

Abstract
Perivascular macrophages (pvMs) are associated with cerebral vasculature and mediate brain drainage and immune regulation. Here, using reporter mouse models, whole brain and section immunofluorescence, flow cytometry, and single cell RNA sequencing, besides the Lyve1+F4/80+CD206+CX3CR1+ pvMs, we identify a CX3CR1– pvM population that shares phagocytic functions and location. Furthermore, the brain parenchyma vasculature mostly hosts Lyve1+MHCII– pvMs with low to intermediate CD45 expression. Using the double Cx3cr1GFP x Cx3cr1-Cre;RosatdT reporter mice for finer mapping of the lineages, we establish that CD45lowCX3CR1– pvMs are derived from CX3CR1+ precursors and require PU.1 during their ontogeny. In parallel, results from the Cxcr4-CreErt2;Rosa26tdT lineage tracing model support a bone marrow-independent replenishment of all Lyve1+ pvMs in the adult mouse brain. Lastly, flow cytometry and 3D immunofluorescence analysis uncover increased percentage of pvMs following photothrombotic induced stroke. Our results thus show that the parenchymal pvM population is more heterogenous than previously described, and includes a CD45low and CX3CR1– pvM population.
Subject terms: Monocytes and macrophages, Lymphatic vessels, Neuroimmunology, Gene regulation in immune cells
Perivascular macrophages (pvMs) are important for brain drainage and immune regulation. Here the authors analyse various reporter mouse strains for finer mapping of pvM subsets and lineage differentiation, and propose CX3CR1negative and CD45low as additional markers of intermediate pvMs for studying this heterogenous population.
Introduction
The central nervous system (CNS) has long been considered immune privileged, devoid of immune cells other than microglia and lacking classical lymphatic vessels. The re-discovery of the dural lymphatic network highlights a route for the drainage of the brain to the periphery1–7. Recent MRI data indicate that drainage from the CNS parenchyma occurs towards lymphatics and draining lymph nodes via the foramen7.
Besides microglia, several myeloid populations have now been characterized, shown to be essential for brain homeostasis and to contribute to brain diseases8. These cells include non-parenchymal or border-associated macrophages (BAMs), which can be subclassified into perivascular, subdural meningeal, and choroid plexus macrophages. These macrophages are established during development by embryonic precursors derived from the yolk sac and with the exception of choroid plexus macrophages, are not replaced by blood monocytes during adulthood9,10. Specifically, perivascular macrophages (pvMs) are located in the perivascular space of the blood vessel, delimited by the vascular basement membrane of blood vessels and the glial basement membranes11. The pvMs have been shown to be involved in many processes in the CNS. Early studies demonstrated that pvMs are capable of scavenging molecules injected into the cerebral ventricles. They were proposed to be involved in blood–brain barrier function and mediate the uptake of macromolecules12–14, and participate in immune regulation15. Data from animal models indicate that these cells are involved in a wide variety of brain related disorders such as cerebrovascular and neurocognitive functions and blood–brain barrier functioning in hypertension16,17, brain infections, immune activation, Alzheimer’s disease18–20, and multiple sclerosis21,22, suggesting that they are a key component of the brain-resident immune system and involved in clearing or draining of the CNS.
The Lymphatic Vessel Endothelial Hyaluronan Receptor (Lyve1) is notably expressed on lymphatic endothelial cells (LEC) and selected macrophages, but not on microglia. In the periphery, macrophage populations lining the blood vessels or nerve bundles have been characterized as Lyve1hi and Lyve1lo, respectively23. In the mouse brain, myeloid cells are a heterogeneous group of cells localized in specific niches and include parenchymal microglia and non-parenchymal pvMs8,24. Previously, brain pvMs were described to be Lyve1+CD45high and were shown to express canonical macrophage markers such as fractalkine receptor (CX3CR1), colony-stimulating factor1 receptor (Csf-1R), CD206, and Iba-1, but also the prototypic macrophage markers CD11b and F4/808,9,25,26.
Recent studies have characterized pvMs and BAM within and outside the CNS, including the peripheral nervous system23,27,28. Particularly, parenchymal Lyve1+ pvMs were shown to be heterogenous by immunofluorescence on sections25.
Here we show, using different reporter and fate-mapping mouse models and employing (whole-mount) immunofluorescence as well as flow cytometry and single-cell RNA sequencing, that the Lyve1+ pvM population in the mouse brain parenchyma is more heterogenous than previously described, and that Lyve1+F4/80+CD206+ pvMs include a previously missed CX3CR1–CD45low population within the parenchyma. Moreover, most parenchymal pvMs lacked MHC-II expression. Using multiple lymphatic reporter models, we exclude the presence of “bona fide” lymphatic endothelial cells within the brain parenchyma, thereby demonstrating that these parenchymal CD45lowLyve1+ cells are not lymphatic endothelial cells. Functionally, these unconventional pvMs are able to phagocytose macromolecules injected into the ventricles. During photothrombotic-induced stroke, pvM populations increase in numbers, without recruitment of cells from bone marrow. Hence, our data describe the heterogeneity of parenchymal pvMs and identify a previously overlooked non-conventional CX3CR1– pvM population.
Results
Identification of a Lyve1+CX3CR1– cell population in the brain parenchyma
Using whole-mount immunofluorescence of the cortex at the dorsal and ventral sides and the cerebellum, we identified Lyve1+ cells adjacent to blood vessels that did not form lumenized vessels (Figs. 1a–d, 3D whole brain in Supplementary Movie 1 and maximum intensity projection in Supplementary Fig. 1a). Morphology of these cells depended on their location. Within the dorsal cortex (Fig. 1a), Lyve1+ cells were small and spread, similar to macrophages in the pia mater. Within the ventral cortex, these cells were elongated and more stretched, as Lyve1+ cells in the hippocampus (Fig. 1b, c). In the olfactory bulb, near the cribriform plate, where cerebrospinal fluid (CSF) lymphatic drainage occurs from the sub-arachnoid space into nasal lymphatics7, numerous parenchymal Lyve1+ cells were also found to be long and stretched (Fig. 1d). Analysis of brain sections of adult Cx3cr1GFP/+ mice revealed that most Lyve1+F4/80+Iba1+CD206+ pvMs expressed as expected the CX3CR1 chemokine receptor9 (white arrows in Fig. 1e–g). However, in addition, we observed a rare population of CX3CR1–Lyve1+F4/80+Iba1+CD206+ pvMs that also display low expression of CD45 (Fig. 1h). Notably, almost all Lyve1+ cells lacked MHC-II expression (red arrow in Fig. 1i), and only few Lyve1–MHC-II+ cells were observed (pink arrows in Fig. 1i). We confirmed lack of CX3CR1GFP expression in this Lyve1+ pvM population in the Cx3cr1GFP/+ adult mouse brain by staining sections with anti-GFP (Supplementary Fig. 1b).
Fig. 1. Identification of a Lyve1+CX3CR1– population in the mouse brain.

Confocal microscopy of brain sections stained with anti-Lyve1 showing the different Lyve1+ cell morphologies in the cortex at the dorsal side (17 µm maximum intensity projection) (a), in the cortex at the ventral side (27 µm maximum intensity projection) (b), the Hippocampal fissure within the hippocampus (15 µm maximum intensity projection) (c), and in the olfactory bulb (61 µm maximum intensity projection) (d) of an adult mouse brain. e–i Immunofluorescence microscopy on sections of Cx3cr1GFP in the dorsal cortex of the mouse brain. e Staining of Cx3cr1GFP sections for Lyve1 (red) and F4/80 (white) (50 µm maximum intensity projection), f Iba1 (29 µm maximum intensity projection), g CD206 (20 µm maximum intensity projection), h CD45 (46 µm maximum intensity projection), i MHC-II (18 µm maximum intensity projection). White arrows point to conventional Lyve1+CX3CR1+ pvMs. The red arrows indicate Lyve1+CX3CR1– cells. The pink arrows indicate Lyve1−MHCII+ cells. (n = 4).
