

Abstract
Secretory preproteins of the Sec pathway are targeted post‐translationally and cross cellular membranes through translocases. During cytoplasmic transit, mature domains remain non‐folded for translocase recognition/translocation. After translocation and signal peptide cleavage, mature domains fold to native states in the bacterial periplasm or traffic further. We sought the structural basis for delayed mature domain folding and how signal peptides regulate it. We compared how evolution diversified a periplasmic peptidyl‐prolyl isomerase PpiA mature domain from its structural cytoplasmic PpiB twin. Global and local hydrogen–deuterium exchange mass spectrometry showed that PpiA is a slower folder. We defined at near‐residue resolution hierarchical folding initiated by similar foldons in the twins, at different order and rates. PpiA folding is delayed by less hydrophobic native contacts, frustrated residues and a β‐turn in the earliest foldon and by signal peptide‐mediated disruption of foldon hierarchy. When selected PpiA residues and/or its signal peptide were grafted onto PpiB, they converted it into a slow folder with enhanced in vivo secretion. These structural adaptations in a secretory protein facilitate trafficking.
Keywords: folding, HDX‐MS, mature domain, secretion, signal peptide
Subject Categories: Translation & Protein Quality
Comparison between a slow‐folding secreted bacterial protein (PpiA) and its fast‐folding cytosolic homologue (PpiB) offers insights into structural features that regulate protein folding.
Introduction
All proteins are synthesized on ribosomes as unstructured polymers. While cytoplasmic proteins fold immediately and become functional (Anfinsen, 1972), most exported proteins delay their folding to insert into or translocate across the membrane bilayer until they reach their final destination (Tsirigotaki et al, 2017a).
The exportome, comprising a third of the bacterial proteome, mainly uses the essential and ubiquitous secretory (Sec) pathway (Tsirigotaki et al, 2017a). In post‐translational export, fully synthesized secretory nascent proteins are released from the ribosome, transit the cytoplasm, reach the Sec translocase while remaining unfolded/soluble and avoiding misfolding/aggregation (Tsirigotaki et al, 2017a; Van Puyenbroeck & Vermeire, 2018). This route is taken by 505 secretory preproteins bearing N‐terminal signal peptides in the Escherichia coli model cell (De Geyter et al, 2016; Tsirigotaki et al, 2017a). Signal peptides and mature domain targeting signals (MTS) are recognized by the SecA translocase subunit and allosterically modulate it to initiate secretion (Gouridis et al, 2009; Chatzi et al, 2017; Krishnamurthy et al, 2021; preprint: Krishnamurthy et al, 2022). Once translocated, signal peptides get cleaved (Auclair et al, 2011), while mature domains fold in functional native states in the cell envelope or beyond (De Geyter et al, 2016).
Intrinsic protein features (Dill, 1999) and their interactions with extrinsic factors (chaperones; Smets et al, 2019) dictate folding in the cytoplasm, ranging from fast folding (micro to low seconds time scale; Mayor et al, 2003) to remaining stably unfolded (i.e. Intrinsically Disordered Proteins (IDPs; Oldfield & Dunker, 2014)). Polar residues, reduced overall hydrophobicity and enhanced backbone dynamics promote disorder in IDPs (Uversky, 2013; Tsirigotaki et al, 2018; Loos et al, 2019). Secretory preproteins display folding behaviours intermediate to those of fast folders and IDPs, by retaining kinetically trapped, loosely folded states due to unique structural/sequence characteristics of their mature domains (Zhou & Dunker, 2018; Tsirigotaki et al, 2018; Loos et al, 2019). They contain fewer, smaller/weaker hydrophobic patches than cytoplasmic proteins but more than IDPs (Tsirigotaki et al, 2018) and smaller, more polar, soluble and disorder‐prone residues (Loos et al, 2019). These differences suffice for the MatureP algorithm to predict secretory proteins with 95% confidence (Orfanoudaki et al, 2017; Loos et al, 2019).
In addition to mature domain features, signal peptides slow down folding (e.g. of Maltose Binding Protein; Park et al, 1988). Fusing various signal peptides to the disordered N terminus of a mature domain differentially modulated disorder across the whole protein (Sardis et al, 2017). In some (but not all) secretory proteins, signal peptides delayed mature domain folding by apparently stabilizing loosely folded intermediates (Tsirigotaki et al, 2018). How this signal peptide effect has co‐evolved with a mature domain's folding properties remains unclear. However, slow folding of secretory chains correlates with their translocation competence and thereby underlies secretability (Tsirigotaki et al, 2018). Secretion‐related chaperones, SecB (Huang et al, 2016) and Trigger Factor (TF; Saio et al, 2014; De Geyter et al, 2020), may stabilize non‐folded states, prevent aggregation and promote translocase targeting but specialize on a small subset of secretory clients (De Geyter et al, 2020) and, therefore, cannot explain the global intrinsic properties of the secretome.
Folding is a complex process, involving multiple topologies and motifs. Two competing models predominate. “Multiple pathways” proposes that proteins fold along multiple, stochastic, microscopic landscapes where the speed of the process is driven by a folding funnel in search of the energetically minimal native state (Onuchic et al, 1997). The “Defined pathway” postulates fixed sequential folding steps with defined intermediates (Gianni et al, 2007; Englander & Mayne, 2017). Here, polypeptide chains fold according to a “stepwise plan”, starting with the gradual assembly of “foldons” through native‐like intermediates (Panchenko et al, 1996; Englander & Mayne, 2014). Foldons, short cooperative folding units (∼15–35 residues), acquire native‐like local structure and mutually stabilize each other hierarchically (Englander & Mayne, 2014, 2017). These “initial” stabilized foldons are extended further to complete folding. Sequences of 5–10 residues (hereafter “early folding regions”) appear structurally primed to intrinsically nucleate foldon formation (Raimondi et al, 2019). Prediction of these linear motifs is unrelated to their 3D context in the protein. They are commonly detected in energetically stable regions of the native structure (Bittrich et al, 2018) and may provide the stepping stones to rapidly trigger the most efficient pathway towards native structure and lead to residue‐residue side chain interactions seen in the native state (Nymeyer et al, 1998). Such early interactions of native residue side chains may bias the formation of native structural elements, thereby making folding efficient and fast (Englander & Mayne, 2017) as seen in small proteins by Molecular Dynamics simulations (Best et al, 2013). In contrast, regions with “frustrated” residues (i.e. with suboptimal stability/interactions in the native structure; Ferreiro et al, 2007; Wolynes, 2015) or inability to create critical β‐turns (Marcelino & Gierasch, 2008; Fuller et al, 2009) could delay folding.
Folding is mainly studied using orthogonal biophysical techniques (circular dichroism, fluorescence, single‐molecule studies; (Schuler & Eaton, 2008; Bornschlogl & Rief, 2011), faster time series (Munoz & Cerminara, 2016) and computer simulations (Chen et al, 2018) etc.) that provide information about the 2D or 3D structure of the whole protein in kinetics and equilibrium studies (Dill & MacCallum, 2012; Braselmann et al, 2013; Hu et al, 2013; Englander & Mayne, 2014; Englander et al, 2016; Munoz & Cerminara, 2016). A powerful tool is Hydrogen (1H) Deuterium (D, 2H) exchange Mass Spectrometry (HDX‐MS). “Global” HDX‐MS detects the different species within the folding population of an intact protein (unfolded, intermediate and folded; Tsirigotaki et al, 2017b, 2018), while “local” HDX‐MS monitors folding of short protein segments at near‐residue resolution (Maity et al, 2005; Walters et al, 2013; Englander & Mayne, 2014; Pancsa et al, 2016). The latter exploits HDX kinetics to observe the transition between the unfolded (i.e. non or weakly H‐bonded) and folded (completely H‐bonded) populations of a single peptide (EX1 kinetics; Ferraro et al, 2004; Englander et al, 2007; Marcsisin & Engen, 2010). H‐bonded regions are “protected” from taking up D and are readily identified.
Delayed folding in most secretory mature domains (Tsirigotaki et al, 2018; Loos et al, 2019) contrasts the fast folding of most cytoplasmic domains. Structural twin pairs (i.e. structural homologues with high sequence identity/similarity and same enzymatic function) display minimal evolutionary “noise” and may allow definition of the structural adaptations needed for each folding behaviour. Such pairs are rare; the one selected here is the secreted peptidyl‐prolyl cis‐trans isomerase PpiA and the cytoplasmic PpiB (Fig 1A; Appendix Fig S1A; Hayano et al, 1991; Ikura et al, 2000). From in vitro refolding (using global/local HDX‐MS; Tsirigotaki et al, 2017b), we identified the folding pathways, foldons and specific residues that promote slow‐ and fast‐folding kinetics. Using structural bioinformatics, we defined native contacts, frustrated regions, early folding regions, suboptimal β‐turns and residues contributing to stability. Both proteins displayed three‐state folding with only modestly different folding pathways and foldons, while PpiA folded more slowly. Folding commenced by the sequential formation of “initial” foldons, located near or interacting with the N‐termini. While foldons were largely shared across the twins, they formed in different order. Moreover, the signal peptide stalled folding of PpiA at an early, little folded intermediate. Few native residues grafted between PpiA and PpiB reciprocally interchanged folding behaviours and in vivo secretability and grafting the PpiA signal peptide to PpiB delayed folding. The signal peptide acted by introducing N‐terminal disorder and disrupted the twins' foldon hierarchy. We propose that delayed‐folding adaptations in secretory mature domains alone leading to altered folding pathways or combined with signal peptide‐driven delayed folding, are universal mechanisms of Sec‐dependent protein secretion.
Figure 1. Structural features of PpiB and PpiA.
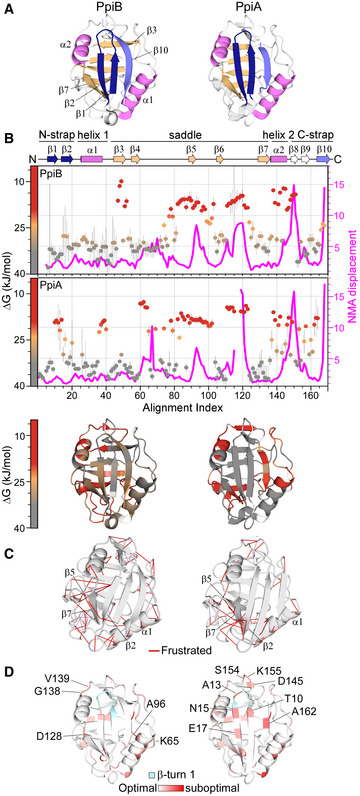
- Structural features are colour‐indicated on 3D structures (top) or linear map of secondary structure (bottom; from Appendix Fig S1D). β‐strands that connect the sheets to form the straps and quasi β‐barrel and α‐helices as annotated.
- Dynamics of native PpiA/B. Top left y‐axis (reversed) displayed as ΔG/residue (from PyHDX analysis of HDX‐MS data at 30°C) colour‐indicated across the linear sequence (top; x‐axis) or on 3D structures (bottom). The apparent rigidity at the extreme N‐tail of PpiA was attributed to high back exchange of this peptide and, therefore, ignored. Dots: grey (stable); orange (flexible); red (unstructured). Grey error bars: variation between subsequent residues (see Fig EV1E for %D‐uptake values; HDX‐MS data in Dataset EV4). n = 3 technical repeats. Top, right y‐axis: normal mode analysis; total displacement of normal modes 7–13 (unweighted sum; magenta) (see Materials and Methods).
- Direct frustrated interactions (red lines) and water‐mediated ones (purple, dashed) are indicated on 3D structures.
- Suboptimal residue/structure compatibility determined by Rosetta scoring analysis coloured using a gradient (see Materials and Methods) on the 3D structures.
Data information: The PDB entries used are as follows: 1LOP for PpiB and 1V9T for PpiA.
Source data are available online for this figure.
Results
Properties of the PpiB and PpiA structures
To define the structural adaptations needed for translocation competence, we studied two twins: the cytoplasmic and the periplasmic peptidyl‐prolyl cis‐trans isomerases PpiB and PpiA. They have practically identical structures (RMSD: 0.37 Å, Appendix Fig S1A) and share 55.6% sequence identity with a further 25.3% high similarity (Appendix Fig S1B).
Both proteins are composed of distinct sub‐structures (Fig 1A): N‐ and C‐terminal straps (β1‐2/β10; dark blue/grey, respectively) assemble from opposite directions to form a β‐sheet on the N‐terminal‐facing half of the structure. The straps perpendicularly overlay a 5‐stranded β‐sheet “saddle” (β3‐7; light orange), which is H‐bonded to each other (via N‐strap/saddle β2/β7 and C‐strap/saddle β10/β3; mainly visible in PpiB; Fig 1A) to complete a quasi‐orthogonal 8‐stranded β‐barrel. On the concave surface of the saddle, opposite the straps, lies the prolyl isomerase catalytic site (Scholz et al, 1997). The N‐/C‐strap β‐sheet docks along a groove on the upper surface of the saddle, while α1 and 2 on either side act as “banisters” (Fig 1A, violet; Appendix Fig S1C). Minor dissimilarities are present; an extra flexible N‐terminal extension in PpiA (1AKGDPH6) and a 3‐residue loop insertion between β6‐β7 in PpiB (Appendix Fig S1D).
Sequence comparison of PpiB/A across 150 bacterial homologues (Dataset EV1A–C; Ashkenazy et al, 2016) revealed a highly conserved saddle/catalytic site (Appendix Fig S1D) with variation in the N‐termini, surface‐exposed residues, connecting loops and the β8‐9 hairpin (Appendix Fig S1B and D). Buried residues retain similar physicochemical properties or form similar hydrophobic cores (Dataset EV1D).
Stability and intrinsic dynamics of native PpiB and PpiA
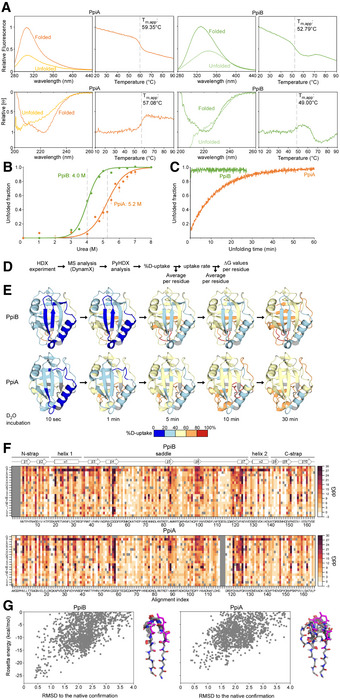
The stability of the native proteins was compared upon thermal or chaotrope denaturation, by monitoring their secondary/tertiary structure using circular dichroism (CD)/intrinsic fluorescence, respectively (Fig EV1A–C). PpiA displayed higher thermal stability (Fig EV1A) and equilibrium unfolding transition point (Fig EV1B) and unfolded > 30 times more slowly in 8 M urea than did PpiB (Fig EV1C).
Figure EV1. Structural dynamics and stability analysis of the native state of PpiB and PpiA (related to Fig 1).
-
ARaw data of thermal denaturation analysis monitored by intrinsic fluorescence (top, in relative units setting the highest value at 1 with excitation at 260 nm and emission at 327 for PpiA and PpiB based on the mainly buried tyrosine residues, as PpiA does not contain Trp and PpiB only contains an outward facing one and circular dichroism (bottom, CD; in relative molar ellipticity ([θ]) with highest value at 1) at 222 nm. The full spectrum of the folded (protein at 25°C) and unfolded state (protein at T m,app + 5°C) is displayed on the left, and apparent melting temperature (T m,app) on the right was determined after smoothening the curves with a Butterworth filter (see Materials and Methods) and plotting the first derivative where the maximum (CD) or minimum (Intrinsic Fluorescence) was determined using a Python script (see Materials and Methods). The T m,app is indicated on the graph with a dotted grey line. n = 3 technical repeats.
-
BChaotrope denaturation analysis in urea monitored by CD at 222 nm (depicted as unfolded fraction calculated from [θ] of the unfolded protein (8 M Urea) set as 1 and that of the natively purified protein (0 M Urea) set as 0). The raw data are shown with dots and fitted using a two‐state transition model ((Lowe et al, 2018), see Materials and Methods) to determine the transition midpoint. n = 3 technical repeats, s.d.
-
CUnfolding of PpiB (green) and PpiA (orange) from their native states in 8 M Urea monitored with CD at 22°C at 222 nm (depicted as the unfolded fraction (as in B.)).
