

Abstract
Genetic diseases cause numerous complex and intractable pathologies. DNA sequences encoding each human’s complexity and many disease risks are contained in the mitochondrial genome, nuclear genome, and microbial metagenome. Diagnosis of these diseases has unified around applications of next-generation DNA sequencing. However, translating specific genetic diagnoses into targeted genetic therapies remains a central goal. To date, genetic therapies have fallen into three broad categories: bulk replacement of affected genetic compartments with a new exogenous genome, nontargeted addition of exogenous genetic material to compensate for genetic errors, and most recently, direct correction of causative genetic alterations using gene editing. Generalized methods of diagnosis, therapy, and reagent delivery into each genetic compartment will accelerate the next generations of curative genetic therapies. We discuss the structure and variability of the mitochondrial, nuclear, and microbial metagenomic compartments, as well as the historical development and current practice of genetic diagnostics and gene therapies targeting each compartment.
Keywords: genetic disease, gene therapies, gene editing, genetic diagnostics, clinical genetics
INTRODUCTION
Early History of Genetic Disease and Therapy
Subtle changes to the genetic code can result in profoundly debilitating and diverse pathologies. The hereditary nature of human traits has been described since classical times. In the history of modern medicine, the first known genetic disorder, alkaptonuria, was described at the turn of the twentieth century, giving rise to the recognition of inborn errors of metabolism (1). Diseases with their basis in mutations and alterations of the human genetic code represent a massive burden, and recognized genetic disorders affect more than 5% of live births and more than two-thirds of miscarriages (2, 3). Beyond highly penetrant monogenic disorders and large-scale chromosomal alterations, the heritability of many common diseases has long suggested a genetic basis for more prevalent disorders such as cardiovascular disease (4). The prospect of passing genetic afflictions on to the next generation adds to the fear of these disorders.
The first heritable alteration in a protein linked to disease was identified in sickle cell anemia in the late 1940s, with the discovery of altered shifts during electrophoresis, a change that corresponded with disease status among tested patients (5, 6). Subsequently, once the DNA code for amino acids was deciphered, scientists recognized the potential for alterations in DNA to cause alterations in enzymes and thus disease. Prior to the advent of DNA sequencing, the cause of Down syndrome, identified in 1959 as the chromosomal abnormality trisomy 21, was the first human genetic alteration found to be associated with disease (7). Beginning in the 1960s, hereditary metabolic disorders such as phenylketonuria could be screened for biochemically without the need to know the causative gene’s location or sequence (8). The advent of Sanger sequencing and recombinant molecular biology in the 1970s and 1980s made the determination of DNA sequences widely accessible for the first time. The following decades, prior to the completion of the Human Genome Project in 2003, saw gene mapping consortia undergo herculean efforts to discover the causative genes in some of the most debilitating diseases, including the first mapped human genetic disorder, Huntington’s disease, in 1983 (9). With the diminishing costs of exome and whole-genome sequencing over the past 2 decades, genetic diagnosis has become increasingly feasible, even for conditions that were not previously recognized as genetic diseases.
Human Genomic Compartments
The genetic material in an adult human can be divided into compartments that differ in size, heritability, and diversity. The mitochondrial genome, the smallest (only 16.5 kb) but by far the most abundant, is inherited maternally and varies little among the human population. The traditional human genome contained in the nucleus is significantly larger and harbors mutations that cause the majority of traditional genetic diseases. The nuclear and mitochondrial genomes are determined at conception, although somatic mutations can drive mosaic disorders, cancer, and even aging.
More broadly, the adaptive immune receptor repertoire, which is a distinctive subset of the nuclear genome, and the microbial metagenome are determined only after conception, and their genetic complexity, at least as measured by the diversity of unique protein coding sequences, dwarfs that of the rest of the nuclear genome. The T and B cells of the adaptive immune system undergo somatic recombination, generating orders of magnitude more unique protein products than are possessed by the other genes of the nuclear genome, and together constitute the adaptive immune receptor repertoire. Finally, the nonhuman cells of the microbiome make up potentially the most dynamic and diverse genomic compartment of all, with a litany of different species, predominantly bacterial and viral, occupying structured niches across human skin, sexual organs, and gastrointestinal and respiratory tracts. Both the adaptive immune receptor repertoire and the microbial metagenome vary significantly more among individuals, and increasing research aims to understand how genetic alterations in immune receptor repertoires and the microbiome contribute to disease pathology.
Modes of Genetic Therapy
Disruptions in any of these compartments, interacting in many cases with environmental factors, can contribute to different classes of genetic disease. The simplicity of the DNA code has enticed researchers and clinicians since the 1960s with the curative promise of correction of genetic alterations, or gene therapy (10). Since the first successful ectopic expression of a foreign gene in human cells in the early 1970s (11), successive generations of gene therapy technology have increased the efficiency, specificity, and safety of gene transfers. These advances led to the first human gene therapy trials at the National Cancer Institute, National Institutes of Health, in the late 1980s (12), following a handful of unregulated and ultimately unpublished gene therapy attempts earlier that decade (13).
The rapid proliferation of gene therapies in the 1990s, with more than 500 trials initiated, came to a halt in the early 2000s following patient deaths in clinical trials for severe combined immunodeficiency (SCID) and a metabolic liver disorder. Further improvements in the safety and efficacy of gene transfer in the 2000s and 2010s ultimately led to the resurgence of new generations of gene therapy approaches and approval by the US Food and Drug Administration (FDA) of the first ex vivo chimeric antigen receptor T cell (CAR T)–based gene therapies in B cell malignancies in 2017 (14, 15), as well as the first in vivo gene therapy for vision loss caused by Leber’s congenital amaurosis in 2017 (16).
In a broad context, genetic mutations can be altered in three general modes: bulk replacement or selection of the entire genomic compartment containing the mutation, nontargeted insertion into the genome of additional genetic material restoring enough functionality to compensate for the genetic defect (nontargeted addition), or specific correction of only the causative mutation or genetic alteration (gene editing). Bulk replacement represents the most basic form of gene therapy (Figure 1). Selective reproduction and, more recently, preimplantation diagnostics offer the chance to preemptively avoid germline mutations in the nuclear genome. Mitochondrial replacement therapy followed by in vitro fertilization can effectively correct mitochondrial mutations by replacing a child’s entire mitochondrial genome with a “third parent.” Similarly, somatic mutations resulting in tumors can be removed in bulk from the body by surgery. Portions of the microbial metagenome are increasingly being therapeutically altered via fecal or microbial community transplants (17).
Figure 1.
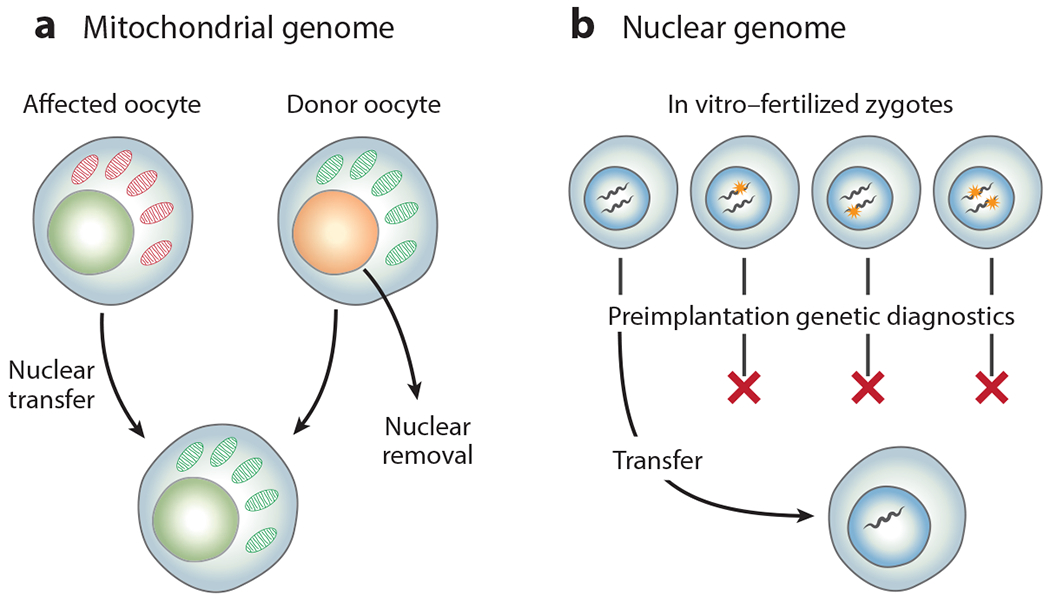
Gene therapies based on bulk replacement or selection of genetic compartments. (a) The mitochondrial genome of an affected mother’s oocyte can be replaced through transfer of its nucleus into a donor mother’s oocyte, which contains mitochondria unaffected by the mutation. (b) The nuclear genome can be selected through preimplantation diagnosis of in vitro–fertilized zygotes.