Lyve1+CX3CR1– cells are non-conventional parenchymal perivascular macrophages
In the light of the recent re-discovery of lymphatic vessels in the meningeal compartment1,6,29, we investigated whether the Lyve1+CX3CR1– population in the parenchyma of adult mice was of lymphatic origin. We observed parenchymal Lyve1+ cells in very close association with blood vessels (Fig. 2a–d, Supplementary Movie 2) outside of a structure called “pia mater cul de sac” (Fig. 2b–d). The pia mater co-migrates with the penetrating arteries into the brain parenchyma during development30, visualized by podoplanin11, surrounding arterioles within the cortex. We observed that the podoplanin+ pia was located between the vascular endothelium and the Lyve1+ cells (Fig. 2b–d, Supplementary Fig. 2a). We also observed that the Lyve1+ cells were positioned within a laminin γ1+ or laminin α1+ basement membrane within the perivascular space31 (Figs. 2e–i, 3D reconstruction movie showing CX3CR1+ and CX3CR1– cells and their nuclei in Supplementary Movies 3–4). This compartment is defined by the vascular basement membrane on the abluminal side of the vessel wall and by the parenchymal basement membrane on the CNS side32. We observed that parenchymal Lyve1+ cells did not express CD31 (Fig. 2a–k), podoplanin (Fig. 2b–d), VEGFR3 (Fig. 2j, k) or the master regulator for lymphatic endothelial identity Prox1 (Fig. 2l–o). We verified the non-lymphatic phenotype using Prox1mOrange2 and Prox1-CreErt2;Rosa26tdT reporter mice33,34 (Supplementary Fig. 2b, c). We analyzed the Prox1-CreErt2;Rosa26tdT brain, 2 weeks after Tamoxifen injection, and did not observe Prox1+ lymphatic endothelial cells, ruling out any LEC identity within the brain parenchyma. While the dura mater contains lymphatic vessels, no conventional lymphatic vessels have been described within the parenchyma or in the leptomeninges. Similarly, while we did not observe lumenized lymphatic vessels within the pia mater, we observed single Lyve1+Prox1+ lymphatic endothelial cells in the pia mater (Supplementary Fig. 2d) as was recently described for mammals35.
Fig. 2. Perivascular Lyve1+ cell location.

a 200 µm Maximum intensity projection of a lightsheet microscope acquisition of the cortex at the dorsal side in a cleared mouse brain labeled for SMA (green), CD31 (red), and Lyve1 (blue) (n = 2). b Confocal microscopy of brain sections stained for podoplanin (green), Lyve1 (red), and CD31 (blue) (n = 4), inserts (c) and d zoom in on Lyve1+ cells lining the blood vessels and placed outside of the « cul-de-sac of the pia mater » defined by the podoplanin labeling. e Confocal microscopy of a dorsal cortex section stained for CD31 (green), laminin-γ1 (red), and Lyve1 (blue) (n = 3) with insert (f) showing a higher magnification showing the Lyve1+ cells in the perivascular space (n = 2). g 55 µm cryosection of the Cx3cr1GFP brain cortex, with the location of Lyve1+(white) pvMs lining the blood vessel (CD31, blue) but within the perivascular space delineated by the laminin α1 staining (red). h CX3XR1– pvMs (red arrow), while i is a CX3XR1+ pvMs (white arrow). Higher resolutions of the inserts are shown to the right, with DAPI (yellow). 3D reconstructions of these cells highlighting the nuclei present within the cells are presented in Supplementary Movies 3 (h) and 4 (i) (n = 2). j Lyve1+ cell characterization by immunofluorescence on adult brain sections for Lyve1 (green), VEGFR3 (red), and CD31 (blue) staining. k The arrowhead points at a blood vessel and the white arrow shows a Lyve1+ cell negative for VEGFR3 and CD31. l Anti-Prox1 staining of a dorsal cortical brain area (39 µm maximum intensity projection). k high magnification of the Lyve1+ and Prox1 cell in l (n = 1). n Also in the ventral cortex Lyve1+ cells (green) did not express Prox1 (red). o High magnification from area in n showing the individual Lyve1 cells without Prox1 expression (50 µm maximum intensity projection) (n = 2).
To further rule out an astrocyte, glia, fibroblast, or pericyte identity of the Lyve1+CX3CR1– cells, we analyzed immunofluorescence staining for aquaporin-4 (AQP4) and GFAP (Supplementary Fig. 2e), ER-TR7 (Supplementary Fig. 2f), PDGFRβ (Supplementary Fig. 2g) and observed the parenchymal Lyve1+ cell population was negative for all these markers. Furthermore, we excluded a neural crest cell origin using the Wnt1-Cre; Rosa26tdT reporter mice (Supplementary Fig. 2h).
We confirmed the presence of the parenchymal CX3CR1– Lyve1+ populations in neonatal up to 1-year-old mice by flow cytometry. Using Cx3cr1GFP brain parenchyma cell suspension devoid of meninges, conventional pvMs were identified as CD64+CD206+F4/80+CX3CR1+ (Fig. 3a, gating strategy in Supplementary Fig. 3a). The CD64+CD206+F4/80+ population was both Ly6C– and Ly6G–, thus excluded monocytes, dendritic cells or neutrophils (Supplementary Fig. 3a). Microglia were Lyve1−CD11b+ and thus not part of the Lyve1+F4/80+CD11b+ populations (Supplementary Fig. 3b). At 2 days after birth (P2) we observed a small CD64+F4/80+CD206+CX3CR1− population which increased at P7, 14, and 21 (Fig. 3a, b) and peaked in adults. Subsequently, we observed a low percentage in 1-year old brain (Fig. 3a, relative CD64+F4/80+CD206+CX3CR1− cell numbers in Fig. 3b). In order to further characterize the parenchymal CD64+F4/80+CD206+CX3CR1− population, we analyzed their expression of macrophage markers compared to the CX3CR1+ population (Fig. 3a) and of microglia (Supplementary Fig. 3b). Notably, the CD64+F4/80+CD206+CX3CR1− population expressed CD45 at low levels. Other known macrophage markers, such as Lyve1 and CD163 were expressed at similar levels as the CD64+CD206+F4/80+CX3CR1+ population (Fig. 3a), indicating a macrophage identity of the CD64+F4/80+CD206+CX3CR1− population. To further confirm the macrophage phenotype, we sorted the parenchymal Lyve1+F4/80+CX3CR1+ and Lyve1+F4/80+CX3CR1− populations and determined that these population had a macrophage morphology36 (Fig. 3c).
Fig. 3. Confirmation of the Lyve1+CX3CR1– population by flow cytometry.
