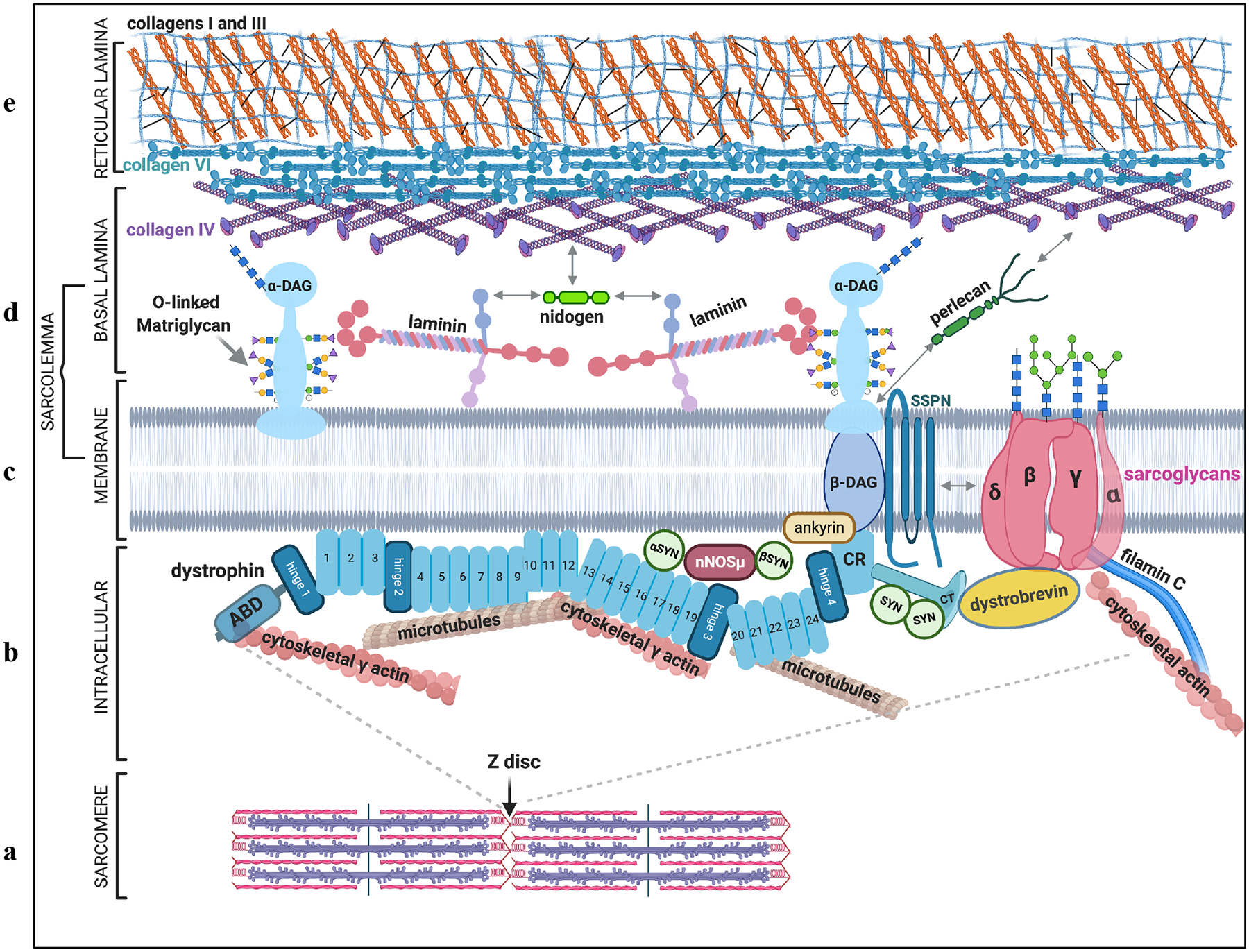
a| The terminal Z disc of the skeletal muscle sarcomere attaches to the membrane at the costamere, where it links to a large protein complex called the DGC. b| On the intracellular side of the membrane, the DGC links to the actin cytoskeleton, microtubules and intermediate filaments via dystrophin protein (aqua). Dystropin is a 427kDa protein comprised of an N-terminal actin binding domain (ABD), 4 hinges, 24 spectrin repeats, a cysteine rich (CR) region that attaches to beta dystroglycan, and a C terminus. Dystrophin is a scaffold for several other molecules including neuronal nitric oxide synthase (nNOS), which attaches vis syntrophin (SYN), as well as ankyrin and dystrobrevin. c| The DGC is made of two membrane-associated subcomplexes, the dystroglycan (DAG) complex and the sarcoglycan complex (consisting of α, β, γ, and δ sarcoglycans), linked together by sarcospan (SSPN). d| Alpha dystroglycan (α-DAG) is the extracellular-facing DGC member in the basal lamina of the ECM. It is an ECM receptor that interacts with laminin, primarily through O-linked glycans on its mucin domain. e| α-DAG also associates with other matrix molecules such as nidogen and perlecan that along with collagens IV and VI, connect the basal lamina to collagens I and III in the reticular lamina.
Dystrophin has four main structural domains: 1) the N terminal region comprised of two actin binding domains17,18; 2) a central rod domain (discussed more below); 3) a cysteine rich region that anchors dystrophin to beta dystroglycan19 and 4) a C terminus that scaffolds molecules like syntrophin20 and dystrobrevin15. It also contains four proline-rich hinges that contribute to dystrophin’s flexibility. The central rod domain is comprised of 24 alpha helical spectrin-like repeats that confer dystrophin’s spring-like properties. The spectrin repeats serve other functions as well, including interaction (via repeats 1–3 and 10–12) with phospholipids in the sarcolemmal membrane21 and binding (via repeats 16–17) the adapter alpha syntrophin22, which help retain neuronal nitric oxide synthase (nNOSμ) at the sarcolemmal membrane22,23. Neuronal NOS also interacts with syntrophin proteins bound to the C terminus of dystrophin. Spectrin repeats 4–15 and 20–23 have been shown to organize transverse microtubules24.
The DGC serves three main roles in skeletal muscle fibers: 1) protecting the sarcolemmal membrane from stresses that arise during muscle contraction; 2) linking the intracellular cytoskeleton to the extracellular matrix and transmitting forces of muscle contraction to the tendon and 3) serving as a scaffold for signaling molecules such as syntrophin and Grb2.25 Other proteins that associate with the DGC include filamin C26, Ankyrin B27, Synemin28, aquaporin29, syncoilin30,31 and keratin 1932. Dystrophin is anchored to the membrane at its cysteine-rich C terminal region via beta dystroglycan, which in turn binds the C-terminus of alpha dystroglycan33. Alpha dystroglycan is an important receptor for many basement membrane proteins such as laminin34, agrin35, nidogen36,37 and perlecan38. These associations help organize the ECM, contribute to the its structural stability and transmit forces of muscle contraction.
Defects in dystrophin and DGC members (dystrophinopathies and LGMDs)
Mutations in a number of DGC-linked genes cause muscular dystrophy or myopathy and are referred to as “dystrophinopathies” with a subset named “dystroglycanopathies”. The best characterized and most prevalent of these is Duchenne muscular dystrophy (DMD) due to mutations in DMD3,39–41. DMD is on the X chromosome, so mutations primarily affect boys, with females primarily as asymptomatic carriers. The majority of pathogenic DMD mutations are deletions that occur within the central rod domain, and lead to frameshift with loss of RNA stability or a introduction of a premature termination codon.
Since the original discovery of the DMD gene42,43, basic science investigations have provided new insights on the role of dystrophin and the DGC on muscle biology. A phenotypically milder disease called Becker muscular dystrophy (BMD) also arises from DMD mutations, but BMD mutations maintain the DMD reading frame and generate internally deleted, but functional dystrophin proteins. In-frame deletions that preserve the phasing of the repeats, as compared to ones that do not, result in dystrophins with enhanced sarcolemmal membrane protection, suggesting the concept that dystrophin protects the sarcolemma through its spectrin repeats44. The majority of studies in the mdx mouse model of Duchenne have shown that loss or reduction of dystrophin leads to impaired membrane integrity and subsequent muscle cell degeneration45–47, following by inflammation48–51, regeneration52 and ultimately, muscle cell replacement by fat and fibrosis. Serum creatine kinase is elevated from birth, due to loss of dystrophin’s contribution to structural integrity of the sarcolemmal membrane.45–47 The clinical progression of the dystrophinopathies has been previously described53–55.
In addition to DMD and BMD, a class of muscular dystrophies referred to as limb girdle muscular dystrophies (LGMDs) result from mutations in genes associated with the DGC. LGMDs are progressive muscle wasting disorders that initially present with symptoms in the proximal musculature; however, weakness progresses to many muscles in the body, depending on the type of LGMD. They can be inherited in autosomal dominant or autosomal recessive fashion, and of the over 30 genetically defined LGMDs, a majority are linked to the DGC. Mutations in genes encoding four of the sarcoglycans (alpha56, beta57, delta58 and gamma59) cause autosomal recessive LGMD. Assembly of the sarcoglycan complex occurs in the golgi, starting with association between delta and beta sarcoglycans, followed by addition of alpha and gamma14. Null mutations in delta sarcoglycan prevent the sarcoglycan complex from forming, suggesting that it is a key organizer of the complex14. Null mutations in the other sarcoglycans tend to lead to a reduction, but not complete loss of the sarcoglycan complex.
Dystroglycan (DAG) is a key component of the skeletal muscle DGC which is also expressed in other tissues, where it assembles DGC-like complexes. In skeletal muscle, αDAG resides on the extracellular surface and serves as a receptor for many ECM proteins, most important of which is laminin (laminin 111/211) in the basement membrane. αDAG binds laminin in the central part of the molecule (mucin domain) which is glycosylated by several glycosyl transferases, each of which adds its unique sugar in order to build the O-linked moieties that are necessary for αDAG’s attachment to laminin. The first glycosyltransferases to add O-mannose to serine and threonine residues on αDAG are POMT1 and POMT2, which reside in the ER60, followed by addition of GlcNAc by POMGNT2 and addition of GalNAc by β3GalNT2. Subsequently, this O-linked glycan is modified by ISPD (which attaches ribitol61). This core structure of three sugars (“M3 core”) is then phosphorylated on the O-linked sugar by a kinase named POMK (SGK196)60,62 and further growth of the sugar structure takes place on this phosphate residue in the golgi. POMGNT1 and other glycosyl transferases such as FKRP, FKTN, TMEM5, and LARGE1 work together to generate the laminin binding motif63, comprised of a repeating di-saccharide (-GlcA-β3-Xyl-α3)n64 referred to as “matriglycan”65. The specific roles of FKRP, FKTN and TMEM5 and the manner in which matriglycan is attached to the phosphate is still not known. The length of matriglycan is modulated by a sulfotransferase, HNK-1, which competes with LARGE1 by 3-O-sulfation of glucuronic acid66. The presence of DAG’s N-terminal “DGN” domain is necessary for proper glycosylation of the mucin domain, which may be linked to N-linked glycosylation; however, further exploration is needed to elucidate the relationship between the presence of DGN and O glycosylation of the mucin domain. For additional review of dystroglycan glycosylation, please see65.
Defects in dystroglycan and dystroglycan glycosylation (dystroglycanopathies)
Dystroglycanopathies share a common feature of impaired or absent αDAG glycosylation and encompass a spectrum of phenotypes, ranging from severe congenital muscular dystrophies with eye and brain involvement to milder “muscle only” presentations including subtypes of LGMD. Since αDAG is not restricted to skeletal muscle, and because it serves an important developmental role, loss or mutations in a subset of glycosyl transferases can result in severe CMDs with multi-organ involvement, affecting the eye and brain, such as Fukuyama CMD, muscle-eye-brain disease, and Walker–Warburg syndrome.67–72 Of note, DAG mutations have been reported as an extremely rare cause of muscular dystrophy; it is assumed that most DAG mutations would not be compatable with life.
Defects in extracellular matrix (ECM) proteins
The sarcolemmal membrane attaches to the basal lamina (comprised of laminin α2, type IV collagen, heparin sulfate proteoglycan, agrin and perlecan), which is in turn is connected to collagen VI, which links the basement membrane to the reticular lamina (comprised mainly of collagens I and III). Mutations in genes encoding the three chains of the collagen VI fibril (COL6A1, COL6A2 or COL6A3) underlie autosomal dominant Bethlem or autosomal recessive Ullrich myopathy, characterized by muscle weakness and contractures. Mutations in COL6A2 can also lead to the milder LGMD R22 or LGMD D5. Most often, these mutations interfere with formation of the trimeric collagen myofibril, with severity dictated by which stage of myofibril assembly is interrupted73–75. Laminins are also trimers, but only mutations in the α2 chain are linked to muscular dystrophy. Mutations in the LAMA2 gene lead to loss or reduction of laminin with corresponding clinical severity from mild (LGMD presentation) to severe (CMD presentation). While laminin 211 and collagen VI are extracellular matrix proteins, mutations share a common pathomechanism of increased myofiber apoptosis. Promoting myocyte survival is one potential treatment strategy for these muscular dystrophies; pre-clinical work has supported the efficacy of this approach, and one drug (omigapil) is currently in early-stage clinical trials.
Gene Therapeutics for DGC-linked diseases
Most myopathies and muscular dystrophies are amenable to gene replacement, antisense oligonucleotide-mediated exon skipping or gene editing approaches to therapy. Much of the focus in therapeutic development for the dystrophies has been on DMD, because this disease is the most prevalent. Because dystrophin can tolerate large, in frame mutations (as evidenced by the clinically milder BMD), most DMD-therapies have focused on altering the reading frame of the mRNA to change DMD mutations into BMD mutations. Modified antisense oligonucleotides (e.g. phosphorodiamidate morpholino oligomers or PMOs), can be systemically administered to skip single exons to reframe the mRNA, and three such therapies have been FDA approved (Exondys 51, Vyondys 53 and Viltepso) for mutations amenable for skipping exons 51 or 53. Additional PMOs are in development for other DMD exons. These exon skipping therapies must be administered by weekly infusion, do not target the heart, and result in only approximately 1% dystrophin re-expression. Adeno associated virus (AAV) has been used to deliver ASOs in murine studies with good success76. New oligonucleotide chemistries are in development (e.g. PPMO), and these new formulations show improved uptake in skeletal muscle and heart77,78. Exon skipping has also shown promise for gamma sarcoglycan deficiency79 and in diseases in which mutations create pseudo-exons that can be skipped80. Gene replacement therapies using AAV to deliver an engineered DMD transgene that retains approximately 30% of the native protein (referred to as mini- or micro-dystrophin) are in phase II and phase III trials, with promising preliminary reports. AAV-mediated gene replacement therapies are also in various stages of development for several of the autosomal recessive LGMDs and CMDs. For reviews of gene replacement therapies please see63. CRISPR/Cas9 gene editing therapies are in preclinical development for DMD and clinical trials may initiate in the next few years81–85. For review of CRISPR gene editing for neuromuscular disorders, please see Young et al.86
Sarcomere pathologies
Introduction to sarcomeric proteins
The sarcomere is the fundamental unit of the myofiber and is the structure that ultimately generates force (Figure 2). It is composed of two primary components, the thin filament and the thick filament, embedded and tethered between two Z discs. The thin filament is composed primarily of skeletal muscle actin (encoded by ACTA1), with the addition of a group of proteins that regulate its length, dynamic interactions with the thick filament, and its stability. The thick filament is composed primarily of myosin, with different myosin isoforms found in distinct muscle fiber subtypes (for example, slow oxidative skeletal muscle vs fast glycolytic muscle). Muscle contraction is achieved when myosin slides along the actin thin filament, made possible by the regulated release of intracellular calcium (released from stores in the sarcoplasmic reticulum), which binds the troponin complex, changing tropomyosin’s confirmation to expose and enable myosin interaction with actin. In keeping with the importance of sarcomeric proteins in muscle contraction, mutations in components of both the thin and thick filaments lead to muscle disease. Also, and perhaps unsurprisingly because of the fact that the sarcomere ultimately performs the “end step” of muscle function, sarcomeric diseases have proven mostly refractory to therapy, and it is likely that gene correction or replacement-based strategies will be necessary to treat the majority of patients with these myopathies87.