In many patients, however, a more practicable gene therapy is the nontargeted introduction of new genetic material to make up for the lost or deleterious function of mutated sequences (Figure 2). This nontargeted addition of genetic material is common in the germline transgenesis of model organisms, but important ethical concerns have, appropriately, prevented additive gene therapies in the human germline. In the more therapeutically relevant somatic cells, successive generations of viral vectors, ranging from SV40 to retroviruses, adenoviruses, and now, prominently, adeno-associated virus (AAV) pseudotypes, have enabled ever greater control over the in vivo cell types receiving new, corrective genetic material, culminating in the recent FDA approval of an AAV2 vector specific for rod and cone photoreceptors (16). A separate line of technical development based on γ-retroviruses and lentiviruses has led to efficient ex vivo manipulation of the nuclear genome of hematopoietic stem cells (HSCs), as well as the adaptive repertoire of T cells, resulting in the similarly recently approved CAR T–based therapies (14, 15).
Figure 2.
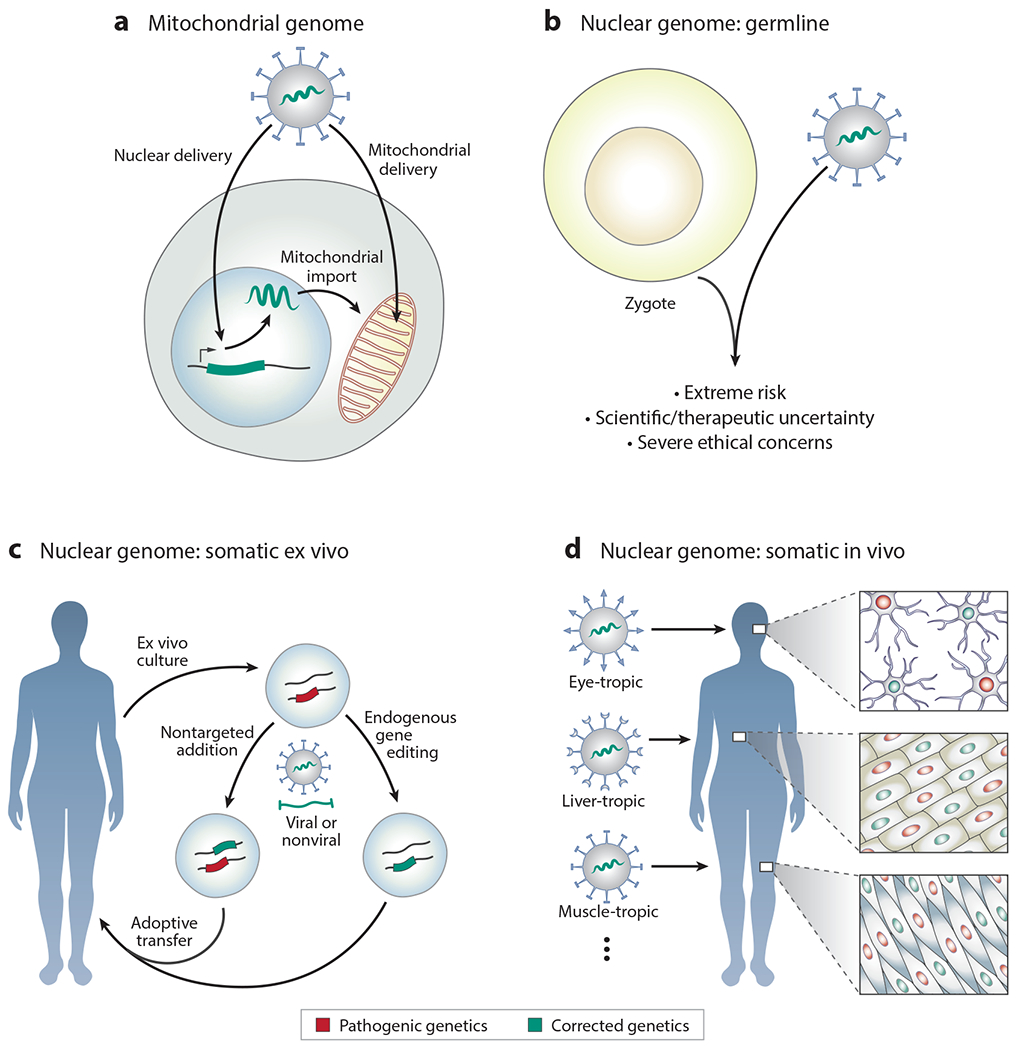
Gene therapies based on nontargeted genetic addition or targeted gene editing. (a) Mutated genes in the mitochondrial genome can be integrated into the nuclear genome, with their protein products targeted for import into the mitochondria. Direct delivery of genetic material to the mitochondrial genome poses a greater challenge. (b) Adding or editing genetic material in the nuclear genome of the human germline poses significant ethical concerns. (c) Nontargeted addition or targeted editing in somatic cells, such as cells cultured ex vivo (e.g., hematopoietic stem cells and T cells). (d) Nontargeted addition or targeted editing in somatic cells in vivo, as in retinal cells, hepatocytes, or myocytes, critically depends on delivery platforms to carry DNA, RNA, and/or protein cargos to the cell type of interest.
However, the history of gene therapies for hemoglobinopathies, the first genetic diseases to be molecularly characterized, reveals the limitations of nontargeted genetic addition. The careful regulatory control of α- and β-hemoglobin during erythropoiesis prevented erythrocyte formation during early trials in patients with sickle cell anemia and other hemoglobinopathies when correct copies of hemoglobin were pseudorandomly added to the genome of their HSCs. The rapid recent development of targetable RNA-guided nucleases such as CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/caspase-9), building upon earlier zinc-finger nuclease (ZFN) and transcription activator–like effector nuclease (TALEN) targetable nucleases, offers a simplified approach to treating such genetic diseases (18). By creating a double-stranded DNA break near the site of a mutation, these nucleases can prompt the cell to fix the damage via templated repair based on a separately provided DNA template containing the desired sequence. Diverse delivery technologies can ferry DNA encoding the ribonucleoprotein (RNA/protein) nuclease complexes (RNPs), or recombinant RNPs themselves, as well as DNA templates to correct specific mutations into target cell populations both ex vivo and increasingly in vivo. These gene editing technologies promise a generalizable ability to correct almost any mutation in the genome (Figure 2).
Genetic disease has haunted families and their clinicians for generations. The unique properties of each human genomic compartment present distinct diagnostic and therapeutic challenges. Significant progress has been made in technologies to sequence DNA, deliver protein and DNA payloads to specific human cell populations ex vivo and in vivo, insert large stretches of new genetic material, and now even directly edit endogenous genetic loci. A growing suite of diagnostic and therapeutic options can target all of the human genomic compartments, and their further development offers the hope of generalized curative genetic therapies (Figure 3).
Figure 3.
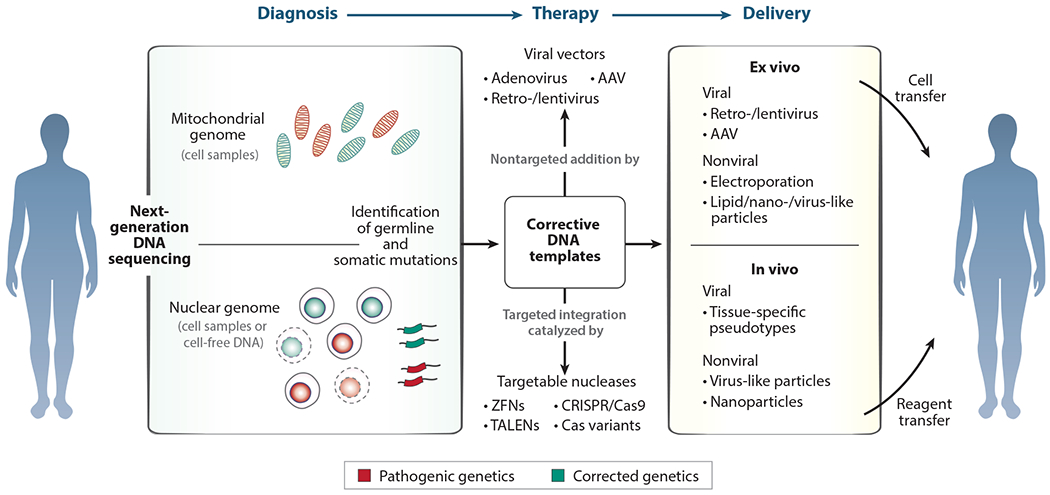
Modular systems for genetic diagnosis and therapy. Gene therapies in all four human genetic compartments depend on the modular process of diagnosis, therapeutic design, and compartment-specific delivery of therapeutic reagents. Diagnosis of genetic disease is now centered around next-generation DNA sequencing to detect errors in the mitochondrial and nuclear genomes. Therapy can be based on bulk replacement or selection of the genetic compartment or the therapeutic nontargeted addition of new genetic material or targeted correction of causative mutations through gene editing. Delivery platforms targeting each genomic compartment in somatic cells, whether in vivo or ex vivo, can carry gene addition and editing reagents with distinct therapeutic sequences or specificities depending on the genetic diagnosis. Abbreviations: AAV, adeno-associated virus; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats/caspase-9; TALEN, transcription activator–like effector nuclease; ZFN, zinc-finger nuclease.
