

Abstract
Clonal haematopoiesis involves the expansion of certain blood cell lineages and has been associated with ageing and adverse health outcomes1–5. Here we use exome sequence data on 628,388 individuals to identify 40,208 carriers of clonal haematopoiesis of indeterminate potential (CHIP). Using genome-wide and exome-wide association analyses, we identify 24 loci (21 of which are novel) where germline genetic variation influences predisposition to CHIP, including missense variants in the lymphocytic antigen coding gene LY75, which are associated with reduced incidence of CHIP. We also identify novel rare variant associations with clonal haematopoiesis and telomere length. Analysis of 5,041 health traits from the UK Biobank (UKB) found relationships between CHIP and severe COVID-19 outcomes, cardiovascular disease, haematologic traits, malignancy, smoking, obesity, infection and all-cause mortality. Longitudinal and Mendelian randomization analyses revealed that CHIP is associated with solid cancers, including non-melanoma skin cancer and lung cancer, and that CHIP linked to DNMT3A is associated with the subsequent development of myeloid but not lymphoid leukaemias. Additionally, contrary to previous findings from the initial 50,000 UKB exomes6, our results in the full sample do not support a role for IL-6 inhibition in reducing the risk of cardiovascular disease among CHIP carriers. Our findings demonstrate that CHIP represents a complex set of heterogeneous phenotypes with shared and unique germline genetic causes and varied clinical implications.
Subject terms: Cancer genetics, Genetics research, Epidemiology, Genome-wide association studies
Exome sequence data from 628,388 individuals was used to identify 24 risk loci in 40,208 carriers of clonal haematopoiesis of indeterminate potential and link them to other conditions including COVID-19, cardiovascular disease and cancer.
Main
As humans age, somatic alterations accrue in the DNA of haematopoietic stem cells (HSCs) due to mitotic errors and DNA damage. Alterations that confer a selective growth advantage can lead to the expansion of particular cell lineages, a phenomenon called clonal haematopoiesis. The presence of clonal haematopoiesis has been associated with an increased risk of haematological neoplasms, cytopaenias, cardiovascular disease (CVD), infection and all-cause mortality1–5. For this reason, identifying germline causes of clonal haematopoiesis has the potential to improve our understanding of initiating events in the development of these common diseases.
Large-scale studies of the germline causes of clonal haematopoiesis have used samples from the UKB and other large cohorts, but those studies have been limited mostly to clonal haematopoiesis phenotypes that can be assessed using single nucleotide polymorphism (SNP) array genotype data, such as mosaic chromosomal alternations (mCA) and mosaic loss of sex chromosomes4,7,8 (mLOX and mLOY). Identifying individuals with CHIP, which is defined by somatic protein-altering mutations in genes that are recurrently mutated in clonal haematopoiesis, requires sequencing of blood1,2. Once a clone has expanded sufficiently, the somatic variants from this clone can be captured along with germline variants by exome sequencing. Since exome sequencing captures protein-altering variants, its large-scale application enables the detection of readily interpretable rare variant association signals, and can elucidate critical genes and pathways and potential therapeutic targeting9,10. So far, the largest genetic association study of CHIP has included 3,831 CHIP mutation carriers in a sample of 65,405 individuals and has identified four common variant loci11.
Here, we use exome sequencing data to characterize CHIP status in 454,803 UKB10 and 173,585 Geisinger MyCode Community Health Initiative (GHS) participants. We then conduct a common variant genome-wide association study (GWAS) and rare variant and gene burden exome-wide association study (ExWAS) of CHIP by leveraging 27,331 CHIP mutation carriers from the UKB. We perform a replication analysis using 12,877 CHIP mutation carriers from the GHS cohort. To identify germline predictors of specific clonal haematopoiesis driver mutations, we also conduct GWAS and ExWAS in carriers of CHIP mutations from individual CHIP genes. We then compare genetic association findings for CHIP to those from analyses of other clonal haematopoiesis phenotypes determined from somatic alterations in the blood, including mCA, mLOX, mLOY and telomere length. Although GWAS of these non-CHIP clonal haematopoiesis phenotypes have been conducted4,7,12, none have evaluated the effect of rare variation. The ExWAS we perform here represents the first systematic large-scale exploration of the effect of rare variants on the genetic susceptibility of these phenotypes. Finally, we examine the clinical consequences of somatic CHIP mutations and germline predictors of CHIP in several ways. We first conduct a PheWAS13 of germline predictors of CHIP to understand their biological functions, and test cross-sectional phenotype associations of CHIP carrier status across 5,194 traits in the UKB. We then test the risk of incident cancer, CVD and all-cause mortality among specific CHIP gene mutation carriers and use Mendelian randomization to test for evidence of causal associations between CHIP and phenotypes of interest.
Calling CHIP
We used exome sequencing data from 454,803 and 173,585 individuals from the UKB and GHS cohorts, respectively, to generate large callsets of CHIP carrier status (Methods). In brief, we called somatic mutations using Mutect2 in a pipeline that included custom QC filtering (Extended Data Fig. 1a), and ultimately restricted our analysis to 23 well defined and recurrent CHIP-associated genes. This focused analysis identified 29,669 variants across 27,331 individuals in the UKB (6%), and 14,766 variants across 12,877 individuals in the GHS (7.4%). DNMT3A, TET2, ASXL1, PPM1D and TP53 were the most commonly mutated genes in both cohorts (Extended Data Fig. 2a). Although the GHS cohort had a wider age range, and therefore a larger number of older individuals, the prevalence by age was similar across cohorts, and reached approximately 15% by 75 years of age (Extended Data Fig. 1b,c). Prevalence of CHIP gene-specific mutations was consistent with recurrence patterns, with mutations in the most commonly mutated CHIP genes beginning to increase in prevalence at younger ages (Extended Data Fig. 1d,e and Supplementary Note 1). Somatic mutations within the IDH2 and SRSF2 genes co-occurred significantly more frequently than expected in both the UKB and GHS cohorts, whereas DNMT3A mutations co-occurred less frequently with other mutations than expected (Extended Data Fig. 2b,c and Supplementary Table 1). Among individuals with multiple CHIP mutations (Supplementary Note 2 and Supplementary Fig. 1), JAK2 mutations consistently had the highest variant allele fraction (VAF) (Supplementary Fig. 1b).
Extended Data Fig. 1. Workflow to Identify CHIP and Prevalence Estimates For Carriers of CHIP Mutations.

A. Graphic depicting at a high-level the workflow used to collect and sequence the exomes of multiple large cohorts and to then identify CHIP mutations from this data. B-C. CHIP prevalence increases with age of donor at time of DNA collection in both the UKB (B, n = 484,629 individuals; one-sided F-test, P < 10−16) and GHS (C, n = 157,724 individuals; one-sided F-test, P < 10−16) cohorts, with the centre line representing the general additive model spline and the shaded region representing the 95% confidence interval. D-E. Similar to B-C, the prevalence of CHIP mutations per CHIP gene for each of the top 8 most common CHIP genes increase with age in the UKB (D, n = 484,629 individuals; one-sided F-test, P < 10−16) and in GHS (E, n = 157,724 individuals; one-sided F-test, P < 10−16).
Extended Data Fig. 2. Count Distribution and Pairwise Enrichments of Clonal Hematopoiesis of Indeterminate Potential (CHIP) Gene Mutations.

A. Total number of individuals with mutations (y axis, log10 scale) in each of the 23 genes that were used to determine CHIP status across the UKB (blue) and GHS (red) CHIP callsets. B-C. Pairwise mutation counts across the UKB (B) and DiscoverEHR (C) callsets across individuals with at least two identified CHIP mutations. The color scale reflects the significance of the p-value for association between mutated CHIP gene pairs as determined by logistic regression. Per CHIP gene pair, these models included CHIP gene 1 mutation carrier status as the outcome, CHIP gene 2 mutation carrier status as the predictor, and age, sex, and smoking status (ever vs never) as covariates. P values are log10 transformed (see Table S1 for complete enrichment results).
CHIP demographics
Compared with controls, CHIP carriers in both the UKB and GHS cohorts were older and more likely to be heavy smokers, consistent with previous studies11 (Table 1). Although our cohorts were predominantly comprised of European ancestry individuals, the prevalence of CHIP was similar across all ancestries (Supplementary Fig. 2). In multivariate logistic regression models, each additional year of age was strongly associated with an increased risk of CHIP in the UKB (odds ratio [range] = 1.08 [1.077–1.082], P < 10−300) and GHS (odds ratio = 1.06 [1.057–1.063], P < 10−300), and heavy smoking was strongly associated with CHIP carrier status in both UKB (odds ratio = 1.17 [1.14–1.21], P = 7.32 × 10−24) and GHS (odds ratio = 1.24 [1.10–1.41], P = 6.3 × 10−4). Overall, our results suggest that the prevalence of CHIP doubles every 9–12 years of life. These associations with age and smoking were stronger when restricting to high-VAF (≥0.1) CHIP carriers. In our multivariate modelling, women were significantly more likely to be CHIP mutation carriers than men in the UKB (odds ratio = 1.08 [1.05–1.11], P = 6.01 × 10−7), but not in the GHS (odds ratio = 1.01 [0.93–1.11, P = 0.77]). These associations were consistent when restricting to high-VAF CHIP carriers, although the risk of high-VAF CHIP was not significantly greater in women in the UKB (odds ratio = 1.035 [0.99–1.08], P = 0.126).
Table 1.
Descriptive statistics for CHIP mutation carriers
| Age (median) | Sex (% female) | Heavy smoking (%) | Previous blood cancer (%) | Any blood cancer (%) | Any cancer (−NMSC, %) | Severe COVID-19 (%) | Ancestry (European, African, South Asian, other (%)) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UKB | CHIP | 62 | 54.0 | 33.9 | 2.18 | 6.49 | 26.6 | 0.52 | 96 | 1.4 | 1.8 | 0.73 |
| No CHIP | 57 | 54.3 | 27.5 | 0.60 | 2.19 | 17.8 | 0.31 | 95 | 2.0 | 2.3 | 0.94 | |
| GHS | CHIP | 73 | 55.2 | 13.7 | 1.59 | 12.7 | 41.7 | 0.45 | 98 | 1.4 | 0.1 | 0.65 |
| No CHIP | 57 | 61.6 | 9.57 | 0.63 | 3.22 | 19.7 | 0.18 | 94 | 3.2 | 0.3 | 1.9 | |
Genetic association with CHIP carrier status
We first conducted genetic association analyses in the UKB cohort to identify germline loci associated with the risk of developing CHIP. In the common variant (minor allele frequency (MAF) > 0.5%) GWAS, which included 25,657 cases and 342,869 controls with European ancestry, we identified 24 loci (21 novel loci) harbouring 57 independently associated variants (Fig. 1 and Supplementary Table 2). To confirm these signals, we conducted a replication analysis in 9,523 CHIP cases and 105,502 controls of European ancestry from the GHS cohort. We estimated that we had sufficient statistical power in the GHS to detect 19.99 true and directionally consistent associations across lead SNPs from the 24 loci we identified in the UKB and achieved nominally significant (P < 0.05) replication for 15 SNPs (Supplementary Table 2). We used conditional analysis and statistical fine-mapping to further evaluate the independence of our genome-wide associations and found results to be consistent across methods (Extended Data Fig. 3, Supplementary Note 3, Supplementary Tables 3–6 and Supplementary Fig. 3).