- D
-
EStructural dynamics of the native state of PpiB and PpiA derived from local HDX‐MS analysis (Fig 1B). The weighted average %D‐uptake at the indicated HDX time was mapped on the 3D structures (PpiB PDB 1LOP, PpiA PDB 1V9T). 0–20%, 20–40%, 40–60%, 60–80% and 80–100% Deuterium uptake intervals are shown in the indicated colour scale. Residues without coverage are in grey. n = 3 technical repeats.
-
FMutational free energy (ddG) predictions for PpiB (PDB 1LOP and PpiA (PDB 1V9T) using in silico mutagenesis displayed as a custom colour map with all substitutions indicated (see scripts on GitHub). Missing residues from alignment and native residues are in dark grey. Increase in ddG values (brown colour) signifies mutations that destabilize the structure or a more stable native residue, while decrease in ddG values (white) signifies the possibility of other residues to fit that same position.
-
GComputed conformation/energy landscape of β‐hairpin 1 of PpiB (left, PDB 1LOP) and PpiA (right, PDB 1V9T). Each point represents one decoy generated with the Rosetta KIC protocol, scored based on Rosetta total_score and aligned to the native structure. The structure of the 10 lowest energy decoys for each protein is presented on the right side of each graph.
Source data are available online for this figure.
The intrinsic dynamics of the native protein state were analysed by local HDX‐MS (Fig 1B, conditions and data in Dataset EV4; Wales & Engen, 2006). Flexible regions are mainly present in “open” states (i.e. high solvent accessibility and D‐uptake; red/orange), while rigid ones remain longer in “closed” states (i.e. low solvent accessibility and D‐uptake; grey). D‐uptake is experimentally determined per peptide, and these differ between structural twins. To allow sequence‐wide comparisons, we used PyHDX to first convert D‐uptake per peptide to D‐uptake per residue (see pipeline in Fig EV1D, Smit et al, 2021) and then to process D‐uptake over multiple HDX times to a single Gibbs free energy (ΔG) value (Fig EV1D and E; Smit et al, 2021) that defines the energy difference between the closed and open state (low for flexible/high for rigid regions). The twins displayed a similar overall dynamics pattern (inversed ΔG y‐axis, Fig 1B): rigid N‐strap, α1 and β7 (grey), flexible saddle (particularly in PpiB; orange) and highly dynamic linker regions (red). Small distinct dynamic islands were detected in the first protein halves, mainly in linkers (one in PpiB; three in PpiA) and the C‐straps were more flexible, particularly in PpiA.
The dynamics of the native states were further probed using normal mode analysis (NMA) that calculates the vibrational movement of atoms by applying harmonic potentials between neighbouring atoms (Fig 1B, magenta; Bahar et al, 2010; Tiwari et al, 2014). The displacements of the lowest frequency normal modes were summed to identify residues with elevated dynamics in the structures. The twins displayed similar patterns, in good agreement with local HDX‐MS (high displacement in flexible regions and low in ordered N‐termini and β7).
The native structures were also screened in silico for frustrated interactions (energetically suboptimal local sequences; Ferreiro et al, 2014; Parra et al, 2016). In both twins, multiple frustrated interactions occurred in loops, the β8‐β9 hairpin and the α‐helices (particularly α1). Distinct differences were observed in the β‐sheet that encompasses the N‐strap and the end of the saddle: Only two frustrated interactions are seen in PpiB (β7 with β1/2) in contrast to the multiple ones in PpiA (e.g. Gly126 and Leu127 of β7 with β5, β2 and the N‐tail, and surface residues like Glu19 and Asp21) that could lead to a suboptimal fit of β1/2 with β5/7 (Fig 1C). Moreover, to evaluate the effect of substitutions on the twin's stability, each residue was examined by in silico deep mutational scanning, using Rosetta (see Materials and Methods; Leman et al, 2020). In both proteins, substitutions highly affected residues located within secondary structure elements, due to their tertiary environment (e.g. in β8), while loops tolerated more mutations (Fig EV1F).
Some suboptimal surface‐exposed polar residues were identified in the first β‐hairpin of PpiA but not in PpiB. The side chains of surface residues typically form less intramolecular contacts than the residues pointing to the core, suggesting that some residue frustrations may arise from intra‐residue energetic contributions rather than suboptimal inter‐residue contacts. Therefore, we probed the local residue/structure compatibility at each position of the PpiA/B structures as a function of the local torsion angles (Rosetta p_aa_pp score per residue; Fig 1D; Dataset EV1E; Alford et al, 2017). Multiple suboptimal residues (Thr10; Ala13; Asn15) were centred around the N‐strap's β‐turn in PpiA, corroborating high flexibility (Fig 1B). To confirm these observations, the conformational energy landscape of this β‐turn was examined in the twins using the Rosetta KIC protocol (Stein & Kortemme, 2013). PpiB's β‐turn produced a funnelled conformation/energy landscape converging to the native structure, indicating good compatibility between the local sequence and structure (Fig EV1G). In contrast, PpiA's β‐turn did not show the same convergence of low‐energy models to the native conformation, consistent with low sequence/structure compatibility (Fig 1D) and higher flexibility (Fig 1B).
The twins have similar overall dynamics, with local differences. Secretory PpiA contains more frustrated and suboptimal residues that may influence its folding pattern.
PpiA displays delayed folding compared with fast‐folding PpiB
The folding kinetics of PpiB and PpiA were probed by global HDX‐MS, at 25 and 4°C (Figs 2A and EV2A; see Materials and Methods). Folding initiated by diluting denatured proteins (in 6 M urea) into aqueous buffer (0.2 M urea, Fig EV2A.i). At distinct refolding timepoints (Fig EV2B, Dataset EV2), protein aliquots were pulse‐labelled in D2O (100 s). Flexible/unfolded proteins (i.e. with no or weak H‐bonds, solvent‐accessible/exchangeable backbone amides) have higher D‐uptake than folded proteins (i.e. H‐bonded secondary structure; Fig EV2A.ii; Wales & Engen, 2006). Pulse‐labelling was quenched at pH 2.5 (Bai et al, 1993), and the polypeptides were analysed with electrospray ionisation MS (see Materials and Methods; Fig EV2A.iii; Ho et al, 2003). Protein folding is visualized as the progressive shift over time of one charged peak, from the high m/z value of the unfolded state (U) towards the lower m/z value of the natively folded state (F; Fig EV2A.iii; reflecting high‐to‐low D‐uptake as D is heavier than H by 1 Da, Dataset EV2). The degree of non‐foldedness (D‐uptake) of the unfolded protein is set as 100%; all other values were expressed relative to this.
Figure 2. Comparison of PpiB and PpiA folding by global HDX‐MS.
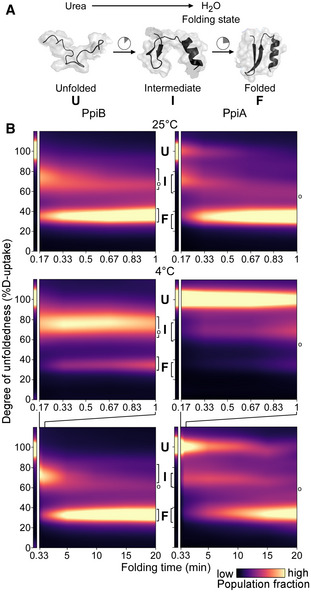
-
ACartoon representation of in vitro refolding protein over time, upon dilution from chaotrope into aqueous buffer.
-
BFolding kinetics of PpiB (left) and PpiA (right), at 25°C (1 min, top) or 4°C (1 and 20 min, bottom). Folding populations are displayed as a continuous colour map of their %D‐uptake (y‐axis) across time (x‐axis). For m/z spectra, see Fig EV2B and C; Dataset EV3. n = 2–6 (biological repeats). Left thin panels: unfolded state (U; 6 M urea); Right main panels: refolding data (0.2 M urea); I, Intermediate; F, Folded populations; o, modifications/adducts, not part of the folding pathway.
Figure EV2. Refolding kinetics analysed with global HDX‐MS of PpiA and PpiB at 25 and 4°C (related to Fig 2).
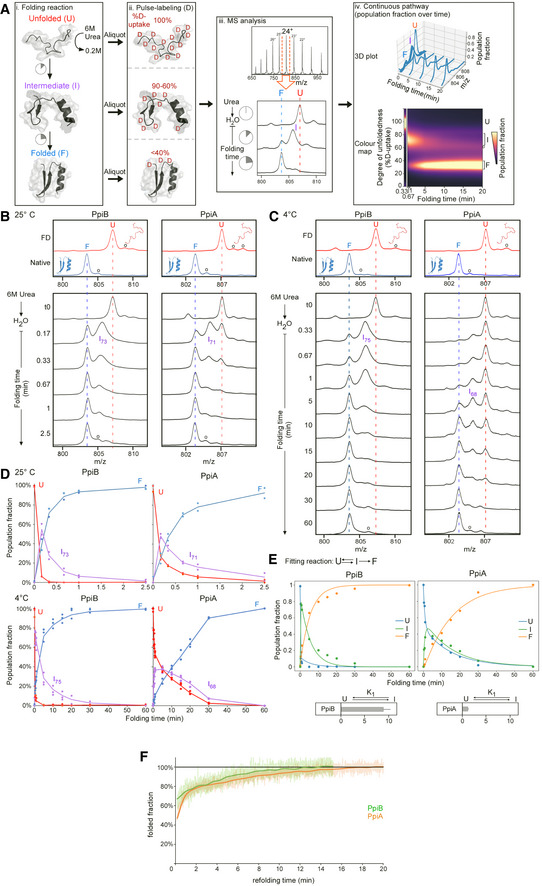
-
APipeline of processing in vitro refolding kinetics of intact proteins using global HDX‐MS analysis and subsequent visualization as a colour map (Fig 2B). (i) Denatured proteins are refolded out of chaotrope (6 M urea) into aqueous buffer where the different folding states are observed. (ii) An aliquot of the refolding reaction is removed at different timepoints and pulse‐labelled in high % D2O where the amount of Deuterium taken up reflects the number of non‐H‐bonded/solvent‐accessible backbone amides and is inversely related to how folded (i.e. stably H‐bonded) the protein is. The unfolded state (6 M Urea) is experimentally defined as a single peak/population with maximum D‐uptake (set as 100%), followed by intermediate D‐uptake and finally the lowest D‐uptake for the folded state. (iii) From the electrospray ionisation MS analysis of each refolding timepoint, an m/z spectrum with multiple charged m/z peaks is obtained. From the latter, a single high‐intensity peak (highest Signal over Noise) is selected and smoothed (Savitzky‐Golay, window: 15, number: 5) to be followed over different refolding timepoints (as depicted in the bottom section). Due to Deuterium being 1 Da heavier than Hydrogen, a shift from a high to lower m/z is observed over time as the protein folds and takes up fewer Deuterium during pulse‐labelling. “o”: Potassium adducts and Urea modification peaks that are visible on the (un)folded state. (iv) The intensities of the folding populations from the single m/z peak at different timepoints are normalized to the integrated area (See Materials and Methods). To observe the conversion of the folding populations over time, a 3D plot was displayed with all the normalized m/z spectra over time. The normalized intensities now reflect the population fractions of each folding state. Linear interpolation was performed between the m/z spectra over time to get a continuous time course of the refolding pathway and used to create a 2D colour map to visualize the interconversion between folding states indicated based on their degree of unfoldedness (%D‐uptake). The colour gradient (“magma” colourmap) reflects increasing population fractions ranging from small (dark) to high (yellow; see Materials and Methods, Dataset EV3C).
-
B, CSmoothed spectra of the 24+ charged m/z peak (highest intensity) of the refolding kinetics of PpiA and PpiB from global HDX‐MS analysis at the indicated timepoints (4 and 25°C) that were used for constructing the continuous colour map (Fig 2B). The denatured protein or the fully deuterated (FD; 6 M Urea‐d4 for 1 h; red line) control and the Native control (i.e. soluble purified native protein; blue line) are marked throughout the folding timepoints. “o” refers to Potassium adducts and Urea modification peaks that are also visible in the colour maps in Fig 2B.
-
DPopulation fraction over time after Lorentzian curve fitting of the 24+ charged m/z peak in (B and C) with the unfolded (red), intermediate (purple) and folded (blue) state (from biological repeats, see below). The relative percentage of D‐uptake of each intermediate state is noted in its subscript. For PpiB at 25°C (n = 2), PpiA at 25°C (n = 2), PpiB at 4°C (n = 7) and PpiA (n = 4), data are shown as dots (up to 3 repeats) and average as line.
-
EFor 4°C, the population fractions were fitted with an ODE model (see equation, see Materials and Methods). The fitted curves are displayed with the different folding states (Unfolded (U), Intermediate (I) and Folded (F)) and the equilibrium constant K1 is displayed below. n = 4 biological repeats.
-
FRefolding of PpiA (orange) and PpiB (green) monitored by CD at 4°C (recorded at 222 nm and shown as the folded fraction over time setting the 6 M Urea state as 0 and the final 0.2 M Urea state as 1).
Both twins displayed three‐state folding (unfolded‐intermediate‐folded; U, I, F) through a single recurring kinetic folding intermediate (Fig EV2B and C). Intermediates were characterized by their %D‐uptake (e.g. I73 for PpiB folding at 25°C). Folding populations were quantified over time by fitting linear combinations of the three folding states, with the intermediate state modelled as a Lorentzian curve of variable position (Fig EV2D). Kinetic parameters were obtained by fitting the interconverting populations to rate equations derived from a model where the unfolded and intermediate states are assumed to be in equilibrium (k1, k−1, equilibrium constant K1) and the folded state is irreversibly formed from the intermediate with a rate constant k2 (see Materials and Methods; Dataset EV3A; Fig EV2E).
We visualized the kinetics of the folding reactions in colour maps (Figs 2B and EV2A.iv), using the experimental timepoints and linearly interpolating the fractions in between (brighter colour indicates more prominent populations; see Materials and Methods; Dataset EV3B and C). Distinct folding populations have different %D‐uptake values (Fig 2B; y‐axis). The starting unfolded state is displayed (U; Fig 2B, thin left panel; 6 M urea) beside the folding reaction (main panel; 0.2 M urea). At 25°C, folding kinetics were fast for both twins (Figs 2B top, and EV2B and D). PpiB immediately formed an I73 intermediate that quickly folded (in ∼1 min). PpiA converted more slowly to an intermediate that folded similarly fast, in agreement with CD analysis (Fig EV2F). At 4°C the folding pathways were similar, occurring via single intermediates, but slower, better resolving the different states (Figs 2B bottom, and EV2C and D). PpiB still folded fast (in ∼5 min). In contrast, unfolded PpiA persisted for 15–20 min in the aqueous solution (sevenfold lower K1 than PpiB, Fig EV2E) and folded slowly (> 30 min to completion; full spectrum in Dataset EV3C; Figs 2B bottom, and EV2C and D).
PpiB and PpiA display similar yet distinct, differently ordered hierarchical foldon pathways
We resolved the folding processes of the twins at near‐residue level using local HDX‐MS. At distinct refolding timepoints (see conditions in Dataset EV4A), proteins were pulse‐labelled in D2O (10 s), quenched, digested and peptides analysed using MS (Fig EV3A; see Materials and Methods). Here, folding of a protein region is seen as bimodal isotope distributions of unfolded (no or weak H‐bonds; high D‐uptake and m/z) and folded derivative peptides (H‐bonded; lower D‐uptake and m/z; EX1 kinetics; Fig EV3A.iii; Englander et al, 2007; Marcsisin & Engen, 2010). The degree of foldedness is described as the folded fraction of each peptide that is equally well determined either by Gaussian fitting of the two distributions and defining the ratio of the folded state or by calculating the centroid of the complete distribution (Fig EV3C; Hodge et al, 2020). In the latter case (used here), the centroid of the unfolded distribution (U; reflecting maximum D‐uptake) and that of the natively folded protein (F; minimum D‐uptake) are set as 0 and 100% folded fraction, respectively (Fig EV3C, left), for all of the generated peptides (> 95% of each twin's sequence; Dataset EV5). Similarly, the centroid masses of all peptides were converted to folded fractions and finally to per‐residue using weighted averaging (per‐residue RFU function of PyHDX, version 0.4.1.; see Materials and Methods; pipeline in Fig EV3B, data in Dataset EV5; Smit et al, 2021). Peptides with minor D‐uptake differences between unfolded/folded states and high standard deviations corresponding to unstructured/loosely folded protein regions (Fig EV3C, Dataset EV5), prolines and residues appearing only in a peptide's N terminus were omitted from analysis.