Figure 2 |. Schematic of a muscle cell and the proteins linked to the sarcomere Figure 2. Sarcomere structure in skeletal muscle.
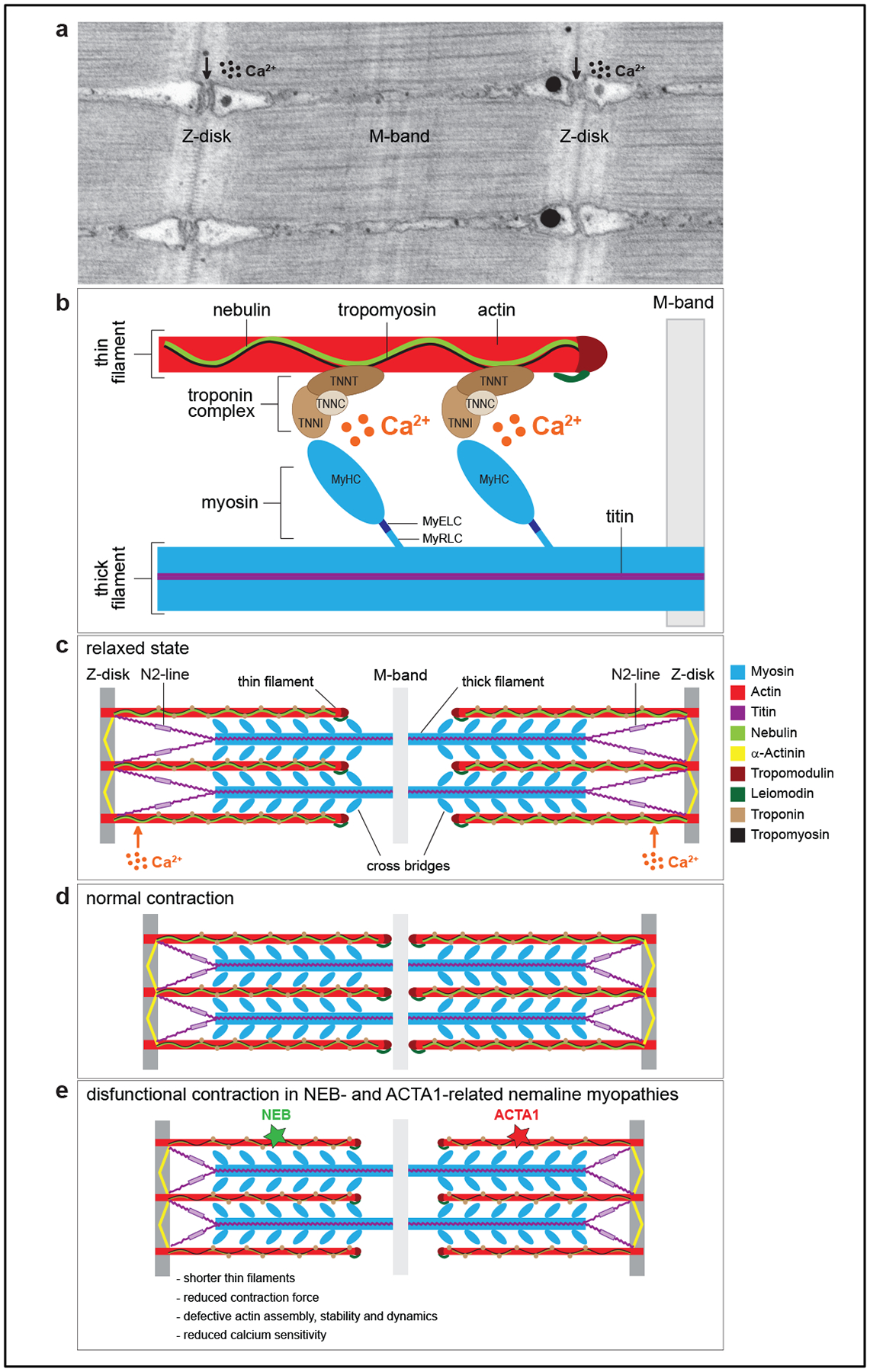
a. Transmission electron micrograph of a longitudinal section of the sarcomere of zebrafish skeletal muscle. Z-disks are visible as electron-dense zig-zag vertical lines, M-band as a smooth dark line in the middle of the sarcomere, and the horizontal striations represent thick and thin filaments. The vacuolated areas are the triads, where excitation contraction coupling takes place. b. Schematic of the major protein components of the sarcomere. Thin filaments are composed of actin, nebulin, tropomyosin and the troponin complex with the following subunits: troponin T (TNNT, binds tropomyosin), troponin I (TNNI, binds actin), and troponin C (TNNC, binds the calcium ions). Thick filaments are composed of myosin and titin. Myosin consists of several domains: a head [two identical myosin heavy chains (MyHC), which bind actin], a neck [one pair of essential light chains (MyELC) and one pair of regulatory light chains (MyRLC)], and a tail. c. Schematic of a sarcomere in a relaxed state. d. Schematic of a sarcomere in contracted state. e. Schematic of a sarcomere during a defective contraction, which is a hallmark of thin-filament related nemaline myopathies. Mutations in nebulin (NEB) and actin (ACTA1) can lead to shorter thin filaments, defective actin assembly and dynamics, reduced force during muscle contraction, and lower sensitivity to calcium ions.
Defects in thin filament proteins that cause myopathies
Nemaline myopathy – the myopathy of the thin filament
Mutations in many components of the thin filament cause a subtype of congenital myopathy called nemaline myopathy88. The most common cause of nemaline myopathy is recessive mutation in the NEB gene89, with heterozygous mutation of the ACTA1 gene being second most common90–92. While there are exceptions, the majority of patients with mutations in components of the thin filament present with a consistent clinical picture (non-progressive diffuse weakness, reduced muscle bulk, and selective involvement of the bulbar and axial musculature) and with shared features on muscle biopsy (myofiber hypotrophy, type I fiber predominance, myofibril disorganization, and the presence of aggregated Z disc material termed nemaline rods or nemaline bodies)93.
Nebulin and NEB related NM
NEB encodes for nebulin, one of the largest proteins in the human genome, the function of which was a mystery for many years, resulting in the unique naming of the gene and its product94. Nebulin is composed primarily of a repeat structure characterized by a series of actin binding domains interspersed with tropomyosin binding domains95. The extreme N terminus extends to the end of the thin filament (where it interacts with tropomodulin), while the C terminus binds and embeds into the Z disc94. A growing body of evidence supports that the main function of nebulin is to serve as a molecular ruler that dictates the length of the thin filament96–99. Key data in this regard include the fact that loss of nebulin results in reduction of thin filament length, that across different species thin filament length correlates with the size of the nebulin protein, and that manipulating nebulin size (making it larger or smaller) correspondingly changes the length of the filament96,100. Since muscle force is generated through the interaction between thick and thin filaments, there is a tight correlation between actin filament length and force generation; thus, loss of nebulin results in reduced contractile force and muscle weakness101. Nebulin has additional functions including regulating actin/myosin cross bridging (independent of its impact on thin filament length102) and participating in signalling pathways critical for actin filament formation103. The importance of these latter functions is less well established, and the consequence of their loss not known.
ACTA1 related NM
ACTA1 encodes skeletal muscle actin; polymers of ACTA1 form the core structure of the thin filament. Actin polymers interact with nebulin, tropomyosins, troponins, elements in the Z disk, and, critically, myosin (to enable muscle contraction)104,105. Mutations in ACTA1 are the commonest dominant mutations found in congenital myopathies, and can produce a range of pathological changes in the muscle (with nemaline myopathy pathology the most common)88. More than 200 different mutations in ACTA1 have been described106. The majority of disease associated variants are missense changes that impair some aspect(s) of actin function, including assembly and stability of the actin filament, as well as potentially altering thin filament dynamics (calcium binding, sliding speed, etc). As with NEB loss of function, the majority of ACTA1 mutations are associated with reduced maximal force generation, which may result from reduced thin filament length in some instances101, and dysfunctional contractility in others107. Of note, some mutations result in hypercontractility that causes a clinical phenotype associated with muscle stiffness108.
There are currently no treatments for ACTA1-related myopathy, and few therapeutic targets. Case report level data has implicated L-tyrosine as a potential therapeutic for several nemaline myopathy subtypes109, primarily for improving bulbar function, but studies in pre-clinical models have not supported its efficacy110,111. Because myofiber smallness is a nearly invariant feature in muscle biopsies from ACTA1 patients, myostatin inhibition has been tested in a transgenic mouse model of the disease, with the result being increased muscle size and muscle force generation112. Conversely, a similar examination of myostatin inhibition in nebulin deficient mice did not produce meaningful improvements113. Lastly, cardiac alpha actin is an intriguing target for ACTA1 disease. In addition to expression in the heart, ACTC is expressed in fetal skeletal muscle, and it functions similarly in terms of thin filament mechanics. Germline replacement of Acta1 with Actc in mice results in normal mice without evidence of muscle abnormalities114. Further, overexpression of Actc1 improves survival in one transgenic mouse model of dominant Acta1 mutation, though for unclear reasons does not promote improvement in a knock-in model harboring a different mutation115.
Other forms of Nemaline Myopathy
Mutations in TPM2, TPM3, TNNT1, TNNT3, TNNI2, and LMOD3 are all associated with nemaline myopathy and/or distal arthrogryposis (a syndrome of congenital joint contractures discussed below) and are regulatory components of the thin filament116–121. TPM2 and TPM3 encode tropomyosin 2 (found in type 1 fibers) and tropomyosin 3 (found in type 2 fibers) respectively. Tropomyosins are coiled-coil proteins that bind along the length of the actin filament and both help stabilize the thin filament and participate in the calcium dependent switch between the relaxed and active state122. Mutations in TPM2 and TPM3, which typically impact tropomyosin dimerization and/or actin binding, generally result in either hypocontraction, associated with reduced calcium sensitivity and decreased actin-myosin sliding speeds, or hypercontraction, with higher calcium sensitivity and increased filament sliding123. Of note, the hypercontraction mutations are more likely to be associated with joint contractures, and can present with a phenotype of arthrogryposis124. TNNT1 encodes the slow skeletal muscle form of troponin; three troponin isoforms (T, C, and I) interact with tropomyosin and together form a complex that regulates the calcium sensitivity of muscle contraction. Interestingly, mutations in the I isoform (i.e. TNNT1) cause congenital myopathy118, while mutations in TNNT3 and TNNI2 are predominantly associated with arthrogryposis125,126. LMOD3 encodes leiomodin 3, a member of the leiomodin protein group that are part of the tropomodulin family119. Tropomodulins in general are found at the barbed end of the actin filament where they interact with tropomyosins and “cap” the end of the thin filament, thus helping establish the length of the filament. LMOD3 in particular is a strong nucleator of actin polymerization, and may have a specific role in promoting and regulating thin filament formation.
The function of the remaining genes associated with nemaline myopathy (CFL2, KBTBD13, KLHL40, and KLHL41)127–130 are less well understood. The latter three genes encode kelch domain containing proteins, and are substrate adaptors for the U3 ubiquitin ligase cullin-3. Evidence is emerging that they participate in the regulation of turnover of the core thin filament proteins. KLHL40, for example, interacts with both NEB and LMOD3, and may reduce their turnover by limiting their polyubiquitination131. KLHL41 binds and targets for ubiquination NRAP (nebulin anchoring protein). Overexpression of NRAP impairs myogenesis, while reducing NRAP levels in zebrafish lacking klhl41 rescues aspects of their myopathic presentation, suggesting a critical interplay between KLHL41 expression and activity, NRAP chaperone function, and muscle development and that KLHL41 mutation driven NM pathology132.
Myopathies of the thick filament
The core unit of the thick filament is the myosin hexamer. It is composed of two units of myosin heavy chains, two essential light chains, and two regulatory light chains. Myosin slides over the thin filament in an ATP dependent manner to generate a muscle contraction. There are 8 myosin heavy chains, 3 essential light chains, and 3 regulatory light chains that predominate in skeletal muscle133. In addition, there are several regulatory proteins are associated with the thick filament, the most important of which from a muscle disease consideration is titin (TTN).
Disorders of the myosin heavy chain
Myosin heavy chains form homodimers that function as the main action component of the thick filament. They have a globular head domain that interacts directly with actin and has ATPase activity, and a distal rod domain important for dimerization and other protein-protein interactions. Of the 8 myosin heavy chains (MyHCs) found in skeletal muscle, mutations in 4 genes (MYH2, MYH3, MYH7, and MYH8) are associated with muscle disease134–137. Mutations in MYH3 and MYH8 are associated with arthrogryposis, a condition characterized by multiple joint contractures present from birth. MYH3 encodes an embryonic form of myosin heavy chain, and dominant mutations cause a spectrum of severe joint contracture disorders that likely result from weakness and lack of limb movement during embryogenesis135. MYH8 encodes a perinatal MyHC isoform, and dominant mutations cause a milder arthrogryposis syndrome called Trismus-pseudocamptydactyly syndrome (jaw contracture and unusual finger bending). MYH2 codes for myosin heavy chain IIA (found exclusively in Type IIA fibers) and mutations cause both dominant and recessive myopathies134,138. Both forms have important involvement of the eye muscles; dominant mutations are associated with myopathy with rimmed vacuoles and a typically milder clinical presentation, while recessive mutations cause a congenital onset myopathy with absent type 2A fibers.
MYH7 mutations are the most common form of myosin related myopathy. MYH7 encodes a “slow” MyHC isoform expressed in type I skeletal myofibers and in cardiomyocytes. More than 200 dominant mutations have been identified that cause either skeletal and cardiac myopathy, with some rare patients manifesting both diseases133. The skeletal myopathy is associated with a range of clinical phenotypes and biopsy findings, including the named syndromes of Laing distal myopathy and Myosin Storage Myopathy137,139, and most commonly occurs with mutations in the distal rod domain. Clinical symptoms most typically start in the distal musculature, with the hanging big toe a common presenting sign, and are often mild. However, severe presentations, including those with congenital onset and failure to achieve ambulation, have been described140.
Titin and TTNopathies
TTN encodes the giant myofilament protein titin, one of the largest and most complex proteins in the human genome. Titin spans from the Z-disc (at its N terminus) to the M line, so is essentially the length of one half of the sarcomere (approximately 1 μM). It is laid down early in development and is believed to be a template for sarcomere formation. Titin has myriad functions in skeletal muscle, the most important and well-studied of which are its roles as a molecular spring, as a generator of passive stiffness for the myofiber, and as a regulator of actin contractile force generation141. It also has kinase functions and has been implicated in signalling pathways. Unsurprisingly, given its large size and importance for muscle physiology, mutations in TTN are a frequent cause of skeletal myopathy, and may be the second most common cause of non-dystrophic muscle disease in childhood142. TTN is also one of the most frequently encountered causes of cardiomyopathy.
TTN mutations affecting skeletal muscle were first described in patients with a rare, mild dominant form of muscular dystrophy called tibial muscular dystrophy143. An additional dominant form of TTNopathy, termed myofibrillar myopathy with early respiratory failure to reflect the clinical and histopathologic features, was subsequently described144. More recently, and reflecting the increased ability to interrogate the TTN genomic locus affording by next generation sequencing, TTN has been identified as a cause of several recessive subtypes of congenital myopathy, including centronuclear myopathy and myopathy with cores. The clinical spectrum of patients with recessive TTNopathy is broad, ranging from early onset with delayed motor milestones and impaired ambulation, to later onset forms with mild/moderate diffuse weakness145. At present, there are no therapies for the skeletal muscle manifestations of TTN mutations.