Fig. 1. GWAS of CHIP.

Manhattan plot showing results from a genome-wide association analysis of CHIP. Twenty-four loci reach genome-wide significance (P ≤ 5 × 10−8, dashed line), and top-associated variants per locus are labelled with biologically relevant genes. Three of these loci have been previously identified (black), whereas 21 represent novel associations (red). Loci with suggestive signal (P ≤ 5 × 10−7) are labelled in grey. Association models were run with age, age2, sex and age × sex, and 10 ancestry-informative principal components as covariates. P-values are uncorrected and are from two-sided tests performed using approximate Firth logistic regression.
Extended Data Fig. 3. Finemapping results at the LY75 locus on chromosome 2.

A. Fine-mapping the summary statistics from our association analysis of CHIP prioritizes the P1247L missense variant (rs78446341-A, AAF = 0.02) as highly likely to be the causal variant driving one of three causal signals at this locus (CPIP = 0.913). At the top of the panel, a locus zoom plot shows marginal association results after inverse variance weighted meta analysis across UKB and GHS (p-values are uncorrected and derive from two-sided tests performed using approximate Firth logistic regression and subsequent meta analysis). Top common variants, including those prioritized by clumping and thresholding and COJO from UKB associations are highlighted with black circles. The rs78446341-A missense variant is highlighted as well and is in low linkage disequilibrium (LD) with the other SNPs. FINEMAP estimated 3 signals were most parsimonious here (PP = 0.55). B. Fine-mapping the summary statistics from our association analysis of DNMT3A-CHIP prioritizes the P1247L missense variant (rs78446341-A, MAF = 0.02, CPIP = 0.20, CS = 4) and the rarer G525E missense variant (rs147820690-T, AAF = 0.002 CPIP = 0.60, CS = 2) as likely to be the causal variants driving the signal at two out of four causal signals at this locus. Here, FINEMAP estimated 3 signals (PP = 0.57) or 4 signals (PP = 0.41) were likely; we report results for K = 3 in Table S6 and show results from K = 4 here. The other prioritized signals are those identified by clumping and thresholding and COJO: rs12472767-C (2-159925824-T-C, CPIP = 0.99, CS = 1) and rs12472767-C (2-159821048-C-T, CPIP = 0.28, CS = 3). CS: Credible Set, PP: Posterior Probability, PIP: Posterior Inclusions Probability, CPIP: Conditional Posterior Inclusion Probability.
We next sought to identify rare germline variants associated with CHIP. Since the CHIP phenotype is based on the presence of rare somatic variants in recurrently mutated genes, rare germline variants potentially misclassified as somatic can lead to false association signals. To address potential misclassification, we evaluated median VAF and association with age for each rare germline variant or gene burden associated with CHIP. We also conditioned these rare variant analyses on independent common variant signals to address confounding due to linkage disequilibrium (LD) (Supplementary Note 4). Ultimately, we identified a single rare germline frameshift variant in the CHEK2 gene that was significantly associated with CHIP (odds ratio = 2.22 [1.89–2.61], P = 8.04 × 10−22; Supplementary Table 7), remained so after conditioning on common variant signals (odds ratio = 2.90 [1.93–4.34], P = 2.40 × 10−7), and replicated in the GHS (odds ratio = 1.56 [1.19–2.04], P = 1.22 × 10−3). The two cancer-associated genes ATM and CHEK2 were associated with an increased risk of CHIP via rare variant gene burden testing (Supplementary Table 8), and we also found a significant gene burden association between rare loss of function (and missense) variants in the telomere maintenance and DNA replication associated gene CTC1 and an increased risk of CHIP (odds ratio = 1.55 [1.32–1.81], P = 5.24 × 10−8). Of these three gene burden associations, the ATM and CHEK2 signals were replicated in the GHS (P = 8.22 × 10−5 and P = 0.03, respectively), and VAF and age-association calculations suggested that all three of these gene burden signals were driven by germline variation. We also performed genome-wide association analyses in individuals of non-European ancestral background (Supplementary Note 5 and Supplementary Table 9).
For each germline variant associated with CHIP and prioritized by clumping and thresholding, conditional analysis or fine-mapping (see Methods), we queried its associations across 937 binary and quantitative health traits from the UKB for which we have previously performed genetic association analysis10 (Supplementary Table 10). Overall, the traits with significant associations consisted predominantly of blood measures (that is, cells counts and biomarker levels), anthropometric measures related to body size, autoimmune phenotypes and respiratory measures. SNPs with the largest number of significant phenotypic associations included those at the HLA, TP53, ZFP36L2 and THADA, CD164 and MYB loci (Extended Data Fig. 4). Whereas associations with blood cell counts and biomarker levels are probably the direct result of expansion of individual cell lineages in blood, association with autoimmune phenotypes could reflect the consequences of disrupted immune system differentiation related to clonal haematopoiesis.
Extended Data Fig. 4. Results from a phenome-wide association analysis.

Results from a phenome-wide association analysis are shown for the thirty SNPs from our GWAS that had the largest number of significant associations (P < 5 x 10−8). Associations are most common among hematological, body mass, and auto-immune traits (seen across the ‘dermatology’, ‘gastroenterology’, and ‘other’ phenotypic categories). For visualization, associations with –log10(P) < 50 were set to 50. Association models were run with age, age2, sex, and age-by-sex, and 10 ancestry-informative principal components (PCs) as covariates. P-values are uncorrected and derive from two-sided tests performed using approximate Firth logistic regression. See Table S10 for full associations results.
Analyses of individual CHIP gene mutations
To identify CHIP subtype-specific risk variants, we defined gene-specific CHIP phenotypes for each of the eight most commonly mutated CHIP genes. For each subtype, we selected individuals with mutations in one of the eight genes and no mutations in any of the other genes used to define CHIP. We then conducted genetic association analyses comparing these single CHIP gene carriers to CHIP-free controls, with replication in the GHS, and observed shared, unique, and opposing effects of associated loci on CHIP subtypes, including 8 genome-wide significant loci that were not significant in our overall analysis of CHIP (Fig. 2a, Extended Data Fig. 5 and Supplementary Tables 11–19).
Fig. 2. Germline effect size comparisons across CHIP and Forest plots of PARP1 and LY75 missense variants.

a, Using results from CHIP gene-specific association analyses, effect sizes of index SNPs are compared across CHIP subtypes. SNPs were chosen as those that were independent on the basis of clumping and thresholding (with some refinement based on our conditionally independent variant list) and genome-wide significant in at least one association with CHIP or a CHIP subtype. Certain loci showed notably different effects across CHIP subtypes, as seen at the CD164 locus, which was associated with DNMT3A CHIP and ASXL1 CHIP but not TET2 CHIP, and the TCL1A locus, which was associated with increased risk of DNMT3A CHIP but reduced risk of other CHIP subtypes (blue rectangles). b, Forest plots are shown reflecting the protective associations of a PARP1 missense variant (rs1136410-G) and two LY75 missense variants (rs78446341-A, rs147820690-T) with our DNMT3A CHIP phenotype in the UKB and GHS cohorts. Centre points represent odds ratios as estimated by approximate Firth logistic regression, with errors bars representing 95% confidence intervals. P-values are uncorrected and reflect two-sided tests. Numbers below the cases and controls columns represent counts of individuals with homozygote reference, heterozygote and homozygous alternative genotypes, respectively.
Extended Data Fig. 5. GWAS of CHIP Subtypes.

Manhattan plot showing results from a genome-wide association analysis of CHIP subtypes. While we ran CHIP subtype analysis for each of the 8 most recurrently mutated CHIP genes (Tables S11–S19), we show Manhattan plots for the 5 CHIP subtypes that had at least 1 genome-wide significant common variant association. These included DNMT3A-CHIP (23 significant loci), TET2-CHIP (6 significant loci), ASXL1-CHIP (2 significant loci), TP53-CHIP (1 significant locus), and JAK2-CHIP (1 significant locus). Novel biologically relevant genes are labeled at each locus, with red denoting novel loci, black identifying previously identified loci and grey identifying loci with suggestive signal (P < 5 x 10−7). Association models were run with age, age2, sex, and age-by-sex, and 10 ancestry-informative principal components (PCs) as covariates. P-values are uncorrected and are from two-sided tests performed using approximate Firth logistic regression.
DNMT3A, which was the most commonly mutated gene in the overall CHIP phenotype, had the largest number of significantly associated loci (n = 23), most of which overlapped with the overall CHIP association signals. Six loci achieved genome-wide significance in our DNMT3A CHIP analysis that were not significant in our overall analysis (RABIF, TSC22D2, ABCC5, MYB, FLT3 and TCL1A; Extended Data Fig. 5). Although most loci harboured variants that increased CHIP risk, two exceptions are noteworthy (Fig. 2b). At the PARP1 locus on chromosome 1, a tightly linked block of around 30 variants (29 in the 95% credible set from fine-mapping; Supplementary Table 6) with an alternate allele frequency (AAF) of 0.15 was associated with reduced risk of DNMT3A CHIP (odds ratio = 0.87 [0.84–0.90], P = 2.70 × 10−17). PARP1 has a role in DNA damage repair, and many variants in this block have been identified across multiple transcriptomic studies of blood as PARP1 expression quantitative trait loci (eQTLs) that associate with reduced PARP1 gene expression14–17. Furthermore, a missense variant (rs1136410-G, V762A) that is predicted as likely to be damaging (combined annotation dependent depletion (CADD) score = 27.9) is a part of this LD block, and has recently been reported to associate with improved prognosis and survival in myelodysplastic syndromes18 (MDS). At a locus on chromosome 2, rs78446341 (P1247L in LY75) was associated with reduced risk of DNMT3A CHIP (odds ratio = 0.78 [0.72–0.84], P = 3.70 × 10−10), and was prioritized by fine-mapping (Extended Data Fig. 3). LY75 features lymphocyte-specific expression (Supplementary Fig. 4a), and is thought to be involved in antigen presentation and lymphocyte proliferation19. We also identified a second rare (AAF = 0.002) missense variant (rs147820690-T, G525E) that associated with reduced risk of DNMT3A CHIP at close to genome-wide significance (odds ratio = 0.48 [0.36–0.63], P = 1.15 × 10−7). This variant was predicted as likely to be damaging (CADD = 23.6) and remains associated (odds ratio = 0.63 [0.51–0.77], P = 4.80 × 10−6) when conditioning on common variant signal in this locus (that is, this rare variant signal is independent of the common variant signal in this locus). This variant was also prioritized by fine-mapping (Extended Data Fig. 3 and Methods for jointly fine-mapping common and rare variants). Finally, these signals in PARP1 and LY75 replicated in the GHS (Fig. 2b).