Figure EV3. Rates and spectra of refolding kinetics analysed with local HDX‐MS of PpiA and PpiB at 25 and 4°C (related to Fig 3).
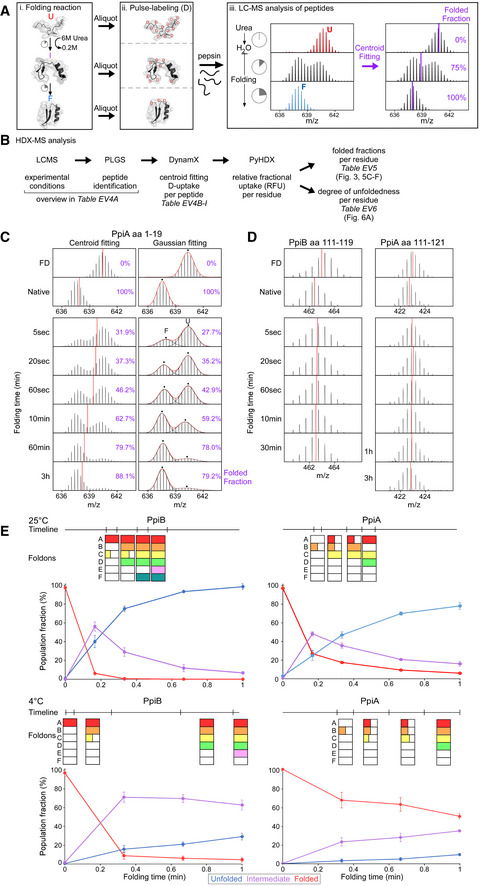
-
APipeline of processing in vitro refolding kinetics of pepsinized proteins using local HDX‐MS analysis (Fig 2B). (i) Denatured proteins are refolded out of chaotrope (6 M urea) into aqueous buffer where the different folding states are observed. (ii) An aliquot of the refolding reaction is removed at different timepoints and pulse‐labelled in high %D2O where the amount of Deuterium taken up reflects the number of non‐H‐bonded/solvent‐accessible backbone amides and is inversely related to how folded (i.e. stably H‐bonded) the protein is. Pulse‐labelled proteins are pepsinized to determine the D‐uptake of each peptide to obtain folding details. (iii) All peptides are identified by their retention time during Liquid Chromatography and their m/z spectrum (Englander et al, 2007; Tsirigotaki et al, 2017b). The unfolded and folded state are a single distribution with the highest and lowest D‐uptake, respectively, where during folding the conversion from a completely unfolded to the folded state is observed (bimodal distributions, EX1 HDX kinetics (Englander et al, 2007; Tsirigotaki et al, 2017b)). The average D‐uptake of each distribution is determined using a centroid that gets converted to the folded fraction using the D‐uptake of the unfolded state as 0% folded and that of the folded state as 100%.
-
BThe schematic pipeline describes the steps of analysis we performed on the local HDX‐MS data using PyHDX, in order to obtain folded fractions per residue or degree of unfoldedness per residue. Data from different steps are presented on separate Datasets (as indicated) and were used on the indicated Figures.
-
CComparison in data processing of a PpiA peptide to calculate folded fractions (results in Dataset EV5 per residue) using centroids vs. Gaussian fitting. Peptide aa1‐19 demonstrates folding with bimodal distributions. Left, the centroid position (red line) of the peptide is used to determine the folded fraction (unfolded m/z value is 0% folded and natively purified protein is 100% folded). Right, the unfolded (U, high m/z) and folded (F, low m/z) distributions are fitted with Gaussian curves (individual Gaussians: dashed lines, fit: red line and dots for the mean of each Gaussian) to determine the % area of the folded one. For both, the folded fractions calculated from centroid and Gaussian curve fitting are shown in purple. Use of the centroid approach avoided the fitting of very broad unfolded Gaussian peaks at later timepoints and was preferred hereafter.
-
DRefolding analysis of two peptides from regions in PpiB and PpiA at 4°C that display small D‐uptake differences between the unfolded and the folded state and only display very minor shift of the whole spectra during refolding. The centroid is depicted as a red line. Both sites did not show any distinct folding and were left out of the analysis (grey bar, Fig 3A and B).
-
EComparing foldons from local HDX‐MS to global HDX‐MS data. The foldons from Fig 3 are displayed on top (based on t 80% or t 50%, Dataset EV5) with their formation timeline where they are coloured after formation in a time interval. If only sections (half or one third) of the foldon are formed this is indicated in the square. These foldons timelines are aligned to the global HDX‐MS data where only 1 min of refolding is shown from Fig EV2D.
The complete folding pathways were visualized as colour maps, with fractions in between experimental timepoints being linearly interpolated (Appendix Fig S2; Dataset EV5). The dynamic range of folding was captured using both high and low temperature (25°C; 4°C). To simplify foldon definition in the twins, the time required (y‐axis) to reach 50% of folded population (t 50% values) was plotted against the aligned linear sequence (x‐axis; Fig 3A and B; colour maps in Appendix Fig S2; Dataset EV5; see Materials and Methods). Both temperatures were considered when assigning foldons, as some resolved better at low temperature, others at high. Foldons were coded in alphabet order as they appear in PpiB (code maintained in PpiA) and are colour‐indicated below a linear secondary map (Fig 3A and B, top) and on 3D structures (Fig 3C). When foldons were formed in distinct segments, numeric subscripts were used (folding times displayed in Fig EV3E, colour maps in Appendix Fig S2).
Figure 3. Initial foldons in PpiB and PpiA using t50% from local HDX‐MS analysis.
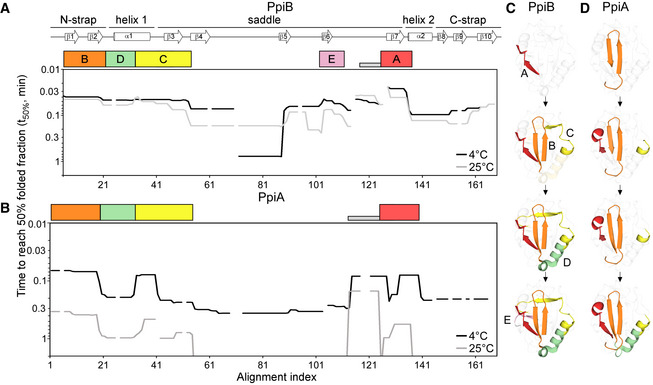
-
A, BFolding kinetics of PpiB (A) and PpiA (B) at 25 or 4°C, monitored by local HDX‐MS (Dataset EV4; n = 3 biological repeats), were analysed by PyHDX to determine the folded fractions per residue (Dataset EV5); see pipeline of analysis in Fig EV3B and folding times in Fig EV3E. For each peptide, 100% folding was set to the D‐uptake of the native protein peptide and 0% folding to the D‐uptake of the same peptide under fully deuterated conditions. Initial foldons were assigned by plotting the time needed to reach 50% of folded fraction (t50%; y‐axis; Dataset EV5) along the linear sequence (x‐axis), at both temperatures (as indicated). Only up to 1 min data are shown here (see extended dataset colour map in Appendix Fig S2; raw data in Dataset EV4). The alignment index is based on the sequence of PpiA (extended N‐tail; missing loop between β6‐β7; Appendix Fig S1D). Gaps: residues absent in one of the twins, prolines or no experimental coverage. Colour boxes below the linear secondary structure map (top) indicate foldons, named in alphabetical order. Grey bar: unstructured fast folding regions (Fig EV3D) omitted from analysis.
-
C, DFoldons, colour‐coded as in the left panels, are indicated relative to their time of formation on the PpiB (1LOP; C) and PpiA (1V9T; D) 3D structures.The indicated time points were as follows: for PpiB, 25°C (t 80% of 0.29‐0.33‐0.42‐0.47 min); for PpiB, 4°C (t 80% of 0.09‐0.29‐0.90‐1.75 min); for PpiA, 25°C (t 80% of 0.24‐0.33‐0.47‐0.51 min); for PpiA, 4°C (t 50% of 0.34‐0.55‐0.79‐0.99 min; Fig EV3E, Dataset EV5).
At either temperature, PpiB started folding with foldon A (β7‐α2; red; Fig 3A and C; Appendix Fig S2A–D) followed by foldon B (N‐strap; orange). The last turn of α1 (that gets extended into β3; foldon C; yellow) formed before the first part of α1 (foldon D; green). The four initial foldons completed the front face of PpiB (Fig 3C) together foldon F (only at 25°C; Appendix Fig S2A) and were followed by foldon E (mauve; β5/6) at the back face.
In PpiA, folding started with foldon B (Fig 3B and D, orange), followed by sequential formation of foldons C (yellow), A (red) and D (green). Some PpiA foldons formed stepwise compared with PpiB (e.g. A, B and C) or were very delayed (E and F; Fig 3; Appendix Fig S2E–H). Here also, the first foldons that were formed completed most of the front protein face (Fig 3D). Corroborating global HDX‐MS analysis, the folding of PpiA at 4°C was significantly delayed; ∼10‐fold slower than at 25°C (Fig 3B).
In summary, the twins each folded via distinct well‐defined consecutive initial foldons (Fig 3) followed by less separable, collective, presumably cooperative, “late” foldons (Appendix Fig S2). The initial foldons may be the main folded components of the intermediates observed with global HDX‐MS (Fig 2B). Foldon location in the primary sequence may be similar in the twins, yet their formation kinetics and hierarchy is distinct (Fig 3, compare C with D).
Hydrophobic islands, considered as main elements of a folding process (Onuchic et al, 1997), are located on the initial foldons but not uniquely; charged and polar residues facing the solvent on the surface of the protein are also included (mainly in foldons D and E; Dataset EV7A). The foldons determined above overlapped well with predicted early folding regions (Raimondi et al, 2019) and similarly aligned islands of minimally frustrated residues (Dataset EV7A, see Materials and Methods; Parra et al, 2016). The latter may guide folding along the energy landscape (Parra et al, 2016; Gianni et al, 2021) forming local stable elements of the folding core (Jenik et al, 2012). Highly frustrated/suboptimal residues in foldons A and B of PpiA (Fig 1C and D) may slow down folding (Figs 2 and 3) by hindering stable interactions (Nymeyer et al, 1998; Gianni et al, 2021).
Grafted residues interconvert PpiB/A folding kinetics
Using the Frustratometer (Parra et al, 2016), we identified the 23 lowest energy native contacts in the two structures (native energy ≤ −5.0 kJ/mol; Fig 4A; Dataset EV7B). Eight of them are dissimilar between PpiB and PpiA (Fig 4B, top), of which six are at the same location in the two 3D structures. Almost all of them are situated on or next to initial foldons (Fig EV4A, top) with invariably bulkier and more branched/hydrophobic side chains in PpiB (Fig 4B, top). Rosetta analysis (see Materials and Methods; Leman et al, 2020) indicated the dissimilar residues to be in the immediate vicinity of residues that are highly optimized or suboptimal in PpiA or PpiB (Figs 4B, bottom and EV4C). Multiple dissimilar native contacts were energetically more optimal in PpiB and incorporating these contacts to the equivalent positions in PpiA was predicted to stabilize the latter (Dataset EV7D). Assuming that the six dissimilar residues underlie foldon formation and/or 3D associations (Fig 4C), it would be anticipated that strengthening or weakening their interactions might modulate folding speed.
Figure 4. Grafting stable native contacts between PpiB and PpiA interconverted folding behaviours.
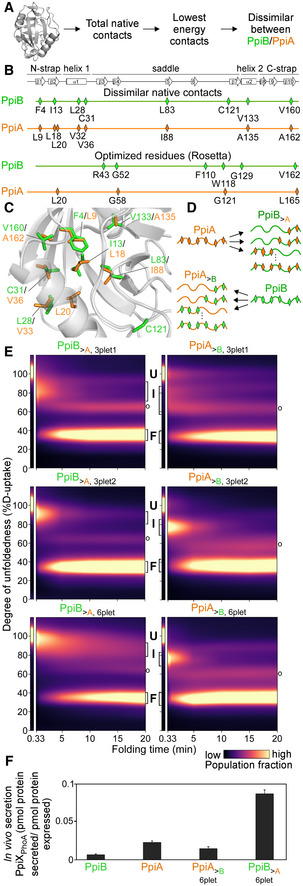
-
APipeline for selecting residues that affect folding behaviour using the Frustratometer and 3D structures of PpiB (PDB 2NUL; 1LOP) and PpiA (PDB 1V9T; 1VAI; 1J2A) to test with grafting (details in Dataset EV7).
-
BHighly stabilized, dissimilar native contacts indicated on a linear map with the secondary structural elements on top.
-
CThe side chains of native contact residues (green: PpiB; orange: PpiA) indicated on their 3D structure.
-
DThe native contact grafting scheme between PpiB and PpiA to test their role on folding behaviour.
- E
-
FIn vivo secretion of the indicated PpiX‐PhoA fusion proteins in MC4100 cells carrying SecYprlA4EG. Secretion is expressed as pmol fusion protein secreted from PhoA activity calculations after removing background (uninduced cells) per pmol protein expressed from western blot analysis in 108 cells (Fig EV4E, Dataset EV9). n = 6 (biological triplicates with 3 technical replicates each, s.d.).
Source data are available online for this figure.
Figure EV4. Grafting of native contacts between PpiA and PpiB (related to Fig 4).
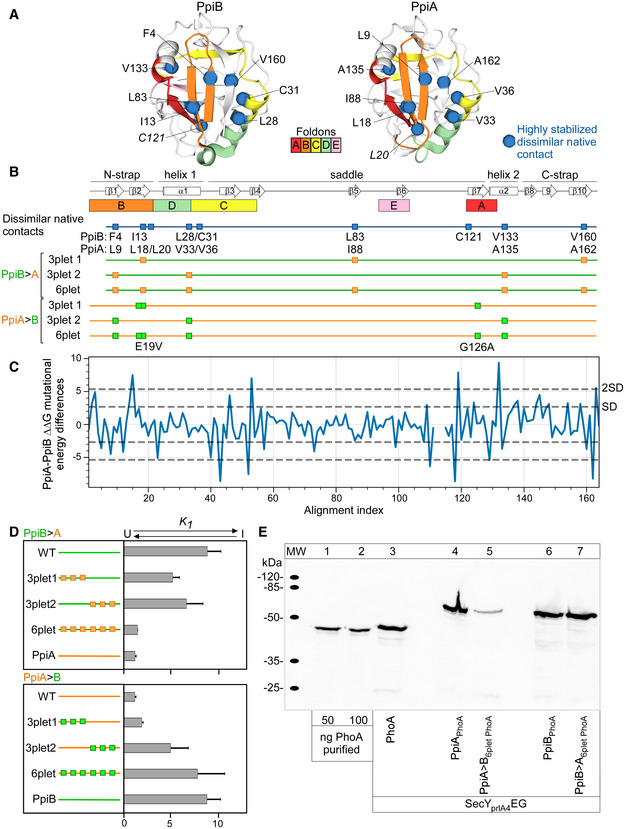
- A
-
BMutant derivatives of PpiA (orange) or PpiB (green) with grafted residues from PpiB and PpiA (labelled PpiA>B and PpiB>A, respectively, Fig 4E) are displayed as mutations as squares below the linear map of secondary structure (PpiB, green; PpiA, orange) with annotations at the bottom.
-
CMutational differences dddG values from in silico mutational scanning using Rosetta cartesian‐ddG application. dddGs are subtracted residue‐wise ddG values of PpiA and PpiB to compare the stability of residues between proteins.
-
DEquilibrium constant K1 of the refolding PpiB>A and PpiA>B derivatives at 4°C between the unfolded and intermediate state are shown as bar plots in rows for the grafted triplets (T) and sixplet (S) mutants compared with the wildtype (WT) proteins (calculated from Fig 4E). n = 2–4 biological repeats, s.d.
-
EIn vivo protein expression in the E. coli strain MC4100 at 30°C during in vivo secretion assay detected by immunostaining with α‐PhoA antibodies on western blots (Fig 4F, See Materials and Methods, PhoA secretion activity in Dataset EV9B). ppiX‐phoA fusions carried on vector pBAD501 (ara promoter) were expressed in the cell (13.3 μM arabinose) to monitor PpiX secretion in the presence of secY prlA4 EG encoded on plasmid pET610 (lac promoter; expressed with 0.05 mM IPTG). Expression of secY prlA4 is required for secretion of proteins that have no signal peptide (Derman et al, 1993). Left, purified PhoA protein loaded at the indicated amounts was used for quantification of protein expression (Dataset EV9).
Source data are available online for this figure.