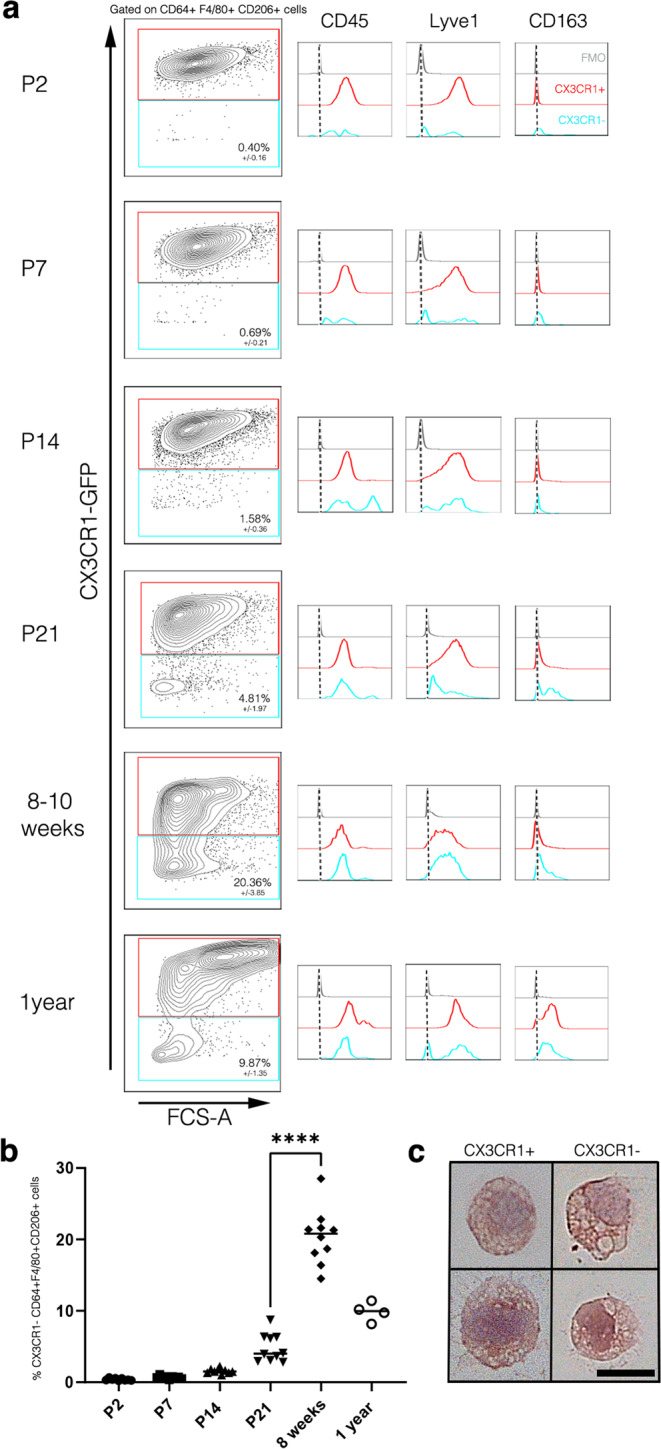
Flow cytometric analysis of Cx3cr1GFP brain parenchyma, pre-gated on living, single cells, CD64+, F4/80+, and CD206+ which excluded monocytes and neutrophils (gating strategy in Supplementary Fig. 3a). Microglia cells were excluded since these are Lyve1–CD11bhigh (Supplementary Fig. 3b). Cx3cr1 GFP/+ mouse brains were analyzed at a P2 (3 individual experiments, n = 9 animals), P7 (3 individual experiments, n = 9 animals), P14 (3 individual experiments, n = 9 animals), P21 (3 individual experiments, n = 10 animals), adult (8–12 weeks after birth, 3 individual experiments, n = 10 animals) and 1-year old brains (1 individual experiment, n = 4 animals) showing CX3CR1+/CX3CR1– gating for CD64+F4/80+CD206+ cells and subsequently on histograms for CD45, Lyve1, and CD163 expression in blue for CX3CR1– and in red for the CX3CR1+ population. FMOs are shown in gray. b Total number of CX3CR1–F4/80+CD64+CD206+ cells at the different stages. **** represent P-value ≤0.0001 (unpaired t-test). c Morphological characteristics of cytospins from CX3CR1+ and CX3CR1– pvMs sorted from brain and analyzed by hematoxylin/eosin staining. Two representative cells are shown for each population. Scale bars represents 10 µm (1 individual experiment, n = 5 animals). Source data are provided as a Source Data file.
Collectively, the cytometry profile plus their morphology confirmed that the parenchymal perivascular macrophage is heterogenous with respect to CX3CR1 expression, and that the CX3CR1– population is a bona-fide macrophage population.
Gene expression profiling of the pvM populations
To molecularly define the pvM subpopulations, we characterized their transcriptomes by single-cell RNA sequencing of the non-neuronal cell population in the cortex of juvenile (10 days), adult (7–11 weeks) and aged (1.5 years) mice (GSE133283) (Fig. 4a). The datasets of the three stages were integrated and did not show changes during aging (data not shown and ref. 37). We focused on the pvM population and identified a single pvM cluster which expressed Lyve1+ (Fig. 4b, c, violin plots of all clusters in Supplementary Fig. 4a). This cluster also expressed the macrophage markers Mrc1 (encoding CD206), Emr1 (encoding F4/80), Fcgr3 (encoding CD64), and others (Fig. 4c). However, this pvM cluster did not express H2-Aa, the genes encoding MHCII in C57Bl/6 mice (Fig. 4b, c). Generally, Cx3cr1 and Ptprc expression levels were lower than in other pvM/BAM populations or in the microglia cluster which was identified by high Siglech expression (Fig. 4b, c). Isolating the cells from the pvM cluster with the highest Lyve1 expression revealed no further unsupervised segregation based on Cx3cr1 or Ptprc (Fig. 4e). However, there appeared to be 2 segregated pvM subclusters, based on e.g., Lyve1, CD209f, CD209g expression in subcluster 1 and Atf3, Fos, Junb in the other (Fig. 4d and Supplementary Fig. 4b).
Fig. 4. Transcriptional profiling of the pvM subsets.
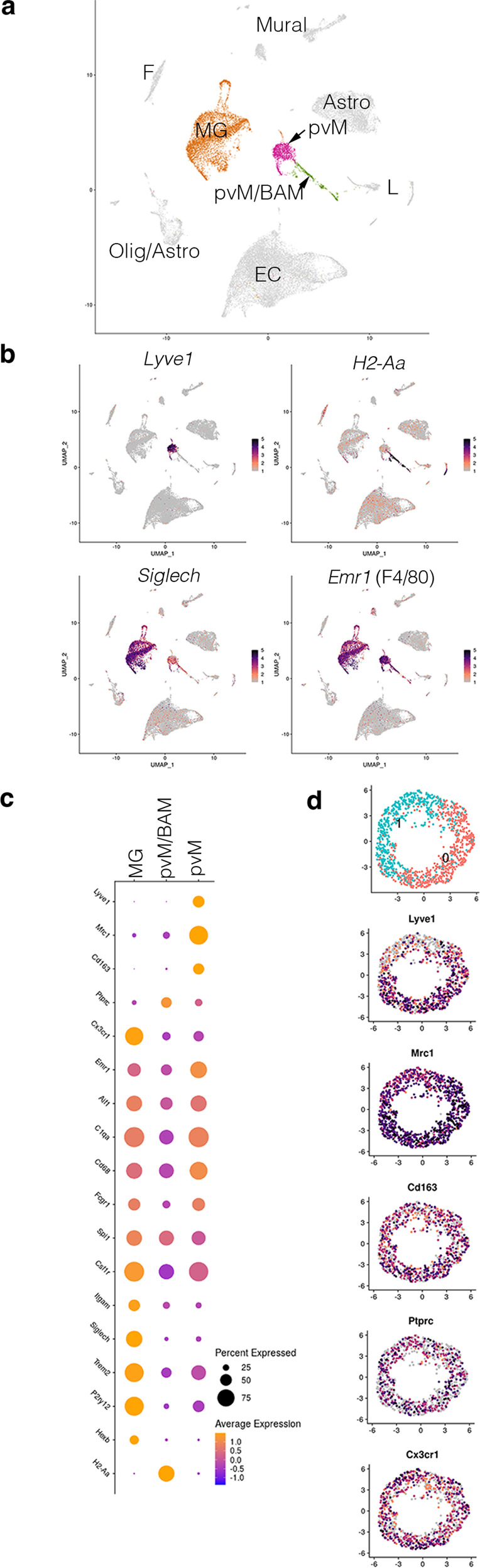
Replicates of mouse brain cortex, excluding neurons and meninges. Shown is the integrated dataset of samples at 10 days (n = 3, 12,843 cells), adult (7–11 weeks, n = 2, 7817 cells), and aged (1.5 years, n = 3, 10,448 cells). Cluster analysis on integrated data (a) focused on the pvM, the pvM/BAM and the microglia clusters. b The pvM cluster was indicated as the sole cluster with Lyve1 expression, but H2-Aa (encoding one of the MHC-II genes) negative. This population was segregated completely from the Siglech expression microglia, which was also Lyve1–. Emr1 (encoding F4/80) is expressed in all 3 clusters. c Dot-plot of the genes associated with macrophage or microglial identity, showing that pvMs is the only cluster which is expressing Lyve1. pvM/BAM is characterized by H2-Aa expression, but lacking Lyve1. The microglia population in cluster 3 was identified by lack of Lyve1, CD163, H2-Aa, and low Mrc1 (encoding CD206) or Ptprc (encoding CD45) expression. EC endothelial cell, MG microglia, pvM perivascular macrophage, BAM border-associated macrophage, L Lymphocytes, F fibroblast, Astro astrocyte, Olig Oligodendrocyte, Mural Mural cells. d Cell from the cluster with the highest Lyve1 expression, the pvM, were isolated to analyze a possible segregation in Cx3cr1+ and Cx3cr1– expression population within this population. Even though we observed 2 subclusters, also based on Lyve1 expression, there was no segregation based on Cx3cr1 or Ptprc.