Among loci associated with multiple CHIP subtypes (Supplementary Note 6), we observed genome-wide significant association signals at the TCL1A locus that were not present in the overall CHIP analysis. This locus is notable because it exhibited genome-wide significant effects in opposing directions across CHIP subtypes (Extended Data Figs. 2a and 5 and Supplementary Table 20), with lead SNPs (for example, rs2887399-T, rs11846938-G and rs2296311-A) at the locus associated with an increased risk of DNMT3A CHIP (odds ratio = 1.14 [1.11–1.17], P = 2.13 × 10−20) but a reduced risk of TET2 CHIP (odds ratio = 0.75 [0.71–0.80], P = 9.14 × 10−22) and ASXL1 CHIP (odds ratio = 0.70 [0.65–0.76], P = 8.59 × 10−18). Effect estimates from the other five CHIP gene-specific association analyses were also consistent with protective effects. This is consistent with findings from a recent genetic association study of CHIP in the TOPMed cohort11, which identified a genome-wide significant positive association of the TCL1A locus and DNMT3A CHIP as well as a nominally significant opposing signal for TET2 CHIP. Additionally, the DNMT3A CHIP-increasing allele has been found to reduce the risk of mLOY in a recent GWAS7. This observation suggests that DNMT3A CHIP is distinct among clonal haematopoietic subtypes with regard to the genetic influence of the TCL1A locus, which may relate to the fact that TCL1A has been reported to directly interact with and inactivate DNMT3A20.
CHIP and mosaic chromosomal alterations
To evaluate the relationship between CHIP and other forms of somatic alterations of the blood, we used phenotype information on other types of clonal haematopoiesis that are available for UKB participants4,7,8,12. We first evaluated the phenotypic overlap between CHIP and mLOY, mLOX and autosomal mosaic chromosomal alterations (mCAaut). CHIP is distinct from mCA phenotypes (mCAaut, mLOX and mLOY), with more than 80% of CHIP carriers having no identified mCAs (Supplementary Fig. 4b). Furthermore, having an mCA is not significantly associated with being a CHIP carrier after adjusting for age, sex and smoking status (odds ratio = 1.02, P = 0.27). Carriers of only a single clonal haematopoiesis driver (that is, CHIP, mLOY, mLOX or mCAaut) were younger on average than those with multiple clonal haematopoiesis lesions, and mCAaut and CHIP carriers were youngest among single clonal haematopoiesis phenotype carriers (Supplementary Fig. 4c).
We then conducted GWAS and ExWAS analyses of these somatic alteration phenotypes and evaluated the germline genetic contributions shared between CHIP and these traits (Supplementary Fig. 5 and Supplementary Tables 21–27). Genome-wide genetic correlation (rg)21,22 was nominally significant between CHIP and mLOY (rg = 0.27, P = 0.014 (uncorrected); Supplementary Table 21). Notably, variants at 4 loci (marked by the genes ATM, LY75, CD164 and GSDMC) showed similar associations with both CHIP and mLOY, whereas variants at the SETBP1 locus were negatively associated with CHIP and positively associated with mLOY. These comparisons suggest that despite being distinct clonal haematopoietic phenotypes, CHIP and mLOY share multiple germline genetic risk factors. Although the common variant association analyses of these other somatic alteration phenotypes were undertaken for the purpose of comparing to CHIP, and our results are consistent with recent published associations for these non-CHIP UKB somatic alteration phenotypes4,7,8, we also identified novel rare variant and gene burden associations via ExWAS analyses (Supplementary Note 7, Supplementary Tables 22–27 and Supplementary Fig. 6). We also extended our ExWAS analysis to telomere length and identified multiple novel rare variant associations (Supplementary Note 8 and Supplementary Tables 28–30).
Phenotypic associations with CHIP
Clonal haematopoiesis has been associated with an increased risk of haematologic malignancy and CVD, as well as other health outcomes including all-cause mortality and susceptibility to infection3,4,23,24. To test for expected as well as potentially novel associations, we performed cross-sectional association analyses across 5,041 traits (2,640 binary and 2,401 quantitative traits) from the UKB, curated as part of our efforts for the UKB Exome Sequencing Consortium. We performed Firth penalized logistic regression using CHIP gene mutation carrier status (that is, whether an individual had a mutation in our callset within a specific CHIP gene) as the binary outcome for 22 of the 23 CHIP genes in our callset (counts were too low for CSF3R; Methods), with age, sex and ten genetic principal components as covariates. Our results are consistent with previous findings, with the majority of associated phenotypes deriving from cardiovascular, haematologic, neoplastic, infectious, renal and/or smoking-related causes (Fig. 3, Supplementary Fig. 7 and Supplementary Table 31).
Fig. 3. Phenome association profiles per CHIP subtype.

Profiles are shown for each CHIP gene subtype reflecting phenome-wide association results. The y-axis (concentric circles) represents the proportion of phenotypes within a trait category that were nominally associated (P ≤ 0.05) with carrier status of the CHIP gene. A CHIP gene had to have at least one disease category with the proportion of associated phenotypes ≥ 0.2 to be included in the figure. As expected, haematological traits show the largest proportion of phenotypic trait associations overall. The largest number of cancer associations are seen for DNMT3A CHIP, whereas JAK2 CHIP shows the highest proportion of cardiovascular associations. Respiratory associations are most pronounced for ASXL1 CHIP. SUZ12 CHIP shows a unique profile across CHIP subtypes, with a higher proportion of ophthalmological and endocrine associations. Association models were run with age, age2, sex and age × sex, and ten ancestry-informative principal components as covariates.
ASXL1 CHIP was associated with the largest number and widest range of traits, and many of these associations traced to correlates of smoking. SUZ12 CHIP showed a distinct association profile amongst CHIP genes, with a larger proportion of associations in endocrine and ophthalmologic traits than other CHIP genes. Many traits showed associations with DNMT3A CHIP and TET2 CHIP that were in opposing directions, including white blood cell count, platelet count and neutrophil count, which were all positively associated with DNMT3A CHIP and negatively associated with TET2 CHIP. These results are consistent with functional differences in the haematopoietic phenotypes of DNMT3A- and TET2-knockout mice25. Notably, body mass index (BMI) and fat percentage were negatively associated with DNMT3A CHIP and other leukaemogenic CHIP mutations (for example, JAK2, CALR and MPL), but are positively associated with other CHIP subtypes (for example, TET2 and ASXL1). We also observed significant associations between JAK2 mutations and gout, which may reflect the increased uric acid production that can accompany haematopoiesis26 and/or renal disease27, or even uric acid-independent associations identified between anaemia and gout28.
Given recent reports that clonal haematopoiesis is associated with an increased risk of COVID-19 and other infections4,29, we also tested for an association between CHIP and COVID-19 infection in the UKB cohort30. When restricting to CHIP carriers with VAF ≥ 10% (Supplementary Note 9), we found that CHIP carrier status was significantly associated with COVID-19 hospitalization (odds ratio = 1.26 [1.07–1.47], P = 4.5 × 10−3) and severe COVID-19 infection (odds ratio = 1.55 [1.19–1.99], P = 8.5 × 10−4) in logistic regression models that excluded individuals with any previous blood cancers and that adjusted for age, sex, smoking, BMI, type 2 diabetes, active malignancy, and five genetic principal components. Analyses at the CHIP subtype level suggested that PPM1D carriers may be at elevated risk of severe COVID-19 (odds ratio = 5.42 [1.89–12.2], P = 2.8 × 10−4; Supplementary Note 9).
Longitudinal disease risk among CHIP carriers
Given the confounding that can bias cross-sectional association analyses, we performed survival analyses to evaluate whether individuals with CHIP at the time of enrolment and blood sampling in the UKB were at an increased risk of subsequent CVD, cancer and all-cause mortality. To do this, we generated aggregate longitudinal phenotypes of CVD, lymphoid cancer, myeloid cancer, lung cancer, breast cancer, prostate cancer, colon cancer and overall survival (that is, any death). Because prior longitudinal studies of CHIP and the risk of many of these outcomes have focused on high-VAF CHIP, we focused on CHIP carriers with VAF ≥ 0.10 for these analyses. To complement these longitudinal analyses, we used Mendelian randomization to evaluate the relationship between CHIP and subsequent disease (Extended Data Fig. 6a, Supplementary Note 10 and Supplementary Table 32).
Extended Data Fig. 6. Results from Mendelian Randomization models and incident risk of death among CHIP carriers.

A. Forest plot of results from Two Sample Mendelian Randomization (MR) modeling of the effect of CHIP on 20 traits of interest (including the two quantitative traits BMI and ALT). Reported p-values are uncorrected, and reflect two-sided Z-tests derived from an inverse variance weighted (IVW) MR procedure. Significant causal association between CHIP and breast cancer, prostate cancer, non-melanoma skin cancer, melanoma, myeloid leukemia, and lung cancer are supported by these models. As expected, estimates of germline effect on CHIP from UKB and GHS are strongly correlated (odds ratio = 1.94 [1.76–2.13], P = 3.2 x 10−42). B. CHIP and its most common subtypes are significantly associated with death from any cause across UKB. Hazard ratio (HR) estimates from cox-proportional hazard models are shown, with error bars that represent a 95% confidence interval. P-values are uncorrected and derive from two-sided Wald tests. Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs.
We observed a significantly increased risk of CVD in CHIP carriers (hazard ratio = 1.11 [1.03–1.19], P = 4.2 × 10−3), which was driven by TET2 CHIP (hazard ratio = 1.31 [1.14–1.51], P = 1.3 × 10−4; Supplementary Fig. 8a). However, this risk estimate is lower than the hazard ratio of 1.59 recently reported by Bick et al.6 in an analysis of CHIP from the first 50,000 UKB participants (hereafter referred to as the 50k UKB subset) with exome sequencing data available. Therefore, we restricted our analysis to the 50,000 individuals from the previous study and found that the estimated hazard ratio is indeed higher in this subset (hazard ratio = 1.30 [1.06–1.59], P = 0.013; Supplementary Fig. 8b). Bick et al. also observed a cardio-protective effect of IL6R rs2228145-C (a genetic proxy for IL-6 receptor inhibition) among CHIP carriers in the 50k UKB subset, so we repeated that analysis in both the 50k UKB subset and the full UKB cohort (n = 430,924 in these analyses). We observed the same CHIP-specific protective IL6R effect in the 50k UKB subset as previously reported (hazard ratio = 0.60 [0.40–0.89], P = 0.012), however we did not find any IL6R effect in the full cohort (hazard ratio = 0.99 [0.91–1.07], P = 0.784, n = 430,924; Extended Data Fig. 7a–d). These results were consistent when varying which CHIP mutations we used to define CHIP case status, as well as when using different VAF thresholds and a variety of CVD endpoint composites (Methods). We did not find any association between CHIP and CVD, nor a CHIP-specific protective IL6R effect, when repeating this analysis in the GHS cohort (Supplementary Figs. 8d and 9a, b). Furthermore, we did not find evidence for a casual association between CHIP and CVD when using a two-sample Mendelian randomization approach (Supplementary Note 10, Supplementary Fig. 10 and Supplementary Table 32).
Extended Data Fig. 7. CVD Incidence in IL6R Mutation Carriers with and without CHIP.

A-B. Survival curves are drawn showing that IL6R p.Asp358Ala mutation carriers (green) are not an elevated risk of CVD incidence (y-axis) compared with non-carriers (blue) in either the first 50K individuals from UKB (A) or the full 450K cohort (B). C-D. In contrast, IL6R p.Asp358Ala mutation carriers are estimated to be at a reduced risk of CVD events (C) (HR = 0.60), but only in the first 50K samples from UKB (D). Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs. Hazard ratios (HR) were estimated using cox-proportional hazard modeling, with p-values uncorrected and derived from two-sided Wald tests.