To test this, we reciprocally grafted the corresponding residues between the two proteins, leaving the rest of the sequences unchanged (Fig 4D). We focused on residues located in or next to foldons A and B, in either twin (Fig EV4B). We generated single, double, triple or multiple mutant derivatives and determined their individual or combined effect on the twins' folding at 4°C, using global HDX‐MS (as in Fig 2B).
First, PpiA residues were grafted onto PpiB (hereafter PpiB>A) to generate slower‐folding derivatives mimicking PpiA that remained longer unfolded before forming an intermediate (Fig 2B, bottom). Only 3plet and 6plet grafts are shown (Fig EV4B); fewer mutations had no discernible effect (all mutants in Dataset EV8). The PpiB>A,3plet1 carried mutations in highly stabilized native contacts (I13L/L83I/V160A). Ile13 is part of foldon B (β2), Val160 (C‐strap) sits between foldons B and D and Leu83 (β5) connects foldon A (β7) to the saddle. The PpiB>A,3plet2 carried mutated native contacts (F4L/L28V/V133A) on foldons B (β1), D (α1) and A (α2), respectively. These residues, belonging to three discontinuous foldons, participate in long‐range hydrophobic contacts and are suspected to be less efficient in PpiA due to their smaller side chains. Neither 3plet derivative slowed down folding significantly but yielded less folded intermediates (higher D‐uptake) compared with the I75 of PpiB (Fig 4E top and middle left; Dataset EV3A). Combining the two 3plets in one derivative delayed folding (> 10 min; Fig 4E, bottom left). The PpiB>A,6plet remained in a broad I85 population and reached the folded state slightly faster than PpiA. Adding more grafted residues blocked PpiB folding at early stages (PpiB>A,Multiplet, Dataset EV8).
Next, PpiB residues were grafted onto PpiA aiming to speed up the latter's folding (hereafter PpiA>B, Fig EV4B). Although single/double grafted residues sped up folding kinetics (Dataset EV8), 3plets and 6plets thoroughly accelerated folding (Fig 4E right). The PpiA>B,3plet1 (E17V/L18I/G126A) carries grafted residues on foldon B1 (β2) and A1 (β7) that are more branched/hydrophobic and in PpiB could promote β‐hairpin formation. While Leu18 is a highly stabilized native PpiA contact in foldon B1, Gly126 has multiple frustrated interactions that are not present in the corresponding PpiB residue (Ala124; Fig 1C) and E17 has a suboptimal sequence/structure compatibility (Fig 1D). The PpiA>B,3plet1 exhibited two modestly sped up intermediates that formed and disappeared simultaneously (I82; I62; Fig 4E top right) but folding still resembled that of PpiA (Fig EV4C). On the contrary, the PpiA>B,3plet2 (L9F/V33L/A135V; the reverse of PpiB>A,3plet2) quickly formed an I76 (Figs 4E middle right; EV4C) with folding kinetics resembling those of PpiB (∼5 min). Either one or two from the 3plet2 mutations increased PpiA's folding (Dataset EV8). The PpiA>B,6plet, (combined 3plets) formed an I76 even faster than PpiA>B,3plet2 (Fig EV4D) and folded slightly faster than PpiB (< 5 min; Fig 4E, bottom right).
We concluded that highly stabilized native contacts on foldons were involved in early folding events and were sufficient to interconvert intermediates and folding behaviours between PpiB and A.
Delayed in vitro folding correlates with improved in vivo secretion
To test whether in vitro slow folding correlated with improved in vivo secretion efficiency, PpiA/B and derivatives were fused N‐terminally to PhoA (alkaline phosphatase; San Millan et al, 1989; Akiyama & Ito, 1993). The PhoA reporter becomes enzymatically active once secreted to the periplasm through the Sec translocase; its secretion now being dependent on the fused N‐terminal PpiX‐partner. Fusions were tested using cells expressing SecYprlA4EG (Fig EV4D), a translocase derivative that allows secretion of signal peptide‐less mature domains (Gouridis et al, 2009). Secretion efficiency was determined from PhoA activity units and normalized on protein amounts (Fig 4F; see Materials and Methods; full analysis in Dataset EV9B; expression levels in Fig EV4E).
The fast‐folding PpiB fusion (Fig 4F) had ∼threefold lower secretion than the slower‐folding PpiA fusion. Accelerating folding reduced secretion by half (compare PpiA>B,6plet with PpiA), while delaying folding significantly enhanced secretion (compare PpiB>A,6plet with PpiB).
These experiments suggested that slow/fast folding correlates with high/low secretion efficiency, respectively.
The signal peptide stalls folding at early intermediates
Mature PpiA is only present in the periplasm. Its pre‐form (signal peptide‐bearing proPpiA; Fig 5A) is cytoplasmic. As the translocase recognizes only unfolded proteins, we anticipated that the signal peptide might have a profound effect on the folding of PpiA as seen for other proteins (Park et al, 1988; Singh et al, 2013; Tsirigotaki et al, 2018).
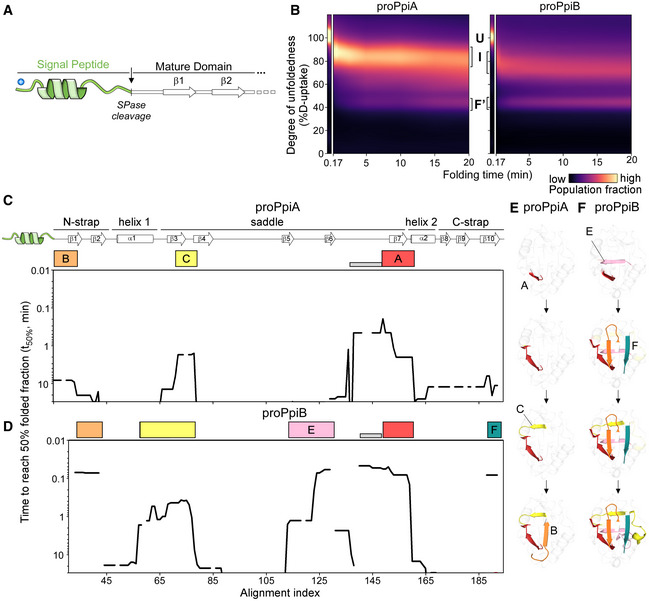
Figure 5. Effect of signal peptide on folding of the twins.
-
ALinear map of the signal peptide/early mature domain region of proPpiA.
- B
-
C, DFolding kinetics of proPpiA and proPpiB, at 25°C, monitored by local HDX‐MS (Dataset EV4; n = 3 biological repeats), were analysed by PyHDX to determine the folded fractions per residue (Dataset EV5). The time needed to reach 50% of folded fraction (t 50% values; only for the mature domains shown here) was plotted as in Fig 3; see extended dataset colour map in Appendix Fig S3.
-
E, FFoldons, coloured (as in C, D) on the PpiA (1V9T; E) and PpiB (1LOP; F) 3D structures. The indicated time points are as follows: for proPpiA (t 50% of 0.9‐2.0‐2.3‐20.8 min) and for proPpiB (t 50% of 0.06‐0.08‐0.44‐1.2 min; Dataset EV5).
Source data are available online for this figure.
Folding of PpiA was compared to that of proPpiA using global HDX‐MS. As slow‐folding kinetics dominated at 4°C and muted the effect of the signal peptide (Fig EV5A), we focused on 25°C. Here, the 3‐state folding behaviour of PpiA (folded in 1 min, Fig 2B) was drastically altered by its signal peptide (Fig 5B). proPpiA remained kinetically trapped for > 20 min in the highly unfolded I87. Folding continued through a second intermediate (I69; Fig EV5B) to an apparent “folded” state (F') that retained higher D‐uptake compared with the corresponding PpiA state (F; Figs 5B vs. 2B, 43 vs. 33% D‐uptake). Within 20 min, only 25% of proPpiA reached an apparent “folded” state (> 250 times more slowly than PpiA based on tFolded,25% between proPpiA and PpiA; Dataset EV3A).
Figure EV5. Refolding kinetics of (pro)PpiA and (pro)PpiB at 25°C analysed with local HDX‐MS followed by secretion efficiency (related to Figs 3, 5 and 6).
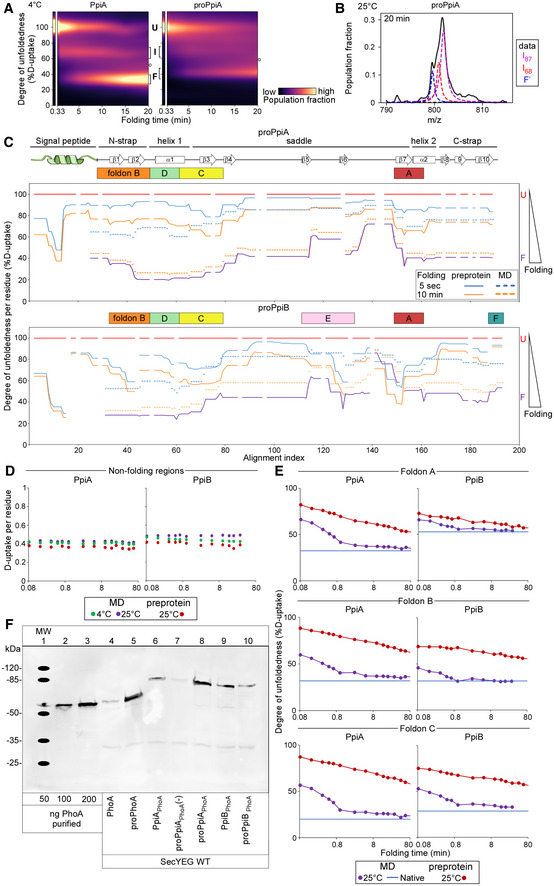
-
ARefolding pathway of PpiA and proPpiA at 4°C. The folding populations are displayed as a continuous colour map over time based on their %D‐uptake (Dataset EV3). The unfolded state (6 M urea, left) is separated from the refolding data in 0.2 M Urea. The Unfolded (U), Intermediate (I) and Folded (F) populations are indicated with brackets. “o” refers to modifications/adducts of the folded state that are not part of the folding pathway. The left panel contains the same data and image as in Fig 2B bottom right panel and is used again here to facilitate comparison.
-
BFitting of the two Lorentzian curves on the broad intermediate of the global HDX‐MS data of proPpiA refolding at 25°C (20 min, Fig 5B). The data were fitted with 3 folding states consisting of the I87, I68 and “folded” (F') state as annotated on the right.
-
CDegree of unfoldedness per residue of proPpiA and proPpiB (signal peptide fused using PpiA N‐terminal tail) during folding (%D‐uptake, data in Dataset EV6) where reduced degree of unfoldedness is related to gain of secondary structure that is shown on the top (based on Appendix Fig S1D). The %D‐uptake during pulse‐labelling is defined by the fully denatured control (FD; 100% D‐uptake) and shown for the preprotein (full line) and mature domain PpiA/PpiB (MD, dashed line). The degree of unfoldedness per residue of the natively folded protein is displayed in purple. Foldons are displayed on top; residues with no coverage as indicated. n = 3 biological repeats.
-
DD‐uptake of peptide in the non‐folding regions during refolding. PpiA or PpiB (green: 4°C; purple: 25°C) vs. their preprotein derivatives (25°C in dark red) display no reduction in D‐uptake during folding and remain disordered and therefore were removed from the analysis (light grey bars, Figs 3E and F, and 5C and D). The peptides of (pro)PpiA (residues 137–145, proPpiA numbering) and (pro)PpiB (residue 140–149, proPpiB numbering) are displayed. n = 3 biological repeats.
-
EComparison of degree of unfoldedness (%D‐uptake) of peptides inside foldons between PpiA/PpiB and their preprotein derivatives (Figs 5C and D vs. 2E and F). Similar to (B), the degree of unfoldedness was determined for the whole peptide and displayed over folding time. Top, refolding of a peptide covering foldon A (β8‐α2) at 25°C for (pro)PpiA (same peptide, residue 146–157, proPpiA numbering) and (pro)PpiB (same peptide, residue 152–160, proPpiB numbering). Middle, refolding of foldon B (N‐strap) at 25°C in (pro)PpiA (same peptide, residue 24–42, proPpiA numbering) and (pro)PpiB (different peptide, residue 30–45 and 24–43, respectively, proPpiB numbering). Bottom, refolding of foldon C (end of α1) at 25°C for (pro)PpiA (same peptide, residue 56–64, proPpiA numbering) and (pro)PpiB (different peptide, residue 43–59 and 44–59, respectively, proPpiB numbering).
-
FIn vivo protein expression in the E. coli strain MC4100 at 30°C detected by immunostaining with α‐PhoA antibodies on western blots (PhoA secretion activity in Dataset EV9B). Transcription of ppiX‐phoA fusions carried on vector pBAD501 was induced in the cell (6.67 μM arabinose) and monitor PpiX secretion monitored (Fig 6A). Lanes 1–3, purified PhoA protein loaded at the indicated amounts used for quantification of protein expression. “‐”: uninduced cells containing the vector with the indicated constructs.
Source data are available online for this figure.
Interestingly, the signal peptide of proPpiA fused to PpiB (hereafter proPpiB) delayed its folding as well. ProPpiB was kinetically trapped in an I76 intermediate, displayed marginal folding in 20 min and reached an apparent folded state (F'; higher %D‐uptake than corresponding PpiB folded state, Fig 2B) that was about > 400‐fold slower than PpiB (based on tFolded,25% between proPpiB and PpiB; Dataset EV3A).
The signal peptide delays folding, not only in a secretory protein but also slows the folding of a protein optimized for cytoplasmic fast folding.
The signal peptide disturbs the initial foldons of the mature domain
To determine the exact effect that the signal peptide had on the folding landscape of the twins, we employed local HDX‐MS (Fig 5C and D, Dataset EV5, colour map in Appendix Fig S3A and C). Foldon formation in proPpiA was significantly slower and altered compared to that in PpiA (Figs 5C compared with 3B and D, and EV5E; foldon spectra in Appendix Fig S4; non‐folding region was removed from analysis; Fig EV5D). In proPpiA, folding started with the slow, partial formation of foldon A (∼11‐times slower than in PpiA; Dataset EV5), followed by partial formation of C (β3), extension of A and partial formation of B (only β1 formed; Fig 5E). These partial initial foldons only formed a limited loose structure presumably corresponding to I87 seen in global HDX‐MS (Fig 5B). At 24 h of incubation, proPpiA reached ∼77% foldedness compared with the native PpiA (Dataset EV5).
Similar effects, albeit less prominent were seen in proPpiB (Fig 5D; colour map in Appendix Fig S3B and D). Some foldons still formed very quickly such as A1 (slightly slower in proPpiB compared with PpiB; Fig EV5E), followed by more extended foldons C1+2, B and F (Fig 5F; Appendix Fig S3B and D) and missing the majority of α1 similar to proPpiA. At 24 h, proPpiB reached ∼89% foldedness compared with native PpiB (Dataset EV5).
The signal peptide modulated the protein folding pathway by obstructing or delaying the formation of critical initial foldons.
Flexibility and stability of the signal peptide during preprotein refolding
Preproteins and primarily signal peptides lack a defined native folded state and cannot be expressed as folded fractions as done above for mature domains. To follow the conformational dynamics of the signal peptide as it disturbs mature domain folding, we examined its degree of unfoldedness per residue (%D‐uptake) over time (defined using the per‐residue RFU function of PyHDX, see pipeline in Fig EV3B). Here, the D‐uptake of the unfolded state for each residue (protein in 6 M urea) was set as 100% (obtained as weighted average of peptides), the non‐deuterated as 0% and all other values of every folding timepoint were expressed relative to this. Hence, any secondary structure acquisition by the signal peptide is seen as a reduction in D‐uptake (Fig 6A; Dataset EV6).
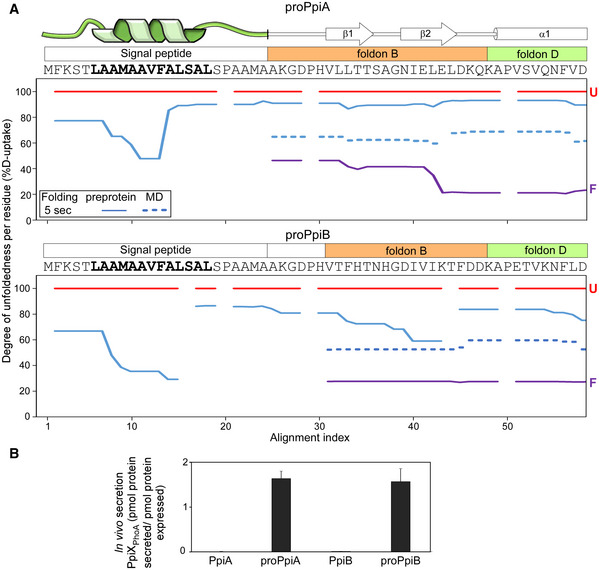
Figure 6. Dynamics of the signal peptide and early mature domain and their effect on in vivo secretion.