MITOCHONDRIAL GENOME
The mitochondrial genome represents both the smallest and the most abundant set of genetic information in the human body. With a conservative estimate of more than a quadrillion copies present within an adult human, different human tissues possess between zero and thousands of mitochondria per cell, and each mitochondrion averages one to two copies of the 16.5-kb mitochondrial genome (19, 20). All mitochondria in the body are generated by repetitive cycles of fission, derived from fewer than 10 mitochondria found in primordial egg cells at the greatest bottleneck during oogenesis (21, 22), and are ultimately the progeny of an unbroken cycle of asexual reproduction going back to the first eukaryotic mitochondrial symbiote. This previously free-living ancestral symbiotic bacterium likely possessed thousands of genes (23). Over hundreds of millions of years, however, mitochondrial genes successively migrated to the relative safety of the nuclear genome through endosymbiotic gene transfer. The 37 mitochondrial genes remaining encode 22 transfer RNAs, 2 mitochondrial ribosomal RNAs, and 13 protein coding genes, which encode primarily quantumly linked members of the electron transport chain. These genes make up more than 90% of the genome, with the noncoding elements of the mitochondrial control region making up the remainder (24).
With its central importance for cellular energy conversion, the mitochondrial genome is highly conserved among humans, although it diverges widely in size and gene content across eukaryotes in general (25). Purifying selection drives the conservation of mitochondrial genomic sequences, as the error rate for mitochondrial DNA replication is approximately 100 times higher than for nuclear replication (26). Larger genetic alterations such as insertions/deletions (indels) and rearrangements are almost invariably nonviable, given the mitochondrial genome’s compactness. Mitochondria undergo strong purifying selection during oogenesis, and approximately one in five children possess de novo mitochondrial mutations, predominantly synonymous mutations (22). In somatic cells, continuous cycles of fission and fusion among mitochondria may enable continuous purifying selection for nonmutated copies of the mitochondrial genome. Extensive division of mitochondria throughout life ultimately leads to genetic changes, and somatic mosaicism within adults’ mitochondria is readily detectable. The mitochondria in an aged adult possess hundreds more genetic changes relative to the adult’s inherited genome (27, 28).
Mitochondrial Genetic Disease
The critical and ubiquitous nature of mitochondrial function ensures that mutations within the mitochondrial genome can have large and deleterious effects on health. While the majority of mitochondrial mutations likely result in nonviable oocytes and are selected against prior to ovulation, mitochondrial disorders such as myopathies and neuropathies are diagnosed in approximately 1 in 5,000 live births (29). Germline mutations in every protein coding gene in the mitochondrial genome are linked to clinical disorders. More speculatively, accumulation of somatic mitochondrial mutations have been implicated in various diseases of aging (30).
The mitochondrial genome was the first human genetic compartment to be completely sequenced, with the entire genome determined by Sanger sequencing announced in 1981 (24). Today, whole–mitochondrial genome sequencing can be rapidly performed by next-generation sequencing, although mitochondrial sequencing is not a common component of newborn screening programs. Recently, the genetic mosaicism shown by mitochondria in adults has even enabled lineage tracing of human clonal cell populations, which may be useful in the diagnosis of age-related disorders (31).
Mitochondrial Gene Therapies
The unique inheritance of mitochondria, the onset of phenotypes quickly after fertilization, their copious numbers per cell, and the physical and chemical barriers surrounding them make gene therapies targeting the mitochondrial genome particularly challenging (32). Similarly, the compactness of mitochondrial genes makes strategies based on nontargeted insertion of new genetic material (e.g., using viral vectors) into the mitochondrial genome impractical. The centrality of mitochondrial gene function means that any genetic therapy likely must correct the majority of the cells in the body, favoring germline correction.
The small size and extremely conserved sequence of the human mitochondrial genome allow for bulk replacement of an oocyte’s mitochondria containing a known genetic disorder with a donor’s wild-type mitochondria, leading to what is popularly known as a three-parent baby (Figure 1a). This bulk mitochondrial genomic replacement actually occurs in reverse: The nuclear genome is removed from a donor’s oocyte and replaced with the nucleus from one of the intended mother’s oocytes, followed by in vitro fertilization (33). This germline genetic therapy was approved in the United Kingdom in 2016 for inherited mitochondrial disorders and results in heritable germline correction in the resulting children.
For patients born with de novo mitochondrial mutations, however, germline bulk mitochondrial replacement is only an option for their own potential children. The correction of mitochondrial mutations in somatic cells, especially in sufficient cells and tissues to be clinically effective, presents a significant challenge (Figure 2a). In some cases, mitochondrial genes can be integrated into the nuclear genome by use of nontargeted viral vectors, a process mirroring the evolutionary nuclear movement of mitochondrial genes. For example, in Leber’s hereditary optic neuropathy (LHON), a mitochondrial disorder resulting in acute vision loss in young adulthood caused by mutations in NADH ubiquinone oxidoreductase subunits (including ND4), nuclear insertion of corrected copies of the ND4 gene resulted in vision improvements in some human patients (34). Nontargeted addition of genetic material directly into mitochondria, although not the mitochondrial genome directly, has also been demonstrated in LHON through the use of a modified AAV capsid engineered to contain an endogenous mitochondrial targeting sequence (35). Viral injection of mitochondrial targeting sequence–modified AAVs containing corrected copies of ND4, the causative gene resulting in acute vision loss in LHON, into the eyes of affected mice similarly resulted in vision improvements.
Targetable nucleases such as CRISPR/Cas9 could enable direct gene editing of the mitochondrial genome, although it is unclear whether microhomology-mediated or homology-directed repair (HDR) is common in mitochondria (36, 37). More directly, when a mutation affects only a portion of mitochondria in a cell, targeted cutting and linearization of affected mitochondrial genome can result in relative loss of mutated compared with healthy mitochondria, as has been demonstrated in vitro using TALENs (38). Overall, however, mitochondrial gene therapies face great challenges in efficient in vivo delivery of genetic editing reagents into the mitochondrial matrix in enough somatic cells to achieve clinical benefit.
The mitochondrial genome contains the living relics of the fundamental symbiotic event at the dawn of eukaryotic life, ubiquitous in multiple copies in all cells of the body, with the exception of mature red blood cells. Its remaining genes play central roles in cellular energy transfers, and the rare mutations within them, affecting only about 0.02% of live births, lead to debilitating disorders. Germline genetic therapy involving the bulk replacement of an affected oocyte’s mitochondria could be an effective cure for mitochondrial genetic diseases, albeit only in subsequent generations. Somatic gene therapies targeting the mitochondrial genome are in early development, but they face great physical and biologic hurdles. In contrast, germline editing of nuclear genes is biologically and ethically much more complex, while somatic gene therapies of the nuclear genome have rapidly proliferated.
NUCLEAR GENOME
The classical human, or nuclear, genome has significantly greater size and complexity than the smaller mitochondrial genome and presents unique challenges for diagnosis and genetic therapies. The human nuclear genome contains approximately three billion base pairs, divided into two copies each of 22 autosomal chromosomes and either XX or XY sex chromosomes, one set inherited from each parent. Only ~2% of the genome encodes directly for 1 of the approximately 20,000 protein coding genes, although noncoding elements, structural elements such as centromeres and telomeres, regulatory elements such as promoters and enhancers, and functional RNA elements such as microRNAs and long noncoding RNAs make up significant portions of non–protein coding genomic space (39). Furthermore, repetitive and selfish genetic elements such as triplet repeats, short interspersed nuclear elements (SINEs), and long interspersed nuclear elements (LINEs) make up more than half of human genomic content (40). Each of the approximately 30 trillion cells in the adult body possesses a single diploid copy of the nuclear genome, with the exception of anuclear red blood cells, certain multinucleated myocytes and osteoclasts, and haploid gametes. On average, two unrelated humans’ genomes differ by approximately 0.1%, with four-fifths of this difference due to individual variability and the remaining fifth due to differences between human population groupings (41).
Germline Genetic Disease
The human genome has evolved at a rate of between 5 and 10 genetic changes per year (42, 43). Human DNA polymerases combined with proofreading enzymes have an overall error rate of approximately 1 bp per 10 billion bp replicated, which can accumulate over the approximately 22 rounds of division an oocyte undergoes before fertilization as well as the hundreds of rounds of division, varying with paternal age, that a sperm cell undergoes (44–46). Small mutations and indels are not the only category of germline genetic changes, and errors in DNA replication and cell division can lead to repeat expansions, large deletions, chromosomal translocations, and autosomal and sex chromosome aneuploidy. However, extensive selective pressures are applied during gametogenesis and following fertilization, an estimated 10–40% of fertilized embryos do not implant, and overall 40–60% of fertilized pregnancies do not result in live birth (47). For example, out of 22 autosomal chromosomes, only aneuploidy of chromosome 21, resulting in Down syndrome, is compatible with prolonged life, whereas aneuploidy of any other autosome is fatal during embryogenesis or soon following birth. Overall, each human newborn contains an average of 10–20 maternally derived and 25–75 paternally derived de novo mutations in a newly fertilized embryo, which have the potential to occur in functional protein coding or noncoding sequences (48, 49). Beyond de novo mutations, though, each newborn possesses inherited germline loss-of-function monoallelic mutations in approximately 100 genes, and even loss-of-function biallelic mutations in an estimated 20 genes (50).