We next tested whether CHIP carriers are at an increased risk of haematologic and solid cancers, and whether risk differed by CHIP mutational subtype for the three most common CHIP genes (that is, DNMT3A, TET2 and ASXL1; Extended Data Figs. 7–9 and Supplementary Figs. 11–14). To control for the possibility that toxic chemotherapeutic treatment for previous cancers might drive the development of CHIP mutations31 and/or otherwise confound association analyses, we performed all analyses after excluding individuals with any diagnoses of cancer prior to DNA collection. As expected, we found CHIP carriers with VAF ≥ 0.10 to be at a significantly elevated risk of developing any blood cancer (hazard ratio = 3.88 [3.46–4.36], P = 9.10 × 10−117; Supplementary Fig. 11a), and we identified similarly elevated risk when replicating these analyses in the GHS (Supplementary Fig. 11d). We also estimated the risk of CHIP on neoplastic myeloid subtypes, including acute myeloid leukaemia (AML), MDS and myeloproliferative neoplasms (MPN), and found that high-VAF CHIP carriers have more than 23-fold increased risk of acquiring an MPN (hazard ratio = 23.11 [17.63–30.29], P = 1.60 × 10−114) (Extended Data Fig. 8). As expected, we identified a significant association between myeloid leukaemia and CHIP by Mendelian randomization (Supplementary Note 10, Supplementary Fig. 12 and Supplementary Table 32).
Extended Data Fig. 9. Incident risk of lung cancer among CHIP carriers from the UKB and GHS cohorts.

A-D. Forest plots and tables featuring hazard ratio (HR) estimates from cox-proportional hazard models are shown, with error bars that represent a 95% confidence interval. CHIP and its most common subtypes are significantly associated with lung cancer in both smokers and non-smokers across UKB (A-B) and GHS (C-D). Here, results are depicted from analyses in which we removed samples that had a diagnosis of malignant cancer prior to DNA collection. Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs. Hazard ratios (HR) were estimated using cox-proportional hazard modeling, with p-values uncorrected and derived from two-sided Wald tests.
Extended Data Fig. 8. Incident risk of myeloid cancer subtypes among CHIP carriers from the UKB.

A-C. Forest plots and tables featuring hazard ratio (HR) estimates from cox-proportional hazard models are shown, with error bars that represent a 95% confidence interval. CHIP and its most common subtypes are significantly associated with acute myeloid leukemia (AML) (A), Myelodysplastic Syndromes (MDS) (B), and myeloproliferative neoplasm (MPN) (C). Here, results are depicted from analyses in which we removed samples that had a diagnosis of malignant cancer prior to sequencing collection. Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs. Hazard ratios (HR) were estimated using cox-proportional hazard modeling, with p-values uncorrected and derived from two-sided Wald tests.
We then tested whether CHIP carriers had an increased risk of developing solid tumours, and found that high-VAF carriers are at significantly increased risk of developing lung cancer (hazard ratio = 1.64 [1.42–1.90], P = 1.10 × 10−11), and more modest increased risk of developing prostate cancer (hazard ratio = 1.18 [1.05–1.32], P = 5.30 × 10−3) and non-melanoma skin cancer (hazard ratio = 1.14 [1.04–1.24], P = 4.7 × 10−3; Fig. 4 and Supplementary Fig. 13). We also observed a non-significant increased risk of developing breast cancer (hazard ratio = 1.14 [0.99–1.31], P = 0.062) and no increase in risk for the development of colon cancer (hazard ratio = 0.95 [0.78–1.15], P = 0.59; Supplementary Fig. 13). Models estimating event risk on the basis of CHIP mutational subtype (for example, DNMT3A CHIP) suggest that these associations with prostate and breast cancer are driven primarily by DNMT3A mutations. Only the association with lung cancer was replicated in the GHS (Fig. 13e), although sample sizes were limited for the analyses in the GHS owing to how the biobank data were ascertained (Methods).
Fig. 4. Increased risk of lung cancer among CHIP carriers.
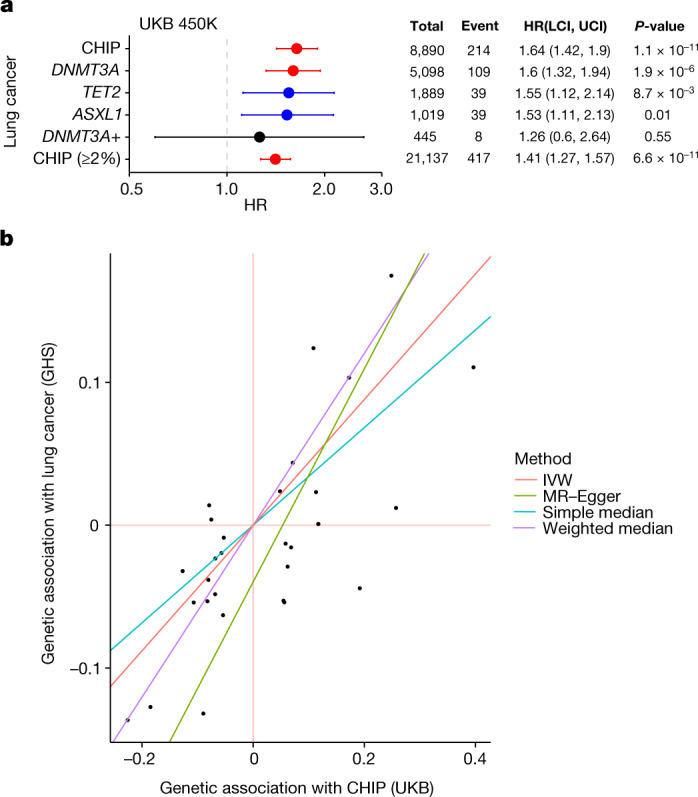
a, Forest plot and table featuring hazard ratio estimates from Cox proportional hazard models of the risk lung cancer among CHIP carriers. Error bars represent a 95% confidence interval. Associations are similar across common CHIP subtypes, as well as among CHIP carriers with lower VAF (≥2%). Models are adjusted for sex, low density lipoprotein, high density lipoprotein, smoking status, pack years, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 genetic principal components specific to a European ancestral background. HR, hazard ratio. UKB 450K, the 450,00-participant full UKB dataset. DNMT3A+ represents subjects with DNMT3A CHIP and at least one other type of CHIP mutation. b, Estimated associations via four Mendelian randomization methods between CHIP and lung cancer. Each point represents one of 29 instrumental variables (that is, conditionally independent SNPs) that were identified in the UKB cohort as associated with CHIP. The x-axis shows the effect estimate (beta) of the SNP on CHIP in the UKB cohort, and the y-axis shows the effect estimate (beta) of the SNP on lung cancer in the GHS cohort. The slope of each regression line represents the effect size estimated by respective methods. IVW, inverse variance weighted.
Given the strong associations between CHIP and both blood and lung cancers, and the associations between smoking and both CHIP and lung cancer, we performed additional analyses stratified by smoking status to test whether these associations were driven by smoking and merely marked by CHIP mutations. Although smoking status is difficult to ascertain, we used an inclusive ‘ever smoker’ definition to minimize the likelihood that individuals labelled as non-smokers had engaged in any smoking (Methods). High-VAF CHIP carriers had an increased risk of developing blood cancers in both smokers (hazard ratio = 3.95 [3.25–4.78], P = 2.80 × 10−44) and non-smokers (hazard ratio = 3.97 [3.43–4.58], P = 1.10 × 10−77; Supplementary Fig. 14a, b). Notably, lung cancer risk for high-VAF CHIP carriers was significantly elevated among both smokers (hazard ratio = 1.67 [1.41–1.97], P = 1.5 × 10−9) and non-smokers (hazard ratio = 2.02 [1.53–2.67], P = 8.30 × 10−7 ; Extended Data Fig. 9a,b). These associations were driven by DNMT3A and ASXL1 CHIP carriers, with both estimated to have elevated lung cancer risk in both smokers and non-smokers. We replicated the association between CHIP carrier status and lung cancer in both smokers and non-smokers in the GHS (Extended Data Fig. 9c,d). Overall, these models suggest that CHIP mutation carriers are at an elevated risk of both blood cancer and lung cancer, independent of smoking status.
We also found support for a causal association between CHIP and lung cancer (inverse variance weighted odds ratio (ORIVW) = 1.55 [1.34–1.80], P = 8.90 × 10−9; Fig. 4 and Extended Data Table 1), as well as more modest support for causal associations between CHIP and melanoma (ORIVW = 1.39 [1.13–1.1.71], P = 0.0021), CHIP and non-melanoma skin cancer (ORIVW = 1.26 [1.13–1.41], P = 5.30 × 10−5), CHIP and prostate cancer (ORIVW = 1.20 [1.03–1.1.39], P = 0.017), and CHIP and breast cancer (1.17 [1.04–1.31], P = 0.01), when performing Mendelian randomization (Extended Data Fig. 6a, Supplementary Note 10 and Supplementary Table 32). Although there is a concern that variants predisposing to CHIP via cancer-associated pathways (for example, telomere biology, DNA damage repair and cell cycle regulation) may confound these associations via horizontal pleiotropy, Egger-based Mendelian randomization methods that account for this bias by fitting a non-zero intercept provided additional support for these associations. Finally, the risk of death from any cause was significantly elevated among high-VAF CHIP carriers (hazard ratio = 1.27 [1.18–1.36], P = 2.70 × 10−11), and was similar across DNMT3A, TET2 and ASXL1 CHIP subtypes (Extended Data Fig. 6b).
Extended Data Table 1.
Results from Mendelian Randomization analysis of CHIP exposure on lung cancer risk

Statistical results are shown from seven MR methods with differing sensitivities to outliers and/or violations of the MR assumptions. P-values are reported uncorrected. The estimated intercept values are shown for the two MR-Egger-based methods that estimate these terms. All models provided support for a casual association between CHIP and lung cancer. Models were significant when run without variants at the TERT locus as instrumental variables, which provides support for a causal association above and beyond any pleiotropic effects at the TERT locus (grey text).
In this study, we present the largest assessment to date of individuals with CHIP mutation carrier information, as well as the use of these calls to identify novel common and rare variant loci associated with CHIP and CHIP subtypes. These loci, which have shared, unique and opposing effects on the risk of developing different types of CHIP and other somatic alterations of the blood, highlight the fact that germline variants can predispose to clonal expansions, and that CHIP encapsulates a complex set of heterogeneous phenotypes. We further show that the genetic aetiology of CHIP is reflected in its clinical consequences, as the risk of various clinical conditions is differentially associated across CHIP gene mutations.
The new loci identified in this study provide a foundation on which to investigate the biological mechanisms that lead to specific features of CHIP. For example, among CHIP-associated loci, variants in the TCL1A locus that are associated with an increase in the risk of DNMT3A CHIP have the opposite effect on the risk of all other CHIP and clonal haematopoiesis subtypes. Coupled with recent findings that link the role of TCL1A in mLOY to lymphocytes7 (for example, B cells), our results further suggest TCL1A as a critical mediator of clonal haematopoiesis as well as clonal haematopoiesis subtype-specific differences.