Introduction to the nuclear envelope
Just as structural proteins are essential to connect the extracellular matrix to the sarcolemma, myonuclei similarly have structural proteins that span the nuclear envelope and anchor the nuclear matrix (Figure 3). Specifically, the protein Lamin A/C (LMNA) connects the nuclear lamina on the inside of myonuclei with the inner nuclear membrane by interacting with emerin (EMD) and the SUN domain proteins, SUN1 and SUN2146. These proteins then connect with the outer nuclear envelope and cytoskeleton via a family of proteins with a KASH domain termed nesprins (SYNE). This complex creates the linker of nucleoskeleton-and-cytoskeleton (LINC) that maintains myonuclear organization and structural integrity147. The nuclear membrane serves to separate proteins in the sarcoplasm and nucleoplasm. While the nucleus requires this protective isolation, it also needs to communicate with the rest of the myofiber, exchanging proteins and RNA, for a variety of nuclear and cytoplasmic processes which act in concert. The nuclear pore complex (NPC), perhaps the largest protein complex in the cell (~125mDa), is responsible for the protected exchange of components between the nucleus and cytoplasm148. The nucleocytoplasmic shuttling of proteins and RNA across the nuclear envelope via nuclear pore complexes (NPCs) further requires a family of nuclear transport receptors called importins to enter the nucleus, and exportins to exit. These receptors target specific cargo and facilitate transport through the NPC utilizing the hydrolysis of GTP within the nucleus via the small ras GTPase, Ran148.
Figure 3: Schematic of a muscle cell and the proteins linked to the nuclear envelope.
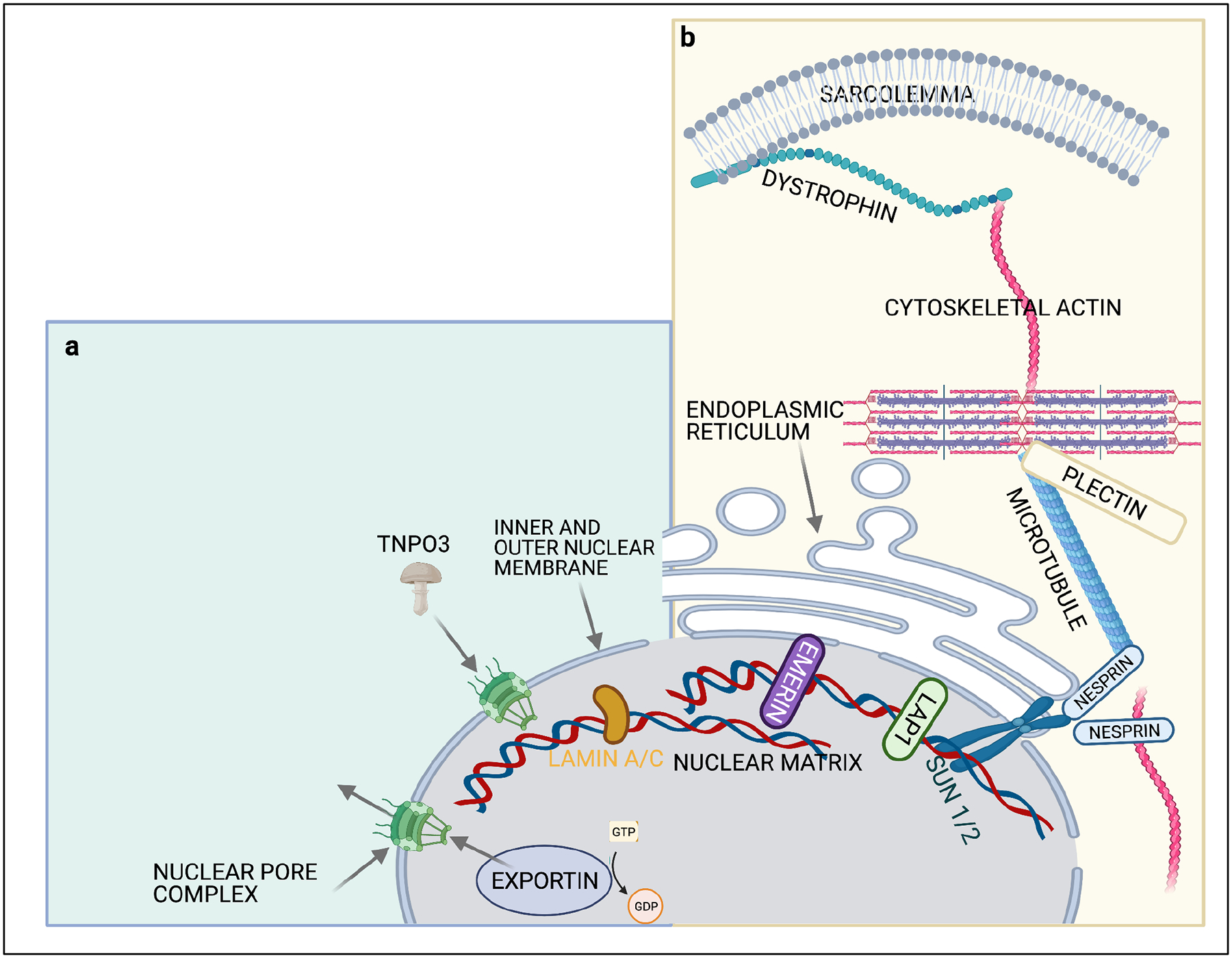
A) Nuclear import and export through nuclear pore complexes requires transport receptors such as importins, exportins and transportins. These proteins shuttle cargo through the NPC along using a GTP gradient generated by RAN GTPase. B) The LINC complex connects lamins that compose the nuclear matrix on the inside of the nuclear envelope with via a family of proteins termed SUN1/2 and the outer nuclear envelope via a family proteins with a KASH domain (Nesprins) that then connect to the actin, intermediate filament and microtubule network.
Disorders linked to the nuclear membrane
Null or missense mutations in genes encoding LINC complex proteins (EMD, LMNA, SYNE1 and SYNE2) lead to a distinct muscular dystrophy syndrome with proximal weakness, joint contractures and cardiomyopathy termed Emery-Dreifuss Muscular Dystrophy (EMD)146. EMD muscle pathology has both myopathic and dystrophic features. Ultrastructural features include altered myonuclear structure with nucleoplasmic extrusion146. The pathophysiology of “nuclear envelopopathies” may be multifactorial. Loss or mutations in LINC complex proteins affects nuclear migration and myonuclear organization that are essential for a multinucleated cell such as skeletal muscle147. Nuclear lamins anchor chromatin and disease mutations alter gene expression via altered epigenetic modifications such as methylation149. A surprising consequence of LINC complex dysfunction is a loss of mechanical stiffness generally thought to be due to dysfunction at the sarcolemma150. This finding emphasizes the necessary connection of the nuclear matrix to the extracellular matrix via the cytoskeletal proteins.
Impaired nucleocytoplasmic transport is seen in motor neuron disease and is associated with the cytosolic accumulation of RNA granules such as stress granules or protein inclusions151. In muscle, a rare form of LGMD, LGMDD2/1F, is due to dominant mutations in the nuclear transport receptor transportin-3 (TNPO3)152. TNPO3 is a cargo receptor for SR domain containing RNA binding proteins and is essential for HIV infection153. Notably, cells from patients with LGMDD2 are immune to HIV infection153.
Therapeutic approaches for disorders linked to myonuclei
Gene replacement strategies may be appropriate for diseases such as EMD associated with the loss of EMD. However other approaches focus on correcting the epigenetic dysregulation that occurs with LMNA mutations. For example, in mice homozygous for the H222P LMNA missense mutation associated with a recessive form of EMD, histone methylation and more specifically H3K4me1 was reduced at key regulatory regions149. Correction of this by inhibiting an H3K4me1 demethylase (LSD1) was demonstrated to improve cardiac dysfunction in Lmna mutant mice. Using this same mouse model, inhibition of mTOR and subsequent activation of autophagy have been demonstrated to improve cardiac function and pathology154. The improvement correlated with a reversal of metabolic deficits such as elevated lipolysis and reduced thermogenesis155. These therapeutic approaches highlight the complicated pathophysiology seen in these disorders.
Heading: Defective Ca2+ handling
Introduction to excitation contraction coupling and the skeletal muscle triad
Skeletal muscle is electrically excitable. It is innervated by motor neurons that project from the spinal cord and synapse on each muscle fiber at the neuromuscular junction (NMJ). An electrical signal, initiated at NMJ following neuronal activation, travels transversely along the sarcolemma and subsequently down into the muscle via transverse tubules (T-tubules), which are invaginations of the sarcolemma (Figure 4). The T-Tubule interacts with the terminal sarcoplasmic reticulum (site of calcium storage) at a specialized junction call the triad. The triad facilitates calcium release from the SR and into the sarcoplasm, which promotes muscle contraction via relaxation of inhibition of actin-myosin cross bridging. The two main proteins of the triad are the skeletal muscle ryanodine receptor (RyR1) and the dihydropyridine receptor (DHPR or Cav1.1). Mutations in RYR1 and CACNA1S (which encodes a Cav1.1 subunit) are associated with a range of skeletal myopathies, including core myopathies, malignant hyperthermia susceptibility, and periodic paralysis91.The triad is composed as well of numerous proteins that support its formation, maintenance and function, and mutations in several of the genes encoding these proteins results in muscle disease156,157.
Figure 4 |. The triad is a muscle-specific substructure that is critical for mediating excitation contraction coupling.
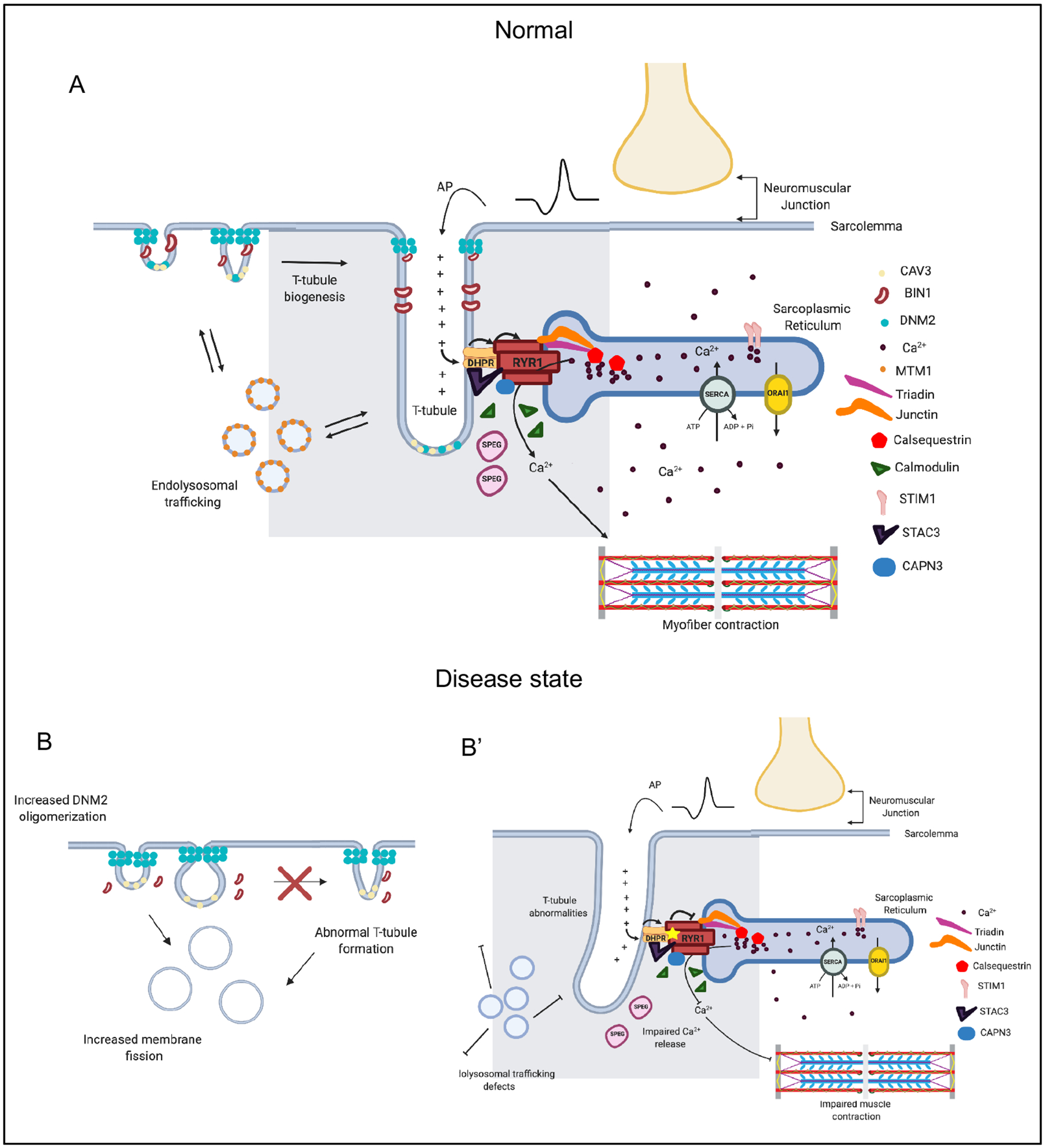
The triad represents the apposition of a Transverse (T)-tubule with two terminal cisternae of the sarcoplasmic reticulum (SR). While many types of proteins and regulatory structures located in or around the triad have been identified, the mechanisms of T-tubule biogenesis and triad formation are still incompletely understood. a | A muscle fiber is excited by the motor neuron at the neuromuscular junction, inducing membrane depolarization, which travels along the sarcolemma and into the t-tubule. The dihydropyridine receptor (DHPR) senses membrane depolarization, and undergoes a conformation change which activates the ryanodine receptor (RYR1), releasing Ca2+ from the SR into the sarcoplasm. Sarcoplasmic Ca2+ then binds to the troponin complex, releasing inhibition and initiating a muscle contraction. Several other proteins are critical for the formation and maintenance of the triad. MTM1 potentially regulates transport of muscle-specific proteins to the triad. BIN1 is involved in membrane remodelling, and in concert with caveole, initiates T tubule formation. DNM2 is speculated to function in parallel (and potentially opposed) to BIN1, via its membrane fission activity, to modulate T-tubule formation and maintenance. Two examples of mutations and their consequences on the triad: b | DNM2 hyperactivity leads to aberrant/premature membrane fission and abnormal t-tubule formation; b’ | RYR1 mutations lead to impaired calcium release and reduced muscle contraction.
RYR1 and RYR1 related myopathies
RYR1 encodes the skeletal muscle ryanodine receptor (RyR1), a massive homo-tetrameric calcium release channel located in the terminal sarcoplasmic reticulum158. RyR1 is the protein that releases calcium from the SR to the sarcoplasm during ECC. Mutations in RYR1 cause a range of myopathies159, which in total represent the commonest group of non-dystrophic pediatric muscle diseases, with prevalence estimates as high as 1:10000160. Mutations can be broadly classified as resulting in either reduced or increased regulated calcium release156. Reduced expression/function mutations are generally associated with static muscle weakness and cause myopathies including central core disease161,162, mini core myopathy163, congenital fibre type disproportion164, and centronuclear myopathy165. These myopathies historically have been named for findings on muscle biopsy, but the field is now evolving toward a “gene first” definition, encompassing all of these entities as RYR1 related myopathies (RYR1 RM)160. Recessive mutations, most typically seen with minicore and centronuclear pathology, tend to result in both reduced function and expression, and are associated with more severe clinical phenotypes, including the need for ventilator and wheelchair supports166,167. Dominant mutations that cause myopathy most typically are missense, reduce function, and result in central core pathology166,167. Dominant/missense mutations that increase calcium release in response to stimuli predispose to dynamic conditions, particularly malignant hyperthermia168,169, though can also include exertional rhabdomyolysis170 and exertional heat illness171. About 30% of patients manifest both these dynamic “hyper-kinetic” conditions as well as static muscle weakness.