-
AFolding kinetics of proPpiA and proPpiB, monitored by local HDX‐MS (Dataset EV4; n = 3 biological repeats), were analysed by PyHDX to determine the degree of unfoldedness per residue (Dataset EV6; Fig EV5C). %D‐uptake for the 5‐s folding time, at 25°C (y‐axis) for the indicated N‐terminal regions (PpiA N‐tail included in proPpiB, predicted signal peptide helix in bold) were plotted along the aligned sequences (x‐axis). Reduced %D‐uptake relative to the U state (red) indicates gain of secondary structure. Top; signal peptide, foldons (B and D) (Appendix Fig S1D; see also Dataset EV6; Fig EV5C). Red: unfolded pre‐forms, purple: native proteins. Gaps: No coverage.
- B
In proPpiA, part of the signal peptide core, specifically the beginning and middle of the predicted α‐helical region, became stabilized within 5 s of folding (48–65% D‐uptake; Fig 6A, top). In contrast, the rest of the helix and the signal peptide's N‐ and C‐regions remained highly flexible. The elevated dynamics continued into the mature domain, destabilizing foldons B and D (Fig 3B; rest of protein in Fig EV5C). This would delay folding of the whole mature domain (Fig 5C).
In proPpiB, the signal peptide displayed similar dynamics but became more rigidified (39–67% D‐uptake), forming a more extensive, stabilized helical structure (Fig 6A, bottom). The rest of signal peptide sequence and early mature domain were flexible but less so than in proPpiA (Fig 6A, top, full protein in Fig EV5C). In proPpiB, segments of foldon B started acquiring stability (particularly β2) similarly to what was seen in PpiB (Fig 6A, bottom, blue dashed line).
The signal peptide allows high secretion efficiency for both PpiA and PpiB
The signal peptide blocked the folding pathway of the twins in vitro. To test whether this is reflected on export, we examined the secretion of the twins' pre‐forms in vivo, using the PhoA reporter system described above (full analysis in Dataset EV9B, expression levels in Fig EV5F).
Signal peptide‐bearing and signal‐less fusions were tested in parallel in cells carrying wildtype SecYEG (Fig 6B). While secretion of signal‐less PpiA and PpiB by the wildtype translocase was negligible, both pre‐forms were secreted equally well.
Discussion
How evolution has manipulated highly efficient protein folding in order to delay it and facilitate translocation remains unclear. Using a structural twin pair, we revealed intrinsic adaptations that slowed down the folding of a secretory mature domain twin. Addition of a secretion‐specific add‐on, a N‐terminal signal peptide, further delayed it.
Folding of both the secretory PpiA and its cytoplasmic homologue PpiB followed a defined three‐stage pathway with a single intermediate (Fig 2B). The process was hierarchical: a small number (4–6) of initial foldons became stabilized in a defined order before collective, rapid, near‐simultaneous, presumably cooperative folding occurred by the remaining foldons (Fig 3; Appendix Fig S2). These initial foldons had features similar to those observed in other studies but were better resolved, in some cases down to three residues (Maity et al, 2005; Walters et al, 2013; Englander & Mayne, 2014). Remarkably, the order of formation of the initial foldons in the twins was similar but not identical (Nickson & Clarke, 2010) following a different order to yield intermediates (Fig 3; Appendix Fig S2). Folding was driven by small differences between the foldons of each twin. Minor side chain changes altered hydrophobicity, bulkiness and degree of residue frustration in the native structure (Fig 1C; 4°C). Changes in loops/β‐turns and increased local flexibility around foldons (e.g. at the N terminus of PpiA) might have restricted or favoured the extent of stochastic collisions between folding segments (Fig 1B–D). Low temperature, presumably by weakening hydrophobic contacts and dynamics, exacerbated the effect of such components in folding (Figs 2 and 3; Baldwin, 1986; Tilton Jr et al, 1992; van Dijk et al, 2015; Tsirigotaki et al, 2018).
Cytoplasmic proteins like PpiB are expected to form multiple foldons with substantial native structure soon after coming out of the ribosome (Figs 2B and 3). Meanwhile, secreted proteins like PpiA would remain longer in minimally folded states, in a signal peptide‐independent manner (Figs 2B, and 3B and D). Their mature domain intrinsic adaptations allow them to slow down, or limit, the formation of initial foldons, enabling secretion compatibility (Huber et al, 2005b; Tsirigotaki et al, 2018). Differences in efficiency of foldons could have major repercussions in facilitating downstream recognition and secretion steps.
Our analysis suggested that even subtle changes would have sufficed to alter the folding fate of a hypothetical primordial ancestor cytoplasmic protein to facilitate its secretion. A grafting experiment clarified that this can be specifically guided by a few highly stabilized, key native contacts that have critical long‐range interactions between or within the initial foldons (Fig 4C). These contacts determined whether an intermediate was quickly formed or delayed (Fig 4E), a key aspect for secretability (Fig 4F).
Secretory mature domains have evolved to display slower folding. Collectively, their sequences bear hallmarks that facilitate this process (Figs 2 and 3; Chatzi et al, 2017; Sardis et al, 2017; Tsirigotaki et al, 2018): enhanced disorder, reduced hydrophobicity, increased number of β‐stranded structures, etc. (Loos et al, 2019). While this enables them to avoid folding during their cytoplasmic and inner membrane crossing, it begs the question of how this inherent property is overcome once across the inner membrane and beyond, when stable final folded structures must be acquired. Interestingly, the native secretome proteins are more stable than their cytoplasmic counterparts (Loos et al, 2019), as exemplified here in the Ppi twins (Fig EV1). This could be the result of higher conformational entropy due to regions with increased flexibility (Fig 1B), requiring more effort to unfold due to the low gain in entropy as observed in thermophilic cytochrome c (Liu et al, 2018). In PpiA, a core initial foldon, such as B, formed rapidly but possibly due to suboptimal residues did not connect well to foldon A (Fig 1C and D) which was very slow to form, leading to differential foldon pathways. Despite delaying folding, this did not prevent PpiA from acquiring a structure similar to its cytoplasmic counterpart PpiB in the end (Fig 1B). Additional means of stabilization of secreted proteins, once at their final location, include use of disulphide bonding, tight binding of prosthetic groups, formation of quaternary complexes and for outer membrane proteins, and embedding in the lipid bilayer (De Geyter et al, 2016).
The evolutionary tinkering towards generating maximally non‐folding states is not uniformly extensive for all secretory proteins (Chun et al, 1993; Tsirigotaki et al, 2018). Over‐optimization of non‐folding in the cytoplasm might yield highly secreted yet non‐folded molecules. Where mature domains could not be tinkered with further, due to penalties in folding or function, the cell relied on signal peptides (Randall & Hardy, 1986). They delay folding of mature domains during their cytoplasmic transit, stabilizing kinetically trapped, loosely folded intermediates (Fig 5B; Randall & Hardy, 1986, 1989; Huber et al, 2005a; Singh et al, 2013; Tsirigotaki et al, 2018) and are proteolytically removed on the trans‐side of the membrane. As revealed here, signal peptides quickly acquire partial α‐helical structure in their core while maintaining disordered C‐terminal ends (Fig 6A) that translates into the early mature domain, preventing some of the crucial initial foldons located there from being stabilized (Figs 5C–F and 6A). As a result, subsequent folding is rendered ineffective.
As an exogenous add‐on, the signal peptide of PpiA also blocked folding of the cytoplasmic PpiB, although less efficiently than proPpiA (Fig 5F vs. E) and led to similar levels of secretion (Fig 6B). This suggested that signal peptide and internal mature domain properties may co‐evolve in secretory proteins so as to optimally stall their cytoplasmic folding, thereby maintaining them translocation‐competent. The signal peptide effect was strongly dominant and able to manipulate the folding features of the cytoplasmic PpiB. However, there are many cases of signal peptides that are inefficient in delaying folding and fail to secrete fast‐folding native E. coli proteins (Huber et al, 2005a, 2005b) or heterologous proteins of biotechnological interest (Zhang et al, 2018; Peng et al, 2019). In addition to a role in cytoplasmic non‐folding, we hypothesize that most secretory mature domains need to remain unfolded in the cell envelope even after their signal peptide has been cleaved. Such proteins need to traffic further, be modified or bind prosthetic groups (De Geyter et al, 2016). How some signal peptides are competent to slow down folding and drive secretion of certain proteins remains unclear and will require future studies.
We assume that the signal peptide's dramatic effect on preventing folding of the succeeding mature domain folding sequence was likely due to its proximity to the initial foldons of the mature domain, primarily B, D and A (Figs 6A and 7, top). Of note, the initial foldons in PpiA, PpiB, MBP (Walters et al, 2013), RNase H (Hu et al, 2013) and Cytochrome c (Hu et al, 2016) whose folding has been dissected in detail to date with local HDX‐MS, are all located at or near the N‐termini of these proteins, according to primary sequence or 3D structure. In this context, it is interesting that Foldon A of PpiB that is located a long way downstream in the linear sequence is not affected by the signal peptide but its interaction with the N‐terminal Foldon B is (Fig 5C and D). An N‐terminal location makes sense as a choice for initial foldons, as these regions exit the ribosome (in cytoplasmic proteins) or/and the Sec translocase (in secretory proteins) first. In either case, these would be the first regions that are available for folding (Raimondi et al, 2019), before the rest of the polypeptide (C terminus) is even synthesized or available for interactions (Jacobs & Shakhnovich, 2017). Hence, it is interesting to speculate that N‐terminal foldons might be a widespread polypeptide feature that can be manipulated by N‐terminal signal peptides or by chaperones during ribosomal exit (Smets et al, 2019). Extensive folding datasets, currently unavailable from most proteins (Pancsa et al, 2016), are required to test this. Secretory chaperones such as SecB, Trigger Factor and SecA might bind to prevent early foldon formation on secretory proteins that would further delay their folding behaviour or ability to be secreted (Saio et al, 2014; Huang et al, 2016).
Figure 7. Model of folding initiation in PpiA and its manipulation by the signal peptide.
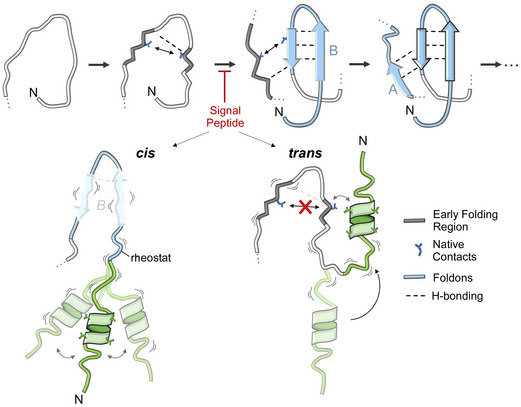
Folding initiation in PpiA using foldons B (from the two N‐terminal β‐strands) and A as suggested by rigidification of early folding regions, H‐bonding and stabilized by native contacts (see text for details). The signal peptide causes disorder in the early mature domain and blocks this process either in “cis” (preventing stable H‐bonding in foldon B) or in “trans” (directly using parts of foldon B).
Finally, to postulate how signal peptides block the first initiating foldons from forming, we considered “cis” and “trans” models (Fig 7, bottom). In the cis model, accommodation of the signal peptide's bulky hydrophobic core in the aqueous environment is frustrated and this leads to high signal peptide mobility, partial helical structure and enhanced disorder (Fig 6A). These effects are translated via the conformational rheostat (Sardis et al, 2017) to enhanced dynamics in the early mature domain and destabilization of the critical initial foldons. In the trans model, the hydrophobic helix of the signal peptide exploits the flexible connecting linker to physically interact with exposed hydrophobic residues on initial foldons (e.g. residues participating in critical highly stabilized native contacts), thus making these residues unavailable for foldon formation. As the folding process is hierarchical and vectorial, that is, N‐terminal foldons must form first, in both cases downstream steps of the folding process are blocked or slowed down. Testing these models will require probing the signal peptide properties and dynamics in parallel to monitoring the folding reaction.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| MC4100 cells (E. coli) | Casadaban (1997) | Prof. Dr. Genevaux, CBI Toulouse, France |
| Recombinant DNA | ||
| Genes (E. coli) | This study unless mentioned otherwise | Appendix Table S4 |
| Antibodies | ||
| Anti‐(pro)PhoA (Rabbit, monoclonal) | Chatzi et al (2017) (Ecolab/Davids) | 1/50,000 dilution |
| Anti‐rabbit (Peroxidase‐conjugated AffiniPure Goat) | Jackson ImmunoResearch Laboratories, Inc. | 111‐007‐003 (1/50,000 dilution) |
| Oligonucleotides and sequence‐based reagents | ||
| Custom oligos | Eurogentec | Appendix Table S2 |
| Chemicals, enzymes and other reagents | ||
| T4 DNA Ligase | Promega | M1801 |
| PFU Ultra Polymerase | Aligent | #600380 |
| Deuteriumoxide | Sigma Aldrich | P/N 151882 |
| Urea‐d4 | Sigma Aldrich | P/N 176087 |
| Formic Acid (MS grade) | Sigma Aldrich | F0507 |
| Acetonitrile (ACN, MS grade) | Merck Millipore | 100030 |
| Leucine Enkephalin (LeuEnk) | Waters | 186006013 |
| para‐Nitrophenolphosphate (PNPP) | Thermo Fisher Scientific | 34045 |
| Software | ||
| Canvas X | 2022 | https://canvasx.net |
| PyHDX | v0.3.3 (e8ea23e) | http://pyhdx.jhsmit.org |
| ImageJ | 1.53g 4 | https://imagej.nih.gov/ij/ |
| Jupyter Notebook (Anaconda, Python) | Python 3.6 | https://jupyter.org |
| AWSEM‐MD Frustratometer | Protein Frustratometer 2 (Parra et al, 2016) | http://frustratometer.qb.fcen.uba.ar |
| MassLynx | v4.1 (Waters) | Waters Corporation |
| ProteinLynx Global Server (PLGS) | v3.0.1 (Waters) | Waters Corporation |
| DynamX | v3.0 (Waters) | Waters Corporation |
| Clustal Omega | Sievers et al (2011) | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| PyMOL | v2.4 | https://pymol.org/2/ |
| Rosetta | 3.13 | https://www.rosettacommons.org/software |
| Other | ||
| Avanti J‐26S XPI, JLA 8.1000 rotor | Beckman | PN B10093AB |
| French Press | Thermo | FA‐078A + FA‐032 (40 k) Standard CELL |
| Sorvall RC 6 plus | Fisher Scientific | NB.81 |
| Ni2+‐NTA Agarose resin | Qiagen | ID: 30210 |
| Dialysis membranes (12–14 kDa MW cut‐off) | Medicell Membranes Ltd. | DTV.12000 |
| Plasmid DNA purification kit (NucleoSpin® Plasmid EasyPure) | Macherey‐Nagel | 740727.50. |
| Wizard SV Gel and PCR Clean‐Up System | Promega | A9281 |
| nanoACQUITY UPLC System with HDX Technology | Waters | Waters Corporation |
| Synapt G2 Mass Spectrometry instrument | Waters | Waters Corporation |
| MassPREP Micro Desalting column | Waters | 186004032 |
| Pepsin column | Sigma (pepsin) + Idex (cartridge) | P0609 + # 5051IP‐M07021‐005‐05TI |
| Nepenthesin‐2 | Affipro | AP‐PC‐004 |
| VanGuard C18 Pre‐column | Waters | 186003975 |
| C18 analytical column | Waters | 186002350 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | 34580 |
| ImageQuant LAS‐4000 (CCD‐camera system) | GE Healthcare Life Sciences | 28‐9610‐74 AC |
| Jasco J‐1500 | Jasco Inc. | J‐1000 series |
| Cary Eclipse Fluorescence Spectrophotometer | Agilent | Agilent Technologies |
| Nanodrop 2000 | Thermo | ND‐2000 |
| Vivaspin centrifugal concentrators (Vivaspin 500) | Viva products | VS0102+ |
Methods and Protocols
Protein preparation
Genes were inserted into the indicated plasmids by restriction enzyme digestion and ligation using T4 DNA Ligase (Promega). Restriction sites for the gene of interest and mutations were added using PCR with PFU Ultra Polymerase (Stratagene) containing templates and primers as indicated (Appendix Tables S1 and S2). Other constructs were designed as synthetic genes cloned in expression vectors (GenScript). To synthesize proteins, E. coli expression cells (Appendix Table S3) were transformed with pET22b vectors carrying the derivative gene (Appendix Table S4) to produce His6‐tagged proteins. The cells were grown in LB medium and induced with 0.1 mM IPTG at 37°C for 3 h or 18°C overnight. In case of preproteins, 5 mM MgCl2 was added to the medium before growth to stabilize the signal peptide and 4 mM sodium azide was added before induction to abolish SecA‐dependent secretion and thus prevent signal peptide cleavage [19]. Cells were collected (4,500 × g; 4°C; 15 min; Avanti J‐26S XPI, JLA 8.1000 rotor; Beckman) and stored at −20°C until purification.