Several novel loci associated with DNMT3A CHIP harbour genes that are potential targets for the development of new treatments to prevent or slow the expansion of CHIP clones. Both PARP1 and LY75 contain missense variants associated with reduced risk of CHIP and of DNMT3A CHIP specifically. The variants in the PARP1 locus are significantly associated with reduced PARP1 gene expression in whole blood32 (P ≤ 1 × 10−13), and the V762A missense variant (rs1136410-G) has been recently reported to associate with improved prognosis and survival in MDS18. Given the well-established role of PARP1 in DNA repair33, and that a recent CRISPR screen study in zebrafish identified PARP1 inhibition as a selective killer of TET2 mutant haematopoietic stem cells34, it seems plausible that a therapeutic strategy that inhibits PARP1 might be viable for the antagonization of CHIP clone expansion. Furthermore, PARP1-inhibiting drugs are already approved for use in the treatment of BRCA-mutant cancers35. Conversely, PARP1 inhibition is known to cause haematologic toxicity and to increase the risk of treatment related haematologic malignancy36. Therefore, further research is needed to test whether PARP1 inhibition may be appropriate for use in antagonizing the expansion of CHIP clones, and whether any effect is clonal haematopoiesis subtype-specific.
The more common LY75 missense variant (rs78446341-A, P1247L) is located in the extracellular domain of lymphocytic antigen 75 (also known as DEC-205 or CD205), and has a role in antigenic capture, processing and presentation37. The rarer LY75 missense variant (rs147820690-T, G525E) is located in a C-type lectin domain and reported to interact directly with this receptor’s ligand. LY75 is expressed predominantly in haematopoietic-derived cells37,38 (and particularly dendritic cells), and its ablation impairs T cell proliferation and response to antigen challenge19. The protective associations with this variant that we identified appear to be most pronounced for DNMT3A CHIP and mLOY, and highlight LY75 as a potential therapeutic target for the antagonization of clonal haematopoiesis in general.
Although most of the phenotypic associations we observe in our cross-sectional analyses are expected associations with haematologic and oncologic traits, the associations we identify with obesity and body mass traits are of particular interest. This relationship between body mass and CHIP may relate to inflammatory or hormonal signalling, and directions of effect that we estimate are consistent with recent findings that DNMT3A CHIP reduces bone mineral density via increases in macrophage-mediated IL-20 signalling39. The fact that the association we report between obesity and body mass and CHIP are in opposing directions across CHIP subtypes (for example, negative in DNMT3A CHIP and positive in TET2 CHIP and ASXL1 CHIP) suggests that the relationship between CHIP and adiposity is complex and requires further investigation.
Perhaps most unexpectedly, we found associations between CHIP and CVD to be more modest than previously reported1–3. DNMT3A mutations do not associate with CVD, which is consistent with the absence of any association between CHIP and CVD when applying Mendelian randomization. However, this pattern is not seen across CHIP associations with solid tumours, which we found to be driven by DNMT3A, and to be supported by Mendelian randomization. Overall, our results further clarify the role of CHIP mutational subtypes in the development of cancer and CVD and emphasize the importance of viewing (and potentially treating) different CHIP subtypes as distinct haematologic preconditions.
Whereas Bick et al. 6. found statistical support for reduced CVD incidence among CHIP carriers with an IL6R coding mutation (rs2228145-C) serving as a genetic proxy for IL-6 inhibition, we do not find any support for this association when extending their analysis from the first 50,000 exomes in the UKB to the full cohort of 450,000 exomes, nor when repeating this analysis in 175,000 exomes from the GHS cohort. The signal identified across the first 50,000 exomes may result from a chance ascertainment bias40. Alternatively, whereas the rs2228145-C variant is thought to mimic IL-6 inhibition, and therefore confer protection from heart disease41, neither our analysis nor Bick et al. found evidence that rs2228145 carriers are protected from CVD in subjects without CHIP. Therefore, it is possible that this mutation is a poor proxy for IL-6 inhibition, and that direct pharmacological inhibition of IL-6 may still antagonize the interplay between CHIP clone expansion and the onset of CVD.
This study benefits from its biobank-scale size, which we leverage to further resolve clonal haematopoiesis subtypes and broadly assess clinical phenotypes associated with CHIP. However, limitations include the potential inclusion in our CHIP callset of a small number of germline variants, a lack of serial sampling, and a lack of experimental data to characterize the mechanisms underpinning the novel associations that we identify. Although we have taken many steps to ensure the quality of our callset and analysis (Supplementary Notes 11 and 12 and Supplementary Figs. 15–18), the misclassification of somatic variants with high VAF as germline variants, and/or the misclassification of true germline variants as somatic clonal haematopoiesis variants (for example, germline variants at genomic positions identified as clonal haematopoiesis hotspots) remain challenges inherent to calling and analysing CHIP and clonal haematopoiesis when using population scale genomic data. Serial sampling would enable the evaluation of changes to CHIP clones over time, and future studies that focus on such serial analysis at large scale will be able to better estimate CHIP subtype-specific clonal changes and clinical risk. Such increased data assets would also likely facilitate the identification of additional genes that show recurrent mutation during clonal haematopoiesis, as well as how such mutations relate to one another (that is, in dependency, mutual exclusivity and temporal order). Nonetheless, we identify many novel common and rare variant associations with CHIP and other clonal haematopoiesis phenotypes, which help to set the stage for future functional, mechanistic and therapeutic studies. On the whole, our analyses emphasize that CHIP is really a composite of somatic mutation-driven subtypes, with shared genetic aetiology and distinct risk profiles.
Methods
Study approval
UKB study: ethical approval for the UKB study was previously obtained from the North West Centre for Research Ethics Committee (11/NW/0382). The work described herein was approved by UKB under application number 26041. GHS study: approval for DiscovEHR analyses was provided by the Geisinger Health System Institutional Review Board under project number 2006-0258.
Exome sequencing and variant calling
Sample preparation and sequencing were done at the Regeneron Genetics Center as previously described10,40. In brief, sequencing libraries were prepared using genomic DNA samples from the UKB, followed by multiplexed exome capture and sequencing. Sequencing was performed on the Illumina NovaSeq 6000 platform using S2 (first 50,000 samples) or S4 (all other samples) flow cells. Read mapping, variant calling and quality control were done according to the Seal Point Balinese (SPB) protocol40, which included the mapping of reads to the hg38 reference genome with BWA MEM, the identification of small variants with WeCall, and the use of GLnexus to aggregate these files into joint-genotyped, multi-sample VCF files. While certain UKB exome analysis efforts have used calls generated with the OQFE pipeline42, this pipeline has only been used to a limited degree for disease association analysis. Therefore, we chose to use calls from the SBP pipeline, which have been used very extensively for disease association analysis, including the largest set of association analyses done with UKB exome data10. Depth and allelic valance filters were then applied, and samples were filtered out if they showed disagreement between genetically determined and reported sex, high rates of heterozygosity or contamination (estimated with the VerifyBamId tool as a FREEMIX score > 5%), low sequence coverage, or genetically determined sample duplication.
Calling CHIP
To call CHIP carrier status, we first used the Mutect2 (GATK v4.1.4.0) somatic caller43 to generate a raw callset of somatic mutations across all individuals. This software aims to use mapping quality measures as well as allele frequency information to identify somatic mutations against a background of germline mutations and sequencing errors. We used data generated from gnomAD v2 as the reference source for germline allele frequency44. We generated a cohort-specific panel of normals, which Mutect2 uses to estimate per-site beta distribution parameters for use in refining somatic likelihood assignment. Since CHIP is strongly associated with age, we chose 100 random UKB samples from 40 year olds and 622 samples from individuals less than 18 years of age in GHS to build these cohort-specific panels of normals. By evaluating the degree to which default Mutect2 filtering excluded known CHIP hotspot mutations, we noted that the default Mutect2 pass/fail filters were too stringent. Therefore, we initially considered all Mutect2 variants (that is, even those that did not pass default Mutect2 filtering), and proceeded to perform our own QC and somatic mutation call refinement. As an initial refinement step, we selected variants occurring within genes that have been recurrently associated with CHIP according to recent reports from the Broad2, the TOPMed Consortium11, and the Integrative Cancer Genomics (IntOGen) project45. We then filtered putative somatic mutations using the outlined functional criteria2. Next, we performed additional QC steps, which consisted of (1) removing multi-allelic somatic calls, (2) applying sequencing depth filters (total depth (DP) ≥ 20; alternate allele depth (AD) ≥ 3, F1R2 and F2R1 read pair depth ≥ 1), (3) removing sites flagged as panel of normals by Mutect2 (unless previously reported), (4) removing indels flagged by the Mutect2 position filter, (5) removing sites within homopolymer runs (a sequence of ≥5 identical bases) if AD < 10 or VAF < 0.08, (6), removing missense mutations in CBL or TET2 inconsistent with somaticism (that is, P-value > 0.001 in a binomial test of VAF = 0.5), (7) removing novel (not previously reported) variants that exhibited characteristics consistent with germline variants or sequencing errors. That is, we excluded variants that had a median VAF ≥ 0.35, since approximately 97% of previously reported variants (that is, from a recent study of CHIP by the TOPMed consortium11) had a median VAF < 0.35. Beyond this, we evaluated the frequency distributions of known variants (stratified by effect—that is, missense or non-missense) to discern thresholds for newly identified variants (that is, AF (allele frequency) of novel variants ≤ AF of previously reported variants). Additionally, novel G>T or C>A SNV calls were evaluated for oxidation artifacts46. Specifically, variants with a maximum alternate allelic depth < 6 (across all samples) and < 2 supportive reads from F1R2 (C>A) or F2R1 (G>T) mate pairs were removed, respectively.
Given that > 90% of mutations belonged to 23 recurrent CHIP-associated genes, we restricted to variants occurring within these genes as a final step to maximize the specificity of our callset. These genes consisted of the 8 most frequent mutated CHIP genes (DNMT3A, TET2, ASXL1, PPM1D, TP53, JAK2, SRSF2 and SF3B1), a collection of CHIP-associated genes containing SNV hotspots (BRAF, CSF3R, ETNK1, GNAS, KRAS, GNB1, IDH2, MPL, NRAS, PHF6 and PRPF8), and CHIP-associated genes of haematological interest (CBL, CALR, RUNX1 and SUZ12). Our final CHIP set of CHIP mutation carriers consisted of 29,669 CHIP mutations across 27,331 unique individuals from UKB, and 14,766 CHIP mutations across 12,877 unique individuals from GHS. Variant allele fraction (VAF) was calculated using AD/(reference allele depth (RD) + AD).