At present, there are no effective therapies for RYR1 RM. Aberrant oxidative stress has been identified in pre-clinical models and patient cells172,173, associated with both gain and loss of function mutations, and is thus an attractive pathway for intervention. The anti-oxidant N-acetylcysteine has shown efficacy in zebrafish and mouse models172,173, but unfortunately failed to meet the primary endpoint in a randomized, placebo controlled clinical trial174. Anti-oxidants with increased muscle penetration are now being examined, as one caveat to the trial result was the fact that oxidative stress in treated patients was not significantly reduced with NAC. Another disease mechanism that is potentially therapeutically tractable relates to mutations that impact RyR1 binding to calstabin, impairment of which causes chronic calcium leak from the RyR1 channel. A class of compounds called Rycals stabilize this interaction and prevent calcium leak, as has been shown in pre-clinical models including ex vivo studies of patient muscle biopsies175. Based on these promising results, Rycals are now being considered for testing in the clinical trial arena.
CACNA1S and CACNA1S related myopathies
Cav1.1, also called the dihydropyridine receptor (or DHPR) is an oligomeric protein complex composed of 5 subunits176,177. Cav1.1 is an L-type calcium channel which functions as a voltage sensor during EC coupling. Membrane depolarization initiated at the NMJ travels down the T tubule until reaching the DHPR, which undergoes a conformation change in response that promotes opening of the RyR1 calcium channel. CACNA1S encodes the alpha 1 subunit of DHPR178, which is the largest subunit and the one primarily responsible for interaction with, and regulation of, RyR1. CACNA1S is the only subunit mutated in skeletal myopathies; mutations are associated with a range of phenotypes including malignant hyperthermia179, exertional heat illness180, periodic paralysis181, and myopathy182. CACNA1S mutations account for approximately 5% of all cases of MH (as opposed to RYR1 mutations, which cause 70%). MH related mutations lie in the domain of CACNA1S that directly interacts with RyR1 and disrupts the negative regulatory function of CACNA1S on RyR1 calcium release. CACNA1S mutations are one of two main causes of hypokalemic periodic paralysis (the other being SCN4A), a condition of muscle inexcitability that manifests as prolonged episodes of flaccid muscle weakness that can last hours to days. Mutations resulting in periodic paralysis reside in the voltage sensing domain, with two alleles accounting for the majority of cases. Individual episodes of paralysis can be aborted with potassium supplementation and reduced by use of acetazolamide and avoidance of known triggers183. However, a progressive myopathy with limb girdle weakness usually emerges in adulthood.
A congenital onset myopathy has recently been described associated with mutations in CACNA1S182. This myopathy resembles recessive RYR1 related myopathy and includes early onset, severe weakness, and involvement of the muscles of eye movement. Histopathology is variable, and may include central nuclei, core or core like lesions, myofibrillar disorganization, and fiber type disproportion, plus the unique feature of a lobular or “alveolar” appearance with oxidative stains. Both dominant and recessive mutations have been identified, with most missense mutations located in regions encoding cytoplasmic loops. Recessive patients have either two nonsense mutations or one missense and one nonsense, while dominant/de novo patients all have missense variants. Interestingly, the majority of patients studied, regardless of mutation type, have reduced CACNA1S protein levels, suggesting loss of expression/function as the main disease pathomechanism. Studies of intracellular calcium dynamics support this, as EC coupling is impaired in the mutations investigated.
STAC3 myopathy
A third essential component of the EC coupling apparatus is encoded by the STAC3 gene. STAC3 is a bridging protein that is required for triad formation (likely by chaperoning Cav1.1 to the triad) and for regulation of the Cav1.1/RyR1 interaction184. Biallelic mutations in STAC3 were first identified in a rare muscle disorder called Native American Myopathy, a condition described in the Lumbee Native Americans of North Carolina and featuring facial weakness with prominent ptosis, extremity weakness and contractures, and MH susceptibility185. All individuals with NAM are homozygous for a recurrent missense variant in STAC3. Additional non-Lumbee individuals with recessive STAC3 mutations have been identified with a similar phenotype that includes congenital onset weakness, facial involvement, short stature, joint contractures, and MH susceptibility186. The same “NAM” mutation has been found in some, and additional variants have also been identified.
Centronuclear myopathy due to mutations in MTM1, DNM2, BIN1, and SPEG
Centronuclear myopathy is a congenital onset muscle disease united by features on muscle biopsy including >25% of fibers with central nuclei, myofiber hypotrophy, and organelle disorganization (as seen with oxidative stains). The genes underlying CNM encode proteins involved in membrane traffic187. A hallmark feature of CNM is disturbance of the formation and/or maintenance of the structure of the triad188,189, the result of which is impaired EC coupling and severe muscle weakness.
Mutations in the X-linked gene MTM1 were the first described cause of CNM190. MTM1 encodes a ubiquitously expressed phosphoinositide phosphatase that is a component of the endolysosome and that is a key regulator of endosomal membrane sorting. Loss of expression/function mutations result in X-linked myotubular myopathy, a devastating myopathy affecting primarily males that is associated with profound weakness, ventilator and feeding tube dependence, and early death in most individuals191–193. The exact function(s) of MTM1 in skeletal muscle, and the reasons why mutations impair triad structure and function, are incompletely understood. MTM1 has been implicated in stabilizing myofiber organization via interaction with desmin and in regulating intermediate filament turnover via interaction with the ubiquitination machinery194,195. How these molecular interactions may impact the triad is unclear. Despite this lack of knowledge, several therapies have been identified that ameliorate pathology and clinical phenotypes of animal models of the disease196. These include gene replacement therapy197, phosphoinositide rebalancing via PIK3C2B inhibition198, and DNM2 reduction (either with antisense oligonucleotide or with tamoxifen)199,200. Building from these targets and robust natural history clinical data, the first clinical trials for XLMTM have been initiated. Preliminary reporting of gene therapy with AAV8-MTM1 are encouraging, though important safety concerns have also emerged201,202.
DNM2 encodes a ubiquitously expressed large GTPase that functions as a molecular scissor to promote membrane fission203. DNM2 protein is implicated in myriad cellular functions, including clathrin-mediated endocytosis and cytokinesis. Dominant/de novo mutations in DNM2 cause a form of autosomal centronuclear myopathy204, similar in characteristics to XLMTM but in general milder in severity. Mutations are thought to result in hyper-functionality of DNM2, either by releasing auto-inhibition or prolonging protein stability205. As with MTM1, the exact mechanism by which DNM2 mutations impact the triad is unclear, though data supports a theory where-by mutations lead to premature or inappropriate fission of the maturing T tubule. Interestingly, increased levels of DNM2 are found in muscle from XLMTM and from BIN1 related CNM206, and overexpression of DNM2 in mice leads to a CNM like phenotype207. Therefore, DNM2 overexpression or hyper-function may be the key aspect that drives CNM pathology. This concept is the basis for the therapeutic strategy of lowering DNM2 levels in the various genetic subtypes of CNM208, and an antisense oligonucleotide based approach has shown efficacy in mouse models of MTM1, DNM2 and BIN1 CNM200,206,209.
BIN1, or amphiphysin 2, is a membrane deforming protein critical for the formation of the T tubule210. It also interacts directly with DNM2211. Recessive mutations, associated primarily with loss of expression, cause CNM211. SPEG encodes a large protein kinase in the obscurin and myosin light chain kinase family. Recessive mutations cause a lethal subtype of CNM also associated with cardiomyopathy212. SPEG interacts directly with MTM1, and loss of SPEG secondarily impacts MTM1 expression212. SPEG also likely phosphorylates key target proteins at the triad (as demonstrated in cardiac myocytes, and including SERCA and potentially RyR1)213,214. Its function, and the consequences of mutation, thus may be a combination of interplay with MTM1 and its role as protein kinase.
SELENON related myopathies
The SELENON gene encodes a cytoplasmic protein (SePN) in the selenocysteine family of proteins215. SEPN appears to function as a critical regulator of cellular stress. Recessive mutations in SELENON are described in patients with a consistent clinical picture (severe and progressive axial weakness, rigid spine with scoliosis, and disproportionately mild extremity weakness) and a range of histotypes (minicore myopathy, congenital fiber type disproportion, Mallory body myopathy, and rigid spine muscular dystrophy)215–219. There is likely an important interplay between SEPN, calcium homeostasis, and regulated calcium release from the triad. As with RYR1 mutations, aberrant oxidative stress has been uncovered in models of SELENON related myopathy, and anti-oxidants such as N-acetylcysteine ameliorate these changes220. Clinical translation of this strategy is in progress.
Store operated calcium entry (SOCE)
While not essential for EC coupling, store operated calcium entry (or SOCE) is critical for maintaining calcium homeostasis in the myofiber221. The main components of the SOCE machinery are the calcium channel ORAI1 and its activator STIM1. Depletion of calcium from the sarcoplasmic reticulum induces a conformational change in STIM1, which, in turn, activates ORAI1, leading to extracellular calcium entry into the sarcoplasm. Mutations in both ORAI1 and STIM cause disease that impacts multiple organ systems222. Dominant, gain of function mutations lead to excessive calcium entry and are most relevant to skeletal muscle, as they are associated with tubular aggregate myopathy and Storkmorken Syndrome223.
Impaired sarcomere remodeling due to impaired Ca2+ handling
Healthy muscle can remodel and change its contractile and metabolic properties to meet the physiological demands placed upon it. This process involves Ca2+ mediated signaling that alters gene expression of slow/oxidative and fast/glycolytic (FG) gene expression programs. During muscle deconditioning (i.e. bed rest), muscle shifts to a more FG phenotype, while during muscle conditioning, it shifts towards the SO type. Studies in the mouse model of LGMD R1 (Capn3 knock out)224 have revealed a role for calpain 3 (Capn3) in Ca2+ handling, muscle remodeling and adaptation224–226. Capn3 is the muscle specific member of a family of cysteine proteases, collectively referred to as calpains227. Like other Capns, the active site aligns through changes in Capn3’s secondary structure, induced by Ca2+-calmodulin binding and autolytic cleavages228,229. Capn3 homodimerizes through its C terminal PEF domains230 and anchors on the giant protein titin231,232. LGMD R1 mutations have been shown to interfere with either Capn3 activity, anchorage to titin or homodimerization233,234. Capn3 proteolytically cleaves titin as well as other sarcomeric proteins such as filamin C (FLNC)235 and myosin light chain 2 (MLC2)236 and these cleavages target sarcomeric proteins to the proteasome.237
Capn3 at the triad
In addition to Capn3’s anchorage on the N2 line of titin (the site where the T tubule enters the myofibril), Capn3 also binds to RyR1 and stabilizes the triad protein complex238. Reductions in RyR1 levels226 are observed in both Capn3 knock out and LGMD R1 patient biopsies and reduced Ca2+ transients are observed in Capn3 KO mice239. Other proteins that are known to be enriched at the triad, such as aldolase238 and calcium calmodulin kinase (CaMKII),226 are also greatly reduced in Capn3 knock out mice. Thus, in addition to its proteolytic role, Capn3 plays a structural role in maintenance of triad complex integrity.
Loss of triad integrity leads to severe reductions in CaMKII levels and activation225,226. Because CaMKII promotes the slow oxidative gene expression program, loss of Capn3/CaMKII leads to impaired expression of slow/oxidative genes and failed skeletal muscle remodelling. The phenotypic features of mitochondrial abnormalities241,242 growth failure224, sarcomere disorganization224,237, and abnormal fat metabolism225 in Capn3 deficient muscles can be explained by reductions in the slow-oxidative gene expression program.
The identification of CAPN3 as the defective gene in LGMD R1 was the first discovery of a gene mutation linked to a LGMD243,244. Pathogenic mutations occur along the entire molecule and approximately 60% are missense mutations. Some missense mutations impact proteolytic activity while others likely impact either secondary structure, titin binding or calmodulin binding229,234,245. Genotype/phenotype correlations have been hindered by the fact that most patients are compound heterozygotes. Additional studies are needed to unravel the relationship between Capn3, titin and CaMKII signalling and the impact of these biochemical changes on disease features.
Skeletal muscle channelopathies
Skeletal muscle is an electrically active tissue, and charge balance between the sarcoplasm, the SR, and the extracellular milieu is critical for maintenance of muscle tone and to ensure proper cycling between the relaxed and contracted state. This balance is regulated by several important ion channels, including the sodium channel SCN4A and the chloride conductor CLCN1. Mutations in these genes, as well as several others, cause a group of muscle disorders collectively referred to a skeletal muscle channelopathies. Phenotypes include periodic paralysis (mentioned above in reference to CACNA1S) and myotonia (prolonged muscle stiffness), as well as some forms of congenital myopathy. An in-depth examination of these important ion channels and the disorders associated with them is presented in these reviews246,247
Heading: Defects of membrane repair
Introduction to skeletal muscle membrane repair and lipid trafficking
The muscle sarcolemma experiences high forces during muscle contraction, and these forces can lead to small sarcolemmal ruptures. These membrane tears cause increased local calcium entry and a change in phosphatidyl serine from the inner leaflet to the outer leaflet of the membrane. Annexin proteins A6, A1 and A2 sense the high calcium and phospholipid breakdown and they traffic to the injury site where they oligomerize and form a “repair cap”248,249. Annexin A6 spreads more broadly across the repair cap than the other annexins250 and it likely contributes to the membrane curvature needed for the cap to form251. The increase in local calcium also activates calcium-dependent proteases (calpains I and II, but not calpain 3252), which have been shown to proteolytically cleave dysferlin, and release a C terminal fragment) that has been referred to as “mini-dysferlin”10. Dysferlin may be a synaptotagmin-like molecule253, functioning to recruit lysosomal vesicles to the injury site250, likely through a process that is similar to mechanisms of neurotransmitter release254. Trim72 (AKA MG53) participates in vesicle trafficking to the injury site, although it is not clear if mini-dysferlin and Trim72 work cooperatively in the repair process.255 Membrane lipids are also recruited from the membrane, lateral to the injury site.248 At the site of repair, mini-dysferlin concentrates at the base or “shoulder” of the repair cap along with Trim72248, EHD proteins, and BIN1248. Annexins, Trim72 and dysferlin have all been shown to associate with phosphatidyl serine, which may be part of the repair signaling.255,256 After the membrane is repaired, the repair cap and excess Ca2+ are shed via exocytosis mediated by Trim72.257,248 The repair process involves microtubules, actin filaments and dynamin248,258–260
Disorders linked to defective membrane repair
Loss-of-function mutations in the DYSF gene, which encodes dysferlin, underlie LGMD2B (LGMD R2) or the allelic distal myopathy called Myoshi Myopathy261,262. Studies in the Dysf knock out mouse have revealed an essential role for dysferlin in skeletal muscle membrane repair263. Defective membrane repair may also underlie LGMD2L due to mutations in ANO5264, which encodes Anoctomin 5 protein. Ano5 knock-out mice show impaired membrane repair, although it is not clear whether Anoctomin 5 directly participates in the repair process.
Treatment of membrane repair disorders
Gene replacement therapies for LGMD2B/Myoshi myopathy are in pre-clinical development, but the large size of the DYSF gene (6.5 kb) prevents its packaging into a single AAV vector. Thus, strategies that utilize two, overlapping vectors have been tested in murine models and have shown some success265. Gene replacement therapies are also in development for LGMD2I (FKRP) and LGMD2L (ANO5).