Taken together, our data rule out a lymphatic identity of the CX3CR1–Lyve1+ cells and establish that these cells are bona-fide pvMs.
The origin of CX3CR1– Lyve1+ perivascular macrophages
PU.1 is considered to be a master regulator of macrophage differentiation. Therefore, we analyzed the Spi1GFP/GFP mice, in which the Spi1 locus (encoding PU.1) is inactive and lack all macrophages and die around birth9,38. In E18.5 Spi1GFP/+ embryos, we identified Lyve1+PU.1+CD45+ and Lyve1+PU.1–CD45low pvMs (Fig. 5a). In contrast, E18.5 Spi1GFP/GFP embryos lacked Lyve1+ cells in the brain parenchyma (Fig. 5b, positive control of Lyve1+ cells in the skull in Supplementary Fig. 5a). This demonstrated that Lyve1+CD45low pvMs depend on PU.1 during their development, but some cells downregulated its expression (Fig. 5c). All Lyve1+ cells within the sections also expressed F4/80 and CD206 (Supplementary Fig. 5b). To confirm lack of PU.1 expression, we stained the PU1-GFP with an anti-GFP and confirmed lack of GFP signal (Supplementary Fig. 5c).
Fig. 5. The origin of CX3CR1– Lyve1+ perivascular macrophages.

a Immunofluorescence on sections of the head of a E18.5 Spi1GFP/+ embryo (PU1-GFP in green), labeled for Lyve1 (red) and CD45 (white) (60 µm maximum intensity projection) shows the presence of the conventional (white arrows) and the non-conventional pvM populations (red arrows) at this stage (n = 3). b No GFP nor Lyve1 fluorescence was observed in the Spi1GFP/GFP E18.5 dorsal cortex (33 µm maximum intensity projection) (n = 2). c Immunofluorescence on Spi1GFP/+ adult mouse brain (48 µm maximum intensity projection) (n = 3). d Immunofluorescence on sections of Cx3cr1GFP/+; Cx3cr1-Cre; Rosa26tdT mouse brain showing that conventional (white arrows) and non-conventional pvM population (red arrows) are expressing tdTomato (n = 3). e Flow cytometry analysis of Cxcr4-CreErt2; Rosa26tdT brain parenchyma, gated on living, single cells and CD64+ F4/80+ CD206+ cells. Subsequently histograms of CD45 and Lyve1 expression of the CX3CR1–tdTomato– population in blue, the CX3CR1+tdTomato− population in red and FMO in gray (n = 3). Source data are provided as a Source Data file.
To establish whether the cells were derived from a CX3CR1 macrophage precursor, we crossed the reporter Cx3cr1GFP animals with mice harboring an inducible Cx3cr1-Cre;Rosa26tdT reporter. In the resulting animals tdTomato is expressed in all cells, which at some point in their ontogeny expressed CX3CR1, while GFP is expressed only in those cells which express CX3CR1 at the time of analysis. As expected, microglia expressed both tdTomato and GFP (Fig. 5d). All Lyve1+ pvMs expressed tdT but not all pvMs expressed GFP (Fig. 5d plus multiple sections in Supplementary Fig. 5d). Therefore, as for PU.1, the CX3CR1– pvMs express CX3CR1 during their ontogeny, but subsequently lose this expression, as has been reported for other tissue macrophages including Kupffer cells39.
The Cxcr4-CreErt2 lineage-tracing model is used to label all embryo derived hematopoietic progenitors, but excludes yolk-sac-derived hematopoietic cells such as microglia and pvMs10,27,28,40. In the brains of naïve Cxcr4-CreErt2;Rosa26tdT mice injected with 4-OHT four weeks before isolation, to label all bone marrow hematopoietic stem cells (HSC), we observed that an average of 97.3% of the CD64+F4/80+CD206+CX3CR1+ pvMs and 96.7% of the CD64+F4/80+CD206+CX3CR1– pvMs were tdTomato– (Fig. 5e, gating strategy in Supplementary Fig. 5e). As validation for the model, and as was previously shown40, microglia were not labeled in this model, while monocytes and neutrophils were labeled (Supplementary Fig. 5e). Collectively, these data establish that as previously suggested10, in naïve animals CX3CR1– and CX3CR1+Lyve1+ parenchymal pvMs are not derived from HSC, but their ontogeny involves a CX3CR1+ macrophage precursor.
Functional analysis brain pvM populations
Besides their role as source for chemokines and growth factors to regulate immune responses, pvMs display phagocytic activity, which could be part of a broader role of these cells in tissue homeostasis23,41. To address a potential role of the CX3CR1–Lyve1+ pvMs in fluid drainage or macromolecule clearance, we injected 2.5 µl 10kD Dextran-AlexaFluor647 or Acetylated-LDL-AlexaFluor594 into the lateral ventricle of Cx3cr1GFP mice. To exclude recirculation through the blood stream, we analyzed the brain 10 min after injection3. We observed that both CX3CR1– and CX3CR1+Lyve1+cells phagocytosed the injected dyes near the ventricle, in the dorsal and in the ventral cortex (Fig. 6a–f). We conclude that like their CX3CR1+ counterparts, CX3CR1– Lyve1+ cells were able to efficiently take up lipoproteins and glycoproteins in the range of at least 3–10 kDa similar to conventional pvMs. The close proximity of the pia mater extending towards the third ventricle (3 V) could explain direct drainage from the injection-site within the lateral and connected 3 V towards the pia mater (Fig. 6g). However, pia dura drainage is unlikely to occur via LECs, since very few Lyve1 and Prox1mOrange2 positive nuclei were observed within the pia mater (Fig. 6g).
Fig. 6. CX3CR1– pvM share phagocytic functioning with CX3CR1+ pvM.

a–c Cx3cr1GFP mouse brains were injected intra-ventricular with Dextran-Alexa647 (37 µm maximum intensity projection) (n = 3) and d–f Acetylated-LDL-Alexa594 (42 µm maximum intensity projection) (n = 3). Ten minutes after injection, mice were euthanized and brains dissected. Confocal analysis on brain sections shows Lyve1 (white), Dyes (red), and CX3CR1 (green). The unconventional pvMs phagocytosed the dyes (red arrows), as well as did conventional pvMs (white arrows) (b, c, e, f). g Tiled confocal acquisition of a Prox1mOrange2 brain section. Lyve1+ cells in green lined blood vessels stained for CD31 in blue. Lyve1+ cells in green penetrated the brain to the hippocampal fissure (hif) and all the way towards the third ventricle (3 V). (n = 4).
Involvement in central nervous system diseases
Emerging data suggested that in stroke pvMs can influence blood–brain barrier function42,43. In patients with intracerebral hemorrhage and focal cerebral ischemia, an accumulation of CD163+ cells around brain blood vessels, which also contained myelin, was observed44. However, it remains unclear whether these cells are pvMs, other BAM or monocytes45.