Defining CHIP and mosaic phenotypes
CHIP phenotypes were derived based on our mutation callset, whereas mosaic chromosomal alteration (mCA) phenotypes were derived based on previously published mCA calls from the UKB4,7,8. First, we used International Classification of Diseases (ICD) codes to exclude 3,596 samples from UKB and 1,222 samples from GHS that had a diagnosis of blood cancer prior to sample collection. We also excluded 13,004 individuals from GHS whose DNA samples were collected from saliva as opposed to blood. For all of the phenotypes we generated and analysed in this study, we used a combination of cancer registry data, hospital inpatient (HESIN) data, and data from general practitioner records to ascertain ICD10 codes. The majority of our cancer data came from the cancer registry, which we supplemented with the other sources. We then defined multiple CHIP and mosaic phenotypes based on whether carriers did (inclusive) or did not (exclusive) have other somatic phenotypes. For example, individuals with at least one CHIP mutation in our callset were defined as carriers for a CHIP_inclusive phenotype, whereas anyone with a CHIP mutation as well as an identified mCA was removed from this inclusive phenotype in order to define a CHIP_exclusive phenotype (20,606 cases and 342,869 controls). Our association analysis with CHIP used this CHIP_inclusive phenotype, which included 25,657 cases and 342,869 controls of European ancestry in UKB, and 11,821 cases and 135,106 controls of European ancestry in GHS. These counts reflect the samples with European ancestral origin that remain in each cohort after removing those with non-CHIP clonal haematopoiesis (60,991 in UKB and 0 in GHS, as we did not call mosaic chromosomal alterations in GHS), and those with missing meta data (348 in UKB and 4,893 in GHS). We defined mLOY carriers as male individuals with a Y chromosome mCA in the UKB mCA callset that had copy change status of loss or unknown, mLOX as individuals with an X chromosome mCA in the UKB mCA callset that had copy change status of loss or unknown, and mCAaut carriers as individuals with autosomal mCAs. We then refined these inclusive phenotypes to define exclusive versions, with mLOY_exclusive consisting of carriers with no X chromosome or autosomal mCAs (36,187 cases and 151,161 controls), mLOX_exclusive consisting of carriers with no Y chromosome or autosomal mCAs (10,743 cases and 364,072 controls), and mCAaut_exclusive consisting of carriers with no Y or X chromosomal alterations of any kind (11,154 cases and 364,072 controls). These exclusive phenotypes were used for all analyses comparing CHIP with mosaic phenotypes, as this approach facilitated the generation of four non-overlapping phenotypes (that is, CHIP, mLOY, mLOX, and mCAaut) that could be compared. We also defined CHIP gene-specific phenotypes by choosing carriers as those with mutations in our callset from a specific gene and no mutations in any other of the 23 CHIP genes defining our callset. For example, CHIP DNMT3A carriers were those with ≥ 1 somatic mutations in our callset within the DNMT3A gene, and no mutations in our callset in any of the other 23 CHIP genes we used for our final callset definition. The set of 364,072 controls used in UKB that had no evidence of any clonal haematopoiesis (that is, no CHIP or mCAs) was considered as our set of healthy controls, and was used across all association analyses in UKB.
Genetic association analyses
To perform genetic association analyses, we used the genome-wide regression approach implemented in REGENIE47, as described10. In brief, regressions were run separately for data derived from exome sequencing as well as data derived from genetic imputation using TOPMed48, and results were combined across these data sources for downstream analysis. Step 1 of REGENIE uses genetic data to predict individual values for the trait of interest (that is, a polygenic risk score), which is then used as a covariate in step 2 to adjust for population structure and other potential confounding. For step 1, we used variants from array data with a MAF > 1%, < 10% missingness, Hardy–Weinberg equilibrium test P-value > 10−15 and LD pruning (1,000 variant windows, 100 variant sliding windows and r2 < 0.9), and excluded any variants with high inter-chromosomal LD, in the major histocompatibility region, or in regions of low complexity. For association analyses in step 2 of REGENIE, we used age, age2, sex and age × sex, and 10 ancestry-informative principal components as covariates. For analyses involving exome data, we also included as covariates an indicator variable representing exome sequencing batch, and 20 principal components derived from the analysis of rare exomic variants (MAF between 2.6 × 10−5 and 0.01). Significance cutoffs and rare variant burden testing were set according to the power calculations and logic outlined by Backman et al.10. In brief, we used P ≤ 5 × 10−8, P ≤ 7.14 × 10−10, P ≤ 3.6 × 10−7, for common, rare and burden associations, respectively. Results were visualized and processed using an in-house version of the FUMA software49. Association analyses were performed separately for different continental ancestries defined based on the array data, as described10.
Replication of associations signals in the GHS cohort
To calculate the power to achieve replication in the GHS cohort, we first adjusted for the effects of ‘winner’s curse’, which are expected when choosing significant associations signals on the basis of a genome-wide threshold50. To do this, we used the conditional likelihood approach described by Ghosh et al.51 as implemented in the winnerscurse R package (version 0.1.1), which adjusts the estimated betas from genome-wide significant associations signals. These adjusted effect estimates are provided in Supplementary Table 2 (column Effect_adj). We then used these adjusted effect estimates to calculate the expected power to detect each lead signal in the GHS replication phase using the GHS sample size, allele frequencies, CHIP prevalence, and an alpha level of 0.05. To summarize our expected power across the replication phase, we summed the power across all lead variants and reported the number of SNPs that replicated at P < 0.05 as a proportion of the cumulative power to detect those variants.
Identifying independent signals from association results
We used three different approaches to identify independent signals across loci that associated with CHIP. First, we used a clumping and thresholding approach (C&T)52 in which index SNPs at each significantly associated locus were defined greedily as those with the lowest P-value. Clumping was then done by extending linkage blocks laterally to include all SNPs that have P < 1 × 10−5 and r2 > 0.1 with the index SNP. Any SNP within a clump was then removed from further analysis. This process was repeated as long as there was ≥ 1 additional SNP in the locus with P ≤ 5 × 10−8. After all clumps were made, we merged any clumps (that is, LD blocks) with overlapping genomic ranges. Since this approach did not feature any iterative conditioning nor model variant effects jointly, we also used conditional joint analysis as implemented in GCTA COJO53 and statistical fine-mapping as implemented in FINEMAP54 to identify independent/causal signals. COJO was run with a subset of 10,000 unrelated European ancestry samples from UKB as an LD references, and with a COJO adjusted P-value threshold of 5 × 10−6, an info score threshold of 0.3, and a MAF cutoff of 0.01. FINEMAP was run with the shotgun stochastic search algorithm using a maximum of 30 causal variants. We included variants in the FINEMAP analysis that had P < 0.1 in inverse variance weighted meta-analysis, and MAF > 0.001. The LD matrices used for the FINEMAP analysis were constructed as weighted meta LD matrices derived from the LD matrices from UKB and GHS. The LD matrices from UKB and GHS were computed independently using the same sets of samples included in each GWAS.
Fine-mapping variants at the LY75 locus
To further evaluate whether the rare variant association at the LY75 locus (rs147820690-T) was independent of other common and rare variant signals, we performed joint fine-mapping (with FINEMAP) on common and rare variants at this locus while including rarer variants then used in our genome-wide fine-mapping. In contrast to the genome-wide fine-mapping described above, this fine-mapping sensitivity analysis was done only in the UKB, was focused on the LY75 locus, and included all variants in our dataset. That is, the fine-mapping analysis was run as described above, but with a MAF > 0.0000000001. While FINEMAP suggests 3 credible sets are most parsimonious at this locus (posterior probability = 0.8), which is consistent with the results we report when preforming genome-wide fine-mapping, the fourth credible set (posterior probability = 0.11) identifies rs147820690-T as the top signal (PIP = 0.133) among 9,417 variants in the 95% credible set. This fine-mapping approach also prioritizes rs78446341-A (CPIP = 0.92, CS = 2). Furthermore, the median pairwise LD between SNPs in this fourth credible set is very low (6.7 × 10−4, compared with 0.995, 0.962, and 0.831 for the first three credible sets, respectively). Therefore, these fine-mapping results provide additional support for both LY75 missense variants, as well as the fact that the rs147820690-T rare variant signal is not driven by the tagging of other rare variants.
PheWAS across CHIP-associated variants
Using 937 traits from the UKB, we queried association results for 171 SNPs from our GWAS of CHIP. These SNPs represent the union of those identified by clumping and thresholding, conditional analysis with GCTA COJO, and fine-mapping with FINEMAP (fine-mapped SNPs were chosen if they had one of the highest two posterior inclusion probabilities—that is, PIPs—in any credible set). While this group of SNPs does include signals with P < 5 × 10−8 in our CHIP GWAS, these SNPs represent signals prioritized as conditionally independent and/or likely to be causal, and we therefore deemed them worthy of exploration via PheWAS. Some of these subthreshold signals featured many significant PheWAS associations (P < 5 × 10−8 in the PheWAS), and likely merit further evaluation (for example, ZFP36L2/THADA locus on chromosome 2, and THRB locus on chromosome 3). The traits used in this PheWAS represent the subset of the 5,041 traits used in our cross-sectional analyses of phenotypic association with CHIP mutations carrier status for which we have previously reported common variant associations10. In brief, for ICD10-based phenotypes, cases were required to have one or more records of diagnosis in the electronic health records, death registry data implicating the disease, or two or more diagnosis in outpatient data mapped to ICD10. For non-ICD10 phenotypes (quantitative measures, clinical outcomes, survey and touchscreen responses, and imaging derived phenotypes), data were derived from the UKB Showcase. Participants who did not meet the case definition for a given ICD10-based phenotype were removed from the analysis if they had one diagnosis code in the outpatient data, and included as controls if they had no diagnosis in the outpatient data. Supplementary Table 10 includes ICD10 codes as well as trait names and descriptions.
Genetic comparisons between CHIP subtypes
For pairwise comparisons between CHIP gene mutation subtypes, we used the union set of index SNPs (that is, independent signals in genome-wide significant loci) from all of our CHIP and CHIP gene subtype associations. This resulted in 93 variants, which we used to compare effect sizes estimates between CHIP subtype pairs. Genetic correlations were calculated using LDSC version 1.0.1 with annotation input version 2.222.
Defining smoking phenotypes
We derived smoking phenotypes from the lifestyle and environment questionnaire in the UKB and from the electronic health records in the GHS. Since smoking is difficult to ascertain and control for, we used a variety of data to code multiple smoking phenotypes for various analyses. These smoking phenotypes consisted of (1) pack years, (2) number of cigarettes smoked per day, (3) age started/stopped smoking (UKB only), (4) former/current smoker, (5) ever smoker and (6) heavy smoker (smoked ≥ 10 cigarettes a day). The ever smoker phenotype was maximally inclusive, and coded as cases all individuals with any evidence of prior smoking across the aforementioned phenotypes. For our longitudinal analyses in UKB, we used the ‘current smoker’ and ‘pack years’ (which captures the cumulative effect of smoking over one’s lifetime) as covariates in all models that did not stratify for smoking status. In the smoking stratified models, we stratified smokers based on the ‘ever smoker’ phenotype and further adjusted for pack years within the smokers subgroup. For our longitudinal analyses in GHS, we used the ‘ever smoker’ and ‘pack years’ phenotypes as covariates in all models that did not stratify for smoking status, and stratified smokers in the same manner as we did in the UKB analyses. For linear models that evaluated the overall relationship between age, sex, and smoking, we used the ‘heavy smoker’ coding. Otherwise, all other analyses used the aforementioned ‘ever smoker’ phenotype as a covariate.
Phenotypic associations with CHIP
To test for known as well as potentially novel associations, we used REGENIE47 to perform Firth-corrected tests for association between our CHIP gene-specific phenotypes and 5,041 traits (2,640 binary traits and 2,401 quantitative traits) from the UKB (version 5). To do this, we coded each CHIP gene-specific phenotype as 1 if an individual had any somatic CHIP mutation in the gene and 0 otherwise and formatted these binary codings as pseudo-genotypes to analyse with REGENIE. Regression models were run as described previously, with age, sex, and genetic principal components as covariates10. After filtering out association tests where the total number of somatic carriers was <5, we were left with 83,779 total association tests (Supplementary Table 31). Only 22 out of 23 CHIP gene subtypes were tested for association across phenotypes as we did not have enough carriers of CSF3R mutations to meet our minimum threshold of 5 somatic carriers that were also disease cases. Quantitative traits were transformed using a reverse inverse normalized transformation (RINT); effect size estimates from these associations are in units of standard deviation. Traits used in this analysis did not exclude any samples on the basis of having a diagnosed haematological disease or malignancy prior to sequencing date. To visualize high-level phenotypic patterns across these CHIP gene-specific phenotypes (Fig. 3), we categorized phenotypes by disease group10, and calculated the proportion of phenotypes per disease group per gene that were associated at a P ≤ 0.05 alpha level (uncorrected). To visualize the most significant of these associations, we plotted effect sizes (Supplementary Fig. 7) by disease category for all associations with P ≤ 1 × 10−5.