Genetic diseases that manifest after birth often affect the nervous, immune, and metabolic systems, which are largely not under selective pressure in the supportive in utero environment. Out of 20,000 protein coding genes, mutations in more than 4,000 have been identified as causative of specific human genetic diseases (51). Approximately 3,000 protein coding genes are essential in human cell lines, and loss-of-function mutations in these genes are likely incompatible with gametogenesis or early embryonic development (52). Estimates of human population size and genetic diversity predict that all possible single-base-pair changes in the protein coding nuclear genome compatible with life are already present in the global human population (53). The complete classification of genotype-phenotype relations in single-protein genetic diseases is a possible, if long-term, goal.
However, the nuclear genome brims with complexity beyond its protein products, and increasingly mutations in noncoding regulatory and functional RNA elements have also been identified as causative for genetic disorders (54). Furthermore, genome-wide association studies (GWASs) conducted in the late 2000s and 2010s have associated numerous inherited genetic alterations with common diseases, although a hallmark finding of GWASs has been the relatively small contribution of each common inherited genetic alteration to the risk of common diseases (55).
Before next-generation sequencing along with the reference human genome became available in the 2000s, careful gene mapping of restriction fragment length polymorphisms (RFLPs) and other traceable areas of genetic variability led to the identification of causative genetic changes in diseases such as Huntington’s disease and muscular dystrophy (9, 56), and even to RFLP-based diagnostic tests for disorders such as sickle cell anemia and thalassemia (57). Today, germline genetic diagnostics are clinically performed through a variety of assays. Common genetic disorders resulting in biochemical deficits can often be diagnosed chemically without sequencing, such as in newborn screening for phenylketonuria and galactosemia. Traditional karyotypes can diagnose large-scale chromosomal abnormalities, and single-nucleotide polymorphism microarrays can detect smaller-scale (but still many-kilobase) deletions (58). Targeted sequencing panels, performed either through exome sequencing with confirmatory Sanger sequencing or by Sanger sequencing directly, are the mainstay diagnostic for genetic diseases with consistent clinical phenotypes (54).
In patients without a clear previously described genetic syndrome, diagnostic whole-exome and, increasingly, whole-genome sequencing of the affected individual and both parents can reveal a causative genetic change in as many as 40% of patients (59). Large-scale chromosomal abnormalities can be diagnosed prior to birth from placental tissue or amniotic fluid, and these samples can also be used for DNA sequencing. Circulating fetal DNA in the mother’s blood offers a less invasive genetic diagnostic modality in individuals with clinical or hereditary suspicion of genetic disease (60). Advances in assisted reproductive technologies even enable genetic sequencing and diagnostics by removing a minimal number of cells during the earliest phases of cell division after in vitro fertilization (61). These preimplantation diagnostics, along with more traditional carrier testing of parents, can accurately diagnose the presence or risk of specific nuclear genetic diseases.
Genetic disease of the germline in many cases is diagnosable, even at the earliest stages of life. Therapeutic correction of these errors in information content can be performed within the germline or later in life in affected somatic tissues. In each case, gene therapy strategies can be divided into three categories: bulk replacement of the entire affected nuclear genome, even though only one base pair out of three billion may be disease causing; nontargeted addition of exogenous genetic material to compensate for the genetic error; and direct correction of the causative mutation by gene editing.
Germline Genetic Therapies
In the broadest sense, the affected nuclear genome of a patient, or future patient, with a germline genetic disease can be addressed preemptively through reproductive decision-making. In communities with known high carrier rates for a specific genetic disease, such as Ashkenazi Jewish communities carrying mutations in HEXA, the cause of Tay–Sachs disease, effective community-based screening and reproductive counseling have reduced the rate of children affected by Tay–Sachs to essentially zero in screened populations (62). Similarly, reproductive decision-making based on genetic diagnostics has enabled individual couples to prevent passing on genetic disorders through in vitro fertilization with sperm or egg donors, as well as through traditional adoption. All of these are, in a sense, bulk nuclear genome strategies to address germline genetic mutations (Figure 1b).
Direct addition of new genetic material to the human germline, or gene editing of endogenous germline sequences, is an ethically momentous step, and as of 2020 the scientific, medical, ethical, religious, and government communities broadly agree that it is inappropriate (Figure 2b) (63, 64). Appropriate scientific outrage at the first publicly announced human germline gene editing attempts in 2018 (65) highlights the need for significant government and scientific oversight and regulation.
Somatic Genetic Disease
Mutations present at fertilization are inherited by every cell in the body, but genetic copying errors continue in each cycle of somatic cell division and continuously accumulate as a result of environmental factors such as ultraviolet light, radiation, and mutagen exposures. The varying accumulation of genetic errors in the final somatic cells of different tissues is emphasized by the linear correlation between the number of cell divisions a mature somatic cell type has undergone and that cell type’s propensity to develop into tumors (66). Cell types such as intestinal epithelial cells, whose stem cells can undergo hundreds of rounds of division (67), are much more likely to cause tumors than are low-division-number osteoblasts or neurons (66). Because mutations in one cell are inherited by all future somatic cells derived from it, changes during early phases of embryogenesis can result in large sections of tissue, and even entire organs, possessing sometimes deleterious mutations. Ultimately, two different somatic cells in an adult human can differ by thousands of base pairs due to this somatic mosaicism (68–70).
Somatic mutations during early development can cause many of the same genetic disorders as germline mutations, with severity depending on the degree of mosaicism and the end organs affected. For example, in ornithine transcarbamylase deficiency caused by X-linked recessive mutations in the OTC gene, patients with germline mutations rarely survive childhood without a liver transplant, but patients with somatic mutations, even those affecting substantial numbers of the affected cell type, hepatocytes, can live relatively normal lives with dietary modifications (71). Somatic mutations undergo many of the same selective pressures as the germline, with strong evidence of selection against missense mutations (70). However, somatic mutations that increase the rate of cellular division, especially in less differentiated stem populations, can proceed along well-defined mutational paths toward oncogenic transformations. In the case of colon cancers, the acquisition of early driver mutations in APC is followed by increased proliferation, mutations in KRAS, and finally mutations in p53, which in turn further decrease DNA copying fidelity and unleash a cascade of DNA changes that ultimately lead to cancer (72). More broadly, accumulation of somatic mutations in normal tissues throughout life may drive age-related declines in function in addition to increasing cancer risk (73, 74).
Advances in DNA sequencing throughput, especially for high-coverage whole exomes and genomes, have similarly enhanced the ability to diagnose oncogenic and other deleterious genetic changes in somatic cells. Since the 1980s, when somatic mutations in tumor suppressor genes such as p53 and oncogenes such as KRAS were identified, ever greater numbers of oncogenic mutations have been cataloged (75, 76). With the conclusion of The Cancer Genome Atlas sequencing project in 2018, thousands of mutations in an estimated 300 cancer driver genes were identified (77), leaving a long tail of only extremely rare mutations to identify by even larger-scale cancer cohort sequencing (78). Clinical somatic genome sequencing of tumor, precancerous, and healthy tissue is becoming increasingly routine, with single-mutation panels being replaced by multigene, exome, or whole-genome sequencing (79). With further development, less invasive methods such as cell-free DNA sequencing to detect DNA sequences and epigenetic alterations derived from early-stage tumors may even extend somatic genome sequencing to large-scale population screening applications (80).
Somatic Genetic Therapies
Gene therapies targeting the somatic genome can correct a genetic deficiency by specific endogenous gene editing and correction back to the normal sequence, nontargeted addition to the genome of exogenous genetic material to compensate for the mutation, or bulk replacement of the somatic genome. Gene therapies in the somatic genome are further differentiated by the need to replace, add, or correct genetic information only in the target tissue of interest, such as tissues of the liver, muscle, or eye. Furthermore, in certain cell types such as HSCs and T cells, which can be cultured outside the body, these gene therapies can be performed ex vivo and the altered cells returned to the patient. However, for the majority of target tissues, somatic gene therapies must overcome the challenge of delivering genetic material and editing reagents in vivo while avoiding rejection by the patient’s own immune system.
The bulk replacement of genetic alterations in the somatic genome is similarly difficult given the large numbers of cells that need to be genetically altered, in comparison to a single manipulated cell in germline therapies. In certain disorders such as polycystic kidney disease, which is caused by mutations in PKD1, PKD2, PKD3, and PKHD1, replacement of the affected kidneys through transplantation effectively removes the disease-causing genetic material, although other organs such as the liver remain affected (81). Similarly, when tumors are operable, surgery in a sense allows for the bulk removal of mutated somatic genomes. Somatic mutational load can be reduced by avoidance of known environmental mutagen sources (73).