Dysferlin deficiency is associated with high levels of intramuscular inflammation, consisting mainly of T cells and macrophages266–270. Given the inflammatory state of dysferlin-deficient muscles, one would expect steroids (an effective therapy for DMD) to be beneficial for LGMD2B, but this is not the case with daily steroid treatment. Weekly treatment may prove to be more beneficial, because the positive effects of steroids on membrane repair outweigh the negative effects on muscle wasting and metabolic reprogramming observed with daily steroids271,272. On the other hand, human biopsies have shown high levels of the complement-associated membrane attack complex (MAC) on the surface of skeletal muscle fibers of LGMD2B pateints273, and studies in the murine model of LGMD2B have demonstrated benefit from interference with the complement pathway274. A small study that used Rituximab (B cell depletion) led to increased muscle strength in two patients who were tested275. Systemic administration of recombinant A6 protein showed benefit in reducing membrane injury in the mouse model of LGMD2C (SGCG knock out mice)248.
Heading: Failure of protein quality control
Introduction to cellular systems that regulate protein quality in muscle
All cells maintain a balance between protein synthesis and protein degradation. However, skeletal muscle in particular utilizes this balance to increase muscle mass via enhanced protein synthesis (hypertrophy) or to decrease muscle mass via enhanced proteolysis (atrophy) under conditions of exercise and inactivity. Moreover, the physiologic stress and strain placed on myofibers and long-lived structural proteins within myofibrils results in their repeated unfolding and refolding that is necessary to prevent protein degradation or aggregation. Additionally, skeletal muscle is exquisitely sensitive to changes in organismal nutrient state and serves as the largest reservoir of free amino acids under conditions of starvation. The protein degradation in skeletal muscle that occurs with disuse, denervation, anorexia or chronic illness is mediated through two principal proteolytic pathways, the ubiquitin proteasome system (UPS) and autophago-lysosomal pathway (ALP).
Myopathies associated with ALP dysfunction
Autophagy and more specifically macroautophagy is the selective sequestration and subsequent engulfment of proteins, organelles and cytoplasm by an expanding phagophore that becomes an autophagosome. Cargo filled autophagosomes are then trafficked to the late endosome/lysosome enabling proteolysis within an acidic milieu. Mutations in both autophagic and lysosomal proteins lead to myopathies with distinctive pathologic features that include autophagic and lysosomal vacuoles. Specifically, inactivating mutations in proteins necessary for autophagosome lysosome fusion such as EPG5 and LAMP2 cause Vici Syndrome and Danon’s disease respectively276,277. These myopathies have the characteristic accumulation of autophagic and lysosomal vacuoles as evidenced by the accumulation of autophagic and lysosomal proteins such as SQSTM1, LC3 and acid phosphatase with associated multisystem involvement including both cardiac and CNS manifestations276,277. Inactivating mutations in proteins necessary for normal lysosomal function can also lead to a vacuolar myopathy. For example, loss of the acidic hydrolase, GAA or a reduction in VMA21, the chaperone necessary for the assembly of the vacuolar-type ATPase both lead to lysosomes with reduced degradative capacity278,279. The pathology in these myopathies termed Pompe’s disease and X-linked myopathy with excessive autophagy (XMEA) is distinctive with the sarcoplasmic accumulation of vacuoles surrounded by sarcolemmal proteins such dystrophin and caveolin-3280.
Myopathies associated with UPS dysfunction
Under conditions of muscle atrophy, the UPS is activated leading to the rapid degradation of myofibrillar proteins and reduction in muscle mass. Proteins are selectively targeted and degraded via the UPS. The selectivity of this process is dictated by a large family of ubiquitin ligases or E3 ligases. During muscle atrophy expression of the E3 ligases muscle RING-finger 1 (MuRF1) and Atrogin1/MAFbx ubiquitinate thick filament proteins such as myosin281. The E3 ligase Trim32 was originally thought to ubiquitinate thin filament proteins282; however, its role is more likely to be control of muscle stem cell283,284 possibly through regulation of Piasy285, an E3 SUMO ligase or NDRG2286. Consistent with this critical role in muscle proteostasis, homozygous loss of function mutations in MURF1 lead to myopathies with vacuoles and sarcoplasmic inclusions. Mutations in TRIM32 underlie LGMD2H, sarcotubular myopathy and the allelic disorder Bardet Beadle Syndrome. The substrate specificity of some E3 ligases require adaptor proteins necessary for efficient ubiquitination. Loss of function mutations in three related Kelch proteins, KLHL40, KLHL41 and KBTBD13 lead to nemaline rod myopathies suggesting their importance in the proteasomal degradation of thin filament proteins287.
Myopathies associated with chaperone dysfunction.
Chaperones are proteins necessary for protein quality control or “proteostasis.” In lower organisms, this group of proteins is critical for cell survival following environmental insults that destabilize proteins such as heat shock and are thus termed heat shock proteins (HSPs). HSPs are a multi-tiered network of proteins that include HSP70s/HSPAs, HSP40s/DNAJs, HSP20s/HSPBs and BAG family proteins. A disruption in the HSP network leads to the accumulation of misfolded and aggregated proteins that can further disrupt protein degradation pathways such as autophagy. Dominant missense mutations in the co-chaperones DNAJB6 (LGMDD1) or BAG3 (MFM6) lead to myopathies with Z-disc disorganization and desmin inclusions288–290. The dominant effect may relate to the mutant proteins’ avidity and sequestration of other HSP proteins (e.g. HSPA1) in aggregates. Mutations in the small HSPs, HSPB5 and HSPB8 also lead to myopathies with prominent myofibrillar disorganization and rimmed vacuole291,292. Finally, disruption of chaperone proteins within the secretory pathway result in myopathies with aggregates and vacuoles. For example, loss of function mutations in Sil1 (Marinesco-sjogren syndrome) a co-chaperone for HSPB5/Bip lead to a congenital myopathy with rimmed vacuoles293,294. Notably therapies aimed at restoring protein homeostasis such as HSP activators may be effective in chaperonopathies.
- Conclusions and perspectives:
Mutations that perturb normal processes in skeletal muscle result in myopathies or muscular dystrophies. Over the past three decades, hundreds of such mutations have been identified. In-depth study of these disease-causing proteins has led to an immense amount of new information regarding the cellular processes necessary to maintain healthy muscle. Understanding the normal function of these proteins has provided valuable insights on why mutations cause disease and will be necessary for development of efficacious therapies. The availability of the underlying genetic causes of these diseases provides a facile path to develop therapies targeting the underlying cause of disease such as gene replacement, gene editing and antisense oligonucleotide-based exon skipping.
Figure 5 |. Skeletal Muscle Membrane Repair.

Skeletal muscle sustains small tears from normal muscle use, which are quickly repaired by cell repair machinery. a | Following a tear, calcium flows down its concentration gradient from outside the cell to inside. Cholesterol is oxidized and phosphatidyl serine changes its conformation from intracellular-facing to extracellular facing. b | Calcium, oxidized cholesterol and phospholipids activate the cellular repair machinery. Annexin proteins bind to calcium and phosphatidyl serine and form a cap that seals the are of the tear.
Figure 6 |. Schematic of a muscle cell and the proteins linked to protein turnover and quality control.
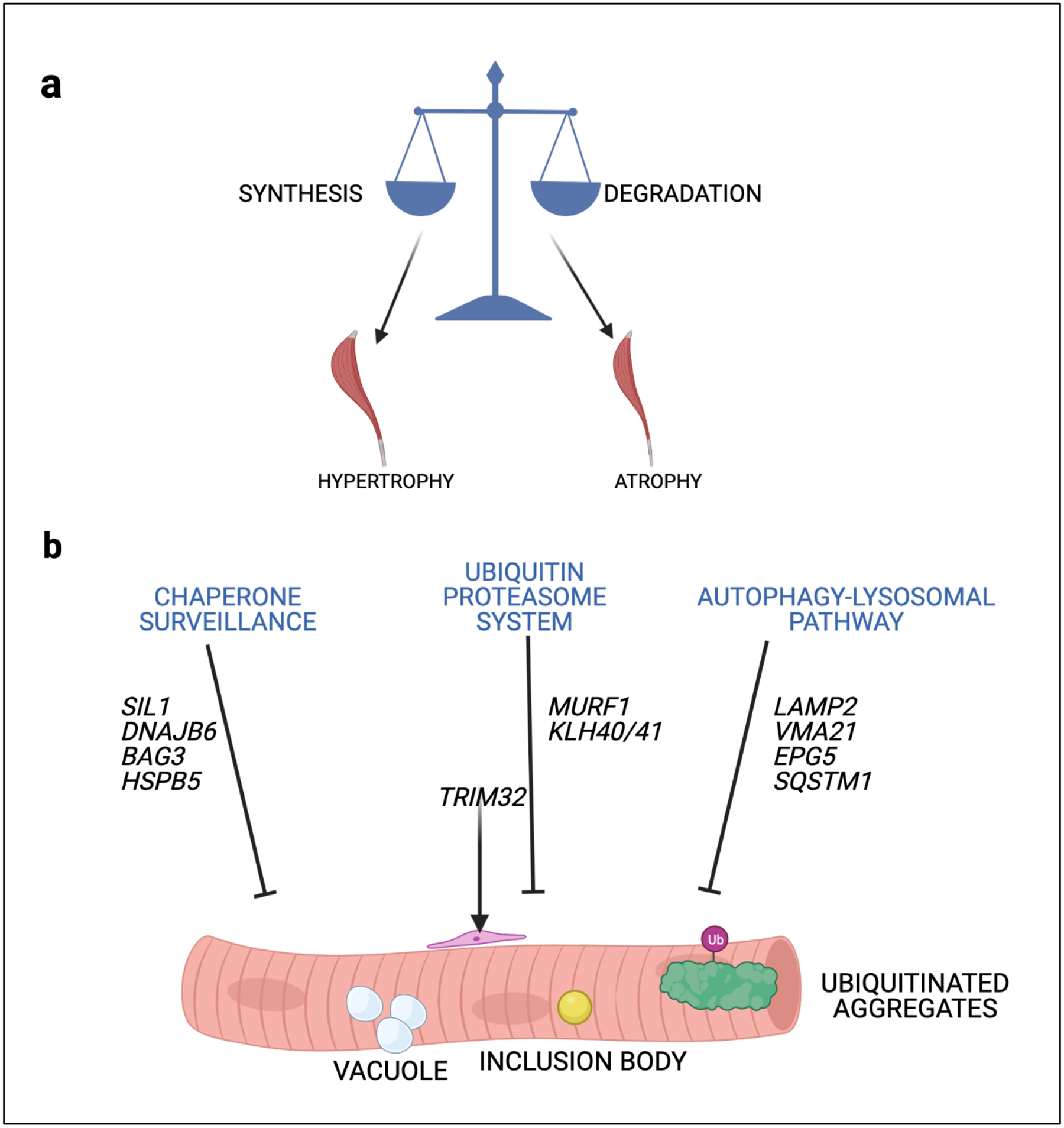
b | Skeletal muscle mass is maintained by the balance of protein synthesis and protein degradation. b | Disruptions in chaperone surveillance, the ubiquitin proteasome system and the autophago-lysosomal pathway lead to pathologic changes in skeletal muscle that include myofibrillar disruption, inclusion bodies, protein aggregates and vacuolation.
Acknowledgements
The authors would like to acknowledge the contributions of individuals who reviewed figures or edited the manuscript: Drs. Elizabeth McNally, Jeffrey Chamberlain, Rachelle Crosbie, Alexis Demonbreun, Elizabeth Gibbs, Courtney Young, Jackie McCort, Kristen Stearnes-Reider. and Joseph O’Brien.
Footnotes
Competing interests
1) MJS is a co-founder of a startup called Myogene Bio 2) MJS and CCW serve on the Research Advisory Board for the Muscular Dystrophy Association 3) JMD is the Chief Medical Officer for Deep Genomics
RELATED LINKS
Resource of genes linked to Neuromuscular Disorders: http://www.musclegenetable.fr/
REFERENCES
- 1.Engel AG, Shen XM, Selcen D & Sine SM Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 14, 420–434, doi: 10.1016/S1474-4422(14)70201-7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monaco AP et al. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 323, 646–650, doi: 10.1038/323646a0 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Monaco AP, Bertelson CJ, Colletti-Feener C & Kunkel LM Localization and cloning of Xp21 deletion breakpoints involved in muscular dystrophy. Hum Genet 75, 221–227, doi: 10.1007/BF00281063 (1987). [DOI] [PubMed] [Google Scholar]
- 4.Hoffman EP, Brown RH & Kunkel LM Dystrophin: the protein product of the Duchene muscular dystrophy locus. 1987. Biotechnology 24, 457–466 (1992). [PubMed] [Google Scholar]
- 5.Thornton CA, Wang E & Carrell EM Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev 44, 135–140, doi: 10.1016/j.gde.2017.03.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyba M et al. Meeting report: the 2020 FSHD International Research Congress. Skelet Muscle 10, 36, doi: 10.1186/s13395-020-00253-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lek A, Rahimov F, Jones PL & Kunkel LM Emerging preclinical animal models for FSHD. Trends Mol Med 21, 295–306, doi: 10.1016/j.molmed.2015.02.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell KP & Kahl SD Association of dystrophin and an integral membrane glycoprotein. Nature 338, 259–262, doi: 10.1038/338259a0 (1989). [DOI] [PubMed] [Google Scholar]; • Discovery of the dystrophin glycoprotein complex.