Risk modelling among CHIP carriers
We performed longitudinal survival analyses using cox proportional hazard models (coxph function) as implemented in the survival R package. Given that CHIP is strongly correlated with age, models used age as the time scale with interval censoring with age at first assessment and age at event or censoring. This allows for an implicit adjustment for age within the proportional hazard models. In UKB, individuals with follow-up time in excess of 13.5 years (3% of the dataset) were censored due to departures from the proportional hazards model. Analyses were performed on individuals of European ancestral background. All models included 10 genetically determined European-specific principal components as covariates, and all analyses excluded individuals genetically determined to be third-degree relatives or closer. In GHS, we had limited sample size with which to perform these longitudinal analyses. This was because GHS samples were collected at later ages (due to the nature of the biobank and the timing of our partnership) and fewer patients had disease onset dates subsequent to sample collection (that is, the time period where the onset of CHIP can be evaluated). Furthermore, in GHS, we could not derive an all-cause mortality phenotype due to the nature of the EHR data available to us. This incomplete ascertainment may also explain why our odds ratio estimates for risk of haematologic malignancy among CHIP carriers are lower in the GHS cohort.
We used a variety of CHIP codings as variables in our models to test for potential differences between high/low VAF CHIP and/or CHIP subtypes. First, we subset CHIP carrier status by gene (DNMT3A, TET2, ASXL1, DNMT3A or TET2) and/or VAF (≥0.1) to test for potential differences between degree of clonal expansion (that is, high/low VAF CHIP) and/or CHIP subtypes. Additional analyses were run restricting CHIP mutation calls to previously reported variants (for example, Jaiswal et al.2), as well as restricting to carriers of DNMT3A mutations with at least one mutation in another CHIP gene. Controls were defined with two approaches: (1) any individual without CHIP mutations (the coding used in the results we report) and (2) those without any genetic evidence of clonal haematopoiesis (that is, healthy controls, as defined above, which did not change our results). The CHIP gene-specific coding described above varies from the phenotypic coding definitions used in our GWAS/ExWAS, which required carriers to have mutations only in the specified CHIP gene and no mutations in any other CHIP genes. Since mutational exclusivity becomes less common as VAF increases (that is, carrying a single mutation with VAF ≥ 0.1 and no other mutations), and substantially lowers sample size, we chose this adjusted definition for these longitudinal analyses of disease incidence. For the composite phenotypes described below, we relied heavily on ICD10 codes from cancer registry data, hospital records and general practitioner records, and supplemented these with self-reported data and procedure codes (OPCS4). We defined prevalent disease on the basis of event codes occurring before sample collection and used this definition to exclude samples from longitudinal analysis of incident disease. For these main analyses, we did not use any minimum number of days to diagnosis from sample collection as an additional filtering criterion (see Supplementary Note 12 for more details).
In UKB, cardiovascular disease was defined with the following ICD10 codes obtained from primary care, HES (hospital episode statistics), or death registry data: I21, I22, I23, I252, I256, Z951, Z955, I248, I249, I241, I251, I255, I258, I259, I630, I631, I632, I633, I634, I635, I637, I638, I639, I651, ICD9 codes: 410, 412, and OPCS codes: K40, K41, K44, K45, K46, K49, K502, K75 and K471. ICD9/ICD10/OPCS diagnoses or procedures recorded prior to enrolment date and self-report codes 1075 (heart attack/myocardial infarction), 1095 (cabg), 1523 (heart bypass), 1070 (coronary angioplasty or stent), 1583 (ischaemic stroke), 1083 (stroke) were used to identify prevalent CVD cases. These were chosen to best reflect the coding use by Bick et al. in their study of CHIP6. In GHS, we used ICD10 codes I20–I25 and I60–I69, CPT codes from 33510–33523 (CABG, not continuous), 33533–33536, 35500, 35572, 35600, and 92920–92975 (PCI, not continuous). We also adjusted the CVD coding in GHS to exclude cerebrovascular events (that is, excluded I60–I69); association results were similar. The CVD coding we used for our Mendelian randomization analysis was comparable to these definitions but did not include ICD10 codes for cerebrovascular events.
For the CVD models, we included sex, LDL, HDL, pack years, smoking status (current vs former, determined by self-reported data), BMI, essential primary hypertension, and type 2 diabetes mellitus as covariates. The results we reported used a composite of myocardial infarction (MI), coronary artery bypass graft (CABG), percutaneous coronary intervention (PCI), and coronary artery disease (CAD), based on the coding described above, and also included death from any of these events. Results were similar when our composite included ischaemic stroke (ISCH.TR), as well as when we repeated analyses with a subset of recurrent CHIP mutations derived from Jaiswal et al.2 or restricting carrier calls to variants in DNMT3A or TET2. We also excluded samples with any diagnosis of malignant blood cancer prior to sequencing (n = 3,596). Missing LDL and HDL values were median imputed, and individuals on cholesterol medication had their raw LDL values increased by a factor of 1/0.68, similar to Bick et al.6. IL6R missense variant (rs2228145-C) genotypes were modelled dominantly (coded as 1 for carriers of any allele and 0 otherwise), and we modelled the effect of this allele in CHIP -stratified proportional hazard models, and also tested for IL6R × CHIP interaction in a full (non-stratified) model. Models considering only the initial 50k UKB individuals restricted to intersection between our unrelated UKB sample set and the samples reported by Bick et al. 6. For visualization, Kaplan–Meier estimates were generated with the survfit function in the aforementioned survival package (version 3.2.13) and plotted using the ggsurvplot function from the survminer package (version 0.4.9).
For models of cancers and overall survival risk tested using all CHIP carriers, high-VAF (VAF ≥ 0.1) CHIP carriers, and carriers of specific CHIP gene mutations, we used unrelated European samples that did not have any cancer diagnoses prior to sample collection (N = 360, 051 after the removal of 33,816 samples with a prior diagnosis of cancer). Results were qualitatively the same when repeating these analyses without excluding samples that had a diagnosis of any malignant cancer prior to sample collection date. Cancer phenotype definitions were derived from medical records indicating the following ICD10 codes: C81–C96, D46, D47.1, D47.3, D47.4 for blood cancers, C81–C86, C91 for lymphoid cancers, C92, C94.4, C94.6, D45, D46, D47.1, D47.3, D47.4 for myeloid cancers, C50 for breast cancers, C34 for lung cancers, C61 for prostate cancers, C44 for non-melanoma skin cancers (NMSC), and C18 for colon cancers (five total solid cancers). Myeloid subtypes were defined as follow: AML (C92), MDS (D46), MPN (D47.1, D47.3, D47.4). Given the rareness and/or non-specificity of myeloid codings C93–95, and that the majority of these codings overlapped with those that we used for the myeloid composite described above (that is, we already captured these samples using the previously described codings), we did not include these codings in our composite. However, we performed sensitivity analyses that used a myeloid definition that did include C93–C95, with findings equivalent to those described in our main results (Supplementary Note 12). For our lymphoid composite, we decided to combine lymphoma with lymphoid leukaemia for multiple reasons. First, in some clinical diagnostic situations (for example, T cell lymphoblastic lymphoma and T cell lymphoblastic leukaemia; Burkitt lymphoma and mature B cell ALL), the distinction between ‘leukaemia’ and ‘lymphoma’ is made on the basis of blast percentage in bone marrow (that is, > 20% blasts diagnosed as leukaemia), and may not reflect meaningful biological differences. Consistently, 22% of C91 codings are already captured in our C81–C86 codings. Moreover, the majority of cases across these codings correspond to tumours derived from mature B cells, namely chronic lymphocytic leukaemia (CLL) and mature non-Hodgkin lymphoma. Given data supporting that mature T cell lymphomas and also some mature non-Hodgkin B cell tumours may arise from hematopoietic stem and progenitor cells55–57, we considered the relationship between a composite of mature lymphoid tumours and CHIP. For blood cancers, we also included cases that self-reported leukaemia, lymphoma, or multiple myeloma. These models included the same covariates as described for CVD (with the exception that we did not adjust cholesterol level based on medication usage). Additionally, models estimating risk for sex-specific cancers (that is, prostate and breast) restricted to individuals of the relevant sex and did not adjust for sex as a covariate. For smoking stratified modelling of blood and lung cancer, we used our stricter definition of smoking (ever vs never) and included pack years as a covariate in models testing risk among smokers. To test a more conservative cutoff for excluding patients with a diagnosis of haematologic malignancy prior to sequencing (that is, exclude individuals with a diagnosis prior to 90 days after DNA collection date rather than prior to the DNA collection date itself), we conducted sensitivity analyses for the longitudinal modelling of the risk among CHIP carriers of acquiring blood cancers (for example, blood cancer, myeloid, lymphoid, AML, MDS and MPN). These results were the same as those reported in our main results (Supplementary Note 12).
Polygenic risk scores
Polygenic risk scores were calculated with Plink58 as a weighted sum of the effects across all conditionally independent variants we identified with GCTA COJO (74 variants, P ≤ 5 × 10−6) We performed association tests using logistic regression, with binary phenotypes of interest (that is, our CHIP subtype phenotypes—for example, TET2 CHIP, and so on) as the dependent variable, this polygenic risk score as the independent variable of interest, and age, sex, smoking status (ever vs never), and 10 genetic principal components as covariates.
Software
The code is publicly available and can be found at https://github.com/rgcgithub/regenie. The REGENIE software for whole-genome regression, which was used to perform all genetic association analysis, is available at https://github.com/rgcgithub/regenie. GCTA v1.91.7 was used for approximate conditional analysis. SHAPEIT4.2.0 was used for phasing of SNP array data. Imputation was completed with IMPUTE5. Somatic calling was done with Mutect2 (GATK v4.1.4.0). We use Plink1.9/2.0 for genotypic analysis as well as for constructing polygenic risk scores. FINEMAP was used for fine-mapping, and genetic correlations were calculated using LDSC version 1.0.1 with annotation input version 2.2. Beyond standard R packages, visualization tools, and data processing libraries (for example, dplyr, ggplot2 and data.table), we used the survival (version 3.2.13) and survminer (version 0.4.9) packages for survival analyses, the MendelianRandomization package for Mendelian randomization (version 0.6.0), and the winnerscurse package (version 0.1.1; https://amandaforde.github.io/winnerscurse/) to adjust GWAS effect size estimates for the effects of Winner’s Curse.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-022-05448-9.
Supplementary information
This file contains supplementary information for numerous analyses, and includes 12 supplementary notes, which are referenced throughout the Article.
Members lists for Regeneron Genetics Center and GHS-RGC DiscovEHR Collaboration.