Nontargeted Addition to the Somatic Genome
The earliest gene therapies to bear that name were based on nontargeted additions to the nuclear genomes of ex vivo-cultured immune cells, particularly T cells and HSCs (Figure 2b). Following the successful engineering of the first replication-deficient retroviruses from Moloney murine leukemia virus (MMLV) in the early 1980s, which presaged later lentiviral vectors adapted from human immunodeficiency virus (82, 83), large multiple-kilobase segments of new DNA could be pseudorandomly introduced into these cell types. The first gene transfer trial used an MMLV-derived retrovirus to add a heterologous tumor necrosis factor (TNF)-α expression cassette to T cells isolated from a metastatic melanoma patient’s tumors and expanded ex vivo (12).
While this first trial did not provide clinical benefit relative to earlier unmodified tumor-infiltrating lymphocyte trials, it precipitated a flood of additive ex vivo gene therapies in T cells and HSCs over the next 3 decades. Immediately following the first T cell trials, the nontargeted addition of a correct copy of the adenosine deaminase enzyme in the ex vivo–cultured HSCs of children with SCID resulted, in some cases, in a durable and so far lifelong cure in these early gene therapy patients (84). These additive ex vivo retroviral technologies have been extended to nontargeted integration of synthetic DNA sequences in current generations of CAR T–based therapies (85).
In parallel to the development of ex vivo gene addition therapies, viral vectors performing the dual function of targeting a specific human tissue type and delivering exogenous DNA have enabled in vivo gene addition to diverse somatic cell types (Figure 2b). Liver hepatocytes have been an active target tissue as protein-generating factories for the addition of missing or dysfunctional blood factors. In the cases of hemophilia A and B, caused by deficiency in circulating clotting factors VIII and IX, respectively, in vivo addition of new factor VIII and IX genes to hepatocytes has resulted in curative gene therapies (86, 87). Multiple generations of viral delivery systems, ranging from adenoviruses to modern engineered AAV serotypes and pseudotypes, have drastically improved the efficacy, specificity, and immunologic safety profiles of in vivo somatic cell gene therapies (88, 89). The retina has also seen a large number of gene therapy trials because of its uniquely accessible location and immunoprivileged tissue status. The first FDA-approved in vivo gene therapy indeed adds a heterologous copy of the RPE65 gene to the retinal cells of patients suffering from Leber’s congenital amaurosis, resulting in durable vision improvements (16).
However, nontargeted additive gene therapies suffer from a variety of constraints. Functionally, the genetic carrying capacity of current generations of commonly used AAV vectors is limited to approximately 4.5 kb, too small to encode cassettes expressing correct copies of large endogenous proteins such as, in an extreme case, the more than 10 kb of complementary DNA required to encode dystrophin, which is mutated in patients with muscular dystrophy (90). Similarly, early trials attempting to insert correct versions of α- or β-hemoglobin into the HSCs of patients with sickle cell anemia and thalassemia resulted in failure of erythropoiesis due to improper regulatory control over hemoglobin expression during erythrocyte development (91).
More importantly, patient deaths in gene therapy clinical trials around the turn of the millennium highlighted numerous safety issues in pseudorandomly integrating viral vectors. First, the 1999 death of a patient with ornithine transcarbamylase deficiency, who possessed a mild form of the disease due to somatic mosaicism, was traced to a massive immunologic response to the viral vector used to deliver the gene cargo in vivo to the patient’s hepatocytes (92, 93). Furthermore, immune responses to newly corrected, and thus recognized as nonself, endogenous gene products may limit therapeutic efficacy in disorders such as Duchenne muscular dystrophy (94). The unintended consequences of the introduction of nontargeted genetic elements, including strong viral promoters to drive the heterologous therapeutic gene, were revealed by a rash of leukemias in early X-linked SCID clinical trials editing HSCs ex vivo (95). Viral vector copies that integrated adjacent to oncogenes such as LMO2 precipitated oncogenic transformation many years after initially successful therapies (96, 97). Finally, the potential dangers of inappropriate regulatory control of pseudorandomly added gene products were underscored by the death of a patient with rheumatoid arthritis from a histoplasmosis fungal infection after successful gene therapy with an anti-TNF decoy receptor (98); on-target toxicity of the gene therapy potentially suppressed the patient’s ability to mount an effective immune response.
Gene Editing in the Somatic Genome
Gene therapies based on correcting the individual causative mutations in somatic cell types of interest could overcome many of these challenges with nontargeted genetic addition. All cells undergoing cell division attempt to repair DNA copying errors though a variety of DNA repair pathways, including HDR, wherein a mutation on one chromosome can be corrected by binding its homologous region on the other chromosome and undergoing templated repair (99). HDR is capable of scarlessly integrating exogenous DNA sequences at specific, user-defined sites in human cell lines in the 1980s, although initially at exceptionally low efficiencies (100). While critical for the generation of genetically modified model organisms (101), therapeutic application in human cells ex vivo or in vivo awaited the development of targetable DNA nucleases that could generate a double-stranded DNA break adjacent to the intended site of genetic correction.
These targeted double-stranded breaks increased the efficiency of HDR in human cells by many orders of magnitude and, in the 2000s, enabled the first targeted gene editing trials using ZFNs to correct SCID-causing mutations in IL2RG (102). The discovery and rapid development of RNA-guided nucleases, most prominently CRISPR/Cas9, in the mid-2010s made these gene editing reagents drastically simpler and cheaper to develop (103, 104). Paired with a corrective exogenous DNA template containing homology arms, reagents to perform endogenous gene editing at almost any site in the human nuclear genome can be rapidly designed and synthesized. Even the DNA and protein components are combinable into a single-component system using a Cas9–reverse transcriptase fusion protein along with a guide RNA with an extended RNA sequence containing the intended mutation correction instead of homologous DNA (105). This process allows templated repair to follow nuclease recognition directly without the need for an additional DNA sequence, potentially simplifying reagent delivery.
Indeed, the challenge of delivering gene editing reagents into target cells represents a great hurdle for wider adoption of corrective gene therapies in somatic cells. For HSC and T cell populations that can be cultured ex vivo, physical delivery methods such as electroporation enable robust delivery of both ribonucleoproteins (e.g., Cas9–guide RNA complexes) and DNA HDR templates into target cells (106). In HSCs, electroporation of Cas9 RNPs targeting the sickle cell mutation in β-hemoglobin, coupled with the delivery of HDR templates containing a correct sequence using AAV6 vectors, has finally resulted in robust correction of the causative mutation in sickle cell anemia, the first molecularly described genetic disease (107). These corrected HSCs were able to fully differentiate and, crucially, undergo erythropoiesis. The ease of generating new editing reagents to target additional mutations may enable this strategy to be broadly applied across the genetic diseases of hematopoiesis (108). Beyond HSCs, we have shown how similar RNP electroporation strategies instead using nonviral DNA can be applied to directly correct causative mutations in differentiated T cell populations such as regulatory T cells (109).
Gene editing to repair endogenous genetic sequences also offers new avenues for in vivo somatic gene therapies. Notably, in conditions such as muscular dystrophy, where the protein product is too large for commonly used viral vectors, gene editing reagents have been designed and successfully delivered in mouse models, among many other rapidly developing preclinical in vivo therapeutics (110, 111). In vivo gene editing further highlights the challenges of editing reagent delivery, as both large protein nucleases, or the DNA sequences encoding them, and corrective DNA HDR templates must be delivered to the somatic tissue of interest (112). Creative solutions involving curative treatments resulting from gene cutting only, without templated repair, may be possible in a handful of conditions. The development of smaller targetable nucleases, single-component templated repair systems (105), and most of all generalized improvements in delivery technology offers great hope for accelerating in vivo corrective gene therapies in somatic tissues.
MANIPULATING OTHER GENOMES
Adaptive Immune Receptor Repertoire
A subset of somatic cells contain a unique addition to the nuclear genome, the T and B cells of the adaptive immune system, which generate new antigen receptor gene products after conception through somatic recombination. The immune receptor repertoire of a young adult contains, conservatively, on the order of 1011 unique antigenic receptors, generating almost six orders of magnitude more individual protein products than the nuclear genome (113–115), although repertoire diversity declines with age (116). The presence or lack of T cell receptor (TCR) genes within the T cell repertoire can contribute to the development of diseases such as type 1 diabetes mellitus and multiple sclerosis, while autoreactive B cell receptors (BCRs) underlie systemic lupus erythematosus, myasthenia gravis, and numerous other conditions (117, 118). In the opposite context, the adaptive immune receptor repertoire sometimes lacks potentially useful TCRs and BCRs, such as those that would respond to liquid and solid tumors, due to regulatory mechanisms to guard against autoimmunity or so-called immune editing in the cancer microenvironment (119).