- 9.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG & Campbell KP Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315–319, doi: 10.1038/345315a0 (1990). [DOI] [PubMed] [Google Scholar]
- 10.Crosbie RH, Heighway J, Venzke DP, Lee JC & Campbell KP Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J Biol Chem 272, 31221–31224 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Crosbie RH et al. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol 145, 153–165 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michele DE & Campbell KP Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem 278, 15457–15460, doi: 10.1074/jbc.R200031200 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Ibraghimov-Beskrovnaya O et al. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702, doi: 10.1038/355696a0 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Hack AA et al. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci 113 (Pt 14), 2535–2544 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Bunnell TM, Jaeger MA, Fitzsimons DP, Prins KW & Ervasti JM Destabilization of the dystrophin-glycoprotein complex without functional deficits in alpha-dystrobrevin null muscle. PLoS One 3, e2604, doi: 10.1371/journal.pone.0002604 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao W et al. Regulation of proteolytic cleavage of retinoid X receptor-α by GSK-3β. Carcinogenesis 34, 1208–1215, doi: 10.1093/carcin/bgt043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keep NH, Norwood FL, Moores CA, Winder SJ & Kendrick-Jones J The 2.0 A structure of the second calponin homology domain from the actin-binding region of the dystrophin homologue utrophin. J Mol Biol 285, 1257–1264, doi: 10.1006/jmbi.1998.2406 (1999). [DOI] [PubMed] [Google Scholar]
- 18.Norwood FL, Sutherland-Smith AJ, Keep NH & Kendrick-Jones J The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure 8, 481–491, doi: 10.1016/s0969-2126(00)00132-5 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Chamberlain JS et al. Interactions between dystrophin and the sarcolemma membrane. Soc Gen Physiol Ser 52, 19–29 (1997). [PubMed] [Google Scholar]
- 20.Adams ME et al. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 11, 531–540, doi: 10.1016/0896-6273(93)90157-m (1993). [DOI] [PubMed] [Google Scholar]
- 21.Zhao J et al. Dystrophin contains multiple independent membrane-binding domains. Hum Mol Genet 25, 3647–3653, doi: 10.1093/hmg/ddw210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams ME, Odom GL, Kim MJ, Chamberlain JS & Froehner SC Syntrophin binds directly to multiple spectrin-like repeats in dystrophin and mediates binding of nNOS to repeats 16–17. Hum Mol Genet 27, 2978–2985, doi: 10.1093/hmg/ddy197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang WJ et al. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci U S A 93, 9142–9147, doi: 10.1073/pnas.93.17.9142 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson DM et al. Rapid, redox-mediated mechanical susceptibility of the cortical microtubule lattice in skeletal muscle. Redox Biol 37, 101730, doi: 10.1016/j.redox.2020.101730 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oak SA, Zhou YW & Jarrett HW Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J Biol Chem 278, 39287–39295, doi: 10.1074/jbc.M305551200 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Thompson TG et al. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J Cell Biol 148, 115–126, doi: 10.1083/jcb.148.1.115 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayalon G, Davis JQ, Scotland PB & Bennett V An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell 135, 1189–1200, doi: 10.1016/j.cell.2008.10.018 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Bhosle RC, Michele DE, Campbell KP, Li Z & Robson RM Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun 346, 768–777, doi: 10.1016/j.bbrc.2006.05.192 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Crosbie RH et al. Characterization of aquaporin-4 in muscle and muscular dystrophy. FASEB J 16, 943–949, doi: 10.1096/fj.01-0327com (2002). [DOI] [PubMed] [Google Scholar]
- 30.Blake DJ & Martin-Rendon E Intermediate filaments and the function of the dystrophin-protein complex. Trends Cardiovasc Med 12, 224–228, doi: 10.1016/s1050-1738(02)00166-4 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Moorwood C Syncoilin, an intermediate filament-like protein linked to the dystrophin associated protein complex in skeletal muscle. Cell Mol Life Sci 65, 2957–2963, doi: 10.1007/s00018-008-8306-9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone MR, O’Neill A, Catino D & Bloch RJ Specific interaction of the actin-binding domain of dystrophin with intermediate filaments containing keratin 19. Mol Biol Cell 16, 4280–4293, doi: 10.1091/mbc.e05-02-0112 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ervasti JM & Campbell KP Membrane organization of the dystrophin-glycoprotein complex. Cell 66, 1121–1131, doi: 10.1016/0092-8674(91)90035-w (1991). [DOI] [PubMed] [Google Scholar]
- 34.Ervasti JM & Campbell KP Dystrophin and the membrane skeleton. Curr Opin Cell Biol 5, 82–87, doi: 10.1016/s0955-0674(05)80012-2 (1993). [DOI] [PubMed] [Google Scholar]
- 35.Bowe MA, Deyst KA, Leszyk JD & Fallon JR Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: a heteromeric complex related to the dystroglycans. Neuron 12, 1173–1180, doi: 10.1016/0896-6273(94)90324-7 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Yurchenco PD, Cheng YS, Campbell K & Li S Loss of basement membrane, receptor and cytoskeletal lattices in a laminin-deficient muscular dystrophy. J Cell Sci 117, 735–742, doi: 10.1242/jcs.00911 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Yurchenco PD & Patton BL Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des 15, 1277–1294, doi: 10.2174/138161209787846766 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaiswal JK et al. Patients with a non-dysferlin Miyoshi myopathy have a novel membrane repair defect. Traffic 8, 77–88, doi: 10.1111/j.1600-0854.2006.00505.x (2007). [DOI] [PubMed] [Google Scholar]
- 39.Hoffman EP, Brown RH & Kunkel LM Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928, doi: 10.1016/0092-8674(87)90579-4 (1987). [DOI] [PubMed] [Google Scholar]
- 40.Koenig M et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517, doi: 10.1016/0092-8674(87)90504-6 (1987). [DOI] [PubMed] [Google Scholar]; • Cloning of the complete DMD gene. This is the first gene mutation linked to a muscular dystrophy.
- 41.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H & Kunkel LM An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Monaco AP et al. Detection of deletions spanning the Duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature 316, 842–845, doi: 10.1038/316842a0 (1985). [DOI] [PubMed] [Google Scholar]
- 43.Monaco AP & Kunkel LM Cloning of the Duchenne/Becker muscular dystrophy locus. Adv Hum Genet 17, 61–98, doi: 10.1007/978-1-4613-0987-1_3 (1988). [DOI] [PubMed] [Google Scholar]
- 44.Harper SQ, Crawford RW, DelloRusso C & Chamberlain JS Spectrin-like repeats from dystrophin and alpha-actinin-2 are not functionally interchangeable. Hum Mol Genet 11, 1807–1815, doi: 10.1093/hmg/11.16.1807 (2002). [DOI] [PubMed] [Google Scholar]; • Demonstration of how spectrin repeats contribute to dystrophin’s spring function
- 45.Petrof BJ, Shrager JB, Stedman HH, Kelly AM & Sweeney HL Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 90, 3710–3714, doi: 10.1073/pnas.90.8.3710 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi YM, Rader EP, Crawford RW & Campbell KP Endpoint measures in the mdx mouse relevant for muscular dystrophy pre-clinical studies. Neuromuscul Disord 22, 34–42, doi: 10.1016/j.nmd.2011.08.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Straub V, Rafael JA, Chamberlain JS & Campbell KP Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol 139, 375–385 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spencer MJ, Montecino-Rodriguez E, Dorshkind K & Tidball JG Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol 98, 235–243, doi: 10.1006/clim.2000.4966 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Capote J et al. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J Cell Biol 213, 275–288, doi: 10.1083/jcb.201510086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ermolova NV et al. Long-term administration of the TNF blocking drug Remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul Disord 24, 583–595, doi: 10.1016/j.nmd.2014.04.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vetrone SA et al. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest 119, 1583–1594, doi: 10.1172/JCI37662 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiaffino S, Gorza L, Dones I, Cornelio F & Sartore S Fetal myosin immunoreactivity in human dystrophic muscle. Muscle Nerve 9, 51–58, doi: 10.1002/mus.880090108 (1986). [DOI] [PubMed] [Google Scholar]
- 53.Soltanzadeh P et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord 20, 499–504, doi: 10.1016/j.nmd.2010.05.010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dowling JJ, D Gonorazky H, Cohn RD & Campbell C Treating pediatric neuromuscular disorders: The future is now. Am J Med Genet A 176, 804–841, doi: 10.1002/ajmg.a.38418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibbs EM et al. Large in-frame 5’ deletions in DMD associated with mild Duchenne muscular dystrophy: Two case reports and a review of the literature. Neuromuscul Disord 29, 863–873, doi: 10.1016/j.nmd.2019.09.009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McNally EM, Bönnemann CG, Kunkel LM & Bhattacharya SK Deficiency of adhalin in a patient with muscular dystrophy and cardiomyopathy. N Engl J Med 334, 1610–1611, doi: 10.1056/NEJM199606133342417 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Bonnemann CG et al. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet 11, 266–273, doi: 10.1038/ng1195-266 (1995). [DOI] [PubMed] [Google Scholar]; • Discovery of a link between sarcoglycan gene mutations and LGMD
- 58.Nigro V et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat Genet 14, 195–198, doi: 10.1038/ng1096-195 (1996). [DOI] [PubMed] [Google Scholar]
- 59.McNally EM et al. Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. Am J Hum Genet 59, 1040–1047 (1996). [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida-Moriguchi T et al. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 341, 896–899, doi: 10.1126/science.1239951 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Praissman JL et al. The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife 5, doi: 10.7554/eLife.14473 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoshida-Moriguchi T et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science 327, 88–92, doi: 10.1126/science.1180512 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willer T et al. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated alpha-dystroglycan functional glycosylation. Elife 3, doi: 10.7554/eLife.03941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Willer T et al. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated α-dystroglycan functional glycosylation. Elife 3, doi: 10.7554/eLife.03941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoshida-Moriguchi T & Campbell KP Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 25, 702–713, doi: 10.1093/glycob/cwv021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sheikh MO et al. HNK-1 sulfotransferase modulates alpha-dystroglycan glycosylation by 3-O-sulfation of glucuronic acid on matriglycan. Glycobiology 30, 817–829, doi: 10.1093/glycob/cwaa024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laverda AM et al. Congenital muscular dystrophy, brain and eye abnormalities: one or more clinical entities? Childs Nerv Syst 9, 84–87, doi: 10.1007/BF00305313 (1993). [DOI] [PubMed] [Google Scholar]
- 68.Kim DS et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in alpha-DG. Neurology 62, 1009–1011, doi: 10.1212/01.wnl.0000115386.28769.65 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Stevens E et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet 92, 354–365, doi: 10.1016/j.ajhg.2013.01.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jensen BS et al. GMPPB-Associated Dystroglycanopathy: Emerging Common Variants with Phenotype Correlation. Hum Mutat 36, 1159–1163, doi: 10.1002/humu.22898 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Inamori K et al. Endogenous glucuronyltransferase activity of LARGE or LARGE2 required for functional modification of α-dystroglycan in cells and tissues. J Biol Chem 289, 28138–28148, doi: 10.1074/jbc.M114.597831 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michele DE, Kabaeva Z, Davis SL, Weiss RM & Campbell KP Dystroglycan matrix receptor function in cardiac myocytes is important for limiting activity-induced myocardial damage. Circ Res 105, 984–993, doi: 10.1161/CIRCRESAHA.109.199489 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baker NL et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 14, 279–293, doi: 10.1093/hmg/ddi025 (2005). [DOI] [PubMed] [Google Scholar]; • Discovery that mutations in ECM-encoding gene can be linked to a muscular dystrophy.
- 74.Lamande SR et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet 7, 981–989, doi: 10.1093/hmg/7.6.981 (1998). [DOI] [PubMed] [Google Scholar]
- 75.Lamande SR, Shields KA, Kornberg AJ, Shield LK & Bateman JF Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem 274, 21817–21822, doi: 10.1074/jbc.274.31.21817 (1999). [DOI] [PubMed] [Google Scholar]
- 76.Gushchina LV et al. Lack of toxicity in non-human primates receiving clinically relevant doses of an AAV9.U7snRNA vector designed to induce DMD exon 2 skipping. Hum Gene Ther, doi: 10.1089/hum.2020.286 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu B et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A 105, 14814–14819, doi: 10.1073/pnas.0805676105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jearawiriyapaisarn N et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol Ther 16, 1624–1629, doi: 10.1038/mt.2008.120 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao QQ et al. Reengineering a transmembrane protein to treat muscular dystrophy using exon skipping. J Clin Invest 125, 4186–4195, doi: 10.1172/JCI82768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blázquez L et al. In vitro correction of a pseudoexon-generating deep intronic mutation in LGMD2A by antisense oligonucleotides and modified small nuclear RNAs. Hum Mutat 34, 1387–1395, doi: 10.1002/humu.22379 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Amoasii L et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362, 86–91, doi: 10.1126/science.aau1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bengtsson NE et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 8, 14454, doi: 10.1038/ncomms14454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hakim CH et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight 3, doi: 10.1172/jci.insight.124297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nelson CE et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat Med 25, 427–432, doi: 10.1038/s41591-019-0344-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Young CS et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 18, 533–540, doi: 10.1016/j.stem.2016.01.021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Young CS, Pyle AD & Spencer MJ CRISPR for Neuromuscular Disorders: Gene Editing and Beyond. Physiology (Bethesda) 34, 341–353, doi: 10.1152/physiol.00012.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qiu B, Ruston J, Granzier H, Justice MJ & Dowling JJ Failure to identify modifiers of. Biol Open 8, doi: 10.1242/bio.044867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sewry CA, Laitila JM & Wallgren-Pettersson C Nemaline myopathies: a current view. J Muscle Res Cell Motil 40, 111–126, doi: 10.1007/s10974-019-09519-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pelin K et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci U S A 96, 2305–2310, doi: 10.1073/pnas.96.5.2305 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Colombo I et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology 84, 28–35, doi: 10.1212/WNL.0000000000001110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gonorazky HD, Bönnemann CG & Dowling JJ The genetics of congenital myopathies. Handb Clin Neurol 148, 549–564, doi: 10.1016/B978-0-444-64076-5.00036-3 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Nowak KJ et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 23, 208–212, doi: 10.1038/13837 (1999). [DOI] [PubMed] [Google Scholar]