For soluble and denaturing purification, cells are resolubilized in buffer S‐A and U‐A (buffers in Appendix Table S5), respectively, containing 50 μg/ml DNase I and 2.5 mM PMSF; and were lysed with a French press (1,000 psi; 5–6 rounds; pre‐cooled cylinder; Thermo). Soluble proteins were separated using centrifugation of lysed cells (26,600 × g; 30 min; 4°C, Sorvall RC 6 plus, Fisher Scientific) to remove the insoluble fractions. The proteins present in inclusion bodies or insoluble fraction were resolubilized in buffer U‐B using a Dounce homogenizer and centrifuged (26,600 × g; 30 min; 4°C, Sorvall RC 6 plus, Fisher Scientific) to remove the insoluble membrane fraction. The urea‐solubilized supernatant was diluted with buffer U‐A to 6 M Urea. Soluble/Urea‐solubilized protein fractions were run through a Ni2+‐NTA Agarose resin (Qiagen) packed in a gravity‐flow column pre‐equilibrated with buffer S‐A/U‐A (gravity flow; 1 ml/min) and washed with buffer S‐A/U‐C and S‐B/U‐D (10 column volumes each). Proteins were eluted with buffer S‐B/U‐E supplemented with 200/100 mM imidazole, incubated with EDTA (10 mM; 10 min, ice) and dialyzed (12–14 kDa MW cut‐off, Medicell Membranes Ltd.); in buffer S‐C/U‐F (overnight, 4°C) followed by buffer S‐D/U‐G (overnight, 4°C). Protein aliquots were stored at −20°C. Protein purity was determined on Coomassie gels using SDS–PAGE and in case of MS analysis, denatured, non‐deuterated proteins were run on global HDX‐MS (see below).
Measuring protein concentration
Protein concentration was determined by spectroscopic measurements (280 nm; Nanodrop 2000; Thermo) in the range of 0.3–3 mg/ml (linear range of the OD measurements; Stoscheck, 1990). The concentration was measured according to the molecular weight and extinction coefficients of each protein, determined using the ExPASy server (http://web.expasy.org/protparam/). Centrifugal ultrafiltration concentrators were used to concentrate protein samples [10 kDa cut‐off, Viva products, Vivaspin 500 for small volumes (12,000 × g; 4°C) and Vivaspin 4 for larger volumes (4,500 × g; 4°C)].
Native state dynamics with Local Hydrogen‐Deuterium exchange (HDX) mass spectrometry (MS)
Local HDX‐MS conditions and analysis routines have been described in detail in Krishnamurthy et al (2021) and preprint: Krishnamurthy et al (2022). Specific conditions used in this study are detailed below.
Labelling experiment
Proteins were dialyzed O/N in buffer B at 4°C. A 100 μM protein stock was prepared and equilibrated at 30°C together with labelling buffers. Labelling buffers were prepared from lyophilized aliquots of buffer A resolubilized in D2O (pD 8.0) with 5 mM DTT and 1 mM EDTA. The protein stock was diluted and labelled in 90% labelling buffer (4 μM protein) for 10 s, 30 s, 1 min, 5 min, 10 min and 30 min at 30°C. The reaction was quenched with pre‐chilled quenching buffer (6 M Urea, 0.1% DDM, 5 mM TCEP, formic acid to pD 2.5) on ice. A fully deuterated control was added, where the protein was labelled O/N at 50°C. n = 3 technical repeats.
MS analysis
This is identical to the analysis of refolding with local HDX‐MS (see below). DynamX data of the defined peptides with average D‐uptake and standard deviations, presented in Dataset EV4 (as suggested in Masson et al, 2019), have been further analysed using PyHDX (see below).
Derivation of ΔG values per residue using PyHDX
ΔG values per residue were derived using PyHDX (v0.4.1 (68624c40) (Smit et al, 2021)). A fully deuterated control sample was used to correct for back exchange. PyHDX settings used for fitting ΔG values: stop_loss: 0.05, stop patience: 50, learning rate: 10, momentum: 0.5. The first and second regularizer values were set at 0.1 and 0.05, respectively, where the latter acts as a damping term for differences between the aligned proteins (Smit et al, 2021).
Refolding kinetics with Global Hydrogen‐Deuterium exchange (HDX) mass spectrometry (MS)
Protein refolding
Proteins dialyzed in buffer C were incubated at 37°C for 40 min for maximal denaturation, diluted to 6 M Urea and pre‐chilled on ice for 40 min. To reduce the proteins to mimic cytoplasmic conditions, they were treated with 100 mM DTT; 5 mM EDTA at 4°C for 20 min and centrifuged (20,000 × g; 15 min; 4°C) prior to refolding. The pre‐treated denatured protein was used as a control for max H/D exchange. The refolding experiment was initiated by diluting the denatured protein in aqueous buffer to 0.2 M urea; 5 mM DTT and 1 mM EDTA (18 μM protein). For refolding at 4°C, samples were pulse‐labelled with an excess of D2O at 20 s, 40 s, 60 s, 5 min, 10 min, 15 min, 20 min, 30 min and 1 h (inc. 24 h if necessary). And for refolding at 25°C, samples were pulse‐labelled at 10 s, 20 s, 40 s, 60 s, 2 min 30 s and 5 min (inc. 10 min, 30 min and 1 h if necessary). In case soluble native protein was purified, this was added as a natively folded control. n = 2 biological repeats.
Deuterium pulse‐labelling
Labelling buffers were made from lyophilized aliquots of buffer A and were directly resolubilized in D2O (99.9% atom D, Sigma Aldrich P/N 151882) or after adding 6 M Urea‐d4 (98% atom D, Sigma Aldrich P/N 176087). Isotope pulse‐labelling during refolding was performed with 0.2 M Urea‐d4 (pD 8.0; 95.52%(v/v) D2O) for 100 s to 0.8 μM protein on ice. Labelling was quenched with pre‐chilled formic acid (to pD 2.5), snap‐frozen in liquid nitrogen and stored at −80°C until MS analysis. Denatured controls were labelled with 6 M Urea‐d4 (pD 8.0; 95.52%(v/v) D2O), 5 mM DTT, 1 mM EDTA on ice for 100 s (t0 control) and 1 h (fully deuterated control). Native controls were prepared in buffer A containing 0.2 M urea; 5 mM DTT; 1 mM EDTA to mimic folding conditions and labelled identical to refolding samples.
MS analysis
For mass determination, unlabelled proteins (0.8 μM) were prepared in buffer A (150 μl) with 0.23% formic acid and analysed with the MS. (Un)labelled samples were manually injected on a nanoACQUITY UPLC System with HDX technology (Waters) online‐coupled with a Synapt G2 ESI‐Q‐TOF instrument (Waters) for intact protein analysis. The UPLC chamber was set at 0.2°C to reduce back exchange and contained solvent A and B (ddH2O + 0.23% (v/v) formic acid and Acetonitrile + 0.23% formic acid, respectively). Proteins were trapped on a MassPREP Micro Desalting column (1,000 Å, 20 mm, 2.1 × 5 mm, Waters) and desalted at 250 μl/min for 2 min with solvent A and subsequently eluted using a linear gradient of solvent B 5–90% over 3 min. The remaining protein was washed from the column with 90% solvent B for 1 min, 5% solvent B for 1 min and again 90% solvent B for 1 min before returning to the initial conditions for re‐equilibration.
Positively charged ions in the range of 50–2,000 m/z were analysed after ionization and desolvation with the following parameters: capillary voltage 3.0 kV, Sampling cone 25V, Extraction cone 3.6V, source temperature 80°C, desolvation gas flow 650 l/h at 175°C. Leucine Enkephalin solution (2 ng/μl in 50:50 ACN:ddH2O with 0.1% formic acid, Waters) was co‐infused at 5 μl/min for accurate mass measurements.
Protein relative D‐uptake determination
Data analysis was performed manually with ESI‐Prot, Excel and Python. Deuterium uptake was normalized to the maximum deuteration control (fully denatured protein) and calculated as follows:
Where M L = mass of the labelled sample, M UNL = mass of the unlabelled sample, M FD = mass of the fully deuterated control (fully denatured protein).
First, the D‐uptake of the different folding states was calculated using the whole m/z spectra that was analysed with ESI‐Prot where the average mass of each peak was calculated (Dataset EV2; Winkler, 2010). Next, a single charged state of the highest intensity was selected for plotting D‐uptake as a function of folding time within a 25 m/z window/range. The highest intensity was set at 100%. First, the mass spectra from every timepoint were smoothed (Savitzky‐Golay, window: 15, number: 5) and baseline corrected by subtracting a polynomial of degree 1 (using PeakUtils (Hermann & Christophe, 2017)). The corrected spectra containing multimodal distributions were integrated into one to express each mode/folding state as population fractions. The m/z values were converted to %D‐uptake by setting the D‐uptake of the FD control as 100% and that of the non‐deuterated control as 0%, reflecting the degree of unfoldedness (see scripts in Data Availability).
Presentation of global HDX‐MS folding spectra as colour maps
The time course of the different folding states (Fig EV2A) of the single charged peak was shown in a folding colour map where we follow the states based on their degree of unfoldedness (%D‐uptake). To create a continuous folding colour map from discrete folding timepoints, the population fractions were linearly interpolated (using NumPy). After which, they were plotted with a “magma” colour map from MatPlotLib using a colour scale from 0 to 0.35 to have a clear visualisation of all folding populations despite their lower fractions (See values in Dataset EV3B). This might give some altered view of the fractions above 0.35 as the bands only broaden after reaching the brightest colour (see comparison in Dataset EV3C) but is the optimal display with the bright colours of the gradient. The unfolded control was displayed as a separate slice on the left where the protein is in 6 M urea before the actual folding pathway is shown in 0.2 M Urea on the right. For the selected charged state, the m/z values were processed to %D‐uptake from the molecular weight determination and with the D‐uptake of the protein in 6 M urea set as 100%. The script is accessible through GitHub (see Data Availability).
Refolding kinetics with Local Hydrogen‐Deuterium exchange (HDX) mass spectrometry (MS)
Refolding kinetics with pulse‐labelling
Proteins dialyzed in buffer C were incubated at 37°C for 20–30 min for complete denaturation, diluted to 6 M Urea and pre‐chilled on ice for 10 min and treated with 100 mM DTT; 5 mM EDTA at 4°C and centrifuged (20,000 × g; 15 min; 4°C) prior to refolding (40 μM protein during refolding). The pre‐treated denatured protein was used as a control for max H/D exchange. For refolding at 4 and 25°C, samples were pulse‐labelled at 5 s, 10 s, 20 s, 30 s, 40 s, 60 s, 2 min 30 s, 5 min, 10 min, 15 min, 20 min and 30 min (inc. 45 min, 1 h, 3 h and 16 h if necessary). An additional tfolding time = 0 control (referred to as t = 0 in Dataset EV4) was added where the denatured protein was added directly to deuterated buffer for the standard HDX time = 10 s, to observe the fastest folding events (H‐bonding faster than D‐uptake). The PpiA and PpiB soluble native proteins were used as natively folded controls. n = 3 biological replicates.
Labelling buffers were prepared as described for global HDX. Isotope pulse‐labelling during folding was performed with 0.2 M Urea‐d4 (pD 8.0; 95.52%(v/v) D2O) for 10 s to 1.8 μM protein at the same temperature as folding. Labelling was quenched with Quenching buffer (7.37 M Urea‐d4, 7.8% FA) to pD 2.5 (final protein concentration of 1.1 μM) and kept for 2 min at 4°C. During this time, samples were centrifuged (20,000 × g; 1.5 min; 4°C). Only supernatants were injected. The denatured controls were labelled with 6 M Urea‐d4 (pD 8.0; 95.52%(v/v) D2O), 5 mM DTT, 1 mM EDTA for 10 s at 4°C (fully deuterated control). Native controls were prepared in buffer A containing 0.2 M urea; 5 mM DTT; 1 mM EDTA to mimic folding conditions and were labelled identically to folding samples.
MS analysis
The same instrument was used as in global HDX‐MS. For local HDX‐MS, the protein was first digested at 16°C through an immobilized pepsin (Sigma) cartridge (2 mm × 2 cm, Idex) or Nepenthesin‐2 (Affipro) cartridge (column‐ 2.1 × 20 mm). The UPLC chamber was set at 2°C to avoid back exchange, and the resulting peptides were trapped onto a VanGuard C18 Pre‐column (130 Å, 1.7 mm, 2.1 × 5 mm, Waters) at 100 μl/min for 3 min using ddH2O with 0.23% (v/v) formic acid. Peptides were subsequently separated on a C18 analytical column (130 Å, 1.7 mm, 1 × 100 mm, Waters) at 40 μl/min. UPLC separation (solvent A: 0.23% v/v formic acid, solvent B: 0.23% v/v formic acidic acetonitrile) was carried out using a 12‐min linear gradient (5–50% solvent B). At the end, solvent B was raised to 90% for 1 min to wash out any remaining protein. The same ionization and desolvation parameters were kept as for intact protein analysis.
The peptide spectrum of the unlabelled protein in buffer B was first determined. Peptide identification was performed using the ProteinLynx Global Server (PLGS v3.0.1, Waters, UK) using the primary sequence of PpiA and PpiB. Peptides were individually assessed for accurate identification and were only considered if they had a signal‐to‐noise ratio above 10 and a PLGS score above 7 and if they appeared in 3 replicates for each protein. Data analysis was carried out using DynamX 3.0 (Waters, Milford MA) software to compile and process raw mass spectral data and generate centroid values to calculate relative deuteration values. DynamX data of the defined peptides with average D‐uptake and standard deviations, presented in Dataset EV4 (as suggested in Masson et al 2019), have been further analysed using PyHDX (see below).
Derivation of folded fraction per residue using PyHDX
Using DynamX, the centroid mass was determined per peptide spectrum to calculate its D‐uptake (Dataset EV4). D‐uptake triplicates from all timepoints and controls were input on PyHDX version 0.4.1; (Smit et al, 2021), and the folded fraction was determined using the “RFU” web application module in PyHDX. To determine the folded fraction, the centroid mass of the fully deuterated control was set as 0 (ND control field in PyHDX) and that of the final folding point as 1 (FD control field in PyHDX). This yields fraction folded per peptide, and these values were transformed to residue‐level folded fractions by weighted averaging (weights are inverse length of the peptides) and were subsequently multiplied by 100 to obtain folded fractions as percentage (Dataset EV5). This final folded state approximates the natively purified protein as the protein reaches a native‐like state with a D‐uptake plateau. The folded fraction was expressed in a colour map plotting the foldedness of residues over time using a custom colour map with a gradient from white with increasing darker blue for 0, 25, 50, 75 and 100% folded fractions. These fractions were determined from interpolation between folded fractions in our discrete experimental timepoints.
Next, time to reach 80 and 50% folded fraction (t 80% and t 50%) was interpolated from the PpiB and PpiA dataset, respectively. The t 80% and t 50% were used to define the size and order of the initial foldons. Each foldon was given a letter (alphabetical order) and colour to show the folding order. The script is accessible through GitHub (see Data Availability).
Derivation of degree of unfoldedness per residue for preproteins using PyHDX
For preproteins, the degree of unfoldedness (%D‐uptake) was determined setting the fully denatured (FD) control as 100% D‐uptake, non‐deuterated as 0% D‐uptake and the D‐uptake resulting from D‐exposure during the labelling pulse after the protein was allowed to fold for a set of timepoints were compared with this control (Dataset EV6).
Circular Dichroism (CD) spectropolarimetry
CD spectra were recorded in the far UV range (190–260 nm) using a J‐1500 spectropolarimeter (Jasco) equipped with a six‐position cuvette holder and a Peltier device to regulate temperature (typically 2–18 μM protein to satisfy −5 to −20 mdeg signal range; 1 mm quartz cuvettes).