The adaptive immune receptor repertoire subset of the nuclear genome can be manipulated genetically through bulk replacement of the entire repertoire or through selective introduction of desired TCR and BCR sequences. The radiation sensitivity of the majority of these cells enables bulk replacement of this genomic compartment by autologous or allogenic bone marrow transplants, which have successfully been used in the treatment of severe autoimmune diseases such as systemic sclerosis (120). Specific introduction of desired antigen receptors can target antigens that are difficult to vaccinate against, such as endogenous peptides or masked pathogenic epitopes. Viral vectors have been clinically used to add new TCR genes at pseudorandom genome sites in primary human T cells since the 2000s (121). More broadly, CARs combining the binding properties of antibodies with the signaling properties of TCRs and costimulatory molecules have been engineered to redirect T cells to self-antigens also expressed on cancers, such as the B cell marker CD19 (85).
Theoretically, one can envision future personalized therapies based on the introduction of antigen receptors specific for various microbes or tumor antigens into the T and B cells of patients with cancer and infectious disease or with the goal of eliminating existing T and B cells specific for self-antigens from patients with autoimmune disease. Practical application of these ideas is challenging because of the enormous diversity of immune receptor repertoires. Identification of TCR, BCR, or synthetic antigen receptor genes that would work against particular microbes or tumors or react against particular self-antigens remains a key challenge. Given the enormous current interest and progress in sequencing T and B cell antigen receptors (114), it is possible that these challenges will be overcome as large-enough databases of receptor sequences are assembled.
Microbial Metagenome
In comparison to the mitochondrial and nuclear genomes, the most complex genetic compartment in terms of protein family diversity within the human body lies not within human cells but rather in the metagenome of the ubiquitous microbiota. Across human tissues, the adult human microbiome contains more than 30 trillion bacterial cells from on the order of 1,000 unique species (122, 123). Different anatomic locations have distinct microbiomes, with varying degrees of diversity according to site, individual, time, and disease state (124). With an average bacterium containing approximately 3,000 protein coding genes, a single human’s overall microbial metagenome likely contains on the order of a million unique gene products (125, 126). The microbial metagenome is not acquired until after birth and is initially inherited primarily from the mother through the vaginal passage, skin-to-skin contact, and breastfeeding. The genetic content of the human metagenome is also much more diverse both across an individual’s life and among individuals than the mitochondrial or nuclear genome. Between two random individuals the microbiome can differ in well over half of its content (although significantly less in cohabiting humans sharing a local environment and diet), in comparison to the average ~0.1% difference in nuclear genetic content between two unrelated individuals (127).
The presence or absence of specific pathogenic microbial components of the microbiome has traditionally been measured by direct culture ex vivo of the organism or detection of a specific disease after introduction to an animal host. Nucleic acid–specific methods such as polymerase chain reaction offer greater speed and flexibility and can detect pathogenic toxin genes differentially from the bacterial species that variably can possess them (128). At greater scale, 16S ribosomal sequencing takes advantage of the evolutionary conservation of ribosomal RNA to directly measure microbial species diversity. First pioneered in the 1970s (129) and since expanded through pairing with next-generation sequencing, 16S metagenomic sequencing can rapidly determine the genus-level diversity of the human microbiome (130). With further decreases in sequencing cost, bulk fragmentation of metagenomic DNA samples and computational assembly into species-specific genomes and transcriptomes increasingly allow definitive sampling of the metagenomic compartment (131).
As molecular techniques have helped identify nonculturable microbes, researchers have hypothesized that alterations in the human microbiome are associated with the development of various diseases, raising the possibility that manipulating the microbiome and its genetic products may be a therapeutic strategy for these diseases. So far, the only clinically widespread microbiome-targeted therapy has been bulk replacement of depleted microbiota (via fecal transplants) in Clostridium difficile–induced pseudomembranous colitis (17). Microbiome abnormalities in inflammatory bowel disease and other inflammatory and metabolic disorders have also been suggested. The broad concept of dysbiosis proposes alterations in the nature and diversity of microbiota as predisposing factors for these disorders. In order to use genetic approaches for restoring the diversity, it will be necessary to identify specific organisms that are perturbed and, furthermore, the genes of the organisms that may contribute to the disease.
Bacteria can readily be made to express exogenous genes on extragenomic plasmids, but direct editing of the bacterial genome may be required to maintain expression with the absence in vivo of traditional laboratory selection pressures. Nontargeted gene addition through bacteriophage vectors or integrative plasmids (132) as well as gene editing with CRISPR/Cas9 and other targetable tools have enabled large heritable changes to be made ex vivo in bacterial species of the human microbiome. In the common nuclear genetic disorder phenylketonuria, a strain of Escherichia coli Nissle engineered to heritably express phenylalanine-metabolizing enzymes showed persistent engraftment into mouse and primate microbiomes after oral introduction and ameliorated blood phenylalanine levels (133). Microbial populations edited ex vivo can be introduced into the normal flora similarly to nonedited therapeutic bacterial species. Engraftment can be further enhanced through targeted nutritional support for engineered strains (134).
With improved delivery methods targeting gene editing reagents to specific microbial species within the body, these therapeutic microbial genetic changes may ultimately be made in vivo, similar to the goals of in vivo gene therapies targeting somatic genomes and the adaptive immune receptor repertoire. The remarkable species specificity of bacteriophages has enabled delivery of gene editing reagents targeting pathogenic strains or even specific antibiotic resistance plasmids for destruction, and may allow for additive gene therapies as well (135). Whether through bulk replacement of the microbiome, gene editing ex vivo or in vivo of specific bacterial species, or recapitulation of physiologic exposures to normal flora, altering the genetic content of the diverse human metagenome could offer promising avenues for future research on genetic therapies.
CONCLUSIONS
The major genetic compartments in humans differ in genomic size, complexity, heritability, and diversity. Germline mutations in the mitochondrial and nuclear genomes often cause developmental disorders, although the great size of the nuclear genome ensures that the thousands of identified monogenic diseases present in diverse contexts. Accumulation of somatic mutations in the nuclear genome underlies the development of cancer, and somatic mutations in mitochondria may contribute to aging. More broadly, the microbial metagenome develops largely after birth and is characterized by much greater diversity among humans and variation over the course of life. Advances in next-generation DNA sequencing have made mitochondrial sequencing, clinical exome and whole-genome sequencing, and 16S and unbiased microbial sequencing widely available.
The genetic alterations revealed by these sequencing approaches are the correctable targets of gene therapies. Entire genomic compartments, such as the mitochondrial and nuclear genomes, can be addressed in bulk by use of modern assisted reproduction technologies, including mitochondrial replacement therapy and preimplantation diagnostics. Additive somatic cell gene therapies began with the development of viral vectors to infect human somatic cells that can be cultured ex vivo, such as T cells, and rapidly developed to include in vivo applications with viral pseudotypes with specific tissue tropisms. Most recently, dramatic advances in CRISPR/Cas9 and related targetable gene editing applications, where the specific causative mutation or gene is corrected at its endogenous locus, have expanded the horizons for more refined ex vivo and in vivo gene therapies.
Overall, the plummeting costs of DNA sequencing over the past 2 decades have accelerated the diagnosis of genetic diseases. Echoing these diagnostic advances, the unexpectedly rapid development of targetable gene editing in the 2010s has now made the design and testing of specific therapeutic reagents to correct genetic changes direct and accessible. One of the great continuing challenges to the widespread application of gene therapies lies in generalized platforms for the delivery of customizable gene editing reagents into the cell type and genomic compartment of interest in a patient’s specific genetic disease (Figure 3). Beyond direct corrections of genetic diseases, new methodologies to rapidly discover synthetic genetic circuits capable of enhancing cellular function in diseases such as cancer and autoimmunity hold the promise of further gene therapy applications in engineered somatic cells (136). Increasingly, genetic diseases across human genetic compartments can be readily diagnosed, and the next generations of gene therapy platforms targeting each compartment are poised to offer flexible and personalized curative treatments.
SUMMARY POINTS.
Humans contain distinct genetic compartments: the mitochondrial genome; the nuclear genome including the adaptive immune receptor repertoire in specialized cells; and the microbial metagenome.
Gene therapies for each of these compartments are based on three broad categories: bulk replacement or selection of affected genomes, nontargeted addition of new genetic information to compensate for genetic errors, and direct gene editing to correct causative genetic alterations.
The mitochondrial and nuclear genomes are determined at conception and are consistent throughout life, with the exception of accumulation of somatic mutations and the adaptive immune receptor repertoire. Genetic disease is driven largely by mutations.
Diagnosis of genetic disease through next-generation sequencing and design of corrective gene therapy reagents are now being widely adopted. Pairing gene addition or gene editing reagents with generalized delivery platforms to target specific genetic compartments in specific somatic cell types in vivo remains a daunting challenge.
ACKNOWLEDGMENTS
We thank members of the Marson lab for helpful discussions and Sarah Pyle for assistance creating figures. T.L.R. was supported by the UCSF Medical Scientist Training Program (T32GM007618), a UCSF Endocrinology Training Grant (T32 DK007418), and a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (F30DK120213). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund; is an investigator at the Chan Zuckerberg Biohub; and has received funding from the Innovative Genomics Institute, the American Endowment Foundation, and the Cancer Research Institute, as well as a Lloyd J. Old STAR award, a gift from the Jordan Family, and a gift from Barbara Bakar. A.M. is a member of the Parker Institute for Cancer Immunotherapy.