To assess the role of the pvMs in cerebrovascular pathologies, we investigated contributions of these cells in a photothrombosis (PT) model of ischemic stroke. PT was induced in 8-week-old male mice and we observed an increase in Lyve1 staining density in whole-mount stained brains (Fig. 7a, quantification of whole-mount staining density as shown in Supplementary Fig. 6, Supplementary Movies 5 and 6). At day 14 after stroke induction (P14), we noticed a significant increase in Lyve1 staining density within the hippocampus (4.2x, p < 0.05), although no significant difference was observed in the dorsal cortex (Fig. 7b). At P30, Lyve1 staining density had normalized to control values (Fig. 7b). We confirmed the increase in the pvMs by flow cytometry at P3, P8, and P14 after stroke using Cx3cr1GFP mice (Fig. 7c, d). After PT, the CX3CR1+Lyve1+F4/80+population increased, while the CX3CR1–Lyve1+F4/80+ population decreased (Fig. 7c, d).
Fig. 7. CX3CR1+ pvM numbers increase after induced ischemic stroke.

a Maximal intensity projections (sagittal view) (2660 µm) of lightsheet acquisitions on cleared brains, stained for Lyve1 (red), control and after stroke (P14 = 14 days after induction of ischemic stroke) (2 individual experiments, n = 7 animals) (Supplementary Movies 5 and 6). b Quantification of Lyve1 in the area containing the whole stroke lesion within the dorsal cortex situated just above the hippocampus, imaged by lightsheet microscopy. * represents P-value ≤0.05 (Ordinary one-way Anova test). c Representative flow cytometry plots from Cx3cr1GFP brain parenchyma 14 days after induced stroke (P14), pre-gated on living, single and showing Lyve1+F4/80+ cells and CX3CR1+/CX3CR1– gating for these cells Lyve1+ F4/80+ and subsequently in blue the CD45 expression of the Lyve1+F4/80+CX3CR1–, in red the CD45 expression of the Lyve1+F4/80+CX3CR1+ and FMO in gray. Microglia cells were excluded since they are Lyve1–. Average percentages for the different subpopulations are shown in the plots (n = 4). d Graphs representing the Lyve1+F4/80+ cell % and Lyve1+F4/80+CX3CR1+ cell % showing an increase of these populations at P3, P8, and P14 after stroke by flow cytometry and Lyve1+F4/80+CX3CR1– cell % showing a decrease of this population at P3, P8, and P14 after stroke. Data are presented as mean value ± SD. Asterisk (*) represents P-value ≤0.05, **P-value ≤0.01, ***P-value ≤0.001, and ****P-value ≤0.0001 (unpaired t-test). e Graphs representing the Lyve1+F4/80+ cell % and Lyve1+F4/80+CXCR4+ cell % showing an increase of the first population at P14 after stroke by flow cytometry. Asterisk (*) represents P-value ≤0.05 (unpaired t-test). Source data are provided as a Source Data file.
Such an increase in pvMs could be due to the migration of bone-marrow-derived monocytes into the lesion and subsequent to differentiation to pvMs, or result from local proliferation of the tissue resident pvMs. Lyve1+ cells in and near the lesion were not positive for Ki67 immunofluorescence (data not shown). To determine if the increase originated from bone marrow HSC, we analyzed Lyve1+F4/80+ pvMs in the brain of Cxcr4-CreErt2;Rosa26tdT male mice, which received 4-OHT four weeks before PT induction40. As observed under naïve conditions (Fig. 5e), most pvMs were tdTomato–, and there was no increase in tdTomato positive cells at 14 days after PT induction (Fig. 7e), suggesting local (progenitor) proliferation of pvMs rather than replacement by bone-marrow-derived monocytes.
Discussion
Here we provide an in-depth study of the perivascular macrophage compartment at the brain borders, including high end imaging, fate mapping as well as phenotypic and transcriptomic profiling.
Detailed analysis for the presence of lymphatic endothelial cells within the CNS using different reporter mice combined with immunostaining for lymphatic endothelial cell markers did not reveal evidence for these cells within the parenchyma. Only few isolated lymphatic endothelial cells were observed within the pial meninges, as described previously35. Using flow cytometry, single-cell RNA sequencing, immunofluorescence and lineage-tracing models, we observed that the pvM population within the CNS was almost exclusively Lyve1+, CD45low-int and lacked MHC-II expression. Low MHC-II expression on peripheral perivascular Lyve1hi macrophages was described before23. How the Lyve1+MHC-II– brain parenchymal pvMs relate to these peripheral Lyve1hiMHC-II– monocytes remains unclear. We observed very few Lyve1–MHC-II+ cells within the parenchyma, and the single-cell RNA sequencing clearly separated the Lyve1+MHC-II– and Lyve1–MHC-II+ pvM subsets. However, we cannot rule out contamination with choroid plexus cells containing BAMs, which might explain the lower CD163 and higher CD45/Ptprc expression of these cells. The Lyve1+ pvMs were located within the perivascular space around arterioles mainly, but not veins. This confirms a recent study in which Lyve1+ pvMs were detected around Cx40 positive arteries and large EphB4 positive veins25.
Our data show that the brain parenchymal pvM population is more heterogenous than previously described and include a non-conventional CX3CR1–Lyve1+ subpopulation. These expressed classical macrophage markers such as F4/80, CD11b, CD206, CD64, and CD163 and clustered together with CX3CR1–Lyve1+ pvMs in single-cell sequencing analyses. The CX3CR1–Lyve1+ population peaked at adulthood reaching around 20% of the Lyve1+ pvM population and its numbers decreased in aged animals. Single-cell RNA sequencing data however did not reveal differences in the gene expression patterns between these stages, indicating that the population did not change drastically over time. Similar to their CX3CR1+ countparts, the CX3CR1–Lyve1+pvMs were situated outside the cul-de-sac of the pia mater and within the Virchow-Robin space, which is likely to be associated with a function in drainage or clearance of brain parenchyma-derived metabolites or breakdown products9,43,46. In adult tissues at steady-state, pvMs are believed to have important functions related to their perivascular location, such as the regulation of vascular permeability, phagocytosis of blood-transmissible pathogens, antigen presentation, or immune regulation9,41. Analysis of phagocytosis activity, revealed that CX3CR1– pvMs displayed similar uptake as CX3CR1+ pvMs.
Fate-mapping studies have established that many tissue macrophages, such as alveolar macrophages, Langerhans’ cells and Kupffer cells originated from CX3CR1+ precursors but cease to express Cx3cr137. In a recent detailed study on the origin and lineage of the pvM, it was shown that the pvMs are derived from the yolk sac hemogenic endothelium and depend on local factors to differentiate10. The use of the Spi1GFP/GFP (PU.1) and Cx3cr1-Cre models showed that the CX3CR1– pvMs are most likely derived from the same lineage as the pvM population, thus, from yolk-sac-derived progenitors. Furthermore, our data suggest that replacement and increase of CX3CR1–Lyve1+ cells is independent from monocytes by using the inducible Cxcr4-CreErt2tdT lineage-tracing model40, as was shown in the naïve situation recently10. We extend these data, by establishing that also the pvMs increase after stroke is mostly due to local proliferation with limited monocyte contribution.
In conclusion, here we demonstrate that the majority of brain parenchymal pvMs are Lyve1+MHCII–CD45low/int and the population is more heterogenous than previously described. The specific function of non-conventional CX3CR1– pvMs as compared to the conventional pvMs remains to be determined. These cells are consistently observed within the brain and may act in drainage or clearance of brain parenchyma-derived metabolites or breakdown products which have been proposed to flow along arterioles into the CSF11. Presently, there are no unique markers available to target these cells specifically, and to study their specific role in brain physiology in the future.
Methods
Mice
C57BL/6 J (stock number 000664) mice were obtained from Charles River (France). Prox1-CreERT2+/− 34, Wnt1-Cre39, and Cx3cr1GFP;Cx3cr1-CretdT mice were kindly provided by Dr. Bajénoff (CIML, Marseille, France). Cx3cr1GFP mice47 were kindly provided by Dr. Lelouard (CIML, Marseille, France). Spi1GFP (PU.1)38, Cxcr4-CreErt2;Rosa26tdT 40, and Prox1mOrange2 48 were maintained at the CIML at SPF conditions (Marseille, France). All mice were co-housed in the same room under similar conditions with water and food ad libitum and 12 h/12 h night/daylight cycle.