For thermal denaturation analysis, native proteins were dialyzed twice in buffer A (1 l; overnight; 4°C followed by 1 l; 1 h; 4°C before measurements). Protein spectra (15 μM) were recorded at 222 nm (minima) from 20 to 90°C with data taken every 0.5°C (CD scale 200 mdeg/1.0 dOD; D.I.T. 0.5 s). Denaturation curves were smoothed with a Butterworth filter (filter order of 3 and cut‐off frequency of 0.1), followed by manually calculating the derivative y = (y n+1−y n )/x + 0.5*(x n+1−x n ) of the curve and defining the x value for the maximum y value (NumPy function) which corresponds to the transition temperature (Python script).
For chemical denaturation analysis using urea, native proteins were diluted 100× in buffer B containing different urea concentrations (final protein concentration 15 μM) and equilibrated, where the time to equilibrate was determined using denaturation kinetics after diluting in 8 M urea. Spectra were measured at 210–260 nm (CD scale 20 mdeg/0.05 dOD; Data pitch 0.5 nm; D.I.T. 0.5 s; 20 accumulations), and the values at 222 nm were plotted. Denaturation curves were fitted using a two‐state transition model to determine the apparent denaturation temperature (Python) using the equation (Clarke & Fersht, 1993; Lowe et al, 2018):
With F as fraction unfolded, m as m‐value (cal*mol−1*M−1), x as denaturation concentration (M), d50 as denaturation midpoint, R as Universal Gas Constant (kcal*mol−1*K−1) and T as Temperature (Kelvin). The script is accessible through GitHub (see Data Availability).
Intrinsic fluorescence
Intrinsic fluorescence of tyrosine residues was recorded for PpiA and PpiB due to the lack of Tryptophane in PpiA. This was performed in a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies) with a 4‐cell holder (15 μM of protein in 1 cm quartz cuvettes; Helma) and cooled with a Peltier device.
For thermal denaturation analysis, native proteins were diluted in buffer B. Protein spectra were recorded with excitation (slit: 2.5 nm) at 260 nm and emission (slit: 20 nm) at 304 nm (PpiA) or 327 nm (PpiB) for 15–90°C in steps of 0.5°C at 1°C/min. Similar to CD data analysis, denaturation curves were smoothed with a Butterworth filter (filter order of 3 and cut‐off frequency of 0.1), followed by plotting the derivative of the curve and defining its minimum which corresponds to the transition temperature. The script is accessible through GitHub (see Data Availability).
Protein sequence and structure analysis
FASTA protein sequences were retrieved from https://www.uniprot.org and aligned using Clustal Omega (Sievers et al, 2011; from https://www.ebi.ac.uk/Tools/msa/clustalo/). Protein structures with PDB codes were obtained from the Protein Data Bank (RCSB, http://www.rcsb.org/), visualized, studied and aligned with PyMOL software.
Bioinformatics tools
Frustratometer‐based analysis
Information about the native energy and frustration of residues in the native structures was derived from existing PDB structures with the AWSEM‐MD (Associative memory, Water mediated, Structure and Energy Model) Frustratometer (Jenik et al, 2012; Parra et al, 2016). An averaged‐out frustration index (Z‐score) was calculated from all the available PDB structures. The Frustratometer calculates empirical native energy based on potential of mean force that depends on the contact counts, type of residue interaction and solvent accessibility. The AWSEM energy function refers to additional incorporation of water‐mediated interactions instead of only hydrophobic ones. Frustration is determined by comparing native to decoy residues at each location and calculating whether the native or other residues are good fits by comparing their energy function in this new environment. We focused on the configurational frustration to define the frustration of each interaction pair in the 3D structures that are a direct output from the Frustratometer with the highly [red; (Fig 1C)] and minimally (green) frustrated contacts displayed as lines between amino acids. Furthermore, the native energy scores per residue (average of all contacts, Dataset EV7) were determined.
Normal mode analysis
This analysis was performed with Webnm@ using existing PDB structures (Tiwari et al, 2014). Total displacement was calculated using the unweighted sum for the first 6 non‐trivial normal modes (modes 7–13).
Rosetta‐based analysis
The residue/structure compatibility scores (p_aa_pp) were calculated using the PpiA (PDB 1V9T) and PpiB (PDB 2NUL) structures (see Dataset EV1E). The PDBs were relaxed in the torsion space with coordinate constraints and coloured using a gradient from white to red (value 0 to 1, optimal to suboptimal) on the structures using PyMOL (Schrödinger & DeLano, 2020).
In silico mutational scanning was computed using the Rosetta cartesian‐ddG application (Frenz et al, 2020). Mutational free energy predictions were computed for every 19 possible substitutions of every residue in PpiA (PDB ID: 1V9T, 3154 substitutions) and PpiB (PDB ID: 1LOP, 3116 substitutions). The PDB structures were relaxed in the cartesian space before the calculations, as required by the cartesian‐ddg protocol (https://www.rosettacommons.org/docs/latest/cartesian‐ddG). For each mutation, three iterations of the Rosetta total_score calculations were carried out for the wildtype and the mutated variant. The computed total_scores were averaged and subtracted (totalscoreMUT ‐ totalscoreWT) to derive the mutational free energy predictions. ddG values of PpiA and PpiB were aligned and subsequently subtracted residue‐wise to obtain mutational differences dddG values. The dddG values were clipped to a symmetric interval containing 95% of datapoints to exclude outlying values. dddG values of all mutations were then averaged to obtain a single per‐residue dddG value.
Stride
Calculating the surface accessibility of each residue in existing PDB structures (Frishman & Argos, 1995) was performed on the Web Stride Server (http://webclu.bio.wzw.tum.de/cgi‐bin/stride/stridecgi.py).
Protein hydrophobicity calculations
The GRAVY index (grand average of hydropathy) of proteins was calculated based on the Kyte‐Doolittle hydrophobicity scale (Kyte & Doolittle, 1982) using the ExPaSy ProtScale server (https://web.expasy.org/protscale/; Wilkins et al, 1999).
Protein polarity calculations
Polarity scores were calculated based on the Grantham scale (Grantham, 1974) using the ExPaSy ProtScale server (Wilkins et al, 1999).
Early folding predictions
The EFoldMine predictor (Raimondi et al, 2017) of early folding regions was trained on residue‐level HDX NMR or MS‐based folding data accumulated in the Start2Fold dataset (Pancsa et al, 2016) to predict the residues with a primed folding confirmation according to their local neighbourhood (primary sequence). Prediction scores above 0.169 were used to define residue groups with high early folding propensity (see Dataset EV7A).
Quantification and statistical analysis
Statistical analysis
Statistical analysis of assays from replicates was performed using Excel and Python. Error bars represent standard error or standard deviation, as indicated.
Fitting folding populations in global HDX‐MS data
Starting from a single charged state of the MS spectra at each refolding time, the folding states (unfolded, intermediate and folded) were defined by fitting a single peak at their proper position. The complete m/z peak for the unfolded and folded state could be experimentally determined by the fully deuterated control and final folded state to include modification and adduct peaks. Intermediates were modelled as a single Lorentzian curve where the position and width were free fit parameters (Dataset EV3).
This fitting procedure resulted in quantified folding population fractions at each timepoint. The script is accessible through GitHub.
Global HDX‐MS ODE model fit
Quantified folding populations were fitted to an ordinary differential equation (ODE) model using python packages symfit (Roelfs & Kroon, 2020) and SciPy (Virtanen et al, 2020). The rate for loss and formation of different folding states was calculated using differential equations. A simple three‐state model seemed to optimally describe the folding kinetics for all refolding behaviours in this study:
With the Unfolded (U), Intermediate (I) and Folded (F) state whose reactions were described with the following equations:
where curves with k1, k−1 and k2 parameters were fitted against the previously defined datapoints. For this study, we focused primarily on the equilibrium constant for the first folding step. The script is accessible through GitHub.
Ιn vivo secretion assay
Protein secretion efficiency was tested in vivo using C‐terminally fused alkaline phosphatase (PhoA). PhoA acts as a secretion reporter as it only becomes an active hydrolase in the periplasm after translocation where it forms disulphide bonds that are necessary to fold and dimerize (Prinz et al, 1996). This will provide information about secretion of the N‐terminally fused target protein that guides translocation. PhoA activity was measured using para‐Nitrophenylphosphate (PNPP, Thermo Fisher Scientific) as hydrolysis results in a yellow substance (para‐Nitrophenol). PhoA fused constructs in pBAD501 were tested in MC4100 cells in combination with SecYprlA4EG in pET610 that can translocate some protein without the need of signal peptide triggering (Derman et al, 1993; Smith et al, 2005). Translocation was confirmed using a negative control condition with the translocation inhibitor sodium azide.
Cells were grown to OD 0.2–0.25, before being induced (6.67–13.3 μM arabinose to express the PhoA fusion constructs and 0.05 mM IPTG to express SecYprlA4EG) for 30 min. One milliliter of cells were transferred on ice and centrifuged (1,500 × g, 8 min), the supernatant was removed, and the cells were redissolved in 1 M Tris–HCl (pH 8.0). The assay was initiated when 0.01 M para‐Nitrophenol phosphate (PNPP) was added to 500 μl cells and put at 37°C for 10 to 40 min. The reaction was stopped by transferring the cells back to ice and adding 0.17 M K2HPO4. The cells were broken with 0.17% Triton X‐100 and removed by centrifugation (15,500 × g; 5 min; 4°C). The supernatant was transferred to ELISA plates to measure the PNPP hydrolysis at OD420 and the cell density at OD600. The OD420 values were divided by the assay time to define the amounts of pmol PhoA secreted using the standard curve and converted to secretion per 108 cells (see Dataset EV9A). Background activity was subtracted from the activity from induction with arabinose (and IPTG) as there was no protein expression from the background as indicated from immunostaining. The amount of protein expressed was determined from analysis of 8*107 cells for each protein with SDS–PAGE (12%), followed by immunostaining with anti‐proPhoA antibody (Chatzi et al, 2017) and secondary peroxidase‐conjugated goat anti‐rabbit antibody (AffiniPure; Jackson ImmunoResearch Laboratories). Staining was visualized using the West Pico kit (ThermoFisher Scientific) and a CCD‐camera system (LAS‐4000; GE Health‐care). The amount of protein was quantified using scanning densitometry [Image J (https://imagej.net)] with each blot containing a standard curve of 50,100 and 200 ng PhoA, which was adjusted to amounts for 108 cells.
Author contributions
Dries Smets: Data curation, software, formal analysis, investigation, visualization, methodology, writing‐original draft, writing‐review and editing. Alexandra Tsirigotaki: Resources, data curation, formal analysis, investigation, methodology. Jochem H Smit: Data curation, software, formal analysis, investigation, visualization, methodology, writing‐review and editing. Srinath Krishnamurthy: Data curation, validation, methodology. Athina G Portaliou: Resources, data curation, investigation. Anastassia Vorobieva: Resources, data curation, software, formal analysis, investigation, visualization, methodology. Wim Vranken: Resources, data curation, software, formal analysis, visualization, methodology, writing‐review and editing. Spyridoula Karamanou: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, visualization, methodology, project administration, writing‐review and editing. Anastassios Economou: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, writing‐original draft, writing‐review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 5
PDF+
Acknowledgements
We are grateful to G. Roussel for discussions and advice on the biophysical refolding experiments, J. De Geyter for help with setting up the in vivo secretion assay, and J. Van den Schilden for the sequence analysis of PpiA and PpiB. Research in our laboratories was funded by grants (to AE): ProFlow EOS, FWO‐FNRS excellence programme (#G0G0818N, FWO‐FNRS), CARBS and DOT3S (#G0C6814N and # G0C9322N; FWO); (to SKa): FWO Research Grant (#G0B4915N, Binamics G094522N and G086222N; FWO); (to AE and SKa): FOscil C1 Basic Research (ZKD4582) and (to WV): (Research grant #G028821N; FWO) and (to AV): (VSC (Flemish Supercomputer Center); FWO and the Flemish Government). JHS was a PDM/KU Leuven fellow (PDM/20/167). SKr was a FWO [PEGASUS]2 MSC fellow and this project has received funding from the Research Foundation—Flanders (FWO) and the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement #665501.
The EMBO Journal (2022) 41: e111344
See also: N McCaul & I Braakman (December 2022)
Contributor Information
Spyridoula Karamanou, Email: lily.karamanou@kuleuven.be.
Anastassios Economou, Email: tassos.economou@kuleuven.be.
Data availability
The Protein Data Bank (RCSB, http://www.rcsb.org/) was used to obtain crystal structures. For PpiA (UniProt P0AFL3), three structures were available from the same study (Konno et al, 2004): PDB 1J2A (K163T, X‐ray, 1.80 Å), 1V9T (K163T, X‐ray, 1.70 Å, 2 chains) and 1VAI (K163T, X‐ray, 1.80 Å, 2 chains). For PpiB (UniProt P23869), two structures were available: PDB 1LOP (E132V, X‐ray, 1.70 Å, Konno et al, 1996) and 2NUL (WT, X‐ray, 2.10 Å, Edwards et al, 1997). For all bioinformatics analysis except frustration index, the most resolved structures (1V9T for PpiA and 1LOP for PpiB) were selected.
Protein sequences were retrieved from UniProt (https://www.uniprot.org). For PpiA, P0AFL3 was used and for PpiB, P23869.
The Python scripts are available on https://github.com/DriesSmets/Non‐folding‐for‐translocation.
The raw Mass Spectrometry data for local and global HDX‐MS can be made accessible from the lead author upon reasonable request.