DISCLOSURE STATEMENT
T.L.R. and A.M. are cofounders of Arsenal Biosciences. T.L.R. served as the Chief Scientific Officer of Arsenal Biosciences. A.M. is a cofounder of Spotlight Therapeutics, served on the scientific advisory board of PACT Pharma, was an advisor to Trizell, and was an advisor to Juno Therapeutics. T.L.R. and A.M. own equity in Arsenal Biosciences, and A.M. owns equity in Spotlight Therapeutics and PACT Pharma. The Marson lab has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead, and Anthem.
Footnotes
The Annual Review of Pathology: Mechanisms of Disease is online at pathol.annualreviews.org
LITERATURE CITED
- 1.Motulsky AG. 2010. History of human genetics. In Vogel and Motulsky’s Human Genetics: Problems and Approaches, ed. Speicher MR, Motulsky AG, Antonarakis SE, pp. 13–29. Heidelberg, Ger.: Springer. 4th ed. [Google Scholar]
- 2.Soler A, Morales C, Mademont-Soler I, Margarit E, Borrell A, et al. 2017. Overview of chromosome abnormalities in first trimester miscarriages: a series of 1,011 consecutive chorionic villi sample karyotypes. Cytogenet. Genome Res 152:81–89 [DOI] [PubMed] [Google Scholar]
- 3.Baird PA, Anderson TW, Newcombe HB, Lowry RB. 1988. Genetic disorders in children and young adults: a population study. Am. J. Hum. Genet 42:677–93 [PMC free article] [PubMed] [Google Scholar]
- 4.Slack J, Evans KA. 1966. The increased risk of death from ischaemic heart disease in first degree relatives of 121 men and 96 women with ischaemic heart disease. J. Med. Genet 3:239–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingram VM. 1956. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature 178:792–94 [DOI] [PubMed] [Google Scholar]
- 6.Pauling L, Itano HA, Singer SJ, Wells IC. 1949. Sickle cell anemia, a molecular disease. Science 110:543–48 [DOI] [PubMed] [Google Scholar]
- 7.Lejeune J, Gautier M,Turpin R. 1959. Etude des chromosomes somatiques de neuf enfants mongoliens. C. R. Hebd. Séances Acad. Sci 248:1721–22 [PubMed] [Google Scholar]
- 8.Guthrie R 1961. Blood screening for phenylketonuria. JAMA 178:863 [Google Scholar]
- 9.Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, et al. 1983. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306:234–38 [DOI] [PubMed] [Google Scholar]
- 10.Friedmann T, Roblin R. 1972. Gene therapy for human genetic disease? Science 175:949–55 [DOI] [PubMed] [Google Scholar]
- 11.Merril CR, Geier MR, Petricciani JC. 1971. Bacterial virus gene expression in human cells. Nature 233:398–400 [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg SA, Aebersold P, Cornetta K, Kasid A,Morgan RA, et al. 1990. Gene transfer into humans—immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N. Engl. J. Med 323:570–78 [DOI] [PubMed] [Google Scholar]
- 13.Cline MJ, Stang H, Mercola K, Morse L, Ruprecht R, et al. 1980. Gene transfer in intact animals. Nature 284:422–25 [DOI] [PubMed] [Google Scholar]
- 14.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, et al. 2017. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med 377:2531–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, et al. 2018. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378:439–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell S, Bennett J,Wellman JA, Chung DC, Yu ZF, et al. 2017. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390:849–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ooijevaar RE, Terveer EM, Verspaget HW, Kuijper EJ, Keller JJ. 2019. Clinical application and potential of fecal microbiota transplantation. Annu. Rev. Med 70:335–51 [DOI] [PubMed] [Google Scholar]
- 18.Carroll D 2014. Genome engineering with targetable nucleases. Annu. Rev. Biochem 83:409–39 [DOI] [PubMed] [Google Scholar]
- 19.Kukat C,Wurm CA, Spåhr H, Falkenberg M,Larsson NG,Jakobs S. 2011. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. PNAS 108:13534–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole LW 2016. The evolution of per-cell organelle number. Front. Cell Dev. Biol 4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jansen RPS. 2000. Germline passage of mitochondria: quantitative considerations and possible embryological sequelae. Hum. Reprod 15(Suppl. 2):112–28 [DOI] [PubMed] [Google Scholar]
- 22.Zaidi AA, Wilton PR, Su MSW, Paul IM, Arbeithuber B, et al. 2019. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. PNAS 50:25172–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martijn J, Vosseberg J, Guy L, Offre P, Ettema TJG. 2018. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 557:101–5 [DOI] [PubMed] [Google Scholar]
- 24.Anderson S, Bankier AT, Barrell BG, De Bruijn MHL, Coulson AR, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature 290:457–65 [DOI] [PubMed] [Google Scholar]
- 25.Gray MW. 2012. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol 4:a011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khrapko K, Coller HA, André PC, Li XC, Hanekamp JS, Thilly WG. 1997. Mitochondrial mutational spectra in human cells and tissues. PNAS 94:13798–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M, Schröder R, Ni S, Madea B, Stoneking M. 2015. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. PNAS 112:2491–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Slone J, Fei L, Huang T. 2019. Mitochondrial DNA variants and common diseases: a mathematical model for the diversity of age-related mtDNA mutations. Cells 8:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. 2004. The epidemiology of mitochondrial disorders. Biochim. Biophys. Acta Bioenerg 1659:115–20 [DOI] [PubMed] [Google Scholar]
- 30.Larsson N-G. 2010. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem 79:683–706 [DOI] [PubMed] [Google Scholar]
- 31.Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, et al. 2019. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176:1325–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patananan AN, Wu T-H, Chiou P-Y, Teitell MA. 2016. Modifying the mitochondrial genome. Cell Metab. 23:785–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, et al. 2009. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 461:367–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Tian Z, Yuan J, Liu C, Liu HL, et al. 2018. The progress of gene therapy for Leber’s optic hereditary neuropathy. Curr. Gene Ther 17:320–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H, Koilkonda RD, Chou T-H, Porciatti V, Ozdemir SS, et al. 2012. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. PNAS 109:E1238–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thyagarajan B, Padua RA, Campbell C. 1996. Mammalian mitochondria possess homologous DNA recombination activity. J. Biol. Chem 271:27536–43 [DOI] [PubMed] [Google Scholar]
- 37.Hagström E, Freyer C, Battersby BJ, Stewart JB, Larsson NG. 2014. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res. 42:1111–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A,et al. 2015. MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther 23:1592–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- 40.de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLOS Genet. 7:e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, et al. 2015. A global reference for human genetic variation. Nature 526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikkelsen TS, Hillier LW, Eichler EE, Zody MC, Jaffe DB, et al. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87 [DOI] [PubMed] [Google Scholar]
- 43.Kuroki Y, Toyoda A, Noguchi H, Taylor TD, Itoh T, et al. 2006. Comparative analysis of chimpanzee and human Y chromosomes unveils complex evolutionary pathway. Nat. Genet 38:158–67 [DOI] [PubMed] [Google Scholar]
- 44.McCulloch SD, Kunkel TA. 2008. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18:148–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crow JF. 2000. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet 1:40–47 [DOI] [PubMed] [Google Scholar]
- 46.Drost JB, Lee WR. 1995. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ. Mol. Mutagen 25:48–64 [DOI] [PubMed] [Google Scholar]
- 47.Jarvis GE. 2017. Early embryo mortality in natural human reproduction: what the data say. F1000Research 5:2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao Z, Moorjani P, Sasani TA, Pedersen BS, Quinlan AR, et al. 2019. Overlooked roles of DNA damage and maternal age in generating human germline mutations. PNAS 116:9491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, et al. 2012. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488:471–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacArthur DG, Balasubramanian S, Frankish A, Huang N,Morris J, et al. 2012. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335:823–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amberger JS, Bocchini CA, Scott AF, Hamosh A. 2019. OMIM.org: leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res. 47:D1038–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartha I, Di Iulio J, Venter JC, Telenti A. 2018. Human gene essentiality. Nat. Rev. Genet 19:51–62 [DOI] [PubMed] [Google Scholar]
- 53.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, et al. 2020. A brief history of human disease genetics. Nature 577:179–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, et al. 2017. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet 101:5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murray JM, Davies KE, Harper PS, Meredith L, Mueller CR, Williamson R. 1982. Linkage relationship of a cloned DNA sequence on the short arm of the X chromosome to Duchenne muscular dystrophy. Nature 300:69–71 [DOI] [PubMed] [Google Scholar]
- 57.Kan YW, Dozy AM. 1978. Polymorphism of DNA sequence adjacent to human β-globin structural gene: relationship to sickle mutation. PNAS 75:5631–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller DT,Adam MP, Aradhya S, Biesecker LG, Brothman AR, et al. 2010. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet 86:749–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark MM, Stark Z, Farnaes L, Tan TY, White SM, et al. 2018. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases.. npj Genom. Med 3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bianchi DW. 2004. Circulating fetal DNA: its origin and diagnostic potential—a review. Placenta 25(Suppl.):S93–101 [DOI] [PubMed] [Google Scholar]
- 61.Treff NR, Zimmerman RS. 2017. Advances in preimplantation genetic testing for monogenic disease and aneuploidy. Annu. Rev. Genom. Hum. Genet 18:189–200 [DOI] [PubMed] [Google Scholar]
- 62.Kaplan F 1998. Tay-Sachs disease carrier screening: a model for prevention of genetic disease. Genet. Test 2:271–92 [DOI] [PubMed] [Google Scholar]