Prox-CreERT2+/− and Wnt1-Cre lines were crossed to homozygosity for the tdTomato reporter using Rosa26tdT mice49. 1.6 mg 4-hydroxy-tamoxifen (4OHT) was injected IP 4 weeks before the isolation of the Cxcr4-CreErt2;Rosa26tdT adult brain and 2 weeks before the isolation of the Prox-CreERT2+/−;Rosa26tdT brain. All animal studies were reviewed and approved by the local ethics committee of Aix-Marseille University and the Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation. We used males for the experiments.
Antibodies in Supplementary Table 1
Cell preparation and flow cytometry
Cx3cr1GFP mice were euthanized with an overdose of ketamine-xylazine anesthetic and perfused with phosphate buffered saline (PBS) plus heparin. Brains were isolated, dura was removed, and the brain cut sagitally in 6–8 pieces, then digested with the Adult Brain Dissociation Kit (Miltenyi, 130-107-677) at 37 °C for 30 min on the gentle MACS dissociator (Miltenyi). Brain cell suspensions were filtered over 70 µm strainers and filters were washed with 10 mL HBSS 2%FBS. After centrifugation (5 min, 400 × g, 4 °C), debris were removed using a 40% Percoll (Sigma–Aldrich, GE17-0891-02) solution in PBS. Cells were centrifuged (30 min, 500 × g, 4 °C) and supernatant was discarded. Red blood cell lysis was performed for 10 min at 4 °C using the Miltenyi RBC lysis buffer from the kit. Lysis was stopped with 9 mL HBSS 2%FBS and cells were centrifuged (5 min, 400 × g, 4 °C). Cells were then blocked (15% normal mouse serum (Jackson Immunoresearch 015-000-120) in FACS buffer (HBSS 2% FBS) for 15 min and subsequently stained for CD45-BUV395 (BD biosciences 564279), Lyve1-eFluor660 (eBioscience 50-0443-82), F4/80-BV421 (Biolegend 123131), CD64-BV711 (Biolegend 139311), CD206-BV785 (Biolegend 141729), CX3CR1-PECy7 (Biolegend 149015), Ly6C-BV510 (Biolegend 128033), LY6G-AF700 (BD Pharmingen 561236), and CD163-SB600 (eBioscience 63-1631-82) diluted in FACS buffer for 30 min on ice. The staining of dead cells with Fixable NIR was performed for another 30 min after the antibody staining in protein free HBSS right after washing the cells. Cells were subsequently washed and resuspended in FACS buffer. Samples were acquired on the LSRFortessa X-20 cytometer (BD Biosciences). Data analyses were done using FlowJo software (version 10, FlowJo, LLC).
Cell sorting
Cells were prepared and stained with Lyve1 and F4/80 antibodies and Fixable NIR as describe above. Live Lyve1+ F4/80+ Cx3cr1−, live Lyve1+ F4/80+ Cx3cr1+ cells were sort-purified separately using the BD FACS ARIA III SORP sorter.
Cell morphology
Sorted cells were spun onto Shandon Cytoslides with a Thermo Shandon Cytospin 4 cytofuge (4 min at 400 × g), fixed with methanol and stained with hematoxylin and eosin.
Gene expression analysis
Cerebral cortices were dissected from brains of perfused 10-day, 8–10-week, and 50-week-old male C57/Bl/6 mice, minced into small pieces in ice cold DMEM (Sigma–Aldrich) and digested at 60 min at 37 °C in an enzyme-cocktail containing 30U/ml papain (LK003153; Worthington), 40 µg/ml DNase type IV (LS006331; Worthington), and 0.125 mg/ml Liberase TM (5401119001; Sigma–Aldrich) in DMEM. Myelin was removed by mixing the cell slurry with 22% BSA in PBS and centrifugation at 1000 × g for 10 min; pellets were collected. Single-cell suspensions were prepared and cells were counted using Luna-II automated cell counter (Logos Biosystems). For scRNASeq, cells were encapsulated in Gel bead emulsion using the 10X Chromium system (10X Genomics). Libraries were prepared according to the manufacturer’s instructions using Chromium Single Cell 3’ Library & Gel Bead Kit v3 (10X Genomics) and sequenced in Illumina NextSeq 500 using High Output Kit v2.5 (150 cycles, Illumina). The Cellranger (10x genomics) pipeline was followed to demultiplex the sequencing data, to align it to the mouse reference genome (mm10), to count the reads, and to aggregate the resulting cell-gene matrices according to sample replicates.
Gene expression data analysis was performed with Seurat R package50 (version 4.1.0). Quality control was performed on each sample replicate from juvenile, adult and aged time-points to remove poor quality cells and genes from the analysis using CreateSeuratObject function. Genes expressed in less than three cells were removed from the analysis as well as cells expressing less than 200 genes. Cells were also removed using an in-house developed R script that filter cells based on their number of detected genes, number of UMIs and percentage of mitochondrial and ribosomal genes using values in the table below. Data from sample replicates from a same time point were merged together and their respective expression matrices were log-normalized using NormalizeData function with a scale factor of 10,000). Their respective variable genes were found by FindVariableFeatures function using the “vst” method and the top 2000 variable genes were selected. Merged datasets from the three time-points (juvenile, adult and aged) were integrated using genes selected by SelectIntegrationFeatures with nfeatures = 2000 then using FindIntegrationAnchors and IntegrateData with default parameters. The first 30 principal components from Principal Component Analysis were used for the UMAP non-linear dimensionality reduction and cells were clustered using Louvain clustering from FindClusters function with a resolution of 0.4. The differentially expressed genes from each cell cluster were identified using the Wilcoxon Rank Sum method from FindAllMarkers function with minimum fraction of cells expressing the gene over 10% and with a log fold-change upper than 0.25. Cellular identity of each cluster was determined comparing those differentially expressed genes with known gene markers specific to cell type from previous studies (Supplementary Table 2).
Whole-mount staining and lightsheet imaging
Anesthetized mice were perfused with PBS-Heparin 5U/ml (Sigma) and subsequently overnight (ON) immersion fixed in paraformaldehyde 4% (PFA-Electron Microscopy Science, ref 15714) in PBS. The iDISCO protocol was used for immunostaining51, prior to which brains were dehydrated in increasing methanol (MetOH) concentrations diluted in PBS (20, 40, 60, 80, and twice 100%) for 30 min for each concentration at RT. Subsequently specimens were incubated ON in a dichloromethane (DCM, Sigma 270997)-MetOH mixture (2 vol DCM:1 vol MetOH) at RT. After two 10 min incubations in absolute MetOH, brains were bleached in 5% H2O2 in MetOH ON at 4 °C and subsequently rehydrated by a decreasing MetOH series (80, 60, 40, 20% in water), followed by PBS and two washes in PBS-Triton X100 (Tx) 0.2% for 1 h each. Bleached brains were permeabilized for 2.5 days at 37 °C (0.4%Tx, 20%DMSO, 2.3% Glycine in PBS) and subsequently blocked with [PTwH (PBS, Tween20, Heparin), 10% DMSO, 6% serum] for 4.5 days at 37 °C. Whole-mount stainings were performed by incubation with primary antibodies for 5 days at 37 °C and subsequently with Alexa-dye coupled secondary antibodies diluted in PTwH containing 3% serum for 5 days at 37 °C. Following each staining step, samples were extensively washed in PTwH (10 min, 15 min, 30 min, 1 h, 2 h, and ON at RT). Finally, the samples were again dehydrated by increasing MetOH concentrations diluted in water (20, 40, 60, 80, 2×100% and ON in absolute MetOH), for 1 h each step at RT and cleared in a MetOH/BABB mix (1:1) [BABB (benzyl alcohol and benzyl benzoate = 1:2) (Sigma 305197 and Fisher Scientific 10654752)] for 8 h at RT and finally placed in BABB ON at RT to complete clearing. All incubations were done under mild agitation. After clearing, brains were imaged using a LaVision Ultramicroscope II (Miltenyi Biotec). Stacks were captured with a step size of 5 µm at ×2.5 magnification using an optic zoom with a NA = 0.144. 3D reconstruction, cell counting and analysis of the sample image stacks were performed using IMARIS software (Version 9.1.0, Bitplane). For Lyve1 quantification, an area of interest was selected (Dorsal cortex, Hippocampus) using the program’s surface function and then a new channel corresponding to the fluorescence to quantify was created in this region. Using this workflow, a quantification of volume (in µm3) of Lyve1 stained material is possible, which was further adapted for volumetric quantifications (Supplementary Fig. 6).