References
- Akiyama Y, Ito K (1993) Folding and assembly of bacterial alkaline phosphatase in vitro and in vivo. J Biol Chem 268: 8146–8150 [PubMed] [Google Scholar]
- Alford RF, Leaver‐Fay A, Jeliazkov JR, O'Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K et al (2017) The Rosetta all‐atom energy function for macromolecular modeling and design. J Chem Theory Comput 13: 3031–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfinsen CB (1972) The formation and stabilization of protein structure. Biochem J 128: 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben‐Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44: W344–W350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair SM, Bhanu MK, Kendall DA (2011) Signal peptidase I: cleaving the way to mature proteins. Protein Sci 21: 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I, Lezon TR, Bakan A, Shrivastava IH (2010) Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem Rev 110: 1463–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Milne JS, Mayne L, Englander SW (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin RL (1986) Temperature dependence of the hydrophobic interaction in protein folding. Proc Natl Acad Sci USA 83: 8069–8072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Hummer G, Eaton WA (2013) Native contacts determine protein folding mechanisms in atomistic simulations. Proc Natl Acad Sci USA 110: 17874–17879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittrich S, Schroeder M, Labudde D (2018) Characterizing the relation of functional and Early Folding Residues in protein structures using the example of aminoacyl‐tRNA synthetases. PLoS ONE 13: e0206369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornschlogl T, Rief M (2011) Single‐molecule protein unfolding and refolding using atomic force microscopy. Methods Mol Biol 783: 233–250 [DOI] [PubMed] [Google Scholar]
- Braselmann E, Chaney JL, Clark PL (2013) Folding the proteome. Trends Biochem Sci 38: 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban MJ (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104: 541–555 [DOI] [PubMed] [Google Scholar]
- Chatzi KE, Sardis MF, Tsirigotaki A, Koukaki M, Sostaric N, Konijnenberg A, Sobott F, Kalodimos CG, Karamanou S, Economou A (2017) Preprotein mature domains contain translocase targeting signals that are essential for secretion. J Cell Biol 216: 1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Liu X, Chen J (2018) Atomistic peptide folding simulations reveal interplay of entropy and long‐range interactions in folding cooperativity. Sci Rep 8: 13668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun SY, Strobel S, Bassford P Jr, Randall LL (1993) Folding of maltose‐binding protein. Evidence for the identity of the rate‐determining step in vivo and in vitro. J Biol Chem 268: 20855–20862 [PubMed] [Google Scholar]
- Clarke J, Fersht AR (1993) Engineered disulfide bonds as probes of the folding pathway of barnase: increasing the stability of proteins against the rate of denaturation. Biochemistry 32: 4322–4329 [DOI] [PubMed] [Google Scholar]
- De Geyter J, Portaliou AG, Srinivasu B, Krishnamurthy S, Economou A, Karamanou S (2020) Trigger factor is a bona fide secretory pathway chaperone that interacts with SecB and the translocase. EMBO Rep 21: e49054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Geyter J, Tsirigotaki A, Orfanoudaki G, Zorzini V, Economou A, Karamanou S (2016) Protein folding in the cell envelope of Escherichia coli . Nat Microbiol 1: 16107 [DOI] [PubMed] [Google Scholar]
- Derman AI, Puziss JW, Bassford PJ Jr, Beckwith J (1993) A signal sequence is not required for protein export in prlA mutants of Escherichia coli . EMBO J 12: 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill KA (1999) Polymer principles and protein folding. Protein Sci 8: 1166–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill KA, MacCallum JL (2012) The protein‐folding problem, 50 years on. Science 338: 1042–1046 [DOI] [PubMed] [Google Scholar]
- Edwards KJ, Ollis DL, Dixon NE (1997) Crystal structure of cytoplasmic Escherichia coli peptidyl‐prolyl isomerase: evidence for decreased mobility of loops upon complexation. J Mol Biol 271: 258–265 [DOI] [PubMed] [Google Scholar]
- Englander SW, Mayne L (2014) The nature of protein folding pathways. Proc Natl Acad Sci USA 111: 15873–15880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Mayne L (2017) The case for defined protein folding pathways. Proc Natl Acad Sci USA 114: 8253–8258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Mayne L, Kan ZY, Hu W (2016) Protein folding‐how and why: by hydrogen exchange, fragment separation, and mass spectrometry. Annu Rev Biophys 45: 135–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Mayne L, Krishna MM (2007) Protein folding and misfolding: mechanism and principles. Q Rev Biophys 40: 287–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro DM, Lazo N, Robertson AD (2004) EX1 hydrogen exchange and protein folding. Biochemistry 43: 587–594 [DOI] [PubMed] [Google Scholar]
- Ferreiro DU, Hegler JA, Komives EA, Wolynes PG (2007) Localizing frustration in native proteins and protein assemblies. Proc Natl Acad Sci USA 104: 19819–19824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro DU, Komives EA, Wolynes PG (2014) Frustration in biomolecules. Q Rev Biophys 47: 285–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenz B, Lewis SM, King I, DiMaio F, Park H, Song Y (2020) Prediction of protein mutational free energy: benchmark and sampling improvements increase classification accuracy. Front Bioeng Biotechnol 8: 558247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D, Argos P (1995) Knowledge‐based protein secondary structure assignment. Proteins 23: 566–579 [DOI] [PubMed] [Google Scholar]
- Fuller AA, Du D, Liu F, Davoren JE, Bhabha G, Kroon G, Case DA, Dyson HJ, Powers ET, Wipf P et al (2009) Evaluating beta‐turn mimics as beta‐sheet folding nucleators. Proc Natl Acad Sci USA 106: 11067–11072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni S, Freiberger MI, Jemth P, Ferreiro DU, Wolynes PG, Fuxreiter M (2021) Fuzziness and frustration in the energy landscape of protein folding, function, and assembly. Acc Chem Res 54: 1251–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni S, Ivarsson Y, Jemth P, Brunori M, Travaglini‐Allocatelli C (2007) Identification and characterization of protein folding intermediates. Biophys Chem 128: 105–113 [DOI] [PubMed] [Google Scholar]
- Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A (2009) Signal peptides are allosteric activators of the protein translocase. Nature 462: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185: 862–864 [DOI] [PubMed] [Google Scholar]
- Hayano T, Takahashi N, Kato S, Maki N, Suzuki M (1991) Two distinct forms of peptidylprolyl‐cis‐trans‐isomerase are expressed separately in periplasmic and cytoplasmic compartments of Escherichia coli cells. Biochemistry 30: 3041–3048 [DOI] [PubMed] [Google Scholar]
- Hermann NL, Christophe V (2017) lucashn/peakutils: v1.1.0 (Version v1.1.0). Zenodo
- Ho CS, Lam CW, Chan MH, Cheung RC, Law LK, Lit LC, Ng KF, Suen MW, Tai HL (2003) Electrospray ionisation mass spectrometry: principles and clinical applications. Clin Biochem Rev 24: 3–12 [PMC free article] [PubMed] [Google Scholar]
- Hodge EA, Benhaim MA, Lee KK (2020) Bridging protein structure, dynamics, and function using hydrogen/deuterium‐exchange mass spectrometry. Protein Sci 29: 843–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Kan ZY, Mayne L, Englander SW (2016) Cytochrome c folds through foldon‐dependent native‐like intermediates in an ordered pathway. Proc Natl Acad Sci USA 113: 3809–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Walters BT, Kan ZY, Mayne L, Rosen LE, Marqusee S, Englander SW (2013) Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA 110: 7684–7689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Saio T, Rossi P, Kalodimos CG (2016) Structural basis for the antifolding activity of a molecular chaperone. Nature 537: 202–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J (2005a) Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle‐dependent translocation. J Bacteriol 187: 2983–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber D, Cha MI, Debarbieux L, Planson AG, Cruz N, Lopez G, Tasayco ML, Chaffotte A, Beckwith J (2005b) A selection for mutants that interfere with folding of Escherichia coli thioredoxin‐1 in vivo. Proc Natl Acad Sci USA 102: 18872–18877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T, Hayano T, Takahashi N, Kuwajima K (2000) Fast folding of Escherichia coli cyclophilin A: a hypothesis of a unique hydrophobic core with a phenylalanine cluster. J Mol Biol 297: 791–802 [DOI] [PubMed] [Google Scholar]
- Jacobs WM, Shakhnovich EI (2017) Evidence of evolutionary selection for cotranslational folding. Proc Natl Acad Sci USA 114: 11434–11439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenik M, Parra RG, Radusky LG, Turjanski A, Wolynes PG, Ferreiro DU (2012) Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res 40: W348–W351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno M, Ito M, Hayano T, Takahashi N (1996) The substrate‐binding site in Escherichia coli cyclophilin A preferably recognizes a cis‐proline isomer or a highly distorted form of the trans isomer. J Mol Biol 256: 897–908 [DOI] [PubMed] [Google Scholar]
- Konno M, Sano Y, Okudaira K, Kawaguchi Y, Yamagishi‐Ohmori Y, Fushinobu S, Matsuzawa H (2004) Escherichia coli cyclophilin B binds a highly distorted form of trans‐prolyl peptide isomer. Eur J Biochem 271: 3794–3803 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy S, Eleftheriadis N, Karathanou K, Smit JH, Portaliou AG, Chatzi KE, Karamanou S, Bondar AN, Gouridis G, Economou A (2021) A nexus of intrinsic dynamics underlies translocase priming. Structure 29: 846–858 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy S, Sardis M‐F, Eleftheriadis N, Chatzi KE, Smit JH, Karathanou K, Gouridis G, Portaliou AG, Bondar AN, Karamanou S et al (2022) Preproteins couple the intrinsic dynamics of SecA to its ATPase cycle to translocate via a catch and release mechanism. BioRxiv 10.1016/j.celrep.2022.110346 [PREPRINT] [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157: 105–132 [DOI] [PubMed] [Google Scholar]
- Leman JK, Weitzner BD, Lewis SM, Adolf‐Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, Barth P et al (2020) Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods 17: 665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Lemmonds S, Huang J, Tyagi M, Hong L, Jain N (2018) Entropic contribution to enhanced thermal stability in the thermostable P450 CYP119. Proc Natl Acad Sci USA 115: E10049–E10058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos MS, Ramakrishnan R, Vranken W, Tsirigotaki A, Tsare EP, Zorzini V, Geyter J, Yuan B, Tsamardinos I, Klappa M et al (2019) Structural basis of the subcellular topology landscape of Escherichia coli . Front Microbiol 10: 1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe AR, Perez‐Riba A, Itzhaki LS, Main ERG (2018) PyFolding: Open‐Source Graphing, Simulation, and Analysis of the Biophysical Properties of Proteins. Biophys J 114: 516–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity H, Maity M, Krishna MM, Mayne L, Englander SW (2005) Protein folding: the stepwise assembly of foldon units. Proc Natl Acad Sci USA 102: 4741–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelino AM, Gierasch LM (2008) Roles of beta‐turns in protein folding: from peptide models to protein engineering. Biopolymers 89: 380–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcsisin SR, Engen JR (2010) Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal Bioanal Chem 397: 967–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, Bou‐Assaf GM, Engen JR, Englander SW, Faber J et al (2019) Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX‐MS) experiments. Nat Methods 16: 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SM, Alonso DO, Daggett V, Fersht AR (2003) The complete folding pathway of a protein from nanoseconds to microseconds. Nature 421: 863–867 [DOI] [PubMed] [Google Scholar]
- Munoz V, Cerminara M (2016) When fast is better: protein folding fundamentals and mechanisms from ultrafast approaches. Biochem J 473: 2545–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickson AA, Clarke J (2010) What lessons can be learned from studying the folding of homologous proteins? Methods 52: 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymeyer H, Garcia AE, Onuchic JN (1998) Folding funnels and frustration in off‐lattice minimalist protein landscapes. Proc Natl Acad Sci USA 95: 5921–5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield CJ, Dunker AK (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 83: 553–584 [DOI] [PubMed] [Google Scholar]
- Onuchic JN, Luthey‐Schulten Z, Wolynes PG (1997) Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem 48: 545–600 [DOI] [PubMed] [Google Scholar]
- Orfanoudaki G, Markaki M, Chatzi K, Tsamardinos I, Economou A (2017) MatureP: prediction of secreted proteins with exclusive information from their mature regions. Sci Rep 7: 3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko AR, Luthey‐Schulten Z, Wolynes PG (1996) Foldons, protein structural modules, and exons. Proc Natl Acad Sci USA 93: 2008–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancsa R, Varadi M, Tompa P, Vranken WF (2016) Start2Fold: a database of hydrogen/deuterium exchange data on protein folding and stability. Nucleic Acids Res 44: D429–D434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Liu G, Topping TB, Cover WH, Randall LL (1988) Modulation of folding pathways of exported proteins by the leader sequence. Science 239: 1033–1035 [DOI] [PubMed] [Google Scholar]
- Parra RG, Schafer NP, Radusky LG, Tsai MY, Guzovsky AB, Wolynes PG, Ferreiro DU (2016) Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res 44: W356–W360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Shi C, Cao X, Li Y, Liu F, Lu F (2019) Factors influencing recombinant protein secretion efficiency in gram‐positive bacteria: signal peptide and beyond. Front Bioeng Biotechnol 7: 139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA, Spiess C, Ehrmann M, Schierle C, Beckwith J (1996) Targeting of signal sequenceless proteins for export in Escherichia coli with altered protein translocase. EMBO J 15: 5209–5217 [PMC free article] [PubMed] [Google Scholar]
- Raimondi D, Orlando G, Pancsa R, Khan T, Vranken WF (2017) Exploring the sequence‐based prediction of folding initiation sites in proteins. Sci Rep 7: 8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi D, Orlando G, Pancsa R, Khan T, Vranken WF (2019) Author correction: exploring the sequence‐based prediction of folding initiation sites in proteins. Sci Rep 9: 12140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall LL, Hardy SJ (1986) Correlation of competence for export with lack of tertiary structure of the mature species: a study in vivo of maltose‐binding protein in E. coli . Cell 46: 921–928 [DOI] [PubMed] [Google Scholar]
- Randall LL, Hardy SJ (1989) Unity in function in the absence of consensus in sequence: role of leader peptides in export. Science 243: 1156–1159 [DOI] [PubMed] [Google Scholar]
- Roelfs M, Kroon PC (2020) symfit 0.5.3. Zenodo
- Saio T, Guan X, Rossi P, Economou A, Kalodimos CG (2014) Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 344: 1250494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Millan JL, Boyd D, Dalbey R, Wickner W, Beckwith J (1989) Use of phoA fusions to study the topology of the Escherichia coli inner membrane protein leader peptidase. J Bacteriol 171: 5536–5541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardis MF, Tsirigotaki A, Chatzi KE, Portaliou AG, Gouridis G, Karamanou S, Economou A (2017) Preprotein conformational dynamics drive bivalent translocase docking and secretion. Structure 25: 1056–1067 [DOI] [PubMed] [Google Scholar]
- Scholz C, Stoller G, Zarnt T, Fischer G, Schmid FX (1997) Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding. EMBO J 16: 54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L, DeLano W (2020) PyMOL.
- Schuler B, Eaton WA (2008) Protein folding studied by single‐molecule FRET. Curr Opin Struct Biol 18: 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J et al (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Sharma L, Kulothungan SR, Adkar BV, Prajapati RS, Ali PS, Krishnan B, Varadarajan R (2013) Effect of signal peptide on stability and folding of Escherichia coli thioredoxin. PLoS ONE 8: e63442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smets D, Loos MS, Karamanou S, Economou A (2019) Protein transport across the bacterial plasma membrane by the sec pathway. Protein J 38: 262–273 [DOI] [PubMed] [Google Scholar]
- Smit JH, Krishnamurthy S, Srinivasu BY, Parakra R, Karamanou S, Economou A (2021) Probing universal protein dynamics using hydrogen‐deuterium exchange mass spectrometry‐derived residue‐level gibbs free energy. Anal Chem 93: 12840–12847 [DOI] [PubMed] [Google Scholar]
- Smith MA, Clemons WM Jr, DeMars CJ, Flower AM (2005) Modeling the effects of prl mutations on the Escherichia coli SecY complex. J Bacteriol 187: 6454–6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Kortemme T (2013) Improvements to robotics‐inspired conformational sampling in rosetta. PLoS ONE 8: e63090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoscheck CM (1990) Quantitation of protein. Methods Enzymol 182: 50–68 [DOI] [PubMed] [Google Scholar]
- Tilton RF Jr, Dewan JC, Petsko GA (1992) Effects of temperature on protein structure and dynamics: X‐ray crystallographic studies of the protein ribonuclease‐A at nine different temperatures from 98 to 320 K. Biochemistry 31: 2469–2481 [DOI] [PubMed] [Google Scholar]
- Tiwari SP, Fuglebakk E, Hollup SM, Skjaerven L, Cragnolini T, Grindhaug SH, Tekle KM, Reuter N (2014) WEBnm@ v2.0: Web server and services for comparing protein flexibility. BMC Bioinformatics 15: 427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigotaki A, Chatzi KE, Koukaki M, De Geyter J, Portaliou AG, Orfanoudaki G, Sardis MF, Trelle MB, Jorgensen TJD, Karamanou S et al (2018) Long‐lived folding intermediates predominate the targeting‐competent secretome. Structure 26: 695–707 [DOI] [PubMed] [Google Scholar]
- Tsirigotaki A, De Geyter J, Sostaric N, Economou A, Karamanou S (2017a) Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15: 21–36 [DOI] [PubMed] [Google Scholar]
- Tsirigotaki A, Papanastasiou M, Trelle MB, Jorgensen TJ, Economou A (2017b) Analysis of translocation‐competent secretory proteins by HDX‐MS. Methods Enzymol 586: 57–83 [DOI] [PubMed] [Google Scholar]
- Uversky VN (2013) The alphabet of intrinsic disorder: II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. Intrinsically Disord Proteins 1: e24684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk E, Hoogeveen A, Abeln S (2015) The hydrophobic temperature dependence of amino acids directly calculated from protein structures. PLoS Comput Biol 11: e1004277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Puyenbroeck V, Vermeire K (2018) Inhibitors of protein translocation across membranes of the secretory pathway: novel antimicrobial and anticancer agents. Cell Mol Life Sci 75: 1541–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J et al (2020) Author correction: SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17: 352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales TE, Engen JR (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25: 158–170 [DOI] [PubMed] [Google Scholar]
- Walters BT, Mayne L, Hinshaw JR, Sosnick TR, Englander SW (2013) Folding of a large protein at high structural resolution. Proc Natl Acad Sci USA 110: 18898–18903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112: 531–552 [DOI] [PubMed] [Google Scholar]
- Winkler R (2010) ESIprot: a universal tool for charge state determination and molecular weight calculation of proteins from electrospray ionization mass spectrometry data. Rapid Commun Mass Spectrom 24: 285–294 [DOI] [PubMed] [Google Scholar]
- Wolynes PG (2015) Evolution, energy landscapes and the paradoxes of protein folding. Biochimie 119: 218–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Lu J, Zhang S, Liu L, Pang X, Lv J (2018) Development an effective system to expression recombinant protein in E. coli via comparison and optimization of signal peptides: expression of Pseudomonas fluorescens BJ‐10 thermostable lipase as case study. Microb Cell Fact 17: 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Dunker AK (2018) Regulating protein function by delayed folding. Structure 26: 679–681 [DOI] [PubMed] [Google Scholar]