- 63.Gyngell C, Douglas T, Savulescu J. 2017. The ethics of germline gene editing.. J. Appl. Philos 34:498–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lander ES, Baylis F, Zhang F, Charpentier E, Berg P, et al. 2019. Adopt a moratorium on heritable genome editing. Nature 567:165–68 [DOI] [PubMed] [Google Scholar]
- 65.Greely HT. 2019. CRISPR’d babies: human germline genome editing in the “He Jiankui affair.” J. Law Biosci 6:111–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tomasetti C, Vogelstein B. 2015. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347:78–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barker N, Van De Wetering M, Clevers H. 2008. The intestinal stem cell. Genes Dev. 22:1856–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, et al. 2015. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350:94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J. 2017. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun 8:15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.García-Nieto PE, Morrison AJ, Fraser HB. 2019. The somatic mutation landscape of the human body. Genome Biol. 20:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin L, Wang J, Tian X, Yu H, Truong C, et al. 2016. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J. Mol. Diagn 18:446–53 [DOI] [PubMed] [Google Scholar]
- 72.Armaghany T, Wilson JD, Chu Q, Mills G. 2012. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res 5:19–27 [PMC free article] [PubMed] [Google Scholar]
- 73.Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, et al. 2019. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364:eaaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steensma DP, Ebert BL. 2020. Clonal hematopoiesis as a model for premalignant changes during aging. Exp. Hematol 83:48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. 1994. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54:4855–78 [PubMed] [Google Scholar]
- 76.Janssen JWG, Steenvoorden ACM, Lyons J, Anger B, Bohlke JU, et al. 1987. RAS gene mutations in acute and chronic myelocytic leukemias, chronic myeloproliferative disorders, and myelodysplastic syndromes. PNAS 84:9228–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, et al. 2018. Comprehensive characterization of cancer driver genes and mutations. Cell 173:371–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, et al. 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505:495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaw KRM, Maitra A. 2019. The status and impact of clinical tumor genome sequencing. Annu. Rev. Genom. Hum. Genet 20:413–32 [DOI] [PubMed] [Google Scholar]
- 80.Corcoran RB, Chabner BA. 2018. Application of cell-free DNA analysis to cancer treatment. N. Engl. J. Med 379:1754–65 [DOI] [PubMed] [Google Scholar]
- 81.Kanaan N, Devuyst O, Pirson Y. 2014. Renal transplantation in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol 10:455–65 [DOI] [PubMed] [Google Scholar]
- 82.Mann R, Mulligan RC, Baltimore D. 1983. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 33:153–59 [DOI] [PubMed] [Google Scholar]
- 83.Naldini L, Trono D, Verma IM. 2016. Lentiviral vectors, two decades later. Science 353:1101–2 [DOI] [PubMed] [Google Scholar]
- 84.Ferrua F, Aiuti A. 2017. Twenty-five years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Hum. Gene Ther 28:9792–81 [DOI] [PubMed] [Google Scholar]
- 85.June CH, Sadelain M. 2018. Chimeric antigen receptor therapy. N. Engl. J. Med 379:64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mannucci PM, Tuddenham EGD. 2001. The hemophilias—from royal genes to gene therapy. N. Engl. J. Med 344:1773–79 [DOI] [PubMed] [Google Scholar]
- 87.Nathwani AC, Reiss UM, Tuddenham EGD, Rosales C, Chowdary P, et al. 2014. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med 371:1994–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cronin J, Zhang X-Y, Reiser J. 2005. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther 5:387–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Asokan A, Schaffer DV, Samulski RJ. 2012. The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther 20:699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chamberlain JS. 2002. Gene therapy of muscular dystrophy. Hum. Mol. Genet 11:2355–62 [DOI] [PubMed] [Google Scholar]
- 91.Orkin SH. 1990. Globin gene regulation and switching: circa 1990. Cell 63:665–72 [DOI] [PubMed] [Google Scholar]
- 92.Marshall E 1999. Gene therapy death prompts review of adenovirus vector. Science 286:2244–45 [DOI] [PubMed] [Google Scholar]
- 93.Wilson JM. 2009. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol. Genet. Metab 96:151–57 [DOI] [PubMed] [Google Scholar]
- 94.Ferrer A, Wells KE, Wells DJ. 2000. Immune responses to dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Gene Ther. 7:1439–46 [DOI] [PubMed] [Google Scholar]
- 95.Kohn DB, Sadelain M, Glorioso JC. 2003. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat. Rev. Cancer 3:477–88 [DOI] [PubMed] [Google Scholar]
- 96.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, et al. 2008. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig 118:3132–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, et al. 2008. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig 118:3143–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frank KM, Hogarth DK, Miller JL, Mandal S, Mease PJ, et al. 2009. Investigation of the cause of death in a gene-therapy trial. N. Engl. J. Med 361:161–69 [DOI] [PubMed] [Google Scholar]
- 99.Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spnng Harb. Perspect. Biol 5:a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. 1985. Insertion of DNA sequences into the human chromosomal β-globin locus by homologous recombination. Nature 317:230–34 [DOI] [PubMed] [Google Scholar]
- 101.Thomas KR, Folger KR, Capecchi MR. 1986. High frequency targeting of genes to specific sites in the mammalian genome. Cell 44:419–28 [DOI] [PubMed] [Google Scholar]
- 102.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, et al. 2005. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–51 [DOI] [PubMed] [Google Scholar]
- 103.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, et al. 2019. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576:149–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, et al. 2015. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. PNAS 112:10437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, et al. 2016. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539:384–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Porteus MH. 2019. A new class of medicines through DNA editing. N. Engl. J. Med 380:947–59 [DOI] [PubMed] [Google Scholar]
- 109.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, et al. 2018. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559:405–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, et al. 2016. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351:407–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, et al. 2016. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351:403–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang D, Zhang F, Gao G. 2020. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell 181:136–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jenkins MK, Chu HH, McLachlan JB, Moon JJ. 2010. On the composition of the preimmune repertoire of T cells specific for peptide–major histocompatibility complex ligands. Annu. Rev. Immunol 28:275–94 [DOI] [PubMed] [Google Scholar]
- 114.Chaudhary N, Wesemann DR. 2018. Analyzing immunoglobulin repertoires. Front. Immunol 9:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mora T, Walczak AM. 2019. How many different clonotypes do immune repertoires contain? Curr. Opin. Syst. Biol 18:104–10 [Google Scholar]
- 116.Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, et al. 2014. Diversity and clonal selection in the human T-cell repertoire. PNAS 111:13139–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Walker LSK, Abbas AK. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol 2:11–19 [DOI] [PubMed] [Google Scholar]
- 118.Nemazee D 2017. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol 17:281–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol 3:991–98 [DOI] [PubMed] [Google Scholar]
- 120.Swart JF, Delemarre EM, Van Wijk F, Boelens JJ, Kuball J, et al. 2017. Haematopoietic stem cell transplantation for autoimmune diseases. Nat. Rev. Rheumatol 13:244–56 [DOI] [PubMed] [Google Scholar]
- 121.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, et al. 2006. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The Human Microbiome Project. Nature 449:804–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 14:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, et al. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, et al. 2015. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom 15:141–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, et al. 2017. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550:61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pimbley DW, Patel PD. 1998. A review of analytical methods for the detection of bacterial toxins. J. Appl. Microbiol. Symp. Suppl 84:S98–109 [DOI] [PubMed] [Google Scholar]
- 129.Woese CR, Fox GE. 1977. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. PNAS 74:5088–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Janda JM, Abbott SL. 2007. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol 45:2761–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, et al. 2019. A new genomic blueprint of the human gut microbiota. Nature 568:499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ronda C, Chen SP, Cabral V, Yaung SJ, Wang HH. 2019. Metagenomic engineering of the mammalian gut microbiome in situ. Nat. Methods 16:167–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Isabella VM, Ha BN, Castillo MJ, Lubkowicz DJ, Rowe SE, et al. 2018. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol 36:857–64 [DOI] [PubMed] [Google Scholar]
- 134.Shepherd ES, Deloache WC, Pruss KM, Whitaker WR, Sonnenburg JL. 2018. An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557:434–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee HL, Shen H, Hwang IY, Ling H, Yew WS, et al. 2018. Targeted approaches for in situ gut microbiome manipulation. Genes 9:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roth TL, Li PJ, Blaeschke F, Roybal K, Shifrut E, Marson A. 2020. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell 181:728–44 [DOI] [PMC free article] [PubMed] [Google Scholar]