Vibratome section immunofluorescence staining and confocal imaging
Anesthetized mice were perfused with PBS/heparin, brains dissected and fixed ON in 4% PFA in PBS at 4 °C and subsequently embedded in 1% low melting agarose for generation of 100 µm vibratome slices (Leica, VT1000S). Sections were blocked in EBT buffer (EBSS, 0.05% Tx, 1% BSA) containing 10% serum for 2 h at RT under agitation. Immunostainings were performed by incubation in primary antibodies for 48 h at 4 °C in EBT, 3% sera and subsequently with Alexa-fluorochrome coupled secondary antibodies diluted in EBT, 3% sera for 24 h at 4 °C. Following each staining step, samples were washed several times in PBS-Tx (0.05% Tx in PBS) and in PBS. Sections were finally cleared in Histodenz (Sigma D2158) medium for 48 h at RT and subsequently mounted in Histodenz medium. Confocal images were acquired at RT on a confocal microscope (LSM880, Zeiss, Germany), with a ×20/0.4 Plan-Apochromat objective and using laser lines at 405, 488, 561, and 633 nm for the excitation of AlexaFluor405/GFP/AlexaFluor555/AlexaFluor647, respectively. Fluorescence was recorded in individual channels acquired in a sequential mode using a highly sensitive 32-channel GaAsP detector. Channels were, respectively, detected using these detection bands: A405 (410–470 nm), A488 (490–540 nm), A555 (565–640 nm), A647 (640–690 nm). The pinhole was set to 1 airy unit. Z stack were acquired with an optical thickness defined for each image in figure legends, satisfying the Nyquist resolution criterion. Image processing (contrast enhancement, scale bars, etc.) was done with ImageJ (National Institutes of Health) without actions modifying image integrity.
Infusion of tracers into lateral ventricle
Mice were anesthetized (150 mg/kg Ketamine and 10 mg/kg Xylazine) and fixed in a stereotaxic frame. The skull was thinned with a dental drill at a location of 0.95 mm lateral and 0.22 mm caudal from the bregma. A 30 G needle with a silica fiber tip (Phymed) was inserted into the right lateral ventricle 2.35 mm ventral to the skull surface as previously described3. Injection of 2.5 μL acetylated-LDL-Alexa594 (ThermoFisher) or Dextran-Alexa647 10 kDa (ThermoFisher) tracer was done at a speed of 0.5 μL/min using a high precision syringe pump. The needle was left in place for 5 min and slowly retracted confirming lack of detectable backflow. To avoid recirculation through the blood stream into the brain, mice were euthanized 10 min after injection3. The mice were perfused with PBS and 4%PFA/PBS, brains isolated and fixed ON in 4% PFA/PBS at 4 °C, subsequently washed and then stored at 4 °C in PBS until further analysis.
Photothrombosis (PT) model of ischemic stroke
Stroke using the PT model was induced in C57BL/6 J, Cx3cr1GFP, and Cxcr4-CreErt2;Rosa26tdT mice. 1.6 mg of 4-OHT was injected 3 consecutive days in the Cxcr4-CreErt2;Rosa26tdT mice 4 weeks before the induction of the lesion, based on a protocol previously used for induction of stroke within the Cxcr4-CreErt2;Rosa26tdT model40. Mice were anesthetized by intraperitoneal Ketamine/Xylazine injection as before and the eyes of the mice were covered with Ocry-gel to protect them from light and dehydration. The skin on the skull was incised from the eyes to the neck and retracted to the edges of the skull. After retro-orbital injection of 100 µL Rose Bengal (Sigma, 330000) a Leica, KL 1600LED cold light source was placed in contact with the skull and the illuminated region was precisely adjusted (using a stereotaxic instrument) to 2.5 mm caudal of the Bregma and 2.0 mm to the Lambda. After illuminating a 1 mm diameter area at power 3 for 15 min, the skin was put back into place, stitched and the animals returned to their home cages. Food was provided in a plate and Buprenorphine (0.4 mg/ml) was added to the drinking water. Mice were euthanized between 14- and 30-days post-induction and brains were analyzed by whole-mount immunofluorescence and flow cytometry.
Statistical analysis
Graphs, average values and standard deviation (SD) shown in all figures were calculated using Prism (version 8.3.0 GraphPad software) software. One-way Anova test was used to determine significance. The number of individual experiments can be found in the legends of all the figures. Photoshop software (CC2015, Adobe) was used to generate figures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We acknowledge the PICSL imaging facility, notably Mathieu Fallet and Sebastian Mailfert, of the CIML (ImagImm), member of the national infrastructure France-BioImaging. Lionel Chasson of the histology platform for the relentless cutting. We thank Toufik Guelmami and Michel Pontier. The flow cytometry core facility, notably Marc Barad, Sylvain Bigot, and Laurence Borge. We thank Hugues Lelouard and Marc Bajénoff for providing us with mouse models. We thank Lucas Bertoia for his help and Sandrine Henri, Marc Bajenoff, and Guillaume Hoeffel for their discussions. We thank Steffen Jung for the discussions and support. The work was supported by the FRM Amorçage jeunes équipes (AJE20150633331), ANR ACHN (ANR-16-ACHN-0011), ANR PRCI (ANR-17-CE13-0029-01), A*midex Chaire d’excellence to SAvdP and institutional grants to the CIML from INSERM, CNRS, and Aix-Marseille University. The project leading to this publication has received funding from the German Research Foundation SO285/11-1, the Marie Sklodowska-Curie International Training Network ENTRAIN (Grant Agreement #813294), the « Investissements d’Avenir » French Government program managed by the French National Research Agency (ANR-16-CONV-0001) and from Excellence Initiative of Aix-Marseille University—A*MIDEX. France-BioImaging is supported by the French National Research Agency (ANR-10-INBS-04). MHS was supported by institutional grants from TU Dresden, ANR-17-CE15-0007-01 and ANR-18-CE12-0019-03, INCa (13-10/405/AB-LC-HS), Fondation ARC pour la Recherche sur le Cancer (PGA1 RF20170205515), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement number 695093 MacAge) and an Alexander von Humboldt Professorship at TU Dresden. R.H.A. and H.W.J. were supported by the Max Planck Society, the Leducq Foundation and the DFG programs SFB1348, SFB1009, SFB1366, and FOR2325. F.K. is funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB1348/1—386797833 and SFB1450/1—431460824, the Cells-in-Motion Cluster of Excellence (EXC 1003—CiM, 194347757), and the IZKF Münster (Kief / 019 / 20).
Source data
Author contributions
C.S., M.L., H.W.J., M.S., S.K., S.K.R.S., K.K., S.W., H.H., L.F., and A.T. performed experiments. S.A.v.d.P. conceived and supervised the study. C.S., M.L., J.B., M.S., and S.A.v.d.P. analyzed data. C.S., M.L., H.W.J., M.S., A.T., M.F., S.S., M.H.S., R.S., L.S., R.H.A., S.S.M., F.K., and S.A.v.d.P. contributed to discussion and wrote, illustrated, reviewed, or edited the manuscript. All authors approved the submitted version of the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
All datasets generated for this study are included in the article/Supplementary Material. Raw data (fastq files) and processed data (gene counts) for single-cell RNA-Seq analysis have been deposited in the Gene Expression Omnibus with the primary accession number, GSE13328337. Source data are provided with this paper.
Code availability
Scripts and detailed instructions to reproduce the NGS analysis for this manuscript are available on GitHub repository https://github.com/JulieBvs/SPlab_PVM2.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-35166-9.