This file contains Supplementary Figs. 1–18.
Supplementary Tables 1–33 - see `Table Descriptions’ worksheet within the file for details.
Extended data figures and tables
Author contributions
M.D.K., A.D., S.O., S.S., N.B., D.L., J.T. and H.M.K. performed bioinformatic and statistical genetic analysis. S.O., K.W., A.M., Y.Z., A.G., C.G., J. Mbatchou, W.S., J.G.R. and H.M.K. contributed to data engineering and bioinformatic pipeline development. J.D.O. developed and oversaw exome sequencing efforts. M.D.K., A.D., S.O., K.W., M.V.M., J.H., J.A.K., V.M.R., Y.H., C.G., R.R.W., N.V., J.B., N.N.P., M.G.L., M.J., D.J.G., L.A.L., M.N.C., G.S.A., A.E.L., M.A.R.F., R.D., G.T., C.P., A.R.S., A.A.F., W.S., J.G.R., J.D.O., J. Marchini, H.M.K., A.B., G.R.A. and E.J. contributed to experimental design, interpretation of results, and genetic programme development. M.D.K. and E.J. drafted the manuscript, with significant input from A.D., S.O., M.V.M., G.T., A.A.F., H.M.K., A.B. and G.R.A. All authors reviewed and approved the final version of the manuscript.
Peer review
Peer review information
Nature thanks Po-Ru Loh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
Individual-level sequence data, CHIP calls and polygenic scores have been deposited with UK Biobank and are freely available to approved researchers, as done with other genetic datasets to date10. Individual-level phenotype data are already available to approved researchers for the surveys and health record datasets from which all our traits are derived. Instructions for access to UK Biobank data is available at https://www.ukbiobank.ac.uk/enable-your-research. Summary statistics from UKB trait are available in the GWAS catalogue (accession IDs are listed in Supplementary Table 33). As described10, the HapMap3 reference panel was downloaded from ftp://ftp.ncbi.nlm.nih.gov/hapmap/, GnomAD v3.1 VCFs were obtained from https://gnomad.broadinstitute.org/downloads, and VCFs for TOPMED Freeze 8 were obtained from dbGaP as described in https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-8. Data used for replication, such as DiscovEHR exome sequencing and genotyping data, and derived CHIP calls, can be made available to qualified, academic, non-commercial researchers upon request via a Data Transfer Agreement with Geisinger Health System (contact person: Lance Adams, ljadams@geisinger.com).
Competing interests
M.D.K., A.D., S.O., N.B., D.L., K.W., A.M., M.V.M., S.S., J.H., J.T., J.A.K., V.M.R., Y.Z., Y.H., A.G., C.G., J. Mbatchou, R.R.W., N.V., J.B., N.N.P., M.G.L., M.J., D.J.G., L.A.L., M.N.C., G.S.A., A.E.L., M.A.R.F., R.D., C.P., A.R.S., G.T., A.A.F., W.S., J.G.R., J.D.O., J. Marchini, H.M.K., A.B., G.R.A. and E.J. are current employees and/or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A list of authors and their affiliations appears at the end of the paper
A full list of members and their affiliations appears in the Supplementary Information
Change history
2/17/2023
A Correction to this paper has been published: 10.1038/s41586-023-05803-4
Extended data
is available for this paper at 10.1038/s41586-022-05448-9.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-022-05448-9.
References
- 1.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. New Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New Engl. J. Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zekavat SM, et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat. Med. 2021;27:1012–1024. doi: 10.1038/s41591-021-01371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niroula A, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 2021;27:1921–1927. doi: 10.1038/s41591-021-01521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bick AG, et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141:124–131. doi: 10.1161/CIRCULATIONAHA.119.044362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson DJ, et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019;575:652–657. doi: 10.1038/s41586-019-1765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loh P-R, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018;559:350–355. doi: 10.1038/s41586-018-0321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbari P, et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. 2021;373:eabf8683. doi: 10.1126/science.abf8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Backman JD, et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature. 2021;599:628–634. doi: 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bick AG, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–768. doi: 10.1038/s41586-020-2819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Codd V, et al. Polygenic basis and biomedical consequences of telomere length variation. Nat. Genet. 2021;53:1425–1433. doi: 10.1038/s41588-021-00944-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denny JC, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene–disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nédélec Y, et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell. 2016;167:657–669. e21. doi: 10.1016/j.cell.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 15.Joehanes R, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017;18:16. doi: 10.1186/s13059-016-1142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhernakova DV, et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 2017;49:139–145. doi: 10.1038/ng.3737. [DOI] [PubMed] [Google Scholar]
- 17.Võsa U, et al. Large-scale cis-and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotoh N, et al. PARP1 V762A polymorphism affects the prognosis of myelodysplastic syndromes. Eurr. J. Haematol. 2020;104:526–537. doi: 10.1111/ejh.13393. [DOI] [PubMed] [Google Scholar]
- 19.Fukaya T, et al. Conditional ablation of CD205+ conventional dendritic cells impacts the regulation of T-cell immunity and homeostasis in vivo. Proc. Natl Acad. Sci. USA. 2012;109:11288–11293. doi: 10.1073/pnas.1202208109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palamarchuk A, et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL) Proc. Natl Acad. Sci. USA. 2012;109:2555–2560. doi: 10.1073/pnas.1200003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer. 2017;17:5. doi: 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawoud AAZ, Gilbert RD, Tapper WJ, Cross NCP. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia. 2022;36:507–515. doi: 10.1038/s41375-021-01382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrander EL, et al. Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Rep. 2020;14:551–560. doi: 10.1016/j.stemcr.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joo SH, Park JK, Lee EE, Song YW, Yoon S-S. Changes in serum uric acid levels after allogeneic hematologic stem cell transplantation: a retrospective cohort study. Blood Res. 2016;51:200–203. doi: 10.5045/br.2016.51.3.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds MD. Gout and hyperuricemia associated with sickle-cell anemia. Semin. Arthritis Rheum. 1983;12:404–413. doi: 10.1016/0049-0172(83)90020-3. [DOI] [PubMed] [Google Scholar]
- 28.McAdams-DeMarco MA, Maynard JW, Coresh J, Baer AN. Anemia and the onset of gout in a population-based cohort of adults: Atherosclerosis Risk in Communities study. Arthritis Res. Ther. 2012;14:R193. doi: 10.1186/ar4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolton KL, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat. Commun. 2021;12:5975. doi: 10.1038/s41467-021-26138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kosmicki JA, et al. Pan-ancestry exome-wide association analyses of COVID-19 outcomes in 586,157 individuals. Am. J. Hum. Genet. 2021;108:1350–1355. doi: 10.1016/j.ajhg.2021.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith MA, McCaffrey RP, Karp JE. The secondary leukemias: challenges and research directions. JNCI. 1996;88:407–418. doi: 10.1093/jnci/88.7.407. [DOI] [PubMed] [Google Scholar]
- 32.Fehrmann RS, et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaudhuri AR, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017;18:610–621. doi: 10.1038/nrm.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jing C-B, et al. Synthetic lethal targeting of TET2-mutant hematopoietic stem and progenitor cells (HSPCs) with TOP1-targeted drugs and PARP1 inhibitors. Leukemia. 2020;34:2992–3006. doi: 10.1038/s41375-020-0927-5. [DOI] [PubMed] [Google Scholar]
- 35.Abbotts R, et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl Acad. Sci. USA. 2019;116:22609–22618. doi: 10.1073/pnas.1903765116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore KN, Mirza MR, Matulonis UA. The poly (ADP ribose) polymerase inhibitor niraparib: management of toxicities. Gynecol. Oncol. 2018;149:214–220. doi: 10.1016/j.ygyno.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Jiang W, et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375:151–155. doi: 10.1038/375151a0. [DOI] [PubMed] [Google Scholar]
- 38.Kato M, et al. Expression of human DEC-205 (CD205) multilectin receptor on leukocytes. Int. Immunol. 2006;18:857–869. doi: 10.1093/intimm/dxl022. [DOI] [PubMed] [Google Scholar]
- 39.Kim PG, et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J. Exp. Med. 2021;218:e20211872. doi: 10.1084/jem.20211872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Hout CV, et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature. 2020;586:749–756. doi: 10.1038/s41586-020-2853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferreira RC, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013;9:e1003444. doi: 10.1371/journal.pgen.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szustakowski JD, et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet. 2021;53:942–948. doi: 10.1038/s41588-021-00885-0. [DOI] [PubMed] [Google Scholar]
- 43.Benjamin, D. et al. Calling Somatic SNVs and Indels with Mutect2. Preprint at bioRxiv10.1101/861054 (2019).
- 44.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pich O, Reyes-Salazar I, Gonzalez-Perez A, Lopez-Bigas N. Discovering the drivers of clonal hematopoiesis. Nat. Commun. 2022;13:4267. doi: 10.1038/s41467-022-31878-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costello M, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mbatchou J, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 2021;53:1097–1103. doi: 10.1038/s41588-021-00870-7. [DOI] [PubMed] [Google Scholar]
- 48.Taliun D, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong H, Prentice RL. Correcting “winner’s curse” in odds ratios from genomewide association findings for major complex human diseases. Genet. Epidemiol. 2010;34:78–91. doi: 10.1002/gepi.20437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh A, Zou F, Wright FA. Estimating odds ratios in genome scans: an approximate conditional likelihood approach. Am. J. Hum. Genet. 2008;82:1064–1074. doi: 10.1016/j.ajhg.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Privé F, Vilhjálmsson BJ, Aschard H, Blum MG. Making the most of clumping and thresholding for polygenic scores. Am. J. Hum. Genet. 2019;105:1213–1221. doi: 10.1016/j.ajhg.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benner C, et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kikushige Y, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20:246–259. doi: 10.1016/j.ccr.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 56.Quivoron C, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 57.Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. New Engl. J. Med. 2012;366:95–96. doi: 10.1056/NEJMc1111708. [DOI] [PubMed] [Google Scholar]
- 58.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains supplementary information for numerous analyses, and includes 12 supplementary notes, which are referenced throughout the Article.
Members lists for Regeneron Genetics Center and GHS-RGC DiscovEHR Collaboration.
This file contains Supplementary Figs. 1–18.
Supplementary Tables 1–33 - see `Table Descriptions’ worksheet within the file for details.
Data Availability Statement
Individual-level sequence data, CHIP calls and polygenic scores have been deposited with UK Biobank and are freely available to approved researchers, as done with other genetic datasets to date10. Individual-level phenotype data are already available to approved researchers for the surveys and health record datasets from which all our traits are derived. Instructions for access to UK Biobank data is available at https://www.ukbiobank.ac.uk/enable-your-research. Summary statistics from UKB trait are available in the GWAS catalogue (accession IDs are listed in Supplementary Table 33). As described10, the HapMap3 reference panel was downloaded from ftp://ftp.ncbi.nlm.nih.gov/hapmap/, GnomAD v3.1 VCFs were obtained from https://gnomad.broadinstitute.org/downloads, and VCFs for TOPMED Freeze 8 were obtained from dbGaP as described in https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-8. Data used for replication, such as DiscovEHR exome sequencing and genotyping data, and derived CHIP calls, can be made available to qualified, academic, non-commercial researchers upon request via a Data Transfer Agreement with Geisinger Health System (contact person: Lance Adams, ljadams@geisinger.com).