- 93.Amburgey K et al. A Cross-Sectional Study of Nemaline Myopathy. Neurology, doi: 10.1212/WNL.0000000000011458 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chu M, Gregorio CC & Pappas CT Nebulin, a multi-functional giant. J Exp Biol 219, 146–152, doi: 10.1242/jeb.126383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Labeit S, Ottenheijm CA & Granzier H Nebulin, a major player in muscle health and disease. FASEB J 25, 822–829, doi: 10.1096/fj.10-157412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kiss B et al. Nebulin and Lmod2 are critical for specifying thin-filament length in skeletal muscle. Sci Adv 6, doi: 10.1126/sciadv.abc1992 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pappas CT, Krieg PA & Gregorio CC Nebulin regulates actin filament lengths by a stabilization mechanism. J Cell Biol 189, 859–870, doi: 10.1083/jcb.201001043 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ottenheijm CA et al. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet 18, 2359–2369, doi: 10.1093/hmg/ddp168 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Witt CC et al. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J 25, 3843–3855, doi: 10.1038/sj.emboj.7601242 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gohlke J, Tonino P, Lindqvist J, Smith JE & Granzier H The number of Z-repeats and super-repeats in nebulin greatly varies across vertebrates and scales with animal size. J Gen Physiol 153, doi: 10.1085/jgp.202012783 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Winter JM et al. Mutation-specific effects on thin filament length in thin filament myopathy. Ann Neurol 79, 959–969, doi: 10.1002/ana.24654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mijailovich SM et al. Nebulin and titin modulate cross-bridge cycling and length-dependent calcium sensitivity. J Gen Physiol 151, 680–704, doi: 10.1085/jgp.201812165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takano K et al. Nebulin and N-WASP cooperate to cause IGF-1-induced sarcomeric actin filament formation. Science 330, 1536–1540, doi: 10.1126/science.1197767 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Nowak KJ, Ravenscroft G & Laing NG Skeletal muscle alpha-actin diseases (actinopathies): pathology and mechanisms. Acta Neuropathol 125, 19–32, doi: 10.1007/s00401-012-1019-z (2013). [DOI] [PubMed] [Google Scholar]
- 105.dos Remedios CG et al. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83, 433–473, doi: 10.1152/physrev.00026.2002 (2003). [DOI] [PubMed] [Google Scholar]
- 106.Laing NG et al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Hum Mutat 30, 1267–1277, doi: 10.1002/humu.21059 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Joureau B et al. Dysfunctional sarcomere contractility contributes to muscle weakness in ACTA1-related nemaline myopathy (NEM3). Ann Neurol 83, 269–282, doi: 10.1002/ana.25144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jain RK et al. Nemaline myopathy with stiffness and hypertonia associated with an ACTA1 mutation. Neurology 78, 1100–1103, doi: 10.1212/WNL.0b013e31824e8ebe (2012). [DOI] [PubMed] [Google Scholar]
- 109.Ryan MM et al. Dietary L-tyrosine supplementation in nemaline myopathy. J Child Neurol 23, 609–613, doi: 10.1177/0883073807309794 (2008). [DOI] [PubMed] [Google Scholar]
- 110.Sztal TE et al. Testing of therapies in a novel nebulin nemaline myopathy model demonstrate a lack of efficacy. Acta Neuropathol Commun 6, 40, doi: 10.1186/s40478-018-0546-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Messineo AM et al. L-tyrosine supplementation does not ameliorate skeletal muscle dysfunction in zebrafish and mouse models of dominant skeletal muscle alpha-actin nemaline myopathy. Sci Rep 8, 11490, doi: 10.1038/s41598-018-29437-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tinklenberg JA et al. Myostatin inhibition using mRK35 produces skeletal muscle growth and tubular aggregate formation in wild type and TgACTA1D286G nemaline myopathy mice. Hum Mol Genet 27, 638–648, doi: 10.1093/hmg/ddx431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tinklenberg JA et al. Myostatin Inhibition Using ActRIIB-mFc Does Not Produce Weight Gain or Strength in the Nebulin Conditional KO Mouse. J Neuropathol Exp Neurol 78, 130–139, doi: 10.1093/jnen/nly120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nowak KJ et al. Rescue of skeletal muscle alpha-actin-null mice by cardiac (fetal) alpha-actin. J Cell Biol 185, 903–915, doi: 10.1083/jcb.200812132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ravenscroft G et al. Cardiac alpha-actin over-expression therapy in dominant ACTA1 disease. Hum Mol Genet 22, 3987–3997, doi: 10.1093/hmg/ddt252 (2013). [DOI] [PubMed] [Google Scholar]
- 116.Donner K et al. Mutations in the beta-tropomyosin (TPM2) gene--a rare cause of nemaline myopathy. Neuromuscul Disord 12, 151–158, doi: 10.1016/s0960-8966(01)00252-8 (2002). [DOI] [PubMed] [Google Scholar]
- 117.Laing NG et al. A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy. Nat Genet 9, 75–79, doi: 10.1038/ng0195-75 (1995). [DOI] [PubMed] [Google Scholar]; • Association between thin filament proteins and nemaline myopathy
- 118.Johnston JJ et al. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet 67, 814–821, doi: 10.1086/303089 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yuen M et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest 124, 4693–4708, doi: 10.1172/JCI75199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sandaradura SA et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat 39, 383–388, doi: 10.1002/humu.23385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A & Tulinius M A mutation in the fast skeletal muscle troponin I gene causes myopathy and distal arthrogryposis. Neurology 67, 597–601, doi: 10.1212/01.wnl.0000230168.05328.f4 (2006). [DOI] [PubMed] [Google Scholar]
- 122.El-Mezgueldi M Tropomyosin dynamics. J Muscle Res Cell Motil 35, 203–210, doi: 10.1007/s10974-014-9377-x (2014). [DOI] [PubMed] [Google Scholar]
- 123.Memo M & Marston S Skeletal muscle myopathy mutations at the actin tropomyosin interface that cause gain- or loss-of-function. J Muscle Res Cell Motil 34, 165–169, doi: 10.1007/s10974-013-9344-y (2013). [DOI] [PubMed] [Google Scholar]
- 124.Marttila M et al. Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies. Hum Mutat 35, 779–790, doi: 10.1002/humu.22554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sung SS et al. Mutations in TNNT3 cause multiple congenital contractures: a second locus for distal arthrogryposis type 2B. Am J Hum Genet 73, 212–214, doi: 10.1086/376418 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sung SS et al. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet 72, 681–690, doi: 10.1086/368294 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ravenscroft G et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet 93, 6–18, doi: 10.1016/j.ajhg.2013.05.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gupta VA et al. Identification of KLHL41 Mutations Implicates BTB-Kelch-Mediated Ubiquitination as an Alternate Pathway to Myofibrillar Disruption in Nemaline Myopathy. Am J Hum Genet 93, 1108–1117, doi: 10.1016/j.ajhg.2013.10.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sambuughin N et al. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum Genet 87, 842–847, doi: 10.1016/j.ajhg.2010.10.020 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Agrawal PB et al. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet 80, 162–167, doi: 10.1086/510402 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Garg A et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J Clin Invest 124, 3529–3539, doi: 10.1172/JCI74994 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jirka C, Pak JH, Grosgogeat CA, Marchetii MM & Gupta VA Dysregulation of NRAP degradation by KLHL41 contributes to pathophysiology in nemaline myopathy. Hum Mol Genet 28, 2549–2560, doi: 10.1093/hmg/ddz078 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tajsharghi H & Oldfors A Myosinopathies: pathology and mechanisms. Acta Neuropathol 125, 3–18, doi: 10.1007/s00401-012-1024-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tajsharghi H et al. Human disease caused by loss of fast IIa myosin heavy chain due to recessive MYH2 mutations. Brain 133, 1451–1459, doi: 10.1093/brain/awq083 (2010). [DOI] [PubMed] [Google Scholar]
- 135.Toydemir RM et al. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet 38, 561–565, doi: 10.1038/ng1775 (2006). [DOI] [PubMed] [Google Scholar]
- 136.Veugelers M et al. Mutation of perinatal myosin heavy chain associated with a Carney complex variant. N Engl J Med 351, 460–469, doi: 10.1056/NEJMoa040584 (2004). [DOI] [PubMed] [Google Scholar]
- 137.Tajsharghi H et al. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 54, 494–500, doi: 10.1002/ana.10693 (2003). [DOI] [PubMed] [Google Scholar]
- 138.Martinsson T et al. Autosomal dominant myopathy: missense mutation (Glu-706 --> Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci U S A 97, 14614–14619, doi: 10.1073/pnas.250289597 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Meredith C et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet 75, 703–708, doi: 10.1086/424760 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lamont PJ et al. Novel mutations widen the phenotypic spectrum of slow skeletal/beta-cardiac myosin (MYH7) distal myopathy. Hum Mutat 35, 868–879, doi: 10.1002/humu.22553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kellermayer D, Smith JE 3rd & Granzier H Titin mutations and muscle disease. Pflugers Arch 471, 673–682, doi: 10.1007/s00424-019-02272-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Savarese M, Sarparanta J, Vihola A, Udd B & Hackman P Increasing Role of Titin Mutations in Neuromuscular Disorders. J Neuromuscul Dis 3, 293–308, doi: 10.3233/JND-160158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hackman P et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 71, 492–500, doi: 10.1086/342380 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pfeffer G et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 135, 1695–1713, doi: 10.1093/brain/aws102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oates EC et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann Neurol 83, 1105–1124, doi: 10.1002/ana.25241 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Heller SA, Shih R, Kalra R & Kang PB Emery-Dreifuss muscular dystrophy. Muscle Nerve 61, 436–448, doi: 10.1002/mus.26782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Janota CS, Calero-Cuenca FJ & Gomes ER The role of the cell nucleus in mechanotransduction. Curr Opin Cell Biol 63, 204–211, doi: 10.1016/j.ceb.2020.03.001 (2020). [DOI] [PubMed] [Google Scholar]
- 148.Paci G, Caria J & Lemke EA Cargo transport through the nuclear pore complex at a glance. J Cell Sci 134, doi: 10.1242/jcs.247874 (2021). [DOI] [PubMed] [Google Scholar]
- 149.Guenantin AC et al. Targeting the histone demethylase LSD1 prevents cardiomyopathy in a mouse model of laminopathy. J Clin Invest 131, doi: 10.1172/JCI136488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Broers JL et al. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet 13, 2567–2580, doi: 10.1093/hmg/ddh295 (2004). [DOI] [PubMed] [Google Scholar]
- 151.Zhang K et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 173, 958–971 e917, doi: 10.1016/j.cell.2018.03.025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Melia MJ et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 136, 1508–1517, doi: 10.1093/brain/awt074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rodriguez-Mora S et al. The mutation of Transportin 3 gene that causes limb girdle muscular dystrophy 1F induces protection against HIV-1 infection. PLoS Pathog 15, e1007958, doi: 10.1371/journal.ppat.1007958 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Choi JC et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med 4, 144ra102, doi: 10.1126/scitranslmed.3003875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liao CY et al. Rapamycin Reverses Metabolic Deficits in Lamin A/C-Deficient Mice. Cell Rep 17, 2542–2552, doi: 10.1016/j.celrep.2016.10.040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jungbluth H et al. Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat Rev Neurol 14, 151–167, doi: 10.1038/nrneurol.2017.191 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Dowling JJ, Lawlor MW & Dirksen RT Triadopathies: an emerging class of skeletal muscle diseases. Neurotherapeutics 11, 773–785, doi: 10.1007/s13311-014-0300-3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zorzato F et al. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem 265, 2244–2256 (1990). [PubMed] [Google Scholar]
- 159.Jungbluth H, Dowling JJ, Ferreiro A, Muntoni F & Consortium RYRM 217th ENMC International Workshop: RYR1-related myopathies, Naarden, The Netherlands, 29–31 January 2016. Neuromuscul Disord 26, 624–633, doi: 10.1016/j.nmd.2016.06.001 (2016). [DOI] [PubMed] [Google Scholar]
- 160.Lawal TA et al. Ryanodine receptor 1-related disorders: an historical perspective and proposal for a unified nomenclature. Skelet Muscle 10, 32, doi: 10.1186/s13395-020-00243-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang Y et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nat Genet 5, 46–50, doi: 10.1038/ng0993-46 (1993). [DOI] [PubMed] [Google Scholar]
- 162.Quane KA et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet 5, 51–55, doi: 10.1038/ng0993-51 (1993). [DOI] [PubMed] [Google Scholar]
- 163.Jungbluth H et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology 59, 284–287, doi: 10.1212/wnl.59.2.284 (2002). [DOI] [PubMed] [Google Scholar]
- 164.Clarke NF et al. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum Mutat 31, E1544–1550, doi: 10.1002/humu.21278 (2010). [DOI] [PubMed] [Google Scholar]
- 165.Wilmshurst JM et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol 68, 717–726, doi: 10.1002/ana.22119 (2010). [DOI] [PubMed] [Google Scholar]
- 166.Amburgey K et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis 8, 117, doi: 10.1186/1750-1172-8-117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Klein A et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat 33, 981–988, doi: 10.1002/humu.22056 (2012). [DOI] [PubMed] [Google Scholar]
- 168.Monnier N et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat 26, 413–425, doi: 10.1002/humu.20231 (2005). [DOI] [PubMed] [Google Scholar]
- 169.MacLennan DH et al. Ryanodine receptor gene is a candidate for predisposition to malignant hyperthermia. Nature 343, 559–561, doi: 10.1038/343559a0 (1990). [DOI] [PubMed] [Google Scholar]
- 170.Dlamini N et al. Mutations in RYR1 are a common cause of exertional myalgia and rhabdomyolysis. Neuromuscul Disord 23, 540–548, doi: 10.1016/j.nmd.2013.03.008 (2013). [DOI] [PubMed] [Google Scholar]
- 171.Gardner L et al. Investigating the genetic susceptibility to exertional heat illness. J Med Genet 57, 531–541, doi: 10.1136/jmedgenet-2019-106461 (2020). [DOI] [PubMed] [Google Scholar]
- 172.Dowling JJ et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain 135, 1115–1127, doi: 10.1093/brain/aws036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Durham WJ et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell 133, 53–65, doi: 10.1016/j.cell.2008.02.042 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Todd JJ et al. Randomized controlled trial of N-acetylcysteine therapy for RYR1-related myopathies. Neurology 94, e1434–e1444, doi: 10.1212/WNL.0000000000008872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kushnir A et al. Intracellular calcium leak as a therapeutic target for RYR1-related myopathies. Acta Neuropathol 139, 1089–1104, doi: 10.1007/s00401-020-02150-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Flucher BE Skeletal muscle CaV1.1 channelopathies. Pflugers Arch 472, 739–754, doi: 10.1007/s00424-020-02368-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Catterall WA Functional subunit structure of voltage-gated calcium channels. Science 253, 1499–1500, doi: 10.1126/science.1654596 (1991). [DOI] [PubMed] [Google Scholar]
- 178.Campbell KP, Leung AT & Sharp AH The biochemistry and molecular biology of the dihydropyridine-sensitive calcium channel. Trends Neurosci 11, 425–430, doi: 10.1016/0166-2236(88)90193-2 (1988). [DOI] [PubMed] [Google Scholar]
- 179.Monnier N, Procaccio V, Stieglitz P & Lunardi J Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet 60, 1316–1325, doi: 10.1086/515454 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fiszer D et al. Next-generation Sequencing of RYR1 and CACNA1S in Malignant Hyperthermia and Exertional Heat Illness. Anesthesiology 122, 1033–1046, doi: 10.1097/ALN.0000000000000610 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Matthews E et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 72, 1544–1547, doi: 10.1212/01.wnl.0000342387.65477.46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schartner V et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol 133, 517–533, doi: 10.1007/s00401-016-1656-8 (2017). [DOI] [PubMed] [Google Scholar]
- 183.Matthews E et al. Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology 77, 1960–1964, doi: 10.1212/WNL.0b013e31823a0cb6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Polster A, Nelson BR, Olson EN & Beam KG Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc Natl Acad Sci U S A 113, 10986–10991, doi: 10.1073/pnas.1612441113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Horstick EJ et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat Commun 4, 1952, doi: 10.1038/ncomms2952 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zaharieva IT et al. STAC3 variants cause a congenital myopathy with distinctive dysmorphic features and malignant hyperthermia susceptibility. Hum Mutat 39, 1980–1994, doi: 10.1002/humu.23635 (2018). [DOI] [PubMed] [Google Scholar]
- 187.Dowling JJ, Gibbs EM & Feldman EL Membrane traffic and muscle: lessons from human disease. Traffic 9, 1035–1043, doi: 10.1111/j.1600-0854.2008.00716.x (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dowling JJ et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet 5, e1000372, doi: 10.1371/journal.pgen.1000372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Al-Qusairi L & Laporte J T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle 1, 26, doi: 10.1186/2044-5040-1-26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Laporte J et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet 13, 175–182, doi: 10.1038/ng0696-175 (1996). [DOI] [PubMed] [Google Scholar]; • Discovery of a function for myotubularin.