References
- 1.Aspelund A, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn, J. H. et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature572, 62–66 (2019). [DOI] [PubMed]
- 3.Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat. Commun. 2017;8:1434. doi: 10.1038/s41467-017-01484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Lessen, M. et al. Intracellular uptake of macromolecules by brain lymphatic endothelial cells during zebrafish embryonic development. Elife6, e25932 (2017). [DOI] [PMC free article] [PubMed]
- 5.Absinta M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017;6:e29738. doi: 10.7554/eLife.29738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature10.1038/nature14432 (2015). [DOI] [PMC free article] [PubMed]
- 7.Decker, Y. et al. Magnetic resonance imaging of cerebrospinal fluid outflow after low-rate lateral ventricle infusion in mice. JCI Insight, 7, e150881 (2022). [DOI] [PMC free article] [PubMed]
- 8.Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017;18:385–392. doi: 10.1038/ni.3703. [DOI] [PubMed] [Google Scholar]
- 9.Goldmann T, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016;17:797–805. doi: 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masuda T, et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature. 2022;604:740–748. doi: 10.1038/s41586-022-04596-2. [DOI] [PubMed] [Google Scholar]
- 11.Zhang ET, Inman CB, Weller RO. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J. Anat. 1990;170:111–123. [PMC free article] [PubMed] [Google Scholar]
- 12.Mato M, Ookawara S, Kurihara K. Uptake of exogenous substances and marked infoldings of the fluorescent granular pericyte in cerebral fine vessels. Am. J. Anat. 1980;157:329–332. doi: 10.1002/aja.1001570308. [DOI] [PubMed] [Google Scholar]
- 13.Mato M, et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc. Natl Acad. Sci. USA. 1996;93:3269 LP–3263274. doi: 10.1073/pnas.93.8.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bechmann I, et al. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci. 2001;14:1651–1658. doi: 10.1046/j.0953-816x.2001.01793.x. [DOI] [PubMed] [Google Scholar]
- 15.Fabriek BO, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia. 2005;51:297–305. doi: 10.1002/glia.20208. [DOI] [PubMed] [Google Scholar]
- 16.Faraco G, et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J. Clin. Invest. 2016;126:4674–4689. doi: 10.1172/JCI86950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santisteban MM, et al. Endothelium-macrophage crosstalk mediates blood-brain barrier dysfunction in hypertension. Hypertension. 2020;76:795–807. doi: 10.1161/HYPERTENSIONAHA.120.15581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of β-amyloid in cerebral amyloid angiopathy. Proc. Natl Acad. Sci. USA. 2009;106:1261 LP–1261266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl Acad. Sci. USA. 2010;107:20816 LP–20820821. doi: 10.1073/pnas.1005888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park L, et al. Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer Aβ peptides. Circ. Res. 2017;121:258–269. doi: 10.1161/CIRCRESAHA.117.311054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polfliet MMJ, et al. The role of perivascular and meningeal macrophages in experimental allergic encephalomyelitis. J. Neuroimmunol. 2002;122:1–8. doi: 10.1016/S0165-5728(01)00445-3. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, et al. Parenchymal accumulation of CD163+ macrophages/microglia in multiple sclerosis brains. J. Neuroimmunol. 2011;237:73–79. doi: 10.1016/j.jneuroim.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Chakarov S, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363:eaau0964. doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 24.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 25.Karam M, et al. Heterogeneity and developmental dynamics of Lyve-1 perivascular macrophages distribution in the mouse brain. J. Cereb. Blood Flow. Metab. 2022;0:1–16. doi: 10.1177/0271678X221101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeisel A, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 27.Utz SG, et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell. 2020;181:557–573.e18. doi: 10.1016/j.cell.2020.03.021. [DOI] [PubMed] [Google Scholar]
- 28.Ydens E, et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat. Neurosci. 2020;23:676–689. doi: 10.1038/s41593-020-0618-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelhardt, B., Vajkoczy, P. & Weller, R. O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 10.1038/ni.3666 (2017). [DOI] [PubMed]
- 30.Hutchings M, Weller RO. Anatomical relationships of the pia mater to cerebral blood vessels in man. J. Neurosurg. 1986;65:316–325. doi: 10.3171/jns.1986.65.3.0316. [DOI] [PubMed] [Google Scholar]
- 31.Hannocks M-J, et al. Molecular characterization of perivascular drainage pathways in the murine brain. J. Cereb. Blood Flow. Metab. 2017;38:669–686. doi: 10.1177/0271678X17749689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 33.Hägerling R, et al. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J. 2013;32:629–644. doi: 10.1038/emboj.2012.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bazigou E, et al. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J. Clin. Invest. 2011;121:2984–2992. doi: 10.1172/JCI58050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibata-Germanos S, et al. Structural and functional conservation of non-lumenized lymphatic endothelial cells in the mammalian leptomeninges. Acta Neuropathol. 2019;139:383–401. doi: 10.1007/s00401-019-02091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baranska A, et al. Unveiling skin macrophage dynamics explains both tattoo persistence and strenuous removal. J. Exp. Med. 2018;215:1115–1133. doi: 10.1084/jem.20171608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong HW, et al. Single-cell transcriptomics reveals functionally specialized vascular endothelium in brain. eLife. 2022;11:e57520. doi: 10.7554/eLife.57520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Back J, Dierich A, Bronn C, Kastner P, Chan S. PU.1 determines the self-renewal capacity of erythroid progenitor cells. Blood. 2004;103:3615–3623. doi: 10.1182/blood-2003-11-4089. [DOI] [PubMed] [Google Scholar]
- 39.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werner Y, et al. Cxcr4 distinguishes HSC-derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nat. Neurosci. 2020;23:351–362. doi: 10.1038/s41593-020-0585-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lapenna, A., De Palma, M. & Lewis, C. E. Perivascular macrophages in health and disease. Nat. Rev. Immunol. 10.1038/s41577-018-0056-9 (2018). [DOI] [PubMed]
- 42.Koizumi T, Kerkhofs D, Mizuno T, Steinbusch HWM, Foulquier S. Vessel-associated immune cells in cerebrovascular diseases: from perivascular macrophages to vessel-associated microglia. Front. Neurosci. 2019;13:1291. doi: 10.3389/fnins.2019.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang T, Guo R, Zhang F. Brain perivascular macrophages: recent advances and implications in health and diseases. CNS Neurosci. Ther. 2019;25:1318–1328. doi: 10.1111/cns.13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kösel S, et al. Long-lasting perivascular accumulation of major histocompatibility complex class II-positive lipophages in the spinal cord of stroke patients: possible relevance for the immune privilege of the brain. Acta Neuropathol. 1997;94:532–538. doi: 10.1007/s004010050747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holfelder K, et al. De novo expression of the hemoglobin scavenger receptor CD163 by activated microglia is not associated with hemorrhages in human brain lesions. Histol. Histopathol. 2011;26:1007–1017. doi: 10.14670/HH-26.1007. [DOI] [PubMed] [Google Scholar]
- 46.Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017;20:136–144. doi: 10.1038/nn.4475. [DOI] [PubMed] [Google Scholar]
- 47.Jung S, et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hägerling R, Pollmann C, Kremer L, Andresen V, Kiefer F. Intravital two-photon microscopy of lymphatic vessel development and function using a transgenic Prox1 promoter-directed mOrange2 reporter mouse. Biochem. Soc. Trans. 2011;39:1674–1681. doi: 10.1042/BST20110722. [DOI] [PubMed] [Google Scholar]
- 49.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao Y, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Renier N, et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell. 2014;159:896–910. doi: 10.1016/j.cell.2014.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material. Raw data (fastq files) and processed data (gene counts) for single-cell RNA-Seq analysis have been deposited in the Gene Expression Omnibus with the primary accession number, GSE13328337. Source data are provided with this paper.
Scripts and detailed instructions to reproduce the NGS analysis for this manuscript are available on GitHub repository https://github.com/JulieBvs/SPlab_PVM2.