- 191.Dowling JJ, Lawlor MW & Das S in GeneReviews((R)) (eds Adam MP et al. ) (1993). [Google Scholar]
- 192.Amburgey K et al. A natural history study of X-linked myotubular myopathy. Neurology 89, 1355–1364, doi: 10.1212/WNL.0000000000004415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Annoussamy M et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology 92, e1852–e1867, doi: 10.1212/WNL.0000000000007319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hnia K et al. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J Clin Invest 121, 70–85, doi: 10.1172/JCI44021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gavriilidis C et al. The MTM1-UBQLN2-HSP complex mediates degradation of misfolded intermediate filaments in skeletal muscle. Nat Cell Biol 20, 198–210, doi: 10.1038/s41556-017-0024-9 (2018). [DOI] [PubMed] [Google Scholar]
- 196.Tasfaout H, Cowling BS & Laporte J Centronuclear myopathies under attack: A plethora of therapeutic targets. J Neuromuscul Dis 5, 387–406, doi: 10.3233/JND-180309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Childers MK et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med 6, 220ra210, doi: 10.1126/scitranslmed.3007523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sabha N et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J Clin Invest 126, 3613–3625, doi: 10.1172/JCI86841 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Maani N et al. Tamoxifen therapy in a murine model of myotubular myopathy. Nat Commun 9, 4849, doi: 10.1038/s41467-018-07057-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tasfaout H et al. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat Commun 8, 15661, doi: 10.1038/ncomms15661 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wilson JM & Flotte TR Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum Gene Ther 31, 695–696, doi: 10.1089/hum.2020.182 (2020). [DOI] [PubMed] [Google Scholar]
- 202.Shieh PB et al. Re: “Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy” by Wilson and Flotte. Hum Gene Ther 31, 787, doi: 10.1089/hum.2020.217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.McNiven MA Dynamin: a molecular motor with pinchase action. Cell 94, 151–154, doi: 10.1016/s0092-8674(00)81414-2 (1998). [DOI] [PubMed] [Google Scholar]
- 204.Bitoun M et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet 37, 1207–1209, doi: 10.1038/ng1657 (2005). [DOI] [PubMed] [Google Scholar]
- 205.Zhao M, Maani N & Dowling JJ Dynamin 2 (DNM2) as Cause of, and Modifier for, Human Neuromuscular Disease. Neurotherapeutics 15, 966–975, doi: 10.1007/s13311-018-00686-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cowling BS et al. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J Clin Invest 127, 4477–4487, doi: 10.1172/JCI90542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Liu N et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J Clin Invest 121, 3258–3268, doi: 10.1172/JCI46267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Demonbreun AR & McNally EM Dynamin 2 the rescue for centronuclear myopathy. J Clin Invest 124, 976–978, doi: 10.1172/JCI74434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Buono S et al. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc Natl Acad Sci U S A 115, 11066–11071, doi: 10.1073/pnas.1808170115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lee E et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196, doi: 10.1126/science.1071362 (2002). [DOI] [PubMed] [Google Scholar]
- 211.Nicot AS et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet 39, 1134–1139, doi: 10.1038/ng2086 (2007). [DOI] [PubMed] [Google Scholar]
- 212.Agrawal PB et al. SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet 95, 218–226, doi: 10.1016/j.ajhg.2014.07.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Campbell HM et al. Loss of SPEG Inhibitory Phosphorylation of Ryanodine Receptor Type-2 Promotes Atrial Fibrillation. Circulation 142, 1159–1172, doi: 10.1161/CIRCULATIONAHA.120.045791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kusumoto D, Yuasa S & Fukuda K SPEG, an Indispensable Kinase of SERCA2a for Calcium Homeostasis. Circ Res 124, 668–670, doi: 10.1161/CIRCRESAHA.119.314678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Moghadaszadeh B et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet 29, 17–18, doi: 10.1038/ng713 (2001). [DOI] [PubMed] [Google Scholar]
- 216.Villar-Quiles RN et al. The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy: A case series. Neurology 95, e1512–e1527, doi: 10.1212/WNL.0000000000010327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Clarke NF et al. SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann Neurol 59, 546–552, doi: 10.1002/ana.20761 (2006). [DOI] [PubMed] [Google Scholar]
- 218.Ferreiro A et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol 55, 676–686, doi: 10.1002/ana.20077 (2004). [DOI] [PubMed] [Google Scholar]
- 219.Ferreiro A et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 71, 739–749, doi: 10.1086/342719 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Arbogast S et al. Oxidative stress in SEPN1-related myopathy: from pathophysiology to treatment. Ann Neurol 65, 677–686, doi: 10.1002/ana.21644 (2009). [DOI] [PubMed] [Google Scholar]
- 221.Lyfenko AD & Dirksen RT Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol 586, 4815–4824, doi: 10.1113/jphysiol.2008.160481 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Silva-Rojas R, Laporte J & Bohm J STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front Physiol 11, 604941, doi: 10.3389/fphys.2020.604941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bohm J & Laporte J Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 76, 1–9, doi: 10.1016/j.ceca.2018.07.008 (2018). [DOI] [PubMed] [Google Scholar]
- 224.Kramerova I, Kudryashova E, Tidball JG & Spencer MJ Null mutation of calpain 3 (p94) in mice causes abnormal sarcomere formation in vivo and in vitro. Hum Mol Genet 13, 1373–1388, doi: 10.1093/hmg/ddh153 (2004). [DOI] [PubMed] [Google Scholar]
- 225.Kramerova I et al. Failure to up-regulate transcription of genes necessary for muscle adaptation underlies limb girdle muscular dystrophy 2A (calpainopathy). Hum Mol Genet 25, 2194–2207, doi: 10.1093/hmg/ddw086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kramerova I et al. Impaired calcium calmodulin kinase signaling and muscle adaptation response in the absence of calpain 3. Hum Mol Genet 21, 3193–3204, doi: 10.1093/hmg/dds144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ono Y & Sorimachi H Calpains: an elaborate proteolytic system. Biochim Biophys Acta 1824, 224–236, doi: 10.1016/j.bbapap.2011.08.005 (2012). [DOI] [PubMed] [Google Scholar]
- 228.Ye Q, Campbell RL & Davies PL Structures of human calpain-3 protease core with and without bound inhibitor reveal mechanisms of calpain activation. J Biol Chem 293, 4056–4070, doi: 10.1074/jbc.RA117.001097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ermolova N, Kramerova I & Spencer MJ Autolytic activation of calpain 3 proteinase is facilitated by calmodulin protein. J Biol Chem 290, 996–1004, doi: 10.1074/jbc.M114.588780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Partha SK, Ravulapalli R, Allingham JS, Campbell RL & Davies PL Crystal structure of calpain-3 penta-EF-hand (PEF) domain - a homodimerized PEF family member with calcium bound at the fifth EF-hand. FEBS J 281, 3138–3149, doi: 10.1111/febs.12849 (2014). [DOI] [PubMed] [Google Scholar]
- 231.Sorimachi H et al. Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J Biol Chem 270, 31158–31162, doi: 10.1074/jbc.270.52.31158 (1995). [DOI] [PubMed] [Google Scholar]
- 232.Sorimachi H et al. Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J Biol Chem 268, 10593–10605 (1993). [PubMed] [Google Scholar]
- 233.Ono Y et al. Functional defects of a muscle-specific calpain, p94, caused by mutations associated with limb-girdle muscular dystrophy type 2A. J Biol Chem 273, 17073–17078 (1998). [DOI] [PubMed] [Google Scholar]
- 234.Ermolova N et al. Pathogenity of some limb girdle muscular dystrophy mutations can result from reduced anchorage to myofibrils and altered stability of calpain 3. Hum Mol Genet 20, 3331–3345, doi: 10.1093/hmg/ddr239 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Guyon JR et al. Calpain 3 cleaves filamin C and regulates its ability to interact with gamma- and delta-sarcoglycans. Muscle Nerve 28, 472–483, doi: 10.1002/mus.10465 (2003). [DOI] [PubMed] [Google Scholar]
- 236.Cohen N et al. Identification of putative in vivo substrates of calpain 3 by comparative proteomics of overexpressing transgenic and nontransgenic mice. Proteomics 6, 6075–6084, doi: 10.1002/pmic.200600199 (2006). [DOI] [PubMed] [Google Scholar]
- 237.Kramerova I, Kudryashova E, Venkatraman G & Spencer MJ Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet 16, 1006, doi: 10.1093/hmg/ddm044 (2007). [DOI] [PubMed] [Google Scholar]
- 238.Kramerova I et al. Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum Mol Genet 17, 3271–3280, doi: 10.1093/hmg/ddn223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.DiFranco M, Kramerova I, Vergara JL & Spencer MJ Attenuated Ca(2+) release in a mouse model of limb girdle muscular dystrophy 2A. Skelet Muscle 6, 11, doi: 10.1186/s13395-016-0081-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gonzalez-Mera L et al. Heterozygous CAPN3 missense variants causing autosomal-dominant calpainopathy in seven unrelated families. Neuropathol Appl Neurobiol, doi: 10.1111/nan.12663 (2020). [DOI] [PubMed] [Google Scholar]
- 241.Kramerova I et al. Mitochondrial abnormalities, energy deficit and oxidative stress are features of calpain 3 deficiency in skeletal muscle. Hum Mol Genet 18, 3194–3205, doi: 10.1093/hmg/ddp257 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.El-Khoury R et al. Divergent Features of Mitochondrial Deficiencies in LGMD2A Associated With Novel Calpain-3 Mutations. J Neuropathol Exp Neurol 78, 88–98, doi: 10.1093/jnen/nly113 (2019). [DOI] [PubMed] [Google Scholar]
- 243.Beckmann JS et al. Identification of muscle-specific calpain and beta-sarcoglycan genes in progressive autosomal recessive muscular dystrophies. Neuromuscul Disord 6, 455–462, doi: 10.1016/s0960-8966(96)00386-0 (1996). [DOI] [PubMed] [Google Scholar]; • First demonstration of an enzyme mutation linked to LGMD
- 244.Beckmann JS et al. A gene for limb-girdle muscular dystrophy maps to chromosome 15 by linkage. C R Acad Sci III 312, 141–148 (1991). [PubMed] [Google Scholar]
- 245.Richard I et al. Calpainopathy-a survey of mutations and polymorphisms. Am J Hum Genet 64, 1524–1540, doi: 10.1086/302426 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cannon SC Sodium Channelopathies of Skeletal Muscle. Handb Exp Pharmacol 246, 309–330, doi: 10.1007/164_2017_52 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cannon SC Channelopathies of skeletal muscle excitability. Compr Physiol 5, 761–790, doi: 10.1002/cphy.c140062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Demonbreun AR et al. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol 213, 705–718, doi: 10.1083/jcb.201512022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Bittel DC et al. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 9, doi: 10.3390/cells9091919 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Croissant C, Gounou C, Bouvet F, Tan S & Bouter A Annexin-A6 in Membrane Repair of Human Skeletal Muscle Cell: A Role in the Cap Subdomain. Cells 9, doi: 10.3390/cells9071742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fischer T, Lu L, Haigler HT & Langen R Annexin B12 is a sensor of membrane curvature and undergoes major curvature-dependent structural changes. J Biol Chem 282, 9996–10004, doi: 10.1074/jbc.M611180200 (2007). [DOI] [PubMed] [Google Scholar]
- 252.Mellgren RL et al. Calcium-dependent plasma membrane repair requires m- or mu-calpain, but not calpain-3, the proteasome, or caspases. Biochim Biophys Acta 1793, 1886–1893, doi: 10.1016/j.bbamcr.2009.09.013 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lek A et al. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J Neurosci 33, 5085–5094, doi: 10.1523/JNEUROSCI.3560-12.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Sudhof TC & Rizo J Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron 17, 379–388, doi: 10.1016/s0896-6273(00)80171-3 (1996). [DOI] [PubMed] [Google Scholar]
- 255.Cai C et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol 11, 56–64, doi: 10.1038/ncb1812 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Weisleder N, Takeshima H & Ma J Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Commun Integr Biol 2, 225–226, doi: 10.4161/cib.2.3.8077 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cai C et al. MG53 regulates membrane budding and exocytosis in muscle cells. J Biol Chem 284, 3314–3322, doi: 10.1074/jbc.M808866200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.McDade JR, Archambeau A & Michele DE Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB J 28, 3660–3670, doi: 10.1096/fj.14-250191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.McDade JR & Michele DE Membrane damage-induced vesicle-vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum Mol Genet 23, 1677–1686, doi: 10.1093/hmg/ddt557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.McDade JR, Naylor MT & Michele DE Sarcolemma wounding activates dynamin-dependent endocytosis in striated muscle. FEBS J 288, 160–174, doi: 10.1111/febs.15556 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Weiler T et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum Mol Genet 8, 871–877, doi: 10.1093/hmg/8.5.871 (1999). [DOI] [PubMed] [Google Scholar]
- 262.Liu J et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 20, 31–36, doi: 10.1038/1682 (1998). [DOI] [PubMed] [Google Scholar]
- 263.Bansal D & Campbell KP Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol 14, 206–213, doi: 10.1016/j.tcb.2004.03.001 (2004). [DOI] [PubMed] [Google Scholar]; • Mechanistic demonstration of a role for dysferlin in membrane repair.
- 264.Hicks D et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 134, 171–182, doi: 10.1093/brain/awq294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lostal W et al. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum Mol Genet 19, 1897–1907, doi: 10.1093/hmg/ddq065 (2010). [DOI] [PubMed] [Google Scholar]
- 266.Kesari A et al. Dysferlin deficiency shows compensatory induction of Rab27A/Slp2a that may contribute to inflammatory onset. Am J Pathol 173, 1476–1487, doi: 10.2353/ajpath.2008.080098 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Nagaraju K et al. Dysferlin deficiency enhances monocyte phagocytosis: a model for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am J Pathol 172, 774–785, doi: 10.2353/ajpath.2008.070327 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Rawat R et al. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am J Pathol 176, 2891–2900, doi: 10.2353/ajpath.2010.090058 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Dillingham BC et al. Inhibition of inflammation with celastrol fails to improve muscle function in dysferlin-deficient A/J mice. J Neurol Sci 356, 157–162, doi: 10.1016/j.jns.2015.06.042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yin X et al. CD4+ cells, macrophages, MHC-I and C5b-9 involve the pathogenesis of dysferlinopathy. Int J Clin Exp Pathol 8, 3069–3075 (2015). [PMC free article] [PubMed] [Google Scholar]
- 271.Quattrocelli M et al. Intermittent Glucocorticoid Dosing Improves Muscle Repair and Function in Mice with Limb-Girdle Muscular Dystrophy. Am J Pathol 187, 2520–2535, doi: 10.1016/j.ajpath.2017.07.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Quattrocelli M et al. Pulsed glucocorticoids enhance dystrophic muscle performance through epigenetic-metabolic reprogramming. JCI Insight 4, doi: 10.1172/jci.insight.132402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Selcen D, Stilling G & Engel AG The earliest pathologic alterations in dysferlinopathy. Neurology 56, 1472–1481, doi: 10.1212/wnl.56.11.1472 (2001). [DOI] [PubMed] [Google Scholar]
- 274.Han R et al. Genetic ablation of complement C3 attenuates muscle pathology in dysferlin-deficient mice. J Clin Invest 120, 4366–4374, doi: 10.1172/JCI42390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Lerario A et al. Effects of rituximab in two patients with dysferlin-deficient muscular dystrophy. BMC Musculoskelet Disord 11, 157, doi: 10.1186/1471-2474-11-157 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Nishino I et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910, doi: 10.1038/35022604 (2000). [DOI] [PubMed] [Google Scholar]
- 277.Cullup T et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 45, 83–87, doi: 10.1038/ng.2497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ramachandran N et al. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol 125, 439–457, doi: 10.1007/s00401-012-1073-6 (2013). [DOI] [PubMed] [Google Scholar]
- 279.Martiniuk F, Bodkin M, Tzall S & Hirschhorn R Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower “affinity” for glycogen (GAA 2) and transient gene expression in deficient cells. Am J Hum Genet 47, 440–445 (1990). [PMC free article] [PubMed] [Google Scholar]
- 280.Sugie K et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J Neuropathol Exp Neurol 64, 513–522, doi: 10.1093/jnen/64.6.513 (2005). [DOI] [PubMed] [Google Scholar]
- 281.Bodine SC et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708, doi: 10.1126/science.1065874 (2001). [DOI] [PubMed] [Google Scholar]
- 282.Cohen S, Zhai B, Gygi SP & Goldberg AL Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J Cell Biol 198, 575–589, doi: 10.1083/jcb.201110067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kudryashova E, Wu J, Havton LA & Spencer MJ Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet 18, 1353–1367, doi: 10.1093/hmg/ddp036 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Nicklas S et al. TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS One 7, e30445, doi: 10.1371/journal.pone.0030445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kudryashova E, Kramerova I & Spencer MJ Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J Clin Invest 122, 1764–1776, doi: 10.1172/JCI59581 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Mokhonova EI et al. The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum Mol Genet 24, 2873–2883, doi: 10.1093/hmg/ddv049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gupta VA & Beggs AH Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet Muscle 4, 11, doi: 10.1186/2044-5040-4-11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Selcen D et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol 65, 83–89, doi: 10.1002/ana.21553 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Sarparanta J et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 44, 450–455, S451–452, doi: 10.1038/ng.1103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Harms MB et al. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol 71, 407–416, doi: 10.1002/ana.22683 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Vicart P et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 20, 92–95, doi: 10.1038/1765 (1998). [DOI] [PubMed] [Google Scholar]
- 292.Ghaoui R et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 86, 391–398, doi: 10.1212/WNL.0000000000002324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Anttonen AK et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet 37, 1309–1311, doi: 10.1038/ng1677 (2005). [DOI] [PubMed] [Google Scholar]
- 294.Senderek J et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet 37, 1312–1314, doi: 10.1038/ng1678 (2005). [DOI] [PubMed] [Google Scholar]